university of groningen organic reactivity in mixed
TRANSCRIPT

University of Groningen
Organic reactivity in mixed aqueous solventsBlokzijl, Wilfried
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:1991
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Blokzijl, W. (1991). Organic reactivity in mixed aqueous solvents: a link between kinetics andthermodynamics. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 18-11-2021

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
CHAPTER 6
Diels-Alder Reactions in Mixed Aqueous Media. Enforced Hydrophobic Interaction
For a wide range of solvent systems, both rate constants and stereospecificities of Diels-Alder reactions are little or only moderately sensitive to changes in the nature of the reaction mediumz3'. In 1980, Breslow et a1.17" showed that, quite surprisingly at that time, Diels-Alder reactions are dramatically accelerated in aqueous solutions. In the past ten years, a number of studies have revealed curious rate effects on different types of cycloaddition reactions in aqueous solutions17ad~1Sae~19a~bbma-C~21a~bb2323262627728. Recently, Grieco et a1.280 reported similar rate enhancements for a Diels-Alder reaction in diethyl ether containing 5 M lithium perchlorate. Not only Diels-Alder reactions are accelerated in aqueous solutions, but also Claisen rearrangernent~'~"~, the benzoin c~ndensation'~", and aldol reactions of silyl en01 ethers '%(see Table 1-1). A common aspect of these reactions is that the reactants are apolar, neutral species. Grieco et a1.18 showed that these accelerations are not just curiosities but that water can be a highly useful solvent for organic transformations of synthetic interest. The high reactivity in aqueous solutions facilitates reaction at low temperatures to prevent decomposition and elimination processes which might occur at high reaction tempera- tures. In some cases products were obtained which hitherto were inaccessible using organic solvents. In addition, for several processes the stereospecificity and regioselec- tivity are significantly chan ed when the reaction is performed in aqueous solutions23. Recently, Waldmann et a15 studied ehiral catalysis of Diels-Alder reactions in mixed aqueous solutions. Unfortunately, these authors were not able to perform the reactions without the presence of a considerable amount of organic cosolvents. In all cases the yield of the reactions was significantly higher than in organic solvents. However, a substantial improvement of the stereoselectivity was not observed. An important disadvantage of water is that the solubility of organic substrates is often small. Although the rate constants do not necessarily decrease dramatically when microheterogenities are present in the reaction medium'7ac, an acceptable high solubility of the reactants is a prerequisite for synthetic applications. This can be achieved by (i) employing organic cosolvents to increase the solubility of the apolar reagents18', or (ii) introducing hydrophilic substituents in the substrate molecule^'^^^^^. Lubineau et a1.19b showed that introduction of carbohydrate substituents in the diene in order to increase the low solubility immediately leads to much smaller rate accelerations of these Diels-Alder reaction in aqueous solutions. It is not known, however, how the reactivity of organic reactions in aqueous media depends on the concentration of the cosolvent(s).
From the previous kinetic and synthetic studies in aqueous solutions, three crucial questions emerge:

6. Diels-Alder reactions in aqueous media Enforced hydrophobic interaction
(i) What is the molecular origin of the curious rate effects on Diels-Alder reactions and related transformations in aqueous solutions?
(ii) How does the reactivity of the above processes in aqueous solutions respond to addition of organic cosolvent(s) or cosolutes?
(iii) Which organic reactions can be accelerated in aqueous media and how do substituents affect these rate accelerations?
A proper understanding of these factors should enable chemists to select organic reactions and appropriate aqueous solvents in order to benefit from the properties of water as a solvent for organic reactions. In this chapter, an attempt is made to answer the questions (i-iii). The chapter describes a quantitative analysis of solvent effects on the intermolecular Diels-Alder reactions of cyclopentadiene (1) with alkyl vinyl ketones (2a,b) and 5-substituted-1,4-naphthoquinones (3a-c) (Scheme 6-1) in water and mixed aqueous solvents containing monohydric alcohols.
Scheme 6-1
Diels-Alder reactions, involving the cloaddition of dienes to quinones have been cornerstones in the synthesis of steroids", cortisonesm2 and many natural productsm. Consequently, much information has been accumulated concerning the regioselectivity and reactivity of Diels-Alder reactions involving quinones, naphthoquinones and anthraq~inones~'~. The Diels-Alder reactions of cyclopentadiene with alkyl vinyl ketones or related acrylic acids and derivatives are also well-known. The latter reactions have been frequently used as models in studies of the effect of external factors on reactivity, stereoselectivity and regioselectivity of Diels-Alder processes20a- c*2332859286. In Section 6.2, some general mechanistic aspects of Diels-Alder reactions will be described. Subsequently, these mechanistic aspects are associated with external factors which govern the reactivity, stereospecificity and regioselectivity of Diels-Alder reactions in solution.
The second-order rate constants for the cycloaddition of dienophiles 2a,b and 3a-c with diene 1 in aqueous solutions are, respectively, about 200 and 5800 times larger

6. Dieh-Alder reactions in aqueous media. Enforced hydrophobic interaction
than those in n-hexane. In comparison to the known rate effects on Diels-Alder reactions in aqueous solutions, the rate effect of water on the Diels-Alder reaction of cyclopentadiene with the substituted naphthoquinones is extremely high. In the past eleven years, several attempts have been made to explain the molecular origin of the high reactivity in aqueous solutions. In Section 6.2, these explanations will be reviewed and critically discussed.
The kinetics of Diels-Alder reactions in mixed aqueous solvents across the mole fraction range, OSx,S1 where x2 is the mole fraction of the cosolvent, cannot be analysed by assuming pairwise interactions of the cosolvent with reactants and activated complex, respectively. This chapter will delineate how the quantitative treatment, developed from the model of Kirkwood and Buff, can be applied to account for the dependence of kinetic parameters on the composition of the mixed aqueous solvent. This analysis is supported by the application of the Kirkwood-Buff treatment for the quantitative analysis of transfer parameters for the transfer of 1, 2a and 2b from 1-propanol to mixed aqueous solvents, containing 1-propanol. The applicability of the theory, developed in Chapter 2, for a quantitative analysis of solvent effects on reactions in mixed solvents in which both solvents are present in comparable amounts, is critically tested. Furthermore, solvent effects are reported on isobaric activation parameters (A*HO and TA*SO) of Diels-Alder reactions in mixed aqueous solvents. The exceptional rate accelerations in aqueous solutions are primarily interpreted in terms of "enforced hydrophobic interaction" between diene and dienophile.
6.2 DieLF-AIder reactions in water. Mechanistic considerations and recent developments
Mechanistic aspects of Diels-Alder reactions. Since the discovery of the Diels-Alder reaction in 1928~~", the mechanism of the reaction has been studied extensively2. Knowledge of the mechanism of Diels-Alder reactions is very important for under- standing the influence of external factors on Diels-Alder reactions. Studies of the mechanisms of Diels-Alder reactions are still strongly dominated by the Woodward- Hoffmann rules. Theoretical predictions, ab initio calculations, approximation methods and frontier molecular orbital theory all contribute to our knowledge of the mecha- nism of Diels-Alder reactions232. Studies of the mechanism of intermolecular Diels- Alder reactions have been characterised by attempts to interpret all Diels-Alder reactions in terms of one universally valid mechanism. Notwithstanding the fact that many [4+2]-cycloadditions involve a symmetry-allowed synchronous bond formation, a two-step mechanism cannot be ignored in all cases. Simultaneous bond formation may be severely hindered by steric or electronic effects and consequently the otherwise energetically less favourable two-step mechanism can become a serious competitor for the one-step process.
An important matter of concern in discussions about the mechanism of Diels- Alder reactions is the occurrence of a charge-tranfer (m) intermediate287. The occurrence of charge-transfer intermediates is plausible for the cycloaddition of very electron-rich dienes to very electron-poor dienophiles. Some evidence, however,

ti DieLr-Alder reactions in aqueous media. Enforced hydrophobic interaction
suggests that charge-transfer is an important step in every cycloaddition reaction287. Claims for the intermediacy of a true CT-intermediate are controversial. A current debate centres on the possibility that the activated complex takes advantage of CT- interactions, even without prior formation of a true CI'-complex, via a process called "electron-transfer activation". Although the activation process must involve the transfer of electrons to the region where new bonds have to be formed, the extent of electron transfer and concomittant formation of radical ions is unclear. Development of some partial charges during the activation process has been suggested by frontier molecular orbital (FMO) ~ a l c u l a t i o n s ~ ~ ~ ~ ~ . Diels-Alder reactions are characterised by small, but significant p-values, indicative of significant electronic substituent effects232. FMO and ab initio calculations have been very useful in the analysis of the rate
constant and regioselectivity of Diels-Alder reactionsmtm. The rate constant and the site of reactivity for reactions of an electrophilic molecule with a nucleophile is dominated by the interaction of the LUMO of the electrophile with the HOMO of the nucleophile. If the orbitals which are involved in the reaction step are brought closer in energy, the overlap is more efficient and the rate constant is enhanced.
The volume of activation, A*VO, has been frequently used as a mechanistic tool289- 291. In extensive review articles, LeNoble et collected volumes of activation for a large series of Diels-Alder reactions. Generally, the volumes of activation are negative. The activated complex resembles the products with respect to spacial requirements, but in some cases, the volume of activation is even more negative than the overall reaction volume. The mechanistic interpretation is twofold. First, the activated complex is considered to be highly ordered, and formed by simultaneous bond formation; secondary orbital overlap causes an additional decrease of the activation volume. Second, part of the volume of activation must be attributed to the contributi- on of the solvent. The formation of partial charges during the activation process leads to a decrease of the volume of activation in polar solvents, due to what has been called "electr~striction"~~.
The effect of external factors on Diels-Alder reactions. A number of external factors, other than temperature, have been identified which may affect the rate constant, regioselectivity and stereospecificity of Diels-Alder reactions in solution. A useful and effective contribution to the question of mechanism was provided by the discove~y of the catalysis of Diels-Alder reactions b Lewis-acids which may accelerate the rate of addition by several powers of ten mm-L. Lewis-acids affect the orbital coefficients of the reactants by interacting with substituents of the dienophile. Recently, Kelly et a1.295 showed that compounds able to form extensive hydrogen bonds to the dienophile, also catalyse Diels-Alder reactions in aprotic media. A less spectacular but very useful extension of the cycloaddition reactions is provided by the fact that Diels-Alder reactions are generally accelerated by applying external pressure2%. The rate effects can be quite dramatic if the volume of activation is strongly negative. The stereospeci- ficity and regioselectivity of the cycloaddition can be significantly altered by changes in pressure if the activated complexes leading to the regio- and stereoisomers are characterised by different volumes of activation286. For Diels-Alder reactions involving cyclopentadiene, the endo-exo product ratio is determined by secondary orbital overlap. An increase in pressure favours one of the two isomeric products297. Recent- ly, it has been reported that also antibodies can accelerate Diels-Alder reactions"'.

