analysis of a role of the lysr-type regulator shvr in ... · virulence of burkholderia cenocepacia...
TRANSCRIPT

Analysis of a role of the LysR-type regulator ShvR invirulence of Burkholderia cenocepacia using zebrafish as a
model
Margarida Castro Gomes
Thesis to obtain the Master of Science Degree in
Biological Engineering
Supervisor(s): Dr. Annette Carolin Vergunst and Professor Jorge Humberto GomesLeitão
Examination CommitteeChairperson: Professor Arsénio do Carmo Sales Mendes FialhoSupervisor: Professor Jorge Humberto Gomes LeitãoMember of the Committee: Professor Leonilde de Fátima Morais Moreira
September 2014

ii

Acknowledgments
First of all, I would like to acknowledge my supervisors, Dr. Annette Vergunst and Professor Jorge
Leitao, for their guidance and supervision of this thesis.
I would like to address a special thanks to Dr. Annette, for introducing me to this interesting and
challenging theme and, most of all, for her support, advice, availability and guidance provided through
these last 7 months. Working with you was a great experience and I hope it will continue for the next 3
years.
To Dr. David O’Callaghan, my future boss, for having received me in the lab and for making everything
look better with his jokes.
Special thanks are also to Joana Feliciano, PhD student and researcher at Biological Sciences Re-
search Group at Instituto Superior Tecnico (and lent to the Zebrafish group for (in)determinate time),
for everything that I could not have imagined to find: invaluable friendship, precious help in all labo-
ratorial experiences and in the thesis, long (Portuguese) talks, infinite patience to listen to me, mental
connection, and support.
To Lily Zhang and Jennifer Mesureur, also members of the Zebrafish group, and Christelle Ngba and
Karellen Mendez for support, advices, laughter, friendship, patience and for making the experience in
the lab (and in Nımes!) memorable.
I would also like to thank all the people in U1047, for receiving me so well and for their support.
To my family and friends, to whom I may not have given full attention for these past months, but I know
that you were always there for me.
Special thanks to my grandmother, who always supported me in every step of my life, and I know that
she will continue to do so from above.
To my mother and my father, who have been my no. 1 supporters, and to my siblings, from whom is
always difficult to be apart, I want to thank them for being so patient, supportive and tireless, for cheering
me up whenever I am down and for their unconditional love. It is because of you that I am here today.
iii

iv

Resumo
Burkholderia cenocepacia e uma bacteria patogenica oportunista pertencente ao complexo Burkholde-
ria cenocepacia (Bcc), causadora de infecoes cronicas em pessoas imunocomprometidas, principal-
mente em pacientes com fibrose quıstica. Estudos demonstram que esta bacteria e capaz de viver
e multiplicar-se dentro de celulas, escapando ao mecanismo de degradacao da celula hospedeira. O
modelo de embrioes de peixe-zebra (Danio rerio) tem sido desenvolvido para estudar infecoes com
bacterias do complexo Bc, tendo sido demonstrada a importancia dos macrofagos na virulencia bacte-
riana.
O referido modelo foi utilizado neste trabalho para compreender um dos papeis do regulador tran-
scricional do tipo LysR, shvR, na virulencia de B. cenocepacia K56-2. Experiencias anteriores em
plantulas de alfalfa e em ratos demonstraram que este regulador e importante para a virulencia e
inflamacao, detectada nos pulmoes dos ratos. Adicionalmente, as bacterias mutantes demonstraram
elevada persistencia nos pulmoes dos ratos.
Neste estudo demonstra-se que o mutante shvR causa uma infecao persistente nos embrioes de
peixes-zebra. Contrastando com a infecao aguda causada por K56-2, o mutante e menos virulento e,
apesar de atingir elevados numeros intracelulares em macrofagos, as bacterias nao sao capazes de
proliferar e causar uma infecao inflamatoria aguda. O fenotipo persistente, residindo as bacterias em
macrofagos, foi confirmado pela analise da resposta fagocıtica das celulas hospedeiras. Foram ainda
desenvolvidas novas ferramentas para melhor compreender o papel do regulador ShvR na virulencia e
persistencia bacteriana.
Palavras-chave: Burkholderia cenocepacia, shvR, peixe-zebra, resposta imunitaria, sobre-
vivencia intracelular, persistencia
v

vi

Abstract
Burkholderia cenocepacia is an opportunistic pathogen, comprised in the Burkholderia cepacia com-
plex, that causes chronic infections in immunocompromised people, mainly in cystic fibrosis patients.
These bacteria have been demonstrated to be capable of living and multiplying inside cells by evading
host cell degradation mechanisms. Zebrafish embryos have been developed as a model to study Bcc
infections and it has been shown that macrophages play an important role in bacterial virulence.
In this study the zebrafish model was used to further understand a role for the LysR-type transcrip-
tional regulator (LTTR) shvR from B. cenocepacia K56-2 in virulence. Previous infection experiments in
alfalfa seedlings and rats have shown that this regulator is important for virulence, and inflammation in
rat lungs. Interestingly, high persistence in the lungs of rats was observed for the shvR mutant.
Here, it is shown that the shvR mutant causes a persistent infection phenotype in zebrafish embryos.
In contrast to the acute infection caused by K56-2, a shvR mutant is less virulent than its parent; although
it survives and replicates inside host macrophages reaching high intracellular numbers, the bacteria
are not able to spread and cause inflammatory acute infection. A persistent phenotype, with bacteria
residing in macrophages, was further confirmed by analysis of host phagocyte response. Furthermore,
new tools were developed to start to better understand the role of ShvR in virulence and persistence.
Keywords: Burkholderia cenocepacia, shvR, zebrafish, immune response, intracellular survival,
persistence
vii

viii

Contents
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . iii
Resumo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vii
List of Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xi
List of Figures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xiii
Acronyms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xvii
1 Introduction 1
1.1 Burkholderia cepacia complex . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.1 Taxonomy and genetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.2 Burkholderia cepacia complex species as a human pathogen . . . . . . . . . . . . 3
1.1.3 Virulence factors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.1.4 Intracellular survival . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.2 Bcc infection models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.2.1 Zebrafish as an infection model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
1.3 ShvR – a global regulator of gene expression . . . . . . . . . . . . . . . . . . . . . . . . . 15
1.4 Motivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2 Materials and Methods 19
2.1 Bacterial strains, plasmids and growth conditions . . . . . . . . . . . . . . . . . . . . . . . 19
2.2 DNA manipulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.2.1 Extraction and purification of plasmid DNA . . . . . . . . . . . . . . . . . . . . . . 20
2.2.2 Polymerase chain reaction (PCR) conditions . . . . . . . . . . . . . . . . . . . . . 20
2.2.3 Agarose gel electrophoresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3 Bacterial transformation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3.1 Electroporation of Burkholderia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3.2 E. coli competent cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.4 Construction of plasmids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.4.1 Construction of a pshvR-mCherry reporter plasmid (pAV209) . . . . . . . . . . . . 22
2.4.2 Construction of shvR complementation plasmids (pMG3 and pMG4) . . . . . . . . 22
2.4.3 Construction of a plasmid expressing an unstable mCherry reporter (pMG5) . . . . 23
ix

2.5 Zebrafish infection model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.5.1 Zebrafish care and maintenance . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.5.2 Transgenic zebrafish lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.5.3 Microinjection of bacteria in zebrafish embryos . . . . . . . . . . . . . . . . . . . . 24
2.6 Statistical analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.7 Microscopic analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.7.1 Computational quantification of fluorescent host immune cells . . . . . . . . . . . . 26
3 Results – A B. cenocepacia shvR mutant is attenuated for virulence in a zebrafish infection
model 27
3.1 Survival assays and bacterial kinetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3.2 Analysis of the behavior of host immune cells in infected embryos . . . . . . . . . . . . . 29
3.2.1 Quantification of host immune cell numbers during infection . . . . . . . . . . . . . 31
3.3 Development of tools to better study the role of ShvR in virulence . . . . . . . . . . . . . . 35
3.3.1 Complementation plasmids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.3.2 Visualizing shvR expression in vivo in zebrafish embryos . . . . . . . . . . . . . . 38
4 Discussion 43
Bibliography 69
x

List of Tables
1.1 Burkholderia cepacia complex species and strains . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Models for Burkhoderia cepacia complex infection studies . . . . . . . . . . . . . . . . . . 12
2.1 Bacterial strains used in this study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.2 Plasmids used in this study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.3 PCR amplification products . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.4 Primers sequence and PCR conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
xi

xii

List of Figures
1.1 Normal and mutant CFTR proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2 Evading mechanisms of intracellular B. cenocepacia . . . . . . . . . . . . . . . . . . . . . 9
1.3 ShvR predicted secondary structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.1 Microinjection site in zebrafish embryos . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.2 Binary conversion of fluorescent image . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
3.1 B. cenocepacia K56-2∆shvR is less virulent than K56-2 in zebrafish embryos . . . . . . . 28
3.2 B. cenocepacia K56-2 and shvR mutant infection in the tail region of zebrafish embryos . 29
3.3 Zebrafish transgenic lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
3.4 B. cenocepacia K56-2 infection in zebrafish embryos . . . . . . . . . . . . . . . . . . . . . 30
3.5 B. cenocepacia K56-2∆shvR infection in zebrafish embryos . . . . . . . . . . . . . . . . . 30
3.6 Macrophage infected with B. cenocepacia K56-2∆shvR . . . . . . . . . . . . . . . . . . . 31
3.7 Macrophage quantification analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
3.8 Representative fluorescence microscopy images used for macrophage quantification anal-
ysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
3.9 Neutrophil quantification analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.10 Representative fluorescence microscopy images used for neutrophil quantification analysis 34
3.11 pUCP28T-shvR almost fully restores virulence to a shvR mutant . . . . . . . . . . . . . . 35
3.12 Cloning scheme for plasmid pMG3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
3.13 Plasmid map of pMG3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
3.14 Cloning scheme for plasmid pMG4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.15 Plasmid map of pMG4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
3.16 Cloning scheme for plasmid pAV209 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.17 Plasmid map of pAV209 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.18 Zebrafish embryos infected with B. cenocepacia K56-2 and the shvR mutant carrying
pVA209 plasmid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.19 Cloning scheme for plasmid pMG5 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.20 Plasmid map of pMG5 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
xiii

xiv

Acronyms
(wt/v) Weight per volume
AHL N-acylated homoserine lactone
AmpR Ampicillin resistance
Amp Ampicillin
BDSF Burkholderia cenocepacia diffusible signal fac-
tor
BP Band pass
BS Beam splitter
BcCV Burkholderia cepacia containing vacuole
Bcc Burkholderia cepacia complex
CFP Cyan fluorescent protein
CFTR Cystic fibrosis transmembrane conductance
regulator
CFU Colony forming unit
CF Cystic fibrosis
CGD Chronic granulomatous disease
CmR Chloramphenicol resistance
Cm Chloramphenicol
DAMP Danger-associated molecular pattern
DIC Differential interference contrast
DNA Deoxyribonucleic acid
Da Dalton
ECM Extracellular matrix
EDTA Ethylenediamine tetraacetic acid
EEA1 Early endosome marker autoantigen 1
EPS Exopolysaccharide
ER Endoplasmic reticulum
ET Electroforetic lineage
FACS Fluorescence-activated cell sorting
GFP Green flourescent protein
xv

HTH Helix-turn-helix motif
IFN Interferon
IL Interleukin
IPTG Isopropyl-β-D-thiogalactopyranoside
LAMP Lysosome-associated membrane protein
LB Luria-Bertani broth
LPS Lipopolysaccharide
LTTR LysR-type transcriptional regulator
MB Methylene blue
NBF Nucleotide-binding fold
PAMP Pathogen-associated molecular pattern
PBS Phosphate buffered saline
PCR Polymerase chain reaction
Par Partitioning gene cassetes
Ptw Plant water soaking
QS Quorum sensing
R domain Regulatory domain
RNAseq RNA sequencing
RNA Ribonucleic acid
RNS Reactive nitrogen species
ROS Reactive oxygen species
Rep Origin of replication
T...SS Type ... Secretion System
TBE Tris/Borate/EDTA buffer
TLR Toll-like receptor
TpR Trimethoprim resistance
Tp Trimethoprim
UV Ultraviolet
WGS Whole genome sequence
X-gal 5-bromo-4cholo-3-indolyl-β-D-
galactopyranoside
Zmp Zinc metalloprotease
°C Degrees Celsius
ptac tac-promoter
trpA Tryptophan terminator
cat Chloramphenicol acetyltransferase
cci Cenocepacia island
hpf Hours post-fertilization
xvi

hpi Hours post-infection
h Hour
kb Thousands of base pairs
min Minute
nt Nucleotide
phox Phagocytic ox idase
rDNA Ribosomic deoxyribonucleic acid
shv Shiny colony variant
xvii

xviii

Chapter 1
Introduction
1.1 Burkholderia cepacia complex
This project focuses on virulence mechanisms of Burkholderia cenocepacia a Gram-negative non-
spore-forming bacillus of the β-proteobacteria subdivision, that belongs to the Burkholderia cepacia
complex (Bcc). This complex currently includes 17 phenotypically similar species that are found ubiqui-
tously in the natural environment, including the rhizosphere of plants, and in industrialized environments,
but importantly, they are life-threatening pathogens of immunocompromised persons, especially those
with cystic fibrosis (CF) (Table 1.1) [1–7].
Here, I will give a general overview of taxonomy and genetics of this complex, two genetic diseases
where B. cenocepacia causes infection and some of the virulence factors that have been found in Bcc
strains. It will also be discussed the importance of intracellular stages and inflammatory responses,
animal models, with a focus on the zebrafish embryo model, and ShvR, a LysR-type global regulator,
which is the basis of this study.
1.1.1 Taxonomy and genetics
Walter Burkholder, in 1949, first described Pseudomonas cepacia as a pathogen causing rot on
onion bulbs [8]. The classification of these bacteria was maintained until molecular taxonomic analyses
were performed and a new genus was created: Burkholderia, with B. cepacia as the genus type species
[9]. In 1997, Vandamme et al. demonstrated that the isolates classified as a single species, consisted
in fact of at least five genetically different species, and they were categorized in genomovars [1]. Since
then, several methods have been used to characterize the different genomovars: 16S rDNA sequences,
DNA-DNA homology values, cellular and fatty acid composition [10]. However, sequence polymorphism
within the recA gene and, more recently, multilocus sequence typing (MLST) allowed further subdivision
in genomovars III A, B, C and D and resulted in the taxonomic classification in species and more defined
sequence types (ST) [11, 12]. Additionally, the results from recA studies confirmed that all species in
the complex could cause opportunistic infections in humans [10].
1

Table 1.1: Burkholderia cepacia complex species and strains. Unfinished genome sequences are indi-cated with a (*). Environmental strains are in bold. Based on [13, 14]
Bacterial species CharacteristicsSequenced/
Unfinished (*)strains
References
B. cepaciaCommon environmental species; contains typestrain ATCC 25416T; consists of two recA lineages;epidemic outbreaks in Portugal
GG4Bu72 (*)
ATCC 25416 (*)
[1, 15, 16]
B. multivorans
Not extensively cultured from natural environment;multireplicon genome first observed for soil strainATCC 17616. Together with B. cenocepacia, is the mostprevalent species in CF; patient-to-patient transmission;epidemic outbreak strains described in the UK and in France
CGD1 (*)CGD2 (*)
CGD2M (*)ATCC 17616
ATCC BAA-247 (*)CF2
[1, 15, 17, 18]
B. cenocepacia
Prevalent in rhizosphere; high genetic diversity; severalphylogenetically distinct groups; genome sequenceof ET-12 strain J2315 ; Major CF pathogen; severalhighly transmissible lineages and often associated withpoor prognosis; epidemic outbreaks in Canada andin the UK
PC184 (*)J2315HI2424
AU 1054MC0-3BC7 (*)
K56-2 Valvano (*)H111 (*)KC-01
[1–3, 10, 15, 19–22]
B. stabilisNot extensively cultured from natural environment;Important, not highly prevalent in CF patients
[1, 15, 23]
B. vietnamiensis
Important bacterium in the rhizosphere; beneficialproperties in rice cultivation; draft genome sequencedetermined for strain G4 (ATCC53617 or R-1808).This strain is being used for bioremediation studies inlaboratory. Limited prevalence as a CF pathogen
G4AU4i (*)
[1, 17]
B. dolosaOne environmental strain described. Almostexclusively cultured in CF infection; patient-to-patienttransmission; strain AUO158 recovered from an outbreak in US
AUO158 (*) [18, 24, 25]
B. ambifaria
Major bacterium in the rhizosphere; type strain AMMD is aneffective biological control strain; two strains registered forbiopesticidal use in the United States. Rarely encounteredas a CF pathogen
AMMDMC40-6
IOP40-10 (*)MEX-5 (*)
[15, 26, 27]
B. anthinaBoth environmental and clinical strains; recently defined species.Prevalence among CF patients is low
[28]
B. pyrrociniaBoth environmental and clinical strains; recently defined species.Prevalence among CF patients is low
CH-67 (*) [28]
B. ubonensis Strains recovered from a nosocomial infection Bu (*) [6]
B. latens Clinical strain recovered from sputum of CF patients [6]
B. diffusa Strains recovered from human infections, soil and water [6]
B. arborisEnvironmental and clinical strains; strains recovered fromsputum of CF patients
[6]
B. seminalisEnvironmentally and clinically recovered strains; strainsrecovered from sputum of CF patients and nosocomial infections
[6]
B. metallica Clinical strain recovered from sputum of CF patients [6]
B. contaminans
Environmentally, industrially and clinically recovered strains;strains have been involved in a widespread outbreak in the USdue to a contaminated nasal spray, contaminants in a waterreservoir supplying a renal dialysis machine in Brazil and in milkof a sheep with mastitis. Major problem in Argentina
[7, 29, 30]
B. lata Environmentally, industrially and soil recovered strains [7]
2

It has been demonstrated that the species in the complex share over 97.5% 16S rDNA sequence
similarity, but only moderate genome-wide similarity with 30 to 60% of DNA–DNA hybridization between
all species, in line with the adaptability and metabolic diversity of the species to extreme variations in
growth and stress conditions [1, 31, 32]. Bcc bacteria are known to resist different (environmental) con-
ditions, including nutrient limitation, toxic compounds [33], antimicrobial peptides [4, 34] and almost all
clinically used antibiotics, thus complicating treatment in infected individuals [35, 36]. In addition to being
an opportunistic human and plant pathogen, Bcc bacteria have a bioremediation potential, demonstrated
by their ability to degrade complex aromatic pollutants [37] and capability of protecting and promoting
growth of plants [26]. However, owing to the virulent nature of the Bcc for immunocompromised hu-
mans, risk assessment studies [26] and extreme care are required for introduction of such strains in the
environment.
The metabolic diversity and flexibility of these species can be explained by their genome variety and
genome plasticity, allowing the bacteria to rapidly adapt to different conditions [38]. Their genomes are
among the largest bacterial genomes known, with sizes ranging from 7 to 9 Mb. Until now 27 strains
belonging to the Bcc complex (see Table 1.1) have their whole genome sequenced (WGS) and pub-
lished, or drafted [13, 14]. Their genomes consist of three replicons: chromosome 1 (c1; 3.3 to 3.9
Mb), chromosome 2 (c2; 2.4 to 3.6 Mb) and chromosome 3 (c3; 0.5 to 1.4 Mb) [26]. However, the latter
has recently been described as a mega virulence plasmid that confers a competitive advantage to the
organism, since it is essential for pathogenicity in various hosts, including rats and zebrafish, and anti-
fungal activity and is involved in stress tolerance [39, 40]. For instance, Bcc strain B. cenocepacia H111
demonstrated to be more resistant to different stress conditions (oxidative, osmotic, high-temperature,
and chlorhexidine-induced stresses) than the cured ∆pC3 strain [40].
Moreover, the B. cenocepacia genome contains many genomic islands and insertion sequence (IS)
elements that confer genomic plasticity, contributing to bacterial virulence and adaptation [40]. However,
the precise mechanisms for adaptation during chronic infection in humans, and for instance rapid clinical
decline, are not known.
1.1.2 Burkholderia cepacia complex species as a human pathogen
Immunocompromised individuals are highly susceptible to infection with Bcc bacteria, and although
only a small percentage of the patients get infected, they can cause serious infections in chronic granulo-
matous disease (CGD) and cystic fibrosis (CF) patients [1, 36, 41], but also other immunocompromised
individuals. In these two diseases the lungs are by far the most affected organs upon infection, and
acute stages of infection with Burkholderia cenocepacia in CF can lead to necrotizing pneumonia and
septicaemia, known as “cepacia syndrome”, often resulting in early death.
It has been reported that the bacteria can be transmitted in hospital settings or through social contact
[20, 42, 43]. Consequently, several control measures have been taken, not only in hospitals, to minimize
the possibility of either infecting a patient or transmitting the bacteria to other patients [4]. Despite the
3

efforts to avoid bacterial infections, some other cases reported direct environmental acquisition of Bcc
bacteria [19].
Chronic granulomatous disease
CGD is characterized by defective generation of a respiratory burst in human phagocytes, such
as neutrophils, mononuclear cells, macrophages and eosinophils [44]. Hohn and Lehrer were able to
demonstrate that the disease was due to defects in the NADPH oxidase complex [45]. These oxidases
are involved in catalyzing the reduction of oxygen into superoxide by electron transfer. Superoxide
spontaneously forms hydrogen peroxide, or can be further metabolized into reactive oxygen species
(ROS), playing an essential role in the immune system [46].
The protein complex consists of cell-membrane-bound gp91phox (phagocytic ox idase) and p22phox,
and of cytoplasmic proteins p40phox, p47phox, and p67phox, that becomes activated by a complex series
of protein recruitment and protein/protein interactions, during the respiratory burst, and assembles in
the plasma or phagosomal membrane [47]. Mutations in the genes encoding gp91phox, p22phox [48],
p67phox [48], and p47phox [49] have been described in CGD disease. Consequently, these defects result
in the inability of the cells to generate ROS, and for that reason they are not able to eliminate certain
pathogens from the organism [50].
Patients with this disease are repeatedly infected with bacteria and fungi, resulting in the formation
of inflammatory granulomas. Using neutrophils from CGD patients, Speert et al. demonstrated that B.
cenocepacia is killed by ROS in healthy neutrophils, and not by non-oxidative mechanisms [51]. Later,
Bylund et al. showed that in the absence of ROS B. cenocepacia can cause necrosis in CGD neutrophils,
which can result in increased inflammation in the patients [52].
Cystic fibrosis
Cystic fibrosis is a lethal recessive human genetic disorder caused by mutations in the gene encoding
the cystic fibrosis transmembrane conductance regulator (CFTR) (reviewed in [53]) (Figure 1.1). The
gene was found to be expressed in epithelial cells, blood cells and in macrophages [53, 54]. The main
function of this regulator is as a chloride channel, however many other functions are associated to
this protein, such as the acidification of intracellular organelles [55] and the enhancement of cytokine
production [56].
The dysfunction of the regulator causes a multi-system pathology that includes the pancreas, the
gastrointestinal tract, the liver and the respiratory system [58].
CF remains the most common lethal inherited disease in the Caucasian population [57] and, accord-
ing to the World Health Organization, in the European Union 1 in 2000-3000 newborns is affected by
CF, having a higher prevalence in Ireland [59]. In the United States of America the incidence of CF is
reported to be 1 in every 3500 births [59].
The mutant protein is predominantly expressed on the apical membrane of epithelial cells [57, 60, 61]
and is associated with defective mucociliary clearance and impaired innate immunity of the airways [62–
4

