chapter- 6 - shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/31128/14/14_chapter6.pdf · 258...
TRANSCRIPT

258
CHAPTER- 6 2.6 DEVELOPMENT AND VALIDATION OF STABILITY INDICAT ING RP-HPLC METHOD FOR THE DETERMINATON OF PRAMIPEXOLE DIHYDROCHLORIDE MONOHYDRATE IN PRAMIPEXOLE DIHYDROCHLORIDE EXTENDED RELEASE TABLETS
CONTENTS
1. Drug profile
2. Review of the past work on the analytical methods for Pramipexole dihydrochloride monohydrate.
3. Experimental and results
a) Material and methods
b) Optimization of chromatographic conditions and method development
c) Validation of the proposed method
4. Summary of the results and Conclusion
5. References

259
1. DRUG PROFILE-PRAMIPEXOLE DIHYDROCHLORIDE
Pramipexole dihydrochloride tablets contain pramipexole1-6, a nonergot dopamine
agonist. The chemical name of pramipexole dihydrochloride is (S)-2-amino-4, 5, 6, 7-
tetrahydro-6-(propylamino) benzothiazole dihydrochloride monohydrate. Its molecular
formula is C10 H17 N3 S · 2HCl · H2O, and its molecular weight is 302.27.
Mechanism of Action
Pramipexole is a nonergot dopamine agonist with high relative in vitro specificity and
full intrinsic activity at the D2 subfamily of dopamine receptors, binding with higher
affinity to D3 than to D2 or D4 receptor subtypes.
Parkinson’s disease
The precise mechanism of action of pramipexole as a treatment for Parkinson’s disease
is unknown, although it is believed to be related to its ability to stimulate dopamine
receptors in the striatum. This conclusion is supported by electrophysiologic studies in
animals that have demonstrated that pramipexole influences striatal neuronal firing rates
via activation of dopamine receptors in the striatum and the substantia nigra, the site of
neurons that send projections to the striatum. The relevance of D3 receptor binding in
Parkinson’s disease is unknown.
Pharmacokinetics
Pramipexole displays linear pharmacokinetics over the clinical dosage range. Its
terminal half-life is about 8 hours in young healthy volunteers and about 12 hours in
elderly volunteers (see CLINICAL PHARMACOLOGY, Pharmacokinetics in
Special Populations). Steady-state concentrations are achieved within 2 days of
dosing.

260
Chemical Structure:
Chemical name (IUPAC ) : (S)-2-amino-4, 5, 6, 7-tetrahydro-6-propylamino)
benzothiazole dihydrochloride monohydrate
Chemical formula : C10H17 N3S-2HCl-H2 O
Molecular weight : 302.27
Physical state : A white to off white powder
Melting point : 296-3010 C
Solubility : Freely Soluble in Methanol,
Official Status of the drug : The drug is official in Merck Index
Table 2.6.1 Important brand names of Pramepexole dihydrochloride monohydrate
Brand Name Formulation Dosage Manufacturer
Mirapex Tablets 0.375,0.75,1.5,3.0 and 4.0 mg
Boehringer Ingelheim International GmbH

261
2. Review of the past work on the analytical methods for Pramipexole dihydrochloridemonohydrate
Panchal JG, et al 7, proposed a GC/MS method for the determination of pramipexole in
rat plasma. Analytes were determined using electron impact ionization in a single
quadrupole mass spectrometer. GC/MS was performed in the selected ion monitoring
mode using target ions at m/z 211, 212 and 152 for pramipexole and m/z 194 and 165
for caffeine as internal standard. A linear calibration curve was plotted over the range of
20-1000�pg/mL for pramipexole (r(2) > 0.996). The LLOQ was 20.0 pg/mL,
respectively, which offered high sensitivity and selectivity enough for bioanalytical
investigation. Inter- and intraday precisions ranged from 0.3 to 8.8% and from 0.9 to
11.33%, respectively.The recovery of pramipexole from plasma ranged from 82.4 ± 7.1
to 87.8 ± 5.7%.
G. Srinubabu et al 8, proposed a RP-HPLC method with UV detection for routine
control of pramipexole in tablets was developed. Chromatography was performed with
mobile phase containing a mixture of acetonitrile/phosphate buffer (60/40; v/v) with a
flow rate of 0.8 mL min−1. Quantitation was accomplished with the internal standard
method; the procedure was validated by linearity (correlation coefficient = 0.99892),
accuracy, robustness and intermediate precision. Limit of quantitation and limit of
detection were found to be 4.5 µg and 1.4 µg respectively.
D. Vijaya Bharathi 9, proposed a LC-MS/MS method for the estimation of pramipexole
(PPX) with 500 µL human plasma using memantine as an internal standard (IS). The
resolution of peaks was achieved with 0.01 M ammonium acetate buffer (pH
4.4):acetonitrile (30:70, v/v) on a Discovery CN column. The total chromatographic run
time was 3.0 min and the elution of PPX and IS occurred at approximately 2.32 and
2.52, respectively. The MS/MS ion transitions monitored were 212.10 → 153.10 for
PPX and 180.20 → 107.30 for IS. The method was proved to be accurate and precise at
linearity range of 20–3540 pg/mL with a correlation coefficient (r) of ≥0.999.
Ramakrishna V. S. Nirogi et al 10, proposed a high-performance liquid
chromatography/electrospray ionization tandem mass spectrometry method for the
quantification of pramipexole in human plasma. Following liquid–liquid extraction, the

