enfermedades oculares geneticas
TRANSCRIPT

Anoftalmia y microftalmia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
Distrofias corneales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
Malformaciones del segmento anterior . . . . . . . . . . . . . . . . . . . . . . . . 39
Catarata congénita. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Glaucoma congénito. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Estrabismo hereditario . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Distrofias retinianas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
Glosario . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43

37
De acuerdo a la Organización Mundial de la Salud (OMS), en el año 2010 existían cerca de 285 millones de personas con discapacidad visual en el mundo; de las cuales, 39 millones presentaban ceguera y 246 millones tenían visión baja. Los factores genéticos tienen una importancia primordial en el origen de las enfermedades que causan ceguera o discapaci-
dad visual. Se ha estimado que en promedio 30% de las enfermedades humanas con una base genética presentan manifestacio-nes oculares de grado variable, por lo que el examen oftalmológico minucioso representa un aspecto de gran importancia en el estudio de pacientes con sospecha de síndromes o padecimientos genéticos. De manera general, se reconocen dos grupos de este tipo de enfermedades: el que incluye padecimientos originados por una combinación de factores genéticos y ambientales (enfer-medades multifactoriales) y el grupo de enfermedades originadas por la mutación de un gen único (enfermedades monogénicas). Dentro de las enfermedades multifactoriales que causan discapacidad visual se encuentran el glaucoma, la degeneración macular relacionada con la edad, la catarata senil, la retinopatía diabética, entre numerosas más. Entre los padecimientos monogénicas que originan falla visual y son de alta prevalencia, se encuentran las degeneraciones retinianas, la catarata congénita, el estrabis-mo congénito, el glaucoma infantil y juvenil, entre muchas otras. En los últimos años se ha logrado un avance considerable en la identificación de las bases genéticas de estas enfermedades (Raca et al., 2010; Sheffield y Stone, 2011), lo que ha llevado a una mejor comprensión de su fisiopatología y ha sentado las bases para el diseño de nuevos esquemas de tratamiento, tal es el caso de la terapia génica en ciertas formas de degeneración retiniana. En este capítulo se revisan de manera general un grupo de enfer-medades y malformaciones oculares de origen genético y se explican de manera breve las alteraciones moleculares que las originan.
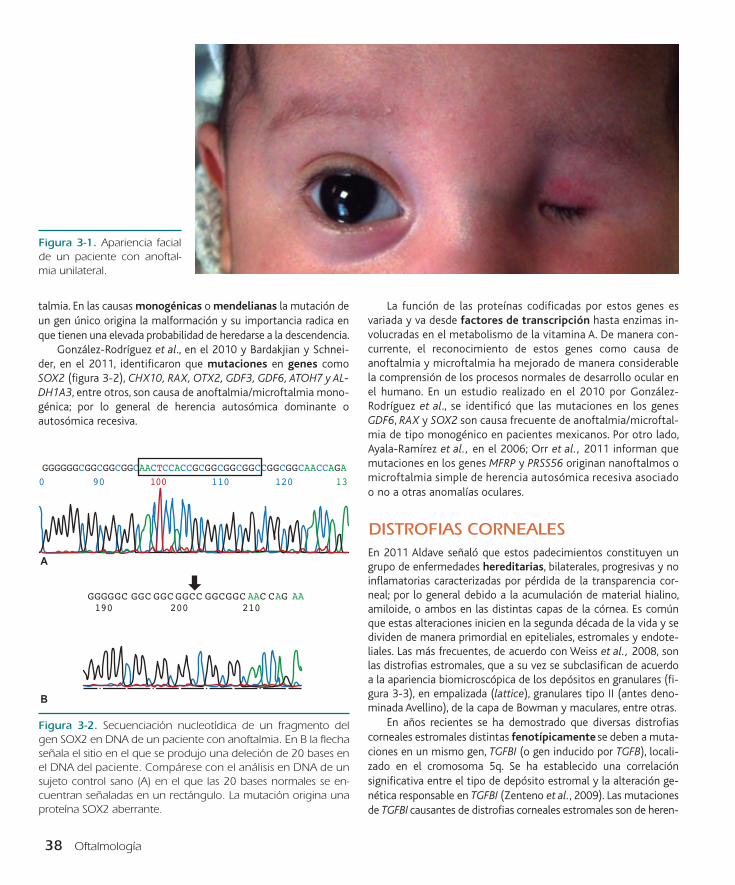
ANOFTALMIA Y MICROFTALMIAEntre los defectos anatómicos oculares más severos se encuentran la anoftalmia y la microftalmia. Bardakjian y Schneider, en 2011, señalan que el primer término se aplica a la ausencia unilateral o bilateral de tejido ocular dentro de la órbita con presen-cia de anexos oculares (figura 3-1), mientras que el segundo se define como un ojo pequeño, con diámetro axial menor a 20 mm y diámetro corneal menor a 10 mm en el adulto.