Figure 1.1: CFTR protein consists of a transmembrane domain, two nucleotide-binding folds (NBF) anda regulatory domain (R domain) [57]. Normal CFTR channel can transport chloride ions to the outsideof the cell, however the mutated protein does not, leading to the formation of a thick mucus layer on theexterior of the cell.
64]. The most common mutation, accounting for 70% of the worldwide CF population, is a deletion of one
amino acid at position 508 in the CFTR protein, which is called ∆F508 [65]. This mutation is associated
with misfolding of the protein in the endoplasmic reticulum, being posteriorly retained and degraded [66].
As a result, patients become more susceptible to chronic respiratory infections and acute exacerbations,
which in turn mediate progressive pulmonary deterioration, causing substantial morbidity and mortality.
The infection of the lungs of CF patients normally occurs during infancy and early childhood with
organisms such as Staphylococcus aureus and Haemophilus influenza [67]. These early colonizers
can damage the epithelial surfaces, creating a more suitable environment for the colonization by Pseu-
domonas aeruginosa [67].
Recent years have seen the emergence of several new pathogens of clinical relevance to CF, which
is the case for Burkholderia cenocepacia [4, 33]. Although only a small percentage of CF patients (3-4%)
become infected with Bcc strains, the bacteria contribute to the worsening of the patient’s condition [68].
The outcome of infections with these bacteria is unpredictable; it can vary from periods of chronic infec-
tion to sudden acute, systemic infection [69], and leaves the physicians with few options for treatment
due to the high intrinsic antibiotic resistance. Moreover, infections with B. cenocepacia are generally
associated with reduced survival and a higher risk to develop “cepacia syndrome” [70, 71]. For patients
in later stages of lung diseases, organ transplantation is an option, however CF individuals infected with
B. cenocepacia are often excluded from the lists, due to the recurrent infections by the bacteria and the
low rate of success of the transplantation [72]. Until now the factors that are involved in the sudden fatal
changes of the infection are still unknown.
For the past 30 years, B. cenocepacia strains belonging to ET12, Midwest and PHDC lineages
caused major epidemic outbreaks with significant mortality, although other strains have also caused
major havoc [73]. In the late 1980s, an epidemic outbreak amongst CF patients throughout the United
5

Kingdom and Canada was caused by highly transmissible strains of B. cenocepacia from the ET12
lineage [71, 74].
The B. cenocepacia isolate J2315 is the index strain of the ET12 epidemic outbreak. Its genome has
been fully sequenced [75]. Highly virulent strains belonging to the ET-12 lineage have also been shown
to be highly virulent in experimental animal models. B. cenocepacia K56-2 is clonally related to J2315,
and even though its draft genome sequence has only recently become available, this strain is often used
in infection studies, as it is more amenable to genetic manipulation [76].
1.1.3 Virulence factors
Although many virulence factors have been identified in Bcc strains, the underlying molecular mech-
anisms are often not fully understood, and not all have been associated with pathogenesis in human
infections. Structures such as flagella, cable pili and 22kDa adhesin structures are considered virulence
factors since they help the bacteria to invade lung epithelial cells [77] and to adhere to the lung surface
[78]; the resistance to antibiotics and to oxidative stress [79] as well as the iron acquisition [80] are also
among these virulence determinants.
Below are described some other virulence factors that have been studied in large detail.
Lipopolysaccharide
Lipopolysaccharide (LPS) is composed of lipid A, core oligosaccharide and O-antigen (see for re-
view [81]). Bcc bacteria have a distinctive LPS since 4-amino-4-deoxyarabinose (Ara4N) residues are
bound to phosphates of the lipid A, the core oligosaccharide has less phosphate and has a disaccha-
ride D-glycero-Dtalo-oct-2-ulosonic acid-(2→4)-3-deoxy-D-manno-oct-2-ulosonic acid (Ko-(2→4)-Kdo)
[82, 83], and the O-antigen structure can have different serotypes [81]. This particular composition
changes the bacterial surface charge inhibiting the binding and successful action of antibiotics [84, 85].
Lipid A is detected by TLR4 and induces the host immune response [86–88]. Between different
Gram-negative bacteria, the composition and structure of lipid-A, and consequent recognition by TLR4,
is variable and can have an effect in the disease in humans. For instance, in E. coli the side chain size
and the fatty acid composition affect the human cells stimulation and the signal intensity, respectively
[89–91], and in other pathogens, as Legionella and Helicobacter, different moieties of lipid A can help
the bacteria to go unnoticed by TLR4 [92, 93].
The expression of O-antigen in Bcc strains has been demonstrated to reduce phagocytosis by
macrophages without interfering with the intracellular survival of the bacteria [94]. Moreover, this struc-
ture is an immunogenic component and it has been used as a basis of serotype, except for strains such
as B. cenocepacia J2315 that do not produce the O-antigen [81].
Studies have demonstrated that when neutrophils interact with Bcc LPS the expression of CD11b
on their surface increases, stimulating neutrophil respiratory burst response [95]; and macrophages and
human blood cells are also stimulated to produce pro-inflammatory cytokines such as TNF-α, IL-6 and
IL-8 [87, 96].
6

Biofilms
Many bacteria species can form biofilms in which they live in communities protected from environ-
mental factors, and, during human infections, from the host immune system and antibiotics. Biofilms
consist of complex matrices of polysaccharides and extracellular products, that can comprise single
or multiple microbial species. Bcc was found to persist in biofilms in vitro, in which they demonstrate
increased resistance to antibiotics [97], and that they form mixed biofilms together with P. aeruginosa
[98]. These structures make it difficult to eradicate the bacteria, and in a recent study, Van Acker et
al. demonstrated that even after the eradication of the biofilm, with different antibiotic concentrations,
persister cells survive and these are the ones that possibly increase the chances of recurrent infections
[99].
The formation and maturation of biofilms is dependent on many factors, including adhesins and
surface proteins, and greatly controlled by gene regulatory systems, such as quorum sensing (QS)
[100], sigma factors [101], and for instance the global regulators ShvR [102] and AtsR [103].
Quorum sensing
Cell-to-cell communication is mediated by the production of diffusible N-acylated homoserine lactone
(AHL) signal molecules, called autoinducers. These molecules accumulate in the external environment
of the bacteria and once their concentration reaches a certain threshold, the bacteria respond by modi-
fication of their gene expression through a response regulator [104].
Burkholderia has a CepIR quorum sensing system that is homologous to the LuxIR system in Vibrio
fischeri (reviewed in [105]). The CepIR system positively influences virulence in many of the infec-
tion models used to study B. cenocepacia virulence, including Caenorhabditis elegans, Galleria mel-
lonella, rodents, zebrafish, alfalfa and onions [106–109]. This QS system regulates the expression of
many genes; it negatively regulates for instance siderophore synthesis and positively regulates the ex-
pression of the genes encoding zinc metalloproteases (Zmp), swarming motility and biofilm formation
[100, 107, 108, 110, 111]. B. cenocepacia, in particular, has four QS systems: CepIR, CciIR (encoded
on the cenocepacia island (cci) found in ET12 lineage strains [112]), CepR2 and BDSF (B. cenocepacia
diffusible signal factor) [113].
Exopolysaccharides
Exopolysaccharides (EPS) are produced by most of the Burkholderia species, and can have different
structures and properties, alone or in mixtures [114]. The particular type and amount of EPS produced
by each Burkholderia strain is probably related to the environment in which that strain usually lives or to
the conditions present during its growth, possibly helping to improve its niche adaptation [115].
The most common EPS, and the characteristic EPS of Bcc, is called cepacian. Studies have shown
that it is produced by Bcc and non-Bcc species, both from clinical and environmental sources [114, 115].
Cepacian biosynthesis has been assigned to two gene clusters, bce-I and bce-II [115, 116]. Cepacian is
structurally characterized by a heptasaccharide repeat-unit backbone (composed by units of D-glucose,
7

D-rhamnose, D-mannose, D-galactose and D-glucuronic acid in the molar ratio of 1:1:1:3:1), three short
lateral chains and 1 to 3 acetyl groups that help bacteria to control EPS properties [114, 117].
This EPS type has been pointed out as contributing to the overall pathogenicity of Bcc bacteria. For
example, cepacian interferes with phagocytosis by human neutrophils, facilitating the bacterial persis-
tence in a mouse model of infection; it has been shown to inhibit the production of ROS by neutrophils
and to scavenge ROS, playing a role in the survival of cepacian-producing strains in different environ-
ments [118–122]. In addition, a B. cepacia IST408-ss3 mutant defective in cepacian production was
found to be less virulent in a mouse model of infection, indicating that the mutated gene is essential for
the virulence of the strain [123].
Protein secretion systems
Bacterial protein secretion systems are important virulence factors for many Gram-negative and
positive bacteria. Secretion systems are used by bacteria to communicate with the environment, to
secrete toxins or other proteins either directly into the environment or into host cells. For instance, type
I and II secretion systems (T1SS, T2SS) have been implicated in the secretion of hemolytic proteins in
ET12 lineage strains and in B. vietnamiensis [124, 125]. ZmpA and ZmpB are two zinc metalloproteases
shown to be important in virulence of B. cenocepacia [108, 126], and are secreted by the T2SS. B.
cenocepacia encodes two T4SS [127], and these secretion systems in other intracellular pathogens,
including Brucella spp and Legionella pneumophila, have been shown to be essential virulence factors
for intracellular survival of the bacteria, by translocating effector proteins directly into host cells, where
these subvert host cell biology in favor of the pathogen [128, 129].
The plasmid encoded T4SS, called Ptw from plant water soaking, was identified in B. cenocepacia
strains as necessary for virulence in onions and intracellular survival in phagocytes [130].
In a mouse agar-bead infection model, the T3SS has been shown to be important for bacterial
survival [131], although it does not seem to play a role in intracellular survival of B. cenocepacia [132].
However, B. cenocepacia also encodes a T6SS, which has been shown to affect the actin cytoskele-
ton of macrophages [103] and the assembly of the NADPH oxidase complex in Burkholderia cepacia
containing vacuoles (BcCVs), by inactivation of Rac1 and Cdc42 [133, 134]. The T6SS, which is neg-
atively regulated by the sensor kinase-response regulator AtsR [103], has been shown to enhance
caspase-1 activation and IL-1β release by activation of the inflammasome, possibly by a yet unchar-
acterized T6SS effector [135]. Gavrilin et al. described that B. cenocepacia efficiently activates the
inflammasome and, consequently, monocytes and THP-1 cells release IL-1β in a pyrin, Asc and T6SS
dependent manner [135]. In addition, a recent paper suggests that the T6SS may be important for the
secretion of T2SS effectors, such as ZmpA and ZmpB into the host cytoplasm [136].
For instance, type I and II secretion systems seem to play an important role in intracellular survival
and replication of B. cenocepacia.
8

1.1.4 Intracellular survival
In higher organisms, the first line of defense against viruses, bacteria and other disease causing or-
ganisms is provided by macrophages and neutrophils, as part of the innate immune response. In most
cases, these cells are capable of eliminating the invading organism through a process called phagolyso-
somal degradation. In this process the microbe is taken up by the cell, forming a phagosome, which
will fuse with early and after with late endosomes. Afterwards, it proceeds to form a phagolysosome in
which the microbe is destroyed by low pH, lysosomal hydrolases and other degrading enzymes [137]
(see Figure 1.2).
B. cenocepacia has been shown to be phagocytized and killed in a ROS-dependent manner in neu-
trophils [51], however in cell culture macrophages, and macrophages in zebrafish embryos, the bacteria
have been shown to escape from the classical endocytic pathway and survive and replicate (reviewed
in [138]). Moreover, B. cenocepacia has been detected inside alveolar macrophages, in lung tissue of
infected patients [139].
Figure 1.2: Representation of the different mechanisms that have been described for B. cenocepaciato evade killing by macrophages. The normal phagocytic process is indicated by the black arrows. B.cenocepacia was found to delay the maturation and acidification of phagolysosomes, others showedB. cenocepacia can end up in ER derived vacuoles. The bacteria have also been shown to use theautophagy process to escape to the cytosol, however other studies have shown that B. cenocepacia canbe killed through this process. Based on the literature described in section 1.1.4.
Schwab et al. have very recently confirmed this by studying lung tissue excised at transplantation or
autopsy, infected with different Bcc bacteria and P. aeruginosa. In fact, they showed that both species
create different niches [140]. Using immunohistochemistry, they were able to show that Bcc bacteria
9

are found in bronchi and, very frequently, they are inside macrophages within mucus, without any evi-
dences of biofilm formation [140]. Contrastingly, P. aeruginosa were found to locate in bronchial luminal
mucopurulent material, forming macro-colonies as in biofilm-like structures [140]. Interestingly, in lungs
co-infected with both species, Bcc bacteria were present in higher numbers and biofilms were not ob-
served [140]. This study emphasizes an important role for intracellular Bcc during human infection.
Studies have been carried out to understand how B. cenocepacia is able to survive inside macrophages
and how the bacteria evade killing by the host immune response. Infection assays using amoeba demon-
strated that B. cenocepacia can survive in an acidified compartment [132, 141]. In murine RAW264.7
macrophages it was subsequently shown that B. cenocepacia J2315 can delay maturation of phagolyso-
somes. It was also demonstrated that the BcCV did not acidify normally, reaching a pH of 6.4, in contrast
to heat killed bacteria that ended up in phagolysosomes with a pH of 4.5 [132]. Hence the bacteria are
capable of altering the acidification of the vacuole. Keith and colleagues observed that intracellular B.
cenocepacia interferes with the formation of an active NADPH oxidase complex in macrophages, delay-
ing by 6 hours the assembly or the recruitment of the NADPH phagocyte oxidase on the BcCV membrane
(more pronounced in CFTR-defective cells), and reducing the production of superoxide [142].
Furthermore, BcCV maturation was demonstrated to be delayed, by assessing fusion with late en-
dosomes. The fusion of the BcCV’s with the early endosomes was analyzed with the early endosome
marker autoantigen 1 (EEA1), and demonstrated that this interaction is achieved shortly after the in-
ternalization of the bacteria. However the fusion with late endosomes, verified by lysosome-associated
membrane protein (LAMP-1), was not achieved before 6 hours post-infection (hpi), in contrast to heat-
killed bacteria that fused with late endosomes at 30 minutes pi [132].
Al-Khodor and colleagues recently demonstrated that B. cenocepacia J2315 only transiently inter-
acted with the endocytic pathway [143]. In contrast to most studies published thus far, they showed
that the bacteria are able to escape rapidly to the cytosol [143]. Their tests were performed in murine
macrophages, and they observed that after 1 hpi only 30% of live bacteria were co-localizing with EEA1.
In time points up to 8 hpi, only 20 to 30% of the vacuoles that contained bacteria were co-localizing with
LAMP-2. They observed that the bacteria resided transiently in single membrane phagosomes, but that
after 3 hpi they were present in membrane damaged vesicles, possibly allowing the bacteria to escape
to the cytosol. Moreover, they verified that the escaped B. cenocepacia localized closely to the ER,
demonstrating that 50% of the bacteria was co-localizing with KDEL marker [143]. They detected that
the escaped bacteria were marked by the ubiquitin conjugation system [143], and at 4 hpi 70 to 80%
of the bacteria co-localized with the autophagy adaptor proteins p62 and NDP52, which are normally
recruited to ubiquitin targeted complexes, as well as with the LC3B autophagy marker [143].
Whether the difference in intracellular bacterial trafficking observed in the different studies is caused by
the use of different cell types or strain differences, or whether B. cenocepacia has multiple mechanisms
to escape its degradation, remains to be elucidated.
10

CFTR-defective cells
To better understand the behavior of the bacteria in CF infected patients, studies have also been
performed in CFTR-defective macrophages. These have demonstrated that the maturation of BcCVs
in those macrophages is delayed to a higher extent than that observed in normal macrophages [144].
Moreover, Lamothe et al. verified that this delay in CFTR-defective macrophages is specific to live
B. cenocepacia, and the malfunction of the CFTR regulator inhibits the clearance of the intracellular
infection [144].
Sajjan et al. described the intracellular trafficking of B. cenocepacia K56-2 in airway CF epithelial
cells, IB3 [145]. Their study demonstrated that the bacteria were able to escape the classical endocytic
pathway, preventing the maturation of lysosomes, shown by the low percentage of co-localization of live
bacteria with cathepsin D, a lysosomal acid hydrolase [145]. They also observed that the autophago-
somes originated in the ER [145]. Therefore, they concluded that, after escaping the endocytic pathway,
the bacteria reside and replicate inside ER-derived autophagosomes [145].
Together with impaired phagolysosomal killing in CFTR-defective cells, dysfunction of this regulator
leads to deficient autophagy [146]. Autophagy is a physiological process that not only helps the cell to
keep metabolic balance but also augments the innate response to intraphagosomal pathogens [147].
Assani et al. studied the influence of IFN-γ, which is used in CGD patients to prevent infections with
Burkholderia, in stimulating the autophagy in CF macrophages [148]. The use of IFN-γ demonstrated
increased clearance of pathogens in macrophages as well as decreased inflammatory cytokine produc-
tion [148].
Based on the capacity of B. cenocepacia to escape normal endocytic degradation, its intracellu-
lar survival has been suggested to be important for virulence and invasiveness of the bacteria [149].
Although B. cenocepacia has been observed in mice in experimental infection models, and in human
alveolar macrophages as described above, a role for an intracellular strategy in virulence is not clear
and difficult to study in animal models.
1.2 Bcc infection models
Through the years many infection models have been developed and used to study Bcc virulence.
These helped not only to characterize the pathogen but also to understand their behavior in a living
system. There is a great variety in the model hosts used: from vertebrates and invertebrates to protozoa
and plants. Each model has its own advantages and disadvantages and it is important to note that there
is no perfect model to study cystic fibrosis airway infections until now. Specific questions are addressed
in individual models, and a combination of different models may help in better understanding complex
virulence factors and their role in virulence [106, 150].
Table 1.2 summarizes the infection models developed for Bcc strains, which were used for assessing
Bcc virulence, and studying host response and intracellular trafficking, giving some examples of studies
performed.
11
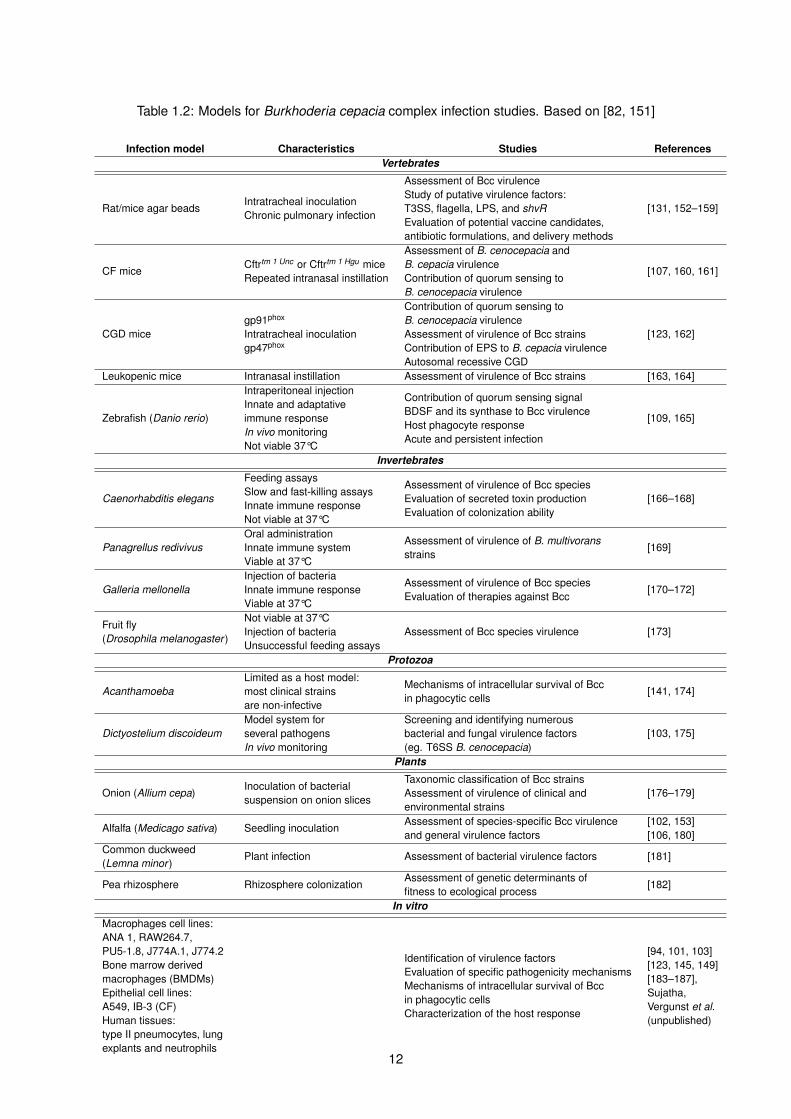
Table 1.2: Models for Burkhoderia cepacia complex infection studies. Based on [82, 151]
Infection model Characteristics Studies ReferencesVertebrates
Rat/mice agar beadsIntratracheal inoculationChronic pulmonary infection
Assessment of Bcc virulenceStudy of putative virulence factors:T3SS, flagella, LPS, and shvREvaluation of potential vaccine candidates,antibiotic formulations, and delivery methods
[131, 152–159]
CF miceCftrtm 1 Unc or Cftrtm 1 Hgu miceRepeated intranasal instillation
Assessment of B. cenocepacia andB. cepacia virulenceContribution of quorum sensing toB. cenocepacia virulence
[107, 160, 161]
CGD micegp91phox
Intratracheal inoculationgp47phox
Contribution of quorum sensing toB. cenocepacia virulenceAssessment of virulence of Bcc strainsContribution of EPS to B. cepacia virulenceAutosomal recessive CGD
[123, 162]
Leukopenic mice Intranasal instillation Assessment of virulence of Bcc strains [163, 164]
Zebrafish (Danio rerio)
Intraperitoneal injectionInnate and adaptativeimmune responseIn vivo monitoringNot viable 37°C
Contribution of quorum sensing signalBDSF and its synthase to Bcc virulenceHost phagocyte responseAcute and persistent infection
[109, 165]
Invertebrates
Caenorhabditis elegans
Feeding assaysSlow and fast-killing assaysInnate immune responseNot viable at 37°C
Assessment of virulence of Bcc speciesEvaluation of secreted toxin productionEvaluation of colonization ability
[166–168]
Panagrellus redivivusOral administrationInnate immune systemViable at 37°C
Assessment of virulence of B. multivoransstrains
[169]
Galleria mellonellaInjection of bacteriaInnate immune responseViable at 37°C
Assessment of virulence of Bcc speciesEvaluation of therapies against Bcc
[170–172]
Fruit fly(Drosophila melanogaster )
Not viable at 37°CInjection of bacteriaUnsuccessful feeding assays
Assessment of Bcc species virulence [173]
Protozoa
AcanthamoebaLimited as a host model:most clinical strainsare non-infective
Mechanisms of intracellular survival of Bccin phagocytic cells
[141, 174]
Dictyostelium discoideumModel system forseveral pathogensIn vivo monitoring
Screening and identifying numerousbacterial and fungal virulence factors(eg. T6SS B. cenocepacia)
[103, 175]
Plants
Onion (Allium cepa)Inoculation of bacterialsuspension on onion slices
Taxonomic classification of Bcc strainsAssessment of virulence of clinical andenvironmental strains
[176–179]
Alfalfa (Medicago sativa) Seedling inoculationAssessment of species-specific Bcc virulenceand general virulence factors
[102, 153][106, 180]
Common duckweed(Lemna minor )
Plant infection Assessment of bacterial virulence factors [181]
Pea rhizosphere Rhizosphere colonizationAssessment of genetic determinants offitness to ecological process
[182]
In vitro
Macrophages cell lines:ANA 1, RAW264.7,PU5-1.8, J774A.1, J774.2Bone marrow derivedmacrophages (BMDMs)Epithelial cell lines:A549, IB-3 (CF)Human tissues:type II pneumocytes, lungexplants and neutrophils
Identification of virulence factorsEvaluation of specific pathogenicity mechanismsMechanisms of intracellular survival of Bccin phagocytic cellsCharacterization of the host response
[94, 101, 103][123, 145, 149][183–187],Sujatha,Vergunst et al.(unpublished)
12

