uric acid as a growth factor for activated b … · uric acid as a growth factor for activated b...
TRANSCRIPT

1
URIC ACID AS A GROWTH FACTOR FOR ACTIVATED B CELLS
by
Hajera Khaja
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Graduate Department of Medical Biophysics
University of Toronto
© Copyright by Hajera Khaja 2009

ii
ABSTRACT
Uric acid as a growth factor for activated B cells
Hajera Khaja
Master of Science 2009
Graduate Department of Medical Biophysics
University of Toronto
Cancer vaccines targeted against tumour cells seek to mimic immune responses against
viral infections. Given the unique properties of B cells that allow them to present antigens
proficiently, activated B cells can be used in a vaccine setting to launch effective anti-tumour T
cell responses. Activation induced cell death, however, presents a major hurdle in the
generation of large numbers of B cells. Since apoptosis is mediated by oxidative stress, uric
acid was used as an antioxidant, enabling increased growth of normal B cells. Further
investigation in TK6 cells revealed that uric acid was mediating its effects extracellularly and
initiating signaling pathways that culminated in the activation of ERK and JNK proteins and
production of IL10, which was necessary, but not sufficient, in mediating the growth effects of
uric acid. The results outlined in this study implicate uric acid as a novel growth factor for
activated B cells.

iii
ACKNOWLEDGEMENTS
First and foremost, my sincerest gratitude goes out to Dr. David Spaner, my research
supervisor, for allowing me to pursue my graduate studies in his laboratory. David is an
exceptional educator and I would have faltered many times over were it not for his constant
guidance and support. Moreover, David‟s enthusiasm for scientific inquiry enabled me to vastly
broaden my areas of research and I greatly appreciate his advice and expertise. Many thanks are
also due to my committee members, Dr. Yaacov Ben-David, Dr. James Booth, and Dr. Richard
Wells for their advice and assistance.
It has been a pleasure to have been a member of the Spaner lab. To my labmates, Jelena,
Yonghong, Caitlin, Dave, and Suchi, your help and guidance is much appreciated. To Jelena,
Yonghong and Caitlin in particular, thank you kindly for your advice and friendship – I could
not have made it through grad school without you.
To my parents, Fareed and Wajeda, I cannot thank you enough for your unconditional
support and love. Your encouragement has always been a blessing and I strive to excel for you.
To my sisters, Javeria and Aisha, thank you for your enthusiasm and your prayers. I also owe
immense gratitude to my parents-in-law, Shabir and Sabitun, for their support and
encouragement. You are truly a blessing. To friends who have cheered from the sidelines:
Aaida, Asmaa, Asma Ali and Safiyyah, I am blessed to have your friendship and am overjoyed
to have been able to celebrate this milestone with you. To Aaida especially, you are an
unrelenting source of encouragement and I value your friendship dearly.
Last, but certainly not the least, I owe an immeasurable amount of gratitude to my
husband, Idrees, for his unwavering support and untiring optimism. Thank you for always being
there when I have needed you and for making this journey worthwhile. This thesis could not
have transpired without you and is in fact, more yours than it is mine.

iv
TABLE OF CONTENTS
Abstract ......................................................................................................................................... ii
Acknowledgements ...................................................................................................................... iii
Table of contents .......................................................................................................................... iv
List of figures................................................................................................................................ vi
List of abbreviations ................................................................................................................... vii
Chapter 1: Introduction and research hypotheses .................................................................... 1
1.1 Introduction........................................................................................................................... 2
1.1.1 Cancer Immunosurveillance .......................................................................................... 2
1.1.2 Immune Evasion: The Seventh Hallmark of Cancer .................................................... 5
1.1.3 Cancer Immunotherapy ................................................................................................. 7
1.1.4 Dendritic Cells and Cancer Vaccines ............................................................................ 9
1.1.5 B Cells as Alternative Antigen Presenting Cells ......................................................... 11
1.1.6 B Cell Expansion Protocol .......................................................................................... 14
1.1.6.1 Toll-like Receptor 7 ............................................................................................. 15
1.1.6.2 Interleukin 2 ......................................................................................................... 18
1.1.6.3 IL2 and TLR7 Stimulated B Cells ....................................................................... 20
1.1.7 Activation-Induced Cell Death in B Lymphocytes ..................................................... 21
1.1.7.1 Reactive Oxygen Species and AICD ................................................................... 22
1.1.8 Oxidative Stress and Uric Acid ................................................................................... 23
1.1.9 Epstein-Barr Virus Cell Lines as a Model System for Studies in Activated B Cells .. 27

v
1.2 Rationale and research hypotheses ..................................................................................... 30
Chapter 2: Materials and methods ........................................................................................... 31
Chapter 3: Results ...................................................................................................................... 39
Chapter 4: Discussion and future directions ............................................................................ 56
4.1 Discussion ........................................................................................................................... 57
4.2 Future directions ................................................................................................................. 63
List of references .......................................................................................................................... 68

vi
LIST OF FIGURES
Figure 1. TLR7 signaling in B cells.
17
Figure 2. Murine B cell cultures stimulated with IL2 and a TLR7 agonist
undergo proliferation and experience AICD which is partially
reversed by uric acid.
41
Figure 3. Growth promoting effects of uric acid in activated human B cell
cultures.
44
Figure 4. Uric acid rescues TK6 cells from death associated with serum
withdrawal and displays functions beyond an antioxidant role.
47
Figure 5. Uric acid does not function as a metabolite as determined by
inhibition of uric acid transporters and G protein-coupled receptors.
50
Figure 6. Activation of signaling pathways and cytokine production by uric
acid-treated TK6 cells.
52
Figure 7. IL10 inhibition leads to loss of uric acid‟s proliferative potential in
TK6 cells.
54
Figure 8. Mechanism of uric acid‟s growth effects on activated B cells.
55

vii
LIST OF ABBREVIATIONS
∆ψm Mitochondrial membrane potential
2‟,5‟-dd-Ado 2‟,5‟-Dideoxyadenosine
2-ME 2-mercaptoethanol
7AAD 7-aminoactinomycin D
AC Adenylate cyclase
Ad D/F Advanced Dulbecco‟s modified Eagle‟s medium: Nutrient Mixture F12
AICD Activation-induced cell death
AIF Apoptosis-inducing factor
AP-1 Activator protein-1
APC Antigen presenting cell
BCR B cell receptor
Ca2+
Calcium
CLL Chronic lymphocytic leukemia
COX Cyclooxygenase
DC Dendritic cell
DPA Dipyridamole
EBV Epstein-Barr Virus
ERK Extracellular signal-regulated kinase
FBS Fetal bovine serum
GM-CSF Granulocyte-macrophage colony stimulating factor
GPCR G protein-coupled receptor
H-89 H-89, Dihydrochloride
HIV Human immunodeficiency virus
HLA Human leukocyte antigen

viii
HPRT Hypoxanthine-guanine phosphoribosyltransferase
IBMX 3-isobutyl-1-methylxanthine
IDO Indoleamine 2,3-dioxygenase
IFN Interferon
Ig Immunoglobulin
IκB Inhibitor of NF-κB
IKK IκB kinase complex
IL Interleukin
IRAK IL1 receptor-associated kinase
ITAG Insulin, Transferrin, Albumin, Glutamine
ITAM Immunoreceptor tyrosine-based activation motifs
JAK Janus kinase
JNK Jun N-terminal kinase
LMP Latent membrane protein
LRR Leucine-rich repeat
mAb Monoclonal antibody
MAPK Mitogen-activated protein kinase
MCP Monocyte chemoattractant protein
M-CSF Macrophage colony-stimulating factor
MDL-12 MDL-12,330A, Hydrochloride
MHC Major histocompatibility complex
MIIC MHC II rich compartment
MSU Monosodium urate
MyD88 Myeloid differentiation primary-response protein
NF-κB Nuclear factor- κB
NK Natural killer

ix
NOS Nitric oxide synthase
OAT Organic anion transporter
PBS Phosphate buffered saline
PDE Phosphodiesterase
PI Phosphatidylinositol
PKA Protein Kinase A
PKC Protein kinase C
PT Permeability transition
RAG Recombination-activating gene
redox Reduction/oxidation
ROS Reactive oxygen species
RPMI Roswell Park Memorial Institute medium
RT-PCR Reverse transcriptase polymerase chain reaction
S2 B cells S28690- and IL2-treated B cells
SAPK Stress-activated protein kinase
STAT Signal transducer and activator of transcription
TAB TAK1-binding protein
TAK TGFβ-activated kinase
TAP Transporter associated with antigen processing
TGF Transforming growth factor
TH T helper cell
TIR Toll/IL1 receptor
TLR Toll-like receptor
TNF tumour necrosis factor
TRAF TNF receptor associated factor
TRAIL TNF-related apoptosis-inducing ligand

x
TReg Regulatory T cell
URAT Uric acid transporter
VEGF Vascular endothelial growth factor
VSMC Vascular smooth muscle cell

1
Chapter 1:
INTRODUCTION AND RESEARCH HYPOTHESES

2
1.1 INTRODUCTION
1.1.1 Cancer Immunosurveillance
The hypothesis of cancer immunosurveillance argues that the immune system can
recognize cancer precursors as foreign entities and eliminate them from the body [1,2]. After
nearly a century of debate, it is now clear that effector cells, from both the innate and adaptive
immune systems, are involved in the recognition and destruction of tumours prior to the clinical
manifestation of cancer.
Several lines of evidence point to the veracity of immunosurveillance. Both occasional
spontaneous regression of tumours in immunocompetent hosts and increased prevalence of
cancer in immunocompromised patients suggest an involvement of the immune system in
keeping tumour progression in check. In a recent study, an examination of the rate of cancer
incidence in heart and/or lung transplant patients revealed that suppression of the immune
system in order to prevent graft rejection led to a dramatic increase in newly diagnosed cancers
in these recipients, and was 7.1 times greater than the rate for the general population [3].
Leukemias and lymphomas were the most common type of cancer detected with a rate that was
26.2 times as many as in the general population. While it is possible that the
immunosuppressive drugs used are mutagenic, another study revealed that the degree of natural
cytotoxicity of blood impacted cancer risk – a study conducted in Japan followed more than
3000 healthy people for 11 years and showed that subjects whose blood lymphocytes had a low
degree of natural cytotoxicity at the start of the investigation had a significantly higher risk of
developing cancer than did subjects whose lymphocytes had a medium or high degree of
cytotoxicity [4].

3
Experimental animal models have also confirmed the existence of anti-tumour
immunity. Studies in mice have shown that when essential parts of the innate or adaptive
immune system are lacking, a corresponding increase in susceptibility to the development of
spontaneous or chemically induced tumours follows. This includes animals in which knockouts
and other experimental manipulations have resulted in the elimination of certain cells of the
immune system, such as T cells, B cells and natural killer T (NKT) cells brought about through
lack of the recombination-activating gene 2 (RAG2), as well as animals missing important
effector genes and molecules, such as interferon (IFN) receptor 1; the transcription factor,
signal transducer and activator of transcription 1 (STAT1), required for IFN-induced signaling;
perforin; or tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [5].
Conversely, a reduction in the development of malignant disease in mouse models can be
brought about by the administration of an immunostimulatory regimen designed to increase the
number of immune effector cells [6].
Another line of evidence indicating that the immune system appears cognizant of
tumours in the body arises from the observation that an accumulation of tumour-infiltrating T
lymphocytes at tumour sites often correlates with improved prognosis [7]. A study of three
multicenter cohorts of colon cancer patients showed that rather than tumour stage and nodal
status, the current gold standards, a more accurate predictor of clinical outcome was the
presence or absence of T cells in the resected tumour [8]. Tumour samples from patients with
cervical cancer [9], breast cancer [10], urothelial carcinoma [11], and follicular lymphoma [12],
revealed similar observations. Likewise, results reported from a colorectal carcinoma study
revealed that when patients were found to express messenger transcripts encoding molecules
made by effector T helper 1 (TH1) and effector memory T cells, a reduced rate of metastasis and
increased survival time was observed compared to patients in which such molecules were absent

4
[13]. On the other hand, an infiltration of regulatory T (TRegs) cells in the tumour predicts
diminished patient survival in the cases of ovarian carcinoma or melanoma [14,15].
As is apparent from the studies cited above, several diverse processes are employed by
the immune system to keep tumour growth in check upon detection. As a consequence of the
genotoxic stress of transformation, tumour growth can stir changes in the surrounding
microenvironment, resulting in the release of “danger” signals. Ideally, cells of the innate
immune system recognize these signals and induce inflammation alongside activating innate
effector cells that possess anti-tumour activity. Further action is taken by professional antigen-
presenting cells (APCs) which engulf tumour-derived antigens and in the context of major
histocompatibility (MHC) proteins, present them to B and T cells in the draining lymph nodes,
thereby triggering the adaptive immune system [16]. Effectors of the adaptive response include
T cells that express clonally distributed antigen receptors. When allied with MHC proteins, the
T cell receptors can recognize either unique tumour antigens, such as those resulting from
mutant or viral-derived proteins, or self-antigens, such as those derived from overexpressed
oncogenic proteins or aberrantly expressed proteins that are normally developmentally or tissue-
restricted [17].
Certain conditions must be met before effector T cells can mediate effective anti-tumour
responses – T cells must be activated by bone marrow-derived APCs that present tumour
antigens and also provide the necessary costimulatory signals; they must be able to migrate with
efficiency and access the tumour microenvironment; and they should be able to overcome
obstacles posed by the tumour and launch an effective attack that will not be abruptly withheld
and result in the induction of tolerance. When effective, T cell activation can lead to cessation
of tumour growth via production of inflammatory cytokines such as IFN and TNF. T cells

5
can also cause direct lysis of malignant cells mediated by perforin and/or Fas and lead to
inhibition of angiogenesis [17].
1.1.2 Immune Evasion: The Seventh Hallmark of Cancer
Despite the presence of a complex surveillance programme, the existence of a tumour in
the body is indicative of its ability to thwart detection and/or evade the immune response
launched against it. In fact, it has been postulated that apart from the six known hallmarks of
cancer (self-sufficiency in growth signals; insensitivity to anti-growth signals; evasion of
apoptosis; limitless replicative potential; sustained angiogenesis; and tissue invasion and
metastasis) [18], evasion of immunosurveillance could be the seventh hallmark of cancer
[19,20]. Due to the selective pressure exerted by the immune system (immunoselection),
tumours have developed a sophisticated array of strategies that allow them to escape the various
devices involved in immunosurveillance [2]. Developing tumours can initiate processes that
encourage evasion from immune recognition [16], suppress the triggering of an inflammatory
response via secretion of immunosuppressive factors [21], or escape death by blocking the
production of pro-inflammatory molecules by effector cells [22]. Even if evasion proves to be
unsuccessful, a cancerous growth can escape elimination via various mechanisms such as
deletion of targeted antigens to prevent immune attack, inducing growth and promotion of
regulatory T cells, and inducing anergy in, or causing deletion of tumour-reactive T cells
[23,24].
Downregulation or loss of expression of MHC class I molecules and disruption of
antigen processing and presentation is a frequent strategy designed to specifically dodge a T-cell
mediated adaptive response. Loss of MHC I expression is often observed in lung cancer and has
also been recorded in several epithelial cell cancers and melanomas. In fact, it has been

6
proposed that the only tumour cells that progress into malignancy are those that have undergone
immunoselection owing to their loss of MHC class I expression, allowing them to escape
immune attack and develop into advanced cancer [25]. In the same vein, tumours frequently
downregulate molecules that partake in the processing and presentation of antigen by MHC I
molecules; for example, during the development of colorectal carcinoma, a progressive loss in
the expression of proteins involved in antigen processing, such as transporter associated with
antigen processing 1 (TAP1), low-molecular-mass protein 2 and 7, and tapasin has been
reported [26].
Immunosubversion, or the active suppression of the immune response, is yet another
tactic whereby tumour cell products can engage in direct repression of immune machinery [2].
For instance, increased arginase-1 activity and overproduction of nitric oxide by some tumours
can lead to inhibition of T cell function [27]. Constitutive overexpression of indoleamine 2,3-
dioxygenase (IDO) in many tumours, in particular prostate, colon and pancreatic carcinomas
[28], can not only block proliferation of CD8+ T cells at the local tumour site [28], but can also
induce apoptosis of CD4+ T cells [29]. The tumour microenvironment also evolves to aid in the
evasion of immunosurveillance and suppression of immune attack [30]. Various
immunosuppressive factors that can inhibit the differentiation, maturation, and function of
dendritic cells (DCs) can be found in tumour beds. These include the vascular endothelial
growth factor (VEGF), Interleukin (IL)6, IL10, transforming growth factor (TGF),
macrophage colony-stimulating factor (M-CSF), nitric oxide synthase (NOS)2, arginase-1, IDO,
and others [30]. Such factors can jeopardize anticancer immunity, which often relies on TH1-
cell responses, by polarizing the T-cell response from a TH1 response to a TH2 response, thus
rendering the immune response against the tumour ineffective [31]. Furthermore, it has been
demonstrated in mouse models that advanced cancer invariably undermines the anti-tumour

7
immune response. At the initial stages of tumour growth, tumour-specific CD8+ T cells are
found to be activated, but eventually lose their cytotoxicity as the tumour expands [32].
Tumour-specific CD4+ T cells also sustain a progressive loss in their anti-tumour activity [33],
coinciding with a rise in the number of regulatory T cells [34]. Taken together, it appears that
induction of tolerance towards the tumour may be a prerequisite for the initial steps of
tumorigenesis [35].
There is hence a ceaseless cycle involved in the advent of cancer with the immune
system recognizing the tumour as a foreign entity and subsequently attacking it, and the tumour
evading and suppressing this response [19]. The progressive selection of weakly immunogenic
and/or immune-resistant malignant cells forms the eventual tumour that can spread in an
uninhibited fashion throughout the body [2]. It is starkly apparent that to win the fight against
cancer, it is necessary not only to develop chemotherapeutic strategies to efficiently kill all
cancer cells, but also to attempt to re-stimulate an immune response so that residual tumour cells
can be kept in check [36].
1.1.3 Cancer Immunotherapy
Despite the versatility of cancer, it is possible to reactivate a defunct immune system into
an anti-tumour state. Studies in both experimental models and cancer patients have shown that
augmenting natural anti-tumour responses can lead to therapeutic benefits [17]. Passive
antibody transfer into cancer patients was one of the first immunotherapeutic strategies
employed to control tumour growth. The initial testing of mouse monoclonal antibodies (mAb)
targeting the IL2 receptor expressed by many T cell leukemias and lymphomas in humans
provided proof of concept for the efficacy of this form of therapy [37]. Various mAbs are now
administered as part of standard treatment regimens. Rituximab, one of the most extensively

8
used mAb, binds CD20 and can induce high remission rates in patients with B cell lymphomas
[38]. Clinical effectiveness has been achieved in other cancers also, such as the administration
of mAbs against tumour antigens in HER2-positive breast cancer (trastuzumab) [39], and head
and neck, lung, and colorectal cancers that express the epidermal growth factor receptor
(cetuximab) [40-42]. A challenge that still needs to be addressed is improving anti-tumour
activity without causing widespread toxicity to normal tissues; selective targeting of the mAb to
tumour sites is one plausible solution that needs to be further explored [43].
Another type of immunotherapy involves the intravenous administration of tumour-
specific autologous T cells that have been grown and expanded in vitro [44]. In a phase 1 study,
patients with metastatic melanoma were treated with infusions of CD8+ T cells specific for the
melanoma antigens MART-1, Melan-A, and glycoprotein 100 (gp100). T cell migration to the
tumour sites and metastatic regression was observed in 8 out of 10 patients [45]. Such studies
reveal that it is possible to harness the body‟s natural defences against cancer by devising
effective immunotherapeutic strategies based on the re-stimulation of an anti-tumour immune
response. As simple as it may sound, exploring this avenue of therapy is challenging for the
reason that most tumour antigens are unable to trigger a strong immune response, mainly due to
tolerance resulting from the fact that the vast majority of tumour-derived antigens are normal
proteins which are aberrantly expressed by cancer cells [43]. While some antigens may be
strongly immunogenic, the lack of a correspondingly strong immune attack reflects the
inconducive environment in which these antigens are presented.
Although administration of pre-formed tumour-specific antibodies or autologous T cells
is beneficial in initiating an immune response, such passive routes to immunotherapy will likely
not lead to the induction of long-lived memory T cells that have the ability to control tumour
outgrowth in the event of a relapse. A more attractive alternative is active immunotherapy,

9
involving the use of cancer vaccines derived from professional APCs that can display tumour
antigens effectively, thereby allowing the vaccine to directly elicit or boost similar anti-tumour
antibodies and T cells in the patient [43]. Such an active approach is crucial to the induction of
productive T cell immunity involving both tumour-specific effector T cells and memory T cells
[46].
1.1.4 Dendritic Cells and Cancer Vaccines
Dendritic cells (DCs), activated macrophages, and activated B cells all possess the
capability to present peptides. DCs however, are considered to be the most efficient APCs as
they are not only exceedingly proficient at antigen capture and processing, but can also migrate
efficiently [47]. Encouraging data from animal models led to promising results in the first
report on DC vaccination in 1996 [48]. Since then, autologous DCs generated ex vivo and
loaded with antigen have been used as vaccines in various trials with melanoma, lymphoma, and
carcinoma patients, and have proven to be safe and effective [48-52]. Many different
approaches to the creation of DC vaccines have culminated in >100 clinical trials to date [53].
The generation of DC vaccines involves ex vivo isolation of DC subtypes from
peripheral blood and subsequent loading of tumour-derived or tumour-specific antigen followed
by reinjection. Clinically applicable protocols for DC differentiation and maturation have been
established for successfully generating immunostimulatory DCs from both proliferating CD34+
precursors and non-proliferating CD14+ monocytes [54,55]. Several tumour-derived and
tumour-associated products have been used as antigenic stimulants, including whole tumour
lysates, apoptotic or necrotic tumour debris, recombinant proteins, synthetic peptides, plasmid
DNA, RNA, and viral vectors [56]. Such mature and differentiated DCs can also successfully
migrate to secondary lymphoid organs [57]. Examples of cancer vaccines designed under such

10
protocols are those generated against breast cancer (the HER2 antigen) [58], melanoma (DCs
loaded with tumour peptides or a mixture of necrotic and apoptotic tumour cells) [59,60], and
prostate cancer (DCs loaded with prostatic acid phosphatase) [61]. Evidence of an immune
response to the vaccines was demonstrated in each of these trials, and clinical responses with
minimal or adverse effects were also observed in a few cases.
Many issues regarding the use of DC vaccines, however, such as optimal method of
generation, delivery and dosage, remain to be resolved [56,62,63]. Furthermore, despite their
ability to induce T cell immunity with high efficiency, the use of DC-based cancer vaccines is
fraught with difficulties. Firstly, DCs are relatively scarce in peripheral blood and comprise
only 0.1 – 0.5% of human peripheral blood mononuclear cells, necessitating isolation via
apheresis [48] or from marrow sources [64]. Consequently, it is considerably difficult and
expensive to obtain adequate numbers of purified mature DCs [65]. Secondly, the use of DCs as
APCs presents a special problem in that the DC population is not homogenous but consists of
several subsets of functionally distinct cell types [66], including tolerogenic DCs [67].
Furthermore, the generation of sufficient numbers of DCs from non-stem cell sources requires
considerable expansion with cytokines like granulocyte M-CSF and IL4 [68]. Such expansion
protocols yield functional DCs, but with limited growth potential – within 2-3 weeks, DCs cease
to proliferate and also become less efficient in their antigen presenting ability [69]. Multiple
phlebotomies are thus needed to obtain fresh DCs with which to repetitively stimulate
autologous T cells.
Although DC-based cancer vaccines can be generated in sufficient numbers and tested in
clinical trials, the complexity and cost of preparing DC precursors and generating functional
DCs makes it difficult to harness their full potential [65]. Especially important is the
requirement for repeated high dose vaccinations to generate sustained T cell responses [63,70].

