xiii curso básico de doenças hereditárias do metabolismo...
TRANSCRIPT

M. Carmo Macário
6 Dez 2016
XIII Curso Básico de Doenças
Hereditárias do Metabolismo
Parte 2
5 a 7 Dezembro 2016|Coimbra

Generalidades
Doenças causam atraso desenvolvimento / regressão
Doenças de tipo intoxicação encefalopatias agudas / comas
Doenças causam AVC /pseudo-AVC Doenças com alterações sugestivas na RM
Paraparésias espásticas

1) Doenças de tipo intoxicação – metabolism intermediário (ciclo
ureia, AA, OA, intolerância dos acúcares), metais
2) Doenças do metabolismo energético (CR, PDH, beta-oxidação
ácidos gordos, biotine)
3) Doenças da sintese ou catabolismo de moleculas complexas
(esfingolipidoses - lisossoma, doenças dos peroxisoma,
cerberotendinous xanthomatosis)
3 grandes grupos de DHM

Historia familiar positiva ou consanguinidade
Atraso de desenvolvimento / regressão
Perda progressiva da visão ou audição
Sintomas neurologicos envolvendo vários sistemas
Alterações imagem (MRI) sugestivas
Envolvimento em simultâneo de outros orgãos

Raramente há DHM com atraso
mental/regressão isolada
Excluir sempre em 1º doenças tratáveis
São os sintomas associados que orientam
o Dg
Só 14 % têm Dg definitivo

Epilepsia • CNL, Alpers MELAS, MERRF
Ataxia • CNL, GM2, gang., Mit, XCT, Refsum, FA, vit E
Distonia • A glutárica, Wilson, GB biotina responsiva, PKAN
Espasticidade • ML, X-ALD, Krabbe, leucodistrofias
Neuropatia • ML, Krabbe, Refsum, FA, Vit E, XCT, Mit
Retinopatia • CLN, PKAN, Refsum, ML IV, Mit
Cataratas • XCT, beta-manosidase, Fabry, Mit
Hepatoesplenomegália • Gaucher, NP, Salla
Disostose multiplex • MPS, Oligossacaridoses, GM1

Encefalopatias agudas / comas
Intoxicação
1. Doenças do ciclo da ureia
2. Doenças da remetilação da homocisteina
3. Porfirias agudas
4. MSUD
Outras:
MCAD
Defeito do metabolismo
energático (Leigh ou
pseudo-stokes)
1. Doença dos gânglios da
base biotina-responsiva)
2. PDH
3. CR (MELAS / Leigh)
outras:
Wilson
Normal RMce Anormal RMce

AP:
Personalidade bizarra
Epilepsia desde A de idade
HDA
Coma inexplicado – Medicina Intensiva
Glasgow 3
Hipotonia
Midriase bilateral
Alguns movimentos expontâneos
RM ce – sem alterações
EEG: encefalopatia metabolica
Amoniemia=276 mM
Deficiencia OTC F. Sedel

AP 6 7, 8, 13 e 17 anos: episodios desencadeados por doença febril: vomitos,
vertigem, ataxia, diplopia, nistagmp, confusão. Diagnostico: neurite vestibular recorrente
HDA
Febre, ptose bilateral, confusão, miose, coma
homem, 22 anos
•Lactatos : 2.1 mmol/l (N:1-1,8), piruvate : 221 µmol/l (N:41- 67), elevação da alanina (681, N=351)
Deficiencia de PDH F. Sedel

AP:
Atraso ligeiro do desenvolvimento psico-motor
7 anos: encefalopatia inexplicada
Desde então: epilepsia, distonia (face e MS)
Homem 33 anos
33 anos
Intercorrência infecciosa benigna
Encefalopatia subaguda (disfunção troco cerebral,
ataxia, coma)
F. Sedel

