documento final xxxxiv eclamc 2012 angra dos reis/rj · ces is a 17-year-old young man with...
TRANSCRIPT

DOCUMENTO FINAL XXXXIV ECLAMC 2012
ANGRA DOS REIS/RJ


X X X X I V E C L A M C ANGRA DOS REIS 2 0 1 2
D O C U M E N T O F I N A L
XXXXIV REUNION ANUAL DEL ESTUDIO COLABORATIVO
LATINOAMERICANO DE MALFORMACIONES CONGENITAS
ANGRA DOS REIS/RJ
14 a 19 de noviembre de 2012
Versión Electrónica
Marzo 2012
Rio deJaneiro

2
INDICE INTRODUCCION
Organizadores y participantes 3
Agradecimientos 5
Programa científico 6
CASOS CLINICOS 7 01 Dysmorphic syndrome, hallux valgus 8 02 Encephalocele, facial cleft, omphalocele 9 03 Triploidy 10 04 Short arms in children with Down syndrome 11 05 Kenny-Caffey syndrome 12 06 Turner syndrome and Langer mesomelic dysplasia 13 07 Skeletal dysplasia 14 08 Diastrophic dysplasia (?) 15 09 Atelosteogenesis type III 16 10 Mosaic trisomy 13 17 11 Double aneuploidy: trisomy X and trisomy 18 18 12 Mother-to-son transmission of Down syndrome 19 13 19p13 deletion 20 14 Nager syndrome 21 15 Goldenhar syndrome 22 16 Dysmorphic syndrome (?) 23 17 Metaphyseal dysplasia, metatarsus varus 24 18 Verma-Naumoff syndrome 25 19 Zimmermann-Laband syndrome (?) 26 20 Femoral hypoplasia, unusual face syndrome (?) 27 21 Dysmorphic syndrome (?) 28 22 Fibrochondrogenesis 29 23 Oral-Facial_Digital spectrum disorder 30 24 Cornelia De Lange syndrome 31 25 Microdeletion 2p and inv(5) 32 26 Macrosomia, ear creases, macrocrania 33 27 Macrosomia, ear creases, maternal t13q;14q 34 28 Oculocerebrocutaneous syndrome 35 29 9p deletion and 16q duplication 36 30 OFD tipo VI (Varadi-Papp) 37

3
Organizadores y participantes
ORGANIZADOR E COMISSÃO ORGANIZADORA Iêda Maria Orioli Rio de Janeiro Eduardo E Castilla Rio de Janeiro PARTICIPANTES HOS NOME ORIGEM Koo Alejandra Mariona Buenos Aires A05 Alexandre Lucidi Rio de Janeiro 228 Amanda Contreras de la Cruz Castro A04 Aurea Gomes Nogueira Florianópolis Koo Belén Comas Buenos Aires F02 Braulio Jattar Senior Coro Klin Camila Melo Saavedra Santiago G19 Carolina Isaza Cali 803 César Manuel Saleme Tucumán Koo Clarice Pagani Savastano Rio de Janeiro Klin Daniela Varela Luquetti Seattle Klin Eduardo Abby Rio de janeiro Koo Eduardo E Castilla Rio de Janeiro Klin Elena Vallespín García Madrid A50 Eliana Ternes Pereira Florianópolis 220 Elisabeth Wettig Santiago Koo Fernando Adrián Poletta Buenos Aires Koo Fernando Regla Vargas Rio de Janeiro Koo Flavia Martinez de Carvalho Rio de Janeiro G26 Gloria Liliana Porras Hurtado Pereira G24 Harry Pachajoa Cali Koo Iêda Maria Orioli Rio de Janeiro G11 Ignacio Zarante Bogotá Koo Jorge López-Camelo Buenos Aires Kon José Carlos Cabral de Almeida Rio de Janeiro Koo Juan Antonio Gili Buenos Aires 907 Juan Carlos Mereb El Bolsón A05 Juan Clinton Llerena Junior Rio de Janeiro Klin Julián Nevado Blanco Madrid A25 Júlio Cesar Loguercio Leite Porto Alegre 201 Julio Nazer Herrera Santiago Klin Lavinia Schüler-Faccini Porto Alegre Klin Leila Cabral de Almeida Cardoso Rio de Janeiro Koo Lucas Gimenez Buenos Aires Kon M. Michael Cohen Jr Halifax Koo Maluah Tostes de Carvalho Rio de Janeiro A39 Marcos José Burle de Aguiar Belo Horizonte 219 Maria Aurora Canessa Tapia Linares A05 Maria Auxiliadora Monteiro Villar Rio de Janeiro Koo Maria da Graça Dutra Rio de Janeiro Klin María Palomares Madrid

4
HOS NOME ORIGEM Koo Mariana Piola Buenos Aires 332 Monica Adriana Jewtuszyk Buenos Aires 416 Mónica Ermini Buenos Aires Koo Mónica Rittler Buenos Aires A05 Nathalia Krause Rio de Janeiro Klin Nidia Triay de Juarez Córdoba Kon Pablo Lapunzina Badía Madrid G22 Paula Hurtado Cali A29 Pedro Antonio Armellini Amparo 227 Pedro Pavez Basualdo Curicó Klin Pilar Guatibonza Bogotá Kon Ricardo Della Coletta Campinas Koo Ricardo Lima do Nascimento Rio de Janeiro A65 Roberta Magalhães de Leite Pinto Angra dos Reis B01 Saul Rueda Arteaga La Paz A05 Sayonara Gonzalez Rio de Janeiro Klin Silvia Castillo Taucher Santiago Klin Tábita Hünemeier Porto Alegre Klin Teresa Aravena Cerda Santiago Klin Victor M. Faúndes G. Santiago Koo Viviana Consentino Buenos Aires Koo Viviane Freitas de Castro Rio de Janeiro G19 Wilmar Saldarriaga Cali HOS= Klin: genética clínica; Koo: Coordenação, Kon: Convidado; resto: código eclamc do hospital.

5
AGRADECIMIENTOS Gracias a las instituciones:
CAPES: Processo nº 23038.006920/2012-51
CNPq: Processo nº 452556/2012-0
FAPERJ: Processo nº E-26/111.062/2012
Laboratório de Epidemiologia de Malformações Congênitas: POM/2012
UFRJ: Processo nº 23079.039869/2012-99

6
44th REUNIÃO ANUAL DO ECLAMC - 14-19 DE NOVEMBRO DE 2012 -
PORTOGALO, ANGRA DOS REIS, RJ
4a Feira 14 19:00-23:00 Recepção, jantar e apresentações
5a Feira 15 08:00-10:30 Oficina 1/3: Análise epidemiológico com a base de dados do ECLAMC 11:00-11:30 Resultados: Fendas orais na Patagônia: Microarranjos (Flávia Martínez) 11:30-12:00 Resultados: Fendas orais na Patagônia: Microarranjos customizados
(M.Palomares) 12:00-13:00 Conferência.1: Síndrome de Microdeleción 22q11.2 y Cardiopatías
Congénitas (Pablo Lapunzina) 15:00-18:30 Consultório: Casos Interessantes 1 a 12 22:00-23:00 The midwife toad (M.Michael Cohen Jr)
6a Feira 16 08:00-10:30 Oficina 2/3: Análise epidemiológico com a base de dados do ECLAMC 11:00-12:00 Conferência.2: Epimutaciones en Enfermedades con Imprinting Genómico
(Pablo Lapunzina) 12:00-13:00 Conferência.3: The Biology of Craniosynostosis (M.Michael Cohen Jr ) 15:00-18:30 Consultório: Casos Interessantes 13 a 24 22:00-23:00 Assembléia das Associações ECLAMC
Sábado 17 08:00-10:30 Oficina 3/3: Análise epidemiológico com a base de dados do ECLAMC. 11:00-12:00 Conferência.4: O Sol e os Defeitos Congênitos (Iêda M Orioli) 12:00-13:00 Conferência.5: Marcadores de Suscetibilidade as Fendas Orais (Ricardo
Coletta) 15:00-18:30 Reunião Satélite do projeto Patarrays Customizado
Domingo 18 08:00-10:30 Consultório: Casos Interessantes 25 a 34 11:00-13:00 Assuntos Operacionais 15:00-16:00 Conferência.6: Do pré-natal ao pós-molecular: cinco exemplos (José
Carlos Cabral de Almeida) 16:30-17:30 Gaúchos e Beduínos (Eduardo Castilla) 17:30-18:30 A Genética Médica Populacional - O Filme (INAGEMP) 20:00-23:00 Jantar de despedida
2a Feira 19 08:00-09:00 Café da manhã 09:00 Primeiro Ônibus para o Rio de Janeiro 15:00 Segundo Ônibus para o Rio de Janeiro
OFICINA EPIDEMIOLÓGICA: Análise epidemiológico com dados da base do ECLAMC.: Conduzida por J.S.López-Camelo, F.Poletta e J.Gilli; dirigida aos contatos do ECLAMC, com dados próprios, e outros interessados, com dados gerais. Tempo total= 7,5 hs.
CASOS: 5' de apresentação e 10' de discussão, total 15' por caso.