6 DieIs-Alder reactions in aqueous media. Enfmed hyirophobic interaction
Generally, solvent effects on Diels-Alder reactions are negligibly Many cycloadditions proceed as fast in the vapour phase as in nonpolar solvents. In a few incidental cases larger solvent effects (accelerations of a factor of 35 on going from an apolar to the most polar solvent) are These solvent effects are attributed to extreme changes in the dipole moment of the reacting species during the activation process. Only in rare cases polar or zwitterionic intermediates are respon- sible for exceptionally large solvent effectsz7. Desimoni et used the FMO approach to link solvent effects on Diels-Alder reactions with catalysis by Lewis acids. Solvent effects on the cycloaddition of 2,3-dimethylbutadiene to 5-substituted-1,4- naphthoquinones were explained by the donor-acceptor interactions between the dienophiles and the solvents. This was confirmed by a correlation between the acceptor numbers of the solvent (see Section 1.2.2) and the rate constant for the Diels-Alder reactions.
Recent developments. Solvent effects on Diels-Alder reactions have been studied frequentl?, though extensive and detailed analyses of solvent effects on Diels-Alder reactions in large series of organic solvents are rare. In 1962, Berson et a1.=' studied solvent effects on the stereoselectivity of Diels-Alder reactions of cyclopentadiene with several dienophiles by means of linear free energy relationships and introduced the 0 solvent polarity scale based on the endo/exo product ratio. In fact, this study reported the first attempt to perform Diels-Alder reactions in water and mixed aqueous solvents: "We made some attempts to conduct experiments in aqueous systems ..............., but the low solubility of cyclopentadiene in the aqueous solvents ....... led us to abandon these." Twenty years later, Breslow et a1.17' reported a first kinetic study of Diels-Alder reactions of cyclopentadiene with several dienophiles in aqueous solutions.
According to Breslow, "hydrophobic packing" of the two reactants is the most likely explanation for the large rate effects of water on Diels-Alder reactions". This idea is mainly based on a series of kinetic experiments on Diels-Alder reactions in aqueous solution in the presence of "salting in" and "salting out" materials. The enhancement or breaking of the water structure by the added cosolutes is used as a criterion for the promotion or inhibition of "hydrophobic packing". Breslow et al.", and later also Schneider et a ~ . ~ , supported this view by performing these Diels-Alder reactions in the presence of P-cyclodextrine. Comparable rate effects were observed and complexation of the reactants in the hydrophobic interior pocket of the polysac- charide was suggested to replace the "hydrophobic packing" in the aqueous solution. The ideas of Breslow, recently enunciated in a review paper17g, seem to be widely a ~ c e p t e d ~ ~ ~ ' ' . As will be shown in Section 6.7, the molecular basis of this "hydrop- hobic packing" is rather ambiguous. The main problem is concerned with the configu- ration of the reactants in the initial state of the reaction.
Alternative explanations were provided by Griecol' and Lubineau19 and cowor- kers. Grieco suggested the occurrence of micellar catalysis for some Diels-Alder reactions, in which large concentrations of a surfactant-like diene were The exceptional rate effects were attributed to hydrophobic binding of the apolar dienop- hile to the micellar aggregate of the diene. For the Diels-Alder reactions of unactiva- ted iminium salts with a series of dienesl& and the effective intramolecular Claisen rearrangementslsg in aqueous solution, this explanation is not appropriate. Unfortuna-

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaciion
tely, Grieco et a1.18 did not report any details about the kinetics of their synthetically very useful organic processes in aqueous solution. Nonetheless, the high yields in spite of ambient pressure, low reaction temperatures and short reaction times are indicative of high rate constants. Also the rate acceleration of the aldol condensation of silyl en01 ethers, reported by Lubineau et al.lk, excluded micellar catalysis. Both authors suggested a possible link between the effects of external pressure on Diels-Alder reactions (see above), performed in conventional organic solvents and the lar e rate enhancements in aqueous medialg1? They refer to publications of Daclc4', who stressed the relation between the activation volume A ' V of a chemical reaction and the cohesive energy density (CED) of the solvent. Generally, reactions with a negative volume of activation are accelerated by solvents with a high CED. The CED of the solvent can be expressed in terms of the Hildebrand solubility parameter (a,*) (see section 1.2.2). Application of the Hildebrand solubility parameter to the analysis of solvent effects on chemical reactions is restricted to reactions of neutral molecules involving a neutral activated complex. However, Dack argued that the ideas can also be applied for reactions involving ionic reactants or a charged activated complex42. The effect of CED of the solvent on rate constants is small. For many organic solvents the CED, which is also called the cohesive pressure, is related to the internal pressure of the solvent. According to a citation of ~ a c k ~ ~ "solvent internal pressure acts on the rate of nonpolar reactions in the same direction as external pressure". The suggestion that the internal pressure of the solvent is the explanation for the exceptional solvent effects on Diels-Alder reactions in aqueous media is supported by the exceptionally large rate acceleration of Diels-Alder reactions in diethyl ether in the presence of 5M of lithium perchlorate (known for its high internal pressure)280. However, the CED is a measure for the enthalpy of evaporation per volume of solvent, whereas the internal pressure is defined as the change in internal energy of a solvent as it undergoes a very small isothermal expansion. The internal pressure results from forces of attraction between solvent molecules exceeding the forces of repulsion (mainly dispersion and dipole-dipole interactions). For highly structured liquids, such as water, the CED deviates strongly from the internal solvent pressure. Compared to organic solvents, the internal pressure of water is even exceptionally small. In fact at 277 K, the internal pressure of water is close to zero. ~ u b i n e a u ' ~ ~ , Grieco"' and Breslowl'g did not discriminate between internal pressure and the CED and erroneously concluded that water, having an extremely large CED, exerts a high internal pressure on organic reactions in this medium.
The exceptionally high reactivity of Diels-Alder reactions in aqueous media has also been related to the solubility of the reactants in aqueous media2092311%. Reaction rate constants and stereospecificities of Diels-Alder reactions in mixed aqueous solvents have been correlated successfully with the solvophobicity parameter Sp6' and q ( 3 0 ) ~ ~ ~ ' ~ . The correlation of the reaction rate constants of Diels-Alder reactions with the CED of the solvent, emphasised by ~r ieco" and ~ u b i n e a u ' ~ was, in fact, also indicative of the im ortance of the solubility of the reactants.
Schneider et a 1 performed a detailed study of the solvent effects on the Diels- Alder reactions of cyclopentadiene with a series of esters of acrylic acid in aqueous solutions as a function of the hydrophobicity of the dienophiles. Following the ideas of Breslow, Schneider implicitly assumed complexation of the reactants in the aqueous solution. The anticipated correlation between the hydrophobiclty of the dienophiles,

6 DieLF-Alder reactions in aqueous media. Enforced hyirophobic interaction
expressed in the Hansch T-increments, and the rate constants for the Diels-Alder reactions in water was, however, not observed'. This forced Schneider to conclude that hydrophobic complexation can be in fact counterproductive due to the fact that this might result either in homotactic complexation of the reactants or in complexes, having the 'bong" geometry for reaction to occur.
Recently, photochemical Diels-Alder reactions have been studied in aqueous s o l ~ t i o n s ~ ' ~ ~ ~ . In some cases the product distribution and the regioselectivity of the process were very different in aqueous solutions as compared with that in organic solvents. This was related to the enhanced photo-efficiency in aqueous media. The interpretation of solvent effects on photochemical reactions is very complex, but the effects have been frequently attributed to stacking of the reactants in aqueous media.
Finally, Diels-Alder reactions have been studied in "organised aqueous media, such as (i) r n i c r ~ e r n u l s i o n s ~ ~ ~ ~ ~ ~ , (ii) micellar systemsw1 and (iii) clay emulsion^^^. These microheterogeneous systems share the following two common features: (i) they provide a hydrophobic pocket in which hydrophobic molecules can be assembled and (ii) they offer an interface between the aqueous exterior and a hydrocarbon-like interior wherein the reactants can be aligned. Frequently, Diels-Alder reactions are significantly accelerated in these systems. Remarkably, the stereospecificity of Diels- Alder reactions in micellar systems and microemulsions and the stereospecificity found in polar environments, are similar. Rate effects have been attributedw1 to (i) cage effects (the ability of microdomains to hold two reactive species together long enough for reaction to occur), (ii) pre-orientation of the reactants (which resembles stacking), (iii) microviscosity and (iv) the local concentration effect. Some organic reactions, involving reagents that are only sparingly soluble in water, have also been performed in heterogeneous aqueous media. The rate constant still was considerably higher than in organic solvent^"^^.
6.3 Kinetic studies of DieIs-Alder reactions in m k d aqueous solvents; theoretical apprwch
Theoq. Transfer parameters for 1, 2a, 2b and the product of the reaction of 1 with 2a have been determined by measuring vapour pressures. We consider a solution of a solute-j at a temperature T. The solvent, or mixed solvent, is liquid-1. The solute is volatile so that at equilibrium the partial pressure of the solute-j above the solution is pj. It is now assumed that the vapour forms a perfect gas. For the gas phase, the chemical potential of the substance-j is given by:
Here, p0 is the standard pressure and Ccj"(g) is the chemical potential of the com- pound-j as a perfect gas at pressure pO. For the compound-j in solution, the chemical
* w e were not able to reproduce the rate constants reported by Schneider et al. for the Diels- Alder reactions of cyclopentadiene with esters of acrylic acid (see Experimental Section).

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
potential is related to molality m, using Equation 6-2:
By definition, limit(mj+O)yj=l.O. At equilibrium, pj(g)=pj(sln,l). Therefore
In dilute solutions of compound-j on a molality scale, according to Henry's Law, we have
where Hj is Henry's constant. So, if mj is very small (yj=l.O),
A similar equation can be derived for a solvent-2 If the solutions are dilute (mj+O), the standard chemical potential of transfer of solute-j from solvent-1 to solvent-2, where solute-j has unit molality, is given by Equation 6-6.
Values of H. were calculated from the slope at infinite dilution of the plot of pj as a function of the molality of j. flsln;2)-lr,D(sln;l) is the transfer parameter.
The vapour pressure of a solute is very sensitive to stacking or clustering of the solute. According to Equation 6-4, a plot of pj against mj is linear for an ideal solution in which yj is unity. Any significant curvature (and deviation of yj from 1) is indicative of pairwise and higher order interactions between the solutes, possibly leading to association.
In Chapter 2, a quantitative treatment was developed, based on the theory of Kirkwood and Buff, which accounts for the dependence of rate constants on the composition of a mixed solvent across a mole fraction 0sx2Sl , where x2 is the mole fraction of the cosolvent. The analysis of the kinetic data is based on the standard chemical potentials of the reactants and the activated complex in a mixed solvent, containing solvents 1 and 2. According to Equation 2-71, a parameter 4 can be
4 is determined by four Kirkwood-Buff integral functions, Gxl, G,, G,, and G,,. These integral functions measure the probability of finding a solvent molecule 1 or 2 in a volume element around reactants X and activated complex (symbol +), respectively. Therefore, 4 measures the change in the relative affinities

6. DieIs-Alder reactions in aqueous media. Enforced hydrophobic interaction
of the reactant, upon going from the initial state to the transition state, for the solvents present in the mixed solvent. In fact, as shown in Section 2.4, six Kirkwood- Buff integral functions are required to determine the magnitude of 4 for bimolecular reactions. To avoid unnecessary complications in the formulae, X represents both diene and dienophile. The quantitative analysis of solvent effects on Diels-Alder reactions in mixed aqueous solutions then involves the calculation of 4 as a function of x,.
For the calculation of 4 three parameters are required that have to be derived from experimental data. 4 is related to the dependence of the rate constant on the composition of the mixed solvent, as expressed by the differential d{ln[k(x2)/k (x,=O)])/dx,. The determination of this first-order derivative involves fitting the dependence of ln[k(x2)k(x2=0)] on the composition of the solvent. It was found to be impossible to formulate a unique mathematical function which accounts for the observed trend. Since the first-order derivative must be a continuous function, the data were approximated by cubic ~plines~'~(see experimental section).
Also important is the property Q, which represents the non-ideal behanour of the mixed solvent. Q is a function of the composition of the mixed solvent (see Equation 2-70). The key step in the determination of Q involves the calculation of the second- order differential d2GmE/d$. Error propagation analysis shows that the calculation of Q is the largest source of error in the evaluation of 4, especially when Q approaches zero (see also Section 6.6)l9'. The excess molar Gibbs energies of mixing were calcula- ted on the basis of activity coefficients or vapour pressures which have been reported in the literature (see experimental part). The dependence of G,' on the mole fraction x, was fitted to the orthogonal polynomial, given in Equation ~ 7 ~ .
where
This orthogonal polynomial is particularly well suited1630435 for analysis of the excess molar Gibbs energy of a binary aqueous mixture where GmE is large and positive. Subsequently, the second-order derivative with respect to the mole fraction of cosolvent was computed. The third parameter, the dependence of the partial molar volume on the composition of the mixed aqueous solvent, was calculated on the basis of densities which were retrieved from the literature.
The change of affinity of the reactants upon going from the initial state to the

