urokinase links plasminogen activation and cell adhesion...
TRANSCRIPT

Article
Urokinase links plasminogen activation and celladhesion by cleavage of the RGD motifin vitronectinValentina De Lorenzi1, Gian Maria Sarra Ferraris1, Jeppe B Madsen2, Michela Lupia3,
Peter A Andreasen2 & Nicolai Sidenius1,*
Abstract
Components of the plasminogen activation system including uroki-nase (uPA), its inhibitor (PAI-1) and its cell surface receptor (uPAR)have been implicated in a wide variety of biological processesrelated to tissue homoeostasis. Firstly, the binding of uPA to uPARfavours extracellular proteolysis by enhancing cell surface plas-minogen activation. Secondly, it promotes cell adhesion andsignalling through binding of the provisional matrix protein vitro-nectin. We now report that uPA and plasmin induces a potentnegative feedback on cell adhesion through specific cleavage ofthe RGD motif in vitronectin. Cleavage of vitronectin by uPAdisplays a remarkable receptor dependence and requires conco-mitant binding of both uPA and vitronectin to uPAR. Moreover, weshow that PAI-1 counteracts the negative feedback and behaves asa proteolysis-triggered stabilizer of uPAR-mediated cell adhesionto vitronectin. These findings identify a novel and highly specificfunction for the plasminogen activation system in the regulationof cell adhesion to vitronectin. The cleavage of vitronectin by uPAand plasmin results in the release of N-terminal vitronectin frag-ments that can be detected in vivo, underscoring the potentialphysiological relevance of the process.
Keywords cell adhesion; extracellular proteolysis; RGD; urokinase-type
plasminogen activator system; vitronectin
Subject Category Cell Adhesion, Polarity & Cytoskeleton
DOI 10.15252/embr.201541681 | Received 30 October 2015 | Revised 23 March
2016 | Accepted 19 April 2016 | Published online 17 May 2016
EMBO Reports (2016) 17: 982–998
Introduction
The urokinase-type plasminogen activator (urokinase, uPA) and its
cell surface receptor (uPAR) are core components of the plasmino-
gen activation system in mammals and orchestrate different cellular
processes, including extracellular proteolysis, cell adhesion,
migration and proliferation [1,2]. These functions of uPA and uPAR
can mechanistically be categorized as either proteolytic or non-
proteolytic, depending on the requirement for the catalytic activity
of uPA in the process.
The proteolytic functions were the first to be characterized.
Cells secrete urokinase as a single-chain zymogen (single-chain
uPA, sc-uPA) that is converted into the active two-chain form
(tc-uPA) by limited proteolysis. The binding of sc-uPA to uPAR,
and plasminogen to one or more of the functionally related plas-
minogen receptors [3], concentrates the two zymogens on the
plasma membrane favouring their reciprocal activation [4]. This
process endows cells with the focalized proteolytic activity
believed to be responsible for the activity of the plasminogen
activation system in tissue invasion. Various protease inhibitors
control the proteolytic cascade; in particular, the primary physio-
logical inhibitors of tc-uPA and plasmin are the two serpins
plasminogen activator inhibitor 1 (PAI-1) and a-2-antiplasmin
(a2AP), respectively. In addition, uPAR has been demonstrated to
act as an endocytosis co-receptor for spent complexes between
uPA and PAI-1 [5,6].
On the non-proteolytic side, overexpression of uPAR and/or
treatment of uPAR-expressing cells with uPA induce signal transduc-
tion resulting in altered cell adhesion, migration, survival and prolif-
eration. Lacking transmembrane and intracellular domains, uPAR
itself is incompetent in signal transduction. A large number of inde-
pendent studies has shown that the uPA/uPAR-induced signal is
relayed into the cells through functional interactions with profes-
sional signalling receptors including several members of the integrin
family of adhesion receptors, receptor tyrosine kinases (RTK) and
G-protein-coupled receptors (GPCRs) (reviewed in [1]). Over the
latest years, it has emerged that the functional coupling between
uPAR and integrins [7–9], RTKs [10] and GPCRs [11], occurs down-
stream of the direct binding of uPAR to the provisional extracellular
matrix (ECM) protein vitronectin (VN). This suggests that the inter-
action between uPAR and the somatomedin B (SMB) domain of VN
may act as the master switch for the signalling activity of uPA/
uPAR. Moreover, a recent in vivo study has documented the
1 Unit of Cell Matrix Signalling, IFOM, The FIRC Institute of Molecular Oncology, Milan, Italy2 Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark3 Department of Experimental Oncology, European Institute of Oncology, Milan, Italy
*Corresponding author. Tel: +39 02 574303 261; Fax: +39 02 574303 231; E-mail: [email protected]
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors982
Published online: May 17, 2016

functional relevance of the uPAR/VN interaction for tumour growth
[12].
Although both the proteolytic and non-proteolytic functions of
uPA/uPAR are well characterized, only little is known about how
plasminogen activation affects the uPAR/VN interaction and how
the uPAR/VN interaction impinges on plasminogen activation. Bind-
ing of sc-uPA to uPAR induces conformational changes in the recep-
tor increasing its affinity for VN [13], but it is not known what
happens when uPA in the sc-uPA/uPAR/VN complex becomes acti-
vated. It has been shown that both tc-uPA and plasmin may cleave
and inactivate uPAR [14,15], suggesting that receptor cleavage
represents a negative feedback mechanism regulating both the
proteolytic and the non-proteolytic functions of uPAR. Nevertheless,
whether such feedback mechanisms are active and have functional
consequences remains to be documented.
We here present direct mechanistic evidence that the proteolytic
and non-proteolytic functions of uPAR are intimately interconnected
through receptor-dependent proteolytic cleavage of the RGD motif
in VN by uPA and plasmin. These findings provide novel conceptual
insight into the biology of the plasminogen activation system,
suggesting that a central function of the plasminogen activation
system is to regulate cell adhesion and signalling through proteolytic
inactivation of VN.
Results
Plasminogen activation exerts a negative feedback on celladhesion to VN
To investigate the possible existence and mechanism of feedback
loops between the function of uPAR in extracellular proteolysis and
cell adhesion, we conducted time-lapse microscopy on HEK293 cells
engineered to overexpress uPAR using the Flp-In system (293/uPAR
[7]). Cells were seeded on VN and exposed to consecutive additions
of sc-uPA and plasminogen to trigger the plasminogen activation
cascade (Fig 1A and B, and Movie EV1). When seeded on VN, 293/
uPAR cells displayed an adherent phenotype characterized by exten-
sive lamellipodia formation that was further enhanced by sc-uPA
addition. Treatment with plasminogen, however, rapidly reversed
the pro-adhesive effect of sc-uPA as evidenced by lamellipodia
retraction and the acquisition of rounded cell morphology, similarly
to what has previously been reported for endothelial cells following
plasminogen activation [16]. To quantitatively analyse the negative
feedback, we utilized a real-time cell analysis (RTCA) instrument
that allows for the continuous and non-invasive evaluation of the
extent and quality of cell matrix interactions by impedance measure-
ments [17]. The data obtained by RTCA analysis of 293/uPAR cells
(Fig 1C) closely paralleled the time-lapse microscopy recordings:
after an initial adhesion phase the addition of sc-uPA caused a
marked increase in cell adhesion that was rapidly reverted upon
subsequent addition of plasminogen. The reduction in cell adhesion
to VN induced by plasminogen activation was also observed using a
plate-and-wash assay (Appendix Fig S1A). The inhibitory effect of
plasminogen activation on cell adhesion to VN was mediated by cell
surface-associated plasmin and/or tc-uPA activity as the addition of
a2AP, which inhibits free but not membrane bound plasmin [18],
had limited effect on the magnitude of the proteolytic feedback
(Fig 1C). Mock-transfected HEK293 cells that do not express
endogenous uPAR did not respond notably to treatments with
sc-uPA and Plg (Appendix Fig S1B). When 293/uPAR cells were
seeded on FN, triggering the plasminogen activation cascade did not
impair cell adhesion, but rather resulted in a delayed and transient
increase (Fig EV1). In contrast to VN, the transient increase in FN
adhesion is mediated by the activity of free plasmin, as it was fully
inhibited by a2AP.These results evidence the existence of both positive and nega-
tive feedback loops between plasminogen activation and cell adhe-
sion to VN and FN.
The catalytic activity of both uPA and plasmin contributes to thenegative feedback
The process of cell surface plasminogen activation is a reciprocal
zymogen activation cascade in which the zymogen sc-uPA is
converted into active tc-uPA and the zymogen plasminogen into
active plasmin, thus resulting in the concomitant generation of two
distinct serine protease activities. To determine which of these
activities is responsible for the negative feedback, we analysed the
cellular response to the isolated proteases (Fig 1D). Both sc-uPA
and tc-uPA stimulated VN adhesion to a similar extent and with
similar kinetics, consistent with the fact that both contain the
receptor binding growth factor-like domain (GFD) responsible for
inducing uPAR binding to VN [13,19]. However, after reaching peak
levels, the adhesion of tc-uPA-treated cells started to decline as
compared to cells treated with sc-uPA, demonstrating that the
catalytic activity of uPA contributes directly to the feedback. Catalyt-
ically active low molecular weight uPA (LMW-uPA), lacking the
GFD domain, failed to induce the feedback demonstrating the
requirement for uPA binding to uPAR (Appendix Fig S1C). Further-
more, control experiments using the plasmin inhibitor aprotinin,
tc-uPA purified from human urine and catalytically inactive uPA
variant (uPAS356A) excluded the possibility that the plasmin used for
activation of recombinant sc-uPA was responsible for the observed
negative feedback (Appendix Fig S1C).
The feedback induced by tc-uPA was not as steep as that
observed with a combination of sc-uPA and plasminogen (see
Fig 1B), indicating that the catalytic activity of uPA is not sufficient
to obtain the full magnitude of the feedback. Consistently, the addi-
tion of plasminogen to tc-uPA-treated cells further accelerated the
feedback. To evaluate the direct contribution of plasmin, we first
treated cells with the GFD domain of uPA to mimic the pro-adhesive
effect of ligand occupancy and then challenged the cells with active
plasmin. Under these conditions, the treatment with plasmin caused
a rapid decrease in cell adhesion showing that the proteolytic activ-
ity of plasmin also contributes directly.