7
CASOS CLÍNICOS No Médico País Resumo
01 Cecília Mellado CHL Dysmorphic syndrome, hallux valgus 02 Cecilia Mellado CHL Encephalocele, facial cleft, omphalocele 03 Julio Nazer CHL Triploidy 04 Nidia Triay ARG Short arms in children with Down syndrome 05 Teresa Aravena CHL Kenny-Caffey syndrome 06 Teresa Aravena CHL Turner syndrome and Langer mesomelic dysplasia 07 Silvia Castillo CHL Skeletal dysplasia 08 Silvia Castillo CHL Diastrophic dysplasia (?) 09 Silvia Castillo CHL Atelosteogenesis type III 10 Harry Pachajoa COL Mosaic trisomy 13 11 Harry Pachajoa COL Double aneuploidy: trisomy X and trisomy 18 12 Harry Pachajoa COL Mother-to-son transmission of Down syndrome 13 Elena Vallespín ESP 19p13 deletion 14 Cesar Saleme ARG Nager syndrome 15 Braulio Jattar VEN Goldenhar syndrome 16 Cesar Saleme ARG Dysmorphic syndrome (?) 17 Braulio Jattar VEN Metaphyseal dysplasia, metatarsus varus 18 Maria Auxiliadora Villar BRS Verma-Naumoff syndrome 19 Maria Auxiliadora Villar BRS Zimmermann-Laband syndrome (?) 20 Maria Auxiliadora Villar BRS Femoral hypoplasia, unusual face syndrome (?) 21 Maria Auxiliadora Villar BRS Dysmorphic syndrome (?) 22 Maria Auxiliadora Villar BRS Fibrochondrogenesis 23 Maria Auxiliadora Villar BRS Oral-Facial_Digital spectrum disorder 24 Maria Auxiliadora Villar BRS Cornelia De Lange syndrome 25 Juan Llerena Jr BRS Microdeletion 2p and inv(5) 26 Fernando Vargas BRS Macrosomia, ear creases, macrocrania 27 Fernando Vargas BRS Macrosomia, ear creases, maternal t13q;14q 28 Marcos Aguiar BRS Oculocerebrocutaneous syndrome 29 Harry Pachajoa COL 9p deletion and 16q duplication 30 Monica Rittler ARG OFD tipo VI (Varadi-Papp)
Posters No Apresenta País Resumo
01 Clarice BRS Detection of common chromosome aneuploidies in holoprosencephaly by QF-PCR
02 Ricardo BRS Cleft palate prevalence with data from the Brazilian Live Births Information System, 2006
03 Maluah BRS Ancestry analysis of normal prevalence regions for cleft lip with or without cleft palate identified
04 Renata BRS TCOF1 gene analysis in a patient with Treacher Collins syndrome
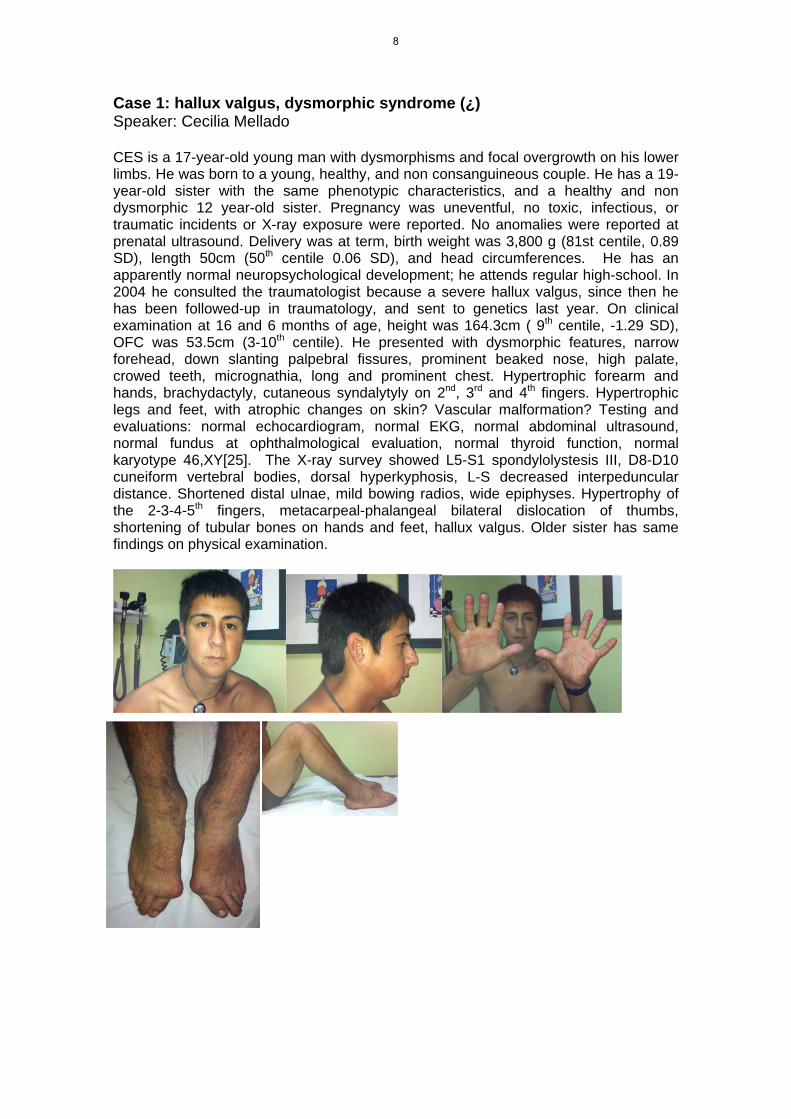
Case 1: hallux valgus, dysmorphic syndrome (¿) Speaker: Cecilia Mellado CES is a 17-year-old young man with dysmorphisms and focal overgrowth on his lower limbs. He was born to a young, healthy, and non consanguineous couple. He has a 19-year-old sister with the same phenotypic characteristics, and a healthy and non dysmorphic 12 year-old sister. Pregnancy was uneventful, no toxic, infectious, or traumatic incidents or X-ray exposure were reported. No anomalies were reported at prenatal ultrasound. Delivery was at term, birth weight was 3,800 g (81st centile, 0.89 SD), length 50cm (50th centile 0.06 SD), and head circumferences. He has an apparently normal neuropsychological development; he attends regular high-school. In 2004 he consulted the traumatologist because a severe hallux valgus, since then he has been followed-up in traumatology, and sent to genetics last year. On clinical examination at 16 and 6 months of age, height was 164.3cm ( 9th centile, -1.29 SD), OFC was 53.5cm (3-10th centile). He presented with dysmorphic features, narrow forehead, down slanting palpebral fissures, prominent beaked nose, high palate, crowed teeth, micrognathia, long and prominent chest. Hypertrophic forearm and hands, brachydactyly, cutaneous syndalytyly on 2nd, 3rd and 4th fingers. Hypertrophic legs and feet, with atrophic changes on skin? Vascular malformation? Testing and evaluations: normal echocardiogram, normal EKG, normal abdominal ultrasound, normal fundus at ophthalmological evaluation, normal thyroid function, normal karyotype 46,XY[25]. The X-ray survey showed L5-S1 spondylolystesis III, D8-D10 cuneiform vertebral bodies, dorsal hyperkyphosis, L-S decreased interpeduncular distance. Shortened distal ulnae, mild bowing radios, wide epiphyses. Hypertrophy of the 2-3-4-5th fingers, metacarpeal-phalangeal bilateral dislocation of thumbs, shortening of tubular bones on hands and feet, hallux valgus. Older sister has same findings on physical examination.
8