11
Repetitive generation of DCs either from cryopreserved precursors or from fresh DC sources
will hence be required [69], not only rendering the process laborious and expensive, but also
limiting the clinic applicability of DC-based cancer vaccines [71].
1.1.5 B Cells as Alternative Antigen Presenting Cells
Owing to the disadvantages associated with DC-based cancer vaccines, effective
alternatives for active immunotherapy need to be explored. A possible approach is to use other
APCs that are available in large numbers, have the potential to be highly immunogenic, and may
have other unique advantages that enable them to elicit strong and effective vaccine responses.
The generation and expansion of such APCs should preferably be relatively straightforward and
should not require large precursor populations that are difficult to obtain or require prior
treatment of donors or patients with chemotherapeutic agents or other drugs. Generation from
small amounts of non-stem cell sources would be ideal, and once ready for vaccination, such
APCs should be capable of priming T cells and generating strong effector responses [72,73].
Furthermore, the production of such APCs should involve the use of recombinant growth factors
without the need to resort to usage of xenogeneic cells [74] or gene-transfer technology [75].
Based on these criteria, activated B cells appear to be a prime candidate for serving as surrogate
DC-based cancer vaccines. Several characteristics confer on B cells the ability to present
antigens efficiently: shortly after antigen encounter, activated B cells can quickly home to T
cells in secondary lymphoid organs; B cell receptor (BCR)-mediated endocytosis allows for
maximal concentration of minimal amounts of specific antigen; and signaling pathways can
guide the antigen processing machinery to give preference to the presentation of antigens
internalized through the BCR [76].

12
After encounter, APCs can internalize antigen via phagocytosis, fluid-phase pinocytosis,
or receptor-mediated endocytosis. In B cells, endocytosis occurs through the BCR, a
membrane-bound immunoglobulin that has high affinity for its given antigen, allowing efficient
antigen presentation through the concentration of very small quantities of the specific antigen.
In fact, BCR affinity has been shown to be directly proportional to the degree to which B cells
are capable of presenting antigen to CD4 T cells. A progressive need for more antigen arises as
the affinity of the BCR-antigen interaction decreases. B cells with a very high affinity BCR (Ka
of 5 x 1010
M-1
) require very low concentrations of soluble antigen (as low as 0.05nM in one
study) to be able to induce CD4 T cell proliferation, whereas cells bearing a BCR of lower
affinity (Ka of 3 x 108 M
-1) require ten times more antigen. In contrast, about 5000 times more
antigen is needed when presentation occurs after uptake via fluid phase pinocytosis [77].
Upon antigen recognition, ligation of the BCR induces its internalization, which leads to
the activation of various signaling cascades essential for activation of the B cell [78-80]. Some
of these signals are directed towards antigen processing and facilitate the traffic of antigen and
MHC class II molecules. Once internalized, the BCR-antigen complex is directed through the
endocytic pathway towards MHC II rich compartments (MIIC) where the formation of peptide-
MHC II complexes occurs [81-84]. Alongside endocytosis, BCR signaling results in the
upregulation of MHC II expression and causes MHC II molecules to be channelled through the
same compartments used by the BCR-antigen complex to travel to the MIIC [85,86]. Moreover,
signals generated from BCR crosslinking also induce the formation of multivesicular bodies and
acidification of MIIC compartments in an effort to create a favourable environment for antigen
processing and peptide loading [87]. The degradation and loss of regions of the antigen bound
to the BCR is also blocked as a result of the strong affinity of the BCR-antigen interaction,
allowing for its preferential presentation [88,89]. B cells are thus able to link antigen

13
recognition with the mechanisms responsible for antigen processing, thereby allowing the
presentation of BCR-internalized antigens preferentially over antigens taken up via other means
such as pinocytosis [76].
Yet another sophisticated aspect of antigen processing in B cells that is absent in other
APCs is the expression of human leukocyte antigen (HLA)-DO, a non-classical MHC II
molecule [90]. Antigen-derived peptide loading occurs in acidic compartments and is mediated
through the activity of another non-classical MHCII molecule, HLA-DM. HLA-DO is an
inhibitor of HLA-DM – HLA-DO activity predominates in compartments with high pH, limiting
HLA-DM activity to low pH compartments [91,92]. This pH-dependent inhibition by HLA-DO
serves to restrict peptide-loading to MHC II in the MIIC, where the acidic conditions are a
requirement for the processing of the stable BCR-antigen complex [93,94]. In this manner, the
processing of other non-BCR derived epitopes is inhibited, thus influencing the immune
response against BCR-internalized antigens.
B cells are thus capable of acquiring characteristics reminiscent of highly proficient
antigen presenting cells: after encountering antigen in secondary lymphoid organs, they can
efficiently internalize antigens specific for their BCR and preferentially process and present
those antigens to T cells. The ensuing response to BCR crosslinking, the first signal for B cell
activation, is proliferation and survival of the B cell. BCR crosslinking also induces expression
of costimulatory molecules required for naïve CD4 T cell activation [95,96]. In addition, B cells
can also present cytoplasmic antigens in the context of MHC class I molecules, thereby leading
to direct activation of CD8 positive T lymphocytes [76,97]. As a result, B cells can be strongly
immunogenic and possess the capability of presenting tumour-derived foreign antigens to other
components of the immune surveillance machinery, allowing for an immediate and speedy
response to cancer cells.

14
Since activated B cells are capable of functioning as potent APCs, cancer vaccines on a
B cell platform may be a more feasible alternative to DC-based vaccines. Unlike their DC
counterparts, B cells possess a greater proliferative capacity and can be easily obtained from
peripheral blood [98]. Consequently, the most apparent and immediate advantage of B cell-
derived cancer vaccines is the potential of generating unlimited cell numbers for repeated
vaccinations required for the induction of a sufficient immune response. Although it unclear
how many activated B cells can replace one activated DC, titration experiments comparing the T
cell stimulatory capability of the two APCs suggest that a T cell:APC ratio of 4:1 is optimal for
activated B cells while a ratio of 20:1 or less is optimal for DCs [99]. However, to generate
about 108 DCs requires roughly 1 litre of blood (or 10
9 peripheral blood mononuclear cells),
while the same number of activated B cells can be generated from only 4-8cc of peripheral
blood [100]. In other words, large numbers of B cells can be obtained and expanded easily from
a small and manageable amount of blood, yielding a pure and homogenous population of
activated B cells [98]. Like DCs, in vitro activation of B cells can be accomplished by treatment
with inflammatory cytokines and toll-like receptor (TLR) ligands [101], leading to enhanced
MHC and costimulatory molecule expression, as well as an increase in cytoskeletal activity and
capacity to stimulate T cells [69,102-104]. Moreover, owing to the deficiencies often
encountered in DCs of patients with advanced cancers [105], activated B cells could substitute
as alternative APCs either by replacing DCs, or boosting T cell responses that have been already
primed by DCs.
1.1.6 B Cell Expansion Protocol
Previous studies in the lab involving the in vitro manipulation of B cells from patients
with chronic lymphocytic leukemia (CLL) had set a precedent for an expansion protocol for

15
normal activated B cells. During the course of investigations on the molecular basis of tumour
immunogenicity, it was found that upon treatment with a TLR7 agonist, S28690 [106,107], and
the inflammatory cytokine IL2 [108], leukemic B cells could be induced to undergo
proliferation in vitro. Subsequent treatment with a Protein Kinase C (PKC) activator caused the
cells to acquire a phenotype resembling that of mature DCs [109]. Although TLR7-stimulated
and IL2-treated cells are less able to elicit an immune response than their PKC-activated
counterparts, normal B cells treated with S28690 and IL2 should induce proliferation in vitro
and generate sufficient quantities of activated B cells to be used as APCs for repeated
administrations in clinical trials of cancer vaccines.
1.1.6.1 Toll-like Receptor 7
Toll-like receptors recognize molecular patterns on infectious agents and play an
important role in the defence initiated against invading pathogens [110]. Stimulation of the toll-
like receptors is an efficient means for the induction of an immune response as TLRs regulate
both innate and adaptive immunity. In fact, activation of the innate immune system is a
prerequisite for the instigation of the adaptive immune response, especially with regards to a
TH1 response, which is central to cancer immunity [36].
The eleven members of the TLR family are type I integral membrane glycoproteins, and
are a subset of a larger superfamily that includes the interleukin 1 receptors (IL1Rs) based on
significant homology in their cytoplasmic regions. The extracellular region of TLRs contain
leucine-rich repeat (LRR) motifs and the cytoplasmic tail has a conserved region of ~200 amino
acids known as the Toll/IL1R (TIR) domain [111]. Although B cells express many members of
the TLR family, they are stimulated most potently by TLR7 and TLR9 ligands [112]. Unlike
other TLRs such as TLR1, TLR2 and TLR4, which are located on the cell surface, TLR3, TLR7

16
and TLR9 are endosomal receptors and are involved in the recognition of nucleic-acid-like
structures [113-115]. TLR7 is primarily expressed by B cells and monocytes and is involved in
the recognition of single-stranded viral RNA in endosomes [116,117]. Oxidized guanosines
[118] and imidazoquinolines [119] are synthetic human TLR7 agonists. Subsequent to ligand
binding, TLRs dimerize and undergo conformational changes necessary for the recruitment of
proteins involved in downstream signaling pathways (Figure 1), which include the adaptor
molecule myeloid differentiation primary-response protein 88 (MyD88), IL1R-associated
kinases (IRAKs), TGF-activated kinase (TAK1), TAK1-binding protein 1 (TAB1), TAB2 and
TNF-receptor-associated factor 6 (TRAF6) [120,121].
Upon receptor engagement, the TIR domain in the intracellular region of TLR7
associates with the adaptor protein, MyD88 [111]. The TLR7-MyD88 complex then recruits
IRAK1 and IRAK4, members of the IRAK family of kinases. IRAK4 is activated first via
MyD88 binding, and then proceeds to activate IRAK1 by phosphorylating specific residues in
its kinase-activation loop. Activation of the intrinsic kinase activity of IRAK1 induces
autophosphorylation of residues in its N-terminus, enabling the recruitment of TRAF6 to the
complex. IRAK1 and TRAF6 then disengage from TLR7 and interact with a plasma
membrane-associated pre-formed complex consisting of TAK1 and two TAK-1 binding
proteins, TAB1 and TAB2. This interaction leads to the phosphorylation of TAB2 and TAK1,
which translocate to the cytoplasm along with TRAF6 and TAB1, leaving IRAK1 to be
degraded at the membrane. TAK1 kinase activity is then triggered in the cytosol upon
ubiquitination of TRAF6, and activated TAK1 phosphorylates components of the inhibitor of
nuclear factor (NF)-B (IB) kinase complex (IKK). Activated IKK then phosphorylates IB,
causing its degradation and permitting release of NF-B dimers, allowing them to enter the
nucleus and function as transcription factors. TAK1 also functions to activate stress-activated

17
Figure 1. TLR7 signaling in B cells. Activation of TLR7 leads to association with MyD88, which in turn allows
for the recruitment of IRAK4 and IRAK1. Activation of IRAK1 allows TRAF6 to associate with the complex,
leading to the activation of TAK1 and TAB2. TAK1 is responsible for the activation of MAPKs, as well as NF-B.

18
protein kinase (SAPK) pathways via activation of mitogen-activated protein kinase kinases such
as MKK4 or MKK3 and MKK6, which lead to phosphorylation and activation of c-Jun-NH2-
kinases (JNK) or p38, respectively. Upon activation, JNK and p38 translocate to the nucleus
and activate c-Jun and c-Fos, components of activator protein-1 (AP-1), resulting in the
transcription of genes encoding molecules such as TNF [111]. TLR engagement also leads to
the activation of transcription factors that modulate the expression of costimulatory molecules
such as CD80 and CD86, and cytokines such as IFN and IL12 that skew the immune response
towards type 1 immunity [122].
Consistent with its capability to induce an immunogenic phenotype, one
immunotherapeutic route may be to manipulate TLR signaling in order to exact an anti-tumour
response. For instance, a recent study demonstrated that treatment with a TLR7 agonist caused
the disappearance of leukemic skin infiltrates in a patient with CLL [106]. In yet another study,
the sensitivity of CLL cells to chemotherapeutic agents was increased with exposure to a TLR7
agonist, Loxoribine [123]. Similarly, if TLR7 stimulation allows for the proliferation of normal
B cells and renders them capable of activating an immune response against tumours, such
activated B cells may have the potential of replacing DC-based vaccines.
1.1.6.2 Interleukin 2
The inflammatory cytokine IL2 is a member of the common cytokine-receptor -chain
(c; also known CD132)-binding family of cytokines, which includes IL4, 7, 9, 15, and 21 [124].
Apart from the common c receptor subunit shared by all members of this family, the IL2
receptor (IL2R) is a heterotrimeric receptor and consists of the IL2 receptor -chain (IL2R)
and the IL2 receptor alpha-chain (IL2R), associated with the high-affinity form of the receptor
[125,126]. IL2R is expressed primarily by activated T and B cells; exposure to IL2 induces

19
expression of IL2R, assisting in further activation [127]. IL2 is a mediator of adaptive
immunity [108] and signaling through the IL2R triggers the activation of multiple pathways,
including stimulation of the phosphatidylinositol-3-kinase (PI3)-AKT pathway, and stimulation
of the RAS-RAF-mitogen-activated-protein-kinase pathway. It also involves the common Janus
kinase (JAK) and STAT signaling molecules [124]. IL2 activation can thus lead to the
expression of various genes mediated by the STAT molecules, and FOS- and JUN-containing
transcription-factor complexes [128]. As a result, exposure to IL2 can lead to a variety of
responses, such as proliferation of T cells, proliferation of and synthesis of immunoglobulin by
activated B cells, and generation of cytotoxic T lymphocytes [129].
Owing to its immunogenic properties, IL2 is being tested as a therapeutic agent for the
treatment of various cancers and has been approved for use in patients with metastatic renal-cell
carcinoma and malignant melanoma [130]. Its anti-tumour effect likely arises from its
proliferation-inducing properties as IL2 stimulation not only leads to expansion of lymphocyte
populations in vivo, including anti-tumour T cells, but also strengthens their effector functions
[130,131]. Rather than have a direct effect on cancer cells, IL2 thus mediates its anti-tumour
activity by altering immune reactions to the tumour.
The growth-stimulating property of IL2 is also exploited by malignant cells of various T
and B cell leukemias. In such situations, one therapeutic strategy aims to develop antibodies
specific for IL2R so as to block the IL2-IL2R interaction, leading to cytokine starvation and
permitting death of IL2 dependent cancer cells directly. The unique IL2R subunit is an
especially attractive component to target as it is constitutively expressed by cancerous T and B
lymphocytes, and is not expressed by any resting cells except TReg cells [125]. Two such
antibodies, basiliximab (Simulect; Novartis AG), a chimeric antibody, and daclizumab (also
known as anti-Tac; Zenapax; F.Hoffman-LaRoche Ltd), the first humanized antibody, have both

20
been approved by the US Food and Drug Administration [132-134]. IL2 thus has a proven
track-record of serving as an immunotherapeutic agent in some forms of cancer, leading to the
hypothesis that its use could allow for proliferation of normal B cells whilst enabling them to
acquire an immunogenic phenotype via simultaneous TLR stimulation.
1.1.6.3 IL2 and TLR7 Stimulated B Cells
As mentioned above, the data from leukemic B cells from CLL patients treated in vitro
with IL2 and the TLR7 agonist S28690 showed that such treatment results in rapid proliferation,
accompanied by IL10 secretion and expression of the costimulatory molecules, CD80 and CD86
[109]. S28690 treatment activated the NF-B, p38 and SAPK pathways, while IL2 induced
activation of extracellular signal-regulated kinase (ERK) family members. STAT signaling was
also apparent 24 hours after treatment. In addition, it was noted that S28690 treatment
sensitized CLL cells to IL2, in part via the increased expression of IL2R. Initially, signaling
from the two stimulants was observed to be merely additive, perhaps due to the difference in
receptor locations with the IL2R on the plasma membrane and TLR7 residing in the endosomal
compartment [114]. Over time however, the signaling patterns became more synergized as
TLR7 activation led to an increase in IL2R expression, allowing for stronger IL2 signaling.
This interaction amplified growth and proliferation, increased costimulatory molecule
expression, and also led to STAT1 and STAT3 activation, as well as cytokine production.
Further stimulation by a PKC agonist allowed for the expression of CD83, a maturation marker,
and led to an increase in the production of TNF and , thus enabling the activated CLL cells to
become strongly immunogenic [109]. Based on these studies, it was hypothesized that normal B
cells procured from healthy individuals would become activated and proliferate in response to
TLR7 stimulation and IL2 treatment. Subsequent activation with a PKC agonist should enable

21
these cells to acquire characteristics reminiscent of potent APCs, allowing for further
investigation of their potential use for cancer vaccines.
1.1.7 Activation-Induced Cell Death in B Lymphocytes
The in vitro activation and proliferation of lymphocytes can prove to be problematic due
to the potential complication of activation-induced cell death (AICD). As the term implies,
AICD refers to apoptosis as an ensuing response to lymphocyte activation. Indeed, after a few
days of robust proliferation, normal B cells stimulated with the TLR7 agonist, S28690 and IL2
seemed to reach the end of their growth potential (elaborated upon in Results below), presenting
a major hurdle in the attempt to generate large numbers of activated B cells.
When B cells encounter their specific antigen, the response that follows can either be
activation and proliferation, or anergy and AICD [135]. In other words, BCR crosslinking and
internalization does not necessarily culminate in an immune response, but can result in tolerance
when other survival signals are lacking. AICD is mechanistically achieved by the regulation of
signaling pathways that promote apoptosis, such as the activation of effector caspases,
expression of pro-apoptotic genes, and inhibition of pro-survival genes. Regulation of the
immune response in mature lymphocytes is thus controlled by AICD, which is an essential
property of lymphocytes [135].
In B cells, AICD was first thought to be primarily restricted to the assembling of the
immune repertoire where elimination of self-reactive immature cells occurs upon crosslinking of
surface IgM receptors [136]. Later studies revealed that mature B cells can also undergo
apoptosis upon stimulation through extensive surface IgM ligation with immobilizing antibodies
[137,138] and MHC class II crosslinking [139]. Also, the finding that antigen-specific B cells
apoptose when they encounter soluble antigen in germinal centres [140], suggests that

22
lymphocytes are affected by AICD throughout their ontogeny, serving as a means for
maintaining peripheral tolerance [141]. Nonetheless, the vast array of signaling processes
involved in the AICD of mature B cells is not entirely understood. IL2 is a likely candidate as it
is known to play a crucial role in AICD in T cells, resulting in the clearance of self-reactive T
lymphocytes [142]. It is possible that cell mortality due to oxidative stress can also lead to
AICD.
1.1.7.1 Reactive Oxygen Species and AICD
The apoptotic process is mediated by intracellular reactive oxygen species (ROS) in
several systems, including excitotoxic neural cell death [143], glucocorticoid-mediated cell
death of thymocytes [144], HIV-induced death of T cells [145], and also in the case of the
immature B cell-line WEHI-231 treated with anti-IgM [146]. In fact, cytotoxicity induced by
increased levels of intracellular ROS results in alterations very similar to those observed in cells
undergoing apoptosis, such as plasma and nuclear membrane blebbing, elevated intracellular
Ca2+
levels, and protease activation [147]. Shortly after the induction of apoptosis,
mitochondrial permeability transition (PT), referring to an increase in the permeability of the
mitochondrial membrane, and loss of mitochondrial membrane potential (m) occur. These
changes allow for the release of the pro-apoptotic proteins, cytochrome C and apoptosis-
inducing factor (AIF), from the mitochondrial intermembrane space into the cytosol, leading to
the activation of effector caspases and thus committing the cell to apoptosis [148,149]. ROS
production is one of the means via which disruption of m occurs [141], and is an early event
upstream of caspase activation [150,151]. Activation of the pro-apoptotic members of the Bcl-2
family, such as Bax, Bad, Bak, Bid, Bik and Bim also leads to loss of m [152].

