x transfusion medicine - quia · 5 hema x transfusion medicine — 2 01/11 unrelated donors are...
TRANSCRIPT

© 2011 Decker Intellectual Properties 5 hema x transfusion medicine — 1DOI 10.2310/7900.1120
01/11
X T R A N S F U S I O N M E D I C I N E
Harvey G. Klein, MD*
Transfusion medicine has evolved from the empirical administration of blood to a laboratory-based clinical discipline.1 The discovery of blood group antigens and the understanding of the host immune response to these antigens, the development of methods of anticoagulation and storage of blood, the creation of plastic bags that allow sterile separation of whole blood into components, and the advent of the automated blood cell separator all contributed to its advancement. The potential of blood to act as an agent of disease transmission has heavily shaped both the dona-tion process and transfusion practice.2 Decisions about whether to transfuse involve weighing the benefi ts against the risks. This chapter provides a basis for these decisions, including indications for blood-component use, complica-tions of transfusion therapy, and methods of reducing risks during the collection, processing, and preparation of blood components. Therapeutic removal of blood (phlebotomy) and components (apheresis) is also discussed.
Blood Donation
The donation process for either whole blood or special products, such as single-donor platelets (SDPs) obtained by apheresis, is designed to protect both the donor and the recipient. Donor qualifi cation includes stringent donor screening, physical examination, sensitive testing, donor tracing, and donor deferral when instances of disease trans-mission are discovered. This system of safeguards has made the US blood supply extremely safe; however, it is not 100% effective. Approximately 2% of volunteer donors still conceal risks that would have led to deferral at the time of donation.3
autologous and directed donation
Autologous donations and directed donations are two strategies adopted by patients seeking to minimize their real or perceived risk of infection from blood components.
Autologous Donation and Bloodless Surgery
In preoperative autologous donation, patients deposit their own blood, which is then available to them should they need transfusion therapy. Autologous blood avoids the risk of new viral infections and sensitization associated with allogeneic blood; however, it does not eliminate bacterial contamination or the risk of receiving the wrong unit of blood because of clerical error.4 Absolute contraindications to autologous donation include tight aortic stenosis, unstabl e angina, and active bacterial infection. Anemia and poor venous access frequently limit the number of units that can be collected; up to half of the collected units are not used. With the increasing safety of allogeneic blood, autologous
* The author and editors gratefully acknowledge the contribu-tions of the previous author, W. Hallowell Churchill, MD, to the development and writing of this chapter.
donation should be limited to selected patients (i.e., those who undergo joint replacement and vascular and cardiotho-racic surgery and who are not anemic at the time of the fi rst donation).5 The advantages and disadvantages of preopera-tive autologous donation need to be weighed for individual patients [see Table 1].
Acute normovolemic hemodilution (ANH) is another form of autologous donation. In ANH, whole blood is removed from the patient immediately before surgery; the patient is infused with crystalloid solution to maintain normovolemia, and the whole blood that was removed is reinfused when needed, often at the conclusion of surgery. ANH can yield modest blood savings and minimize the risk of clerical error.6 For patients who experience massive bleed-ing during surgery, semiautomated collection devices can recover, process, and reinfuse blood lost at the operative site (a process referred to as intraoperative salvage).6,7 Centers that advertise bloodless surgery combine autologous strate-gies, erythropoietic support, and conservative transfusion thresholds to limit exposure to allogeneic blood.8
Directed Donation
Blood donated for a specifi c patient is termed a directed donation; usually, it involves donations made by friends or family members of the intended recipient. Directed dona-tion presumes that the recipient can identify donors who carry lower risk of infections than volunteer donors from the general population. However, prevalence data show that the risk of infectious disease from directed donors is no different from that of fi rst-time donors.9 The current risk of infection via transfusion is so low [see Table 2] that directed donor programs are justifi ed primarily by patient preferences or by the need for a selected donor serving as the only source of blood components to reduce the recipient’s risk from expo-sure to multiple donors. The latter form of directed donation is most appropriate for neonatal transfusions, in which one of the biologic parents may provide all the needed blood components.10 In unusual circumstances, such as cases involving highly immunized patients or patients with rare blood types, directed donations from relatives or matched
Table 1 Advantages and Disadvantages of Preoperative Autologous Donation
AdvantagesReduces the risk of transfusion-transmitted infectionPrevents immune hemolysis and alloimmunizationPrevents many transfusion reactionsProvides compatible blood for patients with antibodiesReassures patients and physicians
DisadvantagesDoes not reduce risk of bacterial contaminationCarries risk of patient reaction during autologous donationMay render patient anemic and iron defi cientCosts more than allogeneic donation and half of collected
units are unused and discarded

5 hema x transfusion medicine — 2
01/11
unrelated donors are medically indicated. However, the use of blood relatives as donors increases the risk of graft versus host disease, unless the blood is gamma-irradiated. In addition, transfusing a woman of childbearing age with blood from her spouse or her spouse’s relatives increases the risk of hemolytic disease of the newborn in subsequent pregnancies.11
Screening Procedures
The combination of improved donor selection and postdo-nation testing has greatly decreased the infectious risks of allogeneic blood [see Table 2]. Ten screening tests are applied to all donated blood, and supplemental assays, such as test-ing for cytomegalovirus, are used for special indications, such as stem cell transplantation.
postdonation testing
Postdonation testing is essential for identifying donors likely to transmit blood-borne infections who are missed in the initial screening process. The risk of transfusion-transmitted viruses is now so low that estimates must be derived from mathematical models rather than from direct measurement of infected blood recipients.12,13
Screening for Hepatitis Viruses
Hepatitis C Screening for hepatitis C began in 1990 with the availability of a serologic enzyme-linked immunosor-bent assay (ELISA). The development of ELISAs with improved sensitivity, associated confi rmatory tests, and nucleic acid testing (NAT) for viral RNA and DNA has led to a reduction in the per-unit risk of hepatitis C virus (HCV) transmission to less than 0.0001% (1:1,149,000).12,13 Before these tests were available, the risk per unit was about 4%. Improved HCV testing has eliminated the need for surro-gate tests, such as the measurement of alanine aminotrans-ferase (ALT) levels and testing for antibody to hepatitis B virus (HBV) core antigen (anti-HBc). However, the test for anti-HBc is still used to detect recently infected donors who
lack measurable circulating hepatitis B virus surface antigen (HBsAg).14
The epidemiology of HCV infection is still poorly under-stood. The majority of transmissions are related to intrave-nous (IV) drug use.15 Sexual transmission is uncommon. However, heterosexual transmission of HCV may be asymp-tomatic and blood donation from a person who was infected with HCV via sexual contact but has not yet developed detectable antibodies is a potential risk to the blood supply. Therefore, persons who are sexual partners of known HCV-infected persons are still excluded from donation. Donors found to be positive for HCV on ELISA should undergo supplemental testing, such as with recombinant immuno-blot assays (RIBAs). Donors with positive supplemental test results are likely to have a chronic HCV infection; such persons are rejected as future donors, and they require further clinical evaluation and treatment.16 Donors with negative supplemental test results probably had false positive screening results and may be eligible for reentry into the allogeneic donor pool after 6 months. The infection status of donors with indeterminate supplemental results is best resolved by testing for HCV RNA; those with only a single band on the most sensitive supplemental test (RIBA-3) have a less than 4% chance of having circulating HCV RNA.17
Gene amplifi cation methods (NAT) for detecting HCV RNA are used on all blood products before those products are released for transfusion. These tests directly detect the presence of virus before antibody development, and their use is responsible for reducing the risk of HCV transmission to the current minuscule level. Correlation studies have shown that only 80% of samples with confi rmed positive results on serologic testing for HCV are also found to be positive on NAT. This fi nding is consistent with previous estimates of the size of the population of persons who were previously HCV positive but who are no longer infected.
Hepatitis B HBV remains a major human pathogen with worldwide distribution that causes acute and chronic
Table 2 Estimated Risks of Blood Transfusion per Unit TransfusedReaction Risk Comment
Fever; allergic reactions 1 in 200 Fever may be > 0.5°C (1°F); allergic reaction may include urticaria
Hemolytic transfusion reaction 1 in 6,000 Most are asymptomatic
Fatal hemolytic transfusion reaction 1 in 1.8 million Most related to ABO errors
HIV infection 1 in 1.9 million
HTLV infection 1 in 3 million
Hepatitis B infection 1 in 180,000
Hepatitis C infection 1 in 1.6 million
Bacterial contamination of platelets 1 in 10,000 Sepsis estimated at 1 in 75,000
Fatalities estimated at 1 in 500,000
Bacterial contamination of red cells 1 in 65,000 Fatality estimated at 1 in 100,000
Transfusion-related acute lung injury 1 in 5,000
Graft versus host disease Uncommon Fatality estimated at 90%
HTLV = human T cell lymphotropic virus.

5 hema x transfusion medicine — 3
01/11
hepatitis, cirrhosis, and hepatocellular carcinoma.18 HBV is highly infectious and is readily transmitted by needle stick and sexual contact. The elimination of the practice of paying whole blood donors together with the development of modern testing methods for HBsAg and anti-HBc has reduced HBV infections to about one in 180,000 units trans-fused.19 However, donors with low levels of virus, especially during the incubation period, still transmit disease. HBV immunization of patients requiring multiple transfusions of blood or components has long been advised, and childhood immunization is now standard in the United States. Although vaccination will dramatically reduce the risk of infected donors in the future, “breakthrough” infections and infections by HBV variants may be a cause for continued vigilance.
Hepatitis A Because the viremic phase of hepatitis A virus infection lasts only about 17 days before signs and symptoms develop, hepatitis A transmission from single-donor components is rare. Pooled products, such as factor concentrates, however, carry a substantially higher risk; plasma pools intended for fractionation are screened for hepatitis A.20
Screening for Retroviruses
All donated blood is screened for HIV-1, HIV-2, human T cell lymphotropic virus type I (HTLV-I), and HTLV-II. Data obtained nationally from American Red Cross donors indicate that the HIV infection risk has been reduced from two per 100 transfusions to about one per 2 million transfu-sions12; improved safety has been achieved by the exclusion of high-risk donors and the postdonation testing for HIV-1 and HIV-2 antibodies and use of NAT for viral RNA or DNA.12 In follow-up studies, 90 to 95% of recipients of blood that is seropositive for HIV become infected.21
To have predictive value, the ELISA screening test for HIV must be confi rmed by some additional assay such as an alternative ELISA, NAT, or repeated testing on donor review. The possibility of a false positive result should be remembered when one is counseling low-risk donors who have had unexplained positive results on ELISA; these false positive results must always be confi rmed by careful clinical follow-up.22
In the United States, the prevalence of HTLV-I or HTLV-II in donors was about 0.03% in 1995. Data from 2001 suggest that the prevalence has been reduced to about 0.01%; about two thirds of these HTLV-positive patients have HTLV-II infection.12 HTLV is transmitted by cellular components but not by cell-free plasma or plasma derivatives. Infectivity of cellular components declines with the length of refrigerated storage. Several longitudinal studies have defi ned the clini-cal consequences of HTLV-I/II infection; they are useful in advising donors who have had positive or indeterminate test results.23 In a prospective, longitudinal study comparing seropositive blood donors with seronegative blood donors, both viruses were associated with an increase in the incidence of some infectious diseases. No cases of adult T cell leukemia or lymphoma were identifi ed; myelopathies, although rare, were associated with both HTLV types.24 The risk of HTLV-I/II transmission by blood components is one per 2,993,000. As with HIV, laboratory studies and
epidemiologic investigations of HTLV-I/II indicate that patients with positive results on screening tests and negative or indeterminate results on supplemental testing are unlikely to have clinical sequelae; positive results in these patients are most likely false positives.25
Screening for Other Agents
West Nile virus West Nile virus (WNV), a fl avivirus imported into the United States in 1999, has become a signifi cant transfusion risk; during periods of epidemics, its transmission rate has been estimated to be 3.02 per 10,000 donations in high-risk metropolitan areas.26 Approximately 80% of patients are asymptomatic, 20% experience a febrile illness, and about one in 150 develop meningoencephalitis. Elderly and immunosuppressed blood recipients are at particular risk. All blood donations are currently tested for WNV by NAT assays. Several thousand potential transmis-sions have been interdicted; however, transmissions of WNV continue by means of blood components that have low levels of virus.27
Chagas disease Chagas disease is caused by infection with the protozoan parasite Trypanosoma cruzi, which is found primarily in Latin America. The risk of severe heart or intestinal complications in infected persons is about 30%; complications usually occur long after the initial infection. Seven cases of transfusion-transmitted T. cruzi and fi ve cases of transmission by organ transplantation have been docu-mented in the United States and Canada, although the mild, nonspecifi c symptoms of early infection make it likely that many cases have gone unrecognized. A serologic blood screening test has been introduced by the major blood collectors. Whereas follow-up of seropositive donors has not shown evidence of disease transmission, bloodstream parasites are detectable—and potentially transmissible—decades after immigration, which strengthens the rationale for donor screening.28
Emerging Infectious Diseases
Sensitive and specifi c testing for known viral agents per-mits surveillance of the changing prevalence and incidence of pathogens that contaminate the blood supply.12 Until either screening tests or sterilization procedures become available, exclusions based on demographic considerations are the only possible protective strategy against newly recognized infections. For example, the recognition that WNV was transmitted by blood components prompted the introduction of screening questions to eliminate donors at risk for this disease; however, the screening questions proved largely ineffective. A nucleic acid–based test for WNV was introduced in June 2003 and is now a standard screening test for all donated units.29
In the United Kingdom, another form of demographic control was instituted to safeguard against possible trans-mission of infectious disease by blood transfusion. The incidence of variant Creutzfeldt-Jakob disease (vCJD), which is the human equivalent of bovine spongiform encephalopa-thy (mad cow disease), and the concern that vCJD may be transmissible by transfusion prompted in the United States the deferral of donors who had lived in or visited countries in which vCJD was reported; this restriction resulted in a

