anexo i relaciÓn de nombres del medicamento,...
TRANSCRIPT

1
ANEXO I
RELACIÓN DE NOMBRES DEL MEDICAMENTO, TITULARES DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN, FORMAS FARMACÉUTICAS, DOSIS, VÍA DE
ADMINISTRACIÓN, PRESENTACIÓN Y TAMAÑOS DEL ENVASE EN LOS ESTADOS MIEMBROS

2
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
AUSTRIA Organon GesmbH Siebenbrunnengasse 21/D/IV A-1050 Wien Austria
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiolComprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiolComprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
AUSTRIA Organon GesmbH, Siebenbrunnengasse 21/D/IV A-1050 Wien Austria
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

3
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
BELGICA Organon Europe B.V. Kloosterstraat 6 5349 AB Oss, Países Bajos
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
BELGICA Organon Europe B.V. Kloosterstraat 6 5349 AB Oss, Países Bajos
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

4
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
DINAMARCA N.V. Organon, Kloosterstraat 6 P.O. Box 20 5340 BH Oss Países Bajos
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
DINAMARCA N.V. Organon, Kloosterstraat 6 P.O. Box 20 5340 BH Oss Países Bajos
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

5
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
FINLANDIA N.V. Organon, Kloosterstraat 6 P.O. Box 20 5340 BH Oss Países Bajos
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
FINLANDIA N.V. Organon, Kloosterstraat 6 P.O. Box 20 5340 BH Oss Países Bajos
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

6
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
ALEMANIA Organon GmbH Mittenheimer Str. 62 D-85764 Oberschleissheim, Munich Alemania
Novial Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
ALEMANIA Organon GmbH Mittenheimer Str. 62 D-85764 Oberschleissheim, Munich Alemania
Novial 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

7
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
GRECIA Organon Hellas SA 122 Vouliagmenis Av. Helliniko 16777 Athens Grecia
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
GRECIA Organon Hellas SA 122 Vouliagmenis Av. Helliniko 16777 Athens Grecia
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

8
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
IRLANDΑ Organon (Ireland) Limited PO Box 2857 Drynam Road Swords Co. Dublin Irlanda
Novial Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
IRLANDA Organon (Ireland) Limited PO Box 2857 Drynam Road Swords Co. Dublin Irlanda
Novial 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos

9
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
LUXEMBURGO Organon Europe B,V, Kloosterstraat 6 5349 AB Oss, Países Bajos
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos
PORTUGAL Organon Portuguesa - Produtos Químicos e Farmacêuticos, Lda. Avenida Conde de Valbom, n.º 30 - 2º 1069-037 Lisboa Países Bajos
Laurina Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos 6 sobres x 1 blister x 21 comprimidos

10
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
Portugal Organon Portuguesa - Produtos Químicos e Farmacêuticos, Lda. Avenida Conde de Valbom, n.º 30 - 2º 1069-037 Lisboa Portugal
Laurina 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos 6 sobres x 1 blister x 28 comprimidos
ESPAÑA Organon Española, S.A Ctra. De Hospitalet, 147-149 Cityparc Ronda de Dalt Edf. Amsterdam 08940 Cornella de Llobregat, Barcelona, España
Novial Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 21 comprimidos 3 sobres x 1 blister x 21 comprimidos

11
Estados Miembros
Titular de la autorización de comercialización
Marca de fantasia
Dosis Forma farmacéutica
Vía de administración
Envase Tamaño del envase
ESPAÑA Organon Española, S.A Ctra. De Hospitalet, 147-149 Cityparc Ronda de Dalt Edf. Amsterdam 08940 Cornella de Llobregat, Barcelona, España
Novial 28 Comprimidos amarillos: 50 microgramos desogestrel, 35 microgramos ethinylestradiol Comprimidos rojos: 100 microgramos desogestrel, 30 microgramos ethinylestradiol Comprimidos blancos: 150 microgramos desogestrel, 30 microgramos ethinylestradiol Placebo comprimidos: (sin principio activo)
Comprimido recubierto con película
Vía oral Blister (PVC/Alu)
1 sobre x 1 blister x 28 comprimidos 3 sobres x 1 blister x 28 comprimidos

12
ANEXO II
CONCLUSIONES CIENTÍFICAS Y MOTIVOS DE LA MODIFICACIÓN DEL RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO PRESENTADAS POR LA EMEA

13
CONCLUSIONES CIENTÍFICAS RESUMEN GENERAL DE LA EVALUACIÓN CIENTÍFICA DE LAURINA Y NOMBRES ASOCIADOS (VÉASE EL ANEXO 1) Laurina (Laurina/Laurina 28 y nombres asociados) es un anticonceptivo oral combinado (AOC) trifásico que contiene etinilestradiol y desogestrel (DSG). La comercialización de Laurina ha sido autorizada en varios países europeos con arreglo a procedimientos nacionales y también mediante el procedimiento de reconocimiento mutuo el 11 de octubre de 2000, siendo Finlandia el Estado miembro de referencia. Los anticonceptivos orales combinados han sido objeto de un detallado escrutinio debido tanto a la preocupación por los efectos adversos de estos productos como a causa de su uso por mujeres fundamentalmente sanas. En 1995, el CPMP creó un grupo de expertos ad hoc sobre anticonceptivos orales tras la publicación de tres estudios que sugerían un mayor riesgo de tromboembolia venosa (TEV) en usuarias de AOC “de tercera generación” que contienen los progestágenos desogestrel o gestodeno, en comparación con los AOC “de la segunda generación” que contienen el progestógeno levonorgestrel. En septiembre de 2001, el CPMP presentó un informe público de evaluación sobre los riesgos relativos de TEV y recomendó el texto de las advertencias sobre la TVE que debía incluirse en el resumen de las características del producto (RCP) de todos los AOC.1 Dicho informe público de evaluación, además de revisar expresamente el riesgo de TEV, contenía también una sección sobre el infarto agudo de miocardio (IAM), ya que en algún momento se pensó que los AOC de tercera generación podrían ser menos propensos a producir IAM que los más antiguos. El informe público de evaluación concluyó que no existían pruebas de que los AOC de tercera generación modificaran significativamente el riesgo de IAM en comparación con los anteriores. El titular de la autorización de comercialización de Laurina solicitó una renovación de la autorización de comercialización en septiembre de 2001, antes de que apareciera el informe público de evaluación, comprometiéndose a añadir, en una variación independiente, las advertencias sobre la TVE según el texto recomendado por el CPMP. En la variación de tipo II resultante, que es el objeto de la presente remisión, se solicitaba un texto que no seguía estrictamente las recomendaciones del CPMP. Además, el titular de la autorización de comercialización solicitaba la inclusión de los resultados de estudios epidemiológicos recientes sobre trombosis arterial y, en particular, sobre el IAM, que en su opinión demostraban la ausencia de aumento significativo del riesgo de IAM en las usuarias de AOC de tercera generación, en comparación con las no usuarias. Durante el procedimiento de reconocimiento mutuo, el titular de la autorización de comercialización revisó el texto para sugerir que, en comparación con las mujeres que no utilizaban AOC, el riesgo de IAM con los AOC de tercera generación podría ser menor que con los AOC de segunda o primera generación. El Estado miembro de referencia estaba dispuesto a aceptar el texto acordado con el titular de la autorización de comercialización al final del procedimiento de modificación, pero Alemania puso firmes objeciones y remitió la cuestión al CPMP el 23 de octubre de 2002, proporcionando información básica adicional el 6 de noviembre de 2002. El CPMP examinó la documentación presentada por el titular de la autorización de comercialización y llegó a las siguientes conclusiones: Infarto agudo de miocardio Las pruebas presentadas por el titular de la autorización de comercialización se basaban principalmente en estudios epidemiológicos publicados, además de algunos otros datos sobre análisis de lípidos procedentes de ensayos clínicos. El artículo fundamental fue un metaanálisis de Spitzer2 con los estudios epidemiológicos que formaron parte del mismo y que proporcionaban datos adicionales; además de un segundo metaanálisis efectuado por Khader3. De los siete estudios que constituían la base del metaanálisis de Spitzer, sólo tres de ellos, el Transnacional (TNS)4, el MICA5 y el Ratio6 fueron diseñados específicamente y tenían potencia