Besides the use of CFTR defective mice and rats, new studies show the production of CFTR-null
and CFTR-∆F508 heterozygous pigs [188] and CFTR-knockout ferrets [189]. These two models share
many similarities with human lungs from its anatomy to cell biology [188, 189]. Zebrafish are another
recent addition to the list of vertebrate infection models of human disease. This model, especially the
young embryos are highly amenable for studies of host-pathogen interactions at the (intra)cellular level
and innate immune response, and will be described in more detail below.
1.2.1 Zebrafish as an infection model
Danio rerio has been studied since the 1930s as a classical developmental and embryological model,
and in recent years it has also become an excellent model in the study of infectious (human) disease
and immunology (reviewed in [190]). One of the great advantages of using this animal as a model
for infection studies is the transparency of its embryos, allowing for real-time observation of infection.
The use of fluorescently labeled bacteria and cell-specific fluorescent reporter fish allows assessing
host-pathogen interactions at the cellular level in vivo using intravital imaging, and study cell biological
events.
Importantly, zebrafish have an innate as well as an adaptive immune system [191], which develops
at 2 to 3 weeks of embryo development. The innate immune response in the young embryos involves
phagocytic cells, including macrophages and neutrophils, as well as innate immune signaling pathways,
acute phase response and complement pathways with high similarity to that of humans [192]. Primitive
macrophages start appearing at 18 hours post fertilization (hpf) from the anterior lateral plate mesoderm,
further differentiating in the yolk sac [193]. The cells then migrate to the head mesenchyme, where they
differentiate into microglial cells, or to the blood circulation [193, 194]. The onset of blood circulation is
around 28 hpf, and circulating macrophages not only remove apoptotic residues but are also capable
of sensing and killing intravenously circulating microbes [193]. Immature neutrophils are also capable
of eliminating microbes at this stage, however they start to form granulocytes at around 33-35 hpf [195]
and are more efficient in scavenging surface-associated bacteria [196].
Many of the components known in mammalian innate immune signaling are conserved in teleost
fish, such as the Toll-like receptors (TLR) and class II cytokine signaling systems [197]. The proteins
involved in downstream signaling like kinases, adaptors, Stats, Trafs and transcriptional regulators are
also conserved [197].
TLR proteins are key components of the innate immune system, recognizing conserved motifs on
pathogens, pathogen-associated molecular patterns (PAMPs) [198, 199]. The proteins are expressed
in the membrane of macrophages recognizing PAMPs, bacterial derived ligands such as LPS, DNA,
flagellin, and other danger-associated pattern molecules (DAMPs) [200]. They have been investigated
in zebrafish, since these are important for recognition of threats to the organism. Key components of
the TLR-signaling pathway in zebrafish have been characterized, for instance the TIR adaptor proteins
MyD88, TIRAP, TRIF, and TRAM, TRAF6, and further downstream signaling factors IRF3 and IRF7 [201].
For TLR2, TLR3 and TLR5 it has been shown that their specificity is conserved between mammals
13

and fish [202–204]; however TLR4 in zebrafish does not seem to be activated in a similar manner to
extracellular LPS because of differences in the extracellular domain [205, 206].
More recently zebrafish has come to light as a tractable vertebrate model system to study various
diseases such as cancer, congenital and hereditary diseases, as well to understand infectious diseases
and the immune system [207]. Different infectious diseases have been studied, focusing on viral, fungal
and bacterial infections [208], and some examples are outlined in more detail below (reviewed in [209]).
Mycobacterium marinum
Studies with M. marinum established zebrafish as a model for human tuberculosis [210]. The infec-
tions were followed in real time, showing that bacteria replicate in macrophages as in human tuberculosis
[211]. Moreover, it was demonstrated that the bacteria controlled granuloma formation, which was not a
host response, but depended on the bacterial secretion system Esx-1 [212]. Recently, Cambier et al. de-
scribed that M. marinum and M. tuberculosis can recruit and infect permissive macrophages other than
microbicidal ones by using cell-surface-associated phthiocerol dimycoceroserate lipids [213]. Through
this mechanism, the bacteria evade being killed by reactive nitrogen species (RNS) [213].
Salmonella Typhimurium
When infected with Salmonella Typhimurium zebrafish are killed by an inflammatory infection, show-
ing an immune response similar to the one in mammals [202]. Transcriptional analysis of Salmonella and
Mycobacterium marinum bacterial infections led to the characterization of several infection-responsive
genes encoding cytokines and chemokines, transcription factors, and complement factors based on the
innate immune response in the embryos [214]. By comparing the results of both infection studies, fish in-
fected with M. marinum showed to have more genes down-regulated [214]. Additionally, by overlapping
the transcriptional results, Ordas et al. found differences in both profiles, but the inflammatory response
was similar, shown by the 206 commonly up-regulated genes associated to host defense (including
apoptosis, complement activation and acute phase response, cytokine and chemokine activity) and cy-
toskeletal structure, and the two only commonly down-regulated genes encoded intermediate filament
protein and a ribosomal protein [214].
Pseudomonas aeruginosa
Experiments with P. aeruginosa demonstrate that at 2 hpi most bacteria are inside cells, which were
identified as neutrophils and macrophages, and the cells are efficient in killing a large part of the mi-
crobes in a short time period [215]. Moreover, Brannon et al. showed that pu.1 morphants1 were more
susceptible to the infection than control embryos, emphasizing that the macrophages are an important
host defense against infection with P. aeruginosa [215]. Infections with a T3SS mutant of P. aeruginosa
1The differentiation and growth of macrophages and neutrophils in zebrafish embryos are dependent on the myeloid transcrip-tion factor gene pu.1 [216, 217]). The injection of a modified antisense oligonucleotide (morpholino) directed against pu.1 in theeggs, can deplete phagocytic cells, creating pu.1 morphants.
14

in pu.1 embryos showed that the secretion system acts to protect the bacteria from the phagocytes
[215].
Staphylocccus aureus
From Staphylocccus aureus studies in zebrafish it was found that these bacteria can also survive and
replicate in phagocytes [218]. In intravenous infection, the bacteria are taken up either by macrophages
or neutrophils (contain much less bacteria), and these cells, most of the time, are capable of controlling
the infection [219]. Using mathematical modeling of pathogen population dynamics, S. aureus was found
to have intracellular reservoirs in neutrophils, from which the bacteria then evades and disseminates
[220].
Burkholderia cepacia complex
In zebrafish embryos, the importance of macrophages for intracellular survival of Bcc strains has also
been demonstrated. The strains J2315 and K56-2 have been previously described as highly virulent in
other infection models, and when injected intravenously in zebrafish embryos an acute inflammatory
infection was observed [109]. The embryos died rapidly after infection (2 to 3 days post-infection (dpi)
with K56-2 and J2315, respectively) with high bacterial replication rates throughout that time [109]. In
contrast to the acute inflammatory infection induced by K56-2 and J2315, infections with B. stabilis
LMG14294 and B. vietnamiensis LMG 18836 had a different outcome: the embryos survived until 8 dpi,
when the experiment was terminated, however the bacteria were able to persist in most embryos and
sometimes, at later stages (5 dpi) caused more severe infection [109].
The zebrafish embryo has proven to be a good model to study different microbial infections, allowing
for the study of the role of phagocytes, intracellular bacterial stages, and immune response, in inflam-
mation and virulence, in large detail. Together with disease modeling, zebrafish can be used for drug
screening helping to determine not only the target of action but also the mechanism of action [221]. In or-
der to increase the efficiency of these studies, automated high-throughput screens are being developed
[222].
1.3 ShvR – a global regulator of gene expression
In this study the main focus is on global regulator of gene expression, the shvR gene in B. cenocepa-
cia K56-2. Bernier and colleagues described the identification of spontaneous shiny variant colonies
(shv ) in B. cenocepacia K56-2 on agar plates with affected virulence [102]. The gene responsible for the
different morphotype, BCAS0225 (shvR, Genbank: AM747722.1), was identified as a gene encoding a
LysR-type transcriptional regulator (LTTR).
LTTRs are a large family of extensively studied regulators, highly conserved in bacteria, with func-
tional orthologs present in archaea and even eukaryotic organisms [223–225]. These regulators activate
15

divergent transcription of linked targets genes on unlinked regulons that are involved in many different
functions: metabolism, virulence factors, biosynthesis of amino acids, nitrogen fixation, oxidative stress
response, quorum sensing, toxin production, among many others (reviewed in [226]). For some LTTRs
the signal that activates the regulator has been identified, as for instance the quinolone signaling path-
way in Pseudomonas, PQS, that activates PqsR (or MvfR) [227], however, for ShvR the activating signal
is not yet known.
By mutagenic studies and amino acid sequence comparisons, three types of binding domains were
found in LTTRs: a DNA-binding domain employing a helix-turn-helix (HTH) motif, domains for co-inducer
response/recognition and a domain required for DNA binding as well as co-inducer response [226].
Figure 1.3 represents the secondary structure prediction of the ShvR protein from B. cenocepacia J2315.
As for many other LTTR, it was shown that ShvR negatively regulates its own expression [228].
Figure 1.3: ShvR predicted secondary structure, from PSIPRED [229]. Pink cylinders represent helices,yellow arrows β-strands and black lines coils.
To further examine the target genes regulated by ShvR, a transcriptome analysis was performed,
comparing a shvR::Tp mutant to its wildtype parent K56-2 grown in LB medium [228]. Over a thousand
genes were found to be expressed differentially in the shvR mutant compared to the wildtype, and
included quorum sensing cepIR, zinc metalloproteases (zmpA and zmpB), type II secretion system
(T2SS) genes, lipase encoding genes and afc genes, important in antifungal activity, among others
[228]. The regulator was shown to negatively regulate both cep and cci QS systems and approximately
40% of the ShvR target genes are co-regulated by either CepIR or CciIR, even though it independently
regulates biofilm formation and rough colony morphology [228].
Bernier et al. observed that shiny variants appeared in less than 1% of the plated population after
growth in shaken cultures [102]. However in static culture conditions the percentage of shv colonies
increased after three passages, showing a higher mutation rate under stress conditions, and higher
adaptability of the mutant to this growth environment [102]. The shvR mutant was found to be defective
in biofilm formation as well as in extracellular matrix (ECM) formation [102].
The shvR mutants were avirulent in an alfalfa seedling infection model, with lower CFU numbers
16

than the wildtype counted after 5 days post-infection [102]. In the same study, experiments using a rat
agar bead model of chronic pulmonary infection demonstrated that not only virulence was reduced, but
also lung inflammation and in vitro biofilm formation were significantly decreased [102]. In an apparent
contrast, the number of CFU from ∆shvR recovered from the infected rats was, in most cases, higher
than in the wild-type [102], suggesting than the mutant has increased persistence.
1.4 Motivation
Infection experiments in zebrafish embryos confirmed that the shvR mutant is attenuated in virulence,
yet able to persist in macrophages (Subramoni, Vergunst et al., unpublished).
The aim of my project is to better understand the role of ShvR in virulence in vivo, by further studying
the persistent phenotype using a zebrafish embryo model of infection, and to develop new tools to aid in
better understanding the role of ShvR in regulating the difference between acute inflammatory infection
and persistence in macrophages. The project is divided in several objectives:
• Analysis of the host immune response to infection:
– Infection phenotype (using survival and kinetics assays, and real time analysis)
– Immune cell behavior (neutrophil and macrophage behavior during infection)
• Development of plasmids and tools to better study the role of ShvR in virulence and persistence:
– Optimization of the complementation of a shvR mutant
– Observation of shvR expression during infection
17

18

Chapter 2
Materials and Methods
2.1 Bacterial strains, plasmids and growth conditions
The bacterial strains and plasmids used in this study are described in Tables 2.1 and 2.2. Escherichia
coli and Bcc strains were cultured at 37°C in Luria-Bertani (LB) broth, with or without 1.6% agar. Both
E. coli and Bcc strains carrying plasmids encoding chloramphenicol resistance (CmR) were grown in
the presence of 30 and 100 µg/mL, respectively, of the antibiotic. For selection of Bcc with trimethoprim
resistance (TpR) a concentration of 50 µg/mL trimethoprim was used and for E. coli 100 µg/mL. Selection
of E. coli colonies using pUC29 plasmid with ampicillin resistance (AmpR) was used at a concentration
of 150 µg/mL of ampicillin.
Table 2.1: Bacterial strains used in this study.
Bacterial strains Description Reference
B. cenocepacia K56-2
(LMG18863)ET12, Toronto, Canada, CF [230]
B. cenocepacia K56-2 ∆shvR Unmarked shvR deletion derivative of K56-2 [228]
B. vietnamiensis FC441
(LMG18836)
9 year old boy with X-linked recessive CGD who
survived septicemia[231]
B. stabilis
(LMG14294)
Belgian CF patient, stable condition; detected in
one other patient[232]
E. coli DH5αϕ80lacZ∆M15 ∆lacU169 endA1 recA1 hsdR17
supE44 thi-1 gyrA96 relA1
19

Table 2.2: Plasmids used in this study.
Plasmids Description Reference
pIN29 oripBBR ∆mob, CmR, tac-DsRed [109]
pIN233 oripBBR ∆mob, CmR, tac-mCherrySubramoni, Vergunstet al., unpublished
pCR11 CmR pMR10 (Mohr and Roberts, unpublished) derivative Gift from M. KovachpAV100 Hind III/XbaI fragment from pIN29 cloned into pIC20R [233] A. Vergunst
pUCP28T-shvRDerivative of pUCPT28T [234] with 1.7kb Pst I-BamHIfragment containing BCAS0225 andupstream region, TpR
[228]
pAV209pIN233 with ∼600bp NdeI-Hind III fragment containingthe upstream region of BCAS0225, CmR This study
pMG1pUC29 with ∼600bp fragment containing the upstreamregion of BCAS0225, AmpR This study
pMG2pUC29 with ∼1.6kb fragment containing BCAS0225 andupstream region, AmpR This study
pMG3pIN29 with ∼1.6kb XhoI-Hind III fragment containingBCAS0225 and upstream region, CmR This study
pMG4pCR11 with 2.473kb SpeI-XbaI fragment containingBCAS0225 and upstream region and DsRed, CmR This study
pMG5 pAV209 with 45bp BsrGI-SacI oligo, CmR This study
pMG6
pUC29 with ∼1.7kb NcoI-Nar I fragment containingBCAS0225 and upstream region from pMG3 and∼800bp XbaI-ClaI fragment containing DsRed frompAV100, AmpR
This study
2.2 DNA manipulations
2.2.1 Extraction and purification of plasmid DNA
Plasmid DNA extraction was performed using QIAprep® Spin Miniprep Kit (QIAGEN) with overnight
cultures of E. coli and Bcc strains, following the manufacturer’s instructions. DNA fragments used in
cloning procedures were purified from agarose gels with a MinElute® Gel Extraction Kit (QIAGEN),
according to the manufacturer’s instructions.
2.2.2 Polymerase chain reaction (PCR) conditions
B. cenocepacia K56-2 genomic DNA was used as a template for PCR amplification of the promoter
of the shvR gene and for the region encompassing both the promoter and the gene. A thermocycler
was used to amplify specific fragments (Table 2.3) using primers described in Table 2.4. The conditions
used for a 25 µL reaction volume: 100 ng of template DNA, 0.5 µM of each primer, 200 µM of each
deoxynucleotide (Invitrogen) and 0.06 U/µL of Pfu DNA Polymerase (Promega). The samples were
subjected to an initial denaturation at 95°C for 3 minutes, followed by 30 cycles of: denaturation (95°C
for 45 seconds), annealing (57°C for 30 seconds) and elongation (72°C for 2 min/kb of expected product).
After a final elongation at 72°C for 5 minutes, the samples were stored at 4°C until use.
20

Table 2.3: PCR amplification products.
Amplification product Product size (nt) Template Primers
shvR promoter 596 B. cenocepacia K56-2pshvR forpshvR rev
shvR promoter and gene (BCAS0225) 1593 B. cenocepacia K56-2pshvRXhoI forshvRXbaI rev
Table 2.4: Primers sequence and PCR conditions. Restriction sites in bold.
Primer/Oligo Sequence (5’–3’)Annealingtemperature
pshvR for 5’-GAACATATGTCTCACATTAGCCATACCGCCGC57°C
pshvR rev 5’-TGCAAGCTTCGAATTCCGCCCGACATGCGpshvRXhoI for 5’-AATTCTCGAGGAATTCCGCCCGACATGCGC
57°CshvRXbaI rev 5’-GTTCTAGACTATCCGACGCGATACATCGGCunst1 5’- GTACAAGCGCCGCAACGACGAATACCGCCTGGTCCGCTAGGAGCTunst2 5’- CCTAGCGGACCAGGCGGTATTCGTCGTTGCGGCGCTT
2.2.3 Agarose gel electrophoresis
Agarose gel electrophoresis was carried out as described by Sambrook et al. [235]. Agarose (Invitro-
gen™, UltraPure™ Agarose) at different concentrations, usually 1% (wt/v) in TBE 0.5X buffer (TBE 10X
– 1.0 M Tris, 0.9 M boric acid, 0.01 M EDTA. Dilution of 1:20 of this buffer was performed to obtain TBE
0.5X), depending on the size of fragments to be separated, were used to migrate the DNA fragments.
As a molecular weight marker the 1 kb Plus DNA Ladder (Invitrogen™) was used. Loading buffer (10X
Orange G DNA loading buffer – 0.5 g/mL of sucrose, 2.5 mg/mL of Orange G) was added to the DNA
samples, prior to separation of DNA fragments by gel agarose electrophoresis in TBE 0.5X buffer at
100V (5.5 V/cm).
The agarose solution was stained with ethidium bromide (final concentration 0.5 µg/ml) and the DNA
was visualized under UV light in a transilluminator, and photos of the gels were taken with a CCD camera
(Vilber Lourmat).
2.3 Bacterial transformation
2.3.1 Electroporation of Burkholderia
For electroporation, the bacteria/DNA mix should not contain high concentrations of salts. The DNA
was previously purified and stored at -20°C. From overnight grown bacterial cultures, 4 mL were washed
with ddH2O at 4°C and a glycerol solution of 10% at 4°C, three times each, by centrifugation at 7,000 rpm
for 1 minute, and gently resuspending the bacterial pellet using 1 mL of water or the glycerol solution.
After the washing steps, aliquots of 40 µL were stored at -80°C.
After the addition of 50-100 ng/µL of DNA, the cells were electroporated using a MicroPulser™
21

Electroporation Apparatus, in a cooled cuvette with a voltage of 2.5 kV for 3.8 ms (with fixed capacitor at
10 µF and 600 Ω resistor in parallel and 30 Ω resistor in series). For cell recovery, 1 mL of SOB medium
(2% bacto-tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10mM MgCl2, 10 mM MgSO4, filter
sterilized) was added rapidly after applying the electric pulse, and the bacteria were transferred to a
shaking incubator, at 100 rpm and 37°C, to let the cells recover.
The cells were then plated on selective agar medium and incubated at 37°C for two days. Uptake of
the plasmids was verified by fluorescence, in case the plasmid encoded a fluorescent reporter protein,
and/or by plasmid isolation and verification on gel.
2.3.2 E. coli competent cells
E. coli cells were prepared according to Inoue et al. [236] and stored at -80°C in 100 µL aliquots.
For transformation, 20 ng of DNA (or 10 µL from ligation mixture) was added and the suspension let to
rest on ice for 10 min. After, the cells were heat-shocked for 90 s at 42°C and 1 mL of SOB medium was
added for cell recovery. The suspension was transferred to a shaking incubator, at 100 rpm and 37°C,
to let the cells recover. The cells were then plated on selective agar medium and incubated at 37°C for
one day.
2.4 Construction of plasmids
The plasmid construction, including cloning schemes, is also described in section 3.
2.4.1 Construction of a pshvR-mCherry reporter plasmid (pAV209)
This plasmid places the reporter gene mCherry under control of the shvR promoter sequence. The
promoter region was obtained by PCR using Pfu polymerase, K56-2 chromosomal DNA as a template
and the primers pshvR for and pshvR rev (Table 2.4), and the blunt-ended fragment was cloned in
EcoRV-digested pUC29. Blue/white screening was performed, selecting white colonies on LB-agar
plates containing 40 µL of X-gal 20 mg/mL and 10 µL of 100 mM IPTG. These were then verified by
sequencing. Next, the fragment was cloned in pIN29, using NdeI and Hind III restriction sites, resulting
in pAV209. Just upstream the pshvR-mCherry cassette there is a strong terminator sequence, trpA, to
prevent read-through from other expression units on the plasmid.
2.4.2 Construction of shvR complementation plasmids (pMG3 and pMG4)
For genetic complementation of a B. cenocepacia K56-2 shvR mutant, chloramphenicol resistant
versions of pBBR (medium copy number) and pMR10 (1-2 copies) derived plasmids, pMG3 and pMG4
respectively, were constructed that contained the shvR promoter and coding region. To achieve the first
construct, pMG3, the shvR cassette was amplified using PCR with Pfu polymerase, K56-2 chromosomal
DNA as a template and the primers pshvRXhoI for and shvRXbaI rev (Table 2.4), and the fragment was
22

cloned into SmaI-digested pUC29. White colonies were selected on LB-agar plates containing 40 µL of
X-gal 20 mg/mL and 10 µL of 100 mM IPTG and sequenced. Then the pshvR-shvR cassette was cloned
in pBBR-derived pIN29, using XhoI and Hind III restriction sites, creating pMG3. This plasmid contains
the pshvR-shvR gene and also a constitutive tac-DsRed reporter gene to be able to follow the bacteria
in real time in zebrafish infection experiments after introduction into the different B. cenocepacia strains.
The second vector was based on pCR11, a Cm-resistant pMR10-derivative (kindly provided by M.
Kovach) as the backbone vector. The shvR gene, isolated from pMG3 using NcoI and Nar I restriction
sites, was cloned in pCR11 (digested with SpeI and XbaI) together with a ptac-DsRedreporter gene,
isolated from plasmid pAV100 using XbaI and ClaI restriction sites. We decided to first clone the two
fragments, containing the shvR and DsRed genes, in cloning vector pUC29, resulting in pMG6, so that
the final cloning in pCR11 would only involve one fragment. The final plasmid, pMG4, is still being
constructed and will be analyzed, together with pMG3 for complementation in infection experiments.
2.4.3 Construction of a plasmid expressing an unstable mCherry reporter (pMG5)
This plasmid used pAV209 as a template. Two complementary DNA oligos encoding the sequence
AANDENYALVA were ordered (Sigma, see Table 2.4), annealed, and cloned into BsrGI/SacI digested
plasmid pAV209, resulting in pMG5.
2.5 Zebrafish infection model
2.5.1 Zebrafish care and maintenance
The zebrafish infections were performed as described in [237]. Briefly, the zebrafish were kept in
3 L (or 8.5 L) tanks containing conditioned water at 28°C [238], with a pH of 7.2 (Zebtec standalone
system from Tecniplast), with a light regime of 10 h of darkness and 14 h of light. Zebrafish (Danio rerio)
were kept and handled according to national regulations for animal welfare (ID 30-189-4 and CEEA-
LR-12186). Experiments were performed using zebrafish embryos and terminated before the larvae
reached the independent feeding stage.
In order to obtain embryos by natural spawning, males and females were put together the evening
before in spawning tanks, which contain an inner tank with holes. These are necessary to avoid that the
parents eat the eggs. The eggs are usually laid half an hour (sometimes longer) after the light in the room
is turned on automatically. On average, a female can lay between 50 to 200 eggs. The eggs are then
removed from the tanks and washed using a very fine fish net to rinse the eggs under running tap water,
and remaining faecal materials, and non-fertilized eggs are removed under a dissecting microscope.
The embryos are then raised in E3 egg water (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33
mM MgSO4, 0.00005% methylene blue (MB)), at 29.5°C. MB is an antiseptic used in aquaria mainly
against fungal growth, however it has low antimicrobial activity. In experiments that required imaging for
fluorescence quantification, and for qRT-PCR analysis the MB was not added, since it can accumulate in
23

the yolk and cause autofluorescence, and has been shown to have a short term effect on the expression
of pro-inflammatory response genes (personal communication with J. Mesureur).
2.5.2 Transgenic zebrafish lines
For infection experiments, Danio rerio AB and the Golden variety [239] were used as wildtypes.
The Golden variety lacks pigmentation due to a mutation in the gene slc24a5, and is preferred for
lower background with imaging (fluorescence, light microscopy, histology). The transgenic reporter lines
Tg(mpeg1::mCherry-F ) and Tg(mpx::GFP) expressing mCherry specifically in macrophages and GFP
specifically in neutrophils, respectively, were used for analysis of host phagocyte behavior [240, 241]. In
some experiments, embryos from crosses between the two transgenic lines were used.
2.5.3 Microinjection of bacteria in zebrafish embryos
Microinjection was performed as described by Mesureur and Vergunst [237]. Briefly, the different
B. cenocepacia strains (described in section 3) were grown overnight in LB broth with appropriate an-
tibiotics on a rotary platform at 37°C. The bacteria were inoculated directly from -80°C glycerol stocks,
except for B. stabilis which was grown on fresh plates overnight, and subsequently inoculated in liquid
culture, grown overnight at 37°C. The next day, bacteria were collected by centrifugation at 7,000 rpm
for 2 min and resuspended in phosphate buffered saline (PBS) (Gibco). The OD600 was measured and
bacterial dilutions of 50 bacteria/nL were prepared in PBS (with 0.05% phenol red to visualize microin-
jection).
The embryos were previously dechorionated about 2 hours before microinjection. Embryos, staged
between 28 and 32 h post-fecundation (pf), were transferred using glass Pasteur pipettes into cell culture
quality dishes (Greiner Cellstar 60-15 mm) to avoid the adherence of the embryo tails to the bottom of
the plastic plate. To avoid wounding the embryos, which can induce the immune response, great care
has to be taken and the embryos were pipetted “head first”.
For injection, the embryos were placed on agar plates with E3 medium containing 0.02% buffered
MS222 (tricaine; ethyl-3-aminobenzoate methanesulfonate salt; Sigma). The injection was performed
with a Femtojet microinjector (Eppendorf) and a micromanipulator with pulled microcapillary pipettes (P-
1000 Micropipette puller, Sutter Instruments), and microcapillaries (Borosilicate Glass Capillary Tubes
with filament (OD 1.0 mm, ID 0.78 mm, 10 cm), Sutter Instrument Company) under a stereo light mi-
croscope (Leica MS5). The needle was loaded with 4 µL of bacterial suspension, and positioned in the
center of the visual field, so that it would be placed above the caudal vein of the blood island region.
About 1 nL of a bacterial suspension was microinjected directly into the blood circulation, either in the
blood island or in the axial vein (see Figure 2.1). In order to achieve those amounts, typically a pressure
of 400 hPa and an injection time of 0.6 seconds are used, adapted with varying needle tip wideness.
To determine the number of colony forming units (CFU) in the inoculum, 5 embryos were individually
plated onto LB selective agar plates immediately after microinjection (T=0). To follow bacterial kinetics,
inoculated embryos were incubated individually in E3 medium in 24- or 48-well plates at 29.5°C and
24

