aprotic and aqueous li−o2 batteries.pdf
TRANSCRIPT

Aprotic and Aqueous Li−O2 BatteriesJun Lu,† Li Li,‡ Jin-Bum Park,§ Yang-Kook Sun,*,§ Feng Wu,*,‡ and Khalil Amine*,†,∥
†Chemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439,United States‡Beijing Key Laboratory of Environmental Science and Engineering, School of Chemical Engineering and the Environment, BeijingInstitute of Technology, Beijing 100081, China§Department of Energy Engineering, Hanyang University, Seoul 133-791, South Korea∥Chemistry Department, Faculty of Science, King Abdulaziz University, 80203 Jeddah, Saudi Arabia
CONTENTS
1. Introduction A2. Overview B3. Aprotic Li−O2 Battery D
3.1. Electrochemical Reactions in the Aprotic Li−O2 System D
3.2. Early-Stage Research on the Aprotic Li−O2
Battery E3.3. Effect of the Electrolyte F
3.3.1. Lessons Learned from Organic Carbo-nate-Based Electrolytes G
3.3.2. Ether-Based Electrolyte H3.3.3. Other Organic Electrolytes K3.3.4. Effect of Lithium Salt M3.3.5. Hard−Soft Acid−Base (HSAB) Theory M3.3.6. Brief Summary N
3.4. Electronic and Magnetic Properties of Li2O2
and Relevance to the Li−O2 Battery N3.5. Air Electrode Architecture O
3.5.1. Porous Carbon P3.5.2. Catalysts on Porous Carbon R
3.6. Lithium Electrode T4. Aqueous Li−O2 Batteries V5. Concluding Remarks and Perspectives WAuthor Information X
Corresponding Authors XNotes XBiographies Y
Acknowledgments ZReferences Z
1. INTRODUCTION
Currently, fossil fuels supply over 85% of the world’s ever-growing energy demand.1 There is an increasing concern aboutthe global climate change resulting from the worldwide use offossil fuels, which release large quantities of CO2 and othergreenhouse gas (GHG) to the atmosphere.1 The petroleumthat is used for automobile and light truck applicationsrepresents 34% of the world’s total primary energy source. Inthe United States, the transportation sector is the single greatestconsumer of imported oil. In 2010, 94% of U.S. transportationenergy was derived from petroleum, nearly half of which camefrom foreign sources.2 In terms of economic impact, petroleumimports represented nearly 41% of the $646 billion U.S. tradedeficit in 2010.3 The CO2 emissions due to the U.S.transportation sector account for 40% of the total CO2emission, which is considered as a major cause of geopoliticalinstability.4 The U.S. transportation sector also representsabout 27% of all U.S. GHG emissions.5 However, even withtoday’s mix of fossil, nuclear, and renewable energy sources forU.S. electric power generation, it is estimated that, on a well-to-wheel basis, an all-electric vehicle will generate 25% less GHGemissions than a conventional gasoline-powered vehicle.6 Evenlower emissions are predicted with increased use of renewableenergy sources. Therefore, it would greatly benefit the UnitedStates and the world to transition to an electrified trans-portation system, which is already beginning with the advent ofhybrid electric vehicles (HEVs) and will accelerate as plug-inhybrid electric vehicles (PHEVs) and ultimately pure electricvehicles (PEVs) gain a larger share of the market. In addition,PEVs have the promise to greatly improve energy efficiency.For example, on a well-to-wheel basis, the all-electric TeslaRoaster charged with electricity generated from natural gas hasan efficiency of 1.14 km/MJ, nearly 2 times as efficient as aToyota Prius hybrid (0.56 km/MJ) and 4 times more efficientthan a typical gasoline-powered car, such as the Toyota Camry(0.28 km/MJ).7 The use of alternative energy sources, such asnuclear, solar, and wind power, would reduce our dependenceon fossil fuels and, thus, also reduce CO2 emissions, but devicesto store the electric energy generated by these power plants aresorely needed. One of the most viable candidates for suchdevices is the rechargeable Li battery.Electrical energy storage is attracting significantly more
interest nowadays, considering the expanding market for
Received: October 16, 2013
Review
pubs.acs.org/CR
© XXXX American Chemical Society A dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXX

portable electronics, the electrification of the transportationsector as a result of the commercialization of battery-poweredvehicles, and the greater use of batteries for grid energystorage.8 The specific energy (energy per unit mass) and energydensity (energy per unit volume) for state-of-the-art Li ionbatteries are much too low for all-electric vehicles, even withthe assumption that the theoretical capacity of the electrodematerials can be achieved.9 Given today’s automobile Li ioncells, the driving range is limited to about 70 miles for a 200 kgbattery pack, assuming a specific energy of 150 W·h/kg at thecell level and 105 W·h/kg at the pack level (70% of a pack’smass is due to the cells) can be realized. With the recentintensive development of high-capacity positive electrodematerials (275 mA·h/g) and high-capacity alloy negativeelectrode materials (2000 mA·h/g), the specific energy of theLi ion batteries could be eventually pushed up to 400 W·h/kg.However, even with such high capacity, the vehicle drivingrange is only doubled at the most (140 miles). The U.S.Advanced Battery Consortium (USABC) set the goals for EVsat a calendar life of 15 years and operating temperature from−30 to +52 °C with a driving range of 300 miles per singlecharge, which is beyond the electrochemical performance oftoday’s lithium ion batteries. To produce electric vehicles with arange comparable to that of today’s vehicles powered by liquidfuels, a battery system that has much higher specific energy andenergy density is required. Therefore, an increasing amount ofrecent research has been devoted to energy storage systems thatcan go beyond the Li ion battery limits.9
One such technology is the Li−air battery, which is based onthe Li−O2 electrochemical couple.10 A lithium−air cell whendischarged to the lithium peroxide composition (Li2O2) at anaverage potential of 3.1 V would provide a theoretical specificenergy of 3623 W·h/kg and when discharged to Li2O at thesame potential would provide a theoretical specific energy of5204 W·h/kg. Originally proposed in the 1970s as a possiblepower source for electric vehicles,11−21 Li−air batteriesrecaptured significant scientific interest in the late 2000s dueto advances in materials technology and an increasing demandfor environmentally safe and oil-independent energy sources.Interest has increased sharply recently, as evidenced by over300 research papers having been published on this topic in thepast 3 years alone. This intense research activity can beattributed to the high energy density of the Li−air battery,potentially up to 2−3 kW·h/kg on the cell level, and the opencell configuration that uses air as the reactant. A fully developedLi−air battery system is expected to truly surpass the currentbattery technology, even that under development for deploy-ment in the medium term (400 W·h/kg), and meet therequirements for the PEV application.Development of a practical Li−air battery will involve
overcoming many formidable challenges, including the need fora fundamental understanding of Li−O2 electrochemistry,development of new and improved cell materials, andinnovation in the critical aspects of cell design. In the pastfew years, dozens of reviews on the topic of Li−airbatteries9,22−43 have been published. These reviews addressthe technical issues and challenges facing Li−air batteries at thecurrent stage from different perspectives, including the stabilityof the electrolytes, importance of the air electrode/electro-catalyst, and oxygen-selective membranes. In this review, wemainly focus on the most critical issues that must be addressed,with the hope that it will help to advance a truly rechargeableLi−air battery toward its practical application.
We start by presenting an overview of the Li−O2 battery insection 2, including operation principles, different cellchemistries, and primary development challenges. The mainfocus of section 3 is the aprotic Li−O2 system, since it has beendemonstrated to possess the greatest potential to meet the PEVrequirements and has dominated the Li−air battery researchefforts in the past decade. This section discusses electro-chemical reactions in the aprotic Li−O2 system, the stability ofaprotic electrolytes and its effect on cell performance, theelectronic and magnetic properties of Li2O2 and their relevanceto the aprotic Li−O2 battery, the importance of the O2-breathing electrode, and the effect of O2 crossover on thestability of the lithium electrode. In section 4, we briefly discussaqueous Li−air systems and the challenges they are facing.Section 5 presents concluding remarks and prospects for thefuture development of Li−air batteries. Covering the immensebody of all the work published in this field is, however, beyondthe scope of this review.
2. OVERVIEWBefore presenting the main topic of this review, we need toclarify some terminology. First, it is not precise to refer to anaprotic solvent as “nonaqueous” in most of the relevantliterature because, strictly speaking, nonaqueous solventsinclude aprotic and protic solvents. The term “aprotic” isused throughout this review since almost all the electrolytesinvestigated so far are based on the aprotic solvent. We do notconsider protic nonaqueous Li−air battery systems because of alack of published work on this topic. Likewise, mixtures ofwater and other protic solvents are beyond the scope of thisreview. From an electrochemical perspective, the protic systemis expected to share general characteristics with the aqueousLi−air chemistry. Also, the term “Li−air battery” with theaprotic system has been widely accepted and used by manyother researchers but does not precisely represent whathappens in these cells at the current time, considering thatmost laboratory work has been performed under a pure oxygenenvironment. This is because other components in air such asH2O and CO2 could interfere with the desired electrochemicalbehavior and, therefore, degrade the overall performance of thebattery. In light of the above concern, we refer to “Li−O2battery” throughout this review when we discuss the aprotic“Li−air” system. This distinction has been recognized in somerecently published papers.44−47 However, it should beemphasized that a true Li−air battery is still the ultimategoal,48,49 as long as a selective membrane can be developed toprevent the permeation of other gases from the air rather thanoxygen gas. In addition, the expression of the specific capacityof the Li−O2 battery is specifically clarified in this review. Theterm “mA·h/g” is commonly used in most of the relevantliterature, some based on the mass of active materials andothers based on the mass of the air electrode support. Thevalue based on the electrode support mass is reliable only whenthe capacity is shown to be proportional to the support mass. Inthis sense, the value based on the active materials is moreappropriate to describe the specific capacity of the Li−O2 cell atthe current moment. Alternatively, the capacity per surface area(mA·h/cm2) of the support is also accepted to describe thespecific capacity of the cell. However, the value based on thesurface area of the electrode could possibly induce improperconclusions if the loading of the active materials is significantlydifferent from case to case. In this paper, we adopt the term“mA·h/g” based on the active materials to describe the specific
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXB

capacity of the cell. Likewise, the rate expression “mA/g” basedon the active materials is used in this paper for the same reason.Practically, the energy density on the cell level would be a morereliable value to demonstrate this promising technology.The Li−O2 battery chemistry uses the oxidation of lithium at
the metal electrode and reduction of oxygen at the air electrodeto induce current flow. The major appeal of the Li−O2 batteryis its extremely high energy density, which rivals that oftraditional gasoline-powered engines. This advantage in energydensity is derived by utilizing the oxygen in air, eliminating theneed to store reactant (oxygen) at the air electrode. On thebasis of the oxidation of 1 kg of lithium metal, the theoreticalenergy density of a Li−O2 cell is calculated to be 11 680 W·h/kg, which is not much lower than that of gasoline (13 000 W·h/kg), as shown in Figure 1.35 Practically, the achievable energy
density for Li−O2 batteries is far less since it also stronglydepends on the porosity of the air electrode and the electrolyteused at the air electrode side.50 In the case of an aprotic system,the amount of insoluble discharge products stored in the airelectrode will ultimately determine the overall energy density,while in the case of an aqueous system, the solubility of LiOHin the base aqueous electrolyte is the limiting factor to theenergy density.34 On the basis of charge balance theory, Zhenget al.50 predicted that the maximum possible gravimetric andvolumetric energy density of a Li−O2 battery in an aqueouselectrolyte is considerably less than that when using an aproticelectrolyte. Note that the usable energy density of gasoline forautomotive applications is approximately 1700 W·h/kg,assuming an average tank-to-wheel efficiency (12.6%) of theU.S. fleet. Fortunately, such energy density accounts only for14.5% of the theoretical energy content of a fully charged Li−O2 battery, so it is not inconceivable that such a high energydensity may be achievable at the cell level, given intensiveresearch effort and long-term development.Currently, four types of Li−O2 batteries are under develop-
ment and are designated by the type of electrolyte employed:aprotic,10,51 aqueous,52 solid-state,53 and hybrid aqueous/
aprotic54 (see Figure 2). For all types of Li−O2 systems, anopen system is required to obtain oxygen from the air becauseoxygen is the active material of the air electrodes. Li metal mustalso be used as the metal electrode to provide the lithiumsource for all the systems at the current stage. In an aprotic Li−O2 cell, porous carbon must be added as the reservoir for theinsoluble discharge products, presumably Li2O2. Most of thetime, electrocatalysts are essential to promote the oxygenreduction and oxygen evolution reactions during the celldischarge and charge. In the case of the aqueous and hybridaqueous/aprotic systems, a protective layer for Li metal, whichprevents the vigorous reaction of lithium with water, isnecessary to enable the desired electrochemistry. The chemistryat the oxygen electrode differs depending on the electrolyte.Aqueous and hybrid systems share the same reactionmechanisms since the air electrodes in both cases are exposedto an aqueous electrolyte. The solid-state Li−O2 battery mayfunction similarly to the aprotic system, although it is notwidely studied in detail yet due to the lack of a solid-stateelectrolyte with sufficient lithium ion conductivity. For thisreason, we only focus on the aprotic and aqueous Li−O2systems, with a particular emphasis on the former since it hasdominated the research effort on Li−O2 batteries for the pastdecade. It is assumed that the knowledge gained from these twosystems could also be applied to the solid-state and hybridsystems due to the similarity of their chemistries.Because oxygen is supplied as a reactant to the cell during
discharge, Li−O2 cells differ from other batteries such as lead−acid, nickel−metal hydride, and lithium ion systems; they canthus be constructed as a part of hybrid battery−fuel cellsystems. During electrochemical discharge, the Li electrode isoxidized by releasing an electron to the external circuit toproduce Li ions in the electrolyte, whereas the oxygen isreduced at a catalytic air electrode surface to form, in the caseof aqueous electrolytes, a lithium hydroxide product and, foraprotic electrolytes, a lithium peroxide (Li2O2) or lithium oxide(Li2O). This process is expected to be reversed on electro-chemical charge in the aprotic system, making the cellrechargeable. The Li−O2 systems must be open with a porousair electrode to allow the diffusion of the oxygen gas to theelectrolyte/electrode interface. This design significantly differsfrom the conventional Li ion cell design with a completelyclosed configuration. Use of a catalytic porous air electrode isparticularly important to aprotic Li−O2 cells since it not onlyfacilitates the oxygen reduction and oxygen evolution reactionsbut also provides the space to store the insoluble dischargedproducts, mainly lithium oxides. A porous air electrode withhighly catalytic activity toward oxygen evolution reaction is alsoa key factor in realizing a secondary aqueous Li−air battery.This promising Li−O2 battery technology is still in its infancy
and, no doubt, will require significant research efforts in avariety of fields to unlock its full potential. However,researchers and industry alike see a great chance in itsdevelopment with the recent progress in advanced electrode,electrolyte, and catalytic materials. Especially, the research effortled by IBM to develop a Li−air battery capable of driving acommercial vehicle 500 miles on a single charge has sharplyaccelerated research interest.35
Currently, many challenges prevent the realization of high-performance Li−O2 batteries for the aprotic system. One of thebiggest is that the current aprotic electrolytes are often unstableagainst several active discharge species (i.e., O2−, O2
2− (Li2O2),LiO2, and LiO2
−).55 Selection of the electrolyte is the key
Figure 1. Gravimetric energy densities (W·h/kg) for various types ofrechargeable batteries compared to gasoline. The theoretical density isbased strictly on thermodynamics and is shown as blue bars, while thepractical achievable density is indicated by orange bars and numericalvalues. For Li−air, the practical value is just an estimate. For gasoline,the practical value includes the average tank-to-wheel efficiency of cars.Reprinted from ref 34. Copyright 2010 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXC

component to improve the electrochemical performance of theaprotic Li−O2 system. Carbonate-based electrolytes have beenproved to be unstable in the aprotic Li−O2 cells, while ether-based electrolyte showed some promise. The electrolytestability at the lithium electrode under the oxygen crossovercondition is also a big issue, necessitating development of newelectrolytes and a redesigned solid−electrolyte interface (SEI)on the lithium electrode. Substantial difficulties are faced inpreparing structures for the storage of insoluble dischargeproducts, presumably lithium oxide or lithium peroxide, at theair electrode, which must also be made electrically conductive.Porous carbon electrodes are the material of choice for mostcurrent research, but pore clogging by the insoluble dischargeproducts in aprotic systems must be balanced with the need foroxygen permeation. Catalysts have been shown to improve airelectrode performance, especially for lowering the chargeoverpotential, with MnO2 being investigated most.56 However,the mechanism of such improvement due to catalytic activity isnot yet clear, which could be the key factor in developing apractical Li−O2 battery. Significant challenges have also beenfaced at the pure lithium electrode: oxygen crossover effects57
and well-known dendritic lithium formation, long a problem inlithium ion batteries, both of which eventually lead to decay ofthe cell. Further complicating the design of Li−O2 batteries isthe degradation of active materials by atmospheric contami-nants, such as H2O and CO2,
58 which require a selectivemembrane that allows O2 permeation only. A difficult challengefor aqueous Li−O2 batteries is the development of good Li ion
conducting membranes to protect the lithium electrode fromreacting vigorously with H2O. In the following sections, wediscuss the recent research to address the aforementionedissues and share our perspectives with readers on how to betterunderstand the chemistries involved in the Li−O2 systems andimprove their electrochemical performance.
3. APROTIC LI−O2 BATTERY
3.1. Electrochemical Reactions in the Aprotic Li−O2 System
As shown in Figure 2a, a typical aprotic Li−O2 cell is composedof a lithium electrode, an electrolyte consisting of dissolvedlithium salt in an aprotic solvent, and a porous O2-breathingelectrode that contains carbon particles and, in some cases, anadded electrocatalyst. Note that the oxygen reduction reaction(ORR) during discharge and oxygen evolution reaction (OER)during charge of a Li−O2 cell occur at a three-phase boundaryinvolving the solid electrode, liquid electrolyte, and oxygen gas,which makes the Li−O2 system more complicated than theconventional Li ion battery.Assuming no parasitic side reactions are taking place, the
fundamental chemistry of a Li−O2 electrochemical coupleduring discharge will entail several possible reactions at the airelectrode via an oxygen reduction process, as described inFigure 3. All the reactions except for reaction 2 (R2), thedisproportionation of lithium superoxide (LiO2), have astandard redox potential close to 3.0 V vs Li/Li+, whichsuggests that these reactions are equally favored thermodynami-cally. However, the oxygen reduction via a one-electron transfer
Figure 2. Schematic cell configurations for the four types of Li−air batteries.
Figure 3. Possible electrochemical and chemical reactions for Li−O2 couples.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXD

(R1) would be most favorable kinetically during discharge,which leads to the formation of LiO2. Subsequently, LiO2 canturn into lithium peroxide (Li2O2) via either the disproportio-nation reaction (R2) due to its chemical instability or anotherone-electron-transfer electrochemical process (R3). Upondischarge, Li2O2 could also form on the air electrode througha direct two-electron transfer, as described in reaction 4. It isunclear at this moment which process is more favorable interms of the Li2O2 formation or whether they coexist andcompete with each other during the oxygen reduction, despite atwo-electron transfer having been reported recently.59 Althoughthe full reduction of O2 to lithium oxide (Li2O) through adirect four-electron transfer (R6) is kinetically least favored forthe substrates with weak binding for oxygen (such as carbon)considering that the oxygen dissociation on the substrates isrequired, it is the most desired reaction for aprotic Li−O2batteries because of its higher specific energy and energydensity. However, if the cutoff voltage is lowered to 2.0 V orbelow (vs Li/Li+), Li2O forms as the discharge product on theair electrode,60−62 so discharge most likely takes multistepreactions, for instance, R1 → R3 → R5, R1 → R2 → R5, or R4→ R5. Due to the thermal stability and insulating nature of thedischarge product, the subsequent charge of Li2O cannotproceed under the test condition.60 Therefore, Li2O2 is thedischarge product observed in most recent cell tests (cutoffvoltage >2.0 V). In addition, from a kinetic point of view, theformation of Li2O2 during discharge may be beneficial to therate performance of the cell, since full cleavage of the O−Obond may not be necessary, given that a suitable catalyst can beidentified. In practice, the reactions that occur on the airelectrode during discharge are much more complicated due tothe strong interaction between the reduced oxygen species(such as O2−, O2
2− (Li2O2), LiO2, and LiO2−) and other
components in the cell, namely, electrolyte, catalyst, and carbonsupport, which directly affect the oxygen reduction reactions aswell as the subsequent oxygen evolution reactions. We furtherreview and discuss these issues in the following sections.To make the Li−O2 cell rechargeable if Li2O2 is the discharge
product, decomposition of Li2O2 to lithium and oxygen(oxygen evolution reaction) is required, which could proceedvia either a two-electron process (Li2O2 → 2Li+ + 2e− + O2) ora one-electron process that involves the formation of LiO2(Li2O2 → Li+ + e− + LiO2). Figure 4 shows a typical discharge−charge profile of an aprotic Li−O2 cell, which shows a largepolarization and poor cycle life.44 One of the big challengeshere is to lower the charge overpotential to improve theroundtrip efficiency of the cell. Electrocatalysts, such asmanganese dioxide56 or noble metals,63 are often found tohave a positive effect on lowering the charge overpotential;however, the exact role they play in the oxygen evolutionreaction is not clear. We discuss the parameters that have beenshown to have a significant impact on the oxygen reduction andevolution reactions and look ahead to how the electrochemicalperformance of the Li−O2 cell can be improved.
3.2. Early-Stage Research on the Aprotic Li−O2 Battery
In 1996, Abraham and Jiang10 reported, for the first time, anaprotic Li−O2 battery using a polymer organic electrolyte inplace of the aqueous electrolyte. In doing so, the battery wasable to be recharged, despite the large overpotential betweenthe discharge and charge as well as a very short cycle life. Adischarge product of Li2O2 was confirmed by Ramanspectroscopy and qualitative analysis; this discharge product
is not formed in aqueous systems. Cobalt pthalocyanine-catalyzed porous carbon was used as the air electrode to lowerthe charge overpotential. Although researchers in this field nowlargely use liquid organic electrolytes in place of the polymer,this work still remains seminal. Abraham and co-workers alsoelucidated the mechanism of O2 reduction in Li−O2 batteriesthrough the application of rotating disk electrodes.64
Read and co-workers65−67 have largely focused on character-izing organic electrolytes for Li−O2 batteries. They have shownthat electrolyte formulation has a very large effect on dischargecapacity and rate capability and that these performancevariables can be correlated to the solubility, diffusioncoefficients, and partial pressure of O2 in the electrolyte.66
The discharge product (Li2O2/Li2O) and location of theproduct in relation to the air electrode are dependent on theelectrolyte type and discharge rate. It was subsequently shownthat the discharge product from oxygen reduction is Li2O if thedischarge potential is allowed to fall below 2.0 V (vs Li/Li+).For this reason, many researchers limit the discharge window toabove 2.0 V, with the expectation that Li2O is toothermodynamically stable to be easily decomposed uponcharging, as mentioned earlier. Read et al.68 have also studiedthe effects of the air electrode porosity, claiming that, at lowcycling rates, the porosity of the air electrode dominates the
Figure 4. (a) Voltage profile of the first cycle for a cathode containingthree-cycle atomic layer deposition (3c ALD) Pd in a carbon matrixcycling in 1 M LiCF3SO3/TEGDME at 100 mA/g. (b) Cell capacity asa function of the cycle number for the same air electrodes cycling in 1M LiCF3SO3/TEGDME at 100 mA/g. The specific capacity andcurrent density were based on the active materials (carbon + Pd).Reprinted from ref 44. Copyright 2010 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXE

battery performance. They developed a model that demon-strates how interparticle micro-, meso-, and macroporositiesdiffer in their importance to air electrode capabilities. Modelcalculations suggest that much improvement can be achievedwith proper tailoring of the air electrode’s physical properties.The research in this field was tremendously boosted in 2006
by Bruce and co-workers,51 who successfully tested a cell formore than 30 cycles using an air electrode loaded withelectrolytic manganese dioxide as the electrocatalyst. Thisperformance was achieved despite the cell having a conven-tional carbonate-based electrolyte, which is now known to beunstable to the oxygen reduction species (discussed in moredetail later). Bruce and co-workers also presented directevidence of oxygen release on cell charging, indicating thereversibility of Li2O2 formation on discharge. In follow-upwork,56 Bruce et al. evaluated an array of different electro-catalysts and showed that the catalyst does not influence thedischarge potential, but greatly affects the charging potentialand capacity retention per cycle. Overall, they unequivocallyshowed that, despite the reactions occurring during discharge
and charge, electrocatalysts are required to achieve multiplesuccessful cycles in Li−O2 batteries.All the aforementioned work on the aprotic Li−O2 batteries
is considered to be pioneering and, therefore, deservesemphasis in this review. Since then, excitement over thepotential of Li−O2 batteries has grown tremendously.Researchers have gained much better understanding of theLi−O2 chemistry, although significant challenges in thedevelopment of advanced materials for the stable electrolytesand electrodes have yet to be overcome.
3.3. Effect of the Electrolyte
One of the biggest challenges in the development ofrechargeable aprotic Li−O2 cells is to develop a stableelectrolyte, in particular one that has an organic solvent andcan survive nucleophilic attack of the superoxide radical (O2
−),an intermediate phase formed from the oxygen reductionreaction upon discharge. Indeed, the choice of the electrolytemight be the most critical factor in determining theelectrochemical performance of the cell. Although variousnonaqueous electrolytes have been investigated and used in Li
Figure 5. Composite electrodes (Super P/α-MnO2/Kynar) that contain the discharge products individually were subjected to charging in 1 M LiPF6in PC under O2. (a) FTIR spectra of the as-prepared electrodes and the charged electrodes for each of the compounds, together with the spectrum ofa pristine electrode. (b) Corresponding charging curves at 70 mA/g. Since the theoretical capacities of the different compounds vary, to aidcomparison, the capacities are all normalized to unity (theoretical capacities: lithium propyl dicarbonate, 1000 mA·h/g, 2e−/mol; Li2CO3, 1500 mA·h/g, 2e−/mol; CH3CO2Li, 750 mA·h/g, 1e−/mol; HCO2Li, 750 mA·h/g, 1e−/mol). (c−e) MS gas analysis at the end of charging under O2 ofCH3CO2Li (c), C3H6(OCO2Li)2 (d), and HCO2Li (e). Note that unmarked peaks arise from fragments of CO2, H2O, O2, and Ar. (f) Gas evolutionmeasured by DEMS on oxidation of a composite electrode containing Li2CO3 in response to a stepwise increased current under Ar. The specificcapacity and current density were based on the mass of the carbon. Reprinted from ref 79. Copyright 2011 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXF

ion batteries in the past few decades,69 these investigations arenormally performed under a closed and oxygen-free environ-ment, which is less aggressive than in the Li−O2 system. Theseelectrolytes should be re-examined for their stability in anoxygen-rich environment, especially in the presence of theoxygen reduced species, before they are tested in Li−O2
systems. Besides stability against the superoxide radical, agood electrolyte for Li−O2 cells should also meet the followingcriteria in addition to the typical requirements69 for electrolytesused in Li ion batteries: (1) stability toward reaction withlithium metal since this is the choice of the metal electrode forLi−O2 cells; (2) high boiling point and low volatility due to theopen cell system; (3) high oxygen solubility and diffusivity tofacilitate the oxygen reduction and oxygen evolution reactionson the air electrode; (4) low viscosity to improve the rateperformance of the oxygen electrode. Unfortunately, no singleelectrolyte investigated so far meets these demanding require-ments, despite extensive efforts having been devoted to thatend in the past few years. Understanding the reaction
mechanisms between the electrolytes and active oxygenreduced species will, no doubt, be the key to developing astable electrolyte for Li−O2 cells.
3.3.1. Lessons Learned from Organic Carbonate-Based Electrolytes. Organic carbonate-based electrolytes, inparticular propylene carbonate (PC), have been widelyinvestigated in aprotic Li−O2 batteries, mainly because theyare widely used in Li ion batteries. Propylene carbonate doespossess several advantages over other types of electrolytes, suchas a wide electrochemical window, low volatility, and a wideliquid-temperature range.69 However, such electrolytes are notstable in the presence of highly reactive species (i.e., O2−, O2
2−,LiO2, and LiO2
−) from the oxygen reduction reaction, despitethe Li2O2 being claimed or assuming to be the dischargeproduct in previous research.56,70,71 In fact, Read et al.72
pointed out the possible instability of carbonate-basedelectrolytes in Li−O2 cells almost a decade ago. Even muchearlier than the first report of the aprotic Li−O2 battery in1996,10 Aurbach et al.73,74 reported that the decomposition of
Figure 6. (a) Reaction free energy profile for nucleophillic attack of O2− at the carbonyl and ethereal carbon atoms of PC and the reaction pathway
toward the formation of allyl alcohol (CH2CHCH2OH) and CO2 calculated within the continuum solvent model based on DFT. Reprinted fromref 81. Copyright 2011 American Chemical Society. (b) Reaction energy profile for O2
− attack on the ethereal carbon atoms of EC based on theDFT and CCSD(T) levels of calculations. Reprinted from ref 80. Copyright 2011 American Chemical Society. (c) DFT results on the barriers foractivation of PC decomposition by other possible reduced O2 species, e.g., LiO2, LiO2
−, and Li2O2, besides O2−. Reprinted from ref 55. Copyright
2011 American Chemical Society. (d) PC decomposition due to the reactivity of PC with a [100] Li2O2 surface through the nucleophilic addition tothe carboxylic carbon. Reprinted with permission from ref 82. Copyright 2011 Wiley-VCH.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXG

propylene carbonate could occur on the intermediates/products of the electrochemical reduction of oxygen.Unfortunately, researchers did not pay much attention to theinstability of carbonate-based electrolytes until 2010, whenMizuno et al.75 presented direct evidence that the dischargeproducts of the PC-based Li−O2 cell were mainly lithiumcarbonate (Li2CO3) instead of the desired Li2O2. Since then, awealth of experimental and theoretical evidence has indicatedthat the organic carbonates (e.g., propylene carbonate, ethylenecarbonate, and dimethyl carbonate) are not stable toward theoxygen reduction products.55,75−81 For example, Bruce and co-workers79 demonstrated that the discharge products from theLi−O2 cell containing alkyl carbonate electrolyte mainly consistof various carbonate species, including lithium formate, lithiumacetate, and lithium carbonate, with no evidence for theformation of Li2O2 from Fourier transform infrared (FTIR)spectra, as shown in Figure 5. These carbonate compounds canbe oxidized on charging (Figure 5),79 leading to the formationof CO2 as the primary charge product, as evidenced by in situdifferential electrochemical mass spectrometry (DEMS)combined with 18O isotope labeling experiments.78 This chargeproduct is responsible for the large overpotential, as widelyobserved by other researchers. As a net effect, the cycleperformance of the Li−O2 cell using carbonate-based electro-lytes is associated with the consumption of the electrolytesrather than reversible discharge/charge of the desired Li2O2.The intermediates from the oxygen reduction reaction during
the discharge of the cell are believed to be responsible for thedecomposition of the carbonate-based electrolytes. To under-stand the nature of the reaction mechanism, a densityfunctional theory (DFT) study and coupled-cluster calculationsby Bryantsev and Blanco81 found that propylene carbonate ishighly susceptible to the nucleophilic attack of superoxideradical (O2
−), as shown in Figure 6a. Subsequent reactions ledto the formation of carbonate species as the discharge products.Bryantsev and Blanco81 also found that similar decompositionpathways are valid for other organic carbonate solvents, e.g.,ethylene carbonate (EC) and dimethyl carbonates (DMC),which have computed activation barriers of about 12.4−15.5kcal/mol. According to Bryantsev and Blanco,81 the nucleo-philic attack by O2
− at ethereal carbon atoms is a commonmechanism of degradation of organic carbonate solvents(Figure 6). However, other oxygen reduced species, such asO2−, O2
2−, LiO2, and LiO2−, may exist in the Li−O2 cell, and
they could also cause the electrolyte decomposition.From the modeling results based on DFT and high-level
quantum chemistry calculation, Zhang et al.55 demonstratedthat the decomposition of propylene carbonate starts from“ring-opening”, i.e., C−O bond breaking, which is consistentwith the findings of Bryantsev et al.81 The calculated energybarriers of the C−O bond breaking for all four oxygen reducedspecies considered (i.e., O2−, O2
2−, LiO2, and LiO2−) are quite
small, with LiO2− surprisingly being the most reactive, as shown
in Figure 6c. Once the ring of PC is opened, the subsequentC−O bond breaking is easier and thermodynamically favorable;this condition could lead to the formation of Li2CO3 or otherproducts such as formaldehyde and acetaldehyde with theassumption that a second electron transfer can occur. Thistheoretical prediction was further confirmed by X-ray photo-electron spectroscopy (XPS),55 which indicated that lithiumcarbonate was the major product formed during discharge, withonly minor amounts of lithium oxides being formed. The XPSdata also indicate that, during the first charge cycle, the lithium
carbonate was decomposed, consistent with previous findingsby Bruce et al.79
In addition, the reactions of the electrolyte at lithiumperoxide surfaces could be responsible for the degradation ofthe solvent. On the basis of a DFT study, Laino et al.82 revealedthat propylene carbonate could easily decompose on a [100]Li2O2 surface with a “barrierless” reaction, as shown in Figure6d. In contrast, the reaction by a Li2O2 molecular unit inpropylene carbonate solution has a barrier as high as 40 kcal/mol among various reaction paths (e.g., hydrogen abstractionand nucleophilic reactions).At present, Li−O2 cells with carbonate-based electrolytes
cannot be considered viable, as the overall reaction is thecontinuous and irreversible oxidation of the electrolytes, andtherefore, the battery is in principle irreversible. The lessonlearned from this case is that direct implantation of a well-established electrolyte for the Li ion battery into the Li−O2system was not successful. On the other hand, the lessonslearned from the study of carbonate-based electrolyte haveprovided a profound understanding of the operation of aproticLi−O2 batteries and resulted in a more reliable process forseeking stable electrolytes. It should be pointed out that the airelectrode catalysts investigated with organic carbonate-basedelectrolyte need to be re-examined, since the claimed catalyticactivities toward the oxygen reduction and oxygen evolutionreactions appear to be more related to the decomposition of theelectrolyte than the reversible formation and decomposition ofLi2O2.
3.3.2. Ether-Based Electrolyte. After realizing that thecarbonate-based electrolytes have suffered from severedecomposition during the operation of Li−O2 cells, researchershave focused much attention on the experimental55,59,60,78,83−91
and theoretical80,81,92−95 search for other solvents that arestable against attack by reduced O2 species and can avoid otherpossible decomposition problems. Ether-based solvents, such as1,2-dimethoxyethane (DME) and tetraethylene glycol dimethylether (“tetraglyme” or TEGDME), have attracted significantattention for the Li−O2 battery very recently, mainly due to therelatively high stability with respect to superoxide radicals andoxidation potentials (>4.5 V versus Li/Li+), inflammability,high thermal stability, and low cost. In fact, Read67 firstreported that ether-based electrolytes perform well in the Li/O2cell, showing both good stability and excellent rate capability,even before the recognition of the instability issue of carbonatesolvents. Read found that the viscosity of ether-basedelectrolytes is lower than that of carbonate-based electrolytes,while still having similar oxygen solubility. Read also pointedout that the viscosity becomes the key factor in determining thedischarge capacity and rate capability of the cell once a certainlevel of oxygen solubility is reached. Unfortunately, Read didnot show any evidence for the formation of Li2O2 upondischarge of the cell, although we know now that the dischargeproduct using ether-based electrolytes is indeed Li2O2.Recently, several groups have found evidence that the major
discharge product is Li2O2 when ether-based solvents are usedin the Li−O2 cell. McClosky et al.88 investigated the stability ofDME in a Li−O2 cell containing different air electrode catalysts,where they confirmed that Li2O2 is the dominant product ofthe electrochemistry. Interestingly, they found no catalyticactivity compared to pure carbon when Au, Pt, or MnO2nanoparticles were used as the air electrode catalyst. In theirfollow-up paper, McClosky et al.96 showed that the amount ofO2 evolved upon charging accounts for only 60% of the
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXH
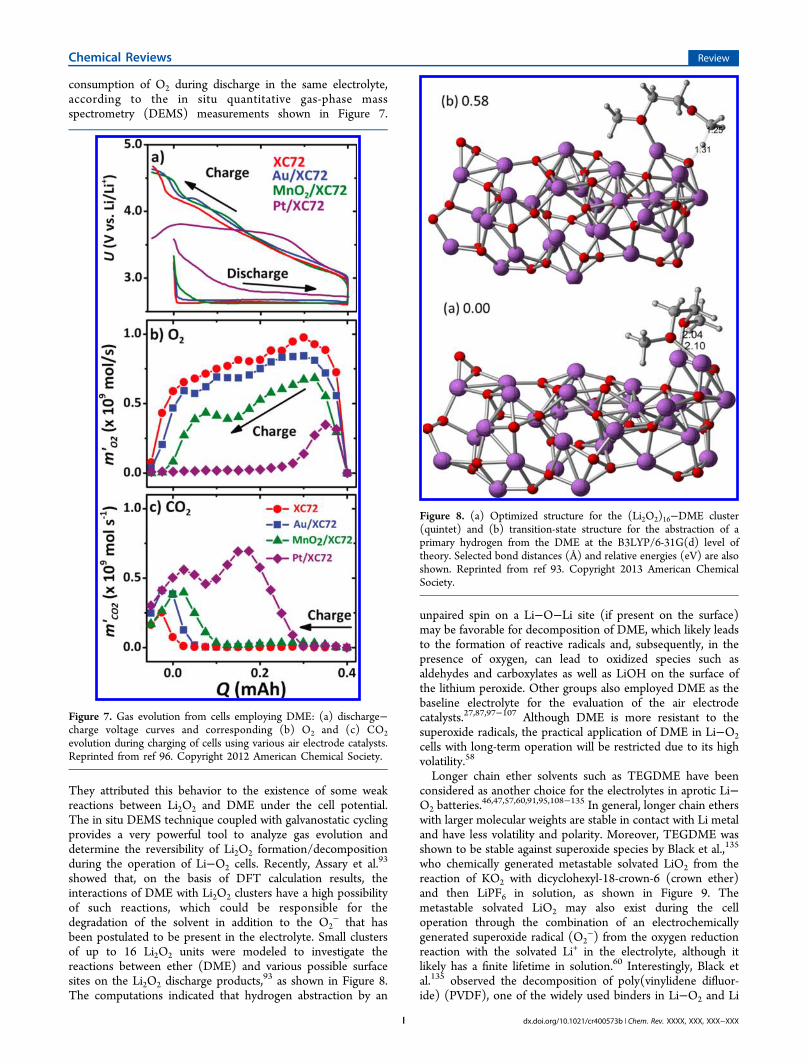
consumption of O2 during discharge in the same electrolyte,according to the in situ quantitative gas-phase massspectrometry (DEMS) measurements shown in Figure 7.
They attributed this behavior to the existence of some weakreactions between Li2O2 and DME under the cell potential.The in situ DEMS technique coupled with galvanostatic cyclingprovides a very powerful tool to analyze gas evolution anddetermine the reversibility of Li2O2 formation/decompositionduring the operation of Li−O2 cells. Recently, Assary et al.93
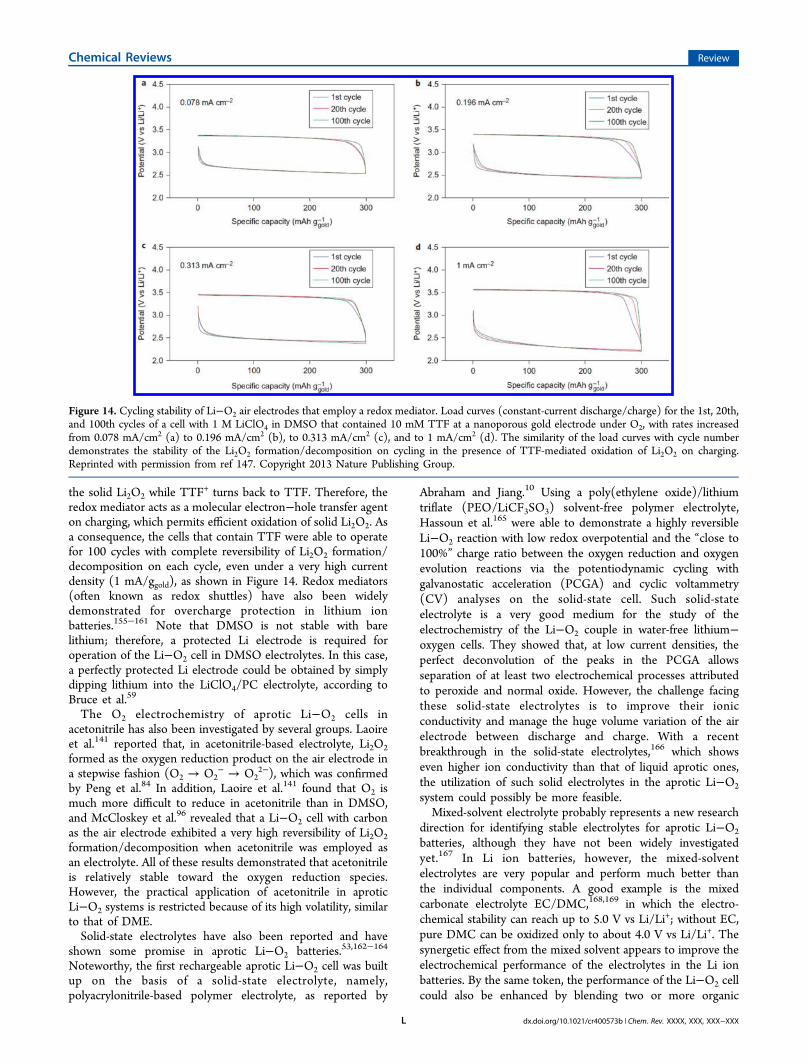
showed that, on the basis of DFT calculation results, theinteractions of DME with Li2O2 clusters have a high possibilityof such reactions, which could be responsible for thedegradation of the solvent in addition to the O2
− that hasbeen postulated to be present in the electrolyte. Small clustersof up to 16 Li2O2 units were modeled to investigate thereactions between ether (DME) and various possible surfacesites on the Li2O2 discharge products,93 as shown in Figure 8.The computations indicated that hydrogen abstraction by an
unpaired spin on a Li−O−Li site (if present on the surface)may be favorable for decomposition of DME, which likely leadsto the formation of reactive radicals and, subsequently, in thepresence of oxygen, can lead to oxidized species such asaldehydes and carboxylates as well as LiOH on the surface ofthe lithium peroxide. Other groups also employed DME as thebaseline electrolyte for the evaluation of the air electrodecatalysts.27,87,97−107 Although DME is more resistant to thesuperoxide radicals, the practical application of DME in Li−O2cells with long-term operation will be restricted due to its highvolatility.58
Longer chain ether solvents such as TEGDME have beenconsidered as another choice for the electrolytes in aprotic Li−O2 batteries.
46,47,57,60,91,95,108−135 In general, longer chain etherswith larger molecular weights are stable in contact with Li metaland have less volatility and polarity. Moreover, TEGDME wasshown to be stable against superoxide species by Black et al.,135
who chemically generated metastable solvated LiO2 from thereaction of KO2 with dicyclohexyl-18-crown-6 (crown ether)and then LiPF6 in solution, as shown in Figure 9. Themetastable solvated LiO2 may also exist during the celloperation through the combination of an electrochemicallygenerated superoxide radical (O2
−) from the oxygen reductionreaction with the solvated Li+ in the electrolyte, although itlikely has a finite lifetime in solution.60 Interestingly, Black etal.135 observed the decomposition of poly(vinylidene difluor-ide) (PVDF), one of the widely used binders in Li−O2 and Li
Figure 7. Gas evolution from cells employing DME: (a) discharge−charge voltage curves and corresponding (b) O2 and (c) CO2evolution during charging of cells using various air electrode catalysts.Reprinted from ref 96. Copyright 2012 American Chemical Society.
Figure 8. (a) Optimized structure for the (Li2O2)16−DME cluster(quintet) and (b) transition-state structure for the abstraction of aprimary hydrogen from the DME at the B3LYP/6-31G(d) level oftheory. Selected bond distances (Å) and relative energies (eV) are alsoshown. Reprinted from ref 93. Copyright 2013 American ChemicalSociety.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXI

ion batteries, through dehydrofluorination along with theformation of H2O2 by the chemically generated LiO2. Black etal.135 also showed that, in the absence of binder and catalyst,the crystallization of Li2O2 is governed by its rate of nucleation/precipitation from LiO2, solubility, and interaction with thecarbon surface. This work provided vital insight into thereactivity of superoxide radical with materials used in the Li−O2system, which should be seriously considered as a screeningmethod in searching for stable electrolytes for Li−O2 batteries.Jung et al.91,136 reported an improved Li−O2 battery using
TEGDME/LiCF3SO3 electrolyte, which exhibited high capacityand stable cycling performance with an impressive ratecapability. The reversible formation of Li2O2 was confirmedby time-of-flight secondary ion mass spectrometry (TOF-SIMS), along with scanning and transmission electronmicroscopy, indicating morphological reversibility of semi-crystalline Li2O2 particles. Moreover, Li2CO3 was not detectedin the discharge and charge states, showing that TEGDME/LiCF3SO3 electrolyte is stable during cell operation. This Li−O2 cell showed very stable cycling performance up to 100cycles with a capacity of 1000 mA·h/gcarbon at both a low rate(50 mA/gcarbon) and a high rate (1000 mA/gcarbon). Even undera very high capacity of 5000 mA·h/gcarbon, this Li−O2 cell iscapable of operating over 30 cycles at a rate of 500 mA/gcarbon,as shown in Figure 10.The reactivity of a new silicon-containing oligo(ethylene
oxide) solvent, (triethylene glycol-substituted methyl)-trimethylsilane (1NM3), toward the oxygen reduced specieshas also been investigated because of its good properties as anelectrolyte, such as low glass transition temperature, effectiveionic transport, low viscosity, and good lithium ionconductivity.57,137−140 On the basis of the DFT calculation,Zhang et al.55 found that this solvent seems to be more stableto the highly active oxygen reduction species when comparedto propylene carbonate. The XPS data collected after a singledischarge show that only lithium oxides are formed when1NM3 is used in place of propylene carbonate in a Li−O2 cell.Moreover, the formed Li2O2 decomposed upon charging at alow charge potential (3.5 V vs Li/Li+), as shown in Figure 11. Itseems that the increased stability of the 1NM3 solvent results
Figure 9. Exploratory reactions of superoxide that parallel those in aLi−O2 cell: (A) KO2 + LiPF6/TEGDME; (B) KO2 + LiPF6/TEGDME + carbon; (C) electrochemical cell (carbon catalyst); (D)KO2 + LiPF6/TEGDME/PVDF; (E) KO2 + LiPF6/TEGDME/PVDF/α-MnO2 catalyst. KO2 = KO2 (crown ether), and PVDF =poly(vinylidene difluoride). Reprinted from ref 135. Copyright 2013American Chemical Society.
Figure 10. Cycling performance of the lithium−TEGDME/LiCF3SO3−O2 battery under various specific capacity limits andcurrent densities: (a) voltage profiles of 100 cycles with a capacity limitof 1000 mA·h/g at a current density of 50 mA/g; (b) voltage profilesof 100 cycles with a capacity limit of 1000 mA·h/g at a current densityof 1000 mA/g; (c) voltage profiles of 30 cycles with a capacity limit of5000 mA·h/g at a current density of 500 mA/g. The specific capacityand current density were based on the mass of the carbon. Reprintedwith permission from ref 91. Copyright 2012 Nature PublishingGroup.
Figure 11. First charge and discharge cycles of a Li−O2 cell with PCand 1NM3 electrolyte. The current density applied in both cases is 100mA/g, based on the mass of the carbon + catalyst. Reprinted from ref55. Copyright 2011 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXJ

in a significantly lower overpotential for the Li−O2 cell, whichcould be due to the oxidation of lithium oxides being easierthan the oxidation of carbonate formed in the propylenecarbonate cells. However, the cycle performance of the cellusing 1NM3 electrolyte is still poor, indicating that the long-term stability of 1NM3 solvent is problematic. The electrolytesolvent stability plays a key role in the performance of Li−O2batteries and will be a key factor in improving their efficiencies.Bruce and co-workers83 investigated the stability of linear or
cyclic ether-based electrolytes (e.g., tetraglyme, triglyme,diglyme, 1,3-dioxolane, and 2-methyl-THF) in Li−O2 cells bycombining electrochemical measurements with powder X-raydiffraction (XRD), FTIR spectroscopy, and nuclear magneticresonance (NMR) spectroscopy. In all cases, Li2O2 was theoxygen reduction product on the first discharge, confirmingthat ether-based electrolytes are more stable than carbonate-based ones against oxygen reduced species. The ether solventsalso decomposed during the operation of the cell accompaniedby the formation of Li2O2. For instance, Bruce et al.83 foundthat the decomposition products for linear-chain ethers consistof a mixture of Li2CO3, HCO2Li, CH3CO2Li, polyethers/esters,CO2, and H2O. Their cycling studies on TEGDME indicatedthat the proportion of Li2O2 diminishes on cycling in favor ofgreater electrolyte decomposition, as evidenced by the XRDand FTIR measurements shown in Figure 12. On the basis ofthese results, they concluded that ether-based electrolytes arenot suitable for Li−O2 cells. We point out, however, that theeffect of the salt was not taken into account in their study. Infact, LiPF6, which was used in their ether-based electrolytes asthe lithium salt, is suspected to be stable to Li2O2. Moreover,LiPF6 has been known to decompose to form HF in thepresence of H2O, even with a trace amount. As a consequence,the observed decomposition of ether solvents may result fromthe active species generated from the side reaction of thelithium salt. When LiCF3SO3 replaces the LiPF6 in theTEGDME electrolyte, the cell performance is significantlyimproved, as demonstrated by Jung et al.91,136 The effect oflithium salt on the cell performance is further discussed later.3.3.3. Other Organic Electrolytes. Several other types of
organic solvents have also been investigated in aprotic Li−O2systems, including acetonitrile (ACN),84,96,141 dimethyl sulf-oxide (DMSO),59,85,99,142−148 dimethylformamide (DMF),149
triethyl phosphate,90 N,N-dimethylacetamide (DMA),150,151 N-methylpyrrolidone (NMP),34 methoxybenzene,152 and ionicliquids,86,153,154 some of which demonstrated relatively highstability toward oxygen reduction intermediates (LiO2) andproducts (Li2O2) in a Li−O2 cell. One of the particularlyinteresting solvents is DMSO. Peng et al.59 reported a verysteady rechargeable Li−O2 cell using LiClO4/DMSO as theelectrolyte coupled with a nanoporous gold (NPG) airelectrode, which is able to cycle up to 100 times with littledecay of the capacity, as shown in Figure 13. The FTIR, Ramanspectroscopy, NMR, and DEMS results indicated that Li2O2 isformed at the air electrode with >99% purity, even on the 100thcycle, and it is completely oxidized on charge. The charge-to-mass ratio on discharge/charge was determined to be 2e−/O2by the DEMS results, suggesting that Li2O2 formation/decomposition is the overwhelming reaction during theoperation of the cell. Note that the charge potential is stillhigh (up to 4.0 V), although a low charge plateau occurs at 3.3V. Charge transfer between the Li2O2 particles and the solidelectrode surface is likely responsible for the voltage polar-ization on charge because of the insulating nature of Li2O2,
which is a significant problem facing the aprotic Li−O2 battery.Very recently, the same group147 seems to have solved the issueof large charge overpotential in their previous cell byintroducing a redox mediator, tetrathiafulvalene (TTF), tothe DMSO electrolyte. During cell charging, TTF is oxidized toTTF+ at the air electrode surface, which subsequently oxidizes
Figure 12. (a) XRD patterns and (b) FTIR spectra of O2 electrodescycled in 1 M LiPF6/TEGDME electrolyte. Reprinted with permissionfrom ref 83. Copyright 2011 Wiley-VCH.
Figure 13. Charge/discharge curves (left) and cycling profile (right)for a Li−O2 cell with a 0.1 M LiClO4/DMSO electrolyte and an NPGair electrode at a current density of 500 mA/g (based on the mass ofAu). Reprinted with permission from ref 59. Copyright 2012 Science.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXK
the solid Li2O2 while TTF+ turns back to TTF. Therefore, the
redox mediator acts as a molecular electron−hole transfer agenton charging, which permits efficient oxidation of solid Li2O2. Asa consequence, the cells that contain TTF were able to operatefor 100 cycles with complete reversibility of Li2O2 formation/decomposition on each cycle, even under a very high currentdensity (1 mA/ggold), as shown in Figure 14. Redox mediators(often known as redox shuttles) have also been widelydemonstrated for overcharge protection in lithium ionbatteries.155−161 Note that DMSO is not stable with barelithium; therefore, a protected Li electrode is required foroperation of the Li−O2 cell in DMSO electrolytes. In this case,a perfectly protected Li electrode could be obtained by simplydipping lithium into the LiClO4/PC electrolyte, according toBruce et al.59
The O2 electrochemistry of aprotic Li−O2 cells inacetonitrile has also been investigated by several groups. Laoireet al.141 reported that, in acetonitrile-based electrolyte, Li2O2formed as the oxygen reduction product on the air electrode ina stepwise fashion (O2 → O2
− → O22−), which was confirmed
by Peng et al.84 In addition, Laoire et al.141 found that O2 ismuch more difficult to reduce in acetonitrile than in DMSO,and McCloskey et al.96 revealed that a Li−O2 cell with carbonas the air electrode exhibited a very high reversibility of Li2O2formation/decomposition when acetonitrile was employed asan electrolyte. All of these results demonstrated that acetonitrileis relatively stable toward the oxygen reduction species.However, the practical application of acetonitrile in aproticLi−O2 systems is restricted because of its high volatility, similarto that of DME.Solid-state electrolytes have also been reported and have
shown some promise in aprotic Li−O2 batteries.53,162−164
Noteworthy, the first rechargeable aprotic Li−O2 cell was builtup on the basis of a solid-state electrolyte, namely,polyacrylonitrile-based polymer electrolyte, as reported by
Abraham and Jiang.10 Using a poly(ethylene oxide)/lithiumtriflate (PEO/LiCF3SO3) solvent-free polymer electrolyte,Hassoun et al.165 were able to demonstrate a highly reversibleLi−O2 reaction with low redox overpotential and the “close to100%” charge ratio between the oxygen reduction and oxygenevolution reactions via the potentiodynamic cycling withgalvanostatic acceleration (PCGA) and cyclic voltammetry(CV) analyses on the solid-state cell. Such solid-stateelectrolyte is a very good medium for the study of theelectrochemistry of the Li−O2 couple in water-free lithium−oxygen cells. They showed that, at low current densities, theperfect deconvolution of the peaks in the PCGA allowsseparation of at least two electrochemical processes attributedto peroxide and normal oxide. However, the challenge facingthese solid-state electrolytes is to improve their ionicconductivity and manage the huge volume variation of the airelectrode between discharge and charge. With a recentbreakthrough in the solid-state electrolytes,166 which showseven higher ion conductivity than that of liquid aprotic ones,the utilization of such solid electrolytes in the aprotic Li−O2
system could possibly be more feasible.Mixed-solvent electrolyte probably represents a new research
direction for identifying stable electrolytes for aprotic Li−O2
batteries, although they have not been widely investigatedyet.167 In Li ion batteries, however, the mixed-solventelectrolytes are very popular and perform much better thanthe individual components. A good example is the mixedcarbonate electrolyte EC/DMC,168,169 in which the electro-chemical stability can reach up to 5.0 V vs Li/Li+; without EC,pure DMC can be oxidized only to about 4.0 V vs Li/Li+. Thesynergetic effect from the mixed solvent appears to improve theelectrochemical performance of the electrolytes in the Li ionbatteries. By the same token, the performance of the Li−O2 cellcould also be enhanced by blending two or more organic
Figure 14. Cycling stability of Li−O2 air electrodes that employ a redox mediator. Load curves (constant-current discharge/charge) for the 1st, 20th,and 100th cycles of a cell with 1 M LiClO4 in DMSO that contained 10 mM TTF at a nanoporous gold electrode under O2, with rates increasedfrom 0.078 mA/cm2 (a) to 0.196 mA/cm2 (b), to 0.313 mA/cm2 (c), and to 1 mA/cm2 (d). The similarity of the load curves with cycle numberdemonstrates the stability of the Li2O2 formation/decomposition on cycling in the presence of TTF-mediated oxidation of Li2O2 on charging.Reprinted with permission from ref 147. Copyright 2013 Nature Publishing Group.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXL

solvents, given that they are stable (at the air electrode andlithium electrode) during the operation of the cell.3.3.4. Effect of Lithium Salt. Besides the stability of
solvents, another source of detrimental electrolyte decom-position in Li−O2 batteries could be from side reactions due tothe decomposition of lithium salt, an indispensable componentof the electrolyte. In contrast to the extensive efforts on solvingthe instability issue of solvents, the effect of lithium salts on thestability of the electrolytes in the aprotic Li−O2 system has notbeen investigated in detail yet, even though the choice oflithium salts is quite limited. The lithium salts that have beenemployed in the aprotic Li−O2 system include LiPF6, lithiumbis((trifluoromethyl)sulfonyl)imide (LiTFSI), LiClO4,LiCF3SO3, LiBF4, LiCl, and lithium bis(oxalato)borate(LiBOB), with the first four being used the most. Analogousto the Li ion battery, a good lithium salt for the Li−O2 batteryhas to meet certain requirements, namely, (1) the salt shouldhave high enough solubility in the solvent to support the iontransport and (2) the anion of the salt should be inert to thesolvent and other cell components, such as current collectorsand separators. In addition to these basic requirements, the saltused in the Li−O2 system should be inert to the aggressiveoxygen reduction species, such as Li2O2 and O2
− radical.Recent evidence has shown that some lithium salts are not
stable toward Li2O2 formed in the Li−O2 cells, with LiPF6being the most obvious one. The XPS results of Oswald etal.170 indicated an almost immediate formation of LiF if theLi2O2 is exposed to a LiPF6-containing electrolyte. Theformation of LiF is partially due to the presence of HF,which is generated by fluorine from fluorinated anion (PF6
−)and protons from water impurities in the electrolyte. The XPSdepth profiles also revealed another possible reason for theformation of LiF, the direct reaction of Li2O2 and LiPF6. Byanalyzing the discharge products of aprotic Li−O2 cells inTEGDME-based electrolytes with various salts (LiPF6, LiClO4,LiTFSI, and LiCF3SO3) using Raman, XPS, and NMRtechniques, Veith et al.171 provided strong evidence that thedischarge product in ether-based electrolytes contains a certainlevel of insoluble Li salt, although the major discharge productis Li2O2. They believe that the discharge chemistries of Li−O2cells appear to undergo similar decomposition of the lithiumsalt, resulting from the strong interaction of the salt and thereactive O2 or reduced oxygen species, such as O2
−.As discussed above, the stability of lithium salt, especially in
the presence of reduced oxygen species (O2−) and a trace
amount of H2O, may significantly affect the cyclability andcapacity of Li−O2 cells. Applying a combined experimental andcomputational approach, Du et al. provided evidence that thestability of the electrolyte in the Li−O2 cell strongly dependson the compatibility of the lithium salt with the solvent.172 Inthe case of the LiPF6/1NM3 electrolyte, they found that HFformed from decomposition of LiPF6 triggers the decom-position of the 1NM3 solvent, as schematized in Figure 15.These reactions lead to severe degradation of the electrolyteand cause the poor cyclability of the cell. The same reactionsare not observed when LiTFSI or LiCF3SO3 is used as thelithium salt in 1NM3 solvent, suggesting that the stability of theelectrolyte in Li−air cells depends on the compatibility of thelithium salt with the solvent.Although not yet having received much attention, the
possible decomposition of the lithium salt and the subsequenteffect on the stability of the electrolytes and performance of theaprotic Li−O2 cells should not be neglected. The challenge in
investigating the stability of lithium salts in Li−O2 cells,however, strongly relies on a stable solvent, which, in turn,should have the capability to dissolve a certain amount ofvarious lithium salts. This situation suggests that progress indeveloping a stable electrolyte for the aprotic Li−O2 system canonly be made in a stepwise fashion.
3.3.5. Hard−Soft Acid−Base (HSAB) Theory. To designa rational approach to the selection of organic electrolyte forthe rechargeable aprotic Li−O2 battery, a more fundamentaltheory or model is required to elucidate the influence ofsolvent/salt on O2 electrochemistry. Laorie et al.
141 successfullyadopted the concept of HSAB theory to explore variouselectrolyte systems to determine the effect of a solvent’s donornumber (DN) and acceptor number (AN) on the dischargeproducts of Li−O2 cells. In Pearson’s HSAB theory,173 Lewisacids and bases are further classified into hard and softsubcategories, where hard acids tend to interact with hard baseswhile soft acids tend to interact with soft bases. In general, hardacids/bases have relatively small ionic radii and are difficult topolarize and vice versa.According to the HSAB theory, Li+ is a hard Lewis acid and
has a high affinity for a hard Lewis base, such as peroxide(O2
2−) or monoxide (O2−). However, in the electrolytesolution, the hard Lewis acid Li+ ions are normally solvatedby the solvents to form a complex (Li+−(solvent)n), which willthen modulate the acidity of Li+ dependent on the DN of thesolvents. Laorie et al.141 showed that the bond strength of Li+−solvent in the complex would follow the solvent DN scale asDMSO > MeCN > DME > TEGDME. On the other hand, thesuperoxide (O2
−), one of the possible oxygen reduction speciesin the Li−O2 system, tends to be a moderately soft Lewis basebecause of its relatively large radius and low charge density.Therefore, the superoxide anion has low affinity for the hardLewis acid Li+ in the solutions, for instance, TEGDME/LiPF6electrolyte. Consequently, the superoxide anion formed as thefirst reduction product, if it does form, will prefer either todecompose via a disproportionation reaction (assuming thesolvent is stable to superoxide anion) or to undergo a fastsecond reduction, both of which will lead to the formation of ahard Lewis base, peroxide anion (O2
2−). Once the peroxideanion forms, it will then combine with the hard Lewis acid Li+
to produce a stable O2 reduction product, Li2O2, on the basis ofthe HSAB theory. This could explain the observation by manyother researchers of Li2O2 formation as the dominant discharge
Figure 15. Schematic reaction mechanism for decomposition ofLiPF6/1NM3 electrolyte in the Li−O2 cell. Reprinted with permissionfrom ref 172. Copyright 2013 PCCP Owner Societies.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXM

product in the Li−O2 cells using TEGDME-based electro-lytes.47,91,120,124,127,174,175 Theoretically, Li2O, the ultimatereduction product of O2, should also be able to form as astable discharge product in a Li−O2 cell, considering that themonoxide (O2−) anion is an even harder Lewis base. However,the charge of Li2O cannot proceed under the test condition dueto the need for 4-electron transfer, which is kineticallyunfavorable in the electrochemical process; therefore, thedischarge product is normally limited to Li2O2 by controllingthe cutoff discharge voltage of the Li−O2 cell to above 2.0 V vsLi/Li+.As mentioned above, the formation of Li+−(solvent)n
complexes would modulate (lower) the acidity of Li+,determined by the DN of the solvent. Therefore, thesuperoxide anions could be stabilized to a certain degreebefore they transform to O2
2− via a chemical or electrochemicalpathway, because of the increased affinity between O2
− (softLewis base) and the solvated Li+ with lower acidity (toward softLewis acid). This could explain the distinct O2/O2
− couple seenin the DMSO/LiPF6 electrolyte.59,85,143−147 Thus, the HSABtheory could provide guidance to further modulate the Li+
acidity in the electrolytes to form different types of dischargeproducts during the oxygen reduction reaction. Successfulimplementation of this theory will be beneficial for futureinvestigation of different electrolytes for optimization of theLi−O2 cell.3.3.6. Brief Summary. In summary, the organic electrolytes
(both solvents and lithium salts) play the most critical role inan aprotic Li−O2 cell and determine whether a trulyrechargeable Li−O2 battery can be realized. Numerous studiesshowed that the stability of the electrolytes during the oxygenreduction and oxygen evolution processes is the key challengefor the aprotic Li−O2 cell. With no doubt, searching for fullystable electrolytes in the oxygen-rich electrochemical environ-ment is the research priority at present. Carbonate-basedelectrolytes have proved to be highly unstable toward theoxygen reduction species. However, there is still a large amountof research work using carbonate-based electrolytes toinvestigate the catalytic activities of the air electrodematerials,126,133,176−216 despite the fact that the severeinstability of these electrolytes has been reported. The catalyticactivity of these air electrode materials needs to be re-examinedin more stable electrolytes. Ether-based electrolytes appear tobe relatively stable in the presence of the reduced oxygenspecies, as evident by the reversible formation/decompositionof Li2O2; however, their electrochemical behavior, in particularon charge and during long-term cycling, remains to bethoroughly investigated for optimization of the Li−O2 cell.Both DMSO59,147,217 and TEGDME91,136 are probably themost promising organic solvents under investigation. The bestelectrochemical performance (including cycle performance) hasbeen achieved with Li−O2 cells having these electrolytesolvents. Lithium salt, an indispensable component of theelectrolyte, deserves much more attention since there isevidence it has an effect on the stability of the electrolytes inLi−O2 cells. Design of a robust strategy for effectively screeningthe stability of various electrolytes would be greatly beneficial tothe development of a Li−O2 battery for practical application. Inaddition, we noticed that there are several different or evenconflicting reports on the reactivity of superoxide anion (O2
−)against ethers, which certainly requires deep fundamentalunderstanding using the state-of-the-art characterizationtechniques. In our opinion, however, the reactivity of the
superoxide anion against ethers is partially dependent on thepotentials applied during cell discharge and charge, which iscertainly related to the electrocatalysts on air electrodes.
3.4. Electronic and Magnetic Properties of Li2O2 andRelevance to the Li−O2 Battery
Another research area that deserves attention is the effect of theelectronic structure and magnetic properties of Li2O2 on theelectrochemical performance (particularly the charge over-potential) and electrolyte stability of Li−O2 cells. Betterunderstanding of these properties could help us elucidate thecharge and discharge chemistries involved in the Li−O2 battery.In general, the most thermodynamically stable structures in
the condensed phase for known stoichiometric LixOycompounds are Li2O (lithia) and Li2O2,
218 both of which aretheoretically predicted to be semiconductors with a highelectronic band gap; for instance, bulk Li2O2 shows a band gapof ∼4.9 eV.219−221 Since high electronic conductivity andcharge transfer are essential for any redox chemistry in thebattery system, such a high band gap fails to explain thereversible formation of Li2O2 on the air electrode duringdischarge/charge of a Li−O2 cell, especially under very highcurrent density.91,136 Recently, Hummelshoj et al.219,222
proposed a theory based on metallicity that shows thepossibility of lowering the electronic band gap by inducingsome Li vacancies into bulk Li2O2. Depending on the vacancyconcentration, the formation energy of Li2O2 could be reducedto ∼3.0 eV, which will enable reasonable electron and chargetransfer in Li2O2. The theory is supported by the finding thatLi2O2 observed as the discharge product of a Li−O2 cell isgenerally in the form of nanoparticles,61,221 which are known tohave vacancies and defects on the surface.Since neither the mechanism of formation and growth of
Li2O2 during discharge nor the mechanism of the decom-position of Li2O2 during charge is well understoodexperimentally, computational studies on this topic will likelyprovide valuable insight in understanding the correlationbetween the reversible formation of Li2O2 and its electronicand magnetic properties, if any. At the current stage, theoreticalcalculation on small Li2O2 clusters is the choice of these studies,considering that computations on large clusters or particleswould be much more complicated and costly. The informationgathered from theoretical computations on small Li2O2 clusterscould provide insight into the properties of larger nanoparticlesas well as their nucleation during cell discharge.In a recent paper, Lau et al.223 reported on a DFT study of
the structure of (Li2O2)n clusters, where many possiblestructures of the clusters and the corresponding electronicstates were considered. They found that, surprisingly, the tripletstate is significantly stabilized relative to the singlet in theseclusters, especially for clusters larger than the dimer. Their DFTcalculations also showed the existence of a high spin state in alarge Li2O2 cluster with n = 16, which can be characterized byO−O pairs protruding from the surface but still chemicallybonded to the Li of the clusters, as shown in Figure 16. TheseLi2O2 clusters have shorter O−O distances and a localizedunpaired spin on the surface compared to a peroxide pair foundin a Li2O2 bulk crystal, which probably suggests the existence ofsuperoxide-like surface structures. This theoretical finding couldexplain the ferromagnetism with magnetic moments of Li2O2,earlier predicted by Radin et al.224 using first-principlescalculations. More importantly, if these superoxide-like surfacestructures indeed exist experimentally, they could have
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXN

important implications for the electrochemistry of reversibleformation of Li2O2 in Li−O2 cells, including surface electronicconductivity and electrolyte surface reactions.Experimentally, Lu et al.225 recently investigated the
magnetic properties of Li2O2 using electron paramagneticresonance (EPR) and superconducting quantum interferencedevice (SQUID) magnetometry. They showed evidence, for thefirst time, that the Li2O2 formed during the discharge of a cellwith an ether-based electrolyte is paramagnetic, as shown inFigure 17. The EPR spectra provided direct evidence that thespin in the discharge Li2O2 product is caused by a superoxide-type structure. The number of spins calculated from magneticmeasurements is also consistent with the predicted one byDFT. More recent work by Yang et al.174 further confirmed thepresence of a superoxide-like species in the Li2O2 dischargeproduct by the presence of a peak at 1125 cm−1 in the Ramanspectra. The presence of magnetism in the discharge product(Li2O2) could play an important role in the charge anddischarge chemistries of Li−O2 cells, since the superoxide-typeoxygen groups with spin on the Li2O2 surface could enable theelectronic conductivity mechanism that is required for thereversible formation and decomposition of Li2O2, thus reducingthe charge overpotential. Yang et al.174 observed two distinctvoltage plateaus during charging, at 3.2−3.5 and 4.2−4.3 V, in aLi−O2 cell that had petroleum coke-based activated carbon(AC) as the air electrode material and a TEGDME/LiCF3SO3electrolyte. The lower plateau corresponds to a form of lithium
peroxide with superoxide-like properties characterized by a low-temperature magnetic phase transition (49.7 K) and a high O−O stretching frequency (Raman peak at 1125 cm−1). A lowcharge potential (3.3 V) was also observed in a Li−O2 cell using1NM3/LiTFSI as the electrolyte, where the Li2O2 dischargeproduct shows magnetic behavior.225 These experimentalfindings suggest that a superoxide surface species lowers thecharge potential, and therefore, control of the forms of Li2O2produced on discharge is important for improving the efficiencyof the Li−O2 cell.
3.5. Air Electrode Architecture
Although electrolyte stability is of paramount importance, airelectrode materials also represent a major technology challengein rechargeable aprotic Li−O2 cell development. The ultimategoal of the investigation is to determine how to effectivelyincrease the specific capacity and power capability of Li−O2cells yet still achieve a long cycle life. Attaining that goalstrongly depends on the materials and their microstructures inthe O2-breathing electrode.62,68,226 Good electronic and ionicconductivity, fast oxygen diffusion, and stable integrity arerequired for air electrode materials to achieve high performancein Li−O2 cells. The specific surface area and porosity of theelectrode material are also critical, since the overall capacity andenergy density of Li−O2 cells are dependent on the quantity ofthe discharge product (Li2O2) that is able to be storeed on orwithin the porous air electrode structure. Typically, a largersurface area provides more surface to uniformly dispersecatalytic particles, if needed, and more active sites to aid theelectrochemical reactions, while a porous structure with anappropriate pore size provides the space to store the dischargeproducts. However, further studies revealed that the volume ofthe pores in the range of 2−50 nm rather than the surface areaof the air electrode material appears to be the limiting factor todetermine the discharge capacity of the cell.227,228 For instance,Cheng and Scott229 showed that Super P carbon with arelatively low surface area (62 m2/g) demonstrated the highestdischarge capacity among the various carbon materialsinvestigated, including AC with a very high surface area up to2100 m2/g. Black et al.23 recently pointed out that, however,the discharge capacity of a Li−O2 cell is not solely determinedby either the surface area or pore size/volume of the airelectrode, but by a more complicated relationship betweenthese components. The surface area in relation to the types ofpores could compromise the air electrode membrane structure,while the nature of the carbon material (especially the defects)may act as an ORR catalyst, both of which will have a significantimpact on the discharge capacity of the cell. Furthermore, anefficient catalyst is often needed in the air electrode structure tofacilitate the ORR, and particularly the OER in Li−O2 cells,similar to that in a proton exchange membrane fuel cell. Insome cases, bare carbon-based materials showed some ability toform Li2O2 when used in conjunction with a relatively stableelectrolyte, since the defects on these carbon-based materialscould serve as the ORR catalyst and the nucleation sites forLi2O2.
61,62
Most of the early studies on catalytic activity toward ORRand OER of the air electrode materials in aprotic Li−O2 cellswere based on the utilization of organic carbonate electro-lytes,33,48,51,56,63,75,226,227,230−242 which have since been shownto be unstable to the oxygen reduced species, as discussedpreviously. Because the discharge products are now known tobe Li2CO3 or lithium carboxylates, not the desired Li2O2 in the
Figure 16. Optimized high-spin (Li2O2)16 cluster (quintet) structuresfrom the PBE/PW (top) and B3LYP/6-31G(d) (bottom) levels oftheory. The distances of the short O−O bonds and Bader charges (inparentheses) of O−O moieties are shown for the PBE/PW calculation.The distances are also shown for the B3LYP/6-31G(d) structure alongwith the computed Mulliken spins and charges in parentheses. Notethat the fourth unpaired electron is delocalized over other oxygenatoms. Reprinted from ref 223. Copyright 2012 American ChemicalSociety.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXO

cell with the carbonate electrolyte, the claimed catalytic activity,especially on the charge reaction, appears to be more related tothe electrolyte decomposition, and their “true” catalytic activitytoward the reversible formation of Li2O2 certainly needs to bere-evaluated in a more stable electrolyte, such as ether- orDMSO-based electrolyte. The knowledge gained from theabove research could be beneficial for identifying efficientelectrocatalysts for the formation of Li2O2 if a stable electrolytecan be developed, since the first step in the formation of eitherLi2CO3 or Li2O2 shares the same reaction, i.e., reduction of O2to O2
−, yet the charge reaction (i.e., lowering the chargeoverpotential) for the Li−O2 cell remains a major challenge.The exact mechanism of the decomposition of Li2CO3 alongwith its derivatives in carbonate electrolytes or Li2O2 in etherelectrolyte is up for debate. It can be foreseen that thedecomposition of lithium carbonate and lithium peroxideduring charging of an electrochemical cell will proceed througha completely different pathway because of the differentcompositions of these compounds. In this sense, the claimedcatalytic activity on charging of a Li−O2 cell in a carbonateelectrolyte will not likely apply to that in ether or otherelectrolytes where formation of Li2O2 occurs. In the followingsections, we discuss the effect of the catalytic air electrode onthe performance of the Li−O2 cell with relatively stableelectrolytes and Li2O2 formation.3.5.1. Porous Carbon. As mentioned previously, one of the
biggest hurdles for the rechargeable Li−O2 battery is the largeoverpotential during discharge and, in particular, charge, even atvery low current density (0.01−0.05 mA/cm2), which results inlow round-trip efficiencies and low power capability. This
performance is strongly believed to depend on the nature of thecatalytic air electrode, in addition to the stability of theelectrolytes. Many catalysts, including metal oxides andnonprecious and precious metals on a porous carbon support,have been examined as the air electrode active material for theoxygen reduction and evolution reactions, showing largedifferences in discharge capacity among different catalysts.Surprisingly, a nearly identical discharge voltage plateau atabout 2.6−2.7 V is observed for different catalysts, similar tothat for the bare carbon without loading of any catalyst. Thiseffect probably implies that either the ORR kinetics in theseLi−O2 air electrodes is limited by the oxygen mass transportresistance toward the catalysts or carbon itself can providesufficient ORR activity. Understanding the role of carbon in theelectrochemical reactions in the Li−O2 cell could provideguidance and a baseline for identifying efficient catalysts tofacilitate ORR and OER and thereby improve the cellperformance.Porous carbon has been chosen as the air electrode material
for almost all the Li−O2 batteries investigated so far, mainlybecause it can provide sufficient charge transfer for theelectrochemical reactions and space for housing the dischargeproducts. Due to the low mass of the carbon-based airelectrode, it is expected to achieve the highest specific capacityin Li−O2 cells using a metallic lithium electrode. Moreover,porous carbon often shows certain catalytic activity towardORR because of the existence of defect sites on the carbonsurface. Commercial carbon black, in particular, Super Pcarbon44,45 and Ketjen Black,48 has been widely used in thecurrent Li−O2 cells. For example, Jung et al.91,136 reported a
Figure 17. Magnetic susceptibility data from SQUID measurements for (a) a pristine carbon air electrode before cycling and a discharged airelectrode with 1NM3/LiCF3SO3 electrolyte and (b) the difference between the pristine carbon air electrode and the initially discharged air electrode.(c) Magnetic moment data from SQUID measurement for Li2O2 bulk powder. (d) EPR spectrum of Li2O2 bulk powder at 4 K. The red trace is theexperimental result. The blue trace is the calculated one-line Lorentzian spectrum to fit the central peak at the g2 value, and the green trace is thedifference between the red and blue traces to reveal peaks at g1 and g3. Reprinted with permission from ref 225. Copyright 2013 Wiley-VCH.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXP

Li−O2 battery with impressive capacity and rate capability usingSuper P carbon as the only air electrode active material incombination with TEGDME/LiCF3SO3 electrolyte.Aside from the commercially available carbon black, recent
studies showed that the performance of the new carbon-basedmaterials could also be very successful when used with a stableelectrolyte. Mitchell et al.61 reported a Li−O2 cell using hollowcarbon fibers as the air electrode material, which directly growon a ceramic porous substrate, eliminating the need for abinder. These all-carbon-fiber electrodes demonstrated anexceptionally high discharge capacity of 7200 mA·h/gcarbon,translating to a gravimetric energy of 2500 W·h/kgcarbon, whichis 4 times higher than that of the state-of-the-art lithiumintercalation compounds such as LiCoO2 (600 W·h/kg). Theyattributed such high capacity of the cell to low carbon packingand highly efficient utilization of the available carbon mass andvoid volume for Li2O2 formation. With the unique structure ofthe carbon fiber, they were able to monitor the Li2O2 formationand morphological evolution during discharge as shown inFigure 18, where the toroid-shape Li2O2 was first reported. Thevisualization of Li2O2 morphologies upon discharge anddisappearance upon charge represents a critical step towardunderstanding key processes that limit the rate capability andlow round-trip efficiencies of Li−O2 batteries. In a follow-uppaper by the same group,101 a more detail mechanism ofmorphological evolution of Li2O2 particles during electro-chemical growth was reported using a freestanding carbonnanotube (CNT) air electrode in a DME-based electrolyte. Themorphology of Li2O2 at the first discharge cycle appears to be afunction of both the discharge rate and capacity (see Figure19), which is related to the nucleation and growth process ofLi2O2 on CNT. At low discharge current density, sparseparticles first nucleate on the CNT sidewalls as stacked thinplates, which spontaneously splay so that secondary nucleationof new plates eventually leads to the development of disk- andtoroid-shaped particles, as shown in Figure 19a,b. The high-resolution TEM images on CNT suggest an instantaneousnucleation of Li2O2 at relatively few sites early in the dischargeprocess. Thin plates of Li2O2 with the large facet of the [001]
plane were found in both disk- and toroid-like particles,consistent with theoretical calculations of the equilibrium Wulffshape for Li2O2 and a layer-by-layer growth mode. In contrast,at higher discharge rates, copious nucleation of equiaxed Li2O2
Figure 18. Evolution of Li2O2 discharge product morphology. Insets show the corresponding discharge voltage profile. (a, b) Galvanostatic dischargeto a capacity of 350 mA·h/g at 68 mA/g. Li2O2 particles appear to first form on the CNT sidewalls as small spheres with <100 nm diameters. (c, d)Intermediate galvanostatic discharge to 1880 mA·h/g at 64 mA/g. Particles appear to develop a toroidal shape as the average particle size increases to400 nm. (e, f) Full discharge to 7200 mA·h/g at 63 mA/g. The discrete particles merge to form a monolithic Li2O2 mass with low porosity. Thecapacity and current density are obtained on the basis of the mass of the CNT. Reprinted with permission from ref 61. Copyright 2011 Royal Societyof Chemistry.
Figure 19. SEM and TEM micrographs of Li2O2 particles onelectrodes consisting of freestanding CNT carpets discharged at lowand intermediate gravimetric rates. (a) SEM and (b) TEMmicrographs of an electrode discharged at 10 mA/gcarbon to 200 mA·h/gcarbon, with disk-shaped particles and bare CNT sidewalls. (c) SEMand (d) TEM micrographs of an electrode discharged at 90 mA/gcarbonto 13 000 mA·h/gcarbon, with a high density of disk particles and a thincoating of discharge product present on the sidewalls of the CNTs.Insets: Higher magnification TEM images of the CNT sidewalls,indicated by a dashed yellow line, showing the (b) absence and (d)presence of small particles (scale bar = 20 nm). Reprinted from ref101. Copyright 2013 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXQ

particles precedes growth of disks and toroids. A much higherdensity of Li2O2 particles with nonuniform shapes wasobserved, and the CNT sidewalls appeared to be coated bynumerous small particles (Figure 19c,d). Their study alsodemonstrated that the coating of the carbon surface with smallirregular particles precedes growth of well-defined larger disk-and toroid-shaped particles with increasing discharge rates.Findings on the toroidal morphology of Li2O2 are interesting
and critical, since it leads to very nonuniform surface coverage,which is beneficial to increasing the discharge capacity of thecell due to easier oxygen diffusion at the late stage of thedischarge. Therefore, understanding and controlling thenucleation and morphological evolution of Li2O2 particlesupon discharge is the key factor to achieving high volumetricenergy density of the Li−O2 cell. Several research groups havealso observed the formation of Li2O2 toroid-shaped particles ondifferent carbon air electrodes with or without additionalcatalysts.44,174,175,243
Graphene-based materials have also been investigated as theair electrode due to their light weight and conductive andcatalytically active surface. For instance, Xiao et al.62 reported aprimary Li−air battery containing hierarchically porousfunctionalized graphene sheets (FGSs) delivers an extremelyhigh capacity of 15 000 mA·h/gcarbon, demonstrating thepotential application of graphene in the Li−O2 system. Suchsignificantly improved performance of the graphene-based airelectrode is attributed to its unique morphology and differentLi2O2 deposition mechanism. The FGS electrode has numerouslarge tunnels facilitating continuous oxygen flow into the airelectrode, while other small “pores” provide ideal triphaseregions for the oxygen reduction, as shown in Figure 20a,b.
Unlike the toroid-shaped particles reported by Mitchel et al.61
as discussed above, the deposited Li2O2 forms isolatednanosized “islands” on FGSs (Figure 20c,d), supported bytheir DFT calculation which suggests that the aggregation ofLi2O2 clusters is energetically unfavorable in the vicinity ofdefective sites of FGSs.
Through the above research work, it is clear that the Li2O2morphology (e.g., shape and characteristic thickness) andstructure (e.g., crystallinity and surface vs bulk composition) areimportant parameters that can influence the discharge capacity,rate capability, and cyclability of a Li−O2 cell. Therefore, it willbe a major contribution to the development of Li−O2 cells ifthe parameters associated with the Li2O2 morphology andstructure can be finely tuned. Understanding the link betweenthe morphology and structure of Li2O2 and the electro-chemistry will be critical for identification of new approaches toreduce the overpotentials on discharge and, more significantly,on charge.27
3.5.2. Catalysts on Porous Carbon. Since the firstdemonstration of a lithium−oxygen system using pyrolyzedcobalt phthalocyanine over carbon as the air electrode,10 manynew air electrode materials have been reported, particularly inthe area of new catalysts for improving the batteryefficiency.28,30,32,59,244 As discussed in section 3.3, most of theearly studies on these catalytic air electrode materials werebased on organic carbonate electrolytes, which were laterproved to catalyze the formation/decomposition of Li2CO3and/or the decomposition of the carbonate electrolyte ratherthan the formation/decomposition of Li2O2. These catalystsinclude transition-metal oxides, nonprecious metals, andprecious metals (and alloys), as reviewed by Shao et al.,42
and their true catalytic activity in Li−O2 systems needs to be re-evaluated. Below, we only discuss some recent research workon catalytic air electrodes that form and decompose Li2O2, i.e.,in more stable electrolytes.To investigate the electrocatalytic activity of any catalyst on
reaction 4 (Figure 3) in an aprotic Li−O2 cell, the formation ofLi2O2 is a prerequisite. As discussed in section 3, the choice ofthe organic electrolyte is the most critical factor determiningwhether Li2O2 will form during cell discharge. Oftentimes, airelectrode materials, including electrocatalysts, influence thedischarge products of the Li−O2 cell, in particular theformation of Li2O2. For example, Freunberger et al.83
investigated three carbons, namely, Super P, Ketjen Black,and Black Pearls, in Li−O2 cells with an ether-based electrolyte,and they identified Li2O2 as the dominating discharge product.However, the electrolyte also decomposed, as was evident intheir FT-IR results (through ν(C−O) bands), which indicatedthe presence of polyethers/esters on the air electrode surface.They also found a much higher concentration of polyethers/esters in Black Pearls than in Super P or Ketjen Black, probablyindicating a higher catalytic activity of Black Pearls toward thedecomposition of the electrolyte. A similar effect on the airelectrode was also reported by Allen et al.153 when theycompared the cyclabilities of a Au electrode and a glassy carbonelectrode. Nevertheless, the catalytic composition of the airelectrode material does significantly impact the dischargeproducts and, therefore, the cyclability of the Li−O2 cell.In general, the electrocatalyst is critical to improve the rate
capability, cyclability, and round-trip efficiency of the Li−O2cell. Earlier studies demonstrated that the catalysts investigatedto date probably did not change the mechanism of thedischarge reaction toward the formation of Li2O2, but they maydecrease the activation energy for oxygen reduction, whichtranslates into a slightly higher discharge voltage in some cases.Various catalysts have been examined to lower the activationbarriers, such as metal oxide, precious metals, and nonpreciousmetals, the mostly widely studied of which is MnO2.
45,216,245
Since MnO2 nanoparticles can exhibit a large variety of
Figure 20. Morphologies of the graphene-based air electrode. (a, b)SEM images of as-prepared FGS (C/O = 14) air electrodes at differentmagnifications. (c, d) Discharged air electrode using FGSs with C/O =14 and C/O = 100, respectively. Reprinted from ref 62. Copyright2011 American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXR

crystallographic structures (e.g., α, β, δ, γ, and λ forms) andmorphologies, controlled synthesis of nanostructured MnO2with high crystalline purity and uniform morphology is criticalto achieving precisely tailored properties and, thus, betterperformance in their applications in Li−O2 batteries. Toaddress this issue, Truong et al.125 produced high-qualitynanosheet-based δ-MnO2 microflowers, α-MnO2 nanowires,and α-MnO2 nanotubes in large quantity through themicrowave-assisted hydrothermal reduction of potassiumpermanganate in the presence of hydrochloric acid, as shownin Figure 21. They found that the chemical reaction determines
the formation of δ-MnO2 microflowers in the earlier stages ofthe process, while the two-step Ostwald ripening process isresponsible for the crystalline and morphological transitionfrom the δ-MnO2 nanosheets to α-MnO2 nanowires andnanotubes in the late stage of the reactions. In terms of theelectrocatalytic activity for these different MnO2 nanoparticles,the single-crystal α-MnO2 nanotubes exhibit much betterperformance to catalyze the electrochemical processes involvedin aprotic Li−O2 cells using TEGDME-based electrolytes,leading to a significant improvement in specific capacity andcycle life. Qin et al.45 developed an in situ wet-chemistryapproach to incorporate MnO2 nanorods into porous carbon.This process avoids any aggressive post-treatment of the carbonmaterials and, thus, preserves the original structure of carbon.As-prepared MnO2/C composites with porous structures andhigh specific surface area provide more active sites for enabling
oxygen reduction and evolution reactions and, therefore, lead tosignificant enhancement of the electrochemical performance ofLi−O2 cells. In this case, a low charge overpotential (3.5 V) wasachieved by using the as-prepared MnO2/C composite as theair electrode material with a TEGDME-based electrolyte.Using a dimethoxyethane-based electrolyte, McCloskey et
al.88 investigated the electrocatalytic role of Au, Pt, and MnO2nanoparticles and found none performed better than carbon.They found the charge potential at the beginning of the chargecycle to be low (∼3.4 V), but it rose to above 4 V during thefirst cycle. Black et al.175 studied the role of catalysts andperoxide oxidation in Li−O2 batteries to address theaforementioned question on the efficacy or necessity ofcatalysis in the aprotic system. They demonstrated that chargeoverpotential could be significantly reduced when a nano-crystalline Co3O4/RGO (reduced graphene oxide) composite isincluded as part of a carbon-based air electrode, as shown inFigure 22. XRD results confirmed the presence of Li2O2 on thefirst discharge, and SEM images showed that these Li2O2particles have a toroid shape. Black et al. indicated thatCo3O4/RGO clearly enhances the kinetics of mass (or surface)transport for both oxygen reduction and evolution rather thanconventional electrocatalytic effects of the particles. However,the significant improvement on cycling appears to be
Figure 21. SEM images of MnO2 products obtained at differentreaction times: (A) 2, (B) 5, (C) 15, (D) 30, (E) 120, and (F) 360min. The temperature of the reaction solutions and the atmospherepressure above the reaction solutions were 150 °C and 70 psi,respectively. The concentrations of KMnO4 and HCl in the reactionsolution were 0.05 and 0.2 M, respectively, before the reaction wasinitiated. The insets are the high-magnification SEM images of thecorresponding structures. The scale bars in (F) also apply to (A)−(E).Reprinted from ref 125. Copyright 2013 American Chemical Society.
Figure 22. (a) First discharge−charge profile for Li−O2 cells withKetjen Black (KB) or Co3O4/RGO/KB at 25 °C at a current rate of140 mA/g. (b) XRD patterns of the cells in (a) on first discharge.Reflections of Li2O2 are marked. (c) SEM micrographs of thedischarged electrodes at 6000 mA·h/g: (i) Co3O4/RGO/KB and (ii)KB. (d) Chronoamperometry showing normalized current evolutionwith time at 2.25 V. (e) Linear sweep voltammetry following a hold at2.25 V for 1 h. (f) Voltage profile on charge for cells containingchemically deposited Li2O2 in the presence of either Co3O4/RGO/KBor KB. The specific capacity and current density were based on themass of the active materials. Reprinted with permission from ref 175.Copyright 2013 Wiley-VCH.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXS
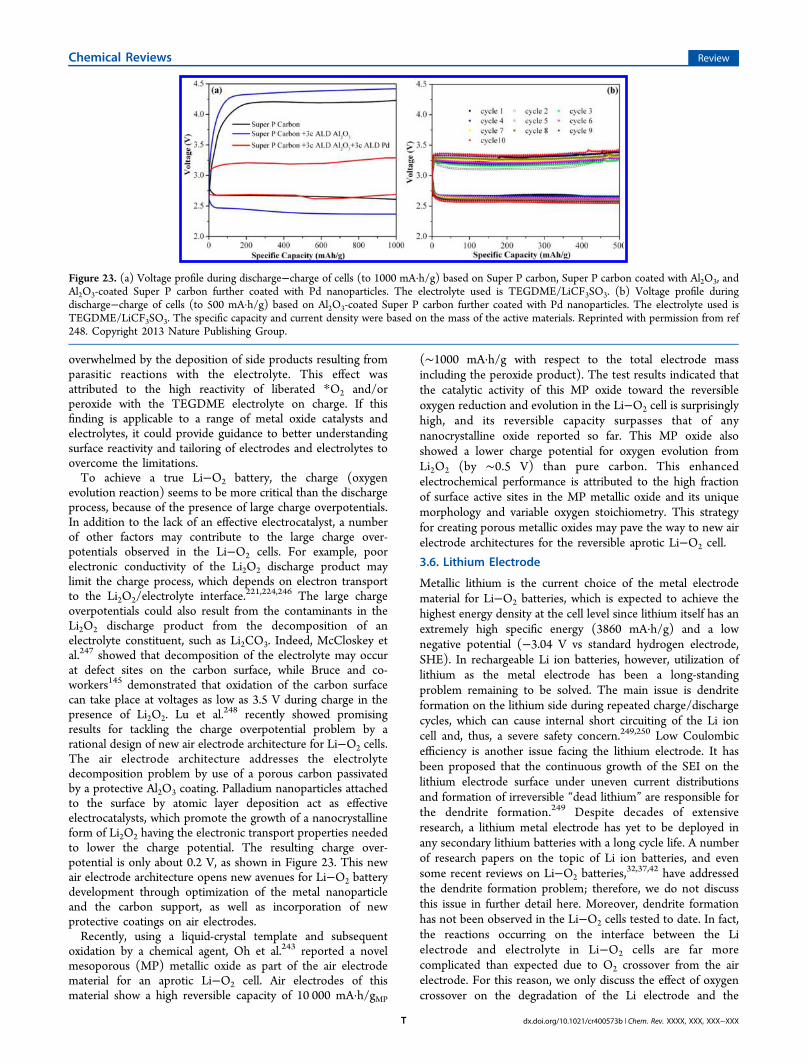
overwhelmed by the deposition of side products resulting fromparasitic reactions with the electrolyte. This effect wasattributed to the high reactivity of liberated *O2 and/orperoxide with the TEGDME electrolyte on charge. If thisfinding is applicable to a range of metal oxide catalysts andelectrolytes, it could provide guidance to better understandingsurface reactivity and tailoring of electrodes and electrolytes toovercome the limitations.To achieve a true Li−O2 battery, the charge (oxygen
evolution reaction) seems to be more critical than the dischargeprocess, because of the presence of large charge overpotentials.In addition to the lack of an effective electrocatalyst, a numberof other factors may contribute to the large charge over-potentials observed in the Li−O2 cells. For example, poorelectronic conductivity of the Li2O2 discharge product maylimit the charge process, which depends on electron transportto the Li2O2/electrolyte interface.221,224,246 The large chargeoverpotentials could also result from the contaminants in theLi2O2 discharge product from the decomposition of anelectrolyte constituent, such as Li2CO3. Indeed, McCloskey etal.247 showed that decomposition of the electrolyte may occurat defect sites on the carbon surface, while Bruce and co-workers145 demonstrated that oxidation of the carbon surfacecan take place at voltages as low as 3.5 V during charge in thepresence of Li2O2. Lu et al.248 recently showed promisingresults for tackling the charge overpotential problem by arational design of new air electrode architecture for Li−O2 cells.The air electrode architecture addresses the electrolytedecomposition problem by use of a porous carbon passivatedby a protective Al2O3 coating. Palladium nanoparticles attachedto the surface by atomic layer deposition act as effectiveelectrocatalysts, which promote the growth of a nanocrystallineform of Li2O2 having the electronic transport properties neededto lower the charge potential. The resulting charge over-potential is only about 0.2 V, as shown in Figure 23. This newair electrode architecture opens new avenues for Li−O2 batterydevelopment through optimization of the metal nanoparticleand the carbon support, as well as incorporation of newprotective coatings on air electrodes.Recently, using a liquid-crystal template and subsequent
oxidation by a chemical agent, Oh et al.243 reported a novelmesoporous (MP) metallic oxide as part of the air electrodematerial for an aprotic Li−O2 cell. Air electrodes of thismaterial show a high reversible capacity of 10 000 mA·h/gMP
(∼1000 mA·h/g with respect to the total electrode massincluding the peroxide product). The test results indicated thatthe catalytic activity of this MP oxide toward the reversibleoxygen reduction and evolution in the Li−O2 cell is surprisinglyhigh, and its reversible capacity surpasses that of anynanocrystalline oxide reported so far. This MP oxide alsoshowed a lower charge potential for oxygen evolution fromLi2O2 (by ∼0.5 V) than pure carbon. This enhancedelectrochemical performance is attributed to the high fractionof surface active sites in the MP metallic oxide and its uniquemorphology and variable oxygen stoichiometry. This strategyfor creating porous metallic oxides may pave the way to new airelectrode architectures for the reversible aprotic Li−O2 cell.
3.6. Lithium Electrode
Metallic lithium is the current choice of the metal electrodematerial for Li−O2 batteries, which is expected to achieve thehighest energy density at the cell level since lithium itself has anextremely high specific energy (3860 mA·h/g) and a lownegative potential (−3.04 V vs standard hydrogen electrode,SHE). In rechargeable Li ion batteries, however, utilization oflithium as the metal electrode has been a long-standingproblem remaining to be solved. The main issue is dendriteformation on the lithium side during repeated charge/dischargecycles, which can cause internal short circuiting of the Li ioncell and, thus, a severe safety concern.249,250 Low Coulombicefficiency is another issue facing the lithium electrode. It hasbeen proposed that the continuous growth of the SEI on thelithium electrode surface under uneven current distributionsand formation of irreversible “dead lithium” are responsible forthe dendrite formation.249 Despite decades of extensiveresearch, a lithium metal electrode has yet to be deployed inany secondary lithium batteries with a long cycle life. A numberof research papers on the topic of Li ion batteries, and evensome recent reviews on Li−O2 batteries,
32,37,42 have addressedthe dendrite formation problem; therefore, we do not discussthis issue in further detail here. Moreover, dendrite formationhas not been observed in the Li−O2 cells tested to date. In fact,the reactions occurring on the interface between the Lielectrode and electrolyte in Li−O2 cells are far morecomplicated than expected due to O2 crossover from the airelectrode. For this reason, we only discuss the effect of oxygencrossover on the degradation of the Li electrode and the
Figure 23. (a) Voltage profile during discharge−charge of cells (to 1000 mA·h/g) based on Super P carbon, Super P carbon coated with Al2O3, andAl2O3-coated Super P carbon further coated with Pd nanoparticles. The electrolyte used is TEGDME/LiCF3SO3. (b) Voltage profile duringdischarge−charge of cells (to 500 mA·h/g) based on Al2O3-coated Super P carbon further coated with Pd nanoparticles. The electrolyte used isTEGDME/LiCF3SO3. The specific capacity and current density were based on the mass of the active materials. Reprinted with permission from ref248. Copyright 2013 Nature Publishing Group.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXT

possible solutions to improve the Li electrode performance inLi−O2 cells.As discussed earlier, the current major focus of studies on
aprotic Li−O2 batteries is the roles that electrolyte decom-position and the catalytic oxygen electrode play in cellperformance. It is well established that electrolyte decom-position in Li−O2 systems mainly arises from chemical orelectrochemical reactions at the electrolyte−air electrodeinterface. The electrolyte decomposition at the electrolyte−metal electrode interface, if any, has not been investigated inmuch detail yet. In a Li ion cell, Aurbach et al. have shown thatlithium metal electrodes fail when carbonate solvent (e.g.,ethylene carbonate) is reduced and decomposes to form surfacelayers of ROCO2Li, ROLi, and Li2CO3 species.
249 Since ethershave reduction potentials similar to those of carbonates, it is ofinterest to investigate the possible decomposition mechanism atthe lithium electrode, especially in the presence of O2 crossingover from the air electrode and the possible effects on theperformance of the Li−O2 cell.Assary et al.57 investigated the stability of an ether-based
electrolyte (TEGDME/LiCF3SO3) at the lithium electrode in aLi−O2 cell where oxygen crossover occurs. In their study, high-energy XRD, FTIR spectroscopy, and DFT were used todetermine the possible reactions that may occur under an O2environment. The DFT calculations indicated that the presenceof O2 at the Li electrode will favor the reaction pathways shownin Figure 24, considering that the binding of O2 with the
TEGDME anion radical (from the electrochemical reduction atthe Li electrode) is exothermic. These reaction pathways willfinally lead to the formation of the LiOH and lithiumcarbonates. As the concentration of these species increases,the insoluble LiOH and Li2CO3 will deposit on the Lielectrode. Assary et al. have also carried out a detailedcharacterization study of the Li electrode during various stagesof discharge and charge using in situ XRD and FTIRmeasurements, which clearly indicate the formation of LiOHand Li2CO3 on cycling. As a consequence, the Li
+ ion migrationwill be significantly blocked by these insoluble products, leadingto poor cell performance. This study indicated that controllingSEI reactions at the lithium electrode through suitablemembranes or passivation films is essential for achievinggood performance of Li−O2 cells. Previous studies alsosuggested the need for an efficient protective layer for metalliclithium to avoid decomposition of the Li electrode due tocontamination by discharge/charge products in the Li−O2cells.37,60,251−253
Recently, Shui et al.108 investigated the reversibility of theanodic lithium inside an operating Li−O2 cell using spatiallyand temporally resolved synchrotron XRD and a 3-dimensionalmicrotomography technique. The combined electrochemicaland in situ XRD results revealed a continuous accumulation ofLiOH on the lithium electrode under both charge anddischarge conditions, along with partial recovery of lithiumduring the charge cycle, as shown in Figure 25. Since LiOH is
Figure 24. Proposed mechanism and energetics for the formation of LiOH from an ether-based solvent (TEGDME) at the electrode upon reductionand oxygen crossover. Also shown in the box is a mechanism for the further formation of lithium alkyl carbonates and carbonates from the resultingaldehydes. All energetics are changes in enthalpies (eV) in solution (ε = 21) at 298 K computed at the B3LYP/6-31+G(d) level of theory Reprintedwith permission from ref 57. Copyright 2013 Wiley-VCH.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXU

neither a Li+ ion nor an electron conductor, a continuoussupply of Li+ from the metallic lithium electrode would beproblematic if LiOH formed on the lithium surface as a densefilm. Using a microtomography technique, Shui et al.demonstrated that a large number of microscopic “tunnels”exist within the LiOH layer. These tunnels provide a pathwayfor sustained ion transport, since they enable the connection ofthe metallic lithium with the electrolyte. Therefore, the batterycan be operated until the total consumption of lithium hasoccurred, assuming that the air electrode and electrolyte cansurvive for that long period.Because the separators currently used in Li−air cells cannot
prevent O2/H2O diffusion to the Li electrode, oxygen crossovercan be a significant problem, leading to fast decay of the Lielectrode, as discussed above. It is critical to develop thin, activemembranes that can be embedded within passive porouspolymeric membranes to effectively eliminate the O2 crossoverto the Li electrode, which is responsible for limiting thereversibility of Li−O2 cells. To address this issue, Truong etal.254 fabricated single-crystal silicon membranes, which arecapable of conducting Li+ ions through their lattices to supportcurrent density as high as 1 mA/cm2. However, compared withthe best NASICON-type lithium ion conducting membranes,the Li+ conductivity of the single-crystal Si membranes is stilltoo low (3−4 orders lower). Their ionic conductivity might beimproved by doping the surface lattices of single-crystal Simembranes, lowering the interfacial energy barriers for Li+
insertion/extraction from the Si lattices, or partially lithiatingthe single-crystal Si membranes to increase the concentration ofLi+ ions in the membranes and thereby reduce their diffusiveresistance. In addition, direct contact of the single-crystal Simembranes with Li metal should be avoided due to the severeelectrochemical reactions, although silicon is stable in manyelectrolytes, including neutral/acidic aqueous electrolytes andaprotic organic electrolytes.
4. AQUEOUS LI−O2 BATTERIES
As presented above, we extensively reviewed the challenges andadvances in the aprotic Li−O2 system, which is garnering themajor fraction of the current research on Li−air batteries.Unlike the aprotic electrolyte, for which numerous types ofliquid or solid ionic conductors can be selected, the aqueouselectrolyte is limited to acidic or basic solutions only. In theaqueous Li−air battery, electrolyte solvent, e.g., H2O, is not alimiting factor in the cell performance, which is its mainadvantage over the aprotic system. In addition, incombustibleaqueous electrolytes circumvent the safety issue,255 which is amajor concern for the organic electrolytes in an open cellconfiguration. However, due to the different electrochemicalreactions involving Li and O2, the gravimetric and volumetriccapacities of an aqueous Li−O2 cell are much lower comparedto those of an aprotic cell.50
A typical aqueous Li−air cell configuration is shown inFigure 2b. During the cell discharge, oxygen is reduced on theair electrode via the ORR, similar to that in the aprotic system.However, water in the electrolyte solution also participates inthe electrochemical reactions for the aqueous Li−air system.On charging of the secondary aqueous Li−air cell, the OERtakes place on the air electrode to produce Li+, O2, andelectrons. Catalysts are often used to promote the 4e− ORRand OER with increasing activities in secondary cells. Tosupport such catalysts, porous carbon along with a possiblebinder has been widely employed as part of the air electrodematerial, which also provides electronic conduction. In somecases, a gas diffusion layer (GDL) is attached to the active airelectrode to allow rapid and uniform diffusion of oxygen to theactive reaction sites. For a rechargeable aqueous Li−air cell, theGDL must have both hydrophobic and hydrophilic pores tosupply O2 during ORR and transport water from the airelectrode to stop flooding, respectively. A Li+ conducting filmmust be present on the lithium electrode to prevent anexcessively vigorous reaction between lithium metal and H2O.
Figure 25. (a) Discharge−charge voltage profile of the operando Li−O2 cell as a function of the cycling time. The stages at which XRD data setswere collected are marked as i, ii, iii, ... on the curve. (b) Four representative XRD sets taken at cycling stages i, iii, vi, and ix; each set consisted ofseven selected XRD patterns collected at various electrode−separator interfacial depths (marked as 1, 2, ..., 7 of different colors on the right of eachset). (c) Expanded XRD data set at stage ix covering the whole electrode through the adjacent separator region. The inset is a representative XRDpattern taken from the position marked by a blue line that includes references of Li and LiOH. Reprinted with permission from ref 108. Copyright2013 Nature Publishing Group.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXV

In general, the discharge products are soluble in the aqueoussolution, and the discharge potentials of the aqueous Li−aircells are slightly higher than those of the aprotic systems,normally in the range of 3.0−3.3 V vs Li/Li+, depending on thepH of the aqueous electrolyte. The typical aqueous electrolyteis a mixture of LiOH−LiCl in water,52,256 where theconcentration of LiCl and/or pH of the solution can vary.Depending on the pH of the electrolyte, several electrochemicalreactions in the aqueous Li−air system could occur at the airelectrode. In a basic aqueous solution, O2 reduction on the airelectrode involves H2O as a reactant, as shown in the followingreaction:
+ + ⇄2Li 12O H O 2LiOH2 2 (4.1)
The discharge product from this reaction is soluble LiOH,which has limited solubility in water at room temperature (12.8g/100 g of H2O). The concentration of OH− rises withincreasing depth of discharge until it reaches the solubility limitof LiOH, which will then precipitate out of the solution as amonohydrate, LiOH·H2O.
257 Such excess precipitates couldclog the pores of the air electrode and, therefore, block thefurther O2 diffusion, if they are not removed mechanically.Considering the standard equilibrium potential, the celldischarge voltage can be slightly higher for an aqueous Li−airbattery in an acidic electrolyte, which also leads to differentdischarge products. For instance, in a mildly acidic medium, thereaction of Li with O2 results in the formation of LiCl,258 asshown in the following reaction:
+ + ⇄ + +2Li 12O 2NH Cl 2LiCl 2NH H O2 4 3 2
(4.2)
Another example of a reaction in an acidic solution is theformation of Li2SO4 in the case in which H2SO4 is used as partof the aqueous electrolyte.258 The main advantage of operatingthe Li−air cell in an acidic solution is that the formation ofinsoluble carbonates, as in basic solution, can be avoided, yetacidic (or neutral) solution will eventually turn into alkalinesolution during the long-term cycling of a cell. Moreimportantly, it is very challenging to develop an effectiveelectrocatalyst for oxygen reduction and evolution reactions inacidic electrolytes; therefore, for the most part, selection of theelectrocatalyst is limited to the expensive noble metals. Inaddition, the specific energy of an aqueous Li−air system inacidic electrolyte is much lower than that in basic electrolyte.The biggest challenge in realizing an aqueous Li−air battery,
primary or secondary, is to prevent the corrosion of metalliclithium by H2O, which is still a major and continuing effort forresearch and development nowadays. As mentioned earlier,preventing H2O from reaching the Li metal electrode requires abarrier layer that allows the transport of Li+ only between thelithium electrode and aqueous electrolyte. A barrier using athick passive KOH film12,13 on the lithium electrode or ahydrophobic polymer membrane between the Li electrode andelectrolyte259 proved to be unsuccessful since water transportthrough these layers could not be completely avoided. In 2004,the PolyPlus Battery Co.260,261 developed a Li ion conductingglass ceramic (LiC-GC) to separate the lithium metal from theelectrolyte, which allows the safe operation of the aqueous Li−air battery by blocking H2O from reaching the lithiumelectrode. Further developed by Fu and patented by OharaCorp.,262−264 the most successful example of such LiC-GCs todate is the single Li+ conducting NASICON-type glass ceramic,
including Li2O/Al2O3/TiO2/P2O5 and Li2O/Al2O3/Ge2O/P2O5. However, while lithium metal is not compatible withLiC-GC, that problem could be overcome by introducing a Li+
conducting interlayer between the Li electrode and LiC-GCmembrane. More details on such an interlayer can be found in arecent review by Crowthe and Salomon.265
It is well established that catalysts are required to improvethe ORR and OER in conventional fuel cells. Since the airelectrodes of the primary and secondary aqueous Li−air cellsoperate in a fashion similar to that of the polymer electrolytefuel cell and unitized regenerative fuel cell, respectively, thecatalysts developed for those fuel cells could serve asbenchmark catalysts for aqueous Li−air cells. As a “proof-of-concept,” Pt is widely used in most initial studies on theaqueous Li−air battery as the electrocatalyst for ORR due to itscatalytic activity being difficult to match, particularly in acidicmedia.251,266−268 Since Pt is relatively rare and expensive, caremust be taken to minimize the Pt loading while stillmaintaining high electroactivity. In an attempt to explorealternatives to the Pt catalyst, Zhou and co-work-ers54,238,255,269,270 recently investigated the electroactivity oftransition metals and metal oxides in the aqueous Li−airsystem. Air electrodes fabricated with Mn3O4/activatedcarbon54 as a catalytic layer coupled with a GDL delivered aspecific capacity of 50 000 mA·h/g based on the total mass ofthe catalytic electrode (carbon + binder + catalyst), a resultindicating that Mn3O4 appears to have high ORR catalyticactivity in basic solution. This is the highest capacity reportedso far for any Li−O2 cells in aprotic or aqueous systems.In general, monofunctional catalysts only increase the activity
of one particular reaction, either the ORR or the OER. Forexample, Pt is a model ORR catalyst as mentioned earlier. Also,IrO2 is known to improve the rate of OER but does notimprove the rate of ORR. For secondary aqueous Li−air cells, abifunctional catalyst with the capability to catalyze both ORRand OER is mandatory. Wang et al.271 studied CoMn2O4nanoparticles on graphene as a catalyst for a secondary Li−air cell in alkaline aqueous electrolyte, which shows a high ORRactivity, comparable to that of a Pt/C catalyst, and asubstantially improved OER activity. They also found thatCoMn2O4 nanoparticles alone are responsible for the OER withgraphene as an inert support and electronic conductor. A recentstudy on perovskite catalyst272 for ORR and OER in aqueoussolution provided further fundamental guidelines to search formore effective catalysts used in the aqueous Li−air batteries.
5. CONCLUDING REMARKS AND PERSPECTIVES
Rechargeable Li−O2 battery technology is still at the infantstage, although significant progress has been achieved, as wehave presented in this review. We mainly reviewed the researchactivities and achievements of the aprotic Li−O2 battery, sinceit has dominated the research efforts for current Li−O2 systems.We have discussed both experimental and modeling resultswith regard to the critical factors that have a significant impacton the sustainable rechargeability of aprotic Li−O2 cells,including the stability of the electrolyte and its influence on theelectrochemical performance of the cell, the structure andmagnetic properties of Li2O2 and their relevance to the Li−O2battery, the design and optimization of the air electrodestructure with different types of catalysts, and the effect ofoxygen crossover on the Li electrode. Without a doubt,substantial challenges exist for each component of the aprotic
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXW

Li−O2 cells. We have also briefly reviewed the recent researchprogress on the aqueous Li−air system.Organic electrolytes (both solvents and lithium salts) play
the most critical role in development of an aprotic Li−O2 celland determine whether a truly rechargeable Li−O2 battery canbe realized. One of the main challenges (probably the greatest)at the current stage for aprotic Li−O2 cells is the search forstable electrolytes. Carbonate-based electrolytes have beenwidely used in most of the initial research work, but it is nowuniversally recognized that these electrolytes decompose in thepresence of the superoxide radicals. Nevertheless, despite thesevere instability of these electrolytes, many research projectsare using them to investigate the catalytic activities of the airelectrode materials. The catalytic activity of the air electrodematerials needs to be re-examined in more stable electrolytes.Ether-based electrolytes seem to be relatively stable in thepresence of the reduced oxygen species; however, their stabilityduring charge, especially at high voltage, remains unclear.Lithium salt, an indispensable component of the electrolyte,deserves much more attention since it may have a positiveeffect on the electrolyte’s stability in aprotic Li−O2 cells.Without question, searching for a fully stable electrolyte in theoxygen-rich electrochemical environment is the researchpriority at present. Design of a robust strategy for effectivelyscreening the stability of various electrolytes would be greatlybeneficial to the development of a Li−O2 battery for practicalapplication.In parallel with the importance of the electrolyte,
investigation on how the porous air electrode architectureaffects the formation of the discharge product, Li2O2, and thespecific capacity of the cell is still of great interest. It isnecessary to understand the key limiting factors to determinethe capacity, rate capability, and cycling efficiency of aproticLi−O2 cells. Porous carbon with or without additional catalystis the current choice of the air electrode material, although afew noncarbon air electrode materials have been re-ported.59,147,273 However, the mechanism of Li2O2 growth onthe porous air electrode during cell discharge and thesubsequent decomposition of Li2O2 on charge is still debatable;this matter needs to be further clarified to develop moreefficient catalysts for the aprotic Li−O2 cell.The lithium electrode has been a historic problem in any of
the Li battery systems, while the long-term cycling of the Lielectrode has yet to be demonstrated. In terms of protecting theLi electrode from oxygen crossover, controlling reactions of theelectrolyte at the Li electrode through suitable membranes orpassivation films will be essential for achieving good perform-ance with aprotic Li−O2 cells. These membranes should meetthe following criteria: (1) block diffusion of oxygen from the airelectrode to the lithium electrode, (2) allow the transport of Li+
to support current flow, and (3) exhibit excellent mechanicalflexibility and stability to be compatible with the mechanicalflexibility of the supporting polymer membranes and batterydesign/processing. Engineering active membranes with ananometer-scale thickness could potentially meet thesecriteria.274
To study the electrolyte stability and electrocatalytic processin the aprotic Li−O2 system, advanced research tools from bothexperimental and theoretical modeling are necessary. Using asingle characterization technique to identify the possiblereaction products in the cell sometimes leads to misleadingresults, simply due to the complexity of these compounds. Inthis sense, the integration of multiple characterization tools,
including FT-IR,51 XPS,45,171 Raman,84,275 NMR,206
DEMS,59,78 and electron microscopy,136,248 becomes important.
Study of the cell during discharge and charge using in situ high-
energy X-ray techniques is expected to provide insight into the
structural changes at the atomic level.276 X-ray absorption fine
structure spectroscopy (XAFS) is another powerful tool for
studies of the electronic and atomic structures of the catalysts in
action,216 due to the penetrating ability of hard X-rays, which
also allows in situ characterization in a suitably designed cell or
reactor. An important advantage of the XAFS technique is the
element-specific nature, which permits investigation of the local
structure of a particular constituent element in a composite
catalyst. In addition, nonresonant inelastic X-ray scattering
(NIXS) using hard X-rays can provide bulk sensitive
information on Li−air battery discharged products with
element specificity.277 NIXS, also known as X-ray Raman
scattering, can be considered as an alternative approach toward
obtaining soft X-ray absorption spectroscopy like information
by using hard X-rays, despite crystalline or amorphous
components being present. Theory and computational
modeling55,278 have already played an integral part in the
research and development of electrolytes and catalysts for the
Li−O2 battery and should be continued for predicting the
stability of the electrolyte and for screening potential catalyst
candidates for optimal Li−O bond breaking and making.In the case of the aqueous Li−air system, a better
understanding of Li−O2 electrocatalysis is required at the
current stage, since the Li−O2 electrochemistry is unique and
different from that of conventional electrocatalysis. The
successful development of any aqueous Li−air batteries severelyrelies on the prevention of direct contact of the lithium metal
electrode with water. The most innovative approach to address
this issue is the introduction of Li ion conducting glass
ceramics. However, these ceramics are generally fragile and
highly resistive at low temperature. Moreover, they may not be
very stable in strong acidic or basic media. Future research and
development of large and more flexible LiC-GC membranes
will be greatly beneficial to the aqueous Li−O2 system.
Searching for effective catalysts, in particular, with respect to
OER, will be a key challenge for rechargeable aqueous Li−aircells.
AUTHOR INFORMATION
Corresponding Authors
*E-mail: [email protected].*E-mail: [email protected].*E-mail: [email protected].
Notes
The authors declare no competing financial interest.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXX

Biographies
Jun Lu is a DOE-EERE postdoctoral fellow in the Vehicles
Technology Program. His research interests focus on electrochemical
energy storage and conversion technology, with the main focus on
beyond Li ion battery technology. Dr. Lu earned his bachelor’s degree
in chemistry physics from the University of Science and Technology of
China (USTC) in 2000. He completed his Ph.D. in the Department of
Metallurgical Engineering at the University of Utah in 2009, with his
major research on metal hydrides for reversible hydrogen storage
application. Dr. Lu has authored/coauthored more than 70 peer-
reviewed research papers and has filed over a dozen patents and patent
applications.
Li Li is an associate professor in the School of Chemical Engineering
and the Environment, Beijing Institute of Technology (BIT), China.
Her research interests focus on electrochemical energy storage and
conversion technology and the recycling technique and life-cycle
analysis for spent secondary batteries. As the principal investigator, Dr.
Li successfully hosted the National Key Program for Basic Research of
China, National High Tech 863 project, National Natural Science
Foundation, etc. Dr. Li earned her Ph.D. from BIT in 2004. She has
authored/coauthored over 60 research papers and filed 18 patents and
patent applications in China.
Jin-Bum Park was born in 1985 in Daegu, Korea. In 2011, he receiveda Bachelor of Science degree in chemical engineering from HanyangUniversity. He is presently a Ph.D. candidate in the Department ofEnergy Engineering at Hanyang University, Korea, under thesupervision of Professor Yang-Kook Sun. His research focuses onmaterials development in the fields of energy conversion and storage,such as air electrodes and electrolyte materials for Li−air batteries.
Yang-Kook Sun received his M.S. degree and Ph.D. degree from theSeoul National University, Korea. In 1996 he was a principalresearcher at Samsung SDI and contributed to the commercializationof the lithium polymer battery. He has worked at the HanyangUniversity in Korea as a professor since 2000. His research interestsare the synthesis of new electrode materials for lithium ion batteries,supercapacitors, and next-generation batteries of Na ion batteries, Li−S batteries, and Li−air batteries. In 2007 and 2011, he was awarded theEnergy Technology Division Research Award and Research Award inBattery Division of the Electrochemical Society. He has been SeniorVisiting Scientist at the Argonne National Laboratory since 2007.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXY

Feng Wu was born in Beijing in 1951 and has been working at theBeijing Institute of Technology (BIT) since graduating in 1982. Prof.Wu is the director of the Professor Commission of the School ofChemical Engineering and the Environment, the director of the GreenEnergy Research Institute, and the vice president of the China BatteryIndustry Association. His research interests are new electrode materialsand electrolytes for green secondary batteries. As the chief scientist, hehas hosted the National Key Program for Basic Research of China(973 projects), Basic Research on New Rechargeable Batteries andRelated Materials, since 2002. He has published over 500 papers andhas been awarded 45 patents. In 2012, he was awarded the IBAResearch Award for outstanding research of battery material andsystems for electric vehicles.
Khalil Amine is a Distinguished Fellow and Manager of the AdvancedBattery Technology programs at Argonne National Laboratory, wherehe is responsible for directing the research and development ofadvanced materials and battery systems for HEV, PHEV, EV, satellite,military, and medical applications. Dr. Amine currently serves as amember of the U.S. National Research Council on battery-relatedtechnologies. Among his many awards, Dr. Amine is a 2003 recipientof Scientific America’s Top Worldwide Research 50 Research Award, a2009 recipient of the U.S. Federal Laboratory Award for Excellence inTechnology Transfer, and a four-time recipient of the R&D 100Award, which is considered as the Oscar of technology and innovation.In addition, he was recently awarded the ECS Battery TechnologyAward and the International Battery Association Award. Dr. Amineholds or has filed over 130 patents and patent applications and hasover 280 publications. From 1998 to 2008, Dr. Amine was the mostcited scientist in the world in the field of battery technology.
ACKNOWLEDGMENTSThis work was supported by the U.S. Department of Energy(DOE) under Contract DE-AC0206CH11357, with the mainsupport provided by the Vehicle Technologies Office, DOEOffice of Energy Efficiency and Renewable Energy (EERE). J.L.was supported by a DOE EERE Postdoctoral Research Awardunder the EERE Vehicles Technology Program administered bythe Oak Ridge Institute for Science and Education (ORISE) forthe DOE. This work was also partially supported by theInternational S&T Cooperation Program of China (Grant2010DFB63370) and the Chinese National 973 Program(Grant 2009CB220100). This work was supported by theHuman Resource s Deve lopment p rog r am (No .20124010203310) of the Korea Institute of Energy TechnologyEvaluation and Planning (KETEP) grant funded by the Koreagovernment Ministry of Trade, Industry and Energy and by aNational Research Foundation (NRF) of Korea grant fundedby the Korean government (MEST) (2009-0092780). We are
also thankful for useful discussions with Dr. David Howell andMr. Tien Duong of the EERE Vehicle Technologies Office.
REFERENCES(1) Metz, B.; Davidson, O.; Coninck, H. C. d.; Loss, M.; Meyer., L. A.IPCC Special Report on Carbon Dioxide Capture and Storage;Cambridge University Press: Cambridge, U.K., New York, 2005.(2) U.S. Energy Information Administration. Annual Energy Review2010; Washington, DC, Oct 19, 2011.(3) U.S. Census Bureau. Foreign Trade Statistics, 2011. http://www.census.gov/foreign-trade/index.html (accessed May 8, 2013).(4) Richter, B.; Goldston, D.; Crabtree, G.; Glicksman, L.; Goldstein,D.; Greene, D.; Kammen, D.; Levine, M.; Lubell, M.; Savitz, M.;Sperling, D. Energy Future: Think Efficiency; American Physical Society:College Park, MD, 2008.(5) U.S. Environmental Protection Agency. Inventory of U.S.Greenhouse Gas Emissions and Sinks 1990−2010; Washington, DC,2012.(6) Elgowainy, A.; Han, J.; Poch, L.; Wang, M.; Vyas, A.; Mahalik, M.;Rousseau, A. Well-to-Wheels Analysis of Energy Use and Greenhouse GasEmissions of Plug-in Hybrid Electric Vehicles; Report No. ANL/ESD/09-2; Argonne National Laboratory: Argonne, IL, February 2009.(7) Eberhard, M.; Tarpenning, M. The 21st Century Electric Car;Tesla Motors: Palo Alto, CA, Oct 6, 2006.(8) Yang, Z.; Zhang, J.; Kintner-Meyer, M. C. W.; Lu, X.; Choi, D.;Lemmon, J. P.; Liu, J. Chem. Rev. 2011, 111, 3577.(9) Bruce, P. G.; Freunberger, S. A.; Hardwick, L. J.; Tarascon, J.-M.Nat. Mater. 2012, 11, 19.(10) Abraham, K. M.; Jiang, Z. J. Electrochem. Soc. 1996, 143, 1.(11) Bennion, D. N.; Littauer, E. L. J. Electrochem. Soc. 1976, 123,1462.(12) Littauer, E. L.; Tsai, K. C. J. Electrochem. Soc. 1976, 123, 771.(13) Littauer, E. L.; Tsai, K. C. J. Electrochem. Soc. 1976, 123, 964.(14) Dey, A. N. Thin Solid Films 1977, 43, 131.(15) Littauer, E. L.; Tsai, K. C. J. Electrochem. Soc. 1977, 124, 850.(16) Littauer, E. L.; Tsai, K. C.; Hollandsworth, R. P. J. Electrochem.Soc. 1978, 125, 845.(17) Blurton, K. F.; Sammells, A. F. J. Power Sources 1979, 4, 263.(18) Gibian, M. J . ; Sawyer, D. T.; Ungermann, T.;Tangpoonpholvivat, R.; Morrison, M. M. J. Am. Chem. Soc. 1979,101, 640.(19) Peled, E. J. Electrochem. Soc. 1979, 126, 2047.(20) Littauer, E. L.; Tsai, K. C. J. Electrochem. Soc. 1980, 127, 521.(21) Momyer, W. R.; Littauer, E. L. Energy to the 21st Century,Proceedings of the 15th Intersociety Energy Conversion EngineeringConference, Seattle, WA, Aug 18−22,1980; American Institute ofAeronautics and Astronautics in cooperation with the IECEC SteeringCommittee: New York, 1980; p 1480.(22) Shao, Y.; Park, S.; Xiao, J.; Zhang, J.-G.; Wang, Y.; Liu, J. ACSCatal. 2012, 2, 844.(23) Black, R.; Adams, B.; Nazar, L. F. Adv. Energy Mater. 2012, 2,801.(24) Cao, R.; Lee, J.-S.; Liu, M.; Cho, J. Adv. Energy Mater. 2012, 2,816.(25) Lee, J.-S.; Kim, S. T.; Cao, R.; Choi, N.-S.; Liu, M.; Lee, K. T.;Cho, J. Adv. Energy Mater. 2011, 1, 34.(26) Park, M.; Sun, H.; Lee, H.; Lee, J.; Cho, J. Adv. Energy Mater.2012, 2, 780.(27) Gallant, B. M.; Kwabi, D. G.; Mitchell, R. R.; Zhou, J.;Thompson, C. V.; Shao-Horn, Y. Energy Environ. Sci. 2013, 6, 2518.(28) Cheng, F.; Liang, J.; Tao, Z.; Chen, J. Adv. Mater. 2011, 23,1695.(29) Ellis, B. L.; Lee, K. T.; Nazar, L. F. Chem. Mater. 2010, 22, 691.(30) Cheng, F.; Chen, J. Chem. Soc. Rev. 2012, 41, 2172.(31) Jeong, G.; Kim, Y.-U.; Kim, H.; Kim, Y.-J.; Sohn, H.-J. EnergyEnviron. Sci. 2011, 4, 1986.(32) Li, F.; Zhang, T.; Zhou, H. Energy Environ. Sci. 2013, 6, 1125.(33) Beattie, S. D.; Manolescu, D. M.; Blair, S. L. J. Electrochem. Soc.2009, 156, A44.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXZ

(34) Christensen, J.; Albertus, P.; Sanchez-Carrera, R. S.; Lohmann,T.; Kozinsky, B.; Liedtke, R.; Ahmed, J.; Kojic, A. J. Electrochem. Soc.2012, 159, R1.(35) Girishkumar, G.; McCloskey, B.; Luntz, A. C.; Swanson, S.;Wilcke, W. J. Phys. Chem. Lett. 2010, 1, 2193.(36) Capsoni, D.; Bini, M.; Ferrari, S.; Quartarone, E.; Mustarelli, P.J. Power Sources 2012, 220, 253.(37) Kraytsberg, A.; Yair, E.-E. J. Power Sources 2011, 196, 886.(38) Padbury, R.; Zhang, X. J. Power Sources 2011, 196, 4436.(39) Song, M.-K.; Park, S.; Alamgir, F. M.; Cho, J.; Liu, M. Mater. Sci.Eng., R 2011, 72, 203.(40) Bruce, P. G.; Hardwick, L. J.; Abraham, K. M. MRS Bull. 2011,36, 506.(41) Kowalczk, I.; Read, J.; Salomon, M. Pure Appl. Chem. 2007, 79,851.(42) Shao, Y.; Ding, F.; Xiao, J.; Zhang, J.; Xu, W.; Park, S.; Zhang, J.-G.; Wang, Y.; Liu, J. Adv. Funct. Mater. 2013, 23, 987.(43) Sun, Y. Nano Energy 2013, 2, 801.(44) Lei, Y.; Lu, J.; Luo, X.; Wu, T.; Du, P.; Zhang, X.; Ren, Y.;Miller, J. T.; Sun, Y.-K.; Elam, J. W. Nano Lett. 2013, 13, 4182.(45) Qin, Y.; Lu, J.; Du, P.; Chen, Z.; Ren, Y.; Wu, T.; Miller, J. T.;Wen, J.; Miller, D. J.; Zhang, Z.; Amine, K. Energy Environ. Sci. 2013, 6,519.(46) Adams, B. D.; Radtke, C.; Black, R.; Trudeau, M. L.; Zaghib, K.;Nazar, L. F. Energy Environ. Sci. 2013, 6, 1772.(47) Lu, J.; Qin, Y.; Du, P.; Luo, X.; Wu, T.; Ren, Y.; Wen, J.; Miller,D. J.; Miller, J. T.; Amine, K. RSC Adv. 2013, 3, 8276.(48) Zhang, J.-G.; Wang, D.; Xu, W.; Xiao, J.; Williford, R. E. J. PowerSources 2010, 195, 4332.(49) Wang, Y.; Zhou, H. Energy Environ. Sci. 2011, 4, 1704.(50) Zheng, J. P.; Liang, R. Y.; Hendrickson, M.; Plichta, E. J. J.Electrochem. Soc. 2008, 155, A432.(51) Ogasawara, T.; Debart, A.; Holzapfel, M.; Novak, P.; Bruce, P.G. J. Am. Chem. Soc. 2006, 128, 1390.(52) He, P.; Wang, Y.; Zhou, H. Electrochem. Commun. 2010, 12,1686.(53) Kumar, B.; Kumar, J. J. Electrochem. Soc. 2010, 157, A611.(54) Wang, Y.; Zhou, H. J. Power Sources 2010, 195, 358.(55) Zhang, Z. C.; Lu, J.; Assary, R. S.; Du, P.; Wang, H. H.; Sun, Y.K.; Qin, Y.; Lau, K. C.; Greeley, J.; Redfern, P. C.; Iddir, H.; Curtiss, L.A.; Amine, K. J. Phys. Chem. C 2011, 115, 25535.(56) Debart, A.; Paterson, A. J.; Bao, J.; Bruce, P. G. Angew. Chem.,Int. Ed. 2008, 47, 4521.(57) Assary, R. S.; Lu, J.; Du, P.; Luo, X.; Zhang, X.; Ren, Y.; Curtiss,L. A.; Amine, K. ChemSusChem 2013, 6, 51.(58) Xu, W.; Xiao, J.; Zhang, J.; Wang, D.; Zhang, J.-G. J. Electrochem.Soc. 2009, 156, A773.(59) Peng, Z.; Freunberger, S. A.; Chen, Y.; Bruce, P. G. Science 2012,337, 563.(60) Laoire, C. O.; Mukerjee, S.; Plichta, E. J.; Hendrickson, M. A.;Abraham, K. M. J. Electrochem. Soc. 2011, 158, A302.(61) Mitchell, R. R.; Gallant, B. M.; Thompson, C. V.; Shao-Horn, Y.Energy Environ. Sci. 2011, 4, 2952.(62) Xiao, J.; Mei, D.; Li, X.; Xu, W.; Wang, D.; Graff, G. L.; Bennett,W. D.; Nie, Z.; Saraf, L. V.; Aksay, I. A.; Liu, J.; Zhang, J.-G. Nano Lett.2011, 11, 5071.(63) Lu, Y.-C.; Xu, Z.; Gasteiger, H. A.; Chen, S.; Hamad-Schifferli,K.; Shao-Horn, Y. J. Am. Chem. Soc. 2010, 132, 12170.(64) Laoire, C. O.; Mukerjee, S.; Abraham, K. M.; Plichta, E. J.;Hendrickson, M. A. J. Phys. Chem. C 2009, 113, 20127.(65) Read, J. J. Electrochem. Soc. 2002, 149, A1190.(66) Read, J.; Mutolo, K.; Ervin, M.; Behl, W.; Wolfenstine, J.;Driedger, A.; Foster, D. J. Electrochem. Soc. 2003, 150, A1351.(67) Read, J. J. Electrochem. Soc. 2006, 153, A96.(68) Zhang, S. S.; Foster, D.; Read, J. J. Power Sources 2010, 195,1235.(69) Xu, K. Chem. Rev. 2004, 104, 4303.(70) Thapa, A. K.; Ishihara, T. J. Power Sources 2011, 196, 7016.
(71) Yang, Y.; Sun, Q.; Li, Y.-S.; Li, H.; Fu, Z.-W. J. Electrochem. Soc.2011, 158, B1211.(72) Read, J.; Motolo, K.; Ervin, M.; Behl, W.; Wolfenstine, J.;Driedger, A.; Foster, D. J. Electrochem. Soc. 2003, 150, A1351.(73) Aurbach, D.; Gofer, Y.; Langzam, J. J. Electrochem. Soc. 1989,136, 3198.(74) Aurbach, D.; Daroux, M.; Faguy, P.; Yeager, E. J. Electroanal.Chem. 1991, 297, 225.(75) Mizuno, F.; Nakanishi, S.; Kotani, Y.; Yokoishi, S.; Iba, H.Electrochemistry (Tokyo, Jpn.) 2010, 78, 403.(76) Xu, W.; Xu, K.; Viswanathan, V. V.; Towne, S. A.; Hardy, J. S.;Xiao, J.; Hu, D.; Wang, D.; Zhang, J.-G. J. Power Sources 2011, 196,9631.(77) Xu, W.; Viswanathan, V. V.; Wang, D.; Towne, S. A.; Xiao, J.;Nie, Z.; Hu, D.; Zhang, J.-G. J. Power Sources 2011, 196, 3894.(78) McCloskey, B. D.; Bethune, D. S.; Shelby, R. M.; Girishkumar,G.; Luntz, A. C. J. Phys. Chem. Lett. 2011, 2, 1161.(79) Freunberger, S. A.; Chen, Y.; Peng, Z.; Griffin, J. M.; Hardwick,L. J.; Barde, F.; Novak, P.; Bruce, P. G. J. Am. Chem. Soc. 2011, 133,8040.(80) Bryantsev, V. S.; Giordani, V.; Walker, W.; Blanco, M.; Zecevic,S.; Sasaki, K.; Uddin, J.; Addison, D.; Chase, G. V. J. Phys. Chem. A2011, 115, 12399.(81) Bryantsev, V. S.; Blanco, M. J. Phys. Chem. Lett. 2011, 2, 379.(82) Laino, T.; Curioni, A. Chem.Eur. J. 2012, 18, 3510.(83) Freunberger, S. A.; Chen, Y.; Drewett, N. E.; Hardwick, L. J.;Barde, F.; Bruce, P. G. Angew. Chem., Int. Ed. 2011, 50, 8609.(84) Peng, Z.; Freunberger, S. A.; Hardwick, L. J.; Chen, Y.;Giordani, V.; Barde, F.; Novak, P.; Graham, D.; Tarascon, J.-M.; Bruce,P. G. Angew. Chem., Int. Ed. 2011, 50, 6351.(85) Xu, D.; Wang, Z.-l.; Xu, J.-j.; Zhang, L.-l.; Zhang, X.-b. Chem.Commun. 2012, 48, 6948.(86) Mizuno, F.; Nakanishi, S.; Shirasawa, A.; Takechi, K.; Shiga, T.;Nishikoori, H.; Iba, H. Electrochemistry (Tokyo, Jpn.) 2011, 79, 876.(87) Lu, Y.-C.; Kwabi, D. G.; Yao, K. P. C.; Harding, J. R.; Zhou, J.;Zuin, L.; Shao-Horn, Y. Energy Environ. Sci. 2011, 4, 2999.(88) McCloskey, B. D.; Scheffler, R.; Speidel, A.; Bethune, D. S.;Shelby, R. M.; Luntz, A. C. J. Am. Chem. Soc. 2011, 133, 18038.(89) Xiao, J.; Hu, J.; Wang, D.; Hu, D.; Xu, W.; Graff, G. L.; Nie, Z.;Liu, J.; Zhang, J.-G. J. Power Sources 2011, 196, 5674.(90) Xu, W.; Hu, J.; Engelhard, M. H.; Towne, S. A.; Hardy, J. S.;Xiao, J.; Feng, J.; Hu, M. Y.; Zhang, J.; Ding, F.; Gross, M. E.; Zhang,J.-G. J. Power Sources 2012, 215, 240.(91) Jung, H.-G.; Hassoun, J.; Park, J.-B.; Sun, Y.-K.; Scrosati, B. Nat.Chem. 2012, 4, 579.(92) Assary, R. S.; Curtiss, L. A.; Redfern, P. C.; Zhang, Z.; Amine, K.J. Phys. Chem. C 2011, 115, 12216.(93) Assary, R. S.; Lau, K. C.; Amine, K.; Sun, Y.-K.; Curtiss, L. A. J.Phys. Chem. C 2013, 117, 8041.(94) Bryantsev, V. S.; Faglioni, F. J. Phys. Chem. C 2012, 116, 7128.(95) Bryantsev, V. S.; Uddin, J.; Giordani, V.; Walker, W.; Addison,D.; Chase, G. V. J. Electrochem. Soc. 2013, 160, A160.(96) McCloskey, B. D.; Bethune, D. S.; Shelby, R. M.; Mori, T.;Scheffler, R.; Speidel, A.; Sherwood, M.; Luntz, A. C. J. Phys. Chem.Lett. 2012, 3, 3043.(97) Zhu, D.; Zhang, L.; Song, M.; Wang, X.; Mei, J.; Lau, L. M.;Chen, Y. J. Solid State Electrochem. 2013, 1.(98) Wang, H.; Xie, K. Electrochim. Acta 2012, 64, 29.(99) Lim, H.-K.; Lim, H.-D.; Park, K.-Y.; Seo, D.-H.; Gwon, H.;Hong, J.; Goddard, W. A.; Kim, H.; Kang, K. J. Am. Chem. Soc. 2013,135, 9733.(100) Ren, X.; Wu, Y. J. Am. Chem. Soc. 2013, 135, 2923.(101) Mitchell, R. R.; Gallant, B. M.; Shao-Horn, Y.; Thompson, C.V. J. Phys. Chem. Lett. 2013, 4, 1060.(102) Gallant, B. M.; Mitchell, R. R.; Kwabi, D. G.; Zhou, J.; Zuin, L.;Thompson, C. V.; Shao-Horn, Y. J. Phys. Chem. C 2012, 116, 20800.(103) Cui, Y.; Wen, Z.; Liang, X.; Lu, Y.; Jin, J.; Wu, M.; Wu, X.Energy Environ. Sci. 2012, 5, 7893.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXAA

(104) Harding, J. R.; Lu, Y.-C.; Tsukada, Y.; Shao-Horn, Y. Phys.Chem. Chem. Phys. 2012, 14, 10540.(105) Lu, Y.-C.; Gasteiger, H. A.; Shao-Horn, Y. J. Am. Chem. Soc.2011, 133, 19048.(106) Lu, Y.-C.; Shao-Horn, Y. J. Phys. Chem. Lett. 2013, 4, 93.(107) Wu, G.; Mack, N. H.; Gao, W.; Ma, S.; Zhong, R.; Han, J.;Baldwin, J. K.; Zelenay, P. ACS Nano 2012, 6, 9764.(108) Shui, J.-L.; Okasinski, J. S.; Kenesei, P.; Dobbs, H. A.; Zhao, D.;Almer, J. D.; Liu, D.-J. Nat. Commun. 2013, 4, 3255.(109) Lei, Y.; Lu, J.; Luo, X.; Wu, T.; Du, P.; Zhang, X.; Ren, Y.;Wen, J.; Miller, D. J.; Miller, J. T.; Sun, Y.-K.; Elam, J. W.; Amine, K.Nano Lett. 2013, 13, 4182.(110) Zhang, L.; Zhang, S.; Zhang, K.; Xu, G.; He, X.; Dong, S.; Liu,Z.; Huang, C.; Gu, L.; Cui, G. Chem. Commun. 2013, 49, 3540.(111) Xu, J.-J.; Xu, D.; Wang, Z.-L.; Wang, H.-G.; Zhang, L.-L.;Zhang, X.-B. Angew. Chem., Int. Ed. 2013, 52, 3887.(112) Trahan, M. J.; Jia, Q.; Mukerjee, S.; Plichta, E. J.; Hendrickson,M. A.; Abraham, K. M. J. Electrochem. Soc. 2013, 160, A1577.(113) Shui, J.-L.; Wang, H.-H.; Liu, D.-J. Electrochem. Commun. 2013,34, 45.(114) Shui, J.; Okasinski, J.; Zhao, D.; ALmer, J.; Liu, D.-J. ESCTrans. 2013, 50, 37.(115) Park, H. W.; Lee, D. U.; Nazar, L. F.; Chen, Z. J. Electrochem.Soc. 2013, 160, A344.(116) Nasybulin, E.; Xu, W.; Engelhard, M. H.; Li, X. S.; Gu, M.; Hu,D.; Zhang, J.-G. Electrochem. Commun. 2013, 29, 63.(117) Meini, S.; Tsiouvaras, N.; Schwenke, K. U.; Piana, M.; Beyer,H.; Lange, L.; Gasteiger, H. A. Phys. Chem. Chem. Phys. 2013, 15,11478.(118) Lim, H.-D.; Park, K.-Y.; Song, H.; Jang, E. Y.; Gwon, H.; Kim,J.; Kim, Y. H.; Lima, M. D.; Robles, R. O.; Lepro, X.; Baughman, R. H.;Kang, K. Adv. Mater. 2013, 25, 1348.(119) Li, F.; Ohnishi, R.; Yamada, Y.; Kubota, J.; Domen, K.;Yamada, A.; Zhou, H. Chem. Commun. 2013, 49, 1175.(120) Jung, H.-G.; Jeong, Y. S.; Park, J.-B.; Sun, Y.-K.; Scrosati, B.;Lee, Y. J. ACS Nano 2013, 7, 3532.(121) Huang, B.-W.; Liao, X.-Z.; Wang, H.; Wang, C.-N.; He, Y.-S.;Ma, Z.-F. J. Electrochem. Soc. 2013, 160, A1112.(122) Elia, G. A.; Park, J.-B.; Scrosati, B.; Sun, Y.-K.; Hassoun, J.Electrochem. Commun. 2013, 34, 250.(123) Barile, C. J.; Gewirth, A. A. J. Electrochem. Soc. 2013, 160, A549.(124) Wu, J.; Park, H. W.; Yu, A.; Higgins, D.; Chen, Z. J. Phys.Chem. C 2012, 116, 9427.(125) Truong, T. T.; Liu, Y.; Ren, Y.; Trahey, L.; Sun, Y. ACS Nano2012, 6, 8067.(126) Fu, Z.; Lin, X.; Huang, T.; Yu, A. J. Solid State Electrochem.2012, 16, 1447.(127) Shui, J.-L.; Karan, N. K.; Balasubramanian, M.; Li, S.-Y.; Liu,D.-J. J. Am. Chem. Soc. 2012, 134, 16654.(128) Bryantsev, V. S. Theor. Chem. Acc. 2012, 131, 1.(129) Oloniyo, O.; Kumar, S.; Scott, K. J. Electron. Mater. 2012, 41,921.(130) Adams, J.; Karulkar, M. J. Power Sources 2012, 199, 247.(131) Lim, H.-D.; Park, K.-Y.; Gwon, H.; Hong, J.; Kim, H.; Kang, K.Chem. Commun. 2012, 48, 8374.(132) Li, Y.; Wang, J.; Li, X.; Geng, D.; Banis, M. N.; Li, R.; Sun, X.Electrochem. Commun. 2012, 18, 12.(133) Lee, S. W.; Gallant, B. M.; Lee, Y.; Yoshida, N.; Kim, D. Y.;Yamada, Y.; Noda, S.; Yamada, A.; Shao-Horn, Y. Energy Environ. Sci.2012, 5, 5437.(134) Hassoun, J.; Jung, H.-G.; Lee, D.-J.; Park, J.-B.; Amine, K.; Sun,Y.-K.; Scrosati, B. Nano Lett. 2012, 12, 5775.(135) Black, R.; Oh, S. H.; Lee, J.-H.; Yim, T.; Adams, B.; Nazar, L. F.J. Am. Chem. Soc. 2012, 134, 2902.(136) Jung, H.-G.; Kim, H.-S.; Park, J.-B.; Oh, I.-H.; Hassoun, J.;Yoon, C. S.; Scrosati, B.; Sun, Y.-K. Nano Lett. 2012, 12, 4333.(137) Zhang, Z. C.; Lyons, L. J.; Jin, J. J.; Amine, K.; West, R. Chem.Mater. 2005, 17, 5646.
(138) Ma, Y.; Li, N.; Li, D.; Zhang, M.; Huang, X. J. Power Sources2011, 196, 2346.(139) Chen, Z.; Wang, H. H.; Vissers, D. R.; Zhang, L.; West, R.;Lyons, L. J.; Amine, K. J. Phys. Chem. C 2008, 112, 2210.(140) Oh, B.; Vissers, D.; Zhang, Z.; West, R.; Tsukamoto, H.;Amine, K. J. Power Sources 2003, 119, 442.(141) Laoire, C. O.; Mukerjee, S.; Abraham, K. M.; Plichta, E. J.;Hendrickson, M. A. J. Phys. Chem. C 2010, 114, 9178.(142) Lopez, N.; Graham, D. J.; McGuire, R.; Alliger, G. E.; Shao-Horn, Y.; Cummins, C. C.; Nocera, D. G. Science 2012, 335, 450.(143) Trahan, M. J.; Mukerjee, S.; Plichta, E. J.; Hendrickson, M. A.;Abraham, K. M. J. Electrochem. Soc. 2013, 160, A259.(144) Sun, B.; Zhang, J.; Munroe, P.; Ahn, H.-J.; Wang, G.Electrochem. Commun. 2013, 31, 88.(145) Ottakam Thotiyl, M. M.; Freunberger, S. A.; Peng, Z.; Bruce,P. G. J. Am. Chem. Soc. 2012, 135, 494.(146) Mozhzhukhina, N.; Mendez De Leo, L. P.; Calvo, E. J. J. Phys.Chem. C 2013, 117, 18375.(147) Chen, Y.; Freunberger, S. A.; Peng, Z.; Fontaine, O.; Bruce, P.G. Nat. Chem. 2013, 5, 489.(148) McCloskey, B. D.; Valery, A.; Luntz, A. C.; Gowda, S. R.;Wallraff, G. M.; Garcia, J. M.; Mori, T.; Krupp, L. E. J. Phys. Chem. Lett.2013, 2989.(149) Chen, Y.; Freunberger, S. A.; Peng, Z.; Barde, F.; Bruce, P. G. J.Am. Chem. Soc. 2012, 134, 7952.(150) Walker, W.; Giordani, V.; Uddin, J.; Bryantsev, V. S.; Chase, G.V.; Addison, D. J. Am. Chem. Soc. 2013, 135, 2076.(151) Giordani, V.; Walker, W.; Bryantsev, V. S.; Uddin, J.; Chase, G.V.; Addison, D. J. Electrochem. Soc. 2013, 160, A1544.(152) Crowther, O.; Meyer, B.; Salomon, M. Electrochem. Solid-StateLett. 2011, 14, A113.(153) Allen, C. J.; Mukerjee, S.; Plichta, E. J.; Hendrickson, M. A.;Abraham, K. M. J. Phys. Chem. Lett. 2011, 2, 2420.(154) De Giorgio, F.; Soavi, F.; Mastragostino, M. Electrochem.Commun. 2011, 13, 1090.(155) Zhang, Z.; Zhang, L.; Schlueter, J. A.; Redfern, P. C.; Curtiss,L.; Amine, K. J. Power Sources 2010, 195, 4957.(156) Zhang, L.; Zhang, Z.; Wu, H.; Amine, K. Energy Environ. Sci.2011, 4, 2858.(157) Zhang, L.; Zhang, Z.; Redfern, P. C.; Curtiss, L. A.; Amine, K.Energy Environ. Sci. 2012, 5, 8204.(158) Qin, Y.; Chen, Z.; Liu, J.; Amine, K. Electrochem. Solid-StateLett. 2010, 13, A11.(159) Chen, Z.; Ren, Y.; Jansen, A. N.; Lin, C.-k.; Weng, W.; Amine,K. Nat. Commun. 2013, 4, No. 1513.(160) Chen, Z.; Qin, Y.; Amine, K. Electrochim. Acta 2009, 54, 5605.(161) Chen, Z.; Liu, J.; Jansen, A. N.; GirishKumar, G.; Casteel, B.;Amine, K. Electrochem. Solid-State Lett. 2010, 13, A39.(162) Kichambare, P.; Rodrigues, S.; Kumar, J. ACS Appl. Mater.Interfaces 2012, 4, 49.(163) Kumar, A.; Ciucci, F.; Morozovska, A. N.; Kalinin, S. V.; Jesse,S. Nat. Chem. 2011, 3, 707.(164) Zhang, D.; Li, R.; Huang, T.; Yu, A. J. Power Sources 2010, 195,1202.(165) Hassoun, J.; Croce, F.; Armand, M.; Scrosati, B. Angew. Chem.,Int. Ed. 2011, 50, 2999.(166) Kamaya, N.; Homma, K.; Yamakawa, Y.; Hirayama, M.; Kanno,R.; Yonemura, M.; Kamiyama, T.; Kato, Y.; Hama, S.; Kawamoto, K.;Mitsui, A. Nat. Mater. 2011, 10, 682.(167) Cecchetto, L.; Salomon, M.; Scrosati, B.; Croce, F. J. PowerSources 2012, 213, 233.(168) Guyomard, D.; Tarascon, J. M. J. Electrochem. Soc. 1993, 140,3071.(169) Tarascon, J. M.; Guyomard, D. Solid State Ionics 1994, 69, 293.(170) Oswald, S.; Mikhailova, D.; Scheiba, F.; Reichel, P.; Fiedler, A.;Ehrenberg, H. Anal. Bioanal. Chem. 2011, 400, 691.(171) Veith, G. M.; Dudney, N. J.; Howe, J.; Nanda, J. J. Phys. Chem.C 2011, 115, 14325.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXAB

(172) Du, P.; Lu, J.; Lau, K. C.; Luo, X.; Bareno, J.; Zhang, X.; Ren,Y.; Zhang, Z.; Curtiss, L. A.; Sun, Y.-K.; Amine, K. Phys. Chem. Chem.Phys. 2013, 15, 5572.(173) Pearson, R. G. J. Am. Chem. Soc. 1963, 85, 3533.(174) Yang, J.; Zhai, D.; Wang, H.-H.; Lau, K. C.; Schlueter, J. A.;Du, P.; Myers, D. J.; Sun, Y.-K.; Curtiss, L. A.; Amine, K. Phys. Chem.Chem. Phys. 2013, 15, 3764.(175) Black, R.; Lee, J.-H.; Adams, B.; Mims, C. A.; Nazar, L. F.Angew. Chem., Int. Ed. 2013, 52, 392.(176) Nelson, R.; Weatherspoon, M. H.; Gomez, J.; Kalu, E. E.;Zheng, J. P. Electrochem. Commun. 2013, 34, 77.(177) Zhang, G. Q.; Hendrickson, M.; Plichta, E. J.; Au, M.; Zheng, J.P. J. Electrochem. Soc. 2012, 159, A310.(178) Lee, B. G.; Nam, S.-C.; Choi, J. Curr. Appl. Phys. 2012, 12,1580.(179) Zhang, Y.; Zhang, H.; Li, J.; Wang, M.; Nie, H.; Zhang, F. J.Power Sources 2013, 240, 390.(180) Kim, K. S.; Park, Y. J. Nanoscale Res. Lett. 2012, 7, 47.(181) Kalubarme, R. S.; Park, C.-J. Chem. Lett. 2012, 41, 612.(182) Adams, J.; Karulkar, M.; Anandan, V. J. Power Sources 2013,239, 132.(183) Meini, S.; Piana, M.; Tsiouvaras, N.; Garsuch, A.; Gasteiger, H.A. Electrochem. Solid-State Lett. 2012, 15, A45.(184) Crowther, O.; Salomon, M. Membranes 2012, 2, 216.(185) Wang, H.; Liao, X.; Jiang, Q.; Yang, X.; He, Y.; Ma, Z. Chin. Sci.Bull. 2012, 57, 1959.(186) Selvaraj, C.; Munichandraiah, N.; Scanlon, L. G. J. PorphyrinsPhthalocyanines 2012, 16, 255.(187) Zheng, D.; Lee, H.-S.; Yang, X.-Q.; Qu, D. Electrochem.Commun. 2013, 28, 17.(188) Garsuch, A.; Badine, D. M.; Leitner, K.; Gasparotto, L. H. S.;Borisenko, N.; Endres, F.; Vracar, M.; Janek, J.; Oesten, R. Z. Phys.Chem. 2012, 226, 107.(189) Crowther, O.; Keeny, D.; Moureau, D. M.; Meyer, B.;Salomon, M.; Hendrickson, M. J. Power Sources 2012, 202, 347.(190) Zhang, L.; Zhang, X.; Wang, Z.; Xu, J.; Xu, D.; Wang, L. Chem.Commun. 2012, 48, 7598.(191) Tsiouvaras, N.; Meini, S.; Buchberger, I.; Gasteiger, H. A. J.Electrochem. Soc. 2013, 160, A471.(192) Yuasa, M.; Matsuyoshi, T.; Kida, T.; Shimanoe, K. J. PowerSources 2013, 242, 216.(193) Younesi, R.; Urbonaite, S.; Edstrom, K.; Hahlin, M. J. Phys.Chem. C 2012, 116, 20673.(194) Wang, H.; Yang, Y.; Liang, Y.; Zheng, G.; Li, Y.; Cui, Y.; Dai,H. Energy Environ. Sci. 2012, 5, 7931.(195) Sun, B.; Wang, B.; Su, D.; Xiao, L.; Ahn, H.; Wang, G. Carbon2012, 50, 727.(196) Sun, B.; Liu, H.; Munroe, P.; Ahn, H.; Wang, G. Nano Res.2012, 5, 460.(197) Shitta-Bey, G. O.; Mirzaeian, M.; Halla, P. J. J. Electrochem. Soc.2012, 159, A315.(198) Mirzaeian, M.; Hall, P. J.; Sillars, F. B.; Fletcher, I.; Goldin, M.M.; Shitta-bey, G. O.; Jirandehi, H. F. J. Electrochem. Soc. 2013, 160,A25.(199) Herranz, J.; Garsuch, A.; Gasteiger, H. A. J. Phys. Chem. C2012, 116, 19084.(200) Yoon, T. H.; Park, Y. J. Nanoscale Res. Lett. 2012, 7, 1.(201) Gao, Y.; Wang, C.; Pu, W.; Liu, Z.; Deng, C.; Zhang, P.; Mao,Z. Int. J. Hydrogen Energy 2012, 37, 12725.(202) Lu, Y.-C.; Crumlin, E. J.; Veith, G. M.; Harding, J. R.; Mutoro,E.; Baggetto, L.; Dudney, N. J.; Liu, Z.; Shao-Horn, Y. Sci. Rep. 2012,2, 715.(203) Li, L.; Zhao, X.; Manthiram, A. Electrochem. Commun. 2012,14, 78.(204) Ke, F.-S.; Solomon, B. C.; Ma, S.-G.; Zhou, X.-D. Electrochim.Acta 2012, 85, 444.(205) Huff, L. A.; Rapp, J. L.; Zhu, L.; Gewirth, A. A. J. Power Sources2013, 235, 87.
(206) Leskes, M.; Drewett, N. E.; Hardwick, L. J.; Bruce, P. G.;Goward, G. R.; Grey, C. P. Angew. Chem., Int. Ed. 2012, 51, 8560.(207) Zhamu, A.; Chen, G.; Liu, C.; Neff, D.; Fang, Q.; Yu, Z.;Xiong, W.; Wang, Y.; Wang, X.; Jang, B. Z. Energy Environ. Sci. 2012, 5,5701.(208) Yin, J.; Fang, B.; Luo, J.; Wanjala, B.; Mott, D.; Loukrakpam,R.; Ng, M. S.; Li, Z.; Hong, J.; Whittingham, M. S.; Zhong, C.-J.Nanotechnology 2012, 23, 305404.(209) Yang, Y.; Shi, M.; Zhou, Q.-F.; Li, Y.-S.; Fu, Z.-W. Electrochem.Commun. 2012, 20, 11.(210) Wang, L.; Ara, M.; Wadumesthrige, K.; Salley, S.; Ng, K. Y. S. J.Power Sources 2013, 234, 8.(211) He, H.; Niu, W.; Asl, N. M.; Salim, J.; Chen, R.; Kim, Y.Electrochim. Acta 2012, 67, 87.(212) Cao, Y.; Wei, Z.; He, J.; Zang, J.; Zhang, Q.; Zheng, M.; Dong,Q. Energy Environ. Sci. 2012, 5, 9765.(213) Yoo, E.; Nakamura, J.; Zhou, H. Energy Environ. Sci. 2012, 5,6928.(214) Thapa, A. K.; Shin, T. H.; Ida, S.; Sumanasekera, G. U.;Sunkara, M. K.; Ishihara, T. J. Power Sources 2012, 220, 211.(215) Ida, S.; Thapa, A. K.; Hidaka, Y.; Okamoto, Y.; Matsuka, M.;Hagiwara, H.; Ishihara, T. J. Power Sources 2012, 203, 159.(216) Trahey, L.; Karan, N. K.; Chan, M. K. Y.; Lu, J.; Ren, Y.;Greeley, J.; Balasubramanian, M.; Burrell, A. K.; Curtiss, L. A.;Thackeray, M. M. Adv. Energy Mater. 2013, 3, 75.(217) Sun, B.; Munroe, P.; Wang, G. Sci. Rep. 2013, 3, 2247.(218) Lau, K. C.; Curtiss, L. A.; Greeley, J. J. Phys. Chem. C 2011,115, 23625.(219) Chen, J.; Hummelshoj, J. S.; Thygesen, K. S.; Myrdal, J. S. G.;Norskov, J. K.; Vegge, T. Catal. Today 2011, 165, 2.(220) Garcia-Lastra, J. M.; Bass, J. D.; Thygesen, K. S. J. Chem. Phys.2011, 135, 121101.(221) Ong, S. P.; Mo, Y.; Ceder, G. Phys. Rev. B 2012, 85,081105(R).(222) Hummelshoj, J. S.; Blomqvist, J.; Datta, S.; Vegge, T.;Rossmeisl, J.; Thygesen, K. S.; Luntz, A. C.; Jacobsen, K. W.; Norskov,J. K. J. Chem. Phy. 2010, 132, 071101.(223) Lau, K. C.; Assary, R. S.; Redfern, P.; Greeley, J.; Curtiss, L. A.J. Phys. Chem. C 2012, 116, 23890.(224) Radin, M. D.; Rodriguez, J. F.; Tian, F.; Siegel, D. J. J. Am.Chem. Soc. 2012, 134, 1093.(225) Lu, J.; Jung, H.-J.; Lau, K. C.; Zhang, Z.; Schlueter, J. A.; Du,P.; Assary, R. S.; Greeley, J.; Ferguson, G. A.; Wang, H.-H.; Hassoun,J.; Iddir, H.; Zhou, J.; Zuin, L.; Hu, Y.; Sun, Y.-K.; Scrosati, B.; Curtiss,L. A.; Amine, K. ChemSusChem 2013, 6, 1196.(226) Xiao, J.; Wang, D.; Xu, W.; Wang, D.; Williford, R. E.; Liu, J.;Zhang, J.-G. J. Electrochem. Soc. 2010, 157, A487.(227) Yang, X.-h.; He, P.; Xia, Y.-y. Electrochem. Commun. 2009, 11,1127.(228) Kuboki, T.; Okuyama, T.; Ohsaki, T.; Takami, N. J. PowerSources 2005, 146, 766.(229) Cheng, H.; Scott, K. Appl. Catal., B 2011, 108, 140.(230) Debart, A.; Bao, J.; Armstrong, G.; Bruce, P. G. J. Power Sources2007, 174, 1177.(231) Jin, L.; Xu, L.; Morein, C.; Chen, C.-h.; Lai, M.; Dharmarathna,S.; Dobley, A.; Suib, S. L. Adv. Funct. Mater. 2010, 20, 3373.(232) Lee, S.; Zhu, S.; Milleville, C. C.; Lee, C.-Y.; Chen, P.;Takeuchi, K. J.; Takeuchi, E. S.; Marschilok, A. C. Electrochem. Solid-State Lett. 2010, 13, A162.(233) Lu, Y.-C.; Gasteiger, H. A.; Crumlin, E.; McGuire, R., Jr.; Shao-Horn, Y. J. Electrochem. Soc. 2010, 157, A1016.(234) Lu, Y.-C.; Gasteiger, H. A.; Parent, M. C.; Chiloyan, V.; Shao-Horn, Y. Electrochem. Solid-State Lett. 2010, 13, A69.(235) Mirzaeian, M.; Hall, P. J. J. Power Sources 2010, 195, 6817.(236) Thapa, A. K.; Saimen, K.; Ishihara, T. Electrochem. Solid-StateLett. 2010, 13, A165.(237) Wang, D.; Xiao, J.; Xu, W.; Zhang, J.-G. J. Electrochem. Soc.2010, 157, A760.(238) Wang, Y.; Zhou, H. Chem. Commun. 2010, 46, 6305.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXAC

(239) Xiao, J.; Xu, W.; Wang, D.; Zhang, J.-G. J. Electrochem. Soc.2010, 157, A294.(240) Yang, X.-h.; Xia, Y.-y. J. Solid State Electrochem. 2010, 14, 109.(241) Zhang, D.; Fu, Z.; Wei, Z.; Huang, T.; Yu, A. J. Electrochem.Soc. 2010, 157, A362.(242) Zhang, G. Q.; Zheng, J. P.; Liang, R.; Zhang, C.; Wang, B.;Hendrickson, M.; Plichta, E. J. J. Electrochem. Soc. 2010, 157, A953.(243) Oh, S. H.; Black, R.; Pomerantseva, E.; Lee, J.-H.; Nazar, L. F.Nat. Chem. 2012, 4, 1004.(244) Ryu, W.-H.; Yoon, T.-H.; Song, S. H.; Jeon, S.; Park, Y.-J.; Kim,I.-D. Nano Lett. 2013, 13, 4190.(245) Trahey, L.; Johnson, C. S.; Vaughey, J. T.; Kang, S. H.;Hardwick, L. J.; Freunberger, S. A.; Bruce, P. G.; Thackeray, M. M.Electrochem. Solid-State Lett. 2011, 14, A64.(246) Viswanathan, V.; Thygesen, K. S.; Hummelshoj, J. S.; Norskov,J. K.; Girishkumar, G.; McCloskey, B. D.; Luntz, A. C. J. Chem. Phys.2011, 135, 214704.(247) McCloskey, B. D.; Speidel, A.; Scheffler, R.; Miller, D. C.;Viswanathan, V.; Hummelshoj, J. S.; Norskov, J. K.; Luntz, A. C. J.Phys. Chem. Lett. 2012, 3, 997.(248) Lu, J.; Lei, Y.; Lau, K. C.; Luo, X.; Du, P.; Wen, J.; Assary, R. S.;Das, U.; Miller, D. J.; Elam, J. W.; Albishri, H. M.; El-Hady, D. A.; Sun,Y.-K.; Curtiss, L. A.; Amine, K. Nat. Commun. 2013, 4, No. 2383.(249) Aurbach, D.; Zinigrad, E.; Cohen, E.; Teller, H. Solid StateIonics 2002, 148, 405.(250) Yamaki, J.; Tobishima, S.; Hayashi, K.; Saito, K.; Nemoto, Y.;Arakawa, M. J. Power Sources 1998, 74, 219.(251) Zhang, T.; Imanishi, N.; Hasegawa, S.; Hirano, A.; Xie, J.;Takeda, Y.; Yamamoto, O.; Sammes, N. J. Electrochem. Soc. 2008, 155,A965.(252) Kumar, B.; Kumar, J.; Leese, R.; Fellner, J. P.; Rodrigues, S. J.;Abraham, K. M. J. Electroanal. Chem. 2010, 157, A50.(253) Armand, M.; Tarascon, J.-M. Nature 2008, 451, 652.(254) Truong, T. T.; Qin, Y.; Ren, Y.; Chen, Z.; Chan, M. K.;Greeley, J. P.; Amine, K.; Sun, Y. Adv. Mater. 2011, 23, 4947.(255) Wang, Y.; He, P.; Zhou, H. Energy Environ. Sci. 2011, 4, 4994.(256) Zhang, J. G.; Wnag, D.; Xu, W.; Xiao, J.; Williford, R. E. J.Power Sources 2010, 195, 4332.(257) Gierszewski, P. J.; Finn, P. A.; Kirk, D. W. Fusion Eng. Des.1990, 13, 59.(258) Visco, S.; Jonghe, L. D.; Nimon, Y.; Petrov, A.; Pridatko, K.Aqueous Lithium/Air Battery Cells. WO Patent Application WO/2010/005, 2010.(259) Welna, D. T.; Stone, D. A.; Allcock, H. R. Chem. Mater. 2006,18, 4486.(260) Visco, S.; Katz, B. D.; Nimon, Y.; Jonghe, L. D. U.S. Patent7,491,458, Feb 17, 2009; see also other patents cited therein.(261) Visco, S.; Katz, B. D.; Nimon, Y.; Jonghe, L. D. U.S. Patent7,282,295, Oct 16, 2007.(262) Fu, J. U. S. Patent 6,485,622, Nov 26, 2002.(263) Fu, J. Solid State Ionics 1997, 96, 195.(264) Fu, J. Solid State Ion. 1997, 104, 191.(265) Crowthe, O.; Salomon, M. In Lithium Batteries; Scrosati, B.,Abraham, K. M., Schalkwijk, W., Hassoum, J., Eds.; John Wiley & SonsInc.: Hoboken, NJ, 2012; Chapter 9.(266) Zhang, T.; Imanishi, N.; Shimonishi, Y.; Hirano, A.; Takeda,Y.; Yamamoto, O.; Sammes, N. Chem. Commun. 2010, 46, 1661.(267) Zhang, T.; Imanishi, N.; Hasegawa, S.; Hirano, A.; Xie, J.;Takeda, Y.; Yamamoto, O.; Sammes, N. Electrochem. Solid-State Lett.2009, 12, A132.(268) Hasegawa, S.; Imanishi, N.; Zhang, T.; Xie, J.; Hirano, A.;Takeda, Y.; Yamamoto, O. J. Power Sources 2009, 189, 371.(269) Yoo, E.; Zhou, H. ACS Nano 2011, 5, 3020.(270) He, P.; Wang, Y.; Zhou, H. Chem. Commun. 2011, 47, 10701.(271) Wang, L.; Zhao, X.; Lu, Y.; Xu, M.; Zhang, D.; Ruoff, R. S.;Stevenson, K. J.; Goodenough, J. B. J. Electrochem. Soc. 2011, 158,A1379.(272) Suntivich, J.; Gasteiger, H. A.; Yabuuchi, N.; Nakanishi, H.;Goodenough, J. B.; Shao-Horn, Y. Nat. Chem. 2011, 3, 546.
(273) Thotiyl, M. M. O.; Freunberger, S. A.; Peng, Z.; Chen, Y.; Liu,Z.; Bruce, P. G. Nat. Mater. 2013, 12, 1050.(274) Sun, Y. G.; Choi, W. M.; Jiang, H. Q.; Huang, Y. G. Y.; Rogers,J. A. Nature Nanotechnol. 2006, 1, 201.(275) Baddour-Hadjean, R.; Pereira-Ramos, J.-P. Chem. Rev. 2009,110, 1278.(276) Ryan, K. R.; Trahey, L.; Okasinski, J. S.; Burrell, A. K.; Ingram,B. J. J. Mater. Chem. A 2013, 1, 6915.(277) Karan, N. K.; Balasubramanian, M.; Fister, T. T.; Burrell, A. K.;Du, P. J. Phys. Chem. C 2012, 116, 18132.(278) Chan, M. K. Y.; Shirley, E. L.; Karan, N. K.; Balasubramanian,M.; Ren, Y.; Greeley, J. P.; Fister, T. T. J. Phys. Chem. Lett. 2011, 2,2483.
Chemical Reviews Review
dx.doi.org/10.1021/cr400573b | Chem. Rev. XXXX, XXX, XXX−XXXAD