Case 2: Encephalocele, facial cleft, omphalocele Speaker: Cecilia Mellado This is the first child of a young, healthy and non consanguineous couple. At 14th weeks GA at prenatal ultrasound was detected an absence of cranial vault/encephalocele, and omphalocele. Prenatal karyotype was normal 46,XY. Delivery was at term by C-Section at 40 week GA. Birth weight was 3,060 g (27th centile, -0.60 DS), length 48 cm (15Th centile -1.00 SD), head circumference (24 cm)? He passed away 10 hours after he was born. On post mortem examination he presented with a big encephalocele covered by broken meningeal membranes, absence of most bones of cranial vault, bilateral microphthalmia, facial clefts more prominent on the right involving nares and eyes, left cleft palate, omphalocele, and club feet. No other testing or evaluations were done.
9

Case 3: Triploidy Speaker: Julio Nazer Male stillborn, born in January 2012, first child of non-consanguineous, mother 33 y.o., father 34, 29 week pregnancy, severe IUCR. Two episodes of metrorragia in the first trimester. Chorionic villus sampling showed 69,XXY [27] karyotype. C-section 21 wk pregnancy. Autopsy findings: macrocrania, asymmetric cranial vault, ventriculomegalia. Microretrognathia. Syndactyly hands and feet. Male genitalia. Adrenal hypoplasia. COMMENT: the karyotype is 69,XXY[25]/46,XY[2]. There were no internal malformations in this case.
10

Case 4: Short arms in children with Down syndrome Speaker: Nidia Juarez The observation that alterations neurosensopsicodesarrollo (having motor impairment leading to decrease in locomotor) is a functional problem manifested in children with Down syndrome and children with bone dysplasias and short arms, was what motivated the following study. Which sample consisted of patients (with down syndrome, and other bone dysplasias with neurological deviations and database hypotonia), clinical and cytogenetic examination, anthropometric measurements and family assessment (genealogy). It follows that these anthropometric deficiencies should be taken into account in re-educational strategies to offset these imbalances with simple equipment like a U-shaped cushion and thus stimulate neurodevelopment.
11

Case 5: Kenny Caffey syndrome Speaker: Teresa Aravena The patient was the first child of healthy non-consanguineous parents. He had a prenatal diagnosis of skeletal dysplasia. He was born at 38 weeks of gestation. His birth weight was 1,580 g (<< 3rd percentile), birth length was 35 cm (<< 3rd percentile), and head circumference was 31 cm (<<3rd percentile). On physical examination, he had a cloverleaf skull with a broad anterior fontanelle, prominent forehead, and bitemporal narrowing; microphtalmia; short upturned nose with a low nasal bridge; and very low set ears. He had a short neck and short limbs without a predominantly short segment. Skull and whole body X-rays showed a poorly ossified skull, thin long bones and a fractured femur. Head CT was “normal”. During his first days of life he developed hypocalcemia with hyperphosphemia, with inappropiately low levels of parathyroid hormone. He had difficulty feeding and required tube feeding. He died at 2 months of
ge because of a bronchopneumonia. Parents declined an autopsy. Tentative iagnosis: Kenny Caffey syndrome?
ad
12

Case 6: Turner syndrome and Langer mesomelic dysplasia Speaker: Teresa Aravena
Patient is a female child born to healthy non-consanguineous parents. Parents' heights are 185 cm for the father and 152 cm for the mother, and the maternal grandmother's height is 154 cm as well. They had a prenatal diagnosis of skeletal dysplasia at 18 weeks. She was born at 38 weeks of gestation. Birth weight was 2840 (p15), birth length was 41,5 cm (<<p3), and OFC was 34 (p50). On physical examination, she has a flat face, low nasal bridge, micrognathia, cup shaped ears, and a broad thorax with widely spaced nipples. Limbs: predominant mesomelic shortening, ulnar deviation of hands, single transverse crease on both hands, bilateral camptodactyly on the 3rd and 4th fingers, edema on both feet, and hypoplastic toe nails. X-rays showed shortening of humeri, hypoplasia of radius and ulna with subluxation of elbows. Radii are bowed and ulnas are broad with a rhomboid shape. The lower extremities have a marked genu varum, mesomelic shortening, marked hypoplasia of fibulas, and broad and slightly bowed tibias. Karyotyping: mosaic Turner Syndrome: 45,X[10]/46,X,del(X)(p11.4[15]. MLPA study and sequencing for SHOX showed a compound heterozygote with deletion and mutation, and confirmed the diagnosis of Langer Syndrome. X-rays of the mother's wrists showed a Madelung deformity, and MLPA showed a heterozygous deletion of SHOX. Her karyotype was normal. The father has a normal phenotype and a normal karyotype. In conclusion, this child has a double condition of Turner syndrome and Langer mesomelic dysplasia. The loss of one of the copies of the SHOX gene can be explained by the findings on the karyotype, and possibly the mutation of the other SHOX gene was inherited from the mother who has Leri Weill dyschondrosteosis.
13

Case 7: Skeletal dysplasia Speaker: Silvia Castillo Patient is a 6 year old boy, born to healthy non-consanguineous parents of normal statures. At 38 weeks of gestation his birth weight was 2,955 g (p25), birth length was 46 cm (p3), and OFC was 35.7 cm (p45). He had postaxial polydactyly on his left foot. He was evaluated at a Genetics Clinic at 1 year and 9 months of age because of short stature and waddling gait. On physical examination he had a prominent forehead, normal facies, shortening of the limbs, and a prominent waddling gait. His height was in the 3rd percentile, weight was in the 15th percentile and head circumference was in the 99th percentile. Skeletal radiological survey showed mild metaphyseal changes mainly on the proximal femurs, distal tibias and distal radius, coxa vara, and short femoral necks. The first lumbar vertebra has a diminished antero-posterior diameter with underdevelopment of its upper segment. Hands appear normal with normal bone age. The left foot has a bifid fifth metatarsian. At 3 years of age the child required correction of the coxa vara because of severe hip pain and waddling gait. After the surgery, the child gained 3 cm in height but remained with a prominent waddling gait and hip pain associated to exercise. His height is still on the third percentile. He has normal intelligence but he developed an expressive speech disorder. He had molecular testing for Metaphyseal Chondrodysplasia Schmid Type which showed no pathologic mutations. Afterwards he was studied for Cartilage Hair Hypoplasia but no pathologic mutations were found either. He had an Array CGH that showed no pathologic copy number variations.
14