14
suficiente para evaluar el riesgo diferencial de los AOC de tercera y segunda generación. De los restantes estudios incluidos en el metaanálisis de Spitzer, el estudio de Lidegaard7 presentaba un resumen de los resultados intermedios, el estudio GPRD8 estaba también en forma de resumen y consistía en una comparación directa entre usuarias de AOC de tercera y segunda generación, el estudio de Dunn9 se centraba en un subconjunto de la población del estudio MICA y hacía una definición diferente de los casos y los controles, mientras que el estudio de la OMS10 contenía un pequeño número de casos, ya que el IAM no era su objetivo principal.
Tabla 1: Razón de posibilidades ajustada (IC del 95%) del riesgo de infarto de miocardio en comparación con mujeres no usuarias de AOC (excepto el estudio GPRD)
TNS4 MICA5 RATIO6 Danés7 OMS10 GPRD8 Dunn9 segunda
generación 2,99 (1,51-5,91)
1,10 (0,52-2,30)
2,5 (1,5-4,1)
1,9 (0,7-4,9)
1,64 (0,49-5,54)
1,0a 2,88 (1,22-6,77)
tercera generación
0,85 (0,30-2,39)
1,96 (0,87-4,39)
1,3 (0,7-2,5)
0,96b (0,4-2,29)
0,97 (0,14-6,96)
0,7 (0,1-8,2)
0,83 (0,25-2,81)
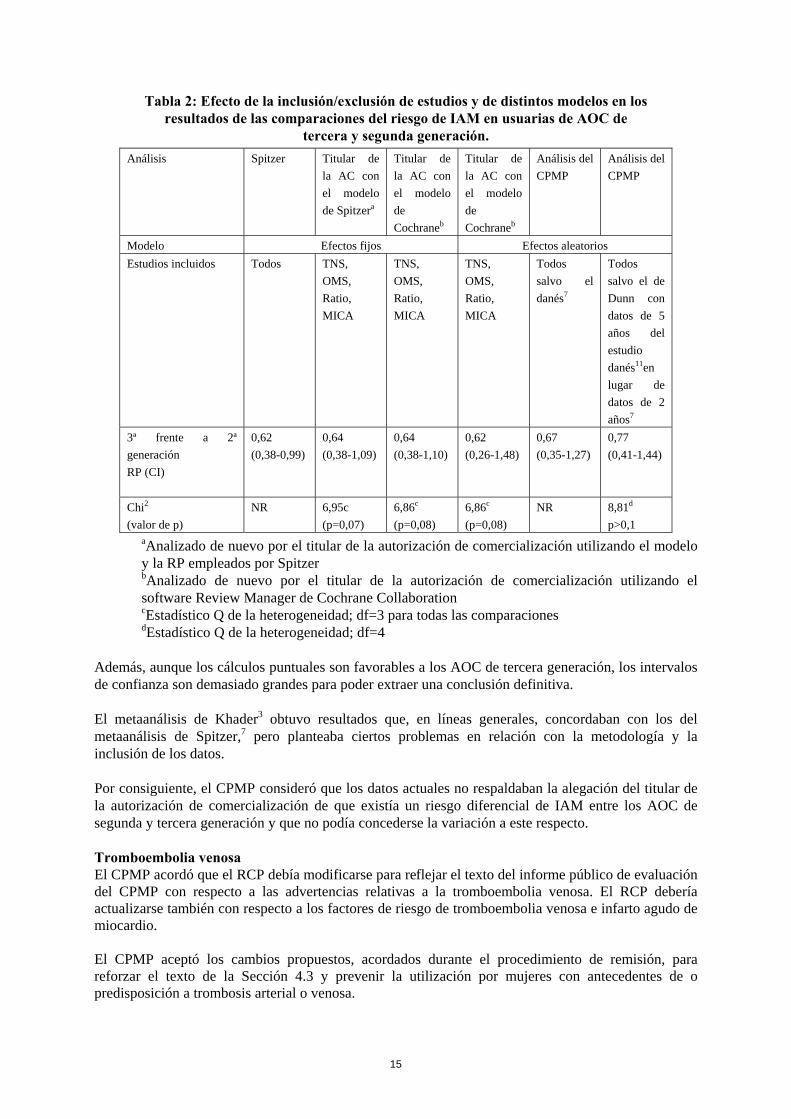
a El estudio GPRD se compararon usuarias de AOC de tercera y segunda generación. b En el artículo publicado, el cálculo de la razón de posibilidades de Spitzer se presenta según la duración del consumo de AOC. El titular de la autorización de comercialización alegó que en todos esos estudios no se apreciaba ninguna diferencia significativa entre usuarias de AOC de tercera generación y no usuarias de AOC y que, salvo en el estudio MICA, el riesgo era siempre menor que con los AOC de la segunda generación. El CPMP consideró que aunque las razones de posibilidades (RP) de las usuarias de AOC de segunda generación era superior a 1, su valor no resultó significativo en ninguno de los tres estudios y que los intervalos de confianza se solapaban con los de los AOC de tercera generación. El CPMP expresó también su preocupación porque los resultados del estudio MICA, que era el que aportaba el mayor número de casos, contradecían los de los otros estudios, sin que este resultado anómalo haya sido debidamente explicado. El CPMP expresó también cierta preocupación respecto al metaanálisis de Spitzer2, en relación con la inclusión y exclusión de estudios en el metaanálisis, la heterogeneidad de los estudios, la posibilidad de un sesgo y la ausencia de ajustes que tuvieran en cuenta los factores de riesgo en algunos de ellos. El CPMP cuestionó la utilización de datos correspondientes a 2 años en lugar de datos correspondientes a 5 años en el estudio danés y consideró también que el estudio de Dunn, realizado en un subgrupo de la población del estudio de MICA, no debería haberse incluido. El CPMP consideró que, dado el grado de heterogeneidad existente, para el análisis debería haberse utilizado un modelo de efectos aleatorios, en lugar de un modelo de efectos fijos. En la Tabla 2 se muestran los efectos de la inclusión/exclusión de los estudios y de la sustitución de un modelo de efectos fijos por otro de efectos aleatorios en los resultados de las comparaciones sobre el riesgo de IAM entre usuarias de AOC de tercera y segunda generación. La comparación entre los AOC de tercera y segunda generación se consideró la de mayor relevancia clínica. Se demuestra así que los datos se ven afectados por la inclusión o exclusión de estudios y por la utilización de un modelo de efectos fijos o aleatorios. Ello sugiere que los resultados no son robustos y que dependen del modelo de análisis utilizado.
15
Tabla 2: Efecto de la inclusión/exclusión de estudios y de distintos modelos en los
resultados de las comparaciones del riesgo de IAM en usuarias de AOC de tercera y segunda generación.
Análisis Spitzer Titular de la AC con el modelo de Spitzera
Titular de la AC con el modelo de Cochraneb
Titular de la AC con el modelo de Cochraneb
Análisis del CPMP
Análisis del CPMP
Modelo Efectos fijos Efectos aleatorios Estudios incluidos Todos TNS,
OMS, Ratio, MICA
TNS, OMS, Ratio, MICA
TNS, OMS, Ratio, MICA
Todos salvo el danés7
Todos salvo el de Dunn con datos de 5 años del estudio danés11en lugar de datos de 2 años7
3ª frente a 2ª generación RP (CI)
0,62 (0,38-0,99)
0,64 (0,38-1,09)
0,64 (0,38-1,10)
0,62 (0,26-1,48)
0,67 (0,35-1,27)
0,77 (0,41-1,44)
Chi2 (valor de p)
NR
6,95c (p=0,07)
6,86c (p=0,08)
6,86c (p=0,08)
NR 8,81d p>0,1
aAnalizado de nuevo por el titular de la autorización de comercialización utilizando el modelo y la RP empleados por Spitzer bAnalizado de nuevo por el titular de la autorización de comercialización utilizando el software Review Manager de Cochrane Collaboration cEstadístico Q de la heterogeneidad; df=3 para todas las comparaciones dEstadístico Q de la heterogeneidad; df=4
Además, aunque los cálculos puntuales son favorables a los AOC de tercera generación, los intervalos de confianza son demasiado grandes para poder extraer una conclusión definitiva. El metaanálisis de Khader3 obtuvo resultados que, en líneas generales, concordaban con los del metaanálisis de Spitzer,7 pero planteaba ciertos problemas en relación con la metodología y la inclusión de los datos. Por consiguiente, el CPMP consideró que los datos actuales no respaldaban la alegación del titular de la autorización de comercialización de que existía un riesgo diferencial de IAM entre los AOC de segunda y tercera generación y que no podía concederse la variación a este respecto. Tromboembolia venosa El CPMP acordó que el RCP debía modificarse para reflejar el texto del informe público de evaluación del CPMP con respecto a las advertencias relativas a la tromboembolia venosa. El RCP debería actualizarse también con respecto a los factores de riesgo de tromboembolia venosa e infarto agudo de miocardio. El CPMP aceptó los cambios propuestos, acordados durante el procedimiento de remisión, para reforzar el texto de la Sección 4.3 y prevenir la utilización por mujeres con antecedentes de o predisposición a trombosis arterial o venosa.

16
MOTIVOS PARA LA MODIFICACIÓN DEL RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO Considerando,
• que el CPMP examinó la remisión realizada conforme al apartado 5 del artículo 7 del Reglamento CE nº 541/95 de la Comisión, para Laurina y nombre asociados (véase el Anexo 1);
• que el CPMP consideró que los datos actuales no demuestran la existencia de un riesgo diferencial de IAM entre las mujeres usuarias de AOC de tercera y segunda generación y que, por tanto, debe rechazar ese aspecto de la modificación;
• que el CPMP acordó la inclusión en el RCP de las advertencias y contraindicaciones referentes a la tromboembolia venosa y los factores de riesgo de TEV e IAM;
el CPMP recomienda modificar la autorización de comercialización cuyo resumen de las características del producto se establece en el Anexo III para Laurina y nombres asociados.

17
Referencias
1. Comité de Especialidades Farmacéuticas de la EMEA (CPMP). Informe público de evaluación del CPMP. Anticonceptivos orales combinados y tromboembolia venosa. EMEA/CPMP/2201/01, 28 de septiembre de 2001.
2. Spitzer WO, Faith J, MacRae K. Myocardial infarction and third generation oral contraceptives: aggregation of recent studies. Hum Reprod 2002; 17: 2307-2314.
3. Khader YS, Rice J, John L, Abueita O. Oral contraceptives use and the risk of myocardial infarction: a meta-analysis. Contraception 2003; 68: 11-17
4. Lewis MA, Heinemann LA, Spitzer WO, MacRae KD, Bruppacher R. The use of oral contraceptives and the occurrence of acute myocardial infarction in young women: results from the Transnational Study on Oral Contraceptives and the Health of Young Women. Contraception 1997; 56: 129-140.
5. Dunn NR, Thorogood M, Faragher B, Caestecker de L, MacDonald TM, McCollum C, Thomas S, Mann R. Oral contraceptives and myocardial infarction: results of the MICA case-control study. Mr Med J. 1999; 318: 1579-1584.
6. Tanis BC, van den Bosch MA, Kemmeren JM, Cats VM, Helmerhorst FM, Algra A, van der Graaf Y, Rosendaal FR. Oral contraceptives and the risk of myocardial infarction. N Engl J Med 2001; 345: 1787-1793.
7. Lidegaard O, Edstrom B. Oral contraceptives and acute myocardial infarction: a case control study. Eur J Contracept Reprod Health Care 1996; 1: 74
8. Jick H, Jick SS, Wald Myers M, Vasilakis C. Risk of acute myocardial infarction and low dose combined oral contraceptives. Lancet 1996; 347: 627-628.
9. Dunn NR, Arscott A, Thorogood M. The relationship between use of oral contraceptives and myocardial infarction in young women with fatal outcome, compared with those who survive: results from the MICA case-control study. Contraception 2001; 63: 65-69.
10. Poulter NR, Meirik O, Chang CL, Farley TMM, Kelaghan J, Marmot MG, et al. WHO Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Acute myocardial infarction and combined oral contraceptives: results of an international multi centre case control study. Lancet 1997; 349: 1202-1209.
11. Lidegaard O, Edstrom B, Kreiner S. Oral contraceptives and myocardial infarction. A five year nation-wide case-control study. Eur J. Contraception Reprod Health Care 2000; 5(supl 1): 29

18
ANEXO III
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO MODIFICADO DEL ESTADO MIEMBRO DE REFERENCIA

19
1. DENOMINACIÓN DEL MEDICAMENTO <Nombre del producto >, comprimidos recubiertos con película 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA <Nombre del producto > es un anticonceptivo oral trifásico en el que: • cada comprimido amarillo contiene 0,050 mg de desogestrel (D.O.E.) y 0,035 mg de
etinilestradiol (D.O.E.); • cada comprimido rojo contiene 0,100 mg de desogestrel (D.O.E.) y 0,030 mg de etinilestradiol
(D.O.E.); • cada comprimido blanco contiene 0,150 mg de desogestrel (D.O.E.) y 0,030 mg de
etinilestradiol (D.O.E.); Lista de excipientes en 6.1. 3. FORMA FARMACÉUTICA Comprimidos recubiertos con película. Los comprimidos son redondos, biconvexos y de 5 mm de diámetro. Están codificados VR4 (comprimidos amarillos), VR2 (comprimidos rojos) o TR5 (comprimidos blancos) en una cara y con Organon y un asterisco en la cara opuesta. 4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas Anticoncepción. 4.2 Posología y forma de administración Cómo tomar <Nombre del producto > Los comprimidos deben tomarse en el orden indicado en el envase cada día aproximadamente a la misma hora con un poco de líquido si se desea. Se toma 1 comprimido al día durante 21 días consecutivos, empezando por los comprimidos amarillos durante 7 días, siguiendo con los rojos durante 7 días y finalmente los blancos durante 7 días. Los envases sucesivos se inician inmediatamente después de dejar 7 días sin tomar los comprimidos, durante los que normalmente se presenta un sangrado por deprivación. Éste empieza generalmente en el 2º ó 3er día después de la toma del último comprimido y puede ocurrir que no haya finalizado antes de empezar el siguiente envase. Cómo empezar <Nombre del producto > Sin empleo previo de anticonceptivos hormonales (en el mes anterior) Se debe empezar a tomar los comprimidos el día 1 del ciclo natural de la mujer (es decir, el primer día de la menstruación). Puede empezarse los días 2 a 5, pero en el primer ciclo se recomienda un método de barrera complementario durante los primeros 7 días de la toma de comprimidos. Cambio desde otro anticonceptivo oral combinado La mujer debe iniciar <Nombre del producto > preferiblemente al día siguiente después del último comprimido activo de su anticonceptivo oral combinado anterior, pero como muy tarde el día después