5 hema x transfusion medicine — 4
01/11
reduction of 4 to 5% in the number of active blood donors. Because no screening assay for vCJD is available and because vCJD infection is uniformly fatal, such epidemio-logic precautions were considered warranted. However, transmissibility of vCJD by blood transfusion is likely but has not been proved, and data are inconclusive and limited.30 Reports from the United Kingdom have identifi ed three probable cases of transfusion-associated vCJD, one subclinical infection in a person who received a transfusion from a vCJD-positive donor, and an infection in a patient with hemophilia that appears to have been transmitted by factor VIII concentrate.31 In known recipients of blood trans-fusions from donors who subsequently developed vCJD, the risk of infection is probably high.31 However, potential blood donors who are rejected on the basis of epidemiologic risk of vCJD should be assured that their risk of having any form of CJD infection is low. A fi lter designed to remove prions from donated blood is currently undergoing trials.32
Models have been constructed to predict the emergence of new infectious pathogens, and a listing of known agents with the potential to invade the blood supply has been developed.31,33 During the past few years, a variety of “agents du jour” have been proposed as presenting risks for blood transmission, including Chikungunya virus, severe acute respiratory syndrome (SARS) virus, the infl uenza viruses H1N1 and H5N1, and even the newly described gamma retrovirus XMRV. Other agents, such as Babesia microti (babesiosis) and the malarial parasites, are readily transmit-ted by transfusion but not easily identifi ed with screening tests. The paradigm of surveillance and testing is gradually being replaced by a strategy of pathogen inactivation of blood components, but it may be decades before all blood components can be rendered safe and effective.
Pretransfusion Testing
antigen phenotyping
Blood recipients are routinely tested to establish their ABO phenotype and Rh type. Establishing ABO type is essential because isoagglutinins (antibodies) against A or B antigens not present on a person’s red cells are acquired during the fi rst 2 years of life. These IgM antibodies can cause an immediate hemolytic reaction if ABO-incompatible red cells are transfused.
The terminal carbohydrate on these antigens determines specifi city in the ABO system, with type A being associated with N-acetylgalactosamine and type B being associated with a terminal galactose. Persons with type O lack both of these terminal sugars. These residues are added by a glyco-syltransferase, which was thought to be either nonfunction-al or absent in type O persons. Yamamoto and colleagues used molecular techniques to prove that glycosyltransferase in type O persons is very similar to the transferase in type A persons.34 The type O glycosyltransferase is nonfunctional because of a single base deletion that produces a frameshift and a downstream stop codon.
All methods of ABO typing depend on demonstrating that the antigens found on the red cells are consistent with the expected isoagglutinins. D antigen specifi city typing in the Rh system is done because of this antigen’s potency as an immunogen. Antibodies to the D antigen are the most
important cause of isoimmune hemolytic disease of new-borns. Rh antigens are membrane glycolipids or glycopro-teins. Antibodies against antigens of this class, which includes the Rh, Duffy, Kell, Kidd, and Lutheran systems, will usually cause shortened red cell survival. In contrast to antigens with carbohydrate-mediated specifi city, glycolipid and glycoprotein antigens do not stimulate antibody forma-tion unless the transfusion recipient was previously exposed to allogeneic red cells, either from transfusion or from fetal red cells during pregnancy or delivery.
D antigen typing is also done using agglutination tech-niques. In some cases, less antigenic forms of the D antigen, called weak D, require an antiglobulin reagent to enhance detection. Structural studies of the complementary DNA associated with the major Rh antigens (D, Cc, and Ee) have provided probes for direct genotyping.35 Molecular methods of prenatal Rh-type determination have revealed that most Rh-negative persons lack the D gene. Some persons with the weak D phenotype have mosaic D genes because of exchange with some of the exons of the CcEe gene.
Because the genotypes of many of the clinically relevant red cell antigens are known, it is now possible to predict red cell phenotype by DNA analysis.36 Although DNA analysis for determining red cell phenotype is not yet widely avail-able, it will be useful for recently transfused patients, for whom circulating allogeneic red cells complicate antigen phenotyping.
screening for antibodies
In addition to identifying patient ABO and D red cell phenotypes, blood banks must screen the patient’s serum for red cell–specifi c antibodies, which can cause serious reactions with transfused red cells. Screening involves test-ing serum against indicator type O red cells displaying all the clinically important red cell antigens. Positive reactions are detected by adding an antiglobulin reagent (i.e., Coombs reagent) to the incubated mixture of type O red cells after it has been washed free of serum. Any observed agglutination is from the reaction of the antiglobulin reagent with anti-body adsorbed on the surface of the indicator red cells. Agglutination of the indicator red cells indicates the pres-ence of other antibodies, which require identifi cation. The absence of agglutination excludes all antibodies except those against antigens so rare that they are not displayed on the indicator red cells. Because the alloantibody concentration may fall below the level detectable by agglutination, a negative screen does not guarantee a compatible blood transfusion.
Use of type-specifi c blood removes the risk of ABO incompatibility; however, residual risk of an immunologic reaction from the antibodies to other red cell antigens remains. Such antibodies are present in about 3 to 5% of a random population; they are also present in 10 to 15% of persons who were recently transfused or women with a history of pregnancy. Screening for antibodies reduces the frequency of reactions to about 0.06%. Performing a full crossmatch, in which the recipient’s serum is tested against the red cells actually being transfused, is of little additional benefi t; a full crossmatch is used primarily to exclude techni-cal errors, confi rm ABO compatibility, and detect the rare antibody that is not detected by the screening.
5 hema x transfusion medicine — 5
01/11
Before receiving allogeneic red cells, patients who have had a transfusion or have become pregnant within the past 3 months must be tested for new antibodies every 3 days. For patients not recently exposed to red cells, there is no consensus concerning the appropriate interval between red cell collection and use of the specimen in pretransfusion test-ing. Commonly, specimens are accepted 14 to 28 days before the date of use. However, one study showed that no new antibodies appeared in paired specimens collected up to 1 year apart, suggesting that a longer acceptance interval may be possible.37
Blood Components
Most blood donations undergo a centrifugal separation process that allows each component to be used for specifi c indications. Whole blood can be separated into red cells (which contain most of the leukocytes), platelet concentrates (which contain some leukocytes), and plasma. Plasma can be further separated into coagulation components and albumin. Each whole blood unit can potentially support many recipients and clinical needs, maximizing use of each donation.
After 24 hours’ storage, whole blood contains no active platelets, and after 2 days, the labile factors V and VIII are in decline. Therefore, except for some autologous blood pro-grams that use whole blood rather than packed red cells, use of whole blood has now been almost completely supplanted by therapy employing specifi c blood components.
red blood cells
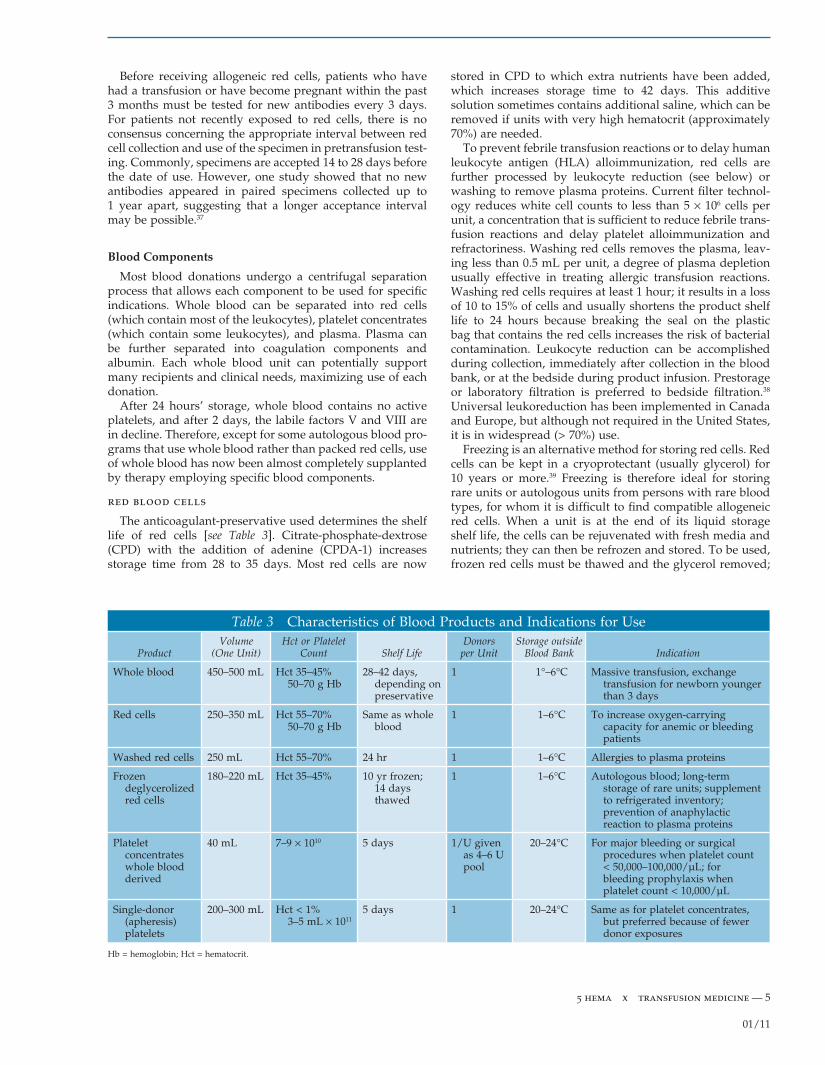
The anticoagulant-preservative used determines the shelf life of red cells [see Table 3]. Citrate-phosphate-dextrose (CPD) with the addition of adenine (CPDA-1) increases storage time from 28 to 35 days. Most red cells are now
stored in CPD to which extra nutrients have been added, which increases storage time to 42 days. This additive solution sometimes contains additional saline, which can be removed if units with very high hematocrit (approximately 70%) are needed.
To prevent febrile transfusion reactions or to delay human leukocyte antigen (HLA) alloimmunization, red cells are further processed by leukocyte reduction (see below) or washing to remove plasma proteins. Current fi lter technol-ogy reduces white cell counts to less than 5 × 106 cells per unit, a concentration that is suffi cient to reduce febrile trans-fusion reactions and delay platelet alloimmunization and refractoriness. Washing red cells removes the plasma, leav-ing less than 0.5 mL per unit, a degree of plasma depletion usually effective in treating allergic transfusion reactions. Washing red cells requires at least 1 hour; it results in a loss of 10 to 15% of cells and usually shortens the product shelf life to 24 hours because breaking the seal on the plastic bag that contains the red cells increases the risk of bacterial contamination. Leukocyte reduction can be accomplished during collection, immediately after collection in the blood bank, or at the bedside during product infusion. Prestorage or laboratory fi ltration is preferred to bedside fi ltration.38 Universal leukoreduction has been implemented in Canada and Europe, but although not required in the United States, it is in widespread (> 70%) use.
Freezing is an alternative method for storing red cells. Red cells can be kept in a cryoprotectant (usually glycerol) for 10 years or more.39 Freezing is therefore ideal for storing rare units or autologous units from persons with rare blood types, for whom it is diffi cult to fi nd compatible allogeneic red cells. When a unit is at the end of its liquid storage shelf life, the cells can be rejuvenated with fresh media and nutrients; they can then be refrozen and stored. To be used, frozen red cells must be thawed and the glycerol removed;
Table 3 Characteristics of Blood Products and Indications for Use
Product Volume
(One Unit) Hct or Platelet
Count Shelf Life Donors
per Unit Storage outside
Blood Bank Indication
Whole blood 450–500 mL Hct 35–45% 50–70 g Hb
28–42 days, depending on preservative
1 1°–6°C Massive transfusion, exchange transfusion for newborn younger than 3 days
Red cells 250–350 mL Hct 55–70% 50–70 g Hb
Same as whole blood
1 1–6°C To increase oxygen-carrying capacity for anemic or bleeding patients
Washed red cells 250 mL Hct 55–70% 24 hr 1 1–6°C Allergies to plasma proteins
Frozen deglycerolized red cells
180–220 mL Hct 35–45% 10 yr frozen; 14 days thawed
1 1–6°C Autologous blood; long-term storage of rare units; supplement to refrigerated inventory; prevention of anaphylactic reaction to plasma proteins
Platelet concentrates whole blood derived
40 mL 7–9 × 1010 5 days 1/U given as 4–6 U pool
20–24°C For major bleeding or surgical procedures when platelet count < 50,000–100,000/µL; for bleeding prophylaxis when platelet count < 10,000/µL
Single-donor (apheresis) platelets
200–300 mL Hct < 1% 3–5 mL × 1011
5 days 1 20–24°C Same as for platelet concentrates, but preferred because of fewer donor exposures
Hb = hemoglobin; Hct = hematocrit.

5 hema x transfusion medicine — 6
01/11
consequently, the product is expensive and preparation time is longer than for products stored in the liquid state.
platelets
Platelets can be provided either as platelet concentrates from a number of blood donors or from a single donor [see Table 3]. SDPs are collected by a continuous apheresis process that removes platelets and returns all other blood components. A single transfusion of platelet concentrates usually consists of platelets derived from four to six units of donated whole blood, which is about the same number of platelets contained in one SDP product. Platelets are suspended in 200 to 300 mL of plasma. The advantage of SDP therapy is the reduced risk of blood-borne infection and antigen exposure because the product is from one donor rather than from four to six; the disadvantages are a longer collection time, greater cost, and, often, limited supply.40 ABO Rh–compatible platelets should be used when possible because signifi cantly better therapeutic results are obtained from compatible transfusions.12,41
plasma
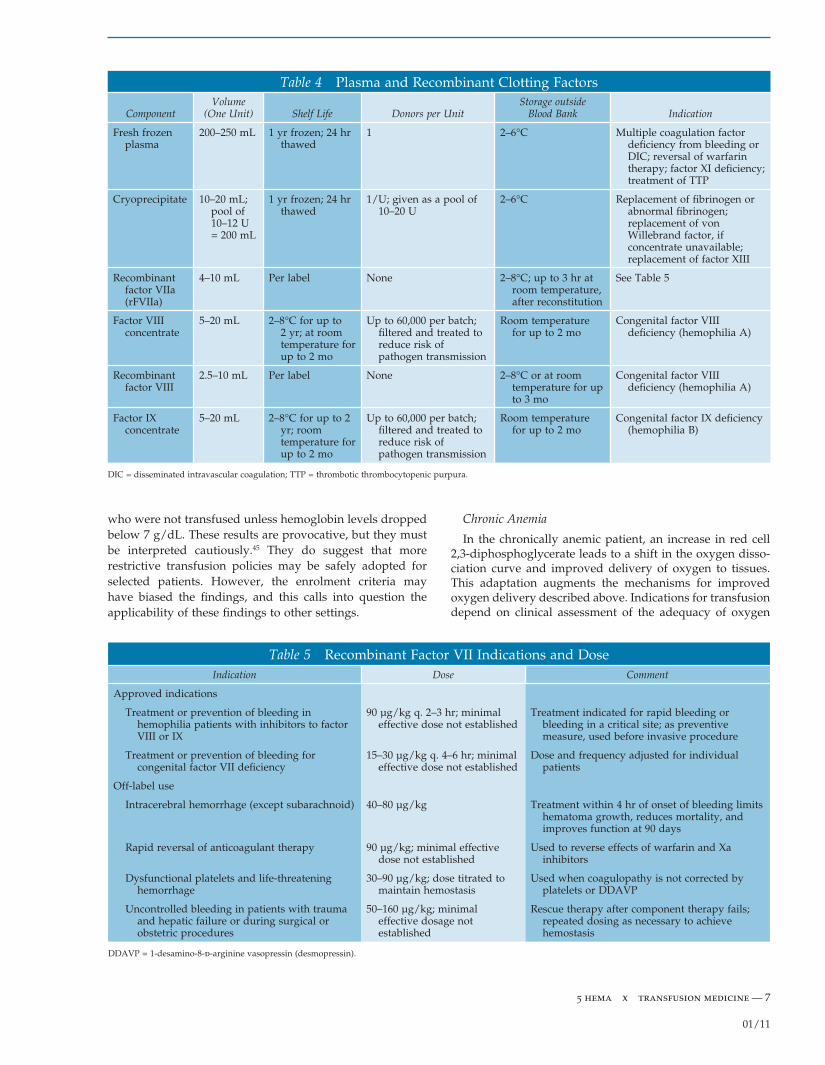
Fresh frozen plasma (FFP), which is plasma that is frozen within 8 hours of collection, contains all the procoagulants at normal plasma concentrations. Units of FFP prepared from whole blood generally contain 300 to 330 mL and virtually all plasma proteins in concentrations equivalent to those of fresh plasma. After thawing, FFP can be kept for 24 hours at 1° to 6°C and will retain 3 to 4 mg/mL of fi brinogen and 1 IU/mL of the other clinically important coagulation proteins. Plasma stored for up to 24 hours before freezing (FP-24) is considered equivalent to FFP for all intents and purposes.
Cryoprecipitate consists of the cryoproteins recovered from FFP when it is rapidly frozen and then allowed to thaw at 2° to 6°C. These cryoproteins include fi brinogen, factor VIII, von Willebrand factor, factor XIII, and fi bronectin. About 40% of the components in FFP are recovered. The cryoproteins are suspended in a small amount of plasma that contains ABO isoagglutinin at the concentration found in normal plasma. A pool of 10 units of cryoprecipitate (each derived from one unit of FFP) contains an amount of fi brin-ogen equivalent to four units of FFP but in one fourth to one fi fth the volume. Consequently, a cryoprecipitate pool permits more rapid replacement of fi brinogen than FFP but has the disadvantage of more donor exposures. After the cryoprecipitate is removed from FFP, the residual product is known as cryopoor plasma. Once frozen, cryopoor plasma has the same shelf life as FFP [see Table 4 and Table 5]. With the exception of a few cell-associated pathogens, such as HTLV I/II and malarial parasites, plasma components present the same risk of infectious disease transmission as does whole blood.
Transfusion of Red Cells
indications for allogeneic transfusion
Acute Blood Loss
The decision whether to use red cells depends on the etiology and duration of the anemia, the rate of change of
the anemia, and assessment of the patient’s ability to compensate for the diminished capacity to carry oxygen that results from the decrease in red cell mass.42 Management of acute anemia caused by bleeding or operative blood loss will differ from management of chronic anemia to which the patient has adapted. However, the question underlying any red cell transfusion is whether there is suffi cient oxygen delivery to tissues for current needs.
Compensatory mechanisms for acute blood loss include adrenergic response, leading to constriction of venous beds, which improves venous return; increased stroke volume, tachycardia, or both; and increased peripheral resistance, which eventually redistributes blood fl ow to essential organs. Also contributing to the maintenance of intravascu-lar volume is the shifting of fl uid to the intravascular space; this shifting occurs relatively rapidly from the extra-vascular space and more slowly from the intracellular to the extravascular space.
A decrease in blood volume has distinct effects on oxygen delivery, depending on the volume of blood lost and the functioning of the compensatory cardiovascular responses. Restoration of intravascular volume, usually with crystal-loid, ensures adequate perfusion of peripheral tissue and is the fi rst treatment goal for a patient with acute blood loss. Whether red cell transfusion is required depends on the extent of blood loss and the presence of comorbid conditions that may limit host response to the blood loss. The American College of Surgeons has correlated blood loss with clinical fi ndings. Loss of up to 15% of total blood volume (class I hemorrhage) usually has little effect; this amount is the maximum permitted in normal blood donation. A class II hemorrhage (15 to 30% loss) results in tachycardia, decreased pulse pressure, and, possibly, restlessness. A class III hemorrhage (30 to 40% loss) leads to obvious signs of hypovolemia; mental status often remains normal. Red cell transfusion is usually indicated when blood loss exceeds 30% in a patient without other signifi cant comorbid condi-tions. However, the presence of serious cardiac, peripheral vascular, or pulmonary disease can lower this threshold. For example, anemic patients with signifi cant coronary artery disease are more likely to have serious postoperative myocardial complications. One unit of red cells will raise the hemoglobin concentration about 1 g/dL in an adult.
The threshold for red cell transfusion has been evaluated in two randomized, controlled trials. In one study of transfu-sion after coronary artery bypass, patients who received transfusions for hemoglobin levels below 8 g/dL did no worse than patients who received transfusions for hemoglo-bin levels below 9 g/dL.43 The other trial compared out-comes in critical care patients who received transfusions when their hemoglobin level fell below either 7 or 10 g/dL.44 Enrolment in this study was limited to patients who were euvolemic at entry and whose hemoglobin levels were from 7 to 9 g/dL; patients who had undergone routine cardiac procedures or who were actively bleeding on entry to the intensive care unit were excluded. There was no statistical difference in 30-day mortality for these two groups. However, in the subgroups of patients younger than 55 years and patients whose illness was less severe, as defi ned by standardized clinical criteria, Kaplan-Meier survival estimates were signifi cantly better in the patients
5 hema x transfusion medicine — 7
01/11
who were not transfused unless hemoglobin levels dropped below 7 g/dL. These results are provocative, but they must be interpreted cautiously.45 They do suggest that more restrictive transfusion policies may be safely adopted for selected patients. However, the enrolment criteria may have biased the fi ndings, and this calls into question the applicability of these fi ndings to other settings.
Chronic Anemia
In the chronically anemic patient, an increase in red cell 2,3-diphosphoglycerate leads to a shift in the oxygen disso-ciation curve and improved delivery of oxygen to tissues. This adaptation augments the mechanisms for improved oxygen delivery described above. Indications for transfusion depend on clinical assessment of the adequacy of oxygen
Table 4 Plasma and Recombinant Clotting Factors
Component Volume
(One Unit) Shelf Life Donors per Unit Storage outside
Blood Bank Indication
Fresh frozen plasma
200–250 mL 1 yr frozen; 24 hr thawed
1 2–6°C Multiple coagulation factor defi ciency from bleeding or DIC; reversal of warfarin therapy; factor XI defi ciency; treatment of TTP
Cryoprecipitate 10–20 mL; pool of 10–12 U = 200 mL
1 yr frozen; 24 hr thawed
1/U; given as a pool of 10–20 U
2–6°C Replacement of fi brinogen or abnormal fi brinogen; replacement of von Willebrand factor, if concentrate unavailable; replacement of factor XIII
Recombinant factor VIIa (rFVIIa)
4–10 mL Per label None 2–8°C; up to 3 hr at room temperature, after reconstitution
See Table 5
Factor VIII concentrate
5–20 mL 2–8°C for up to 2 yr; at room temperature for up to 2 mo
Up to 60,000 per batch; fi ltered and treated to reduce risk of pathogen transmission
Room temperature for up to 2 mo
Congenital factor VIII defi ciency (hemophilia A)
Recombinant factor VIII
2.5–10 mL Per label None 2–8°C or at room temperature for up to 3 mo
Congenital factor VIII defi ciency (hemophilia A)
Factor IX concentrate
5–20 mL 2–8°C for up to 2 yr; room temperature for up to 2 mo
Up to 60,000 per batch; fi ltered and treated to reduce risk of pathogen transmission
Room temperature for up to 2 mo
Congenital factor IX defi ciency (hemophilia B)
DIC = disseminated intravascular coagulation; TTP = thrombotic thrombocytopenic purpura.
Table 5 Recombinant Factor VII Indications and DoseIndication Dose Comment
Approved indications
Treatment or prevention of bleeding in hemophilia patients with inhibitors to factor VIII or IX
90 µg/kg q. 2–3 hr; minimal effective dose not established
Treatment indicated for rapid bleeding or bleeding in a critical site; as preventive measure, used before invasive procedure
Treatment or prevention of bleeding for congenital factor VII defi ciency
15–30 µg/kg q. 4–6 hr; minimal effective dose not established
Dose and frequency adjusted for individual patients
Off-label use
Intracerebral hemorrhage (except subarachnoid) 40–80 µg/kg Treatment within 4 hr of onset of bleeding limits hematoma growth, reduces mortality, and improves function at 90 days
Rapid reversal of anticoagulant therapy 90 µg/kg; minimal effective dose not established
Used to reverse effects of warfarin and Xa inhibitors
Dysfunctional platelets and life-threatening hemorrhage
30–90 µg/kg; dose titrated to maintain hemostasis
Used when coagulopathy is not corrected by platelets or DDAVP
Uncontrolled bleeding in patients with trauma and hepatic failure or during surgical or obstetric procedures
50–160 µg/kg; minimal effective dosage not established
Rescue therapy after component therapy fails; repeated dosing as necessary to achieve hemostasis
DDAVP = 1-desamino-8-d-arginine vasopressin (desmopressin).

