apol1 mediated cell injury involves disruption of...
TRANSCRIPT

BASIC RESEARCH www.jasn.org
APOL1–Mediated Cell Injury Involves Disruption ofConserved Trafficking Processes
Etty Kruzel-Davila,* Revital Shemer,† Ayala Ofir,* Ira Bavli-Kertselli,* Ilona Darlyuk-Saadon,†
Pazit Oren-Giladi,* Walter G. Wasser,*‡ Daniella Magen,*† Eid Zaknoun,† Maya Schuldiner,§
Adi Salzberg,† Daniel Kornitzer,| Zvonimir Marelja,¶ Matias Simons,¶ and Karl Skorecki*†
*Department of Nephrology, Rambam Health Care Campus, Haifa, Israel; Departments of †Genetics andDevelopmental Biology and |Microbiology and Inflammation, Rappaport Faculty of Medicine and Research Institute,Technion—Israel Institute of Technology, Haifa, Israel; ‡Department of Nephrology, Mayanei HaYeshua MedicalCenter, Bnei Brak, Israel; §Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel; and¶Imagine Institute, Paris Descartes University–Sorbonne Paris Cité, Paris, France
ABSTRACTAPOL1 harbors C–terminal sequence variants (G1 andG2), which account formuchof the increased risk forkidney disease in sub–Saharan African ancestry populations. Expression of the risk variants has also beenshown to cause injury to podocytes and other cell types, but the underlying mechanisms are not under-stood.WeusedDrosophilamelanogaster and Saccharomyces cerevisiae to help clarify thesemechanisms.Ubiquitous expression of the human APOL1 G1 and G2 disease risk alleles caused near-complete lethalityinD. melanogaster, with no effect of the G0 nonrisk APOL1 allele, corresponding to the pattern of humandisease risk. We also observed a congruent pattern of cellular damage with tissue-specific expressionof APOL1. In particular, expression of APOL1 risk variants in D. melanogaster nephrocytes caused cell-autonomous accumulation of the endocytic tracer atrial natriuretic factor-red fluorescent protein at early stagesand nephrocyte loss at later stages.We also observeddifferential toxicity of theAPOL1 risk variants comparedwiththe APOL1 nonrisk variants in S. cerevisiae, including impairment of vacuole acidification. Yeast strains defective inendosomal trafficking or organelle acidification but not those defective in autophagy displayed augmented APOL1toxicitywith all isoforms. This pattern of differential injury by theAPOL1 risk alleles comparedwith thenonrisk allelesacross evolutionarily divergent species is consistent with an impairment of conserved core intracellular endosomaltrafficking processes. This finding should facilitate the identification of cell injury pathways and corresponding ther-apeutic targets of interest in these amenable experimental platforms.
J Am Soc Nephrol 28: ccc–ccc, 2016. doi: 10.1681/ASN.2016050546
Two sets of DNA–derived sequence variants (termedG1 and G2 in contrast to the ancestral G0) at thehuman APOL1 gene are associated with a markedlyincreased risk of progressive CKD in sub–Saharan Af-rican ancestry populations.1–7 The G1 haplotypecomprises two nonsynonymous coding variantsrs73885319 (S342G) and rs60910145 (I384M),whereas the G2 (rs71785313) allele contains a 6-bpdeletion (del.N388/Y389).1,2 APOL1 is one memberof theAPOL1–6 cluster of innate immunity genes andthe onlymember of that cluster with a secretory signalpeptide and a prominent circulating protein prod-uct.8–10 Circulating APOL1 can lyse Trypanosomabrucei species and protects humans against African
sleeping sickness.11–13 TheG1 andG2 risk alleles con-fer trypanolytic activity against the T. brucei rhode-siense subspecies that has developed resistance to the
Received May 16, 2016. Accepted October 5, 2016.
E.K.-D., R.S., A.O., and I.B.-K. contributed equally to this work.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Prof. Karl Skorecki, Annie Chutick Professor inMedicine (Nephrology), Technion—Israel Institute of Technol-ogy, Director of Medical and Research Development, RambamHealth Care Campus, 8 Ha’Aliyah Street, Haifa 31096, Israel.Email: [email protected]
Copyright © 2016 by the American Society of Nephrology
J Am Soc Nephrol 28: ccc–ccc, 2016 ISSN : 1046-6673/2804-ccc 1

ancestral G0 allelic version ofAPOL1, explaining their rise to highfrequency in at–risk parent populations. Genetic epidemiologystudies show that kidney disease risk association is significantprimarily under a recessive inheritance mode (two APOL1 riskalleles) or that there is a very marked step up in disease riskfrom one to two risk alleles.1–7,14–16 These studies and othersalso point to an important role for nongenetic second risktriggers in transforming disease risk to clinical disease.3,7,17–19
The recessive inheritance mode might suggest that the G1 andG2 mutations cause a loss of function essential to kidney func-tional integrity. However, APOL1 is absent and therefore, dis-pensable for kidney health inmost species, including nonhumanprimates,9,10,13,20 and the null state was not found to cause akidney disease phenotype in at least one human individual.21
Correspondingly, transient transfection or induced expressionin a variety of human cells in culture (including human podo-cytes and human embryonic kidney [HEK] 293 cells) and in vivomodels caused transfection dose– and time–dependent cell in-jury, with a significantly greater effect of the G1 and G2 variantscompared with G0 but still some cytotoxic effect of even the G0allele.13,19,22–29 Taken together, these studies point rather to a gainof function of G1 and G2 compared with G0 and a very markedgene dose or expression effect, with several formulations havingbeen proposed for the marked step up, such as the occurrence ofmultimeric structures.30 APOL1 protein is expressed in the cir-culation and Trypanosome Lytic Factor (TLF) particles (TLF1and TLF2)31–33 as well as intracellularly in a variety of cell types,including the podocyte, endothelial cells, and other tissues, suchas brain, placenta, and a variety of tumors.8,9,19,34–36 Althoughcirculating APOL1 is responsible for trypanolysis, studies in thesetting of kidney transplantation point to intracellular APOL1 asresponsible for kidneydisease risk, because reduced kidney trans-plant allograft survival tracks with the donor and not the recip-ient APOL1 genotype in the African ancestry population.37–40
These studies reinforce the importance of continuing to investi-gate gain of function mechanisms and pathways in experimentalsystems displaying differentially greater cell injury of G1 and G2compared with G0 variants of APOL1.
Although human cells in culture and murine models pro-vide the relevant complement of pathways to study mecha-nisms of human disease risk, simpler organisms offer theopportunity for efficient and potentially informative geneticinterrogation. Accordingly, we turned to model eukaryoticorganisms that are readily amenable to genetic interrogationof conserved pathways. Here, we show that the G1 and G2allelic variants, which confer human disease risk, also conferenhanced eukaryotic cell toxicity, even in species with diver-gent evolutionary histories that do not naturally expressAPOL1 (Drosophila melanogaster and Saccharomyces cerevi-siae), suggesting that APOL1 engages core pathways that arehighly conserved in evolution. Genetic interrogation of thefly and yeast systems points to a local intracellular (includingfly nephrocytes) rather than systemic effect of APOL1, withimpairment of intracellular acidification and endosomaltrafficking.
RESULTS
APOL1 G1 and G2 Differential Toxicity Is Conserved inthe Fruit FlyWe examined the effect of expressing human APOL1 G0, G1,and G2 gene products as well as the artificial C–terminaltruncated (C-tr) construct that lacks the Serum Resistance–Associated (SRA) interacting domain (Figure 1A) in D. mela-nogaster. The expression of APOL1 G0 under the ubiquitousdriver daughterless (da)-GAL4 caused no evident phenotypecompared with the wild type (WT) crossed with da-GAL4. Incontrast, the G1 and G2 variants consistently led to signifi-cantly higher rates of pupal–pharate adult lethality (Figure 1Band Table 1). The C-tr construct, which lacks trypanolyticactivity and toxicity in an in vivo murine model,13,41,42 wasinnocuous. The da-GAL4 driver induced high levels of APOL1protein in the hemolymph (Supplemental Figure 1). To dis-tinguish between systemic and cell-specific effects of APOL1,we used Glass multiple reporter (GMR)-GAL4 to drive APOL1expression in the eye. As shown in Figure 1, C–E and Table 2,significantly greater percentages with a rough eye phenotypeand severely disrupted ommatidia were evident in flies ex-pressing the G1 or G2 human transgene compared with G0.As shown in Figure 1E, the observed eye phenotypes were notcaused by systemic distribution of APOL1, which was re-stricted to the heads of adult flies when expressed under theGMR-GAL4 driver. This is consistent with an APOL1 injurymechanism originating at a local intracellular rather than sys-temic circulating level.
APOL1 G1 and G2 Differential Toxicity Is Conserved inD. melanogaster Pericardial Nephrocytes and Leads toAtrial Natriuretic Factor-Red Fluorescent ProteinAccumulationDrosophila pericardial nephrocytes share remarkable similar-ity with podocytes of the mammalian nephron. The fly neph-rocyte diaphragm regulates filtration in a manner similar tothe mammalian slit diaphragm.43–45 After passage throughthis filtration barrier, the main function of nephrocytes is toregulate hemolymph composition by endocytosis and metab-olism or recycling of filtrate constituents. Therefore, the flynephrocyte system enables filtration and endocytic uptakecomponents to be monitored in a manner that is not readilyrecapitulated in human podocytes in cell culture. We usedDot-GAL4 to drive APOL1 expression in nephrocytes in fliesexpressing the Red Fluorescent Protein (RFP) reporter fusedwith atrial natriuretic factor (ANF) secretion peptide drivenby the myosin heavy chain enhancer.44 ANF-RFP is endocy-tosed from the hemolymph by nephrocytes and degraded,and therefore, it serves as a marker for these nephrocytefunctions.44 One-day-old APOL1 G1 and G2 but not G0transgenic flies showed a striking increase of vesicularANF-RFP in the pericardial nephrocytes (Figure 2). Knock-down of Kirre (fly ortholog of human NEPH1), which isknown to disrupt filtration, led to a significant reduction of
2 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org

ANF-RFP uptake (negative control). At later time points, weobserved nephrocyte toxicity, characterized by condensed orabsent nuclei as well as surrounding cell fragments in G1 andG2 but not G0 transgenic flies. Taken together, the ectopicexpression of G1 and G2 in nephrocytes caused nephrocyte-specific defects. The accumulation of ANF-RFP by nephro-cytes followed by their death is consistent with well preserveduptake but impaired degradation, suggesting disruption ofthe endolysosomal processes. This led us to examine the ef-fects of APOL1 and its risk variants on intracellular traffick-ing in S. cerevisiae.