Figure 2.1: The microinjection site in the blood island is indicated (30 hpf zebrafish embryo).
sampled at 24 and 48 h post-infection (hpi), five embryos at each time point. To determine bacterial
numbers, embryos were rinsed in E3 medium and transferred to a 1.5 ml Eppendorf tube in 50 µL
(total volume) of tissue-culture-grade trypsin-EDTA (2%), followed by disruption by vigorous pipetting
(40 times) with a 200 µL yellow tip and incubated for precisely 20 min for T=0. For the other time
points, 50 µL of 2% Triton X-100 (in H2O) was then added, the tubes were gently “flicked”, and the
preparations were incubated for precisely 30 min at room temperature, which was followed by additional
homogenization by pipetting (20 times). Depending on the expected number of bacteria (an indication
can be obtained by using fluorescence microscopy to detect the fluorescent bacteria), the complete
mixture could be plated on LB selective agar plates or serial dilutions were prepared in PBS and plated
as 10 µL droplets, essentially as described by [237]. For K56-2 at 24 hpi 10 µL drops of 10-1, 10-2
and 10-3 dilutions, and at 48 hpi 10 µL drops of 10-1, 10-2, 10-3 and 10-4dilutions of infected embryos
are plated. As for K56-2∆shvR at 24 hpi dilutions up to 10-2 were done, plating the remaining 80 µL of
mixture, and at 48 hpi the dilutions went up to 10-3. Bcc strains are generally resistant to treatment with
trypsin and Triton-X 100 at the indicated concentration.
For survival assays, embryos were maintained individually in 24-well plates in E3 medium at 28°C.
At regular time points after infection, the number of dead embryos was determined visually based on the
absence of a heartbeat.
2.6 Statistical analysis
Statistical analyses were performed using Prism 6.0c (GraphPad) and are detailed in each figure
legend. Embryo survival data were presented in Kaplan-Meier survival plot, and a log-rank (Mantel-
Cox) test was used for statistical analysis. Kinetics data were represented in scatter plots, and t-tests
were performed to determine the p-value between different conditions.
2.7 Microscopic analysis
For microscopy, a Leica DM IRB inverted microscope equipped for bright-field, differential interfer-
ence contrast (DIC), and fluorescence imaging was used. GFP, DsRed/mCherry and m2-Turquoise were
excited using a 100 W mercury lamp, and fluorescence was detected using filter sets L5 (band pass [BP]
480/40; beam splitter [BS] 505; emission BP 527/30), N2.1 (515 to 560; BS 580; emission long pass
[LP] 590), A (340 to 380; BS 400; emission LP 425) and CFP (BP 436/20; BS 455; emission BP 480/40),
25

respectively. For imaging we used a Coolsnap fx (Roper Scientifique). Embryos were transferred to E3
medium containing MS222 in glass-bottom dishes (MatTek Corp., Ashland, MA) for direct visualization
using 40x and 63x oil objectives.
A Nikon AZ100 equiped for bright-field and appropriated filter for red and green fluorescence imaging,
and coupled with Coolsnap HQ2 (Roper Scientifique), were used for imaging embryos for further fluo-
rescence quantification. MetaVue software was used for imaging, and images were processed further
using Adobe Photoshop, or Image J.
2.7.1 Computational quantification of fluorescent host immune cells
The analysis of host immune cell numbers was performed according to [242] and will be briefly
described. The images of the transgenic infected embryos were obtained using the Nikon AZ100 micro-
scope and processed using Adobe Photoshop®, as mentioned above. The resulting RGB color images
were further processed in ImageJ 1.47v software, where they were converted to binary images (see
Figure 2.2). Once the black and white image was obtained, the total fluorescent area was measured as
well as 5 randomly selected individual cells per embryo to determine pixel size per phagocyte. The mea-
surements were processed using Excel®, where the total fluorescent area was divided by the average
of the 5 individual cells resulting in the total number of fluorescent cells in the embryo.
Figure 2.2: Binary conversion of fluorescent image of a zebrafish embryo expressing GFP specifically inneutrophils, Tg(mpx::GFP). (A) Fluorescent image of the embryo. (B) Binary image obtained from Im-ageJ. The black background is converted into white background, whereas the green cells are convertedto black pixels to be further analyzed.
26

Chapter 3
Results – A B. cenocepacia shvR
mutant is attenuated for virulence in a
zebrafish infection model
The zebrafish embryo model has been developed to study infections with Bcc strains. Whereas B.
cenocepacia K56-2 causes acute fatal infection with embryo death occurring in 2 days, other strains,
including B. stabilis LMG14294, induce a persistent infection with stable bacterial numbers, but the host
is unable to eradicate the bacteria that reside within macrophages [109].
Virulence in zebrafish embryos can be assessed by survival assays, in which generally 20 infected
embryos are followed throughout the experiment and the time of death is registered; bacterial multipli-
cation, in which for each experiment 5 infected embryos per time point are disrupted and the lysate is
plated for colony forming unit (CFU) determination; and in real time using fluorescently labeled bacte-
ria visualized with fluorescence microscopy. Embryo survival and bacterial kinetics studies, performed
previously in the lab, have shown that a shvR mutant is attenuated in virulence in zebrafish embryos
(Subramoni, Vergunst et al., unpublished). Here I confirmed these results, further studied the role of
host phagocytes during infection with the shvR mutant compared to its parent K56-2, and developed
new tools to be able to better study the role of this regulator in virulence.
3.1 Survival assays and bacterial kinetics
Firstly, the shvR mutant was analyzed in survival and kinetics assays. To this end, thirty-five em-
bryos (30 hours post fertilization (hpf)) were microinjected with B. cenocepacia K56-2, harboring re-
porter plasmid pIN29 (Table 2.2) to fluorescently label the bacteria, and a K56-2 shvR (pIN29) mutant.
Five embryos were used immediately after microinjection to determine the inoculum size (T=0). Twenty
randomly picked embryos were used for survival assays, in which the time of death was determined as
the moment the embryos have no more heartbeat. The remaining embryos were sacrificed at different
27
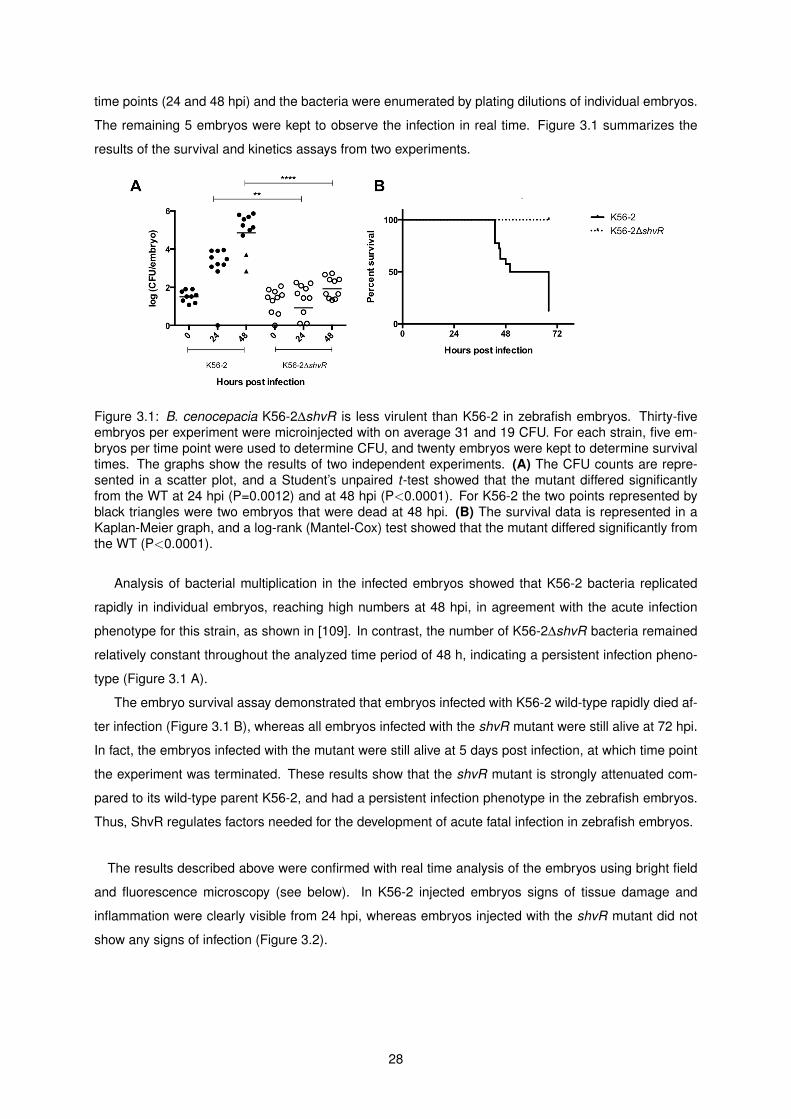
time points (24 and 48 hpi) and the bacteria were enumerated by plating dilutions of individual embryos.
The remaining 5 embryos were kept to observe the infection in real time. Figure 3.1 summarizes the
results of the survival and kinetics assays from two experiments.
Figure 3.1: B. cenocepacia K56-2∆shvR is less virulent than K56-2 in zebrafish embryos. Thirty-fiveembryos per experiment were microinjected with on average 31 and 19 CFU. For each strain, five em-bryos per time point were used to determine CFU, and twenty embryos were kept to determine survivaltimes. The graphs show the results of two independent experiments. (A) The CFU counts are repre-sented in a scatter plot, and a Student’s unpaired t-test showed that the mutant differed significantlyfrom the WT at 24 hpi (P=0.0012) and at 48 hpi (P<0.0001). For K56-2 the two points represented byblack triangles were two embryos that were dead at 48 hpi. (B) The survival data is represented in aKaplan-Meier graph, and a log-rank (Mantel-Cox) test showed that the mutant differed significantly fromthe WT (P<0.0001).
Analysis of bacterial multiplication in the infected embryos showed that K56-2 bacteria replicated
rapidly in individual embryos, reaching high numbers at 48 hpi, in agreement with the acute infection
phenotype for this strain, as shown in [109]. In contrast, the number of K56-2∆shvR bacteria remained
relatively constant throughout the analyzed time period of 48 h, indicating a persistent infection pheno-
type (Figure 3.1 A).
The embryo survival assay demonstrated that embryos infected with K56-2 wild-type rapidly died af-
ter infection (Figure 3.1 B), whereas all embryos infected with the shvR mutant were still alive at 72 hpi.
In fact, the embryos infected with the mutant were still alive at 5 days post infection, at which time point
the experiment was terminated. These results show that the shvR mutant is strongly attenuated com-
pared to its wild-type parent K56-2, and had a persistent infection phenotype in the zebrafish embryos.
Thus, ShvR regulates factors needed for the development of acute fatal infection in zebrafish embryos.
The results described above were confirmed with real time analysis of the embryos using bright field
and fluorescence microscopy (see below). In K56-2 injected embryos signs of tissue damage and
inflammation were clearly visible from 24 hpi, whereas embryos injected with the shvR mutant did not
show any signs of infection (Figure 3.2).
28

Figure 3.2: B. cenocepacia K56-2 and shvR mutant infection in the tail region of zebrafish embryos(Tg(mpx::GFP)). At 42 hpi embryos infected with WT show severe signs of tissue damage, whereasthe embryo infected with the mutant bacteria shows no visible tissue damage. Photo of B. cenocepaciaK56-2 courtesy of J. Mesureur.
3.2 Analysis of the behavior of host immune cells in infected em-
bryos
The transparent nature of the zebrafish embryos makes it possible to follow the infection progress in
real time using fluorescent bacteria, and embryos expressing different reporter proteins in specific host
cells. To better understand the behavior of macrophages and neutrophils during infection, reporter fish
lines expressing GFP in neutrophils Tg(mpx::GFP) and mCherry in macrophages Tg(mpeg1::mCherry-
F ) (Figure 3.3) were used.
Figure 3.3: Zebrafish embryos expressing (A) GFP specifically in neutrophils, Tg(mpx::GFP), and (B)mCherry specifically in macrophages, Tg(mpeg1::mCherry-F ).
In embryos injected intravenously with B. cenocepacia K56-2, bacteria have been shown to be
phagocytized mainly by macrophages, in which they start to replicate about 6 to 7 hours later. The
bacteria start to spread from such infected macrophages around 10-12 hpi, first locally, again replicating
inside cells, and resulting in local infection sites, sometimes forming visible cell aggregates [109]. At
such sites neutrophils are visibly recruited to infection sites (Mesureur et al., manuscript in preparation,
Figure 3.4). Then at about 16-24 hpi, depending on the infectious dose, the bacteria re-enter the blood
circulation resulting in a systemic infection. At 24 hpi, most embryos show signs of severe tissue de-
terioration and necrosis – some embryos lose parts of the body (cell aggregates) – and the blood flow
rate is severely reduced or has even stopped at this time point (Figure 3.4). At this time point, most
neutrophils are recruited to infection sites, and their numbers have started to decline, with often hardly
any macrophages and neutrophils detectable in the embryo, compared to non-infected embryos, which
have a steadily increasing neutrophil number during their development (Mesureur et al., manuscript in
preparation). The embryos succumb to K56-2 infection, depending on the infection dose, from 40-48
29

hpi.
Figure 3.4: B. cenocepacia K56-2 infection in zebrafish embryos (Tg(mpx::GFP x mpeg1::mCherry-F )and Tg(mpx::GFP)). Fluorescence images of different time points after infection. The smaller insetsshow the corresponding bright field image. After injection (2 hpi), bacteria (blue) co-localize mainly withmacrophages (red) but not with neutrophils (green). At 24 hpi massive neutrophil (green) recruitment toinfection sites (bacteria in red) can be observed. This recruitment causes tissue damage, being visiblein the bright field image (white arrow). At 42 hpi the infection has spread and almost no neutrophils arefound, and as seen in the bright light image, the tissue damage is extensive. The image at 42 hpi, is alsoshown enlarged in Figure 3.2. Photos at 2 and 24 hpi courtesy of A. Vergunst and 42 hpi of J. Mesureur.
In contrast to the acute fatal infection seen with K56-2, shvR-infected embryos, at 24 hpi, show no
signs of external deterioration even though bacteria can be seen in macrophages. After phagocytizing
shvR rapidly after microinjection, macrophages full of bacteria can already be observed before 24 hpi
(Figure 3.5). Often the whole cell was filled with bacteria, possibly in one large vacuole, but this has
to be further determined. ShvR-infected macrophages seemed larger than the ones infected with K56-
2, although this has to be further quantified. The results suggest that the bacteria replicate inside
macrophages and are able to establish a niche inside the macrophages. The shvR mutant bacteria
were not able to spread from the infected macrophages as wild-type bacteria, resulting in a (intracellular)
persistent phenotype.
Further microscopic observations indicated that although neutrophils are present in high numbers,
they are not recruited massively to the site of infected macrophages, as in K56-2 infected embryos.
Figure 3.5: B. cenocepacia K56-2∆shvR infection in zebrafish embryos (Tg(mpx::GFP)). Fluorescenceimages of different time points after infection. The smaller insets show the corresponding bright lightimage. Throughout the infection time, macrophages become fuller with bacteria (red, indicated by whitearrows) and generally no neutrophil recruitment is observed, although their numbers increase.
30

At 3 and 5 days post-infection macrophages full of bacteria were still observed. Occasionally,
macrophage and neutrophil recruitment was seen around infected cells (Figure 3.6). Currently we do
not know the reason for this, whether the phagocytes were recruited to bacteria that were liberated in
the host due to the collapse of the sometimes highly infected host cells, or whether, after longer infection
periods, the infected cells started to secrete immune signals resulting in the recruitment of macrophages
and neutrophils.
Figure 3.6: Macrophage infected with B. cenocepacia K56-2∆shvR in zebrafish embryos(Tg(mpx::GFP)). At 48 hpi the macrophages are full with bacteria and occasionally neutrophils are seenaround the infected cells. Scale bar 25 µm.
3.2.1 Quantification of host immune cell numbers during infection
In young embryos, at 30 hpf, immature macrophages and neutrophils are already present, and have
been shown to be able to phagocytose and kill bacteria, including Escherichia coli and Staphylococcus
aureus [193, 219]. In non-infected embryos, phagocyte numbers increase steadily during development
(see Figures 3.7 and 3.9). As mentioned briefly above, in embryos infected with K56-2, neutropenia
is observed at later stages of infection. In addition, macrophages also become depleted from K56-2-
infected embryos.
Since the shvR mutant induces persistent infection in the embryos, we were interested in the fate of
the host immune cells during infection. In order to quantify neutrophils and macrophages, mpx::GFP and
mpeg1::mCherry reporter fish, respectively, were used. Ten embryos (per strain) were injected with wild-
type K56-2 and the shvR mutant and the numbers of neutrophils and macrophages were determined.
At different time points after infection fluorescent images were taken of the individual embryos using
identical camera and exposure settings, followed by computational quantification of fluorescent cells as
described in the materials and methods section using ImageJ [242].
Macrophage numbers are not affected in shvR-infected embryos
Tg(mpx::GFP x mpeg1::mCherry-F ) embryos were injected with 10 to 30 CFU of B. cenocepacia
K56-2 and shvR mutant (Figure 3.7 B). In control embryos the number of macrophages increased
throughout the 72h of the experiment, as expected (Figure 3.7). In contrast, embryos infected with
K56-2 showed significantly lower numbers of macrophages, which reduced during the infection in num-
bers, consistent with earlier real time observations (Mesureur et al., manuscript in preparation, and this
31
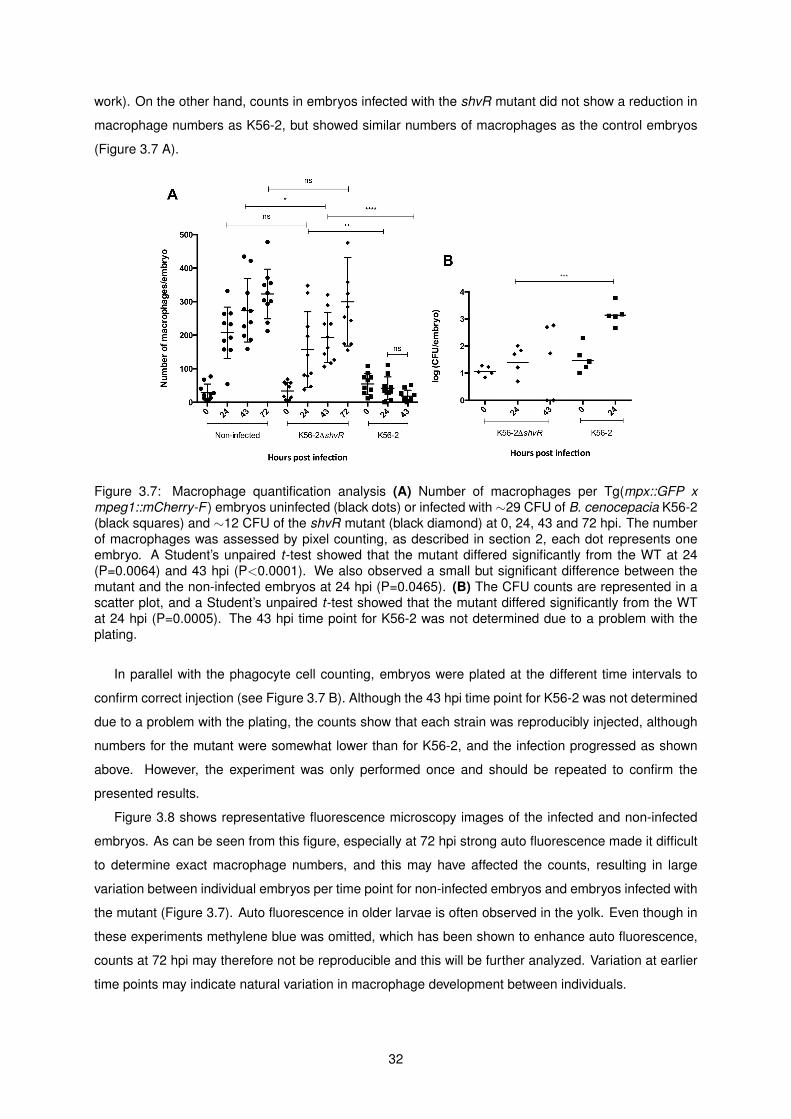
work). On the other hand, counts in embryos infected with the shvR mutant did not show a reduction in
macrophage numbers as K56-2, but showed similar numbers of macrophages as the control embryos
(Figure 3.7 A).
Figure 3.7: Macrophage quantification analysis (A) Number of macrophages per Tg(mpx::GFP xmpeg1::mCherry-F ) embryos uninfected (black dots) or infected with ∼29 CFU of B. cenocepacia K56-2(black squares) and ∼12 CFU of the shvR mutant (black diamond) at 0, 24, 43 and 72 hpi. The numberof macrophages was assessed by pixel counting, as described in section 2, each dot represents oneembryo. A Student’s unpaired t-test showed that the mutant differed significantly from the WT at 24(P=0.0064) and 43 hpi (P<0.0001). We also observed a small but significant difference between themutant and the non-infected embryos at 24 hpi (P=0.0465). (B) The CFU counts are represented in ascatter plot, and a Student’s unpaired t-test showed that the mutant differed significantly from the WTat 24 hpi (P=0.0005). The 43 hpi time point for K56-2 was not determined due to a problem with theplating.
In parallel with the phagocyte cell counting, embryos were plated at the different time intervals to
confirm correct injection (see Figure 3.7 B). Although the 43 hpi time point for K56-2 was not determined
due to a problem with the plating, the counts show that each strain was reproducibly injected, although
numbers for the mutant were somewhat lower than for K56-2, and the infection progressed as shown
above. However, the experiment was only performed once and should be repeated to confirm the
presented results.
Figure 3.8 shows representative fluorescence microscopy images of the infected and non-infected
embryos. As can be seen from this figure, especially at 72 hpi strong auto fluorescence made it difficult
to determine exact macrophage numbers, and this may have affected the counts, resulting in large
variation between individual embryos per time point for non-infected embryos and embryos infected with
the mutant (Figure 3.7). Auto fluorescence in older larvae is often observed in the yolk. Even though in
these experiments methylene blue was omitted, which has been shown to enhance auto fluorescence,
counts at 72 hpi may therefore not be reproducible and this will be further analyzed. Variation at earlier
time points may indicate natural variation in macrophage development between individuals.
32