Tratamento com biotina
• Biotina 600 mg/d
• Recuperação em 24h
Mutação SLC19A3
« Biotin responsive basal ganglia disease »
F. Sedel
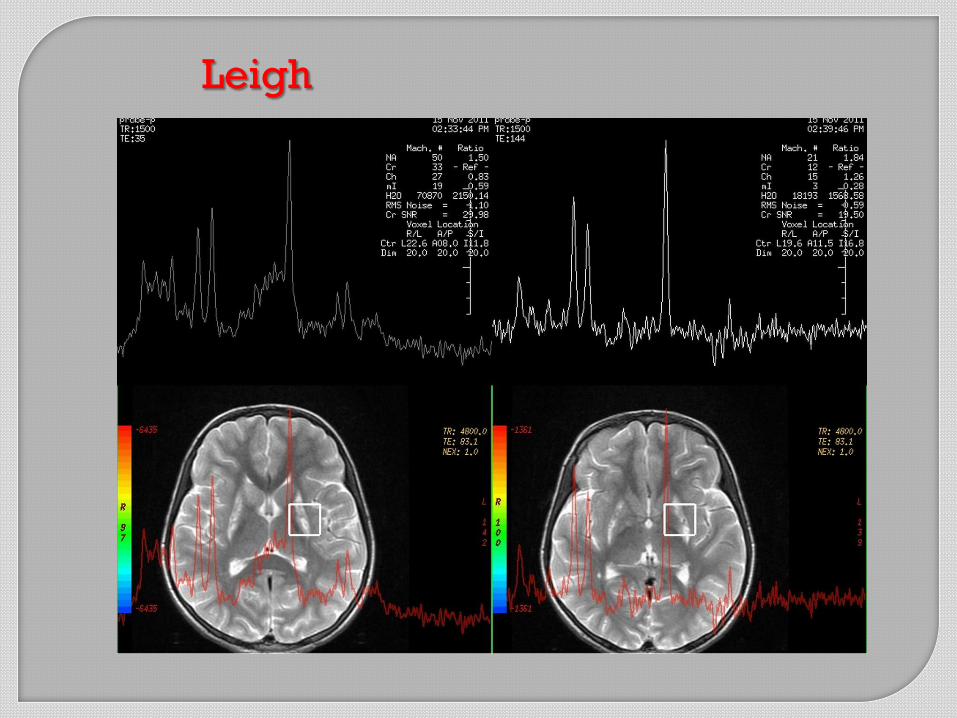
Leigh

• AVC: • Doenças Fabry
• Hiperhomocisteinemia
• MPS (direto /indireto) • Pseudo-AVC
• Doenças metabolismo energético
• Doenças ciclo ureia

HDA; 2013
Defice motor direito – AVC
Protese valvular mecânica
Acenocumarol
INR 2,93
AP
Stenose aórtica e mitral com
substituição mecânica em 2008,
hipocoagulado desde então
Hérnia umbilical corrigida em 2000
Hérnia inguinal corrigida em 2000
Contractura bilateral dos flexores
dos dedos mãos
Opacidade corneana bilateral
Tiroidite autoimune
HF
• Pais primos em primeiro grau
• 5 irmãos
• 1 irmão, 50A: patologia valvular,
mão em garra bilateral e atraso do
desenvolvimento sem diagnóstico

João Irmão
Altura
160 cm
Peso
67 kg
P. cefálico
54 cm

João
MPS I

irmão, 50A

leucodistrofias
Doença do armazenamento lipidos
- Polineuropatia
- leucodistrofia
1) Leucodistrofia metacromatica
2) Doença de Krabbe
3) X-Adrenoleucodistrofia
4) Doença de Refum
5) Xantomatose cerebrotendinosa
Intoxicação (AA e OA)
- normal EMG
- Envolvimento fibra em U –
sub branca
1. Organic acidurias
1. Aciduria L2OH
glutarica
2. Aminoacidopathies
outras
1) CR
2) Doença dos poliglucosanos
do adulto

Doenças
progressivas,
debilitantes,
incapacitantes e
que levam à morte
prematura

Doença dos lisossomas AR; gene ARSA 22q13,33
• P.I179S – quadros psiq; p.P426L mais comum 1/40000 Deficiência arilsulfatase A (excluir pseudo def) Diagnostico: atividade arilsulfatase e
sulfatideos urinarios Clinica (adulto): deterioração cognitiva,
demência, problemas psiquiátricos (alucinações, esquizofrenia, psicose), polineuropatia desmielinizante, paraparesia espática, ataxia, sint. extrapiramidais
Diagnóstico: actividade da arilsulfatase A nos leuc. ou fibroblastos (excluir psedodeficiência) ou excreção de sulfatídeos urinários (def. do activador)
Tratamento: transplante MO, enzima ASA em estudo
RMN: leucodistrofia “Aspeto tigróide”
Leucodistrofia difusa, poupando fibras em U e aspecto tigróide,
de predomínio frontal, sem realce com contraste

