52930 protein informatics liisa holm. organization lectures – wednesdays 6 september to 14 october...
TRANSCRIPT

52930 Protein informatics
Liisa Holm

Organization
• Lectures – Wednesdays 6 September to 14 October– Exam Friday 16 October
• Essay type question• Numerical problems
• Textbook– DW Mount: Bioinformatics. Sequence and genome analysis.
2nd edition. Chapters 3-7,10-11• Web site
– http://ekhidna.biocenter.helsinki.fi/teaching/winter2009/proteiinianalyysi

Aims & scope
• Expose biology students to background of methods
• Related practical course– Practical course in protein informatics
(Proteiinianalyysin harjoitustyöt)– Hands-on practice in using web servers that
implement methods– Neither course required for the other

Topics
• Pairwise alignment• Probability and statistical analysis of sequence
alignments• Multiple sequence alignment• Database searching• Phylogenetic prediction• Protein classification and structure prediction• Genome annotation

Pairwise alignment

Why align sequences?
• Common ancestor• Infer common evolutionary
origin from similarity– Then can infer function and
structure• Similarity can be due to
– Gene duplication + speciation– Horizontal gene transfer– Gene fusion– Convergence (similarity without
homology)
Ancestral sequence
Sequence A Sequence B
x steps y steps

Similar sequences are likely homologous
• Dissimilar sequences are less likely to be homologous

4-letter word example
• This is not the usual substitution model
• WORD (d=0, p=1/N^4)• WORE (d=1, p=4/N^3)• GORE (d=2, p=6/N^2)• GONE (d=3, p=4/N)• GENE (d=4, p=1)

Optimal alignment
• Assuming independence between scores for each position, the optimal alignment can be determined using dynamic programming
• Setup: scoring matrix, gap penalties

Dynamic programming
A
B
C
BEGINEND
D
Maximal path sum BEGIN END ?(a) Enumerate every path brute force(b) Use induction: only one optimal path up to any node in graph.
1
30
31
2
4

Example: all paths leading to B
A
B
C
BEGINEND
D
1
30
31
2
43
1
3
8
7

Global alignment
• Needleman-Wunsch algorithm• Maximal trace from beginning to end• Global alignment score may be negative

Local alignment
• Aligned region truncated to segment giving the largest positive contribution

Scoring alignments
• Substitution matrices– Gap penalties
• Significance– Aligning two sequences, would you expect the
same level of similarity by chance alone?

Conversion between odds score, log odds and bit scores
• Odds score = ratio of likelihoods of two events or outcomes. E.g. observed frequency of aligned A and B in related sequences divided by the frequency with which A and B align by chance– f(A and B) / [ f(A) * f(B)]
• Odds scores are often converted to logarithms to create log odds scores.
• Log odds scores are additive.• Bit score = log odds score converted to a logarithm
to the base 2

Bit-scores
• The score needed to distinguish an MSP from chance is approximately the number of bits needed to specify where the MSP starts in each of the two sequences being compared– MSP = maximally scoring pair– Ungapped alignment case
• Log2 N bits are needed to distinguish among N possibilities– Two proteins of 250 residues: 16 bits– Database of 4M residues: 30 bits [160 M: 34 bits]

Dayhoff model
• Markov chain: mutations independent of previous mutations
• Data: 71 groups of closely related sequnces (>85 % similarity), yielding 1572 substitution events
• Mutability of amino acid types (per 100 accepted point mutations)

PAM1 and PAM250 for Phe -> XX PAM1 PAM250 X PAM1 PAM250
Ala 0.0002 0.04 Leu 0.0013 0.13
Arg 0.0001 0.01 Lys 0.0000 0.02
Asn 0.0001 0.02 Met 0.0001 0.02
Asp 0.0000 0.01 Phe 0.9946 0.32
Cys 0.0000 0.01 Pro 0.0001 0.02
Gln 0.0000 0.01 Ser 0.0003 0.03
Glu 0.0000 0.01 Thr 0.0001 0.03
Gly 0.0001 0.03 Trp 0.0001 0.01
His 0.0002 0.02 Tyr 0.0021 0.15
Ile 0.0007 0.05 Val 0.0001 0.05
These are mutation probabilities!