The negative feedback is partially acting through uPAR cleavage
It has been recognized for a long time that the linker region connect-
ing domain 1 (D1) and domains 2 and 3 (D2D3) of uPAR is highly
susceptible to proteolysis by various proteases including uPA and
plasmin [20]. Since the binding site for VN in uPAR involves deter-
minants in both D1 and D2D3 [7,13], cleavage of the linker region
connecting these domains could possibly account for the negative
feedback. To address this possibility, we generated HEK293 cells
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
983
Published online: May 17, 2016

expressing a uPAR variant (uPARR83/89A) in which the reported uPA
and plasmin cleavage sites (83R↓A and 89R↓S) have been disrupted
by alanine substitutions. Analysis by flow cytometry confirmed that
the different receptor variants were equally expressed on the cell
surface (Appendix Fig S1D) and induced largely comparable cell
adhesion to VN (Appendix Fig S1E). In cells expressing the
uPARR83/89A receptor (Fig 1E), the onset of the negative feedback
induced by tc-uPA was markedly delayed as compared to cells
expressing uPARWT (Fig 1D), indicating that uPAR cleavage indeed
contributes to the feedback. Notably, however, the impairment of
receptor cleavage did not fully phenocopy the effect of treatment
with catalytically inactive sc-uPA showing that other proteolytic
events must be involved. In support of this, the treatment of 293/
uPARR83/89A cells with GFD followed by plasmin also resulted in a
pronounced reduction in VN adhesion. Western blotting analysis
using domain-specific antibodies confirmed that the treatment with
tc-uPA, as well as sc-uPA plus plasminogen, resulted in efficient
cleavage of uPARWT, but not uPARR83/89A (Fig 1F). Importantly,
despite its pronounced effect on cell adhesion, plasmin treatment
did not result in marked uPAR cleavage.
These data thus demonstrate that cleavage of uPAR is only
partially responsible for the negative feedback and that other func-
tionally relevant uPA/plasmin substrates must be involved in the
process.
A B C
D E F
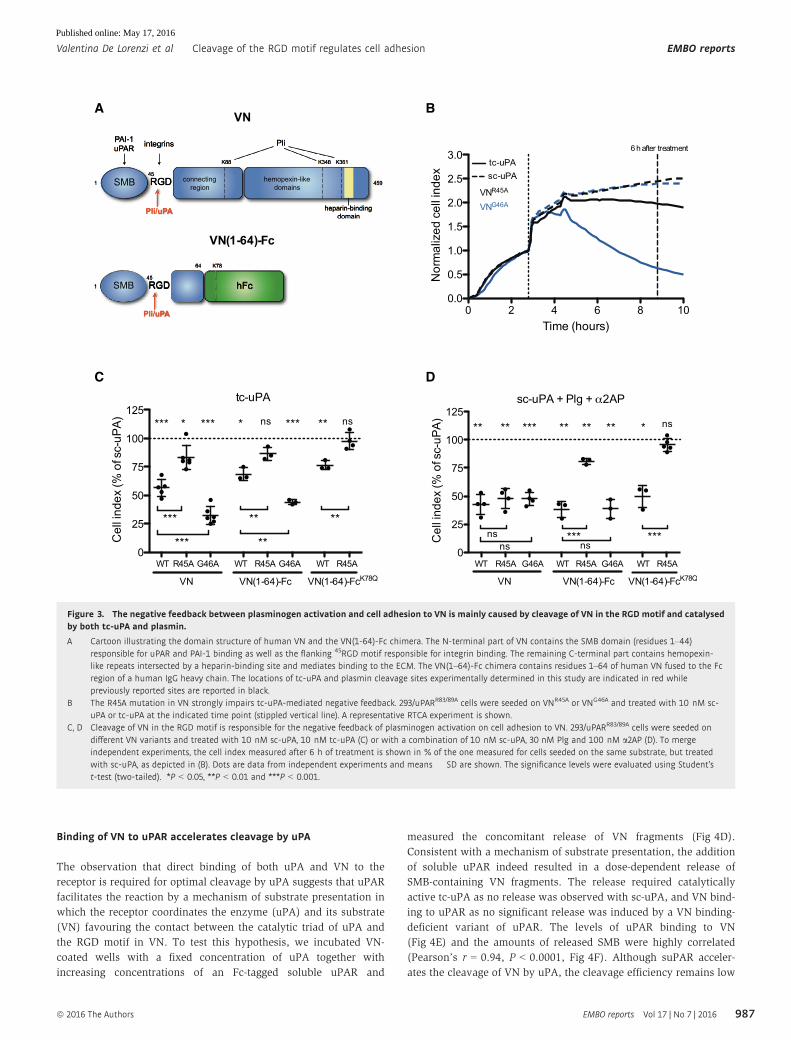
Figure 1. Plasminogen activation imposes a negative feedback on uPAR-mediated cell adhesion to VN.
A Effect of plasminogen activation on 293/uPAR cells morphology. 293/uPAR cells were seeded on VN and imaged by time-lapse microscopy. Cells were treated withsubsequent additions of 10 nM sc-uPA and 30 nM plasminogen (Plg), and representative phase contrast images, taken just before and 2 h after addition ofplasminogen, are shown. The complete time-lapse recording can be found in Movie EV1. Scale bar, 20 lm.
B Effect of plasminogen activation on 293/uPAR cells F-actin cytoskeleton. 293/uPAR cells were seeded on VN and treated with 10 nM sc-uPA (upper panel) or with acombination of 10 nM sc-uPA and 30 nM Plg (lower panel). After 2 h, cells were fixed, permeabilized and stained with phalloidin. Representative images are shown.
C Negative feedback between plasminogen activation and cell adhesion to VN. RTCA experiment of 293/uPAR cells seeded in VN-coated E-plates is shown. The data arefrom a representative experiment.
D The catalytic activity of both uPA and plasmin (Pli) contributes to the negative feedback between plasminogen activation and cell adhesion. RTCA experiment wasconducted with 293/uPARWT cells seeded in VN-coated E-plates and treated with 10 nM sc-uPA, tc-uPA and GFD, and 30 nM Plg and Pli as shown in the figure.A representative experiment is shown.
E The cleavage of uPAR partially contributes to the negative feedback. 293/uPARR83/89A cells were seeded on VN and treated as in (D). A representative experiment isshown.
F uPARR83/89A is not cleaved by uPA and plasmin. 293/uPARWT and 293/uPARR83/89A cells were seeded on FN-coated tissue culture plates and allowed to adhere beforetreatment with the indicated reagents: 10 nM sc-uPA and tc-uPA, 30 nM Plg and Pli. After 1 h, cells were lysed, resolved by SDS–PAGE and analysed byimmunoblotting using monoclonal antibodies recognizing different epitopes in uPAR [51]. R2 binds an epitope in D3 of uPAR and recognizes the full-length receptor(uPAR) as well as the truncated form lacking D1 (D2D3). R3 binds an epitope in D1 and only recognizes full-length uPAR. The blot is from a representative experimentand the mobility of molecular weight standards is shown.
Data information: (C–E) Stippled vertical lines indicate the time points at which the cells were treated. Each condition was recorded in quadruplicates and the curvesrepresent the average cell index as a function of time. Cell index values are normalized to the cell index measured before the first treatment.
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
984
Published online: May 17, 2016

Plasmin cleaves the RGD motif in VN
Plasmin cleaves purified VN at multiple locations [21,22], and
the treatment of VN-coated surfaces with plasmin has been
shown to attenuate subsequent avb5-dependent adhesion of
keratinocytes [23]. Consistent with these observations, we found
that pre-treatment of VN-coated surfaces with plasmin, but not
with tc-uPA, resulted in a dose-dependent reduction of sub-
sequent cell adhesion (Fig 2A). The inhibitory effect was not
specific for uPAR-mediated cell adhesion as the integrin-mediated
adhesion of HeLa cells was also impaired (Fig 2A). The inhibi-
tory effect of plasmin was, however, specific for VN as identical
treatments of FN failed to modulate subsequent cell adhesion
(Fig 2B).
To investigate whether the reported cleavage sites for plasmin
in VN were responsible for the negative feedback, we seeded
293/uPARR83/89A cells on a recombinant form of VN (VN(1-64)-Fc)
containing the N-terminal SMB domain responsible for uPAR bind-
ing and the flanking 45RGD motif interacting with integrin adhe-
sion receptors, but none of the previously reported plasmin
cleavage sites located in the more C-terminal regions (88K↓G,348K↓K and 361R↓S [21,22]). Treatment of cells seeded on this
substrate with tc-uPA, or sc-uPA plus plasminogen, still resulted
in a robust negative feedback suggesting that the responsible
cleavage site(s) are located within the N-terminal region of VN
(Appendix Fig S1F). To identify these novel cleavage sites, we
treated intact VN with plasmin and analysed the reactions by
MALDI-TOF mass spectrometry (Fig 2C). The molecular weight of
the main peak observed in the resulting spectra closely matched
that of an N-terminal VN fragment (VN(1–45)) generated by cleav-
age within the RGD motif (45R↓GD). A peak with the same mass,
but lower intensity, was also observed in spectra of non-treated
A B
C
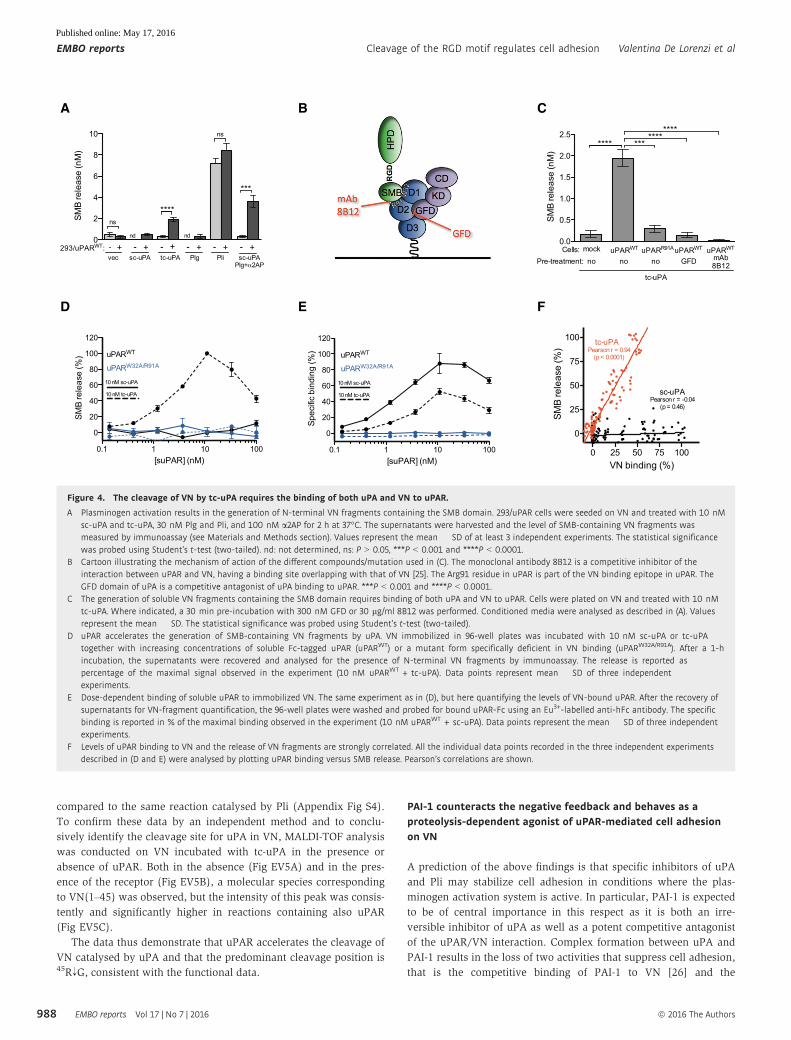
Figure 2. Plasmin cleaves VN in the RGD motif.