APOL1 G1 and G2 Differential Toxicity Is AlsoConserved in S. cerevisiae and Is a Manifestation ofCell LethalityIn the S. cerevisiae drop titration assay, we observed toxicity ofthe G0, G1, and G2 variants of APOL1 but not the C-tr version(Figure 3A). For G1, we also used constructs with S342G orI384Mmutations separately and found that S342G rather thanthe I384M mutation suffices to recapitulate toxicity of theAPOL1 G1 missense haplotype (Figure 3B). Genetic studieshave also shown this to be the sequence variant in the APOL1G1missense haplotype that is strongly associated with kidney
Figure 1. APOL1 G1 and G2 are toxic to D. melanogaster. (A) Schematic representation of the protein domains in APOL1. A full–lengthAPOL1 protein with a schematic representation of the protein domains: BH3 Domain (BH3), Membrane Addressing Domain (MAD), PoreForming Domain (PFD), Signal Peptide (SP), and SRA-interacting domain. Lower panel shows the amino acid composition at the SRA-bindingdomain of APOL1 G0, G1, and G2 variants. (B) D. melanogaster expressing APOL1 kidney risk variants under the regulation of da-GAL4 shows a lethal phenotype and wing malformations. (B, a) A da-GAL4 . APOL1 G1 fly that failed to emerge from its pupal case(representing 98% of the flies of this genotype). (B, b) A da-GAL4 . APOL1 G1 fly that succeeded in eclosing but displays a severewing phenotype. (C) Expression of APOL1 G1 and G2 gene products under the regulation of the eye–specific driver GMR-GAL4results in an abnormal eye phenotype. Rough eye phenotypes were observed in 81% of G1, 48% of G2, and 11% of G0 flies. Therough eyes were characterized by abnormalities in architecture, often leaving a concavity. (D) Hematoxylin and eosin staining of eyesof GMR-GAL4 . APOL1 flies reveals abnormal eye histology with APOL1 G1 and G2 variants. (D, a and b) WT eye shows normalommatidia structure. (D, c) Intact ommatidia in eye expressing APOL1 G0. (D, d) Eyes expressing APOL1 G1 and G2 (D, e) showingseverely disrupted ommatidial structure (rectangle compared with D, a); black arrows show concave eye structure. (D, f) ControlGMR-GAL4 eye. The ommatidial structure is abnormally condensed compared with the WT structure; however, the severe structuralchanges shown in the G1- and G2-expressing eyes are absent (compressed structure is shown in the rectangle). Scale bars, 100 mmin D, a, b, and d; 50 mm in D, c, e, and f. (E) Western analysis of APOL1 and b-actin in D. melanogaster head and body protein lysatesfrom flies expressing APOL1 G0, G1, and G2 gene products under the eye driver GMR-GAL4. Protein loading is as described in ConciseMethods to account for hemolymph protein in body lysates. APOL1 is expressed in the heads but not the bodies of the flies, emphasizing therole of endogenous APOL1 versus circulating APOL1.
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 3
www.jasn.org BASIC RESEARCH

disease.3,46 Because in yeast, unlikeD.melanogaster but similarto mammalian cells in culture,13,19,22–29 even the G0 nonriskvariant was toxic compared with vector or the C-tr construct,it was necessary to quantitatively compare the relative toxic-ities. To quantify and determine whether APOL1 simply in-hibits yeast growth or actually increases lethality, we conducted asurvival assay by counting yeast CFU after 0 or 8 hours of growth(G0, G1, G2, and C-tr) under inducing conditions for APOL1expression followed by restoration to noninducing conditions inoptimal growth media (conditions that would allow any yeastcells that had not been irreversibly injured or killed to establishcountable colonies). As shown in Tables 3 and 4, expression ofvector or the C-tr variant shows a similar percentage of survival.In contrast, induction of APOL1 G0 caused lethality with fewersurvivors, and APOL1 G1 or G2 caused a significantly greaterdegree of lethality compared with G0.
S. cerevisiae Strains Deleted in Components of theClass 3 Phosphatidylinositol 3-Kinase Complex AreHypersensitive to APOL1 ToxicityThe recapitulation of the significantly greater APOL1G1 andG2toxicity in S. cerevisiae along with the suggestive endocytic traf-ficking perturbation leading to fly nephrocyte toxicity promptedus to explore endosomal pathways in yeast. APOL1 has beenreported to be involved in autophagy regulation26,47 and inducelysosomal injury in human cells, including podocytes.22 Thecontributions of autophagy and endosomal trafficking to thefunctional integrity of podocytes have been well character-ized.48–54 The class 3 phosphatidylinositol (PI) 3–kinase phos-phorylates position 3 of PI, producing PI 3-phosphate, and it is akey signal for both endosomal trafficking and autophagosomeformation.55–59Wemonitored growth of S. cerevisiae strains de-leted for class 3 PI 3–kinase complex geneswith expression of theAPOL1 variants. Yeast strains deleted for vacuole protein sorting34 (VPS34), VPS30 (ATG6/human BECLIN1), or VPS15 werehypersensitive to APOL1 toxicity compared with the WTstrain
(Figures 3 and 4, A–C). In these strains, even the G0 version ofAPOL1 was toxic and indistinguishable from the G1 and G2alleles. Notably, however, the artificial C-tr construct remainedinnocuous in all strains.
Loss of the Endocytic Vps34 Complex II but Not theAutophagy–Specific Vps34 Complex I Confers APOL1HypersensitivityVps34 together with Vps30 and Vps15 form multiple com-plexes that are responsible for its diverse cellular functions.56,58
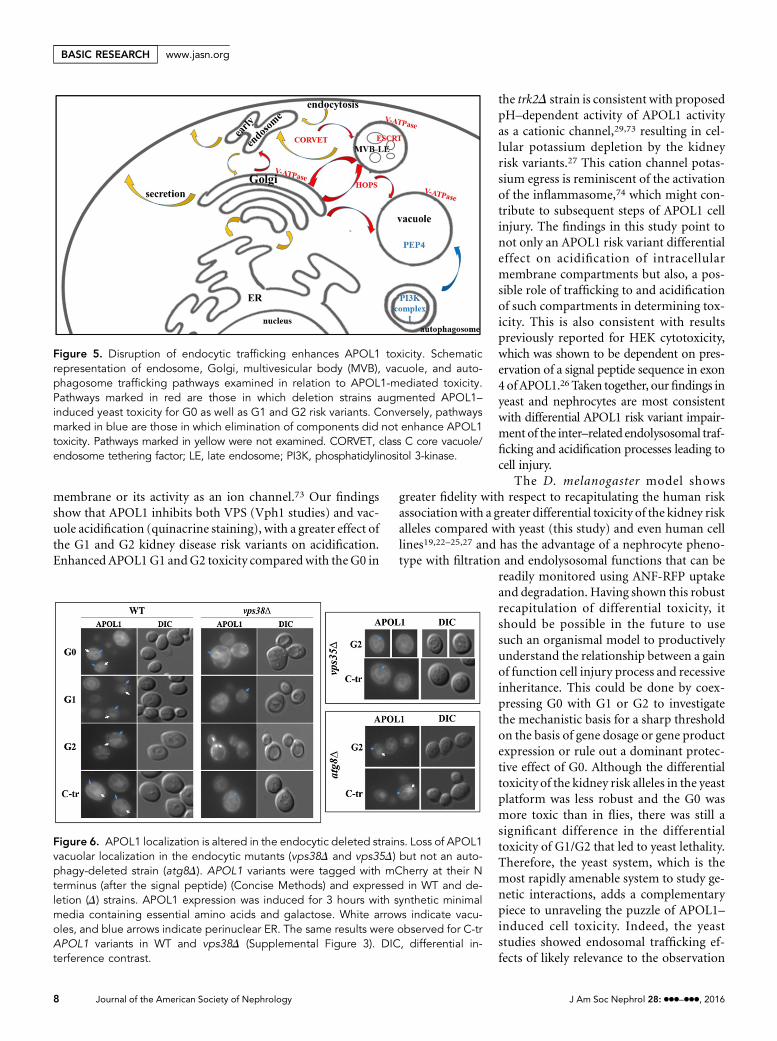
The autophagy–specific Vps34 complex I comprises Vps34,Vps15, Atg6/Vps30, and Atg14. The endocytic Vps34 complexII is formed by interaction with Vps38 and required for endo-some to Golgi retrograde trafficking and late Golgi/TGN toendosome-VPS.55–59 To differentiate between these pathways,we investigated the viability of ATG14 or VPS38 deletionstrains. Only vps38D showed augmented APOL1 toxicity, sim-ilar to the deletion of the VPS34, VPS30, or VPS15 componentof the core complex (Figure 4), an effect that could not beattributed to elevated APOL1 protein levels (SupplementalFigure 2). Notably, the survival assay using the vps38D strainalso confirmed irreversible injury/lethality that was significantlyhigher in the vps38D compared with the WT (Supplemental Ta-ble 1). In contrast, atg14D showed no such augmented APOL1–mediated toxicity and preserved the differential toxicity betweenthe G0 and G1, G2 variants (Figure 4E). Similar results wereobtained for atg8D (Figure 4F). These results prompted us tosystematically investigate the role of anterograde and retrogradeendosomal trafficking inAPOL1 toxicity (Figure 5, SupplementalTable 2). Strains that enhance APOL1 toxicity have essential rolesin the anterograde and retrograde Golgi–endosomal traffickingpathways,55 the endosomal sorting complex required for trans-port (ESCRT), the sorting of the ubiquitinated multivesicularbody cargoes,60 and vacuole trafficking. This is consistent withthe phenotypes of the class 3 PI 3–kinase complex II–deletedstrains described above, and here, the C-tr APOL1 was also nottoxic.