5 hema x transfusion medicine — 8
01/11
delivery and are also guided by the etiology of the anemia.42 In patients for whom the anemia can be reversed with iron, folic acid, or vitamin B12, transfusion therapy is indicated only when clinical conditions cannot be tolerated during the period in which the endogenous red cell mass is being regenerated. Patients with chronic renal disease are typically defi cient in erythropoietin. Replacement therapy with exogenous erythropoietin often obviates transfusion. It is rarely necessary to return the hematocrit to “normal” levels to provide safe, clinically effective therapy.46 Patients with anemia that is a result of chronic disease such as rheumatoid arthritis, chemotherapy for malignancy, myelodysplasia, or AIDS may also respond to erythropoietin, but caveats regarding the rate of hemoglobin elevation and the tolerable hemoglobin concentration remain.47
Relatively little is known about transfusion thresholds in specifi c medical illnesses. Observational analyses have suggested that the “transfusion trigger” should be more liberal for patients with cardiovascular disease and that correction of anemia improves mortality in elderly patients hospitalized with myocardial infarction and for patients with congestive heart failure. No single laboratory measure-ment or combination of physiologic markers predicts the need for red cell transfusion; the decision to transfuse red cells continues to rely on evaluation of the individual patient by skilled clinicians at the bedside who use hemoglobin concentration as no more than a helpful guide.42
indications for autologous transfusion
Whether the criteria for autologous transfusion should be the same as that for allogeneic transfusion remains unre-solved. Although the risk associated with autologous blood is less than that associated with allogeneic blood, it is not zero. Errors in labeling, storage, and processing can still occur. For these reasons, many argue that uniform standards based on oxygen delivery should apply, regardless of the blood source. Others, citing the reduced risk, advocate returning most or all of the predeposited units to the patient. There is no clinical evidence that either transfusion policy is associated with better or worse patient outcomes.
Transfusion of Platelets
In general, the decision to transfuse platelets rests on the answers to two questions: (1) Is thrombocytopenia the result of underproduction or increased consumption of platelets? and (2) Do the existing platelets function normally?
indications for transfusion
Low Platelet Count
Thrombocytopenia can result from decreased production caused by marrow hypoplasia or from increased consump-tion caused by conditions such as disseminated intravascu-lar coagulation (DIC) or a combination of both as in immune thrombocytopenic purpura (ITP).48 In a patient with ITP, surviving platelets are larger and younger and function better than would be expected given the platelet count; platelet transfusion is largely avoided or minimized for such a patient, although in life-threatening situations, the tran-sient increment from a platelet transfusion can prove vital.
In contrast, with hypoplasia, hemostasis is more severely impaired, and the risk of bleeding is relatively higher. Plate-let transfusions should be given to patients with clinically signifi cant hemorrhage and severe thrombocytopenia. The decision to transfuse patients who have hypoproliferative thrombocytopenia prophylactically is generally initiated when the platelet count drops below a certain threshold. Published consensus guidelines provide an excellent summary of all aspects of platelet therapy.49
The time-honored transfusion threshold of 20,000/µL used for platelet prophylaxis was established on the basis of studies of patients receiving aspirin; the results of more recent controlled trials indicate that this threshold is high. The prevalence of bleeding increases signifi cantly below a threshold of about 10,000 platelets/µL in otherwise asymptomatic patients.50 Transfusion at levels above 10,000 platelets/µL may be necessary in newborns; in patients with signs of hemorrhage, high fever, precipitous decline in platelet count, and additional hemostatic defects; and in patients undergoing invasive procedures.49
Dysfunctional Platelets
Platelet function is the second criterion for the transfusion of platelets. Transfusion is appropriate in a bleeding patient whose platelet count is adequate but whose platelets are dysfunctional as a result of medications such as aspirin or thienopyridines or as a result of bypass surgery. In a bleed-ing patient, if platelet dysfunction is the result of inherited or acquired defects, transfusion is indicated to provide a minimum number of normal platelets. Platelet function is abnormal in uremic patients, and defi nitive treatment requires correction of the uremia. Some studies suggest that interventions that increase von Willebrand factor levels, such as desmopressin (1-desamino-8-d-arginine vasopressin [DDAVP]), conjugated estrogen, or cryoprecipitate, may favorably infl uence platelet function in uremia.44 In vitro evidence suggests that DDAVP may improve platelet dysfunction caused by glycoprotein IIb or glycoprotein IIIa (GPIIb/IIIa) inhibitors (e.g., eptifi batide, abciximab, tirofi ban) or aspirin.49
contraindications to platelet transfusion
Proper investigation of the causes of thrombocytopenia will identify clinical situations in which platelets are traditionally withheld because they may contribute to the evolution of the illness. These disorders include thrombotic microangiopathies, such as thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome, and HELLP syndrome (hemolysis, elevated liver enzymes, and a low platelet count). Patients with these disorders rarely bleed; when hemorrhage occurs, platelet transfusion may prove lifesaving. Posttransfusion purpura is usually unresponsive to platelet transfusions that are not matched to avoid the platelet-specifi c antigen, but it may respond to intravenous immune globulin (IVIg) or plasma exchange. Transfused platelets are short-lived in patients with immune thrombo-cytopenia (e.g., ITP), but they cause no harm and may effect hemostasis temporarily when hemorrhage occurs. Platelet infusions should be considered in high-risk patients with ITP, such as children with bleeding signs beyond petechiae and purpura who may be at particular danger of intercranial hemorrhage.51

5 hema x transfusion medicine — 9
01/11
response to platelet transfusions
Both platelet and host factors infl uence the response to platelet transfusions. Length of in vitro storage, storage temperature, adequacy of oxygenation, and extent of pretransfusion manipulation all infl uence in vivo survival. Important host factors that infl uence survival are body temperature, underlying disease, splenomegaly, ABO compatibility, and immune status.
A transfusion of appropriately stored fresh platelets—whether pooled concentrates or SDPs—should contain about 6,000 to 10,000/µL platelets per unit (5.5 × 1011 platelets). Thus, in an unsensitized 75 kg (165 lb) recipient, each unit should yield an increment of about 60,000 platelets/µL. A posttransfusion count is usually obtained after 1 hour; how-ever, a count can be obtained as early as 10 minutes after transfusion. Smaller doses of prophylactic platelet transfu-sions result in a decreased number of platelets transfused per patient and no effect on the incidence of bleeding but an increased number of transfusions.52 A case is considered refractory to platelet transfusions when the 1-hour post-transfusion increment is less than 10,000 platelets/µL after the patient is given 3.3 × 1011 freshly stored (< 48 hours) platelets.
platelet transfusions in refractory cases
There is no evidence that repeated administration of plate-let concentrates in the absence of a measurable increment improves hemostasis.
Poor response (refractoriness) to platelets may be either immune or nonimmune. Platelets express platelet-specifi c antigens, HLAs, and blood group antigens. Immune response to any of these can contribute to platelet unrespon-siveness. Platelet surfaces have only class I HLA antigens, of which only HLA-A and HLA-B are clinically important. Polymorphic antigens are found in association with each of the major platelet proteins: HPA1a/2a (formerly called PlA1/A2) and HPA4 (Pen) on glycoprotein IIIa, HPA3a/b (Bak system) on glycoprotein IIb, and HPA 2a/b (Sib and Ko) on glycoproteins Ia and Ib. Each of these antigen groups is associated with isoimmune neonatal thrombocytopenia. The prevalence of antibodies to platelet-specifi c antigens is increased for patients sensitized to HLA antibodies; there-fore, antibodies to both sets of epitopes may contribute to refractoriness in patients who fail to respond to HLA-matched platelets.53
For patients who are refractory to platelet transfusions, treatment involves addressing nonimmune causes (e.g., fever, sepsis, bleeding, and DIC) and providing recently collected ABO-compatible components. If these strategies fail, minimization of the effects of HLA antibodies or plate-let antigens through HLA typing, platelet crossmatching, or both is indicated.48,52 Selecting platelets matched at the HLA-A and HLA-B loci may improve responsiveness in about half of patients with positive HLA antibody screens. Computed best-match selection programs prove useful when identical matches are unavailable.54 Unless contraindi-cated because of transplant considerations, an empirical trial of donations from family members may also be helpful.
Platelets can be selected for alloimmunized patients by HLA matching or by platelet crossmatching. If a patient has a high titer antibody to a specifi c HLA antigen, platelets
lacking the cognate antibody can be selected. Otherwise, closely HLA-matched platelet preparations provide the best chance of an effective transfusion. Crossmatched platelets provide equivalent platelet increments that may be indepen-dent of the grade of HLA match.55 Although these results are promising, the effectiveness of selection either by HLA and crossmatching or by crossmatching alone is often limited by nonimmune host factors.
Modifying the effects of alloimmunization is diffi cult. IVIg can improve platelet increments but not platelet survival. In some circumstances, response refl ects an under-lying autoantibody in addition to alloantibodies. Plasma exchange is of limited value because it is diffi cult to remove IgG antibodies. In some patients, the HLA antibodies responsible for refractoriness may regress over time; it is therefore important to periodically retest for the presence of HLA antibodies. If the HLA antibody screen becomes negative, a trial of non–HLA-matched (i.e., from random donors) platelets is warranted.
All in all, the best strategy is prevention, which can be achieved by avoiding unnecessary transfusions and using only leukocyte-depleted components. A randomized, prospective trial examined how best to prevent alloimmuni-zation in newly diagnosed patients with acute myeloid leukemia. The study compared leukocyte reduction by fi ltration and by ultraviolet B irradiation of platelets; both methods were equally effective.56 In addition, the study found that platelets obtained from single random donors provided no additional benefi t over pooled platelet concen-trates from random donors.56 Although leukocyte reduction signifi cantly reduced the occurrence of alloimmunization, it did not prevent secondary immune responses in patients already sensitized through either pregnancy or transfusion.56,57
Transfusion of Fresh Frozen Plasma, Plasma Derivatives, and Recombinant Products
fresh frozen plasma
Despite a paucity of indications for FFP use, roughly 4 million units are transfused annually.12,41,58 FFP is most appropriate for replacing the multiple coagulation defi cien-cies that result from massive transfusion, liver disease, warfarin toxicity, or acute or chronic DIC. In addition, FFP can be used to treat thrombotic microangiopathies and specifi c factor defi ciencies when factor concentrates are not available.59 After one blood volume exchange using only red cells, plasma components are diluted to about 40% of their original concentration; after two blood volume exchanges, plasma components are diluted to 15%. Prothrombin time (PT) and partial thromboplastin time (PTT) become pro-longed when coagulation components are lower than 30%, but abnormal bleeding from dilution usually does not occur until these values are less than 17% of normal. Microvascu-lar bleeding associated with a PT and a PTT greater than 1.5 times normal is an indication for FFP. Whether FFP replacement is needed when PT and PTT are over 1.5 times normal but are not associated with bleeding is less clear-cut; paracentesis and thoracentesis did not cause increased bleeding in patients with PT and PTT that were up to twice