6 Diels-Alder reactions in aqueous media Enforced hyirophobic interaction
6.4 Properties of alcohd-water mirtures.
For many years, aqueous solutions of low-molecular weight alcohols have been of interest to many scientists and technologists. The complete miscibility of alcohols with water and low costs make mixtures of water and alcohols an attractive class of solvents for industrial applications. Although chemists have been intrigued by the remarkable properties of these mixtures in the highly aqueous concentration range, their properties have usually been determined over the whole concentration range (see, for an extensive review, ref.15). High precision microcalorimetry, nuclear magnetic relaxation studies, scattering methods and new theories of liquids and solutions have contributed to a better understanding of the structure and dynamics of aqueous solutions of alcohols. Properties of alcohol-water mixtures are discussed considering separately several concentration ranges. The demarcation between these concentration ranges is determined by more or less abrupt changes in the properties of alcohol-water mixtures. The following concentration ranges have successively been distinguished (i) infinitely dilute aqueous solutions and dilute aqueous solutions, (ii) water-like mixed aqueous solutions, and (iii) alcohol-like mixed aqueous soIutions.
In Section 3.2, the properties have been discussed of infinitely dilute and dilute solutions. The thermodynamic properties of these solutions can be accurately descri- bed by concentration expansions, involving pairwise and higher-order terms. The alcohol molecules are fully solvated by water molecules. At higher concentration of alcohol, partial molar quantities, such as partial molar volumes and heat capacities, exhibit extrema. These extrema are very sensitive to temperature and pressure and depend on the nature of the alcohol molecule. Light scattering data, ultrasound measurements, kinetics and SAXS indicate the onset of aggregation-like processes (defined by Franks as typically aqueou~'~). The increased affinity of alcohol molecules for other alcohol molecules is elegantly shown by extrema in the Kirkwood-Buff integral functions representing the mutual affinity of alcohol molecules. Beyond these extrema the mixed aqueous solvent is characterised by short-lived water-rich and alcohol-rich clusters. The ~ar t i a l molar quantities exhibit a second, less pronounced change at still higher alcohol content. Beyond this concentration, the properties of the mixed aqueous solvent start to resemble those of the pure alcohol.
6.5 Dieb-Alder readions in water and in mired aqueous solvents; erperimental re&
The dependences of the rate constants for Diels-Alder reactions on the composition of the binary water-alcohol mixtures are characterised by two critical mole fractions of cosolvent. These mole fractions almost coincide with the critical mole fractions that characterise the thermodynamic properties of the alcohol-water mixtures (see Section 6.4). Following the subdivision of mixed aqueous solvents, containing monohydric alcohols, described in Section 6.4, the dependences are reported of (i) rate constants, (ii) A'HO and TA'SO and (iii) standard chemical potentials of reactants and activated complexes on the composition of the solvent. Four separate concentration ranges are identified.

6 Dielr-Alder reactions in aqueous media. Enforced hydrophobic interaction
Water and organic solvents. Second-order rate constants and isobaric activation parameters for the intermolecular Diels-Alder reactions of diene 1 with dienophiles 2a, 2b and 3 a c in water and in 1-propanol, are listed in Table 6-1. The rate constants for the Diels-Alder reactions of 3a-c with cyclopentadiene in 1-propanol decrease in the followin order: 3cc3ac3b. In aqueous solutions, however, we find 3aSb<3c. Houk et a 1 5 used frontier molecular orbital approaches to calculate substituent effects on rate constants and regioselectivities of Diels-Alder reactions, involving a large series of quinones. Electron-donating substituents in the quinones raise the orbital coefficients and decrease the rate constants, whereas electron-accepting substituents lower the orbital coefficients and lead to an increase of the rate constant. When the hydroxy substituent of compound 3b (also known as juglone) adopts the syn-conformation, the hydroxy group binds to the peri carbonyl via intramolecular hydrogen bonding. In this case, the hydroxy substituent acts as an acceptor. If the hydroxy group is constrained to the anti-conformation, intramolecular hydrogen bonding with the peri carbonyl is impossible and the hydroxy group acts as a donor. The syn-conformation is more stable in all solvents284. The FMO calculations predict rate constants in the order of 3cc3a<3b. This order was only found in aqueous solutions.
Table 6-1. Second-order Rate Constants and Isobaric Activation Parameters for the Diels-Alder Reactions of Cyclopentadiene (1) with Dienophiles 2a, 2b, and 3a-c in Aqueous Solutions and Organic Solvents, at 25OC.
Da Solvent k2 A*GO, A *HO, -TA 'So, dm3 mol-' s-' kJ morl kJ mol-' kJ mol-I
2a Methanol Ethanol 1-Propanol Water W a t e r + S ~ S ~ Water +SDSC water+CI'ABb Water+CTABC water +CHPd
2b I-Propanol Water
3a 1Propanol Water
3b 1Propanol Water
3c 1-Propanol Water
a Dienophile. 50 mmol in 1 kg of water. 100 mmol in 1 kg of water 750 mmol N-cyclohexyl- pyrrolidone in 1 kg of water.
6. Diek-AUer reactions in aqueous media. Enforced hydrophobic interaction
Desimoni et a1.299 found the following order of rate constants of Diels-Alder reactions of 5-substituted-naphthoquinones with 2,3-dimethylbutadiene in organic solvents: 3a<3c<3b. In summary, the order of reactivity is sensitive to the solvent and cannot be predicted accurately by FA40 calculations.
The rate constants for all Diels-Alder reactions studied are spectacularly enhanced in aqueous solutions. The magnitude of the rate acceleration is dependent on the nature of the dienophile. The Gibbs energies of activation, found in 1-propanol, are reduced by 10-14 kJ mol-' in aqueous solutions. The magnitude of this reduction is dependent on the size andlor hydrophobicity of the dienophiles. The substituent effects are moderate but significant. Both entropies and enthalpies of activation are more favourable for reactions in water than in 1-propanol and contribute cooperati- vely to the rate acceleration. Generally though, the entropy term is the dominating factor.
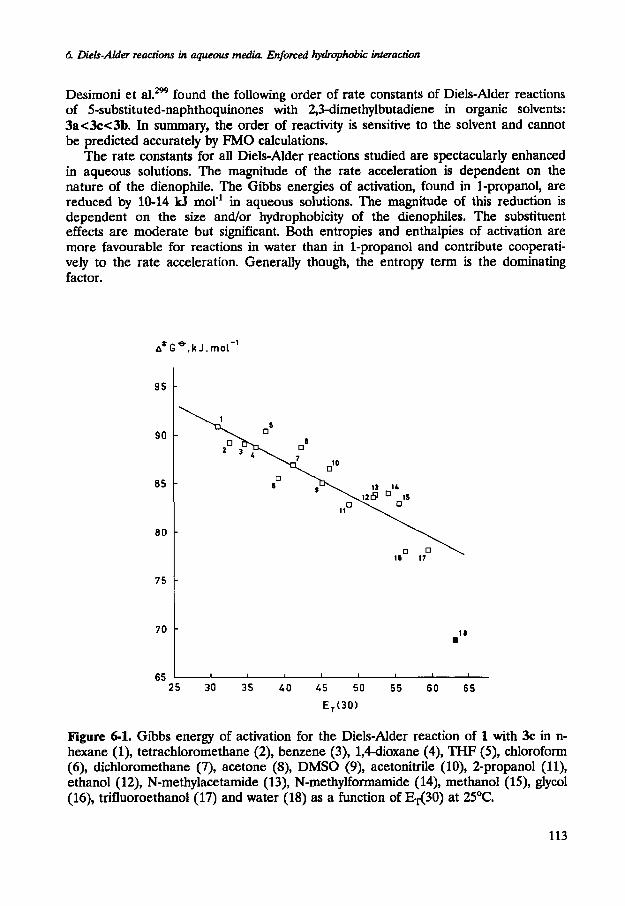
Figure 6-1. Gibbs energy of activation for the Diels-Alder reaction of 1 with 3c in n- hexane (I), tetrachloromethane (2), benzene (3), 1,4-dioxane (4), THF (5), chloroform (6), dichloromethane (7), acetone (8), DMSO (9), acetonitrile (lo), 2-propanol (ll), ethanol (12), N-methylacetarnide (13), N-methylformarnide (14), methanol (IS), glycol (16), trifluoroethanol (17) and water (18) as a function of q 3 0 ) at 25°C.
6 DM-Alder reactions in aqueous media. Enforced hydrophobic interaction
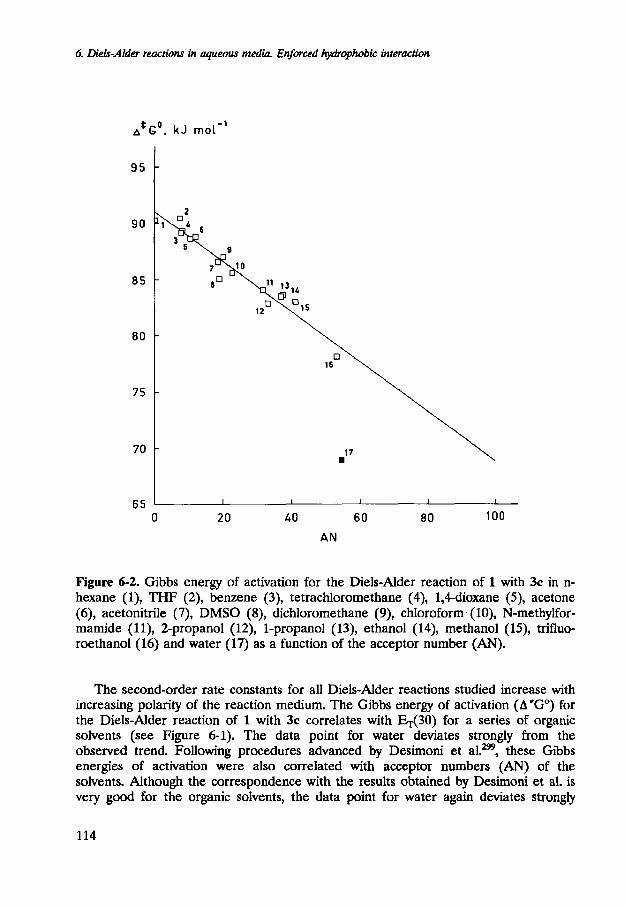
Figure 6-2. Gibbs energy of activation for the Diels-Alder reaction of 1 with 3c in n- hexane (I), THE: (2), benzene (3), tetrachloromethane (4), 14-dioxane ( 9 , acetone (6), acetonitrile (7), DMSO (81, dichloromethane (9), chloroform. (lo), N-methylfor- marnide ( l l ) , Zpropanol (12), 1-propanol (13), ethanol (14), methanol (15), trifluo- roethanol (16) and water (17) as a function of the acceptor number (AN).
The second-order rate constants for all Diels-Alder reactions studied increase with increasing polarity of the reaction medium. The Gibbs energy of activation (A*GO) for the Diels-Alder reaction of 1 with 3c correlates with w 3 0 ) for a series of organic solvents (see Figure 6-1). The data point for water deviates strongly from the observed trend. Following procedures advanced by Desimoni et a1.299, these Gibbs energies of activation were also correlated with acceptor numbers (AN) of the solvents. Although the correspondence with the results obtained by Desimoni et al. is very good for the organic solvents, the data point for water again deviates strongly
6 DieIs-Alder reactions in aqueous media. Enforced hydrophobic interaction
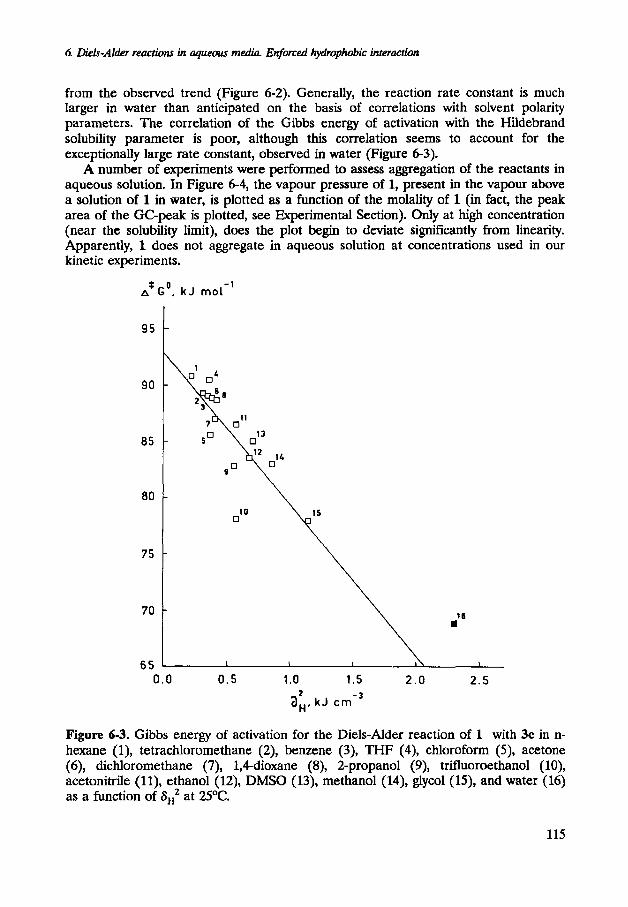
from the observed trend (Figure 6-2). Generally, the reaction rate constant is much larger in water than anticipated on the basis of correlations with solvent polarity parameters. The correlation of the Gibbs energy of activation with the Hildebrand solubility parameter is poor, although this correlation seems to account for the exceptionally large rate constant, observed in water (Figure 6-3).
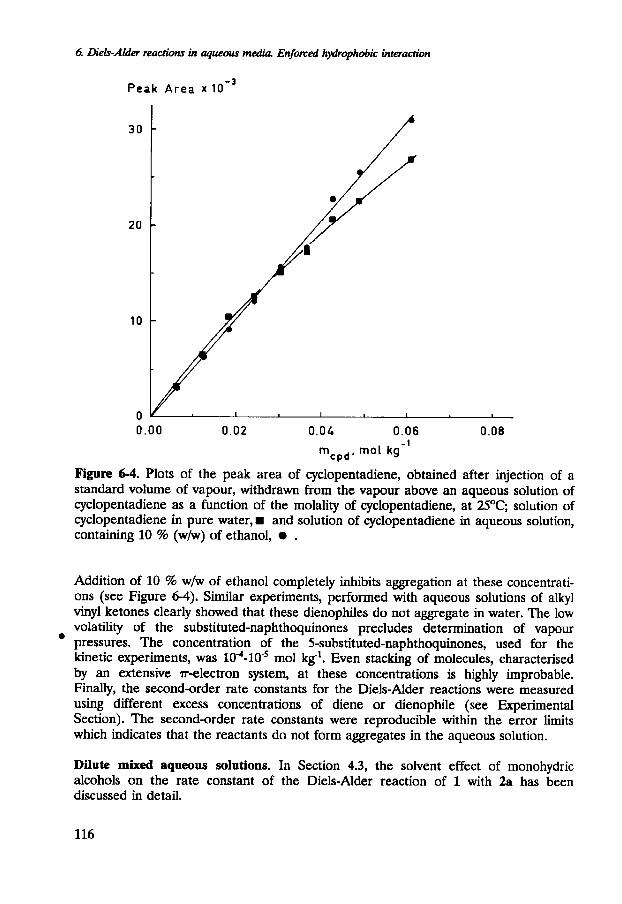
A number of experiments were performed to assess aggregation of the reactants in aqueous solution. In Figure 6-4, the vapour pressure of 1, present in the vapour above a solution of 1 in water, is plotted as a function of the molality of 1 (in fact, the peak area of the GC-peak is plotted, see Experimental Section). Only at high concentration (near the solubility limit), does the plot begin to deviate significantly from linearity. Apparently, 1 does not aggregate in aqueous solution at concentrations used in our kinetic experiments.
Figure 6-3. Gibbs energy of activation for the Diels-Alder reaction of 1 with 3c in n- hexane (I), tetrachloromethane (2), benzene (3), THF (4), chloroform (S), acetone (6), dichloromethane (7), 1,Cdioxane (8), 2-propanol (9), trifluoroethanol (lo), acetonitrile (ll), ethanol (12), DMSO (13), methanol (14), glycol (IS), and water (16) as a function of aH2 at 2S°C.
4 Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
Peak A r e a x 1 0 " ~
0 . 0 0 0 . 0 2 0 . 0 4 0 . 0 6 0 .08 - 1
mcpd. mol kg
Figure 6-4. Plots of the peak area of cyclopentadiene, obtained after injection of a standard volume of vapour, withdrawn from the vapour above an aqueous solution of cyclopentadiene as a function of the molality of cyclopentadiene, at 2S°C; solution of cyclopentadiene in pure water, w and solution of cyclopentadiene in aqueous solution, containing 10 % (wh) of ethanol, .
Addition of 10 % w h of ethanol completely inhibits aggregation at these concentrati- ons (see Figure 6-4). Similar experiments, performed with aqueous solutions of alkyl vinyl ketones clearly showed that these dienophiles do not aggregate in water. The low volatility of the substituted-naphthoquinones precludes determination of vapour pressures. The concentration of the 5-substituted-naphthoquinones, used for the kinetic experiments, was 10~-10~' mol kg-'. Even stacking of molecules, characterised by an extensive T-electron system, at these concentrations is highly improbable. Finally, the second-order rate constants for the Diels-Alder reactions were measured using different excess concentrations of diene or dienophile (see Experimental Section). The second-order rate constants were reproducible within the error limits which indicates that the reactants do not form aggregates in the aqueous solution.
Dilute mixed aqueous solutions. In Section 4.3, the solvent effect of monohydric alcohols on the rate constant of the Diels-Alder reaction of 1 with 2a has been discussed in detail.
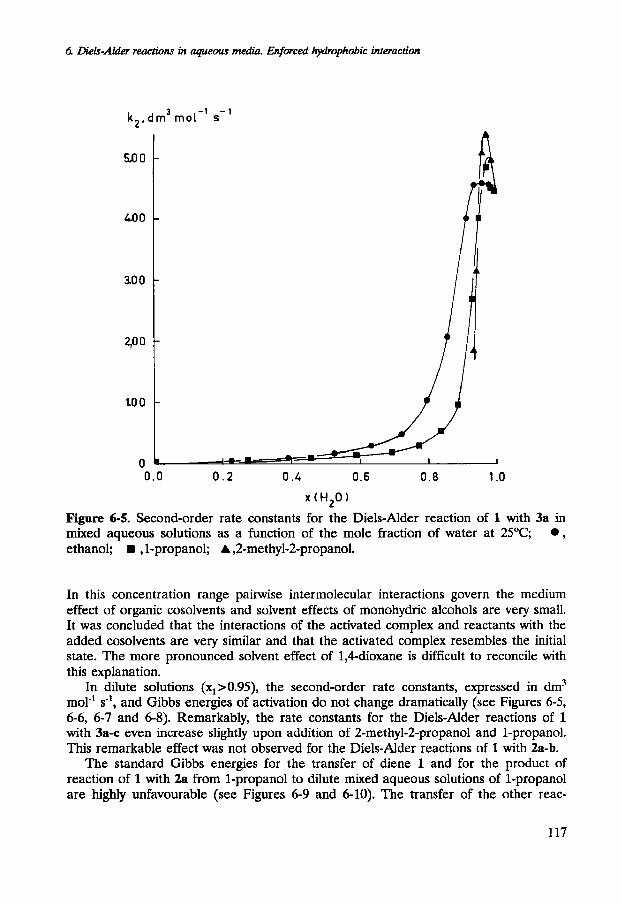
6 DieIs-Alder reactions in aqueous media. Enforced hydrophobic interaction
3 - 1 - 1 k2 ,dm m o l s
Figure 6-5. Second-order rate constants for the Diels-Alder reaction of 1 with 3a in mixed aqueous solutions as a function of the mole fraction of water at 25°C; , ethanol; , 1-propanol; A ,2-methyl-2-propanol.
In this concentration range pairwise intermolecular interactions govern the medium effect of organic cosolvents and solvent effects of monohydric alcohols are very small. It was concluded that the interactions of the activated complex and reactants with the added cosolvents are very similar and that the activated complex resembles the initial state. The more pronounced solvent effect of 1,4-dioxane is difficult to reconcile with this explanation.
In dilute solutions (x1>0.95), the second-order rate constants, expressed in dm3 mol-' s-', and Gibbs energies of activation do not change dramatically (see Figures 6-5, 6-6, 6-7 and 6-8). Remarkably, the rate constants for the Diels-Alder reactions of 1 with 3s-c even increase slightly upon addition of 2-methyl-Zpropanol and 1-propanol. This remarkable effect was not observed for the Diels-Alder reactions of 1 with 2a-b.
The standard Gibbs energies for the transfer of diene 1 and for the product of reaction of 1 with 2a from 1-propanol to dilute mixed aqueous solutions of 1-propanol are highly unfavourable (see Figures 6-9 and 6-10). The transfer of the other reac-

