3. aromatic chemistry
DESCRIPTION
Benzene, Aromaticity, Aromatic HydrocarbonsTRANSCRIPT

Reactions of Aromatic Compounds Simple alkenes tend to undergo addition reactions:
HBrBr2Br
Br Br
C3H6C3H6Br2 C3H7Br The elements of the reagent (HBr or Br2) are simply added to the starting material. This is called, unsurprisingly, and addition reaction. Aromatic compounds do not react in this manner. A look at a simple aromatic bromination:
Br2 / FeBr3
Br
C6H6 C6H5Br What we have done in this case is substitute a bromine for a hydrogen. Hence the term “aromatic substitution.” Because the benzene ring is quite electron-rich, it almost always behaves as the nucleophile in a reaction - which means that the substitution on benzene occurs by the addition of an electrophile -> electrophilic aromatic substitution. There is basically one simple mechanism for all electrophilic aromatic substitutions:
E+ H
EH
EH
EB-
E
The benzene acts as a nucleophile, attacking the electrophile with a pair of its π-electrons. This initial step destroys the aromaticity of the molecule! The resulting positive charge is delocalized over the ortho and para positions. The conjugate base of the initial electrophile then assists in removing the now-extraneous proton, and restores aromaticity. Because all electrophilic aromatic substitutions proceed in this way, the only thing that matters is the preparation of a “hot’ electrophile. Why a “hot” electrophile? As you can see, the first step of the reaction involves destroying aromaticity. In order to do this, there must be a significant energetic driving force. This driving force comes in the form of a very reactive (unhappy) electrophile. How are such electrophiles generated? Halogenation As you can imagine, halogens bearing a positive charge are particularly reactive. I will focus on preparing halogen electrophiles from Br, Cl and I. Bromine: Allowing bromine to react with iron metal first generates FeBr3, which then interacts with the remaining Br2 to form a highly polarized system:
FeBr2
FeBr3Br3Fe Br Br
!-
!+
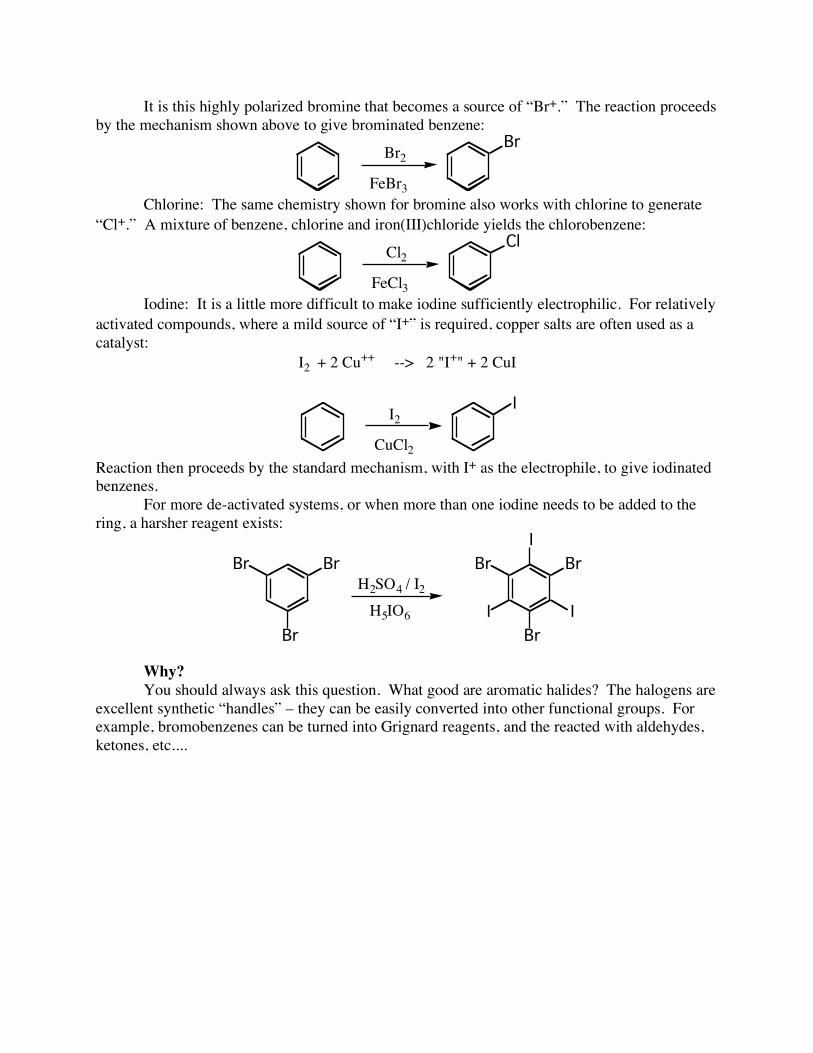
It is this highly polarized bromine that becomes a source of “Br+.” The reaction proceeds by the mechanism shown above to give brominated benzene:
Br2
FeBr3
Br
Chlorine: The same chemistry shown for bromine also works with chlorine to generate “Cl+.” A mixture of benzene, chlorine and iron(III)chloride yields the chlorobenzene:
Cl2
FeCl3
Cl
Iodine: It is a little more difficult to make iodine sufficiently electrophilic. For relatively activated compounds, where a mild source of “I+” is required, copper salts are often used as a catalyst:
I2
CuCl2
I
I2 + 2 Cu++
--> 2 "I+" + 2 CuI
Reaction then proceeds by the standard mechanism, with I+ as the electrophile, to give iodinated benzenes. For more de-activated systems, or when more than one iodine needs to be added to the ring, a harsher reagent exists:
Br Br
Br
H2SO4 / I2
H5IO6
Br Br
Br
I I
I
Why? You should always ask this question. What good are aromatic halides? The halogens are excellent synthetic “handles” – they can be easily converted into other functional groups. For example, bromobenzenes can be turned into Grignard reagents, and the reacted with aldehydes, ketones, etc....