23
If apoptosis is mediated by ROS, it is worth investigating whether B cell activation
creates an intracellular environment that encourages ROS production, leading to AICD. This
hypothesis is quite plausible since cellular activation necessitates increased oxidative
phosphorylation. The increased rate of respiration in the mitochondria of activated cells could
lead to ROS generation, which is also a by-product of the mitochondrial electron transport
chain. In fact, a recent study demonstrated that activation of B cells with soluble antigen leads
to the generation of intracellular ROS, resulting in the disturbance of the reduction/oxidation
(redox) potential [141], which controls mitochondrial PT [153]. The intracellular redox
potential regulates the proclivity of cells to undergo apoptosis [154,155]; an oxidative shift in
the cellular redox state can hence modify the nature of the stimulatory signal received, resulting
in death instead of proliferation [156]. Moreover, it has been reported that BCR engagement on
activated B cells induces m breakdown, leading to caspase 9 activation, followed by further
activation of downstream effector caspases [157]. Elevated intracellular ROS levels could result
in the activation of genes known to mediate apoptosis, perhaps through an oxidative stress-
responsive nuclear transcription factor such as NF-B [141]. A strong case can hence be made
for the role of intracellular ROS generation in serving as the missing link in the mechanism
behind the induction of AICD in mature B cells. In light of this, it was postulated that the
introduction of strong antioxidants in a cell culture system may allow for decreased ROS
generation, thereby enabling prolonged growth.
1.1.8 Oxidative Stress and Uric Acid
Oxidative damage to basic cellular constituents – DNA and RNA, protein, lipids, and
carbohydrates – is an inevitable consequence of fuel-efficient aerobic metabolism [158]. While
low levels of ROS are a typical occurrence under physiological conditions, their effect becomes

24
detrimental once their rate of production rises enough to overpower the protective measures
cells have in place to combat the harmful consequences of oxidative stress. Notably, a role for
oxidative stress has been implicated in more than a hundred physiological disorders, including
ageing, cancer, and cardiovascular disease [159]. In response, evolution in aerobic organisms
has led to the amassing of a variety of defence mechanisms for inhibiting oxidant formation and
lipid peroxidation, as well as for counteracting and repairing oxidative damage. These
protective measures include enzymes, such as superoxide dismutase [160] and the selenium-
containing glutathione peroxidase, as well as antioxidants and radical scavengers, such as -
tocopherol (vitamin E) and -carotene confined to the lipid portion of the cell, and glutathione
and ascorbic acid restricted to the aqueous phase [161]. Another defence mechanism against
ROS is increased levels of the antioxidant uric acid in the plasma [161].
Uric acid is derived from adenine- and guanine-based compounds and is a product of
purine catabolism. Xanthine oxidoreductase is the sole enzyme responsible for its production
[162]. Whereas most mammals express the enzyme uricase (urate oxidase), which converts uric
acid to allantoin and urea, it is not expressed in humans and higher primates due to a non-
transcribed defective gene as a result of a nonsense mutation, hence making uric acid the final
product of purine degradation in these species [163-165]. As a result, the plasma level of uric
acid in humans is more than 10 times the amount found in most mammals [162]. Uric acid is
also found within cells and in all body fluids at lower concentrations than in plasma, where it
exists mostly in its ionized form, urate, at physiological pH [159].
Prior to the knowledge of its antioxidant and radical scavenging properties [166-168],
uric acid was traditionally viewed as being biologically dispensable, perhaps owing to the
harmful consequences arising from its enhanced retention or abnormally high production,
manifesting in crystal formation (due to its limited solubility in water) and leading to conditions

25
such as gout, where urate accumulates in joints and causes arthritis. This presumption was not
an unfounded one given that with regards to the human purine balance, uric acid is truly a waste
product. Unlike other purines, no further utilization of xanthine or uric acid is possible [169].
Whereas compounds such as adenosine, inosine, hypoxanthine, adenine, and guanine can be
salvaged from blood and added back to the cellular purine pool, any purine component that is
degraded to uric acid must be resynthesized anew with considerable energy expenditure, or
replaced by salvaging the appropriate precursors [169]. Upon ingestion, however, purines are
converted to uric acid via mucosal xanthine dehydrogenase, and instead of being reabsorbed in
the gastro-intestinal tract, uric acid is the only purine derivative that appears in the systemic
circulation [170]. Moreover, although it is filtered in the kidneys and then secreted, about 90%
of uric acid is typically reabsorbed and returned to the blood [161].
If the physiological handling of uric acid is any indication, it seems unlikely that it is an
expendable waste product. In fact, the idea that uric acid is one of the most powerful
antioxidants in the body was first put forth nearly 30 years ago, when in vitro studies
demonstrated uric acid to be a powerful scavenger of peroxyl and hydroxyl radicals, as well as
singlet oxygen [161]. About 30-65% of the peroxyl radical-scavenging capacity of plasma is
ascribed to uric acid [171,172], and in severe preeclampsia, a spike in uric acid levels accounts
for more than 90% of the increase in the radical-scavenging capabilities of the plasma [162]. It
was also shown that uric acid can serve as an electron donor, in particular, for heme
protein/H2O2 systems [159]. Beyond its role as a radical scavenger, uric acid also has the ability
to chelate metal ions such as iron and copper, rendering them incapable of catalyzing free
radical reactions [173-175]. More recently, it was discovered that uric acid acts as a specific
inhibitor of radicals resulting from the breakdown of peroxynitrite (ONOO-) [176,177], a
powerful oxidant that can inflict a variety of injuries by damaging essential cellular components

26
[178,179]. Peroxynitrite can also erode the structure and function of proteins by causing
nitration of their tyrosine residues [179,180]. In fact, given that uric acid is the strongest
antioxidant in blood plasma [161], it may prove to be useful in an in vitro setting for curtailing
ROS generation and relieving oxidative stress.
The initial studies on uric acid starting in the early 80s led a few investigators to
speculate that the antioxidant properties of uric acid may have allowed it to be a contributor to
the increased life span in humans [161,162]. An interspecies comparison of uric acid levels in
primates and other mammals reveals that there is a positive association between plasma uric
acid concentration and species longevity [162]. The majority of mammals have less than 0.5mg
uric acid/100ml of blood, while the levels in humans are close to its maximum solubility limit,
about 5mg/100ml of blood [161]. This stark difference arose over the course of about 60
million years of evolution and correlated with a substantial increase in life span as well as brain
size. Increased uric acid levels may also correlate with superior health. In patients diagnosed
with multiple sclerosis, for instance, serum uric acid levels are generally lower than those of
healthy individuals. Given that oxidative stress plays an important role in multiple sclerosis, it
is interesting to note that very seldom do patients with hyperuricaemia develop the disease
[181]. In addition, in patients with acute stroke, uric acid concentration in the plasma correlates
inversely with neurological impairments at the onset of stroke and size of the infarct upon
follow-up [182]. Similar studies reporting confirmatory results have led to the suggestion that
uric acid administration may be beneficial to patients suffering from conditions of heightened
oxidative stress, such as acute ischemic stroke [183].
Given the antioxidant properties of uric acid and the possible involvement of ROS in
AICD, the role of uric acid in the minimization of AICD in B cells was investigated. To this

27
end, a model system in which to study the effects of uric acid was required, and an Epstein-Barr
Virus (EBV) cell line was used.
1.1.9 Epstein-Barr Virus Cell Lines as a Model System for Studies in Activated B Cells
EBV is a human herpes virus that infects B cells primarily, but can infect other cell types
as well, especially epithelial cells. Given its ability to remain in a transcriptionally repressed
state indefinitely, almost all individuals (>95%) test positive for the virus and are infected for
life [184]. In certain circumstances however, at times involving even a single mutated protein,
this seemingly innocuous virus can prove to be deadly [185,186]. In fact, EBV was first
detected in Burkitt‟s lymphoma and is now known to be associated with various lymphomas,
carcinomas and other neoplastic disorders [187]. Of more significance is the fact that EBV can
infect resting B cells in vitro and compel them into activation, thereby causing their
transformation into proliferating lymphoblasts [188,189].
It is possible for EBV to achieve this dual status of benign and malignant infection by
altering its programme of gene expression based on the site and differentiation state of infected
cells. In vivo, EBV-infected B cells express one of four different gene expression programmes,
three of which are associated with latent infection and one of which is used for the manufacture
of infectious virus. The three latency transcription programmes are: the growth programme,
which demands expression of all nine latent proteins; the default programme, which prescribes
expression of only three latent proteins; and the latency programme, which is associated with
the expression of few, if any, latent genes [184]. Infected resting memory B cells circulating in
the peripheral blood express the latency programme, thereby allowing the virus to persist in the
infected individual for life [190-192]. Latent infection however, does not involve a state of
simple passivity but is associated with an immune response against the virus – a stable number

28
of infected B cells constantly circulate through the blood [193], along with stable levels of
cytotoxic T cells and serum antibodies to lytic and latent-stage proteins, leading to consistent
shedding of infectious EBV into the saliva [194,195]. The virus uses the growth gene
expression programme to drive resting B cells into an activated state, the same programme that
enables development of lymphoblastoid cell lines in vitro, such as TK6 [196]. Two important
proteins expressed in the growth programme enable this transformation – Latent membrane
protein 1 (LMP1) and LMP2A [197,198].
LMP1 and LMP2A are both multiple membrane-spanning proteins that lack functional
extracellular domains but signal in a constitutive fashion by acting as ligand-independent
receptors [199,200]. LMP1 is a functional homologue of CD40 [201,202], a member of the
TNF receptor (TNFR) family of receptors, and interacts with a host of proteins known as
TRAFs [203]. CD40 is an essential receptor on germinal centre B cells and interacts with
CD154 on T helper cells to deliver a costimulatory survival signal [204]. The signaling domains
of LMP1 and CD40 are comparable [200]; both proteins interact with JAK3 and STAT
molecules and lead to AP-1 activation via the JNK signaling pathways [205-207]. Signaling
through LMP1 thus serves to propel B cells into a proliferative state by mimicking signals
induced by the CD40 ligand via activation of the NF-B, MAPK, PI3K, and JAK3/STAT
pathways [207-211]. In this fashion, LMP1 is indispensable for the immortalization of B cells
in vitro [212]. LMP2A on the other hand, does not cause B cells to grow in vivo, but delivers an
important survival signal [213]. The amino-terminal domain of LMP2A contains the same
immunoreceptor tyrosine-based activation motifs (ITAMs) found in the - and -chains of the B
cell receptor [214], and associates with Lyn, a member of the Src family of tyrosine kinases
[215,216]. LMP2A mimics the non-proliferative BCR-initiated signal that is delivered in the
absence of antigen and is vital for the survival of all B cells [217]. Signaling via LMP2A

29
culminates in Protein Kinase C activation, leading to the resultant changes in gene expression
[199].
Between them, LMP1 and LMP2A bear the signaling capabilities necessary to mimic
signals from the BCR and T-helper cells – signals that are essential for the growth and survival
of activated B cells [218,219]. It was recently recognized that TLR signaling may serve as a
third signal for optimal B-cell activation [220]. There is evidence to indicate that this third
proliferative signal is also induced by EBV infection – EBV infection results in enhanced
expression of TLR7 and MyD88, an adaptor molecule that is an important constituent in TLR
signal transduction, which also plays a role in the initial B cell growth response [221].
EBV transformed cell lines thus mimic the pro-proliferative effects of TLR7 activation
as well, thereby rendering the collective signaling processes in the EBV-transformed line, TK6
analogous to those of normal activated B cells resulting from S28690 and IL2 treatment. TK6
cells can thus be used as a model in which to study the role that uric acid plays in overcoming
AICD of activated B cells.

30
1.2 RATIONALE AND RESEARCH HYPOTHESES
Although dendritic cell-based cancer vaccines have demonstrated potential as a useful
form of cancer immunotherapy, their production is fraught with many difficulties. One of the
main challenges that needs to be addressed is the difficulty involved in generating large numbers
of DCs for repeated vaccinations, required for sustained T cell responses against tumours. A
promising alternative is the use of other professional antigen presenting cells such as B cells,
which possess unique qualities that can make them potent APCs upon activation. Healthy
activated B cells can hypothetically be generated in large numbers via IL2 and TLR7
stimulation; however, AICD presents a major obstacle. Since oxidative stress is a mediator of
apoptosis, it appeared quite conceivable that the activated B cells were undergoing apoptosis
due to ROS-induced oxidative stress resulting from consistent stimulation over a prolonged
period of time. If true, the introduction of strong antioxidants into the culture media could tip
the redox balance and reduce ROS generation. Given that uric acid is the most powerful
antioxidant in plasma, its addition to the growth media should curtail oxidative stress and allow
for a prolonged proliferative capacity. To study in more detail the role of uric acid, and in
particular, to explore its possible function as a growth factor for activated B cells, the EBV-
transformed cell line, TK6, was used as a model system.

31
Chapter 2:
MATERIALS AND METHODS

32
Antibodies and Reagents
Clinical grade IL2 (Chiron Corp., San Francisco, CA) was purchased from the
Sunnybrook Health Sciences Centre hospital pharmacy. The TLR7 agonist S28690 was
obtained from 3M Pharmaceuticals (St. Paul, MN). The powder was dissolved in a mixture of
AIM-V medium (Invitrogen Life Technologies, Carlsbad, CA) with 33% DMSO at 1.3 mg/ml
and stored at 4oC in the dark. Phycoerythrin- or FITC-labelled CD19 and CD3 mouse
antibodies were purchased from BD Pharmingen (Mississauga, ON). Advanced D-MEM/F12,
Knockout Serum Replacement and Trypan blue were purchased from Invitrogen Life
Technologies. Thy1.2 antibody was purchased from the Sunnybrook Research Institute
Antibody Core Facility and Rabbit Complement was from Cedarlane Laboratories Ltd.
(Burlington, ON). Transferrin, 2-mercaptoethanol (2-ME), 7-aminoactinomycin D (7AAD),
dipyridamole (DPA), uric acid, Probenecid and Benzbromarone were from Sigma-Aldrich
Canada Ltd. (Oakville, ON). 2‟,5‟-Dideoxyadenosine (2‟,5‟-dd-Ado), MDL-12,330A,
Hydrochloride (MDL-12), Pentoxyfylline, 3-isobutyl-1-methylxanthine (IBMX), 14-22 Amide,
and H-89, Dihydrochloride (H-89) were from Calbiochem (San Diego, CA). Insulin (Humulin
RTM
, Eli Lilly Canada Inc., Toronto, ON) and 25% Human Albumin (Talecris Biotherapeutics
Ltd., Mississauga, ON) were obtained from the hospital pharmacy and blood bank. Phosphate
buffered saline (PBS) and Glutamine was purchased from Wisent Bioproducts (St. Bruno, QC).
Abs against phosphorylated JNK, phosphorylated ERK, and phosphorylated Akt were obtained
from Cell Signaling Technology (Beverly, MA). -Actin Ab was obtained from Sigma-Aldrich.
Recombinant human IL10 was obtained from R&D Systems, Inc. (Minneapolis, MN) and anti-
human IL10 antibody was purchased from eBioscience (San Diego, CA), along with IL15 and
IL7. TK6 cells were a generous gift from Dr. H. Liber (Colorado State University, Fort Collins,
CO).

33
Human blood samples and cell purification
Heparinized blood (30 ml) was collected from consenting healthy volunteers with
approval from the Research Ethics Board, Sunnybrook Health Sciences Centre. B cells were
isolated immediately after collection from each sample through negative selection using the
RosetteSep Human B Cell Enrichment Cocktail (StemCell Technologies, Vancouver, BC)
according to the manufacturer‟s instructions.
Isolation of murine B Cells
Spleens and lymph nodes were obtained from C57BL/6J wild type mice and
lymphocytes were obtained via ficoll gradient density centrifugation (Lympholyte-M, Cedarlane
Laboratories Ltd.). The isolated cells were T cell depleted with Thy1.2 antibody and Rabbit
complement. Briefly, cells were suspended at 107cells/ml in RPMI 1640 medium with 10%
FBS. 10g/ml of Thy1.2 antibody was added to the lymphocyte suspension and incubated for
30 minutes on ice. After two washes in PBS at 1000 rmp, the supernatant was discarded and
pelleted cells were resuspended in rabbit complement (diluted to 1:7 with media) for 30 minutes
at 37oC. Complement-treated cells were washed with PBS twice and depletion of T cells was
confirmed via flow cytometric analysis of the B and T cell marker, CD19 and CD3,
respectively.
Cell culture
Murine T cell-depleted lymphocytes were cultured in RPMI 1640 (Invitrogen Life
Technologies) with 5% Fetal Bovine Serum (FBS) (Lot No. 115615, Wisent) in flat bottom 96
well plates (BD Labware, Franklin Lakes, NJ) at 37oC in 5% CO2. 100 U/ml IL2 and 0.1 g/ml
S28690, the TLR7 agonist were added to 5 x 105 cells/ml at the start of the culture period. Cells

34
were re-stimulated with the two growth factors, along with addition of fresh media every two
days for the duration of the culture period. When uric acid was included in the media, the
appropriate amount was weighed (so as to obtain 0.1 mg/ml) and added to RPMI + 5% FBS.
The mixture was warmed at 37oC for about 15 minutes, or until completely dissolved, and then
filter sterilized via sterile 0.20m filters. The process was repeated every time cells were re-fed.
Human B cells were also plated at 5 x 105 cells/ml and cultured in 24 well plates (BD
Labware) in the indicated media. The same concentrations of S28690 and IL2 were used as for
mouse cells, and cells were maintained in the same manner as the murine B cell cultures.
TK6 cells were harvested every 3 days and re-cultured at approximately 1 x 105 cells/ml
in RPMI + 5% FBS. After a 1-2 month culture period, cells were discarded and a freshly
thawed aliquot from the original TK6 stock was used to set up a new culture. For experiments
assessing proliferation and/or growth inhibition, TK6 cells were plated at 5 x 104 cells/ml in 24
well plates in serum-free media consisting of RPMI plus 1 g/ml Insulin, 5 g/ml Transferrin,
25% Human Albumin, and 10mM Glutamine (ITAG).
Determination of live and dead cell populations
When simple cell counts were employed, live cells were determined by Trypan Blue
exclusion and counted in a hemocytometer. To determine percentages of live vs. dead cells,
approximately 1 x 106 cells were collected into a 100-200 l volume and 4 l of 7AAD solution
was added to the cell suspension. The cells were then analyzed for 7AAD staining on a
FACScan flow cytometer (Beckton Dickinson, San Jose, CA) and at least 10 000 events were
collected in each experiment.
When utilizing a colorimetric assay for proliferation, 20 l of Alamar blue (Biosource
International, Camarillo, CA) was added to cells cultured in 96-well round-bottomed plates (in

35
200 l volumes) and incubated at 37oC for 6-8 hours [222]. An absorbance colorimeter
microplate reader (Molecular Devices, Menlo Park, CA) was used to determine absorbance at
wavelengths of 540 nm (reduced state) and 595 nm (oxidized state).
Flow Cytometry
Flow cytometric analyses of surface CD19 and CD3 expression on murine lymphocytes
was performed after T cell depletion. Isotype-matched irrelevant antibodies were used as
negative controls (Pharmingen, Franklin Lakes, NJ). At least 10 000 events were collected in
each experiment and viable nucleated cells were determined by gating on 7AAD negative cells.
Before each experiment, SpheroParticles (Spherotech Inc., Chicago, IL) were used to
standardize measurements obtained from a FACScan flow cytometer. Data was analyzed using
CELLQUEST software (Becton Dickinson).
Cytokine Measurements
Cytokine levels in culture supernatants from TK6 were measured via a multi-analyte
fluorescent bead assay with a Luminex-100 system (Luminex Corp, Austin, TX). A kit
allowing measurements for IL5, IL6, IL10, Granulocyte-macrophage colony stimulating factor
(GM-CSF) and TNF was used according to the manufacturer‟s instructions (R&D Systems,
Inc.). Individual cytokine concentrations were ascertained from standard curves using Bio-Plex
2.0 software (BioRad, Mississauga, ON). For each cytokine, assays were linear between 3 and
10 000 pg/ml.