APOL1 Localization Is Altered in vps38D and OtherEndocytic MutantsWe next examined the localization of APOL1 variants in theWTversus vps38D. In the WT strain, expression of APOL1 was ob-served in the vacuole and the endoplasmic reticulum (ER) andpossibly, also separately in the juxtaposed plasma membrane,
Table 1. APOL1 G1 and G2 differential toxicity inD. melanogaster: reduced viability of D. melanogasterexpressing APOL1 kidney risk variants under da-GAL4.
Cross n Survival, %
♀UAS APOL1 G0 3 ♂da-GAL4 1727 97a,b
♀UAS APOL1 G1 3 ♂da-GAL4 2113 5a,c,d
♀UAS APOL1 G2 3 ♂da-GAL4 1891 6b,e,f
♀ WT 3 ♂da-GAL4 1028 98c,e
♀UAS APOL1 C truncated 3 ♂da-GAL4 1795 98d,f
aPairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: G1-G0.bPairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: G2-G0.cPairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: WT-G1.dPairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: C truncated-G1.ePairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: WT-G2.fPairs with a statistically significant (P,0.001 for each pair) difference usingone-way ANOVA. Comparison: C truncated-G2.
Table 2. APOL1 G1 and G2 differential toxicity in D.melanogaster: structural eye abnormality (rough eye) in D.melanogaster expressing APOL1 kidney risk variants underGMR-GAL4
Cross n Rough Eye, %
♀UAS APOL1 G0 3 ♂GMR-GAL4 535 11♀UAS APOL1 G1 3 ♂GMR-GAL4 443 81♀UAS APOL1 G2 3 ♂GMR-GAL4 337 48♀ WT 3 ♂GMR-GAL4 529 0
All pairwise differences are statistically significant (P,0.001 for each pair).
4 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org

with no difference evident in localization between APOL1 G0,G1, andG2 (Figure 6, Supplemental Figure 3). It should be notedthat juxtaposition of the ER to the plasmamembrane inyeast cellsmakes it difficult to determine plasma membrane localizationwith complete certainty. Notably, the nontoxic C-tr mutantshowed the same localization pattern, more readily discernedin this case because of the absence of yeast cell damage. In
contrast, in vps38D, APOL1 did not accumu-late in the vacuole and appears most promi-nently in the ER. Loss of vacuole localizationwas also observed with deletions of the Ret-romer complex responsible for retrogradeendosomal to Golgi trafficking (vps35D inFigure 6) or the ESCRT complex responsiblefor endosomal sorting required for transport(vps25D in Supplemental Figure 4), whichalso enhancedAPOL1 toxicity. This is in con-trast to strains that did not affect APOL1 tox-icity, such as atg8D (Figure 6) and atg14D(Supplemental Figure 4), whichwere also in-distinguishable from the WT strain with re-gard to APOL1 localization. Because therewas no difference in cellular localizationamong APOL1 variants, including the non-toxic C-tr construct, it seems that differencesin localization per se do not account fordifferential APOL1 toxicity. Rather, thesefindings point to a role for the vacuole ingoverning the differential toxicity of APOL1and its risk variants in yeast and by extrapo-lation, endolysosomal compartments inhigher eukaryotes.
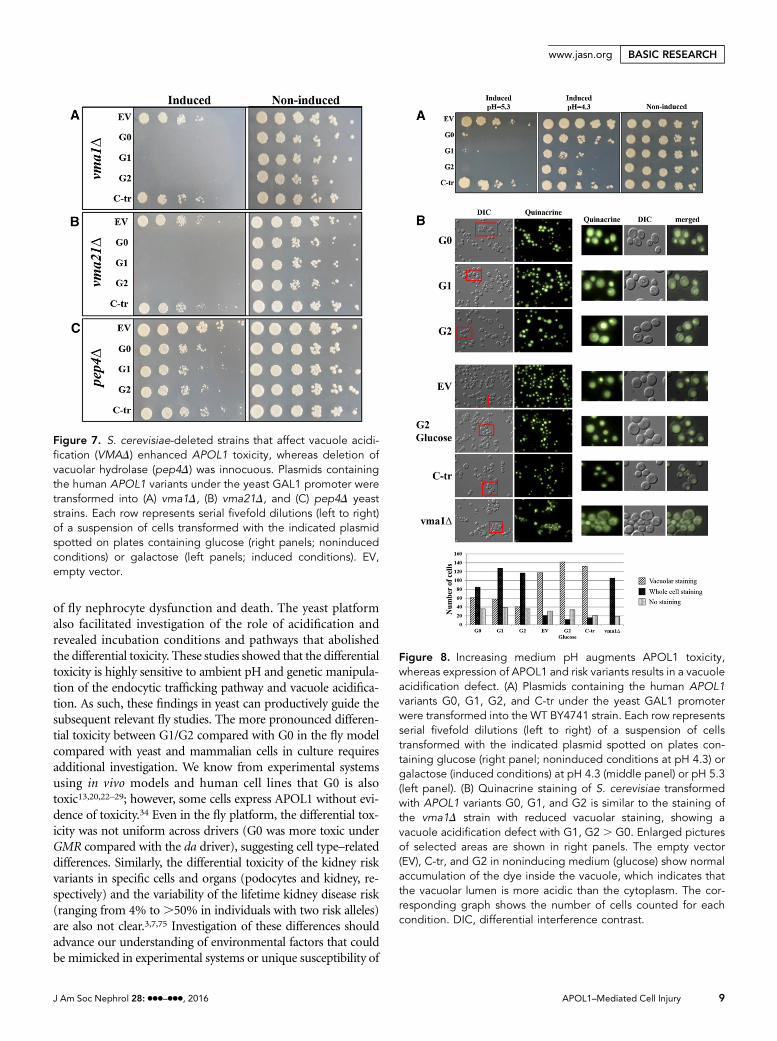
Reciprocal Interactions betweenAPOL1 and pHFunctional integrity of the vacuole in yeastand lysosomes in higher eukaryotic cellsenables their acidification, without whichcells, tissues, and organs undergo cell injuryand death.61 Because many of the proteinsaffecting APOL1 toxicity are involved intrafficking to the vacuole, we used pep4D,vma1D, and vma21D strains to investigatethe involvement of vacuolar function inAPOL1 toxicity. VMA1 encodes one ofthe V1 subunits of the vacuolar-ATPase,an ATP–dependent proton pump respon-sible for the acidification of the vacuole andother endocytic pathway components.62,63
Vma21 is needed for assembly of the V0subunit, whereas Pep4 encodes the vacuoleprotein hydrolase–activating enzyme; in itsabsence, vacuolar proteolysis is impaired.In the vma1D and vma21D but not pep4Dstrains, the pattern of APOL1 cytotoxicity
was similar to that observed for deletion of any one of thegenes encoding components of endosomal trafficking (Figure7). This suggests that it is the acidification status of the vacuolethat is affecting toxicity.
The augmented lethality in the vmaD strains led us to ex-plore the relationship between vacuole acidification andAPOL1 toxicity. S. cerevisiae strains lacking vacuolar ATPase
Figure 2. APOL1 G1 and G2 affect Drosophila pericardial nephrocyte function and leadto cell death during aging. Pericardial nephrocytes of adult flies were dissected, andANF-RFP uptake and nuclear GFP were analyzed by confocal microscopy. (A) ANF-RFPuptake by pericardial nephrocytes in 1-day-old flies. (B and C) ANF-RFP uptake bypericardial nephrocytes in 15-day-old flies. In B, the maximal intensity projection of 20focal planes was applied to visualize fragments of dead cells (arrowheads). Close-upimages are shown in C9 (corresponding to the white frames in C). Genotypes used in thisexperiments were (from top to bottom) with myosin heavy chain (MHC)-ANF-RFP, Hand-GFP, Dot-GAL4/+ (control); with MHC-ANF-RFP, Hand-GFP; Dot-GAL4/UAS-KirreRNAi(Dot . KirreRNAi); with MHC-ANF-RFP, Hand-GFP; Dot-GAL4/+;UAS-APOL1-G0 (Dot .APOL1-G0); with MHC-ANF-RFP, Hand-GFP; Dot-GAL4/+;UAS-APOL1-G1 (dot . APOL1-G1), and withMHC-ANF-RFP, Hand-GFP;Dot-GAL4/+; UAS-APOL1-G2 (dot. APOL1-G2).Scale bars, 50 mm in A–C; 10 mm in C9.
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 5
www.jasn.org BASIC RESEARCH

activity show a conditional lethal phenotype that manifests asgrowth arrest at elevated pH (pH.5.0).64 Increasing pH from4.3 to 5.3 mimicked the effect of endocytic mutants, with aug-mented toxicity of the G0, G1, and G2 variants of APOL1, buthadno effect on theC-trAPOL1 (Figure 8A).Weusedquinacrineto visualize acidification of the vacuole in the absence and pres-ence of APOL1 variants (Figure 8B). Expression of APOL1 var-iants resulted in an acidification defect, similar to the pattern ofquinacrine staining observed in the vma1D strain. AlthoughAPOL1 G0 also displayed reduced vacuole acidification, this ef-fect was more marked with the G1 and G2 and not evident at allfor the nontoxic C-tr APOL1. These findings suggest that APOL1risk variant toxicity is accompanied by impaired vacuole acidifi-cation. Because impaired vacuole acidification requires accom-panying processes to balance electroneutrality, such as anion orcation influx or egress,65 we examined APOL1 toxicity in trk2D.The TRK2 gene product is required for low–affinity K+ influx inS. cerevisiae.66 Indeed, trk2D exhibited moderately enhancedsensitivity to APOL1 G0, G1, and G2 but in this case, with pres-ervation of differential hierarchy of toxicity in the G1 and G2variants compared with G0 and again, no toxicity of C-tr APOL1compared with vector alone (Figure 9).