5 hema x transfusion medicine — 10
01/11
normal values.60 No data support the use of FFP to correct slight prolongation of PT and PTT. In a prospective audit, transfusion of 1,091 units of FFP to correct mild abnormali-ties in coagulation values resulted in partial normalization of PT in a minority of patients and failed to correct the PT in 99% of patients.61
The FFP dose depends on whether a consumptive process, such as TTP or DIC, is being treated concurrently with the use of hemodilution. For a patient requiring hemodilution alone, 15 mL/kg will usually be suffi cient. However, if a thrombotic microangiography is present, the dose is best guided by the effect of treatment on PT and PTT. If fi brino-gen is lower than 80 mg/dL, cryoprecipitate may be required to rapidly increase fi brinogen; PT and PTT deter-minations are inaccurate at this level. However, four units of FFP can be used in most cases to provide the same amount of fi brinogen as one pooled unit of cryoprecipitate. Urgent reversal of the effects of warfarin can usually be accomplished with about 5 to 10 mL/kg of FFP.
Thrombotic microangiopathies are treated with either FFP transfusions or, more often, plasma exchange with either FFP or cryopoor plasma.57 The dose of either product is usually equal to a plasma volume exchange of 1.0 to 1.5, which is carried out daily until clinical improvement occurs.62
Early plasma infusion, often at a fi xed 1:1 ratio with red cells, has become increasingly common for the subset of trauma patients who are coagulopathic on presentation (acute traumatic coagulopathy) and require massive transfu-sion, often including platelets on a fi xed ratio protocol.63 The optimal trigger for initiation of a protocol for aggressive plasma infusion warrants prospective evaluation. However, patients receiving uncrossmatched blood in the emergency department are more than three times more likely to receive early massive transfusion of red cells and are more likely to receive six units or more of plasma and two or more apher-esis platelet transfusions. Given these fi ndings, emergency transfusion of uncrossmatched red cells is a potential trigger for activation of an institution’s massive transfusion proto-col.64 For nonmassively transfused trauma patients, plasma administration has been associated with a substantial increase in complications, in particular acute respiratory dis-tress syndrome (ARDS), with no improvement in survival. An increase in multiple organ dysfunction, pneumonia, and sepsis was likewise seen with increasing volumes of transfused plasma.65
No factor XI concentrates are licensed in the United States. Therefore, FFP is the treatment of choice for factor XI defi ciency. FFP is no longer used to replace antithrombin III, because a purifi ed concentrate is available.
factor viia
Recombinant activated factor VII (rFVIIa) was approved in 1999 for the treatment of bleeding episodes in patients with hemophilia A or B who have antibodies (inhibitors) to factor VIII or IX, respectively.66 Recently, rFVIIa was approved by the Food and Drug Administration (FDA) for use as replacement therapy in factor VII defi ciency, whether the defi ciency is acquired (e.g., as a consequence of liver disease) or inherited. In addition, rFVIIa is useful in the activation of the coagulation tissue factor pathway. For
patients with inhibitors, rFVIIa is given at a dosage of 90 µg/kg as a slow IV push over 2 to 5 minutes; the dosage is repeated every 2 hours, as needed. For factor VII defi -ciency, the dosage is 20 to 30 µg/kg given as a slow IV push over 10 minutes; given in this manner, rFVIIa treatment will reduce the PT to normal within 20 minutes after administra-tion. Depending on the clinical setting, the PT will become prolonged again 3 to 4 hours after treatment.
Off-label use of rFVIIa as a universal hemostatic agent has become increasingly common in the treatment of uncon-trolled hemorrhage in patients who do not have a preexist-ing bleeding disorder and who are unresponsive to FFP.8,67–69 Because rFVIIa is believed to act primarily on the platelet surface, it is important that severe thrombocytopenia be corrected before administration of rFVIIa. Furthermore, rFVIIa has been associated with thromboembolic events and should be used cautiously in patients at increased risk, such as those with cardiovascular disease, cerebrovascular disease, or DIC.70 Its high cost and potential for contributing to the development of DIC should limit its use to carefully selected patients for whom other alternatives are not available [see Table 4 and Table 5].
factor viii concentrates
The introduction of plasma-derived factor VIII concen-trates in the 1960s brought a signifi cant improvement in the treatment of hemophilia A. Unfortunately, these concen-trates were derived from large pools of donor plasma, and contamination of the factor with HBV, HCV, and, especially, HIV resulted in the widespread transmission of these infections in the hemophilia community. Since 1980, new methods of heat sterilization, solvent-detergent treatment, and immunoaffi nity purifi cation have yielded an array of factor concentrates that are highly purifi ed and free from these infectious agents.71 The effi cacy of these viral inactiva-tion methods has been validated by molecular testing for the presence of these pathogens and by longitudinal epidemio-logic surveillance. There have been no documented trans-missions since 1985. Recombinant factor VIII concentrates free of any human blood–derived proteins and of ultrahigh purity (> 3,000 IU of clotting factor activity per milligram of protein) are available and are widely used for newly diagnosed hemophilia patients [see Table 4]. The dosage is calculated on the assumption that that 1 U/kg body weight of factor VIII will raise the plasma activity by about 2%. The circulating half-life of factor VIII is 8 to 12 hours.
The factor VIII preparation Humate-P is also rich in von Willebrand factor and is approved for the treatment of von Willebrand disease. This product has the major advantage of being free of the risks of infection associated with cryopre-cipitate. If Humate-P is not available, the factor VIII prepara-tions Alphanate or Koate-DVI may be used, but they are not approved for this purpose and their effi cacy is uncertain.
The advances in the safety and purity of factor VIII con-centrates, especially in the case of the recombinant products, have increased the cost per unit fi vefold to 10-fold.
factor ix concentrates
Factor IX complex concentrates contain about equal amounts of the vitamin K–dependent factors II, VII, IX, and X. These preparations are available in several degrees of

5 hema x transfusion medicine — 11
01/11
purity, but all have the disadvantage of being thrombogenic when used for extended periods or in patients with liver disease. These concentrates can be used for urgent reversal of warfarin anticoagulation. Highly purifi ed plasma-derived and genetically engineered factor IX preparations are free of this complication and are the products of choice in treating factor IX defi ciency [see Table 4].72 One unit per kilogram of factor IX concentrate raises the plasma activity by about 1%; the half-life is approximately 18 hours.
Activated prothrombin complex concentrates (i.e., Autoplex-T and FEIBA) have been used to bypass the need for factor VIII in selected patients with hemophilia A and acquired inhibitors. This provides an alternative for patients who do not respond to rFVIIa.
Transfusion of Granulocytes
A direct relationship between the number of circulating granulocytes and bacterial infection has been recognized for more than 40 years.73 Granulocyte transfusion can be effective in the treatment of severely neutropenic patients (absolute neutrophil count < 500/µL) with bacterial or fungal infections. Transfusion of granulocytes in doses in the range of 4 × 1010 to 8 × 1010 can be obtained by apheresis of donors who have been pretreated with granulocyte colony-stimulating factor (G-CSF) and a single dose of dexamethasone.74 Granulocyte transfusions at these dose levels have been shown to produce measurable, sustained increments in neutrophils, even into the normal range. The indications and clinical benefi ts of granulocyte transfusion at these higher doses are still being determined. Random-ized trials are required to fully defi ne the clinical effi cacy of granulocyte transfusions. After collection, granulocytes must be stored at room temperature and irradiated to prevent transfusion-associated graft versus host disease. Granulocyte concentrates contain large numbers of erythrocytes; crossmatching between the specimen and the potential recipient should be performed to ensure red cell compatibility. Administration of granulocytes from random donors to alloimmunized patients is inadvisable because improvement will usually be negligible and severe reactions may result.75
Transfusion of Immune Globulin
Many human immune globulin preparations are avail-able. Immune serum globulin, administered intramuscularl y, is used to treat chronic immunodefi ciency disease and for the prevention or alleviation of measles, tetanus, and rabies. Hepatitis A and B can now be prevented by vaccination. Alternatively, a traveler who will spend less than 3 months in an endemic area can receive 0.02 mL/kg of immune serum globulin. Hepatitis B immune globulin (HBIg) is used for postexposure prophylaxis against HBV infection. HBIg is prepared from plasma with high titers of antibody to hepatitis B surface antigen. RhO(D) immune globulin is used to prevent the development of anti-RhO (anti-D) antibodies in Rh-negative women who have just given birth, under-gone amniocentesis, or aborted if the biologic father is thought to be Rh positive. Intramuscular preparations must not be administered intravenously.
Several IV preparations of immune globulin are available, with concentrations ranging from 3 to 12%. Whereas the numerous commercially available IVIg products do not differ appreciably in terms of effi cacy, different manufactur-ing processes and the fi nal composition of IVIg products have resulted in different safety and tolerability profi les. Patients receiving an IVIg product should be carefully monitored at the initial exposure and switched to an alternative brand if the product is not well tolerated. IV administration of human immune globulin promptly elevates circulating IgG levels and is preferable to intramus-cular administration. The half-life is about 21 days. IVIg is used to treat congenital or acquired chronic immunodefi -ciency disease. The IV dosage for such defi ciency syndromes is 0.2 g/kg/month; however, the dosage can be raised to 0.3 g/kg/month, or the agent can be given more often if needed. IVIg is widely used to treat autoimmune disorders such as ITP, Guillain-Barré syndrome, and chronic infl am-matory demyelinating polyneuropathy.76 Burgeoning off-label use has resulted in repeated product shortages.
The most common side effects of IVIg therapy—headache, nausea, and fever—usually respond to symptomatic treat-ment and a reduction in the infusion rate. Less common and potentially more severe side effects are anaphylactic reactions, hemolysis from anti-A and anti-B antibodies, and acute renal failure.76 Renal failure has been attributed to osmotic nephrosis caused by the high sucrose concentration in some IgG preparations. In one study, aseptic meningitis was the most common of the serious side effects, with a frequency of 11%; patients with a history of migraine had a signifi cantly higher incidence of aseptic meningitis.77 Aseptic meningitis usually occurs within 24 hours after administration and does not respond to a reduction in the infusion rate. Patients may be required to stay in the hospital for symptomatic treatment; if further treatment is needed, changing the lot or preparation of IVIg may alleviate this side effect. Thromboembolism of unknown cause has also been reported, and manufacturers have called specifi c attention to this complication.78 As with most plasma frac-tionation products, current screening and manufacturing practices eliminate transfusion-transmitted viruses from IVIg preparations.
Transfusion of Stem Cells
Stem cell transplantation, initially pioneered for use in leukemia, is used to treat a number of life-threatening, malignant, hereditary, and immunologic disorders.
Complications of Transfusions
hemolytic transfusion reactions
Hemolytic transfusion reactions are classifi ed as immedi-ate or delayed, depending on their pathophysiology. Imme-diate hemolytic reactions may be caused by a preexisting antibody in the recipient that was not detected during pretransfusion testing or, more commonly, by transfusion of ABO-incompatible blood in error.79 Delayed hemolytic reac-tions are the result of an anamnestic response to an antigen to which the recipient is already sensitized. The renewed

5 hema x transfusion medicine — 12
01/11
antigenic exposure in a person already sensitized to an anti-gen can result in stimulation of antibody to levels that can cause hemolysis. This is in contrast to an immune response during primary sensitization, which seldom causes hemoly-sis because antibody levels develop at a much slower rate. Not all antibodies are clinically signifi cant; the most common and important ones are in the Rh, Kell, Duffy, and Kidd systems.
Patients with sickle cell disease appear more likely than others to become alloimmunized and to have delayed hemo-lytic transfusion reactions, which often occur in association with occlusive pain crisis. These reactions are occasionally associated with severe hyperhemolysis involving autolo-gous, as well as allogeneic, red cells; these episodes can be life-threatening. The cause of these episodes is unknown, but they have been attributed to so-called bystander hemo-lysis associated with abnormal function of CD59 (membrane inhibitor of reactive lysis [MIRL]), transfusion-associated marrow suppression, or both.80
Diagnosis of Hemolytic Reactions
The pathophysiologic differences between immediate and delayed hemolytic transfusion reactions account for some of their differences in clinical fi ndings. Fever is a common sign associated with both immediate and delayed hemolytic transfusion reactions.
Clinically, hemolysis is likely to be more severe in imme-diate hemolytic reactions; clinical fi ndings may include back pain, pain along the vein into which the blood is being transfused, diaphoresis, changes in vital signs, evidence of acute renal failure, respiratory compromise, and signs of developing DIC. Red or brown plasma points to hemoglobi-nemia from intravascular hemolysis, whereas red urine in the absence of red cells indicates that hemoglobin from lysed red cells is being cleared by the kidney (hemoglobinuria). Immune-mediated acute hemolytic reactions refl ect a sys-temic infl ammatory response that involves multiple organ systems. These fi ndings are probably caused by immune complexes activating the complement and kinin systems, by the direct effects of red cell stroma on kidney function, and possibly by the release of infl ammatory cytokines such as interleukin-1a (IL-1a), IL-6, and tumor necrosis factor (TNF).81
In delayed hemolytic reactions, hemolysis with hemoglo-binemia and hemoglobinuria (sometimes associated with renal failure) also occurs but is less common and generally less severe. In many delayed hemolytic transfusion reac-tions, the only clinical fi ndings may be mild anemia, a newly positive Coombs test result, and the appearance of a new antibody against red cell antigens. Many such reactions go undetected because they occur 5 days or more after trans-fusion, at a time when some patients have already been dis-charged from the hospital. When hemolysis is absent, these reactions are sometimes called delayed serologic transfusion reactions.82 At the Mayo Clinic, two surveys sought to identify the incidence of both kinds of delayed transfusion reactions. The most recent survey, covering the period from 1993 to 1998, revealed a relative increase in delayed serologic transfusion reactions and an associated decrease in delayed hemolytic reactions, with overall increases in the incidence of these reactions. The earlier survey, which
covered the period from 1980 to 1992, revealed an associa-tion between delayed transfusion reactions and the presence of antibodies to Jka and Fya or antibodies with multiple spec-ifi city; this association was not found in the later survey. These changes probably result from improved systems for identifying clinically signifi cant nonhemolytic antibodies.83
In some cases, antiglobulin testing may yield positive results after all the transfused cells have been cleared, often with only complement being detected on the red cells. This fi nding has been attributed to autoimmune hemolysis that sometimes accompanies the delayed transfusion reaction.
Treatment of Hemolytic Reactions
As soon as a hemolytic transfusion reaction is suspected, the transfusion should be immediately discontinued. The diagnosis can be confi rmed or excluded by sending the remaining blood product, together with a freshly drawn posttransfusion specimen, to the blood bank. The blood bank rechecks all records, confi rms the patient’s type and antibody screen, checks for evidence of hemoglobin in the plasma, and rechecks the crossmatch and antiglobulin test results. These tests will confi rm or disprove the diagnosis and identify the antibody causing the immediate hemolytic reaction, when present. Until these studies have been completed, any further blood products should be given only with great caution.
Acute hemolytic reactions Acute hemolytic transfusion reactions constitute a medical emergency and should be managed with aggressive supportive care in an intensive care setting. Circulatory and ventilatory support may be required. Mannitol has been infused traditionally to encour-age tubular urine fl ow. Little evidence supports the use of diuretics to increase renal blood fl ow or heparinization to treat DIC, although platelets and FFP may be needed to manage the coagulopathy. Until the antibody causing the immune hemolysis is identifi ed, only type O red cells and AB plasma should be used.
Delayed hemolytic reactions Managing delayed trans-fusion reactions is simpler because of the slower tempo at which these reactions develop. The diagnosis requires identifying a new antibody against red cell antigens and searching for clinical evidence of hemolysis. Treatment involves replacement with the appropriate antigen-negative blood components when transfusion is necessary. Acute renal failure and DIC are unlikely but would be managed as described for immediate hemolytic reactions. The severe, atypical delayed transfusion reactions sometimes found in patients with sickle cell disease may require aggressive transfusion support.
Prevention of Hemolytic Reactions
Prevention of immediate and delayed hemolytic transfu-sion reactions depends on recognizing their respective proximate causes. Immediate hemolytic reactions are usually caused by errors made during the procurement or processing of blood specimens, during pretransfusion testing, or during product infusion.79 In a review of transfusion-related deaths reported to the FDA between 1990 and 1998, approximately 50% were caused by clerical