36
Western Blots
TK6 cells growing in RPMI and 5% FBS were washed twice in PBS and cultured in 6
well plates in 4ml volumes at 5 x 105 cells/ml. Cells were collected at the indicated time points,
and after an initial wash in cold PBS, they were lysed on ice for 30 minutes in lysis buffer (0.5%
Triton X-100, 25mM MES, 150mM NaCl, 1mM Na3VO4, 2mM EDTA, 1mM PMSF, 1 g/ml
aprotinin), followed by 10 minutes of high speed centrifugation. Protein extracts were
quantified [223] after collection and prepared for Western blot analysis by 1:4 dilution in 5X
sample buffer (8% [wt/vol] SDS, 8% [vol/vol] 2-ME, 250mM Tris, 40% glycerol, 2%
bromophenol blue in dd-H2O) and denaturation at 95oC for 5 minutes. Protein samples were
then loaded on a discontinuous polyacrylamide gel consisting of 7.5% resolving and 4%
stacking gels. PVDF membranes were then preactivated with 100% methanol and separated
proteins from the resolving gel were transferred onto the membranes. Phosphorylation status of
ERK and JNK proteins were determined through the use of antibodies listed in Antibodies and
Reagents. After overnight incubation at 4oC with the primary antibodies, blots were incubated
with anti-rabbit antibody conjugated with horseradish peroxidase for 1 hour, according to the
manufacturer‟s instructions. Chemiluminescence signals were detected with Supersignal West
Pico Luminol Enhancer and Stable Peroxide Solution (Pierce, Rockford, IL). -actin was used
as a loading control.
Isolation of total RNA and cDNA synthesis
Total RNA was extracted from 4 ml volumes of TK6 cells plated at 5 x 105 cells/ml at
the indicated time points. RNA was isolated using the RNeasy kit (Qiagen) according to the
manufacturer‟s instructions and concentration was measured in a spectrophotometer at 260nm.

37
cDNA was synthesized using the Superscript First Strand Synthesis System (Invitrogen
Life Technologies) in a 20 l reaction volume comprising 3 g of total collected RNA, 20 mM
Tris-HCl (pH 8.4), 50 mM KCl, 2.5 mM MgCl2, 10 mM DTT, 0.5 g oligo dT18, 0.5 mM
dATP, dGTP, dCTP, dTTP, and 200 U of Superscript II Reverse Transcriptase. The mixture
was incubated at 70oC for 5 minutes and cooled to 4
oC to anneal the priming oligonucleotide to
the total RNA. Reverse transcription was performed at 42oC for 2 hours and the cDNA obtained
from the reaction was stored at -20oC.
Real-time polymerase chain reaction
cDNA was amplified using the following primers: IL-10 forward, 5'-
CATCGATTTCTTCCCTGTGA-3', IL-10 reverse, 5'-CGTATCTTCATTGTCATGTAGGC-3';
hypoxanthine-guanine phosphoribosyltransferase (HPRT) forward, 5'-
GAGGATTTGGAAAGGGTGTT-3' HPRT reverse, 5'-ACAATAGCTCTTCAGTCTGA -3'.
PCR was performed on a DNA engine Opticon System (MJ Research Inc, Waltham, MA) using
SYBR Green I as a double-strand DNA-specific binding dye. PCR reactions were cycled 34
times after initial denaturation (95°C, 15 minutes) with the following parameters: denaturation
at 95°C for 15 seconds, primer annealing at 54°C and 52°C for IL10 and HPRT, respectively,
for 20 seconds, and extension at 72°C for 20 seconds. Fluorescent data were acquired during
each extension phase. After each PCR reaction, a melting curve analysis of the amplification
products was performed. Rapid fluorescence loss was uniquely observed at the
denaturing/melting temperature of the amplified DNA fragment. Standard curves were
generated with serial 10-fold dilutions of cDNAs obtained using the above primers. Expression
values of IL10 were normalized to those of HPRT.

38
Statistical analysis
Data are presented as mean standard error, unless stated otherwise. P values for
differences between sample means were determined using the Student t test, and P values for
assessing the significance of differences between two measurements were determined using the
paired t test. Statistical significance was set at P 0.05.

39
Chapter 3:
RESULTS

40
Murine B cells activated with IL2 and S28690 proliferate and then die
Mouse cells were first used as a model system to study B cell expansion with IL2 and
TLR stimulation. Cells isolated from the spleen and lymph nodes of B6 mice were T cell-
depleted using Thy 1.2 antibody, which binds T cells, and complement lysis of the antibody-
coated cells to obtain a predominantly B cell population. The negligible heterogeneity of the
population was confirmed by high CD19 staining, as determined via flow cytometry, and
staining for the T cell marker CD3, which decreased dramatically upon T cell depletion (Figure
2a). At the start of the culture period, and every two days thereafter, the isolated cells were
stimulated with 0.1 g/ml of the TLR7 agonist, S28690, and 100 U/ml IL2 (subsequently
referred to as S2 B cells), and re-fed with fresh media to prevent death associated with nutrient
exhaustion. Upon doubling, the culture was split and allowed to re-grow starting at the initial
cell concentration. In this manner, a roughly 5X expansion was achieved in one week from
mouse S2 B cells before the culture disintegrated as a result of large amounts of cell death
(Figure 2b).
IL2 as a stimulator of AICD
AICD was observed to be a major limiting factor in the expansion of S2 B cells and
evidence of apoptosis from cell counts was apparent in the cell culture starting as early as two
days after initial stimulation. IL2 is known to cause AICD in T cells [142] and could be
functioning in the same manner in activated B cells. Using other cytokines with similar
proliferative effects may allow the cells to stay alive without causing AICD. To test this
possibility, IL2 was replaced with other members of the c binding family of cytokines, which
comprises IL2, IL15, IL4, IL7, IL9, and IL21 [124]. IL15 seemed likely to be the most obvious

41
(a) (d)
(b)
0
5
10
15
20
25
30
35
40
45
day 0 day 2 day 5 day 7 day 9
Cell
co
un
ts (
x10^
5)
untreated
s28 only
s28/IL2
s28/IL15
s28/IL7
(c)
0
10
20
30
40
50
60
day 0 day 2 day 5 day 7
Cell
co
un
ts (
x10^
5)
untreated
s28/IL2
s28/IL2 & UA
Figure 2. Murine B cell cultures stimulated with IL2 and a TLR7 agonist undergo proliferation and
experience AICD which is partially reversed by uric acid. (a) B cells were isolated from the spleens and lymph
nodes of healthy B6 mice. After T cell depletion, an aliquot from the remaining lymphocytes was stained with B
and T cell markers to assess the purity of the population. A representative example shows the percentages of CD19
and CD3 expressing cells immediately before (left panel) and after (right panel) T cell depletion, as determined by
flow cytometric analysis. (b) After undergoing T cell depletion, B cells were plated at 5 x 105 cells/ml in RPMI +
5% FBS. Cells were treated with 0.1 g/ml of the TLR7 agonist, S28690 (abbreviated s28), and either 100 U/ml
IL2, 1 ng/ml IL15, or 1 g/ml IL7. Live cells, as assessed by lack of trypan blue uptake, were counted in a
hemocytometer on the indicated days of the culture period. Cell counts are an average of at least two separate
experiments with error bars indicating standard error of the means. (c) In another set of experiments, B cells were
stimulated with either S28690 and IL2 alone, or with S28690, IL2 and 0.1 mg/ml uric acid (UA). Live cells were
determined and plotted as above. P < 0.05 for the difference in cell counts between s28/IL2 and s28/IL2 & UA on
day 5. (d) After seven days of culture, mouse B cells were harvested and stained with 7AAD and analyzed via flow
cytometry. Cell size (forward scatter) is shown on the x-axis and the percentage of live cells is determined by
7AAD negative staining (shown in the gated regions).
18% live cells 52% live cells
S28/IL2
S28/IL2 & UA
CD3
CD
19
CD19: 38%
CD3: 53%
CD19: 82%
CD3: 2%

42
replacement candidate as it is known to be functionally similar to IL2 owing to the unique
heterotrimeric construction of their receptors. Moreover, IL15 serves an anti-apoptotic factor in
many systems and was shown to inhibit IL2-induced AICD in some circumstances [224]. IL2
was replaced with 1ng/ml of IL15, and although there was an initial significantly large increase
in cell growth observed 5 days into the culture period, IL15 treatment was unable to overcome
AICD (Figure 2b). Similar to S2 B cells, cells treated with S28690 and IL15 all died after a
week in culture. IL15 was also used in conjunction with IL2 to determine if the dual cytokine
combination would affect cells differently, but identical results were obtained, and in fact, there
seemed to be no difference between the S2+IL15 cultures and the S2 B cell cultures (data not
shown). Similar observations were made when IL7, a pre-pro-B cell growth factor that plays a
role in the survival and expansion of lymphoid precursors [225], was used in place of, or in
addition to IL2 (Figure 2b; data not shown). As with IL15 treatment, despite increased
proliferation when S26890 was used with 1 g/ml IL7 instead of IL2, after 7 days there were
very few live cells left in the culture, and within 2 more days, all remaining cells died. Due to
lack of significant differences with the use of other cytokines, the IL2 and S28690 combination
was used for B cell stimulation in all subsequent experiments as it stimulated B cell proliferation
as best as seemed possible and allowed for the highest amount of sustained growth.
Oxidative stress as a mediator of AICD
Since cellular apoptosis is mediated by ROS [143], it is highly plausible that due to
repeated stimulation over a prolonged period of time, oxidative stress led to increased ROS
production in S2 B cells, resulting in AICD [135,141]. If true, the introduction of strong
antioxidants into the cell culture medium should curtail oxidative stress and reduce the amount
of cell death observed. Uric acid, a product of purine degradation, is known to be the most

43
potent antioxidant in plasma [161] and was added to the S2 B cell growth media. Indeed, it
appeared that treatment with 0.1 mg/ml uric acid not only reduced death, but also enabled
increased proliferation, acting as a growth supplement for activated B cells (Figure 2c). In a
representative experiment for instance, after about one week, only 18% of cells in the S2 culture
were alive (Figure 2d), as confirmed by 7AAD staining (a dye that penetrates cell membranes of
dying or dead cells and intercalates into double stranded nucleic acids). In contrast, 52% of S2
B cells growing in medium that contained uric acid were still alive at the end of 7 days,
demonstrating that uric acid could partially rescue S2 activated B cells from AICD.
Human B cell expansion cultures
Based on the positive preliminary results from mouse B lymphocytes, the expansion
protocol for murine B cells was extended to human B cells isolated from peripheral blood
collected from healthy volunteers. Human B cells were cultured in the same manner as were
murine B cells with the exception of the culture medium: serum-free AIM-V with the
antioxidant 2-ME (5 x 10-5
M) was observed to be a superior media than RPMI supplemented
with FBS (data not shown). Apart from increased amino acid and vitamin concentrations, this
difference in growth may be attributed to the inclusion of serum replacing components in AIM-
V, such as transferrin and lipids, as well as trace elements and antioxidants (Patent No.
WO/1988/002774) [226]. Initial experiments with IL2 and S28690 treated human B cells
yielded results that were almost identical to their murine counterparts. After several days in
culture, S2 B cells began undergoing apoptosis and AICD was reduced when 2-ME was
replaced by the more powerful antioxidant, uric acid (Figure 3). Replacement with uric acid
also allowed for the generation of larger numbers of S2 B cells. However, as with mouse
cultures, uric acid was unable to fully rescue cells from AICD and after about 12 days in culture,
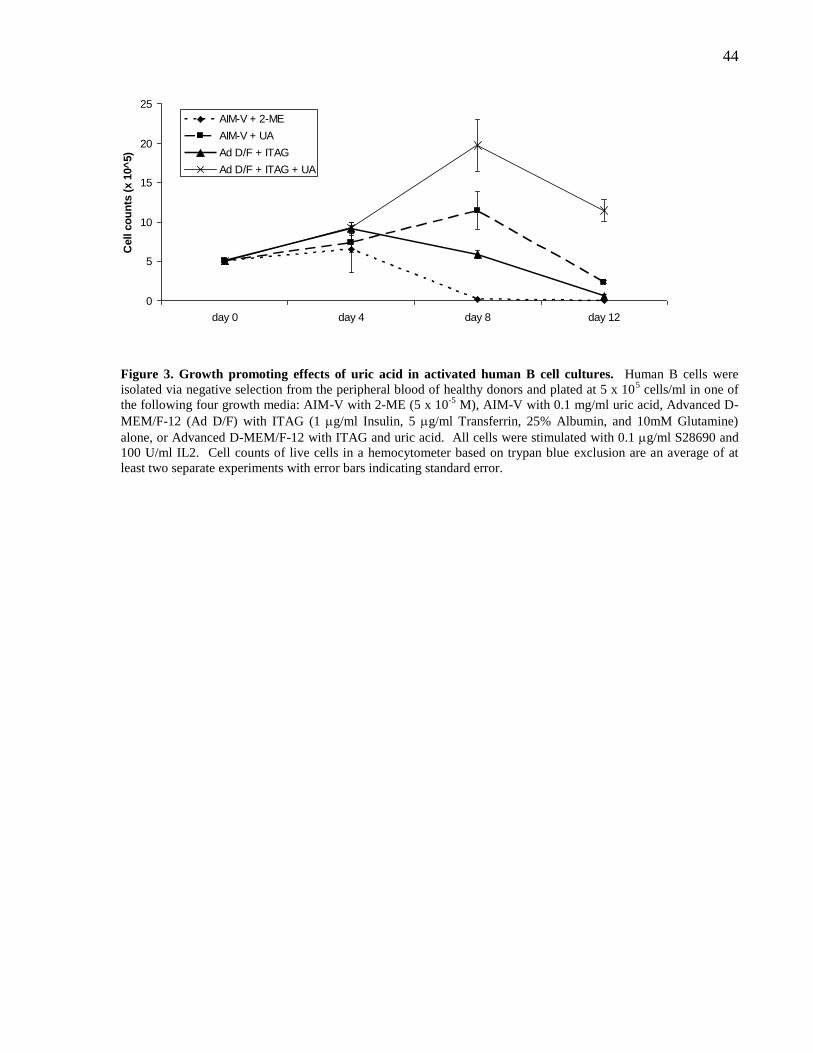
44
0
5
10
15
20
25
day 0 day 4 day 8 day 12
Cell c
ou
nts
(x 1
0^
5)
AIM-V + 2-ME
AIM-V + UA
Ad D/F + ITAG
Ad D/F + ITAG + UA
Figure 3. Growth promoting effects of uric acid in activated human B cell cultures. Human B cells were
isolated via negative selection from the peripheral blood of healthy donors and plated at 5 x 105 cells/ml in one of
the following four growth media: AIM-V with 2-ME (5 x 10-5
M), AIM-V with 0.1 mg/ml uric acid, Advanced D-
MEM/F-12 (Ad D/F) with ITAG (1 g/ml Insulin, 5 g/ml Transferrin, 25% Albumin, and 10mM Glutamine)
alone, or Advanced D-MEM/F-12 with ITAG and uric acid. All cells were stimulated with 0.1 g/ml S28690 and
100 U/ml IL2. Cell counts of live cells in a hemocytometer based on trypan blue exclusion are an average of at
least two separate experiments with error bars indicating standard error.

45
most, if not all activated B cells succumbed to death, despite the use of a rich growth medium
supplemented with antioxidants to stabilize culture conditions.
Further examination of the AIM-V patent revealed that it contained Indomethacin, a
non-selective inhibitor of cyclooxygenase (COX)1 and COX2. COX1 is required for tissue
homeostasis and is constitutively expressed, whereas COX2 is expressed in response to various
stimuli ranging from cytokines and growth factors to stress inducers such as free radicals and
hypoxia [227]. COX2 is expressed at high levels in activated human B cells and possesses
growth-promoting properties that are exploited in many cancers including B cell malignancies
[228], while its inhibition leads to decreased proliferation and survival of CLL cells [229].
Moreover, it has been shown that inhibitors like Indomethacin severely restrict the ability of
human B cells to produce IgG and IgM in vitro and negatively affect proliferation as well [229].
In light of this information, it was apparent that the full proliferative potential of activated B
cells could not be realized in the AIM-V medium. Instead, Advanced D-MEM/F-12 (Ad D/F), a
medium rich in strong antioxidants such as ascorbic acid and glutathione seemed more likely to
provide an improved environment conducive to the continued growth of activated human B cells
and was used in subsequent experiments. In an effort to keep culture conditions free of animal-
derived components, it was also essential to replace the bovine-derived serum growth
requirement. More than three decades ago, Iscove and Melchers discovered that when growth
media for B cells is supplemented with additional amino acids and vitamins, the growth-
inducing properties of serum can be completely recovered with the use of albumin, transferrin
and lipids, thus rendering the requirement of FBS redundant [226]. Ad D/F was thus
supplemented with 1 g/ml Insulin, 5 g/ml Transferrin, 25% lipid-rich Albumin [230], and
10mM Glutamine (subsequently referred to as ITAG), and these components together were

46
found to be an adequate and desirable serum replacement given the lack of FBS-associated
xenogeneic components.
Cell counts of live cells revealed that S2 human B cells cultured in Ad D/F + ITAG were
able to stay alive longer than cells cultured in AIM-V medium (Figure 3). Addition of uric acid
to the new media enabled acquisition of the highest yield of cells with the least amount of death
compared to all other alternatives that were tested (Figure 3). Notably, despite the fact that Ad
D/F is rich in antioxidants, a significantly greater number of cells were attained with the
addition of uric acid (P < 0.05), indicating that perhaps uric acid was possibly playing a role
beyond that of an antioxidant.
Uric acid plays an additional role above that of an antioxidant in supporting proliferation of TK6
cells
To study the role that uric acid plays in the minimization of AICD in S28690 and IL2
treated B cells, the spontaneous EBV transformed lymphoblastoid B cell line, TK6, was used as
a model system for activated B cells. The cell line is derived from a paediatric patient with non-
malignant hereditary haemolytic anemia [231,232]. TK6 cells can serve as a model for S2 B
cells in particular, given that the transformation of these cells is achieved via signaling pathways
analogous to those activated upon IL2 stimulation and TLR7 activation [184,221].
While TK6 cells are typically grown in FBS supplemented media, serum starvation
causes these cells to alter their signaling and undergo a death process (Figure 4a) that is
morphologically similar to the AICD observed in S2 B cell cultures. When uric acid is added to
the serum-free TK6 growth media (RPMI + ITAG) however, cell death is inhibited and robust

47
(a)
(b)
Figure 4. Uric acid rescues TK6 cells from death associated with serum withdrawal and displays functions
beyond an antioxidant role. (a) TK6 cells growing in RPMI + 5% FBS were washed twice in PBS and plated at 5
x 104 cells/ml in RPMI plus ITAG alone, with 2-ME (5 x 10
-5 M), or with uric acid (0.1 mg/ml). Cells were
harvested after 3-4 days and viability was assessed by 7AAD exclusion on a flow cytometer. Average percentages
of the 7AAD negative populations are shown along with standard errors of the indicated number of experiments.
(b) TK6 cells were plated in RPMI plus ITAG with either uric acid, DPA (0.3 g/ml), or both, and percentage of
live cells was determined 3-4 days after start of the culture period via flow cytometric analysis of 7AAD staining.
The left panel depicts differences between the percentage of viable cells in uric acid-treated and DPA-treated TK6
cells and P value, the significance of the differences between the two treatments, was determined via a paired t test
for the indicated number of experiments. The right panel depicts differences between DPA- and uric acid plus
DPA-treated cells, and P value was also determined via a paired t test for the indicated number of experiments.
65
70
75
80
85
90
UA DPA
% L
ive
ce
lls
65
70
75
80
85
90
DPA UA + DPA
% L
ive c
ells
P < 0.05 P < 0.01
(n=4) (n=4) (n=4) (n=4)
0
10
20
30
40
50
60
70
80
90
ITAG 2-ME UA
% L
ive c
ell
sP < 0.05
(n=3) (n=2) (n=2)

48
growth is exhibited by these cells (Figure 4a), akin to the observation that uric acid partially
rescues normal activated B cells from AICD. TK6 is thus a valid and applicable system in
which to investigate the relieving effects of uric acid and to determine if it has a previously
underappreciated mechanism of action in activated B cells.
Other antioxidants besides uric acid such as dipyridamole (DPA) [233] and 2-ME were
also able to rescue TK6 cells in a serum-free setting from death, although uric acid delivered the
most amount of growth (Figure 4a and b). When DPA was added along with uric acid, an
additive effect was observed and an increased percentage of cells in the culture were viable,
implying that uric acid was perhaps acting as more than just an antioxidant (Figure 4b). Taken
together, the observations that uric acid had a consistent growth effect in activated mouse B
cells, in activated human B cells, and in TK6 cells; that uric acid enabled a more pronounced
growth effect than other antioxidants; and that an additive effect was observed when uric acid
was used in combination with DPA, led to the hypothesis that uric acid was acting as a growth
factor for activated B cells.
Uric acid does not function intracellularly as a metabolite
Since uric acid is a purine derivative, and given that the dose of uric acid which seemed
most effective in all three systems (activated murine B cell cultures, activated human B cell
cultures, and TK6 cells) was 0.1 mg/ml (600 μM), falling within the dose range in which purine
metabolites are known to exert their effects [234], it was possible that uric acid was functioning
as a metabolite, rather than a growth factor acting via a receptor. If this were to be the case,
uric acid would need entry into the cell to be functional, and exposure to transport inhibitors
would abrogate its survival effect. To test this hypothesis, two uric acid transport inhibitors
were used –Probenecid, an inhibitor of the organic anion transporter (OAT), which transports