Log odds form of PAM250
• Unit is 10 * logarithm to the base 10 of ratio • S(A,B) = ½ * (10 * log10(p(A->B)/f(A)) + 10 *
log10 (p(B->A)/f(B))• Range -8 … +17• Local alignment scores are maximal, when
PAM distance corresponds to the similarity of the target sequences

BLOSUM matrices
• The BLOSUM matrix assigns a probability score for each residue pair in an alignment based on:–the frequency with which that pairing is
known to occur within conserved blocks of related proteins.
• BLOSUM matrices are constructed from observations which lead to observed probabilities

BLOSUM substitution matrices
BLOSUM matrices are used in ‘log-odds’ form based on actually observed substitutions.This is because:
Ease of use: ‘Scores’ can be just added (the raw probabilities would have to be multiplied) Ease of interpretation:
S=0 : substitution is just as likely to occur as random S<0 : substitution is more likely to occur randomly than observed S>0 : substitution is less likely to occur randomly than observed
Unit is half-bits (odds ratio to logarithm base 2, multiplied by 2)

Information content• Using a standard measure for overall amino acid frequencies gives the
information content of a random protein sequence as 4.19 bits/residue.• Thus, for an average size protein domain (150 residues), the message length
is ~630 bits and the probability that 2 random sequences would specify the same message is 2-630 (10-190).> Database searching for protein similarities is doable, even for fairly short sequences
• BUT, for a transcription binding site of 8-10 bp, the odds of 2 random sequences arriving at the same message is 10-5.> Database searching for regulatory elements does not work well as
databases get larger

Relative entropy H of target and background distributions
• Scale score matrix s to bits qij
• H = S qij sij = S qij log ----------- pi pj
q = target frequencies of amino acidsp = background frequencies
H measures the average information available per position to distinguish the alignment from chance

qij
• Score = S fij sij ~ S fij ln -----------
pi pj
Optimal scoring matrix: target distribution q = frequencies in alignment f

Affine gap penalties
• Gap opening penalty (g)• Gap extension penalty (r)
• W(x) = g + rx• X is the length of the gap
• Well working gap penalties:• BLSOUM62 (-11,-1)

Statistical Significance
• A good way to determine if an alignment score has statistical meaning is to compare it with the score generated from the alignment of two random sequences
• A model of ‘random’ sequences is needed. The simplest model chooses the amino acid residues in a sequence independently, with background probabilities (Karlin & Altschul (1990) Proc. Natl. Acad. Sci. USA, 87 (1990) 2264-2268)

Alignment score
• Optimal alignment scores follow extreme value distribution– Exact theory for ungapped local alignments
• There is at least one positive score sij
• Average score is negative
– Results hold empirically for gapped alignments

Probability and statistics

The need for statistics
• Statistics is very important for bioinformatics. – It is very easy to have a computer analyze the data
and give you back a result. – Problem is to decide whether the answer the
computer gives you is any good at all. • Questions:
– How statistically significant is the answer?– What is the probability that this answer could
have been obtained by random? What does this depend on?

Nn
X
S
Population Sample
Basics

Nn
X
Population Sample
Basics
Descriptive statistics
Probability

Substitution matrices
ba
ab
ffpbaS log),( 1
Lambda is a scaling factor equal to 0.347, set so that the scores can be rounded off to sensible integers
Pab is the observed frequency that residues a and b are correlated because of homology
fafb is the expected frequency of seeing residues a and b paired together, which is just the product of the frequency of residue a multiplied by the frequency of residue b
Source: Where did the BLOSUM62 alignment score matrix come from? Eddy S., Nat. Biotech. 22 Aug 2004
Score of amino acid a with amino acid b

Substitution matrices
Sff
p eba
ab
Lambda is a scaling factor equal to 0.347, set so that the scores can be rounded off to sensible integers
Pab is the observed frequency that residues a and b are correlated because of homology
fafb is the expected frequency of seeing residues a and b paired together, which is just the product of the frequency of residue a multiplied by the frequency of residue b