5 hema x transfusion medicine — 13
01/11
errors that led to transfusion of ABO-incompatible blood, a rate virtually unchanged since reporting began in 1976.84 Prevention of immediate transfusion reactions is best accomplished by following protocols for obtaining speci-mens from patients in adequate time before transfusion and checking to see that blood products are appropriate for the intended recipient.
Delayed transfusion reactions are the result of an anam-nestic response of antibodies from a previous transfusion (or pregnancy) that are not present in detectable levels at the time the specimen is crossmatched. A careful transfusion history can best prevent delayed hemolytic reactions. Many patients will know whether there were diffi culties involving blood obtained for transfusion. If a patient has a history of diffi culty with crossmatches, the blood bank can obtain the details from the institution responsible for the previous transfusion. A proper transfusion history can uncover patients likely to have antibodies that the blood bank would not detect. For example, antibodies to Jka and Fya are typically hard to identify because they are quick to rise on stimulation and fall equally rapidly, making later detection diffi cult.
febrile transfusion reactions
Febrile nonhemolytic transfusion reactions occur in 0.5 to 2% of all transfusions and are more likely to occur after platelet transfusions. Until recently, most febrile transfusion reactions were attributed to recipient antibody reactions against HLA antigens on donor leukocytes in the transfused product.85 It is now apparent that cytokines produced during storage may also contribute to these reactions.86 This conclusion is based on observations that platelet products associated with transfusion reactions have higher levels of infl ammatory cytokines (e.g., IL-1a, TNF, IL-6, and IL-8) in the supernatant than are found in platelets that do not cause febrile transfusion reactions.
Diagnosis of Febrile Reactions
Febrile reactions are characterized by the development of fever during transfusion or within 5 hours after transfusion. These reactions may be limited to an increase in body temperature of 1° to 2°F but are often associated with chills and rigors. Febrile nonhemolytic reactions are a diagnosis of exclusion.
The differential diagnosis for a patient who develops fever in the setting of transfusion always includes hemolysis and unrecognized sepsis. When a febrile reaction is observed, immediate management consists of discontinuing the transfusion, obtaining appropriate cultures, and return-ing the component to the blood bank. The blood bank checks for evidence of incompatibility, obtains cultures from the product, and verifi es that no errors have occurred in its preparation or administration. The probability that a febrile transfusion reaction has occurred is infl uenced by the type of product, the number of white cells contained therein, and the transfusion history of the recipient. Febrile reactions to products that have few or no white cells (e.g., leukoreduced red cells, frozen-deglycerolized red cells, or FFP) are unusua l. Unmodifi ed whole blood and red cells contain between 1.3 × 109 and 3 × 109 white cells and are much more likely to cause febrile reactions. In the case of transfused platelets,
reactions can be from cytokines made during in vitro room temperature storage or from bacterial contamination.
Treatment of Febrile Transfusion Reactions
Febrile transfusion reactions are usually self-limited and respond to symptomatic management with antipyretics. However, symptoms may be of suffi cient magnitude to require the use of 50 to 75 mg of meperidine by IV bolus. To prevent further occurrences, leukocyte-depleted components or premedication are indicated.
Prevention of Febrile Reactions
Newer designs of fi lters for leukocyte reduction decrease the white cell content to below the threshold for febrile transfusion reactions.87 Because infl ammatory cytokines may be involved in febrile transfusion reactions, methods are being implemented to accomplish leukocyte reduction either during or after collection but before storage. In a study comparing products that underwent leukocyte reduction either before storage or at the bedside, signifi cantly fewer febrile reactions occurred in patients receiving prestorage leukocyte-depleted products; there was no difference in the number of allergic reactions.88
Prestorage leukocyte reduction is particularly important for platelets because platelets are stored at room tempera-ture and accumulate signifi cantly more cytokines than do red cells, which are refrigerated. Febrile transfusion reac-tions are also more likely with older components. Platelets that were used after they were in storage for 3 days or less have been found to cause signifi cantly fewer febrile transfusion reactions than platelets that were used after longer storage periods.89 Unfortunately, testing for infec-tious diseases often takes 2 to 3 days, during which time the product cannot be used; it is therefore impractical to rely on younger products to reduce the risk of febrile transfusion reactions. Other benefi ts of leukocyte reduction include reduction of HLA alloimmunization; decreased transmis-sion of leukocyte-associated viruses such as cytomegalovi-rus (CMV), Epstein-Barr virus, HTLV-I, and HTLV-II; and, possibly, reduction of immune modulation.
Whether these advantages justify leukocyte reduction for all blood products remains controversial; some physicians argue that the benefi ts do not justify the associated increased costs.90 Managing patients who continue to have febrile reac-tions after receiving leukocyte-depleted products is a clinical problem for which there are no clear solutions. In addition to premedication with antipyretics and steroids, use of HLA-matched components for patients who are known to have HLA antibodies may be helpful. Occasionally, use of washed products is benefi cial, although 10 to 20% of the cells are lost in the washing process.
transfusion-related acute lung injury
Transfusion-related acute lung injury (TRALI) is a clinical syndrome that presents as respiratory distress and hypox-emia with bilateral pulmonary infi ltrates within 6 hours of transfusion.91 The clinical and radiographic picture is that of normal-pressure acute respiratory distress syndrome (ARDS); however, fever, hypotension, or hypertension may occur. The differential diagnosis is suffi ciently broad to

5 hema x transfusion medicine — 14
01/11
make the possible causal role of transfusion often go unnoticed. There is no diagnostic test for TRALI. Current evidence suggests that TRALI is associated with the interaction of antibodies (HLA class 1 or class 2 antibodies, monocyte antibodies, or granulocyte antibodies), with the corresponding antigens on leukocytes92; a recent study found such associations in 14 of 16 TRALI patients.93 These interac-tions cause endothelial injury, alveolar exudation, and the associated clinical fi ndings of ARDS.
A second form of TRALI that involves two clinical events has been proposed.94 In this two-event model, which is based on clinical fi ndings and rat lung studies, the fi rst step is the priming of neutrophils by mediators that arise in certain clinical settings (e.g., recent surgery, massive transfusion, cytokine therapy, or infection).95 The primed neutrophils adhere to pulmonary endothelium and are activated by a second event, such as exposure to biologically active lipids from blood products.
Diagnosing TRALI depends on excluding cardiac and other causes of ARDS. Demonstration of antileukocyte anti-bodies helps confi rm the diagnosis, but their absence does not exclude TRALI in the appropriate clinical setting. Early diagnosis is important because most patients will improve within 24 hours after supportive treatment is initiated. Unfortunately, a minority of patients develop TRALI in association with severe pulmonary edema and the fi lling of the trachea with fl uid, for which no effective therapy exists. Mortality approaches 10%. Because multiparous women are more likely to have developed HLA antibodies, most blood collectors have policies to prepare FFP from male donors and plateletpheresis concentrates from men or nulliparous women. National hemovigilance statistics suggest that this strategy has reduced the frequency of TRALI.
transfusion-associated circulatory overload
Transfusion, particularly in patients with compromised cardiovascular function, may result in hydrostatic pulmo-nary edema and is commonly referred to as transfusion-associated circulatory overload (TACO).96 The clinical and radiologic manifestations of TACO and TRALI are similar. Although echocardiography and B-type natriuretic peptide measurements have been proposed to aid in the differential diagnosis, invasive techniques such as right heart catheter-ization and the sampling of alveolar fl uid protein are some-times necessary.97 Differentiation is particularly diffi cult in critically ill patients with multiple comorbidities. The cause of edema may only be determined post hoc based on the clinical course and response to therapy. Unlike TRALI, TACO in critically ill patients is not associated with decreased long-term survival.98
allergic transfusion reactions
Allergic transfusion reactions are more common than febrile nonhemolytic transfusion reactions, occurring in 3 to 4% of transfusions. Allergic transfusion reactions usually present as pruritus and urticaria. A small percentage of patients have anaphylactoid symptoms, including wheez-ing, bronchospasm, and, occasionally, true anaphylaxis.99 These reactions have been attributed to an immune response to plasma proteins. However, one study suggested that they may instead be provoked by increased levels of RANTES
(regulated on activation, normal T cell expressed and secreted), an infl ammatory chemokine that is stored in plate-let alpha granules and that accumulates during storage.100 This is an intriguing hypothesis because RANTES is known to affect eosinophil and basophil function.
In most cases, symptoms of allergic reactions are local and do not require discontinuance of the transfusion if they are controlled with antihistamines. There is, however, no means as yet to identify the rare patient who will experience anaphylaxis. Certain protein defi ciency states, such as haptoglobin and IgA defi ciency, are associated with an increased likelihood of anaphylaxis, especially if the patient has a preformed antibody; however, many patients who are IgA defi cient never have any diffi culty.101
For most patients with urticaria, which seldom progresses to anaphylaxis, management is symptomatic. However, patients known to be IgA defi cient should receive cells that have been washed thoroughly to remove plasma. When plasma products are required, they should be administered in a facility equipped to manage anaphylactic reactions. Using IgA-defi cient plasma can minimize the risk, but such plasma is diffi cult to obtain and may require drawing from a rare donor pool, testing family members, or both.
atypical transfusion reactions
Occasionally, patients have reactions that do not fi t the categories already defi ned but clearly seem related to blood transfusion. These reactions have mainly consisted of severe hypotension after platelet infusions. No allergic features are present. The reactions are associated with blood-product infusions through bedside leukocyte reduction fi lters and often occur in patients who are receiving angiotensin-converting enzyme (ACE) inhibitors. Such reactions may be caused by excessive accumulation of des-Arg9-bradykinin. This metabolite of bradykinin is known to be vasoactive and to be metabolized by ACE.102 Clinical observations suggest that atypical hypotensive reactions are more likely to occur in patients receiving ACE inhibitors during plasma exchange using albumin replacement solutions, hemodialysis, low-density lipoprotein (LDL) apheresis, IgG-affi nity column apheresis, and desensitization immunotherapy. These fi ndings have led to the suggestion that ACE inhibitors be withheld for 24 hours before any of these procedures are initiated. Such reactions are suffi ciently rare that it may be adequate to limit this restriction to patients who have already experienced one of these reactions.103
transfusion-associated graft versus host disease
Transfusion-associated graft versus host disease (TA-GVHD) is a feared consequence of transfusion therapy because mortality approaches 90%. TA-GVHD results from transfusing immunocompetent lymphocytes into a recipient who is unable to reject the allogeneic cells. The transfused lymphocytes engraft and proliferate, attacking host tissue antigens on the target organs.
Diagnosis and Identifying Patients at Risk
The diagnosis of TA-GVHD should be considered in any patient who presents after transfusion with fever, skin rash, and diarrhea and has pancytopenia and abnormal results on liver function testing.104 Signs and symptoms in neonates are

5 hema x transfusion medicine — 15
01/11
similar to those in adults, but fever and rash develop later: in adults, fever occurs after a median of 10 days after trans-fusion; in neonates, fever occurs after 28 days, with rash appearing 1 to 2 days later. The diagnosis is generally made on the basis of clinical fi ndings and can be confi rmed with a biopsy that demonstrates cytogenetic evidence of donor lymphoid engraftment. TA-GVHD is best prevented by identifying potentially susceptible recipients. Patients who are at signifi cant risk for TA-GVHD include premature infants receiving large doses of fresh allogeneic lympho-cytes, patients with congenital defects in cellular immunity or immunity resulting from illness or chemotherapy, and patients who are unable to reject infused cells because of shared antigens with the allogeneic lymphocytes. Patients undergoing autologous or allogeneic bone marrow trans-plantation are particularly at risk. Many case reports docu-ment the association of Hodgkin disease with TA-GVHD, which occurs presumably as a result of acquired defects in T cell immunity. The intensive chemotherapy that is used to treat leukemia, high-grade lymphomas, and solid tumors may also set the stage for TA-GVHD. However, no cases have been identifi ed in AIDS patients. One hypothesis explaining this surprising fi nding is that the HIV-mediated injury to CD4+ T cells blocks the development of TA-GVHD.105
Patients who are at risk for TA-GVHD because of receiv-ing transfusions from a homozygous donor of a shared haplotype are the hardest to identify a priori. Donor lym-phocytes are not rejected by the recipient but do respond to the nonshared recipient haplotype. This mechanism proba-bly accounts for the majority of cases of TA-GVHD. The chances of receiving haplotype-homozygous blood from an unrelated donor vary with different populations. In Japan, the risk for adults may be as high as 1 in 874; it is estimated to be 1 in 102 in neonates because of the use of fresh whole blood from family members.106 In the United States, the risk for the white population is thought to be about 1 in 7,147. The risk increases if fi rst-degree relatives are donors.
Preventive Measures
Once patients at risk are identifi ed [see Table 6], pretreat-ment of all cellular transfused products with gamma radia-tion is indicated. No cases associated with FFP have been
documented. On the basis of in vitro studies, the current recommended dose is 2,500 cGy, which does not affect red cell function or platelet survival if administered imme-diately before transfusion.107 However, irradiated red cells stored for 42 days show signifi cant increases in plasma potassium and hemoglobin and a small but signifi cant decrease in cell survival. Consequently, it is recommended that red cells be stored for no longer than 28 days after irradiation; however, most institutions prefer to irradiate immediately before product release. Platelets normally survive in storage for 5 days after being irradiated; they can be irradiated at regional centers before distribution.
In addition to irradiation, leukocyte reduction may pro-vide some protection against TA-GVHD, which is related to the dose of lymphocytes. However, fi ltration alone is not preventive and must never be used as a substitute for gamma irradiation. Because of the risk associated with a one-way HLA match, blood-bank standards require that family members’ blood and the blood of directed donors be irradiated.
Treatment
Treatment of TA-GVHD remains ineffective. Prevention by providing irradiated blood components to all recipients—the current practice in Japan—may become the most practical solution to this complication.106
bacterial and protozoan infections
Platelets are associated with the majority of cases of transfusion-related sepsis because the platelets are stored at room temperature, which is conducive to bacterial prolifera-tion.108 All platelet concentrates are currently screened for bacterial contamination; however, even the best available assays fail to detect low-level contamination. Controlling this problem requires improved disinfection of the donor’s phlebotomy site, better detection of subclinical infection, and the development of methods for storage at lower temperatures or postcollection component sterilization. If sepsis is suspected in patients who have been given red cells, the possibility of Yersinia enterocolitica infection should be considered.109 This organism can grow in the cold, iron-rich environment provided by stored red cells. When such infections occur, the blood is almost always at least 2 weeks old; this period corresponds to the time needed for the usually small initial inoculum to reach clinically signifi cant amounts. Malaria infections have been almost completely eliminated by predonation screening. T. cruzi can cause a chronic parasitic infection; concern for blood-borne trans-mission has increased to the point that pretransfusion test-ing for it has been introduced. Spirochetes do not tolerate refrigerated temperatures for longer than 80 hours, and syphilis is no longer considered a clinically signifi cant source of blood-borne infection.110
cytomegalovirus infection
CMV is a common blood-borne infection of no clinical consequence to healthy, immunocompetent recipients, but it can be a lethal problem for patients with either acquired or congenital immunodefi ciency [see Table 7]. Judged on the basis of screening for antibody to CMV, more than 40% of healthy donors may have the potential to transmit CMV.111
Table 6 Patients for Whom Irradiated Blood Products Are Recommended
Fetuses and neonates Patients with congenital immunodefi ciencyAllogeneic and autologous bone marrow transplantation
patientsRecipients of some solid-organ transplants*Patients with hematologic malignancies† Patients with nonhematologic malignancies, especially if
undergoing intensive chemotherapy‡ Recipients who may share haplotypes with donor§
*Heart, liver, and lung recipients should receive irradiated products, whereas recipients of renal allografts do not require irradiated blood.†Patients with low-grade lymphomas and leukemias in remission may not require irradiated products. Applying restriction to all lymphoma and leukemia patients avoids mistakes.‡No consensus, but patients undergoing intensive chemotherapy are at risk.§Donors in this group include directed donors and fi rst- and second-degree relatives.