49
uric acid from urine into plasma [235], and Benzbromarone, a uricosuric agent that inhibits the
reabsorption of uric acid regulated by the uric acid transporter, URAT1 [236]. Neither of these
two inhibitors caused a reduction in uric acid-mediated TK6 proliferation as assessed by a
colorimetric assay (Figure 5a), implying that uric acid was exerting its effects from within the
extracellular milieu and did not require entry into the cell, but was acting at the cell surface.
Proliferative effects of uric acid are not mediated by a G protein-coupled receptor
Many metabolites are known to signal via G protein-coupled receptors (GPCRs), such as
amino acid receptors, glutamate receptors, purinergic receptors, etc. [237]. If uric acid was
acting as a metabolite at the cell surface, it would be possible that its effects were mediated
through a G-protein and subsequent Protein Kinase A (PKA) activation. To test this theory,
various inhibitors in the PKA activation pathway were employed and their effects on uric acid-
mediated proliferation were tested (a brief illustration of this pathway in relation to the sites of
inhibitor action can be found in Figure 5b). As a control, the inhibitors were also used on TK6
cells growing with the support of the antioxidant DPA to rule out nonspecific growth inhibitory
or toxicity effects. An examination of cell viability through flow cytometric analysis revealed
that the adenylate cyclase (AC) inhibitors, 2‟,5‟-Dideoxyadenosine (2‟,5‟-dd-Ado) and MDL-
12,330A Hydrochloride (MDL-12), decreased the viability of uric acid-treated TK6 cells, but
also decreased the viability of DPA-treated TK6 cells, precluding interpretation of its inhibitory
effects (Figure 5c). In accordance with these results, phosphodiesterase (PDE) inhibitors,
Pentoxyfylline and 3-isobutyl-1-methylxanthine (IBMX) did not increase viability in the uric
acid culture (DPA was not used as a control since it has been characterized as a PDE inhibitor as
well [238]; Figure 5c). Treatment with 14-22 Amide and H-89 Dihydrochloride (H-89), two
PKA inhibitors, also did not decrease the viability of these cells. Taken together, results from

50
(a) (b)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
UA Probenecid + UA Benzbromarone +
UA
Re
lati
ve
Gro
wth
(c)
Figure 5. Uric acid does not function as a metabolite as determined by inhibition of uric acid transporters
and G protein-coupled receptors. (a) TK6 cells were plated in a 96 well plate in RPMI plus ITAG with uric acid
alone, or with either Probenecid (1mM) or Benzbromarone (1 M). Doses were based on toxicity experiments on
cells growing in FBS and the highest non-toxic dose was used (data not shown). After 3-4 days in culture,
proliferation was assessed by Alamar blue reduction in a colorimetric assay as described in Materials and Methods.
Absorbance measures were normalized to the proliferation readout for uric acid-treated TK6 cells. The experiment
was repeated three times with similar results. (b) G protein-coupled receptor (GPCR) activation by ligand binding
induces the translocation of G proteins (Gs) associated with the receptor, leading to activation of adenylate cyclases
(ACs), which catalyze conversion of ATP to 3‟5‟-cyclic AMP (cAMP) and pyrophosphate. Phosphodiesterases
(PDEs) degrade cAMP to AMP, thereby inhibiting Protein Kinase A (PKA) activation. While AC and PKA
inhibitors should decrease PKA-dependent growth, PDE inhibitors should allow for enhanced growth of cells that
rely on PKA activity. (c) TK6 cells were plated at 5 x 104 cells/ml in RPMI plus ITAG alone, or with 2‟,5‟-
Dideoxyadenosine (2‟,5‟-dd-Ado) (1 M), MDL-12,330A Hydrochloride (MDL-12) (20 M), Pentoxyfylline (250
M), 3-isobutyl-1-methylxanthine (IBMX) (25 M), 14-22 Amide (1 M), or H-89 Dihydrochloride (H-89) (1
M). Cells were also plated in RPMI + ITAG plus uric acid with or without all the listed inhibitors, as well as in
RPMI + ITAG with DPA either with or without all the listed inhibitors except for pentoxyfylline and IBMX. Doses
for all inhibitors were based on toxicity experiments on cells growing in FBS and the highest non-toxic dose was
used (data not shown). After 3-4 days in culture, viability was assessed by flow cytometry using 7AAD exclusion.
The experiments have been repeated at least three times with similar results.
0
10
20
30
40
50
60
70
80
90
100
Controls 2'5'-dd-Ado MDL-12 Pentoxyfylline IBMX 14-22 Amide H-89
% L
ive
ce
lls
ITAG
UA
DPA
AC inhibitors PDE inhibitors PKA inhibitors
ligand
GPCR
Gs Gs AC
ATP cAMP AMP
PDE
inactive PKA
active PKA
2’5’-dd-Ado
MDL-12
Pentoxyfylline
IBMX
14-22 Amide
H-89

51
the GPCR pathway inhibitor studies led to the conclusion that uric acid does not act as a
metabolite and exerts its effect through a mechanism other than PKA activation.
Uric acid activates signaling pathways and leads to cytokine production
To determine if uric acid was acting via signal transduction pathway(s) other than PKA,
proteins from various signaling pathways were examined. Immunoblotting revealed that uric
acid treated TK6 cells had enhanced pERK and pJNK activation five minutes after stimulation,
compared to cells growing in non-supplemented media with ITAG alone (Figure 6a). Both
signals quickly diminished and were barely visible when examined ten minutes after
stimulation, suggestive of receptor activation and ensuing signaling. Notably, one hour after
stimulation, the state of both ERK and JNK activation in uric acid-treated cells was similar to
that of cells grown in media supplemented with serum, indicative of the stable state of the cells
in both cultures (Figure 6a). TK6 cells in serum-free ITAG supplemented media, on the other
hand, exhibit what seemed to be an aberrant signaling pattern with highly enhanced phospho-
ERK expression after one hour, likely a reflection of the stress-inducing conditions in the
culture. The activation statuses of other signaling molecules, namely, pAkt, pIB, pIRAK1,
pMARCKS, pVASP, pSTAT3, and pSTAT1, were also examined and did not reveal any
differences (data not shown).
In addition to protein activation, uric acid-treated TK6 cells also engaged in cytokine
production. While an increase in both IL6 and IL10 was observed, the level of IL10 in uric
acid-treated cells was more than 100X greater than those in cells growing in FBS or serum-free
media (ITAG alone), both of which had negligible IL10 production (Figure 6b). RT-PCR was
also performed to confirm increased IL10 message production in uric acid-treated cells. Indeed,
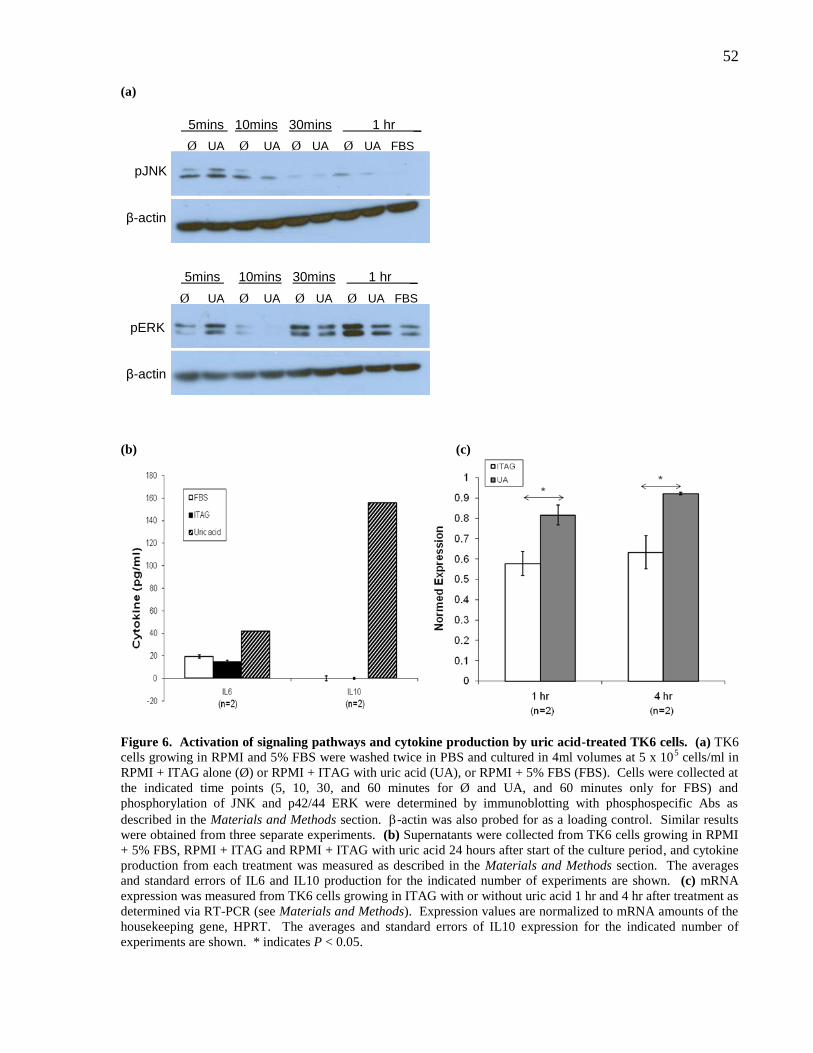
52
(a)
(b) (c)
Figure 6. Activation of signaling pathways and cytokine production by uric acid-treated TK6 cells. (a) TK6
cells growing in RPMI and 5% FBS were washed twice in PBS and cultured in 4ml volumes at 5 x 105 cells/ml in
RPMI + ITAG alone (Ø) or RPMI + ITAG with uric acid (UA), or RPMI + 5% FBS (FBS). Cells were collected at
the indicated time points (5, 10, 30, and 60 minutes for Ø and UA, and 60 minutes only for FBS) and
phosphorylation of JNK and p42/44 ERK were determined by immunoblotting with phosphospecific Abs as
described in the Materials and Methods section. -actin was also probed for as a loading control. Similar results
were obtained from three separate experiments. (b) Supernatants were collected from TK6 cells growing in RPMI
+ 5% FBS, RPMI + ITAG and RPMI + ITAG with uric acid 24 hours after start of the culture period, and cytokine
production from each treatment was measured as described in the Materials and Methods section. The averages
and standard errors of IL6 and IL10 production for the indicated number of experiments are shown. (c) mRNA
expression was measured from TK6 cells growing in ITAG with or without uric acid 1 hr and 4 hr after treatment as
determined via RT-PCR (see Materials and Methods). Expression values are normalized to mRNA amounts of the
housekeeping gene, HPRT. The averages and standard errors of IL10 expression for the indicated number of
experiments are shown. * indicates P < 0.05.
β-actin
pERK
Ø UA Ø UA Ø UA Ø UA FBS
5mins 10mins 30mins 1 hr _
β-actin
pJNK
Ø UA Ø UA Ø UA Ø UA FBS
5mins 10mins 30mins 1 hr _

53
1 and 4 hours after stimulation, a greater amount of IL10 expression at the mRNA level was
observed in these cells (Figure 6c).
IL10 inhibition partially blocks the proliferative effects of uric acid
In order to determine if increased IL10 production was the mechanism via which uric
acid was exerting its proliferative effects, recombinant human IL10 was added to serum-free
media to test if IL10 could mimic the growth effects observed in uric acid-treated TK6 cells. No
proliferation was observed in cells treated with IL10, and in fact, these cells behaved exactly
like those cultured in ITAG alone and died within 2-3 days (data not shown). Although
enhanced IL10 levels appeared insufficient in explaining its growth effects, it was possible that
IL10 was partly responsible for uric acid-induced proliferation. This did indeed prove to be the
case as inhibition of IL10 resulted in a significant decrease in the number of live uric acid-
treated TK6 cells (Figure 7, left panel). As assessed by cell counts based on trypan blue uptake,
both treatments, however, exhibited similar amounts of cell death, implying that in the absence
of IL10 production, uric acid still allowed TK6 cells to grow, but at a significantly slower pace
(Figure 7). These results indicate that uric acid, at least in part, exerts its proliferative effects via
production of the IL10 cytokine, and that IL10, although necessary, is insufficient to fully
recapitulate the growth effects of uric acid (Figure 8).

54
Figure 7. IL10 inhibition leads to loss of uric acid-dependent proliferative potential in TK6 cells. 5 x 104
TK6 cells were plated in RPMI + ITAG, RPMI + ITAG + uric acid, with or without anti-IL10 antibody (1 g/ml).
After 3-4 days of growth in culture, the number of live (left panel) and dead cells (right panel) were assessed by
trypan blue uptake and counted in a hemocytometer. Proliferation is depicted as differences in cell counts between
uric acid-treated and uric acid plus IL10-treated cells and differences in the number of dead cells in the two
treatments is also shown. P value was determined by a paired t test for the indicated number of experiments.
0
10
20
30
40
50
UA UA + anti-IL10
Liv
e c
ells
(x
10
^4
)
0
10
20
30
40
50
UA UA + anti-IL10D
ea
d c
ells
(x
10
^4
) (n=3) (n=3) (n=3) (n=3) *
P < 0.05

55
Figure 8. Mechanism of uric acid-dependent growth effects on activated B cells. The results from this study
implicate uric acid as a growth factor for activated B cells that likely functions at the cell surface via a receptor.
Upon receptor engagement, various signaling pathways appear to be activated, including those involving ERK and
JNK, as demonstrated by increased expressions of the phosphorylated forms of the two proteins. Cytokine
production, particularly that of IL10, and to a lesser extent, that of IL6 is also observed. Inhibition studies
involving anti-IL10 have revealed that uric acid does, in fact, exert some of its proliferative effects via enhanced
levels of IL10 production.
ERK JNK
IL6 IL10
Cell Growth
Uric acid
Cytosol
Nucleus

56
Chapter 4:
DISCUSSION AND FUTURE DIRECTIONS

57
4.1 DISCUSSION
The idea that activated B cells can serve as APCs and replace DC-based cancer vaccines
is not novel. In fact, many researchers in the last decade have been studying B cells in a quest to
find viable alternatives to generate autologous vaccines [69,71,98,99,239]. This search is
especially relevant to cancer patients in whom large volume leukapheresis is not practical, such
as pediatric patients or breast cancer patients that have had to undergo mastectomy and axillary
lymph node dissection. While clinical trials have yet to take place, most recently, activated B
cells generated from canine lymphoma patients with CD40 and IL4 stimulation and loaded with
antigen-specific mRNA showed T cell responses in vitro [239].
Earlier this year, Wiesner et al. also reported that they were successfully able to generate
long-term activated human B cell cultures that have the capacity to undergo proliferation
indefinitely via activation of the cell surface CD40 receptor in the presence of IL4 [240]. The
CD40/IL4 combination (CD40-B cells) mimics B cell activation by T helper cells, and these
cells were originally used as an in vitro model system to study B cell differentiation into plasma
cells [103,241]. CD40-B cells exhibited uninhibited CD40 ligand (CD40L)- and IL4-dependent
growth, with more than 370 population doublings, thereby justifying characterization as
„conditionally immortalized‟ [240,242]. Moreover, CD40-B cells could also activate and induce
expansion of antigen-specific CD8+ T cells, demonstrating their potential as effective APCs in a
vaccine setting.
Weisner‟s results provide evidence for the ability to generate long term activated B-cell
cultures. Such cells however, may not serve as adequate DC-based vaccine replacements due to
problematic generation protocols that rely on the use of xenogeneic components like FBS and
murine fibroblast feeder cells (NIH3T3) transfected with human CD40L as a source of CD40

58
stimulation. NIH3T3 cells in particular, cannot be used under good manufacturing practice
(GMP) conditions due to their xenogeneic origins. In addition, regulations concerning the
therapeutic use of such activated B cells for the purpose of creating cancer vaccines would be
similar to those for xenotransplantion and gene therapy, resulting in restricted clinical
applicability [243]. The use of a soluble recombinant human CD40L is preferable in that regard
[71]. Alternatively, Mason et al. described a novel method which involves transfection of a
human erythroleukemic cell line with a CD40L expression plasmid, resulting in the generation
of feeder cells that are able to provide stimulation to the CD40 receptor [239]. The main
concern in instances where the use of non-human components is employed is the potential
danger to patients as a result of contact with foreign entities, which has the possibility of causing
widespread inflammation in the body. Detrimental effects may result upon exposure to animal
proteins in the cells or infections caused by animal microbes. The concern is relevant to other
situations as well, most notably that of human embryonic stem cell generation and expansion,
where the search for xenogeneic-free culture components is a dominant topic of discussion in
the field [244-247].
The need to replace serum from bovine origins is also crucial. Not only is FBS
potentially harmful due its xenogeneic nature, but its use also entails the drawback of including
an unknown and uncontrolled set of substances in the expansion protocol, making it harder to
interpret growth requirements [226]. Although the use of autologous human serum is a
possibility, it may not be the best alternative given the added procedural requirements. In
addition, it makes for the generation of long-term activated B cell cultures more cumbersome
and potentially inefficient if several phlebotomies are needed for the purposes of serum
derivation. A better solution is to replace FBS with its crucial active ingredients and supplement
growth media with such components.

59
The above-mentioned considerations led to the use of an antioxidant rich medium
(Invitrogen‟s Advanced D-MEM/F-12) supplemented with insulin, transferrin, albumin, and
glutamine for the generation of human activated B cells. AICD was still a major concern and
uric acid demonstrated an enhanced ability to alleviate oxidative stress and allowed for
increased cell proliferation. The use of uric acid thus presents a new approach for the
cultivation of B cells as APCs. In fact, the data presented herein not only confirmed the
antioxidant properties of uric acid, but also pointed towards a novel function beyond its
ostensible role as an antioxidant, affording new insights into B cell growth. Apart from
addressing the challenge of AICD, uric acid activated ERK signaling in TK6 cells and increased
IL10 production, which was shown to be necessary for its proliferative effects. Taken together,
the results from this study led to the proposition that uric acid functions as a growth factor for
activated B cells, and also implicated the presence of a cell surface receptor that mediates uric
acid signaling.
Until recently, uric acid was for the most part viewed simply as an antioxidant and
interest in the molecule was marginally sustained due to its role in causing gout [248], and also
due to a longstanding correlation between hyperuricemia and cardiovascular disease [249,250].
Lately, however, uric acid has garnered renewed interest because of its recent identification as a
danger signal that is released by injured cells, enabling it to function as a stimulus for activating
immune responses [251]. The “danger model” [252,253] posits that APCs such as DCs can be
activated by endogenously produced molecules released by injured cells into the extracellular
environment. Such signals serve to promote DC activation and engender subsequent T cell
responses alongside antigen presentation. Employing a host of in vitro as well as in vivo studies,
Shi et al. identified uric acid to be a primary immunological danger signal [251]. A significant
increase in uric acid levels was observed when mouse lymphoma cells (EL4 cells) were forced

60
into an injurious state, and exposure to uric acid led to a strong increase in the expression of
costimulatory molecules in cultured DCs from healthy mice. In vivo significance of the
molecule was demonstrated by showing that mice injected with uric acid and antigen displayed
enhanced CD8+ T cell priming. Furthermore, when injured cells treated with allopurinol (a uric
acid analogue that reduces production of uric acid) and uricase were injected into mice that were
also exposed to the two inhibitors, significantly reduced plasma uric acid concentrations were
observed and priming of effector T cells was also reduced by almost 90% in the mice.
Subsequent investigations by other groups have further demystified this danger-alerting role of
uric acid. Increased levels of uric acid were observed in tumour cells undergoing death
processes, either due to responsiveness to chemotherapy, or as a result of immune rejection
[254]. Its withdrawal delayed the rejection process, whereas uric acid administration enhanced
it. Also, in the absence of T cell expansion, augmented antibody immunity was shown to be
induced by uric acid [255].
Given that uric acid is the end product of purine catabolism, it is likely that when cells
get injured and degrade their nucleic acids, the resultant purine build up allows for increased
accumulation of uric acid [251]. Dying cells are thus able to produce greater amounts of danger
signal without relying on protein synthesis [256]. With relation to immune surveillance, Shi et
al. propose a model which comprises dying cells simultaneously releasing antigens that are
picked up by DCs upon ingestion of cellular debris, as well as uric acid. The uric acid „danger
signal‟ allows DCs to mature and become immunostimulatory, thereby allowing the immune
system to respond even to tumours and infections that may not be associated with adjuvant
stimulation [251].
Although seemingly incongruent upon first glance, the newly identified role of uric acid
as a growth factor of activated B cells is not inconsistent with the recent reports describing uric

61
acid as an immunostimulatory danger signal. The key difference lies in the dose – the
concentration of uric acid at which DCs are stimulated corresponds to the point at which uric
acid precipitates into crystals. Unlike preformed monosodium urate (MSU) crystals, soluble
concentrations of uric acid (such as that used in the described experiments (0.1mg/ml)) cannot
activate DC responses [251]. Though uric acid is relatively insoluble in biological fluids, its
concentration in the cytosol is much higher (> 4 mg/ml) and increases dramatically in dying
cells as a result of DNA and RNA degradation. Upon lysis, the extracellular environment then
becomes flooded with high levels of uric acid, thus favouring MSU formation. These crystals
can also stimulate other immunological constituents like monocytes, leading to cytokine
production and further activation of the immune system [257]. In the same vein, along with
other endogenous danger signals, hyperuricemia may inspire autoimmunity in genetically
predisposed individuals [251]. In fact, it was recently reported that uric acid in the context of
gout can activate the NALP3 inflammasome [258], which is a known sensor of other
endogenous danger signals like extracellular ATP or hypotonic stress [259]. Soluble uric acid
on the other hand, seems to function as a separate molecule and possesses activity that appears
to be distinct from that of its crystalline form. The proliferative advantage of uric acid in
activated B cell cultures was observed only at the soluble dose of 0.1 mg/ml and this effect
could not be replicated at higher doses (data not shown). When used at 0.75 mg/ml, uric acid
precipitated out into the culture medium as crystals and was incapable of rescuing cells from
AICD.
That uric acid can possess growth factor activity at soluble levels has also been reported
in rat vascular smooth muscle cells (VSMCs) in an in vitro setting [260]. Uric acid was shown
to stimulate DNA synthesis, and as seems to be the case with activated B cells, this proliferative
capacity was independent of its antioxidant function. In addition, uric acid-induced growth in

62
VSMCs also implicated the involvement of signaling pathways as confirmed by the activation
of ERK1/2 [261], and an increase in the expression levels of platelet-derived growth factor
[260]. Further investigations led to the discovery that uric acid at the same soluble doses can
also directly exert proinflammatory effects on VSMCs [262]. The chemokine, monocyte
chemoattractant protein-1 (MCP-1), which is a major mediator of inflammation and is known to
play an active role in the initiation of atherosclerosis-associated lesions [263], was produced at
much higher levels upon exposure to uric acid. MCP-1 production was mediated via p38
MAPK, as well as activation of the nuclear transcription factors NF-B and AP-1. Uric acid
stimulation also resulted in the production of other inflammatory molecules such as IL1, IL6
and TNF [262]. Given that uric acid was found to stimulate IL10 production in TK6 cells, it
may be that it also possesses similar immunogenic capabilities in activated B cells as in VSMCs.
The results emerging from VSMC studies not only point to a potential mechanism
through which uric acid mediates vascular disease, but also highlight the many facets of uric
acid stimulation in different settings, as illustrated by the results presented here as well. In fact,
in light of the deleterious effects on vascular cells, and its role in cardiovascular disease,
atherosclerosis and kidney disease, many researchers are beginning to question the previously
unchallenged role of uric acid as an antioxidant [264-266]. How this information plays out with
regards to the role of uric acid in B cells and the distinction between its soluble and insoluble
crystalline form remains to be ascertained.