La prevalencia general de anoftalmia y o microftalmia, o ambas es de 1 en 3 300; en el 2007, Verma y Fitzpatrick, 2007, esti-maron que hasta 11% de los casos de ceguera infantil se deben a estas malformaciones congénitas. Sundin et al., en el 2008, determinaron que el término nanoftalmia o microftalmia simple se refiere a un ojo de tamaño pequeño, por lo general en el adulto tiene una longitud axial menor a 18 mm, sin malformaciones oculares concurrentes y con hipermetropía severa. La mi-croftalmia puede acompañarse de otras alteraciones oculares como esclerocórnea, anomalía de Peters, cataratas, persistencia de vítreo hiperplásico y displasia retinianas, entre otras. Cerca de un tercio de los casos de microftalmia se presenta en individuos con otras alteraciones a nivel extra-ocular y conforman síndromes genéticos específicos. La etiología de la anoftalmia/microf-talmia es compleja multifactorial) e incluye factores ambientales y factores genéticos. Dentro de las causas genéticas se en-cuentran las anomalías cromosómicas detectables con un cariotipo, como la trisomía 13 y los síndromes de deleción de los cromosomas 4p y 13q, y las anomalías monogénicas que se presentan en cerca de 10-20% de los sujetos con anoftalmia/microf-
Enfermedades oculares genéticas
Capítulo 3
38 Oftalmología
talmia. En las causas monogénicas o mendelianas la mutación de un gen único origina la malformación y su importancia radica en que tienen una elevada probabilidad de heredarse a la descendencia.
González-Rodríguez et al., en el 2010 y Bardakjian y Schnei-der, en el 2011, identificaron que mutaciones en genes como SOX2 (figura 3-2), CHX10, RAX, OTX2, GDF3, GDF6, ATOH7 y AL-DH1A3, entre otros, son causa de anoftalmia/microftalmia mono-génica; por lo general de herencia autosómica dominante o autosómica recesiva.
La función de las proteínas codificadas por estos genes es variada y va desde factores de transcripción hasta enzimas in-volucradas en el metabolismo de la vitamina A. De manera con-currente, el reconocimiento de estos genes como causa de anoftalmia y microftalmia ha mejorado de manera considerable la comprensión de los procesos normales de desarrollo ocular en el humano. En un estudio realizado en el 2010 por González-Rodríguez et al., se identificó que las mutaciones en los genes GDF6, RAX y SOX2 son causa frecuente de anoftalmia/microftal-mia de tipo monogénico en pacientes mexicanos. Por otro lado, Ayala-Ramírez et al., en el 2006; Orr et al., 2011 informan que mutaciones en los genes MFRP y PRSS56 originan nanoftalmos o microftalmia simple de herencia autosómica recesiva asociado o no a otras anomalías oculares.
DISTROFIAS CORNEALESEn 2011 Aldave señaló que estos padecimientos constituyen un grupo de enfermedades hereditarias, bilaterales, progresivas y no inflamatorias caracterizadas por pérdida de la transparencia cor-neal; por lo general debido a la acumulación de material hialino, amiloide, o ambos en las distintas capas de la córnea. Es común que estas alteraciones inicien en la segunda década de la vida y se dividen de manera primordial en epiteliales, estromales y endote-liales. Las más frecuentes, de acuerdo con Weiss et al., 2008, son las distrofias estromales, que a su vez se subclasifican de acuerdo a la apariencia biomicroscópica de los depósitos en granulares (fi-gura 3-3), en empalizada (lattice), granulares tipo II (antes deno-minada Avellino), de la capa de Bowman y maculares, entre otras.
En años recientes se ha demostrado que diversas distrofias corneales estromales distintas fenotípicamente se deben a muta-ciones en un mismo gen, TGFBI (o gen inducido por TGFB), locali-zado en el cromosoma 5q. Se ha establecido una correlación significativa entre el tipo de depósito estromal y la alteración ge-nética responsable en TGFBI (Zenteno et al., 2009). Las mutaciones de TGFBI causantes de distrofias corneales estromales son de heren-
GGGGGGCGGCGGCGGCAACTCCACCGCGGCGGCGGCCGGCGGCAACCAGA0 90 110
190
A
B
200 210
120 13100
GGGGGC GGC GGCGGCC GGCGGC AAC CAG AA
Figura 3-2. Secuenciación nucleotídica de un fragmento del gen SOX2 en DNA de un paciente con anoftalmia. En B la flecha señala el sitio en el que se produjo una deleción de 20 bases en el DNA del paciente. Compárese con el análisis en DNA de un sujeto control sano (A) en el que las 20 bases normales se en-cuentran señaladas en un rectángulo. La mutación origina una proteína SOX2 aberrante.
Figura 3-1. Apariencia facial de un paciente con anoftal-mia unilateral.

Capítulo 3 Enfermedades oculares genéticas 39
cia dominante (figura 3-4), por lo que es frecuente observar fami-lias con varias generaciones afectadas por la alteración corneal.