Francisco


Molecular genetic testing
Compound heterozygous state
p.N295T Already described
p.I552T Novel mutation
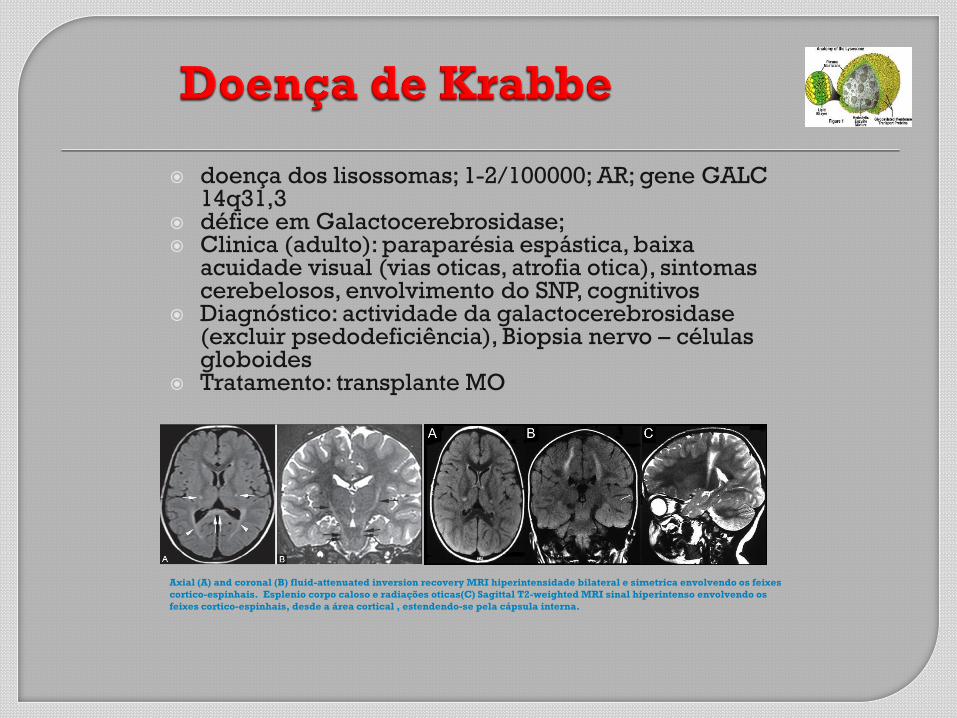
doença dos lisossomas; 1-2/100000; AR; gene GALC 14q31,3
défice em Galactocerebrosidase; Clinica (adulto): paraparésia espástica, baixa
acuidade visual (vias oticas, atrofia otica), sintomas cerebelosos, envolvimento do SNP, cognitivos
Diagnóstico: actividade da galactocerebrosidase (excluir psedodeficiência), Biopsia nervo – células globoides
Tratamento: transplante MO
Axial (A) and coronal (B) fluid-attenuated inversion recovery MRI hiperintensidade bilateral e simetrica envolvendo os feixes
cortico-espinhais. Esplenio corpo caloso e radiações oticas(C) Sagittal T2-weighted MRI sinal hiperintenso envolvendo os
feixes cortico-espinhais, desde a área cortical , estendendo-se pela cápsula interna.

