effect of imatinib mesylate on neuroblastoma tumorigenesis and
TRANSCRIPT

Effect of Imatinib Mesylate on NeuroblastomaTumorigenesis and Vascular Endothelial GrowthFactor Expression
Kiichiro Beppu, Jerry Jaboine, Melinda S. Merchant, Crystal L. Mackall,Carol J. Thiele
Background: Alternative treatment options are needed foradvanced neuroblastoma patients because their prognosisremains poor after intensive chemotherapy. Neuroblastomacells express platelet-derived growth factor (PDGF), stemcell factor (SCF), and vascular endothelial growth factor(VEGF) and their respective receptors, PDGFR, c-Kit, andFlk-1. We therefore evaluated the effects of imatinib mesy-late (imatinib), a selective inhibitor of the tyrosine kinaseactivities of c-Kit and PDGFR, on the growth of neuroblas-toma cells in vivo and in vitro. Methods: We tested sevenhuman neuroblastoma cell lines for their sensitivity to imatinib.Cell viability was assessed by trypan blue dye exclusion. Apo-ptosis was evaluated by nuclear staining, flow cytometry,and western blotting. Protein assays included immunopre-cipitation, western blotting, enzyme-linked immunosorbentassays, and immunohistochemistry. mRNA expression wasassessed by northern blotting. We used a xenograft model inSCID mice (10 mice per group) to evaluate the effects ofimatinib oral therapy (50 or 100 mg/kg every 12 hours for14 days) on neuroblastoma tumor growth. All statisticaltests were two-sided. Results: All seven neuroblastomacell lines treated with imatinib displayed concentration-dependent decreases in cell viability, which coincided with aninduction of apoptosis, and with ligand-stimulated phos-phorylation of c-Kit and PDGFR. The imatinib concen-trations that caused 50% inhibition of growth and 50%inhibition of ligand-induced phosphorylation of these recep-tors were 9–13 �M and 0.1–0.5 �M, respectively. Expres-sion of VEGF, but not phosphorylation of Flk-1, its receptor,was reduced in neuroblastoma cells treated with imatinib at10 �M or higher. Mice treated with imatinib at 50 mg/kg or100 mg/kg had statistically significantly smaller tumors thancontrol mice treated with vehicle (mean tumor volume inmice treated with imatinib at 50 mg/kg � 1546 mm3, incontrol mice � 2954 mm3; difference � 1408 mm3, 95%confidence interval [CI] � 657 to 2159 mm3; P<.001; meantumor volume in mice treated with imatinib at 100 mg/kg �463 mm3; difference � 2491 mm3, 95% CI � 1740 to 3242mm3; P<.001). Conclusions: Imatinib inhibited the growthof neuroblastoma cells in vitro and in vivo. This inhibitionwas associated with suppression of PDGFR and c-Kit phos-phorylation and inhibition of VEGF expression. [J Natl Can-cer Inst 2004;96:46–55]
Neuroblastoma is the most common solid extracranial tumorof early childhood (1). Several tumor characteristics, such asamplification of the N-myc oncogene and loss of heterozygosityat chromosome 1p, are statistically significant indicators of poorprognosis for neuroblastoma patients (3). Neuroblastoma pa-tients with high-risk or disseminated disease continue to have a
poor prognosis, even after they have undergone intensive che-motherapy and autologous bone marrow transplantation (2).Alternative treatments are therefore needed to improve the prog-nosis of neuroblastoma patients with high-risk disease.
Receptor tyrosine kinases have been proposed as potentialtargets for antitumor therapy. For example, imatinib mesylate(also known as STI571 or Gleevec, and hereafter called ima-tinib) was initially shown to inhibit in vitro growth of Abl-transformed cells and Bcr-Abl–positive chronic myelogenousleukemia (CML) cells through selective inhibition of the Abltyrosine kinase (4). Imatinib is also a highly selective inhibitorof the tyrosine kinase activities of c-Kit, the receptor for stemcell factor (SCF), and platelet-derived growth factor receptor(PDGFR) (5). Results of recent clinical studies (7) have shownthat imatinib therapy is well tolerated and leads to remission inpatients with Bcr-Abl–positive CML or c-Kit–positive gastroin-testinal stromal tumor (GIST). Preclinical data suggest thatimatinib has cytotoxic and cytostatic activities and effectivelyinhibits the growth of tumor cell lines that contain gain-of-function mutations in Abl and c-Kit as well as tumor cells thatexpress autocrine growth loops (5). Imatinib has also been reportedto inhibit the growth of glioblastoma, dermatofibrosarcoma protu-berans, and small-cell lung cancer, all of which may express thePDGF/PDGFR or SCF/c-Kit autocrine growth loops (8–10). Re-cently, we have shown that imatinib interferes with the in vitro andin vivo growth of Ewing’s sarcoma which, like neuroblastoma, is atumor of peripheral neuroectodermal origin (11).
SCF and c-Kit mRNAs are expressed in neuroblastoma celllines and in neuroblastoma tumor samples (12). The SCF/c-Kitautocrine growth loop is thought to be important for cell survivalbecause neuroblastoma cells treated with an antibody to c-Kitundergo increased levels of apoptosis (13). Neuroblastoma celllines also express functional PDGFR-�, PDGFR-�, and theirrespective ligands, PDGF-AA and PDGF-BB (14). These obser-vations led us to hypothesize that imatinib might inhibit neuro-blastoma cell growth and survival via inhibition of the SCF/c-Kit and PDGF/PDGFR autocrine growth loops.
Tyrosine kinase receptors also play a role in angiogenesis, anessential step for tumor growth and metastasis. Tumor cellgrowth and metastasis require perturbation of the local balanceof pro-angiogenic and anti-angiogenic factors, which is referred
Affiliation of authors: Pediatric Oncology Branch, Center for Cancer Re-search, National Cancer Institute, National Institutes of Health, Bethesda, MD.
Correspondence to: Carol J. Thiele, PhD, Pediatric Oncology Branch, Na-tional Cancer Institute, National Institutes of Health, 10 Center Dr., MSC 1298,Bldg. 10/13N240, Bethesda, MD 20892 (e-mail: [email protected]).
See “Notes” following “References.”
DOI: 10.1093/jnci/djh004Journal of the National Cancer Institute, Vol. 96, No. 1, © Oxford UniversityPress 2004, all rights reserved.
46 ARTICLES Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

to as the angiogenic switch (15). In addition, vascular endothe-lial growth factor (VEGF) plays a key role in tumor angiogen-esis. Most neuroblastoma cell lines and tumors express VEGF(16,17), and the degree of VEGF expression and angiogenesis inneuroblastoma tumors is associated with both disease progres-sion and poor prognosis (18). Results of recent studies (20) haveshown that VEGF expressed by neuroblastoma cells contributesto the growth of endothelial cells in vitro and to angiogenesis invivo. Blockade of VEGF function is associated with the suppres-sion of neuroblastoma growth (21,22), suggesting the potentialutility of VEGF-targeted therapies in neuroblastoma treatment.
Here, we examined the effects of imatinib on the growth ofneuroblastoma cells in vitro and in vivo, on VEGF production,and on c-Kit and PDGFR phosphorylation.