262
analytes were separated using an isocratic mobile phase on a reverse-phase column and
analyzed by MS/MS in the multiple reaction monitoring mode using the respective [M +
H]+ions, m/z 212/152 for pramipexole and m/z 409/228 for the IS. The method exhibited
a linear dynamic range of 200–8000 pg/mL for pramipexole in human plasma. The
lower limit of quantification was 200 pg/mL with a relative standard deviation of less
than 8%.
D.B. Pathare et al 11 , proposed a chiral liquid chromatographic method for the
enantiomeric resolution of Pramipexole dihydrochloride monohydrate, (S)-2-amino-
4,5,6,7-tetra-hydro-6-(propylamino) benzothiazole dihydrochloride monohydrate, a
dopamine agonist in bulk drugs. The enantiomers of Pramipexole dihydrochloride
monohydrate were resolved on a Chiralpak AD (250 mm × 4.6 mm, 10 µm) column
using a mobile phase system containing n-hexane:ethanol:diethylamine (70:30:0.1,
v/v/v). The resolution between the enantiomers was found not less than eight. The limit
of detection and limit of quantification of (R)-enantiomer are 300 and 900 ng/ml,
respectively for 20 µl injection volume. The percentage recovery of (R)-enantiomer was
ranged from 97.3 to 102.0 in bulk drug samples of Pramipexole dihydrochloride
monohydrate.
Yadav Manish 12, proposed a UPLC-MS-MS method for the determination of
pramipexole, a dopamine agonist, in human plasma. The chromatographic separation is
achieved on a Waters Acquity UPLC BEH C18 (100 mm × 2.1 mm, 1.7 µm) analytical
column using an isocratic mobile phase, consisting of 10 mM ammonium formate (pH
7.50)-acetonitrile (15:85, v/v), at a flow-rate of 0.5 mL/min. The precursor → product
ion transition for pramipexole (m/z 212.1 → 153.0) and IS (m/z 315.0 → 176.1) were
monitored on a triple quadrupole mass spectrometer, operating in the multiple reaction
monitoring (MRM) and positive ion mode.
B.M. Gurupadayya et al 13, proposed two methods A and B for the quantitative
estimation of pramipexole dihydrochloride drug and its formulations. Method A is
based on the diazotization of primary amine group of pramipexole with sodium nitrate
and hydrochloric acid followed by coupling with N-(1-napthyl) ethylene diamine
hydrochloride (BM Reagent) to form a colored chromogen with a characteristic
absorption maximum at 616 nm. Method B is based on the reaction of the drug in

263
methanolic solution with paradimethylaminobenzaldehyde (PDAB) in acidic condition
producing Schiff’s base having a 8 at 474.5nm. Beer’s law is obeyed in concentrations
ranging from 4-20 µg/ml for method max A and 50-150 µg/ml for method B.
Petikam Lavudu1 et al 14, proposed three vis-spectrophotometric methods for the
determination of pramipexole dihydrochloride in bulk and tablet dosage forms. The
methods (A & B) are based on the oxidation of pramipexole dihydrochloride with
iron(III) and the subsequent formation of an intensive orange red complex between the
liberated iron(II) and 1,10- phenanthroline (method A) or 2,2'-bipyridyl (method B)
reagents. Method C is based on the fact that iron(III) is reduced to iron(II) by
pramipexole dihydrochloride, then the in situ formed iron(II) reacts with potassium
ferricyanide to give the soluble Prussian blue in acidic conditions. By measuring the
absorbance of iron(II)-1,10-phenanthroline complex, iron(II)-2,2'-bipyridyl and soluble
Prussian blue at the absorption maximum of 530, 530 and 735 nm, respectively, the
indirect determination of pramipexole dihydrochloride can be obtained. A good linear
relationship of the concentration of pramipexole dihydrochloride versus absorbance is
observed with a linear range of 4-40, 3-30 and 2-20 µg mL-1 for methods A, B and C,
respectively.
C.vinodhini et al 15, proposed two spectrophotometric methods in ultraviolet and
visible region for the estimation of Pramipexole dihydrochloride monohydrate in
pharmaceutical dosage forms. Method I was based on Pramipexole dihydrochloride
monohydrate showing absorption maximum at 265nm in methanol. Method II was
based on reaction of Pramipexole dihydrochloride monohydrate with ferric nitrate under
acidic condition to yield a yellowish green color. This color has a characteristics light
absorption in the visible region with the absorption maximum at 435nm. Method I and
II obeyed Beer’s law in the concentration range of 10 - 80µg/ml and 5 – 25µg/ml
respectively.
The present investigation by the author describes the development of a rapid, accurate
and precise RP-HPLC method for the determination of Pramipexole dihydrochloride
monohydrate in tablet dosage forms.

264
3) EXPERIMENTAL AND RESULTS a) MATERIALS AND METHODS Instrumentation
The author had attempted to develop a liquid chromatographic method for
simultaneous estimation of Pramipexole dihydrochloride. The separation of the analyte
was done by using a Gradient Waters Allaince HPLC instrument, on a Inertsil C8
column (250 x 4.6mm; 5µm). The instrument was equipped with a pump (2695),
injector, PDA Detector (2996) and column oven. Data acquisition was done by using
Empower software
Degassing of the mobile phase was done by using a Spectra lab model DGA
20A3 ultrasonic bath sonicator. A Sartorious electronic balance was used for weighing
the materials. Class ‘A’ Borosil glassware was employed for volumetric and general
purpose in the study.
Drugs
The reference sample of Pramipexole dihydrochloride was gifted by M/s
Wockhardt Pharmaceutical limited. The Branded formulations of Pramipexole
dihydrochloride (Mirapex tablets of Boehringer Ingelheim International GmbH) were
procured from the local market.
Reagents
Potassium dihydrogen phosphate : AR/GR grade
Octane-1 sulphonic acid sodium salt : AR/GR grade
Orthophosphoric acid (min 88%) : AR/GR grade
Concentrated hydrochloric acid (min. 35%) : AR/GR grade
Acetonitrile : HPLC grade
Methanol : HPLC grade
Water : Milli-Q / HPLC grade
Preparation of mobile phase A
9.1 g of potassium dihydrogen phosphate and about 5.0 g of Octane-1 sulphonic acid
sodium salt was dissolved in 1000 mL of water. The pH was adjusted to 3.0 (+0.05)
with orthophosphoric acid. This solution was filtered through a 0.45µm membrane
filter.

265
Preparation of mobile phase B
Acetonitrile used as a mobile phase B
Preparation of diluent-1
8.5 mL concentrated hydrochloric acid was diluted to 1000 mL with methanol and
mixed well.
Preparation of diluent-2
8.5 mL concentrated hydrochloric acid was diluted to 1000 mL with water and mixed
well.
Chromatographic conditions
Column : Inertsil C 8-3 (250 x 4.6mm; 5µm)
Flow rate : 1.5 mL/min
Wavelength : 264 nm
Injection volume : 50µL
Column Oven Temperature : 40°C
Runtime : 20 min
Gradient Program
Time (min) % Mobile phase A % Mobile phase B
0 80 20
8 67 33
11 67 33
12 80 20
20 80 20
Preparation of Blank
5.0 mL of diluent-1 was taken into 20 mL volumetric flask; volume made up to the
mark with diluent -2 and mixed well. The solution was filtered through a 0.45µ PTFE
membrane filter by discarding 5 mL of filtrate.