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
tants, 2a and 2b is significantly less unfavourable. Remarkably, the standard chemical potentials of the activated complexes of the reactions of 1 with both 2a and 2b in 1- propanol are almost identical to the standard chemical potentials in water and dilute aqueous solutions of 1-propanol. The standard chemical potentials of reactants as well as activated complexes do not change dramatically as a function of the mole fraction of cosolvent in dilute aqueous solution.
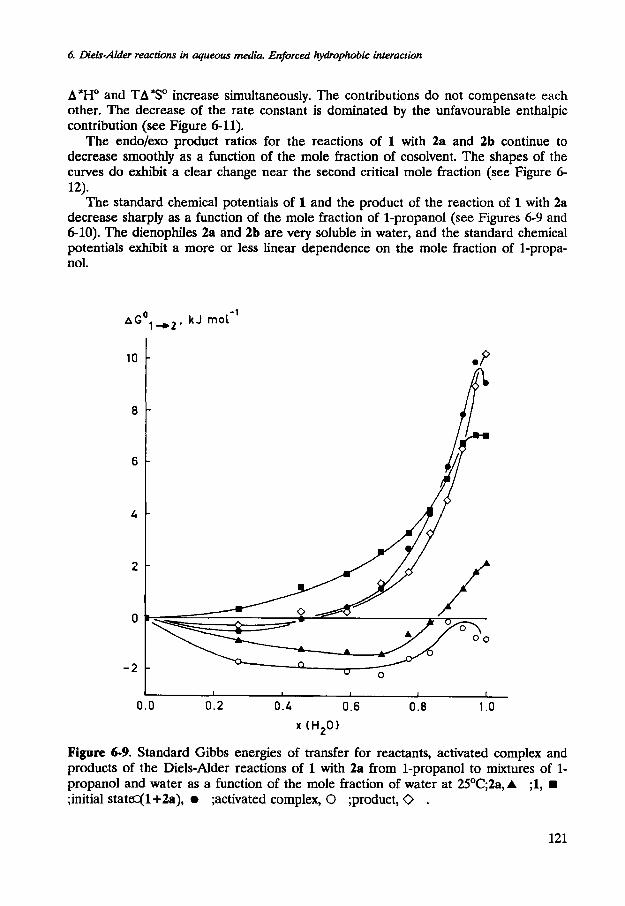
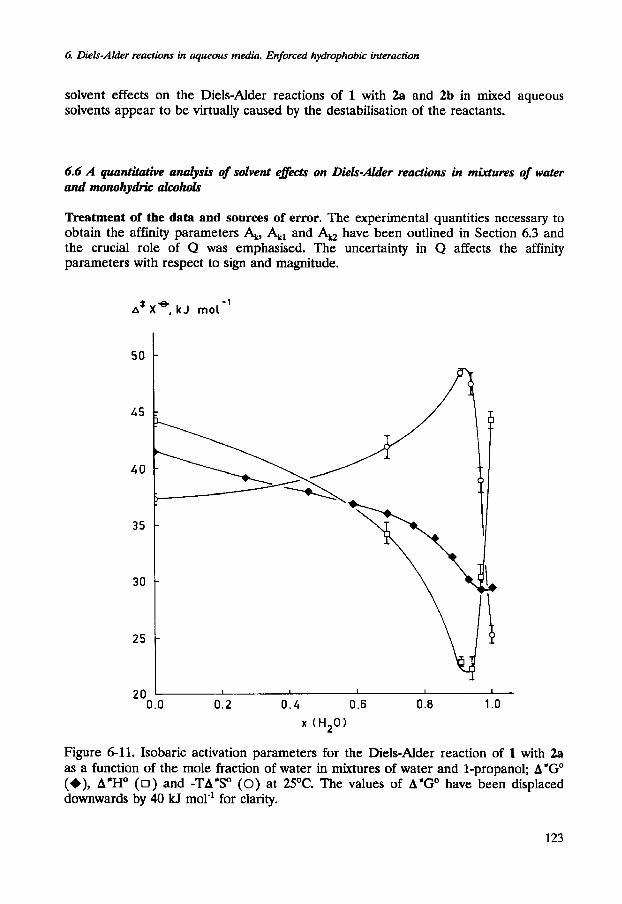
The dependence of A*HO and TA*SO (at 298.15 K) for the reaction of 1 with 3b on the mole fraction of 1-propanol in dilute aqueous solutions is dramatic (see Figure 6- 11). The changes of A'HO and TA*SO across the mole fraction range of 0<x2<0.07 are spectacular and amount to 24 kJ mol-'. The enthalpy of activation decreases sharply upon addition of 1-propanol. Simultaneously, the entropy of activation exhibits an equally dramatic decrease. The effects almost fully compensate each other. In the absence of an organic cosolvent, the rate acceleration of the Diels-Alder reaction of 1 with 3a is almost completely due to a more favourable entropy of activation. It is interesting to note that a similar acceleration can be achieved in a mixed aqueous solution of 1-propanol, x2=0.07, which is due entirely to a more favourable enthalpy of activation.
Figure 6-6. Gibbs energy of activation for the Diels-Alder reaction of 1 with 2a in mixed aqueous containing methanol, A ;ethanol, ;l-propanol, o ;2-methyl-2- propanol, 0 as a function of the mole fraction of water at 2S°C.
4 Diek-Alder reactions in aqueous media. Enforced hydrophobic interaction
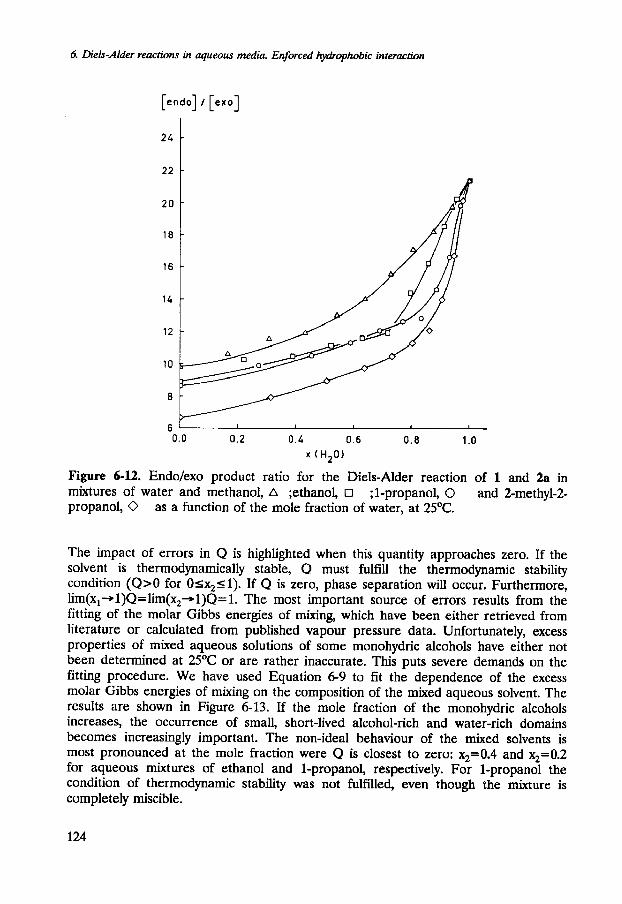
The dependence of the endolexo ratio of the products on the molality of cosol- vents for the reaction of 1 with 2a and 2b in dilute mixed aqueous solvents is similar (see Figure 6-12 for the endolexo product-ratio of the reaction of 1 with 2a). The endo product, which is already favoured in organic solvents, is almost exclusively formed in aqueous solutions. The endolexo ratio decreases smoothly with the addition of monohydric alcohols.
In Table 6-1, second-order rate constants are given for the Diels-Alder reaction of 1 with 2a in the presence of a cationic (cetyItrimethylammonium bromide) and an anionic (sodium dodecylsulphate) surfactant. The critical micellar concentrations are 0.95 mM and 8.3 mM, respectively. In addition, the second-order rate constants are given for the Diels-Alder reaction of 1 with 2a in the presence of N-cyclohexyl yrroli- done. The latter cosolvent forms microdomains in dilute aqueous solutions%'. All additives induce a sharp decrease of the rate constant.
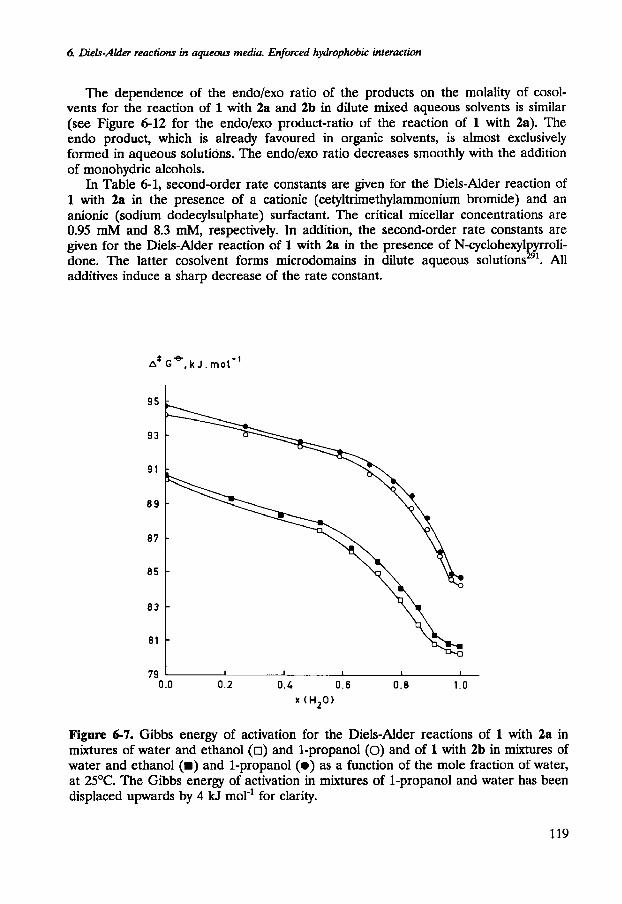
Figure 6-7. Gibbs energy of activation for the Diels-Alder reactions of 1 with 2a in mixtures of water and ethanol (0) and 1-propanol (0) and of 1 with 2b in mixtures of water and ethanol (m) and 1-propanol (e) as a function of the mole fraction of water, at 2S°C. The Gibbs energy of activation in mixtures of 1-propanol and water has been displaced upwards by 4 kJ mol-' for clarity.
6. Dieb-Alder reactions in aqueous media. Enforced hydrophobic interactbn
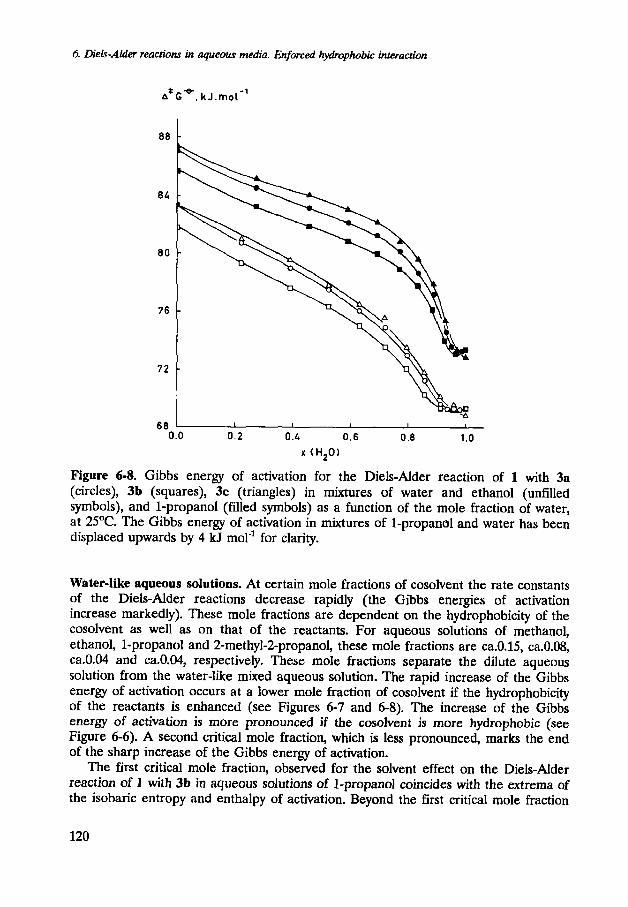
Figure 6-8. Gibbs energy of activation for the Diels-Alder reaction of 1 with 3a (circles), 3b (squares), 3c (triangles) in mixtures of water and ethanol (unfilled symbols), and 1-propanol (filled symbols) as a function of the mole fraction of water, at 2S°C. The Gibbs energy of activation in mixtures of 1-propano1 and water has been displaced upwards by 4 kJ mol-' for clarity.
Water-like aqueous solutions. At certain mole fractions of cosolvent the rate constants of the Diels-Alder reactions decrease rapidly (the Gibbs energies of activation increase markedly). These mole fractions are dependent on the hydrophobicity of the cosolvent as well as on that of the reactants. For aqueous solutions of methanol, ethanol, 1-propanol and 2-methyl-2-propanol, these mole fractions are ca.0.15, ca.0.08, ca.0.04 and ca.0.04, respectively. These mole fractions separate the dilute aqueous solution from the water-like mixed aqueous solution. The rapid increase of the Gibbs energy of activation occurs at a lower mole fraction of cosolvent if the hydrophobicity of the reactants is enhanced (see Figures 6-7 and 6-8). The increase of the Gibbs energy of activation is more pronounced if the cosolvent is more hydrophobic (see Figure 6-6). A second critical mole fraction, which is less pronounced, marks the end of the sharp increase of the Gibbs energy of activation.
The first critical mole fraction, observed for the solvent effect on the Diels-Alder reaction of 1 with 3b in aqueous solutions of 1-propanol coincides with the extrema of the isobaric entropy and enthalpy of activation. Beyond the first critical mole fraction
6 DieLF-Alder reactions in aqueous media. Enforced hydrophobic interaction
A*HO and TA*SO increase simultaneously. The contributions do not compensate each other. The decrease of the rate constant is dominated by the unfavourable enthalpic contribution (see Figure 6-11).
The endolexo product ratios for the reactions of 1 with 2a and 2b continue to decrease smoothly as a function of the mole fraction of cosolvent. The shapes of the curves do exhibit a clear change near the second critical mole fraction (see Figure 6- 12).
The standard chemical potentials of 1 and the product of the reaction of 1 with 2a decrease sharply as a function of the mole fraction of 1-propanol (see Figures 6-9 and 6-10). The dienophiles 2a and 2b are very soluble in water, and the standard chemical potentials exhibit a more or less linear dependence on the mole fraction of l-propa- nol.
Figure 6-9. Standard Gibbs energies of transfer for reactants, activated complex and products of the Diels-Alder reactions of 1 with 2a from 1-propanol to mixtures of 1- propanol and water as a function of the mole fraction of water at 25OC;2a, A ;I, rn ;initial stata=(1+2a), a ;activated complex, 0 ;product, 0 .

