university of groningen wnt signaling in airway remodeling ... · wnt pathway is altered in a mouse...
TRANSCRIPT

University of Groningen
WNT signaling in airway remodeling in asthmaKumawat, Kuldeep
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2015
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Kumawat, K. (2015). WNT signaling in airway remodeling in asthma: novel roles for WNT-5A in airwaysmooth muscle. [S.l.]: [S.n.].
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 13-08-2019

173 | P a g e
WNT signaling pathway is altered in
a mouse model of chronic allergic
airway inflammation
Kuldeep Kumawat
Mark H. Menzen
Nienke De Haas
Marieke Smit
Andrew J. Halayko
Reinoud Gosens
6

Chapter 6
174 | P a g e

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
175 | P a g e
Abstract
Asthma is a chronic obstructive disease of the airways characterized by bronchial
hyperresponsiveness, airway inflammation and remodeling. The underlying mechanisms
that initiate, drive and maintain asthma are not completely understood. We have recently
reported increased abundance of the WNT ligand, WNT-5A, in asthmatic airway smooth
muscle (ASM) cells and have identified its novel role in TGF-β-induced extracellular matrix
(ECM) expression. The WNT pathway is composed of a multitude of signaling components
and participates in a myriad of biological functions in health and disease including
pulmonary disorders. We have undertaken a comprehensive analysis of expression of WNT
signaling members using an animal model of allergen-induced chronic inflammation to
identify potential mediators of asthma pathophysiology. We here show a wide ranging
modulation of most of the WNT ligands, Frizzled (FZD) receptors and various intracellular
and extracellular mediators and modulators in the lungs of mice after ovalbumin (OVA)
exposure. Notably, WNT-7A, -9A and -10B and FZD4 and FZD6 were significantly
downregulated along with a modest but significant reduction in β-catenin mRNA levels post-
OVA challenge. Of note, WIF1, a WNT signaling antagonist, showed strong downregulation
in OVA-challenged lungs as compared to saline-challenged controls. Moreover, TGF-β
reduced WIF1 mRNA in ASM cells. Owing to its antagonistic effect on WNT signaling, WIF1
downregulation may have key effects. In conclusion, allergen-induced modulation of the
WNT signaling pathway with suppression of WIF1 may indicate a significant pathological
event.
Introduction
Asthma is a heterogeneous chronic obstructive disease of the airways inflicting
approximately 300 million people worldwide and imposing a substantial burden on the
patients and healthcare system [1]. It is characterized by the presence of chronic
inflammation, bronchial hyperresponsiveness and extensive structural changes in the
airways, termed as airway remodeling [1,2].
Exposure to inhaled stimuli such as allergens or respiratory viruses triggers an exaggerated
response in asthmatic airways inducing airway constriction leading to episodes of
breathlessness and wheezing. Asthma can be effectively managed by β2 adrenoreceptor-
agonists and/ or corticosteroids in most patients providing substantial relief from the
episodic breathlessness. However, despite the most effective current therapies, a subset of
asthma patients remain poorly controlled even at the highest doses of the asthma medication
[3,4]. Asthma is a complex manifestation of (epi)genetic and environmental factors that
contribute to the evolution of the disease from childhood and often actively regulate the
course of disease and its management in later stages [1,5]. However, the factors and
mechanisms that govern the initiation, progression and maintenance of asthma remain
poorly understood hampering the efforts to develop new and more effective therapeutic
strategies targeting asthma.
Airway remodeling is a key pathological feature of individuals with asthma and is associated
with airway obstruction [6] and bronchial hyperresponsiveness [7]. It is characterized by

Chapter 6
176 | P a g e
extensive structural changes in the airway wall which include airway smooth muscle (ASM)
cell hypertrophy and hyperplasia, subepithelial fibrosis, mucus hypersecretion,
neovascularization and increased and altered extracellular matrix (ECM) expression, leading
to airway wall thickening [8]. Airway remodeling is associated with the severity of disease.
For instance, in fatal asthma, the entire airway tree is massively remodeled whereas in non-
fatal asthma, remodeling is less prominent and afflicts mainly small airways [8]. Similarly,
the thickness of the remodeled airway wall also correlates with the severity of the disease [8-
11].
WNT signaling is an important developmental pathway which plays critical roles in
embryonic morphogenesis and regulates biological functions in post-natal life. The term
WNT is derived from a combination of two homologues genes integrase 1 (int1) and wingless
(wg) [12]. The Int1 gene was first identified as a locus for the integration of mouse mammary
tumor virus DNA leading to its activation and involvement in the development of virally-
induced breast tumors in mice [12,13]. Wg, which was identified for its role in development
of wing tissue in Drosophila and regulation of larval segment polarity, was later found to be
a homologue of Int1 [12]. The WNT signaling family has grown multifold since then both in
the number of its members and its complexity. In humans, the WNT family is comprised of
19 WNT ligands, 10 Frizzled (FZD) receptors, low-density lipoprotein receptor-related
protein (LRP) 5/6 coreceptors, several non-frizzled receptors such as RYK, ROR2, PTK7
along with intracellular mediators, several extracellular and intracellular antagonists and a
range of modulators [14]. These WNT ligands can function through signaling mechanisms
broadly categorized on the basis of the requirement of an intracellular mediator- β-catenin.
The β-catenin-dependent WNT signaling pathway is termed as canonical WNT signaling
whereas all the WNT ligands activated signaling cascades functioning independent of β-
catenin are collectively described as noncanonical WNT signaling.
Accumulating evidence point to a key role of WNT signaling pathway in asthma pathology.
A study has highlighted an indirect link between the expression of various WNT ligands such
as WNT-5A, -3A and 10A and the presence of Th2 inflammation in asthmatic subjects [15].
Moreover, we have recently identified significantly increased abundance of WNT-5A mRNA
and protein in the airway smooth muscle (ASM) cells of asthma patients as compared to the
healthy subjects (Chapter 3). We demonstrated that TGF-β, a growth factor implicated
extensively in airway remodeling and asthma pathobiology [16], induces WNT-5A
expression in ASM cells where it mediates ECM expression, thus, providing a possible link
between WNT signaling and features of asthma pathobiology (Chapter 3).
WNT is an inherently complex pathway comprised of large number of members. In view of
the crucial roles assigned to some of its members in lung development and diseases including
asthma, we evaluated WNT signaling pathway expression patterns in lungs during asthma
using a mouse model of allergen-induced chronic airway inflammation. Here, we present
evidence of extensive modulation of WNT signaling components in ovalbumin (OVA)-
challenged lungs and demonstrate that WNT inhibitory factor 1 (WIF1), a WNT signaling
antagonist, was significantly attenuated on OVA exposure.