266
Preparation of working standard solution
About 60 mg of Pramipexole dihydrochloride monohydrate standard was accurately
weighed and transferred into a 100 mL volumetric flask, about 60 mL of the diluent-1
was added and the contents were sonicated to dissolve and further diluted to volume
with the diluent-1 and mixed well. 5.0 mL of this solution was diluted into a 50 mL
volumetric flask and volume made up to the mark with the the diluent-1 and mixed well.
Further 5.0 mL of this solution was diluted into a 20 mL volumetric flask and volume
made up with the diluent-2 and mixed well. The solution was filtered through a 0.45µ
PTFE membrane filter by discarding 5 mL of filtrate.
Preparation of Formulation sample solution
16 intact tablets of Pramipexole dihydrochloride monohydrate were taken into (Mirapex
tablets of Boehringer Ingelheim International GmbH) a 200 mL dry volumetric flask,
about 100.0 mL of the diluent-1was added by using 50 mL pipette and the contents were
stirred on magnetic stirrer for about 30 min. The solution was sonicated for 10 min with
intermittent vigorous shaking. The flask was cooled to room temperature. A portion of
this solution was centrifuged at about 5000 rpm for 5 min in stoppered centrifuge tube.
5.0 mL of this supernatant solution was diluted into a 20 mL volumetric flask with the
diluent-2 and mixed well. The solution was filtered through a 0.45µ PTFE membrane
filter by discarding 5 mL of filtrate.
b) OPTIMIZATION OF THE CHROMATOGRAPHIC
CONDITIONS AND METHOD DEVELOPMENT
For developing the HPLC method, a systematic study of the effect of various
factors for ideal separation of the drugs was undertaken. This was done by varying one
parameter at a time and keeping all other conditions constant. The following studies
were conducted for this purpose. A non-polar C8 column was chosen as the stationary
phase for this study.

267
The mobile phase and the flow rate
In order to get sharp peaks and good base line separation of the components,
the author carried out a number of experiments by varying the commonly used solvents,
their compositions and flow rate.
To find out the most suitable mobile phase to effect ideal separation of the drugs under
Gradient conditions, mixtures of commonly used solvents like water, methanol and
acetonitrile with or without different buffers in different combinations were tested as
mobile phases on a C8 stationary phase.
Mobile phase A: 9.1 g of potassium dihydrogen phosphate and about 5.0 g of Octane-1
sulphonic acid sodium salt was dissolved in 1000 mL of water. The pH of this solution
was adjusted to 3.0 (+0.05) with orthophosphoric acid. This solution was filtered
through a 0.45µm (or fine porosity) membrane filters and degassed.
Mobile phase B: Acetonitrile was used as a mobile phase B
A mobile phase flow rate of 1.5 mL/min was found to be suitable in the study
range of 0.5 -2.0 mL/min.
Detection wave length
The UV absorption spectrum of the drug was taken in methanol and the λ max
found to be at 264 nm. Hence, detection of the drug was made at 264nm.
Retention time of Pramipexole
A model chromatogram showing the separation of Pramipexole is presented in
Fig 2.6.1 Under the above optimized conditions a retention time of Pramipexole about
8.53 min.
After a thorough study of the various parameters the following optimized
conditions mentioned in Table 2.6.2 were followed for the determination of
Pramipexole bulk samples and Pharmaceutical formulations.

268
Fig 2.6.1 A Model Chromatogram showing the separation of Pramipexole
Table 2.6.2 Optimized Chromatographic Conditions
Parameter Value
Column Inertsil C8, (250 x 4.6mm; 5µm)
Mobile Phase Mobile phase A and Mobile phase B
(Gradient)
Flow Rate 1.5 mL/min
Run Time 20 min
Column Temperature 30±1 ˚C
Volume Of Injection 20 µL
Detection Wave Length 264 nm
Retention Time 8.53 min

269
c) VALIDATION OF THE PROPOSED METHOD The method was validated in compliance with ICH guidelines16-19. The
parameters determined for validation were specificity, precision, accuracy, robustness,
Linearity, Forced Degradation, Limit of Quantification and Limit of Detection, system
suitability and stability of analytical solution.
1. Specificity
The method specificity was assessed by comparing the chromatograms obtained from a
placebo solution containing a mixture of most commonly used excipients without the
drug and another solution containing the excpeints with the drug. These solutions were
prepared in the diluent. The drug to excipient ratio used was similar to that in the
commercial formulation. The commonly used excipients in formulations like lactose,
starch, microcrystalline cellulose, ethyl cellulose, hydroxyl propyl methylcellulose,
magnesium stearate and colloidal silicon dioxide were taken up for the study. The
mixtures were filtered through 0.45µ membrane filter before injection. The placebo
solution and the sample solution (placebo and the drug) were injected into HPLC
system separately in triplicate and the relevant chromatograms observed. There was no
interference from blank and placebo at the retention time of analyte peak. The absence
of additional peaks in the chromatogram indicates non interference of the commonly
used excipients in the tablets and hence the method is specific. The relevant
chromatograms are given in Fig 2.6.2, 2.6.3 and 2.6.4 for chromatograms of blank,
placebo and placebo with the drug sample solutions respectively.
Acceptance criteriaon
a) No interfering peak should appear at the retention time of Pramipexole peak from
blank and placebo. Peak purity for analyte peak should pass.
Conclusion
The proposed method is specific for determination of Pramipexole dihydrochloride
monohydrate in its formulation tablet dosage forms as method meets acceptance
criterion.