20
del periodo de descanso o del intervalo de comprimidos inactivos de su anticonceptivo oral combinado anterior. Cambio a partir de un método con progestágeno solo (píldora con progestágeno solo, inyectable, implante) La mujer puede cambiar cualquier día en el caso de la píldora con progestágeno solo (el día de su extracción si lleva un implante, cuando corresponda la siguiente inyección en el caso del inyectable), pero en todos estos casos se debe recomendar la utilización de un método de barrera complementario durante los primeros 7 días de toma de comprimidos. Después de un aborto en el primer trimestre del embarazo La mujer puede empezar inmediatamente. En este caso, no necesita tomar medidas anticonceptivas complementarias. Tras el parto o un aborto en el segundo trimestre del embarazo En el caso de mujeres que estén dando el pecho ver el Apartado 4.6. Debe aconsejarse a la mujer que empiece entre los días 21 y 28 después del parto o de un aborto en el segundo trimestre del embarazo. Si empieza después, debe recomendarse a la mujer que utilice un método de barrera complementario durante los primeros 7 días de toma de comprimidos. Sin embargo, si ya ha tenido relaciones sexuales, debe excluirse un embarazo antes de que empiece a tomar el anticonceptivo oral combinado o bien la mujer tendrá que esperar a su primer periodo menstrual. Recomendaciones en caso de olvido de la toma de algún comprimido Si se produce un retraso menor de 12 horas en la toma de algún comprimido, la protección anticonceptiva no se ve reducida. La usuaria debe tomar el comprimido olvidado en cuanto se acuerde y tomar los siguientes de la forma y a la hora habitual. Si el retraso es mayor de 12 horas la protección anticonceptiva puede verse reducida. Las pautas de actuación para mantener la protección anticonceptiva en este caso se deben regir por las dos reglas básicas siguientes: 1. Nunca se puede dejar de tomar comprimidos durante más de 7 días. 2. Es necesario tomar los comprimidos durante 7 días consecutivos para lograr una supresión
adecuada del eje hipotálamo hipófiso ovárico. Según estas reglas, en la práctica diaria se pueden seguir las siguientes recomendaciones según se trate de: • Semana 1 (comprimidos amarillos) La usuaria debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos al mismo tiempo y continuar tomando el resto de los comprimidos a su hora habitual. Además, debe utilizar un método de barrera (el preservativo) durante los 7 días siguientes. Si mantuvo relaciones sexuales en los 7 días previos al olvido se debe tener en cuenta la posibilidad de embarazo. Este riesgo será mayor cuantos más comprimidos se hayan olvidado y cuanto más próximo esté el día del olvido al intervalo libre de toma. • Semana 2 (comprimidos rojos) La usuaria deberá tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos al mismo tiempo y continuar tomando los comprimidos a su hora habitual. Siempre que la mujer haya tomado sus comprimidos correctamente durante los 7 días previos al comprimido olvidado, no es necesario utilizar precauciones anticonceptivas adicionales. Sin embargo, si éste no es el caso, o si olvidó más de un comprimido, se debe recomendar a la mujer la utilización de precauciones adicionales durante 7 días. • Semana 3 (comprimidos blancos) El riesgo de que se reduzca la fiabilidad es elevado dada la proximidad del intervalo libre de toma. No obstante, si se modifica la pauta de administración de los comprimidos siguiendo una de las dos opciones siguientes aún se puede evitar que se reduzca la protección anticonceptiva. Además, se debe tener en cuenta que siempre que la usuaria haya tomado correctamente todos los comprimidos en los 7 días previos al primer comprimido olvidado, no es necesario adoptar medidas anticonceptivas adicionales. De

21
no ser así, se debe recomendar a la usuaria que siga la primera de estas dos opciones y tome además precauciones anticonceptivas adicionales durante los 7 días siguientes al último comprimido olvidado. 1ª. La usuaria debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si
esto significa tomar dos comprimidos al mismo tiempo y continuar tomando el resto de los comprimidos a su hora habitual. Además, debe comenzar el siguiente envase tan pronto como termine el envase actual, es decir, sin dejar un intervalo libre de toma. En este caso, no es de esperar que la usuaria presente hemorragia por deprivación hasta el final del segundo envase, pero sí puede presentar manchado o hemorragia por disrupción durante este tiempo.
2ª. También se puede aconsejar a la usuaria que deje de tomar los comprimidos del envase actual y dejar un intervalo libre de toma como máximo de 7 días contando los días en que olvidó comprimidos. Posteriormente, continuará con el siguiente envase.
Siempre que la usuaria olvide la toma de comprimidos activos y posteriormente no presente hemorragia por disrupción en el primer intervalo normal libre de toma, la posibilidad de un embarazo debe ser considerada. Recomendaciones en caso de vómito Si se produjera vómito dentro de las 3 ó 4 horas posteriores a la ingestión del comprimido, la absorción del preparado puede no ser completa. En este caso, se deben seguir las recomendaciones del apartado anterior “Recomendaciones en caso de olvido de la toma de algún comprimido”. Si la mujer no desea modificar su pauta normal de toma puede tomar los comprimidos adicionales necesarios de otro envase distinto del actual. Cómo retrasar un periodo El retraso de un periodo no es una indicación del producto. Sin embargo, si en casos excepcionales se requiere retrasar un periodo, la mujer deberá continuar con los comprimidos blancos de un envase diferente de <Nombre del producto > sin intervalo libre de toma. La prolongación puede continuarse durante un máximo de 7 días hasta el final del segundo envase. Durante la prolongación la mujer puede experimentar sangrado o manchado por disrupción. Entonces, tras el intervalo habitual de 7 días sin comprimidos se reanuda la toma regular de <Nombre del producto >. Para cambiar el periodo a otro día de la semana al que la mujer esté acostumbrada con su pauta actual, se le puede aconsejar que acorte el siguiente intervalo sin comprimidos en tantos días como desee. Cuanto más corto sea el intervalo, mayor es el riesgo de que no presente sangrado por deprivación y experimentará manchado y sangrado por disrupción durante el segundo envase (al igual que para retrasar un periodo). 4.3 Contraindicaciones Los anticonceptivos orales combinados no se deben emplear en presencia de cualquiera de las circunstancias que se indican a continuación. El tratamiento se debe interrumpir inmediatamente si aparece cualquiera de estas circunstancias durante el empleo de anticonceptivos orales combinados. • Presencia o antecedentes de trombosis venosa (trombosis venosa profunda; embolismo
pulmonar). • Presencia o antecedentes de trombosis arterial (infarto de miocardio, accidente cerebrovascular)
o pródromos de trombosis (p. ej.: trastorno isquémico agudo, angina de pecho). • Predisposición conocida a la trombosis venosa o arterial, como resistencia a la Proteína C
Activada (PCA), déficit de antitrombina III, déficit de proteína C, déficit de proteína S, hiperhomocisteinemia y anticuerpos antifosfolípidos.
• Diabetes mellitus con afectación vascular. • La presencia de un factor o factores de riesgo graves de padecer trombosis venosa o arterial
también puede constituir una contraindicación (véase el apartado “Advertencias y precauciones especiales de empleo”).
• Existencia o antecedentes de hepatopatía grave, siempre que los parámetros de función hepática no se hayan normalizado.
• Existencia o antecedentes de tumores hepáticos (benignos o malignos).

22
• Existencia o sospecha de neoplasias malignas de los órganos genitales o de la mama que sean dependientes de esteroides sexuales
• Hiperplasia endometrial. • Hemorragia vaginal sin diagnosticar. • Existencia o sospecha de embarazo. • Hipersensibilidad a alguno de los principios activos o excipientes de <Nombre del producto >. 4.4 Advertencias y precauciones especiales de empleo Advertencias Si está presente cualquiera de las circunstancias o factores de riesgo mencionados más adelante se deben sopesar en cada caso concreto los efectos beneficiosos del empleo de anticonceptivos orales combinados frente a los posibles riesgos, y comentar estos aspectos con la mujer antes de que decida comenzar a emplearlos. Si se agrava o agudiza alguna de estas circunstancias o factores de riesgo durante el empleo de anticonceptivos orales combinados, o si aparece por primera vez, la usuaria debe comunicárselo a su médico. El médico debe ser quien decida si el empleo de anticonceptivos orales combinados debe ser interrumpido. 1. Alteraciones de la circulación • El uso de cualquier anticonceptivo oral combinado lleva asociado un incremento del riesgo de
tromboembolismo venoso (TEV), comparado con la no utilización. La incidencia de TEV se considera de 5-10 por 100.000 mujeres-año en no usuarias de anticonceptivos orales. El riesgo adicional de TEV es mayor durante el primer año de uso. Este incremento del riesgo es menor que el riesgo de TEV asociado con el embarazo, que se estima en 60 casos por 100.000 embarazos. El TEV es fatal en 1-2% de los casos.
• En varios estudios epidemiológicos se ha encontrado que las mujeres que utilizan anticonceptivos orales combinados que contienen etinilestradiol (generalmente a dosis de 30 µg) y un progestágeno como el desogestrel tienen un mayor riesgo de TEV comparado con las que utilizan anticonceptivos orales combinados que contienen menos de 50 µg de etinilestradiol y el progestágeno levonorgestrel.
• Para los preparados que contienen 30 µg de etinilestradiol asociado a desogestrel o gestodeno en comparación con los preparados que contienen menos de 50 µg de etinilestradiol y levonorgestrel, el riesgo relativo global de TEV se ha estimado entre 1,5 y 2,0. La incidencia de TEV en los preparados que contienen levonorgestrel combinado con menos de 50 µg de etinilestradiol es de aproximadamente de 20 casos por 100.000 mujeres-año de uso. Para <Nombre del producto > la incidencia será aproximadamente de 30-40 casos por 100.000 mujeres-año de uso, lo que significa 10-20 casos adicionales por 100.000 mujeres-año de uso. El impacto del riesgo relativo en el número de casos adicionales es mayor en las mujeres que utilizan anticonceptivos orales combinados por primera vez, cuando el riesgo de TEV con todos los anticonceptivos orales combinados es más alto.
• El riesgo de padecer tromboembolismo venoso aumenta con: • edad avanzada; • existencia de antecedentes familiares (es decir, algún tromboembolismo venoso en un hermano
o en uno de los padres a edad relativamente joven). Si se sospecha la existencia de una predisposición hereditaria se debe remitir a la mujer a un especialista para recibir asesoramiento antes de decidir sobre el empleo de cualquier anticonceptivo hormonal;
• obesidad (índice de masa corporal mayor de 30 kg/m2); • inmovilización prolongada; cirugía mayor; cualquier cirugía de miembros inferiores o
traumatismos importantes. En estas situaciones es recomendable suspender el empleo de anticonceptivos orales combinados (al menos con cuatro semanas de anticipación en el caso de cirugía programada) y no volver a emplearlos hasta dos semanas después de recuperar la movilidad por completo;

