trombofilias congÉnitas
DESCRIPTION
V Congreso Cubano de Patología Clinica CONAPAC 2004. TROMBOFILIAS CONGÉNITAS. Dr. Ariel Colina Rodríguez. INTRODUCCIÓN. HEMOSTASIA. FACTORES DE RIEGO. DEFICIENCIAS FUNCIONALES O ANTIGÉNICAS.. POLIMORFISMOS GENÉTICOS. ESTUDIOS DE LABORATORIO SELECCIÓN DE PRUEBAS. NIVEL DE EVIDENCIA. - PowerPoint PPT PresentationTRANSCRIPT

TROMBOFILIAS CONGÉNITAS.
Dr. Ariel Colina Rodríguez
V Congreso Cubano de Patología Clinica
CONAPAC 2004

INTRODUCCIÓN.
HEMOSTASIA.
FACTORES DE RIEGO.
DEFICIENCIAS FUNCIONALES O ANTIGÉNICAS..
POLIMORFISMOS GENÉTICOS.
ESTUDIOS DE LABORATORIO
SELECCIÓN DE PRUEBAS. NIVEL DE EVIDENCIA.
RECOMENDACIONES.

Vasoconstricción refleja
Lesión Endotelial
Músculo liso
Membrana Basal
Endotelio
Subendotelio Colágeno
ETAPA 1: VASOCONSTRICCIÓN

4Lesión Endotelial
Músculo liso
Membrana Basal
Endotelio
Subendotelio
ETAPA 2. HEMOSTASIA PRIMARIA
1
F vW
Colágeno
22 3 3
2. Cambio de Forma
3
3 Liberación
1. Adhesión
4. Reclutamiento

Músculo liso
Membrana Basal
Endotelio
Subendotelio
ETAPA 3. HEMOSTASIA SECUNDARIA
F vW
Colágeno
2
Factor TisularExpresión de Fosfoíípidos
TROMBINA FIBRINA

Músculo liso
Membrana Basal
Endotelio
Subendotelio
ETAPA 4: TROMBO Y MECANISMOS ANTITROMBÓTICOS
2
TROMBINA FIBRINA
1. Liberación del Activador Tisular del plasminogéno
1
2
2. Expresión de Trombomodulina y activación de la proteína C.
Plasmina
Degradación del Coágulo de fibrina
PLASMINÓGENO
3. Activación de la coagulación3

Péptido señal
Dominio GLA
Domino A
Dominio EGF
Región de activación
Dominio catalítico
Dominio Kringle
Domino de secuencias repetitivas
Propéptido
Dominio C
Dominio B
Factor VII
Proenzimas
Factor IX
Factor X
Protrombina
Factor XI
Procofactores
Factor Tisular
Factor VIIIFactor V
Proteína S
Proteína C
Inhibidores