Una de las pocas distrofias estromales que no es de transmisión dominante es la de tipo macular, entidad caracterizada por opacidad corneal bilateral, adelgazamiento central, depósitos blanquecinos irregulares y erosiones epiteliales. Klintworth et al., 2006 encon-traron que ésta se hereda de manera autosómica recesiva y es causada por mutaciones en CHST6, un gen que codifica una enzi-ma con actividad de sulfotransferasa de carbohidratos.
La distrofia corneal endotelial más frecuente es la distrofia de Fuchs, caracterizada por degeneración del endotelio corneal, pérdi-da progresiva de células endoteliales, cambios morfológicos en la hexagonalidad endotelial, formación de depósitos de matriz extra-celular llamados gutas y edema corneal progresivo. En el 2006, Darlington JK. et al., señalaron que esta enfermedad afecta a más del 4% de individuos mayores de 40 años en los EUA y es una de las indicaciones más frecuentes de trasplante corneal. Por su parte, en 2012, Schmedt T. et al., señalaron que bien la mayoría de casos de distrofia endotelial de Fuchsse se presenta de manera esporá-dica, existen casos familiares con herencia autosómica dominante que son causados por mutaciones en los genes TCF8, COL8A2 y SLC4A11.
La distrofia endotelial congénita hereditaria (CHED) se presen-ta en una forma autosómica recesiva de origen congénito y de rápi-da evolución; se caracteriza por edema corneal, nistagmo, baja visual severa y en ocasiones por hipoacusia (cuando va acompañada con este último dato, conforma el síndrome de Harboyan). La causa de esta variante, denominada CHED2, es la mutación homocigota (en
ambos alelos) en el gen SLC4A11, el cual codifica una proteína transportadora de sodio/borato en el endotelio corneal. Ciralsky y Colby en 2007 demostraron que la distrofia endotelial congénita tipo 1 (CHED1) se hereda de manera autosómica dominante, tiene un curso clínico menos severo que la tipo 2 y a la fecha no se ha identificado al gen responsable. Otras distrofias endoteliales inclu-yen la polimórfica posterior caracterizada por la presencia de varias capas de células endoteliales en lugar de una monocapa y la distro-fia corneal amorfa posterior en la que se observa pérdida de la mo-nocapa endotelial. En 2005 Krafchack et al., señalaron que mutaciones en el gen TCF8 son una causa relativamente frecuente de distrofia endotelial polimórfica posterior..
MALFORMACIONES DEL SEGMENTO ANTERIOREntre las malformaciones que se presentan en esta parte del ojo se encuentra un grupo, también llamado disgenesias del segmento anterior, caracterizadas por malformaciones de la córnea, el iris, el cristalino y el ángulo iridocorneal. Son un grupo frecuente de ano-malías del desarrollo ocular que pueden presentarse de manera esporádica o de forma hereditaria. En este último caso, el tipo de herencia más común es el autosómico dominante. Idrees F. et al., en 2006 encontraron que dentro de este grupo se encuentran al-teraciones como la esclerocórnea, la anomalía de Peters, la anomalía de Axenfeld-Rieger, la aniridia (figura 3-5), la afaquia, y la microes-ferofaquia, entre otras.
De manera particular, las mutaciones en el gen PAX6 son causa de aniridia congénita (figura 3-6). La esclerocórnea se define como
A G G G G G CNA
160 17
A A A A
Figura 3-3. Imagen biomicroscópica corneal en un paciente con distrofia corneal estromal de tipo granular. Se observan numero-sas opacidades stromales blanquecinas que interfieren con la transparencia corneal.
Figura 3-4. Secuenciación nucleotídica de un fragmento del gen TGFBI en un sujeto con distrofia corneal estromal de tipo granular. La flecha señala el sitio de la mutación representada por dos señales: un pico azul que corresponde a la base o nu-cleótido citosina normal y un pico rojo que corresponde a la mutación hacia la base timina. Este cambio predice una muta-ción del aminoácido arginina (Arg, triplete CGG) hacia el ami-noácido triptófano (Trp, triplete TTG) en la posición 555 de la proteína TGFBI.

40 Oftalmología
una falla en la delimitación del tejido escleral y corneal, que origina que la esclera cubra la porción periférica de la córnea (escleraliza-ción) o en casos severos que la cubra en su totalidad (esclerocor-nea totalis, figura 3-7).
Esta forma severa se hereda de manera autosómica recesiva y puede ser causada por mutaciones en el gen FOXE3 (figura 3-8).
La anomalía de Peters está caracterizada por opacidad central de la córnea con alteraciones correspondientes en el estroma pos-terior, la membrana de Descemet y el endotelio, acompañada de adherencias iridocorneales o entre la córnea y el cristalino. Se ori-gina por una falla en la separación entre la vesícula del cristalino y el ectodermo de superficie durante el desarrollo ocular. Mutacio-nes en genes como PITX2, CYP1B1 y PAX-6 pueden ocasionar ano-malía de Peters de tipo hereditario. Estos genes codifican proteínas que aseguran una correcta separación de la vesícula del cristalino durante el desarrollo ocular por lo que su disfunción, en caso de mutación, se traduce en anomalía de Peters.