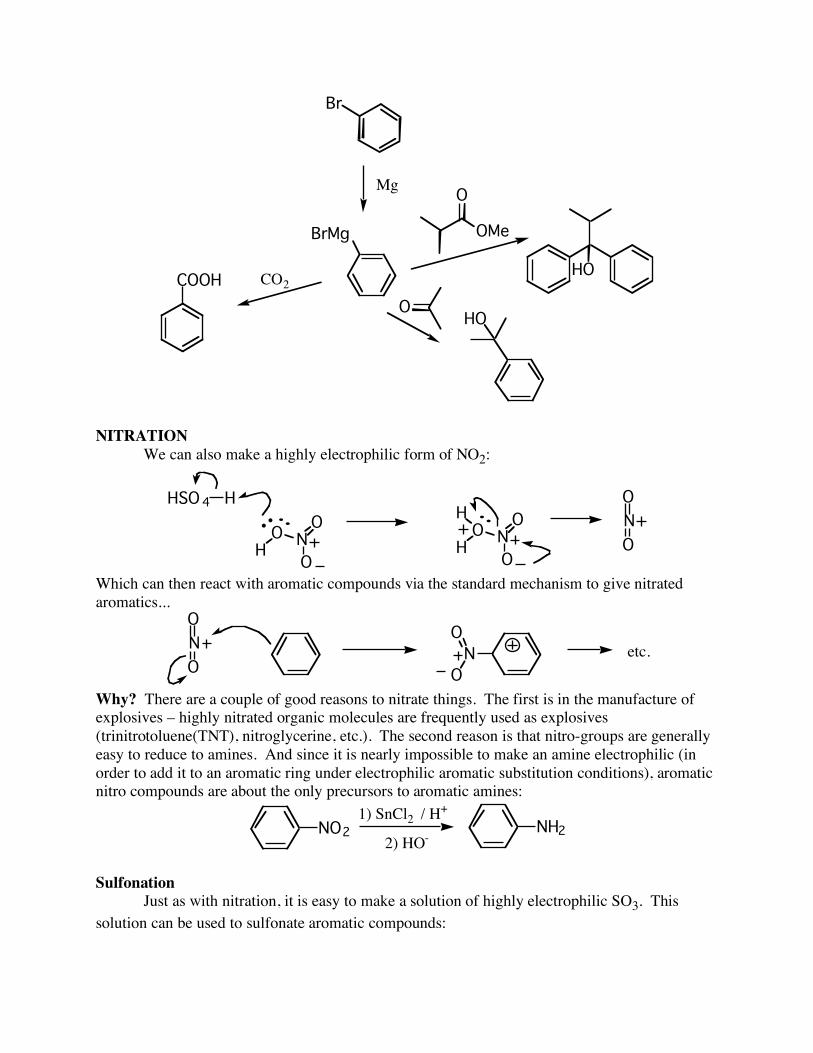
Br
Mg
BrMg
OHO
OMe
O
HOCO2COOH
NITRATION We can also make a highly electrophilic form of NO2:
N
O
O
O
H
HSO4 H
N
O
O
O
H
HO
N
O
Which can then react with aromatic compounds via the standard mechanism to give nitrated aromatics...
O
N
ON
O
O
etc.
Why? There are a couple of good reasons to nitrate things. The first is in the manufacture of explosives – highly nitrated organic molecules are frequently used as explosives (trinitrotoluene(TNT), nitroglycerine, etc.). The second reason is that nitro-groups are generally easy to reduce to amines. And since it is nearly impossible to make an amine electrophilic (in order to add it to an aromatic ring under electrophilic aromatic substitution conditions), aromatic nitro compounds are about the only precursors to aromatic amines:
NO2
1) SnCl2 / H+
2) HO-NH2
Sulfonation Just as with nitration, it is easy to make a solution of highly electrophilic SO3. This solution can be used to sulfonate aromatic compounds:
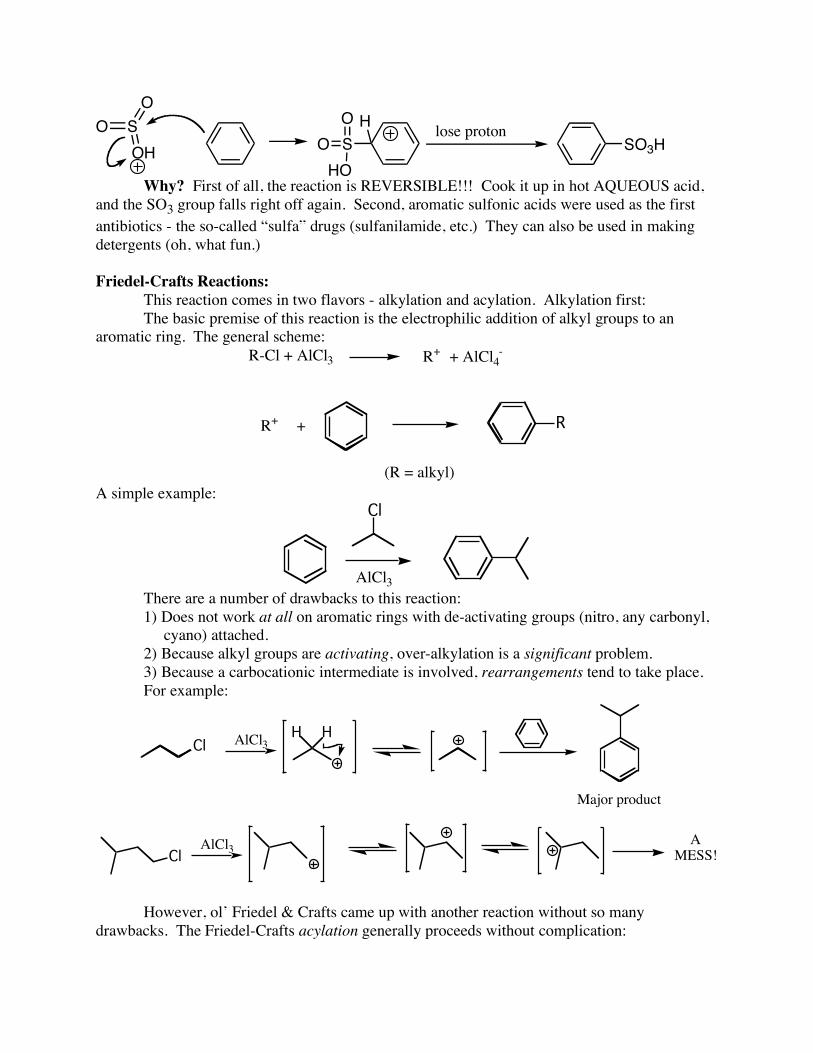
O
S
OHS
HO
OO
O
H
SO3Hlose proton
Why? First of all, the reaction is REVERSIBLE!!! Cook it up in hot AQUEOUS acid, and the SO3 group falls right off again. Second, aromatic sulfonic acids were used as the first antibiotics - the so-called “sulfa” drugs (sulfanilamide, etc.) They can also be used in making detergents (oh, what fun.) Friedel-Crafts Reactions: This reaction comes in two flavors - alkylation and acylation. Alkylation first: The basic premise of this reaction is the electrophilic addition of alkyl groups to an aromatic ring. The general scheme:
R-Cl + AlCl3 R+ + AlCl4-
R+ + R
(R = alkyl) A simple example:
Cl
AlCl3 There are a number of drawbacks to this reaction: 1) Does not work at all on aromatic rings with de-activating groups (nitro, any carbonyl, cyano) attached. 2) Because alkyl groups are activating, over-alkylation is a significant problem. 3) Because a carbocationic intermediate is involved, rearrangements tend to take place. For example:
ClH H
Major product
Cl
AlCl3
AlCl3A
MESS!