Case 8: skeletal dysplasia, diastrophic dysplasia (?) Speaker: Silvia Castillo Patient is a male child born to healthy non-consanguineous parents. The pregnancy was a dichorial-diamniotic twin pregnancy. Prenatal sonograms diagnosed a skeletal dysplasia on Twin 1. He was born at 36 weeks of gestation. His birth weight was 2812 g (50th percentile), birth length was 40 cm (<<3rd percentile) and head circumference was 34 cm (60th percentile). Twin 2 is a healthy boy. On physical examination he has a flat face, mid-face hypoplasia, short upturned nose, cleft palate and micrognathia. Upper limbs are short, with the hands having a radial deviation. Fingers are short and stubby. Lower limbs are also shortened and he has bilateral club foot. Joints are stiff and he appears to have bilateral hip dislocation. X-rays show the following anomalies: upper limbs have bilateral radial and humeri hypoplasia, medial curvature of ulnae which appear of normal size, bilateral elbow dislocation, absence of medial phalanges on both fifth fingers, phalanges appear broad and metacarpals are short and broad; lower limbs have bilateral hip dislocation, severe fibular hypoplasia, minimal curvature of the tibias which appear of normal size, shortened femurs, bilateral instability of the knees, bilateral club foot and absence of medial phalanges on the 2nd, 3rd, 4th and 5th toes; spine X-ray shows platyspondyly; skull appears to be normal. Echocardiogram was normal, and brain sonogram showed grade 1 intracranial hemorrhage. He has had several episodes of fever without a clear origin and he has also presented episodes of cyanosis without hemodynamic repercussion, which is why he is still hospitalized. Without diagnosis, some similarities with diastrophic dysplasia.
15

Case 9: Atelosteogenesis type III Speaker: Silvia Castillo Patient is a male child born to healthy non-consanguineous parents. The pregnancy was a dichorial-diamniotic twin pregnancy. Prenatal sonograms diagnosed a skeletal dysplasia on Twin 1. He was born at 36 weeks of gestation. His birth weight was 2812 g (50th percentile), birth length was 40 cm (<<3rd percentile) and head circumference was 34 cm (60th percentile). Twin 2 is a healthy boy. On physical examination he has a flat face, mid-face hypoplasia, short upturned nose, cleft palate and micrognathia. Upper limbs are short, with the hands having a radial deviation. Fingers are short and stubby. Lower limbs are also shortened and he has bilateral club foot. Joints are stiff and he appears to have bilateral hip dislocation. X-rays show the following anomalies: Upper limbs have distal hypoplasia of humeri (club shaped humeri), radial hypoplasia, medial curvature of ulnae which appear of normal size, bilateral elbow dislocation, absence of medial phalanges on both fifth fingers, phalanges appear broad and metacarpals are short and broad; Lower limbs have bilateral hip dislocation, severe tibial hypoplasia which have a triangular shape, minimal curvature of the fibulas which appear of normal size, shortened femurs, bilateral instability of the knees, bilateral club foot and absence of medial phalanges on the 2nd, 3rd, 4th and 5th toes; Spine X-ray show mild platyspondyly; Skull appears to be normal. Echocardiogram was normal, and brain sonogram showed grade 1 intracranial hemorrhage. He has had several episodes of fever without a clear origin and he has also presented episodes of cyanosis without hemodynamic repercussion, which is why he is still hospitalized. The phenotype and skeletal findings on the X-rays point to a diagnosis of Atelosteogenesis type III, the autosomal dominant disease caused by mutations on FLNB. Molecular confirmation is still pending.
16

Case 10: Mosaic trisomy 13 Speaker: Harry Pachajoa Patient is a 2.5 months old boy born from a 28 year old G2P1C1 mother at 36 weeks gestation via cesarean section due to a nonreassuring fetal status. The parents were healthy and unrelated. Apgar scores were 9 at 1 min and 10 at 5 min. There was no family history for any other genetic condition. The mother denied any exposure to medications or teratogens during pregnancy. Birth weight was 2.4 kg (2-5th centile) and height 48 cm (25-50th centile). On physical examination, weight was 3.4 kg (<2th centile), height 53 cm (<5th centile), head circumference 35.5 cm (<2nd centile), external intercanthal distance 7.2 cm (75-97th centile), internal intercanthal distance 1.6 cm (<5th centile), and interpupillary distance 4 cm (<5th centile). She had bilateral cleft lip and palate, a left preauricular tag, a hemangioma in the occipital region and a sacral appendage. Results of routine laboratory studies were normal. Chromosome analysis from peripheral blood leukocytes using GTG banding technique at 100 metaphase demonstrated trisomy 13 mosaicism; 46 XX [80] / 47, XX, +13 [20]. Transfontanelar ultrasound, abdominal ultrasound and echocardiogram were normal. The lumbosacral ultrasound showed a small thickening and an increased echogenicity of the filum terminale, which could correspond to a lipoma.
17

Case 11: Double aneuploidy, trisomy X and trisomy 18 Speaker: Harry Pachajoa Patient is a girl born from a 39 years old G3P3 mother and a non consanguineous father at 34 weeks gestation. Intrauterine growth restriction was diagnosed during pregnancy. The girl was delivered at a level 1 hospital in a rural community. At birth, the weight was 1600 gr (< 3rd centile), the height 44 cm (< 3rd centile) and the head circumference 30.5 cm (< 3rd centile). Due to respiratory distress and intrauterine growth restriction, the patient was admitted to a high complexity level 4 institution. On physical examination, we found short palpebral fissures, bilateral low set, dysplastic and posterior rotated ears with a prominent cross on each side. The chest examination showed mammary hypertelorism and a holosystolic “clenched-like” grade IV/VI heart murmur. Club foot, dimples in the joints, rocker bottom feet, and dermatoglyphic anomalies were found in the lower extremities. G-banding karyotype analysis showed 48, XXX+18. Echocardiogram showed perimembranous interventricular septal defect, moderate to severe pulmonary stenosis, and an adequate biventricular function.
18

Case 12: Fertility in Down syndrome, mother to son transmission Speaker: Harry Pachajoa Down syndrome represents the main genetic cause of mental retardation. It is characterized by mental retardation, typical facies, and caused by an extra copy of chromosome 21. Case reports of fertile Down syndrome female patients are rare. We present here a male baby with trisomy 21 who is the son of a woman with Down syndrome. Available information on reproduction of women with Down syndrome is scanty in the literature.
19

Case 13: 19p13 deletion Speaker: Elena Vallespín Three year-old girl born to non-consanguineous parents. Developmental delay, speech delay and some degree of intellectual disability. No congenital malformations. Physical examination: Macrocephaly (> p 97), frontal bossing (deep set eyes) that remember Sotos’ syndrome but not the rest of the characteristics. Normal stature (p 10). Normal feet and hands (p 50). Computed tomography and magnetic resonance imaging normal. Karyotype: 45, XX,der(13;14)(q10;q10) inherited pat. Normal father phenotype.
20

Case 14: Nager syndrome (?) Speaker: Saleme
Male, non-consanguineous parents, mother 27 y.o., Gesta III, father 30 y.o. Pregnancy 36 wk, polyhydramnios, mild ventriculomegaly, oligodactyly hands, and single umbilical artery seen at 24th wk. C-section, birth weight 1,685 g, height 28.5 cm, Apgar 7/8. Physical exam: upslanting palpebral fissures, hypertelorism, dysplastic, low set left ear. Micrognathia, microglossia, cleft palate. Bilateral agenesis of thumbs, second finger clinodactyly. Echocardiogram, brain, and abdominal ultrasound normal. X-ray: normal forearm bones.
COMMENT: Dr Cohen agrees with diagnosis of Nager syndrome.
21