La anomalía de Axenfeld-Rieger se caracteriza por embrio-toxón posterior (prominencia de la línea de Schwalbe), hipoplasia del estroma del iris y corectopia. Cuando a estas anomalías ocula-res se agregan alteraciones cráneo-faciales, dentales y umbilicales, se aplica el término de síndrome de Axenfeld-Rieger. Mutaciones en el gen FOXC1 pueden originar la anomalía ocular aislada o el síndrome de Axenfeld-Rieger completo. La aniridia es una altera-ción definida por grados variables de hipoplasia o ausencia de tejido iridiano; por lo regular está asociada a otras alteraciones oculares como glaucoma, queratitis, alteraciones del disco óptico e hipopla-sia foveal. La mayoría de los casos de aniridia se debe a mutaciones dominantes del gen PAX-6. Una característica en los casos fami-
liares es la variabilidad de expresión de la anomalía. Debe consi-derarse que algunos ejemplos de aniridia se deben a una deleción del brazo corto del cromosoma 11 que elimina al gen PAX-6 y a otros genes adyacentes; en estos casos se presenta el síndrome WAGR o síndrome de genes contiguos, el cual está definido por el tumor de Wilms, aniridia, anomalías genitourinarias y discapaci-
A
B
G
G A T C C C C C C C CT T T T T TA AG G G G
A A A A A AG C C C C C C CC C C CC CT T T T T TT T T T TG G G G G40 150 170160
220 230
Figura 3-5. Imagen biomicroscópica en un paciente con aniridia congénita. Se observa ausencia total de tejido iridiano.
Figura 3-7. Apariencia fenotípica de un paciente con esclerocór-nea total en ojo derecho. La córnea se encuentra cubierta por una extensión de la esclera.
Figura 3-6. Secuenciación nucleotídica de un fragmento del gen PAX6 en DNA de un paciente con aniridia congénita. En B la flecha señala el sitio en el que se produjo una deleción de 14 bases en el DNA del paciente. Compárese con el análisis en DNA de un sujeto sano (A) en el que las 14 bases normales se encuen-tran señaladas en un rectángulo. Esta mutación origina una proteína PAX6 aberrante.

Capítulo 3 Enfermedades oculares genéticas 41
dad intelectual. La afaquia y la microesferofaquia son alteraciones caracterizadas por ausencia congénita de cristalino y presencia de un cristalino pequeño con diámetro anteroposterior aumentado, de manera respectiva. En 2002, Chan y Collin, 2002 encontraron mutaciones en el gen FOXE-3 y las relacionaron con afaquia con-génita, así como que la microesferofaquia es una anomalía que puede presentarse en síndrome genéticos como los síndromes de Marfán y de Weill-Marchesani.
CATARATA CONGÉNITAEste padecimiento se define como cualquier opacidad en el crista-lino presente al nacimiento o durante los primeros meses de vida y se considera la causa más común de pérdida visual permanente en niños. Su prevalencia se estima en 1 a 6 casos por cada 10 000 nacimientos y cerca de 30 a 40% de ellas tienen una etiología genética con transmisión dominante en casi en todos los casos (figura 3-9).
Existen más de 25 genes cuyas mutaciones son causa demos-trada de catarata congénita y entre éstos se encuentran los genes de cristalinas, que codifican ciertas proteínas solubles del cristali-no, en específico CRYA, CRYB, CRYGD (figura 3-10) y CRRYG, ade-más de genes de conexinas como Cx43, Cx46 y Cx50. Huang y He, en 2010 descubrieron que otras mutaciones causantes de catarata congénita pueden ocurrir en genes como GALK (galactoquinasa), PITX3, MAF y HSF4.
GLAUCOMA CONGÉNITOEl glaucoma congénito primario es el tipo más común de este pa-decimiento en la población infantil; constituye una alteración ge-nética autosómica recesiva debida a un desarrollo anormal de la malla trabecular y del ángulo de la cámara anterior. En la clínica se caracteriza por aumento de la presión intraocular, megalocórnea y estrías de Haab asociados a fotofobia, epifora y blefaroespasmo. Ha sido demostrado que esta anomalía se debe a mutaciones en el gen del citocromo P4501B1 (CYP1B1) en cerca de 30% a 50% de los enfermos, de acuerdo con la población estudiada. En estos casos, los padres de los afectados son portadores sanos. De ma-nera reciente, en 2011, Liu & Allingham, lograron identificar que mutaciones en el gen LTBP2 son también una causa de glaucoma congénito autosómico recesivo, en una proporción menor que las de CYP1B1.
ESTRABISMO HEREDITARIOEl estrabismo se define como la desviación de un ojo en relación con el otro, lo que origina una incapacidad para la visión binocular. Este padecimiento afecta a entre 2 a 4% de la población general y puede ocasionar ambliopía. Dentro de las formas genéticas más comunes de estrabismo se encuentra el síndrome de Duane, diag-nosticado en hasta 5% de todos los pacientes con este padeci-miento. Esta anomalía se caracteriza por limitación o ausencia de
Figura 3-9. Apariencia biomicroscópica de una catarata congé-nita de tipo aculeiforme. La opacidad del cristalino presenta una apariencia de agujas y espículas que se proyectan radialmente desde el núcleo.