23
• y posiblemente también con la tromboflebitis superficial y las venas varicosas. No existe acuerdo acerca del posible papel de estos trastornos en la etiología del tromboembolismo venoso.
• El uso de anticonceptivos orales combinados en general se ha asociado a un incremento del riesgo de infarto agudo de miocardio (IAM) o accidente cerebrovascular, un riesgo que está muy influido por la presencia de otros factores de riesgo (como por ejemplo el tabaquismo, la hipertensión y la edad, ver también más adelante). Estas patologías se producen raramente. No se ha estudiado como modifica <Nombre del producto > el riesgo de IAM.
• El riesgo de padecer complicaciones tromboembólicas arteriales aumenta con: • edad avanzada; • tabaquismo (el riesgo se incrementa con la cantidad de tabaco y con la edad, especialmente en
mujeres mayores de 35 años); • dislipoproteinemia; • obesidad (índice de masa corporal mayor de 30 kg/m2); • hipertensión; • valvulopatía cardiaca; • fibrilación auricular; • antecedentes familiares de trombosis arterial (es decir, trombosis arterial en un hermano o en
uno de los padres a edad relativamente joven). Si se sospecha la existencia de una predisposición hereditaria se debe remitir a la mujer a un especialista para recibir asesoramiento antes de decidir sobre el empleo de cualquier anticonceptivo hormonal.
• En usuarias de anticonceptivos orales combinados se han comunicado casos extraordinariamente raros de trombosis en otros vasos sanguíneos como arterias y venas hepáticas, mesentéricas, renales o retinianas. No existe acuerdo sobre si la aparición de estos casos está asociada con el empleo de anticonceptivos orales combinados.
• Los síntomas de trombosis venosa o arterial pueden ser: dolor y/o hinchazón unilateral en miembros inferiores; dolor intenso y de aparición brusca en el tórax con o sin irradiación a miembro superior izquierdo; dificultad respiratoria súbita; tos de comienzo brusco; cefalea no habitual de cualquier tipo intensa o prolongada; pérdida repentina de visión parcial o completa; diplopía; lenguaje ininteligible o afasia; vértigo; síncope con o sin epilepsia focal; pérdida de fuerza o entumecimiento marcado de aparición brusca que afecta a un hemicuerpo o a una extremidad; trastornos motores; abdomen agudo.
• Otras patologías que se han asociado con la aparición de acontecimientos adversos vasculares son: diabetes mellitus; lupus eritematoso sistémico; síndrome hemolítico urémico; enfermedad intestinal inflamatoria crónica (enfermedad de Crohn y colitis ulcerosa); y anemia de células falciformes.
• Debe tenerse en cuenta que el riesgo de padecer enfermedades tromboembólicas puede estar aumentado en el puerperio (véase el apartado 4.6 “Embarazo y lactancia”).
• El aumento de la frecuencia o de la intensidad de las migrañas durante el empleo de anticonceptivos orales combinados pueden ser pródromos de un accidente cerebrovascular, y por tanto, pueden ser una razón para suspender de forma inmediata el tratamiento.
• Los factores bioquímicos que pueden indicar una predisposición hereditaria o adquirida a padecer trombosis venosa o arterial son: resistencia a la proteína C activada (PCA); hiperhomocisteinemia; déficit de antitrombina III; déficit de proteína C; déficit de proteína S; y presencia de anticuerpos antifosfolípidos (anticuerpos anticardiolipina o anticoagulante lúpico).
• A la hora de considerar la relación riesgo/beneficio del empleo de anticonceptivos orales combinados el médico debe tener en cuenta que el tratamiento adecuado de una situación puede reducir el riesgo asociado de trombosis, y que el riesgo de trombosis asociado con el embarazo es mayor que el asociado con el empleo de anticonceptivos orales combinados.
2. Tumores • Algunos estudios epidemiológicos han informado de un aumento del riesgo de padecer cáncer
cervical en usuarias de anticonceptivos orales combinados a largo plazo. No obstante, continúa existiendo controversia acerca del grado en que este hallazgo es atribuible al efecto producido

24
por factores de confusión como son los hábitos sexuales y otros factores como el virus del papiloma humano (VPH).
• Un metanálisis realizado sobre 54 estudios epidemiológicos ha informado que existe un ligero incremento del riesgo relativo (RR=1,24) de que se diagnostique cáncer de mama entre mujeres que emplean anticonceptivos orales. Este incremento desaparece gradualmente durante los 10 años posteriores a haber dejado de emplear anticonceptivos orales. El cáncer de mama es raro entre mujeres con edad menor de 40 años, por lo que el aumento en el número de cánceres de mama diagnosticados entre mujeres usuarias actuales o recientes de anticonceptivos orales es pequeño en relación con el riesgo global de padecer cáncer de mama. Estos estudios no constituyen una evidencia de relación causal. El incremento observado puede ser debido: a un diagnóstico más precoz entre las usuarias de anticonceptivos orales; al efecto biológico de los anticonceptivos orales; o a una combinación de ambos factores. Por otro lado, los cánceres de mama diagnosticados entre usuarias de anticonceptivos orales tienden a ser menos avanzados clínicamente que los cánceres diagnosticados entre las mujeres que no los han empleado nunca.
• En raros casos se han observado tumores hepáticos benignos y, aún más raramente malignos en usuarias de anticonceptivos orales combinados. En casos aislados, estos tumores han originado hemorragia intraabdominal con peligro de muerte. Si se presentase dolor epigástrico intenso, aumento del tamaño del hígado o signos de hemorragia intraabdominal, el diagnóstico diferencial debe contemplar la posibilidad de existencia de un tumor hepático.
3. Otras patologías • En las mujeres con hipertrigliceridemia o antecedentes familiares de la misma puede existir un
aumento del riesgo de padecer pancreatitis durante el empleo de anticonceptivos orales combinados.
• Durante el empleo de anticonceptivos orales combinados se ha observado que muchas usuarias presentan pequeños aumentos de la tensión arterial, aunque raramente estos incrementos de la tensión tienen una repercusión clínica importante. No se ha establecido una relación entre el empleo de anticonceptivos orales combinados y el desarrollo de hipertensión arterial clínica. Sin embargo, si durante el empleo de un anticonceptivo oral combinado se presenta hipertensión mantenida de importancia clínica es prudente que el médico recomiende suspender la toma del anticonceptivo y tratar la hipertensión. Cuando se considere oportuno se puede reanudar el empleo de anticonceptivos orales combinados si se han alcanzado valores de tensión arterial normales con el tratamiento antihipertensivo.
• Los siguientes procesos pueden aparecer o agravarse en el curso del embarazo y durante el empleo de anticonceptivos orales combinados: ictericia y/o prurito por colestasis; formación de cálculos biliares; porfiria; lupus eritematoso sistémico; síndrome urémico hemolítico; corea de Sydenham; herpes gestacional; pérdida de audición por otosclerosis. No obstante, la evidencia no es concluyente respecto a la asociación entre estos procesos y el empleo de anticonceptivos orales combinados.
• Las alteraciones agudas o crónicas de la función hepática pueden requerir la interrupción del empleo de anticonceptivos orales combinados hasta que los marcadores de la función hepática se normalicen. La reaparición de una ictericia colestática que se presentó por primera vez durante un embarazo o coincidiendo con el empleo previo de esteroides sexuales requiere la suspensión del empleo de los anticonceptivos orales combinados.
• Los anticonceptivos orales combinados pueden alterar la resistencia periférica a la insulina y la tolerancia a la glucosa. Sin embargo, no existe evidencia de que sea necesario modificar los requerimientos de antidiabéticos orales o de insulina en las mujeres diabéticas que emplean anticonceptivos orales combinados. No obstante, estas mujeres requieren supervisión médica estricta durante el empleo de anticonceptivos orales combinados.
• El desarrollo de la enfermedad de Crohn y de la colitis ulcerosa se ha asociado con el empleo de anticonceptivos orales combinados.
• Ocasionalmente, durante el empleo de anticonceptivos orales combinados se puede presentar cloasma sobre todo en las mujeres con antecedentes personales de cloasma gravídico. Las

25
mujeres con tendencia a presentar cloasma deben evitar la exposición al sol o a las radiaciones ultravioleta mientras estén empleando anticonceptivos orales combinados.
Toda esta información debe tenerse en cuenta al prescribir este anticonceptivo oral combinado y al recomendar la elección de método(s) anticonceptivos. Examen médico y revisiones Antes de iniciar o de reanudar el uso de <Nombre del producto > se debe realizar una historia clínica completa (incluyendo antecedentes familiares) y excluir el embarazo. Se valorará la presión arterial y se realizará una exploración física orientada según las contraindicaciones (apartado 4.3) y las advertencias (apartado 4.4). Asimismo, se recomendará a la mujer que lea con atención el prospecto y seguir las recomendaciones que se dan en el mismo. La frecuencia y naturaleza de los siguientes controles periódicos deberá basarse en los protocolos establecidos y adaptándolos a las necesidades clínicas de cada caso. Se debe recordar a las usuarias de anticonceptivos orales que éstos no protegen de la infección por el VIH (SIDA) ni de otras enfermedades de transmisión sexual. Reducción de la eficacia La eficacia de los anticonceptivos orales combinados puede verse reducida si la usuaria se olvida de tomar algún comprimido (véase el apartado 4.2), presenta vómitos (véase el apartado 4.2) o toma algún medicamento concomitante (véase el apartado 4.5). No deben tomarse preparaciones a base de plantas medicinales con Hierba de San Juan (Hypericum perforatum) simultáneamente con <Nombre del producto > debido al riesgo de disminución de los niveles plasmáticos y reducción de los efectos clínicos de <Nombre del producto > (véase el apartado 4.5 Interacciones). Irregularidades en el control del ciclo Durante el empleo de cualquier anticonceptivo oral combinado puede ocurrir que la usuaria presente sangrado intermenstrual (manchado o hemorragia por disrupción) sobre todo durante los primeros meses de empleo. Por tanto, para la evaluación de cualquier sangrado irregular se debe tener en cuenta que sólo es relevante si éste ocurre después de un intervalo de adaptación de unos tres ciclos. Si las irregularidades en el sangrado persisten o se presentan después de ciclos regulares previos deben considerarse las causas no hormonales y está indicado aplicar medidas diagnósticas adecuadas, para descartar una enfermedad maligna o un embarazo, lo que puede incluir legrado. En algunas mujeres puede no presentarse la hemorragia por deprivación durante el intervalo sin comprimidos. Si se ha tomado el anticonceptivo oral combinado según las instrucciones del apartado 4.2 “Posología y forma de administración” es improbable que la usuaria esté embarazada. Sin embargo, si el anticonceptivo no se ha tomado siguiendo estas instrucciones antes de producirse la primera falta, o si se han producido dos faltas seguidas, se debe descartar la existencia de embarazo antes de continuar con el empleo del anticonceptivo oral combinado. 4.5 Interacción con otros medicamentos y otras formas de interacción Interacciones Las interacciones farmacológicas que producen un aumento del aclaramiento plasmático de las hormonas sexuales pueden dar lugar a que se presente hemorragia por disrupción y a fallo del anticonceptivo. Este hecho ha quedado demostrado con los siguientes fármacos: hidantoínas; barbitúricos; primidona; carbamazepina; y rifampicina. También se sospecha de esta forma de interacción con: oxcarbazepina; topiramato; felbamato; ritonavir y griseofulvina. El mecanismo de esta interacción parece basarse en la propiedad inductora que poseen estos fármacos sobre los enzimas