5 hema x transfusion medicine — 16
01/11
Although few institutions test directly for the virus, two approaches are used to prevent CMV transmission. The fi rst is to use CMV antibody–negative components. The second, more practical approach is to use leukocyte-reduced compo-nents because CMV is highly cell associated. On the basis of a prospective, randomized study of more than 500 trans-plant patients, products that have undergone leukocyte reduction to the current standard of fewer than 0.5 × 106 leukocytes per milliliter are considered to be as effective as seronegative products in preventing CMV infection.112 It is unclear which approach provides the best protection against transfusion-associated CMV infection; however, both prepa-rations should be considered CMV safe rather than CMV negative.113 Direct comparisons between seroconversion rates after transfusion of prestorage leukocyte-depleted components and CMV-negative products are required to settle this issue.
immune modulation as a result of transfusion
Evidence that transfusions result in modulation of host immunity has come from studies of transplantation, cancer recurrence, and posttransfusion infection rates.114 The effect was fi rst observed in cadaver-kidney transplantation; patient survival was shown to increase with increased trans-fusions. Although this benefi t became less important with the introduction of cyclosporine, Opelz and colleagues found increased cadaver-graft survival in transfused recipients whose immunosuppression regimen included cyclosporine.115
The hypothesis that immune modulation is related to infused white cells has been supported by studies in animal models and by clinical observations of tumor recurrence and posttransfusion infection rates. This effect of allogeneic blood is abrogated by prestorage leukocyte reduction but not by poststorage reduction. Randomized clinical studies of posttransfusion infection and cancer recurrence have pro-duced confl icting results.116 The confl icting data concerning the magnitude and clinical relevance of transfusion-induced immunomodulation have yet to be resolved. If leukocyte reduction is shown to reduce posttransfusion infections and cancer recurrence, the argument for universal leukocyte reduction of cellular blood products, which is already strong, would become irrefutable. Until this matter is settled, the possible immunomodulatory effect of blood transfusion is another reason to limit the use of allogeneic blood transfusion.
Apheresis
Apheresis therapy is the converse of transfusion therapy; it entails treating disease by removing plasma, specifi c antibodies, or cells. Apheresis has been applied to a broad spectrum of diseases [see Table 8].117 Therapeutic apheresis has real risks and may provide little benefi t. It is usually an acute intervention that is transiently effective, unless the underlying problem is being treated effectively. Conse-quently, establishing criteria for both starting and stopping such treatment is critical. Evidence-based recommendations for apheresis that are approved by the American Society for Apheresis have been summarized.117
red cell exchange
Red cell exchange has been used primarily to treat or prevent complications of sickle cell disease. Although the molecular defect of sickle hemoglobin is simple, the pathophysiology of the vaso-occlusive crises is complex, involving hemoglobin polymerization, change in cell shape, adhesion to endothelial cells, scavenging of nitric oxide (NO), and release of infl ammatory cytokines. Clinical mani-festations vary from patient to patient. Exchange transfusion aims to improve tissue oxygenation and prevent microvas-cular sickling by diluting the patient’s abnormal red cells, which results in improved whole blood viscosity and rheol-ogy; red cell exchange may also be used to correct anemia and reduce chronic hemolysis. No clinical data support a single optimal level of hemoglobin A (HbA); however, as few as 30% of transfused cells markedly decrease blood viscosity; at mixtures of 50% or greater, resistance to membrane fi lterability approaches normal.118 Raising the level of HbA to between 60 and 70% while lowering the level of hemoglobin S (HbS) to 30% is generally effi cacious.
Red cell exchange has been used to treat stroke, acute chest crises, and priapism. In patients with these disorders, the indications for red cell exchange—compared with simple transfusion—are poorly defi ned. For example, in a randomized study of patients with sickle cell disease who are undergoing surgery, a conservative simple transfusion regimen was as effective as an aggressive exchange regimen with respect to perioperative non–transfusion-related complications. The patients in the aggressive regimen group received twice as many units of blood, had a proportionally increased red blood cell alloimmunization rate, and had more hemolytic transfusion reactions.119
Exchange transfusion has also been used for prophylaxis during pregnancy and before surgery. The only randomized trial of transfusion during pregnancy has shown that prophylactic transfusion suffi cient to reduce the incidence of painful crises did not reduce maternal morbidity from other causes, nor did it reduce perinatal mortality.120
Long-term transfusion prophylaxis is unquestionably indicated for children at high risk for stroke, as determined by transcranial Doppler ultrasonography. A randomized controlled study demonstrated a risk reduction of 90% in the patients who were maintained at levels of 30% or less HbS by simple or exchange transfusion.121 In this group of children, transfusion therapy should begin before the fi rst event and should be continued indefi nitely. Red cell exchange leads to less iron accumulation than simple
Table 7 Patients for Whom Cytomegalovirus-Negative Blood Products Are Recommended
Premature infants and neonates, especially if weight is less than 1,200 g
Seronegative pregnant women, as a means of preventing primary intrauterine infections
Recipients of solid-organ transplants, especially when the recipient and the organ donor are both CMV negative
Patients with severe combined immunodefi ciency*
CMV = cytomegalovirus.*Most AIDS patients are CMV positive and do not require CMV-safe components.
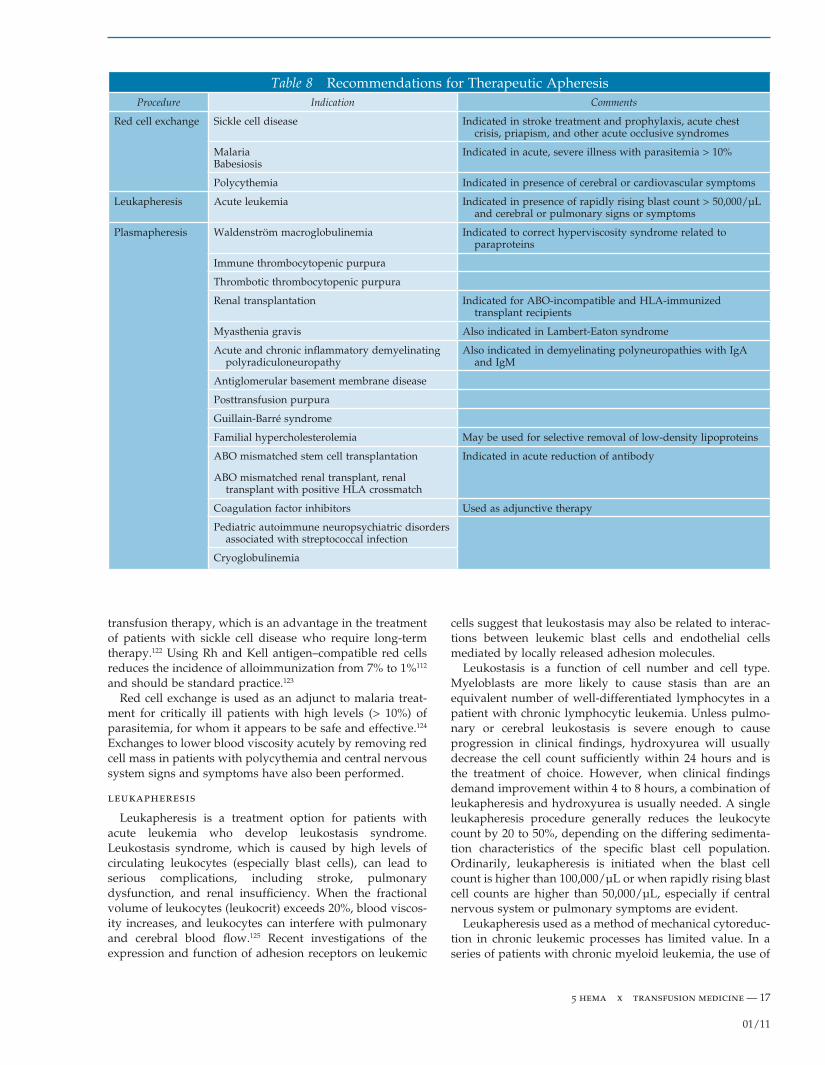
5 hema x transfusion medicine — 17
01/11
transfusion therapy, which is an advantage in the treatment of patients with sickle cell disease who require long-term therapy.122 Using Rh and Kell antigen–compatible red cells reduces the incidence of alloimmunization from 7% to 1%112 and should be standard practice.123
Red cell exchange is used as an adjunct to malaria treat-ment for critically ill patients with high levels (> 10%) of parasitemia, for whom it appears to be safe and effective.124 Exchanges to lower blood viscosity acutely by removing red cell mass in patients with polycythemia and central nervous system signs and symptoms have also been performed.
leukapheresis
Leukapheresis is a treatment option for patients with acute leukemia who develop leukostasis syndrome. Leukostasis syndrome, which is caused by high levels of circulating leukocytes (especially blast cells), can lead to serious complications, including stroke, pulmonary dysfunction, and renal insuffi ciency. When the fractional volume of leukocytes (leukocrit) exceeds 20%, blood viscos-ity increases, and leukocytes can interfere with pulmonary and cerebral blood fl ow.125 Recent investigations of the expression and function of adhesion receptors on leukemic
cells suggest that leukostasis may also be related to interac-tions between leukemic blast cells and endothelial cells mediated by locally released adhesion molecules.
Leukostasis is a function of cell number and cell type. Myeloblasts are more likely to cause stasis than are an equivalent number of well-differentiated lymphocytes in a patient with chronic lymphocytic leukemia. Unless pulmo-nary or cerebral leukostasis is severe enough to cause progression in clinical fi ndings, hydroxyurea will usually decrease the cell count suffi ciently within 24 hours and is the treatment of choice. However, when clinical fi ndings demand improvement within 4 to 8 hours, a combination of leukapheresis and hydroxyurea is usually needed. A single leukapheresis procedure generally reduces the leukocyte count by 20 to 50%, depending on the differing sedimenta-tion characteristics of the specifi c blast cell population. Ordinarily, leukapheresis is initiated when the blast cell count is higher than 100,000/µL or when rapidly rising blast cell counts are higher than 50,000/µL, especially if central nervous system or pulmonary symptoms are evident.
Leukapheresis used as a method of mechanical cytoreduc-tion in chronic leukemic processes has limited value. In a series of patients with chronic myeloid leukemia, the use of
Table 8 Recommendations for Therapeutic ApheresisProcedure Indication Comments
Red cell exchange Sickle cell disease Indicated in stroke treatment and prophylaxis, acute chest crisis, priapism, and other acute occlusive syndromes
MalariaBabesiosis
Indicated in acute, severe illness with parasitemia > 10%
Polycythemia Indicated in presence of cerebral or cardiovascular symptoms
Leukapheresis Acute leukemia Indicated in presence of rapidly rising blast count > 50,000/µL and cerebral or pulmonary signs or symptoms
Plasmapheresis Waldenström macroglobulinemia Indicated to correct hyperviscosity syndrome related to paraproteins
Immune thrombocytopenic purpura
Thrombotic thrombocytopenic purpura
Renal transplantation Indicated for ABO-incompatible and HLA-immunized transplant recipients
Myasthenia gravis Also indicated in Lambert-Eaton syndrome
Acute and chronic infl ammatory demyelinating polyradiculoneuropathy
Also indicated in demyelinating polyneuropathies with IgA and IgM
Antiglomerular basement membrane disease
Posttransfusion purpura
Guillain-Barré syndrome
Familial hypercholesterolemia May be used for selective removal of low-density lipoproteins
ABO mismatched stem cell transplantation Indicated in acute reduction of antibody
ABO mismatched renal transplant, renal transplant with positive HLA crossmatch
Coagulation factor inhibitors Used as adjunctive therapy
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection
Cryoglobulinemia

5 hema x transfusion medicine — 18
01/11
repeated leukapheresis procedures adequately reduced cell count; however, the median patient survival rate was not signifi cantly different from that achieved with conventional chemotherapy.126
plateletpheresis
Therapeutic plateletpheresis is generally reserved for pa-tients with myeloproliferative disorders and hemorrhage or thrombosis associated with increased numbers of circulating abnormal platelets. Many centers consider using platelet-pheresis when the patient’s peripheral platelet count is greater than 106/µL, although no consistent relation between the level of platelet elevation and the occurrence of symp-toms has been found, and no generally accepted assay of platelet dysfunction predicts which patients are at risk.127,128 A single procedure can lower the platelet count by 30 to 50%. Attempts to maintain thrombocythemic patients at normal platelet counts by cytapheresis alone have not been successful; aspirin and cytoreductive chemotherapy should be instituted concurrently. Most patients with thrombocyto-sis, including patients with myeloproliferative disorders, do not develop symptoms; prophylactic plateletpheresis in an asymptomatic patient is unwarranted regardless of the platelet count.128
plasmapheresis (plasma exchange)
Indications for Plasma Exchange
Neurologic diseases Neurologic diseases whose patho-genesis may be antibody mediated are now the most common indications for plasma exchange. Myasthenia gravis occurs when antibodies to acetylcholine receptors cause abnormal neuromuscular transmission. Reductions in these antibody titers from plasma exchange are associated with clinical improvement. A randomized trial compared the use of plasma exchange with the use of IVIg therapy in the treatment of myasthenia gravis. The investigators noted a trend toward better results with plasma exchange.129
Similar fi ndings were reported from much larger studies of Guillain-Barré syndrome, which is thought to be caused by antibodies to myelin. Two large series comparing plasma exchange with current best therapy showed faster improve-ment with the addition of plasma exchange. Randomized comparisons of plasma exchange and IVIg in the treatment of Guillain-Barré syndrome have shown these approaches to be equivalent; no additional benefi t from using both therapies was shown.130,131
Chronic infl ammatory demyelinating polyneuropathy (CIDP) is an autoimmune disorder that causes proximal and distal weakness; CIDP has a progressive or relapsing course and is sometimes associated with monoclonal gammopa-thies. CIDP responds to plasma exchange, except in patients with distal weakness and associated IgM monoclonal gammopathies132,133; such patients respond poorly to all modalities of therapy. IVIg therapy and plasma exchange have been shown to be comparably effective in CIDP.134
The use of plasma exchange in multiple sclerosis remains controversial. Meta-analysis of six controlled trials of plasma exchange provided some evidence of benefi t, but the authors concluded that the subgroups of patients likely to benefi t need further defi nition.135 A randomized study of
plasma exchange in patients with acute infl ammatory demy-elinating central nervous system disease showed a signifi -cant benefi t from the therapy. However, patients continued to experience relapse.136
Hematologic diseases The hematologic diseases that require plasma apheresis are those associated with obstruc-tion of vascular fl ow by proteins as a result of increased viscosity or cryoprecipitation; antibody-mediated diseases that lead to destruction of the formed elements of the blood; and thrombotic microangiopathies.
In patients with TTP, plasma exchange with FFP replace-ment has been estimated to improve survival rates from 10% to more than 75%; TTP is the only hematologic condition in which a specifi c replacement solution seems to make a difference, possibly because reduction of an antibody is accompanied by replacement of a missing plasma factor.137 Comprehensive reviews of the clinical and laboratory evaluation and treatment of patients with suspected TTP, including management with plasma exchange therapy, have been published.138 Treatment usually involves daily single-volume plasma exchange; the frequency and duration of treatment are guided by clinical response, an increase in platelet count (i.e., to 100,000/L or more), and evidence of decline in hemolysis (as measured by normalization of serum l-lactate dehydrogenase [LDH] and decline in the number of schistocytes on the peripheral blood smear). Despite promising initial reports, the use of cryoprecipitate-poor plasma may not be more effective than the use of stan-dard FFP as a specifi c replacement fl uid for plasma exchange in patients with TTP.138,139
The effectiveness of plasma exchange in patients with TTP may derive from the removal of antibody to von Willebrand factor, replacement of the von Willebrand factor–cleaving zinc metalloprotease (ADAMTS13 [a disintegrin and metal-loproteinase with a thrombospondin type 1 motif, member 13]), or both.137,139 However, patients with clinical features of TTP and moderate ADAMTS13 defi ciency—or even normal activity—may respond to plasma exchange. The striking response of some of these cases to the administration of rituximab lends further support to the fi nding that some cases of TTP and hemolytic-uremic syndrome (HUS) share an autoimmune mechanism. The results of plasma exchange in hematopoietic stem cell transplant recipients with clinical features of TTP, commonly referred to as transplant-associated thrombotic microangiopathy (TAP), have been disappointing at best.64 These syndromes are often associate d with high levels of calcineurin inhibitors and are best managed by discontinuing the offending drug.138
In patients with Waldenström disease, plasma exchange has produced clinical improvement; however, it does not affect the disease process. The benefi t of plasma change is the result of rapid reduction of paraprotein concentrations and normalization or a signifi cant decrease in serum viscos-ity in patients with hyperviscosity syndrome.140 The concen-tration of paraprotein infl uences plasma protein viscosity, as does its heavy-chain class. IgM is the largest plasma protein and is nearly 100% intravascular; it is most likely to cause hyperviscosity. IgA and IgG3 are more likely to aggregate and are associated with hyperviscosity more often than other IgG subclasses. As in leukostasis, the choice between