270
2. Forced degradation study
Forced degradation study was carried out by exposing the placebo and the formulation
sample of to the following conditions.
1. Treatment with hydrochloric acid.
2. Treatment with sodium hydroxide.
3. Treatment with hydrogen peroxide.
4. Thermal exposure.
5. Photolytic exposure.
6. Exposure to humidity
a) Acid degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg into two
separate 200 mL volumetric flasks. 96 mL of diluent-1was added to each flask and
stir on magnetic stirrer for about 30 min, this solution was sonicated for 10 min with
intermittent shaking. To each flask added 2.0 mL of 10N hydrochloric acid and kept
at room temperature for about 3 hrs. After 3 hrs the resulting solution were
neutralized using 2.0 mL of 10N sodium hydroxide and mixed well. Centrifuged the
portion of solution at 5000 rpm for 5 min. 5.0 mL of this supernant solution was
diluted into a 20 mL with diluent-2 and mixed well. Filtered the solutions through a
0.45µ PTFE filter by discarding 5.0 mL of the filtrate.
b) Alkali degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg into two
separate 200 mL volumetric flasks. 94 mL of diluent-1was added to each flask and stir
on magnetic stirrer for about 30 min, this solution was sonicated for 10 min with
intermittent shaking. To each flask added 3.0 mL of 10 N Sodium hydroxide solution
and kept at room temperature for about 3 hrs. After 3 hrs the resulting solution was
neutralized using 3.0 mL of 10N hydrochloric acid and mixed well. Centrifuged the
portion of solution at 5000 rpm for 5 min. 5.0 mL of this supernant solution was diluted
into a 20 mL with diluent-2 and mixed well. Filtered the solutions through a 0.45µ
PTFE filter by discarding 5.0 mL of the filtrate.
c) Peroxide degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg in to two
separate 200 mL volumetric flasks. 99 mL of diluent-1 1was added to each flask and
stir on magnetic stirrer for about 30 min. This solution was sonicated for 10 min with
intermittent shaking. To each flask added 1.0 mL of 30% w/v Hydrogen peroxide

271
solution and mixed well. Immediately centrifuged the portion of solution at 5000 rpm
for 5 min. 5.0 mL of this supernant solution was diluted into a 20 mL with diluent-2
and mixed well. Filtered the solutions through a 0.45µ PTFE filter by discarding first
5.0 mL of the filtrate.
d) Thermal degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg in to two
separate 200 mL volumetric flasks. Placebo and sample were exposed to heat at 80°C
for about 24 hr. 100 mL of diluent-1 1was added to each flask and stir on magnetic
stirrer for about 30 min.This solution was sonicated for 10 min with intermittent
shaking. Centrifuged the portion of solution at 5000 rpm for 5 min. 5.0 mL of this
supernant solution to 20 mL with diluent-2 and mixed well. Filtered the solutions
through 0.45µ PTFE filter by discarding 5.0 mL of the filtrate.
e) Photolytic degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg into two
separate 200 mL volumetric flasks. Placebo and sample were exposed to photolytic
treatment for about 22 hrs (1.2 million lux hrs).100 mL of diluent-1 1was added to each
flask and stir on magnetic stirrer for about 30 min.This solution was sonicated for 10
min with intermittent shaking. Centrifuged the portion of solution at 5000 rpm for 5
min. 5.0 mL of this supernant solution was diluted into a 20 mL with diluent-2 and
mixed well. Filtered the solutions through a 0.45µ PTFE filter by discarding 5.0 mL of
the filtrate.
f) Humidity degradation
Transferred 16 tablets of sample and Placebo powder equivalent to 6 mg in to two
separate 200 mL volumetric flask. Placebo and sample were exposed to humidity at
40°C/75%RH for about 86 hrs 100 mL of diluent-1 1was added to each flask and stir on
magnetic stirrer for about 30 min, this solution was sonicated for 10 min with
intermittent shaking. Centrifuged the portion of solution at 5000 rpm for 5 min. 5.0 mL
of this supernant solution was diluted into 20 mL with diluent-2 and mixed well.
Filtered the solutions through a 0.45µ PTFE filter by discarding 5.0 mL of the filtrate.
The % Assay values with respect to untreated sample and peak purity data of
Pramipexole at each condition are tabulated in Table 2.6.3. Refer Fig 2.6.5 for
chromatograms and purity plots of untreated sample. Refer in Fig
2.6.6,2.6.7,2.6.8,2.6.9,2.6.10 and 2.6.11 for chromatograms and purity plots of Acid,
Alkali, Peroxide, Thermal, Photolytic and humidity treated sample solutions.

272
Acceptance criterion
Peak purity of analyte peak should pass.
Table 2.6.3 Forced degradation data
Sr. No.
Sample condition
%
Assay
% Degradation
w.r.t. Untreated
sample
Peak purity data for Pramipexole peak
Purity Angle
Purity Threshold
Purity Flag
1. Untreated sample* 103.60 - 0.077 0.283 No
2. Acid Treated
Treated with 2.0 mL of 10N HCl at room temperature for 3 hr.
102.38 1.18 0.262 0.678 No
3. Alkali Treated
Treated with 3.0 mL of 10N NaOH at room temperature for 3 hr.
104.06 0.00 0.260 0.668 No
4. Peroxide Treated
Treated with 1.0mL of 30% w/v hydrogen peroxide solution and immediately prepared
94.94 8.36 0.211 0.509 No
5. Thermal Treated
Exposed in oven at 80°C for 24 hr.
101.95 1.59 0.239 0.534 No
6. Photolytic Treated
Exposed to photolytic treatment for 22 hr. (1.2 million lux hr.)
101.61 1.92 0.268 0.605 No
7. Humidity Treated
Exposed at 40°C/75%RH for about 86 hr.
104.22 0.00 0.327 0.675 No
*Untreated sample data taken from method precision experiment. Conclusion
As the method meets acceptance criteria, it is concluded that the method is stability
indicating for determination of Pramipexole dihydrochloride monohydrate in
Pramipexole dihydrochloride extended release tablets.

273
3. Precision
3.1 System precision
Six replicate injections of standard solution were injected into HPLC system. Mean, SD
and % RSD were calculated for peak area counts of Pramipexole. The results are
tabulated in Table 2.6.4.
Acceptance criterion
% RSD should not be more than 2.0 for Pramipexole peak area counts.
Table 2.6.4 System precision data
.
Sr. No. Pramipexole peak area
counts (µV*sec)
1. 752200
2. 751520
3. 752795
4. 752618
5. 751847
6. 752348
Mean 752221
SD 476.4
% RSD 0.06

274
3.2 Method precision
Six sample preparations were made from a single batch and analyzed as per the
proposed method. % Assay of Pramipexole dihydrochloride monohydrate for six
samples was calculated. The results are tabulated in Table 2.6.5.
Acceptance criterion
% RSD for % Assay of six preparations should not be more than 2.0
Table 2.6.5 Method precision data
Sr. No.
Assay of Pramipexole
dihydrochloride monohydrate
1. 102.52
2. 103.07
3. 104.29
4. 103.74
5. 103.68
6. 104.30
Mean 103.60
SD 0.698
% RSD 0.67