26
hepáticos. Generalmente, la inducción enzimática máxima no se observa hasta transcurridas 2 ó 3 semanas de la utilización de estos fármacos, pero después puede mantenerse durante al menos 4 semanas una vez suspendido el tratamiento. También se han comunicado fallos anticonceptivos por la administración concomitante de ciertos antibióticos como las ampicilinas y las tetraciclinas. El mecanismo de esta interacción todavía no se conoce con certeza. Las usuarias en tratamiento de corto plazo con los tipos de fármacos o con uno cualquiera de los fármacos antes indicados deben continuar la toma del anticonceptivo oral combinado y además, utilizar temporalmente un método de barrera (el preservativo), es decir, durante el tiempo que se administre concomitantemente el fármaco y durante los 7 días posteriores a su suspensión. Las usuarias en tratamiento con rifampicina deben continuar la toma del anticonceptivo oral combinado y además, utilizar un método de barrera (el preservativo) durante el tiempo en que se administre la rifampicina y durante los 28 días siguientes a su suspensión. Si la administración del fármaco concomitante continuara después de terminar los comprimidos del envase actual, se debe iniciar el siguiente envase sin dejar el periodo de descanso habitual. Algunos expertos han recomendado aumentar la dosis de los esteroides anticonceptivos en las usuarias que reciben tratamiento a largo plazo con fármacos inductores de los enzimas hepáticos. Si no es apropiado administrar una dosis elevada de anticonceptivo, o este aumento resulta insatisfactorio o ineficaz (p. ej.: en caso de presentarse sangrado irregular), se debe recomendar a la usuaria utilizar otro método de anticoncepción. Los preparados a base de plantas medicinales con Hierba de San Juan (Hypericum perforatum) no deben tomarse de forma concomitante con anticonceptivos orales, ya que potencialmente podría causar una pérdida del efecto anticonceptivo. Se han citado sangrado por disrupción y embarazos no previstos. Este efecto es debido a la inducción de enzimas que metabolizan fármacos por la Hierba de San Juan. Este efecto inductor puede persistir durante al menos 2 semanas después de dejar el tratamiento con Hierba de San Juan. Pruebas analíticas La utilización de anticonceptivos orales puede influir sobre los resultados de ciertas pruebas de laboratorio, incluyendo parámetros bioquímicos de función hepática, tiroidea, suprarrenal y renal, niveles plasmáticos de proteínas transportadoras (p. ej.: globulina transportadora de corticosteroides o fracciones lipídicas y lipoproteicas), parámetros del metabolismo de carbohidratos y parámetros de la coagulación y fibrinolisis. Por lo general, estas variaciones se mantienen dentro de los rangos de normalidad de laboratorio. 4.6 Embarazo y lactancia Los anticonceptivos orales combinados no están indicados durante el embarazo. En la mayoría de estudios epidemiológicos sobre anticonceptivos orales combinados no se ha encontrado un aumento del riesgo de padecer defectos congénitos en los hijos de mujeres que han empleado anticonceptivos orales antes de la gestación. Tampoco se ha informado de ningún efecto teratogénico por la toma de anticonceptivos orales combinados de forma inadvertida durante las fases precoces del embarazo. La lactancia puede verse influida por el empleo de anticonceptivos orales combinados ya que éstos pueden reducir la cantidad de la leche materna y modificar su composición. Por lo general, no se debe recomendar el empleo de anticonceptivos orales combinados hasta finalizar completamente el periodo de lactancia materna. Con la leche materna pueden excretarse pequeñas cantidades de esteroides anticonceptivos y/o de sus metabolitos, sin embargo, no se ha demostrado que esto influya de forma adversa sobre la salud del lactante. 4.7 Efectos sobre la capacidad de conducir y utilizar máquinas

27
No se han observado efectos sobre la capacidad para conducir y utilizar máquinas. 4.8 Reacciones adversas Reacciones adversas graves Existe un incremento del riesgo de tromboembolismo venoso en todas las mujeres que toman un anticonceptivo oral combinado. En el apartado 4.4 se proporciona información sobre las diferencias de riesgo entre anticonceptivos orales combinados, y sobre otras reacciones adversas graves. Otras reacciones adversas posibles Se han citado las siguientes reacciones adversas en usuarias de anticonceptivos orales combinados: Mamas hipersensibilidad, dolor y secreción; Sistema Nervioso Central cefalea, jaqueca, cambios en la libido, estado de ánimo depresivo; Ojos intolerancia a las lentes de contacto Tracto gastrointestinal náuseas, vómitos; Tracto urogenital cambios en la secreción vaginal; Piel alteraciones cutáneas varias (por ej. eritema nudoso, eritema
multiforme, fotosensibilidad, urticaria); Otras alteraciones retención de líquidos, modificaciones del peso corporal, reacciones de
hipersensibilidad. 4.9 Sobredosis No se ha informado de efectos adversos graves tras sobredosis. Los síntomas que pueden aparecer en este caso son náuseas o vómitos. En niñas puede producirse una ligera hemorragia vaginal. No existen antídotos y el tratamiento debe ser sintomático. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Clasificación ATC G03AB05 El efecto anticonceptivo de los anticonceptivos orales combinados se basa en la interacción de diversos factores de los que los más importantes son la inhibición de la ovulación y las modificaciones de la secreción cervical. Además de proteger contra el embarazo, los anticonceptivos orales combinados producen efectos beneficiosos no anticonceptivos que, aún considerando los posibles efectos indeseables (véanse los apartados: “Advertencias” y “Reacciones adversas”), pueden ser útiles a la hora de decidir sobre el método de control de la fertilidad. Durante el empleo de anticonceptivos orales combinados los ciclos son más regulares y la menstruación suele ser menos dolorosa y con menor cantidad de sangrado. Esto último puede dar lugar a una menor incidencia de casos de mujeres con deficiencia de hierro. Al margen de esto, con el empleo de los anticonceptivos orales combinados de mayor dosis (50 µg de etinilestradiol) se ha demostrado una reducción del riesgo de padecer: tumores fibroquísticos de mama; quistes ováricos; enfermedad inflamatoria pélvica; embarazo ectópico; cáncer de endometrio; y cáncer de ovario. Todavía no se ha confirmado si estas conclusiones pueden aplicarse a los anticonceptivos orales combinados con menor dosificación. En estudios clínicos se ha demostrado que <Nombre del producto > redujo significativamente los parámetros androgénicos 3-α androstenodiol-glucurónido, androstenodiona, DHEA-S y testosterona libre. 5.2 Propiedades farmacocinéticas Desogestrel Absorción

28
El desogestrel administrado por vía oral se absorbe y convierte en etonogestrel de forma rápida y completa. Los niveles séricos máximos del fármaco son aproximadamente de 1,5 ng/ml (primera semana) a 5 ng/ml (tercera semana) y se alcanzan aproximadamente al cabo de 1,5 horas. La biodisponibilidad es del 62 – 81%. Distribución El etonogestrel se une a la albúmina y a la globulina transportadora de hormonas sexuales (SHBG). Tan sólo de un 2 a 4% de los niveles séricos totales del fármaco están presentes como esteroide libre, pero el 40 - 70% se une específicamente a la SHBG. La distribución en las proteínas plasmáticas depende del incremento de las concentraciones de SHBG inducidas por etinilestradiol, causando un aumento de la fracción unida a la SHBG, mientras que disminuye la fracción unida a la albúmina. El volumen de distribución aparente del desogestrel es 1,5 l/kg. Metabolismo El etonogestrel se metaboliza por completo a través de las rutas metabólicas conocidas para los esteroides. La velocidad de aclaramiento metabólico sérico es de aproximadamente 2 ml/min/kg. No se ha encontrado interacción alguna con la administración concomitante de etinilestradiol. Eliminación Las concentraciones séricas de etonogestrel disminuyen en dos fases. La fase de disposición final se caracteriza por una semivida de aproximadamente 30 horas. El desogestrel y sus metabolitos se excretan por vía urinaria y biliar con un cociente de aproximadamente 6:4. Estado de equilibrio La farmacocinética del etonogestrel está influida por los niveles de SHBG, que se incrementan 3 veces con el etinilestradiol. Con la administración diaria, las concentraciones séricas del fármaco aumentan aproximadamente de 2 a 3 veces, alcanzando un estado de equilibrio durante la segunda mitad del ciclo de tratamiento. Etinilestradiol Absorción El etinilestradiol administrado por vía oral se absorbe de forma rápida y completa. Las concentraciones séricas máximas en torno a 80 pg/ml se alcanzan al cabo de 1 a 2 horas. La biodisponibilidad absoluta resultante de la conjugación presistémica y del metabolismo de primer paso es de aproximadamente 60%. Distribución El etinilestradiol se une en gran medida, pero inespecíficamente, a la albúmina sérica (aproximadamente 98,5%) e induce un incremento de las concentraciones séricas de SHBG. Se ha determinado un volumen de distribución aparente del etinilestradiol de aproximadamente 5 l/kg. Metabolismo El etinilestradiol experimenta conjugación presistémica en la mucosa del intestino delgado y en el hígado. Se metaboliza primero por hidroxilación aromática, formándose gran variedad de metabolitos metilados e hidroxilados, que se presentan como metabolitos libres y como conjugados con glucurónidos y sulfatos. La velocidad de aclaramiento metabólico es de aproximadamente 5 ml/min/kg. Eliminación Los niveles séricos de etinilestradiol disminuyen en dos fases. La fase de disposición final se caracteriza por una semivida de aproximadamente 24 horas. El fármaco inalterado no se excreta. Los metabolitos del etinilestradiol se excretan por vía urinaria y biliar con un cociente de 4:6 y con una semivida de aproximadamente 1 día. Estado de equilibrio

29
Las concentraciones de estado de equilibrio se alcanzan al cabo de 3 a 4 días cuando los niveles séricos de fármaco son de un 30% a un 40% más altos que si se administra una dosis única. 5.3 Datos preclínicos sobre seguridad Los datos preclínicos no han revelado ningún peligro especial para los humanos si se usan los anticonceptivos orales combinados de la forma recomendada. Esto se basa en estudios convencionales de toxicidad a dosis repetidas, toxicidad génica, potencial carcinogénico y toxicidad en la reproducción. Sin embargo, hay que tener presente que los esteroides sexuales podrían estimular el crecimiento de ciertos tejidos y tumores dependientes de hormonas. 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Núcleo α-tocoferol, lactosa monohidrato, almidón de patata, povidona, dióxido de silicio coloidal anhidro y ácido esteárico. Recubrimiento Para todos los comprimidos: Hipromelosa, macrogol 400, talco, dióxido de titanio (E171) y además: Óxido férrico amarillo (E172) para los comprimidos con 0,050 mg de desogestrel/0,035 mg de etinilestradiol. Óxido férrico rojo (E172) para los comprimidos con 0,100 mg de desogestrel/0,030 mg de etinilestradiol. 6.2 Incompatibilidades No aplicable. 6.3 Periodo de validez 3 años. 6.4 Precauciones especiales de conservación No conservar a temperatura superior a 30ºC. Conservar en el envase original. 6.5 Naturaleza y contenido del recipiente El blister es de aluminio/PVC y consiste en una lámina de aluminio con una capa para sellado por calor y una película de PVC. Cada blister contiene 21 comprimidos y se acondiciona en un sobre de aluminio impreso. El sobre se envasa en cajas de cartón impresas con 1, 3 ó 6 sobres por caja, junto con un prospecto. No todas las presentaciones se encuentran comercializadas. 6.6 Instrucciones de uso, manipulación y eliminación Ninguna especial. 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN 8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN

30
9. FECHA DE LA REVALIDACIÓN DE LA AUTORIZACIÓN 10. FECHA DE LA REVISIÓN DEL TEXTO

31
1. DENOMINACIÓN DEL MEDICAMENTO <Nombre del producto >, comprimidos recubiertos con película 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA <Nombre del producto > es un anticonceptivo oral trifásico en el que: • cada comprimido amarillo contiene 0,050 mg de desogestrel (D.O.E.) y 0,035 mg de
etinilestradiol (D.O.E.); • cada comprimido rojo contiene 0,100 mg de desogestrel (D.O.E.) y 0,030 mg de etinilestradiol
(D.O.E.); • cada comprimido blanco contiene 0,150 mg de desogestrel (D.O.E.) y 0,030 mg de
etinilestradiol (D.O.E.); • los comprimidos verdes no contienen principios activos (comprimidos inactivos). Lista de excipientes en 6.1. 3. FORMA FARMACÉUTICA Comprimidos recubiertos con película. Los comprimidos son redondos, biconvexos y de 5 mm de diámetro. Están codificados VR4 (comprimidos amarillos), VR2 (comprimidos rojos), TR5 (comprimidos blancos) o KH sobre 2 (comprimidos verdes) en una cara y con Organon y un asterisco en la cara opuesta. 4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas Anticoncepción. 4.2 Posología y forma de administración Cómo tomar <Nombre del producto > Los comprimidos deben tomarse en el orden indicado en el envase cada día aproximadamente a la misma hora con un poco de líquido si se desea. Se toma 1 comprimido al día durante 28 días consecutivos, empezando por los comprimidos amarillos durante 7 días, siguiendo con los rojos durante 7 días, los blancos durante 7 días y finalmente los verdes (inactivos) durante 7 días. Los envases sucesivos se inician inmediatamente después del último comprimido inactivo. Durante los días de toma de comprimidos inactivos normalmente se presenta un sangrado por deprivación. Éste empieza generalmente en el 2º ó 3er día después de la toma del último comprimido activo y puede ocurrir que no haya finalizado antes de empezar el siguiente envase. Cómo empezar <Nombre del producto > Sin empleo previo de anticonceptivos hormonales (en el mes anterior) Se debe empezar a tomar los comprimidos el día 1 del ciclo natural de la mujer (es decir, el primer día de la menstruación). Puede empezarse los días 2 a 5, pero en el primer ciclo se recomienda un método de barrera complementario durante los primeros 7 días de la toma de comprimidos. Cambio desde otro anticonceptivo oral combinado La mujer debe iniciar <Nombre del producto > preferiblemente al día siguiente después del último comprimido activo de su anticonceptivo oral combinado anterior, pero como muy tarde el día después