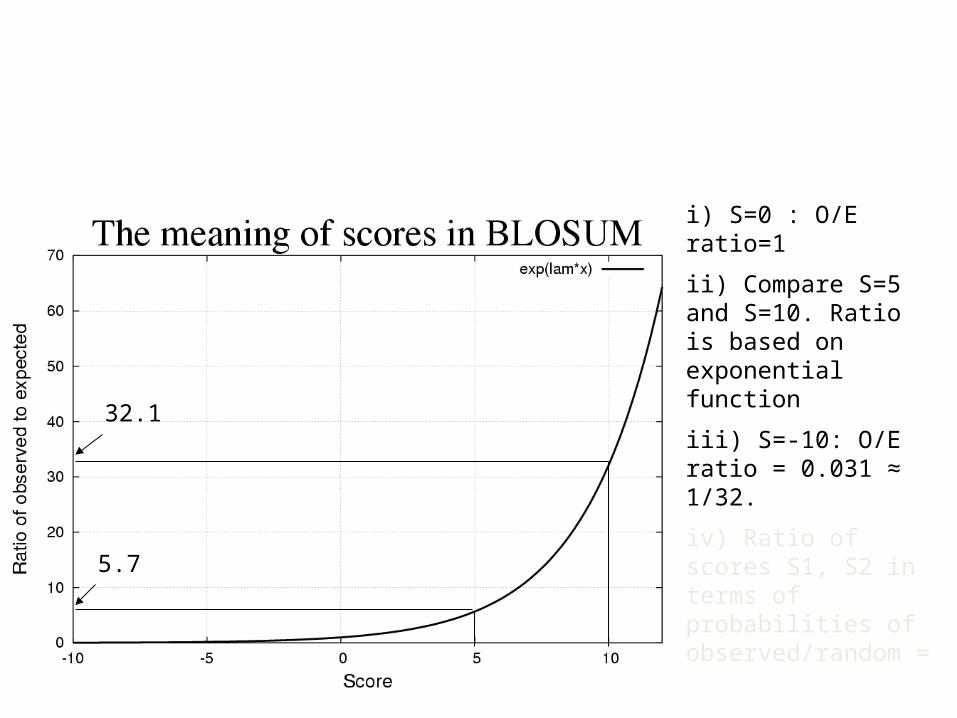
i) S=0 : O/E ratio=1
ii) Compare S=5 and S=10. Ratio is based on exponential function
iii) S=-10: O/E ratio = 0.031 ≈ 1/32.
iv) Ratio of scores S1, S2 in terms of probabilities of observed/random =

5.7
32.1
i) S=0 : O/E ratio=1
ii) Compare S=5 and S=10. Ratio is based on exponential function
iii) S=-10: O/E ratio = 0.031 ≈ 1/32.
iv) Ratio of scores S1, S2 in terms of probabilities of observed/random =
5.7
32.1
i) S=0 : O/E ratio=1
ii) Compare S=5 and S=10. Ratio is based on exponential function
iii) S=-10: O/E ratio = 0.031 ≈ 1/32.
iv) Ratio of scores S1, S2 in terms of probabilities of observed/random =

5.7
32.1
i) S=0 : O/E ratio=1
ii) Compare S=5 and S=10. Ratio is based on exponential function
iii) S=-10: O/E ratio = 0.031 ≈ 1/32.
iv) Ratio of scores S1, S2 in terms of probabilities of observed/random =
)( 2121 / SSSS eee

Example: BLAST
• Motivations–Exact algorithms are exhaustive but
computationally expensive.–Exact algorithms are impractical for comparing
a query sequence to millions of other sequences in a database (database scanning),
–and so, database scanning requires heuristic alignment algorithm (at the cost of optimality).

Interpret BLAST results - Description
ID (GI #, refseq #, DB-specific ID #) Click to access the record in GenBank
Bit score – higher, better. Click to access the pairwise alignment
Expect value – lower, better. It tells the possibility that this is a random hit
Gene/sequence Definition
Links

Problems with BLAST
• Why do results change?• How can you compare results from different
BLAST tools which may report different types of values?
• How are results (eg evalue) affected by query• There are _many_ values reported in the
output – what do they mean?

Example: Importance of Blast statistics
But, first a review.