275
3.3 Intermediate precision (Ruggedness)
Ruggedness of the method was verified by analyzing six sample preparations of same
batch used under method precision as per proposed method by different analysts using
different instrument and different column on different day.
The amount of Pramipexole dihydrochloride monohydrate for six samples was
determined. The %RSD for Assay and overall %RSD for above results of the method
precision were calculated. The results are tabulated in Table 2.6.6.
Acceptance criteria
% RSD for % Assay of six preparations should not be more than 2.0 and overall %RSD
should not be more than 2.0
Table 2.6.6 Ruggedness data
Sr. No.
Assay of Pramipexole dihydrochloride monohydrate
Method precision
Ruggedness
1. 102.52 101.87
2. 103.07 102.85
3. 104.29 103.67
4. 103.74 101.43
5. 103.68 102.43
6. 104.30 104.20
Mean 103.60 102.74
SD 0.698 1.056
% RSD 0.67 1.03
Overall Mean 103.17
Overall SD 0.964
Overall %RSD
0.93
Conclusion:
The proposed analytical method meets the acceptance criteria for precision. Hence, the
method is precise.
276
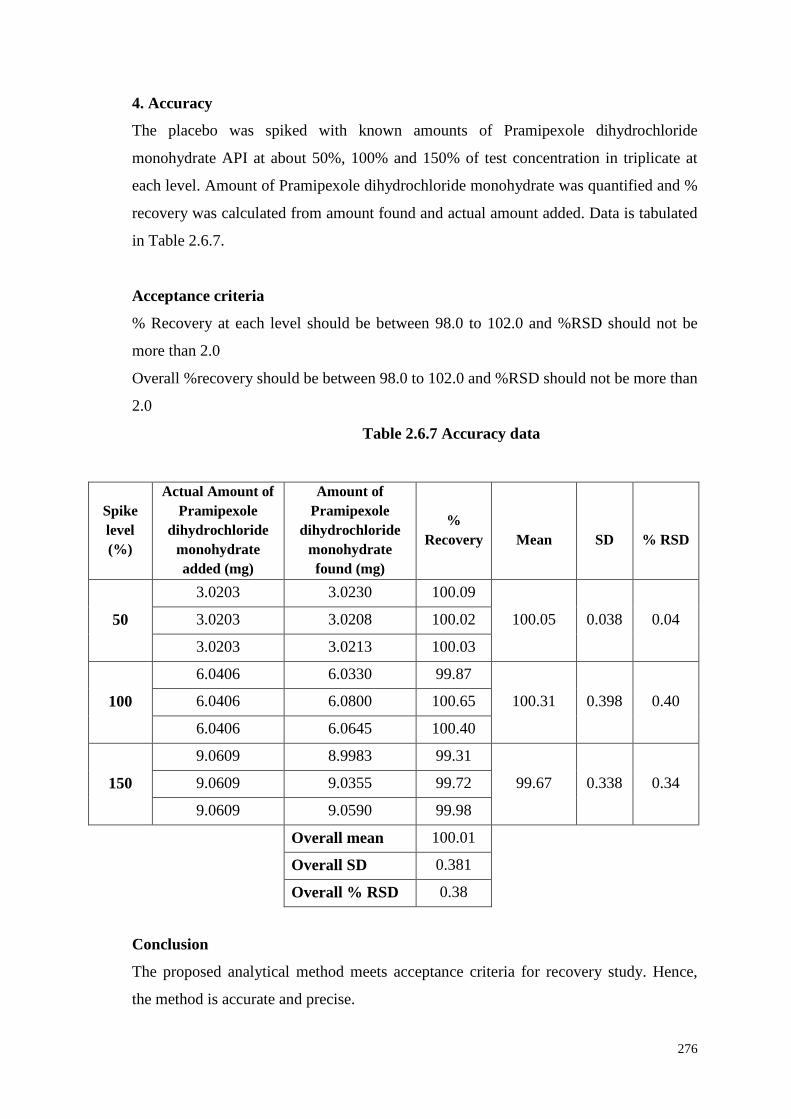
4. Accuracy
The placebo was spiked with known amounts of Pramipexole dihydrochloride
monohydrate API at about 50%, 100% and 150% of test concentration in triplicate at
each level. Amount of Pramipexole dihydrochloride monohydrate was quantified and %
recovery was calculated from amount found and actual amount added. Data is tabulated
in Table 2.6.7.
Acceptance criteria
% Recovery at each level should be between 98.0 to 102.0 and %RSD should not be
more than 2.0
Overall %recovery should be between 98.0 to 102.0 and %RSD should not be more than
2.0
Table 2.6.7 Accuracy data
Conclusion
The proposed analytical method meets acceptance criteria for recovery study. Hence,
the method is accurate and precise.
Spike level (%)
Actual Amount of Pramipexole
dihydrochloride monohydrate added (mg)
Amount of Pramipexole
dihydrochloride monohydrate found (mg)
% Recovery
Mean
SD
% RSD
50
3.0203 3.0230 100.09
100.05 0.038 0.04 3.0203 3.0208 100.02
3.0203 3.0213 100.03
100
6.0406 6.0330 99.87
100.31 0.398 0.40 6.0406 6.0800 100.65
6.0406 6.0645 100.40
150
9.0609 8.9983 99.31
99.67 0.338 0.34 9.0609 9.0355 99.72
9.0609 9.0590 99.98
Overall mean 100.01
Overall SD 0.381
Overall % RSD 0.38

277
5. Linearity
Linearity of response for Pramipexole dihydrochloride monohydrate was performed
using the standard solution in a range of 7.705 mcg/mL to 23.115 mcg/mL[about 50%
to 150% of the test concentration]. Results are tabulated in Table 2.6.8 and represented
graphically in Fig 2.6.12.
Acceptance criterion
Correlation coefficient (r) value should not be less than 0.99
Table 2.6.8 Linearity data
Conclusion
Response of Pramipexole dihydrochloride monohydrate was found linear in mentioned
range of 7.705 to 23.115 mcg/mL.
Linearity level (%)
Concentration in mcg/mL
Pramipexole average peak area counts
(µV*sec)
50 7.705 395416
60 9.246 471957
70 10.787 545932
80 12.328 632844
90 13.869 708402
100 15.410 787282
110 16.951 865870
120 18.492 941393
130 20.033 1024589
140 21.574 1099292
150 23.115 1180302
Slope 50996
Intercept 816
CC 0.99996