A
B
A T C C C C G CA
A AT C C C CGT
Figura 3-8. Secuenciación nucleotídica de un fragmento del gen FOXE3 en un sujeto con esclerocórnea bilateral. En (A) se señala el triplete CAC normal observado en DNA de un sujeto normal; en B se muestra la mutación de la priemra base del tri-plete, representada por un pico rojo que corresponde a la base mutante timina. Esta mutación predice una cambio del aminoá-cido tirosina (Tir) hacia el aminoácido histidina (His) en la posi-ción 98 de la proteína FOXE3.

42 Oftalmología
abducción y, o aducción ocular, o ambas; acompañada de retrac-ción del globo y cierre de la fisura palpebral al intentar la aducción. La gran mayoría de casos de síndrome de Duane son esporádicos, pero existen casos familiares con transmisión autosómica domi-nante y variabilidad de expresión. Con frecuencia, los casos fami-liares son bilaterales y de mayor severidad que los esporádicos. En fechas recientes, el estudio de diversas familias (incluyendo una originaria de México) con síndrome de Duane dominante permi-tió identificar que el gen responsable de esta forma hereditaria es CHN1, el cual codifica la alfa-quimerina, proteína que tiene como función guiar los axones de los nervios hacia su músculo corres-pondiente durante el desarrollo del sistema ocular (Miyake et al., 2008). Otras formas de estrabismo con componente genético in-cluyen a la fibrosis congénita de los músculos extraoculares, enti-dad caracterizada por alteraciones variables de los movimientos oculares horizontales y, o verticales, o ambos y ptosis. Existen tres tipos de acuerdo a las características clínicas y se ha demostrado que el tipo 1 y algunos casos del tipo 3 se deben a mutaciones en el gen KIF21A. El gen KIF21A codifica una proteína que tiene la función de transportar moléculas a lo largo de los axones que iner-van a los músculos extraoculares.
DISTROFIAS RETINIANASLas distrofias retinianas incluyen un amplio grupo de enfermeda-des hereditarias que tienen en común la pérdida de células de las distintas capas de la retina. Son una de las causas más frecuentes
de ceguera hereditaria en el mundo y tienen una amplia variabilidad en sus características clínicas aún entre sujetos afectados en una misma familia. Por lo general se clasifican en centrales o periféricas y también de acuerdo al tipo celular o capa retiniana afectada. El hecho de que una alteración localizada en un inicio a un tipo celu-lar o capa retiniana pueda afectar de manera tardía también a otros tipos celulares complica el cuadro clínico y la clasificación de las distrofias retinianas.
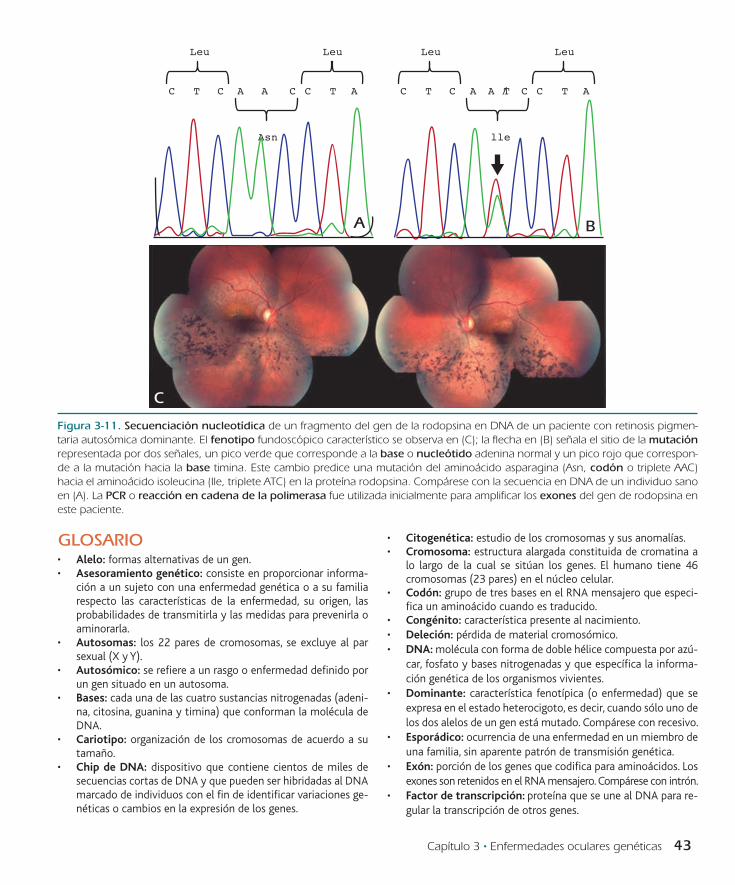
La distrofia retiniana más frecuente es la retinosis pigmentaria (en ocasiones llamada retinitis pigmentosa, a pesar de no estar asociada a un proceso inflamatorio en la retina). La frecuencia de la retinosis pigmentaria es de 1 en 3 500 individuos y el cuadro clínico, aunque variable, inicia con mala visión nocturna o en con-diciones de poca luminosidad (nictalopía), pérdida de los campos visuales periféricos y baja visual en estadios avanzados. Los datos clínicos fundoscópicos típicos son adelgazamiento de la vasculatu-ra retiniana, palidez del disco óptico y presencia de pigmento en la periferia con morfología de “espículas óseas” (figura 3-11, panel C). La retinosis pigmentaria es el prototipo de enfermedad con hete-rogeneidad genética, lo que significa que una misma enfermedad hereditaria tiene más de un patrón de herencia. Ferrari et al., en 2011 encontraron que de hecho existen casos de retinosis pigmen-taria autosómicos dominantes, autosómicos recesivos, ligados al cromosoma X e incluso de herencia mitocondrial. La mayoría de los casos familiares tiene un patrón autosómico recesivo. Existe una correlación entre la gravedad de la enfermedad y el tipo de herencia (vínculo genotipo-fenotipo) ya que los casos autosómi-cos dominantes son de inicio tardío y de progresión lenta, mien-tras que los casos ligados al X son muy graves y de progresión acelerada. Las mutaciones del gen de la rodopsina son causa de hasta 25% de las formas autosómicas dominantes (figura 3-11).