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
177 | P a g e
Material and Methods
Reagents- Recombinant human TGF-β1, recombinant human WIF1 and mouse anti-WIF1
antibody were from R&D systems (Abingdon, UK). Human WIF1 siRNA and mouse anti-
GAPDH, mouse anti-β-actin antibodies were purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Mouse anti-total β-catenin antibody was from BD Biosciences (San
Jose, CA, USA) and mouse anti-active β-catenin antibody (clone 8E7) was obtained from
Millipore (Amsterdam, the Netherlands). HRP-conjugated goat anti-mouse antibody and
human Interleukin-1β (IL-1β) was obtained from Sigma (St. Louis, MO, USA) and
Fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit antibody was procured
from Jackson Immunoresearch Europe (Suffolk, UK). Non-targeting siRNA was procured
from Qiagen (Venlo, The Netherlands) and rabbit- anti-α-smooth muscle-actin and rabbit
anti-WIF1 antibody were obtained from Abcam (Cambridge, UK). X-tremeGENE siRNA and
X-tremeGENE DNA HP transfection reagents were purchased from Roche Applied Science
(Mannheim, Germany). All other chemicals were of analytical grade.
Animals- Female BALB/c mice about 8-12 weeks old, were procured from Charles River
Laboratories (Leiden, the Netherlands). Animals were housed under a conventional 12-hour
light/dark cycle and received food and water ad libitum. All animal studies were conducted
according to the national guidelines and were approved by the University of Groningen
Committee for Animal Experimentation (no. 5912A).
Animal Model- Animals were sensitized to OVA (Sigma-Aldrich, Zwijndrecht, the
Netherlands) on Days 1, 14, and 21 by intraperitoneal administration of 10 µg OVA
emulsified in 1.5 mg aluminum hydroxide (Aluminject; Pierce Chemical, Etten-Leur, The
Netherlands) in a final volume of 200 µl in PBS. Subsequently, mice were divided into two
groups (n=10-12) and challenged with either saline or OVA aerosols (1% in PBS) for 20
minutes twice weekly on consecutive days for 4 weeks (Figure 1). The aerosol was delivered
to a Perspex exposure chamber (9 liter) by a De Vilbiss nebulizer (type 646; De Vilbiss,
Somerset, PA) driven by an airflow of 40 L/min, providing an aerosol with an output of 0.33
ml/min, as described previously (19).
Figure 1. Animal model of allergen-
induced chronic inflammation. Female
BALB/c mice (n=10 per group) were sensitized
to ovalbumin (OVA) on Day 1, 14 and 21.
Animals were subsequently challenged with
saline or OVA aerosols twice weekly for 4 weeks
as described in the material and methods
section. 24 hour following the last challenge,
animals were sacrificed and lungs were
harvested for further analysis.
Sensitization Challenge Sacrifice
1 14 21 26 33 40 47 49 27 34 41 48
Day

Chapter 6
178 | P a g e
Tissue collection- 24 hours following the last challenge, animals were anesthetized with
~2% (v/v) Isoflurane in oxygen and sacrificed by exsanguination. Right lung was inflated
with 50% Tissue-Tek in PBS (Sakura Finetek, Alphen aan de Rijn, the Netherlands) and
lungs were subsequently harvested. The smallest lower and the upper right lung lobes were
separately snap-frozen for mRNA analysis, the two middle right lung lobes were snap frozen
for immunohistochemistry. Uninflated left lung was snap frozen for protein analysis.
Immunohistochemistry- Morphometric analyses were performed on the transverse
tissue cryosections of 5µM thickness. Eosinophils were visualized by staining cryosections
with diaminobenzidine (Sigma-Aldrich, Zwijndrecht, the Netherlands) for cyanide-resistant
endogenous peroxidase activity. The number of eosinophils around the airways was counted
and expressed as number of cells per mm basement membrane.
Cryosections were stained for α-smooth muscle-actin using a rabbit anti-α-smooth muscle-
actin antibody (Abcam, Cambridge, UK) which was visualized by staining with horseradish-
peroxidase-linked secondary antibody and diaminobenzidine (Sigma-Aldrich, Zwijndrecht,
the Netherlands). Airways within sections were digitally photographed and α-smooth
muscle-actin around the airway was quantified using Image J (National Institute of Health).
The surface of positively stained tissue was expressed as mm2 per mm2 basement membrane.
Immunofluorescence- For immunofluorescence, cryosections were air-dried, fixed in
acetone for 10 minutes and blocked with a mix of 2% donkey serum and 1% BSA for 1 hour.
WIF1 was detected using rabbit anti-WIF1 antibody (Abcam, Cambridge, UK) and FITC-
conjugated donkey anti-rabbit antibody.
β-Catenin was visualized by mouse anti-β-catenin antibody using mouse-on-mouse (MOM)
elite peroxidase kit (Vector Laboratories, Burlingame, USA) as per manufacture’s instruction
with minor modifications. Briefly, following the blocking with a mix of 2% donkey serum and
1% BSA as mentioned above, cryosections were further incubated with the mouse Ig blocking
reagent for 1 hour and with MOM diluent for 5 minutes. Subsequently, cryosections were
incubated with anti-β-catenin antibody for 30 minutes and with a secondary rabbit anti-
mouse IgG for another 30 minutes. β-Catenin was visualized by using a tertiary FITC-
conjugated donkey anti-rabbit antibody.
Nuclei were stained with Hoechst diluted in ddH2O. After staining, cryosections were
mounted in ProLong Gold Antifade reagent (Life Technologies, Bleiswijk, the Netherlands).
Immunofluorescence was analyzed using Olympus AX70 microscope.
Cell culture- Immortalized human airway smooth muscle cell lines (ASM) cells and human
bronchial epithelial cell lines (HBE) were used for in vitro experiments. Cell lines were
maintained and used for experiments as described previously (Chapter 3 and [17]).
RNA isolation and real time PCR- Lung homogenates were prepared by pulverizing
tissues under liquid nitrogen. Total RNA was extracted from the lung homogenates and cell
lines using the Nucleospin RNAII kit (Macherey-Nagel, Duren, Germany) as per
manufacturer’s instructions. Equal amounts of total RNA were reverse transcribed using the

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
179 | P a g e
Reverse Transcription System (Promega, Madison, USA). 1 µl of 1:2 diluted cDNA was
subjected to real-time PCR using FastStart Universal SYBR Green Master (Rox) from Roche
Applied Science (Mannheim, Germany) in an Illumina Eco Personal QPCR System
(Westburg, Leusden, the Netherlands). Real time PCR was performed with denaturation at
94°C for 30 seconds, annealing at the mentioned temperatures (Table 1) for 30 seconds and
extension at 72°C for 30 seconds for 40 cycles followed by 10 minutes at 72°C. Data was
analyzed using the comparative cycle threshold (Cq: amplification cycle number) method.
The amount of target gene was normalized to the endogenous reference gene 18S ribosomal
RNA (∆Cq). Where appropriate, relative differences were determined using the equation 2(-
∆∆Cq). Primers used for PCR amplification are listed in Table 1.
Western Blotting- Lung homogenates were prepared by pulverizing tissues under the
liquid nitrogen with subsequent sonication in SDS lysis buffer (62.5 mM Tris, 2% w/v SDS,
1 mM NaF, 1 mM Na3VO4, 1µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml pepstatin A, 1 mM
β-glycerophosphate, pH 6.8). The hTERT and HBE cells were also lysed in SDS lysis buffer
supplemented with protease inhibitors.
Protein concentration was determined using Pierce BCA protein assay kit (Thermo
Scientific, Rockford, IL, USA). Equal amounts of protein were separated by electrophoresis,
electro-transferred to nitrocellulose membranes and analyzed for proteins of interest using
specific primary and HRP-conjugated secondary antibodies. Westerns were subsequently
visualized using the G-box gel documentation system (Syngene, Cambridge, UK) using
enhanced chemiluminescence reagents and were quantified by densitometry using
Genetools software where necessary.
siRNA transfection- ASM cells were grown to ~90% confluence in 6-well cluster plates
and transfected with 200 pmol of WIF1-specific in serum and antibiotic free DMEM with X-
tremeGENE siRNA transfection reagent (Roche). Control transfections were performed
using a non-targeting control siRNA (Qiagen). After 6 hours of transfection, medium was
replaced with DMEM supplemented with antibiotics and ITS for a period of 42 hours before
TGF-β stimulation.
DNA transfection- ASM cells or HBE cells were grown to ~90% confluence in 6-well or
24-well cluster plates and were subsequently transfected with 1 µg of Myc-DDK-tagged
human WIF1 plasmid (Myc-WIF1) (Origene, Rockville, MD, USA) in serum and antibiotic
free medium using X-tremeGENE HP DNA transfection reagent. 1 µg of Green Fluorescent
Protein (GFP) expression vector was transfected as control. After 6 hours of transfection,
medium was replaced with DMEM or MEM supplemented with antibiotics and 10% (v/v)
fetal bovine serum (FBS) for 18 hours. Cells were then serum-deprived for 24 hours before
stimulation(s).
IL-8 ELISA- ASM cells were stimulated for 24 hours with IL-1β. Supernatants were
collected and IL-8 concentrations were determined by ELISA as per manufacturer's
instructions (Sanquin, the Netherlands).