MATERIALS AND METHODS
Cell Culture and Reagents
Seven human neuroblastoma cell lines, SMS-KCNR (23),NGP (24), SK-N-BE2 (25), LAN-1 (24), LAN-5 (24), SH-SY5Y(24), SK-N-AS (26), mouse NIH3T3 fibroblast cells (providedby J. Bolen, National Cancer Institute), and human CCL-243cells, a Philadelphia chromosome–positive K562 CML cell line(American Type Culture Collection, Manassas, VA) were usedin this study. The cells were cultured in RPMI-1640 medium and10% fetal bovine serum as described previously (27). Imatinibwas provided by Dr. Elisabeth Buchdunger (Novartis Pharma-ceuticals, Basel, Switzerland). For growth factor stimulationassays, SMS-KCNR cells and SH-SY5Y cells (1 106) wereplated on 60-mm dishes in medium containing 10% serum. Thecells were shifted to 0.1% serum-containing medium for theindicated time and then incubated for 30 minutes with either 1�M imatinib or media control. The cells were then incubated for24 hours in medium containing 0.1% fetal bovine serum towhich either human recombinant SCF at 50 ng/mL (Endogen,Woburn, MA) or human recombinant PDGF-BB at 50 ng/mL(R&D Systems, Minneapolis, MN) and human recombinantVEGF (R&D Systems) or an equivalent volume of medium wasadded. Cells were stained with propidium iodide (PI) and sub-jected to fluorescence-activated cell sorting using a FACScanapparatus and CellQuest software (BD Biosciences, San Jose,CA) to quantify the percentage of apoptotic cells (i.e., those inthe sub-G1 fraction).
Cell Viability and Cell Growth Assays
We used the trypan blue dye exclusion assay to evaluate cellnumber and viability. Cells (1 105) were plated on 60-mmdishes and incubated with various concentrations of imatinib for4 days. The cells were then trypsinized and stained with 0.2%trypan blue, and viable cells (i.e., those that excluded the dye) werecounted under low-power microscopy. In some experiments, cellgrowth was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (SigmaChemical Company, St. Louis, MO) after 4 days of imatinib treat-ment, as previously described (11).
Apoptosis Assays
We detected morphologic changes in the nuclear chromatinof cells undergoing apoptosis by staining with Hoechst 33342
(Molecular Probes, Eugene, OR). Neuroblastoma cells weregrown in 96-well plates and cultured for 48 hours in the presenceor absence of 15 �M imatinib. After treatment, the cells wereincubated in the dark with Hoechst 33342 for 30 minutes at 37°C and were then observed with a fluorescence microscope todetect morphologic changes in the nuclear chromatin of cellsundergoing apoptosis. To assess cell cycle changes as well as theproportion of the cell population with less than a G1 content ofDNA, cells were trypsinized, washed with ice-cold phosphate-buffered saline (PBS), and fixed in 70% ethanol. After anotherwash with PBS, cells were incubated with RNase at 100 �g/mLand PI at 50 �g/mL (Sigma Chemical Company). DNA–PIfluorescence was measured with a FACScan apparatus.
Northern Blot Analysis
Cells were harvested by trypsinization, and total RNA wasextracted from 106 cells with the use of an RNeasy kit (Qiagen,Santa Clarita, CA). Northern blot analysis for VEGF RNA wasperformed according to a previously described protocol (27).Briefly, total RNA (25 �g) was resolved on 1% agarose–6%formaldehyde gels. The gels were stained with ethidium bromideat 2 mg/mL to allow inspection of the quantity and quality of theRNA. The RNA was transferred to Nytran membranes (Schlei-cher & Schuell, Keene, NH) and then hybridized with a 32P-labeled VEGF or glyceraldehyde-3-phosphate dehydrogenase(GAPDH) cDNA.
Protein Assays
Western blotting and immunoprecipitation were performed asdescribed previously (28). Briefly, cells were harvested bytrypsinization, resuspended in cold protein lysis buffer (0.5%Nonidet P-40, 50 mM Tris [pH 7.4], 150 mM NaCl, 5 mMEDTA [pH 8.0], 10 mM NaF, 1 mM phenylmethylsulfonylfluo-ride, aprotinin at 10 �g/mL, leupeptin at 1 �g/mL, and 500 �Msodium orthovanadate), and incubated for 30 minutes on ice.Insoluble material was removed from the lysates by centrifuga-tion at 10 000g for 15 minutes at 4 °C. For immunoprecipitation,we incubated 1 mg of protein lysate with 1 �g of an anti-PDGFR-� (Santa Cruz Biotechnology, Santa Cruz, CA), 1�g ofan anti-PDGFR-� antibody (Santa Cruz Biotechnology), 1 �g ofan anti-c-Kit antibody (Lab Vision-NeoMarkers, Fremont, CA),or 1 mg of immunoglobulin G as a negative control (data notshown) overnight at 4 °C. We then added 20 �L of 10%(vol/vol) protein A–agarose beads (Santa Cruz Biotechnology)to each reaction and incubated the mixture, with gentle rocking,for 2 hours at 4 °C. The immune complexes were then collectedby brief centrifugation (i.e., 500g for 2 minutes) and washedtwice with lysis buffer followed by a single wash with water.Precipitates were resuspended in 20 �L of 2 Tris–glycine–sodium dodecyl sulfate (SDS) buffer (Invitrogen, Carlsbad, CA).The lysates and immunoprecipitates were separated by SDS–polyacrylamide gel electrophoresis, and the separated proteinswere electrophoretically transferred to nitrocellulose membranes(Schleicher & Schuell). Where indicated, the gels were electro-phoresed under nonreducing conditions (i.e., the gel and runningbuffer did not contain SDS). The membranes were incubated in5% powdered milk in Tris-buffered saline/Tween-20 (TBS–Tween; 20 mM Tris–HCl [pH 7.4], 150 mM NaCl, and 0.5%Tween-20) to block nonspecific antibody binding and then in-cubated at room temperature for 2 hours or at 4 °C overnight in
Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004 ARTICLES 47
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

the same blocking solution to which one of the following anti-bodies was added (at a 1:1000 dilution, except where indicated):anti-phospho-tyrosine (pTy) antibody, anti-PDGFR-�or anti-PDGFR-� antibody, anti-caspase-3 antibody, anti-VEGF anti-body, and anti-Flk-1 antibody (1:500 dilution) (all from SantaCruz Biotechnology); anti-c-Kit antibody (DAKO, Carpinteria,CA); anti-poly(ADP-ribose) polymerase (PARP) antibody(Pharmingen, San Diego, CA); anti-Akt antibody, anti-phos-pho-Akt antibody, anti-extracellular signal-regulated kinase(ERK)1/2 antibody, and anti-phospho-ERK1/2 antibody (allfrom Cell Signaling Technology, Beverly, MA); or anti-actinantibody (1:2000 dilution) (Oncogene Research Products,Darmstadt, Germany). The membranes were washed with TBS–Tween and incubated with horseradish peroxidase (HRP)–con-jugated secondary antibodies (1:2000 dilution; Santa Cruz Bio-technology) for 1 hour at room temperature. Bound antibodieswere detected by chemiluminescence with the use of a LumiGlodetection system (Kirkegaard and Perry Laboratories, Gaithers-burg, MD).
We measured the concentration of VEGF in conditionedmedium obtained from 2 105 cells that were incubated for 24hours at 80% confluence in 12-well plates containing completemedium (27). The medium was collected and centrifuged at1000g for 10 minutes, and the supernatants were stored at80 °C. The concentration of VEGF protein was measuredusing enzyme-linked immunosorbent assay Quantikine kits(R&D Systems) with a mouse monoclonal antibody againsthuman VEGF (R&D Systems), according to the manufacturer’sinstructions.
Xenograft Tumor Study
Subconfluent SMS-KCNR cells were harvested by trypsiniza-tion and resuspended in Hank’s balanced salt solution at 4 107
cells/mL. We injected 2 106 cells in 50 �L of Hank’sbalanced salt solution into the left gastrocnemii of 5-week-oldfemale SCID mice (Taconic, Germantown, NY). One week afterthe injections, the mice (10 mice in each group) were randomlyassigned to receive either imatinib at 50 mg/kg in PBS, imatinibat 100 mg/kg in PBS, or vehicle (PBS) alone every 12 hours for14 days by oral gavage. We measured the dimensions of theresulting tumors at 2 weeks after the initiation of injections withthe use of a digital caliper and determined tumor volume (V) byusing the formula V � �/6 2a b, where a is the shorterdiameter and b is the longer diameter, as described previously(29). All mice were killed by asphyxiation with CO2, and theirtumors were excised and immediately frozen at 80 °C. Thesexenograft studies were approved by the Animal Care and UseCommittee of the National Cancer Institute, and all animaltreatments, including their housing, were in accordance withinstitutional guidelines and as described in protocol numberPB-023.