63
4.2 FUTURE DIRECTIONS
In summary, the results from this study suggest a novel role for soluble uric acid as a
growth stimulant for activated B cells. This conclusion was arrived at primarily from
experiments conducted in TK6 cells. In order to validate these results and ensure the soundness
of their implications, it is essential to perform these studies in normal human B cells, and
secondarily, it is also of interest to validate these results in murine B cells. In particular,
experiments involving uric acid transport inhibitors, confirmation of ERK and JNK activation,
confirmation of IL10 production, and IL10 inhibitor studies need to be repeated in normal B
cells so that it can be assessed if the results observed in TK6 cells are an aberrant response due
to the malignant nature of the cell line, or if they hold true for both normal and transformed B
cells. The applicability of the uric acid response to healthy B cells is especially crucial if the
results are to be interpreted in the context of normal B cell growth and function. Eventually, the
effects of uric acid on T cells, macrophages, and DCs can also be studied to determine if a
functional pattern can be ascribed to the activity of uric acid in immune cells.
Along with repeating the experiments in normal B cells, better characterization of the
response induced by uric acid in TK6 cells is needed. The experiments described here only
surveyed those proteins involved in major signaling pathways (as determined by their
phosphorylation status) and cytokine production was also only minimally examined. A more
large scale study involving gene arrays assessing activation of transcription factors and
production of messenger RNA needs to be undertaken in order to fully assess the signaling
pathways activated in TK6 cells by uric acid. A study of this magnitude would be particularly
beneficial in specifying the nature of the immunostimulatory (or not) response to uric acid.
Likewise, an examination of the changes in gene expression upon IL10 inhibition will help

64
further elucidate the role of this cytokine and shed light on the mechanism(s) via which IL10
mediates the proliferative effects of uric acid.
Although it seems likely that a receptor at the cell surface is activated upon exposure to
uric acid, further studies need to be conducted before the issue of receptor involvement can be
conclusively settled. A dose response study of uric acid as it relates to ERK activation, for
instance, would be helpful in this regard as the determination of the precise dose of uric acid that
leads to ERK activation would enable more fine-tuned studies to be performed, particularly as it
pertains to receptor identification. Blocking experiments involving ERK inhibitors and how the
uric acid response changes on a genome wide scale as observed in gene expression studies
would also be beneficial in elucidating the precise role of ERK and whether its activation is
sufficient and/or necessary in mediating the effects of uric acid. Studying the effects of JNK
inhibitors along with ERK inhibitor studies would also be helpful in determining if multiple
contradictory or confirmatory pathways are activated upon stimulation via uric acid.
If it becomes more apparent that uric acid functions extracellularly via engagement with
a cell surface receptor, a strategy to identify and clone the receptor needs to be determined.
Since there are no known receptors of uric acid, it is difficult to pinpoint likely candidates. In
terms of receptor class, given that uric acid is a purine derivative, it is possible that it activates a
purine receptor and mediates its effects through one of the known purinergic receptors. The
adenosine or P1 receptors, all of which couple to G proteins, and P2 receptors that primarily
engage ATP, ADP, UTP, and UDP are the two main families of purine receptors [267].
Although it would be worthwhile to examine P1 receptors, it is likely that uric acid does not act
via this family given that its members are GPCRs and lack of PKA activity (normally induced
upon engagement of GPCRs) did not abrogate the survival effects of uric acid in TK6 cells. P2
receptors, on the other hand, can be subdivided into two families, one of which consists of

65
GPCRs (P2Y receptors), and the other, the P2X receptors, which are ligand-gated ion channels
[267]. In fact, the P2X subclass of purine receptors are known to be expressed on VSMCs
[268], and it may be that uric acid exerts its effects on these cells via the activation of a P2X
receptor. The P2X7 receptor (P2X7R), in particular, is an attractive candidate given its
involvement in mediating immune responses in macrophages and leukemic lymphocytes [267].
In fact, it has been reported that IL1 secretion in murine macrophages as a response to
extracellular stimuli is not possible without the autocrine activation of P2X7R by ATP [269].
More recently, it has been reported that danger signals such as MSU crystals induce endogenous
ATP release and that antagonists of P2X7R can block the MSU-induced IL1 and IL18 secretion
in human monocytes [270]. While this information may prove to be irrelevant, it may be a
pragmatic starting point to determine if soluble non-crystalline uric acid can engage any of the
P2X receptors, in particular, P2X7, via use of receptor antagonists [267]. For the sake of
thoroughness, a quick screening of the results obtained by blocking P2Y receptors and P1
receptors in uric acid-treated TK6 cells should also be undertaken.
If uric acid is unable to act as a ligand for purine receptors, other strategies for
identification of its receptor will be required. Although it may be cumbersome, the activation of
ERK upon uric acid treatment can be taken advantage of for the development of a more direct
approach to receptor identification via functional screening of ERK activation and subsequent
cloning strategies. A reporter cell line that displays activation of the AP1 transcription factor,
which is a downstream activator of the ERK protein, can be used in an assay to monitor ERK
activity. Such a reporter line can be transfected with subsets of the TK6 gene library and ERK
activation can be assessed via luciferase or GFP activity upon treatment with uric acid. Isolation
of GFP/luciferase-positive clones in subsequent phases involving smaller and smaller gene
regions would eventually lead to the isolation of the uric acid receptor gene, which can then be

66
identified by cDNA cloning. Reporter cell lines are widely available and can be purchased
directly. Panomics Inc. (Fremont, CA) for instance, produces an AP1 reporter line derived from
human 293T cells that induces luciferase-tagged AP1 activity upon treatment with known
activators like phorbol myristate acetate. Such a reporter line, if easily transfectable, may be
ideal for a screening-based identification strategy.
Discovery of the uric acid receptor will shed further light on the intracellular
mechanisms via which uric acid exerts its effects, and perhaps reveal more information
pertaining to the disparity between the functions of soluble and crystalline uric acid, which
needs to be further explored. Moreover, based on the nature of the identified receptor, it can be
determined if the growth-inducing properties of uric acid are unique to certain cell types or not.
The implication arising from the antioxidant properties of uric acid and its apparent
function as a growth factor is that uric acid can be used in the generation of large numbers of
activated B cells that can serve as antigen presenting platforms in cancer vaccines. Further
studies characterizing the immunogenicity of uric acid treated human S2 B cells will be required
before investigations related to the development of vaccines can be initiated. Based on
previously reported results with CLL cells [109], it is plausible that normal activated B cells will
also require further exposure to PKC agonists before they are able to achieve maximal
upregulation of costimulatory molecules and produce inflammatory cytokines that can mediate
sufficiently strong CD8+ T cell responses. However, it may well be that soluble uric acid alone
can induce such immunostimulatory changes, as reported in VSMCs.
It is also worth investigating whether the two „danger signal‟ and growth factor roles of
uric acid can be married into an effective immunotherapeutic strategy for cancer treatment.
Perhaps soluble uric acid can be used to induce proliferation of S2 B cells and exposure to the
crystalline form can subsequently enable the acquisition of a DC-like phenotype. Such a

67
protocol will be more efficient and save time and money with regards to vaccine development.
Even if this is not the case, the implications for immunotherapy are still exciting given that uric
acid crystals could potentially be administered with B cell-based cancer vaccines for the
induction of potent DC activation, thereby dispensing the need for an initial T cell boost with
DC-based vaccines.
As further progress is made with regards to its mechanisms of action, a cohesive picture
of the seemingly disparate roles of uric acid in maintaining health and disease will hopefully
emerge. The data presented in this thesis have furthered this endeavour and have shed light on
the diverse nature of this molecule that is only now beginning to be appreciated.

68
LIST OF REFERENCES
[1] M Burnet. Cancer; a biological approach. I. The processes of control, Br.Med.J. 1 (1957)
779-786.
[2] L Zitvogel, A Tesniere, G Kroemer. Cancer despite immunosurveillance: immunoselection
and immunosubversion, Nat.Rev.Immunol. 6 (2006) 715-727.
[3] S Roithmaier, AM Haydon, S Loi, D Esmore, A Griffiths, P Bergin, et al. Incidence of
malignancies in heart and/or lung transplant recipients: a single-institution experience, J.Heart
Lung Transplant. 26 (2007) 845-849.
[4] K Imai, S Matsuyama, S Miyake, K Suga, K Nakachi. Natural cytotoxic activity of
peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general
population, Lancet. 356 (2000) 1795-1799.
[5] GP Dunn, LJ Old, RD Schreiber. The immunobiology of cancer immunosurveillance and
immunoediting, Immunity. 21 (2004) 137-148.
[6] MJ Smyth, ME Wallace, SL Nutt, H Yagita, DI Godfrey, Y Hayakawa. Sequential activation
of NKT cells and NK cells provides effective innate immunotherapy of cancer, J.Exp.Med. 201
(2005) 1973-1985.
[7] L Zhang, JR Conejo-Garcia, D Katsaros, PA Gimotty, M Massobrio, G Regnani, et al.
Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer, N.Engl.J.Med. 348
(2003) 203-213.

69
[8] J Galon, A Costes, F Sanchez-Cabo, A Kirilovsky, B Mlecnik, C Lagorce-Pages, et al. Type,
density, and location of immune cells within human colorectal tumors predict clinical outcome,
Science. 313 (2006) 1960-1964.
[9] SJ Piersma, ES Jordanova, MI van Poelgeest, KM Kwappenberg, JM van der Hulst, JW
Drijfhout, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is
associated with the absence of lymph node metastases in patients with large early-stage cervical
cancer, Cancer Res. 67 (2007) 354-361.
[10] HE Kohrt, N Nouri, K Nowels, D Johnson, S Holmes, PP Lee. Profile of immune cells in
axillary lymph nodes predicts disease-free survival in breast cancer, PLoS Med. 2 (2005) e284.
[11] P Sharma, Y Shen, S Wen, S Yamada, AA Jungbluth, S Gnjatic, et al. CD8 tumor-
infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma,
Proc.Natl.Acad.Sci.U.S.A. 104 (2007) 3967-3972.
[12] SS Dave, G Wright, B Tan, A Rosenwald, RD Gascoyne, WC Chan, et al. Prediction of
survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells,
N.Engl.J.Med. 351 (2004) 2159-2169.
[13] F Pages, A Berger, M Camus, F Sanchez-Cabo, A Costes, R Molidor, et al. Effector
memory T cells, early metastasis, and survival in colorectal cancer, N.Engl.J.Med. 353 (2005)
2654-2666.
[14] TJ Curiel, G Coukos, L Zou, X Alvarez, P Cheng, P Mottram, et al. Specific recruitment of
regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival,
Nat.Med. 10 (2004) 942-949.

70
[15] M Viguier, F Lemaitre, O Verola, MS Cho, G Gorochov, L Dubertret, et al. Foxp3
expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic
melanoma lymph nodes and inhibit the function of infiltrating T cells, J.Immunol. 173 (2004)
1444-1453.
[16] FM Marincola, EM Jaffee, DJ Hicklin, S Ferrone. Escape of human solid tumors from T-
cell recognition: molecular mechanisms and functional significance, Adv.Immunol. 74 (2000)
181-273.
[17] JN Blattman, PD Greenberg. Cancer immunotherapy: a treatment for the masses, Science.
305 (2004) 200-205.
[18] D Hanahan, RA Weinberg. The hallmarks of cancer, Cell. 100 (2000) 57-70.
[19] GP Dunn, LJ Old, RD Schreiber. The three Es of cancer immunoediting,
Annu.Rev.Immunol. 22 (2004) 329-360.
[20] MJ Smyth, GP Dunn, RD Schreiber. Cancer immunosurveillance and immunoediting: the
roles of immunity in suppressing tumor development and shaping tumor immunogenicity,
Adv.Immunol. 90 (2006) 1-50.
[21] DI Gabrilovich, HL Chen, KR Girgis, HT Cunningham, GM Meny, S Nadaf, et al.
Production of vascular endothelial growth factor by human tumors inhibits the functional
maturation of dendritic cells, Nat.Med. 2 (1996) 1096-1103.
[22] T Wang, G Niu, M Kortylewski, L Burdelya, K Shain, S Zhang, et al. Regulation of the
innate and adaptive immune responses by Stat-3 signaling in tumor cells, Nat.Med. 10 (2004)
48-54.

71
[23] K Staveley-O'Carroll, E Sotomayor, J Montgomery, I Borrello, L Hwang, S Fein, et al.
Induction of antigen-specific T cell anergy: An early event in the course of tumor progression,
Proc.Natl.Acad.Sci.U.S.A. 95 (1998) 1178-1183.
[24] EY Woo, H Yeh, CS Chu, K Schlienger, RG Carroll, JL Riley, et al. Cutting edge:
Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation,
J.Immunol. 168 (2002) 4272-4276.
[25] T So, M Takenoyama, M Mizukami, Y Ichiki, M Sugaya, T Hanagiri, et al. Haplotype loss
of HLA class I antigen as an escape mechanism from immune attack in lung cancer, Cancer Res.
65 (2005) 5945-5952.
[26] D Atkins, A Breuckmann, GE Schmahl, P Binner, S Ferrone, F Krummenauer, et al. MHC
class I antigen processing pathway defects, ras mutations and disease stage in colorectal
carcinoma, Int.J.Cancer. 109 (2004) 265-273.
[27] V Bronte, P Zanovello. Regulation of immune responses by L-arginine metabolism,
Nat.Rev.Immunol. 5 (2005) 641-654.
[28] C Uyttenhove, L Pilotte, I Theate, V Stroobant, D Colau, N Parmentier, et al. Evidence for
a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-
dioxygenase, Nat.Med. 9 (2003) 1269-1274.
[29] P Terness, TM Bauer, L Rose, C Dufter, A Watzlik, H Simon, et al. Inhibition of allogeneic
T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of
suppression by tryptophan metabolites, J.Exp.Med. 196 (2002) 447-457.

72
[30] W Zou. Immunosuppressive networks in the tumour environment and their therapeutic
relevance, Nat.Rev.Cancer. 5 (2005) 263-274.
[31] G Stassi, M Todaro, M Zerilli, L Ricci-Vitiani, D Di Liberto, M Patti, et al. Thyroid cancer
resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-
10, Cancer Res. 63 (2003) 6784-6790.
[32] Y Huang, N Obholzer, R Fayad, L Qiao. Turning on/off tumor-specific CTL response
during progressive tumor growth, J.Immunol. 175 (2005) 3110-3116.
[33] G Zhou, Z Lu, JD McCadden, HI Levitsky, AL Marson. Reciprocal changes in tumor
antigenicity and antigen-specific T cell function during tumor progression, J.Exp.Med. 200
(2004) 1581-1592.
[34] S Sakaguchi. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and
negative control of immune responses, Annu.Rev.Immunol. 22 (2004) 531-562.
[35] G Willimsky, T Blankenstein. Sporadic immunogenic tumours avoid destruction by
inducing T-cell tolerance, Nature. 437 (2005) 141-146.
[36] L Zitvogel, L Apetoh, F Ghiringhelli, G Kroemer. Immunological aspects of cancer
chemotherapy, Nat.Rev.Immunol. 8 (2008) 59-73.
[37] M von Mehren, GP Adams, LM Weiner. Monoclonal antibody therapy for cancer,
Annu.Rev.Med. 54 (2003) 343-369.
[38] BD Cheson. Monoclonal antibody therapy for B-cell malignancies, Semin.Oncol. 33 (2006)
S2-14.

73
[39] MJ Piccart-Gebhart, M Procter, B Leyland-Jones, A Goldhirsch, M Untch, I Smith, et al.
Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer, N.Engl.J.Med. 353
(2005) 1659-1672.
[40] J Mendelsohn. Epidermal growth factor receptor inhibition by a monoclonal antibody as
anticancer therapy, Clin.Cancer Res. 3 (1997) 2703-2707.
[41] G Giaccone. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell
lung cancer, J.Clin.Oncol. 23 (2005) 3235-3242.
[42] ES Kim, EE Vokes, MS Kies. Cetuximab in cancers of the lung and head & neck,
Semin.Oncol. 31 (2004) 61-67.
[43] OJ Finn. Cancer immunology, N.Engl.J.Med. 358 (2008) 2704-2715.
[44] CH June. Adoptive T cell therapy for cancer in the clinic, J.Clin.Invest. 117 (2007) 1466-
1476.
[45] C Yee, JA Thompson, D Byrd, SR Riddell, P Roche, E Celis, et al. Adoptive T cell therapy
using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic
melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells,
Proc.Natl.Acad.Sci.U.S.A. 99 (2002) 16168-16173.
[46] AK Palucka, H Ueno, JW Fay, J Banchereau. Taming cancer by inducing immunity via
dendritic cells, Immunol.Rev. 220 (2007) 129-150.
[47] RM Steinman, M Witmer-Pack, K Inaba. Dendritic cells: antigen presentation, accessory
function and clinical relevance, Adv.Exp.Med.Biol. 329 (1993) 1-9.

74
[48] FJ Hsu, C Benike, F Fagnoni, TM Liles, D Czerwinski, B Taidi, et al. Vaccination of
patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells, Nat.Med. 2
(1996) 52-58.
[49] FO Nestle, S Alijagic, M Gilliet, Y Sun, S Grabbe, R Dummer, et al. Vaccination of
melanoma patients with peptide- or tumor lysate-pulsed dendritic cells, Nat.Med. 4 (1998) 328-
332.
[50] B Thurner, I Haendle, C Roder, D Dieckmann, P Keikavoussi, H Jonuleit, et al.
Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands
specific cytotoxic T cells and induces regression of some metastases in advanced stage IV
melanoma, J.Exp.Med. 190 (1999) 1669-1678.
[51] J Banchereau, AK Palucka, M Dhodapkar, S Burkeholder, N Taquet, A Rolland, et al.
Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-
derived dendritic cell vaccine, Cancer Res. 61 (2001) 6451-6458.
[52] L Fong, Y Hou, A Rivas, C Benike, A Yuen, GA Fisher, et al. Altered peptide ligand
vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy,
Proc.Natl.Acad.Sci.U.S.A. 98 (2001) 8809-8814.
[53] D Ridgway. The first 1000 dendritic cell vaccinees, Cancer Invest. 21 (2003) 873-886.
[54] E Gatti, MA Velleca, BC Biedermann, W Ma, J Unternaehrer, MW Ebersold, et al. Large-
scale culture and selective maturation of human Langerhans cells from granulocyte colony-
stimulating factor-mobilized CD34+ progenitors, J.Immunol. 164 (2000) 3600-3607.

75
[55] J Banchereau, B Schuler-Thurner, AK Palucka, G Schuler. Dendritic cells as vectors for
therapy, Cell. 106 (2001) 271-274.
[56] RM Steinman, M Dhodapkar. Active immunization against cancer with dendritic cells: the
near future, Int.J.Cancer. 94 (2001) 459-473.
[57] T Luft, M Jefford, P Luetjens, T Toy, H Hochrein, KA Masterman, et al. Functionally
distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2)
regulates the migratory capacity of specific DC subsets, Blood. 100 (2002) 1362-1372.
[58] BJ Czerniecki, GK Koski, U Koldovsky, S Xu, PA Cohen, R Mick, et al. Targeting HER-
2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst
secretion, Cancer Res. 67 (2007) 1842-1852.
[59] JW Fay, AK Palucka, S Paczesny, M Dhodapkar, DA Johnston, S Burkeholder, et al. Long-
term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed
CD34(+) progenitor-derived dendritic cells, Cancer Immunol.Immunother. 55 (2006) 1209-
1218.
[60] AK Palucka, H Ueno, J Connolly, F Kerneis-Norvell, JP Blanck, DA Johnston, et al.
Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical
responses and MART-1 specific CD8+ T-cell immunity, J.Immunother. 29 (2006) 545-557.
[61] P Gould. Sipuleucel-T shows partial advantage in prostate cancer, Lancet Oncol. 7 (2006)
710.
[62] M Jefford, E Maraskovsky, J Cebon, ID Davis. The use of dendritic cells in cancer therapy,
Lancet Oncol. 2 (2001) 343-353.