Chapter 6
180 | P a g e
Table 1: Primers used for WNT pathway gene expression analysis
Gene Forward 5’ ����3’ Reverse 5’ ����3’
WNT-1 ATTTTGCGCTGTGACCTCTT AGCAACCTCCTTTCCCACTT
WNT-2 GAAAGGAAGTGCCAAGGACA CCTTCCTTCCAGCTCTGTTG
WNT-2B CGAGGTGGCAAACATCCTAT CTTTGAAGGCTCCACTCCTG
WNT-3 CTCGAGGCTTACCTTTGCAC AGGCAGCTTGGACACAGAAT
WNT-3A GTGCACACCTGCAAGTAGGA TCTCCGCCCTCAAGTAAGAA
WNT-4 CTGGAGAAGTGTGGCTGTGA CAGCCTCGTTGTTGTGAAGA
WNT-5A CAAATAGGCAGCCGAGAGAC CTCTAGCGTCCACGAACTCC
WNT-5B GGTTCCACTGGTGTTGCTTT AGACTTTTGTGAGGCGGAGA
WNT-6 TTCGGGGATGAGAAGTCAAG AAAGCCCATGGCACTTACAC
WNT-7A GGTCAGATCACAGGCAGGAT TGCAGGAAACCCAGAATACC
WNT-7B TCCTTGCAGAACTCGAGGAT GCCTGACACAAGGGACATTT
WNT-8A AACGGTGGAATTGTCCTGAG GGTGACTGCGTACATGATGG
WNT-8B GTAGGCAGGTTGGCAATCAT TCCTTCCTATGCCCTTCCTT
WNT-9A TCGTGGGTGTGAAGGTGATA TGGCTTCATTGGTAGTGCTG
WNT-9B CTTCTCTGGGACGACTTTGC GGTTGCCAAGGAACACATCT
WNT-10A ATTGACATTCCTCCGCTCAC TGGGGAAGGGAAGAAGAGAT
WNT-10B CCACTGGTGCTGTTATGTGC AGCCCTCACACAGTGCTTCT
WNT-11 ACCCCTACACGGACAGAGTG AGGGAAGTCCACCAAGGTCT
WNT-16 GAGCTGTGCAAGAGGAAACC GCGACCATACAGTTCCACCT
FZD1 CAAGGTTTACGGGCTCATGT GTAACAGCCGGACAGGAAAA
FZD2 CCGTCTCTGCATCCTCACAT TAGCAGCCGGACAGAAAGAT
FZD3 GAAGCAAAGCAGGGAGTGTC ATGCTGCCGTGAGGTAGTCT
FZD4 CTGCAGCATGCCTAATGAGA CGTCTGCCTAGATGCAATCA
FZD5 AGCCAAATGAGGCACATACC TCTCCTTTTGCGAGCGTTAT
FZD6 TCCGACGCTTGAAGAAAACT CAACCCCAGGTCCTCAAGTA
FZD7 ATCATCTTCCTGTCGGGTTG AAGCACCATGAAGAGGATGG
FZD8 TCCGTTCAGTCATCAAGCAG CGGTTGTGCTGCTCATAGAA
FZD9 CCAGCTGTCAAGGTCAGACA CACTCCCTGCATGAGACAGA
FZD10 TGGTACGCATAGGGGTCTTC TCAGGCAGTCAGGTGTCTTG
SFRP1 ATCCCCCTCTTTCTGCCTTA GGCCAGTTCAAAAGCTGAAG
SRFP2 ACGACAACGACATCATGGAA ACGCCGTTCAGCTTGTAAAT
WIF1 GAGTGTCCGGATGGGTTCTA AGCAGGTGGTTGAGCAGTTT
DKK1 CAGCTCAATCCCAAGGATGT CAGGGGAGTTCCATCAAGAA
CTNNBIP1 (ICAT) TTGAACCCAAGCAGATACCC TCCCCCTTCTATGCTGACAC
β-catenin AGGGTGGGAATGGTTTTAGG GTGGCAAAAACATCAACGTG
Axin2 GTGGAGAGGATCGACTGAGC GCCCAGAGTGGTCATGTTTT
Wisp1 GGGTCTTCCCAGGTATCCAT TTCCTTGAACCAGGCCATA
18S AAACGGCTACCACATCCAAG CCTCCAATGGATCCTCGTTA
WNT pathway is altered in a mouse model of chronic allergic airway inflammation
181 | P a g e
Data Analysis- Values reported for all data are represented as mean ± SEM. The statistical
significance of differences between means was determined on log transformed data by
Student’s t-test or by 1-way ANOVA, followed by Student-Newman Keuls multiple
comparisons test. Differences were considered to be statistically significant when p<0.05.
Results
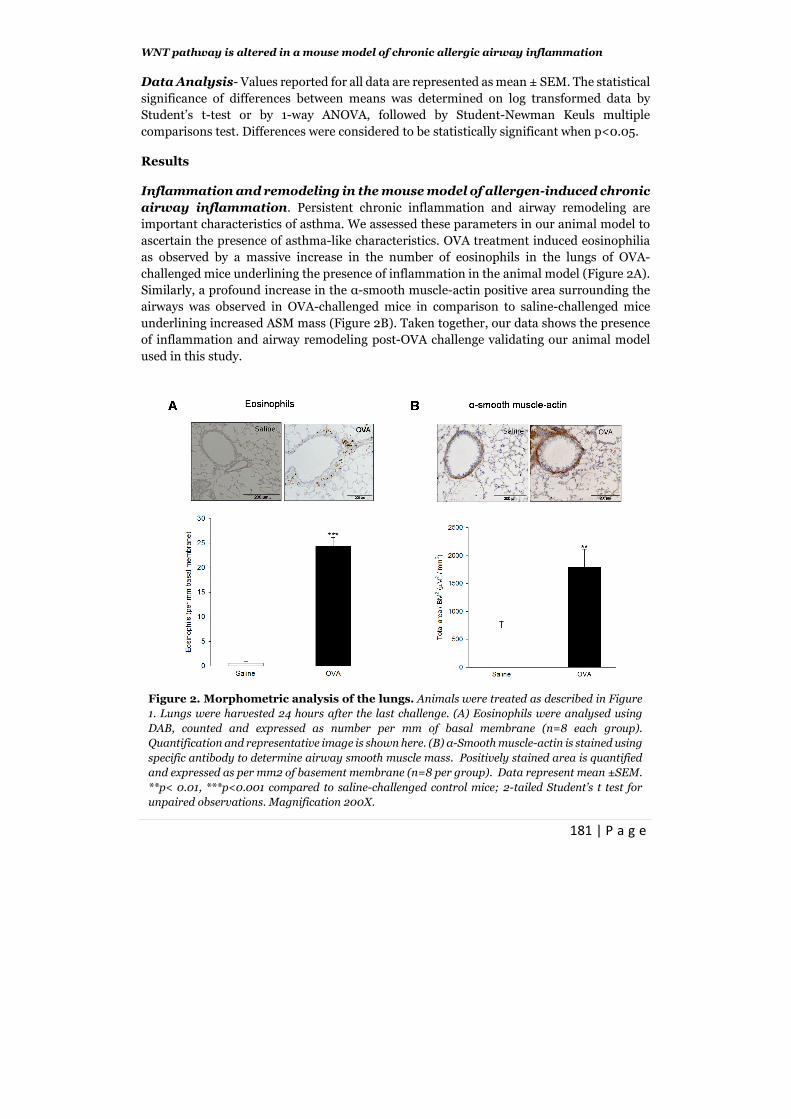
Inflammation and remodeling in the mouse model of allergen-induced chronic
airway inflammation. Persistent chronic inflammation and airway remodeling are
important characteristics of asthma. We assessed these parameters in our animal model to
ascertain the presence of asthma-like characteristics. OVA treatment induced eosinophilia
as observed by a massive increase in the number of eosinophils in the lungs of OVA-
challenged mice underlining the presence of inflammation in the animal model (Figure 2A).
Similarly, a profound increase in the α-smooth muscle-actin positive area surrounding the
airways was observed in OVA-challenged mice in comparison to saline-challenged mice
underlining increased ASM mass (Figure 2B). Taken together, our data shows the presence
of inflammation and airway remodeling post-OVA challenge validating our animal model
used in this study.
Figure 2. Morphometric analysis of the lungs. Animals were treated as described in Figure
1. Lungs were harvested 24 hours after the last challenge. (A) Eosinophils were analysed using
DAB, counted and expressed as number per mm of basal membrane (n=8 each group).
Quantification and representative image is shown here. (B) α-Smooth muscle-actin is stained using
specific antibody to determine airway smooth muscle mass. Positively stained area is quantified
and expressed as per mm2 of basement membrane (n=8 per group). Data represent mean ±SEM.
**p< 0.01, ***p<0.001 compared to saline-challenged control mice; 2-tailed Student’s t test for
unpaired observations. Magnification 200X.