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
0 .O 0.2 0 . 4 0.6 0 .8 1 .O
x ( HZO)
Figure 6-10. Standard Gibbs energies of transfer for reactants and activated complex for the Diels-Alder reaction of 1 with 2b from 1-propanol to aqueous mixtures of 1- propanol as a function of the mole fraction of water;2b, A 1 ;initial state (1+2b), ;activated complex, 0 .
Interestingly, the standard chemical potential of 2a is even lower in a mixed aqueous solution of 1-propanol than in the pure solvents. Most striking, however, is the fact that the standard chemical potentials of the activated complexes for the reactions of 1 with 2a and 2b are hardly affected by addition of 1-propanol.
Alcohol-like mixed aqueous solutions. Beyond the second critical mole fraction, kinetic parameters and stereospecificities of Diels-Alder reactions and standard chemical potentials of reactants, products and activated complexes show only a modest dependence on the solvent composition.
Two observations are particularly interesting. First, the rate acceleration of the Diels-Alder reaction of 1 with 3b in mixed aqueous solutions is, except in dilute aqueous solutions, governed by a more favourable enthalpy of activation. Second, the
6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
solvent effects on the Diels-Alder reactions of 1 with 2a and 2b in mixed aqueous solvents appear to be virtually caused by the destabilisation of the reactants.
6.6 A quallritative analysis of solvent effects on Dieh-Alder reactions in mixtures of water and monohydric nlcohoIs
Treatment of the data and sources of error. The experimental quantities necessary to obtain the affinity parameters 4, and & have been outlined in Section 6.3 and the crucial role of Q was emphasised. The uncertainty in Q affects the affinity parameters with respect to sign and magnitude.
Figure 6-11. Isobaric activation parameters for the Diels-Alder reaction of 1 with 2a as a function of the mole fraction of water in mixtures of water and 1-propanol; A*GO (+), A*HO ( 0 ) and -TA'SO (0) at 25OC. The values of A'GO have been displaced downwards by 40 kJ mol-' for clarity.
6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
[endo] / [exo]
Figure 6-12. Endo/exo product ratio for the Diels-Alder reaction of 1 and 2a in mixtures of water and methanol, A ;ethanol, ;I-propanol, 0 and 2-methyl-2- propanol, 0 as a function of the mole fraction of water, at 25OC.
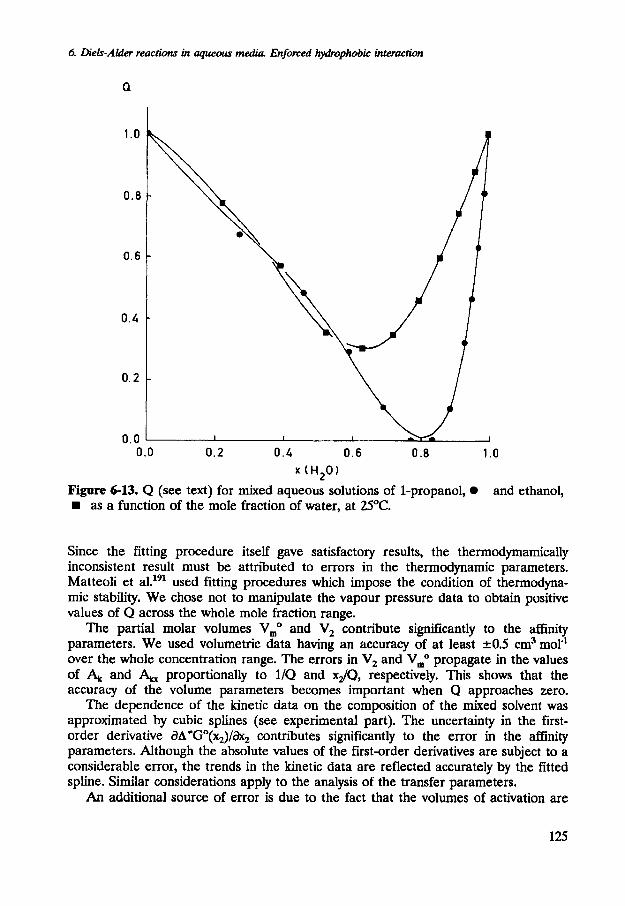
The impact of errors in Q is highlighted when this quantity approaches zero. If the solvent is thermodynamically stable, Q must fulfill the thermodynamic stability condition (Q>O for 0 1 ~ ~ 1 1 ) . If Q is zero, phase separation will occur. Furthermore, lim(x,-+1)Q=lim(x2~l)Q=l. The most important source of errors results from the fitting of the molar Gibbs energies of mixing, which have been either retrieved from literature or calculated from published vapour pressure data. Unfortunately, excess properties of mixed aqueous solutions of some monohydric alcohols have either not been determined at 25OC or are rather inaccurate. This puts severe demands on the fitting procedure. We have used Equation 6-9 to fit the dependence of the excess molar Gibbs energies of mixing on the composition of the mixed aqueous solvent. The results are shown in Figure 6-13. If the mole fraction of the monohydric aIcohols increases, the occurrence of small, short-lived alcohol-rich and water-rich domains becomes increasingly important. The non-ideal behaviour of the mixed solvents is most pronounced at the mole fraction were Q is closest to zero: x2=0.4 and x2=0.2 for aqueous mixtures of ethanol and 1-propanol, respectively. For 1-propanol the condition of thermodynamic stability was not fulfilled, even though the mixture is completely miscible.
4 DieIs-Alder reactions in aqueous media Enfmed hydrophobic interaction
. .
0.0 0.2 0.4 0.6 0.8 1 .O
x (H,O)
Figure 6-13. Q (see text) for mixed aqueous solutions of 1-propanol, and as a function of the mole fraction of water, at 25°C.
ethanol,
Since the fitting procedure itself gave satisfactory results, the thermodyrnamically inconsistent result must be attributed to errors in the thermodynamic parameters. Matteoli et a1.191 used fitting procedures which impose the condition of thermodyna- mic stability. We chose not to manipulate the vapour pressure data to obtain positive values of Q across the whole mole fraction range.
The partial molar volumes VmO and V, contribute significantly to the affinity parameters. We used volumetric data having an accuracy of at least k0.5 cm3 morl over the whole concentration range. The errors in V, and VmO propagate in the values of A, and A, proportionally to 1/Q and xJQ, respectively. This shows that the accuracy of the volume parameters becomes important when Q approaches zero.
The dependence of the kinetic data on the composition of the mixed solvent was approximated by cubic splines (see experimental part). The uncertainty in the first- order derivative aA*G0(x2)/ax, contributes significantly to the error in the affinity parameters. Although the absolute values of the first-order derivatives are subject to a considerable error, the trends in the kinetic data are reflected accurately by the fitted spline. Similar considerations apply to the analysis of the transfer parameters.
An additional source of error is due to the fact that the volumes of activation are