A Pre-treatment of VN with plasmin, but not uPA, inhibits subsequent cell adhesion mediated by uPAR and integrins. E-plates wells coated with VN were incubatedwith a dilution curve of plasmin or tc-uPA for 1 h at 37°C prior to cell seeding. After washing, 293/uPARWT or HeLa cells were seeded and their adhesion was followedby RTCA. The cell indexes recorded 1 h after seeding are shown. Data are reported as percentage of the cell index measured in non-pre-treated wells and representthe mean � SD of three independent experiments.
B Pre-treatment of FN with plasmin or tc-uPA does not affect subsequent cell adhesion. RTCA experiments were performed as described for (A), but with FN-coatedwells. Data are reported as percentage of the cell index measured in non-pre-treated wells and represent the mean � SD of three independent experiments.
C Plasmin cleaves VN at the 45RG peptide bond. Urea-purified VN was incubated with plasmin (substrate/enzyme ratio: 10:1 w/w) for 2 h at 37°C and the reactionswere analysed by MALDI-TOF mass spectrometry.
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
985
Published online: May 17, 2016

VN (Fig EV2A), but not in spectra of plasmin alone (Fig EV2B)
suggesting that VN may be particularly prone to hydrolysis at this
position and already partially cleaved. The same VN fragment was
also observed upon plasmin treatment of VN(1-64)-Fc (Fig EV2C),
but not in a variant of this chimera (VN(1-64)R45A-Fc) containing
an alanine substitution of 45R. The treatment of this VN variant
rather resulted in a peak corresponding to a molecular species
generated by cleavage after a lysine (78K) located in the Fc tag
(Fig EV2D). Taken together, these data conclusively identify the
RGD motif as a cleavage site for plasmin in VN.
Cleavage of the RGD motif in VN is responsible for the negativefeedback between plasminogen activation and cell adhesion
To determine whether the identified target site is also functionally
responsible for the feedback between the plasminogen activation
cascade and cell adhesion on VN, we performed RTCA experi-
ments using 293/uPARR83/89A cells seeded on VN and different
VN variants in which cleavage has been prevented by alanine
substitution of 45R in the RGD motif (see cartoon in Fig 3A).
Since the 45R?A substitution employed to prevent cleavage also
results in impaired integrin binding, we generated a second
control variant carrying a 46G?A substitution that also abrogates
integrin binding, but is expected to have only little effect on
cleavage. A representative experiment illustrating the effect of the
two substitutions on the proteolytic feedback is shown in Fig 3B
and the quantified data from multiple experiments in Fig 3C and D.
Introduction of the cleavage-blocking 45R?A substitution in both
intact VN and the shorter VN(1–64)-Fc chimera significantly and
almost completely impaired the proteolytic feedback mediated by
tc-uPA (Fig 3C), demonstrating that the 45R cleavage site in the
RGD motif of VN is indeed functionally responsible for the bulk
of the negative feedback mediated by active tc-uPA. The minor
residual feedback observed on VN(1-64)R45A-Fc was mediated by
cleavage at a lysine residue (78K) located in the Fc part of the
chimera. The control mutation used in these experiments (46G?A) gave rise to a negative feedback significantly more potent than
that of the wild-type RGD motif (Fig 3C). This finding may be
explained in at least two ways: firstly, the RAD sequence may be
a better substrate for uPA than the RGD sequence; secondly,
integrin binding to the RGD motif may protect it from proteolysis.
In vitro cleavage experiments using purified components indicated
that the 45RA peptide bond is indeed more susceptible to proteo-
lytic cleavage than the wild-type 45RG sequence (Appendix
Fig S2A and B). Considering the small size of the
RGD motif, it is also plausible that integrin occupancy may
prevent proteases from assessing the cleavage site by steric
hindrance, but we did not test this experimentally. When cells
were treated with a combination of sc-uPA, plasminogen and
a2AP (Fig 3D), the 45R?A substitution had little effect on the
proteolytic feedback on intact VN indicating that other cleavage
sites for plasmin in VN are also important, or become important
when the primary cleavage site has been inactivated. Neverthe-
less, the 45R?A substitution did significantly impair the feedback
on the shorter VN(1-64)-Fc chimera documenting that 45R↓GDcleavage does contribute to the feedback. Additional experiments
confirmed that the 45R?A substitution also impaired Pli-induced
cell detachment (Appendix Fig S2C and D).
To more accurately quantify the contribution of uPAR cleavage
to the feedback, we utilized the cleavage refractory VN substrate
(VN(1–64)R45A-FcK78Q) in combination with cell lines expressing
uPAR mutants in which the known cleavage sites ((83R↓A and89R↓S)) have been mutated individually or in combination
(Fig EV3A and B). This analysis revealed that cleavage at both loca-
tions contributes to the negative feedback and that these sites are
the only important ones in uPAR as alanine substitution of both
completely abolished the negative feedback.
The data thus demonstrate that the proteolytic feedback between
plasminogen activation and cell adhesion to VN is functionally
mediated by cleavage of both VN and uPAR and catalysed by both
tc-uPA and plasmin. In the process, the cleavage of VN appears to
be relatively more important than cleavage of uPAR. The cleavage
site in VN responsible for the negative feedback is 45R↓G in the RGD
motif even if other functionally relevant plasmin cleavage sites may
exist.
uPAR accelerates the cleavage of matrix VN by uPA and plasmin
Proteolytic cleavage of matrix bound VN in the RGD motif is
expected to result in the release of an N-terminal VN fragment
(i.e. the SMB domain) from the connecting region and C-terminal
hemopexin-like domains, which mediate the binding of VN to
other ECM components [24]. To quantify this release, we treated
immobilized VN with different forms of uPA and plasminogen in
the absence or presence of 293/uPAR cells and measured the
levels of SMB-containing VN fragments released to the conditioned
medium (Fig 4A). In the absence of cells, only the treatment with
plasmin was found to release VN fragments, consistent with the
pre-treatment experiments (Fig 2A), whereas the presence of 293/
uPAR cells resulted in the release of VN fragments also by tc-uPA
and the combination of sc-uPA, plasminogen and a2AP. The plas-
min inhibitor aprotinin did not inhibit the release mediated by
uPA excluding the possibility that trace amounts of plasmin in the
uPA preparation were responsible for the proteolysis (Appendix
Fig S3A). The release of VN fragments was caused by cleavage
within the RGD motif as it was impaired by the R45A substitution
(Appendix Fig S3B). Furthermore, the release mediated by tc-uPA
requires the following: (i) uPAR expression, as mock-transfected
cells did not induce release; (ii) uPA binding to uPAR, as the
release was impaired by the GFD domain of uPA; (iii) uPAR bind-
ing to VN, as cells expressing a VN binding-deficient uPAR mutant
(uPARR91A) failed to promote the release and because an antibody
(8B12) binding to the VN epitope in uPAR [25] also prevented
release (Fig 4C). The mode of action of the different compounds/
mutations utilized is depicted in Fig 4B. Similar results were
observed for the treatment with sc-uPA, plasminogen and a2AP(Fig EV4), even if the inhibition by GFD and the impairment by
the uPAR 91R?A substitution in this case were only partial.
The more likely explanation for this difference is that the cleavage
of matrix VN by cell surface plasmin is less dependent upon the
direct uPAR/VN interaction and that the concentration of GFD used
for competition was insufficient to block plasminogen activation.
These data show that VN fragments containing the SMB domain
are released from matrix VN upon plasminogen activation in a
highly specific process that is accelerated by the binding of both
uPA and VN to uPAR.
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
986
Published online: May 17, 2016
Binding of VN to uPAR accelerates cleavage by uPA
The observation that direct binding of both uPA and VN to the
receptor is required for optimal cleavage by uPA suggests that uPAR
facilitates the reaction by a mechanism of substrate presentation in
which the receptor coordinates the enzyme (uPA) and its substrate
(VN) favouring the contact between the catalytic triad of uPA and
the RGD motif in VN. To test this hypothesis, we incubated VN-
coated wells with a fixed concentration of uPA together with
increasing concentrations of an Fc-tagged soluble uPAR and
measured the concomitant release of VN fragments (Fig 4D).
Consistent with a mechanism of substrate presentation, the addition
of soluble uPAR indeed resulted in a dose-dependent release of
SMB-containing VN fragments. The release required catalytically
active tc-uPA as no release was observed with sc-uPA, and VN bind-
ing to uPAR as no significant release was induced by a VN binding-
deficient variant of uPAR. The levels of uPAR binding to VN
(Fig 4E) and the amounts of released SMB were highly correlated
(Pearson’s r = 0.94, P < 0.0001, Fig 4F). Although suPAR acceler-
ates the cleavage of VN by uPA, the cleavage efficiency remains low
C D
BA
Figure 3. The negative feedback between plasminogen activation and cell adhesion to VN is mainly caused by cleavage of VN in the RGD motif and catalysedby both tc-uPA and plasmin.
A Cartoon illustrating the domain structure of human VN and the VN(1-64)-Fc chimera. The N-terminal part of VN contains the SMB domain (residues 1–44)responsible for uPAR and PAI-1 binding as well as the flanking 45RGD motif responsible for integrin binding. The remaining C-terminal part contains hemopexin-like repeats intersected by a heparin-binding site and mediates binding to the ECM. The VN(1–64)-Fc chimera contains residues 1–64 of human VN fused to the Fcregion of a human IgG heavy chain. The locations of tc-uPA and plasmin cleavage sites experimentally determined in this study are indicated in red whilepreviously reported sites are reported in black.
B The R45A mutation in VN strongly impairs tc-uPA-mediated negative feedback. 293/uPARR83/89A cells were seeded on VNR45A or VNG46A and treated with 10 nM sc-uPA or tc-uPA at the indicated time point (stippled vertical line). A representative RTCA experiment is shown.
C, D Cleavage of VN in the RGD motif is responsible for the negative feedback of plasminogen activation on cell adhesion to VN. 293/uPARR83/89A cells were seeded ondifferent VN variants and treated with 10 nM sc-uPA, 10 nM tc-uPA (C) or with a combination of 10 nM sc-uPA, 30 nM Plg and 100 nM a2AP (D). To mergeindependent experiments, the cell index measured after 6 h of treatment is shown in % of the one measured for cells seeded on the same substrate, but treatedwith sc-uPA, as depicted in (B). Dots are data from independent experiments and means � SD are shown. The significance levels were evaluated using Student’st-test (two-tailed). *P < 0.05, **P < 0.01 and ***P < 0.001.
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
987
Published online: May 17, 2016
compared to the same reaction catalysed by Pli (Appendix Fig S4).
To confirm these data by an independent method and to conclu-
sively identify the cleavage site for uPA in VN, MALDI-TOF analysis
was conducted on VN incubated with tc-uPA in the presence or
absence of uPAR. Both in the absence (Fig EV5A) and in the pres-
ence of the receptor (Fig EV5B), a molecular species corresponding
to VN(1–45) was observed, but the intensity of this peak was consis-
tently and significantly higher in reactions containing also uPAR
(Fig EV5C).