APOL1 expression induced a loss of vacuole acidificationsimilar to that observed in the absence of the vacuolar ATPasesubunit Vma1. However, growth impairment is not evident invmaD strains. Therefore, we reasoned that additional mecha-nisms beyond disruption of VMA1–mediated vacuole acidifi-cation are involved in APOL1-mediated loss of viability. Wesought to determine whether APOL1 has a broader effect onvesicle trafficking. We used Green Fluorescent Protein (GFP)–tagged Vph1, a vacuolar ATPase, as a marker for the VPS path-way.67 In the WTstrain as well in the C-tr APOL1, Vph1-GFPlocalized normally to the vacuole, whereas APOL1 expression(G0, G1, and G2) inhibited Vph1 trafficking to the vacuole butwithout a differential effect of the risk versus nonrisk variants(Figure 10, Supplemental Figure 5).
DISCUSSION
Population genetics studies provide powerful but circumstan-tial evidence that the APOL1 G1 and G2 variant kidney diseaserisk association reflects causality,68 whereas the mechanismunderlying kidney injury has not been resolved at this stage.The cardinal feature of these studies is the finding that theAPOL1 toxicity displays a parallel pattern of differential tox-icity between the human disease risk and nonrisk alleles inD. melanogaster and S. cerevisiae. We were able to dissect thetwo G1 mutations and show that it is the S342G mutation inthe APOL1 G1 risk missense haplotype that is toxic to yeast,consistent with the finding that it is this amino acid change that isresponsible for the association with increased risk for humankidney disease.3,46 This finding also dissociates the APOL1 re-quirement for APOL1 eukaryotic cell injury from themodulationof the APOL1 trypanosomal SRA interaction.13 The results in D.melanogaster indicate that cell injury does not require the presenceof the gene product in the hemolymph, supporting an intracellu-lar rather than circulating source of APOL1 in pathogenesis, con-sistent with human transplantation studies.37–40 Expression ofAPOL1 G1 and G2 but not G0 in pericardial nephrocytes led tothe accumulation of constitutively secretedANF-RFP invesicles atearlier stages, suggesting an endolysosomal defect that may
Figure 3. Expression of human APOL1 in the yeast S. cerevisiaecauses reduced yeast growth. (A) Differential growth defectbetween APOL1 G0 versus APOL1 G1 and G2 expression de-termined by drop titration growth assay. (B) APOL1 S342G mu-tant shows reduced growth compared with the I384M APOL1mutant. Plasmids containing the human APOL1 variants G0, G1,and G2 and the artificial APOL1 constructs C-tr, S342G, andI384M under the yeast GAL1 promoter were transformed into theWT BY4741 yeast strain. Each row represents serial fivefold di-lutions (left to right) of a suspension of cells transformed with theindicated plasmid spotted on plates containing glucose (rightpanels; noninduced conditions) or galactose (left panels; inducedconditions). The boxes highlight the dilutions where the differ-ences are well shown. For S342G and I384M, two independenttransformants were tested, and all titration assays were con-ducted at least twice with similar results. EV, empty vector.
Table 3. S. cerevisiae survival assay: survival ratios at 0 and8 hours
APOL1 Variant0-h CFUCount
8-h CFUCount
8 h-to-0 hRatio (95% CI)
EV 5716 4935 0.86 (0.78 to 0.95)G0 5220 3710 0.71 (0.64 to 0.78)G1 5850 3245 0.55 (0.50 to 0.61)G2 5926 2680 0.45 (0.41 to 0.50)C truncated 5160 4250 0.82 (0.74 to 0.91)
Survival assays were conducted as described in Concise Methods, withmeasurement of surviving cells determined as CFU expressed as ratios at0 and 8 hours of induction for each experimental condition. Each pair ofexperiments was conducted in parallel for all conditions three times, and Pvalues for differences were determined as described in Concise Methods.95% CI, 95% confidence interval; EV, empty vector.
6 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org

contribute to nephrocyte loss seen in older adult flies. The yeastexperimental platform strengthens the hypothesis that APOL1kidney risk variants impair endosomal trafficking, an effect me-diated at least in part by impaired acidification, which may alsoexplain the accumulation of ANF-RFP in nephrocytes.
The yeast deletion strains revealed that cell injury is especiallysensitive to the integrity of PI 3-phosphate–related intracellularendosomal trafficking rather than autophagy. However, becausein mammalian cells, the VPS34 regulators ATG14 and UVRAG(VPS38) are less partitioned than in yeast,69,70 a role for impair-ment of autophagy in human disease cannot be completely ruledout by these studies. This increased toxicity also applied to G0
(but not the C-tr construct), such that differences comparedwithG1 and G2 were no longer apparent. This is consistent with anaction of the C-terminal domain that disrupts plasma or organ-ellar membrane integrity, which has been recently proposed.13,41
Diversion of APOL1 from the vacuolar membrane was shown inthe vps38D and other endocytic mutants and was not differentbetween the G0, G1, or G2 APOL1 variant gene products or theC-tr construct. Similarly, in previous studies, attenuation ofAPOL1 toxicity by coexpression with MCL-1 in Xenopus oocyteshas been attributed to decreased cell surface abundance.29 Theseyeast studies do not show a C–terminal sequence dependency oflocalization. Rather, diversion of APOL1 in the endocytic mu-tants augmented toxicity of all APOL1 variants, whereas the C-trremained innocuous. Thus, the extent of injury induced byAPOL1 depends on (1) the extent of its localization to its siteof injury, which is not a function of the C terminus, and (2) thepresence and protein conformation of the C terminus.71
The similar effect of perturbations in endosomal traffickingand acidification processes to increase APOL1 toxicitysuggests a model wherein acidifying conditions, such as inthe yeast vacuole, attenuate APOL1 activity. The pH depen-dence of APOL1 activity as an ion channel has been studiedin trypanosomes, lipid bilayers, and indirectly, HEKcells.12,27,29,72 Although the detailed findings in these studiesdiffer depending on the experimental system and conditions,pH dependency is observed for APOL1 insertion into the
Figure 4. S. cerevisiae strains deleted in components of the endocytic Vps34 complex II but not the autophagic–specific Vps34complex I confer APOL1 hypersensitivity. Plasmids containing the human APOL1 variants under the yeast GAL1 promoter weretransformed into (A) atg6D/Beclin, (B) vps34D, and (C) vps15D strains. (D) vps38D, (E) atg14D, and (F) atg8D yeast strains. Each rowrepresents serial fivefold dilutions (left to right) of a suspension of cells transformed with the indicated plasmid spotted on platescontaining glucose (right panels; noninduced conditions) or galactose (left panels; induced conditions). EV, empty vector.
Table 4. S. cerevisiae survival assay: comparisons of survivalratios
APOL1 Variant Comparison P Value for Difference
G0-EV ,0.01G1-G0 ,0.001G2-G0 ,0.001G2-G1 ,0.01C truncated-EV 0.51
Survival assays were conducted as described in Concise Methods, withmeasurement of surviving cells determined as CFU expressed as ratios at0 and 8 hours of induction for each experimental condition. Each pair ofexperiments was conducted in parallel for all conditions three times, and Pvalues for differenceswere determined as described inConciseMethods. EV,empty vector.
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 7
www.jasn.org BASIC RESEARCH
membrane or its activity as an ion channel.73 Our findingsshow that APOL1 inhibits both VPS (Vph1 studies) and vac-uole acidification (quinacrine staining), with a greater effect ofthe G1 and G2 kidney disease risk variants on acidification.Enhanced APOL1G1 andG2 toxicity compared with the G0 in
the trk2D strain is consistent with proposedpH–dependent activity of APOL1 activityas a cationic channel,29,73 resulting in cel-lular potassium depletion by the kidneyrisk variants.27 This cation channel potas-sium egress is reminiscent of the activationof the inflammasome,74 which might con-tribute to subsequent steps of APOL1 cellinjury. The findings in this study point tonot only an APOL1 risk variant differentialeffect on acidification of intracellularmembrane compartments but also, a pos-sible role of trafficking to and acidificationof such compartments in determining tox-icity. This is also consistent with resultspreviously reported for HEK cytotoxicity,which was shown to be dependent on pres-ervation of a signal peptide sequence in exon4 ofAPOL1.26 Taken together, ourfindings inyeast and nephrocytes are most consistentwith differential APOL1 risk variant impair-ment of the inter–related endolysosomal traf-ficking and acidification processes leading tocell injury.
The D. melanogaster model showsgreater fidelity with respect to recapitulating the human riskassociationwith a greater differential toxicity of the kidney riskalleles compared with yeast (this study) and even human celllines19,22–25,27 and has the advantage of a nephrocyte pheno-type with filtration and endolysosomal functions that can be
readily monitored using ANF-RFP uptakeand degradation. Having shown this robustrecapitulation of differential toxicity, itshould be possible in the future to usesuch an organismal model to productivelyunderstand the relationship between a gainof function cell injury process and recessiveinheritance. This could be done by coex-pressing G0 with G1 or G2 to investigatethe mechanistic basis for a sharp thresholdon the basis of gene dosage or gene productexpression or rule out a dominant protec-tive effect of G0. Although the differentialtoxicity of the kidney risk alleles in the yeastplatform was less robust and the G0 wasmore toxic than in flies, there was still asignificant difference in the differentialtoxicity of G1/G2 that led to yeast lethality.Therefore, the yeast system, which is themost rapidly amenable system to study ge-netic interactions, adds a complementarypiece to unraveling the puzzle of APOL1–induced cell toxicity. Indeed, the yeaststudies showed endosomal trafficking ef-fects of likely relevance to the observation
Figure 6. APOL1 localization is altered in the endocytic deleted strains. Loss of APOL1vacuolar localization in the endocytic mutants (vps38D and vps35D) but not an auto-phagy-deleted strain (atg8D). APOL1 variants were tagged with mCherry at their Nterminus (after the signal peptide) (Concise Methods) and expressed in WT and de-letion (D) strains. APOL1 expression was induced for 3 hours with synthetic minimalmedia containing essential amino acids and galactose. White arrows indicate vacu-oles, and blue arrows indicate perinuclear ER. The same results were observed for C-trAPOL1 variants in WT and vps38D (Supplemental Figure 3). DIC, differential in-terference contrast.