However, ol’ Friedel & Crafts came up with another reaction without so many drawbacks. The Friedel-Crafts acylation generally proceeds without complication:
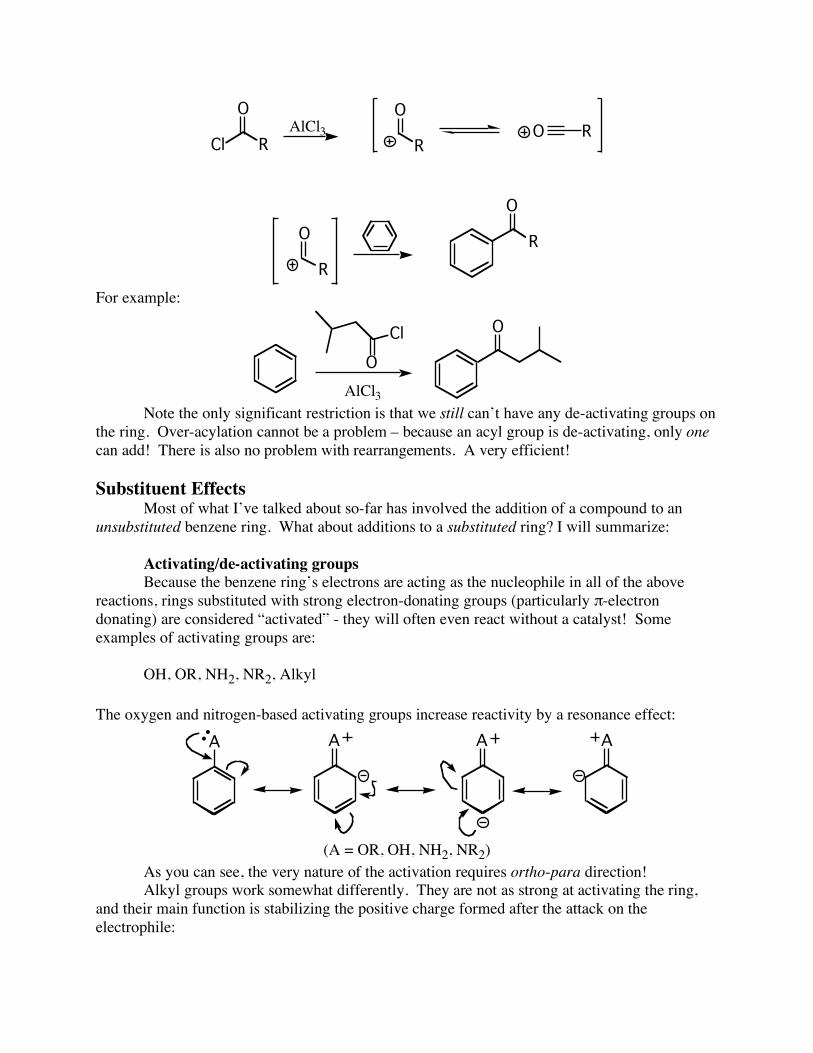
Cl R
O
AlCl3
R
O
O R
R
OR
O
For example:
Cl
O
AlCl3
O
Note the only significant restriction is that we still can’t have any de-activating groups on the ring. Over-acylation cannot be a problem – because an acyl group is de-activating, only one can add! There is also no problem with rearrangements. A very efficient! Substituent Effects Most of what I’ve talked about so-far has involved the addition of a compound to an unsubstituted benzene ring. What about additions to a substituted ring? I will summarize: Activating/de-activating groups Because the benzene ring’s electrons are acting as the nucleophile in all of the above reactions, rings substituted with strong electron-donating groups (particularly π-electron donating) are considered “activated” - they will often even react without a catalyst! Some examples of activating groups are: OH, OR, NH2, NR2, Alkyl The oxygen and nitrogen-based activating groups increase reactivity by a resonance effect:
A A A A
(A = OR, OH, NH2, NR2)
As you can see, the very nature of the activation requires ortho-para direction! Alkyl groups work somewhat differently. They are not as strong at activating the ring, and their main function is stabilizing the positive charge formed after the attack on the electrophile:

R
E+
R
E H
R
E H
Stable tertiary carbocation!
(R = alkyl)
Note well: All activating groups direct ortho-para!
The Halogens: The halogens are in a class by themselves, and behave in an unusual manner. They are VERY electron withdrawing, and thus DE-ACTIVATE the ring towards electrophilic substitution. However, their multiple lone-pairs are able to stabilize the cation formed after electrophilic addition, and thus direct ortho-para:
Br
E+
E H E H
Br Br
Meta-Directing De-activators. Strongly electron-withdrawing groups de-activate the ring towards electrophilic substitution. Examples of such groups are: Ester (COOR), Acid (COOH), Aldehyde (CHO), Nitro (NO2), Ketone (or acyl, COR), Cyano (CN) Acid Halide (COCl) However, with a strong enough electrophile, the rings often will react. However, in order to avoid putting the charge that develops (after attacking an electrophile) on the carbon attached to the electron-withdrawing group, the incoming electrophile must attach to the meta carbons (see below). Note that the charge cannot be delocalized onto the carbon containing the acyl group (also note - the charge delocalizes over all of the ortho and para positions!)
O
E+
O
E
H
O
E
H
O
E
H
Other considerations:

Sterics, Sterics, Sterics. This is particularly a problem with bulky ortho-para directors - the ortho positions are blocked, forcing addition to the para positions:
Br2 / Fe
BrNO ORTHO!