4 Die&-Alder reactions in aqueous media. Enforced hyirophobic interaction
not known for the reactions studied in this chapter. Volumes of activation for Diels- Alder reactions in water have, to our knowledge, not been determined. The error in the estimated volumes of activation may lead to a considerable error in the affinity parameters only when 4, and A, are small. In addition, the volume of activation of organic reactions in aqueous solutions depends on the composition of the mediumu1. Generally, the effect of these errors is negligibly small if the affinity parameters are large.
Results and discussion. The kinetic solvent effects are understood in terms of preferential solvation of both the reactants and the activated complex by one or the other of the components of the solvent mixture. The term "preferential solvation" means that the composition of the cosphere of the reactant and activated complex differs from that of the bulk solvent. The values of 4,, A, and 4 are determined by Kirkwood-Buff integral functions GIs and G, which are determined by the composi- tion of the solvent in close proximity of the solute S. In Figure 6-14, values of 4, Akl and k, are plotted as a function of the mole fraction of water for the Diels-Alder reactions of 1 with 2a and 2b in ethanol-water mixtures. The solvent dependences of the affinity parameters for the Diels-Alder reaction of 1 with 3b in mixed aqueous solutions of ethanol, are shown in Figure 6-15. For the cycloadditions of 1 with 3a and 3c, the solvent dependences of Ak, 4. and Ala are very similar. Analysis of solvent effects on the rate constant of Diels-Alder reactions in aqueous solutions of 1- propanol is seriously hampered by the fact that Q becomes zero near x2=0.2. This thermodynamic inconsistency leads to asymptotic results for the affinity parameters (&-.a, or -00 for x2+0.2). An example is given in Figure 6-16, where the affinity parameters are plotted for the Diels-Alder reaction of 1 with 2a in aqueous solutions of ethanol and 1-propanol.
In all plots, 4 increases dramatically with increasing x2, which points to a marked difference between the composition of the solvation shells and the bulk solvent. It is difficult to interpret the solvent dependence of 4. In particular for a bimolecular process 4 is determined by a series of integral functions. Further insight into the factors, affecting the solvent dependence of the rate constant on x2 is provided by 4, and &. Recalling that and A, are given by [GI,-GI,-GI,] and [G,,-G,-G,], respectively, where A and B are the diene and the dienophile, the solvent dependence of 4 can be interpreted in terms of a decreasing preference of the reactants for the cosolvent upon forming the activated complex.
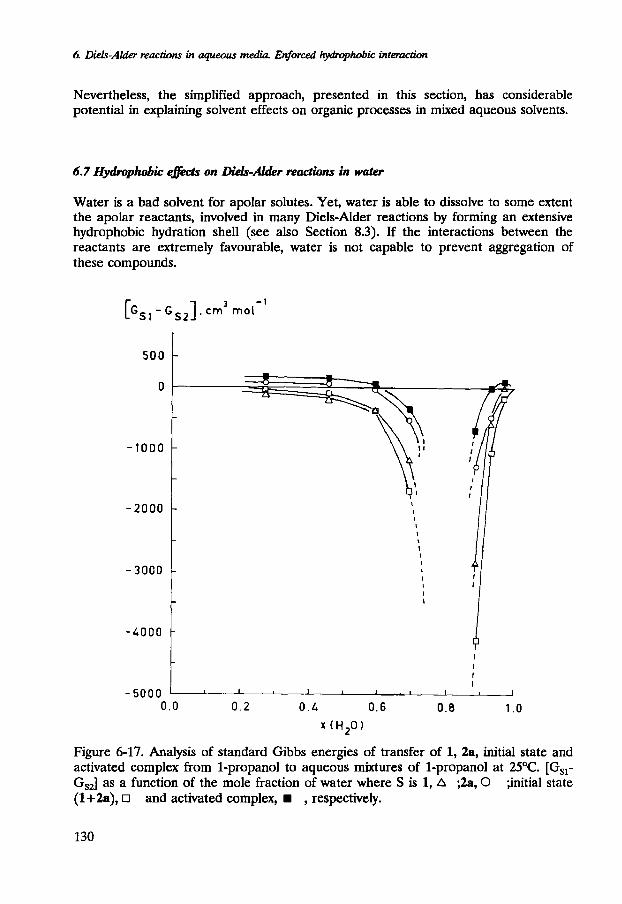
More information was obtained by analysing the standard chemical potentials of 1, 2a, 2b and the corresponding activated complexes in aqueous mixtures of 1-propanol. In Figure 6-17, the dependence is analysed of the standard chemical potentials of (i) 1, (ii) 2a, (iii) the initial state (1+2a) and (iv) the corresponding activated complex on the composition of the aqueous solution of 1-propanol by plotting [GIs-G,] as a function of the mole fraction of water. Unfortunately, the standard chemical potentials could not be analysed near x2=0.2. Clearly, however, reactants as well as activated complex are preferentially solvated by 1-propanol when the mixed aqueous solvent approaches x2=0.2.
An important point to note is that the magnitude of the Kirkwood-Buff integral function, G,, is strongly determined by the tendency of the cosolvents to aggregate. p2G, (p2 is the number density of the cosolvent) represents the average excess (or

6. Diels-Alder reactions in aqueous media Enforced hydrophobic interaction
deficiency) number of cosolvent molecules in the whole space around molecule S with respect to the bulk. Therefore, G, does not only reflect the presence of the cosolvent in the first solvation shell but in the whole cosphere. When the mixed solvent is characterised by the presence of short-lived alcohol-rich and water-rich domains, the composition of the cosphere of the apolar solute is largely determined by clustering of the organic cosolvent. Hence, if Q of a mixed aqueous solvent approaches zero, and the solute S has a slight preference for an apolar environment, [GI,-G,] becomes strongly negative (see Figure 6-17). Nevertheless, the solute is not solely solvated by the organic cosolvent and still experiences the presence of water.
The activated complexes of the Diels-Alder reactions of 1 with 2a and 2b exhibit chameleon-behaviour and are indifferent to the composition of the mixed aqueous solvent (see Figure 6-9 and 6-10).
Figure 6-14. Affinity parameters 4, 4, and A, for the Diels-Alder reactions of 1 with 2a, and of 1 with 2b, 0 in mixed aqueous solutions of ethanol as a function of the mole fraction of water, at 25OC.