Case 15: Goldenhar syndrome (?) Speaker: Braulio Jatar
Male, DOB: May 15, 2012, pregnancy 35 weeks, cephalic presentation, C-section, died on day 1 with prenatal diagnosis of left diaphragmatic hernia. Mother, 24 y.o., Gesta II, used Ponstan® for headache during the first and second trimesters. Father 30 y.o., non-consanguineous. Physical exam: bilateral epicanthal folds, telecanthus, anteverted nares, cupid. Low set ears. Microtia I, external auditory canal permeable. One preauricular tag on left cheek, 10 mm diameter. Short neck. Bilateral chriptorchidism. Chest X-ray: 11 horizontalized, slender ribs. Thoracic hemivertebrae.
COMMENTS: Goldenhar syndrome is improbable. Consider possibilities of Pallister‐Killian syndrome or Fryns syndrome.
22

Case 16: Dysmorphic syndrome (?) Speaker: Cesar Saleme
DOB: July 15, 2012. Healthy parents; non consanguineous; no family antecedents of malformations. Mother age 26. G: 2. Complete primary education. Father age 32. Complete primary education. Skilled worker. Pregnancy history: Uncertain LMP, 13 prenatal visits, from the 2nd month, 5 prenatal ultrasounds, Pregnancy-induced hypertension, without treatment. Drugs received: Ranitidine in 1st and 2nd trimester; Norfloxacin in 3rd trimester. Spontaneous labor. Cephalic presentation. Female sex. Weight: 3755 grams; Height: 51 cm; HC: 36 cm; GA: 39 weeks; Apgar: 8/9, adequate for GA. Physical exam: Short and wide nose. Anteverted narines. Long philtrum. Thin lips. Short neck. Recto vulvar fistula. Wide thorax. Bilateral diastasis of 1st finger and toe, with abnormal implantation. Clinodactyly of 2nd finger. Complementary studies. Short and wide first metacarpal and metatarsal. Echocardiography: Normal. Brain and abdominal ultrasound: Normal. Released from hospital in good conditions.
COMMENTS: Dr. Lapunzina considered the possibility of Oto‐Palato‐Digital syndrome, and Dr. Orioli suggested Townes‐Brocks syndrome. Both suggested better radiographs, and wait for karyotype results.
23
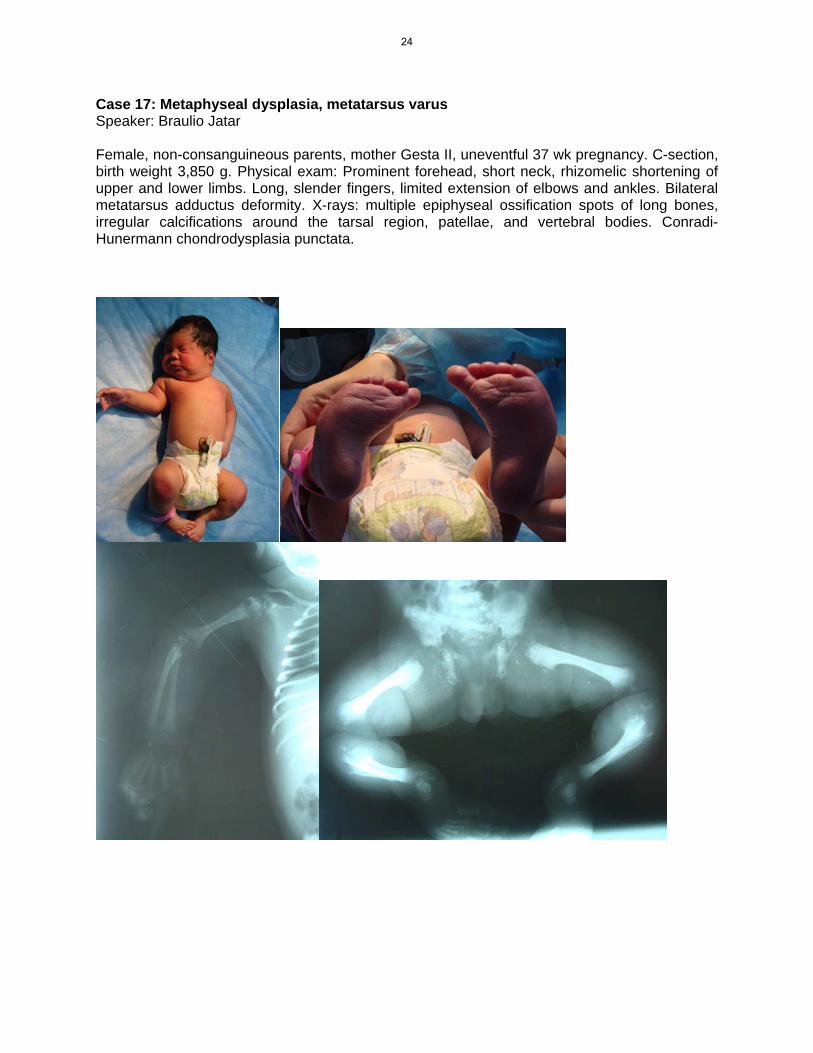
Case 17: Metaphyseal dysplasia, metatarsus varus Speaker: Braulio Jatar Female, non-consanguineous parents, mother Gesta II, uneventful 37 wk pregnancy. C-section, birth weight 3,850 g. Physical exam: Prominent forehead, short neck, rhizomelic shortening of upper and lower limbs. Long, slender fingers, limited extension of elbows and ankles. Bilateral metatarsus adductus deformity. X-rays: multiple epiphyseal ossification spots of long bones, irregular calcifications around the tarsal region, patellae, and vertebral bodies. Conradi-Hunermann chondrodysplasia punctata.
24

Case 18: Verma-Naumoff syndrome Speaker: Dorita
This is a newborn with 36 weeks of gestational age, male, weighed 3100g and died a few hours after birth due to pulmonary hypoplasia. Product of the first pregnancy of non consanguineous couple, having the mother 34 years and the father 50 years, at birth of propositus. Gestational ultrasound: ocular hypertelorism, flat nasal bridge, generalized edema, mild bilateral pulmonary hypoplasia, barrel-shaped short chest, bilateral brachydactyly, shortening of long bones (humeri, radii, ulna, femura, tibia, fibula) bilaterally, suggesting the diagnosis of skeletal dysplasia probably lethal. At birth the following characteristics were observed: short limbs, narrow thorax, protuberant abdomen, postaxial polydactyly in the right hand, and male genitalia with small penis. Radiological study: very short ribs, horizontal trident-shaped acetabular margins. The metaphyses of long bones are widened, especially that of the tibia, and longitudinal spurring is found at the margin of the metaphyses of the distal humeri and proximal and distal radii and femora. The autopsy confirmed the findings observed at physical exam and shows bilateral hydronephrosis. COMMENT: probably Jeune thoracic dystrophy syndrome, a ciliopathy.
25