Review
• What is a distribution?– A plot showing the frequency of a given variable or
observation.

Review
• What is a distribution?– A plot showing the frequency of a given variable or
observation.

Features of a Normal Distribution
m = meanSymmetric DistributionHas an average or mean value at the centreHas a characteristic width called the standard deviation (S.D. = σ)Most common type of distribution known

Standard Deviations (Z-score)
± 1.0 S.D. 0.683 > + 1.0 S.D. 0.158
± 2.0 S.D. 0.954 > + 2.0 S.D. 0.023
± 3.0 S.D. 0.9972 > + 3.0 S.D. 0.0014
± 4.0 S.D. 0.99994 > + 4.0 S.D. 0.00003
± 5.0 S.D. 0.999998 > + 5.0 S.D. 0.000001

Mean, Median & ModeMode
Median
Mean

Mean, Median, Mode
• In a Normal Distribution the mean, mode and median are all equal
• In skewed distributions they are unequal• Mean - average value, affected by extreme
values in the distribution• Median - the “middlemost” value, usually half
way between the mode and the mean• Mode - most common value

Different Distributions
Unimodal Bimodal

Other Distributions
• Binomial Distribution
• the discrete probability distribution of the number of successes in a sequence
of n independent yes/no experiments, each of which yields success with
probability p.
• Poisson Distribution• expresses the probability of a number of events occurring in a fixed period of time if these
events occur with a known average rate and independently of the time since the last event.
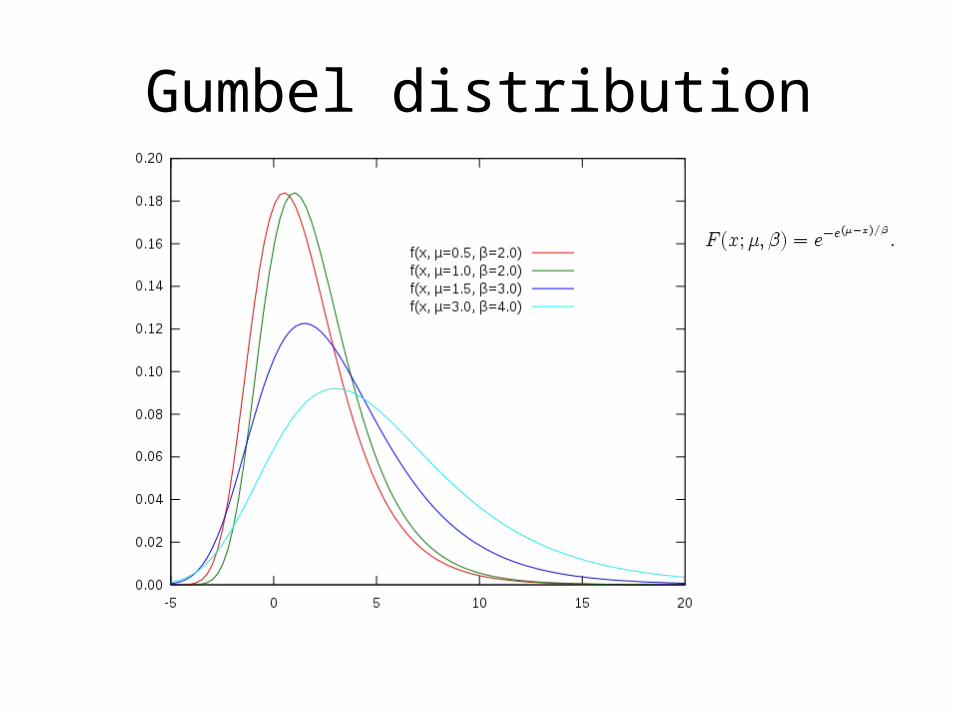
• Extreme Value Distribution
– Gumbel distribution
– used to model the distribution of the maximum (or the minimum)
of a number of samples of various distributions.