Chapter 6
182 | P a g e
Extensive modulation of WNT signaling pathway by allergen challenge.
Having validated our animal model, we next screened the lung homogenates for mRNA
expression of various components of the WNT signaling pathway. OVA challenge led to a
wide ranging modulation in gene expression of WNT ligands. WNT-1, -3, -3A, -5A, -5B, -7B,
-9B, -10A, -11 and -16 showed varying degree of up or downregulation following OVA
challenge but the observed changes failed to achieve statistical significance. On the other
hand, WNT-7A, -9A and -10B were significantly downregulated upon OVA exposure in
comparison to the saline-challenged group (Figure 3A, 3B). Similarly, FZD4 and FZD6
expression were significantly downregulated by OVA exposure whereas other FZD receptors
remained either unaltered or showed minimum modulation (Figure 3C, 3D). The canonical
WNT signaling mediator-β-catenin, showed modest but significant reduction following OVA
challenge whereas Axin2, a member of the β-catenin destruction complex, remained
unchanged (Figure 3E). The expression of secreted FZD related protein (SFRPs) and
Dickkopf1 (DKK1) which function as extracellular inhibitors of WNT signaling. SFRP1
showed a trend of increased expression upon OVA challenge whereas SFRP2 and DKK1
didn’t change (Figure 3F). Similarly, the intracellular inhibitor of β-catenin-mediated
transcription- inhibitor of β-catenin and TCF (ICAT), also remained unaltered. One of the
most important observations came from the analysis of WIF1 which showed a significant
decline in response to OVA challenge (Figure 3F). WIF1 is an extracellular WNT inhibitor
which antagonizes WNT signaling by binding to various WNT ligands.
Since the reduction in WIF1 was among the most significant changes, we further expanded
the mRNA observations at protein level. We performed immunofluorescence microscopy on
lung cryosections using WIF1-specific antibody which indicated a reduction in WIF1
abundance in the OVA-challenged airways as compared to saline-challenged group (Figure
3G).
Thus, our data suggest a widespread modulation of WNT signaling pathway components in
the lungs of OVA-challenged animals with a significant decline in expression of the WNT
antagonist WIF1 among others.
Functional significance of WIF1 downregulation in asthma. We directed our
focus on WIF1 which was significantly downregulated by OVA-challenge. We hypothesized
that reduced abundance of WIF1 releases its target WNT ligands from inhibition and allows
enhanced intracellular WNT signaling, irrespective of their expression levels. To this aim,
we first analyzed the activation of β-catenin signaling. As revealed by immunofluorescence
staining, β-catenin shows a clear localization to the lateral membrane of airway epithelial
cells in the saline-challenged group, in line with its established role as a component of
adherens junctions (Figure 4A). Interestingly, OVA challenge completely altered the
membrane localization of β-catenin in the airway epithelial cells with the lateral staining
completely lost and replaced by a less clear apical staining pattern (Figure 4A). Of note, we
also failed to detect the nuclear localization of β-catenin, a marker of active β-catenin
signaling, post-OVA challenge. We also performed western blot analysis to detect any
changes in β-catenin signaling. As shown in Figure 4B, there was no difference in abundance

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
183 | P a g e
Frizzled receptors
1 2 3 4 5 6 7 8 9 10
Re
lativ
e m
RN
A e
xpre
ssi
on
(fo
ld o
f re
spe
ctiv
e s
alin
e)
0
1
2
3
4
5 Saline
Ova
** *
WNT Ligand
1 2 2B 3 3A 4 5A 5B 7A 7B 8A 8B 9A 9B 10A 10B 11 16
Re
lative
mR
NA
exp
ressi
on
(fo
ld o
f re
sp
ective
sa
line
)
0
1
2
3
4
5
6 Saline Ova
* *** **
WNT Ligand
1 2 2B 3 3A 4 5A 5B 7A 7B 8A 8B 9A 9B 10A 10B 11 16
Re
lative
mR
NA
exp
ress
ion
(fold
of W
NT
-9A
)
0
2
4
6
8
10 Saline Ova
*
***
**
Frizzled receptors
1 2 3 4 5 6 7 8 9 10
Re
lative
mR
NA
exp
ressio
n(f
old
of F
ZD
5)
0
1
2
3
4
15
20
25 Saline
Ova
** *
A
B
C D