The data thus demonstrate that uPAR accelerates the cleavage of
VN catalysed by uPA and that the predominant cleavage position is45R↓G, consistent with the functional data.
PAI-1 counteracts the negative feedback and behaves as aproteolysis-dependent agonist of uPAR-mediated cell adhesionon VN
A prediction of the above findings is that specific inhibitors of uPA
and Pli may stabilize cell adhesion in conditions where the plas-
minogen activation system is active. In particular, PAI-1 is expected
to be of central importance in this respect as it is both an irre-
versible inhibitor of uPA as well as a potent competitive antagonist
of the uPAR/VN interaction. Complex formation between uPA and
PAI-1 results in the loss of two activities that suppress cell adhesion,
that is the competitive binding of PAI-1 to VN [26] and the
A B C
D E F
Figure 4. The cleavage of VN by tc-uPA requires the binding of both uPA and VN to uPAR.
A Plasminogen activation results in the generation of N-terminal VN fragments containing the SMB domain. 293/uPAR cells were seeded on VN and treated with 10 nMsc-uPA and tc-uPA, 30 nM Plg and Pli, and 100 nM a2AP for 2 h at 37°C. The supernatants were harvested and the level of SMB-containing VN fragments wasmeasured by immunoassay (see Materials and Methods section). Values represent the mean � SD of at least 3 independent experiments. The statistical significancewas probed using Student’s t-test (two-tailed). nd: not determined, ns: P > 0.05, ***P < 0.001 and ****P < 0.0001.
B Cartoon illustrating the mechanism of action of the different compounds/mutation used in (C). The monoclonal antibody 8B12 is a competitive inhibitor of theinteraction between uPAR and VN, having a binding site overlapping with that of VN [25]. The Arg91 residue in uPAR is part of the VN binding epitope in uPAR. TheGFD domain of uPA is a competitive antagonist of uPA binding to uPAR. ***P < 0.001 and ****P < 0.0001.
C The generation of soluble VN fragments containing the SMB domain requires binding of both uPA and VN to uPAR. Cells were plated on VN and treated with 10 nMtc-uPA. Where indicated, a 30 min pre-incubation with 300 nM GFD or 30 lg/ml 8B12 was performed. Conditioned media were analysed as described in (A). Valuesrepresent the mean � SD. The statistical significance was probed using Student’s t-test (two-tailed).
D uPAR accelerates the generation of SMB-containing VN fragments by uPA. VN immobilized in 96-well plates was incubated with 10 nM sc-uPA or tc-uPAtogether with increasing concentrations of soluble Fc-tagged uPAR (uPARWT) or a mutant form specifically deficient in VN binding (uPARW32A/R91A). After a 1-hincubation, the supernatants were recovered and analysed for the presence of N-terminal VN fragments by immunoassay. The release is reported aspercentage of the maximal signal observed in the experiment (10 nM uPARWT + tc-uPA). Data points represent mean � SD of three independentexperiments.
E Dose-dependent binding of soluble uPAR to immobilized VN. The same experiment as in (D), but here quantifying the levels of VN-bound uPAR. After the recovery ofsupernatants for VN-fragment quantification, the 96-well plates were washed and probed for bound uPAR-Fc using an Eu3+-labelled anti-hFc antibody. The specificbinding is reported in % of the maximal binding observed in the experiment (10 nM uPARWT + sc-uPA). Data points represent the mean � SD of three independentexperiments.
F Levels of uPAR binding to VN and the release of VN fragments are strongly correlated. All the individual data points recorded in the three independent experimentsdescribed in (D and E) were analysed by plotting uPAR binding versus SMB release. Pearson’s correlations are shown.
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
988
Published online: May 17, 2016

proteolytic degradation of VN and uPAR described here. We there-
fore reasoned that the uPA•PAI-1 complex, in contrast to its molecu-
lar precursors, would be a stable agonist of cell adhesion to VN as:
(i) it is proteolytically inactive and consequently unable to cleave
uPAR and VN, (ii) does not bind to VN and therefore cannot
compete with uPAR binding, (iii) maintains high-affinity binding to
uPAR [27] and promotes receptor binding to VN [19]. To address
this hypothesis, we first tested the biological activity of PAI-1, a
combination of PAI-1 and tc-uPA (1:1) as well as purified uPA•PAI-1
complex in modulating uPAR-mediated cell adhesion to VN. Consis-
tent with its high-affinity binding to the SMB domain, PAI-1, but not
a mutant PAI-1 deficient in VN binding [28], acted as a potent antag-
onist of uPAR-mediated cell adhesion to VN (Fig 5A and
Appendix Fig S5A). On the contrary, the combination of tc-uPA and
PAI-1, as well as the purified uPA•PAI-1 complex, displayed strong
agonistic activity. Morphological analysis of cells subjected to the
different treatments (Fig 5B) evidenced that treatment with PAI-1
induced cell rounding, while treatment with uPA•PAI-1 complex
induced cell spreading. The quantification of these observations is
presented in Appendix Fig S5B. Since the strength of cell adhesion
and extent of spreading are major determinants of cell migration,
we also evaluated the biological effect of the treatments on the 2D
migration velocity (Fig 5C and Movies EV2, EV3, EV4 and EV5).
Both the treatment with PAI-1 and uPA•PAI-1 strongly reduced
cell migration, indicating that both the reduction in cell adhesion
caused by PAI-1 and the increase in cell adhesion induced by the
uPA•PAI-1 complex impair cell migration. While the inhibitory
effect of the uPA•PAI-1 complex on cell migration is persistent
throughout the observation time, inhibition by PAI-1 is transient,
possibly due to latency transition of PAI-1 releasing it from VN [29].
Although not evident in the experiment presented in Fig 5A, also
the anti-adhesive activity of PAI-1 was frequently found to be tran-
sient (Appendix Fig S5A).
Together, these data suggest that the reaction of PAI-1 with tc-
uPA promotes cell adhesion to VN by dual mechanisms. Firstly,
inactivation of the catalytic activity of tc-uPA by PAI-1 halts VN and
uPAR cleavage and therefore also the negative proteolytic feedback
between plasminogen activation and cell adhesion. Secondly,
complex formation between uPA and PAI-1 impairs the VN binding
activity of PAI-1 and therefore its antagonistic effect on cell adhe-
sion. As PAI-1 reacts extremely slowly with sc-uPA [30], the interac-
tion between uPA and PAI-1 is triggered by zymogen activation of
sc-uPA, possibly by cell surface-generated plasmin. To verify the
occurrence of this complex series of reactions, we seeded 293/uPAR
cells on VN and treated them with a combination of sc-uPA and
PAI-1 (Fig 5D). As expected from the agonistic activity of sc-uPA
and the antagonistic activity of PAI-1 on cell adhesion, the effect of
the combined treatment was intermediate to the effect of the indi-
vidual treatments. Remarkably, the subsequent treatment with Plg
resulted in a potent induction of cell adhesion that was not observed
in the absence of PAI-1.
These data clearly identify a central role for PAI-1 in the process.
In the absence of proteolysis, PAI-1 acts as a suppressor of cell
adhesion through its high-affinity binding to VN. In conditions of
plasminogen activation, the activity of PAI-1 is reverted into
pro-adhesive as it is released from VN, protects VN and uPAR from
proteolysis by inhibition of tc-uPA (Appendix Fig S5C) and enforces
the uPAR/VN interaction.
VN cleavage correlates with the expression of uPA in cancercell lines
To probe the importance of the plasminogen activation system in
matrix VN cleavage as well as to determine the relative importance
of the individual plasminogen activation system components in the
process, we extended the analysis to the 38 epithelial cell lines of
the NCI-60 panel [31] representing cancers of the breast, colon,
lung, ovary, prostate and kidney. The different cell lines were
seeded in VN-coated wells and incubated in the absence or presence
of exogenous Plg and a2AP. In the absence of Plg and a2AP, 23
(61%) of the cell lines released levels of SMB above background (no
cells) and this number increased to 35 (92%) in the presence of Plg
and a2AP (Fig 6A). MALDI-TOF analysis of conditioned medium
from selected cell lines (HOP92 and OVCAR_5) evidenced the cell-
dependent generation of a VN(1–45) fragment corresponding to
cleavage within the RGD motif (Appendix Fig S6) as observed for
purified tc-uPA and plasmin. Across all cell lines, a highly signifi-
cant sevenfold increase in released SMB was measured in the pres-
ence of Plg and a2AP and this increase was almost entirely cell
dependent as the treatment with Plg and a2AP in the absence of
cells did not result in any significant increase. Comparing the
release measured for the different cell lines in the absence and in
the presence of Plg and a2AP did not evidence any strong associa-
tions between the aetiology of the cell lines and the levels of VN
fragments released (Appendix Fig S7A and B). A wealth of baseline
“omics” data is available for the NCI-60 panel of cell lines through
the CellMiner web tool [32] and we next queried this information
using the concentrations of VN fragments released by the different
cell lines. In this analysis, the transcript encoding uPA (PLAU) was
found to correlate strongly with SMB release (r = 0.569, P = 0.002).
The absolute strength of this correlation is underscored by the fact
that the PLAU transcript ranks 3 out of the 25,722 transcripts in the
database (Fig 6B and Dataset EV1). Transcripts encoding other core
components of the plasminogen activation system, including uPAR
(PLAUR), tPA (PLAT), PAI-1 (SERPINE1), PAI-2 (SERPINB2) and
Plg-RKT (PLGRKT) did not correlate significantly with SMB release
(Dataset EV1), suggesting that the expression level of uPA repre-
sents the rate-limiting step of the process.
These data demonstrate that most cancer cell lines are harnessed
to degrade matrix VN, the process is strongly potentiated by exo-
genous Plg and the expression level of uPA is rate-limiting.
Plasminogen activation attenuates the adhesion of cancer celllines to VN
The SMB release data document that cancer cells employ cell
surface plasminogen activation to process matrix VN, but they do
not demonstrate whether this proteolytic event has any functional
consequences for cell adhesion. To address this point directly, we
conducted RTCA experiments for the same 38 cell lines seeded on
VN and incubated with Plg and a2AP or vehicle control (Fig 6C).
The basal cell adhesion to VN was found to correlate with the
expression levels of the canonical VN receptor aVb3 integrin, while,
among the components of the plasminogen activation system, only
PAI-1 expression levels displayed a weak but significant correlation
(Fig 6D and Dataset EV2). The treatment with Plg and a2APresulted in a highly significant (P = 0.0005) 17% reduction in
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
989
Published online: May 17, 2016
average cell adhesion measured 16 h after treatment (Fig 6C).
Importantly, the magnitude of the reduction recorded for the indi-
vidual cell lines (Appendix Fig S7C) was strongly correlated with
the concentrations of SMB released by the same cell lines (Fig 6E).
As for SMB release, also the reduction in cell adhesion correlated
(r = 0.47, P = 0.003) with PLAU expression levels (Fig 6D and
Dataset EV3). Despite the strong correlation between uPA expres-
sion levels and reduction in cell adhesion, the exogenous addition
of tc-uPA to selected cell lines plated on VN did not result in a
decrease in cell adhesion (Appendix Fig S8). This may indicate that
the proteolytic activity of plasmin has a predominant role in the
process or that endogenously expressed, but inactive, sc-uPA
competes for uPAR binding with the exogenously added enzyme.