Binomial Distribution1
1 1
1 2 1
1 3 3 1
1 4 6 4 1
1 5 10 10 5 1
P(x) = (p + q)n

Poisson DistributionPr
opor
tion
of s
ampl
es
m = 10
m=0.1
m = 1
m = 2
m = 3
P(x)
x
!)( xexxP

Review
• What is a distribution?– A plot showing the frequency of a given variable or observation.
• What is a null hypothesis?– A statistician’s way of characterizing “chance.” – Generally, a mathematical model of randomness with respect to
a particular set of observations.– The purpose of most statistical tests is to determine whether the
observed data can be explained by the null hypothesis.

Review
• What is a distribution?– A plot showing the frequency of a given variable or observation.
• What is a null hypothesis?– A statistician’s way of characterizing “chance.” – Generally, a mathematical model of randomness with respect to
a particular set of observations.– The purpose of most statistical tests is to determine whether the
observed data can be explained by the null hypothesis.

Review
• Examples of null hypotheses:– Sequence comparison using shuffled sequences.– A normal distribution of log ratios from a
microarray experiment.– LOD scores from genetic linkage analysis when the
relevant loci are randomly sprinkled throughout the genome.

Empirical score distribution
The picture shows a distribution of scores from a real database search using BLAST.This distribution contains scores from non-homologous and homologous pairs.
High scores from homology.

Empirical null score distribution
This distribution is similar to the previous one, but generated using a randomized sequence database.

Review
• What is a p-value?– The probability of observing an effect as strong or
stronger than you observed, given the null hypothesis. I.e., “How likely is this effect to occur by chance?”
– Pr(x > S|null)

Review
What is the name of the distribution created by sequence similarity scores, and what does it look like?Extreme value distribution,
or Gumbel distribution.It looks similar to a normal
distribution, but it has a larger tail on the right.

Review
What is the name of the distribution created by sequence similarity scores, and what does it look like?
Extreme value distribution, or Gumbel distribution.It looks similar to a normal distribution, but it has a larger tail on the right.
0
1000
2000
3000
4000
5000
6000
7000
8000
<20 30 40 50 60 70 80 90 100 110 >120

Statistics• BLAST (and also local i.e. Smith-Waterman and BLAT scores)
between random, unrelated sequences follow the Gumbel Extreme Value Distribution (EVD)
• Pr(s>S) = 1-exp(-Kmn e-lS)– This is the probability of randomly encountering a score greater than S.– S alignment score– m,n query sequence lengths, and length of database resp.– K, l parameters depending on scoring scheme and sequence composition
• Bit score : S’ = lS – log(K) log(2)

BLAST output revisited
K
S’ S E
nm
From: Expasy BLAST

Review
EVD for random blastUpper tail behaviour:
Pr( s > S ) ~ Kmn e-lS
0
1000
2000
3000
4000
5000
6000
7000
8000
<20 30 40 50 60 70 80 90 100 110 >120
This is the EXPECT value = Evalue

P-value in Sequence Matching
• P(s > S) = .01 – P-value of .01 occurs at score threshold S (392 below) where score s
from random comparison is greater than this threshold 1% of the time
• Likewise for P=.001 and so on.

What Distribution Really Looks Like• N Dependence• True Positives

A most important caveat...
• For database searches, the ONLY criteria available to judge the likelihood of a structural or evolutionary relationship between 2 sequences is an estimate of statistical significance
• For a medium-sized protein using default parameters (Blosum62, E = 10), the cut-off for statistical significance is P =10-7-10-5
• Statistical significance and biological significance are NOT necessarily the same

P-value
P<=10-100 exact match
10-100 < P < 10-50 sequences very nearly identical, e.g., alleles or SNPs
10-50 < P < 10-10 closely related sequences, homology certain
10-5 < P < 10-1 usually distant relatives
P > 0.1 match probably insignificant

Significance Dependson Database Size
• The Significance of Similarity Scores Decreases with Database Growth – The score between any pair of sequence pair is constant– The number of database entries grows exponentially– The number of non-homologous entries >> homologous entries– Greater sensitivity is required to detect homologies
Greater s• Score of 100 might rank as best in database of 1000 but only in top-100 in
database of 1000000– expectation value
DB-1 DB-2

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score–S’ = lS – log(K)
log(2)

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score–S’ = lS – log(K)
log(2)
Score and bit score grow linearly with the length of the alignment

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score– S’ = lS – log(K)
log(2)• E-value of bit score
– E = mn2-S’
Score and bit score grow linearly with the length of the alignment

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score– S’ = lS – log(K)
log(2)• E-value of bit score
– E = mn2-S’
Score and bit score grow linearly with the length of the alignment
E-Value shrinks really fast as bit score grows

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score– S’ = lS – log(K)
log(2)• E-value of bit score
– E = mn2-S’
Score and bit score grow linearly with the length of the alignment
E-Value shrinks really fast as bit score grows
E-Value grows linearly with the product of target and query sizes.