HNO3
H2SO4
NO2
Multi-functional compounds: What if the ring has several substituents? Your text has a rather detailed description of what to do, so here I’ll just supplement that data. 1) If the effects of two substituents point to reactivity at one particular carbon, you’re in luck! That’s where the electrophile will go (providing the sterics are not horrible). 2) The strongest directing group almost always prevails (e.g. OH over alkyl, NR2 over Br) 3) The order of precedence is a) Strong o,p directors (OR, NR2) b) Alkyl groups and halogen c) All meta-directors. 4) And of course, keep a close eye on the steric environment. Nucleophilic Aromatic Substitution Your text has a good description of this phenomenon (Sec. 16.8). This sort of reaction is not very common, but it would be a good idea to be familiar with the mechanism. Benzyne Benzyne was discovered when some crazed scientists mixed chlorobenzene and sodium hydroxide at high temperature and pressure. The resulting phenol was found to have arisen from an elimination/re-addition reaction:
H
Cl
HO-
H2OOH
Because benzyne is such a useful intermediate, a method has been discovered which allows it to be prepared by much milder means. It starts with anthranilic acid, diazotizes the amine, which then decomposes to benzyne, carbon dioxide and nitrogen. The benzyne can then be used in many ways:
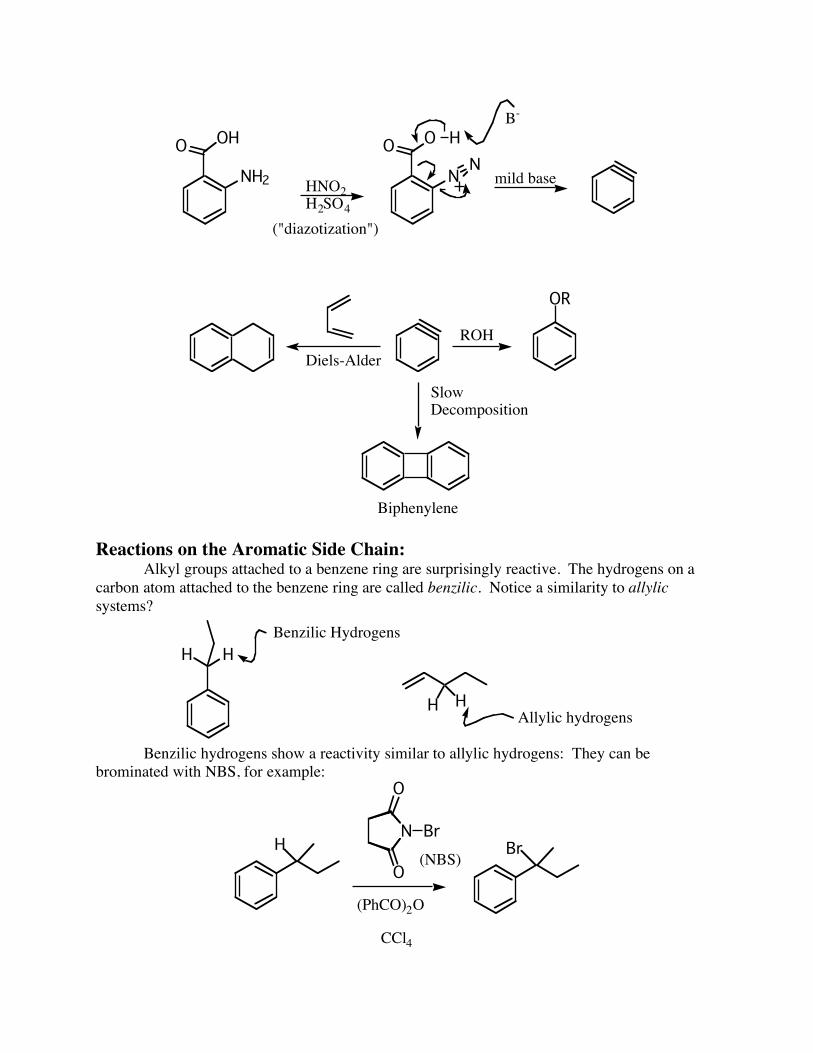
NH2
OOH
N
OO
N
H
HNO2H2SO4
("diazotization")
B-
OR
mild base
ROH
Diels-Alder
SlowDecomposition
Biphenylene
Reactions on the Aromatic Side Chain: Alkyl groups attached to a benzene ring are surprisingly reactive. The hydrogens on a carbon atom attached to the benzene ring are called benzilic. Notice a similarity to allylic systems?
H H
Benzilic Hydrogens
H H
Allylic hydrogens
Benzilic hydrogens show a reactivity similar to allylic hydrogens: They can be brominated with NBS, for example:
HN
O
O
Br
(NBS)
(PhCO)2O
CCl4
Br
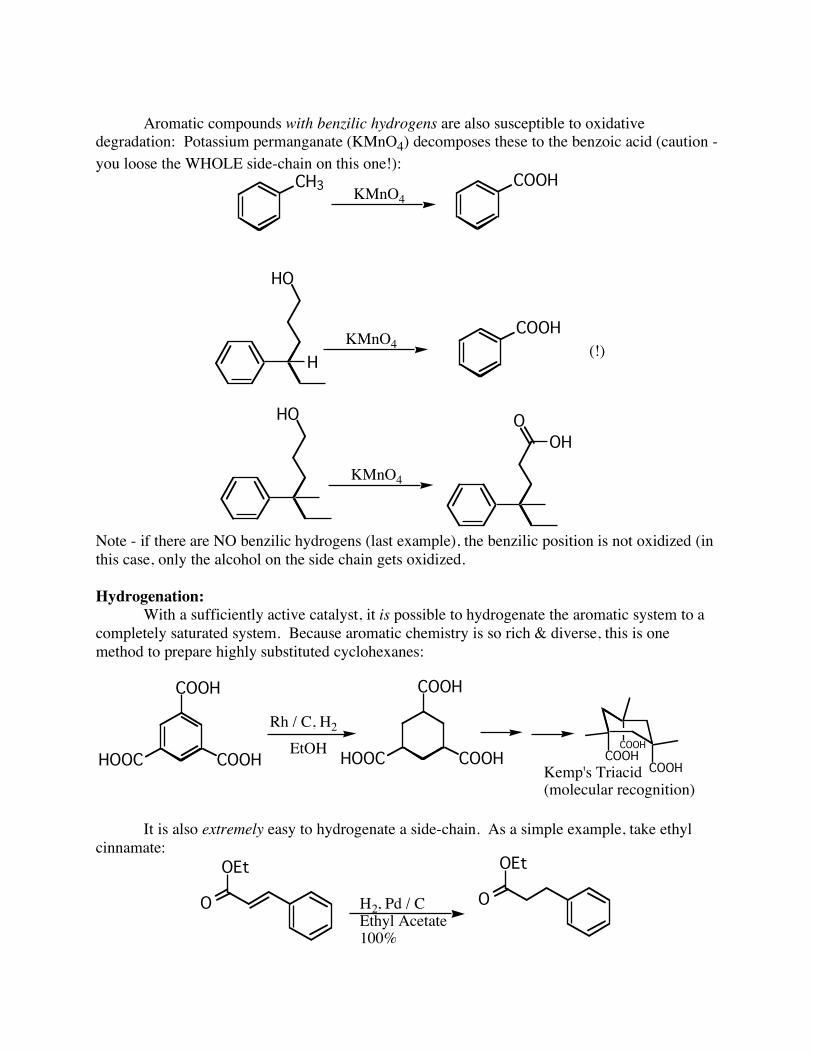
Aromatic compounds with benzilic hydrogens are also susceptible to oxidative degradation: Potassium permanganate (KMnO4) decomposes these to the benzoic acid (caution - you loose the WHOLE side-chain on this one!):
CH3KMnO4
COOH
HO
KMnO4COOH
(!)