278
6. Stability in analytical solution
Stability of Pramipexole dihydrochloride monohydrate in analytical solution was
verified by analyzing sample solution initially and also at different time intervals up to
25 hrs and 46 min by storing sample solution at room temperature. Cumulative % RSD
for Pramipexole peak area counts was calculated. The results are tabulated in Table
2.6.9.
Acceptance criteria
Cumulative %RSD should not be more than 2.0 for Pramipexole peak area counts at
each time interval.
Table 2.6.9 Stability in analytical solution data
Time Pramipexole peak
area counts (µV*sec) Cumulative
% RSD
Initial 780458 -
22 Min 780744 0.03
41 Min 780736 0.02
02 Hr 06 Min 780756 0.02
03 Hr 52 Min 780594 0.02
07 Hr 24 Min 781658 0.05
11 Hr 38 Min 781822 0.07
15 Hr 52 Min 782302 0.09
21 Hr 49 Min 782257 0.10
25 Hr 46 Min 781627 0.09
Conclusion
It was found that the solution is stable up to 25 hrs at room temperature and hence, it is
concluded that the proposed analytical method meets the pre-established acceptance
criteria,
279
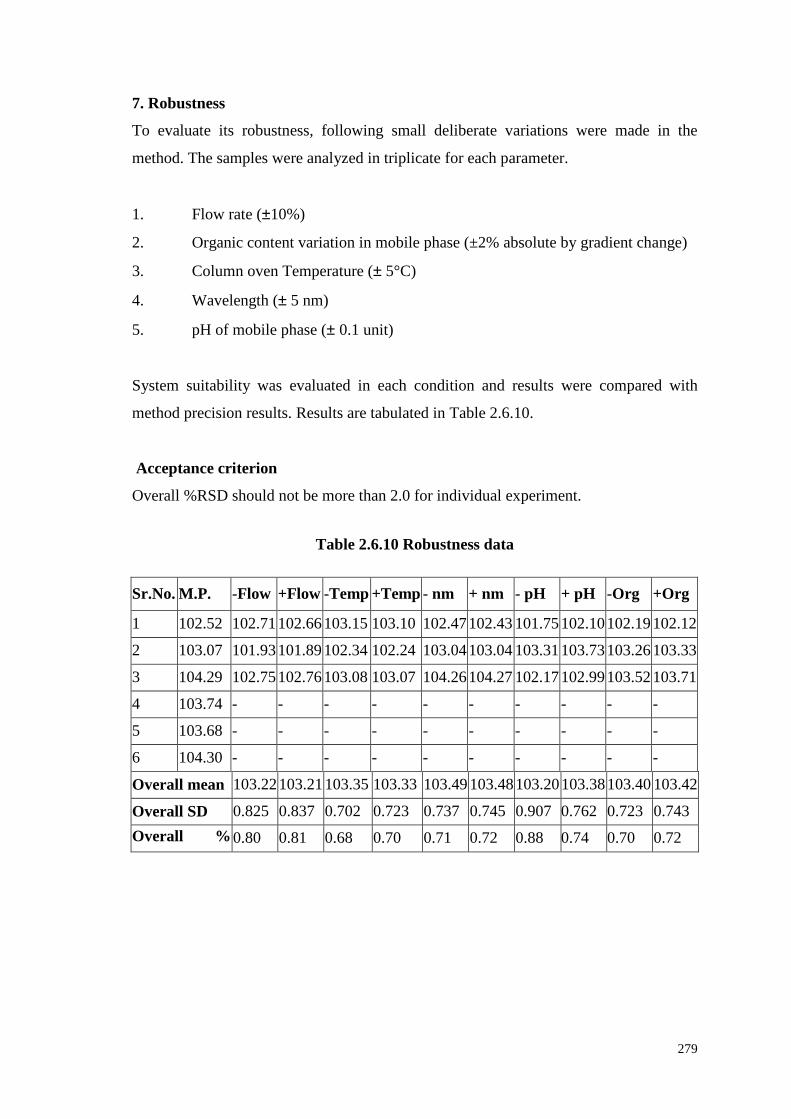
7. Robustness
To evaluate its robustness, following small deliberate variations were made in the
method. The samples were analyzed in triplicate for each parameter.
1. Flow rate (±10%)
2. Organic content variation in mobile phase (±2% absolute by gradient change)
3. Column oven Temperature (± 5°C)
4. Wavelength (± 5 nm)
5. pH of mobile phase (± 0.1 unit)
System suitability was evaluated in each condition and results were compared with
method precision results. Results are tabulated in Table 2.6.10.
Acceptance criterion
Overall %RSD should not be more than 2.0 for individual experiment.
Table 2.6.10 Robustness data
Sr.No. M.P. -Flow +Flow -Temp +Temp - nm + nm - pH + pH -Org +Org
1 102.52 102.71 102.66 103.15 103.10 102.47 102.43 101.75 102.10 102.19 102.12
2 103.07 101.93 101.89 102.34 102.24 103.04 103.04 103.31 103.73 103.26 103.33
3 104.29 102.75 102.76 103.08 103.07 104.26 104.27 102.17 102.99 103.52 103.71
4 103.74 - - - - - - - - - -
5 103.68 - - - - - - - - - -
6 104.30 - - - - - - - - - -
Overall mean 103.22 103.21 103.35 103.33 103.49 103.48 103.20 103.38 103.40 103.42
Overall SD 0.825 0.837 0.702 0.723 0.737 0.745 0.907 0.762 0.723 0.743
Overall % RSD
0.80 0.81 0.68 0.70 0.71 0.72 0.88 0.74 0.70 0.72

280
M.P. Method precision data
-Flow Flow rate (1.35 mL/minute)
+Flow Flow rate (1.65 mL/minute)
-Temp Column oven temperature (35°C)
+Temp Column oven temperature (45°C)
-nm Wavelength (259 nm)
+nm Wavelength (269 nm)
-Org Organic content variation in mobile phase (- 2% absolute by gradient change)
+Org Organic content variation in mobile phase (+ 2% absolute by gradient change)
- pH pH of buffer in mobile phase (2.90)
+ pH pH of buffer in mobile phase (3.10)
Conclusion
As method meets acceptance criteria, the method considered to be robust for changes in
flow rate, column oven temperature, wavelength, pH of buffer in mobile phase and
organic content variation in mobile phase.
8. Limit of Detection and Limit of Quantification
Limit of detection (LOD) is defined as the lowest concentration of analyte that gives a
measurable response. LOD is determined based on signal to noise ratio (S/N) of three
times typically for HPLC methods.The limit of quantification (LOQ) is defined as the
lowest concentration that can be quantified reliably with a specified level of accuracy
and precision. It is the lowest concentration at which the precision expressed by an RSD
of less than 2%. In this study the analyte response is 10 times greater than the noise
response. For this study six replicates of the analyte at lowest concentration in the
calibration range were measured and quantified. The LOD and LOQ of Pramipexole
obtained by the proposed method were 0.035 and 0.122 µg/mL respectively