Muchos casos de la enfermedad se presentan en la consulta como los primeros en la familia y en estos casos, que son denomina-dos tipos simples desde el punto de vista hereditario, es compli-cada la identificación del probable patrón de herencia con el que se transmitirá la enfermedad a generaciones subsecuentes. Para el primer semestre del año 2013 se habían identificado cerca de 70 genes cuyas mutaciones originan distintas formas de retinosis pig-mentaria. Este dato permite comprender la dificultad que existe para el diagnóstico a nivel de DNA (diagnóstico molecular) en los afectados. Por fortuna, en 2013 Shanks et al., han desarrollado diversas estrategias que permiten un mejor estudio genético de estos pacientes utilizando metodologías novedosas de análisis del genoma como los microarreglos (chips) de DNA y la secuencia-ción de DNA de siguiente generación. La aplicación de estas tec-nologías en un número cada vez mayor de hospitales y laboratorios del mundo ha permitido no sólo aumentar el número de pacientes que tienen un diagnóstico molecular sino también identificar ge-nes nuevos causantes de retinosis pigmentaria. En 2010 Stieger & Lorenz, mostraron que la identificación de la mutación responsa-ble en un paciente es de gran trascendencia no nada más porque confirma el diagnóstico, permite conocer el pronóstico y mejora el asesoramiento genético al afectado y su familia, sino también porque es la base para los protocolos de terapia génica para algu-nas distrofias retinianas que ya se desarrollan en varios países.
A
B
Figura 3-10. Secuenciación nucleotídica de un fragmento del gen CRYGD en un sujeto con catarata congénita aculeiforme. La flecha en (B) señala el sitio de la mutación representada por dos señales, un pico negro que corresponde a la base o nucleótido guanina y un pico verde que corresponde a la mutación hacia la base adenina. Este cambio predice una mutación del aminoáci-do arginina (Arg) hacia el aminoácido histidina (His) en la posi-ción 58 de la proteína cristalina gamma D. Compárese con la secuencia en DNA de un individuo sano en (A), en donde se observa solamente la señal correspondiente a la base guanina (pico negro) normal.
Capítulo 3 Enfermedades oculares genéticas 43
GLOSARIOAlelo: formas alternativas de un gen.Asesoramiento genético: consiste en proporcionar informa-ción a un sujeto con una enfermedad genética o a su familia respecto las características de la enfermedad, su origen, las probabilidades de transmitirla y las medidas para prevenirla o aminorarla. Autosomas: los 22 pares de cromosomas, se excluye al par sexual (X y Y).Autosómico: se refiere a un rasgo o enfermedad definido por un gen situado en un autosoma.Bases: cada una de las cuatro sustancias nitrogenadas (adeni-na, citosina, guanina y timina) que conforman la molécula de DNA.Cariotipo: organización de los cromosomas de acuerdo a su tamaño.Chip de DNA: dispositivo que contiene cientos de miles de secuencias cortas de DNA y que pueden ser hibridadas al DNA marcado de individuos con el fin de identificar variaciones ge-néticas o cambios en la expresión de los genes.