Statistical Analysis
A one-way analysis of variance (ANOVA) was performed ondata for SMS-KCNR and SH-SY5Y cell lines treated with SCF,PDGF-BB, and all combinations of the two factors. Residualswere examined for normality and homogeneity of variance.Because the P value for the ANOVA F statistic was less than.001, we calculated the 95% confidence intervals (CIs) for thedifferences between the groups using the post-test Bonferroni
multiple comparisons method to reduce the overall chance of atype I error (30). We analyzed xenograft tumor volume data byANOVA with the use of Prism 3.0 software (GraphPad Soft-ware, San Diego, CA). Tumor volumes were compared using apost-test Bonferroni comparison of groups to reduce the overallchance of a type I error (30). We used Student’s unpaired t testsfor the other statistical analyses. All statistical analyses weretwo-sided, and results were considered to be statistically signif-icant at P�.05.
RESULTS
Effect of Imatinib on the Growth of Neuroblastoma CellLines
We studied the effects of imatinib on the growth of sevenneuroblastoma cell lines: five cell lines [SMS-KCNR, NGP,SK-N-BE2, LAN-1, and LAN-5] had amplified N-myc genesand expressed N-Myc protein, one cell line (SH-SY5Y) ex-pressed N-Myc protein but did not have an amplified N-mycgene, and one cell line (SK-N-AS) neither expressed N-Mycprotein nor had an amplified N-myc gene (Table 1). We treatedthe neuroblastoma cells with 0–15 �M imatinib for 4 days andused trypan blue dye exclusion to determine cell viability at theend of the treatment period. All of the neuroblastoma cells linestreated with imatinib displayed a concentration-dependent de-crease in cell viability (Fig. 1, A). The concentration of imatinibthat was associated with 50% inhibition of growth of the neu-roblastoma cell lines (biologic IC50) ranged from 9 to 13 �M(Fig. 1, A). We also used the MTT assay to assess relative cellnumber and found that the biologic IC50 for imatinib in theSMS-KCNR cell line was 10 �M, identical to the IC50 in thesecells as determined by using trypan blue dye exclusion, buthigher than the IC50 for imatinib in CCL-243 cells (IC50 � 0.1�M) (Fig. 1, A, inset graph). SMS-KCNR and SH-SY5Y cellstreated with 10 �M imatinib displayed morphologic changes,such as becoming rounded, denser, more refractive, and de-tached, which were suggestive of the induction of cell death(Fig. 1, B). However, the growth of NIH3T3 cells was inhibitedby only 25% when cultured in 10 �M imatinib (Fig. 1, A), andsuch treatment did not affect the morphologic appearance ofNIH3T3 cells (Fig. 1, B).
Effect of Imatinib on Apoptosis in Neuroblastoma Cells
We next investigated one possible mechanism by whichimatinib might inhibit the growth of neuroblastomas. Because
Table 1. Expression of c-Kit, platelet-derived growth factor receptor(PDGFR)-�, and PDGFR-� in neuroblastoma cell lines*
Cell linestested
N-mycamplification/
expression c-Kit PDGFR PDGFR Ref.
SMS-KCNR �/� � � (23)NGP �/� � � (24)SK-N-BE2 �/� � � (25)LAN-1 �/� � � (24)LAN-5 �/� � � (24)SH-SY5Y /� � � � (24)SK-N-SH / � � (25)
*Expression of the receptors was analyzed by immunoprecipitation and west-ern blotting. � � detected; � not detected.
48 ARTICLES Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

we previously showed that Ewing’s sarcoma cells treated withimatinib undergo apoptosis (IC50 � 10–12 �M) (11), we eval-uated the effect of imatinib on the induction of apoptosis inneuroblastoma cells. SMS-KCNR cell cultures treated for 48hours with 15 �M imatinib (the concentration of imatinib thatwas associated with 80% inhibition of growth of the SMS-KCNR cells) and stained with Hoechst 33342 contained apopto-tic cells that displayed nuclear fragmentation and condensation,
whereas cells treated with 1 �M imatinib did not (Fig. 2, A).Similar results were found for the six other neuroblastoma celllines treated with imatinib (data not shown).
Fang et al. (31) reported that imatinib-induced apoptosis inhuman leukemia cells is mediated by activation of caspases,cysteine proteases that are key mediators of apoptosis. Proteo-lytic cleavage of caspase-3 leads to its activation and ability toactivate other caspases and degrade target proteins such asPARP. We therefore examined the effect of imatinib on caspaseactivation. SMS-KCNR cells displayed cleaved caspase-3 after18 hours of treatment with 15 �M imatinib (Fig. 2, B). We alsofound that SMS-KCNR cells displayed cleavage of PARP, asubstrate of caspase-3, after 24 hours of treatment with 15 �Mimatinib (Fig. 2, C). These data suggest that imatinib inducesapoptosis via activation of the caspase cascade.
Effect of Imatinib on PDGFR and c-Kit Phosphorylation
To address the pharmacologic effect of imatinib on PDGFRand c-Kit, we examined PDGFR and c-Kit expression in theseven neuroblastoma cell lines and the effect of imatinib onligand-induced PDGFR and c-Kit phosphorylation in three rep-resentative cell lines. PDGFR-� and c-Kit were expressed in allseven neuroblastoma cell lines, whereas PDGFR-� was ex-pressed only in SH-SY5Y cells (Table 1). In SMS-KCNR,SH-SY5Y, and SK-N-BE2 cells cultured in serum-free media,the basal level of PDGFR-� and c-Kit phosphorylation were, atmost, barely detectable (Fig. 3, A). Cells pretreated with
Fig. 1. Effect of imatinib on the growth of neuroblastoma cell lines. A) Dose-dependent inhibition of the growth of neuroblastoma cell lines by imatinib.Seven neuroblastoma cell lines (solid lines) and the control cell line NIH3T3(dashed line) were plated at 1 105 cells per well on six-well plates andincubated for 4 days with imatinib at the indicated concentrations. Viable cellswere counted by trypan blue exclusion and plotted as a percentage of the valueobtained for medium-treated control neuroblastoma cells. Each point representsthe mean value for each of the three wells. Similar results were obtained in threeseparate experiments. Inset: Human chronic myelogenous leukemia CCL-243(K562) cells and human neuroblastoma SMS-KCNR cells were plated at 5 103
cells per well in a 96-well microtiter plate and incubated for 4 days withimatinib at the indicated concentrations. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess relative cellnumbers, which were plotted as a percentage of cells treated with controlmedium. Each point is the mean for six replicate wells and is representative ofone of two independent experiments performed. B) Representative morphologicchanges observed in SMS-KCNR, SH-SY5Y, and NIH3T3 cells after 4 days oftreatment with 10 �M imatinib or media control (Control). Scale bar � 50 �m.
Fig. 2. Effect of imatinib on apoptosis in neuroblastoma cells. A) SMS-KCNRcells seeded on 96-well plates were treated with media control (0 �M) or theindicated concentration of imatinib for 48 hours, stained with Hoechst 33342 for30 minutes at 37 °C in the dark, and observed with a fluorescence microscope.Scale bar � 50 �m. B and C) SMS-KCNR cells (2 106) were plated on100-mm dishes, treated with 15 �M imatinib for the indicated times, and thenused to make protein lysates. The protein lysates were resolved by electrophore-sis and immunoblotted with antibodies to caspase-3, poly(ADP-ribose)-polymerase (PARP), and actin. Active (i.e., cleaved) caspase-3 is indicated in Bby the two arrows.
Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004 ARTICLES 49
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

PDGF-BB (50 ng/mL) displayed an increase in PDGFR-� phos-phorylation, and cells pretreated with SCF (50 ng/mL) displayedan increase in c-Kit phosphorylation. Ligand-induced phosphor-ylation of PDGFR-� and c-Kit was inhibited in cells pretreatedwith imatinib (Fig. 3, A). The concentrations of imatinib thatcaused 50% inhibition of ligand-induced phosphorylation ofthese receptor tyrosine kinases (pharmacologic IC50) rangedfrom 0.1 to 0.5 �M (Fig. 3, B), consistent with results of aprevious report (32). The pharmacologic IC50 of imatinib forligand-treated cells cultured in media containing 10% serum wasless than 1 �M (data not shown).
We and others have shown that the phosphatidylinositol3-kinase (PI3K)/Akt and mitogen-activated protein kinase(MAPK) pathways, which are activated by phosphorylation ofreceptor tyrosine kinases, play key roles in the growth andsurvival of neuroblastoma cells (34). To assess in more detail theconsequences of imatinib inhibition of activation of a tyrosinekinase in a representative neuroblastoma cell line, we evaluated
Akt and ERK1/2 phosphorylation. We found that SMS-KCNRcells treated with imatinib had less PDGF-BB–induced Akt andERK1/2 phosphorylation than SMS-KCNR cells not treated withimatinib (Fig. 3, C). The pharmacologic IC50 of imatinib forinhibition of Akt and ERK1/2 phosphorylation in these cells wasless than 1 �M (Fig. 3, C), similar to the pharmacologic IC50 ofimatinib for inhibition of PDGFR autophosphorylation in thesecells. Imatinib treatment, at similar concentrations, also inhibitedSCF-induced Akt and ERK1/2 phosphorylation in SMS-KCNRcells (data not shown).
We next examined the effect of imatinib on the ability ofPDGF and SCF to rescue SMS-KCNR and SH-SY5Y cells fromserum starvation–induced apoptosis (Fig. 3, D). SMS-KCNRcells cultured in serum-free medium had a statistically signifi-cantly higher percentage of apoptotic cells than SMS-KCNRcells cultured in medium containing 10% serum (50.2% versus10.3%; difference � 39.9%, 95% CI � 33.9% to 46.0%;P�.001). Similarly, SH-SY5Y cells cultured in serum-free me-
Fig. 3. Effect of imatinib on c-Kit and platelet-derived growth factor receptor(PDGFR) phosphorylation. A and B) Neuroblastoma SMS-KCNR, SH-SY5Y,and SK-N-BE2 cells (2 106) were plated on 100-mm dishes, incubated for 24hours in medium containing 10% fetal calf serum, and then incubated in mediumcontaining 0.1% fetal calf serum for 6 hours (i.e., serum starvation conditions).The cells were then incubated for an additional 30 minutes in the absence (–) orpresence (�) of imatinib at 1 �M (A) or at the indicated concentrations (B),followed by treatment with either SCF at 50 ng/mL, 50 ng/mL PDGF-BB, ormedia control for 5 minutes. Protein lysates were then prepared, and 1 mg ofeach lysate was immunoprecipitated (IP) with either anti-c-Kit antibody oranti-PDGFR-� antibody, resolved by sodium dodecyl sulfate–polyacrylamidegel electrophoresis, and immunoblotted (IB) with an anti-phospho-tyrosine an-tibody (pTy), an anti-c-Kit antibody, or an anti-PDGFR-� antibody. C) Afterserum starvation, cells were pre-incubated for 30 minutes with imatinib at the
indicated concentrations or media control, followed by treatment with 50 ng/mLPDGF-BB for 5 minutes. Protein lysates were prepared, and 40 �g of each lysatewas resolved by electrophoresis, transferred to membranes, and probed with theindicated antibodies. D) SMS-KCNR cells (solid bars) and SH-SY5Y cells(open bars) (1 106) were plated on 60-mm dishes. The medium was changedto 0.1% serum-containing medium for 24 hours, and the cells were pre-incubatedfor 30 minutes with either 1 �M imatinib (�) or medium (–) followed bytreatment for 24 hours in medium containing 0.1% fetal bovine serum eitheralone or supplemented with SCF (50 ng/mL) or PDGF-BB (50 ng/mL). Cellswere stained with propidium iodide and subjected to fluorescence-activated cellsorter analysis to quantify the percentage of apoptotic cells (i.e., those in thesub-G1 fraction). Each experimental point was performed in duplicate, and theexperiment was performed twice. Data are presented as the mean value (n � 4),and error bars represent 95% confidence intervals.
50 ARTICLES Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021
dia had a statistically significantly higher percentage of apopto-tic cells than SH-SY5Y cells cultured in media containing 10%serum (45.6% versus 6.6%; difference � 39.0%, 95% CI �32.7% to 45.3%; P�.001). SMS-KCNR cells cultured in thepresence of SCF, PDGF-BB, or SCF and PDGF-BB combinedhad statistically significantly lower percentages of apoptoticcells than SMS-KCNR cells cultured in serum-free media (SCFversus serum-free media: 36.9% versus 50.2%; difference �13.3%, 95% CI � 7.3% to 19.4%; P�.001; PDGF-BB versusserum-free media: 32.5% versus 50.2%; difference � 17.7%,95% CI � 11.7 % to 23.8%; P�.001; SCF and PDGF-BB versusserum-free media: 25.9% versus 50.2%; difference � 24.3%,95% CI � 18.3% to 30.4%; P�.001). Similarly, SH-SY5Y cellscultured in the presence of SCF, PDGF-BB, or SCF andPDGF-BB combined had statistically significantly lower per-centages of apoptotic cells than SH-SY5Y cells cultured inserum-free media (SCF versus serum-free media: 34.5% versus45.6%; difference � 11.1%, 95% CI � 4.7% to 17.4%; P�.001;PDGF-BB versus serum-free media: 27.5% versus 45.6%; dif-ference � 18.1%, 95% CI � 11.8% to 24.4%; P�.001; SCF andPDGF-BB versus serum-free media: 19.2% versus 45.6%; dif-ference � 26.4%, 95% CI � 20.1% to 32.7%; P�.001). How-ever, SMS-KCNR cells cultured in the presence of SCF, PDGF-BB, and 15 �M imatinib had a statistically significantly higherpercentage of apoptotic cells than SMS-KCNR cells cultured inthe presence of SCF and PDGF-BB (48.2% versus 25.9%;difference � 22.3%, 95% CI � 13.7% to 31%; P � .004) but didnot have a higher percentage than SMS-KCNR cells cultured inserum-free media (48.2% versus 50.2%; difference � 2.0%;95% CI � 1.3% to 2.7%; P � .39). Similarly, SH-SY5Y cellscultured in the presence of SCF, PDGF-BB, and 15 �M imatinibhad a statistically significantly higher percentage of apoptoticcells compared with SH-SY5Y cells cultured in the presence ofSCF and PDGF-BB (40.9% versus 19.2%; difference � 21.7%,95% CI � 12.1% to 31.4%; P � .006) but did have a statisticallysignificantly lower percentage of apoptotic cells than SH-SY5Ycells cultured in serum-free media (40.9% versus 46.5%; differ-ence � 5.6%, 95% CI � 2.1% to 7.2%; P � .01) (Fig. 3, D).These results suggest that the signaling initiated by eitherPDGFR or c-Kit contributes to the survival of neuroblastomacells and that imatinib blocks SCF- or PDGF-BB–induced sur-vival of neuroblastoma cells.