281
9. Summary of system suitability
System suitability was evaluated by injecting standard solution during different days of
validationand monitoring tailing factor and theoretical plates for different parameters.
The % Relative standard deviation for the peak area counts of Pramipexole from five
replicate injections of standard solution were verified during every experiment. The
results are tabulated in Table 2.6.11.
Acceptance criteria
1. Column efficiency determined for the Pramipexole peak in standard solution should
be not less than 15000 theoretical plates and tailing factor for the same peak should be
not more than 2.0
2. Percentage relative standard deviation for peak area counts of Pramipexole from
five replicate injection of standard solution should be not more than 2.0
Table 2.6.11 Summary of system suitability data
Sr. No.
Name of experiment
Theoretical Plate counts of Pramipexole
peak
Tailing factor of Pramipexole
peak % RSD
1 System precision, Method precision
58292 1.0 0.07
2 Robustness (- Wavelength) 58262 1.0 0.09
3 Robustness (+ Wavelength) 58310 1.0 0.07
4 Solution stability 58979 1.0 0.04
5 Linearity 80377 1.0 0.32
6 Specificity 55419 1.0 0.34
7 Recovery 78255 1.0 0.07
8 Robustness (- Flow Rate) 63765 1.0 0.10
9 Robustness (+ Flow Rate) 53835 1.0 0.03
10 Robustness (- Temperature) 56499 1.0 0.05
11 Robustness (+ Temperature) 59090 1.0 0.06
12 Ruggedness 72940 1.0 0.10
13 Robustness (- pH) 55263 1.0 0.06
14 Robustness (+ pH) 55358 1.0 0.10
15 Robustness (-Organic) 94705 1.0 0.04
16 Robustness (+Organic) 53807 1.0 0.03
17 Forced Degradation-1 55590 1.0 0.15
18 Forced Degradation-2 54932 1.0 0.19

282
4) SUMMARY OF THE RESULTS AND CONCLUSION
The present study was aimed at developing a simple, precise and accurate HPLC
method for the analysis of pramipexole dihydrochloride monohydrate from tablet
dosage forms. A non-polar C8 analytical chromatographic column was chosen as the
stationary phase for the separation and determination of Pregabalin. For the selection of
the mobile phase a number of eluting systems were examined. Mixtures of commonly
used solvents like water, and acetonitrile with or without different buffers in different
combinations were tested as mobile phases on a C8 stationary phase. The choice of the
optimum composition is based on the chromatographic response factor, a good peak
shape with minimum tailing. A buffer mixture containing 9.1 g of potassium dihydrogen
phosphate and about 5.0 g of Octane-1 sulphonic acid sodium salt was dissolved in
1000 mL of water. The pH was adjusted to 3.0 (+0.05) with orthophosphoric acid and
acetonitrile with the gradient program was proved to be the most suitable of all the
combinations since the chromatographic peak obtained was better defined and resolved
and almost free from tailing. The retention time of the drug was found at 8.53 min.
Summary of Validation:
Specificity
No interfering peak was observed at the retention time of Pramipexole dihydrochloride
monohydrate from blank and placebo samples. Thus, the Peak purity for the analyte
passed.
Hence, it is concluded that method is specific for determination of Pramipexole
dihydrochloride monohydrate in Pramipexole dihydrochloride extended release tablets.
Forced Degradation
Forced degradation study was carried out by subjecting the placebo and sample to the
following conditions.
1. Treatment with hydrochloric acid.
2. Treatment with sodium hydroxide.
3. Treatment with hydrogen peroxide.
4. Thermal exposure.
5. Photolytic exposure.
6. Exposure to humidity

283
In all the above conditions, met the acceptance criterion for peak purity. Hence, the
method can be considered as stability indicating for determination of Pramipexole
dihydrochloride monohydrate in Pramipexole dihydrochloride extended release tablets.
Observation: During Force Degradation study it was observed that Pramipexole
dihydrochloride monohydrate is sensitive for Peroxide degradation.
System Precision
The % RSD of Pramipexole peak area from six replicate injections of Standard solution
was less than 2.0 and it meets the acceptance criterion.
Method Precision
Six samples from a single batch were prepared and analyzed as per test method. % RSD
for % assay of Pramipexole dihydrochloride monohydrate was calculated for six
preparations. The % RSD for % assay of Pramipexole dihydrochloride monohydrate
was less than 2.0 and meets the acceptance criteria.
Ruggedness
Six samples from a single batch (same batch used under Method precision) were
prepared and analysed as per test method by different analyst by using different column,
different HPLC system on different day.
% RSD for % assay of Pramipexole dihydrochloride monohydrate was calculated for six
preparations.
% RSD for % assay of Pramipexole dihydrochloride monohydrate was less than 2.0 and
meets the acceptance criteria.
Overall % RSD for % assay of Pramipexole dihydrochloride monohydrate obtained
from ruggedness and method was less than 2.0 and meets the acceptance criteria.
The proposed analytical method meets acceptance criteria for precision. Hence the
method is precise.
Accuracy (Recovery)
The sample solutions were prepared at each level by spiking the placebo with
Pramipexole dihydrochloride monohydrate API with at about 50%, 100 % and 150 % of
test concentration. The % Recovery at each level was calculated.