Doença do armazenamento de glicogénio tipo IV
Deficiência na enzima ramificadora do glicogénio
AR; gene GBE1 3p12,2 (também causa forma infantil multissistémica)
Clinica: Alterações da memória/demência, paraparésia espática,
bexiga neurogénica, Sintomas motores de primeiro e segundo
neurónio (ALS), Polineuropatia sensitiva
DG: clinica + RMNCE + biopsia nervo ou pele axilar + actividade
enzima do glicogénio (GBE) pele + genético
Não há tratamento
Corpos de poliglucosanos intra
neuronais
Leucodistrofia difusa supra e infratentorial;
Confluente periventricular ,
poupa fibras U


Doença dos peroxissomas; Ligada ao X; Xq28; gene ABCD1, Homens: 1/42 000
acumulação da ácidos gordos de cadeia muito longa; • Formas clinicas:
XALD inicio na infância;
XALD início no adulto
AMN Adrenomieloneuropatia – 50% das Mulheres portadoras; homens 30-40A
Clínica adulto XALD / infância: cérebro + adrenal (doença Addison): alteração comportamento (agressivo), deterioração cognitiva, paraparésia espástica, atrofia otica, hipogonadismo
Clinica AMN: paraparésia espástica progressiva ou remitente, alterações da sensibilidades profundas por polineuropatia, disfunção urinária, hipogonadismo, infertilidade, calvice: nesta forma clinica podem ter RMNCE normal ou evoluir para a forma cerebral (em 60 %, após 10 A e é menos severa)
Mulheres portadoras, cerca 50%: sinais de paraparésia espástica sem incapacidade significativa
Agravamento severo após TCE ou stroke
DG: doseamento ácidos gordos de cadeia muito longa/ estudo genético
Tratamento: Transplante; óleo de Lorenzo (ácidos gordos monoinsaturados) – “efeito cosmético”
Predominio posterior
Gradação de desmieliniz.
Realce com contraste

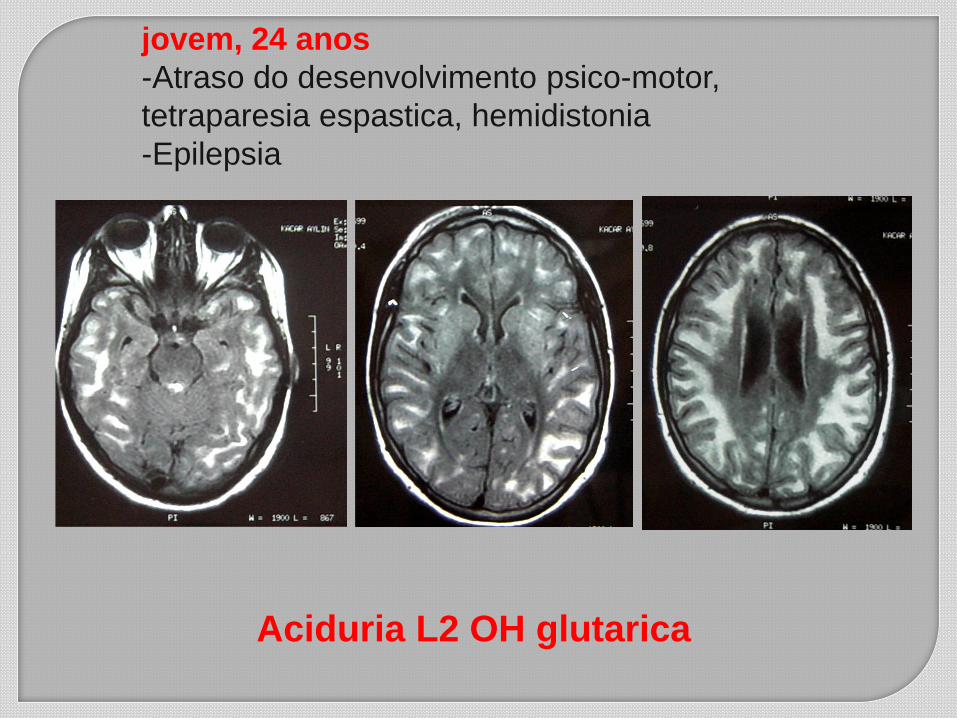
jovem, 24 anos
-Atraso do desenvolvimento psico-motor,
tetraparesia espastica, hemidistonia
-Epilepsia
Aciduria L2 OH glutarica

• Pensar DHM quando: • Lesões bilaterais e simétricas envolvendo grupos
específicos de fibras
• Quando associada com envolvimento SNP
• Que doenças: • Doenças do armazenamento lipídico
Localização seletiva
Sem envolvimento das fibras em U
Com envolvimento SNP
• Aminoacidopatias / acidúrias orgânicas Com envolvimento das fibras em U
Sem envolvimento SNP