Case 19: Zimmermann-Laband syndrome (?) Speaker: Dorita Newborn male, 35weeks of gestational age, weighting 1950g. Non-consanguineous parents. Gestational diabetes. Alcohol ingestion until the second trimester, without other complications. Fetal karyotype: 46, XY. Clinical Findings: hirsutism, brachycephaly, low-set ears, small palpebral fissures, bilateral coloboma of iris, prominent nose, high nasal bridge, anteverted nostrils, high palate, thick lower lip. Small thorax, nipple hypertelorism, hyperdynamic precordium. Terminal phalanges of fingers and toes markedly shortened, absence of nails on all toes, hypoplasia or absence of nails in fingers, proximal implantation of thumbs. Male genitalia with large falus. Bilateral cryptorchidism. Complementary exams: Transfontanelle ultrasonography suggestive of agenesis/dysgenesis of the corpus callosum. Abdominal ultrasound: bilateral hydronephrosis. Echocardiogram: ductus arteriosus aneurysm with right to left shunt. Clinical Course: dependent of mechanical ventilation; patient complicated with sepsis and died at 20 days of life due to septic shock. Necropsy: cornea dystrophy, bilateral ureteral stenosis and complex cardiopathy (aneurysmal dilatation of the ductus arteriosus, aortic hypoplasia (variant of coarctation of the aorta) and right ventricular hypertrophy. Diagnosis: Zimmermanp-Laband (?). COMMENT: Zimmermann‐Laband is not a consensus diagnosis.
26
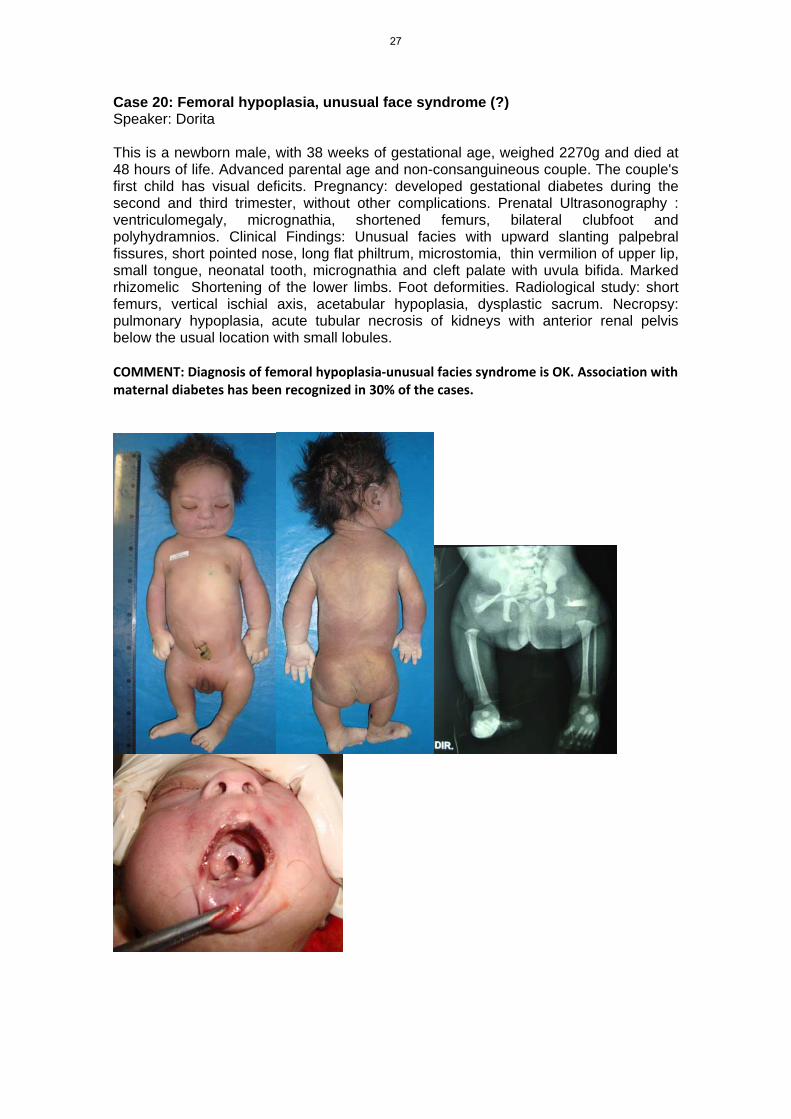
Case 20: Femoral hypoplasia, unusual face syndrome (?) Speaker: Dorita This is a newborn male, with 38 weeks of gestational age, weighed 2270g and died at 48 hours of life. Advanced parental age and non-consanguineous couple. The couple's first child has visual deficits. Pregnancy: developed gestational diabetes during the second and third trimester, without other complications. Prenatal Ultrasonography : ventriculomegaly, micrognathia, shortened femurs, bilateral clubfoot and polyhydramnios. Clinical Findings: Unusual facies with upward slanting palpebral fissures, short pointed nose, long flat philtrum, microstomia, thin vermilion of upper lip, small tongue, neonatal tooth, micrognathia and cleft palate with uvula bifida. Marked rhizomelic Shortening of the lower limbs. Foot deformities. Radiological study: short femurs, vertical ischial axis, acetabular hypoplasia, dysplastic sacrum. Necropsy: pulmonary hypoplasia, acute tubular necrosis of kidneys with anterior renal pelvis below the usual location with small lobules. COMMENT: Diagnosis of femoral hypoplasia‐unusual facies syndrome is OK. Association with maternal diabetes has been recognized in 30% of the cases.
27
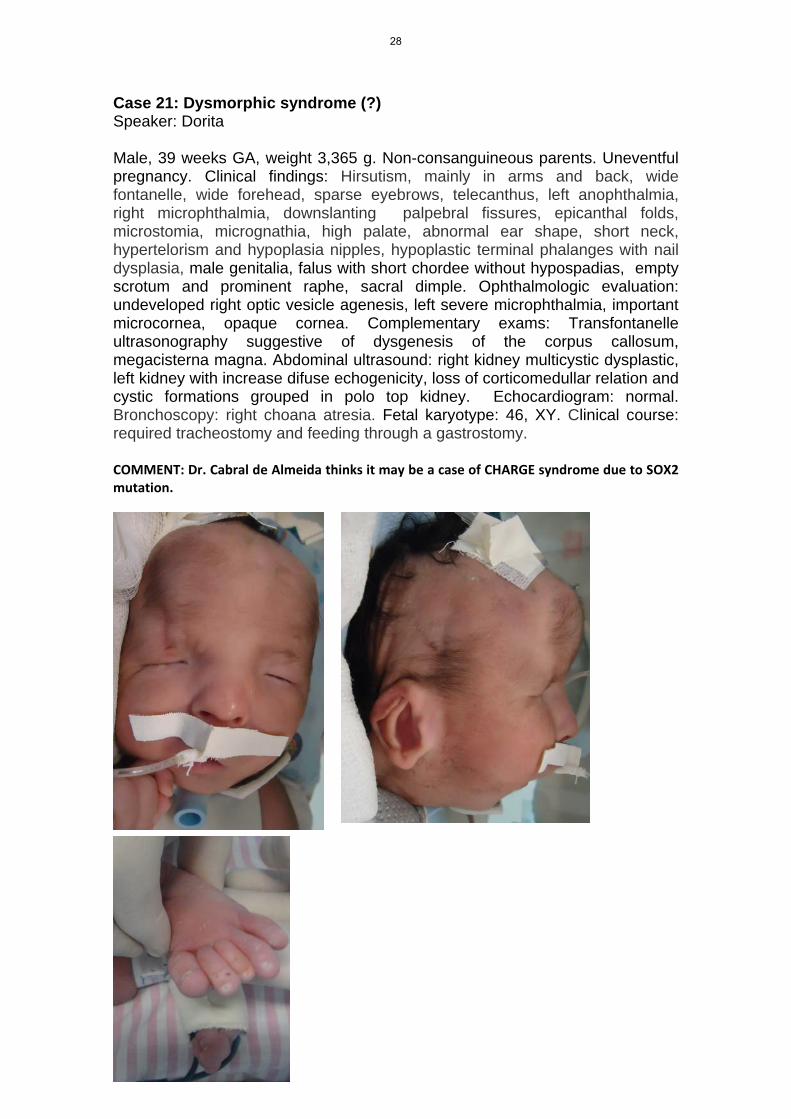
Case 21: Dysmorphic syndrome (?) Speaker: Dorita Male, 39 weeks GA, weight 3,365 g. Non-consanguineous parents. Uneventful pregnancy. Clinical findings: Hirsutism, mainly in arms and back, wide fontanelle, wide forehead, sparse eyebrows, telecanthus, left anophthalmia, right microphthalmia, downslanting palpebral fissures, epicanthal folds, microstomia, micrognathia, high palate, abnormal ear shape, short neck, hypertelorism and hypoplasia nipples, hypoplastic terminal phalanges with nail dysplasia, male genitalia, falus with short chordee without hypospadias, empty scrotum and prominent raphe, sacral dimple. Ophthalmologic evaluation: undeveloped right optic vesicle agenesis, left severe microphthalmia, important microcornea, opaque cornea. Complementary exams: Transfontanelle ultrasonography suggestive of dysgenesis of the corpus callosum, megacisterna magna. Abdominal ultrasound: right kidney multicystic dysplastic, left kidney with increase difuse echogenicity, loss of corticomedullar relation and cystic formations grouped in polo top kidney. Echocardiogram: normal. Bronchoscopy: right choana atresia. Fetal karyotype: 46, XY. Clinical course: required tracheostomy and feeding through a gastrostomy. COMMENT: Dr. Cabral de Almeida thinks it may be a case of CHARGE syndrome due to SOX2 mutation.
28