The identified cleavage site in VN and the highly significant
correlation between the reduction in cell adhesion and the release of
VN fragments evidence that proteolysis of matrix VN by compo-
nents of the plasminogen activation system indeed regulates cell
adhesion.
Identification of N-terminal VN fragments in vivo
Cleavage of VN in the RGD motif generates small N-terminal SMB-
containing fragments that are released from the extracellular matrix
to the surrounding fluid phase. To investigate whether such
fragments are also generated in vivo, we analysed two liquid
biopsies (ascites fluid) of ovarian cancer patients by MALDI-TOF
A B
C D
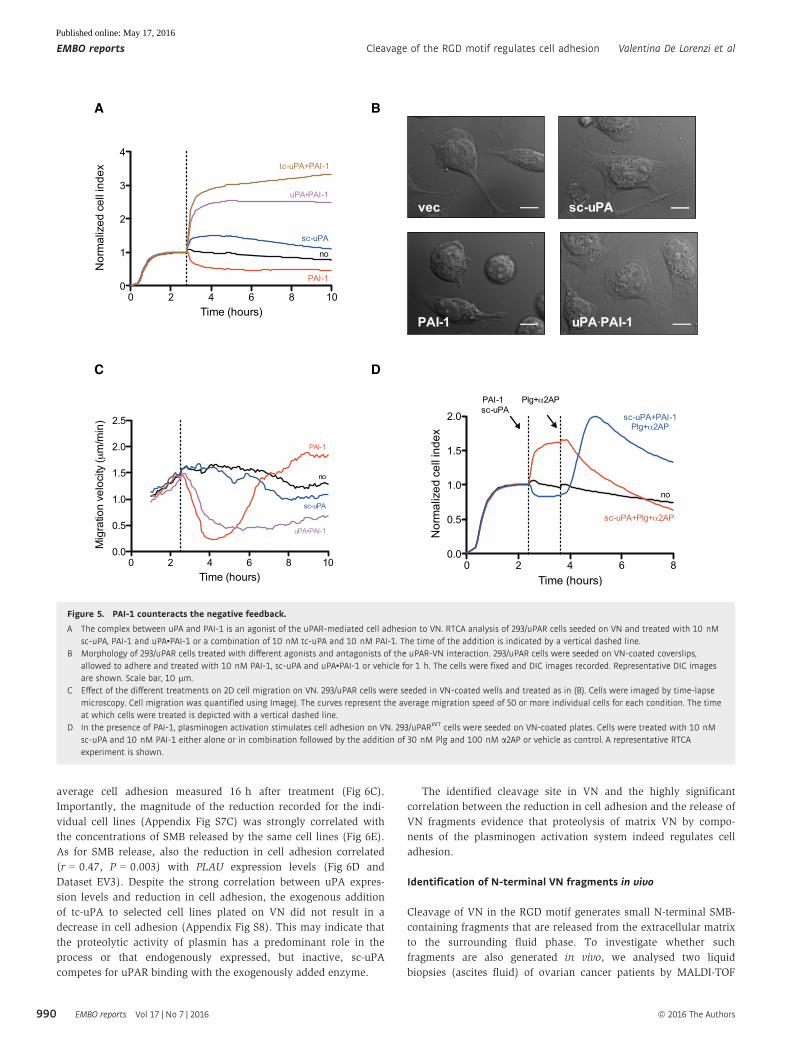
Figure 5. PAI-1 counteracts the negative feedback.
A The complex between uPA and PAI-1 is an agonist of the uPAR-mediated cell adhesion to VN. RTCA analysis of 293/uPAR cells seeded on VN and treated with 10 nMsc-uPA, PAI-1 and uPA•PAI-1 or a combination of 10 nM tc-uPA and 10 nM PAI-1. The time of the addition is indicated by a vertical dashed line.
B Morphology of 293/uPAR cells treated with different agonists and antagonists of the uPAR-VN interaction. 293/uPAR cells were seeded on VN-coated coverslips,allowed to adhere and treated with 10 nM PAI-1, sc-uPA and uPA•PAI-1 or vehicle for 1 h. The cells were fixed and DIC images recorded. Representative DIC imagesare shown. Scale bar, 10 lm.
C Effect of the different treatments on 2D cell migration on VN. 293/uPAR cells were seeded in VN-coated wells and treated as in (B). Cells were imaged by time-lapsemicroscopy. Cell migration was quantified using ImageJ. The curves represent the average migration speed of 50 or more individual cells for each condition. The timeat which cells were treated is depicted with a vertical dashed line.
D In the presence of PAI-1, plasminogen activation stimulates cell adhesion on VN. 293/uPARWT cells were seeded on VN-coated plates. Cells were treated with 10 nMsc-uPA and 10 nM PAI-1 either alone or in combination followed by the addition of 30 nM Plg and 100 nM a2AP or vehicle as control. A representative RTCAexperiment is shown.
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
990
Published online: May 17, 2016

mass spectrometry (Fig 7A–C). The resulting spectra documented
the presence of the VN-derived peptides VN(1–45) and VN(1–44).
The first of these peptides is identical to the fragment generated by
uPA or plasmin cleavage described above, while the second is likely
to represent a processed form generated by the removal of the
C-terminal arginine residue by one or more carboxypeptidases well
known to be abundant in biological fluids. Also in urine N-terminal
VN fragments could be readily identified. The VN(1–45) fragment
was not detected, but shorter forms of VN(1–44), VN(1–42) and
VN(1–41) were abundant (Fig 7D).
These data demonstrate that VN fragments compatible with a
cleavage by uPA/plasmin in the RGD motif can indeed be found
in vivo.
Discussion
Since the discovery of the importance of the RGD sequence for the
adhesive properties of FN more than three decades ago [33], the
central importance of this sequence motif has been extensively
substantiated for a variety of ECM proteins [34]. The provisional ECM
component VN contains a single RGD motif that is both required and
sufficient for the cell adhesive activity of the protein [35]. We here
demonstrate that the adhesive properties of VN are intimately regu-
lated by the proteases of the plasminogen activation system, which
inactivate the protein through specific cleavage of the RGD motif.
The serine protease uPA is endowed with very narrow substrate
specificity, and only few substrates have been extensively
A B
C D E
Figure 6. Plasminogen activation induces VN cleavage and reduces cell adhesion in several human cancer cell lines.
A The SMB domain is released by a wide variety of cancer cell lines. The cancer cell lines of epithelial origin belonging to the NCI-60 panel (n = 38) were seeded on VNin the absence or presence of 30 nM Plg and 100 nM a2AP. The supernatants were harvested after 16 h and the level of SMB-containing VN fragments wasmeasured by immunoassay. Dots represent the concentration measured for the individual cell lines or the value obtained analysing control wells (no cells) from 10independent experiments. Means � SD are shown. The statistical significance was probed using paired Student’s t-test (two-tailed). *P < 0.05 and ****P < 0.0001.
B PLAU mRNA expression levels correlate with the SMB release. The levels of SMB released in the presence of Plg and a2AP were used as a query pattern and correlatedwith the expression levels of about 26,000 genes across the NCI-60 panel, using the CellMiner web tool [32]. The table shows the 10 most strongly correlated genes.The rank, gene name and Pearson’s correlation coefficient (r) are reported.
C Plasminogen activation induces a reduction in cell adhesion of human cancer cell lines. The epithelial cancer cell lines of the NCI panel were seeded on VN-coatedE-plates and allowed to adhere for 4 h before the treatment with 30 nM Plg and 100 nM a2AP or vehicle. The graph shows the average normalized cell index of the38 cell lines. The time of the addition (4 h) and the time at which the reduction of cell adhesion has been quantified (20 h) are depicted by dashed vertical lines.
D Correlation between gene expression and basal cell adhesion to VN and reduction in cell adhesion upon Plg treatment. Adhesion 4 h after seeding the cells on VN(expressed as cell index) was measured in the experiment described in (C). The extent of cell adhesion reduction was calculated at the indicated time point in (C) aspercentage of the cell index value measured in control wells treated with vehicle. Both parameters were correlated with the expression levels of the indicated genes.Gene expression data were downloaded from the CellMiner web tool. The table shows gene name, Pearson’s correlation coefficient (r) and P-value.
E The negative regulation of cell adhesion correlates with the amount of released SMB. Scatter plot showing the correlation between the reduction in cell adhesion(panel C and Appendix Fig S7C) and SMB release (panel A and Appendix Fig S7B). The extent of cell adhesion reduction was calculated at the indicated time point in(C) as percentage of the cell index value measured in control wells treated with vehicle. Samples size (n), Pearson’s correlation coefficient (r) and P-value are reported.
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
991
Published online: May 17, 2016

A
B
C
D
Figure 7. Identification of SMB-containing VN fragments in vivo.
A–D Human ascites and urine samples were immunoprecipitated with HU3-conjugated beads, and eluted material was analysed by MALDI-TOF mass spectrometry.Representative spectra are shown.
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
992
Published online: May 17, 2016

characterized (i.e. plasminogen, PAI-1 and uPAR). We here identify
VN as a novel direct substrate for uPA and demonstrate that the
cleavage reaction is enhanced by a unique mechanism of substrate
presentation in which uPAR directly binds and constructively coor-
dinates both the enzyme (uPA) and the substrate (VN) as illustrated
in the cartoon in Fig 8. Although the ternary structure of the sc-
uPA/uPAR/VN complex still has to be determined, experiments
employing small-angle X-ray scattering have demonstrated that acti-
vation of sc-uPA to tc-uPA is associated with increased interdomain
flexibility [36], possibly permitting an optimal positioning of the
catalytic domain relative to the RGD motif in VN. It is well described
that cell surface uPAR accelerates plasminogen activation by uPA
[4], but in this case, the receptor does so by concentrating the
enzyme to the cell surface where also the substrate (plasminogen)
binds. In fact, in contrast to the cleavage of VN presented here, puri-
fied soluble uPAR does not accelerate plasminogen activation by
uPA [4]. Similarly, the cleavage of cell surface uPAR by uPA
requires binding of the protease to the receptor [14], while the much
less efficient cleavage in solution is entirely independent of receptor
binding [20]. We therefore believe that VN represents the first
substrate for uPA to display a marked uPAR dependence.
Although uPA directly cleaves the RGD motif in VN, the process
is greatly accelerated when the plasminogen activation cascade is
also triggered (i.e. in the presence of plasminogen). Indeed, we
show that also cell surface-associated and free plasmin cleaves the
RGD motif in VN and that the reaction is even more efficient than
the catalysis by uPA. Although we have here sought to dissect the
relative contribution of uPA and plasmin to VN cleavage, it is impor-
tant to note that these two proteases are likely to act together physi-
ologically as they are also responsible for each other activation.