Paraparésia espástica
Doenças do armazenamento de lipidos
- Polineuropatia – axonal /
desmilinizantes
- Leucodistrofia
1. Leucodistrofia metacromática
2. Doença de Krabbe
3. Adrenomieloneuropatia
4. Xantomatose cérebro-tendinosa
Intoxicação
1. Defeitos da remetilação da
homocisteina
2. Fenilcetonuria
3. Deficiência de argininase (UCD)
4. Síndrome HHH -
hiperornitinémia, hiperamoniémia
e homocitrulinúria (UCD)

Gene: CYP27A1
Enz: esterol 27 hidroxilase
Prev < 5/100,000

AR; Distúrbio hereditário do metabolismo ácidos biliares
Neonatal: hepatite colestática; dificuldades escolares;
diarreias inexplicadas
Cataratas juvenis, litíase vesicular precoce
Quadro neurológico em adulto jovem: paraparésia espática,
ataxia cerebelosa e cordões posteriores, distonia,
polineuropatia (a/d), sinais bulbares e pseudobulbares;
xantomas tendinosos (por vezes só 20 A depois dos sintomas
neurológicos); aterosclerose prematura, osteoporose
Diagnóstico: valores ligeiramente elevados de colesterol e
muito elevados de colestanol
Diagnostico: doseamento do colestanol palmático
Estudo genético gene CYP27A1
Tratamento: ácido quenodesoxicólico (750 mg/dia)
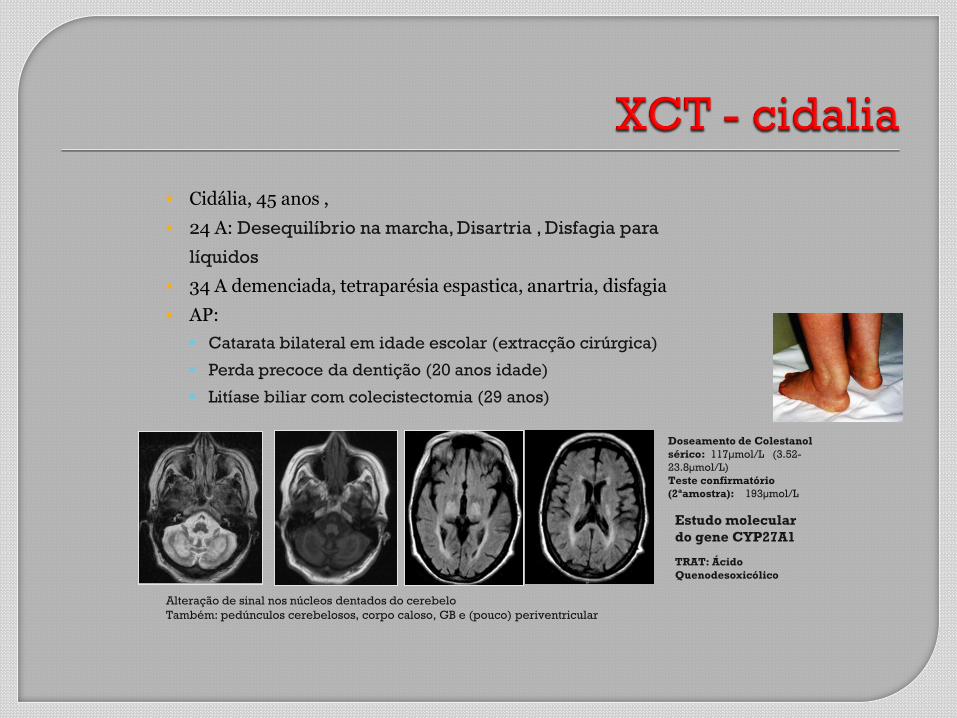
Cidália, 45 anos ,
24 A: Desequilíbrio na marcha, Disartria , Disfagia para
líquidos
34 A demenciada, tetraparésia espastica, anartria, disfagia
AP:
Catarata bilateral em idade escolar (extracção cirúrgica)
Perda precoce da dentição (20 anos idade)
Litíase biliar com colecistectomia (29 anos)