6 DieLr-Alder reactions in aqueous media. Enfotced hydrophobic interaction
Figure 6-15. Affinity parameters 4 , 0 and A, for the Diels-Alder reaction of 1 with 3b in mixed aqueous solutions of ethanol at 25°C.
Although the activated complex is preferentially solvated by 1-propanol in the water- like aqueous solution of 1-propanol its standard chemical potential is hardly affected (see Figure 6-16). Consequently, the solvent dependence of [GI,-G2,] is much smaller than that of [Gl~-GZIs]. Hence, the increase in the Gibbs energy of activation for the Diels-Alder reaction of 1 with 2a in the water-like aqueous solutions of 1-propanol is determined by preferential solvation of the reactants by the organic cosolvent.
Interestingly, the dependences of the affinity parameters 4, 4, and k, on the composition of the aqueous solution of ethanol show extrema which are less pronoun- ced if more hydrophobic dienophiles are involved in the Diels-Alder reaction (see Figure 6-14). This can be explained by the fact that the affinity parameters are determined by the quotient [i3A*G0(xz)l&z]lQ. Preferential solvation of the reactants can lead to a pronounced increase in the Gibbs energy of activation before Q has reached its lowest value. Subsequently, the affinity of the organic cosolvents for the reactants is more pronounced in dilute aqueous solutions if the reactants are more hydrophobic (see Figure 6-14). It is not inconceivable that the apolar reactants even induce the formation of microdomains.

6. Dieh-Alder reactions in aqueous media. Enforced hydrophobic interaction
Figure 6-16. Affinity parameters 4 (closed symbols) and A, (open symbols) for the Diels-Alder reaction of 1 with 2a in mixed aqueous solutions, containing ethanol, and 1-propanol W as a function of the mole fraction of water, at 25OC.
Unfortunately, the applied method for quantitative analysis of solvent effects on Diels- Alder reactions is rather inaccurate in dilute aqueous solutions.
The most serious problem in extracting the affinity parameters is that one needs several sets of thermodynamic and kinetic data to obtain these parameters. The accuracy of the analysis can be significantly improved if Kirkwood-Buff integrals could be determined directly from an experiment. A considerable improvement might be provided by a method, reported recently, to directly measure the property Q332. An interesting development is reported by Bot et who advance methods to determi- ne Kirkwood-Buff integrals on the basis of Rayleigh-Brillouin light scattering data.
The cubic spline approximation of the dependence of the Gibbs energy of activation on the composition of the medium can be improved by the determination of more data points. Finally, it is desirable to determine the dependence of the volume of activation of the reaction on the compositon of the mixed aqueous solvent.
4 --Alder reactions in aqueous media. Enfwed hydrophobic interactton
Nevertheless, the simplified approach, presented in this section, has considerable potential in explaining solvent effects on organic processes in mixed aqueous solvents.
6.7 Hydmphobic e w on DieIs-Alder readions in water
Water is a bad solvent for apolar solutes. Yet, water is able to dissolve to some extent the apolar reactants, involved in many Diels-Alder reactions by forming an extensive hydrophobic hydration shell (see also Section 8.3). If the interactions between the reactants are extremely favourable, water is not capable to prevent aggregation of these compounds.
Figure 6-17. Analysis of standard Gibbs energies of transfer of 1, 2a, initial state and activated complex from 1-propanol to aqueous mixtures of 1-propanol at 25OC. [G,,- G,,] as a function of the mole fraction of water where S is 1, A ;2a, 0 ;initial state (1+2a), R and activated complex, w , respectively.

6. Diet%-Alder reacriom in aqueous media. Enfoced hydrophobic interaction
Stacking of organic solutes in aqueous solutions is a well-known phenomenon and has been described extensively for organic dyes and nucleosides. These molecules are characterised by extended .rr-sy~tems~~'. Generally, association of solutes in organic solutions as well as in water is counteracted by an unfavourable entropy factor. In water, hydrophobic hydration shells also counteract aggregation at room temperature (see Section 8.3). The present results (Section 6.5) provided no indication of homotac- tic aggregation or stacking of the reactants at the concentrations used for the kinetic experiments. Heterotactic stacking of the reactants is also highly unlikely. Apparently, the pairwise hydrophobic interactions do not lead to aggregation of the reactants, because the Gibbs energy of this process is unfavourable. This immediately explains the ambiguity of the term "hydrophobic packing" as the explanation of the rate enhancement of Diels-Alder reactions in water. An additional argument against "hydrophobic packing" of the reactants is provided by the fact that intramolecular Diels-Alder reactions are accelerated by a similar large factor in water and aqueous solutions (see Chapter 7). A further point of concern is the fact that pairwise hydrop- hobic interactions do not necessarily lead to a complex that possesses the right geometry to undergo a cycloaddition reaction. Inhibition of the Diels-Alder reaction by hydrophobic interactions, leading to homotactic complexes or heterotactic com- plexes of the wrong geometry for cycloaddition, as suggested by Schneider et a ~ . ~ ~ , is not confirmed by the present results (Section 6.5).
In spite of the fact that water is capable of preventing aggregation of apolar solutes when their concentration is sufficiently low, the apolar solutes do sense each others presence by pairwise and higher-order interactions. Pairwise and higher order hydrophobic interactions between the apolar reacting species might lead to a concen- tration dependence of the rate constant (a similar dependence was observed for the keto-en01 equilibrium quotient of 2,4-pentanedione). The frequency of these hydro- phobic "encounters" is proportional to the concentration of the reactants and the molar Gibbs energy, associated with these interactions. However, the concentrations of the diene and the dienophiles, used for the kinetic experiments, are low. Conse- quently, pairwise interactions and, for certain, higher-order interactions are rare and the concentration dependence of the rate constant can be neglected.
A bimolecular reaction necessarily involves the formation of a direct, solvent- unseparated complex of diene and dienophile during the activation process. When two relatively apolar reactants are involved in this process and the medium is highly aqueous, we might speak of an "enforced hydrophobic interaction". The term "enfor- ced" is introduced here (i) to emphasise the fact that the complexation of the diene and the dienophile is not necessarily favourable in terms of Gibbs energy and (ii) to reflect the forced geometry of the association process. In order to account for the exceptionally large rate acceleration of Diels-Alder reactions in aqueous solutions, the "enforced hydrophobic interaction" of diene and dienophile has to be more favourable (or more appropriately, less unfavourable) in water than in organic solvents where we might speak of an "enforced solvophobic interaction".
The Gibbs energy, associated with "enforced hydrophobic (solvophobic) interacti- ons'' is given by1!

6 Diels-Alder reactions in aqueous me& Enforced hydrophobic interaction
The gradient of AG(R) provides the average force between the solutes S. The first term represents the direct diene-dienophile pair potential, which is the work required in the process of bringing diene and dienophile from an infinite separation to a distance R in vacuum. The second term takes account of the solvent effect on the same associative process. The first term is similar in every solvent. The exceptionally large acceleration of Diels-Alder reactions in water must therefore be fully attributed to the second term. This solvent effect has to be explained in terms of the transfer of the reactants and activated complex from an organic solvent to water. The standard Gibbs energy of transfer of 1, 2a and 2b from 1-propanol to water is highly unfavou- rable (Section 6.5, Figures 6-9 and 6-10). The standard chemical potentials of the activated complexes are almost independent of the composition of the mixed aqueous solvent and are similar in monohydric alcohols and in water. Apparently, the "enfor- ced hydrophobic interaction", involving (i) the enforced pairwise interaction between the reactants (the complex might, tentatively, be identified as the CT-complex), (ii) the volume contraction during the bond formation step and (iii) an increase in polarisability, dramatically reduces the hydrophobic surface of the reactants. The chameleon-like behaviour of the activated complex in mixed aqueous solvents (see Figures 6-9 and 6-10) suggests that water induces a more polar activated complex that can be optimally accomodated in the aqueous medium. Recently, Jorgensen et a1.352 reported computer simulations of the Diels-Alder reaction of 1 with 2a. This study supports the idea that enhanced polarisation of the activated complex in aqueous solution contributes strongly to the acceleration of aqueous Diels-Alder reactions.
This process has important implications for the isobaric activation parameters of Diels-Alder reactions in water. The "enforced hydrophobic interaction" of the reactants is accompanied by destructive overlap of the hydrophobic hydration shells of the reactants. This phenomenon clearly distinguishes water from organic solvents and leads, at room temperature, to a favourable entropic contribution, which is almost fully canceled by an equally unfavourable enthalpy term (see Section 8.3). Conse- quently, destructive overlap of hydrophobic hydration shells causes both TA'SO and A'HO to be equally increased in water with respect to organic solvents. The solvent effect, associated with the desctructive overlap of hydrophobic hydration shells is negligibly small. The actual solvent effect is due to the fact that the London dispersion interactions which stabilise the initial state (both reactants) in organic solvents, are absent in water. During the activation process the hydrophobic surface of the reactants is dramatically reduced. In water, the interactions between water and the activated complex might be enhanced by induced polarity of the activated complex. Consequently, interactions of the activated complex with water and organic solvents are similar. Hence, the A'HO is more favourable (or, more correctly, less unfavoura- ble) in water than in organic solvents. In summary, the enthalpy of activation of Diels- Alder reactions in aqueous solutions is affected by destructive overlap of the hydro- phobic hydration shells of the reactants as well as by the lack of interactions between the reactants and water. Both contributions are counteractive. Whether the enthalpies of activation of Diels-Alder reactions in organic solvents are more or less favourable than in water depends on the magnitude of both contributions. Our results (see Table 6-1) suggest that the contribution of the unfavourable enthalpy term, due to the destruction of hydrophobic hydration shells, is surpassed by the favourable enthalpy contribution. The entropy of activation of Diels-Alder reactions in aqueous solutions

6 Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
differs from that in organic solvents mainly due to overlap of hydrophobic hydration shells and is therefore more favourable in water than in organic solvents (see Table 6- 1). The increased preference for the endo-product of Diels-Alder reactions of 1 with 2a and 2b can be explained by the fact that formation of the endo-product is charac- terised by a more negative volume of activation and a concomittantly more pronoun- ced reduction of the hydrophobic surface area.
Recently, Diederich et a1.= showed that complexation of organic compounds in large, apolar guest-molecules was more effective in water than in organic solvents. As for the isobaric activation parameters of Diels-Alder reactions, the enthalpy as well as the entropy of complexation were found to be more favourable in water than in organic solvents. In this case, the enthalpy term was shown to dominate the overall solvent effect. Apparently, the favourable enthalpy contribution is so large that it not only outweighs the favourable entropic contribution inspite of the competing unfavou- rable enthalpic term. This is not inconceivable considering the large decrease of solute-solvent interactions.
Interestingly, a superficial analysis of the isobaric activation parameters suggests a major role of hydrophobic hydration in the exceptional acceleration of Diels-Alder reactions in aqueous solutions. A more sophisticated analysis reveals that the molecu- lar origin of the exceptional rate acceleration is provided by the enthalpically less favourable interactions between the reactants and water, which are relieved during the activation process. The large rate constants for Diels-Alder reactions in aqueous solutions are an example of "hydrophobic acceleration" (see chapter 5). Section 8.3 will be devoted to a detailed discussion of hydrophobic effects and hydrophobic interactions in particular.
6.8 Hydrophobic effects on Diels-Alder reactions in mired aqueous solutions
Solvent effects on the rate constants of Diels-Alder reactions in mixed aqueous solvents reflect the interactions of cosolvents with the diene, the dienophile and the activated complex, respectively. In dilute aqueous solvents (Section 6.5), pairwise hydrophobic interactions govern the interactions of the cosolvent with the reacting species and the activated complex. The extreme dependence of the entropy and enthalpy of activation for the Diels-Alder reaction of 1 with 3b on the molality of 1- propanol (Figure 6-11) is governed by two effects. First, at the so-called "magic mole fraction" of cosolvent, which depends on the size and hydrophobicity of the cosolvent, all water molecules are part of an extended hydrophobic hydration shell (see Section 4.2). Beyond this mole fraction the overlap of the hydrophobic hydration shells of the reactants, accompanying the activation process, cannot lead to release of "hydrophobic hydration water" to the bulk solution. Consequently, A'HO and TA*SO will be dramati- cally decreased at the magic mole fraction. A second contribution is due to the fact that the hydrophobic surface of the reactants decreases significantly during the activation process. Pairwise interactions between the organic cosolvent and the reactants involve destructive overlap of the hydrophobic hydration shells. Since the activated complex is less hydrophobic, the pairwise interactions also result in a gradual decrease of A*HO and TA*SO (see also Section 4.3).