Chapter 6
184 | P a g e
of total β-catenin in lung homogenates obtained from saline- and OVA-challenged groups.
Interestingly, the nonphospho β-catenin which represents the active fraction of β-catenin
showed a modest increase in the lung homogenates of OVA-challenged group as revealed by
quantification of the westerns. Though the data indicate a possible activation of β-catenin
signaling, they remain inconclusive due to lack of any statistical significance in the effects.
Figure 3. Expression analysis of WNT family members. Animals were treated as described
in Figure 1. Lungs were harvested 24 hours after the last challenge. Total RNA was isolated from
whole lung homogenates and mRNA expression was determined using qRT-PCR. Expression of
genes is represented as Cq-values corrected for 18S rRNA. WNT ligands mRNA abundance as fold
of respective saline-challenged control (A) or relative to WNT-9A (B). FZD receptor mRNA
expression as fold of respective saline-challenged control mice (C) or relative to FDZ5 abundance
(D). (E, F) Expression of various WNT pathway components relative to saline-challenged control
mice as depicted in the respective panels. Data represent mean ±SEM; n=10 each group. *p<0.05,
**p<0.01, ***p<0.001 compared to respective saline-challenged control mice; 2-tailed Student’s t
test for unpaired observations. (G) Animals were treated as described in Figure 1. Lungs were
harvested 24 hours after the last challenge. Cryosections were prepared and immunofluorescence
microscopy was performed as described in material and methods. Representative image showing
WIF1 staining in saline- and OVA-challenged mice. Magnification 40X.
β-catenin Axin2
Re
lative
mR
NA
exp
ress
ion
(fo
ld o
f sa
line
)
saline Ova
0.0
0.5
1.0
1.5
2.0
*
E
SFRP1 SFRP2 WIF-1 WISP ICAT DKK-1
Re
lative
mR
NA
exp
ressio
n(f
old
of salin
e)
0
1
2
3saline
Ova
***
F

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
185 | P a g e
Figure 4. Effect of OVA exposure on β-catenin protein levels. (A) Animals were treated as
described in Figure 1. Lungs were harvested 24 hours after the last challenge. Cryosections were
prepared and immunofluorescence microscopy was performed as described in material and methods.
Representative image showing β-catenin staining in saline- and OVA-challenged mice. Magnification
40X. (B) Total and active β-catenin expression levels were analysed in whole lung extracts by western
blotting. Equal protein loading was verified by analysis of β-actin. Abundance of total and active β-
catenin bands is quantified and expressed as percentage of saline-challenged controls. Data represent
mean ±SEM; n=4 each group.
We expanded our investigation in vitro using ASM and HBE cell lines to identify which
compartments of lungs are contributing to the observed reduction in WIF1 expression in
OVA-challenged group. TGF-β is the most prominent cytokine which is increased in
asthmatic airways and regulates various aspects of asthma pathophysiology [18-21].
Interestingly, TGF-β significantly reduced WIF1 mRNA expression in ASM cells but not in
HBE cells underlining the cell-dependent effects (Figure 5A, 5B). Additionally, HBE cells
were treated with house dust mite extract (HDM), a common allergen, which also failed to
alter WIF1 mRNA levels (Figure 5B).

Chapter 6
186 | P a g e
Our previous study has shown that TGF-β induces expression of WNT-5A in ASM cells where
it mediates expression of TGF-β-induced ECM proteins (Chapter 3). As WIF1 can
antagonize WNT-5A and as observed, TGF-β decreases WIF1 expression in ASM cells (Figure
5A), we investigated the possible role of WIF1 in TGF-β-induced ECM expression. We
observed that the presence of recombinant WIF1 had no effect on TGF-β-induced collagen
1α1 and fibronectin expression in ASM cells (Figure 6A). Similarly, exogenous WIF1 failed to
affect other key TGF-β responses in ASM cells such as upregulation of WNT-5A and
reduction of SMAD3 expression (Figure 6B, 6C). An increase in WNT-5A message in ASM
cells appears only after 12 hours of TGF-β stimulation (Chapter 3), so we decided to
coincide WIF1 treatment with the time of TGF-β-induced WNT-5A expression. ASM cells
were first stimulated with TGF-β for 12 hours following which recombinant WIF1 was added
and cells were further incubated for 12 hours. However, we failed to observe any effect of
exogenous recombinant WIF1 on TGF-β-induced expression of collagen and fibronectin
(Figure 6D). Considering that complete absence of serum might have some effect on the
stability or function of WIF1 directly or indirectly, we decided to tweak our experimental
conditions. Instead of serum-free media, stimulations were carried out using a medium
supplemented with 0.5% serum and ASM cells treated with or without TGF-β and/ or
recombinant WIF1. However, presence of exogenous WIF1 didn’t change the expression of
TGF-β-induced ECM (Figure 6E).
Using a loss-of-function approach, we observed that knock-down of WIF1 using specific
siRNA had no effect on collagen Iα1 or fibronectin expression in response to TGF-β in ASM
cells (Figure 7A). In a reciprocal approach, we overexpressed WIF1 in ASM cells and
evaluated expression of TGF-β-induced ECM proteins. Following transfection with Myc-
Figure 5. WIF1 expression in ASM and HBE cells. (A) ASM cells were stimulated with TGF-
β (2 ng/ml) and (B) HBE cells were stimulated with TGF-β (2 ng/ml) or HDM extract (30 µg/ml)
for 24 hours. Expression of WIF1 mRNA was determined by qRT-PCR. Data represents mean ±
SEM of 4 independent experiments. *p<0.05 compared to untreated control; 2-tailed Student’s t-
test for paired observations.
A
0.0
0.5
1.0
1.5
2.0
Basal TGF-β
WIF
1 m
RN
A e
xp
ress
ion
(fo
ld o
f b
asa
l)
*
ASM cells
B
0.0
0.5
1.0
1.5
2.0
Basal TGF-β HDM
WIF
1 m
RN
A e
xpre
ssio
n (
fold
of
ba
sal)
HBE cells

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
187 | P a g e
WIF1, the WIF1 message showed ~26000 fold increase at basal level (Figure 7B). However,
similar to knock-down, WIF1 overexpression failed to alter collagen or fibronectin
expression in response to TGF-β (Figure 7C).
WNT signaling has recently been linked to Th2 inflammation in asthma subjects [15]. So, we
investigated whether WIF1 participates in the inflammatory responses in ASM cells. IL-1β
induces IL-8 secretion in ASM cells [22,23]. However, WIF1 overexpression had no effect on
IL-1β-induced IL-8 release in ASM cells (Figure 8).
Taken together, our comprehensive analysis revealed that WIF1 had no effect on TGF-β-
induced ECM expression and doesn’t participate in regulating IL-1β-induced inflammatory
responses in ASM cells.
Discussion
In the current study, we evaluated alterations in the expression of WNT pathway
components using an animal model of allergen-induced chronic airway inflammation. We
demonstrate a broad modulation of WNT family gene expression levels post-OVA challenge.
Most importantly, we identified a significant decrease in WIF1 in OVA-challenged lungs
compared to the saline-challenged controls. Moreover, functional studies using gain- and
loss-of-function approaches revealed that WIF1 doesn’t participate in TGF-β-induced ECM
production or IL-1β-induced IL-8 release in ASM cells.
Aberrant WNT signaling activation has been reported in several fibroproliferative disorders.
For instance, increased expression of WNT signaling pathway genes and enhanced nuclear
abundance of β-catenin is observed in fibroproliferative diseases of kidney, liver and bone
[24-27]. Similarly, expression of several WNT signaling pathway genes such as WNT-1,
WNT-7B, WNT-10B, FZD2, FZD3, β-catenin and LEF1 and nuclear localization of β-catenin
are augmented in idiopathic pulmonary fibrosis patients [28,29], supporting a
comprehensive role for WNT signaling in fibroproliferative disorders, including those of the
lung. We have recently shown that WNT-5A expression is increased in ASM cells derived
from asthmatic patients in comparison to healthy subjects. WNT-5A is also overexpressed in
fibroblasts of patients with usual interstitial pneumonia [30]. Here, we present evidence for
a wide range modulation of WNT signaling pathway in asthmatic lungs obtained from the
mouse model of allergen-induced chronic airway inflammation. WNT ligands exhibit
extensive up and downregulation in expression, with significant downregulation of WNT-
7A, WNT-9A and WNT-10B in OVA-challenged group in comparison to the saline-
challenged control group. Similarly, FZD receptors and other components of WNT pathway
examined also showed widespread modulation in expression post-allergen challenge.
Importantly, basal expression of WNT-5A was one of the highest in mice lungs with a trend
towards upregulation post-allergen challenge. WNT-5A along with WNT-5B and WNT-11
and FZD8 are targets of TGF-β in ASM cells as we reported previously. However, lack of a
specific TGF-β signature on WNT ligands and FZD receptor expression in the lungs obtained
from mouse model could be attributed to the use of whole lungs instead of isolated
compartments in this analysis. The effect of TGF-β on WNTs in ASM might be different from
its effect in epithelial cells or fibroblasts.