32
del periodo de descanso o del intervalo de comprimidos inactivos de su anticonceptivo oral combinado anterior. Cambio a partir de un método con progestágeno solo (píldora con progestágeno solo, inyectable, implante) La mujer puede cambiar cualquier día en el caso de la píldora con progestágeno solo (el día de su extracción si lleva un implante, cuando corresponda la siguiente inyección en el caso del inyectable), pero en todos estos casos se debe recomendar la utilización de un método de barrera complementario durante los primeros 7 días de toma de comprimidos. Después de un aborto en el primer trimestre del embarazo La mujer puede empezar inmediatamente. En este caso, no necesita tomar medidas anticonceptivas complementarias. Tras el parto o un aborto en el segundo trimestre del embarazo En el caso de mujeres que estén dando el pecho ver el Apartado 4.6. Debe aconsejarse a la mujer que empiece entre los días 21 y 28 después del parto o de un aborto en el segundo trimestre del embarazo. Si empieza después, debe recomendarse a la mujer que utilice un método de barrera complementario durante los primeros 7 días de toma de comprimidos. Sin embargo, si ya ha tenido relaciones sexuales, debe excluirse un embarazo antes de que empiece a tomar el anticonceptivo oral combinado o bien la mujer tendrá que esperar a su primer periodo menstrual. Recomendaciones en caso de olvido de la toma de algún comprimido Si se produce un retraso menor de 12 horas en la toma de algún comprimido, la protección anticonceptiva no se ve reducida. La usuaria debe tomar el comprimido olvidado en cuanto se acuerde y tomar los siguientes de la forma y a la hora habitual. Si el retraso es mayor de 12 horas la protección anticonceptiva puede verse reducida. Las pautas de actuación para mantener la protección anticonceptiva en este caso se deben regir por las dos reglas básicas siguientes: 1. Nunca se puede dejar de tomar comprimidos activos durante más de 7 días. 2. Es necesario tomar los comprimidos activos durante 7 días consecutivos para lograr una
supresión adecuada del eje hipotálamo hipófiso ovárico. Según estas reglas, en la práctica diaria se pueden seguir las siguientes recomendaciones según se trate de: • Semana 1 (comprimidos amarillos) La usuaria debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos al mismo tiempo y continuar tomando el resto de los comprimidos a su hora habitual. Además, debe utilizar un método de barrera (el preservativo) durante los 7 días siguientes. Si mantuvo relaciones sexuales en los 7 días previos al olvido se debe tener en cuenta la posibilidad de embarazo. Este riesgo será mayor cuantos más comprimidos se hayan olvidado y cuanto más próximo esté el día del olvido al intervalo de comprimidos inactivos. • Semana 2 (comprimidos rojos) La usuaria deberá tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos al mismo tiempo y continuar tomando los comprimidos a su hora habitual. Siempre que la mujer haya tomado sus comprimidos correctamente durante los 7 días previos al comprimido olvidado, no es necesario utilizar precauciones anticonceptivas adicionales. Sin embargo, si éste no es el caso, o si olvidó más de un comprimido, se debe recomendar a la mujer la utilización de precauciones adicionales durante 7 días. • Semana 3 (comprimidos blancos) El riesgo de que se reduzca la fiabilidad es elevado dada la proximidad del intervalo de comprimidos inactivos. No obstante, si se modifica la pauta de administración de los comprimidos siguiendo una de las dos opciones siguientes aún se puede evitar que se reduzca la protección anticonceptiva. Además, se debe tener en cuenta que siempre que la usuaria haya tomado correctamente todos los comprimidos en los 7 días previos al primer comprimido olvidado, no es necesario adoptar medidas anticonceptivas adicionales. De no ser así, se debe recomendar a la usuaria que siga la primera de estas dos opciones y

33
tome además precauciones anticonceptivas adicionales durante los 7 días siguientes al último comprimido olvidado. 1a. La usuaria debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si
esto significa tomar dos comprimidos al mismo tiempo y continuar tomando el resto de los comprimidos a su hora habitual. Además, debe comenzar el siguiente envase en cuanto termine los comprimidos activos del envase actual, es decir, sin tomar los comprimidos inactivos. En este caso, no es de esperar que la usuaria presente hemorragia por deprivación hasta el intervalo de comprimidos inactivos del segundo envase, pero sí puede presentar manchado o hemorragia por disrupción durante los días que tome comprimidos activos.
2a. También se puede aconsejar a la usuaria que deje de tomar los comprimidos activos del envase actual y continuar con los comprimidos inactivos. El número total de comprimidos olvidados más los comprimidos inactivos nunca debe exceder de 7. Posteriormente, continuará con el siguiente envase.
• Semana 4 (comprimidos verdes) La protección anticonceptiva no se ve reducida. La mujer deberá tomar los siguientes comprimidos a la hora habitual. Siempre que la usuaria olvide la toma de comprimidos activos y posteriormente no presente hemorragia por disrupción en el primer intervalo normal de comprimidos inactivos, la posibilidad de un embarazo debe ser considerada. Recomendaciones en caso de vómito Si se produjera vómito dentro de las 3 ó 4 horas posteriores a la ingestión del comprimido, la absorción del preparado puede no ser completa. En este caso, se deben seguir las recomendaciones del apartado anterior “Recomendaciones en caso de olvido de la toma de algún comprimido”. Si la mujer no desea modificar su pauta normal de toma puede tomar los comprimidos adicionales necesarios de otro envase distinto del actual. Cómo retrasar un periodo El retraso de un periodo no es una indicación del producto. Sin embargo, si en casos excepcionales se requiere retrasar un periodo, la mujer deberá continuar con los comprimidos blancos de un envase diferente de <Nombre del producto > sin intervalo de comprimidos inactivos. La prolongación puede continuarse durante un máximo de 7 días (los comprimidos blancos del segundo envase). Durante la prolongación la mujer puede experimentar sangrado o manchado por disrupción. Entonces, tras el intervalo habitual de 7 días de comprimidos inactivos se reanuda la toma regular de <Nombre del producto >. Para cambiar el periodo a otro día de la semana al que la mujer esté acostumbrada con su pauta actual, se le puede aconsejar que acorte el siguiente intervalo de comprimidos inactivos en tantos días como desee. Cuanto más corto sea el intervalo, mayor es el riesgo de que no presente sangrado por deprivación y experimentará manchado y sangrado por disrupción durante el segundo envase (al igual que para retrasar un periodo). 4.3 Contraindicaciones Los anticonceptivos orales combinados no se deben emplear en presencia de cualquiera de las circunstancias que se indican a continuación. El tratamiento se debe interrumpir inmediatamente si aparece cualquiera de estas circunstancias durante el empleo de anticonceptivos orales combinados. • Presencia o antecedentes de trombosis venosa (trombosis venosa profunda; embolismo
pulmonar). • Presencia o antecedentes de trombosis arterial (infarto de miocardio, accidente cerebrovascular)
o pródromos de trombosis (p. ej.: trastorno isquémico agudo, angina de pecho). • Predisposición conocida a la trombosis venosa o arterial, como resistencia a la Proteína C
Activada (PCA), déficit de antitrombina III, déficit de proteína C, déficit de proteína S, hiperhomocisteinemia y anticuerpos antifosfolípidos.
• Diabetes mellitus con afectación vascular.

34
• La presencia de un factor o factores de riesgo graves de padecer trombosis venosa o arterial también puede constituir una contraindicación (véase el apartado “Advertencias y precauciones especiales de empleo”).
• Existencia o antecedentes de hepatopatía grave, siempre que los parámetros de función hepática no se hayan normalizado.
• Existencia o antecedentes de tumores hepáticos (benignos o malignos). • Existencia o sospecha de neoplasias malignas de los órganos genitales o de la mama que sean
dependientes de esteroides sexuales • Hiperplasia endometrial. • Hemorragia vaginal sin diagnosticar. • Existencia o sospecha de embarazo. • Hipersensibilidad a alguno de los principios activos o excipientes de <Nombre del producto >. 4.4 Advertencias y precauciones especiales de empleo Advertencias Si está presente cualquiera de las circunstancias o factores de riesgo mencionados más adelante se deben sopesar en cada caso concreto los efectos beneficiosos del empleo de anticonceptivos orales combinados frente a los posibles riesgos, y comentar estos aspectos con la mujer antes de que decida comenzar a emplearlos. Si se agrava o agudiza alguna de estas circunstancias o factores de riesgo durante el empleo de anticonceptivos orales combinados, o si aparece por primera vez, la usuaria debe comunicárselo a su médico. El médico debe ser quien decida si el empleo de anticonceptivos orales combinados debe ser interrumpido. 1. Alteraciones de la circulación • El uso de cualquier anticonceptivo oral combinado lleva asociado un incremento del riesgo de
tromboembolismo venoso (TEV), comparado con la no utilización. La incidencia de TEV se considera de 5-10 por 100.000 mujeres-año en no usuarias de anticonceptivos orales. El riesgo adicional de TEV es mayor durante el primer año de uso. Este incremento del riesgo es menor que el riesgo de TEV asociado con el embarazo, que se estima en 60 casos por 100.000 embarazos. El TEV es fatal en 1-2% de los casos.
• En varios estudios epidemiológicos se ha encontrado que las mujeres que utilizan anticonceptivos orales combinados que contienen etinilestradiol (generalmente a dosis de 30 µg) y un progestágeno como el desogestrel tienen un mayor riesgo de TEV comparado con las que utilizan anticonceptivos orales combinados que contienen menos de 50 µg de etinilestradiol y el progestágeno levonorgestrel.
• Para los preparados que contienen 30 µg de etinilestradiol asociado a desogestrel o gestodeno en comparación con los preparados que contienen menos de 50 µg de etinilestradiol y levonorgestrel, el riesgo relativo global de TEV se ha estimado entre 1,5 y 2,0. La incidencia de TEV en los preparados que contienen levonorgestrel combinado con menos de 50 µg de etinilestradiol es de aproximadamente de 20 casos por 100.000 mujeres-año de uso. Para <Nombre del producto > la incidencia será aproximadamente de 30-40 casos por 100.000 mujeres-año de uso, lo que significa 10-20 casos adicionales por 100.000 mujeres-año de uso. El impacto del riesgo relativo en el número de casos adicionales es mayor en las mujeres que utilizan anticonceptivos orales combinados por primera vez, cuando el riesgo de TEV con todos los anticonceptivos orales combinados es más alto.
• El riesgo de padecer tromboembolismo venoso aumenta con: • edad avanzada; • existencia de antecedentes familiares (es decir, algún tromboembolismo venoso en un hermano
o en uno de los padres a edad relativamente joven). Si se sospecha la existencia de una predisposición hereditaria se debe remitir a la mujer a un especialista para recibir asesoramiento antes de decidir sobre el empleo de cualquier anticonceptivo hormonal;
• obesidad (índice de masa corporal mayor de 30 kg/m2);