Figure 3.8: Representative fluorescence microscopy images used for macrophage quantification anal-ysis. The embryos line used was Tg(mpx::GFP x mpeg1::mCherry-F ) for the counts in Figure 3.7 fornon-infected (control embryo) or embryos infected by B. cenocepacia K56-2 and the shvR mutant at 0,24, 43 and 72 hpi. Macrophages are represented by red fluorescence. In control (data not shown) andinfected embryos with K56-2∆shvR, specially at later time points strong auto fluorescence can affect theanalysis.
Neutrophil numbers increase in shvR-infected embryos
Tg(mpx::GFP) embryos were injected with 20 to 80 CFU of K56-2∆shvR, K56-2∆shvR+pUCP28T-
shvR (complemented strain) and K56-2. Figure 3.9 A, shows the number of neutrophils at different
time points in infected and non-infected embryos. During infection with wild-type bacteria, neutrophil
numbers decreased drastically, already at 24 hpi, causing neutropenia, confirming results obtained by
Mesureur et al. (manuscript in preparation, and Figure 3.9). However, infection with the shvR mutant
resulted in an increase in the production of new neutrophils compared to non-infected control embryos.
This neutrophilia was evident at 48 hpi. Additionally, the results obtained with complemented strain K56-
2∆shvR+pUCP28T-shvR demonstrated complementation in most embryos to neutrophil numbers found
after infection with wild-type K56-2, showing the shvR mutation was almost fully complemented by the
overexpression of the shvR gene in trans. In contrast to the macrophage quantification, embryos at
72 hpi did not show high levels of auto fluorescence, indicating natural variation in neutrophil numbers
between embryos. As for the macrophage assay, a bacterial enumeration assay was performed in
parallel as a control to verify correct injection (Figure 3.9 B). This experiment should also be repeated to
analyze a more significant number of embryos.
As for the macrophage assays,
33
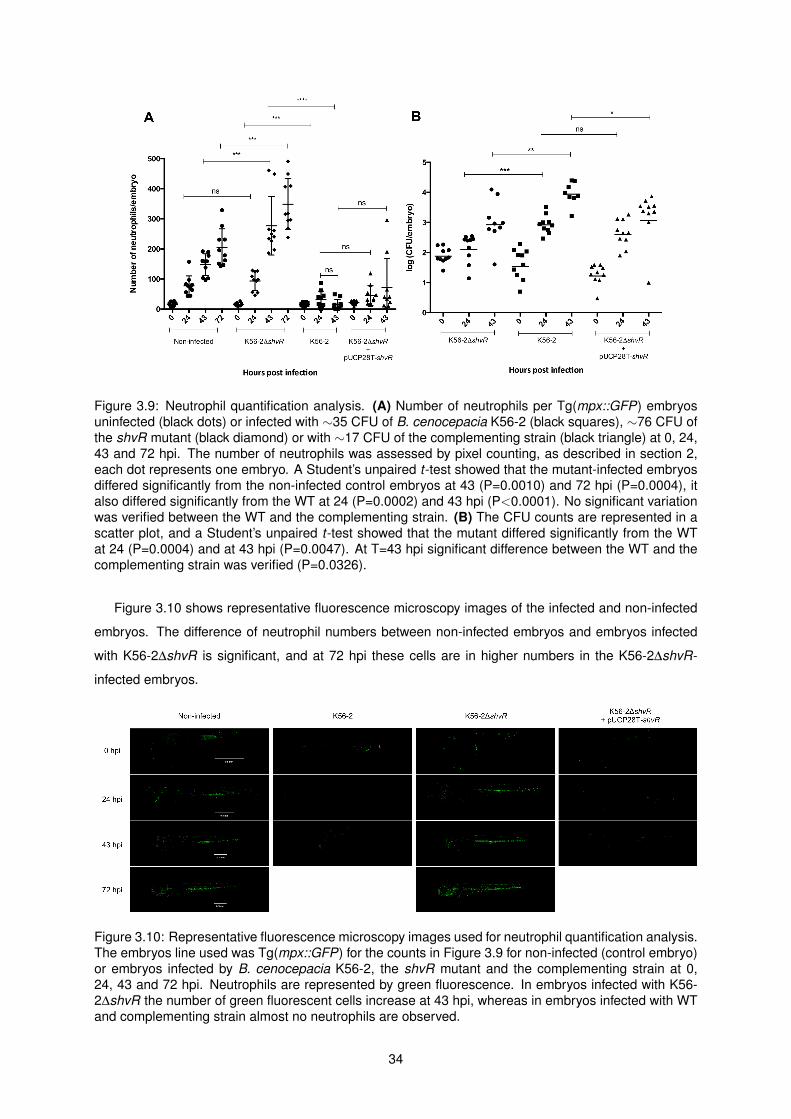
Figure 3.9: Neutrophil quantification analysis. (A) Number of neutrophils per Tg(mpx::GFP) embryosuninfected (black dots) or infected with ∼35 CFU of B. cenocepacia K56-2 (black squares), ∼76 CFU ofthe shvR mutant (black diamond) or with ∼17 CFU of the complementing strain (black triangle) at 0, 24,43 and 72 hpi. The number of neutrophils was assessed by pixel counting, as described in section 2,each dot represents one embryo. A Student’s unpaired t-test showed that the mutant-infected embryosdiffered significantly from the non-infected control embryos at 43 (P=0.0010) and 72 hpi (P=0.0004), italso differed significantly from the WT at 24 (P=0.0002) and 43 hpi (P<0.0001). No significant variationwas verified between the WT and the complementing strain. (B) The CFU counts are represented in ascatter plot, and a Student’s unpaired t-test showed that the mutant differed significantly from the WTat 24 (P=0.0004) and at 43 hpi (P=0.0047). At T=43 hpi significant difference between the WT and thecomplementing strain was verified (P=0.0326).
Figure 3.10 shows representative fluorescence microscopy images of the infected and non-infected
embryos. The difference of neutrophil numbers between non-infected embryos and embryos infected
with K56-2∆shvR is significant, and at 72 hpi these cells are in higher numbers in the K56-2∆shvR-
infected embryos.
Figure 3.10: Representative fluorescence microscopy images used for neutrophil quantification analysis.The embryos line used was Tg(mpx::GFP) for the counts in Figure 3.9 for non-infected (control embryo)or embryos infected by B. cenocepacia K56-2, the shvR mutant and the complementing strain at 0,24, 43 and 72 hpi. Neutrophils are represented by green fluorescence. In embryos infected with K56-2∆shvR the number of green fluorescent cells increase at 43 hpi, whereas in embryos infected with WTand complementing strain almost no neutrophils are observed.
34

3.3 Development of tools to better study the role of ShvR in viru-
lence
3.3.1 Complementation plasmids
Studies using genetic mutants require complementation of the mutant phenotype by introducing a
plasmid expressing the gene, to verify that the phenotype is due to the specific gene deletion. In the
laboratory the plasmid pUCP28T-shvR, providing trimethoprim resistance (kindly provided by P. Sokol),
has thus far been used for complementation studies of the shvR mutation in virulence in zebrafish
embryos. The complemented strain almost fully restores virulence, measured as embryo survival (Figure
3.11), and bacterial multiplication (Figure 3.9 B and Subramoni, Vergunst et al., unpublished) but the
complementation does not reach 100% wild-type levels. As shown above (Figure 3.9 A), this is also true
for the effect on neutrophil numbers, although the complementation for neutrophil numbers is significant.
This partial complementation could be explained by the fact that ShvR is a global regulator controlling
over 1000 genes, and complementation to wild-type virulence requires wild-type level of expression
of shvR that regulates the expression of different targets of ShvR involved in virulence. The plasmid
pUCP28T-shvR has the shvR gene under control of its own shvR promoter. Although the exact copy
number of pUCT28 in B. cenocepacia is not known to our knowledge, in B. gladioli there are over 100
copies per cell [243]. Thus, overexpression of shvR from plasmid pUCP28T-shvR may prevent full
complementation for virulence.
We decided to clone the pshvR-shvR cassette on two different plasmids, a derivative of pBBR, a
medium copy plasmid, and a derivative of the low copy plasmid pMR10 (1-2 copies per cell), called
pCR11. In addition, we generally use CmR resistant plasmids in our assays, and therefore used CmR
resistant versions of these plasmids. A third option, to integrate the gene back into the mutant strain, will
be performed if these two plasmids do not fully restore the mutant phenotype.
Figure 3.11: pUCP28T-shvR almost fully restores virulence to a shvR mutant. The survival data isrepresented in a Kaplan-Meier graph, and a log-rank (Mantel-Cox) test showed that the mutant differedsignificantly from the WT (P<0.0001) and from the complementing strain (P<0.0001).
Thus two plasmids were constructed in order to attempt to fully restore shvR expression in the mutant
to wild-type levels.
To construct pMG3 (see cloning scheme Figure 3.12), the shvR gene, including the promoter se-
35

quence (∼1.6 kb), was amplified by PCR using Pfu polymerase, B. cenocepacia K56-2 DNA as a tem-
plate, and primers pshvRXhoI for and shvRXbaI rev (sequence indicated in Table 2.4 in section 2 re-
striction sites indicated in bold). The blunt end amplification product was first ligated into SmaI-digested
pUC29 (pMG2), and transformed into competent E. coli DH5α using heat shock. White colonies were
selected by a blue/white screening, on LB-agar plates containing X-gal and IPTG. After the insertion was
confirmed by restriction digests, the samples were verified by sequencing, the plasmid was digested with
XhoI and Hind III enzymes (XhoI site was previously added upstream of the promoter region by PCR)
and the fragment was inserted in pIN29, a pBBR derived vector containing a chloramphenicol resistance
marker, creating pMG3 (Figure 3.13). The new plasmid also contains a ptac-DsRed reporter gene for
simultaneous real time visualization of bacteria, thus avoiding the need to transfer two compatible plas-
mids, each with a different selectable marker, to make the complementing mutant strain.
Figure 3.12: Cloning scheme for plasmid pMG3.
Figure 3.13: Plasmid map of pMG3. Rep stands for the origin of replication, cat is for chloramphenicolacetyltransferase, trpA is for tryptophan terminator and ptac is for tac-promoter.
The plasmid pMG3 was introduced into B. cenocepacia K56-2∆shvR by electroporation, and the
strain was compared to the original complementing strain, K56-2∆shvR (pUCP28-shvR) described for
the neutrophil counting experiment, using zebrafish infections.
Preliminary experiments demonstrated that the new plasmid, pMG3, was not able to fully restore the
36

wild-type virulence levels of the shvR mutant, as seen with pUCP28-shvR (not shown). Although by real
time microscopy analysis the infection progression showed similar phenotypes as previously observed,
due to problems in plating the dilutions of the embryo lysates, inconsistent results were obtained for
bacterial kinetics. These experiments will be repeated.
To construct the second plasmid (see cloning scheme Figure 3.14), a low copy plasmid, the pshvR-
shvR casette (∼1.7 kb) was isolated from pMG3 using NcoI and Nar I restriction sites and the DsRed
reported gene was obtained from pAV100 plasmid (∼800bp, see Table 2.1 in section 2), using XbaI and
ClaI (compatible with Nar I) restriction sites. These two fragments were ligated (∼2.5 kb) and extracted
from gel. The plasmid pUC29 was digested with NcoI and XbaI and ligated to the extracted fragment
(pMG6). After, white colonies were selected by a blue/white screening, on LB-agar plates containing
X-gal and IPTG. pMG6 was digested with SpeI and XbaI and the fragment corresponding to shvR gene
and DsRed was extracted from gel. Afterwards, the fragment was ligated to digested pCR11, a low copy
plasmid with a chloramphenicol resistance marker, with XbaI and SpeI creating pMG4 (Figure 3.15).
Figure 3.14: Cloning scheme for plasmid pMG4.
37

Figure 3.15: Plasmid map of pMG4. Par stand for partitioning gene cassettes, ptac is for tac-promoterand trpA is for tryptophan terminator.
3.3.2 Visualizing shvR expression in vivo in zebrafish embryos
In order to analyze the expression of the shvR gene in vivo, during infection in zebrafish embryos, and
other experimental growth conditions in vitro, a plasmid was constructed that places the reporter gene
mCherry under control of the shvR promoter sequence. To construct this plasmid (see cloning scheme
Figure 3.16) the upstream region of the shvR gene, encompassing the promoter sequence (596 kb), was
amplified by PCR using Pfu polymerase, B. cenocepacia K56-2 DNA as a template, and primers pshvR
for and pshvR rev (sequence indicated in Table 2.4 section 2, restriction sites indicated in bold). The
blunt end amplification product was first ligated into EcoRV-digested pUC29 (pMG1), and transformed
into competent E. coli DH5α using heat shock. White colonies were selected by a blue/white screening,
on LB-agar plates containing X-gal and IPTG. After insertion was confirmed by restriction digests, the
samples were verified by sequencing (Eurofins MWG Operon, Germany). pMG1 was digested with
NdeI and Hind III enzymes (both sites previously added by PCR), the fragment was purified from gel,
and inserted in equally digested pIN233, a pBBR based vector containing a chloramphenicol resistance
marker, creating pAV209 (Figure 3.17). This resulted in replacement of the tac promoter region in
pIN233 with the shvR promoter sequence, placing the mCherry reporter gene under control of shvR
regulatory signals. Read through expression from plasmid encoded sequences was prevented by the
presence of a strong termination signal upstream the shvR-mCherry cassette.
Despite the trpA sequence, which has been shown to prevent possible read-through expression
in E. coli and K56-2 (A. Vergunst personal communication), we observed low fluorescence levels of
mCherry from the shvR promoter in E. coli colonies, confirmed with preliminary FACS analysis. This
observation will be further analyzed using FACS, and might give some more insight into shvR expression
and regulation, of which no ortholog has been detected in E. coli.
Wild-type K56-2 and the shvR mutant strains were electroporated with the pAV209 plasmid. In fact,
the colonies on LB-agar plates showed bright red fluorescent by fluorescence microscopy, indicating
that shvR is expressed under these growth conditions. Also in liquid cultures the bacteria appeared red
38

Figure 3.16: Cloning scheme for plasmid pAV209.
Figure 3.17: Plasmid map of pAV209. Rep stands for the origin of replication, cat is for chloramphenicolacetyltransferase and trpA is for tryptophan terminator.
fluorescent. Further experiments will be performed to compare the expression levels from the pshvR-
mCherry reporter gene in E. coli and B. cenocepacia under these in vitro conditions with strains con-
taining a plasmid that allows strong constitutive expression (ptac-mCherry ).
A preliminary zebrafish infection experiment showed that K56-2 and the shvR mutant, carrying plas-
mid pAV209, strongly expressed mCherry throughout the infection (Figure 3.18). The mCherry protein
is very stable, and will not allow analysis of changes in expression levels.
Therefore, to be better able to verify if shvR expression is repressed, or activated during infection,
including the intracellular stages, an unstable version of the mCherry protein was made. Campbell-
Valois et al. recently described the use of a fast maturing GFP to study Type 3 Secretion System (T3SS)
activity in Shigella flexneri [244]. Therefore, we created a small peptide tag based on the studies of
unstable GFP variants in Pseudomonas putida [245]. Andersen and colleagues demonstrated that the
sequence AANDENYALVA besides being degraded by intracellular tail-specific proteases [246], is the
one that leads to a faster destabilization of GFP in P. putida [245].
39

Figure 3.18: Zebrafish embryos (Tg(mpx::GFP)) infected with B. cenocepacia K56-2 and the shvR mu-tant carrying pVA209 plasmid at 24 hpi. The strong expression of mCherry protein is visible after 24post-infection for both bacterial strains.
This plasmid has as backbone pAV209, in which we included the C-terminal peptide tag AAN-
DENYALVA to mCherry. Two complementary oligos (indicated in Table 2.4, in section 2), codon opti-
mized for B. cenocepacia and including a TAG terminator codon, were designed and ordered (Sigma)
(see cloning scheme Figure 3.19). On each 5’ and 3’ end of the oligos, additional nucleotides were
included that would result in cohesive ends of the enzymes BsrGI and SacI after annealing (sequence
in Table 2.4). To anneal the oligos, 25 µM of each oligo was added in a total volume of 20 µL of H2O,
containing 1X restriction buffer (NEB2, New England Biolabs). The suspension was heated to 95°C for 5
minutes and left for 3 h to slowly reduce the temperature until 30°C. Plasmid pAV209 was digested with
BsrGI and SacI enzymes and the fragment was inserted by ligation, resulting in pMG5 (Figure 3.20).
The plasmid is now being verified by sequencing, and will be introduced into wild-type K56-2, and
the shvR mutant for further analysis.
Figure 3.19: Cloning scheme for plasmid pMG5. AANDENYALVA is the peptide tag sequence used todestabilize the mCherry protein.
40

Figure 3.20: Plasmid map of pMG5. Rep stands for the origin of replication, cat is for chloramphenicolacetyltransferase and trpA is for tryptophan terminator.
41

42

Chapter 4
Discussion
Burkholderia cenocepacia belongs to the Burkholderia cepacia compIex (Bcc) and has been de-
scribed as a pathogen for immunocompromised people, mainly for cystic fibrosis (CF) patients. The
complex comprises 17 bacterial species that can be found in different environments, from natural, as
in rhizosphere and water, to industrialized, in pharmaceutical products and polluted soils [1–7]. These
bacteria can adapt and resist to different stress conditions, which medically constitutes a great concern
to physicians since the bacteria are resistant to many of the antibiotics used. The species from the
Bc complex are also known for their metabolic diversity. Its large genome size and genomic plastic-
ity, conferred by the many genomic islands and IS elements, may contribute to the high flexibility and
adaptability of this complex.
B. cenocepacia has been suggested to resist and survive in biofilms in lungs of CF patients [247],
which are often co-colonized with P. aeruginosa and other pathogens. The formation of these structures
complicates antibiotic treatment, can cause persistent infections, and may be a source for recurrent
infections, due to persister cells [99]. As shown by cell culture experiments and in vivo in zebrafish em-
bryos, B. cenocepacia is capable of surviving intracellularly in host macrophages by evading phagolyso-
somal degradation, and creating a replication niche. B. cenocepacia has been described not to form
biofilms in lung samples of CF patients [140], and this has posed the question to what extent an intra-
cellular strategy of the Bcc may be involved in enhancing inflammation, and causing persistent infection,
as well as its invasiveness.
Recent studies on B. cenocepacia K56-2, an ET12 strain recovered during an epidemic outbreak,
described the appearance of shiny variant colonies that encoded a mutation in a global regulator, named
ShvR. This regulator belongs to the LysR-type transcriptional regulator family (LTTR), which is a highly
conserved family of regulators in bacteria [223–225]. The gene is highly conserved in B. cenocepacia
and B. cepacia strains, and from a BLAST analysis, using the PATRIC bioinformatics tool [13], orthologs
of ShvR were not detected in most of the other species in the complex (data not shown). ShvR is
encoded on pC3, a non-essential megaplasmid in B. cenocepacia demonstrated to be needed for full
virulence in different models [40], and, together with previous experiments with a K56-2 shvR mutant,
43

the bacteria were shown to be less virulent in alfalfa seedlings and showed reduced inflammation in rat
lung [102], suggesting that ShvR regulates virulence factors involved in establishing an acute infection.
Previous studies using a chronic rat agar bead model, showed that the shvR mutant had reduced
virulence and reduced lung inflammation; however, the bacteria were able to persist sometimes better
than the wildtype in the lungs [102]. In addition, the shvR mutant showed low biofilm formation in
vitro. Thus ShvR might be important in regulating factors involved in the excessive inflammation and
dissemination of the bacteria, and inversely, factors needed for a persistent infection.
The aim of this study was to start to better understand the behavior of the shvR mutant and the
immune response during infection, using the zebrafish embryo model. The zebrafish has recently been
developed as a model for Bcc infections allowing us to detect differences in virulence caused by different
strains, for example B. cenocepacia K56-2 can develop an acute and fatal infection whereas B. stabilis
LMG14294 causes a persistent infection [109]. My results show that in the absence of ShvR the lethal
K56-2 can only cause persistent infection in zebrafish. This is seen by reduced bacterial growth, reduced
inflammation and virulence, and absence of host phagocyte cell death, compared to K56-2. Instead the
mutant somehow activates the host immune response, observed by the increased number of neutrophils,
similar to the response seen to infection with B. stabilis (Mesureur et al., manuscript in preparation).
Embryos infected with B. cenocepacia K56-2 die after 2 dpi with a high number of bacteria, in line with
published data that this strain creates an acute fatal infection [109]. In contrast, embryos infected with
K56∆shvR are all still alive at 5 dpi, when the experiment is terminated, with a relatively constant number
of bacteria, consistent with a persistent phenotype, as shown for B. stabilis LMG14294 by Vergunst et
al. [109]. In agreement with the findings in experimental rat infections, these results suggest that ShvR
regulates factors involved in the virulence of K56-2 needed to develop acute inflammatory infection.
Microscopic observations showed that shvR mutant bacteria survived inside host macrophages, and
were still observed 5 dpi. The bacteria are able to reach high intracellular numbers, and occasionally
small infection sites can be observed, with macrophage recruitment, although the exact reason is not
known yet. Such infected macrophages sometimes had extreme sizes, but further analysis will be per-
formed to estimate bacterial numbers inside the macrophages, in order to affirm a possible difference
between shvR-infected and K56-infected macrophages. Moreover, the importance of the macrophages
for the bacterial replication and survival should be assessed by performing a macrophage ablation as-
say. Earlier, Bernier et al. found that shvR mutant bacteria reached numbers that could be equal or even
higher than the wildtype in rats lungs, yet, the mutant shows reduced capacity to form biofilms [102].
This raises the question whether the shvR bacteria could better persist inside rat lung cells than the
wild type, such as macrophages and/or epithelial cells. Experimental evidence for this could change the
current idea of persistence, and show a more important role for intracellular bacteria in vivo.
Additionally, the number of host immune cells was analyzed in order to further demonstrate a persis-
tent infection phenotype. Concerning macrophages, Mesureur et al. (manuscript in preparation) found
that in K56-2 infected embryos, the number of non-infected macrophages decreases rapidly during infec-
tion, reaching very low numbers, whereas infected macrophages were kept alive by the bacteria. Similar
44

results were obtained in this study for K56-infected embryos, however, in embryos infected with the shvR
mutant the macrophage numbers increased in a similar manner as in control embryos. Although we do
not know the reasons for the disappearance of macrophages during K56-2 infection, this shows that the
shvR mutant lacks the regulation of the factors involved in the macrophage killing, or induces altered
innate immune responses leading to the difference in host phagocyte response.
When neutrophil numbers during infection were analyzed, it was found that in K56-2 infected em-
bryos neutrophils were reduced to very low numbers, as found also by Mesureur et al. (manuscript in
preparation). Thus, these embryos are not able to resolve the inflammation caused by the bacteria. As
for the mutant, it showed that the bacteria induced neutrophilia in the embryos, again similar to an in-
fection with B. stabilis, where Mesureur et al. (manuscript in preparation) verified that the strain induced
neutrophil production at higher levels even at 5 dpi.
The analyses of host immune cell numbers suggest once more that the infection with the shvR mutant
follows a persistent phenotype, similar to B. stabilis infection and strikingly different from K56-2 infection.
During this study, tools were developed to better understand the role of ShvR in virulence in vivo: new
complementing plasmids were created to optimize complementation studies, and two other plasmids
were created to study the expression of shvR in vivo during infection.
In this study, as in Subramoni, Vergunst et al. (unpublished), genetic complementation of the shvR
mutant for virulence in zebrafish embryos was performed using the plasmid pUCP28T-shvR. Although
this plasmid that expresses the shvR gene from its own promoter sequence restored virulence to the
mutant in zebrafish infections, the complementation was not 100%. As mentioned in Results section,
it is possible that full complementation is not achieved because, as ShvR regulates over 1000 genes,
reaching the correct expression levels of ShvR is important; although copy number of the pUCP28T-
shvR plasmid in B. cenocepacia has not been determined exactly, in B. gladioli there are over 100
copies per cell [243]. It is thus likely that ShvR is produced at higher levels than from its genomic
position. Therefore two plasmids were constructed: pMG3 with a pBBR backbone vector (CmR), which
has a medium number of copies per cell, and pMG4 with a pMR10 backbone vector (CmR), that only has
1-2 copies. Preliminary experiments with plasmid pMG3 indicated that also this plasmid was not able
to fully restore virulence to the shvR mutant. Although the construction of pMG4 is still underway, it will
be interesting to find out if the further reduction in copy number will allow full complementation. It has
been shown previously for instance for a virB5 mutant of Brucella, that VirB5 over expression affected
expression and/or stability of other VirB proteins, resulting in attenuation, even of the wild type strain
[248]. Future experiments include transforming wild type K56-2 with pMG3, the pBBR-based plasmid,
to verify if overexpression reduces its virulence [248].
To better understand a role for ShvR in regulating target genes involved in acute, or persistent infec-
tion, it is important to know when during infection the shvR gene is expressed. Therefore, a new plasmid
that allows for observation of the regulator’s expression in vivo was constructed. The plasmid pAV209
encoding a mCherry fluorescent protein under control of the shvR promoter, was constructed. Although
not brightly expressed in E. coli (data not shown), experiments, using K56-2 and the mutant bacteria
transformed with this plasmid, indicated that the expression of ShvR started at some time point during
45

bacterial growth in LB medium, since the bacteria were bright red fluorescent in overnight grown cul-
tures. During infection experiments in zebrafish, the bacteria were visible with fluorescence microscopy
throughout the experimental time. Due to the stability of the mCherry protein (although the half-life of
mCherry is not known in Bcc, in E. coli the half-life of GFPmut3, which is also very stable, has been
shown to be more than 24 h [245]), we cannot study any changes in the expression levels of shvR. To
be able to better analyze a possible regulation of ShvR expression in vivo, also in other Bcc strains,
an unstable m-Cherry reporter gene was created. In studies with Shigella flexneri, a GFP reporter was
created that matured faster in order to assess T3SS activity [244]. In our study we included a small
peptide tag (AANDENYALVA) to the C-terminal end of mCherry to allow its rapid degradation by intracel-
lular tail-specific proteases [245]. Further experiments, including infection and fluorescence extinction
experiments, will be performed to assess the efficiency of this plasmid.
In conclusion, in agreement with other infection models, ShvR has a role in virulence of B. cenocepacia
in the zebrafish embryo model. The techniques used in this study contributed to confirm and better
understand the persistent infection phenotype of B. cenocepacia K56-2∆shvR. More detailed studies
have to be performed to determine a more precise role for ShvR in regulating factors that determine
the difference between acute and persistent infection, using not only the created tools but also other
techniques, including proteomic and transcriptional studies (dual RNAseq to analyze both bacterial and
host transcriptome) that could give more insights in ShvR regulation.
46