Plasmin has previously been reported to cleave VN at multiple
locations in the central and C-terminal regions of the protein
[21,22]. However, we have here demonstrated that the functional
proteolytic feedback between plasminogen activation and cell adhe-
sion can be recapitulated using a short recombinant form of VN that
contains none of the previously reported target sites, suggesting that
cleavage of the RGD motif is the functionally relevant event. Cleav-
age of VN within or close to the RGD motif has also been reported
for the serine protease granzyme B [37] and the cancer-related
serine protease tissue kallikrein 14 [38].
The overlapping binding sites for uPAR and PAI-1 in the small
N-terminal SMB domain in VN, immediately proximal to the RGD-
motif, strongly suggest the existence of relevant functional interplay
between the components of the plasminogen activation system and
cell adhesion. Consistently, such crosstalk has been documented.
Firstly, PAI-1 has been shown to act as an antagonist of cell adhe-
sion to VN through its competitive inhibition of adhesion receptors
binding to the RGD motif [39]. Secondly, both uPAR and uPA have
been characterized as agonist of cell adhesion to VN, with uPAR
acting as a bona fide VN adhesion receptor [40] and uPA as a soluble
factor increasing the affinity and avidity of uPAR for VN [13,41].
The findings presented here, starting from the discovery of cleavage
of the RGD motif by uPA and plasmin, clearly suggest a novel and
radically different view on the function of the plasminogen activa-
tion system components in the regulation of cell adhesion to VN.
Firstly, our data document that uPA and uPAR may act as negative
regulators of cell adhesion by targeting the proteolytic activity of the
plasminogen activation system towards the RGD motif. Secondly,
the data show that PAI-1 may act as a positive regulator of cell adhe-
sion by silencing the proteolytic activity of the plasminogen activa-
tion system and through stabilization of the uPAR/VN interaction.
Do uPA and uPAR promote or reduce cell adhesion to VN?
Extensive evidence demonstrates that uPAR interacts directly with
VN through a mechanism that is stimulated by concomitant ligand
occupation by uPA or its derivatives containing the N-terminal
GFD domain including sc-uPA, tc-uPA [13] and the uPA•PAI-1
complex [19]. When overexpressed by transfection, it is equally
well demonstrated that the uPAR/VN interaction leads to
increased cell adhesion, spreading and migration by mechanisms
that have been carefully investigated by others and us and have
lead to a significantly better understanding of the mechanisms by
which non-integrin adhesion receptors signal [7–9,42]. Neverthe-
less, little is known/published about the contribution of uPAR and
uPA to cell adhesion when expressed at pathophysiological levels.
Our analysis of the adhesive properties of the 38 epithelial cell
lines of the NCI-60 panel demonstrates that the mRNA expression
levels of both uPA and uPAR display non-significant correlation with
baseline cell adhesion to VN (r = 0.10; P > 0.5), suggesting that
neither of these proteins contributes significantly to baseline cell
adhesion to VN in vitro. Across the same cell lines, the expression
levels of mRNAs encoding proteins well established to be involved in
cell adhesion to VN, such as the a and b chains of the canonical VN
integrin (ITGAV/ITGB3), are strongly correlated with cell adhesion
(r = 0.5 and 0.6; P < 0.001), confirming the power of the analysis.
Importantly, however, when analysing the impact of plasminogen
activation on cell adhesion to VN, the expression level of uPA is
among the absolute strongest correlates with reduced cell adhesion.
Taken together, these findings thus suggest that the more impor-
tant function of uPA and uPAR, at least in vitro, is to enhance the
proteolytic turnover of VN and consequently to limit cell adhesion
on this substrate.
Is PAI-1 an agonist or an antagonist of cell adhesion to VN? To
act as an antagonist of cell adhesion, PAI-1 needs to saturate the
extracellular matrix VN and this activity is therefore expected in
conditions of high PAI-1 expression. Quantitative analysis of breast
carcinoma tissue protein extracts has documented a 10- to 1,000-
fold excess of VN over uPA and PAI-1 (both present at comparable
levels) [43], suggesting that the anti-adhesive activity of PAI-1 is
globally disfavoured. The concentrations of PAI-1 may locally be
sufficiently high to impact cell adhesion, but even under these
conditions, the inhibitory activity of PAI-1 is limited by its latency
conversion and complex formation with tc-uPA, both of which lead
to the dissociation of the molecule from VN. We here document that
complex formation between PAI-1 and uPA may enhance cell adhe-
sion to VN by at least three different mechanisms. Firstly, the reac-
tion of PAI-1 with uPA halts plasminogen activation and therefore
the proteolytic inactivation of VN. Secondly, we show that the
uPA•PAI-1 complex is a strong and irreversible agonist of uPAR
binding to VN. Thirdly, complex formation with uPA results in the
release of PAI-1 from matrix VN, liberating binding sites for adhe-
sion receptors. It is difficult to estimate which of these mechanisms
is dominant, in particular in vivo, but importantly, they are all
potentially active at concentrations of PAI-1 equal to or even below
those of tc-uPA. Notably, the three mechanisms by which PAI-1
may promote cell adhesion are only operational under conditions in
which active tc-uPA is generated and PAI-1 therefore behaves a
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
993
Published online: May 17, 2016

proteolysis-triggered promoter of cell adhesion. Correlation analysis
employing the NCI-60 panel of cell lines demonstrates a weak, but
significant, positive correlation between PAI-1 mRNA levels and cell
adhesion to VN, indicating that the pro-adhesive properties of PAI-1
are likely to be the most important at least in vitro.
The data presented here may have important physiological impli-
cations. It is well documented that uPAR accelerates plasminogen
activation and localizes the generated proteases on the cell surface
and we now demonstrate that the provisional matrix protein VN is a
highly specific substrate for these proteolytic activities. While plas-
min is crucial for fibrin surveillance in vivo [44], the only process
that has been well documented to require the binding of uPA to
uPAR in vivo is the suppression of inflammation secondary to fibrin
deposition [45]. We propose that while plasmin is responsible for
the resolution of fibrin deposits in the provisional matrix, the main
function of uPA/uPAR may be to bind and promote the focused
degradation of VN. Also in the context of cancer, our findings
provide novel mechanistic insight that might explain the peculiar
prognostic values of uPA and PAI-1 in certain types of tumours. The
well-documented observation that high levels of both uPA (an
enzyme) and PAI-1 (its specific inhibitor) correlate with poor prog-
nosis is known as the PAI-1 paradox [46]. We here provide mecha-
nistic evidence demonstrating that together these molecules, even if
endowed with opposite biochemical activities, cooperate in driving
the same biological process (cell adhesion to VN). This conclusion
would support a scenario in which high expression of the compo-
nents of the plasminogen activation system, when expressed
together, might act in concert to promote cancer progression
through increased fibrosis, cell adhesion and tissue stiffening, rather
than through excessive ECM degradation and turnover.
We show that small N-terminal fragments of VN consistent with
processing of the RGD motif are present in liquid biopsies of cancer
patients as well as in the urine of healthy individuals. These data
show that proteolytic processing of VN indeed occurs in vivo provid-
ing support for a physiological role of this cleavage. Although further
studies are required to determine to which extent these fragments are
A
B
Figure 8. Functional interactions between uPAR-mediated cell adhesion to VN, plasminogen activation and PAI-1.
A Binding of sc-uPA to uPAR induces conformational changes in the receptor increasing its avidity and affinity for matrix-associated VN (I) leading to increasedmechanical cell matrix coupling and subsequent cellular signalling. When at the plasma membrane, sc-uPA and Plg activate each other (II) and cleave VN in theRGD motif (IIIa). Cleavage of VN at this position inactivates the pro-adhesive properties of the molecule by destroying integrin binding and through release of theSMB domain from the extracellular matrix, thereby attenuating the mechanical coupling between cells and the extracellular matrix.
B In conditions where PAI-1 is also present, the activity of sc-uPA in promoting cell adhesion is counterbalanced by PAI-1 binding to an overlapping epitope in VN.When proteolysis is triggered, tc-uPA reacts with VN-associated PAI-1 (IIIb) resulting in the formation of a transient quaternary uPAR:uPA•PAI-1:VN complex (IV). Thecomplex formation between uPA and PAI-1 permanently inactivates the proteolytic activity of uPA and leads to major conformational changes in PAI-1 that release itfrom VN (V) thereby liberating the SMB domain for interaction with uPAR (VI).
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
994
Published online: May 17, 2016

generated by uPA and/or plasmin, it is tempting to speculate that the
levels of these fragments in liquid biopsies may be used as surrogate
biomarkers for the activity of the plasminogen activation system.
Materials and Methods
Material
Human urea-purified VN was obtained from Promega and human
FN from Trimital. Human Glu Plg, human Pli and human a2AP were
purchased from Molecular Innovations. Aprotinin was purchased
from SIGMA. Anti-uPAR monoclonal antibody R2 and R3 are a kind
gift of Dr. Gunilla Hoyer-Hansen (Finsen Laboratory, Denmark).
GFD was kindly provided by Dr. Steve Rosenberg. sc-uPA was kindly
provided by Dr. Jack Henkin (Abbott Laboratories, Abbott Park, IL).
To obtain active tc-uPA either WT or S356A, the single-chain form
was incubated with Pli (100:1, w:w ratio) for 30 min at 37°C and Pli
was subsequently inactivated with an excess of a2AP. Human tc-
uPA prepared from human urine and human recombinant LMW-uPA
were purchased from Molecular Innovations.
Expression and purification of recombinant proteins
The expression vectors described in the Appendix Supplementary
Methods were transfected into CHO Flip-In cells (Invitrogen Corp.)
and the recombinant proteins expressed under serum-free condi-
tions as previously described [7]. Recombinant Fc-tagged proteins
were purified from the conditioned media by standard Protein A
affinity chromatography and dialysed extensively against PBS. His-
tagged proteins were purified by immobilized metal-affinity chro-
matography and dialysed extensively against PBS.
Human PAI-1WT and PAI-1R103A/M112A/Q125A, extended at the
N-terminus with a His6 tag and recognition motif for heart muscle
kinase, were expressed in Escherichia coli and purified by nickel affin-
ity chromatography and size-exclusion chromatography as previously
described [47]. A fully active fraction was generated by hydrophobic
chromatography using phenyl-Sepharose and an ammonium sulphate
gradient [48]. The N-terminal extension did not affect the specific
inhibitory activity of PAI-1, its second-order rate constant for reaction
with uPA, its VN binding or its rate of latency transition [47]. The
uPA•PAI-1 complex was prepared by allowing the reaction to proceed
for 3 h at 37°C. Purification of the complex was achieved by affinity
chromatography using, in order, Sepharose-bound anti-PAI-1 Mab-2
and anti-uPA Mab-6 (monoclonal anti-uPA antibody from hybridoma
clone 6), essentially as described previously [49].
Generation of monoclonal anti-VN antibodies
C57Bl/6 VN�/� mice were immunized with VN(1–66)-Fc [7] and
splenocytes were fused to the mouse SP2/0 myeloma cell line by the
polyethylene glycol method using standard procedures [50].
Selected hybrids were subcloned by limiting dilution.