Chapter 6
188 | P a g e
Figure 6. Effects of exogenous WIF1 on TGF-β cellular responses. (A-C) ASM cells were
stimulated with TGF-β (2 ng/ml) in the presence or absence of recombinant WIF1 (3 µg/ml) for 24
hours. Expression of collagen 1α1, fibronectin and PAI1 mRNA (A), WNT-5A mRNA (B) and
SMAD3 mRNA (C) was determined by qRT-PCR, corrected for 18S rRNA and expressed relative to
the untreated control. Data represent mean ±SEM of 3-7 independent expriments. *p<0.05,
**p<0.01, ***p<0.001 compared to untreated control; 1-way ANOVA followed by Newman-Keuls
multiple comparisons test. (D) ASM cells were stimulated with TGF-β (2 ng/ml) for 12 hours
following which recombinant WIF1 (3 µg/ml) was added and cells were further incubated for 12
hours. Expression of collagen 1α1, fibronectin mRNA was determined by qRT-PCR, corrected for
18S rRNA and expressed relative to the untreated control. Data represent mean ±SD of 2
Basal TGF-β Basal TGF-β Basal TGF-β
Extr
ace
llula
r m
atr
ix g
en
e e
xpre
ssi
on
(fo
ld o
f b
asa
l)
0
2
4
6
8
10
12 Vehicle
Rec. WIF1 (3 µg/ml)
Collagen 1α1 Fibronectin PAI-1
*** ***
*** ***
***
***
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Basal TGF-β
WN
T-5
A m
RN
A e
xpre
ssio
n (
fold
of
ba
sal)
Vehicle
Rec. WIF1 (3 µg/ml)
**
**
0.0
0.5
1.0
1.5
2.0
Basal TGF-β
SM
AD
3 m
RN
A e
xpre
ssio
n (
fold
of
ba
sal)
Vehicle
Rec. WIF1 (3 µg/ml)
* **
B
C D
E
Basal TGF-β Basal TGF-β
Extr
acellu
lar
matr
ix g
en
e e
xpre
ssio
n(f
old
of b
asal)
0
1
2
3
4
5
6 Vehicle
Rec. WIF1 (3 µg/ml; 12 hours)
Collagen 1α1 Fibronectin
Basal TGF-β Basal TGF-β
Ext
race
llula
r m
atr
ix g
ene
expre
ssio
n(f
old
of
ba
sal)
0
1
2
3
4 Vehicle
Rec. WIF1 (3 µg/ml)
Collagen 1α1 Fibronectin

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
189 | P a g e
independent experiments. (E) ASM cells were stimulated with TGF-β (2 ng/ml) in the presence or
absence of recombinant WIF1 (3 µg/ml) for 24 hours. DMEM supplemented with 0.5% FBS is used
for all treatment conditions. Expression of collagen 1α1, fibronectin mRNA was determined by
qRT-PCR, corrected for 18S rRNA and expressed relative to the untreated control. Data represent
mean ±SD of 2 independent experiments.
Figure 7. Effects of WIF1 loss- or gain-
of-function in ECM production in ASM
cells. (A) ASM cells were transfected with
WIF1-specific siRNA or a negative (control)
siRNA as control. Subsequently, cells were
stimulated with TGF-β (2 ng/ml) for 24
hours. Expression of collagen IαI and
fibronectin mRNA was analyzed by qRT-
PCR and expressed relative to negative
siRNA-transfected, untreated control. Data
represent mean ±SD of 2 independent
experiments. (B, C) ASM cells were
transfected with Myc-WIF or GFP as
control. Subsequently, cells were stimulated
with TGF-β (2 ng/ml) for 24 hours. Expression of WIF1 mRNA (B) and collagen IαI and fibronectin mRNA (C) was analyzed by qRT-
PCR and expressed relative to GFP-transfected, untreated control. Data represent mean ±SD of 2
independent experiments.
Earlier studies have provided some evidence about the possible involvement of WNT
signaling in asthma. Airway remodeling is a critical feature of the asthmatic airway which
involves extensive structural changes in the affected airway [1,2]. These changes include
ASM hypertrophy and hyperplasia, epithelial damage, basement membrane thickening and
altered and augmented ECM deposition [1,2]. A study from our lab showed that β-catenin
mediates TGF-β-induced expression of ECM expression in ASM cell, implicating cross-talk
between TGF-β and canonical WNT signaling mediator to regulate airway remodeling [31].
A
Basal TGF-β Basal TGF-β
Ext
racellu
lar
ma
trix
ge
ne
exp
ress
ion
(fold
of
no
nta
rge
ting
siR
NA
)
0
1
2
3
4
5
6 Negative siRNA
WIF1 siRNA
Collagen 1α1 Fibronectin
GFP Myc-WIF1
WIF
1 m
RN
A e
xpre
ssio
n
(fold
of
GF
P-t
ransfe
cted
)
0
1
2
20000
30000
40000
B
Basal TGF-β Basal TGF-β
Ext
racellu
lar
ma
trix
ge
ne e
xpre
ssio
n(f
old
of
GF
P-t
ran
sfe
cte
d b
asal)
0
2
4
6
8
10 GFP
Myc-WIF1
Collagen 1α1 Fibronectin
C