Case 22: Fibrochondrogenesis Speaker: Dorita Born at term and small for gestational age, the proband is the first son of young and non consanguineous couple. At prenatal ultrasound: flat nasal bridge, hemivertebrae (at cervical, lumbar and sacral spine), long bones and thoracic cage under fifth percentile. At birth, were noticed macrocrania, large anterior and posterior fontanelles, prominent eyes, midface hypoplasia, microretrognathia, rhizomelic, and mesomelic shortening of arms and legs. Small, short thorax with protuberant abdomen. Also, there was bilateral hip dysplasia. Radiological findings included: shortening of ribs, small thoracic cage, flattening vertebrae, lumbar spine with apparent absence and poor definition of vertebrae; pseudoarthrosis of humerus and ulna, bilateral dislocation of radium; large ilia, bilateral acetabular dysplasia, pseudoarthrosis of left femur with tibia and fibulae; dislocation of left tibia and fibulae; bilateral shortening with bowing and enlargement of femur, tibia and fibulae. Diagnostic hypothesis: fibrochondrogenesis COMMENT: Fibrochondrogenesis is ok.
29
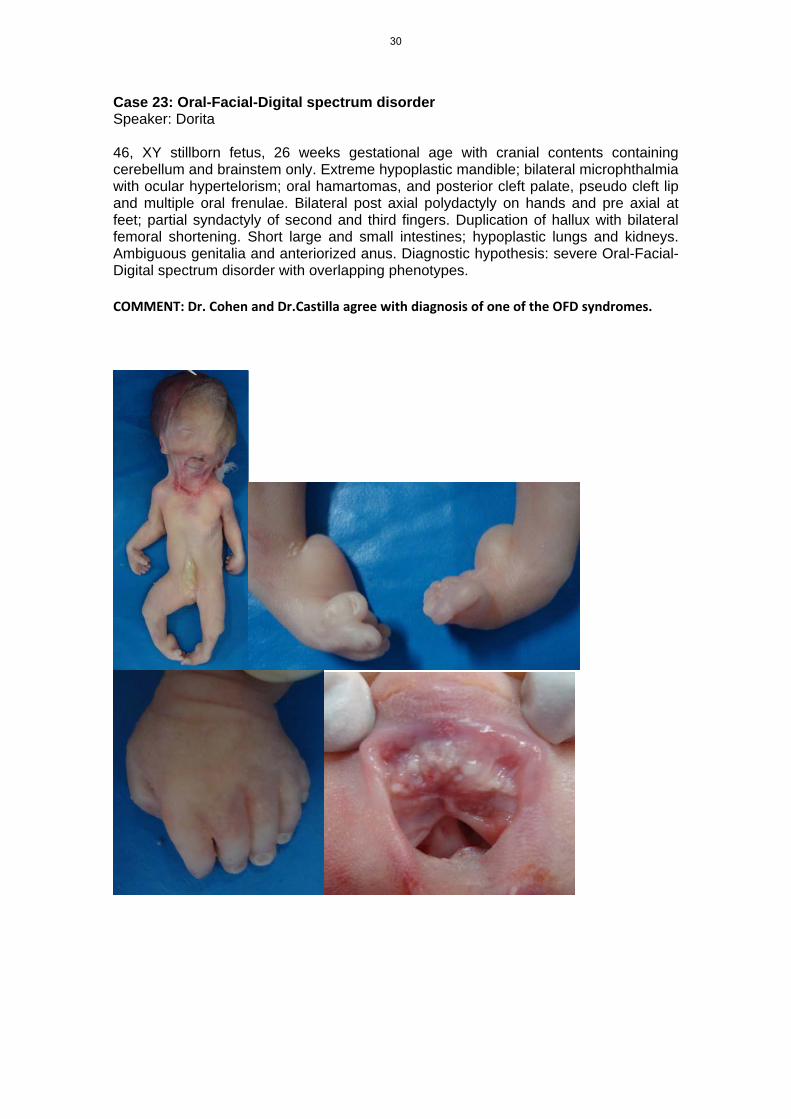
Case 23: Oral-Facial-Digital spectrum disorder Speaker: Dorita 46, XY stillborn fetus, 26 weeks gestational age with cranial contents containing cerebellum and brainstem only. Extreme hypoplastic mandible; bilateral microphthalmia with ocular hypertelorism; oral hamartomas, and posterior cleft palate, pseudo cleft lip and multiple oral frenulae. Bilateral post axial polydactyly on hands and pre axial at feet; partial syndactyly of second and third fingers. Duplication of hallux with bilateral femoral shortening. Short large and small intestines; hypoplastic lungs and kidneys. Ambiguous genitalia and anteriorized anus. Diagnostic hypothesis: severe Oral-Facial-Digital spectrum disorder with overlapping phenotypes.
COMMENT: Dr. Cohen and Dr.Castilla agree with diagnosis of one of the OFD syndromes.
30
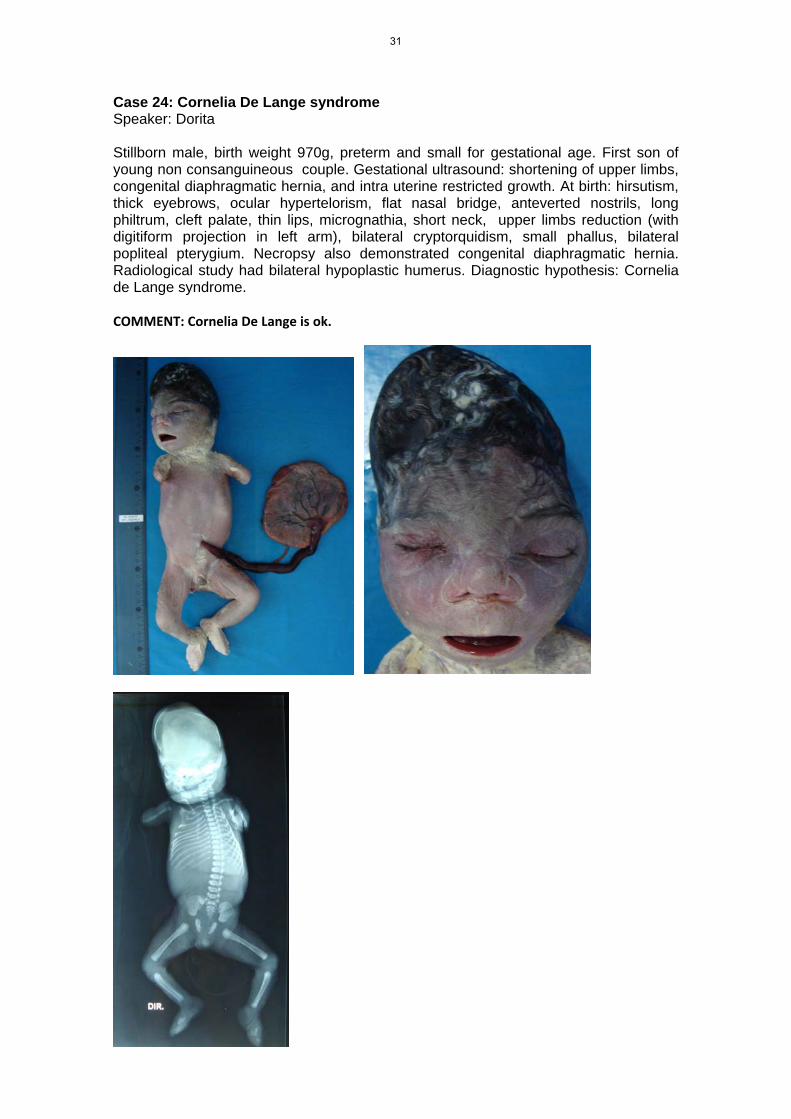
Case 24: Cornelia De Lange syndrome Speaker: Dorita Stillborn male, birth weight 970g, preterm and small for gestational age. First son of young non consanguineous couple. Gestational ultrasound: shortening of upper limbs, congenital diaphragmatic hernia, and intra uterine restricted growth. At birth: hirsutism, thick eyebrows, ocular hypertelorism, flat nasal bridge, anteverted nostrils, long philtrum, cleft palate, thin lips, micrognathia, short neck, upper limbs reduction (with digitiform projection in left arm), bilateral cryptorquidism, small phallus, bilateral popliteal pterygium. Necropsy also demonstrated congenital diaphragmatic hernia. Radiological study had bilateral hypoplastic humerus. Diagnostic hypothesis: Cornelia de Lange syndrome. COMMENT: Cornelia De Lange is ok.
31