HO
KMnO4
H
O
OH
Note - if there are NO benzilic hydrogens (last example), the benzilic position is not oxidized (in this case, only the alcohol on the side chain gets oxidized. Hydrogenation: With a sufficiently active catalyst, it is possible to hydrogenate the aromatic system to a completely saturated system. Because aromatic chemistry is so rich & diverse, this is one method to prepare highly substituted cyclohexanes:
COOH
COOHHOOC
Rh / C, H2
EtOH
COOH
COOHHOOC COOHCOOH
COOHKemp's Triacid(molecular recognition)
It is also extremely easy to hydrogenate a side-chain. As a simple example, take ethyl cinnamate:
O
OEt
H2, Pd / CEthyl Acetate100%
O
OEt
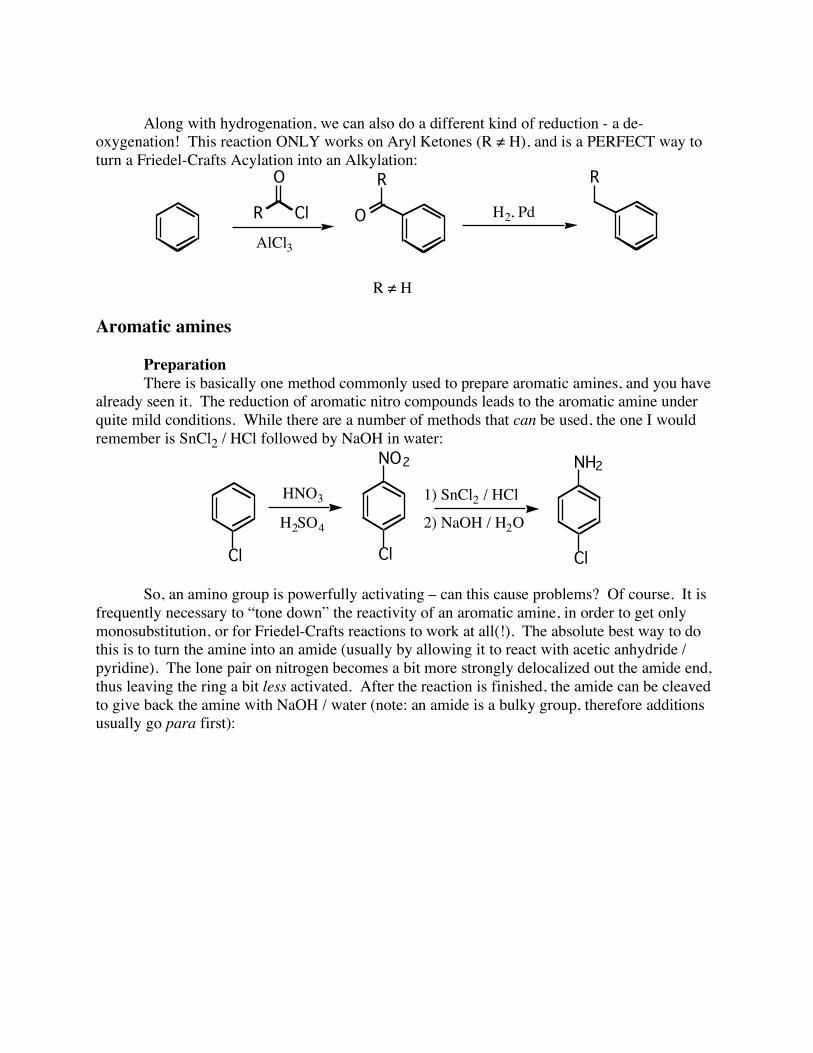
Along with hydrogenation, we can also do a different kind of reduction - a de-oxygenation! This reaction ONLY works on Aryl Ketones (R ≠ H), and is a PERFECT way to turn a Friedel-Crafts Acylation into an Alkylation:
O H2, Pd
R
R ! H
R
R Cl
O
AlCl3
Aromatic amines Preparation There is basically one method commonly used to prepare aromatic amines, and you have already seen it. The reduction of aromatic nitro compounds leads to the aromatic amine under quite mild conditions. While there are a number of methods that can be used, the one I would remember is SnCl2 / HCl followed by NaOH in water:
Cl
HNO3
H2SO4
Cl
NO2
1) SnCl2 / HCl
2) NaOH / H2O
Cl
NH2
So, an amino group is powerfully activating – can this cause problems? Of course. It is frequently necessary to “tone down” the reactivity of an aromatic amine, in order to get only monosubstitution, or for Friedel-Crafts reactions to work at all(!). The absolute best way to do this is to turn the amine into an amide (usually by allowing it to react with acetic anhydride / pyridine). The lone pair on nitrogen becomes a bit more strongly delocalized out the amide end, thus leaving the ring a bit less activated. After the reaction is finished, the amide can be cleaved to give back the amine with NaOH / water (note: an amide is a bulky group, therefore additions usually go para first):