Contacto
HMWKXII XII
aKAL
XI XIa
IX IXaPL, Ca++
VIIIa + Ca++
XXa
Va
Protrombina Trombina
Fibrinógeno Fibrina
Factor VII
Ca++Factor
tisular
Factor VIIa
Lesión tisular
Ca++
Vía Extrínseca Vía Intrínseca
FOSFOLÍPIDOS
Contacto
HMWKXII XII
aKAL
XI XIa
IX IXaPL, Ca++
VIIIa + Ca++
XXa
Va
Protrombina Trombina
Fibrinógeno Fibrina
Factor VII
Ca++Factor
tisular
Factor VIIa
Lesión tisular
Ca++
Vía Extrínseca Vía Intrínseca
FOSFOLÍPIDOS
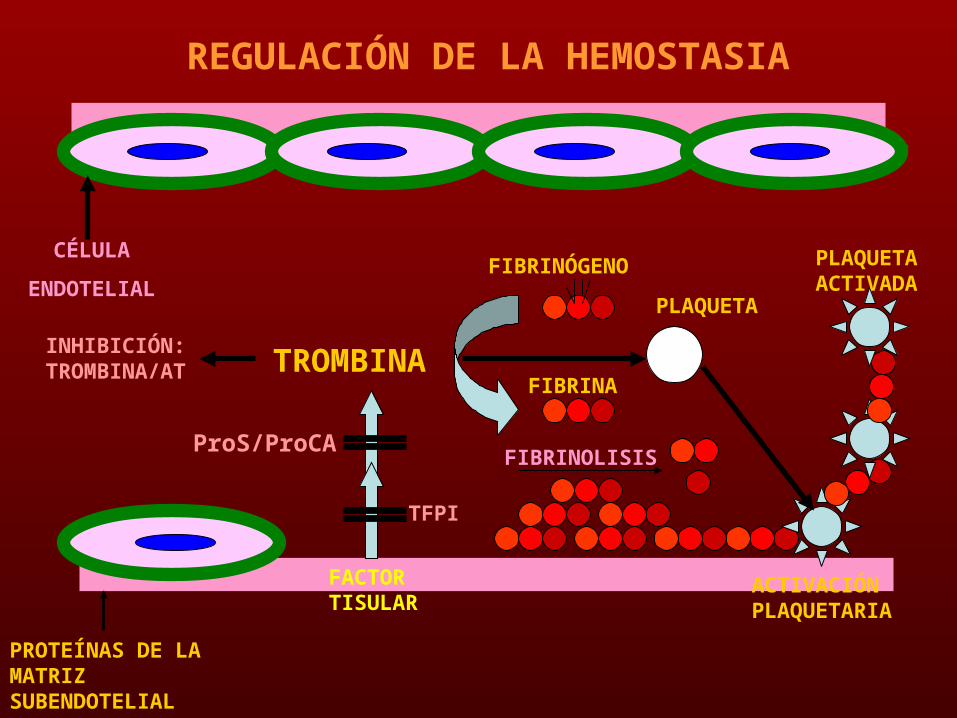
REGULACIÓN DE LA HEMOSTASIA
PROTEÍNAS DE LA MATRIZ SUBENDOTELIAL
CÉLULA
ENDOTELIAL
TFPI
ProS/ProCA
TROMBINA
INHIBICIÓN: TROMBINA/A
T
FIBRINÓGENO
FIBRINA
PLAQUETA
PLAQUETA ACTIVADA
FACTOR TISULAR
FIBRINOLISIS
ACTIVACIÓN PLAQUETARIA

BALANCE DE LA COAGULACION VS ANTICOAGULACION
PLAQUETAS
FACTORES
PLASMATICOS
PLASMINOGENO
T-PA, FCTOR XII, FIBRINA
COAGULACIONFIBRINOLISIS
INHIBIDORESAT IIIPC—PSHCIIEPI
INHIBIDORESPAIalfa 2-APH R G P

FIBRINOLISIS
PÉRDIDA DE FUNCIONES
COAGULACIÓN PLAQUETAS
INHIBIDORES PAI, -2APL
FACTORES tPA, PLG
INCREMENTO DE LAS FUNCIONES
HIPERACTIVIDAD PLAQUETARIA, TROMBOCITOSIS.
AUMENTO DE FACTORES FACTOR V LEIDEN PROTROMBINA G20210A
INHIBIDORES PLAQUETARIOS
PGI2DISMINUCIÓN DE
ANTICOAGULANTES NATURALES Pro C, Prot S, AT