Effect of Imatinib on VEGF Expression in NeuroblastomaCells
Despite the ability of PDGFR and c-Kit to promote thesurvival of some neuroblastoma cells (Fig. 3, D), there was adifference between the biologic IC50 (9–13 �M) of imatinib andthe pharmacologic IC50 (0.1–0.5 �M) of imatinib. The differ-ence between the biologic IC50 (9–13 �M) and the pharmaco-logic IC50 (0.1–0.5 �M) of imatinib suggests the presence ofadditional mechanism(s) by which imatinib treatment mightaffect of the growth and survival of neuroblastoma cells. Resultsof a recent study (35) suggest that imatinib inhibits VEGF-stimulated phosphorylation of Flk-1 at an IC50 of 11 �M. Inaddition, VEGF and Flk-1 are co-expressed in many neuroblas-toma cells lines and tumors (17), suggesting the possible pres-ence of a VEGF/Flk-1 autocrine growth loop. For these reasons,we evaluated the effect of imatinib on Flk-1 phosphorylation andVEGF expression in several neuroblastoma cell lines that had
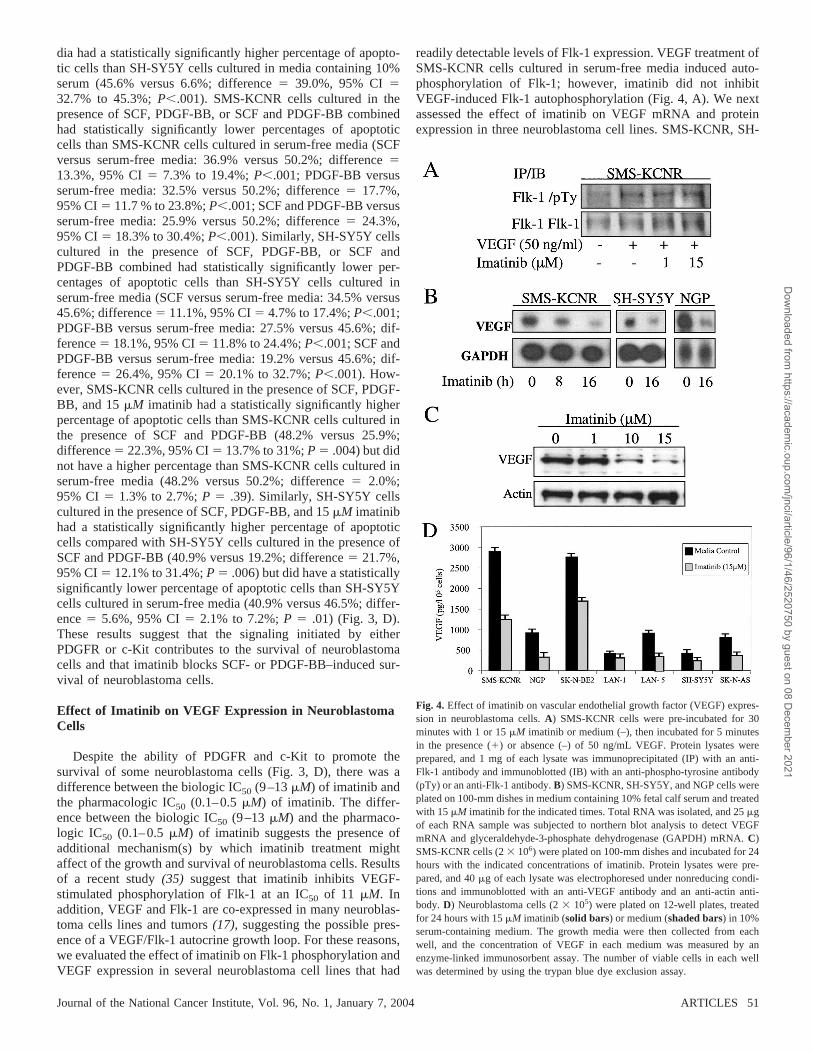
readily detectable levels of Flk-1 expression. VEGF treatment ofSMS-KCNR cells cultured in serum-free media induced auto-phosphorylation of Flk-1; however, imatinib did not inhibitVEGF-induced Flk-1 autophosphorylation (Fig. 4, A). We nextassessed the effect of imatinib on VEGF mRNA and proteinexpression in three neuroblastoma cell lines. SMS-KCNR, SH-
Fig. 4. Effect of imatinib on vascular endothelial growth factor (VEGF) expres-sion in neuroblastoma cells. A) SMS-KCNR cells were pre-incubated for 30minutes with 1 or 15 �M imatinib or medium (–), then incubated for 5 minutesin the presence (�) or absence (–) of 50 ng/mL VEGF. Protein lysates wereprepared, and 1 mg of each lysate was immunoprecipitated (IP) with an anti-Flk-1 antibody and immunoblotted (IB) with an anti-phospho-tyrosine antibody(pTy) or an anti-Flk-1 antibody. B) SMS-KCNR, SH-SY5Y, and NGP cells wereplated on 100-mm dishes in medium containing 10% fetal calf serum and treatedwith 15 �M imatinib for the indicated times. Total RNA was isolated, and 25 �gof each RNA sample was subjected to northern blot analysis to detect VEGFmRNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. C)SMS-KCNR cells (2 106) were plated on 100-mm dishes and incubated for 24hours with the indicated concentrations of imatinib. Protein lysates were pre-pared, and 40 �g of each lysate was electrophoresed under nonreducing condi-tions and immunoblotted with an anti-VEGF antibody and an anti-actin anti-body. D) Neuroblastoma cells (2 105) were plated on 12-well plates, treatedfor 24 hours with 15 �M imatinib (solid bars) or medium (shaded bars) in 10%serum-containing medium. The growth media were then collected from eachwell, and the concentration of VEGF in each medium was measured by anenzyme-linked immunosorbent assay. The number of viable cells in each wellwas determined by using the trypan blue dye exclusion assay.
Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004 ARTICLES 51
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021
SY5Y, and NGP cells treated with 15 �M imatinib had lowerlevels of VEGF mRNA and protein (Fig. 4, B and C, respec-tively) than untreated cells. The inhibitory effect of imatinibon VEGF protein expression in SMS-KCNR cells wasconcentration-dependent (Fig. 4, C), and the concentration ofimatinib that was associated with a 50% reduction in VEGFwas similar to the biologic IC50 for imatinib in those cells. Inaddition, culture media collected from each of the sevenneuroblastoma cell lines treated with 15 �M imatinib hadlower concentrations of VEGF than culture media collectedfrom the corresponding untreated cells (Fig. 4, D). BecauseVEGF has been shown to prevent neural cells from undergo-ing apoptosis (36), we examined whether VEGF promotes thegrowth and survival of neuroblastoma cells. SMS-KCNR andSH-SY5Y cells treated with VEGF still underwent serumstarvation–induced apoptosis (data not shown). In addition,incubation of SMS-KCNR or SH-SY5Y cells with an anti-VEGF blocking antibody that would neutralize endogenousVEGF produced by cells did not inhibit cell growth (data notshown). These results suggest that it is unlikely that VEGFcontributes to in vitro growth and survival of neuroblastomacells.