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
We anticipated a reduction of the Gibbs energy of activation due to these pairwise interactions. However, the present results show that in some cases Diels-Alder reactions are even accelerated due to the addition of organic cosolvents (see, e.g. Figure 6-5). The interpretation of this effect must be that addition of 1-propanol also has a competing favourable effect on the "enforced hydrophobic interaction". Indeed, ~ e n - ~ a i m ~ ~ showed that pairwise hydrophobic interactions are more favourable in the presence of small concentrations of an organic cosolvent. Consequently, the overall solvent effect on Diels-Alder reactions in dilute aqueous solutions will be small and it depends on the magnitude of the competing contributions whether addition of organic cosolvent will increase or decrease the rate constant.
The onset of preferential solvation of the reactants in water-like mixed aqueous solvents (Section 6.6) cancels the driving-force for the "hydrophobic acceleration" of Diels-Alder reactions in dilute aqueous solvents and the Gibbs energy of activation increases dramatically. A'HO and TA'SO gradually approach values, found in the pure alcohol. The complete disappearance of aqueous phases inaugurates the alcohol-like mixed aqueous solvents.
Aqueous solutions of surfactants and N-cyclohexylpyrrolidone are, already at very low concentrations, characterised by the presence of micelles and small clusters, respectively. Although the diene and the dienophile are preferentially solvated in these phases and the local concentration of the reactants is considerably enhanced, the reduced rate constant in these apolar environments reverses this concentration effect. Hence, rate constants of Diels-Alder reactions decrease sharply due to addition of surfactants above the CMC or organic cosolvents that tend to aggregate at low concentration.
6.9 Experimental part
Materiat. 1,6Naphthoquinone (3s) and 5-hydroxy-1,4-naphthoquinone (3b) were commercially available (Aldrich) and were recrystallised from methanol; mp 121- 122OC and 162-163OC, respectively (litm 123°C and 163OC, respectively). 5-Methoxy- 1,4,-naphthoquinone (3c) was prepared from 3 4 as reported in the literature299, and was recrystallised from methanol; mp 185-186°C (lit.m mp 187'C). Methyl vinyl ketone (2a, bp 81°C) and ethyl vinyl ketone (2b, bp 102-103°C) (Janssen) were freshly distilled before use. Cyclopentadiene (1) was prepared from its dimer immediately before use.
Dimineralised water was distilled twice in an all-quartz distillation unit. All cosolvents were of the highest purity available.
Kinetic measurements. Pseudo-first-order rate constants were determined by following the change in absorbance at appropriate wavelengths in a quartz UV cell (1 cm) that was placed in a thermostated cell compartment of a Perkin Elmer A2 spectrophoto- meter, equipped with a standard personal computer. For reactions in highly aqueous media with half-lives longer than about ten minutes, the cuvettes were carefully sealed to prevent evaporation of cyclopentadiene. Evaporation of cyclopentadiene may otherwise seriously hamper the kinetic measurements and can lead to dramatic errors

6. Die&-Alder reactions in aqueous media. Enforced hydrophobic interaction
in the determined rate constants, because the evaporation process is also approxi- mately first-order. A recent example of the danger of measuring evaporation of cyclo- pentadiene instead of a rate constant for the Diels-Alder process are the exceptionally high rate constants of Diels-Alder reactions of acrylic acids with cyclopentadiene, reported by Schneider et a ~ . ~ .
Reactions of 1 with 2a and 2b were followed by monitoring the decrease of absorbance of cyclopentadiene at 250-255 nm (dependent on the composition of the reaction mixture), using an excess of the alkyl vinyl ketone. About 5-8 pL of a concentrated stock solution of cyclopentadiene in 1-propanol was added to the reaction medium (3 cm3) containing a known concentration of alkyl vinyl ketone. Typical concentrations, used for the determination of rate constants were 1x10~ mol dms of cyclopentadiene and 3.5x10-~ mol dm" of alkyl vinyl ketone.
Reactions of dienophiles 3a-c with 1 were followed by monitoring the decrease of absorbance of the dienophiles at, respectively, 333, 426 and 400 nm. An excess of 1 was used. Especially in highly aqueous solutions it was appropriate to add 1, dissolved in a very concentrated stock solution in the cosolvent, just before the measurement, followed by addition of 5-8 pL of a stock solution of the dienophile. Stock solutions were made in the cosolvent. After addition of the two reactants, the solution was stirred briefly with an external micro-stirrer. Typical concentrations, used in the kinetic runs were 6-8x10'~ mol dm" of 1 and 5x10" mol dm" of 3a-c.
All reactions were followed for at least 5 half-lives and were perfectly first-order. A total of 200-600 data points were collected and pseudo-first-order rate constants were calculated by fitting the data points to an exponential function. Rate constants were reproducible to within 1%, except for those Diels-Alder reactions of cyclopent- adiene with naphthoquinones in highly aqueous solutions, which were reproducible to within 3%. Isobaric activation parameters were calculated with the Eyring equation, for kinetic data obtained at at least six different temperatures. To prevent evaporation of 1, rate constants were measured in the temperature range 10-35°C. Plots of ln[k/T] against 1E were perfectly linear, and A*HO was determined by least-squares regres- sion. Finally, pseudo-first-order rate constants of all Diels-Alder reactions were also determined by employing different excess concentrations of 2a, 2b and 1, respectively. Second-order rate constants were reproducible to within 3%.
Product analysis. The Diels-Alder reaction of 1 with 2a and 2b were performed in aqueous solutions on a preparative scale. 0.01 moles of 1 and 0.01 moles of 2a and 2b were dissolved in 1 kg of water and stirred during 15 minutes at room temperature. The aqueous solution was extracted with ether (2x75 ml). After evaporation of the ether, the norbornene products was obtained almost quantitatively. The reaction products were analysed by GC (Hewlett-Packard 5890 instrument, equipped with a 15 m wide bore HP1 fused silica column), GC-MS (Ribermag R-10-10C) and NMR (Varian VXR-300 (300 MHz) instrument). The NMR spectra matched spectra, that were reported in the literature. The endolexo ratios of the products were determined by NMR and GC, following procedures, reported by Segushi et a~.~". The retention times of the exo and endo products of the reaction of 1 with 2b were 4.6 min. and 5.0 min., respectively, at 150°C. The retention times of the exo and endo products of the reaction of 1 with 2a were 4.5 min. and 5.0 rnin., respectively, at 120°C. The endolexo product ratios were also determined by NMR. The characteristic signals for the

6. Diels-Alder reactions in aqueous media. Enforced hydrophobic interaction
products of the reaction of 1 with 2a are: 'H-NMR (CDCI,) 2.00 ppm (CH, endo) and 2.10 ppm (CH,, exo), and for the reaction of 1 with 2b: 'H-NMR (CDCl,) 1.01 ppm (CH, endo) and 1.06 (CH, exo). The results obtained by GC and by NMR were identical. No side reactions were observed.
Transfer parameters. Vapour pressures of 1, 2a, 2b and the product of the reaction of 1 with 2a in mixed aqueous solutions, containing 1-propanol, were determined by analysing the composition of the vapour above the solution. The concentration of the compound in the vapour phase is linearly related to the vapour pressure. The composition was determined by GLC (Hewlett-Packard 5890 instrument, equipped with a 15 m wide bore HP1 fused silica column). Solutions were prepared by weight. To ensure that there was a large volume of vapour above the solution, special wide flasks, sealed with self-sealing septum caps were used.
(i) Determination of homotactic association of the dime and the dienophiles. The flasks, containing one compound in increasing concentrations, were simultaneously placed in a thermostated water bath at 25 -cO.l°C and allowed to equilibrate for an hour. To prevent condensation of the solvent (particularly of 1-propanol) on the inside of the septum cap, the flasks were submersed as far as possible in the water bath and the water was covered with an insulating sheet. Atmospheric pressure inside each flask was maintained by piercing a very narrow hypodermic needle through the septum cap. Vapour samples were taken using a sealed gas-syringe, that was kept at 2S°C. After withdrawal of the vapour, the samples were expanded quickly to prevent condensa- tion and immediately analysed by chromatography. The vapour above each solution was chromatographed three times. After each run, the syringe was cleaned carefully. The peak area was determined by integration. Peak areas were reproducible to within 3%. In a typical experiment, 200 pL of vapour was chromatographed. The peak area was plotted as a function of the molality of the compounds in water and in mixed aqueous solutions, containing 10 and 20 mole percent of ethanol. The applied concentration of the compounds in solution was selected on the basis of the volatility of the compound. Typical concentration ranges for solutions of 2a, 2b and 1 were lo4- 5x10-~ mol kg". For cyclopentadiene in aqueous solution small deviations from linearity could be observed only at higher concentrations (>0.04 mol kg-'). However, linear plots were obtained in mixed aqueous solvents containing small amounts of ethanol. For the alkyl vinyl ketones no deviations from linearity were observed.
(ii) Determination of the standard Gibbs energies of transfer. Appropriate concentrati- ons were choosen to determine peak areas as a function of the composition of the mixed aqueous solvent. Concentrations were selected as such that (i) the peak areas, obtained for each run throughout the whole composition range, were similar, and (ii) the solutions could be treated as ideal. In this way, the dependence of the GLC-signal on the concentration of the compound did not affect the measurement. In addition, clustering or stacking of the solute did not hamper the analysis. In a typical experi- ment, ten solutions with increasing mole fractions of 1-propanol were analysed simultaneously. The peak areas were determined as described above. The peak areas were divided by the concentration of the solute, defined in mol dm". The data were then used to calculate the transfer parameters, as described in-section 6.3, for the

6 &Is-Alder reactions in aqueous media. Enforced hydrophobic interaction
transfer of the solute from 1-propanol to mixtures of water and 1-propanol.
Quantitative analysis. Excess Gibbs energies of mixing of 1-pro an01 and water were calculated based on data and methods reported by Dawe et a1.k The G~~ data are fitted as a function of temperature and mole fraction. This fitting method was used to estimate the excess Gibbs energies of mixing at 25OC. Excess molar Gibbs energies of mixing of ethanol and water were calculated using the vapour pressure measurements reported by ~ o b s o n ~ ' ~ . The dependence of the excess molar Gibbs energy of mixing on the composition of the medium was fitted by applying Equation 6-7 (see also ref.303). Q was calculated by evaluating the second-order derivative.
Volumetric parameters for the aqueous mixtures of 1-pro an01 and water were calculated by using the density data reported by Dawe et al.' and Benson et al?lO. Volumetric parameters for the aqueous mixtures of ethanol and water were calcula- ted, using the data reported by Benson et al.310.
The volumes of activation for the reactions of cyclopentadiene and alkyl vinyl ketones were estimated, using volumes of activation reported by Segushi et al.311 for the Diels-Alder reaction of cyclopentadiene with acrylic acid:A'Vo= -30 cm3 mol-'. The volumes of activation for the Diels-Alder reaction of cyclopentadiene with the 5- substituted-naphthoquinones were estimated, using volumes of activation reported by LeNoble et ~ I . ~ ~ ' : A * V O = - ~ ~ cm3 mol-'.
The dependence of ln[A*G0(x2)] and $(x,)-pr(x2=1) on x2 was fitted using a spline appro~imation~'~. A spline approximation is based on the generalisation of a function f (x,) by partitioning the mole fraction range O l x , ~ 1 into subintervals with common endpoints, called nodes (O=a,,<al, ........ <a,=l). In each subinterval, the function f(x) is defined by a polynomial. Therefore, instead of approximating f(x) by one single function (as was tried, without satisfactory results), f(x) is approximated by a series of n polynomials. The functions f(x) thus obtained are called splines. Different splines can be defined. We have approximated the dependence of the kinetic data on the composition of mixed solvent by cubic splines. A cubic spline f(x) on the interval 01x2Sl is, by definition, a continuous function which has continuous first-order and second-order derivatives everywhere in that interval and is represented by a series of polynomials that, in each subinterval, do not exceed a degree of three. Generally, the data were accurate enough to be used as nodes, forcing the cubic spline to cross all data points. The occurrence of unrealistic ripples in the fitted spline were avoided by deleting strongly deviating data points. The approximation as well as the calculation of the first-order derivative were performed using a commercially available program.