Binding assays
Binding assays were performed in black 96-well immunoplates
coated with VN (5 lg/ml) in coating buffer (50 mM sodium
carbonate, pH 9.6) at 4°C overnight (O/N). Plates were washed with
wash buffer (PBS containing 0.1% Tween-20, PBS-T) and remaining
binding sites saturated with blocking buffer (PBS containing 2%
BSA) for > 2 h at room temperature (RT). After washing with PBS-T,
wells were incubated with the indicated concentrations of uPAR-Fc
or uPARW32A/R91A-Fc diluted in PBS-T in the presence of 10 nM
sc-uPA or tc-uPA. After 1 h at RT, unbound reagents were removed
by rinsing with wash buffer. The binding of the different uPAR-Fc
variants was detected by incubation with a Eu3+-labelled anti-
human Fc antibody (1:1,000, Perkin Elmer Corp.). The Eu3+-label
was detected by measuring time-resolved fluorescence intensity
using an Envision Xcite plate reader (Perkin Elmer Corp.). Back-
ground binding was measured in wells incubated with no receptor
and was subtracted from the total binding.
Label-Free Real-Time Cell-based Assay (RTCA) experiments
Ninety-six-well E-plates (Acea Biosciences) were coated with FN
(10 lg/ml), VN (5 lg/ml) or recombinant VN variants (5 lg/ml)
O/N at 4°C and then blocked with 5% BSA in PBS for 1 h at 37°C.
HEK293 cells (15,000 cells/well) in 100 ll serum-free Opti-MEM
medium (Life Technologies Corp.) supplemented with 100 U/ml
penicillin, 100 U/ml streptomycin and 1 lg/ml tetracycline were
seeded in each well. After 15 min of pre-incubation at RT to settle
the cells, the plate was transferred to a real-time cell analyser instru-
ment (RTCA, xCELLigence SP, Roche Corp.) located in a humidified
cell culture incubator (37°C and 5% CO2). At regular intervals, the
impedance (termed cell index, CI) was recorded. Cells were
subjected to one or more treatments as reported in the figures and
the times at which treatments were performed are indicated in the
graphs by vertical lines. Final reagent concentrations were the
following: 10 nM sc-uPA, tc-uPA, GFD, PAI-1 and uPA•PAI-1, 30 nM
Plg and Pli, 100 nM a2AP and 300 nM aprotinin. To calculate the
normalized cell index, all the measured cell indexes were normalized
to the cell index recorded in the same well prior to the first
treatment.
For experiments conducted using the NCI-60 cell lines, plates
were coated with VN and blocked as described above. Cells were
seeded at 25,000 cells/well except for HS578T, HOP92, NCI H23,
NCI H522, IGROV1, OVCAR3, OVCAR4, OVCAR8, NCI ADR RES,
DU145, A498, ACHN, RXF393, SN12C, TK10 and UO31 that were
seeded at 12,500 cells/well and 786-0 and CAKI1 that were seeded
at 6,250 cells/well. The treatment was performed adding Plg and
a2AP (final concentration 30 and 100 nM, respectively), tc-uPA
(final concentration 10 nM) or vehicle diluted in Opti-MEM.
Time-lapse imaging
Time-lapse live cell imaging was performed at 37°C, 5% CO2 with
an inverted microscope (IX80, Olympus) equipped with an incuba-
tion chamber (OKOlab). Twelve-well plates were coated with VN
(5 lg/ml) O/N at 4°C and then blocked with 5% heat-inactivated
BSA in PBS for 1 h at 37°C. 293/uPAR cells (250,000 cells/well)
were plated in Opti-MEM supplemented with 100 U/ml penicillin,
100 U/ml streptomycin and 1 lg/ml tetracycline. Two hours after
seeding, sc-uPA was added to a final concentration of 10 nM and
about 1 h later Plg was added to 30 nM. Cells were imaged at regu-
lar intervals (2 min) using a 20× objective.
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
995
Published online: May 17, 2016

For cell migration experiments, 293/uPAR cells were plated and
allowed to adhere as described above. After about 2 h, cells were
treated with 10 nM sc-uPA, PAI-1, uPA•PAI-1 or vehicle and imaged
every 5 min using a 10× objective. Cell migration was quantified
using the “manual tracking” plugin of ImageJ. To obtain a time-
resolved quantification of cell speed, we calculated the average
migration speed over the 30 min preceding every time point and
followed at least 50 individual cells per condition.
Differential interference contrast (DIC) microscopy
Adherent cells were fixed with 4% paraformaldehyde in PBS for
10 min at RT and then washed with PBS. DIC imaging was
performed using an inverted microscope Olympus IX81. Cells were
viewed through a high-aperture 60× objective (UIS2 60× TIRFM
PlanApo N, NA 1.45; Olympus). Images were acquired using Hama-
matsu Orca-ER digital camera with the software Metamorph 7.5.6.0.
Cell matrix contact area was quantified using ImageJ.
Cell lysis and Western blotting
Cells were lysed directly on the culture dish in Laemmli buffer. Equal
volumes were separated by SDS–PAGE and probed as indicated.
Release experiments
For VN fragment release experiments, 12-well plates were coated
with VN or VN variants (5 lg/ml) O/N at 4°C and residual binding
sites saturated with blocking buffer (5% heat-inactivated BSA in
PBS) for 1 h at 37°C. Cells (500,000/well) were seeded in Opti-MEM
and allowed to adhere for about 2 h before further treatments. The
final concentrations of reagents used in the release experiments
were: 10 nM sc-uPA or tc-uPA, 30 nM plasminogen or plasmin,
100 nM a2AP and 300 nM aprotinin. The GFD domain (300 nM)
and mAb 8B12 (30 lg/ml) were added to the cells 30 min prior to
the addition of proteases. The conditioned media were harvested
2 h after treatment, filtered and added PMSF to 1 mM. For release
experiments conducted using NCI-60 cell lines, 50,000 cells/well
were seeded in VN-coated 48-well plates in the presence or absence
of 30 nM Plg and 100 nM a2AP. Supernatants were collected after
16 h and processed as described above. Quantification of SMB-
containing VN fragments in supernatants was performed by
immunoassay. Black 96-well immunoplates were coated with the
anti-SMB monoclonal antibody HU3 (0.5 lg/ml) diluted in coating
buffer at 4°C O/N. Plates were washed with PBS-T and blocked with
The Blocking Solution for ≥ 2 h at RT shaking. After washing with
PBS-T, wells were incubated with samples properly diluted in PBS-T.
The binding was allowed to occur 1–2 h at RT shaking. Bound
VN fragments were detected by sequential incubations with a
biotinylated anti-SMB monoclonal antibody HF6 (0.5 lg/ml) and
Eu3+-labelled streptavidin (1:10,000, Perkin Elmer Corp.) for 1 h at
RT shaking. The Eu3+-label was detected as described above. Back-
ground binding was measured in wells incubated with PBS-T and
was subtracted from the total binding. VN(1–61) was used as stan-
dard. The standard curve was fitted to the experimental data and
the unknown concentrations were interpolated by nonlinear regres-
sion using the log[agonist] vs. response—variable slope (four
parameter) algorithm and the GraphPad Prism (V6.0b) software.
MALDI-TOF mass spectrometry
For in vitro fragments generation, VN (Promega) or recombinant VN
variants were subjected to proteolytic digestion with plasmin or tc-
uPA for 2 h at 37°C. The reaction was carried out in PBS with a 10:1
(weight/weight, w/w) ratio of VN to protease. Two picomoles of
digested VN were deposited onto a MALDI plate and allowed to air-
dry. Matrix (5 mg/ml a-cyano-hydroxy-cinnamic acid (CHCA) in
50% acetonitrile/0.1% TFA) was spotted directly on top. For the
analysis of cell supernatants and human samples, a pull-down assay
using HU3-conjugated beads was performed. The ascites were
subjected to size-exclusion filtration (30 kDa cut-off) before the
immunoprecipitation. Samples were supplemented with Tris–HCl pH
7.5 (final concentration, 50 mM) and PMSF (final concentration,
1 mM), and SMB-containing VN fragments were immunoprecipitated
using the conjugated beads. Eluted material was desalted using C8
zip-tip before spotting it on the MALDI plate. Mass spectra were
acquired in linear mode on a 4800 MALDI-TOF/TOF mass spectro-
meter (Applied Biosystems, Foster City, CA) equipped with a 336-nm
nitrogen laser. Ions were accelerated with a 20-kV pulse. Spectra
were generated in the mass range 2–20 kDa by averaging 40
subspectra for each of 25 randomized positions within the spot (1,000
spectra/spot). Laser intensity was set at 4,000 V to optimize the
signal-to-noise ratio and the resolution of mass peaks of the analyte.
Human samples
Fresh ascites were obtained upon informed consent from patients
with high-grade serous ovarian carcinoma who underwent paracen-
tesis. Peritoneal fluid was centrifuged at 16.6 g for 5 min and the
supernatant was collected and stored at �80°C.
The urine sample was obtained from a healthy volunteer. The
sample was snap-frozen and stored at �80°C. Before analysis, the
sample was thawed at 37°C and centrifuged.
Expanded View for this article is available online.
AcknowledgementsThis work was supported by research grants from Associazione Italiana per la
Ricerca sul Cancro (AIRC) hold by NS (grant number IG 14466) and research
fellowships from Fondazione Italiana per la Ricerca sul Cancro (FIRC) and AIRC
to GMSF and VDL, respectively.
Author contributionsVDL and NS conceived the study and analysed the data. VDL, GMSF, JBM and
ML conducted the experimental work and collected human samples. VDL, PAA
and NS wrote the manuscript.
Conflict of interestThe authors declare that they have no conflict of interest.
References
1. Smith HW, Marshall CJ (2010) Regulation of cell signalling by uPAR. Nat
Rev Mol Cell Biol 11: 23 – 36
2. Ferraris GM, Sidenius N (2013) Urokinase plasminogen activator
receptor: a functional integrator of extracellular proteolysis, cell
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
996
Published online: May 17, 2016

adhesion, and signal transduction. Semin Thromb Hemost 39:
347 – 355
3. Miles LA, Parmer RJ (2013) Plasminogen receptors: the first quarter
century. Semin Thromb Hemost 39: 329 – 337
4. Ellis V, Behrendt N, Danø K (1991) Plasminogen activation by receptor-
bound urokinase. A kinetic study with both cell-associated and isolated
receptor. J Biol Chem 266: 12752 – 12758
5. Cubellis MV, Wun TC, Blasi F (1990) Receptor-mediated internalization
and degradation of urokinase is caused by its specific inhibitor PAI-1.
EMBO J 9: 1079 – 1085
6. Nykjaer A, Conese M, Christensen EI, Olson D, Cremona O, Gliemann J,
Blasi F (1997) Recycling of the urokinase receptor upon internalization
of the uPA:serpin complexes. EMBO J 16: 2610 – 2620
7. Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N (2007)
uPAR-induced cell adhesion and migration: vitronectin provides the key.