Bibliography
[1] Peter Vandamme, B. Holmes, Marc Vancanneyt, Tom Coenye, Bart Hoste, Renata Coopman,
H. Revets, S. Lauwers, Monique Gillis, Karel Kersters, and others. Occurrence of multiple
genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia mul-
tivorans sp. nov. International Journal of Systematic Bacteriology, 47(4):1188–1200, 1997.
[2] Peter Vandamme, Barry Holmes, Tom Coenye, Johan Goris, Eshwar Mahenthiralingam, John J.
LiPuma, and John R. W. Govan. Burkholderia cenocepacia sp. nov.– a new twist to an old story.
Research in Microbiology, 154(2):91–96, 2003.
[3] Tom Coenye and John J. LiPuma. Population structure analysis of Burkholderia cepacia
genomovar III: varying degrees of genetic recombination characterize major clonal complexes.
Microbiology, 149(Pt 1):77–88, 2003.
[4] Eshwar Mahenthiralingam, Teresa A. Urban, and Joanna B. Goldberg. The multifarious, multi-
replicon Burkholderia cepacia complex. Nature Reviews Microbiology, 3(2):144–156, 2005.
[5] E. Mahenthiralingam, A. Baldwin, and C.g. Dowson. Burkholderia cepacia complex bacte-
ria: opportunistic pathogens with important natural biology. Journal of Applied Microbiology,
104(6):1539–1551, 2008.
[6] E. Vanlaere, J. J. LiPuma, A. Baldwin, D. Henry, E. De Brandt, E. Mahenthiralingam, D. Speert,
C. Dowson, and P. Vandamme. Burkholderia latens sp. nov., Burkholderia diffusa sp. nov.,
Burkholderia arboris sp. nov., Burkholderia seminalis sp. nov. and Burkholderia metallica sp. nov.,
novel species within the Burkholderia cepacia complex. International Journal of Systematic and
Evolutionary Microbiology, 58(7):1580–1590, 2008.
[7] E. Vanlaere, A. Baldwin, D. Gevers, D. Henry, E. De Brandt, J. J. LiPuma, E. Mahenthiralingam,
D. P. Speert, C. Dowson, and P. Vandamme. Taxon k, a complex within the Burkholderia cepacia
complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholde-
ria lata sp. nov. International Journal of Systematic and Evolutionary Microbiology, 59(1):102–111,
2009.
[8] W. H. Burkholder. Sour skin, a bacterial rot of onion bulbs. Phytopathology, 40(1):115–117 pp.,
1950.
47

[9] E. Yabuuchi, Y. Kosako, H. Oyaizu, I. Yano, H. Hotta, Y. Hashimoto, T. Ezaki, and M. Arakawa.
Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas
homology group II to the new genus, with the type species Burkholderia cepacia (palleroni and
holmes 1981) comb. nov. Microbiology and Immunology, 36(12):1251–1275, 1992.
[10] Eshwar Mahenthiralingam, Jocelyn Bischof, Sean K. Byrne, Christopher Radomski, Julian E.
Davies, Yossef Av-Gay, and Peter Vandamme. DNA-based diagnostic approaches for identifi-
cation of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans,
Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. Journal of clinical microbi-
ology, 38(9):3165–3173, 2000.
[11] Adam Baldwin, Eshwar Mahenthiralingam, Kathleen M. Thickett, David Honeybourne, Martin C. J.
Maiden, John R. Govan, David P. Speert, John J. LiPuma, Peter Vandamme, and Chris G. Dow-
son. Multilocus sequence typing scheme that provides both species and strain differentiation for
the Burkholderia cepacia complex. Journal of Clinical Microbiology, 43(9):4665–4673, 2005.
[12] Theodore Spilker, Adam Baldwin, Amy Bumford, Chris G. Dowson, Eshwar Mahenthiralingam,
and John J. LiPuma. Expanded multilocus sequence typing for Burkholderia species. Journal of
Clinical Microbiology, 47(8):2607–2610, 2009.
[13] A. R. Wattam, D. Abraham, O. Dalay, T. L. Disz, T. Driscoll, J. L. Gabbard, J. J. Gillespie, R. Gough,
D. Hix, R. Kenyon, D. Machi, C. Mao, E. K. Nordberg, R. Olson, R. Overbeek, G. D. Pusch,
M. Shukla, J. Schulman, R. L. Stevens, D. E. Sullivan, V. Vonstein, A. Warren, R. Will, M. J. C.
Wilson, H. S. Yoo, C. Zhang, Y. Zhang, and B. W. Sobral. PATRIC, the bacterial bioinformatics
database and analysis resource. Nucleic Acids Research, 42(D1):D581–D591, 2014.
[14] Geoffrey L. Winsor, Bhavjinder Khaira, Thea Van Rossum, Raymond Lo, Matthew D. Whiteside,
and Fiona S. L. Brinkman. The Burkholderia genome database: facilitating flexible queries and
comparative analyses. Bioinformatics, 24(23):2803–2804, 2008.
[15] Eshwar Mahenthiralingam, Adam Baldwin, and Peter Vandamme. Burkholderia cepacia complex
infection in patients with cystic fibrosis. Journal of medical microbiology, 51(7):533–538, 2002.
[16] Karen Vermis, Tom Coenye, Eshwar Mahenthiralingam, Hans J. Nelis, and Peter Vandamme.
Evaluation of species-specific recA-based PCR tests for genomovar level identification within the
Burkholderia cepacia complex. Journal of medical microbiology, 51(11):937–940, 2002.
[17] Thomas G. Lessie, William Hendrickson, Brendan D. Manning, and Richard Devereux. Genomic
complexity and plasticity of Burkholderia cepacia. FEMS Microbiology Letters, 144(2-3):117–128,
1996.
[18] Rhiannon Biddick, Theodore Spilker, Alissa Martin, and John J LiPuma. Evidence of transmission
of Burkholderia cepacia , Burkholderia multivorans and Burkholderia dolosa among persons with
cystic fibrosis. FEMS Microbiology Letters, 228(1):57–62, 2003.
48

[19] John J. LiPuma, Theodore Spilker, Tom Coenye, and Carlos F. Gonzalez. An epidemic Burkholde-
ria cepacia complex strain identified in soil. Lancet, 359(9322):2002–2003, 2002.
[20] David P. Speert, Deborah Henry, Peter Vandamme, Mary Corey, and Eshwar Mahenthiralingam.
Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis, canada. Emerging
Infectious Diseases, 8(2):181–187, 2002.
[21] E. Mahenthiralingam, P. Vandamme, M. E. Campbell, D. A. Henry, A. M. Gravelle, L. T. Wong,
A. G. Davidson, P. G. Wilcox, B. Nakielna, and D. P. Speert. Infection with Burkholderia cepacia
complex genomovars in patients with cystic fibrosis: virulent transmissible strains of genomovar
III can replace Burkholderia multivorans. Clinical infectious diseases, 33(9):1469–1475, 2001.
[22] L. Sun, R. Z. Jiang, S. Steinbach, A. Holmes, C. Campanelli, J. Forstner, U. Sajjan, Y. Tan,
M. Riley, and R. Goldstein. The emergence of a highly transmissible lineage of cbl+ Pseu-
domonas (Burkholderia) cepacia causing CF centre epidemics in North America and Britain. Na-
ture medicine, 1(7):661–666, 1995.
[23] P. Vandamme, E. Mahenthiralingam, B. Holmes, T. Coenye, B. Hoste, P. De Vos, D. Henry, and
D. P. Speert. Identification and population structure of Burkholderia stabilis sp. nov. (formerly
Burkholderia cepacia genomovar IV). Journal of clinical microbiology, 38(3):1042–1047, 2000.
[24] Tom Coenye, John J. LiPuma, Deborah Henry, Bart Hoste, Katrien Vandemeulebroecke, Monique
Gillis, David P. Speert, and Peter Vandamme. Burkholderia cepacia genomovar VI, a new member
of the Burkholderia cepacia complex isolated from cystic fibrosis patients. International journal of
systematic and evolutionary microbiology, 51(2):271–279, 2001.
[25] Karen Vermis, Tom Coenye, John J. LiPuma, Eshwar Mahenthiralingam, Hans J. Nelis, and Pe-
ter Vandamme. Proposal to accommodate Burkholderia cepacia genomovar VI as Burkholderia
dolosa sp. nov. International journal of systematic and evolutionary microbiology, 54(Pt 3):689–
691, 2004.
[26] J. L. Parke and D. Gurian-Sherman. Diversity of the Burkholderia cepacia complex and implica-
tions for risk assessment of biological control strains. Annual Review of Phytopathology, 39:225–
258, 2001.
[27] T. Coenye, E. Mahenthiralingam, D. Henry, J. J. LiPuma, S. Laevens, M. Gillis, D. P. Speert,
and P. Vandamme. Burkholderia ambifaria sp. nov., a novel member of the Burkholderia cepacia
complex including biocontrol and cystic fibrosis-related isolates. International journal of systematic
and evolutionary microbiology, 51(Pt 4):1481–1490, 2001.
[28] Peter Vandamme, Deborah Henry, Tom Coenye, Sazini Nzula, Marc Vancanneyt, John J. LiPuma,
David P. Speert, John R. W. Govan, and Eshwar Mahenthiralingam. Burkholderia anthina sp.
nov. and Burkholderia pyrrocinia, two additional Burkholderia cepacia complex bacteria, may con-
found results of new molecular diagnostic tools. FEMS immunology and medical microbiology,
33(2):143–149, 2002.
49

[29] Pavel Drevinek, Sarka Vosahlikova, Klara Dedeckova, Ondrej Cinek, and Eshwar Mahenthi-
ralingam. Direct culture-independent strain typing of Burkholderia cepacia complex in sputum
samples from patients with cystic fibrosis. Journal of Clinical Microbiology, 48(5):1888–1891,
2010.
[30] Pablo Martina, Marisa Bettiol, Cecilia Vescina, Patricia Montanaro, M. Constanza Mannino, Clau-
dia I. Prieto, Carlos Vay, Dieter Naumann, Juergen Schmitt, Osvaldo Yantorno, Antonio Lagares,
and Alejandra Bosch. Genetic diversity of Burkholderia contaminans isolates from cystic fibrosis
patients in Argentina. Journal of clinical microbiology, 51(1):339–344, 2013.
[31] Tom Coenye, Peter Vandamme, John R. W. Govan, and John J. LiPuma. Taxonomy and identifi-
cation of the Burkholderia cepacia complex. Journal of Clinical Microbiology, 39(10):3427–3436,
2001.
[32] Peter Vandamme and Peter Dawyndt. Classification and identification of the Burkholderia cepacia
complex: Past, present and future. Systematic and Applied Microbiology, 34(2):87–95, 2011.
[33] Tom Coenye and Peter Vandamme. Diversity and significance of Burkholderia species occupying
diverse ecological niches. Environmental Microbiology, 5(9):719–729, 2003.
[34] Slade A. Loutet, Ronald S. Flannagan, Cora Kooi, Pamela A. Sokol, and Miguel A. Valvano. A
complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholde-
ria cenocepacia to antimicrobial peptides and bacterial survival in vivo. Journal of Bacteriology,
188(6):2073–2080, 2006.
[35] S. D. Aaron, W. Ferris, D. A. Henry, D. P. Speert, and N. E. Macdonald. Multiple combination
bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia.
American Journal of Respiratory and Critical Care Medicine, 161(4 Pt 1):1206–1212, 2000.
[36] Sazini Nzula, Peter Vandamme, and John R. W. Govan. Influence of taxonomic status on the in
vitro antimicrobial susceptibility of the Burkholderia cepacia complex. The Journal of Antimicrobial
Chemotherapy, 50(2):265–269, 2002.
[37] L. A. O’Sullivan and E. Mahenthiralingam. Biotechnological potential within the genus Burkholde-
ria. Letters in Applied Microbiology, 41(1):8–11, 2005.
[38] Pavel Drevinek, Adam Baldwin, Laurens Lindenburg, Lovleen Tina Joshi, Angela Marchbank,
Sarka Vosahlikova, Christopher G. Dowson, and Eshwar Mahenthiralingam. Oxidative stress of
Burkholderia cenocepacia induces insertion sequence-mediated genomic rearrangements that
interfere with macrorestriction-based genotyping. Journal of Clinical Microbiology, 48(1):34–40,
2010.
[39] K. Agnoli, S. Schwager, S. Uehlinger, A. Vergunst, D. F. Viteri, D. T. Nguyen, P. A. Sokol, A. Car-
lier, and L. Eberl. Exposing the third chromosome of Burkholderia cepacia complex strains as a
virulence plasmid. Molecular Microbiology, 83(2):362–378, 2012.
50

[40] Kirsty Agnoli, Carmen Frauenknecht, Roman Freitag, Stephan Schwager, Christian Jenul, An-
nette Vergunst, Aurelien Carlier, and Leo Eberl. The third replicon of members of the Burkholderia
cepacia complex, plasmid pC3, plays a role in stress tolerance. Applied and Environmental Mi-
crobiology, 80(4):1340–1348, 2014.
[41] Scott A. Bernhardt, Theodore Spilker, Todd Coffey, and John J. LiPuma. Burkholderia cepacia
complex in cystic fibrosis: frequency of strain replacement during chronic infection. Clinical Infec-
tious Diseases: An Official Publication of the Infectious Diseases Society of America, 37(6):780–
785, 2003.
[42] J. J. LiPuma, S. E. Dasen, D. W. Nielson, R. C. Stern, and T. L. Stull. Person-to-person trans-
mission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet, 336(8723):1094–
1096, 1990.
[43] J. R. Govan, P. H. Brown, J. Maddison, C. J. Doherty, J. W. Nelson, M. Dodd, A. P. Greening,
and A. K. Webb. Evidence for transmission of Pseudomonas cepacia by social contact in cystic
fibrosis. Lancet, 342(8862):15–19, 1993.
[44] E. Song, Gayatri Bala Jaishankar, Hana Saleh, Warit Jithpratuck, Ryan Sahni, and Guha Krish-
naswamy. Chronic granulomatous disease: a review of the infectious and inflammatory complica-
tions. Clinical and Molecular Allergy, 9(10), 2011.
[45] D. C. Hohn and R. I. Lehrer. NADPH oxidase deficiency in X-linked chronic granulomatous dis-
ease. The Journal of Clinical Investigation, 55(4):707–713, 1975.
[46] Karen Bedard and Karl-Heinz Krause. The NOX family of ROS-generating NADPH oxidases:
physiology and pathophysiology. Physiological Reviews, 87(1):245–313, 2007.
[47] Caroline Kannengiesser, Benedicte Gerard, Jamel El Benna, Dominique Henri, Yolande
Kroviarski, Sylvie Chollet-Martin, Marie-Anne Gougerot-Pocidalo, Carole Elbim, and Bernard
Grandchamp. Molecular epidemiology of chronic granulomatous disease in a series of 80 kin-
dreds: identification of 31 novel mutations. Human Mutation, 29(9):E132–149, 2008.
[48] T. Umei, K. Takeshige, and S. Minakami. NADPH-binding component of the superoxide-generating
oxidase in unstimulated neutrophils and the neutrophils from the patients with chronic granuloma-
tous disease. The Biochemical Journal, 243(2):467–472, 1987.
[49] J. T. Curnutte, R. Kuver, and P. J. Scott. Activation of neutrophil NADPH oxidase in a cell-free
system. Partial purification of components and characterization of the activation process. Journal
of Biological Chemistry, 262(12):5563–5569, 1987.
[50] S. D. Rosenzweig. Inflammatory manifestations in chronic granulomatous disease (CGD). Journal
of Clinical Immunology, 28 Suppl 1:S67–72, 2008.
51

[51] David P. Speert, Mason Bond, Richard C. Woodman, and John T. Curnutte. Infection with Pseu-
domonas cepacia in chronic granulomatous disease: Role of nonoxidative killing by neutrophils in
host defense. Journal of Infectious Diseases, 170(6):1524–1531, 1994.
[52] Johan Bylund, Paul A. Campsall, Rebecca C. Ma, Barbara-Ann D. Conway, and David P. Speert.
Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. The
Journal of Immunology, 174(6):3562–3569, 2005.
[53] Brian P. O’Sullivan and Steven D. Freedman. Cystic fibrosis. Lancet, 373(9678):1891–1904, 2009.
[54] Paola Del Porto, Noemi Cifani, Simone Guarnieri, Enea Gino Di Domenico, Maria A. Mariggio,
Francesca Spadaro, Silvia Guglietta, Marco Anile, Federico Venuta, Serena Quattrucci, and
Fiorentina Ascenzioni. Dysfunctional CFTR alters the bactericidal activity of human macrophages
against Pseudomonas aeruginosa. PLoS ONE, 6(5):e19970, 2011.
[55] J. Barasch, B. Kiss, A. Prince, L. Saiman, D. Gruenert, and Q. al Awqati. Defective acidification of
intracellular organelles in cystic fibrosis. Nature, 352(6330):70–73, 1991.
[56] Nina Reiniger, Jeffrey K. Ichikawa, and Gerald B. Pier. Influence of cystic fibrosis transmembrane
conductance regulator on gene expression in response to Pseudomonas aeruginosa infection of
human bronchial epithelial cells. Infection and Immunity, 73(10):6822–6830, 2005.
[57] John R. Riordan, Johanna M. Rommens, Bat-sheva Kerem, Noa Alon, Richard Rozmahel,
Zbyszko Grzelczak, Julian Zielenski, Si Lok, Natasa Plavsic, Jia-Ling Chou, Mitchell L. Drumm,
Michael C. Iannuzzi, Francis S. Collins, and Lap-Chee Tsui. Identification of the cystic fibrosis
gene: Cloning and characterization of complementary DNA. Science, 245(4922):1066–1073,
1989.
[58] Nico Derichs. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator
modulators in cystic fibrosis. European Respiratory Review, 22(127):58–65, 2013.
[59] WHO | genes and human disease.
[60] Richard C. Boucher. Cystic fibrosis: a disease of vulnerability to airway surface dehydration.
Trends in Molecular Medicine, 13(6):231–240, 2007.
[61] S. H. Cheng, R. J. Gregory, J. Marshall, S. Paul, D. W. Souza, G. A. White, C. R. O’Riordan, and
A. E. Smith. Defective intracellular transport and processing of CFTR is the molecular basis of
most cystic fibrosis. Cell, 63(4):827–834, 1990.
[62] M. J. Goldman, G. M. Anderson, E. D. Stolzenberg, U. P. Kari, M. Zasloff, and J. M. Wilson.
Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell,
88(4):553–560, 1997.
[63] H. Matsui, B. R. Grubb, R. Tarran, S. H. Randell, J. T. Gatzy, C. W. Davis, and R. C. Boucher.
Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis
of cystic fibrosis airways disease. Cell, 95(7):1005–1015, 1998.
52

[64] J. J. Smith, S. M. Travis, E. P. Greenberg, and M. J. Welsh. Cystic fibrosis airway epithelia fail to
kill bacteria because of abnormal airway surface fluid. Cell, 85(2):229–236, 1996.
[65] C. Castellani, H. Cuppens, M. Macek, J.J. Cassiman, E. Kerem, P. Durie, E. Tullis, B.M. Assael,
C. Bombieri, A. Brown, T. Casals, M. Claustres, G.R. Cutting, E. Dequeker, J. Dodge, I. Doull,
P. Farrell, C. Ferec, E. Girodon, M. Johannesson, B. Kerem, M. Knowles, A. Munck, P.F. Pignatti,
D. Radojkovic, P. Rizzotti, M. Schwarz, M. Stuhrmann, M. Tzetis, J. Zielenski, and J.S. Elborn.
Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice.
Journal of Cystic Fibrosis, 7(3):179–196, 2008.
[66] M Nissim-Rafinia, BATSHEVA Kerem, EITAN Kerem, M Hodson, D Geddes, and A Bush. Molec-
ular biology of cystic fibrosis: CFTR processing and functions, and classes of mutations. Cystic
Fibrosis. London: Hodder Arnold, pages 49–58, 2007.
[67] Jeffrey B. Lyczak, Carolyn L. Cannon, and Gerald B. Pier. Lung infections associated with cystic
fibrosis. Clinical Microbiology Reviews, 15(2):194–222, 2002.
[68] John J. LiPuma. The changing microbial epidemiology in cystic fibrosis. Clinical Microbiology
Reviews, 23(2):299–323, 2010.
[69] D. D. Frangolias, E. Mahenthiralingam, S. Rae, J. M. Raboud, A. G. Davidson, R. Wittmann, and
P. G. Wilcox. Burkholderia cepacia in cystic fibrosis. Variable disease course. American Journal
of Respiratory and Critical Care Medicine, 160(5 Pt 1):1572–1577, 1999.
[70] Lisa Saiman and Jane Siegel. Infection control in cystic fibrosis. Clinical Microbiology Reviews,
17(1):57–71, 2004.
[71] J. R. Govan, J. E. Hughes, and P. Vandamme. Burkholderia cepacia: medical, taxonomic and
ecological issues. Journal of Medical Microbiology, 45(6):395–407, 1996.
[72] Robert M. Aris, Jonathan C. Routh, John J. LiPuma, David G. Heath, and Peter H. Gilligan. Lung
transplantation for cystic fibrosis patients with Bukholderia cepacia complex. American Journal of
Respiratory and Critical Care Medicine, 164(11):2102–2106, 2001.
[73] P. Drevinek and E. Mahenthiralingam. Burkholderia cenocepacia in cystic fibrosis: epidemiology
and molecular mechanisms of virulence. Clinical Microbiology and Infection, 16(7):821–830, 2010.
[74] W. M. Johnson, S. D. Tyler, and K. R. Rozee. Linkage analysis of geographic and clinical clusters
in Pseudomonas cepacia infections by multilocus enzyme electrophoresis and ribotyping. Journal
of Clinical Microbiology, 32(4):924–930, 1994.
[75] Matthew T. G. Holden, Helena M. B. Seth-Smith, Lisa C. Crossman, Mohammed Sebaihia,
Stephen D. Bentley, Ana M. Cerdeno-Tarraga, Nicholas R. Thomson, Nathalie Bason, Michael A.
Quail, Sarah Sharp, Inna Cherevach, Carol Churcher, Ian Goodhead, Heidi Hauser, Nancy Hol-
royd, Karen Mungall, Paul Scott, Danielle Walker, Brian White, Helen Rose, Pernille Iversen,
53