284
Analytical method meets acceptance criteria for recovery study. Hence, the method is
accurate and precise.
Linearity
Linearity range for Pramipexole dihydrochloride monohydrate was determined using
solutions containing about 50% to 150% of test concentration. It was found that
response for Pramipexole dihydrochloride monohydrate was linear in the range of 7.705
mcg/mL to 23.115 mcg/mL and the relevant correlation coefficient value is more than
0.99.
Stability in analytical solution
By analyzing the sample solution, at different time intervals, stability of the drug in
analytical solution was carried out. It was found that sample solution was stable up to
25hrs at room temperature.
Robustness
Robustness of analytical method was carried out by deliberately varying optimized
chromatographic conditions of flow rate (+10%), column oven temperature (±5°C),
wavelength (±5nm) and organic content of mobile phase (+2% absolute by gradient
change), change in pH of buffer in mobile phase (± 0.1unit). The method is robust for
change in organic content in mobile phase, change in flow rate, change in column oven
temperature, change in pH of mobile phase and change in wavelength, as method meets
acceptance criteria
Limit of detection and Limit of Quantification
The lowest values of LOD and LOQ as obtained by the proposed method indicate the
method is sensitive.
Conclusion
The validation data proves that the proposed method for determination of Pramipexole
dihydrochloride monohydrate in Pramipexole dihydrochloride extended release tablets
is specific, precise, accurate, robust and linear under the given conditions of
methodology and is suitable for use.

285
Fig 2.6.2: HPLC Chromatogram of blank

286
Fig 2.6.3: HPLC Chromatogram of placebo solution

287
Fig 2.6.4: HPLC Chromatogram and purity plot of placebo with drug sample
solution

288
Fig 2.6.5: HPLC Chromatogram and purity plot of untreated sample

289
Fig 2.6.6: HPLC Chromatogram and purity plot of acid treated sample

290
Fig 2.6.7: HPLC Chromatogram and purity plot of alkali treated sample

291
Fig 2.6.8: HPLC Chromatogram and purity plot of peroxide treated sample

292
Fig 2.6.9: HPLC Chromatogram and purity plot of thermal treated sample

293
Fig 2.6.10: HPLC Chromatogram and purity plot of photolytic treated sample

294
Fig 2.611: HPLC Chromatogram and purity plot of humidity treated sample

295
Fig 2.6.12: Linearity plot for Pramipexole dihydrochloride monohydrate
Linearity level (%)
Concentration (mcg/mL)
Pramipexole average peak area counts (µV*sec)
50 7.705 395416
60 9.246 471957
70 10.787 545932
80 12.328 632844
90 13.869 708402
100 15.410 787282
110 16.951 865870
120 18.492 941393
130 20.033 1024589
140 21.574 1099292
150 23.115 1180302
Slope 50996
Intercept 816
CC 0.99996

296
5. REFERENCES
1. Merck Index, P.No.: 1755(7705).
2. USP 32 NF 27, United States Phamacopoeia, The United States Pharmacopoeial
Convention, Rockville, MD, 2009.
3. http://www.drugbank.ca/drugs/DB00413
4. http://www.rxlist.com/cgi/generic/Pramipexole.htm
5. http://www.wikipedia.com.
6. Sweetman SC. Martindale, The Complete Drug Reference. 34rd ed. London:
Pharmaceutical Press, 2005.
7. Panchal JG, Patel RV, Menon SK.
Development and validation of GC/MS method for determination of pramipexole
in rat plasma.
Biomed Chromatogr. 25(4)524-30(2011).
8. G. Srinubabu, K. Jaganbabu, B. Sudharani, K. Venugopal, G.
Girizasankar and J. V. L. N. S. Rao.
Development and Validation of a LC Method for the Determination of
Pramipexole Using an Experimental Design.
CHROMATOGRAPHIA. 64(1-2), 95-100.
.
9. D. Vijaya Bharathi, Kishore Kumar Hotha, P. V. Vidya Sagar, S. Sirish
Kumar, A. Naidu and Ramesh Mullangi.
Development and validation of a sensitive LC-MS/MS method with electrospray
ionization for quantitation of pramipexole in human plasma: application to a
clinical pharmacokinetic study.
Biomedical Chromatography, 23(2 )212–218(2009).

297
10. V.Ramakrishna, S.Nirogi, Vishwottam, Kandikere, Wishu Shrivastava,
Koteshwara Mudigonda, Santosh Maurya and Devender Ajjala.
Quantification of pramipexole in human plasma by liquid chromatography
tandem mass spectrometry using tamsulosin as internal standard.
Biomedical Chromatography, 21(11), 1151–1158, (2007).
11. D.B. Pathare, A.S. Jadhav, and M.S. Shingare.
Validated chiral liquid chromatographic method for the enantiomeric separation
of Pramipexole dihydrochloride monohydrate.
Journal of Pharmaceutical and Biomedical Analysis, 41(4), 1152-1156(2006).
12. Yadav, Manish; Rao, Rajasekhar; Kurani, Hemal; Rathod, Jaysukh; Patel,
Rakesh; Singhal, Puran; Shrivastav, S.Pranav
Validated Ultra-Performance Liquid Chromatography Tandem Mass
Spectrometry
Method for the Determination of Pramipexole in Human Plasma.
Journal of Chromatographic Science, 48(10),811-818(2010).
13. B.M. Gurupadayya, V. Vishwajith and N. Srujana.
Spectrophotometric Methods for the Estimation of Pramipexole Dihydrochloride
in Pharmaceutical Formulations.
World Journal of Chemistry 4 (2), 157-160( 2009).
14. Petikam Lavudu, Chandra Bala Sekaran, Nallani Gayathri , Tumpara Srinu, and
Avula Prameela Rani.
Analysis of Pramipexole dihydrochloride by Vis-Spectrophotometric method
Development and Validation.
Journal of Pharmacy Research 4(5), 1493-1496(2011).

298
15. C.vinodhini, v.n.d. Malladi sravani, Mangam, Bhanuprakash, Mantena, sudha
devi, Mohamad Imran, Mohamed Omer Abdelaziz Osman, K.Chitra, C. and
Uma Maheshwara reddy.
Method Development and Validation of Pramipexole Dihydrochloride
Monohydrate in Tablet Dosage Form by UV and Visible Spectrophotometric
Methods.
International Journal of Research in Pharmaceutical and Biomedical Sciences 2
(2), 680-686(2011).
16. ICH Guidelines Q2B, Validation of Analytical Procedure: Definitions, published
in March 1996, Geneva, Switzerland.
17. Validation of analytical procedures: Text and Methodology, ICH Hormonised
Tripartite Guideline Q2 (R1), Commission of the European Communities
2005.
18. ICH (2003) Stability Testing of New Drug Substances and Products,
International Conference on Harmonization, Q1A (R2), IFPMA, Geneva,
Switzerland.
19. ICH, Q2A Validation of Analytical Procedure: Methodology International
Conference on Harmonization, Geneva October 1994.