Figure 5. Disruption of endocytic trafficking enhances APOL1 toxicity. Schematicrepresentation of endosome, Golgi, multivesicular body (MVB), vacuole, and auto-phagosome trafficking pathways examined in relation to APOL1-mediated toxicity.Pathways marked in red are those in which deletion strains augmented APOL1–induced yeast toxicity for G0 as well as G1 and G2 risk variants. Conversely, pathwaysmarked in blue are those in which elimination of components did not enhance APOL1toxicity. Pathways marked in yellow were not examined. CORVET, class C core vacuole/endosome tethering factor; LE, late endosome; PI3K, phosphatidylinositol 3-kinase.
8 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org
of fly nephrocyte dysfunction and death. The yeast platformalso facilitated investigation of the role of acidification andrevealed incubation conditions and pathways that abolishedthe differential toxicity. These studies showed that the differentialtoxicity is highly sensitive to ambient pH and genetic manipula-tion of the endocytic trafficking pathway and vacuole acidifica-tion. As such, these findings in yeast can productively guide thesubsequent relevant fly studies. The more pronounced differen-tial toxicity between G1/G2 compared with G0 in the fly modelcompared with yeast and mammalian cells in culture requiresadditional investigation. We know from experimental systemsusing in vivo models and human cell lines that G0 is alsotoxic13,20,22–29; however, some cells express APOL1 without evi-dence of toxicity.34 Even in the fly platform, the differential tox-icity was not uniform across drivers (G0 was more toxic underGMR compared with the da driver), suggesting cell type–relateddifferences. Similarly, the differential toxicity of the kidney riskvariants in specific cells and organs (podocytes and kidney, re-spectively) and the variability of the lifetime kidney disease risk(ranging from 4% to.50% in individuals with two risk alleles)are also not clear.3,7,75 Investigation of these differences shouldadvance our understanding of environmental factors that couldbe mimicked in experimental systems or unique susceptibility of
Figure 7. S. cerevisiae-deleted strains that affect vacuole acidi-fication (VMAD) enhanced APOL1 toxicity, whereas deletion ofvacuolar hydrolase (pep4D) was innocuous. Plasmids containingthe human APOL1 variants under the yeast GAL1 promoter weretransformed into (A) vma1D, (B) vma21D, and (C) pep4D yeaststrains. Each row represents serial fivefold dilutions (left to right)of a suspension of cells transformed with the indicated plasmidspotted on plates containing glucose (right panels; noninducedconditions) or galactose (left panels; induced conditions). EV,empty vector.
Figure 8. Increasing medium pH augments APOL1 toxicity,whereas expression of APOL1 and risk variants results in a vacuoleacidification defect. (A) Plasmids containing the human APOL1variants G0, G1, G2, and C-tr under the yeast GAL1 promoterwere transformed into the WT BY4741 strain. Each row representsserial fivefold dilutions (left to right) of a suspension of cellstransformed with the indicated plasmid spotted on plates con-taining glucose (right panel; noninduced conditions at pH 4.3) orgalactose (induced conditions) at pH 4.3 (middle panel) or pH 5.3(left panel). (B) Quinacrine staining of S. cerevisiae transformedwith APOL1 variants G0, G1, and G2 is similar to the staining ofthe vma1D strain with reduced vacuolar staining, showing avacuole acidification defect with G1, G2 . G0. Enlarged picturesof selected areas are shown in right panels. The empty vector(EV), C-tr, and G2 in noninducing medium (glucose) show normalaccumulation of the dye inside the vacuole, which indicates thatthe vacuolar lumen is more acidic than the cytoplasm. The cor-responding graph shows the number of cells counted for eachcondition. DIC, differential interference contrast.
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 9
www.jasn.org BASIC RESEARCH

specific cells, organs, organisms, or even individuals to APOL1variant–mediated deleterious effect.
Perturbations in endosomal trafficking are especially rele-vant to podocyte function, with functional integrity that iscrucially dependent on these pathways for appropriate regu-lation of slit diaphragm proteins, proper functioning of theactin cytoskeletal meshwork, and protein clearance to preventfiltration barrier clogging.51,53,54 Several genes implicated inhuman nephrotic syndrome directly or indirectly associatewith the endocytic pathway.52 The role of VPS34 as a majorregulator of endocytic pathways in podocytes had been clari-fied,49,50 with rapid onset kidney disease observed in micelacking podocyte Vps34 compared with late onset kidney dis-ease occurring in Atg5 knockout mice.48
The findings of endocytic trafficking impairment by APOL1may also be of importance with respect to mechanisms, wherebynongenetic second hits may trigger APOL1–associated kidneydisease.3,6,7,17–19 HIV infection constitutes the most robust sec-ond hit that transforms APOL1 genotypic risk into a form ofkidney disease designated as HIV-associated nephropathy.3,7
Multiple enveloped viruses use the ESCRTmachinery for bud-ding. Taylor et al.76 have reported the increased secretion of HIVVif protein intomicrovesicles from cells transfected withAPOL1.Moreover, the HIV-1–encoded Gag protein has been shown tointeract with the mammalian ortholog of Vps23 (tsg101), redi-recting ESCRT-1 to the plasma membrane to execute viral bud-ding.77,78 In addition to the expectation of increased expressionof APOL1 under the high IFNg induction state that characterizesHIV infection and has been shown to induce APOL1 gene ex-pression,19 a Gag protein–mediated redirection mechanism
could also contribute to the especially powerful second hit effectof HIV infection.
The yeast and flymodels described in this study have severallimitations. Both of these models lack endogenousAPOL1 andtherefore, do not normally engage pathways that interact,transport, and degrade APOL1 protein. Our data point towell conserved endocytic trafficking and endolysosomal acid-ification pathways involved in APOL1-mediated toxicity;however, other pathways with specific interacting proteins de-veloped in humans as an adaptation to APOL1 deleteriouseffects may have a role. These would not be discerned usingnonhuman reductionist models. More complex experimentalsystems might show that the core conserved pathways that aredifferentially engaged by the different APOL1 moieties mighttrigger and then, be amplified or augmented by engagement ofinflammatory or other mechanisms. These latter mechanismsmay have a major role in human disease risk and will requiremoving up from simple models amenable to genetic interro-gation to models with a full complement of cell injury mech-anisms to understand the pathobiology of APOL1-mediatednephropathy. However, the results of this study point to theinvolvement of core conserved processes that distinguish thenonrisk from the risk APOL1 variants. Although fly nephro-cytes share many features of mammalian podocytes,43,45
nephrocytes differ from human podocytes in several poten-tially relevant physiologic and molecular features. For exam-ple, nephrocytes are not exposed to high-filtrate fluxes, andnephrocytes also possess proximal tubule attributes.44,79 Inmammals, the filtration system is composed of three layers:fenestrated endothelium, glomerular basement membrane,and podocyte foot processes. In Drosophila, the filtration sys-tem is composed of two: the nephrocyte basement membraneand nephrocyte diaphragm. Taken together, these limitationsindicate that, although the findings in this study point to thetwo experimental systems that are readily amenable to genetic
Figure 10. APOL1 inhibits the VPS pathway. Defective transportof Vph1 in yeast expressing G2 but not C-tr APOL1. Plasmidscontaining the human APOL1 variants under the yeast GAL1promoter were transformed into S. cerevisiae expressing GFP-Vph1. In the C-tr APOL1, Vph1-GFP localized normally to thevacuole, whereas G2 APOL1 expression inhibited Vph1 traffick-ing to the vacuole, which was mis-sorted to multiple punctatecompartments. DIC, differential interference contrast.
Figure 9. Deletion of a component of the potassium transportsystem (trk2D) enhances APOL1 toxicity. Plasmids containing thehuman APOL1 variants under the yeast GAL1 promoter weretransformed into the WT and trk2D strains. Each row representsserial fivefold dilutions (left to right) of a suspension of cellstransformed with the indicated plasmid spotted on plates con-taining glucose (right panel; noninduced conditions) or galactose(left panel; induced conditions). EV, empty vector.
10 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org

interrogation as valuable in uncovering core conserved mecha-nisms differentially engaged by the APOL1 variants found toconfer risk for kidney disease, insights gleaned will require ex-tension and validation in more complex mammalian systems.
In conclusion, we have shown that differential toxicity ofAPOL1 kidney disease risk and nonrisk variants is conservedacross eukaryotic evolution and may be mediated by pertur-bation of endosomal trafficking and acidification. Becausethese species evolved without a known endogenous APOL1homolog, the differential toxicity of G0 compared with theG1 and G2 risk alleles reflects an inherent difference in themode of interaction of the respective gene products with corebiologic processes without respect to evolutionary history. Tothe extent that the parallel pattern of differential toxicitypoints to mechanisms that are relevant to the increased riskof kidney disease, then this also renders the phenotypes thatare readily observable in these simple experimental platformsuseful for genetic and compound screens in a phenotype–based high–throughput discovery process.