J Cell Biol 177: 927 – 939
8. Smith HW, Marra P, Marshall CJ (2008) uPAR promotes formation of the
p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol 182:
777 – 790
9. Ferraris GM, Schulte C, Buttiglione V, De Lorenzi V, Piontini A, Galluzzi
M, Podesta A, Madsen CD, Sidenius N (2014) The interaction between
uPAR and vitronectin triggers ligand-independent adhesion signalling by
integrins. EMBO J 33: 2458 – 2472
10. Eastman BM, Jo M, Webb DL, Takimoto S, Gonias SL (2012)
A transformation in the mechanism by which the urokinase receptor
signals provides a selection advantage for estrogen receptor-expressing
breast cancer cells in the absence of estrogen. Cell Signal 24:
1847 – 1855
11. Bifulco K, Longanesi-Cattani I, Franco P, Pavone V, Mugione P, Di Carluccio
G, Masucci MT, Arra C, Pirozzi G, Stoppelli MP et al (2012) Single amino
acid substitutions in the chemotactic sequence of urokinase receptor
modulate cell migration and invasion. PLoS One 7: e44806
12. Pirazzoli V, Ferraris GM, Sidenius N (2013) Direct evidence of the impor-
tance of vitronectin and its interaction with the urokinase receptor in
tumor growth. Blood 121: 2316 – 2323
13. Gardsvoll H, Ploug M (2007) Mapping of the Vitronectin-binding Site on
the Urokinase Receptor: involvement of a coherent receptor interface
consisting of residues from both domain I and the flanking interdomain
linker region. J Biol Chem 282: 13561 – 13572
14. Hoyer-Hansen G, Ploug M, Behrendt N, Ronne E, Dano K (1997) Cell-
surface acceleration of urokinase-catalyzed receptor cleavage. Eur J
Biochem/FEBS 243: 21 – 26
15. Sidenius N, Blasi F (2000) Domain 1 of the urokinase receptor (uPAR) is
required for uPAR-mediated cell binding to vitronectin. FEBS Lett 470:
40 –46
16. Conforti G, Dominguez-Jimenez C, Ronne E, Hoyer-Hansen G, Dejana E
(1994) Cell-surface plasminogen activation causes a retraction of
in vitro cultured human umbilical vein endothelial cell monolayer. Blood
83: 994 – 1005
17. Atienza JM, Zhu J, Wang X, Xu X, Abassi Y (2005) Dynamic monitoring of
cell adhesion and spreading on microelectronic sensor arrays. J Biomol
Screen 10: 795 – 805
18. Hall SW, Humphries JE, Gonias SL (1991) Inhibition of cell surface recep-
tor-bound plasmin by alpha 2-antiplasmin and alpha 2-macroglobulin.
J Biol Chem 266: 12329 – 12336
19. Kanse SM, Kost C, Wilhelm OG, Andreasen PA, Preissner KT (1996) The
urokinase receptor is a major vitronectin-binding protein on endothelial
cells. Exp Cell Res 224: 344 – 353
20. Hoyer-Hansen G, Ronne E, Solberg H, Behrendt N, Ploug M, Lund LR,
Ellis V, Dano K (1992) Urokinase plasminogen activator cleaves its cell
surface receptor releasing the ligand-binding domain. J Biol Chem 267:
18224 – 18229
21. Kost C, Benner K, Stockmann A, Linder D, Preissner KT (1996) Limited
plasmin proteolysis of vitronectin. Characterization of the adhesion
protein as morpho-regulatory and angiostatin-binding factor. Eur J
Biochem 236: 682 – 688
22. Chain D, Kreizman T, Shapira H, Shaltiel S (1991) Plasmin cleavage of
vitronectin. Identification of the site and consequent attenuation in
binding plasminogen activator inhibitor-1. FEBS Lett 285: 251 – 256
23. Reinartz J, Schäfer B, Batrla R, Klein CE, Kramer MD (1995)
Plasmin abrogates alpha v beta 5-mediated adhesion of a human kerati-
nocyte cell line (HaCaT) to vitronectin. Exp Cell Res 220: 274 – 282
24. Ishikawa-Sakurai M, Hayashi M (1993) Two collagen-binding domains of
vitronectin. Cell Struct Funct 18: 253 – 259
25. Zhao B, Gandhi S, Yuan C, Luo Z, Li R, Gardsvoll H, de Lorenzi V, Sidenius
N, Huang M, Ploug M (2015) Stabilizing a flexible interdomain hinge
region harboring the SMB binding site drives uPAR into its closed
conformation. J Mol Biol 427: 1389 – 1403
26. Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ (1996) Is plas-
minogen activator inhibitor-1 the molecular switch that governs uroki-
nase receptor-mediated cell adhesion and release? J Cell Biol 134:
1563 – 1571
27. Jensen PH, Christensen EI, Ebbesen P, Gliemann J, Andreasen PA (1990)
Lysosomal degradation of receptor-bound urokinase-type plasminogen
activator is enhanced by its inhibitors in human trophoblastic
choriocarcinoma cells. Cell Regul 1: 1043 – 1056
28. Jensen JK, Durand MK, Skeldal S, Dupont DM, Bodker JS, Wind T,
Andreasen PA (2004) Construction of a plasminogen activator
inhibitor-1 variant without measurable affinity to vitronectin but other-
wise normal. FEBS Lett 556: 175 – 179
29. Lawrence DA, Palaniappan S, Stefansson S, Olson ST, Francis-Chmura
AM, Shore JD, Ginsburg D (1997) Characterization of the binding of dif-
ferent conformational forms of plasminogen activator inhibitor-1 to
vitronectin. Implications for the regulation of pericellular proteolysis. J
Biol Chem 272: 7676 – 7680
30. Behrendt N, List K, Andreasen PA, Dano K (2003) The pro-urokinase plas-
minogen-activation system in the presence of serpin-type inhibitors and
the urokinase receptor: rescue of activity through reciprocal pro-enzyme
activation. Biochem J 371: 277 – 287
31. Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C,
Langley J, Cronise P, Vaigro-Wolff A et al (1991) Feasibility of a high-flux
anticancer drug screen using a diverse panel of cultured human tumor
cell lines. J Natl Cancer Inst 83: 757 – 766
32. Reinhold WC, Sunshine M, Varma S, Doroshow JH, Pommier Y (2015)
Using Cell Miner 1.6 for Systems Pharmacology and Genomic Analysis of
the NCI-60. Clin Cancer Res 21: 3841 – 3852
33. Pierschbacher MD, Ruoslahti E (1984) Cell attachment activity of fibro-
nectin can be duplicated by small synthetic fragments of the molecule.
Nature 309: 30 – 33
34. Ruoslahti E (1996) RGD and other recognition sequences for integrins.
Annu Rev Cell Dev Biol 12: 697 – 715
35. Cherny RC, Honan MA, Thiagarajan P (1993) Site-directed mutagenesis
of the arginine-glycine-aspartic acid in vitronectin abolishes cell adhe-
sion. J Biol Chem 268: 9725 – 9729
36. Behrens MA, Botkjaer KA, Goswami S, Oliveira CL, Jensen JK, Schar CR,
Declerck PJ, Peterson CB, Andreasen PA, Pedersen JS (2011) Activation of
ª 2016 The Authors EMBO reports Vol 17 | No 7 | 2016
Valentina De Lorenzi et al Cleavage of the RGD motif regulates cell adhesion EMBO reports
997
Published online: May 17, 2016

the zymogen to urokinase-type plasminogen activator is associated with
increased interdomain flexibility. J Mol Biol 411: 417 – 429
37. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, Froelich CJ,
Nice EC, Bird PI (2005) Extracellular matrix remodeling by human gran-
zyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem
280: 23549 – 23558
38. Borgono CA, Michael IP, Shaw JL, Luo LY, Ghosh MC, Soosaipillai A, Grass
L, Katsaros D, Diamandis EP (2007) Expression and functional characteri-
zation of the cancer-related serine protease, human tissue kallikrein 14.
J Biol Chem 282: 2405 – 2422
39. Stefansson S, Lawrence DA (1996) The serpin PAI-1 inhibits cell migra-
tion by blocking integrin alpha V beta 3 binding to vitronectin. Nature
383: 441 – 443
40. Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA
(1994) Identification of the urokinase receptor as an adhesion receptor
for vitronectin. J Biol Chem 269: 32380 – 32388
41. Caiolfa VR, Zamai M, Malengo G, Andolfo A, Madsen CD, Sutin J, Digman
MA, Gratton E, Blasi F, Sidenius N (2007) Monomer dimer dynamics and
distribution of GPI-anchored uPAR are determined by cell surface
protein assemblies. J Cell Biol 179: 1067 – 1082
42. Kjøller L, Hall A (2001) Rac mediates cytoskeletal rearrangements and
increased cell motility induced by urokinase-type plasminogen activator
receptor binding to vitronectin. J Cell Biol 152: 1145 – 1157
43. Aaboe M, Offersen BV, Christensen A, Andreasen PA (2003) Vitronectin in
human breast carcinomas. Biochim Biophys Acta 1638: 72 – 82
44. Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL
(1996) Loss of fibrinogen rescues mice from the pleiotropic effects of
plasminogen deficiency. Cell 87: 709 – 719
45. Connolly BM, Choi EY, Gardsvoll H, Bey AL, Currie BM, Chavakis T, Liu S,
Molinolo A, Ploug M, Leppla SH et al (2010) Selective abrogation of the
uPA-uPAR interaction in vivo reveals a novel role in suppression of fib-
rin-associated inflammation. Blood 116: 1593 – 1603
46. Kwaan HC, Mazar AP, McMahon BJ (2013) The apparent uPA/PAI-1 para-
dox in cancer: more than meets the eye. Semin Thromb Hemost 39:
382 – 391
47. Jensen JK, Wind T, Andreasen PA (2002) The vitronectin binding area of
plasminogen activator inhibitor-1, mapped by mutagenesis and protec-
tion against an inactivating organochemical ligand. FEBS Lett 521:
91 – 94
48. Kvassman JO, Lawrence DA, Shore JD (1995) The acid stabilization of
plasminogen activator inhibitor-1 depends on protonation of a single
group that affects loop insertion into beta-sheet A. J Biol Chem 270:
27942 – 27947
49. Skeldal S, Larsen JV, Pedersen KE, Petersen HH, Egelund R, Christensen
A, Jensen JK, Gliemann J, Andreasen PA (2006) Binding areas of uroki-
nase-type plasminogen activator-plasminogen activator inhibitor-1
complex for endocytosis receptors of the low-density lipoprotein recep-
tor family, determined by site-directed mutagenesis. FEBS J 273:
5143 – 5159
50. Galfre G, Howe SC, Milstein C, Butcher GW, Howard JC (1977) Antibodies
to major histocompatibility antigens produced by hybrid cell lines.
Nature 266: 550 – 552
51. Rønne E, Behrendt N, Ellis V, Ploug M, Danø K, Høyer-Hansen G (1991)
Cell-induced potentiation of the plasminogen activation system is abol-
ished by a monoclonal antibody that recognizes the NH2-terminal
domain of the urokinase receptor. FEBS Lett 288: 233 – 236
EMBO reports Vol 17 | No 7 | 2016 ª 2016 The Authors
EMBO reports Cleavage of the RGD motif regulates cell adhesion Valentina De Lorenzi et al
998
Published online: May 17, 2016