5 hema x transfusion medicine — 19
01/11
plasma exchange and chemotherapy is guided mainly by the clinical symptoms and their rate of progression. Plasma exchange can lower viscosity within hours, whereas most chemotherapy requires days.141
Despite the role that antibody and immune complexes play in hematologic cytopenia, there are no well-controlled studies supporting the use of plasma exchange. The available case reports usually describe the role of plasma exchange as being that of backup after failure of more established therapies. FFP or cryopoor plasma is used in replacement therapy for thrombotic microangiopathies. Case reports suggest that patients with severe preeclampsia, HELLP syndrome, or both may benefi t from plasma exchange with FFP replacement if they fail to improve after delivery.142
Antibody-mediated renal, muscular, and cutaneous diseases Despite promising reports from case studies, controlled trials of patients with pemphigus vulgaris,143 polymyositis, dermatomyositis,144 and Goodpasture syn-drome145 have raised doubts concerning the value of plasma exchange. However, plasma exchange does appear to be valuable in stopping pulmonary hemorrhage in Goodpasture syndrome.
Immune complex diseases The only indication for plas-ma exchange in rheumatoid arthritis and systemic lupus erythematosus is severe vasculitis that does not respond to other therapies. Plasma exchange is usually requested as an intervention of last resort.
Metabolic diseases Plasma exchange and selective removal of LDLs have both been used to treat familial hypercholesterolemia. Selective removal of LDLs can be accomplished by immunoadsorption, heparin precipitation, or dextran sulfate cellulose absorption, whereas plasma exchange causes signifi cant reduction of both LDLs and high-density lipoproteins (HDLs). Other rare genetic storage diseases, such as Refsum disease (characterized by the accumulation of phytanic acid), benefi t from mobilization of the toxic metabolite by plasmapheresis.
Complications of Plasma Exchange
The complications associated with plasma exchange are best divided into problems related to apheresis machines and problems related to venous access, type of replacement fl uids, and anticoagulant.146 Apheresis machines accomplish cell and plasma separation by either centrifugation or mem-brane fi ltration. All systems monitor air and access pressure, allowing air emboli to be eliminated and access problems to be promptly recognized. Excess transmembrane pressure may cause red cell hemolysis, which leads to increased hemoglobin in the separated plasma. The majority of com-plications associated with plasma exchange result from the replacement fl uid and anticoagulant used. Plasma removed by exchange is commonly replaced with 5% albumin, which carries no risk of infection and does not increase the citrate return but does dilute coagulation factors, causing mild coagulopathy for 24 to 48 hours. On an every-other-day treatment schedule, coagulation abnormalities are usually
not clinically signifi cant, but they may become signifi cant if the patient is on a daily treatment schedule. Using FFP prevents dilutional coagulopathy but increases the risks of blood-borne infection and allergic reactions. Peripheral venous access is often inadequate to maintain the required fl ow rates of 45 to 80 mL/min, necessitating central venous access with a large, double-lumen catheter; life-threatening or fatal complications from central catheter placement have been reported.147 Catheter malfunction should always be considered when a patient shows clinical evidence of hypo-volemia, shock, or both while undergoing plasma exchange. The majority of complications, however, are side effects of the citrate anticoagulant.148 These can include paresthesias, abdominal cramps, tetany, and, in rare instances, cardiac arrhythmias or seizures. Citrate toxicity is usually managed easily by slowing the return rate and providing extra calcium, either orally or sometimes intravenously. Patients with renal failure who receive large amounts of citrate may develop a profound metabolic alkalosis.149
Future Prospects for Transfusion Therapy
The evolution of transfusion practice has been a steady progression from whole blood to components to fractionate d and recombinant products designed for specifi c therapies. The search for a practical replacement for red cells that would allow stable storage, provide adequate oxygen deliv-ery, and be free of signifi cant toxins has been long and fi lled with substantial obstacles. No red cell substitutes are close to licensure.150 In the case of coagulation components and other plasma proteins, recombinant products are beginning to provide highly specifi c treatment of clinical problems that are poorly managed by current therapy. Bioengineering holds the promise of improving the effectiveness of current recombinant products. Methods to remove or mask red cell antigens provide the promise of a so-called universal red cell.151 Pathogen reduction technologies seem likely to rende r cellular components as free of infectious transmission as their plasma counterparts.152
A major change in transfusion practice may evolve from the availability of cytokines that can modify endogenous production. Erythropoietin has changed the treatment of anemia associated with chronic renal disease; as a result, many dialysis patients no longer require transfusions. Eryth-ropoietin can also facilitate patients’ self-banking their blood for anticipated surgical needs. In some cases, erythropoietin use is accepted by Jehovah’s Witness patients, thereby allowing such patients to undergo surgical procedures that would otherwise not be possible. The availability of myeloid growth factors has contributed substantially to the develop-ment of methods for collection, and it is possible that mobi-lizing leukocytes with growth factors can increase the effectiveness of granulocyte transfusions. Thrombopoietin has proved effective in treating some patients with ITP, abrogating the need for IVIg and emergency platelet infu-sions, and may in time be used to enhance platelet apheresis collections.153 In very early studies, recombinant cytokines have been combined with hematopoietic progenitor cells to grow blood cells in culture; however, the prospects of making available millions of units of cultured red cells and platelets remain more a hope than a promise.154

5 hema x transfusion medicine — 20
01/11
Even in an era of accelerating change, certain aspects of transfusion medicine will remain constant. Despite claims of “bloodless surgery” and total blood management, blood transfusion remains, when indicated, a lifesaving and irre-placeable therapy. The blood donor remains a linchpin that cannot be replaced by recombinant methodology. Transfu-sion practice has improved in safety, but there will always be residual risks. Each transfusion will require careful assessment of whether the risks to the recipient outweigh the benefi ts.
The author has no commercial relationships with manufacturers of products or providers of services discussed in this chapter.
Recombinant activated factor VII (rVIIa) has not been approved by the FDA for uses described in this chapter.
References
1. Klein HG. Transfusion medicine: the evolution of a new discipline. JAMA 1987;258:2108–9.
2. Dodd RY. Current safety of the blood supply in the United States. Int J Hematol 2004;80:301–5.
3. Glynn SA, Schreiber GB, Busch MP, et al. Demographic characteristics, unreported risk behaviors, and the preva-lence and incidence of viral infections: a comparison of apheresis and whole-blood donors. The Retrovirus Epidemiology Donor Study. Transfusion 1998;38:350–8.
4. Domen RE. Adverse reactions associated with autologous blood transfusion: evaluation and incidence at a large academic hospital. Transfusion 1998;38:296–300.
5. Brecher ME, Goodnough LT. The rise and fall of preopera-tive autologous blood donation [editorial]. Transfusion 2002;42:1618–22.
6. Segal JB, Blasco-Colmenares E, Norris EJ, et al. Preopera-tive acute normovolemic hemodilution: a meta-analysis. Transfusion 2004;44:632–44.
7. Goodnough LT, Monk TG, Sicard G, et al. Intraoperative salvage in patients undergoing elective abdominal aortic aneurysm repair: an analysis of cost and benefi t. J Vasc Surg 1996;24:213–8.
8. Goodnough LT, Shander A. Blood management. Arch Pathol Lab Med 2007;131:695–701.
9. Starkey JM, MacPherson JL, Bolgiano DC, et al. Markers for transfusion-transmitted disease in different groups of blood donors. JAMA 1989;262:3452–4.
10. Strauss RG, Burmeister LF, Johnson K, et al. Randomized trial assessing the feasibility and safety of biologic parents as RBC donors for their preterm infants. Transfusion 2000;40:450–6.
11. Kanter MH, Hodge SE. Risk of hemolytic disease of the newborn as a result of directed donations from relatives. Transfusion 1989;29:620–5.
12. Zou S, Dorsey KA, Notari EP, et al. Prevalence, incidence, and residual risk of human immunodefi ciency virus and hepatitis C virus infections among United States blood donors since the introduction of nucleic acid testing. Transfusion 2010;50:1495–504.
13. Kleinman SH, Lelie N, Busch MP. Infectivity of human immunodefi ciency virus-1, hepatitis C virus, and hepatitis B virus and risk of transmission by transfusion. Transfusion 2009;49:2454–89.
14. Hennig H, Puchta I, Luhm J, et al. Frequency and load of hepatitis B virus DNA in fi rst-time blood donors with antibodies to hepatitis B core antigen. Blood 2002;100:2637–41.
15. Bialek SR, Terrault NA. The changing epidemiology and natural history of hepatitis C virus infection. Clin Liver Dis 2006;10:697–715.
16. Alter MJ, Kuhnert WL, Finelli L. Guidelines for laboratory testing and result reporting of antibody to hepatitis C virus. Centers for Disease Control and Prevention. MMWR Recomm Rep 2003;52(RR-3):1–13.
17. Lemaire JM, Courouce AM, Defer C, et al. HCV RNA in blood donors with isolated reactivities by third-generation RIBA. Transfusion 2000;40:867–70.
18. Ganem D, Prince AM. Hepatitis B virus infection: natural history and clinical consequences. N Engl J Med 2004;350:1118–29.
19. Biswas R, Tabor E, Hsia CC, et al. Comparative sensitivity of HBV NATs and HBsAg assays for detection of acute HBV infection. Transfusion 2003;43:788–98.
20. Bower WA, Nainan OV, Han X, et al. Duration of viremia in hepatitis A virus infection. J Infect Dis 2000;182:12–7.
21. Donegan E, Lee H, Operskalski EA, et al. Transfusion transmission of retroviruses: human T-lymphotropic virus types I and II compared with human immunodefi ciency virus type 1. Transfusion 1994;34:478–83.
22. Sayre KR, Dodd RY, Tegtmeier G, et al. False-positive human immunodefi ciency virus type 1 Western blot tests in noninfected blood donors. Transfusion 1996;36:45–52.
23. Murphy EL, Glynn SA, Fridey J, et al. Increased preva-lence of infectious diseases and other adverse outcomes in human T lymphotropic virus types I– and II–infected blood donors. Retrovirus Epidemiology Donor Study (REDS) Study Group. J Infect Dis 1997;176:1468–75.
24. Orland JR, Engstrom J, Fridey J, et al. Prevalence and clinical features of HTLV neurologic disease in the HTLV Outcomes Study. Neurology 2003;61:1588–94.
25. Busch MP, Switzer WM, Murphy EL, et al. Absence of evi-dence of infection with divergent primate T-lymphotropic viruses in United States blood donors who have seroinde-terminate HTLV test results. Transfusion 2000;40:443–9.
26. Biggerstaff BJ, Petersen LR. Estimated risk of transmission of the West Nile virus through blood transfusion in the United States, 2002. Transfusion 2003;43:1007–17.
27. Busch MP, Caglioti S, Robertson EF, et al. Screening the blood supply for West Nile virus RNA by nucleic acid amplifi cation testing. N Engl J Med 2005;353:460–7.
28. Leiby DA, Herron RM Jr, Garratty G, Herwaldt BL. Trypanosoma cruzi parasitemia in US blood donors with serologic evidence of infection. J Infect Dis 2008;198:609–13.
29. Orton SL, Stramer SL, Dodd RY. Self-reported symptoms associated with West Nile virus infection in RNA-positive blood donors. Transfusion 2006;46:272–7.
30. Chohan G, Llewelyn C, Mackenzie J, et al. Variant Creutzfeldt-Jakob disease in a transfusion recipient: coincidence or cause? Transfusion 2010;50:1003–6.
31. Cahill MR, Murphy T, Khan M, et al. Phase I/II safety study of transfusion of prion-fi ltered red cell concentrates in transfusion-dependent patients. Vox Sang 2010;99:174–6.

5 hema x transfusion medicine — 21
01/11
32. Kleinman S, Cameron C, Custer B, et al. Modeling the risk of an emerging pathogen entering the Canadian blood supply. Transfusion 2010 Jun 10. [Epub ahead of print]
33. Stramer SL, Hollinger FB, Katz LM, et al. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009;49 Suppl 2:1S–29S.
34. Yamamoto F, Clausen H, White T, et al. Molecular genetic basis of the histo-blood group ABO system. Nature 1990;345:229–33.
35. Flegel WA, von Zabern I, Wagner FF. Six years’ experience performing RHD genotyping to confi rm D- red blood cell units in Germany for preventing anti-D immunizations. Transfusion 2009;49:465–71.
36. Avent ND. Large-scale blood group genotyping: clinical implications. Br J Haematol 2009;144:3–13.
37. Marosszeky S, McDonald S, Sutherland J, et al. Suitability of preadmission blood samples for pretransfusion testing in elective surgery. Transfusion 1997;37:910–2.
38. Seftel MD, Growe GH, Petraszko T, et al. Universal prestorage leukoreduction in Canada decreases platelet alloimmunization and refractoriness. Blood 2004;103:333–9.
39. Valeri CR, Ragno G, Pivacek LE, et al. An experiment with glycerol-frozen red blood cells stored at –80 degrees C for up to 37 years. Vox Sang 2000;79:168–74.
40. Chambers LA, Herman JH. Considerations in the selection of a platelet component: apheresis versus whole blood–derived. Transfus Med Rev 1999;13:311–22.
41. Lee EJ, Schiffer CA. ABO compatibility can infl uence the results of platelet transfusion: results of a randomized trial. Transfusion 1989;29:384–9.
42. Klein HG, Spahn DR, Carson JL. Red cell transfusion in clinical practice. Lancet 2007;370:415–26.
43. Bracey AW, Radovancevic R, Riggs SA, et al. Lowering the hemoglobin threshold for transfusion in coronary artery bypass procedures: effect on patient outcome. Transfusion 1999;39:1070–7.
44. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion require-ments in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999;340:409–17.
45. Deans KJ, Minneci PC, Klein HG, Natanson C. The rele-vance of practice misalignments to trials in transfusion medicine. Vox Sang 2010;99:16–23.
46. Lankhorst CE, Wish JB. Anemia in renal disease: diagnosis and management. Blood Rev 2010;24:39–47.
47. Testa U. Erythropoietic stimulating agents. Expert Opin Emerg Drugs 2010;15:119–38.
48. Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol 2009;146:585–96.
49. Schiffer CA, Anderson KC, Bennett CL, et al. Platelet trans-fusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2001;19:1519–38.
50. Wandt H, Frank M, Ehninger G, et al. Safety and cost effec-tiveness of a 10 × 10(9)/L trigger for prophylactic platelet transfusions compared with the traditional 20 × 10(9)/L trigger: a prospective comparative trial in 105 patients with acute myeloid leukemia. Blood 1998;91:3601–6.
51. Psaila B, Petrovic A, Page LK, et al. Intracranial hemor-rhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
52. Slichter SJ, Kaufman RM, Assmann SF, et al. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med 2010;362:600–13.
53. Kickler T, Kennedy SD, Braine HG. Alloimmunization to platelet-specifi c antigens on glycoproteins IIb-IIIa and Ib/IX in multiply transfused thrombocytopenic patients. Transfusion 1990;30:622–5.
54. Nambiar A, Duquesnoy RJ, Adams S, et al. HLAMatchmaker-driven analysis of responses to HLA-typed platelet transfusions in alloimmunized thrombocy-topenic patients. Blood 2006;107:1680–7.
55. Friedberg RC, Donnelly SF, Mintz PD. Independent roles for platelet crossmatching and HLA in the selection of platelets for alloimmunized patients. Transfusion 1994;34:215–20.
56. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. The Trial to Reduce Alloimmuniza-tion to Platelets Study Group. N Engl J Med 1997;337:1861–9.
57. Novotny VM, van Doorn DR, Witvliet MD, et al. Occur-rence of allogeneic HLA and non-HLA antibodies after transfusion of prestorage fi ltered platelets and red blood cells: a prospective study. Blood 1995;85:1736–41.
58. The 2005 Nationwide Blood Collection and Utilization Survey Report. 2007. Available at: http://www.hhs.gov/bloodsafety/2005NBCUS.pdf (accessed Oct 13, 2010).
59. O’Shaughnessy DF, Atterbury C, Bolton MP, et al. Guide-lines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol 2004;126:11–28.
60. McVay PA, Toy PT. Lack of increased bleeding after paracentesis and thoracentesis in patients with mild coagulation abnormalities. Transfusion 1991;31:164–71.
61. Abdel-Wahab OI, Healy B, Dzik WH. Effect of fresh-frozen plasma transfusion on prothrombin time and bleeding in patients with mild coagulation abnormalities. Transfusion 2006;46:1279–85.
62. George JN. Clinical practice: thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
63. Phan HH, Wisner DH. Should we increase the ratio of plasma/platelets to red blood cells in massive transfusion: what is the evidence? Vox Sang 2010;98(3 Pt 2):395–402.
64. Nunez TC, Dutton WD, May K, et al. Emergency depart-ment blood transfusion predicts early massive transfusion and early blood component requirement. Transfusion 2010 May 7. [Epub ahead of print]
65. Inaba K, Branco BC, Rhee P, et al. Impact of plasma trans-fusion in trauma patients who do not require massive transfusion. J Am Coll Surg 2010;210:957–65.
66. Astermark J, Donfi eld SM, DiMichele DM, et al. A ran-domized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) Study. Blood 2007;109:546–51.
67. Hedner U, Lee CA. First 20 years with recombinant FVIIa (NovoSeven). Haemophilia 2010 Jun 29. [Epub ahead of print]
68. Grounds RM, Seebach C, Knothe C, et al. Use of recombi-nant activated factor VII (Novoseven) in trauma and