35
• inmovilización prolongada; cirugía mayor; cualquier cirugía de miembros inferiores o traumatismos importantes. En estas situaciones es recomendable suspender el empleo de anticonceptivos orales combinados (al menos con cuatro semanas de anticipación en el caso de cirugía programada) y no volver a emplearlos hasta dos semanas después de recuperar la movilidad por completo;
• y posiblemente también con la tromboflebitis superficial y las venas varicosas. No existe acuerdo acerca del posible papel de estos trastornos en la etiología del tromboembolismo venoso.
• El uso de anticonceptivos orales combinados en general se ha asociado a un incremento del riesgo de infarto agudo de miocardio (IAM) o accidente cerebrovascular, un riesgo que está muy influido por la presencia de otros factores de riesgo (como por ejemplo el tabaquismo, la hipertensión y la edad, ver también más adelante). Estas patologías se producen raramente. No se ha estudiado como modifica <Nombre del producto > el riesgo de IAM.
• El riesgo de padecer complicaciones tromboembólicas arteriales aumenta con: • edad avanzada; • tabaquismo (el riesgo se incrementa con la cantidad de tabaco y con la edad, especialmente en
mujeres mayores de 35 años); • dislipoproteinemia; • obesidad (índice de masa corporal mayor de 30 kg/m2); • hipertensión; • valvulopatía cardiaca; • fibrilación auricular; • antecedentes familiares de trombosis arterial (es decir, trombosis arterial en un hermano o en
uno de los padres a edad relativamente joven). Si se sospecha la existencia de una predisposición hereditaria se debe remitir a la mujer a un especialista para recibir asesoramiento antes de decidir sobre el empleo de cualquier anticonceptivo hormonal.
• En usuarias de anticonceptivos orales combinados se han comunicado casos extraordinariamente raros de trombosis en otros vasos sanguíneos como arterias y venas hepáticas, mesentéricas, renales o retinianas. No existe acuerdo sobre si la aparición de estos casos está asociada con el empleo de anticonceptivos orales combinados.
• Los síntomas de trombosis venosa o arterial pueden ser: dolor y/o hinchazón unilateral en miembros inferiores; dolor intenso y de aparición brusca en el tórax con o sin irradiación a miembro superior izquierdo; dificultad respiratoria súbita; tos de comienzo brusco; cefalea no habitual de cualquier tipo intensa o prolongada; pérdida repentina de visión parcial o completa; diplopía; lenguaje ininteligible o afasia; vértigo; síncope con o sin epilepsia focal; pérdida de fuerza o entumecimiento marcado de aparición brusca que afecta a un hemicuerpo o a una extremidad; trastornos motores; abdomen agudo.
• Otras patologías que se han asociado con la aparición de acontecimientos adversos vasculares son: diabetes mellitus; lupus eritematoso sistémico; síndrome hemolítico urémico; enfermedad intestinal inflamatoria crónica (enfermedad de Crohn y colitis ulcerosa); y anemia de células falciformes.
• Debe tenerse en cuenta que el riesgo de padecer enfermedades tromboembólicas puede estar aumentado en el puerperio (véase el apartado 4.6 “Embarazo y lactancia”).
• El aumento de la frecuencia o de la intensidad de las migrañas durante el empleo de anticonceptivos orales combinados pueden ser pródromos de un accidente cerebrovascular, y por tanto, pueden ser una razón para suspender de forma inmediata el tratamiento.
• Los factores bioquímicos que pueden indicar una predisposición hereditaria o adquirida a padecer trombosis venosa o arterial son: resistencia a la proteína C activada (PCA); hiperhomocisteinemia; déficit de antitrombina III; déficit de proteína C; déficit de proteína S; y presencia de anticuerpos antifosfolípidos (anticuerpos anticardiolipina o anticoagulante lúpico).
• A la hora de considerar la relación riesgo/beneficio del empleo de anticonceptivos orales combinados el médico debe tener en cuenta que el tratamiento adecuado de una situación puede reducir el riesgo asociado de trombosis, y que el riesgo de trombosis asociado con el embarazo es mayor que el asociado con el empleo de anticonceptivos orales combinados.

36
2. Tumores • Algunos estudios epidemiológicos han informado de un aumento del riesgo de padecer cáncer
cervical en usuarias de anticonceptivos orales combinados a largo plazo. No obstante, continúa existiendo controversia acerca del grado en que este hallazgo es atribuible al efecto producido por factores de confusión como son los hábitos sexuales y otros factores como el virus del papiloma humano (VPH).
• Un metanálisis realizado sobre 54 estudios epidemiológicos ha informado que existe un ligero incremento del riesgo relativo (RR=1,24) de que se diagnostique cáncer de mama entre mujeres que emplean anticonceptivos orales. Este incremento desaparece gradualmente durante los 10 años posteriores a haber dejado de emplear anticonceptivos orales. El cáncer de mama es raro entre mujeres con edad menor de 40 años, por lo que el aumento en el número de cánceres de mama diagnosticados entre mujeres usuarias actuales o recientes de anticonceptivos orales es pequeño en relación con el riesgo global de padecer cáncer de mama. Estos estudios no constituyen una evidencia de relación causal. El incremento observado puede ser debido: a un diagnóstico más precoz entre las usuarias de anticonceptivos orales; al efecto biológico de los anticonceptivos orales; o a una combinación de ambos factores. Por otro lado, los cánceres de mama diagnosticados entre usuarias de anticonceptivos orales tienden a ser menos avanzados clínicamente que los cánceres diagnosticados entre las mujeres que no los han empleado nunca.
• En raros casos se han observado tumores hepáticos benignos y, aún más raramente malignos en usuarias de anticonceptivos orales combinados. En casos aislados, estos tumores han originado hemorragia intraabdominal con peligro de muerte. Si se presentase dolor epigástrico intenso, aumento del tamaño del hígado o signos de hemorragia intraabdominal, el diagnóstico diferencial debe contemplar la posibilidad de existencia de un tumor hepático.
3. Otras patologías • En las mujeres con hipertrigliceridemia o antecedentes familiares de la misma puede existir un
aumento del riesgo de padecer pancreatitis durante el empleo de anticonceptivos orales combinados.
• Durante el empleo de anticonceptivos orales combinados se ha observado que muchas usuarias presentan pequeños aumentos de la tensión arterial, aunque raramente estos incrementos de la tensión tienen una repercusión clínica importante. No se ha establecido una relación entre el empleo de anticonceptivos orales combinados y el desarrollo de hipertensión arterial clínica. Sin embargo, si durante el empleo de un anticonceptivo oral combinado se presenta hipertensión mantenida de importancia clínica es prudente que el médico recomiende suspender la toma del anticonceptivo y tratar la hipertensión. Cuando se considere oportuno se puede reanudar el empleo de anticonceptivos orales combinados si se han alcanzado valores de tensión arterial normales con el tratamiento antihipertensivo.
• Los siguientes procesos pueden aparecer o agravarse en el curso del embarazo y durante el empleo de anticonceptivos orales combinados: ictericia y/o prurito por colestasis; formación de cálculos biliares; porfiria; lupus eritematoso sistémico; síndrome urémico hemolítico; corea de Sydenham; herpes gestacional; pérdida de audición por otosclerosis. No obstante, la evidencia no es concluyente respecto a la asociación entre estos procesos y el empleo de anticonceptivos orales combinados.
• Las alteraciones agudas o crónicas de la función hepática pueden requerir la interrupción del empleo de anticonceptivos orales combinados hasta que los marcadores de la función hepática se normalicen. La reaparición de una ictericia colestática que se presentó por primera vez durante un embarazo o coincidiendo con el empleo previo de esteroides sexuales requiere la suspensión del empleo de los anticonceptivos orales combinados.
• Los anticonceptivos orales combinados pueden alterar la resistencia periférica a la insulina y la tolerancia a la glucosa. Sin embargo, no existe evidencia de que sea necesario modificar los requerimientos de antidiabéticos orales o de insulina en las mujeres diabéticas que emplean anticonceptivos orales combinados. No obstante, estas mujeres requieren supervisión médica estricta durante el empleo de anticonceptivos orales combinados.

37
• El desarrollo de la enfermedad de Crohn y de la colitis ulcerosa se ha asociado con el empleo de anticonceptivos orales combinados.
• Ocasionalmente, durante el empleo de anticonceptivos orales combinados se puede presentar cloasma sobre todo en las mujeres con antecedentes personales de cloasma gravídico. Las mujeres con tendencia a presentar cloasma deben evitar la exposición al sol o a las radiaciones ultravioleta mientras estén empleando anticonceptivos orales combinados.
Toda esta información debe tenerse en cuenta al prescribir este anticonceptivo oral combinado y al recomendar la elección de método(s) anticonceptivos. Examen médico y revisiones Antes de iniciar o de reanudar el uso de <Nombre del producto > se debe realizar una historia clínica completa (incluyendo antecedentes familiares) y excluir el embarazo. Se valorará la presión arterial y se realizará una exploración física orientada según las contraindicaciones (apartado 4.3) y las advertencias (apartado 4.4). Asimismo, se recomendará a la mujer que lea con atención el prospecto y seguir las recomendaciones que se dan en el mismo. La frecuencia y naturaleza de los siguientes controles deberá basarse en los protocolos establecidos y adaptándolos a las necesidades clínicas de cada caso. Se debe recordar a las usuarias de anticonceptivos orales que éstos no protegen de la infección por el VIH (SIDA) ni de otras enfermedades de transmisión sexual. Reducción de la eficacia La eficacia de los anticonceptivos orales combinados puede verse reducida si la usuaria se olvida de tomar algún comprimido (véase el apartado 4.2), presenta vómitos (véase el apartado 4.2) o toma algún medicamento concomitante (véase el apartado 4.5). No deben tomarse preparaciones a base de plantas medicinales con Hierba de San Juan (Hypericum perforatum) simultáneamente con <Nombre del producto > debido al riesgo de disminución de los niveles plasmáticos y reducción de los efectos clínicos de <Nombre del producto > (véase el apartado 4.5 Interacciones). Irregularidades en el control del ciclo Durante el empleo de cualquier anticonceptivo oral combinado puede ocurrir que la usuaria presente sangrado intermenstrual (manchado o hemorragia por disrupción) sobre todo durante los primeros meses de empleo. Por tanto, para la evaluación de cualquier sangrado irregular se debe tener en cuenta que sólo es relevante si éste ocurre después de un intervalo de adaptación de unos tres ciclos. Si las irregularidades en el sangrado persisten o se presentan después de ciclos regulares previos deben considerarse las causas no hormonales y está indicado aplicar medidas diagnósticas adecuadas, para descartar una enfermedad maligna o un embarazo, lo que puede incluir legrado. En algunas mujeres puede no presentarse la hemorragia por deprivación durante el intervalo de comprimidos inactivos. Si se ha tomado el anticonceptivo oral combinado según las instrucciones del apartado 4.2 “Posología y forma de administración” es improbable que la usuaria esté embarazada. Sin embargo, si el anticonceptivo no se ha tomado siguiendo estas instrucciones antes de producirse la primera falta, o si se han producido dos faltas seguidas, se debe descartar la existencia de embarazo antes de continuar con el empleo del anticonceptivo oral combinado. 4.5 Interacción con otros medicamentos y otras formas de interacción Interacciones Las interacciones farmacológicas que producen un aumento del aclaramiento plasmático de las hormonas sexuales pueden dar lugar a que se presente hemorragia por disrupción y a fallo del anticonceptivo. Este hecho ha quedado demostrado con los siguientes fármacos: hidantoínas; barbitúricos; primidona; carbamazepina; y rifampicina. También se sospecha de esta forma de