Effect of Imatinib on Neuroblastoma Tumor Growth InVivo
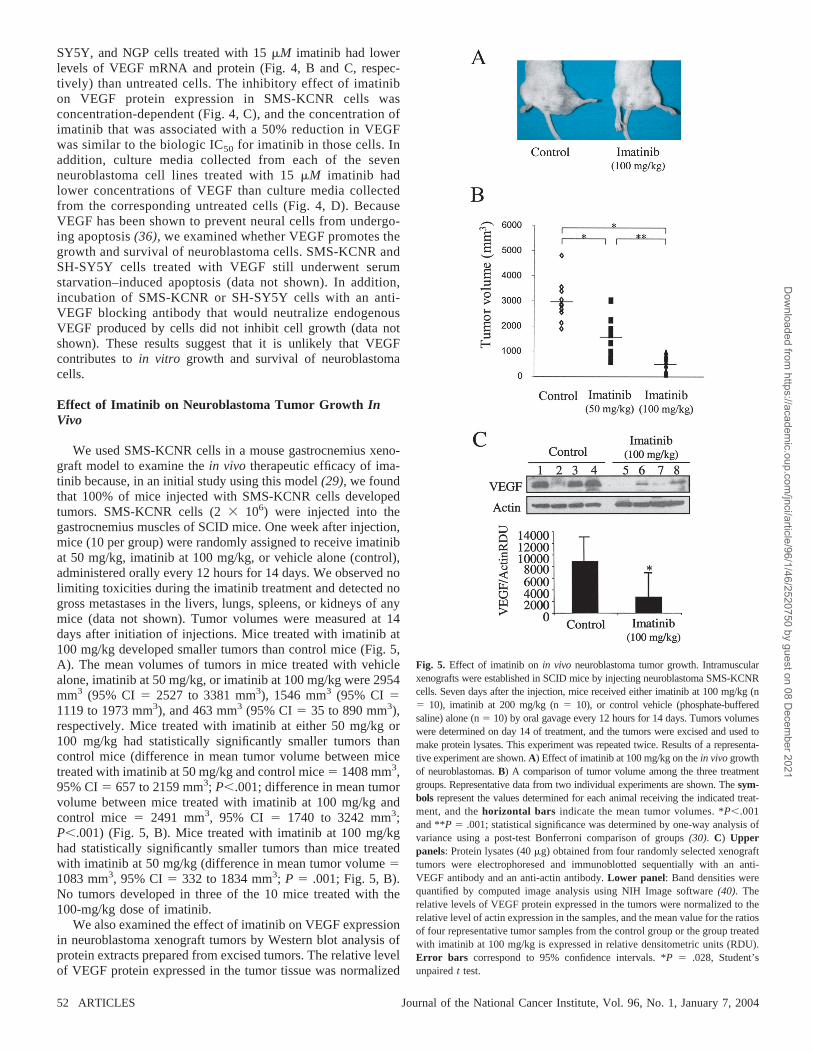
We used SMS-KCNR cells in a mouse gastrocnemius xeno-graft model to examine the in vivo therapeutic efficacy of ima-tinib because, in an initial study using this model (29), we foundthat 100% of mice injected with SMS-KCNR cells developedtumors. SMS-KCNR cells (2 106) were injected into thegastrocnemius muscles of SCID mice. One week after injection,mice (10 per group) were randomly assigned to receive imatinibat 50 mg/kg, imatinib at 100 mg/kg, or vehicle alone (control),administered orally every 12 hours for 14 days. We observed nolimiting toxicities during the imatinib treatment and detected nogross metastases in the livers, lungs, spleens, or kidneys of anymice (data not shown). Tumor volumes were measured at 14days after initiation of injections. Mice treated with imatinib at100 mg/kg developed smaller tumors than control mice (Fig. 5,A). The mean volumes of tumors in mice treated with vehiclealone, imatinib at 50 mg/kg, or imatinib at 100 mg/kg were 2954mm3 (95% CI � 2527 to 3381 mm3), 1546 mm3 (95% CI �1119 to 1973 mm3), and 463 mm3 (95% CI � 35 to 890 mm3),respectively. Mice treated with imatinib at either 50 mg/kg or100 mg/kg had statistically significantly smaller tumors thancontrol mice (difference in mean tumor volume between micetreated with imatinib at 50 mg/kg and control mice � 1408 mm3,95% CI � 657 to 2159 mm3; P�.001; difference in mean tumorvolume between mice treated with imatinib at 100 mg/kg andcontrol mice � 2491 mm3, 95% CI � 1740 to 3242 mm3;P�.001) (Fig. 5, B). Mice treated with imatinib at 100 mg/kghad statistically significantly smaller tumors than mice treatedwith imatinib at 50 mg/kg (difference in mean tumor volume �1083 mm3, 95% CI � 332 to 1834 mm3; P � .001; Fig. 5, B).No tumors developed in three of the 10 mice treated with the100-mg/kg dose of imatinib.
We also examined the effect of imatinib on VEGF expressionin neuroblastoma xenograft tumors by Western blot analysis ofprotein extracts prepared from excised tumors. The relative levelof VEGF protein expressed in the tumor tissue was normalized
Fig. 5. Effect of imatinib on in vivo neuroblastoma tumor growth. Intramuscularxenografts were established in SCID mice by injecting neuroblastoma SMS-KCNRcells. Seven days after the injection, mice received either imatinib at 100 mg/kg (n� 10), imatinib at 200 mg/kg (n � 10), or control vehicle (phosphate-bufferedsaline) alone (n � 10) by oral gavage every 12 hours for 14 days. Tumors volumeswere determined on day 14 of treatment, and the tumors were excised and used tomake protein lysates. This experiment was repeated twice. Results of a representa-tive experiment are shown. A) Effect of imatinib at 100 mg/kg on the in vivo growthof neuroblastomas. B) A comparison of tumor volume among the three treatmentgroups. Representative data from two individual experiments are shown. The sym-bols represent the values determined for each animal receiving the indicated treat-ment, and the horizontal bars indicate the mean tumor volumes. *P�.001and **P � .001; statistical significance was determined by one-way analysis ofvariance using a post-test Bonferroni comparison of groups (30). C) Upperpanels: Protein lysates (40 �g) obtained from four randomly selected xenografttumors were electrophoresed and immunoblotted sequentially with an anti-VEGF antibody and an anti-actin antibody. Lower panel: Band densities werequantified by computed image analysis using NIH Image software (40). Therelative levels of VEGF protein expressed in the tumors were normalized to therelative level of actin expression in the samples, and the mean value for the ratiosof four representative tumor samples from the control group or the group treatedwith imatinib at 100 mg/kg is expressed in relative densitometric units (RDU).Error bars correspond to 95% confidence intervals. *P � .028, Student’sunpaired t test.
52 ARTICLES Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

to the relative level of actin expressed in the same sample, andthe mean values for the ratios of four representative tumorsamples from the control group and the group treated withimatinib at 100 mg/kg were expressed in relative densitometricunits (RDU) and graphed in the lower panel of Fig. 5, C. Therelative levels of VEGF to actin in tumors from mice treatedwith imatinib at 100 mg/kg were statistically significantly lowerthan the relative levels of VEGF to actin in tumors from controlmice (mean ratio of VEGF to actin for imatinib-treated mice �2560 RDU; mean ratio of VEGF to actin for control mice �8673 RDU; difference � 6113 RDU, 95% CI � 193 to 12 419RDU; P � .028; Fig. 5, C).
DISCUSSION
Results of recent clinical studies (7) have suggested thepromising therapeutic impact of imatinib in the treatment ofCML and GIST. In the present study, we found that imatinibtreatment was associated with the inhibition of in vitro growth ofall neuroblastoma cell lines tested and of in vivo neuroblastomatumor growth. The biologic IC50 for imatinib in neuroblastomacells was higher than that for imatinib in both the CML cell lineused in this study and in other CML cell lines (36). The biologicIC50s for imatinib in neuroblastoma cell lines are two- to three-fold higher than the serum imatinib concentration achieved inearly clinical trials (4.6 �M) (6). However, it is not clear whetherthe maximally tolerated doses of imatinib were achieved in thosetrials. In addition to the known effects of imatinib on c-Abl,c-Kit, and PDGFR kinase activities (36), we propose that theinhibitory effect of imatinib on the in vivo growth of neuroblas-toma cells may also be through the decreased expression ofneuroblastoma-derived VEGF. We found that imatinib was cy-totoxic in all seven neuroblastoma cell lines tested and that thecytotoxic effect was associated with the induction of apoptosis,mediated by PARP cleavage, which was evident by 24 hoursafter exposure to imatinib. All neuroblastoma cell lines testedexpressed both PDGFR and c-Kit, and both receptors werefunctional because pretreatment with their respective ligands,PDGF and SCF, rescued neuroblastoma cells from serum star-vation–induced apoptosis. Our finding that imatinib treatmentwas associated with the inhibition of PDGFR and c-Kit phos-phorylation in these neuroblastoma cells at a pharmacologic IC50
of less than 0.5 �M is consistent with results from previousstudies (32). However, although the pharmacologic IC50s ofimatinib for PDGFR and c-Kit phosphorylation were in a similarrange (�0.5 �M), approximately 20-fold higher concentrationsof imatinib (9–13 �M) were required to induce apoptosis inneuroblastoma cells in vitro. This finding is consistent with ourpreviously reported observation that the biologic IC50 of ima-tinib in Ewing’s sarcoma cells (10–12 �M) (11) is much higherthan the biologic IC50s (1 �M) reported for CML and GIST cells(5).