Dalila Mil-Homens, Eduardo P. C. Rocha, Arsenio M. Fialho, Adam Baldwin, Christopher Dowson,
Bart G. Barrell, John R. Govan, Peter Vandamme, C. Anthony Hart, Eshwar Mahenthiralingam,
and Julian Parkhill. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of
cystic fibrosis patients. Journal of Bacteriology, 191(1):261–277, 2009.
[76] S. A. Loutet and M. A. Valvano. A decade of Burkholderia cenocepacia virulence determinant
research. Infection and Immunity, 78(10):4088–4100, 2010.
[77] Mladen Tomich, Christine A. Herfst, Joseph W. Golden, and Christian D. Mohr. Role of flagella in
host cell invasion by Burkholderia cepacia. Infection and Immunity, 70(4):1799–1806, 2002.
[78] U. S. Sajjan and J. F. Forstner. Role of a 22-kilodalton pilin protein in binding of Pseudomonas
cepacia to buccal epithelial cells. Infection and Immunity, 61(8):3157–3163, 1993.
[79] M. Lefebre and M. Valvano. In vitro resistance of Burkholderia cepacia complex isolates to reactive
oxygen species in relation to catalase and superoxide dismutase production. Microbiology, 147(Pt
1):97–109, 2001.
[80] Marcus Miethke and Mohamed A. Marahiel. Siderophore-based iron acquisition and pathogen
control. Microbiology and molecular biology reviews: MMBR, 71(3):413–451, 2007.
[81] Arlene D. Vinion-Dubiel and Joanna B. Goldberg. Lipopolysaccharide of Burkholderia cepacia.
Journal of Endotoxin Research, 9(4):201–213, 2003.
[82] Jorge H. Leitao, Sılvia A. Sousa, Ana S. Ferreira, Christian G. Ramos, Ines N. Silva, and
Leonilde M. Moreira. Pathogenicity, virulence factors, and strategies to fight against Burkholde-
ria cepacia complex pathogens and related species. Applied Microbiology and Biotechnology,
87(1):31–40, 2010.
[83] Anthony De Soyza, Alba Silipo, Rosa Lanzetta, John R. Govan, and Antonio Molinaro. Chemical
and biological features of Burkholderia cepacia complex lipopolysaccharides. Innate Immunity,
14(3):127–144, 2008.
[84] A. D. Cox and S. G. Wilkinson. Ionizing groups in lipopolysaccharides of Pseudomonas cepacia
in relation to antibiotic resistance. Molecular Microbiology, 5(3):641–646, 1991.
[85] Hirofumi Shimomura, Motohiro Matsuura, Shinji Saito, Yoshikazu Hirai, Yasunori Isshiki, and
Kazuyoshi Kawahara. Unusual interaction of a lipopolysaccharide isolated from Burkholderia
cepacia with polymyxin B. Infection and Immunity, 71(9):5225–5230, 2003.
[86] Masahiro Yamamoto and Shizuo Akira. Lipid A receptor TLR4-mediated signaling pathways. Ad-
vances in Experimental Medicine and Biology, 667:59–68, 2010.
[87] Michael L Hutchison, Emma C Bonell, Ian R Poxton, and John R.W Govan. Endotoxic activity of
lipopolysaccharides isolated from emergent potential cystic fibrosis pathogens. FEMS Immunol-
ogy & Medical Microbiology, 27(1):73–77, 2000.
54

[88] Samuel I. Miller, Robert K. Ernst, and Martin W. Bader. LPS, TLR4 and infectious disease diversity.
Nature Reviews Microbiology, 3(1):36–46, 2005.
[89] A. B. Schromm, K. Brandenburg, H. Loppnow, U. Zahringer, E. T. Rietschel, S. F. Carroll, M. H.
Koch, S. Kusumoto, and U. Seydel. The charge of endotoxin molecules influences their confor-
mation and IL-6-inducing capacity. Journal of Immunology, 161(10):5464–5471, 1998.
[90] A. B. Schromm, K. Brandenburg, H. Loppnow, A. P. Moran, M. H. Koch, E. T. Rietschel, and
U. Seydel. Biological activities of lipopolysaccharides are determined by the shape of their lipid A
portion. European Journal of Biochemistry / FEBS, 267(7):2008–2013, 2000.
[91] J. E. Somerville, L. Cassiano, and R. P. Darveau. Escherichia coli msbB gene as a virulence
factor and a therapeutic target. Infection and Immunity, 67(12):6583–6590, 1999.
[92] A. P. Moran, B. Lindner, and E. J. Walsh. Structural characterization of the lipid A component
of Helicobacter pylori rough- and smooth-form lipopolysaccharides. Journal of Bacteriology,
179(20):6453–6463, 1997.
[93] Robert Girard, Thierry Pedron, Satoshi Uematsu, Viviane Balloy, Michel Chignard, Shizuo Akira,
and Richard Chaby. Lipopolysaccharides from Legionella and Rhizobium stimulate mouse bone
marrow granulocytes via Toll-like receptor 2. Journal of Cell Science, 116(Pt 2):293–302, 2003.
[94] M. Soledad Saldıas, Ximena Ortega, and Miguel A. Valvano. Burkholderia cenocepacia O anti-
gen lipopolysaccharide prevents phagocytosis by macrophages and adhesion to epithelial cells.
Journal of Medical Microbiology, 58(12):1542–1548, 2009.
[95] J E Hughes, J Stewart, G R Barclay, and J R Govan. Priming of neutrophil respiratory burst
activity by lipopolysaccharide from Burkholderia cepacia. Infection and Immunity, 65(10):4281–
4287, 1997.
[96] Hirofumi Shimomura, Motohiro Matsuura, Shinji Saito, Yoshikazu Hirai, Yasunori Isshiki, and
Kazuyoshi Kawahara. Lipopolysaccharide of Burkholderia cepacia and its unique character to
stimulate murine macrophages with relative lack of Interleukin-1β-inducing ability. Infection and
Immunity, 69(6):3663–3669, 2001.
[97] E. Caraher, G. Reynolds, P. Murphy, S. McClean, and M. Callaghan. Comparison of antibiotic
susceptibility of Burkholderia cepacia complex organisms when grown planktonically or as biofilm
in vitro. European Journal of Clinical Microbiology & Infectious Diseases, 26(3):213–216, 2007.
[98] K. L. Tomlin, O. P. Coll, and H. Ceri. Interspecies biofilms of Pseudomonas aeruginosa and
Burkholderia cepacia. Canadian Journal of Microbiology, 47(10):949–954, 2001.
[99] Heleen Van Acker, Andrea Sass, Silvia Bazzini, Karen De Roy, Claudia Udine, Thomas Messi-
aen, Giovanna Riccardi, Nico Boon, Hans J. Nelis, Eshwar Mahenthiralingam, and Tom Coenye.
Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding produc-
tion of reactive oxygen species. PLoS ONE, 8(3):e58943, 2013.
55

[100] Kerry L. Tomlin, Rebecca J. Malott, Gordon Ramage, Douglas G. Storey, Pamela A. Sokol, and
H. Ceri. Quorum-sensing mutations affect attachment and stability of Burkholderia cenocepacia
biofilms. Applied and Environmental Microbiology, 71(9):5208–5218, 2005.
[101] M. Soledad Saldias, Julie Lamothe, Robert Wu, and Miguel A. Valvano. Burkholderia ceno-
cepacia requires the RpoN sigma factor for biofilm formation and intracellular trafficking within
macrophages. Infection and Immunity, 76(3):1059–1067, 2008.
[102] S. P. Bernier, D. T. Nguyen, and P. A. Sokol. A LysR-type transcriptional regulator in Burkholderia
cenocepacia influences colony morphology and virulence. Infection and Immunity, 76(1):38–47,
2008.
[103] Daniel F. Aubert, Ronald S. Flannagan, and Miguel A. Valvano. A novel sensor kinase-response
regulator hybrid controls biofilm formation and type VI secretion system activity in Burkholderia
cenocepacia. Infection and Immunity, 76(5):1979–1991, 2008.
[104] C. Fuqua, S. C. Winans, and E. P. Greenberg. Census and consensus in bacterial ecosystems:
the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annual Review of Microbiology,
50:727–751, 1996.
[105] Vittorio Venturi, Arianna Friscina, Iris Bertani, Giulia Devescovi, and Claudio Aguilar. Quorum
sensing in the Burkholderia cepacia complex. Research in Microbiology, 155(4):238–244, 2004.
[106] S. Uehlinger, S. Schwager, S. P. Bernier, K. Riedel, D. T. Nguyen, P. A. Sokol, and L. Eberl.
Identification of specific and universal virulence factors in Burkholderia cenocepacia strains by
using multiple infection hosts. Infection and Immunity, 77(9):4102–4110, 2009.
[107] P. A. Sokol, U. Sajjan, M. B. Visser, S. Gingues, J. Forstner, and C. Kooi. The CepIR quorum-
sensing system contributes to the virulence of Burkholderia cenocepacia respiratory infections.
Microbiology (Reading, England), 149(Pt 12):3649–3658, 2003.
[108] C. Kooi, B. Subsin, R. Chen, B. Pohorelic, and P. A. Sokol. Burkholderia cenocepacia ZmpB is a
broad-specificity zinc metalloprotease involved in virulence. Infection and Immunity, 74(7):4083–
4093, 2006.
[109] A. C. Vergunst, A. H. Meijer, S. A. Renshaw, and D. O’Callaghan. Burkholderia cenocepacia
creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissem-
ination and establishment of systemic infection. Infection and Immunity, 78(4):1495–1508, 2010.
[110] B. Huber, K. Riedel, M. Hentzer, A. Heydorn, A. Gotschlich, M. Givskov, S. Molin, and L. Eberl.
The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and
swarming motility. Microbiology, 147(Pt 9):2517–2528, 2001.
[111] S. Lewenza, B. Conway, E. P. Greenberg, and P. A. Sokol. Quorum sensing in Burkholderia
cepacia: identification of the LuxRI homologs CepRI. Journal of Bacteriology, 181(3):748–756,
1999.
56

[112] Adam Baldwin, Pamela A. Sokol, Julian Parkhill, and Eshwar Mahenthiralingam. The Burkholderia
cepacia epidemic strain marker is part of a novel genomic island encoding both virulence and
metabolism-associated genes in Burkholderia cenocepacia. Infection and Immunity, 72(3):1537–
1547, 2004.
[113] Sujatha Subramoni and Pamela A. Sokol. Quorum sensing systems influence Burkholderia ceno-
cepacia virulence. Future Microbiology, 7(12):1373–1387, 2012.
[114] Yury Herasimenka, Paola Cescutti, Carlos E. Sampaio Noguera, Jose R. Ruggiero, Ranieri Ur-
bani, Giuseppe Impallomeni, Flavio Zanetti, Stephane Campidelli, Maurizio Prato, and Roberto
Rizzo. Macromolecular properties of cepacian in water and in dimethylsulfoxide. Carbohydrate
Research, 343(1):81–89, 2008.
[115] A. S. Ferreira, J. H. Leitao, I. N. Silva, P. F. Pinheiro, S. A. Sousa, C. G. Ramos, and L. M. Mor-
eira. Distribution of cepacian biosynthesis genes among environmental and clinical Burkholderia
strains and role of cepacian exopolysaccharide in resistance to stress conditions. Applied and
Environmental Microbiology, 76(2):441–450, 2010.
[116] Leonilde M. Moreira, Paula A. Videira, Sılvia A. Sousa, Jorge H. Leitao, Monica V. Cunha, and
Isabel Sa-Correia. Identification and physical organization of the gene cluster involved in the
biosynthesis of Burkholderia cepacia complex exopolysaccharide. Biochemical and Biophysical
Research Communications, 312(2):323–333, 2003.
[117] Paola Sist, Paola Cescutti, Silvia Skerlavaj, Ranieri Urbani, Jorge H. Leitao, Isabel Sa-Correia,
and Roberto Rizzo. Macromolecular and solution properties of cepacian: the exopolysaccharide
produced by a strain of burkholderia cepacia isolated from a cystic fibrosis patient. Carbohydrate
Research, 338(18):1861–1867, 2003.
[118] Johan Bylund, Lee-Anna Burgess, Paola Cescutti, Robert K. Ernst, and David P. Speert. Ex-
opolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge re-
active oxygen species. The Journal of Biological Chemistry, 281(5):2526–2532, 2006.
[119] Barbara-Ann D. Conway, Karen K. Chu, Johan Bylund, Eleonora Altman, and David P. Speert.
Production of exopolysaccharide by Burkholderia cenocepacia results in altered cell-surface inter-
actions and altered bacterial clearance in mice. The Journal of Infectious Diseases, 190(5):957–
966, 2004.
[120] James E. A. Zlosnik, Trevor J. Hird, Monica C. Fraenkel, Leonilde M. Moreira, Deborah A.
Henry, and David P. Speert. Differential mucoid exopolysaccharide production by members of
the Burkholderia cepacia complex. Journal of Clinical Microbiology, 46(4):1470–1473, 2008.
[121] James E. A. Zlosnik and David P. Speert. The role of mucoidy in virulence of bacteria from the
Burkholderia cepacia complex: a systematic proteomic and transcriptomic analysis. The Journal
of Infectious Diseases, 202(5):770–781, 2010.
57

[122] Bruno Cuzzi, Paola Cescutti, Linda Furlanis, Cristina Lagatolla, Luisa Sturiale, Domenico
Garozzo, and Roberto Rizzo. Investigation of bacterial resistance to the immune system response:
cepacian depolymerisation by reactive oxygen species. Innate Immunity, 18(4):661–671, 2012.
[123] Silvia A. Sousa, Martina Ulrich, Alessandra Bragonzi, Margaret Burke, Dieter Worlitzsch, Jorge H.
Leitao, Christoph Meisner, Leo Eberl, Isabel Sa-Correia, and Gerd Doring. Virulence of Burkholde-
ria cepacia complex strains in gp91phox-/- mice. Cellular Microbiology, 9(12):2817–2825, 2007.
[124] Christine C. Fehlner-Gardiner, Tammy M.-H. Hopkins, and Miguel A. Valvano. Identification of a
general secretory pathway in a human isolate of Burkholderia vietnamiensis (formerly B. cepa-
cia complex genomovar V) that is required for the secretion of hemolysin and phospholipase C
activities. Microbial Pathogenesis, 32(5):249–254, 2002.
[125] Paul W. Whitby, Timothy M. VanWagoner, Ashlee A. Taylor, Thomas W. Seale, Daniel J. Mor-
ton, John J. LiPuma, and Terrence L. Stull. Identification of an RTX determinant of Burkholderia
cenocepacia J2315 by subtractive hybridization. Journal of Medical Microbiology, 55(Pt 1):11–21,
2006.
[126] C. R. Corbett, M. N. Burtnick, C. Kooi, D. E. Woods, and P. A. Sokol. An extracellular zinc metallo-
protease gene of Burkholderia cepacia. Microbiology (Reading, England), 149(Pt 8):2263–2271,
2003.
[127] Ruifu Zhang, John J. LiPuma, and Carlos F. Gonzalez. Two type IV secretion systems with different
functions in Burkholderia cenocepacia K56-2. Microbiology, 155(12):4005–4013, 2009.
[128] David O’Callaghan, Chantal Cazevieille, Annick Allardet-Servent, Maria Laura Boschiroli, Gisele
Bourg, Vincent Foulongne, Patrice Frutos, Youri Kulakov, and Michel Ramuz. A homologue of the
Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essen-
tial for intracellular survival of Brucella suis. Molecular Microbiology, 33(6):1210–1220, 1999.
[129] Gil Segal, James J. Russo, and Howard A. Shuman. Relationships between a new type IV secre-
tion system and the icm/dot virulence system of Legionella pneumophila. Molecular Microbiology,
34(4):799–809, 1999.
[130] S. Umadevi Sajjan, Lisa A. Carmody, Carlos F. Gonzalez, and John J. LiPuma. A type IV secretion
system contributes to intracellular survival and replication of Burkholderia cenocepacia. Infection
and Immunity, 76(12):5447–5455, 2008.
[131] Mladen Tomich, Adam Griffith, Christine A. Herfst, Jane L. Burns, and Christian D. Mohr. Attenu-
ated virulence of a Burkholderia cepacia type III secretion mutant in a murine model of infection.
Infection and Immunity, 71(3):1405–1415, 2003.
[132] Julie Lamothe, Kassidy K. Huynh, Sergio Grinstein, and Miguel A. Valvano. Intracellular survival of
Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-
containing vacuoles. Cellular Microbiology, 9(1):40–53, 2007.
58

[133] Ronald S. Flannagan, Valentin Jaumouille, Kassidy K. Huynh, Jonathan D. Plumb, Gregory P.
Downey, Miguel A. Valvano, and Sergio Grinstein. Burkholderia cenocepacia disrupts host cell
actin cytoskeleton by inactivating Rac and Cdc42. Cellular Microbiology, 14(2):239–254, 2012.
[134] Roberto Rosales-Reyes, Alexander M. Skeldon, Daniel F. Aubert, and Miguel A. Valvano. The type
VI secretion system of Burkholderia cenocepacia affects multiple Rho family GTPases disrupting
the actin cytoskeleton and the assembly of NADPH oxidase complex in macrophages. Cellular
Microbiology, 14(2):255–273, 2012.
[135] Mikhail A. Gavrilin, Dalia H. A. Abdelaziz, Mahmoud Mostafa, Basant A. Abdulrahman, Jayku-
mar Grandhi, Anwari Akhter, Arwa Abu Khweek, Daniel F. Aubert, Miguel A. Valvano, Mark D.
Wewers, and Amal O. Amer. Activation of the pyrin inflammasome by intracellular Burkholderia
cenocepacia. Journal of Immunology, 188(7):3469–3477, 2012.
[136] Roberto Rosales-Reyes, Daniel F. Aubert, Jennifer S. Tolman, Amal O. Amer, and Miguel A. Val-
vano. Burkholderia cenocepacia type VI secretion system mediates escape of type II secreted
proteins into the cytoplasm of infected macrophages. PLoS ONE, 7(7):e41726, 2012.
[137] Carrie M. Rosenberger and B. Brett Finlay. Phagocyte sabotage: disruption of macrophage sig-
nalling by bacterial pathogens. Nature Reviews Molecular Cell Biology, 4(5):385–396, 2003.
[138] M. S. Saldias and M. A. Valvano. Interactions of Burkholderia cenocepacia and other Burkholderia
cepacia complex bacteria with epithelial and phagocytic cells. Microbiology, 155(9):2809–2817,
2009.
[139] U. Sajjan, M. Corey, A. Humar, E. Tullis, E. Cutz, C. Ackerley, and J. Forstner. Immunolocalisation
of Burkholderia cepacia in the lungs of cystic fibrosis patients. Journal of Medical Microbiology,
50(6):535–546, 2001.
[140] U. Schwab, L. H. Abdullah, O. S. Perlmutt, D. Albert, C. W. Davis, R. R. Arnold, J. R. Yankaskas,
P. Gilligan, H. Neubauer, S. H. Randell, and R. C. Boucher. Localization of Burkholderia cepacia
complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic
mucus. Infection and Immunity, 2014.
[141] Julie Lamothe, Sandra Thyssen, and Miguel A. Valvano. Burkholderia cepacia complex isolates
survive intracellularly without replication within acidic vacuoles of Acanthamoeba polyphaga. Cel-
lular Microbiology, 6(12):1127–1138, 2004.
[142] Karen E. Keith, Daniel W. Hynes, Judith E. Sholdice, and Miguel A. Valvano. Delayed association
of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen
Burkholderia cenocepacia. Microbiology, 155(4):1004–1015, 2009.
[143] Souhaila Al-Khodor, Kimberly Marshall-Batty, Vinod Nair, Li Ding, David e. Greenberg, and Iain
D. C. Fraser. Bukholderia cenocepacia J2315 escapes to the cytosol and actively subverts au-
tophagy in human macrophages. Cellular Microbiology, 16(3):378–395, 2014.
59

[144] Julie Lamothe and Miguel A. Valvano. Burkholderia cenocepacia-induced delay of acidification and
phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective
macrophages. Microbiology, 154(Pt 12):3825–3834, 2008.
[145] Umadevi S. Sajjan, Jeffrey H. Yang, Marc B. Hershenson, and John J. LiPuma. Intracellular
trafficking and replication of Burkholderia cenocepacia in human cystic fibrosis airway epithelial
cells. Cellular Microbiology, 8(9):1456–1466, 2006.
[146] Alessandro Luciani, Valeria Rachela Villella, Speranza Esposito, Nicola Brunetti-Pierri, Diego
Medina, Carmine Settembre, Manuela Gavina, Laura Pulze, Ida Giardino, Massimo Pettoello-
Mantovani, Maria D’Apolito, Stefano Guido, Eliezer Masliah, Brian Spencer, Sonia Quaratino,
Valeria Raia, Andrea Ballabio, and Luigi Maiuri. Defective CFTR induces aggresome formation
and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature Cell
Biology, 12(9):863–875, 2010.
[147] Danielle Glick, Sandra Barth, and Kay F. Macleod. Autophagy: cellular and molecular mecha-
nisms. The Journal of Pathology, 221(1):3–12, 2010.
[148] Kaivon Assani, Mia F. Tazi, Amal O. Amer, and Benjamin T. Kopp. IFN-γ stimulates autophagy-
mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS
ONE, 9(5):e96681, 2014.
[149] Umadevi Sajjan, Shaf Keshavjee, and Janet Forstner. Responses of well-differentiated airway
epithelial cell cultures from healthy donors and patients with cystic fibrosis to Burkholderia ceno-
cepacia infection. Infection and Immunity, 72(7):4188–4199, 2004.
[150] David O’Callaghan and Annette Vergunst. Non-mammalian animal models to study infectious
disease: worms or fly fishing? Current Opinion in Microbiology, 13(1):79–85, 2010.
[151] Tom Coenye and Peter Vandamme. Burkholderia: Molecular Microbiology and Genomics. Horizon
Scientific Press, 2007.
[152] J. R. Starke, M. S. Edwards, C. Langston, and C. J. Baker. A mouse model of chronic pul-
monary infection with Pseudomonas aeruginosa and Pseudomonas cepacia. Pediatric Research,
22(6):698–702, 1987.
[153] Steve P. Bernier, Laura Silo-Suh, Donald E. Woods, Dennis E. Ohman, and Pamela A. Sokol.
Comparative analysis of plant and animal models for characterization of Burkholderia cepacia
virulence. Infection and Immunity, 71(9):5306–5313, 2003.
[154] Martin V. Cieri, Nicole Mayer-Hamblett, Adam Griffith, and Jane L. Burns. Correlation between an
In Vitro invasion assay and a murine model of Burkholderia cepacia lung infection. Infection and
Immunity, 70(3):1081–1086, 2002.
[155] Luisa Pirone, Alessandra Bragonzi, Alessio Farcomeni, Moira Paroni, Cristina Auriche, Mas-
simo Conese, Luigi Chiarini, Claudia Dalmastri, Annamaria Bevivino, and Fiorentina Ascenzioni.
60

Burkholderia cenocepacia strains isolated from cystic fibrosis patients are apparently more inva-
sive and more virulent than rhizosphere strains: Rhizosphere and CF B. cenocepacia pathogenic-
ity. Environmental Microbiology, 10(10):2773–2784, 2008.
[156] Teresa A. Urban, Adam Griffith, Anastasia M. Torok, Mark E. Smolkin, Jane L. Burns, and
Joanna B. Goldberg. Contribution of Burkholderia cenocepacia flagella to infectivity and inflam-
mation. Infection and Immunity, 72(9):5126–5134, 2004.
[157] Tracey A. Hunt, Cora Kooi, Pamela A. Sokol, and Miguel A. Valvano. Identification of Burkholderia
cenocepacia genes required for bacterial survival in vivo. Infection and Immunity, 72(7):4010–
4022, 2004.
[158] Jean-Francois Marier, Jean Lavigne, and Murray P. Ducharme. Pharmacokinetics and effica-
cies of liposomal and conventional formulations of tobramycin after intratracheal administration
in rats with pulmonary Burkholderia cepacia infection. Antimicrobial Agents and Chemotherapy,
46(12):3776–3781, 2002.
[159] P. A. Sokol, P. Darling, D. E. Woods, E. Mahenthiralingam, and C. Kooi. Role of ornibactin biosyn-
thesis in the virulence of Burkholderia cepacia: characterization of pvdA, the gene encoding L-
ornithine N(5)-oxygenase. Infection and Immunity, 67(9):4443–4455, 1999.
[160] U. Sajjan, G. Thanassoulis, V. Cherapanov, A. Lu, C. Sjolin, B. Steer, Y. J. Wu, O. D. Rotstein,
G. Kent, C. McKerlie, J. Forstner, and G. P. Downey. Enhanced susceptibility to pulmonary infec-
tion with Burkholderia cepacia in Cftr(-/-) mice. Infection and Immunity, 69(8):5138–5150, 2001.
[161] D. J. Davidson, J. R. Dorin, G. McLachlan, V. Ranaldi, D. Lamb, C. Doherty, J. Govan, and D. J.
Porteous. Lung disease in the cystic fibrosis mouse exposed to bacterial pathogens. Nature
Genetics, 9(4):351–357, 1995.
[162] M. Mardiney, S. H. Jackson, S. K. Spratt, F. Li, S. M. Holland, and H. L. Malech. Enhanced
host defense after gene transfer in the murine p47phox-deficient model of chronic granulomatous
disease. Blood, 89(7):2268–2275, 1997.
[163] Karen K. Chu, Donald J. Davidson, T. Keith Halsey, Jacqueline W. Chung, and David P. Speert. Dif-
ferential persistence among genomovars of the Burkholderia cepacia complex in a murine model
of pulmonary infection. Infection and Immunity, 70(5):2715–2720, 2002.
[164] Jacqueline W. Chung, Eleonora Altman, Terry J. Beveridge, and David P. Speert. Colonial mor-
phology of Burkholderia cepacia complex genomovar III: Implications in exopolysaccharide pro-
duction, pilus expression, and persistence in the mouse. Infection and Immunity, 71(2):904–909,
2003.
[165] Yinyue Deng, Calvin Boon, Leo Eberl, and Lian-Hui Zhang. Differential modulation of Burkholde-
ria cenocepacia virulence and energy metabolism by the quorum-sensing signal BDSF and its
synthase. Journal of Bacteriology, 191(23):7270–7278, 2009.
61