TROMBOFILIA
TENDENCIA INCREMENTADA A LA ENFERMEDAD TROMBOEMBÓLICA VENOSA ADQUIRIDA O CONGÉNITA.
MULTIFACTORIAL Y MULTIGÉNICA, PENETRANCIA INCOMPLETA, EXPRESIVIDAD VARIABLE, INTERACCIÓN GENÉTICA Y
AMBIENTAL.
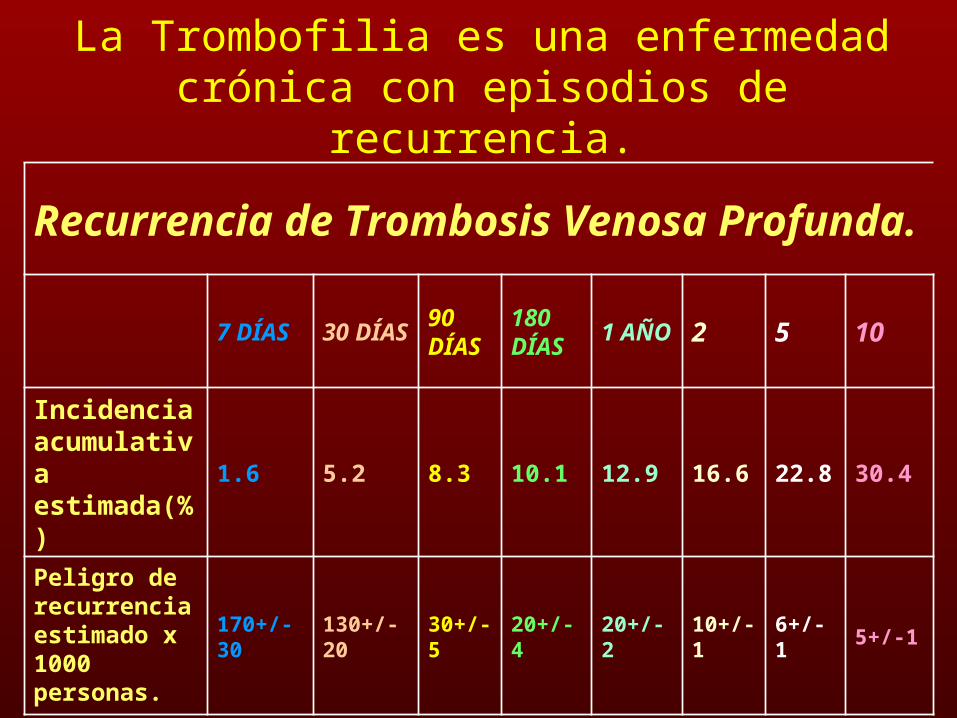
La Trombofilia es una enfermedad crónica con episodios de recurrencia.
Recurrencia de Trombosis Venosa Profunda.
7 DÍAS30 DÍAS
90 DÍAS
180 DÍAS
1 AÑO 2 5 10
Incidencia acumulativa estimada(%)
1.6 5.2 8.3 10.1 12.9 16.6 22.8 30.4
Peligro de recurrencia estimado x 1000 personas.
170+/-30 130+/-20 30+/-5 20+/-4 20+/-2 10+/-1 6+/-1 5+/-1

FACTORES DE RIESGO DE TROMBOSIS
VARIABLES QUE SE PRESENTAN EN DETERMINADOS INDIVIDUOS Y LOS PREDISPONEN A UNA MAYOR INCIDENCIA DE FENÓMENOS TROMBÓTICOS, QUE LAS PERSONAS QUE NO LOS TIENEN.
LOS FACTORES DE RIESGO INCLUYEN:
CARACTERÍSTICAS PERSONALES, CARACTERÍSTICAS FAMILIARES,FACTORES CIRCUNSTANCIALES, CONDICIONES CLÍNICAS ADQUIRIDAS Y TRASTORNOS HEREDITARIOS.

CARACTERÍSTICAS INDIVIDUALES.CARACTERÍSTICAS INDIVIDUALES.
•ANTECEDENTES FAMILIARES DE TEV.
•ANTECEDENTES PERSONALES DE TEV.
•RAZA O SEXO.
•OBESIDAD
•HÁBITOS: TABAQUISMO, SEDENTARISMO, ESTRÉS.

CONDICIONES CLÍNICAS.CONDICIONES CLÍNICAS.
•TRASTORNOS MIELOPROLIFERATIVOS.
•TROMBOCITOPENIA INDUCIDA POR HEPARINA.
•SÍNDROME ANTIFOSFOLÍPIDOS.
•SÍNDROME NEFRÓTICO.
•CID, PTT, HPN.
•CÁNCER.
•INFECCIONES.
•TRAUMATISMOS.
•CIRUGÍA.
•PARÁLISIS.
•EMBARAZO Y PUERPERIO.
•COLITIS ULCERATIVA.

TROMBOFILIAS CONGÉNITAS.TROMBOFILIAS CONGÉNITAS.
DEFICIENCIA PROTEÍNAS ANTICOAGULANTES: (AT, PC, PS) 5-20%.
POLIMORFISMOS GENÉTICOS (40%):
•RESISTENCIA A LA PROTEÍNA C ACTIVADA (FACTOR V LEIDEN).
•PROTROMBINA G20210A.