Chapter 6
190 | P a g e
Figure 8. Functional analysis of WIF1 in
inflammation in ASM cells. ASM cells were
transfected with Myc-WIF or GFP as control.
Subsequently, cells were stimulated with IL-1β
(1 ng/ml) for 24 hours. IL-8 abundance was
measured in cell supernatants by ELISA. Data
represent mean ±SEM of 3 independent
expriments. ***p<0.001 compared to GFP-
transfected, untreated control; 1-way ANOVA
followed by Newman-Keuls multiple
comparisons test.
TGF-β is a pleiotropic cytokine and highly increased in asthmatic lung tissue and broncho-
alveolar lavage (BAL) fluid and functions as master regulator of airway remodelling in
asthma [16,19-21]. While accumulating evidence have key roles for β-catenin in airway
remodeling (Chapter 2), a direct link between canonical or noncanonical WNT signaling
with asthma is not known. Increased expression of WNT signaling pathway members in
airway epithelium and endobronchial biopsies of asthma patients, including WNT-5A, have
been reported although the cellular localization and functional roles of these WNT ligands
were not determined [15,32]. Here, we demonstrate a direct link with WNT signaling family
modulation using our animal model.
Furthermore, we demonstrate a significant decrease in WIF1 mRNA levels in the lungs of
OVA-challenged mice as compared to saline-challenged mice. WIF1 is an evolutionary
conserved protein that can bind to and antagonize WNT signaling. WIF1 has been shown to
bind to WNT-3A, -4, -5A, -7A, -9B and -11 via its WIF domain with varying affinities (WNT-
5A>WNT-9B>WNT-11>WNT-4>WNT-7A>WNT-3A) [33]. WIF1 binds to WNT ligands via
its WIF domain and tethers this WIF1-WNT complex in the ECM by its interaction with the
heperan sulphate proteoglycan- glypican via epidermal growth factor-like domains [34].
Indeed, glypicans can have modulatory effects on WNT signaling [35] and formation of this
WNT-WIF1-glypican complex is required for complete WNT antagonizing activity of WIF1
[34]. Of note, the observed suppression of WIF1 is particularly important as single nucleotide
polymorphisms in WIF1 associate with lung function in two cohorts of asthma patients (14).
Moreover, WIF1 is a target of bone morphogenetic protein 4 (BMP4)-SMAD1 signaling in
developing lungs where it antagonizes WNT/β-catenin signaling and regulates lung
morphogenesis by fine tuning the WNT and BMP signaling. Abrogation of WIF1 expression
leads to severe fetal lung abnormalities, in part, due to hyperactivation of WNT/β-catenin
signaling [36]. In line with this, a study has demonstrated downregulation of SMAD1 and
WIF1 expression during the saccular stage of lung development in mouse model of
congenital diaphragmatic hernia, leading to the retardation of lung morphogenesis and
appearance of hypoplastic lung [37]. Owing to the crucial role of WIF1 in WNT signaling
Basal IL-1β
IL-8
secre
tion (
pg/m
l)
0
150
300
450
100000
150000
200000GFP
Myc-WIF1
******

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
191 | P a g e
regulation, suppression of WIF1 is often associated with malignancies such as prostate,
breast, lung, and bladder cancer [38]. Thus, WIF1 downregulation could lead to disruption
of lung homeostasis both during embryogenic and adult life.
Lungs are highly specialized organ composed of many different compartments. Epithelium
and ASM represent important structural and functional units of the lung. Epithelium
constitutes the barrier between airways and the external environment and also serves as the
first line of defense. ASM cells, on the other hand, provide mechanical properties to the
airways and maintain bronchial tone. Both epithelium and ASM cells can respond to a range
of growth factors and other substances present in the lung and as such actively participate,
not only in maintenance of lung homeostasis but also in respiratory disorders such as
asthma. In fact, anomalies in epithelial and ASM bundle are prominent features of a
remodeled airway in asthma [1,2,39]. We attempted to identify the lung compartment(s)
contributing to the reduction WIF1 abundance in OVA-challenged lung. We here
demonstrate that TGF-β suppresses WIF1 expression in ASM cells but not in HBE cells. Since
epithelial cells are exposed to external environmental allergens, we treated HBE cells with
HDM extract, a common aeroallergen. HDM extract alone or in combination with TGF-β
failed to convincingly reduce WIF1 mRNA levels in HBE cells (Figure 3B; data not shown)
suggesting a highly context dependent regulation of WIF1 expression. Moreover, basal
abundance of WIF1 transcripts is much higher in ASM cells as compared to HBE cells. In
view of these observations, reduced expression of WIF1, as seen in OVA-challenged lungs,
might be attributed to the ASM cells.
We have recently demonstrated that WNT-5A regulates TGF-β-induced ECM expression in
ASM cells (Chapter 3). We hypothesized that downregulation of WIF1 by TGF-β in ASM
cells could be an important mechanism for allowing maximum WNT-5A signaling to drive
ECM expression. However, the presence of exogenous WIF1 didn’t affect TGF-β-induced
ECM expression in ASM cells. Similarly, loss of WIF1 using siRNA or overexpression of WIF1
didn’t show substantial effects on TGF-β-induced ECM expression negating our preliminary
hypothesis about the role of WIF1 in TGF-β responses. However, the observations from WIF1
loss-of-function and gain-of-function investigation should be viewed with caution owing to
insufficient replicates for statistical analysis. Further experiments are warranted to
determine the functional significance of WIF1 downregulation in ASM cells.
Epigenetic mechanisms and posttranscriptional mechanisms can regulate WIF1 expression
levels. DNA methylation is an epigenetic modification that renders target gene inactive.
Promoter hypermethylation is associated with WIF1 suppression in lung [40], breast cancer
[41] and astrocytomas [42]. Further, a study demonstrated that miRNA-374a can target
WIF1 transcripts therby limiting its expression in breast cancer cells [43]. Interestingly TGF-
β has been shown to induce expression of DNA methyltransferases (DNMTs), enzymes
involved in DNA methylation, in prostate cancer cells to suppress its own signaling [44]. On
the contrary, TGF-β downregulates DNMTs in cardiac fibroblasts thereby reducing collagen
type I promoter methylation and allowing its subsequent expression [45] highlighting the
context-dependent regulation of DNA methylation by TGF-β. How TGF-β induces WIF1

Chapter 6
192 | P a g e
suppression in ASM cells and whether any epigenetic mechanisms are involved or not needs
further investigation.
Taken together, the data presented in current study establish that the WNT signaling
pathway is extensively modulated in response to allergen challenge. Of note, downregulation
of WIF1 by TGF-β may represent a detrimental event which may allow increased WNT
signaling in the lungs driving asthma pathological manifestations. The inference, however,
be taken with caution as more experiments are needed to substantiate the findings.
Acknowledgements
This study was supported by a Vidi Grant (grant nr. 016.126.307) from the Dutch
Organization for Scientific Research (NWO) to R. Gosens. The funders had no role in study
design, data collection and analysis, decision to publish, or preparation of the manuscript.