CONCISE METHODS
Fly StrainsThe following strainswere used in this study (described in FlyBase; http://
flybase.bio.indiana.edu/80): w1118 served as WT control, da-GAL4 was
used to drive transgene expression as a model for systemic APOL1 ex-
pression, and the eye–specificGMR-GAL4 driver was used as amodel for
tissue-specific expression. For adult pericardial nephrocyte uptake/filtra-
tion studies, virgin females myosin heavy chain-ANF-RFP, Hand-GFP;
Dot-GAL4 (a gift from Zhe Han and as reported in the work by Zhang
et al.44) were crossed to male UAS-APOL1 WT (G0) and UAS-APOL1
variant (G1 and G2) transgenic lines. Hand-GFP was used to label peri-
cardial nephrocytes at all developmental stages. The Kirre RNAi line
(P[mw, UAS-kirre-RNAi]GD14476) was obtained from the Vienna Dro-
sophila RNAi Center. For the generation of APOL1 transgenic flies,
APOL1 G0, G1, G2, and C-tr were cloned into the pUASTattB vector81
at NotI and BglII, and transgenic strains were generated by FC31
integrase–based transgenesis. All transgenes were inserted into the attP2
chromosomal landing site (Genetic Services Inc., Cambridge, MA and
BestGene Inc., ChinoHills, CA). Newly eclosedflies from each cross were
collected on the same day and transferred to fresh vials. Adult flies were
flipped every secondday, grownon standard cornmealfly food, yeast, and
molasses at 24°C and incubated at 29°C after crossing with GAL4 drivers.
In Vivo Nephrocyte Filtration AssayPericardial nephrocytes of 1- and 15-day-old female adult flies were
dissected in cold PBS and fixed in 4% paraformaldehyde diluted in
PBS for 20 minutes at room temperature. Nephrocytes were washed
twice with PBS andmounted in Roti-Mount FluorCare (Roth). Images
for GFP and ANF-RFP fluorescence were taken with a confocal micro-
scope Leica TCS SP8 SMD equipped with an HC PL APO CS2 403 oil
objective with a numerical aperture of 1.3, and the imaging software
LAS X from Leica was used for operating the system and image ac-
quisition. The GFP and RFP were excited sequentially (sequential
scan) at 488 nm (PMT detector; 1% laser intensity) and 561 nm
(HyD detector; 30% sensitivity and 2% laser intensity), respectively.
Images were taken with a depth of 12 bit in the spectral ranges of the
emission at 498–552 nm (for GFP) and 571–684 nm (for RFP). Images
were processed with ImageJ/Fiji.82
Extraction of Larval HemolymphLarval hemolymph extractionwas performed as described inMusselman
et al.83
Fly Eye PathologyFlieswere anesthetized, and their headsweredetached.Theheadswere
fixed with 4% paraformaldehyde and embedded in paraffin. Four–
micrometer paraffin sections were mounted on Super FrostPlus Mi-
croscope Slides (Menzel-Glaser, Braunschweig, Germany) and
stainedwith hematoxylin and eosin.Digital presentationswere generated
using an Olympus BX51 Microscope equipped with an Olympus DP70
Camera. Pictures were processed using the analySIS 5 software (Soft
Imaging Systems, Muenster, Germany).
Generation of DNA ConstructsAPOL1 WT full–length (isoform B1: 414 amino acids; transcript
variant 2; NM_145343), G1, and G2 cDNAs were purchased from
Hy-labs. The terminology of APOL1 domains is on the basis of the
APOL1 A isoform (398 amino acids). The C-tr APOL1 was cloned to
remove the SRA binding domain at amino acid 354 (for isoform B1).
For expression in yeast under the Gal1 promoter, APOL1 variants
were cloned to high–copy p426-GAL1 (2 m; URA3) vector using
BamHI and EcoRI restriction sites or high–copy p425-GAL1 (2 m;
LEU2) using BamHI and XhoI sites. The S342G and I384M APOL1
mutated versions were generated by site-directed mutagenesis. The
N–terminal mCherry-APOL1 vectors were cloned by fusion PCR to
introduce the mCherry after the signal peptide (1–47 amino acids).
The C–terminal mCherry constructs were cloned by introducing the
mCherry at the C terminus.
All primers are listed in Supplemental Table 3.
Yeast Strains and MediaThe S. cerevisiae strains used in this study are listed in Supplemental
Table 4.84 The strains were grown at 30°C in standard yeast extract/
peptone/dextrose (1% yeast extract, 2% peptone, and 2% dextrose),
complete yeast nitrogen base medium (1.5 g yeast nitrogen base per
1 L, 5 g ammonium sulfate per 1 L, 2% glucose or galactose, and 0.1 g/L
each amino acid with the appropriate amino acids removed as re-
quired for plasmid selection), or minimal medium (1.5 g yeast nitro-
gen base per 1 L, 5 g ammonium sulfate per 1 L, 2% glucose or
galactose, and 0.1 g/L essential amino acids).
Yeast Survival AssayYeast cells were grown in minimal nitrogen base media with 2% raf-
finose overnight. For APOL1 induction, cells were diluted to OD=0.4
and grown in minimal nitrogen base media with 2% galactose for
0 (t=0) or 8 hours (t=8). Equal aliquots of cells (normalized to
OD=2∙1023) were then plated on complete nitrogen base plates with-
out uracil. CFU were counted after 48 hours at 30°C.
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 11
www.jasn.org BASIC RESEARCH

Western Blotting and AntibodiesLysate protein samples were separated by SDS-PAGE and blotted onto
WesternBright NCNitrocelluloseMembranes (Advansta). Themem-
branes were blocked with 5% nonfat dry milk (Santa Cruz) in Tris-
buffered saline with Tween 20 and incubated with primary antibodies
as indicated below. The membranes were then incubated with the
appropriate secondaryantibodies (as indicatedbelow).Afterextensive
washing in Tris-buffered saline with Tween 20, the proteins were
visualized using chemiluminescence reagents.
Western Blot of APOL1 and b-Actin of Fly Heads and BodiesBecause protein lysates of bodies show an extensive representation of
secreted hemolymph proteins (along with cellular protein), whereas
protein lysates for heads predominantly represent cellular proteins with
littleornosecretedhemolymphproteins, cellularproteins, suchasb-actin,
are less abundant in the body comparedwith the head. Therefore, protein
loading per lane was as follows: head: 20 mg and body: 50 mg.
Antibodies UsedThe antibodies used were anti-APOL1 (1:1000; HPA018885; Sigma-
Aldrich) and anti–b-actin (1:5000; ab8224; Abcamor 69100MP for flies).
Anti–rabbit HRP (1:10,000; 111–035–144; Jackson) and anti–mouseHRP
(1:10,000; 115–035–166; Jackson) were used as secondary antibodies.
Quinacrine StainingThe yeast cells were grown for 2 hours on YEP-HEPES (pH 7.5) with 2%
galactose media; 1.53107 cells were incubated with 200 mMQuinacrine
for 10 minutes, washed twice with cold YEP-HEPES, and micrographed
at 3100 magnification, and images show both bright field and corre-
sponding software pseudocolor of intensity. For the accompanying
graph,.150 cells from at least four fields were counted.
Statistical AnalysesDrosophila StudiesDifferences between proportions were tested using one-way ANOVA
with post hoc Tukey HSD test. P values,0.05 were considered statis-
tically significant.
Yeast Survival AssayDifferences between 0- and 8-hour survival ratios were tested using
pairwise two–sample t tests for comparisons identified as being of
primary biologic interest. Ratio SEMs were obtained by running a
simulation of 10,000 runs for each APOL1 group (each with three re-
peats) incorporating measurement error inherent to the experiment.
Measurement errors (estimated at 5%) are considered to be random
(no systematic trend to over- or undercounting on the basis of experi-
mental condition) and independent of other variables. A Bonferroni
correction for multiple comparisons was applied, unadjusted P values
are shown, and thresholds for significance were determined using Bon-
ferroni correction for the number of comparisons tested (P,0.01).
ACKNOWLEDGMENTS
The authors acknowledge the expert and thoughtful input provided
by Dr. Sara Selig. We also acknowledge M. Garfa-Traoré, N. Goudin
(Necker Cell Imaging Facility), and Gwenn Le Meur for technical
assistance as well as AmyGalick for assistance with statistical analyses.
We acknowledge the support provided by Israel Science Founda-
tion grant 182/15, an academic research grant from GlaxoSmithKline,
and from the Ernest and Bonnie Beutler Research Grant Program in
Genomic Medicine.
Part of this research work was presented as an abstract at the 11th
International Podocyte Conference (Haifa, Israel, April 3–6, 2016).
DISCLOSURESNone.