•DEFICIENCIA DE AT (1965)
•DEFICIENCIA DE PROTEÍNA C (1981)
•DEFICIENCIA DE PROTEÍNA S (1984)
•RESISTENCIA A LA PROTEÍNA C (1993)
•FACTOR V LEIDEN (1994)
•HIPERHOMOCISTEINEMIA (1994)
•GEN DE LA PROTROMBINA G20210A (1996)

MÉTODOS PARA ESTUDIAR LAS PROTEÍNAS DE LA COAGULACIÓN.
FUNCIONALES:
COAGULOMÉTRICOS O CRONOMÉTRICOS.
CROMOGÉNICOS.
INMUNOLÓGICOS:
INMUNODIFUSIÓN RADIAL.
ELECTROINMUNODIFUSIÓN.
INMUNOELECTROFORESIS CRUZADA.
ELISA, OTROS.

DEFICIENCIA DE ANTITROMBINA (AT)-127 mutaciones diferentes.
TIPO I:
NIVELES DISMINUIDOS DE LA PROTEÍNA DETERMINADA FUNCIONALMENTE Y ANTIGÉNICAMENTE (50% DEL VALOR NORMAL).TIPO II:
PRODUCCIÓN DE UNA VARIANTE DE LA PROTEÍNA ANORMAL.
II RS (ALTERACIÓN EN EL SITIO DE REACCIÓN)
II HBS (ALTERACIÓN EN LA UNIÓN CON LAS HEPARINAS)
II PE (ALTERACIÓN EN AMBOS)

AT FUNCIONAL: MIDE LA HABILIDAD PARA INHIBIR LA TROMBINA O EL FACTOR Xa.
LA TROMBINA RESIDUAL O EL X ACTIVADO SON DETERMINADOS ACTUALMENTE CON SUSTRATOS
CROMÓGENIICOS. LOS ENSAYOS QUE MIDEN LA ACTIVIDAD PROGRESIVA HAN SIDO REMPLAZADOS POR ENSAYOS DE
COFACTOR DE HEPARINAS.
AT ANTIGÉNICA: SE DETERMINA LA PRESENCIA DE LA AT MEDIANTE ANTICUERPOS MONOCLONALES QUE RECONOCEN DIFERENTES EPÍTOPES DE LA PROTEÍNA.
SON UTILIZADOS PARA EL DIAGNÓSTICO DE LAS VARIANTES TIPO II

CAUSAS DE DEFICIENCIAS ADQUIRIDAS DE DEFICIENCIA DE AT.
TRATAMIENTO CON HEPARINAS.
CID.
PRE-ECPLAMSIA.
HEPATOPATÍAS.
SÍNDROME NEFRÓTICO.
CIRUGÍA MAYOR.
NEOPLASIAS.
LMA-M3.
TRATAMIENTO CON L-ASPARGINASA.

DEFICIENCIA DE PROTEÍNA C.
161 mutaciones diferentes.
TIPO I: REDUCCIÓN DE LA ACTIVIDAD Y DEL ANTÍGENO.
TIPO II: DEFECTO CUALITATIVO DEBIDO A UNA VARIANTE DE PROTEÍNA C. REDUCIDA LA ACTIVIDAD Y NORMAL LA CONCENTRACIÓN DE ANTÍGENO.

SENSIBILIDAD DE ENSAYOS DE PROTEÍNA C EN PLASMA PARA PROTEÍNA C DISFUNCIONAL
DEFECTOFUNCI ONAL
ENSAYOCOAGULABLE
ENSAYOCROMOGÉNI CO.
ACTI VACI ÓN DELA PC
SENSI BLE SENSI BLE
SI TI O ACTI VO SENSI BLE SENSI BLE
UNI ÓN ALSUSTRATO
SENSI BLE I NSENSI BLE
UNI ÓN A LA PS SENSI BLE I NSENSI BLE
UNI ÓN ASUPERFI CI ES
SENSI BLE I NSENSI BLE
UNI ÓN ALCALCI O
SENSI BLE I NSENSI BLE

CAUSAS DE DEFICIENCIA DE PROTEÍNA C ADQUIRIDAS.
TRATAMIENTO CON ANTICOAGULANTES ORALES.
ENFERMEDAD HEPÁTICA.
CID.
DEFICIENCIA DE VITAMINA K.