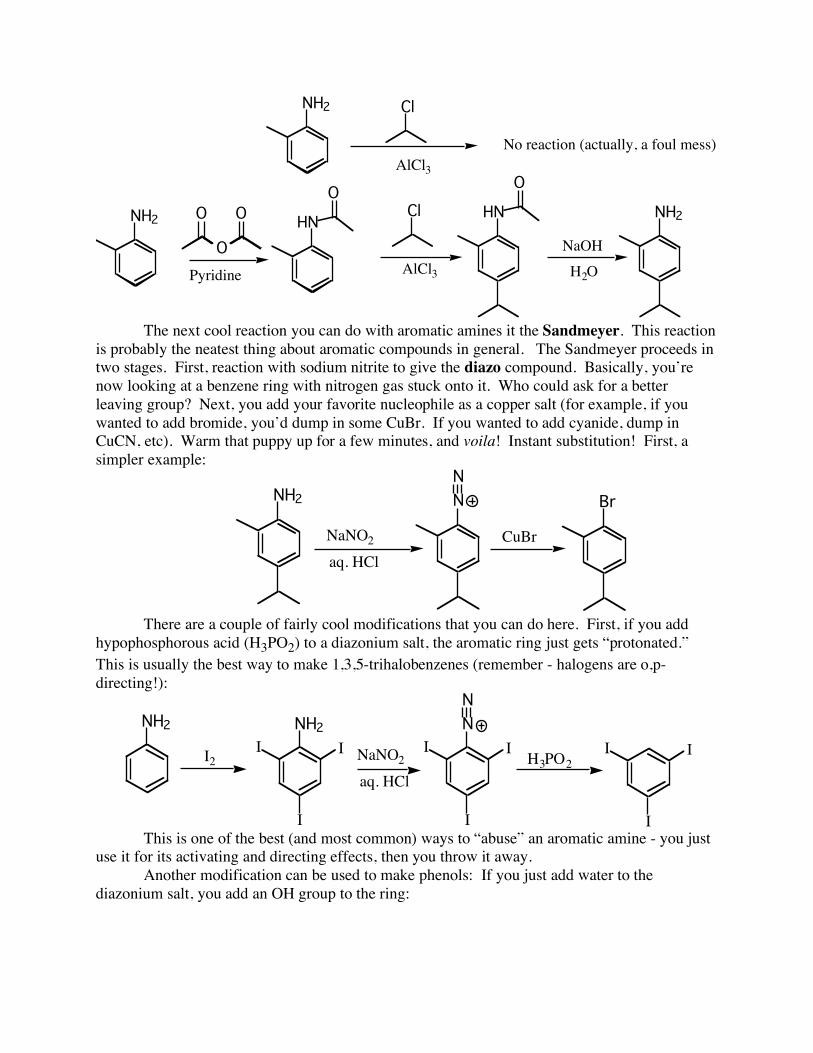
NH2 Cl
AlCl3
No reaction (actually, a foul mess)
NH2O
O
O
Pyridine
HN
O
Cl
AlCl3
HN
O
NaOH
H2O
NH2
The next cool reaction you can do with aromatic amines it the Sandmeyer. This reaction is probably the neatest thing about aromatic compounds in general. The Sandmeyer proceeds in two stages. First, reaction with sodium nitrite to give the diazo compound. Basically, you’re now looking at a benzene ring with nitrogen gas stuck onto it. Who could ask for a better leaving group? Next, you add your favorite nucleophile as a copper salt (for example, if you wanted to add bromide, you’d dump in some CuBr. If you wanted to add cyanide, dump in CuCN, etc). Warm that puppy up for a few minutes, and voila! Instant substitution! First, a simpler example:
NH2
NaNO2
aq. HCl
N
N
CuBr
Br
There are a couple of fairly cool modifications that you can do here. First, if you add hypophosphorous acid (H3PO2) to a diazonium salt, the aromatic ring just gets “protonated.” This is usually the best way to make 1,3,5-trihalobenzenes (remember - halogens are o,p-directing!):
NH2
I2
NH2
I I
I
NaNO2
aq. HCl
N
I I
I
N
H3PO2
I I
I This is one of the best (and most common) ways to “abuse” an aromatic amine - you just use it for its activating and directing effects, then you throw it away. Another modification can be used to make phenols: If you just add water to the diazonium salt, you add an OH group to the ring:
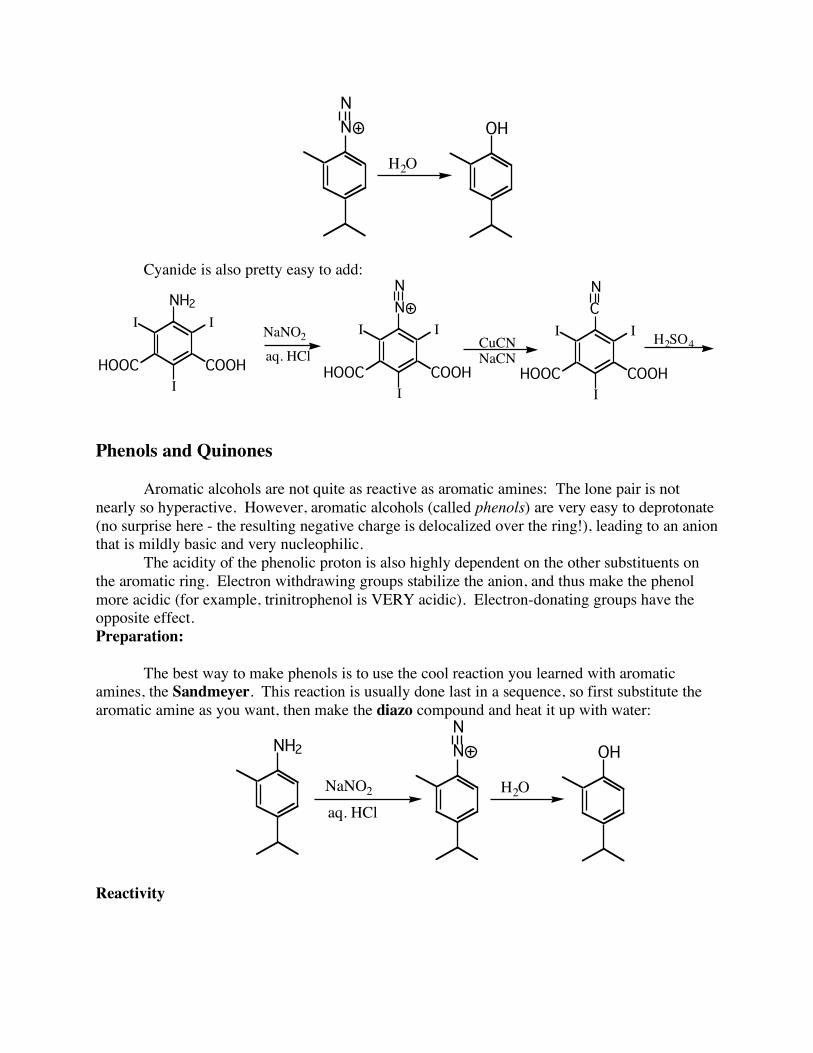
N
N
H2O
OH
Cyanide is also pretty easy to add:
NH2
COOH
I
I
HOOC
INaNO2
aq. HCl
N
COOH
I
I
HOOC
I
N