38
interacción con: oxcarbazepina; topiramato; felbamato; ritonavir y griseofulvina. El mecanismo de esta interacción parece basarse en la propiedad inductora que poseen estos fármacos sobre los enzimas hepáticos. Generalmente, la inducción enzimática máxima no se observa hasta transcurridas 2 ó 3 semanas de la utilización de estos fármacos, pero después puede mantenerse durante al menos 4 semanas una vez suspendido el tratamiento. También se han comunicado fallos anticonceptivos por la administración concomitante de ciertos antibióticos como las ampicilinas y las tetraciclinas. El mecanismo de esta interacción todavía no se conoce con certeza. Las usuarias en tratamiento de corto plazo con los tipos de fármacos o con uno cualquiera de los fármacos antes indicados deben continuar la toma del anticonceptivo oral combinado y además, utilizar temporalmente un método de barrera (el preservativo), es decir, durante el tiempo que se administre concomitantemente el fármaco y durante los 7 días posteriores a su suspensión. Las usuarias en tratamiento con rifampicina deben continuar la toma del anticonceptivo oral combinado y además, utilizar un método de barrera (el preservativo) durante el tiempo en que se administre la rifampicina y durante los 28 días siguientes a su suspensión. Si la administración del fármaco concomitante continuara después de terminar los comprimidos del envase actual, se debe iniciar el siguiente envase sin tomar los comprimidos inactivos. Algunos expertos han recomendado aumentar la dosis de los esteroides anticonceptivos en las usuarias que reciben tratamiento a largo plazo con fármacos inductores de los enzimas hepáticos. Si no es apropiado administrar una dosis elevada de anticonceptivo, o este aumento resulta insatisfactorio o ineficaz (p. ej.: en caso de presentarse sangrado irregular), se debe recomendar a la usuaria utilizar otro método de anticoncepción. Los preparados a base de plantas medicinales con Hierba de San Juan (Hypericum perforatum) no deben tomarse de forma concomitante con anticonceptivos orales, ya que potencialmente podría causar una pérdida del efecto anticonceptivo. Se han citado sangrado por disrupción y embarazos no previstos. Este efecto es debido a la inducción de enzimas que metabolizan fármacos por la Hierba de San Juan. Este efecto inductor puede persistir durante al menos 2 semanas después de dejar el tratamiento con Hierba de San Juan. Pruebas analíticas La utilización de anticonceptivos orales puede influir sobre los resultados de ciertas pruebas de laboratorio, incluyendo parámetros bioquímicos de función hepática, tiroidea, suprarrenal y renal, niveles plasmáticos de proteínas transportadoras (p. ej.: globulina transportadora de corticosteroides o fracciones lipídicas y lipoproteicas), parámetros del metabolismo de carbohidratos y parámetros de la coagulación y fibrinolisis. Por lo general, estas variaciones se mantienen dentro de los rangos de normalidad de laboratorio. 4.6 Embarazo y lactancia Los anticonceptivos orales combinados no están indicados durante el embarazo. En la mayoría de estudios epidemiológicos sobre anticonceptivos orales combinados no se ha encontrado un aumento del riesgo de padecer defectos congénitos en los hijos de mujeres que han empleado anticonceptivos orales antes de la gestación. Tampoco se ha informado de ningún efecto teratogénico por la toma de anticonceptivos orales combinados de forma inadvertida durante las fases precoces del embarazo. La lactancia puede verse influida por el empleo de anticonceptivos orales combinados ya que éstos pueden reducir la cantidad de la leche materna y modificar su composición. Por lo general, no se debe recomendar el empleo de anticonceptivos orales combinados hasta finalizar completamente el periodo de lactancia materna. Con la leche materna pueden excretarse pequeñas cantidades de esteroides anticonceptivos y/o de sus metabolitos, sin embargo, no se ha demostrado que esto influya de forma adversa sobre la salud del lactante.

39
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas No se han observado efectos sobre la capacidad para conducir y utilizar máquinas. 4.8 Reacciones adversas Reacciones adversas graves Existe un incremento del riesgo de tromboembolismo venoso en todas las mujeres que toman un anticonceptivo oral combinado. En el apartado 4.4 se proporciona información sobre las diferencias de riesgo entre anticonceptivos orales combinados, y sobre otras reacciones adversas graves. Otras reacciones adversas posibles Se han citado las siguientes reacciones adversas en usuarias de anticonceptivos orales combinados: Mamas hipersensibilidad, dolor y secreción; Sistema Nervioso Central cefalea, jaqueca, cambios en la libido, estado de ánimo depresivo; Ojos intolerancia a las lentes de contacto; Tracto gastrointestinal náuseas, vómitos; Tracto urogenital cambios en la secreción vaginal; Piel alteraciones cutáneas varias (por ej. eritema nudoso, eritema
multiforme, fotosensibilidad, urticaria); Otras alteraciones retención de líquidos, modificaciones del peso corporal, reacciones de
hipersensibilidad. 4.9 Sobredosis No se ha informado de efectos adversos graves tras sobredosis. Los síntomas que pueden aparecer en este caso son náuseas o vómitos. En niñas puede producirse una ligera hemorragia vaginal. No existen antídotos y el tratamiento debe ser sintomático. 5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas Clasificación ATC G03AB05 El efecto anticonceptivo de los anticonceptivos orales combinados se basa en la interacción de diversos factores de los que los más importantes son la inhibición de la ovulación y las modificaciones de la secreción cervical. Además de proteger contra el embarazo, los anticonceptivos orales combinados producen efectos beneficiosos no anticonceptivos que, aún considerando los posibles efectos indeseables (véanse los apartados: “Advertencias” y “Reacciones adversas”), pueden ser útiles a la hora de decidir sobre el método de control de la fertilidad. Durante el empleo de anticonceptivos orales combinados los ciclos son más regulares y la menstruación suele ser menos dolorosa y con menor cantidad de sangrado. Esto último puede dar lugar a una menor incidencia de casos de mujeres con deficiencia de hierro. Al margen de esto, con el empleo de los anticonceptivos orales combinados de mayor dosis (50 µg de etinilestradiol) se ha demostrado una reducción del riesgo de padecer: tumores fibroquísticos de mama; quistes ováricos; enfermedad inflamatoria pélvica; embarazo ectópico; cáncer de endometrio; y cáncer de ovario. Todavía no se ha confirmado si estas conclusiones pueden aplicarse a los anticonceptivos orales combinados con menor dosificación. En estudios clínicos se ha demostrado que <Nombre del producto > redujo significativamente los parámetros androgénicos 3-α androstenodiol-glucurónido, androstenodiona, DHEA-S y testosterona libre.

40
5.2 Propiedades farmacocinéticas Desogestrel Absorción El desogestrel administrado por vía oral se absorbe y convierte en etonogestrel de forma rápida y completa. Los niveles séricos máximos del fármaco son aproximadamente de 1,5 ng/ml (primera semana) a 5 ng/ml (tercera semana) y se alcanzan aproximadamente al cabo de 1,5 horas. La biodisponibilidad es del 62 – 81%. Distribución El etonogestrel se une a la albúmina y a la globulina transportadora de hormonas sexuales (SHBG). Tan sólo de un 2 a 4% de los niveles séricos totales del fármaco están presentes como esteroide libre, pero el 40 - 70% se une específicamente a la SHBG. La distribución en las proteínas plasmáticas depende del incremento de las concentraciones de SHBG inducidas por etinilestradiol, causando un aumento de la fracción unida a la SHBG, mientras que disminuye la fracción unida a la albúmina. El volumen de distribución aparente del desogestrel es 1,5 l/kg. Metabolismo El etonogestrel se metaboliza por completo a través de las rutas metabólicas conocidas para los esteroides. La velocidad de aclaramiento metabólico sérico es de aproximadamente 2 ml/min/kg. No se ha encontrado interacción alguna con la administración concomitante de etinilestradiol. Eliminación Las concentraciones séricas de etonogestrel disminuyen en dos fases. La fase de disposición final se caracteriza por una semivida de aproximadamente 30 horas. El desogestrel y sus metabolitos se excretan por vía urinaria y biliar con un cociente de aproximadamente 6:4. Estado de equilibrio La farmacocinética del etonogestrel está influida por los niveles de SHBG, que se incrementan 3 veces con el etinilestradiol. Con la administración diaria, las concentraciones séricas del fármaco aumentan aproximadamente de 2 a 3 veces, alcanzando un estado de equilibrio durante la segunda mitad del ciclo de tratamiento. Etinilestradiol Absorción El etinilestradiol administrado por vía oral se absorbe de forma rápida y completa. Las concentraciones séricas máximas en torno a 80 pg/ml se alcanzan al cabo de 1 a 2 horas. La biodisponibilidad absoluta resultante de la conjugación presistémica y del metabolismo de primer paso es de aproximadamente 60%. Distribución El etinilestradiol se une en gran medida, pero inespecíficamente, a la albúmina sérica (aproximadamente 98,5%) e induce un incremento de las concentraciones séricas de SHBG. Se ha determinado un volumen de distribución aparente del etinilestradiol de aproximadamente 5 l/kg. Metabolismo El etinilestradiol experimenta conjugación presistémica en la mucosa del intestino delgado y en el hígado. Se metaboliza primero por hidroxilación aromática, formándose gran variedad de metabolitos metilados e hidroxilados, que se presentan como metabolitos libres y como conjugados con glucurónidos y sulfatos. La velocidad de aclaramiento metabólico es de aproximadamente 5 ml/min/kg. Eliminación Los niveles séricos de etinilestradiol disminuyen en dos fases. La fase de disposición final se caracteriza por una semivida de aproximadamente 24 horas. El fármaco inalterado no se excreta. Los

41
metabolitos del etinilestradiol se excretan por vía urinaria y biliar con un cociente de 4:6 y con una semivida de aproximadamente 1 día. Estado de equilibrio Las concentraciones de estado de equilibrio se alcanzan al cabo de 3 a 4 días cuando los niveles séricos de fármaco son de un 30% a un 40% más altos que si se administra una dosis única. 5.3 Datos preclínicos sobre seguridad Los datos preclínicos no han revelado ningún peligro especial para los humanos si se usan los anticonceptivos orales combinados de la forma recomendada. Esto se basa en estudios convencionales de toxicidad a dosis repetidas, toxicidad génica, potencial carcinogénico y toxicidad en la reproducción. Sin embargo, hay que tener presente que los esteroides sexuales podrían estimular el crecimiento de ciertos tejidos y tumores dependientes de hormonas. 6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes Núcleo comprimidos activos α-tocoferol, lactosa monohidrato, almidón de patata, povidona, dióxido de silicio coloidal anhidro y ácido esteárico. Núcleo comprimidos inactivos lactosa monohidrato, estearato de magnesio y almidón de maíz. Recubrimiento Para todos los comprimidos: Hipromelosa, macrogol 400, talco, dióxido de titanio (E171) y además: Óxido férrico amarillo (E172) para los comprimidos con 0,050 mg de desogestrel/0,035 mg de etinilestradiol. Óxido férrico rojo (E172) para los comprimidos con 0,100 mg de desogestrel/0,030 mg de etinilestradiol. Óxido férrico amarillo (E172) e indigotina laca (E132) para los comprimidos inactivos. 6.2 Incompatibilidades No aplicable. 6.3 Periodo de validez 3 años. 6.4 Precauciones especiales de conservación No conservar a temperatura superior a 30ºC. Conservar en el envase original. 6.5 Naturaleza y contenido del recipiente El blister es de aluminio/PVC y consiste en una lámina de aluminio con una capa para sellado por calor y una película de PVC. Cada blister contiene 28 comprimidos y se acondiciona en un sobre de aluminio impreso. El sobre se envasa en cajas de cartón impresas con 1, 3 ó 6 sobres por caja, junto con un prospecto. No todas las presentaciones se encuentran comercializadas.

42
6.6 Instrucciones de uso, manipulación y eliminación Ninguna especial. 7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN 8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN 9. FECHA DE LA REVALIDACIÓN DE LA AUTORIZACIÓN 10. FECHA DE LA REVISIÓN DEL TEXTO