76
[63] AF Ochsenbein, S Sierro, B Odermatt, M Pericin, U Karrer, J Hermans, et al. Roles of
tumour localization, second signals and cross priming in cytotoxic T-cell induction, Nature. 411
(2001) 1058-1064.
[64] S Siena, M Di Nicola, M Bregni, R Mortarini, A Anichini, L Lombardi, et al. Massive ex
vivo generation of functional dendritic cells from mobilized CD34+ blood progenitors for
anticancer therapy, Exp.Hematol. 23 (1995) 1463-1471.
[65] G Girolomoni, P Ricciardi-Castagnoli. Dendritic cells hold promise for immunotherapy,
Immunol.Today. 18 (1997) 102-104.
[66] J Banchereau, RM Steinman. Dendritic cells and the control of immunity, Nature. 392
(1998) 245-252.
[67] FD Finkelman, A Lees, R Birnbaum, WC Gause, SC Morris. Dendritic cells can present
antigen in vivo in a tolerogenic or immunogenic fashion, J.Immunol. 157 (1996) 1406-1414.
[68] KM Ardeshna, AR Pizzey, NS Thomas, S Orr, DC Linch, S Devereux. Monocyte-derived
dendritic cells do not proliferate and are not susceptible to retroviral transduction,
Br.J.Haematol. 108 (2000) 817-824.
[69] JL Schultze, S Michalak, MJ Seamon, G Dranoff, K Jung, J Daley, et al. CD40-activated
human B cells: an alternative source of highly efficient antigen presenting cells to generate
autologous antigen-specific T cells for adoptive immunotherapy, J.Clin.Invest. 100 (1997) 2757-
2765.

77
[70] A MartIn-Fontecha, S Sebastiani, UE Hopken, M Uguccioni, M Lipp, A Lanzavecchia, et
al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte
traffic and priming, J.Exp.Med. 198 (2003) 615-621.
[71] MS von Bergwelt-Baildon, RH Vonderheide, B Maecker, N Hirano, KS Anderson, MO
Butler, et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently
induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical
application, Blood. 99 (2002) 3319-3325.
[72] RM Steinman. Dendritic cells and immune-based therapies, Exp.Hematol. 24 (1996) 859-
862.
[73] W Brugger, P Brossart, S Scheding, G Stuhler, K Heinrich, V Reichardt, et al. Approaches
to dendritic cell-based immunotherapy after peripheral blood stem cell transplantation,
Ann.N.Y.Acad.Sci. 872 (1999) 363-371.
[74] JB Latouche, M Sadelain. Induction of human cytotoxic T lymphocytes by artificial
antigen-presenting cells, Nat.Biotechnol. 18 (2000) 405-409.
[75] WF Anderson. Human gene therapy, Nature. 392 (1998) 25-30.
[76] D Rodriguez-Pinto. B cells as antigen presenting cells, Cell.Immunol. 238 (2005) 67-75.
[77] FD Batista, MS Neuberger. Affinity dependence of the B cell response to antigen: a
threshold, a ceiling, and the importance of off-rate, Immunity. 8 (1998) 751-759.
[78] A Stoddart, ML Dykstra, BK Brown, W Song, SK Pierce, FM Brodsky. Lipid rafts unite
signaling cascades with clathrin to regulate BCR internalization, Immunity. 17 (2002) 451-462.

78
[79] MA Putnam, AE Moquin, M Merrihew, C Outcalt, E Sorge, A Caballero, et al. Lipid raft-
independent B cell receptor-mediated antigen internalization and intracellular trafficking,
J.Immunol. 170 (2003) 905-912.
[80] H Niiro, A Allam, A Stoddart, FM Brodsky, AJ Marshall, EA Clark. The B lymphocyte
adaptor molecule of 32 kilodaltons (Bam32) regulates B cell antigen receptor internalization,
J.Immunol. 173 (2004) 5601-5609.
[81] VR Aluvihare, AA Khamlichi, GT Williams, L Adorini, MS Neuberger. Acceleration of
intracellular targeting of antigen by the B-cell antigen receptor: importance depends on the
nature of the antigen-antibody interaction, EMBO J. 16 (1997) 3553-3562.
[82] D Lankar, V Briken, K Adler, P Weiser, S Cassard, U Blank, et al. Syk tyrosine kinase and
B cell antigen receptor (BCR) immunoglobulin-alpha subunit determine BCR-mediated major
histocompatibility complex class II-restricted antigen presentation, J.Exp.Med. 188 (1998) 819-
831.
[83] K Siemasko, BJ Eisfelder, C Stebbins, S Kabak, AJ Sant, W Song, et al. Ig alpha and Ig
beta are required for efficient trafficking to late endosomes and to enhance antigen presentation,
J.Immunol. 162 (1999) 6518-6525.
[84] K Siemasko, BJ Skaggs, S Kabak, E Williamson, BK Brown, W Song, et al. Receptor-
facilitated antigen presentation requires the recruitment of B cell linker protein to Igalpha,
J.Immunol. 168 (2002) 2127-2138.
[85] PC Cheng, CR Steele, L Gu, W Song, SK Pierce. MHC class II antigen processing in B
cells: accelerated intracellular targeting of antigens, J.Immunol. 162 (1999) 7171-7180.

79
[86] VS Zimmermann, P Rovere, J Trucy, K Serre, P Machy, F Forquet, et al. Engagement of B
cell receptor regulates the invariant chain-dependent MHC class II presentation pathway,
J.Immunol. 162 (1999) 2495-2502.
[87] K Siemasko, BJ Eisfelder, E Williamson, S Kabak, MR Clark. Cutting edge: signals from
the B lymphocyte antigen receptor regulate MHC class II containing late endosomes,
J.Immunol. 160 (1998) 5203-5208.
[88] HW Davidson, C Watts. Epitope-directed processing of specific antigen by B lymphocytes,
J.Cell Biol. 109 (1989) 85-92.
[89] C Watts, A Lanzavecchia. Suppressive effect of antibody on processing of T cell epitopes,
J.Exp.Med. 178 (1993) 1459-1463.
[90] L Karlsson, CD Surh, J Sprent, PA Peterson. A novel class II MHC molecule with unusual
tissue distribution, Nature. 351 (1991) 485-488.
[91] LK Denzin, DB Sant'Angelo, C Hammond, MJ Surman, P Cresswell. Negative regulation
by HLA-DO of MHC class II-restricted antigen processing, Science. 278 (1997) 106-109.
[92] M van Ham, M van Lith, B Lillemeier, E Tjin, U Gruneberg, D Rahman, et al. Modulation
of the major histocompatibility complex class II-associated peptide repertoire by human
histocompatibility leukocyte antigen (HLA)-DO, J.Exp.Med. 191 (2000) 1127-1136.
[93] C Alfonso, M Liljedahl, O Winqvist, CD Surh, PA Peterson, WP Fung-Leung, et al. The
role of H2-O and HLA-DO in major histocompatibility complex class II-restricted antigen
processing and presentation, Immunol.Rev. 172 (1999) 255-266.

80
[94] X Chen, PE Jensen. The expression of HLA-DO (H2-O) in B lymphocytes, Immunol.Res.
29 (2004) 19-28.
[95] DJ Lenschow, AI Sperling, MP Cooke, G Freeman, L Rhee, DC Decker, et al. Differential
up-regulation of the B7-1 and B7-2 costimulatory molecules after Ig receptor engagement by
antigen, J.Immunol. 153 (1994) 1990-1997.
[96] TO Nashar, JR Drake. The pathway of antigen uptake and processing dictates MHC class
II-mediated B cell survival and activation, J.Immunol. 174 (2005) 1306-1316.
[97] ME Skelsey, E Mayhew, JY Niederkorn. Splenic B cells act as antigen presenting cells for
the induction of anterior chamber-associated immune deviation, Invest.Ophthalmol.Vis.Sci. 44
(2003) 5242-5251.
[98] JL Schultze, S Grabbe, MS von Bergwelt-Baildon. DCs and CD40-activated B cells:
current and future avenues to cellular cancer immunotherapy, Trends Immunol. 25 (2004) 659-
664.
[99] CM Coughlin, BA Vance, SA Grupp, RH Vonderheide. RNA-transfected CD40-activated
B cells induce functional T-cell responses against viral and tumor antigen targets: implications
for pediatric immunotherapy, Blood. 103 (2004) 2046-2054.
[100] CM Coughlin, RH Vonderheide. Targeting adult and pediatric cancers via cell-based
vaccines and the prospect of activated B lymphocytes as a novel modality, Cancer.Biol.Ther. 2
(2003) 466-470.
[101] D Verthelyi, RA Zeuner. Differential signaling by CpG DNA in DCs and B cells: not just
TLR9, Trends Immunol. 24 (2003) 519-522.

81
[102] JL Schultze, S Michalak, J Lowne, A Wong, MH Gilleece, JG Gribben, et al. Human non-
germinal center B cell interleukin (IL)-12 production is primarily regulated by T cell signals
CD40 ligand, interferon gamma, and IL-10: role of B cells in the maintenance of T cell
responses, J.Exp.Med. 189 (1999) 1-12.
[103] C Arpin, J Dechanet, C Van Kooten, P Merville, G Grouard, F Briere, et al. Generation of
memory B cells and plasma cells in vitro, Science. 268 (1995) 720-722.
[104] C van Kooten, J Banchereau. Functions of CD40 on B cells, dendritic cells and other cells,
Curr.Opin.Immunol. 9 (1997) 330-337.
[105] P Lissoni, L Vigore, R Ferranti, R Bukovec, S Meregalli, M Mandala, et al. Circulating
dendritic cells in early and advanced cancer patients: diminished percent in the metastatic
disease, J.Biol.Regul.Homeost.Agents. 13 (1999) 216-219.
[106] DE Spaner, RL Miller, J Mena, L Grossman, V Sorrenti, Y Shi. Regression of
lymphomatous skin deposits in a chronic lymphocytic leukemia patient treated with the Toll-like
receptor-7/8 agonist, imiquimod, Leuk.Lymphoma. 46 (2005) 935-939.
[107] DE Spaner, Y Shi, D White, J Mena, C Hammond, J Tomic, et al. Immunomodulatory
effects of Toll-like receptor-7 activation on chronic lymphocytic leukemia cells, Leukemia. 20
(2006) 286-295.
[108] DE Spaner, C Hammond, J Mena, Y Shi. Effect of IL-2R beta-binding cytokines on
costimulatory properties of chronic lymphocytic leukaemia cells: implications for
immunotherapy, Br.J.Haematol. 127 (2004) 531-542.

82
[109] J Tomic, D White, Y Shi, J Mena, C Hammond, L He, et al. Sensitization of IL-2
signaling through TLR-7 enhances B lymphoma cell immunogenicity, J.Immunol. 176 (2006)
3830-3839.
[110] CA Janeway Jr, R Medzhitov. Innate immune recognition, Annu.Rev.Immunol. 20 (2002)
197-216.
[111] S Akira, K Takeda. Toll-like receptor signalling, Nat.Rev.Immunol. 4 (2004) 499-511.
[112] C Pasare, R Medzhitov. Control of B-cell responses by Toll-like receptors, Nature. 438
(2005) 364-368.
[113] P Ahmad-Nejad, H Hacker, M Rutz, S Bauer, RM Vabulas, H Wagner. Bacterial CpG-
DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments,
Eur.J.Immunol. 32 (2002) 1958-1968.
[114] F Heil, P Ahmad-Nejad, H Hemmi, H Hochrein, F Ampenberger, T Gellert, et al. The
Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within
the TLR7, 8 and 9 subfamily, Eur.J.Immunol. 33 (2003) 2987-2997.
[115] M Matsumoto, K Funami, M Tanabe, H Oshiumi, M Shingai, Y Seto, et al. Subcellular
localization of Toll-like receptor 3 in human dendritic cells, J.Immunol. 171 (2003) 3154-3162.
[116] A Iwasaki, R Medzhitov. Toll-like receptor control of the adaptive immune responses,
Nat.Immunol. 5 (2004) 987-995.
[117] TH Chuang, RJ Ulevitch. Cloning and characterization of a sub-family of human toll-like
receptors: hTLR7, hTLR8 and hTLR9, Eur.Cytokine Netw. 11 (2000) 372-378.

83
[118] J Lee, TH Chuang, V Redecke, L She, PM Pitha, DA Carson, et al. Molecular basis for
the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor
7, Proc.Natl.Acad.Sci.U.S.A. 100 (2003) 6646-6651.
[119] H Hemmi, T Kaisho, O Takeuchi, S Sato, H Sanjo, K Hoshino, et al. Small anti-viral
compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway,
Nat.Immunol. 3 (2002) 196-200.
[120] K Takeda, T Kaisho, S Akira. Toll-like receptors, Annu.Rev.Immunol. 21 (2003) 335-
376.
[121] A Dunne, LA O'Neill. The interleukin-1 receptor/Toll-like receptor superfamily: signal
transduction during inflammation and host defense, Sci.STKE. 2003 (2003) re3.
[122] T Kawai, S Akira. Pathogen recognition with Toll-like receptors, Curr.Opin.Immunol. 17
(2005) 338-344.
[123] P Tosi, PL Zinzani, A Pellacani, E Ottaviani, M Magagnoli, S Tura. Loxoribine affects
fludarabine activity on freshly isolated B-chronic lymphocytic leukemia cells, Leuk.Lymphoma.
26 (1997) 343-348.
[124] K Sugamura, H Asao, M Kondo, N Tanaka, N Ishii, K Ohbo, et al. The interleukin-2
receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell
development in XSCID, Annu.Rev.Immunol. 14 (1996) 179-205.
[125] TA Waldmann. The interleukin-2 receptor, J.Biol.Chem. 266 (1991) 2681-2684.
[126] T Taniguchi, Y Minami. The IL-2/IL-2 receptor system: a current overview, Cell. 73
(1993) 5-8.

84
[127] TA Waldmann. The biology of interleukin-2 and interleukin-15: implications for cancer
therapy and vaccine design, Nat.Rev.Immunol. 6 (2006) 595-601.
[128] T Miyazaki, ZJ Liu, A Kawahara, Y Minami, K Yamada, Y Tsujimoto, et al. Three
distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic
cell proliferation, Cell. 81 (1995) 223-231.
[129] TA Waldmann, S Dubois, Y Tagaya. Contrasting roles of IL-2 and IL-15 in the life and
death of lymphocytes: implications for immunotherapy, Immunity. 14 (2001) 105-110.
[130] SA Rosenberg. Interleukin-2 and the development of immunotherapy for the treatment of
patients with cancer, Cancer J.Sci.Am. 6 Suppl 1 (2000) S2-7.
[131] SA Rosenberg, JC Yang, SL Topalian, DJ Schwartzentruber, JS Weber, DR Parkinson, et
al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using
high-dose bolus interleukin 2, JAMA. 271 (1994) 907-913.
[132] B Nashan, R Moore, P Amlot, AG Schmidt, K Abeywickrama, JP Soulillou. Randomised
trial of basiliximab versus placebo for control of acute cellular rejection in renal allograft
recipients. CHIB 201 International Study Group, Lancet. 350 (1997) 1193-1198.
[133] RL Kirkman, ME Shapiro, CB Carpenter, DB McKay, EL Milford, EL Ramos, et al. A
randomized prospective trial of anti-Tac monoclonal antibody in human renal transplantation,
Transplantation. 51 (1991) 107-113.
[134] F Vincenti, R Kirkman, S Light, G Bumgardner, M Pescovitz, P Halloran, et al.
Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal
transplantation. Daclizumab Triple Therapy Study Group, N.Engl.J.Med. 338 (1998) 161-165.

85
[135] D Donjerkovic, DW Scott. Activation-induced cell death in B lymphocytes, Cell Res. 10
(2000) 179-192.
[136] GJ Nossal. Negative selection of lymphocytes, Cell. 76 (1994) 229-239.
[137] SL Parry, J Hasbold, M Holman, GG Klaus. Hypercross-linking surface IgM or IgD
receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and
anti-CD40, J.Immunol. 152 (1994) 2821-2829.
[138] SL Parry, MJ Holman, J Hasbold, GG Klaus. Plastic-immobilized anti-mu or anti-delta
antibodies induce apoptosis in mature murine B lymphocytes, Eur.J.Immunol. 24 (1994) 974-
979.
[139] MK Newell, J VanderWall, KS Beard, JH Freed. Ligation of major histocompatibility
complex class II molecules mediates apoptotic cell death in resting B lymphocytes,
Proc.Natl.Acad.Sci.U.S.A. 90 (1993) 10459-10463.
[140] B Pulendran, G Kannourakis, S Nouri, KG Smith, GJ Nossal. Soluble antigen can cause
enhanced apoptosis of germinal-centre B cells, Nature. 375 (1995) 331-334.
[141] T Hamano, T Iwasaki, A Ogata, N Hashimoto, E Kakishita. The molecular mechanism in
activation-induced cell death of an Ag-reactive B cell clone, Clin.Exp.Immunol. 128 (2002)
436-443.
[142] MJ Lenardo. Fas and the art of lymphocyte maintenance, J.Exp.Med. 183 (1996) 721-724.
[143] M Patel, BJ Day, JD Crapo, I Fridovich, JO McNamara. Requirement for superoxide in
excitotoxic cell death, Neuron. 16 (1996) 345-355.

86
[144] TM Buttke, PA Sandstrom. Redox regulation of programmed cell death in lymphocytes,
Free Radic.Res. 22 (1995) 389-397.
[145] TS Dobmeyer, S Findhammer, JM Dobmeyer, SA Klein, B Raffel, D Hoelzer, et al. Ex
vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte
loss in HIV infection, Free Radic.Biol.Med. 22 (1997) 775-785.
[146] W Fang, JJ Rivard, JA Ganser, TW LeBien, KA Nath, DL Mueller, et al. Bcl-xL rescues
WEHI 231 B lymphocytes from oxidant-mediated death following diverse apoptotic stimuli,
J.Immunol. 155 (1995) 66-75.
[147] TM Buttke, PA Sandstrom. Oxidative stress as a mediator of apoptosis, Immunol.Today.
15 (1994) 7-10.
[148] X Liu, CN Kim, J Yang, R Jemmerson, X Wang. Induction of apoptotic program in cell-
free extracts: requirement for dATP and cytochrome c, Cell. 86 (1996) 147-157.
[149] SA Susin, HK Lorenzo, N Zamzami, I Marzo, BE Snow, GM Brothers, et al. Molecular
characterization of mitochondrial apoptosis-inducing factor, Nature. 397 (1999) 441-446.
[150] PF Li, R Dietz, R von Harsdorf. p53 regulates mitochondrial membrane potential through
reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2,
EMBO J. 18 (1999) 6027-6036.
[151] NS Chandel, PT Schumacker, RH Arch. Reactive oxygen species are downstream
products of TRAF-mediated signal transduction, J.Biol.Chem. 276 (2001) 42728-42736.
[152] DR Green, JC Reed. Mitochondria and apoptosis, Science. 281 (1998) 1309-1312.

87
[153] G Kroemer, JC Reed. Mitochondrial control of cell death, Nat.Med. 6 (2000) 513-519.
[154] K Polyak, Y Xia, JL Zweier, KW Kinzler, B Vogelstein. A model for p53-induced
apoptosis, Nature. 389 (1997) 300-305.
[155] AG Hall. Review: The role of glutathione in the regulation of apoptosis, Eur.J.Clin.Invest.
29 (1999) 238-245.
[156] DW Voehringer, DL Hirschberg, J Xiao, Q Lu, M Roederer, CB Lock, et al. Gene
microarray identification of redox and mitochondrial elements that control resistance or
sensitivity to apoptosis, Proc.Natl.Acad.Sci.U.S.A. 97 (2000) 2680-2685.
[157] M Berard, P Mondiere, M Casamayor-Palleja, A Hennino, C Bella, T Defrance.
Mitochondria connects the antigen receptor to effector caspases during B cell receptor-induced
apoptosis in normal human B cells, J.Immunol. 163 (1999) 4655-4662.
[158] IF Benzie. Evolution of antioxidant defence mechanisms, Eur.J.Nutr. 39 (2000) 53-61.
[159] GK Glantzounis, EC Tsimoyiannis, AM Kappas, DA Galaris. Uric acid and oxidative
stress, Curr.Pharm.Des. 11 (2005) 4145-4151.
[160] I Fridovich. The biology of oxygen radicals, Science. 201 (1978) 875-880.
[161] BN Ames, R Cathcart, E Schwiers, P Hochstein. Uric acid provides an antioxidant defense
in humans against oxidant- and radical-caused aging and cancer: a hypothesis,
Proc.Natl.Acad.Sci.U.S.A. 78 (1981) 6858-6862.
[162] BF Becker. Towards the physiological function of uric acid, Free Radic.Biol.Med. 14
(1993) 615-631.

88
[163] CC Lee, XW Wu, RA Gibbs, RG Cook, DM Muzny, CT Caskey. Generation of cDNA
probes directed by amino acid sequence: cloning of urate oxidase, Science. 239 (1988) 1288-
1291.
[164] K Motojima, S Goto. A human genomic sequence highly homologous to the 3'-
untranslated region of rat uricase mRNA, Biochem.Biophys.Res.Commun. 155 (1988) 1266-
1270.
[165] AV Yeldandi, YD Patel, M Liao, FT Kao, MS Rao, JK Reddy, et al. Localization of the
human urate oxidase gene (UOX) to 1p22, Cytogenet.Cell Genet. 61 (1992) 121-122.
[166] M Grootveld, B Halliwell. Measurement of allantoin and uric acid in human body fluids.
A potential index of free-radical reactions in vivo? Biochem.J. 243 (1987) 803-808.
[167] BF Becker, N Reinholz, T Ozcelik, B Leipert, E Gerlach. Uric acid as radical scavenger
and antioxidant in the heart, Pflugers Arch. 415 (1989) 127-135.
[168] H Kaur, B Halliwell. Action of biologically-relevant oxidizing species upon uric acid.
Identification of uric acid oxidation products, Chem.Biol.Interact. 73 (1990) 235-247.
[169] JP Manfredi, EW Holmes. Purine salvage pathways in myocardium, Annu.Rev.Physiol.
47 (1985) 691-705.
[170] W Loffler, W Grobner, R Medina, N Zollner. Influence of dietary purines on pool size,
turnover, and excretion of uric acid during balance conditions. Isotope studies using 15N-uric
acid, Res.Exp.Med.(Berl). 181 (1982) 113-123.