DEFICIENCIA DE PROTEÍNA S.
131 mutaciones diferentes.
TIPO I: DISMINUCIÓN DEL ANTÍGENO TOTAL Y LIBRE Y DE LA ACTIVIDAD.
TIPO III: REDUCIDA LA ACTIVIDAD Y LA CONCENTRACIÓN DE ANTIGÉNO LIBRE, NORMAL LA CONCENTRACIÓN DE ANTÍGENO TOTAL.
TIPO II: DEFECTO CUALITATIVO DEBIDO A UNA VARIANTE DE PROTEÍNA S. LA ACTIVIDAD ESTÁ REDUCIDA Y ES NORMAL LA CONCENTRACIÓN DE ANTIGÉNO LIBRE.

LAS FRACCIONES TOTAL Y LIBRE SE MIDEN MEDIANTE MÉTODOS INMUNOENZIMÁTICOS.
LA FRACCIÓN LIBRE SE DETERMINA, PREVIA PRECIPITACIÓN DEL COMPLEJO FORMADO POR LA PS Y LA PROTEÍNA QUE UNE C4b, UTILIZANDO POLIETILENGLICOL.
LOS NIVELES DE PS TOTAL SE SOBRELAPAN EN PERSONAS NORMALES Y HETEROCIGÓTICOS Y SE INCREMENTAN CON LA EDAD, POR TANTO LA MEDIDA DE PS LIBRE TIENE MAYOR SENSIBILIDAD Y ESPECIFICIDAD DIAGNÓSTICA QUE LA TOTAL.

CAUSAS DE DEFICIENCIAS ADQUIRIDAS DE PROTEÍNA S.
EMBARAZO.
PUERPERIO.
USO DE CONTRACEPTIVOS ORALES..
ANTICOAGULACIÓN ORAL.
ANTICOAGULANTE LÚPICO.
ENFERMEDADES HEPÁTICAS.
CID..
DEFICIENCIA DE VITAMINA K.

EN LAS DEFICIENCIAS DE AT,PC Y PS LOS ESTUDIOS DE ADN SE UTILIZAN PARA DETERMINAR LAS ALTERACIONES GENÓMICAS QUE PRODUCEN EL FENOTIPO DEFICIENTE.
POR LA GRAN CANTIDAD DE EVENTOS ENCONTRADOS SON ÚNICAMENTE UTILIZADAS CON FINES INVESTIGATIVOS Y NO DIAGNÓSTICOS.

POLIMORFISMOS GENÉTICOS
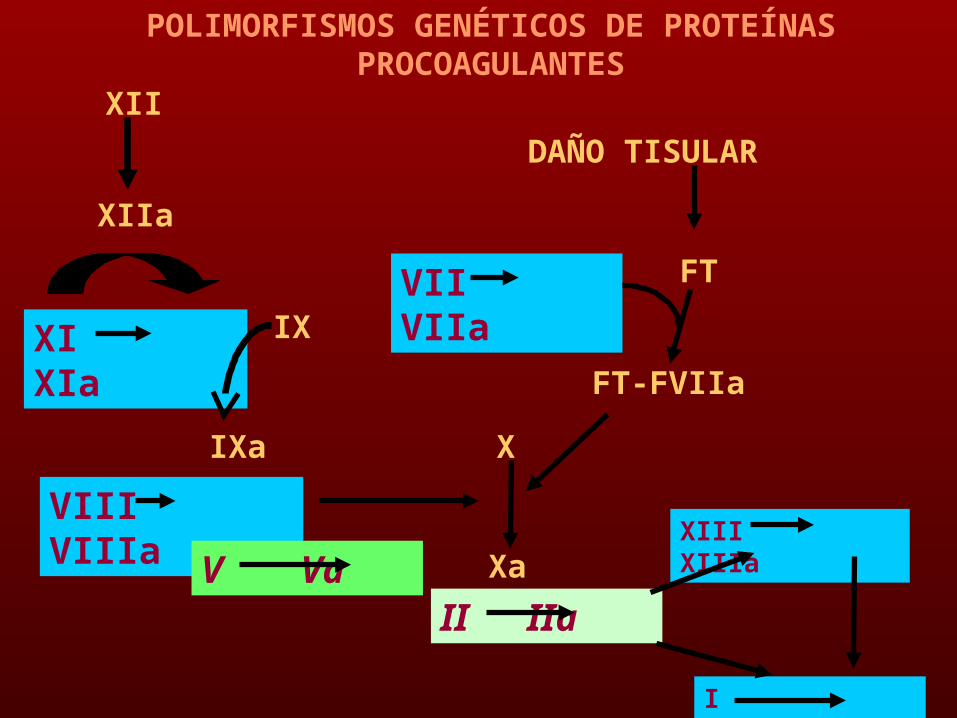
XIII XIIIa
I Ia
XII
XIIa
XI XIa
IXa
IX
VIII VIIIa
V Va
X
Xa
II IIa
FT-FVIIa
DAÑO TISULAR
FTVII VIIa
POLIMORFISMOS GENÉTICOS DE PROTEÍNAS PROCOAGULANTES