Summary
• Want to be able to compare scores in sequences of different compositions or different scoring schemes
• Score: S = sum(match) – sum(gap costs)
• Bit score– S’ = lS – log(K)
log(2)• E-value of bit score
– E = mn2-S’
Score and bit score grow linearly with the length of the alignment
E-Value shrinks really fast as bit score grows
E-Value grows linearly with the product of target and query sizes.
Doubling target set size and doubling query length have the same effect on e-value

Conclusion
• You should now be able to compare BLAST results from different databases, converting values if they are reported differently (which happens frequently)
• You should now know why BLAST results might change from one day to the next, even on the same server
• You should understand also the dependence of query length on E-value.• Statistical rankings are reported for (almost) every database search
tool. When making comparisons between databases, between sequences it is useful to know how the statistics are derived to know if comparisons are meaningful.

Exercises

Exercise 1: Calculation of Log Odds and Odds Scores by the BLOSUM Method
• In one column of an alignment of a set of related, similar sequences, amino acid D changes to amino acid E at a frequency of 0.10, and the number of times this change is expected based on the number of occurrences of D and E in the column is 0.05.
• What is the odds score of finding a D-to-E substitution in an alignment? • What is the log odds score for the D-to-E substitution in bits? (Note: log to
base 2 = natural log / 0.693.) • What would be the entry in the BLOSUM amino acid scoring matrix for
this substitution? Compare your result to the actual entry in the BLOSUM62 matrix (D to E in BLOSUM62 : +2).
• In the same column, D does not change at all at a frequency of 0.80, and the expected frequency of D not changing is 0.10. Calculate the corresponding log odds score and the BLOSUM62 entry for D not changing (D to D in BLOSUM62: +6).

Exercise 2: USING THE DYNAMIC PROGRAMMING METHOD TO CALCULATE THE LOCAL ALIGNMENT OF TWO SHORT SEQUENCES
BY HAND
• The BLASTP algorithm performs a local alignment between a query sequence and a matching database sequence using the dynamic programming algorithm with the BLOSUM62 scoring matrix, a gap opening penalty of –11, and a gap extension penalty of –1 (i.e., a gap of length 1 has a penalty of –11, one of length 2, –12, etc.). Align the sequences MDPW and MEDPW using the Smith–Waterman algorithm described in the dynamic programming notes by following the global alignment example given in the notes, but using the Smith–Waterman algorithm.
• Make a matrix for keeping track of best scores and a second matrix to keep track of the moves that give the best scores. (Hint: The alignment of M's, P's, and W's all give high scores, so the problem boils down to how to align D with ED and is actually quite a trivial problem.)
• Use the BLOSUM62 matrix and BLASTP gap penalties of –11,–1. What is the optimal alignment and score between these two sequences?

Exercise 3 • What is the odds score and log odds score of the following alignment?
Blosum (D,D)=+6, (D,E)=+2 DEDEDEDE DDDDDDDD
• Using the approximation S ~ log2(nm) and assuming that the above alignment was found by aligning two sequences of length 250, is the alignment significant at the 0.05 level? (That is, could an alignment of two random sequences of the same length achieve such a score with a probability of 0.05?)
• If the gap penalty was very high, e.g., gap opening of 8 and gap extension of 8, so that no gaps were produced, and the BLOSUM62 scoring matrix was used, calculate the significance of the alignment using the equation P(S>x) ~ 1-exp(-Kmn exp(- λ x)). Use K=0.060 and λ=0.270; note that this λ assumes that the alignment score is in half-bits so that the alignment score must be in these units also.