[166] Manuela Kothe, Melanie Antl, Birgit Huber, Kilian Stoecker, Doreen Ebrecht, Ivo Steinmetz, and
Leo Eberl. Killing of Caenorhabditis elegans by Burkholderia cepacia is controlled by the cep
quorum-sensing system. Cellular Microbiology, 5(5):343–351, 2003.
[167] Silvia T. Cardona, Julia Wopperer, Leo Eberl, and Miguel A. Valvano. Diverse pathogenicity of
Burkholderia cepacia complex strains in the Caenorhabditis elegans host model. FEMS Microbi-
ology Letters, 250(1):97–104, 2005.
[168] Sılvia A. Sousa, Christian G. Ramos, Filipe Almeida, Luıs Meirinhos-Soares, Julia Wopperer,
Stephan Schwager, Leo Eberl, and Jorge H. Leitao. Burkholderia cenocepacia J2315 acyl carrier
protein: a potential target for antimicrobials’ development? Microbial Pathogenesis, 45(5-6):331–
336, 2008.
[169] Thomas R. Laws, Simon A. Smith, Martin P. Smith, Sarah V. Harding, Timothy P. Atkins, and
Richard W. Titball. The nematode Panagrellus redivivus is susceptible to killing by human
pathogens at 37 °c. FEMS microbiology letters, 250(1):77–83, 2005.
[170] Kimberley D. Seed and Jonathan J. Dennis. Development of Galleria mellonella as an alternative
infection model for the Burkholderia cepacia complex. Infection and Immunity, 76(3):1267–1275,
2008.
[171] Kimberley D. Seed and Jonathan J. Dennis. Experimental bacteriophage therapy increases sur-
vival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepa-
cia complex. Antimicrobial Agents and Chemotherapy, 53(5):2205–2208, 2009.
[172] Dalila Mil-Homens, Eduardo P. C. Rocha, and Arsenio M. Fialho. Genome-wide analysis of
DNA repeats in Burkholderia cenocepacia J2315 identifies a novel adhesin-like gene unique to
epidemic-associated strains of the ET-12 lineage. Microbiology, 156(Pt 4):1084–1096, 2010.
[173] Josee Castonguay-Vanier, Ludovic Vial, Julien Tremblay, and Eric Deziel. Drosophila
melanogaster as a model host for the Burkholderia cepacia complex. PLoS ONE, 5(7):e11467,
2010.
[174] C. L. Marolda, B. Hauroder, M. A. John, R. Michel, and M. A. Valvano. Intracellular survival
and saprophytic growth of isolates from the Burkholderia cepacia complex in free-living amoebae.
Microbiology (Reading, England), 145 ( Pt 7):1509–1517, 1999.
[175] Can Unal and Michael Steinert. Dictyostelium discoideum as a model to study host-pathogen in-
teractions. In Ludwig Eichinger and Francisco Rivero, editors, Dictyostelium discoideum Protocols,
number 346 in Methods in Molecular Biology, pages 507–515. Humana Press, 2006.
[176] Carlos F. Gonzalez and Anne K. Vidaver. Bacteriocin, plasmid and pectolytic diversity in Pseu-
domonas cepacia of clinical and plant origin. Journal of general microbiology, 110(1):161–170,
1979.
62

[177] C. F. Gonzalez, E. A. Pettit, V. A. Valadez, and E. M. Provin. Mobilization, cloning, and sequence
determination of a plasmid-encoded polygalacturonase from a phytopathogenic Burkholderia
(Pseudomonas) cepacia. Molecular plant-microbe interactions: MPMI, 10(7):840–851, 1997.
[178] P. Wigley and N. F. Burton. Genotypic and phenotypic relationships in Burkholderia cepacia
isolated from cystic fibrosis patients and the environment. Journal of Applied Microbiology,
86(3):460–468, 1999.
[179] D. S. Yohalem and J. W. Lorbeer. Intraspecific metabolic diversity among strains of Burkholderia
cepacia isolated from decayed onions, soils, and the clinical environment. Antonie Van Leeuwen-
hoek, 65(2):111–131, 1994.
[180] Steve P. Bernier and Pamela A. Sokol. Use of suppression-subtractive hybridization to identify
genes in the Burkholderia cepacia complex that are unique to Burkholderia cenocepacia. Journal
of Bacteriology, 187(15):5278–5291, 2005.
[181] Euan L. S. Thomson and Jonathan J. Dennis. Common duckweed (Lemna minor ) is a versa-
tile high-throughput infection model for the Burkholderia cepacia complex and other pathogenic
bacteria. PLoS ONE, 8(11):e80102, 2013.
[182] Louise A. O’Sullivan, Andrew J. Weightman, T. Hefin Jones, Angela M. Marchbank, James M.
Tiedje, and Eshwar Mahenthiralingam. Identifying the genetic basis of ecologically and biotech-
nologically useful functions of the bacterium Burkholderia vietnamiensis. Environmental Microbi-
ology, 9(4):1017–1034, 2007.
[183] L. S. Saini, S. B. Galsworthy, M. A. John, and M. A. Valvano. Intracellular survival of Burkholderia
cepacia complex isolates in the presence of macrophage cell activation. Microbiology (Reading,
England), 145 ( Pt 12):3465–3475, 1999.
[184] Michael L. Hutchison, Ian R. Poxton, and John R. W. Govan. Burkholderia cepacia produces
a hemolysin that is capable of inducing apoptosis and degranulation of mammalian phagocytes.
Infection and Immunity, 66(5):2033–2039, 1998.
[185] K.-John Cheung, Gang Li, Teresa A. Urban, Joanna B. Goldberg, Adam Griffith, Fuqu Lu, and
Jane L. Burns. Pilus-mediated epithelial cell death in response to infection with Burkholderia
cenocepacia. Microbes and Infection / Institut Pasteur, 9(7):829–837, 2007.
[186] J L Burns, M Jonas, E Y Chi, D K Clark, A Berger, and A Griffith. Invasion of respiratory epithelial
cells by Burkholderia (Pseudomonas) cepacia. Infection and Immunity, 64(10):4054–4059, 1996.
[187] Paula M. Keig, Eileen Ingham, and Kevin G. Kerr. Invasion of human type II pneumocytes by
Burkholderia cepacia. Microbial Pathogenesis, 30(3):167–170, 2001.
[188] Christopher S. Rogers, Yanhong Hao, Tatiana Rokhlina, Melissa Samuel, David A. Stoltz, Yuhong
Li, Elena Petroff, Daniel W. Vermeer, Amanda C. Kabel, Ziying Yan, Lee Spate, David Wax,
63

Clifton N. Murphy, August Rieke, Kristin Whitworth, Michael L. Linville, Scott W. Korte, John F.
Engelhardt, Michael J. Welsh, and Randall S. Prather. Production of CFTR-null and CFTR-∆F508
heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear
transfer. The Journal of Clinical Investigation, 118(4):1571–1577, 2008.
[189] Xingshen Sun, Hongshu Sui, John T. Fisher, Ziying Yan, Xiaoming Liu, Hyung-Ju Cho, Nam Soo
Joo, Yulong Zhang, Weihong Zhou, Yaling Yi, Joann M. Kinyon, Diana C. Lei-Butters, Michelle A.
Griffin, Paul Naumann, Meihui Luo, Jill Ascher, Kai Wang, Timothy Frana, Jeffrey J. Wine, David K.
Meyerholz, and John F. Engelhardt. Disease phenotype of a ferret CFTR-knockout model of cystic
fibrosis. The Journal of Clinical Investigation, 120(9):3149–3160, 2010.
[190] Annemarie H. Meijer and Herman P. Spaink. Host-pathogen interactions made transparent with
the zebrafish model. Current Drug Targets, 12(7):1000–1017, 2011.
[191] Masanori Kasahara, Takashi Suzuki, and Louis Du Pasquier. On the origins of the adaptive im-
mune system: novel insights from invertebrates and cold-blooded vertebrates. Trends in Immunol-
ogy, 25(2):105–111, 2004.
[192] Michiel van der Vaart, Herman P. Spaink, and Annemarie H. Meijer. Pathogen recognition and
activation of the innate immune response in zebrafish. Advances in Hematology, 2012:e159807,
2012.
[193] P. Herbomel, B. Thisse, and C. Thisse. Ontogeny and behaviour of early macrophages in the
zebrafish embryo. Development, 126(17):3735–3745, 1999.
[194] P. Herbomel, B. Thisse, and C. Thisse. Zebrafish early macrophages colonize cephalic mes-
enchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent inva-
sive process. Developmental Biology, 238(2):274–288, 2001.
[195] D. Le Guyader, M. J. Redd, E. Colucci-Guyon, E. Murayama, K. Kissa, V. Briolat, E. Mordelet,
A. Zapata, H. Shinomiya, and P. Herbomel. Origins and unconventional behavior of neutrophils in
developing zebrafish. Blood, 111(1):132–141, 2008.
[196] Emma Colucci-Guyon, Jean-Yves Tinevez, Stephen A. Renshaw, and Philippe Herbomel. Strate-
gies of professional phagocytes in vivo: unlike macrophages, neutrophils engulf only surface-
associated microbes. Journal of Cell Science, 124(18):3053–3059, 2011.
[197] Cornelia Stein, Mario Caccamo, Gavin Laird, and Maria Leptin. Conservation and divergence of
gene families encoding components of innate immune response systems in zebrafish. Genome
Biology, 8(11):R251, 2007.
[198] Annemarie H Meijer, S. F Gabby Krens, Indira A Medina Rodriguez, Shuning He, Wilbert Bitter,
B Ewa Snaar-Jagalska, and Herman P Spaink. Expression analysis of the Toll-like receptor and
TIR domain adaptor families of zebrafish. Molecular Immunology, 40(11):773–783, 2004.
64

[199] null Janeway and null Medzhitov. Introduction: the role of innate immunity in the adaptive immune
response. Seminars in Immunology, 10(5):349–350, 1998.
[200] G. M. Barton and R. Medzhitov. Toll-like receptors and their ligands. Current Topics in Microbiology
and Immunology, 270:81–92, 2002.
[201] Maureen K. Purcell, Kelly D. Smith, Leroy Hood, James R. Winton, and Jared C. Roach. Con-
servation of toll-like receptor signaling pathways in teleost fish. Comparative biochemistry and
physiology. Part D, Genomics & Proteomics, 1(1):77–88, 2006.
[202] Oliver W. Stockhammer, Anna Zakrzewska, Zoltan Hegedus, Herman P. Spaink, and An-
nemarie H. Meijer. Transcriptome profiling and functional analyses of the zebrafish embryonic
innate immune response to Salmonella infection. Journal of Immunology, 182(9):5641–5653,
2009.
[203] Carla M. S. Ribeiro, Trudi Hermsen, Anja J. Taverne-Thiele, Huub F. J. Savelkoul, and Geert F.
Wiegertjes. Evolution of recognition of ligands from Gram-positive bacteria: similarities and differ-
ences in the TLR2-mediated response between mammalian vertebrates and teleost fish. Journal
of Immunology, 184(5):2355–2368, 2010.
[204] Aya Matsuo, Hiroyuki Oshiumi, Tadayuki Tsujita, Hiroshi Mitani, Hisae Kasai, Mamoru Yoshimizu,
Misako Matsumoto, and Tsukasa Seya. Teleost TLR22 recognizes RNA duplex to induce IFN and
protect cells from birnaviruses. Journal of Immunology, 181(5):3474–3485, 2008.
[205] Marıa P. Sepulcre, Francisca Alcaraz-Perez, Azucena Lopez-Munoz, Francisco J. Roca, Jose
Meseguer, Marıa L. Cayuela, and Victoriano Mulero. Evolution of lipopolysaccharide (LPS) recog-
nition and signaling: Fish TLR4 does not recognize LPS and negatively regulates NF-κB activa-
tion. The Journal of Immunology, 182(4):1836–1845, 2009.
[206] Con Sullivan, Jeremy Charette, Julian Catchen, Christopher R. Lage, Gregory Giasson, John H.
Postlethwait, Paul J. Millard, and Carol H. Kim. The gene history of zebrafish tlr4a and tlr4b is
predictive of their divergent functions. Journal of immunology, 183(9):5896, 2009.
[207] Graham J. Lieschke and Peter D. Currie. Animal models of human disease: zebrafish swim into
view. Nature Reviews Genetics, 8(5):353–367, 2007.
[208] John D. Lambris and George Hajishengallis, editors. Current Topics in Innate Immunity II, volume
946 of Advances in Experimental Medicine and Biology. Springer New York, New York, NY, 2012.
[209] Vincenzo Torraca, Samrah Masud, Herman P. Spaink, and Annemarie H. Meijer. Macrophage-
pathogen interactions in infectious diseases: new therapeutic insights from the zebrafish host
model. Disease Models & Mechanisms, 7(7):785–797, 2014.
[210] Lalita Ramakrishnan. Looking within the zebrafish to understand the tuberculous granuloma.
Advances in Experimental Medicine and Biology, 783:251–266, 2013.
65

[211] J. Muse Davis, Hilary Clay, Jessica L. Lewis, Nafisa Ghori, Philippe Herbomel, and Lalita Ramakr-
ishnan. Real-time visualization of Mycobacterium-macrophage interactions leading to initiation of
granuloma formation in zebrafish embryos. Immunity, 17(6):693–702, 2002.
[212] J. Muse Davis and Lalita Ramakrishnan. The role of the granuloma in expansion and dissemina-
tion of early tuberculous infection. Cell, 136(1):37–49, 2009.
[213] C. J. Cambier, Kevin K. Takaki, Ryan P. Larson, Rafael E. Hernandez, David M. Tobin, Kevin B.
Urdahl, Christine L. Cosma, and Lalita Ramakrishnan. Mycobacteria manipulate macrophage
recruitment through coordinated use of membrane lipids. Nature, 505(7482):218–222, 2013.
[214] Anita Ordas, Zoltan Hegedus, Christiaan V. Henkel, Oliver W. Stockhammer, Derek Butler, Hans J.
Jansen, Peter Racz, Matyas Mink, Herman P. Spaink, and Annemarie H. Meijer. Deep sequencing
of the innate immune transcriptomic response of zebrafish embryos to Salmonella infection. Fish
& Shellfish Immunology, 31(5):716–724, 2011.
[215] Mark K. Brannon, J. Muse Davis, Jonathan R. Mathias, Chris J. Hall, Julia C. Emerson, Philip S.
Crosier, Anna Huttenlocher, Lalita Ramakrishnan, and Samuel M. Moskowitz. Pseudomonas
aeruginosa type III secretion system interacts with phagocytes to modulate systemic infection of
zebrafish embryos. Cellular microbiology, 11(5):755–768, 2009.
[216] Hilary Clay, J. Muse Davis, Dana Beery, Anna Huttenlocher, Susan E. Lyons, and Lalita Ramakr-
ishnan. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the
zebrafish. Cell Host & Microbe, 2(1):29–39, 2007.
[217] Jennifer Rhodes, Andreas Hagen, Karl Hsu, Min Deng, Ting Xi Liu, A. Thomas Look, and John P.
Kanki. Interplay of pu.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish.
Developmental Cell, 8(1):97–108, 2005.
[218] Kevin M. Rigby and Frank R. DeLeo. Neutrophils in innate host defense against Staphylococcus
aureus infections. Seminars in Immunopathology, 34(2):237–259, 2012.
[219] Tomasz K. Prajsnar, Vincent T. Cunliffe, Simon J. Foster, and Stephen A. Renshaw. A novel
vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of
zebrafish to non-host specialized pathogens. Cellular Microbiology, 10(11):2312–2325, 2008.
[220] Tomasz K. Prajsnar, Ruth Hamilton, Jorge Garcia-Lara, Gareth McVicker, Alexander Williams,
Michael Boots, Simon J. Foster, and Stephen A. Renshaw. A privileged intraphagocyte niche is
responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cellular
Microbiology, 14(10):1600–1619, 2012.
[221] Jonathan Margolis and Greg D. Plowman. Overcoming the gridlock in discovery research. Nature
Biotechnology, 22(5):522–524, 2004.
[222] Herman P. Spaink, Chao Cui, Malgorzata I. Wiweger, Hans J. Jansen, Wouter J. Veneman, Ruben
Marın-Juez, Jan de Sonneville, Anita Ordas, Vincenzo Torraca, Wietske van der Ent, William P.
66

Leenders, Annemarie H. Meijer, B. Ewa Snaar-Jagalska, and Ron P. Dirks. Robotic injection of
zebrafish embryos for high-throughput screening in disease models. Methods, 62(3):246–254,
2013.
[223] E. Perez-Rueda and J. Collado-Vides. Common history at the origin of the position-function cor-
relation in transcriptional regulators in archaea and bacteria. Journal of Molecular Evolution,
53(3):172–179, 2001.
[224] Junsong Sun and Albrecht Klein. A lysR-type regulator is involved in the negative regulation of
genes encoding selenium-free hydrogenases in the archaeon Methanococcus voltae. Molecular
Microbiology, 52(2):563–571, 2004.
[225] Emilia Stec, Malgorzata Witkowska-Zimny, Monika M. Hryniewicz, Piotr Neumann, Anthony J.
Wilkinson, Andrzej M. Brzozowski, Chandra S. Verma, Jolanta Zaim, Stanislaw Wysocki, and
Grzegorz D. Bujacz. Structural basis of the sulphate starvation response in E. coli : crystal struc-
ture and mutational analysis of the cofactor-binding domain of the Cbl transcriptional regulator.
Journal of Molecular Biology, 364(3):309–322, 2006.
[226] M. A. Schell. Molecular biology of the LysR family of transcriptional regulators. Annual Review of
Microbiology, 47:597–626, 1993.
[227] John M. Farrow, Zoe M. Sund, Matthew L. Ellison, Dana S. Wade, James P. Coleman, and Ev-
erett C. Pesci. PqsE functions independently of PqsR-Pseudomonas quinolone signal and en-
hances the rhl quorum-sensing system. Journal of Bacteriology, 190(21):7043–7051, 2008.
[228] E. P. O’Grady, D. T. Nguyen, L. Weisskopf, L. Eberl, and P. A. Sokol. The burkholderia cenocepacia
LysR-type transcriptional regulator ShvR influences expression of quorum-sensing, protease, type
II secretion, and afc genes. Journal of Bacteriology, 193(1):163–176, 2011.
[229] Daniel W. A. Buchan, Federico Minneci, Tim C. O. Nugent, Kevin Bryson, and David T. Jones.
Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Research,
41:W349–357, 2013.
[230] Patricia Darling, Maria Chan, Andrew D. Cox, and Pamela A. Sokol. Siderophore production by
cystic fibrosis isolates of Burkholderia cepacia. Infection and Immunity, 66(2):874–877, 1998.
[231] Eshwar Mahenthiralingam, Tom Coenye, Jacqueline W. Chung, David P. Speert, John R. W. Go-
van, Peter Taylor, and Peter Vandamme. Diagnostically and experimentally useful panel of strains
from the Burkholderia cepacia complex. Journal of Clinical Microbiology, 38(2):910–913, 2000.
[232] H. Revets, P. Vandamme, A. Van Zeebroeck, K. De Boeck, M. J. Struelens, J. Verhaegen, J. P. Ursi,
G. Verschraegen, H. Franckx, A. Malfroot, I. Dab, and S. Lauwers. Burkholderia (Pseudomonas)
cepacia and cystic fibrosis: the epidemiology in Belgium. Acta Clinica Belgica, 51(4):222–230,
1996.
67

[233] J. L. Marsh, M. Erfle, and E. J. Wykes. The pIC plasmid and phage vectors with versatile cloning
sites for recombinant selection by insertional inactivation. Gene, 32(3):481–485, 1984.
[234] H. P. Schweizer, T Klassen, and T. Hoang. Improved methods for gene analysis and expression
in Pseudomonas spp. Molecular Biology of Pseudomonads, 1996.
[235] J. Sambrook, E.F. Fritsch, and T. Maniatis. Molecular cloning: a laboratory manual. Number vol.
1 in Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, 1989.
[236] H. Inoue, H. Nojima, and H. Okayama. High efficiency transformation of Escherichia coli with
plasmids. Gene, 96(1):23–28, 1990.
[237] Annette C. Vergunst and David O’Callaghan. Host-Bacteria Interactions - Methods and Protocols.
Health and Medicine-Medical Specialties. Humana Press, 2014.
[238] C. Nusslein-Volhard and R. Dahm. Zebrafish: A Practical Approach. Number n.º 975 in Practical
approach series. Oxford University Press, 2002.
[239] Rebecca L. Lamason, Manzoor-Ali P. K. Mohideen, Jason R. Mest, Andrew C. Wong, Heather L.
Norton, Michele C. Aros, Michael J. Jurynec, Xianyun Mao, Vanessa R. Humphreville, Jasper E.
Humbert, Soniya Sinha, Jessica L. Moore, Pudur Jagadeeswaran, Wei Zhao, Gang Ning, Izabela
Makalowska, Paul M. McKeigue, David O’donnell, Rick Kittles, Esteban J. Parra, Nancy J. Mangini,
David J. Grunwald, Mark D. Shriver, Victor A. Canfield, and Keith C. Cheng. SLC24A5, a puta-
tive cation exchanger, affects pigmentation in zebrafish and humans. Science (New York, N.Y.),
310(5755):1782–1786, 2005.
[240] Mai Nguyen-Chi, Quang Tien Phan, Catherine Gonzalez, Jean-Francois Dubremetz, Jean-Pierre
Levraud, and Georges Lutfalla. Transient infection of the zebrafish notochord with E. coli induces
chronic inflammation. Disease Models & Mechanisms, 7(7):871–882, 2014.
[241] Stephen A. Renshaw, Catherine A. Loynes, Daniel M. I. Trushell, Stone Elworthy, Philip W. Ing-
ham, and Moira K. B. Whyte. A transgenic zebrafish model of neutrophilic inflammation. Blood,
108(13):3976–3978, 2006.
[242] Felix Ellett and Graham J. Lieschke. Computational quantification of fluorescent leukocyte num-
bers in zebrafish embryos. In Methods in Enzymology, volume 506, pages 425–435. Elsevier,
2012.
[243] Nawarat Somprasong, Ian McMillan, RoxAnn R. Karkhoff-Schweizer, Skorn Mongkolsuk, and Her-
bert P. Schweizer. Methods for genetic manipulation of Burkholderia gladioli pathovar cocovene-
nans. BMC Research Notes, 3(1):308, 2010.
[244] Francois-Xavier Campbell-Valois, Pamela Schnupf, Giulia Nigro, Martin Sachse, Philippe J. San-
sonetti, and Claude Parsot. A fluorescent reporter reveals on/off regulation of the shigella type
III secretion apparatus during entry and cell-to-cell spread. Cell Host & Microbe, 15(2):177–189,
2014.
68

[245] Jens Bo Andersen, Claus Sternberg, Lars Kongsbak Poulsen, Sara Petersen Bjørn, Michael
Givskov, and Søren Molin. New unstable variants of green fluorescent protein for studies of tran-
sient gene expression in bacteria. Applied and environmental microbiology, 64(6):2240–2246,
1998.
[246] K. C. Keiler, P. R. Waller, and R. T. Sauer. Role of a peptide tagging system in degradation of
proteins synthesized from damaged messenger RNA. Science (New York, N.Y.), 271(5251):990–
993, 1996.
[247] Kathrin Riedel, Morten Hentzer, Otto Geisenberger, Birgit Huber, Anette Steidle, Hong Wu, Niels
Høiby, Michael Givskov, Søren Molin, and Leo Eberl. N-Acylhomoserine-lactone-mediated com-
munication between Pseudomonas aeruginosa and Burkholderia cepacia in mixed biofilms. Mi-
crobiology, 147(12):3249–3262, 2001.
[248] Nicolas Sprynski, Christine Felix, David O’Callaghan, and Annette C. Vergunst. Restoring vir-
ulence to mutants lacking subunits of multiprotein machines: functional complementation of a
Brucella virB5 mutant. FEBS open bio, 2:71–75, 2012.
69

70