Citogenética: estudio de los cromosomas y sus anomalías.Cromosoma: estructura alargada constituida de cromatina a lo largo de la cual se sitúan los genes. El humano tiene 46 cromosomas (23 pares) en el núcleo celular. Codón: grupo de tres bases en el RNA mensajero que especi-fica un aminoácido cuando es traducido.Congénito: característica presente al nacimiento.Deleción: pérdida de material cromosómico.DNA: molécula con forma de doble hélice compuesta por azú-car, fosfato y bases nitrogenadas y que específica la informa-ción genética de los organismos vivientes.Dominante: característica fenotípica (o enfermedad) que se expresa en el estado heterocigoto, es decir, cuando sólo uno de los dos alelos de un gen está mutado. Compárese con recesivo.Esporádico: ocurrencia de una enfermedad en un miembro de una familia, sin aparente patrón de transmisión genética.Exón: porción de los genes que codifica para aminoácidos. Los exones son retenidos en el RNA mensajero. Compárese con intrón.Factor de transcripción: proteína que se une al DNA para re-gular la transcripción de otros genes.
Leu
Asn lle
A B
C CC A A A AA A /T T T T TC C C C C
Leu Leu Leu
C
Figura 3-11. Secuenciación nucleotídica de un fragmento del gen de la rodopsina en DNA de un paciente con retinosis pigmen-taria autosómica dominante. El fenotipo fundoscópico característico se observa en (C); la flecha en (B) señala el sitio de la mutación representada por dos señales, un pico verde que corresponde a la base o nucleótido adenina normal y un pico rojo que correspon-de a la mutación hacia la base timina. Este cambio predice una mutación del aminoácido asparagina (Asn, codón o triplete AAC) hacia el aminoácido isoleucina (Ile, triplete ATC) en la proteína rodopsina. Compárese con la secuencia en DNA de un individuo sano en (A). La PCR o reacción en cadena de la polimerasa fue utilizada inicialmente para amplificar los exones del gen de rodopsina en este paciente.

44 Oftalmología
Fenotipo: características visibles de un individuo. Se aplica también a presencia o ausencia de enfermedad.Gen: unidad fundamental de la herencia. Secuencia de DNA que codifica para una proteína o para un RNA funcional.Genoma: la totalidad del contenido de DNA de un organismo.Genotipo: constitución alélica de un individuo en un locus específico.Hereditario: rasgo fenotípico que se trasmite en una familia a través del material genético Herencia mitocondrial: características o enfermedades cau-sadas por mutaciones en los genes localizados en el cromoso-ma de la mitocondria.Heterogeneidad genética: se aplica cuando una misma en-fermedad genética presenta más de un patrón de transmisión hereditaria.Intrón: secuencia de DNA situada entre dos exones. Al inicio se transcribe en el RNA mensajero (RNAm) pero luego es eli-minada en el RNAm maduro.Ligado al X: fenotipos o enfermedades originadas por muta-ciones en genes localizados en el cromosoma X. Mendeliano: referente a herencia mendeliana o atribuida a un gen único. Sinónimo de monogénico.Multifactorial: rasgo o enfermedad causada por una combi-nación de factores genéticos y factores ambientales.Mutación: alteración permanente y heredable en la secuencia de bases del DNA.Nucleótido: unidad estructural fundamental del DNA consti-tuida por un azúcar, un grupo fosfato y una base nitrogenada.
PCR (reacción en cadena de la polimerasa): Técnica para producir un gran número de copias de un fragmento específico de DNA.Portador: sujeto heterocigoto para una mutación recesiva. No expresa la enfermedad ya que se requiere que ambos alelos tengan mutación para manifestar el fenotipo.RNA (ácido ribonucleico): ácido nucléico de cadena sencilla. Existen tres tipos básicos: RNA mensajero, RNA ribosomal y RNA de transferencia.Síndrome: un patrón de diversas anomalías o malformaciones debidas a una causa única subyacente (ej. Síndrome de Mar-fan, síndrome de Down).Síndrome de genes contiguos: enfermedad causada por la deleción o duplicación de varios genes consecutivos en un cro-mosoma.Recesivo: característica fenotípica (o enfermedad) que se ex-presa en el estado homocigoto, es decir, cuando los dos alelos de un gen están mutados. Compárese con dominante.Secuenciación nucleotídica: identificación del ordenamiento de las bases a lo largo de un fragmento de DNA.Terapia génica: procedimiento de inserción o modificación de un gen para corregir una enfermedad genética.Transcripción: proceso por el cual una molécula de RNA men-sajero es sintetizada a partir de una secuencia de DNA. Varia-bilidad de expresión: se aplica cuando un mismo genotipo (una mutación) origina fenotipos de gravedad variable.