CuCNNaCN
C
COOH
I
I
HOOC
I
N
H2SO4
Phenols and Quinones Aromatic alcohols are not quite as reactive as aromatic amines: The lone pair is not nearly so hyperactive. However, aromatic alcohols (called phenols) are very easy to deprotonate (no surprise here - the resulting negative charge is delocalized over the ring!), leading to an anion that is mildly basic and very nucleophilic. The acidity of the phenolic proton is also highly dependent on the other substituents on the aromatic ring. Electron withdrawing groups stabilize the anion, and thus make the phenol more acidic (for example, trinitrophenol is VERY acidic). Electron-donating groups have the opposite effect. Preparation: The best way to make phenols is to use the cool reaction you learned with aromatic amines, the Sandmeyer. This reaction is usually done last in a sequence, so first substitute the aromatic amine as you want, then make the diazo compound and heat it up with water:
NH2
NaNO2
aq. HCl
N
N
H2O
OH
Reactivity
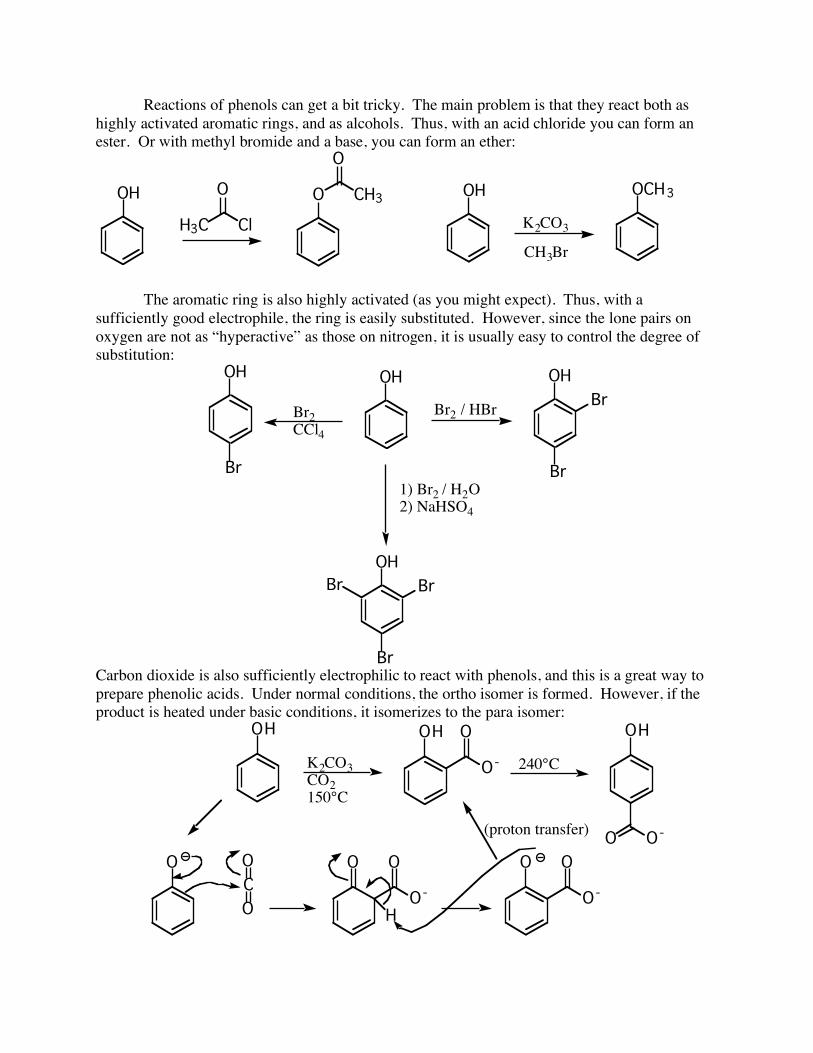
Reactions of phenols can get a bit tricky. The main problem is that they react both as highly activated aromatic rings, and as alcohols. Thus, with an acid chloride you can form an ester. Or with methyl bromide and a base, you can form an ether:
OH
K2CO3
CH3Br
OCH3OH
H3C
O
Cl
O
O
CH3
The aromatic ring is also highly activated (as you might expect). Thus, with a sufficiently good electrophile, the ring is easily substituted. However, since the lone pairs on oxygen are not as “hyperactive” as those on nitrogen, it is usually easy to control the degree of substitution:
OH OHOH
OH
Br
BrBr
Br
Br
Br
Br2
CCl4
Br2 / HBr
1) Br2 / H2O
2) NaHSO4
Carbon dioxide is also sufficiently electrophilic to react with phenols, and this is a great way to prepare phenolic acids. Under normal conditions, the ortho isomer is formed. However, if the product is heated under basic conditions, it isomerizes to the para isomer:
OH
K2CO3CO2150°C
OH
O-
O
240°C
OH
O O-
O O
C
O
O
O-
O
H
O
O-
O
(proton transfer)
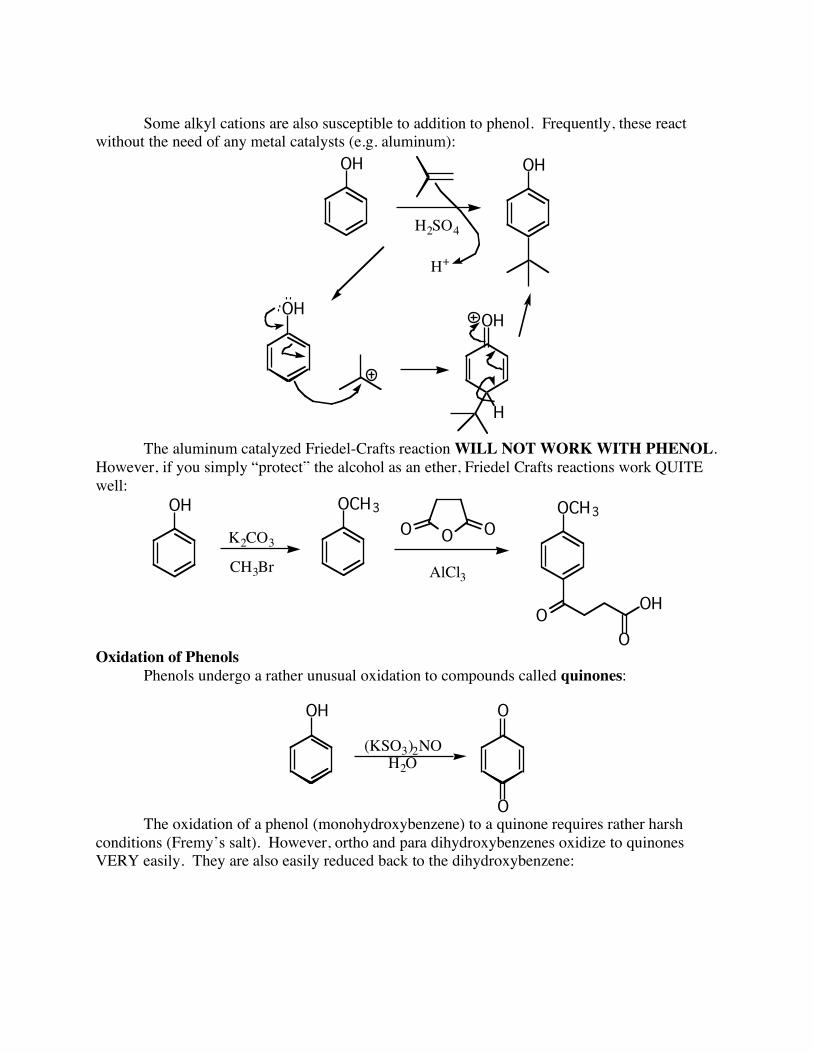
Some alkyl cations are also susceptible to addition to phenol. Frequently, these react without the need of any metal catalysts (e.g. aluminum):
OH OH
H2SO4
H+
OHOH
H
The aluminum catalyzed Friedel-Crafts reaction WILL NOT WORK WITH PHENOL. However, if you simply “protect” the alcohol as an ether, Friedel Crafts reactions work QUITE well:
OH
K2CO3
CH3Br
OCH3
OOO
AlCl3
OCH3
OOH
O Oxidation of Phenols Phenols undergo a rather unusual oxidation to compounds called quinones:
OH
(KSO3)2NO
H2O
O
O The oxidation of a phenol (monohydroxybenzene) to a quinone requires rather harsh conditions (Fremy’s salt). However, ortho and para dihydroxybenzenes oxidize to quinones VERY easily. They are also easily reduced back to the dihydroxybenzene:
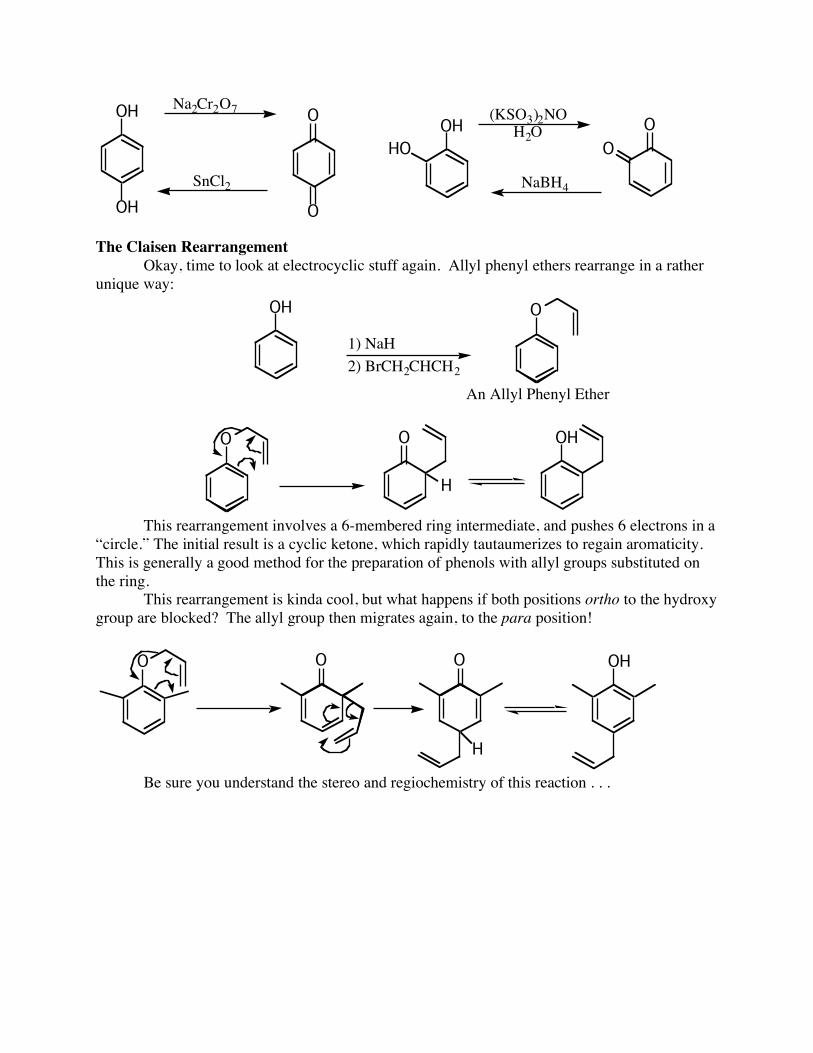
OH O
O
OH(KSO3)2NO
H2OO
HO
OH
O
Na2Cr2O7
SnCl2 NaBH4
The Claisen Rearrangement Okay, time to look at electrocyclic stuff again. Allyl phenyl ethers rearrange in a rather unique way:
O
O
An Allyl Phenyl Ether
O
H
OH
OH
1) NaH
2) BrCH2CHCH2
This rearrangement involves a 6-membered ring intermediate, and pushes 6 electrons in a “circle.” The initial result is a cyclic ketone, which rapidly tautaumerizes to regain aromaticity. This is generally a good method for the preparation of phenols with allyl groups substituted on the ring. This rearrangement is kinda cool, but what happens if both positions ortho to the hydroxy group are blocked? The allyl group then migrates again, to the para position!
O O O
H
OH
Be sure you understand the stereo and regiochemistry of this reaction . . .