The reason for the discrepancy between the pharmacologicIC50 for imatinib and the biologic IC50 for imatinib is unclear. Itseems likely that an additional target or targets for imatinib-mediated inhibition of in vitro growth are expressed in neuro-blastoma cells. There is precedent in the literature for suchadditional targets. Results of previous studies (10) suggest thatimatinib inhibits epithelial growth factor–dependent growth ofmouse epithelial BALB/MK cells and insulin-like growth factor-I–dependent growth of human small-cell lung cancer cells at
concentrations similar to the biologic IC50 for neuroblastomacells, even though imatinib does not directly inhibit the insulin-like growth factor or epithelial growth factor receptors (4).Taken together, these observations suggest that imatinib mighthave other effects on growth factor–induced intracellular signal-ing pathways.
To examine the possible mechanism by which neuroblastomacells are susceptible to imatinib-mediated apoptosis at 15 �M,we assessed phosphorylation of the downstream mediators Aktand ERK1/2, which play key roles in the growth and survivalsignaling pathway in neuroblastoma cells (34). We found thatboth PDGF and SCF were individually able to induce phosphor-ylation of Akt and ERK1/2 in neuroblastoma cells cultured inserum-starved conditions and that treatment with 1 �M imatinibinhibited this phosphorylation. However, imatinib treatment didnot affect the basal levels of Akt and ERK1/2 phosphorylation inneuroblastoma cells cultured in media containing 10% serum.Therefore, in 10% serum-containing media, other growth factorsmay also activate the PI3K/Akt and MAPK pathways and, thus,contribute to the growth and survival of neuroblastoma cells. Bycontrast with the gain-of-function mutations in c-Abl and c-Kit,other serum factors may aid in the support of the unregulatedgrowth and survival of neuroblastoma cells.
We found that treatment with imatinib was associated withthe suppression of growth of the neuroblastoma xenograft tu-mors in a mouse model. Although we did not measure plasmalevels of imatinib in the treated mice, a dosing regimen similarto ours resulted in an average plasma level of 11.8 �M imatinib(37), which is similar to the biologic IC50 that we obtained forneuroblastoma cell lines in vitro. Our in vitro data showing thatimatinib inhibits VEGF production in neuroblastoma cells sug-gest that imatinib may have anti-angiogenic activity in vivo inaddition to its cytotoxic effects. VEGF is secreted by neuroblas-toma cells (16–18) and, as such, is thought to play a role in theangiogenic environment of the growing neuroblastoma tumor.Results of in vitro studies have shown that the neuroblastomacell–conditioned media-stimulated growth of endothelial cellscan be inhibited by treatment with a monoclonal antibody toVEGF (20). In addition, results of recent animal studies (22)showed that blockade of VEGF function by an anti-VEGFmonoclonal antibody or expression of an Flk-1 gene lacking thekinase domain, which interferes with Flk-1 signaling, was asso-ciated with the inhibition of angiogenesis and tumor growth in aneuroblastoma xenograft model, suggesting the important role ofVEGF in development of neuroblastoma. Our in vitro datarevealed that treatment with imatinib was associated with theinhibition of VEGF expression and secretion in neuroblastomacells. Our in vivo data suggest that imatinib also inhibits VEGFexpression in neuroblastoma xenografts. The decrease in VEGFexpression was associated with suppression of in vivo tumorgrowth. These results suggest that imatinib may have anti-angiogenic potential via its effects on the expression of neuro-blastoma cell–derived VEGF, which is known to stimulate pro-liferation of endothelial cells (21).
The potential anti-angiogenic role of imatinib is not likely tobe limited to its effects on VEGF expression in tumor cellsbecause imatinib may also inhibit the vascular formation activ-ities of c-Kit and PDGFR (35) that are expressed on vascularendothelial cells. Results of a recent in vivo study showed thatimatinib, especially in combination with paclitaxel, inhibits thegrowth of prostate cancer bone metastases and tumor angiogen-
Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004 ARTICLES 53
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

esis by blocking PDGFR (38). Another study (39) reported thatSCF secreted by colon tumor cells induces in vitro and in vivoangiogenesis. Therefore, it is possible that in a tumor environ-ment imatinib could inhibit angiogenesis by inhibiting tumorcell–secreted PDGF- or SCF-stimulated phosphorylation ofPDGFR and c-Kit on endothelial cells.
VEGF as a potential target for imatinib-associated apoptosisof neuroblastoma cells is intriguing for several reasons. First,recent evidence (36) suggests that VEGF promotes the growthand survival of neural cells. Second, VEGF and its receptorFlk-1 are co-expressed in neuroblastoma cell lines and tumorspecimens (17). Together, these findings suggest that a VEGF/Flk-1 autocrine growth loop exists in neuroblastoma cells. Be-cause the concentrations of imatinib required to inhibit VEGFexpression were similar to the biologic IC50 of imatinib inneuroblastoma cells, we hypothesized that VEGF inhibitionmight be involved in imatinib inhibition of in vitro growth ofneuroblastoma cells. However, exogenously added VEGF didnot substantially promote the proliferation or survival of serum-starved neuroblastoma cells, nor did a VEGF-blocking antibodyinhibit the growth of neuroblastoma cells. Thus, although adecrease in VEGF expression may be associated with the inhi-bition of in vivo growth of neuroblastoma cells, it is unlikely tobe the sole mechanism by which imatinib inhibits in vitrogrowth of neuroblastoma cells.
In summary, our results suggest that the mechanism by whichimatinib suppresses the growth of neuroblastoma may includethe inhibition of VEGF expression in addition to the inhibitionof PDGFR and c-Kit phosphorylation. Our data showing thatimatinib inhibited the in vivo growth of neuroblastoma cellssuggest that imatinib should be considered for the treatment ofneuroblastoma. Evidence for the potential of imatinib to de-crease expression of VEGF in neuroblastoma cells also suggeststhat it may act against a broader range of tumors than initiallypredicted.
REFERENCES
(1) Crist WM, Kun LE. Common solid tumors of childhood. N Engl J Med1991;324:461–71.
(2) Brodeur GM, Maris JM. Neuroblastoma. In: Pizzo PA, Poplack DG,editors. Principles and practice of pediatric oncology. 4th ed. Philadelphia(PA): Lippincott; 2002. p. 895–937.
(3) Brodeur GM, Nakagawara A. Molecular basis of clinical heterogeneity inneuroblastoma. Am J Pediatr Hematol Oncol 1992;14:111–6.
(4) Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al.Effects of a selective inhibitor of the Abl tyrosine kinase on the growth ofBcr-Abl positive cells. Nat Med 1996;2:561–6.
(5) Buchdunger E, O’Reilly T, Wood J. Pharmacology of imatinib (STI571).Eur J Cancer 2002;38 Suppl 5:S28–36.
(6) Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al.Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinasein chronic myeloid leukemia. N Engl J Med 2001;344:1031–7.