Autoevaluación
1. Las enfermedades genéticas mendelianas que afectan el ojo son causadas por:a) Alteraciones cromosómicas b) Interacción genes-ambientec) Mutación de un gend) Agentes teratogénicos
2. La anomalía de Peters es:a) Un tipo de malformación del segmento anterior b) Un tipo de microftalmiac) Un tipo de glaucoma del desarrollod) Un tipo de distrofia retiniana
3. Es la distrofia retiniana más frecuente:a) Enfermedad de Stargardtb) Enfermedad de Bestc) Ceguera nocturna congénita estacionariad) Retinosis pigmentaria
Respuestas: 1. c, 2. a, 3. d

Capítulo 3 Enfermedades oculares genéticas 45
Referencias
Aldave AJ: The genetics of the corneal dystrophies. Dev Ophthalmol. 2011; 48:51-66.Bardakjian TM, Schneider A: The genetics of anophthalmia and microphthalmia. Curr Opin Ophthalmol. 2011 Sep; 22(5):309-313.Ciralsky J, Colby K: Congenital corneal opacities: a review with a focus on genetics. Semin Ophthalmol. 2007 Oct-Dic;22(4):241-246.Chan RT, Collin HB: Microspherophakia. Clin Exp Optom. 2002 Sep;85(5):294-299.Darlington JK, Adrean SD, Schwab IR. Trends of penetrating keratoplasty in the United States from 1980 to 2004. Ophthalmology 2006 Dic;113(12):2171-
2175.Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F: Retinitis pigmentosa: genes and disease mechanisms. CurrGenomics 2011
Jun; 12(4):238-249.Gonzalez-Rodriguez J, Pelcastre EL, Tovilla-Canales JL, Garcia-Ortiz JE, Amato-Almanza M, Villanueva-Mendoza C, Espinosa-Mattar Z, Zenteno
JC. Mutational screening of CHX10, GDF6, OTX2, RAX and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br J Ophthalmol. 2010 Ag;94(8):1100-1104.
Huang B, He W: Molecular characteristics of inherited congenital cataracts. Eur J Med Genet. 2010 Nov-Dic; 53(6):347-357. Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT: A review of anterior segment dysgeneses. Surv Ophthalmol. 2006 May-Jun; 51(3):213-231.Klintworth GK, Smith CF, Bowling BL: CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular
genetic review. Mol Vis. 2006 Mar 10; 12:159-176.Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, Mian S, Nairus T, Elner V, Schteingart MT, Downs CA, Kijek TG, Johnson JM,
Trager EH, Rozsa FW, Mandal MN, Epstein MP, Vollrath D, Ayyagari R, Boehnke M, Richards JE: Mutations in TCF8 cause posterior polymor-phous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005 Nov; 77(5):694-708.
Liu Y, Allingham RR: Molecular genetics in glaucoma. Exp Eye Res. 2011 Oct; 93(4):331-339.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan WM, Law K, Crosier M, Lindsay S, Cheung M, Allen J, Gutowski NJ, Ellard S, Young
E, Iannaccone A, Appukuttan B, Stout JT, Christiansen S, Ciccarelli ML, Baldi A, Campioni M, Zenteno JC, Davenport D, Mariani LE, Sahin M, Guthrie S, Engle EC: Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science 2008 Ag; 321(5890):839-843.
Orr A, Dubé MP, Zenteno JC, Jiang H, Asselin G, Evans SC, Caqueret A, Lakosha H, Letourneau L, Marcadier J, Matsuoka M, Macgillivray C, Nightingale M, Papillon-Cavanagh S, Perry S, Provost S, Ludman M, Guernsey DL, Samuels ME: Mutations in a novel serine protease PRSS56 in families with nanophthalmos. Mol Vis. 2011; 17:1850-1861.
Raca G, Jackson C, Warman B, Bair T, Schimmenti LA: Next generation sequencing in research and diagnostics of ocular birth defects. Mol Genet Metab. 2010 Jun; 100(2):184-192.
Sheffield VC, Stone EM: Genomics and the eye. N Engl J Med. 2011 May ; 364(20):1932-1942.Schmedt T, Silva MM, Ziaei A, Jurkunas U: Molecular bases of corneal endothelial dystrophies. Exp Eye Res. 2012 Feb; 95(1):24-34.Shanks ME, Downes SM, Copley RR, Lise S, Broxholme J, Hudspith KA, Kwasniewska A, Davies WI, Hankins MW, Packham ER, Clouston P, Seller
A, Wilkie AO, Taylor JC, Ragoussis J, Németh AH: Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet 2013 Mar; 21(3):274-280.
Stieger K, Lorenz B: Gene therapy for vision loss -- recent developments. Discov Med 2010 Nov; 10(54):425-433.Sundin OH, Dharmaraj S, Bhutto IA, Hasegawa T, McLeod DS, Merges CA, Silva ED, Maumenee IH, Lutty GA: Developmental basis of nanophthal-
mos: MFRP Is required for both prenatal ocular growth and postnatal emmetropization. Ophthalmic Genet 2008 Mar; 29(1):1-9.Verma AS, Fitzpatrick DR: Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007 Nov; 2:47.Weiss JS, Møller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW, Kivelä T, Busin M, Munier FL, Seitz B, Sutphin J, Bredrup C, Mannis MJ, Rapuano
CJ, Van Rij G, Kim EK, Klintworth GK: The IC3D classification of the corneal dystrophies. Cornea 2008 Dic; 27 Suppl 2:S1-83.Zenteno JC, Correa-Gomez V, Santacruz-Valdez C, Suarez-Sanchez R, Villanueva-Mendoza C: Clinical and genetic features of TGFBI-linked corneal
dystrophies in Mexican population: description of novel mutations and novel genotype-phenotype correlations. ExpEye Res 2009 Ag;89(2):172-177.