REFERENCES
1. GenoveseG, FriedmanDJ, RossMD, Lecordier L, Uzureau P, FreedmanBI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL,Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA,Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variantswith kidney disease in African Americans. Science 329: 841–845, 2010
2. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E,BradmanN,WasserWG,BeharDM, Skorecki K:Missensemutations in theAPOL1 gene are highly associated with end stage kidney disease riskpreviously attributed to theMYH9 gene.HumGenet 128: 345–350, 2010
3. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P,Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL,Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, SmithMC, Trachtman H,Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA: APOL1 ge-netic variants in focal segmental glomerulosclerosis andHIV-associatednephropathy. J Am Soc Nephrol 22: 2129–2137, 2011
4. Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N,Boerwinkle E, Parekh RS, Kao WH: APOL1 variants associate with in-creased risk of CKD among African Americans. J Am Soc Nephrol 24:1484–1491, 2013
5. Parsa A, KaoWH, Xie D, Astor BC, Li M, Hsu CY, Feldman HI, Parekh RS,Kusek JW, Greene TH, Fink JC, Anderson AH, Choi MJ, Wright JT Jr.,Lash JP, Freedman BI, Ojo A, Winkler CA, Raj DS, Kopp JB, He J,JensvoldNG, Tao K, LipkowitzMS, Appel LJ; AASK Study Investigators;CRIC Study Investigators: APOL1 risk variants, race, and progression ofchronic kidney disease. N Engl J Med 369: 2183–2196, 2013
6. Kruzel-Davila E, Wasser WG, Aviram S, Skorecki K: APOL1 nephropa-thy: From gene to mechanisms of kidney injury. Nephrol Dial Trans-plant 31: 349–358, 2016
7. Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T,Limou S, Sezgin E, Nelson GW, Fogo AB, Goetsch S, Kopp JB, Winkler CA,Naicker S: APOL1 risk variants are strongly associated with HIV-associatednephropathy inblackSouthAfricans.JAmSocNephrol26:2882–2890,2015
8. Page NM, Butlin DJ, Lomthaisong K, Lowry PJ: The human apolipo-protein L gene cluster: Identification, classification, and sites of distri-bution. Genomics 74: 71–78, 2001
9. Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJ: The apolipo-protein L gene cluster has emerged recently in evolution and is ex-pressed in human vascular tissue. Genomics 79: 539–546, 2002
10. Smith EE, Malik HS: The apolipoprotein L family of programmed celldeath and immunity genes rapidly evolved in primates at discrete sitesof host-pathogen interactions. Genome Res 19: 850–858, 2009
11. Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, VanDen Abbeele J, Pays A, Tebabi P, Van XongH, Jacquet A,Moguilevsky N,DieuM, Kane JP, De Baetselier P, Brasseur R, Pays E: Apolipoprotein L-I isthe trypanosome lytic factor of human serum. Nature 422: 83–87, 2003
12. Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L,Homblé F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A,
12 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org

Brasseur R, Pays E: Apolipoprotein L-I promotes trypanosome lysis byforming pores in lysosomal membranes. Science 309: 469–472, 2005
13. Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK,CarringtonM,Winkler CA, Kopp J, Rotimi C, Adeyemo A, Doumatey A,Ayodo G, Alper SL, Pollak MR, Friedman DJ, Raper J: Evolution of theprimate trypanolytic factor APOL1. Proc Natl Acad Sci USA 111:E2130–E2139, 2014
14. Papeta N, Kiryluk K, Patel A, Sterken R, Kacak N, Snyder HJ, Imus PH,Mhatre AN, Lawani AK, Julian BA,Wyatt RJ, Novak J,Wyatt CM, RossMJ,Winston JA, Klotman ME, Cohen DJ, Appel GB, D’Agati VD, Klotman PE,Gharavi AG: APOL1 variants increase risk for FSGS andHIVAN but not IgAnephropathy. J Am Soc Nephrol 22: 1991–1996, 2011
15. Kanji Z, Powe CE, Wenger JB, Huang C, Ankers E, Sullivan DA,Collerone G, Powe NR, Tonelli M, Bhan I, Bernhardy AJ, Dibartolo S,Friedman D, Genovese G, Pollak MR, Thadhani R: Genetic variation inAPOL1 associates with younger age at hemodialysis initiation. J Am
Soc Nephrol 22: 2091–2097, 201116. Tzur S, Rosset S, Skorecki K, Wasser WG: APOL1 allelic variants are
associated with lower age of dialysis initiation and thereby increased di-alysis vintage in African and Hispanic Americans with non-diabetic end-stage kidney disease.Nephrol Dial Transplant 27: 1498–1505, 2012
17. Divers J, Núñez M, High KP, Murea M, Rocco MV, Ma L, Bowden DW,Hicks PJ, Spainhour M, Ornelles DA, Kleiboeker SB, Duncan K,Langefeld CD, Turner J, Freedman BI: JC polyoma virus interacts withAPOL1 in African Americans with nondiabetic nephropathy. Kidney Int84: 1207–1213, 2013
18. Freedman BI, Skorecki K: Gene-gene and gene-environment interac-tions in apolipoprotein L1 gene-associated nephropathy.Clin J AmSoc
Nephrol 9: 2006–2013, 201419. NicholsB, JogP, LeeJH,BlacklerD,WilmotM,D’AgatiV,MarkowitzG,Kopp
JB,Alper SL, PollakMR,FriedmanDJ: Innate immunitypathways regulate thenephropathy gene Apolipoprotein L1. Kidney Int 87: 332–342, 2015
20. Capewell P, Cooper A, Clucas C, Weir W, Macleod A: A co-evolutionaryarms race: Trypanosomes shaping the human genome, humans shapingthe trypanosome genome. Parasitology 142[Suppl 1]: S108–S119, 2015
21. JohnstoneDB, Shegokar V,NihalaniD, RathoreYS,Mallik L, Ashish, ZareV,Ikizler HO, Powar R, Holzman LB: APOL1 null alleles from a rural village inIndia do not correlate with glomerulosclerosis. PLoS One 7: e51546, 2012
22. Lan X, Jhaveri A, Cheng K,WenH, SaleemMA,Mathieson PW,MikulakJ, Aviram S, Malhotra A, Skorecki K, Singhal PC: APOL1 risk variantsenhance podocyte necrosis through compromising lysosomal mem-brane permeability. Am J Physiol Renal Physiol 307: F326–F336, 2014
23. Lan X,Wen H, Lederman R, Malhotra A, Mikulak J, PopikW, Skorecki K,Singhal PC: Protein domains of APOL1 and its risk variants. Exp Mol
Pathol 99: 139–144, 201524. Lan X, Wen H, Saleem MA, Mikulak J, Malhotra A, Skorecki K, Singhal
PC: Vascular smooth muscle cells contribute to APOL1-induced po-docyte injury in HIV milieu. Exp Mol Pathol 98: 491–501, 2015
25. ChengD,Weckerle A, Yu Y,Ma L, Zhu X,MureaM, Freedman BI, Parks JS,Shelness GS: Biogenesis and cytotoxicity of APOL1 renal risk variant pro-teins in hepatocytes and hepatoma cells. J Lipid Res 56: 1583–1593, 2015
26. Khatua AK, Cheatham AM, Kruzel ED, Singhal PC, Skorecki K, PopikW:Exon 4 encoded sequence is a major determinant of cytotoxicity ofapolipoprotein L1. Am J Physiol Cell Physiol 309: C22–C37, 2015
27. Olabisi OA, Zhang JY, VerPlank L, Zahler N, DiBartolo S III, HeneghanJF, Schlöndorff JS, Suh JH, Yan P, Alper SL, Friedman DJ, Pollak MR:APOL1 kidney disease risk variants cause cytotoxicity by depletingcellular potassium and inducing stress-activated protein kinases. ProcNatl Acad Sci USA 113: 830–837, 2015
28. Olabisi O, Al-Romaih K, Henderson J, Tomar R, Drummond I, MacRaeC, Pollak M: From man to fish: What can Zebrafish tell us about ApoL1nephropathy? [published online ahead of print August 10, 2016]. ClinNephrol doi: 10.5414/CNP86S116
29. HeneghanJF,VandorpeDH,ShmuklerBE,GiovinazzoJA,RaperJ,FriedmanDJ,PollakMR,AlperSL:BH3domain-independentapolipoproteinL1toxicity
rescued by BCL2 prosurvival proteins. Am J Physiol Cell Physiol 309: C332–C347, 2015
30. Limou S, Dummer PD, Nelson GW, Kopp JB,Winkler CA: APOL1 toxin,innate immunity, and kidney injury. Kidney Int 88: 28–34, 2015
31. Raper J, Nussenzweig V, Tomlinson S: The main lytic factor of Trypa-nosoma brucei brucei in normal human serum is not high density li-poprotein. J Exp Med 183: 1023–1029, 1996
32. Weckerle A, Snipes JA, Cheng D, Gebre AK, Reisz JA, Murea M,Shelness GS, Hawkins GA, Furdui CM, Freedman BI, Parks JS, Ma L:Characterization of circulating APOL1 protein complexes in AfricanAmericans. J Lipid Res 57: 120–130, 2015
33. VanhollebekeB, Pays E: The trypanolytic factor of human serum:Manywaysto enter the parasite, a single way to kill.Mol Microbiol 76: 806–814, 2010
34. Hu CA, Klopfer EI, Ray PE: Human apolipoprotein L1 (ApoL1) in cancerand chronic kidney disease. FEBS Lett 586: 947–955, 2012
35. Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, BruggemanLA, Sedor JR: APOL1 localization in normal kidney and nondiabetickidney disease. J Am Soc Nephrol 22: 2119–2128, 2011
36. Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D,Saleem MA, Satchell SC, Banas B, Mathieson PW, Kretzler M, HemalAK, Rudel LL, Petrovic S, Weckerle A, Pollak MR, Ross MD, Parks JS,Freedman BI: Localization of APOL1 protein and mRNA in the humankidney: Nondiseased tissue, primary cells, and immortalized cell lines. JAm Soc Nephrol 26: 339–348, 2015
37. Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M,Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, Lin JJ,Kiger DF, Gautreaux MD, Divers J, Freedman BI: The APOL1 gene andallograft survival after kidney transplantation. Am J Transplant 11:1025–1030, 2011
38. Lee BT, Kumar V, Williams TA, Abdi R, Bernhardy A, Dyer C, Conte S,Genovese G, Ross MD, Friedman DJ, Gaston R, Milford E, Pollak MR,Chandraker A: The APOL1 genotype of African American kidneytransplant recipients does not impact 5-year allograft survival. Am J
Transplant 12: 1924–1928, 201239. Freedman BI, Julian BA, Pastan SO, Israni AK, Schladt D, Gautreaux