Case 25: del2p microdeletion disguised by familial inv(5) Speaker: Juan We describe the case of a patient followed up from birth with a number of congenital malformations at the time suggested as CHARGE due to the presence of choanal atresia and coloboma of iris. After the emergence of single upper central incisor, the diagnosis of holoprosencephaly (HPE) was suggested on its microform. Conventional cytogenetic studies revealed the presence of an inv peric of 5 (p15.1q21) inherited from the father and present on her brother with severe hyperactivity and attention deficit; but both without birth defects. The patient is currently on hers 18 years, normal mensis and normal puberty; important short stature (<< 3rd PC); microcephaly (<< 3rd PC); hypotelorism; prominent ears; broad nose with a transverse depression in the middle third; left iris coloboma; single upper incisor. Anosmia and corpus callosum agenesis were present. She has mental retardation (WISC) with language difficulty in the areas of content and structure. The patient presents with a very introverted behavior and uses anti-depressive agents. GH deficiency was excluded. The molecular cytogenetic technique MCB5 (multicolor banding 5) (http://www.med.uni-jena.de/fish/mFISH/ mFISHlit.htm) and number of BACs, including 5p14.3, 5p13.2-3, 5q21.1, 5q22.2, 5q23.1, 5q23.2 identified the possible breakpoints as 5p14.3 and 5q23.1. No cytogenetic imbalance, especially with MCB technique was identified comparing the chromosomes of patient, brother and father. The molecular study using MLPA (Salsa MLPA kit P187-B1) excluded subtle chromosomal abnormalities within eight (SHH-7q36, 13q32-ZIC2; SIX3-2p21; TGIF1-18p11.31; TRAPPC10 - 21q22.3; GLI2 - 2q14; PTCH1 - 9q22. 1-31; FBXW11 - 5q35.1) of 12 known genes associated with HPE. Furthermore, sequencing of SHH; ZIC2; SIX3 were considered normal. A CGH-array identified a deletion of 2.82Mb at 2p22.1-p21.
32

Case 26: macrosomia, ear creases, macrocrania Speaker: Fernando Proband is the female child of non-consanguineous parents, birth weight > 4.000 g, no history of familial or gestational diabetes. Father’s height is 168 cm and mother’s is 158 cm. Physical exam at age four months showed macrosomia, deep creases on the lobules of both ears. She does not have macroglossia. Normal abdominal ultrasound and slightly advanced bone age. Child began with non-febrile seizures at age 2, controlled with phenobarbital. In spite of mild motor delay in the first year of life, development and speech are now normal. Dx: macrosomia and ear creases similar to BWS but without other findings of this disease.
33

Case 27: Macrosomia, ear creases, maternal t14q;21q Speaker: Fernando Male, only child of non-consanguineous couple. Uneventful pregnancy, C-section, weight 4,850 g, height 57 cm. Mild developmental delay, walked 16 months, first words 36 months. Physical exam shows cow-lick, large bifid ear lobules with posterior indentations, mild hemangioma on the glabella. Mouth, tongue, abdomen ok. Left accessory nipple. Square fingers with fetal pads, pes planus. Karyotype: 45,XY,-14,-21,+t14;21, inherited from the mother. Normal echocardiogram and abdominal ultrasound.
34

Case 28: Oculocerebrocutaneous syndrome Speaker: Marcos Aguiar Female, non-consanguineous parents, uneventful pregnancy until 34th week, when severe hydrocephalus and cleft palate were disclosed. C-section, birth weight 4,100 g, gestational age 36 weeks + 4 days, adequate for GA. Physical exam: macrocrania, plagiocephaly; microphthalmia; bifid, proboscis-like nose; cleft palate (0/3 1/3 2/3 0/3), low set ears, left preauricular tag. Ring constrictions in fingers in both hands. Brain ultrasound: hydranencephalus (?). Brain CT: severe hydrocephalus, cerebellar hemispheres and vermis visualized. Clinical findings suggestive of oculocerebrocutaneous syndrome with severe phenotypic presentation.
35

Case 29: 9p deletion and 16q duplication Speaker: Harry Pachajoa Male, Mother 32 years old, father 32 years old, consanguineous parents. Birth weight 3580, size 52 cm. Patient with a history of hypotonia, psychomotor developmental delay, facial abnormalities, craniosynostosis of metopic suture. G-banding karyotype showed a deletion 9p (46, XX, del (9) (p23), MLPA showed a deletion of 9p more than 5 MB and duplication 16q less than 1MB. Normal echocardiogram. Auditory evoked potential suggesting moderate bilateral endocochlear damage. Physical examination shows trigonocephaly, epicanthal folds, dysplastic ears, high palate, rigid interphalangeal joints. Bilateral cryptorchidism.
36

Case 30: OFD tipo VI (Varadi-Papp) - CASO NO ECLAMC Speaker: Monica Rittler Caso 1: Primer hijo de pareja joven, sana, no consanguínea. Sin antecedentes familiares ni prenatales relevantes. Recién nacido masculino de 30 semanas, con peso en percentilo 3. Falleció a los pocos minutos. Microcefalia, cefalocele parietooccipital, hipertelorismo, puente nasal alto, labio leporino bilateral y paladar hendido completos, polidactilia axial bilateral por bifurcación del tercer metacarpiano, primer metatarsiano ancho y duplicación de falanges del hallux bilaterales, tibia derecha corta y ancha, varoequino derecho. Fémur derecho levemente acortado. Holoprosencefalia semilobar, hipoplasia cerebelosa, suprarrenal y renal. Caso 2: Hermano del anterior. Recién nacido masculino de 38 semanas, peso adecuado. Falleció a los 3 días. Hidrocefalia, cefalocele occipital, hipertelorismo, puente nasal alto, labio leporino derecho y paladar hendido completos, polidactilia axial izquierda por bifurcación del tercer metacarpiano y postaxial derecha, primer metatarsiano ancho, duplicación de falanges del hallux y equinovaro bilaterales. Tibias algo cortas y gruesas. Dandy Walker, hipófisis, suprarrenales y riñones hipoplásicos. OFD tipo VI (Varadi-Papp).
37

38