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
193 | P a g e
References
1. Cohn L, Elias JA, Chupp GL. (2004) Asthma: Mechanisms of disease persistence and
progression. Annu Rev Immunol 22: 789-815.
2. Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. (2000) Asthma. from
bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med
161: 1720-1745.
3. Barnes PJ. (2012) Severe asthma: Advances in current management and future therapy. J
Allergy Clin Immunol 129: 48-59.
4. Barnes PJ. (2004) New drugs for asthma. Nat Rev Drug Discov 3: 831-844.
5. Holgate ST, Davies DE, Powell RM, Howarth PH, Haitchi HM, et al. (2007) Local genetic and
environmental factors in asthma disease pathogenesis: Chronicity and persistence
mechanisms. Eur Respir J 29: 793-803.
6. Bumbacea D, Campbell D, Nguyen L, Carr D, Barnes PJ, et al. (2004) Parameters associated
with persistent airflow obstruction in chronic severe asthma. Eur Respir J 24: 122-128.
7. Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. (2007) Remodeling and airway
hyperresponsiveness but not cellular inflammation persist after allergen challenge in
asthma. Am J Respir Crit Care Med 175: 896-904.
8. Homer RJ, Elias JA. (2005) Airway remodeling in asthma: Therapeutic implications of
mechanisms. Physiology (Bethesda) 20: 28-35.
9. Awadh N, Muller NL, Park CS, Abboud RT, FitzGerald JM. (1998) Airway wall thickness in
patients with near fatal asthma and control groups: Assessment with high resolution
computed tomographic scanning. Thorax 53: 248-253.
10. Little SA, Sproule MW, Cowan MD, Macleod KJ, Robertson M, et al. (2002) High resolution
computed tomographic assessment of airway wall thickness in chronic asthma:
Reproducibility and relationship with lung function and severity. Thorax 57: 247-253.
11. Niimi A, Matsumoto H, Amitani R, Nakano Y, Mishima M, et al. (2000) Airway wall thickness
in asthma assessed by computed tomography. relation to clinical indices. Am J Respir Crit
Care Med 162: 1518-1523.
12. Clevers H, Nusse R. (2012) Wnt/beta-catenin signaling and disease. Cell 149: 1192-1205.
13. Nusse R, Varmus HE. (1982) Many tumors induced by the mouse mammary tumor virus
contain a provirus integrated in the same region of the host genome. Cell 31: 99-109.
14. Baarsma HA, Konigshoff M, Gosens R. (2013) The WNT signaling pathway from ligand
secretion to gene transcription: Molecular mechanisms and pharmacological targets.
Pharmacol Ther 138: 66-83.
15. Choy DF, Modrek B, Abbas AR, Kummerfeld S, Clark HF, et al. (2011) Gene expression
patterns of Th2 inflammation and intercellular communication in asthmatic airways. J
Immunol 186: 1861-1869.
16. Makinde T, Murphy RF, Agrawal DK. (2007) The regulatory role of TGF-beta in airway
remodeling in asthma. Immunol Cell Biol 85: 348-356.
17. Heijink IH, de Bruin HG, van den Berge M, Bennink LJ, Brandenburg SM, et al. (2013) Role
of aberrant WNT signalling in the airway epithelial response to cigarette smoke in chronic
obstructive pulmonary disease. Thorax 68: 709-716.
18. Duvernelle C, Freund V, Frossard N. (2003) Transforming growth factor-beta and its role in
asthma. Pulm Pharmacol Ther 16: 181-196.

Chapter 6
194 | P a g e
19. Chu HW, Trudeau JB, Balzar S, Wenzel SE. (2000) Peripheral blood and airway tissue
expression of transforming growth factor beta by neutrophils in asthmatic subjects and
normal control subjects. J Allergy Clin Immunol 106: 1115-1123.
20. Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, et al. (1997) Transforming
growth factor-beta 1 in asthma. measurement in bronchoalveolar lavage fluid. Am J Respir
Crit Care Med 156: 642-647.
21. Vignola AM, Chanez P, Chiappara G, Merendino A, Pace E, et al. (1997) Transforming growth
factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir
Crit Care Med 156: 591-599.
22. John M, Au BT, Jose PJ, Lim S, Saunders M, et al. (1998) Expression and release of
interleukin-8 by human airway smooth muscle cells: Inhibition by th-2 cytokines and
corticosteroids. Am J Respir Cell Mol Biol 18: 84-90.
23. Oenema TA, Kolahian S, Nanninga JE, Rieks D, Hiemstra PS, et al. (2010) Pro-inflammatory
mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir Res 11:
130-9921-11-130.
24. Gregory CA, Gunn WG, Reyes E, Smolarz AJ, Munoz J, et al. (2005) How wnt signaling affects
bone repair by mesenchymal stem cells from the bone marrow. Ann N Y Acad Sci 1049: 97-
106.
25. Iwano M, Neilson EG. (2004) Mechanisms of tubulointerstitial fibrosis. Curr Opin Nephrol
Hypertens 13: 279-284.
26. Thompson MD, Monga SP. (2007) WNT/beta-catenin signaling in liver health and disease.
Hepatology 45: 1298-1305.
27. Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, et al. (2002) A role for the beta-catenin/T-
cell factor signaling cascade in vascular remodeling. Circ Res 90: 340-347.
28. Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, et al. (2003) Aberrant Wnt/beta-catenin
pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162: 1495-1502.
29. Konigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, et al. (2008) Functional wnt
signaling is increased in idiopathic pulmonary fibrosis. PLoS One 3: e2142.
30. Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, et al. (2009) WNT5A
is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol
Biol 41: 583-589.
31. Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, et al. (2011) Beta-catenin
signaling is required for TGF-beta1-induced extracellular matrix production by airway
smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 301: L956-65.
32. Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, et al. (2010) Decreased fibronectin
production significantly contributes to dysregulated repair of asthmatic epithelium. Am J
Respir Crit Care Med 181: 889-898.
33. Banyai L, Kerekes K, Patthy L. (2012) Characterization of a wnt-binding site of the WIF-
domain of wnt inhibitory factor-1. FEBS Lett 586: 3122-3126.
34. Avanesov A, Honeyager SM, Malicki J, Blair SS. (2012) The role of glypicans in wnt inhibitory
factor-1 activity and the structural basis of Wif1's effects on wnt and hedgehog signaling.
PLoS Genet 8: e1002503.
35. Song HH, Shi W, Xiang YY, Filmus J. (2005) The loss of glypican-3 induces alterations in wnt
signaling. J Biol Chem 280: 2116-2125.
36. Xu B, Chen C, Chen H, Zheng SG, Bringas P,Jr, et al. (2011) Smad1 and its target gene Wif1
coordinate BMP and wnt signaling activities to regulate fetal lung development.
Development 138: 925-935.

WNT pathway is altered in a mouse model of chronic allergic airway inflammation
195 | P a g e
37. Fujiwara N, Doi T, Gosemann JH, Kutasy B, Friedmacher F, et al. (2012) Smad1 and WIF1
genes are downregulated during saccular stage of lung development in the nitrofen rat
model. Pediatr Surg Int 28: 189-193.
38. Wissmann C, Wild PJ, Kaiser S, Roepcke S, Stoehr R, et al. (2003) WIF1, a component of the
wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J Pathol 201:
204-212.
39. Al-Muhsen S, Johnson JR, Hamid Q. (2011) Remodeling in asthma. J Allergy Clin Immunol
128: 451-62; quiz 463-4.
40. Mazieres J, He B, You L, Xu Z, Lee AY, et al. (2004) Wnt inhibitory factor-1 is silenced by
promoter hypermethylation in human lung cancer. Cancer Res 64: 4717-4720.
41. Ai L, Tao Q, Zhong S, Fields CR, Kim WJ, et al. (2006) Inactivation of wnt inhibitory factor-1
(WIF1) expression by epigenetic silencing is a common event in breast cancer.
Carcinogenesis 27: 1341-1348.
42. Yang Z, Wang Y, Fang J, Chen F, Liu J, et al. (2010) Downregulation of WIF-1 by
hypermethylation in astrocytomas. Acta Biochim Biophys Sin (Shanghai) 42: 418-425.
43. Cai J, Guan H, Fang L, Yang Y, Zhu X, et al. (2013) MicroRNA-374a activates Wnt/beta-
catenin signaling to promote breast cancer metastasis. J Clin Invest 123: 566-579.
44. Zhang Q, Chen L, Helfand BT, Jang TL, Sharma V, et al. (2011) TGF-beta regulates DNA
methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and
predicts disease recurrence. PLoS One 6: e25168.
45. Pan X, Chen Z, Huang R, Yao Y, Ma G. (2013) Transforming growth factor beta1 induces the
expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS One 8:
e60335.

196 | P a g e