MD, Hauptfeld V, Bray RA, Gebel HM, Kirk AD, Gaston RS, Rogers J,Farney AC, Orlando G, Stratta RJ, Mohan S, Ma L, Langefeld CD, HicksPJ, Palmer ND, Adams PL, Palanisamy A, Reeves-Daniel AM, Divers J:Apolipoprotein L1 gene variants in deceased organ donors are asso-ciatedwith renal allograft failure.Am JTransplant 15: 1615–1622, 2015
40. Freedman BI, Pastan SO, Israni AK, Schladt D, Julian BA, GautreauxMD, Hauptfeld V, Bray RA, Gebel HM, Kirk AD, Gaston RS, Rogers J,Farney AC, Orlando G, Stratta RJ, Mohan S, Ma L, Langefeld CD,Bowden DW, Hicks PJ, Palmer ND, Palanisamy A, Reeves-Daniel AM,Brown WM, Divers J: APOL1 genotype and kidney transplantationoutcomes from deceased African American donors. Transplantation100: 194–202, 2016
41. Molina-Portela MP, Samanovic M, Raper J: Distinct roles of apolipo-protein components within the trypanosome lytic factor complex revealedin a novel transgenic mouse model. J Exp Med 205: 1721–1728, 2008
42. Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F,Andris F, Lins L, Pays E: C-terminal mutants of apolipoprotein L-I efficientlykill bothTrypanosomabrucei brucei andTrypanosomabrucei rhodesiense.PLoS Pathog 5: e1000685, 2009
43. Weavers H, Prieto-Sánchez S, Grawe F, Garcia-López A, Artero R,Wilsch-Bräuninger M, Ruiz-Gómez M, Skaer H, Denholm B: The insectnephrocyte is a podocyte-like cell with a filtration slit diaphragm. Na-
ture 457: 322–326, 200944. Zhang F, Zhao Y, Han Z: An in vivo functional analysis system for renal
gene discovery in Drosophila pericardial nephrocytes. J Am Soc
Nephrol 24: 191–197, 201345. Zhuang S, Shao H, Guo F, Trimble R, Pearce E, Abmayr SM: Sns and
Kirre, theDrosophila orthologs of Nephrin andNeph1, direct adhesion,fusion and formation of a slit diaphragm-like structure in insect neph-rocytes. Development 136: 2335–2344, 2009
J Am Soc Nephrol 28: ccc–ccc, 2016 APOL1–Mediated Cell Injury 13
www.jasn.org BASIC RESEARCH

46. Ito K, Bick AG, Flannick J, Friedman DJ, Genovese G, Parfenov MG,Depalma SR, Gupta N, Gabriel SB, Taylor HA Jr., Fox ER, Newton-ChehC, Kathiresan S, Hirschhorn JN, Altshuler DM, Pollak MR, Wilson JG,Seidman JG, Seidman C: Increased burden of cardiovascular disease incarriers of APOL1 genetic variants. Circ Res 114: 845–850, 2014
47. Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA: ApolipoproteinL1, a novel Bcl-2 homology domain 3-only lipid-binding protein, in-duces autophagic cell death. J Biol Chem 283: 21540–21549, 2008
48. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S,Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD,Pavenstädt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB:Autophagy influences glomerular disease susceptibility and maintainspodocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010
49. Chen J, Chen MX, Fogo AB, Harris RC, Chen JK: mVps34 deletion inpodocytes causes glomerulosclerosis by disrupting intracellular vesicletrafficking. J Am Soc Nephrol 24: 198–207, 2013
50. Bechtel W, Helmstädter M, Balica J, Hartleben B, Kiefer B, Hrnjic F,Schell C, Kretz O, Liu S, Geist F, Kerjaschki D, Walz G, Huber TB: Vps34deficiency reveals the importance of endocytosis for podocyte ho-meostasis. J Am Soc Nephrol 24: 727–743, 2013
51. Akilesh S, Huber TB, Wu H, Wang G, Hartleben B, Kopp JB, Miner JH,Roopenian DC, Unanue ER, Shaw AS: Podocytes use FcRn to clear IgGfrom theglomerular basementmembrane.ProcNatl Acad Sci USA 105:967–972, 2008
52. Soda K, Ishibe S: The function of endocytosis in podocytes. Curr OpinNephrol Hypertens 22: 432–438, 2013
53. Sampogna RV, Al-Awqati Q: Taking a bite: Endocytosis in the mainte-nance of the slit diaphragm. J Clin Invest 122: 4330–4333, 2012
54. Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S,Volpicelli-Daley L, Tian X, Wu Y, Ma H, Son SH, Zheng R, Moeckel G,Cremona O, Holzman LB, De Camilli P, Ishibe S: Role of dynamin,synaptojanin, and endophilin in podocyte foot processes. J Clin Invest122: 4401–4411, 2012
55. Backer JM: The regulation and function ofClass III PI3Ks:Novel roles forVps34. Biochem J 410: 1–17, 2008
56. Kihara A, Noda T, Ishihara N, Ohsumi Y: Two distinct Vps34 phospha-tidylinositol 3-kinase complexes function in autophagy and carboxy-peptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152: 519–530, 2001
57. Kim J, KimYC, FangC, Russell RC, Kim JH, FanW, Liu R, ZhongQ,GuanKL: Differential regulation of distinct Vps34 complexes by AMPK innutrient stress and autophagy. Cell 152: 290–303, 2013
58. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y: Dynamics and diversityin autophagy mechanisms: Lessons from yeast. Nat Rev Mol Cell Biol10: 458–467, 2009
59. Obara K, Sekito T, Ohsumi Y: Assortment of phosphatidylinositol 3-kinase complexes–Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell17: 1527–1539, 2006
60. Katzmann DJ, Babst M, Emr SD: Ubiquitin-dependent sorting into themultivesicular body pathway requires the function of a conserved en-dosomal protein sorting complex, ESCRT-I. Cell 106: 145–155, 2001
61. Eskelinen EL, Tanaka Y, Saftig P: At the acidic edge: Emerging functionsfor lysosomal membrane proteins. Trends Cell Biol 13: 137–145, 2003
62. Corbacho I, Teixidó F, Olivero I, Hernández LM: Dependence of Sac-charomyces cerevisiae Golgi functions on V-ATPase activity. FEMSYeast Res 12: 341–350, 2012
63. Smardon AM, Diab HI, Tarsio M, Diakov TT, Nasab ND,West RW, KanePM: The RAVE complex is an isoform-specific V-ATPase assembly factorin yeast. Mol Biol Cell 25: 356–367, 2014
64. Kane PM: The where, when, and how of organelle acidification by theyeast vacuolar H+-ATPase. Microbiol Mol Biol Rev 70: 177–191, 2006
65. Huotari J,HeleniusA:Endosomematuration.EMBOJ30: 3481–3500, 201166. Ko CH, Gaber RF: TRK1 and TRK2 encode structurally related K+ trans-
porters in Saccharomyces cerevisiae.Mol Cell Biol 11: 4266–4273, 1991
67. Toshima JY, Nishinoaki S, Sato Y, Yamamoto W, Furukawa D, SiekhausDE, Sawaguchi A, Toshima J: Bifurcation of the endocytic pathway intoRab5-dependent and -independent transport to the vacuole. NatCommun 5: 3498, 2014
68. Genovese G, Friedman DJ, Pollak MR: APOL1 variants and kidneydisease in people of recent African ancestry. Nat Rev Nephrol 9: 240–244, 2013
69. Kim HJ, Zhong Q, Sheng ZH, Yoshimori T, Liang C, Jung JU: Beclin-1-interacting autophagy protein Atg14L targets the SNARE-associatedprotein Snapin to coordinate endocytic trafficking. JCell Sci 125: 4740–4750, 2012
70. Liang C, Lee JS, Inn KS, GackMU, Li Q, Roberts EA, Vergne I, Deretic V,Feng P, Akazawa C, Jung JU: Beclin1-binding UVRAG targets the classC Vps complex to coordinate autophagosome maturation and endo-cytic trafficking. Nat Cell Biol 10: 776–787, 2008
71. Sharma AK, Friedman DJ, Pollak MR, Alper SL: Structural characterizationof the C-terminal coiled-coil domains of wild-type and kidney disease-associated mutants of apolipoprotein L1. FEBS J 283: 1846–1862, 2016
72. Vanwalleghem G, Fontaine F, Lecordier L, Tebabi P, Klewe K, NolanDP, Yamaryo-Botté Y, Botté C, Kremer A, Burkard GS, Rassow J, RoditiI, Pérez-Morga D, Pays E: Coupling of lysosomal and mitochondrialmembrane permeabilization in trypanolysis by APOL1.Nat Commun 6:8078, 2015
73. Thomson R, Finkelstein A: Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: Relevance totrypanosome lysis. Proc Natl Acad Sci USA 112: 2894–2899, 2015
74. Stutz A, Golenbock DT, Latz E: Inflammasomes: Too big to miss. J ClinInvest 119: 3502–3511, 2009
75. Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, WinklerCA, Kopp JB: APOL1 kidney disease risk variants: An evolving land-scape. Semin Nephrol 35: 222–236, 2015
76. Taylor HE, Khatua AK, Popik W: The innate immune factor apolipo-protein L1 restricts HIV-1 infection. J Virol 88: 592–603, 2014
77. Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH,Wang HE, Wettstein DA, Stray KM, Côté M, Rich RL, Myszka DG,Sundquist WI: Tsg101 and the vacuolar protein sorting pathway areessential for HIV-1 budding. Cell 107: 55–65, 2001
78. Martin-Serrano J, Zang T, Bieniasz PD: HIV-1 and Ebola virus encodesmall peptide motifs that recruit Tsg101 to sites of particle assembly tofacilitate egress. Nat Med 7: 1313–1319, 2001
79. Zhang F, Zhao Y, Chao Y, Muir K, Han Z: Cubilin and amnionless me-diate protein reabsorption in Drosophila nephrocytes. J Am SocNephrol 24: 209–216, 2013
80. St Pierre SE, Ponting L, Stefancsik R, McQuilton P; FlyBase Consortium:FlyBase 102–advanced approaches to interrogating FlyBase. NucleicAcids Res 42: D780–D788, 2014
81. Bischof J, Maeda RK, Hediger M, Karch F, Basler K: An optimizedtransgenesis system for Drosophila using germ-line-specific phiC31integrases. Proc Natl Acad Sci USA 104: 3312–3317, 2007
82. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, PietzschT, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ,Hartenstein V, Eliceiri K, Tomancak P, Cardona A: Fiji: An open-sourceplatform for biological-image analysis. Nat Methods 9: 676–682, 2012
83. Musselman LP, Fink JL, Narzinski K, Ramachandran PV, Hathiramani SS,Cagan RL, Baranski TJ: A high-sugar diet produces obesity and insulinresistance in wild-type Drosophila. Dis Model Mech 4: 842–849, 2011
84. Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M,Kaganovich D: Confinement to organelle-associated inclusion struc-tures mediates asymmetric inheritance of aggregated protein in bud-ding yeast. Cell Rep 2: 738–747, 2012
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016050546/-/DCSupplemental.
14 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BASIC RESEARCH www.jasn.org