Alteração de sinal nos núcleos dentados do cerebelo
Também: pedúnculos cerebelosos, corpo caloso, GB e (pouco) periventricular
Doseamento de Colestanol
sérico: 117µmol/L (3.52-
23.8µmol/L)
Teste confirmatório
(2ªamostra): 193µmol/L
Estudo molecular
do gene CYP27A1
TRAT: Ácido
Quenodesoxicólico
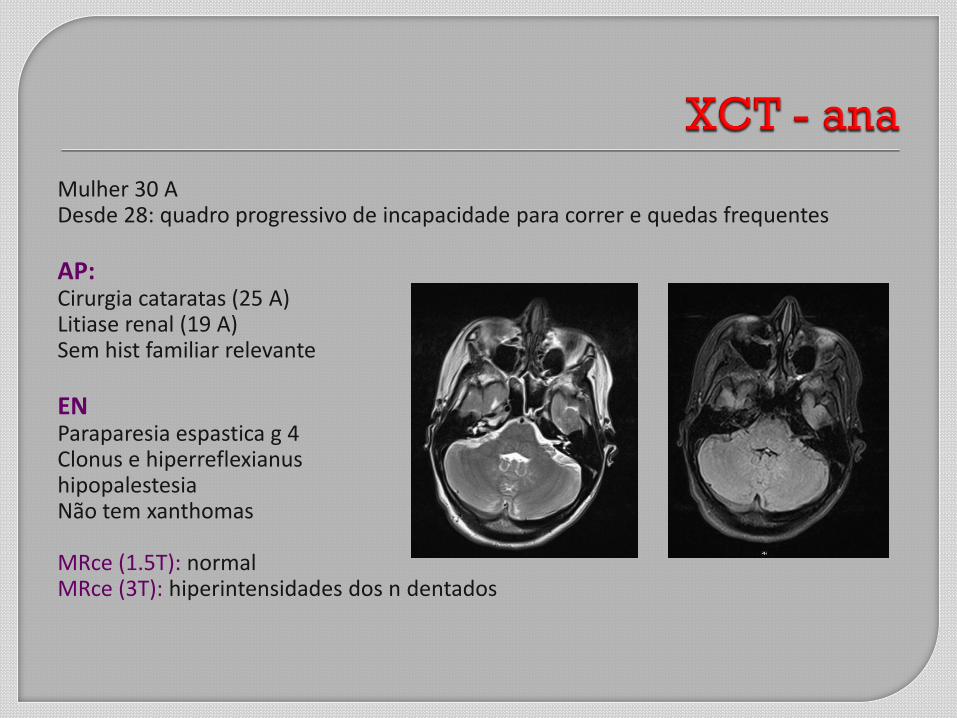
Mulher 30 A Desde 28: quadro progressivo de incapacidade para correr e quedas frequentes AP: Cirurgia cataratas (25 A) Litiase renal (19 A) Sem hist familiar relevante EN Paraparesia espastica g 4 Clonus e hiperreflexianus hipopalestesia Não tem xanthomas MRce (1.5T): normal MRce (3T): hiperintensidades dos n dentados

Cholestanol serico: 83.6 umol/L (3.52 – 23,8 umol/L) Clestanol serico: 82 umol/L Tratamento Ácido quenodesoxicolico 750 mg/day Follow up Melhoria; sem progressão de incapacidade CYP27A1 mutação homozigotica c.1016C>T; p.T339M

Mensagens
• DHM ocorrem em qualquer idade
• Formas de inicio mais precoce são mais severas,
os adultos podem ter formas relativamente
monossintomáticas
• Atenção à associação de sintomas, história
familiar
• Os doentes necessitam de equipas
multidisciplinares para acompanhamento e
seguimento do doente

obrigada

SPDM Portuguese Society of Inherited Metabolic Diseases
leukodystrophies in clinical pratice
Formação pós graduada Post graduate education
Coimbra, 13 Janeiro 2017 13 January 2017 Auditório do Hospital Pediátrico de Coimbra
Inscrições: http://www.spdm.org.pt