(7) Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P,Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patientwith a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344:1052–6.
(8) Kilic T, Alberta JA, Zdunek PR. Intracranial inhibition of platelet-derivedgrowth factor-mediated glioblastoma growth by an orally active kinaseinhibitor of the 2-phenylaminopyrimidine class. Cancer Res 2000;60:5143–50.
(9) Sjoblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P, Buchdunger E,et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by theplatelet-derived growth factor receptor antagonist STI571 through induc-tion of apoptosis. Cancer Res 2001;61:5778–83.
(10) Krystal GW, Honsawek S, Litz J, Buchdunger E. The selective tyrosinekinase inhibitor STI571 inhibits small cell lung cancer growth. Clin CancerRes 2000;6:3319–26.
(11) Merchant MS, Woo CW, Mackall CL, Thiele CJ. Potential use of imatinibin Ewing’s sarcoma: evidence for in vitro and in vivo activity. J NatlCancer Inst 2002;94:1673–9.
(12) Cohen PS, Chan JP, Lipkunskaya M, Biedler JL, Seeger RC. Expression ofstem cell factor and c-kit in human neuroblastoma. Blood 1994;84:3465–72.
(13) Timeus F, Crescenzio N, Valle P, Pistamiglio P, Piglione M, Garelli E, etal. Stem cell factor suppresses apoptosis in neuroblastoma cell lines. ExpHematol 1997;25:1253–60.
(14) Matsui T, Sano K, Tsukamoto T, Ito M, Takaishi T, Nakata H, et al. Humanneuroblastoma cells express alpha and beta platelet-derived growth factorreceptors coupling with neurotrophic and chemotactic signaling. J ClinInvest 1993;92:1153–60.
(15) Folkman J. Role of angiogenesis in tumor growth and metastasis. SeminOncol 2002;29(6 Suppl 16):15–8.
(16) Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP.High-level expression of angiogenic factors is associated with advancedtumor stage in human neuroblastomas. Clin Cancer Res 2000;6:1900–8.
(17) Meister B, Grunebach F, Bautz F, Brugger W, Fink FM, Kanz L, et al.Expression of vascular endothelial growth factor (VEGF) and its receptorsin human neuroblastoma. Eur J Cancer 1999;35:445–9.
(18) Meitar D, Crawford SE, Rademaker AW, Cohn SL. Tumor angiogenesiscorrelates with metastatic disease, N-myc amplification, and poor outcomein human neuroblastoma. J Clin Oncol 1996;14:405–14.
(19) Ribatti D, Alessandri G, Vacca A, Iurlaro M, Ponzoni M. Human neuro-blastoma cells produce extracellular matrix-degrading enzymes, induceendothelial cell proliferation and are angiogenic in vivo. Int J Cancer1998;77:449–54.
(20) Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM.Expression of the neurotrophin receptor TrkA down-regulates expressionand function of angiogenic stimulators in SH-SY5Y neuroblastoma cells.Cancer Res 2002;62:1802–8.
(21) Kim ES, Serur A, Huang J, Manley CA, McCrudden KW, Frischer JS, etal. Potent VEGF blockade causes regression of coopted vessels in a modelof neuroblastoma. Proc Natl Acad Sci U S A 2002;99:11399–404.
(22) Davidoff AM, Leary MA, Ng CY, Vanin EF. Gene therapy-mediatedexpression by tumor cells of the angiogenesis inhibitor flk-1 results ininhibition of neuroblastoma growth in vivo. J Pediatr Surg 2001;36:30–6.
(23) Reynolds CP, Biedler JL, Spengler BA, Reynolds DA, Ross RA, FrenkelEP, et al. Characterization of human neuroblastoma cell lines establishedbefore and after therapy. J Natl Cancer Inst 1986;76:375–87.
(24) Brodeur GM, Green AA, Hayes FA, Williams KJ, Williams DL, TsiatisAA. Cytogenetic features of human neuroblastomas and cell lines. CancerRes 1981;41:4678–86.
(25) Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, et al.Human neuroblastomas and abnormalities of chromosomes 1 and 17.Cancer Res 1984;44:5444–9.
(26) White PS, Maris JM, Beltinger C, Sulman E, Marshall HN, Fujimori M,et al. A region of consistent deletion in neuroblastoma maps withinhuman chromosome 1p36.2-36.3. Proc Natl Acad Sci U S A 1995;92:5520 – 4.
(27) Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-mycprecedes retinoic acid-induced morphological differentiation of humanneuroblastoma. Nature 1985;313:404–6.
(28) Kim CJ, Matsuo T, Lee KH, Thiele CJ. Up-regulation of insulin-likegrowth factor-II expression is a feature of TrkA but not TrkB activation inSH-SY5Y neuroblastoma cells. Am J Pathol 1999;155:1661–70.
(29) Khanna C, Jaboin J, Drakos E, Tsokos M, Thiele CJ. Biologically relevantorthotopic neuroblastoma xenograft models: primary adrenal tumor growthand spontaneous distant metastasis. In Vivo 2002;16:77–86.
(30) Miller RG. Simultaneous statistical inference. 2nd ed. New York (NY):Springer Verlag; 1981. p. 129–271.
(31) Fang G, Kim CN, Perkins CL, Ramadevi N, Winton E, Wittmann S, et al.CGP57148B (STI-571) induces differentiation and apoptosis and sensitizesBcr-Abl-positive human leukemia cells to apoptosis due to antileukemicdrugs. Blood 2000;96:2246–53.
54 ARTICLES Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021

(32) Krystal GW. Mechanisms of resistance to imatinib (STI571) and prospectsfor combination with conventional chemotherapeutic agents. Drug Resis-tance Updates 2001;4:16–21.
(33) Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factoractivation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3�-kinase pathway. Cancer Res2002;62:6756–63.
(34) Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulatedkinases and phosphatidylinositol 3-kinase are involved in brain-derivedneurotrophic factor-mediated survival and neuritogenesis of the neuroblas-toma cell line SH-SY5Y. J Neurochem 1999;73:1409–21.
(35) Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, etal. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signaltransduction mediated by c-kit and platelet-derived growth factor receptors.J Pharmacol Exp Ther 2000;295:139–45.
(36) Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor:direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U SA 2000;97:10242–7.
(37) Wolff NC, Richardson JA, Egorin M, Ilaria RL Jr. The CNS is a sanctuaryfor leukemic cells in mice receiving imatinib mesylate for Bcr/Abl-inducedleukemia. Blood 2003;101:5010–3.
(38) Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, et al.Effects of blocking platelet-derived growth factor-receptor signaling in amouse model of experimental prostate cancer bone metastases. J NatlCancer Inst 2003;95:458–70.
(39) Attoub S, Rivat C, Rodrigues S, Van Bocxlaer S, Bedin M, Bruyneel E, etal. The c-kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy.Cancer Res 2002;62:4879–83.
(40) Vodovotz Y, Hsing A, Cook JA, Miller RW, Wink DA, Ritt DM, et al.Qualitative and quantitative analysis of DNA fragmentation using digitalimaging. Anal Biochem 1997;250:147–52.
NOTES
We thank Dr. Elisabeth Buchdunger, Novartis Pharmaceuticals, for pro-viding imatinib. We also thank Xuezhong Yang and Chan-Wook Woo forhelpful discussions and Kim Mott for editorial assistance. We thank Drs. SethSteinberg and David Liewehr for assistance on aspects of the statisticalanalysis and the members of the Cell and Molecular Biology Section,National Cancer Institute, for their thoughtful review of the studies.
Manuscript received April 9, 2003; revised October 22, 2003; acceptedNovember 6, 2003.
Journal of the National Cancer Institute, Vol. 96, No. 1, January 7, 2004 ARTICLES 55
Dow
nloaded from https://academ
ic.oup.com/jnci/article/96/1/46/2520750 by guest on 08 D
ecember 2021