5 hema x transfusion medicine — 22
01/11
surgery: analysis of outcomes reported to an international registry. J Intensive Care Med 2006;21:27–39.
69. Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2005;352:777–85.
70. O’Connell KA, Wood JJ, Wise RP, et al. Thromboembolic adverse events after use of recombinant human coagula-tion factor VIIa. JAMA 2006;295:293–8.
71. Kingdon HS, Lundblad RL. An adventure in biotechnol-ogy: the development of haemophilia A therapeutics: from whole-blood transfusion to recombinant DNA to gene therapy. Biotechnol Appl Biochem 2002;35:141–8.
72. Kasper CK. Concentrate safety and effi cacy. Haemophilia 2002;8:161–5.
73. Bodey GP, Buckley M, Sathe YS, et al. Quantitative rela-tionships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med 1966;64:328–40.
74. Stroncek DF, Yau YY, Oblitas J, et al. Administration of G-CSF plus dexamethasone produces greater granulocyte concentrate yields while causing no more donor toxicity than G-CSF alone. Transfusion 2001;41:1037–44.
75. Stroncek DF, Leonard K, Eiber G, et al. Alloimmunization after granulocyte transfusions. Transfusion 1996;36:1009–15.
76. Knezevic-Maramica I, Kruskall MS. Intravenous immune globulins: an update for clinicians. Transfusion 2003;43:1460–80.
77. Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Ann Intern Med 1994;121:259–62.
78. Dalakas MC, Clark WM. Strokes, thromboembolic events, and IVIg: rare incidents blemish an excellent safety record. Neurology 2003;60:1736–7.
79. Linden JV, Wagner K, Voytovich AE, et al. Transfusion errors in New York State: an analysis of 10 years’ experience. Transfusion 2000;40:1207–13.
80. Garratty G. Severe reactions associated with transfusion of patients with sickle cell disease. Transfusion 1997;37:357–61.
81. Capon SM, Goldfi nger D. Acute hemolytic transfusion reaction, a paradigm of the systemic infl ammatory response: new insights into pathophysiology and treat-ment. Transfusion 1995;35:513–20.
82. Ness PM, Shirey RS, Thoman SK, et al. The differentiation of delayed serologic and delayed hemolytic transfusion reactions: incidence, long-term serologic fi ndings, and clinical signifi cance. Transfusion 1990;30:688–93.
83. Pineda AA, Vamvakas EC, Gorden LD, et al. Trends in the incidence of delayed hemolytic and delayed serologic transfusion reactions. Transfusion 1999;39:1097–103.
84. Sazama K. Transfusion errors: scope of the problem, consequences, and solutions. Curr Hematol Rep 2003;2:518–21.
85. Brittingham TR, Chaplin H Jr. Febrile transfusion reactions caused by sensitivity to donor leukocytes and platelets. J Am Med Assoc 1957;165:819–25.
86. Heddle NM, Klama L, Singer J, et al. The role of the plasma from platelet concentrates in transfusion reactions. N Engl J Med 1994;331:625–8.
87. Hebert PC, Fergusson D, Blajchman MA, et al. Clinical outcomes following institution of the Canadian universal leukoreduction program for red blood cell transfusions. JAMA 2003;289:1941–9.
88. Federowicz I, Barrett BB, Andersen JW, et al. Characteriza-tion of reactions after transfusion of cellular blood components that are white cell reduced before storage. Transfusion 1996;36:21–8.
89. Kelley DL, Mangini J, Lopez-Plaza I, et al. The utility of < or = 3-day-old whole-blood platelets in reducing the incidence of febrile nonhemolytic transfusion reactions. Transfusion 2000;40:439–42.
90. Leukoreduction: the techniques used, their effectiveness, and costs. Report from the Canadian Coordinating Offi ce for Health Technology Assessment (CCOHTA). Int J Technol Assess Health Care 1998;14:586–8.
91. Kleinman S, Caulfi eld T, Chan P, et al. Toward an under-standing of transfusion-related acute lung injury: state-ment of a consensus panel. Transfusion 2004;44:1774–89.
92. Toy P, Popovsky MA, Abraham E, et al. Transfusion-related acute lung injury: defi nition and review. Crit Care Med 2005;33:721–6.
93. Kopko PM, Paglieroni TG, Popovsky MA, et al. TRALI: correlation of antigen-antibody and monocyte activation in donor-recipient pairs. Transfusion 2003;43:177–84.
94. Silliman CC, Boshkov LK, Mehdizadehkashi Z, et al. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood 2003;101:454–62.
95. Silliman CC, Paterson AJ, Dickey WO, et al. The associa-tion of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion 1997;37:719–26.
96. Gajic O, Gropper MA, Hubmayr RD. Pulmonary edema after transfusion: how to differentiate transfusion-associated circulatory overload from transfusion-related acute lung injury. Crit Care Med 2006;34(5 Suppl):S109–13.
97. Li G, Daniels CE, Kojicic M, et al. The accuracy of natriu-retic peptides (brain natriuretic peptide and N-terminal pro-brain natriuretic) in the differentiation between trans-fusion-related acute lung injury and transfusion-related circulatory overload in the critically ill. Transfusion 2009;49:13–20.
98. Li G, Kojicic M, Reriani MK, et al. Long-term survival and quality of life after transfusion-associated pulmonary edema in critically ill medical patients. Chest 2010;137:783–9.
99. Sandler SG, Mallory D, Malamut D, et al. IgA anaphylactic transfusion reactions. Transfus Med Rev 1995;9:1–8.
100. Kluter H, Bubel S, Kirchner H, et al. Febrile and allergic transfusion reactions after the transfusion of white cell-poor platelet preparations. Transfusion 1999;39:1179–84.
101. Gilstad CW. Anaphylactic transfusion reactions. Curr Opin Hematol 2003;10:419–23.
102. Cyr M, Hume HA, Champagne M, et al. Anomaly of the des-Arg9-bradykinin metabolism associated with severe hypotensive reactions during blood transfusions: a preliminary study. Transfusion 1999;39:1084–8.
103. Owen HG, Brecher ME. Atypical reactions associated with use of angiotensin-converting enzyme inhibitors and apheresis. Transfusion 1994;34:891–4.

5 hema x transfusion medicine — 23
01/11
104. Ohto H, Anderson KC. Survey of transfusion-associated graft-versus-host disease in immunocompetent recipients. Transfus Med Rev 1996;10:31–43.
105. Ammann AJ. Hypothesis: absence of graft-versus-host disease in AIDS is a consequence of HIV-1 infection of CD4+ T cells. J Acquir Immune Defi c Syndr 1993;6:1224–7.
106. Klein HG. Transfusion-associated graft-versus-host dis-ease: less fresh blood and more gray (Gy) for an aging population. Transfusion 2006;46:878–80.
107. Moroff G, Luban NL. The irradiation of blood and blood components to prevent graft-versus-host disease: technical issues and guidelines. Transfus Med Rev 1997;11:15–26.
108. Fang CT, Chambers LA, Kennedy J, et al. Detection of bacterial contamination in apheresis platelet products: American Red Cross experience, 2004. Transfusion 2005;45:1845–52.
109. Red blood cell transfusions contaminated with Yersinia enterocolitica—United States, 1991–1996, and initiation of a national study to detect bacteria-associated transfusion reactions. Centers for Disease Control and Prevention. JAMA 1997;278:196–7.
110. Orton SL, Liu H, Dodd RY, et al. Prevalence of circulating Treponema pallidum DNA and RNA in blood donors with confi rmed-positive syphilis tests. Transfusion 2002;42:94–9.
111. Preiksaitis JK, Brennan DC, Fishman J, et al. Canadian Society of Transplantation consensus workshop on cyto-megalovirus management in solid organ transplantation fi nal report. Am J Transplant 2005;5:218–27.
112. Bowden RA, Slichter SJ, Sayers M, et al. A comparison of fi ltered leukocyte-reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfu-sion-associated CMV infection after marrow transplant. Blood 1995;86:3598–603.
113. Nichols WG, Price TH, Gooley T, et al. Transfusion-transmitted cytomegalovirus infection after receipt of leukoreduced blood products. Blood 2003;101:4195–200.
114. Klein HG. The immunomodulatory effects of blood transfusion. Tumori 2001;87(2):S17–9.
115. Opelz G, Vanrenterghem Y, Kirste G, et al. Prospective evaluation of pretransplant blood transfusions in cadaver kidney recipients. Transplantation 1997;63:964–7.
116. Blajchman MA. Transfusion immunomodulation or TRIM: what does it mean clinically? Hematology 2005;10:208–14.
117. Szczepiorkowski ZM, Winters JL, Bandarenko N, et al; Apheresis Applications Committee of the American Soci-ety for Apheresis. Guidelines on the use of therapeutic apheresis in clinical practice—evidence-based approach from the Apheresis Applications Committee of the Ameri-can Society for Apheresis. J Clin Apher 2010;25:83–177.
118. Lessin LS, Kurantsin-Mills J, Klug PP, et al. Determination of rheologically optimal mixtures of AA and SS erythro-cytes for transfusion. Prog Clin Biol Res 1978;20:123–37.
119. Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995;333:206–13.
120. Koshy M, Burd L, Wallace D, et al. Prophylactic red-cell transfusions in pregnant patients with sickle cell disease:
a randomized cooperative study. N Engl J Med 1988;319:1447–52.
121. Adams RJ, McKie VC, Hsu L, et al. Prevention of a fi rst stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasono-graphy. N Engl J Med 1998;339:5–11.
122. Kim HC, Dugan NP, Silber JH, et al. Erythrocytapheresis therapy to reduce iron overload in chronically transfused patients with sickle cell disease. Blood 1994;83:1136–42.
123. Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and fi nal results. Blood 2006;108:847–52.
124. van Genderen PJ, Hesselink DA, Bezemer JM, et al. Effi -cacy and safety of exchange transfusion as an adjunct therapy for severe Plasmodium falciparum malaria in nonimmune travelers: a 10-year single-center experience with a standardized treatment protocol. Transfusion 2010;50:787–94.
125. Lichtman MA, Rowe JM. Hyperleukocytic leukemias: rheological, clinical, and therapeutic considerations. Blood 1982;60:279–83.
126. Hester JP, McCredie KB, Freireich EJ. Response to chronic leukapheresis procedures and survival of chronic myelog-enous leukemia patients. Transfusion 1982;22:305–7.
127. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythe-mia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
128. Kessler CM, Klein HG, Havlik RJ. Uncontrolled thrombo-cytosis in chronic myeloproliferative disorders. Br J Haematol 1982;50:157–67.
129. Gajdos P, Tranchant C, Clair B, et al. Treatment of myas-thenia gravis exacerbation with intravenous immunoglob-ulin: a randomized double-blind clinical trial. Arch Neurol 2005;62:1689–93.
130. Randomised trial of plasma exchange, intravenous immu-noglobulin, and combined treatments in Guillain-Barre syndrome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Lancet 1997;349:225–30.
131. van der Meche FG, Schmitz PI. A randomized trial com-paring intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. N Engl J Med 1992;326:1123–9.
132. Dyck PJ, Low PA, Windebank AJ, et al. Plasma exchange in polyneuropathy associated with monoclonal gammopa-thy of undetermined signifi cance. N Engl J Med 1991;325:1482–6.
133. Hahn AF, Bolton CF, Pillay N, et al. Plasma-exchange therapy in chronic infl ammatory demyelinating polyneu-ropathy: double-blind, sham-controlled, cross-over study. Brain 1996;119:1055–66.
134. Dyck PJ, Litchy WJ, Kratz KM, et al. A plasma exchange versus immune globulin infusion trial in chronic infl am-matory demyelinating polyradiculoneuropathy. Ann Neurol 1994;36:838–45.
135. Vamvakas EC, Pineda AA, Weinshenker BG. Meta-analysis of clinical studies of the effi cacy of plasma exchange in the treatment of chronic progressive multiple sclerosis. J Clin Apher 1995;10:163–70.

5 hema x transfusion medicine — 24
01/11
136. Weinshenker BG, O’Brien PC, Petterson TM, et al. A ran-domized trial of plasma exchange in acute central nervous system infl ammatory demyelinating disease. Ann Neurol 1999;46:878–86.
137. Tsai HM, Raoufi M, Zhou W, et al. ADAMTS13-binding IgG are present in patients with thrombotic thrombocyto-penic purpura. Thromb Haemost 2006;95:886–92.
138. Moake J. Thrombotic thrombocytopenia purpura (TTP) and other thrombotic microangiopathies. Best Pract Res Clin Haematol 2009;22:567–76.
139. Rock G, Shumak KH, Sutton DM, et al. Cryosupernatant as replacement fl uid for plasma exchange in thrombotic thrombocytopenic purpura. Members of the Canadian Apheresis Group. Br J Haematol 1996;94:383–6.
140. Gertz MA, Anagnostopoulos A, Anderson K, et al. Treatment recommendations in Waldenstrom’s macro-globulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroblobulinemia. Semin Oncol 2003;30:121–6.
141. Zarkovic M, Kwaan HC. Correction of hyperviscosity by apheresis. Semin Thromb Hemost 2003;29:535–42.
142. Martin JN Jr, Files JC, Blake PG, et al. Postpartum plasma exchange for atypical preeclampsia-eclampsia as HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. Am J Obstet Gynecol 1995;172:1107–25.
143. Guillaume JC, Roujeau JC, Morel P, et al. Controlled study of plasma exchange in pemphigus. Arch Dermatol 1988;124:1659–63.
144. Miller FW, Leitman SF, Cronin ME, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med 1992;326:1380–4.
145. Johnson JP, Moore J Jr, Austin HA III, et al. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic signifi cance of clinical, pathologic and treatment factors. Medicine (Baltimore) 1985;64:219–27.
146. McLeod BC, Sniecinski I, Ciavarella D, et al. Frequency of immediate adverse effects associated with therapeutic apheresis. Transfusion 1999;39:282–8.
147. Rizvi MA, Vesely SK, George JN, et al. Complications of plasma exchange in 71 consecutive patients treated for clinically suspected thrombotic thrombocytopenic purpura-hemolytic-uremic syndrome. Transfusion 2000;40:896–901.
148. Bolan CD, Cecco SA, Wesley RA, et al. Controlled study of citrate effects and response to I.V. calcium administration during allogeneic peripheral blood progenitor cell donation. Transfusion 2002;42:935–46.
149. Pearl RG, Rosenthal MH. Metabolic alkalosis due to plasmapheresis. Am J Med 1985;79:391–3.
150. Kluger R. Red cell substitutes from hemoglobin—do we start all over again? Curr Opin Chem Biol 2010;14:538–43.
151. Garratty G. Modulating the red cell membrane to produce universal/stealth donor red cells suitable for transfusion. Vox Sang 2008;94:87–95.
152. Bryant BJ, Klein HG. Pathogen inactivation: the defi nitive safeguard for the blood supply. Arch Pathol Lab Med 2007;131:719–33.
153. Ghanima W, Bussel JB. Thrombopoietic agents in immune thrombocytopenia. Semin Hematol 2010;47:258–65.
154. Timmins NE, Nielsen LK. Blood cell manufacture: current methods and future challenges. Trends Biotechnol 2009;27:415–22.