89
[171] DD Wayner, GW Burton, KU Ingold, LR Barclay, SJ Locke. The relative contributions of
vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity
of human blood plasma, Biochim.Biophys.Acta. 924 (1987) 408-419.
[172] T Hasegawa, M Kuroda. A new role of uric acid as an antioxidant in human plasma,
Rinsho Byori. 37 (1989) 1020-1027.
[173] KJ Davies, A Sevanian, SF Muakkassah-Kelly, P Hochstein. Uric acid-iron ion
complexes. A new aspect of the antioxidant functions of uric acid, Biochem.J. 235 (1986) 747-
754.
[174] H Einsele, MR Clemens, U Wegner, HD Waller. Effect of free radical scavengers and
metal ion chelators on hydrogen peroxide and phenylhydrazine induced red blood cell lipid
peroxidation, Free Radic.Res.Commun. 3 (1987) 257-263.
[175] T Miura, S Muraoka, T Ogiso. Inhibitory effect of urate on oxidative damage induced by
adriamycin-Fe3+ in the presence of H2O2, Res.Commun.Chem.Pathol.Pharmacol. 79 (1993)
75-85.
[176] GL Squadrito, R Cueto, AE Splenser, A Valavanidis, H Zhang, RM Uppu, et al. Reaction
of uric acid with peroxynitrite and implications for the mechanism of neuroprotection by uric
acid, Arch.Biochem.Biophys. 376 (2000) 333-337.
[177] M Whiteman, U Ketsawatsakul, B Halliwell. A reassessment of the peroxynitrite
scavenging activity of uric acid, Ann.N.Y.Acad.Sci. 962 (2002) 242-259.

90
[178] PT Doulias, A Barbouti, D Galaris, H Ischiropoulos. SIN-1-induced DNA damage in
isolated human peripheral blood lymphocytes as assessed by single cell gel electrophoresis
(comet assay), Free Radic.Biol.Med. 30 (2001) 679-685.
[179] AJ Gow, CR Farkouh, DA Munson, MA Posencheg, H Ischiropoulos. Biological
significance of nitric oxide-mediated protein modifications, Am.J.Physiol.Lung
Cell.Mol.Physiol. 287 (2004) L262-8.
[180] SA Greenacre, H Ischiropoulos. Tyrosine nitration: localisation, quantification,
consequences for protein function and signal transduction, Free Radic.Res. 34 (2001) 541-581.
[181] DC Hooper, S Spitsin, RB Kean, JM Champion, GM Dickson, I Chaudhry, et al. Uric
acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and
multiple sclerosis, Proc.Natl.Acad.Sci.U.S.A. 95 (1998) 675-680.
[182] A Chamorro, V Obach, A Cervera, M Revilla, R Deulofeu, JH Aponte. Prognostic
significance of uric acid serum concentration in patients with acute ischemic stroke, Stroke. 33
(2002) 1048-1052.
[183] A Chamorro, AM Planas, DS Muner, R Deulofeu. Uric acid administration for
neuroprotection in patients with acute brain ischemia, Med.Hypotheses. 62 (2004) 173-176.
[184] DA Thorley-Lawson. Epstein-Barr virus: exploiting the immune system,
Nat.Rev.Immunol. 1 (2001) 75-82.
[185] JK Hamilton, LA Paquin, JL Sullivan, HS Maurer, FG Cruzi, AJ Provisor, et al. X-linked
lymphoproliferative syndrome registry report, J.Pediatr. 96 (1980) 669-673.

91
[186] DT Purtilo, CK Cassel, JP Yang, R Harper. X-linked recessive progressive combined
variable immunodeficiency (Duncan's disease), Lancet. 1 (1975) 935-940.
[187] MA EPSTEIN, BG ACHONG, YM BARR. Virus Particles in Cultured Lymphoblasts
from Burkitt's Lymphoma, Lancet. 1 (1964) 702-703.
[188] P Aman, B Ehlin-Henriksson, G Klein. Epstein-Barr virus susceptibility of normal human
B lymphocyte populations, J.Exp.Med. 159 (1984) 208-220.
[189] DA Thorley-Lawson, KP Mann. Early events in Epstein-Barr virus infection provide a
model for B cell activation, J.Exp.Med. 162 (1985) 45-59.
[190] AM Joseph, GJ Babcock, DA Thorley-Lawson. EBV persistence involves strict selection
of latently infected B cells, J.Immunol. 165 (2000) 2975-2981.
[191] GJ Babcock, LL Decker, M Volk, DA Thorley-Lawson. EBV persistence in memory B
cells in vivo, Immunity. 9 (1998) 395-404.
[192] EM Miyashita, B Yang, GJ Babcock, DA Thorley-Lawson. Identification of the site of
Epstein-Barr virus persistence in vivo as a resting B cell, J.Virol. 71 (1997) 4882-4891.
[193] G Khan, EM Miyashita, B Yang, GJ Babcock, DA Thorley-Lawson. Is EBV persistence
in vivo a model for B cell homeostasis? Immunity. 5 (1996) 173-179.
[194] QY Yao, AB Rickinson, MA Epstein. A re-examination of the Epstein-Barr virus carrier
state in healthy seropositive individuals, Int.J.Cancer. 35 (1985) 35-42.

92
[195] LC Tan, N Gudgeon, NE Annels, P Hansasuta, CA O'Callaghan, S Rowland-Jones, et al.
A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers,
J.Immunol. 162 (1999) 1827-1835.
[196] JH Pope, MK Horne, W Scott. Transformation of foetal human keukocytes in vitro by
filtrates of a human leukaemic cell line containing herpes-like virus, Int.J.Cancer. 3 (1968) 857-
866.
[197] L Brooks, QY Yao, AB Rickinson, LS Young. Epstein-Barr virus latent gene transcription
in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts,
J.Virol. 66 (1992) 2689-2697.
[198] R Fahraeus, HL Fu, I Ernberg, J Finke, M Rowe, G Klein, et al. Expression of Epstein-
Barr virus-encoded proteins in nasopharyngeal carcinoma, Int.J.Cancer. 42 (1988) 329-338.
[199] RG Caldwell, JB Wilson, SJ Anderson, R Longnecker. Epstein-Barr virus LMP2A drives
B cell development and survival in the absence of normal B cell receptor signals, Immunity. 9
(1998) 405-411.
[200] O Gires, U Zimber-Strobl, R Gonnella, M Ueffing, G Marschall, R Zeidler, et al. Latent
membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule,
EMBO J. 16 (1997) 6131-6140.
[201] E Kilger, A Kieser, M Baumann, W Hammerschmidt. Epstein-Barr virus-mediated B-cell
proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40
receptor, EMBO J. 17 (1998) 1700-1709.

93
[202] U Zimber-Strobl, B Kempkes, G Marschall, R Zeidler, C Van Kooten, J Banchereau, et al.
Epstein-Barr virus latent membrane protein (LMP1) is not sufficient to maintain proliferation of
B cells but both it and activated CD40 can prolong their survival, EMBO J. 15 (1996) 7070-
7078.
[203] G Mosialos, M Birkenbach, R Yalamanchili, T VanArsdale, C Ware, E Kieff. The
Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis
factor receptor family, Cell. 80 (1995) 389-399.
[204] J Banchereau, F Bazan, D Blanchard, F Briere, JP Galizzi, C van Kooten, et al. The CD40
antigen and its ligand, Annu.Rev.Immunol. 12 (1994) 881-922.
[205] Y Hsing, BS Hostager, GA Bishop. Characterization of CD40 signaling determinants
regulating nuclear factor-kappa B activation in B lymphocytes, J.Immunol. 159 (1997) 4898-
4906.
[206] SH Hanissian, RS Geha. Jak3 is associated with CD40 and is critical for CD40 induction
of gene expression in B cells, Immunity. 6 (1997) 379-387.
[207] O Gires, F Kohlhuber, E Kilger, M Baumann, A Kieser, C Kaiser, et al. Latent membrane
protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins, EMBO J. 18
(1999) 3064-3073.
[208] CW Dawson, G Tramountanis, AG Eliopoulos, LS Young. Epstein-Barr virus latent
membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to
promote cell survival and induce actin filament remodeling, J.Biol.Chem. 278 (2003) 3694-
3704.

94
[209] AG Eliopoulos, NJ Gallagher, SM Blake, CW Dawson, LS Young. Activation of the p38
mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane
protein 1 coregulates interleukin-6 and interleukin-8 production, J.Biol.Chem. 274 (1999)
16085-16096.
[210] DS Huen, SA Henderson, D Croom-Carter, M Rowe. The Epstein-Barr virus latent
membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via
two effector regions in its carboxy-terminal cytoplasmic domain, Oncogene. 10 (1995) 549-560.
[211] M Luftig, T Yasui, V Soni, MS Kang, N Jacobson, E Cahir-McFarland, et al. Epstein-Barr
virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent
noncanonical NF-kappaB activation, Proc.Natl.Acad.Sci.U.S.A. 101 (2004) 141-146.
[212] KM Kaye, KM Izumi, E Kieff. Epstein-Barr virus latent membrane protein 1 is essential
for B-lymphocyte growth transformation, Proc.Natl.Acad.Sci.U.S.A. 90 (1993) 9150-9154.
[213] P Speck, KA Kline, P Cheresh, R Longnecker. Epstein-Barr virus lacking latent
membrane protein 2 immortalizes B cells with efficiency indistinguishable from that of wild-
type virus, J.Gen.Virol. 80 ( Pt 8) (1999) 2193-2203.
[214] P Beaufils, D Choquet, RZ Mamoun, B Malissen. The (YXXL/I)2 signalling motif found
in the cytoplasmic segments of the bovine leukaemia virus envelope protein and Epstein-Barr
virus latent membrane protein 2A can elicit early and late lymphocyte activation events, EMBO
J. 12 (1993) 5105-5112.
[215] CL Miller, AL Burkhardt, JH Lee, B Stealey, R Longnecker, JB Bolen, et al. Integral
membrane protein 2 of Epstein-Barr virus regulates reactivation from latency through dominant
negative effects on protein-tyrosine kinases, Immunity. 2 (1995) 155-166.

95
[216] T Kurosaki. Genetic analysis of B cell antigen receptor signaling, Annu.Rev.Immunol. 17
(1999) 555-592.
[217] KP Lam, R Kuhn, K Rajewsky. In vivo ablation of surface immunoglobulin on mature B
cells by inducible gene targeting results in rapid cell death, Cell. 90 (1997) 1073-1083.
[218] IC MacLennan. Germinal centers, Annu.Rev.Immunol. 12 (1994) 117-139.
[219] YJ Liu, C Arpin. Germinal center development, Immunol.Rev. 156 (1997) 111-126.
[220] CR Ruprecht, A Lanzavecchia. Toll-like receptor stimulation as a third signal required for
activation of human naive B cells, Eur.J.Immunol. 36 (2006) 810-816.
[221] HJ Martin, JM Lee, D Walls, SD Hayward. Manipulation of the toll-like receptor 7
signaling pathway by Epstein-Barr virus, J.Virol. 81 (2007) 9748-9758.
[222] SA Ahmed, RM Gogal Jr, JE Walsh. A new rapid and simple non-radioactive assay to
monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine
incorporation assay, J.Immunol.Methods. 170 (1994) 211-224.
[223] MM Bradford. A rapid and sensitive method for the quantitation of microgram quantities
of protein utilizing the principle of protein-dye binding, Anal.Biochem. 72 (1976) 248-254.
[224] TA Waldmann, Y Tagaya. The multifaceted regulation of interleukin-15 expression and
the role of this cytokine in NK cell differentiation and host response to intracellular pathogens,
Annu.Rev.Immunol. 17 (1999) 19-49.

96
[225] C Wei, R Zeff, I Goldschneider. Murine pro-B cells require IL-7 and its receptor complex
to up-regulate IL-7R alpha, terminal deoxynucleotidyltransferase, and c mu expression,
J.Immunol. 164 (2000) 1961-1970.
[226] NN Iscove, F Melchers. Complete replacement of serum by albumin, transferrin, and
soybean lipid in cultures of lipopolysaccharide-reactive B lymphocytes, J.Exp.Med. 147 (1978)
923-933.
[227] T Hla, K Neilson. Human cyclooxygenase-2 cDNA, Proc.Natl.Acad.Sci.U.S.A. 89 (1992)
7384-7388.
[228] RA Soslow, AJ Dannenberg, D Rush, BM Woerner, KN Khan, J Masferrer, et al. COX-2
is expressed in human pulmonary, colonic, and mammary tumors, Cancer. 89 (2000) 2637-
2645.
[229] EP Ryan, SJ Pollock, TI Murant, SH Bernstein, RE Felgar, RP Phipps. Activated human
B lymphocytes express cyclooxygenase-2 and cyclooxygenase inhibitors attenuate antibody
production, J.Immunol. 174 (2005) 2619-2626.
[230] FR Garcia-Gonzalo, JC Belmonte. Albumin-associated lipids regulate human embryonic
stem cell self-renewal, PLoS ONE. 3 (2008) e1384.
[231] HL Liber, WG Thilly. Mutation assay at the thymidine kinase locus in diploid human
lymphoblasts, Mutat.Res. 94 (1982) 467-485.
[232] JA Levy, M Virolainen, V Defendi. Human lymphoblastoid lines from lymph node and
spleen, Cancer. 22 (1968) 517-524.

97
[233] SE Farinelli, LA Greene, WJ Friedman. Neuroprotective actions of dipyridamole on
cultured CNS neurons, J.Neurosci. 18 (1998) 5112-5123.
[234] MJ Bours, EL Swennen, F Di Virgilio, BN Cronstein, PC Dagnelie. Adenosine 5'-
triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation,
Pharmacol.Ther. 112 (2006) 358-404.
[235] PH Hsyu, LG Gisclon, AC Hui, KM Giacomini. Interactions of organic anions with the
organic cation transporter in renal BBMV, Am.J.Physiol. 254 (1988) F56-61.
[236] F Perez-Ruiz, A Alonso-Ruiz, M Calabozo, A Herrero-Beites, G Garcia-Erauskin, E Ruiz-
Lucea. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A
pathogenic approach to the treatment of primary chronic gout, Ann.Rheum.Dis. 57 (1998) 545-
549.
[237] WK Kroeze, DJ Sheffler, BL Roth. G-protein-coupled receptors at a glance, J.Cell.Sci.
116 (2003) 4867-4869.
[238] T Nakamizo, J Kawamata, K Yoshida, Y Kawai, R Kanki, H Sawada, et al.
Phosphodiesterase inhibitors are neuroprotective to cultured spinal motor neurons,
J.Neurosci.Res. 71 (2003) 485-495.
[239] NJ Mason, CM Coughlin, B Overley, JN Cohen, EL Mitchell, TA Colligon, et al. RNA-
loaded CD40-activated B cells stimulate antigen-specific T-cell responses in dogs with
spontaneous lymphoma, Gene Ther. 15 (2008) 955-965.
[240] M Wiesner, C Zentz, C Mayr, R Wimmer, W Hammerschmidt, R Zeidler, et al.
Conditional immortalization of human B cells by CD40 ligation, PLoS ONE. 3 (2008) e1464.

98
[241] J Banchereau, P de Paoli, A Valle, E Garcia, F Rousset. Long-term human B cell lines
dependent on interleukin-4 and antibody to CD40, Science. 251 (1991) 70-72.
[242] M Sugimoto, H Tahara, T Ide, Y Furuichi. Steps involved in immortalization and
tumorigenesis in human B-lymphoblastoid cell lines transformed by Epstein-Barr virus, Cancer
Res. 64 (2004) 3361-3364.
[243] U.S. Public Health Service Guideline on Infectious Disease Issues in Xenotransplantation.
Centers for Disease Control and Prevention, MMWR Recomm Rep. 50 (2001) 1-46.
[244] H Skottman, S Narkilahti, O Hovatta. Challenges and approaches to the culture of
pluripotent human embryonic stem cells, Regen.Med. 2 (2007) 265-273.
[245] JM Fletcher, PM Ferrier, JO Gardner, L Harkness, S Dhanjal, P Serhal, et al. Variations in
humanized and defined culture conditions supporting derivation of new human embryonic stem
cell lines, Cloning Stem Cells. 8 (2006) 319-334.
[246] LG Chase, MT Firpo. Development of serum-free culture systems for human embryonic
stem cells, Curr.Opin.Chem.Biol. 11 (2007) 367-372.
[247] T Lei, S Jacob, I Ajil-Zaraa, JB Dubuisson, O Irion, M Jaconi, et al. Xeno-free derivation
and culture of human embryonic stem cells: current status, problems and challenges, Cell Res.
17 (2007) 682-688.
[248] FD Suttenfield. Arthritis after intrasynovial injection of sodium urate crystals, Lancet. 2
(1962) 1380-1381.
[249] AP Hall. Correlations among hyperuricemia, hypercholesterolemia, coronary disease and
hypertension, Arthritis Rheum. 8 (1965) 846-852.

99
[250] WJ Fessel. High uric acid as an indicator of cardiovascular disease. Independence from
obesity, Am.J.Med. 68 (1980) 401-404.
[251] Y Shi, JE Evans, KL Rock. Molecular identification of a danger signal that alerts the
immune system to dying cells, Nature. 425 (2003) 516-521.
[252] P Matzinger. The danger model: a renewed sense of self, Science. 296 (2002) 301-305.
[253] P Matzinger. Tolerance, danger, and the extended family, Annu.Rev.Immunol. 12 (1994)
991-1045.
[254] DE Hu, AM Moore, LL Thomsen, KM Brindle. Uric acid promotes tumor immune
rejection, Cancer Res. 64 (2004) 5059-5062.
[255] MD Behrens, WM Wagner, CJ Krco, CL Erskine, KR Kalli, J Krempski, et al. The
endogenous danger signal, crystalline uric acid, signals for enhanced antibody immunity, Blood.
111 (2008) 1472-1479.
[256] Y Shi, W Zheng, KL Rock. Cell injury releases endogenous adjuvants that stimulate
cytotoxic T cell responses, Proc.Natl.Acad.Sci.U.S.A. 97 (2000) 14590-14595.
[257] RC Landis, DR Yagnik, O Florey, P Philippidis, V Emons, JC Mason, et al. Safe disposal
of inflammatory monosodium urate monohydrate crystals by differentiated macrophages,
Arthritis Rheum. 46 (2002) 3026-3033.
[258] F Martinon, V Petrilli, A Mayor, A Tardivel, J Tschopp. Gout-associated uric acid crystals
activate the NALP3 inflammasome, Nature. 440 (2006) 237-241.

100
[259] M Yamamoto, K Yaginuma, H Tsutsui, J Sagara, X Guan, E Seki, et al. ASC is essential
for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor
molecules, Genes Cells. 9 (2004) 1055-1067.
[260] GN Rao, MA Corson, BC Berk. Uric acid stimulates vascular smooth muscle cell
proliferation by increasing platelet-derived growth factor A-chain expression, J.Biol.Chem. 266
(1991) 8604-8608.
[261] S Watanabe, DH Kang, L Feng, T Nakagawa, J Kanellis, H Lan, et al. Uric acid,
hominoid evolution, and the pathogenesis of salt-sensitivity, Hypertension. 40 (2002) 355-360.
[262] J Kanellis, S Watanabe, JH Li, DH Kang, P Li, T Nakagawa, et al. Uric acid stimulates
monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-
activated protein kinase and cyclooxygenase-2, Hypertension. 41 (2003) 1287-1293.
[263] L Boring, J Gosling, M Cleary, IF Charo. Decreased lesion formation in CCR2-/- mice
reveals a role for chemokines in the initiation of atherosclerosis, Nature. 394 (1998) 894-897.
[264] CX Santos, EI Anjos, O Augusto. Uric acid oxidation by peroxynitrite: multiple reactions,
free radical formation, and amplification of lipid oxidation, Arch.Biochem.Biophys. 372 (1999)
285-294.
[265] M Bagnati, C Perugini, C Cau, R Bordone, E Albano, G Bellomo. When and why a water-
soluble antioxidant becomes pro-oxidant during copper-induced low-density lipoprotein
oxidation: a study using uric acid, Biochem.J. 340 ( Pt 1) (1999) 143-152.
[266] PM Abuja. Ascorbate prevents prooxidant effects of urate in oxidation of human low
density lipoprotein, FEBS Lett. 446 (1999) 305-308.

101
[267] V Ralevic, G Burnstock. Receptors for purines and pyrimidines, Pharmacol.Rev. 50
(1998) 413-492.
[268] SP Kunapuli, JL Daniel. P2 receptor subtypes in the cardiovascular system, Biochem.J.
336 ( Pt 3) (1998) 513-523.
[269] L Franchi, TD Kanneganti, GR Dubyak, G Nunez. Differential requirement of P2X7
receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular
bacteria, J.Biol.Chem. 282 (2007) 18810-18818.
[270] A Piccini, S Carta, S Tassi, D Lasiglie, G Fossati, A Rubartelli. ATP is released by
monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18
secretion in an autocrine way, Proc.Natl.Acad.Sci.U.S.A. 105 (2008) 8067-8072.