RESISTENCIA A LA PROTEÍNA C ACTIVADA.
Dahlbäck B, Carlsson M, Svensoon P. Familial thrombophilia due to a previously unrecognizae mechanism charaterized by poor anticoagulant response to activated protein C: Predition of a cofactor to activated protein C. Proc Natl Acad Sci.1993;90:1004-1008.

RESISTENCIA A LA PROTEINA C ACTIVADA.DIAGNÓSTICO DE LABORATORIO.
PLASMA NO DILUIDO . EL TIEMPO DE COAGULACIÓN (TP, TPTA, Xa) ES MEDIDO EN PRESENCIA Y AUSENCIA DE PCA (CANTIDAD CALIBRADA) Y EL RESULTADO SE EXPRESA COMO LA RAZÓN TCP CON PCA/ TC PLASMA SIN PCA O UNA RAZON PARA EL PACIENTE/RAZÓN PLASMA CONTROL NORMAL.
PLASMA DILUIDO CON PLASMA SUSTRATO DEFICITARIO DE FACTOR V. EL TIEMPO DE COAGULACIÓN (TP, TPTA, Xa) ES MEDIDO EN PRESENCIA Y AUSENCIA DE PCA (CANTIDAD CALIBRADA) Y EL RESULTADO SE EXPRESA COMO LA RAZÓN TCP CON PCA/ TC PLASMA SIN PCA.

DIFERENTES COAGULÓMETROS PRODUCEN DIFERENTES RESULTADOS.
LA FRECUENCIA DE FV LEIDEN ES ELEVADA POR LO QUE LOS VALORES DE REFERENCIA DEBEN OBTENERSE EXCLUYENDO LA PRESENCIA DE LA MUTACIÓN.
EL PLASMA DEBE SER POBRE EN PLAQUETAS, ESTAS REDUCEN LA RAZÓN.
SEPARADOS RANGOS SON NECESITADOS PARA MUESTRAS FRESCAS Y CONGELADAS.

FACTOR V LEIDEN. Bertina RM, Koeleman BPC, Koster T, Rosendaal FR, et al. Mutation in blood coagulation factor V associated with resistence to activated protein C. Nature. 1994;369:64-67.
PROTROMBINA G20210A.Poort SR, Rosendaal FR, Reitsman PH, Bertina RM. A common genetic variation in the 3´-untraslated region of the prothrombin gene is associated with elevated plasma prothrombin level and an increase in venous thrombosis. Blood. 1996;88:3698-3703.

FACTOR V INACTIVADO
Arg 306 Arg 506 Arg 679 Arg 306 Arg 506 Arg 679
FACTOR V ACTIVADO
PC PCA
TROMBINA
TM
PROTEÍNA S
CÉLULA ENDOTELIAL

POLIMORFISMO FENOTIPO ASOCIACIÓN TV ASOCIACIÓN EA FACTOR V LEIDEN 1691G- A(Arg506Gln)
RPCA Factor de riesgo Poco probable
FACTOR V CAMBRIDGE (Arg306Thr)
RPCA EN ESTUDIO NO PROBABLE
FACTOR V HAPLOTIPO HR2
Ligera RPCA EN ESTUDIO NO ESTUDIADO
FACTOR V HONG KONG (Arg306Gly)
RPCA EN ESTUDIO NO ESTUDIADO
TROMBOMODULINA 1418C- T(Ala455Val)
NO POCO PROBABLE DESCONOCIDO
TROMBOMODULINA 33G- A
NO POSIBLE DESCONOCIDO
EPCR 23 bp INSERCIÓN EN EL EXÓN 3.
NO SUGESTIVO DESCONOCIDO
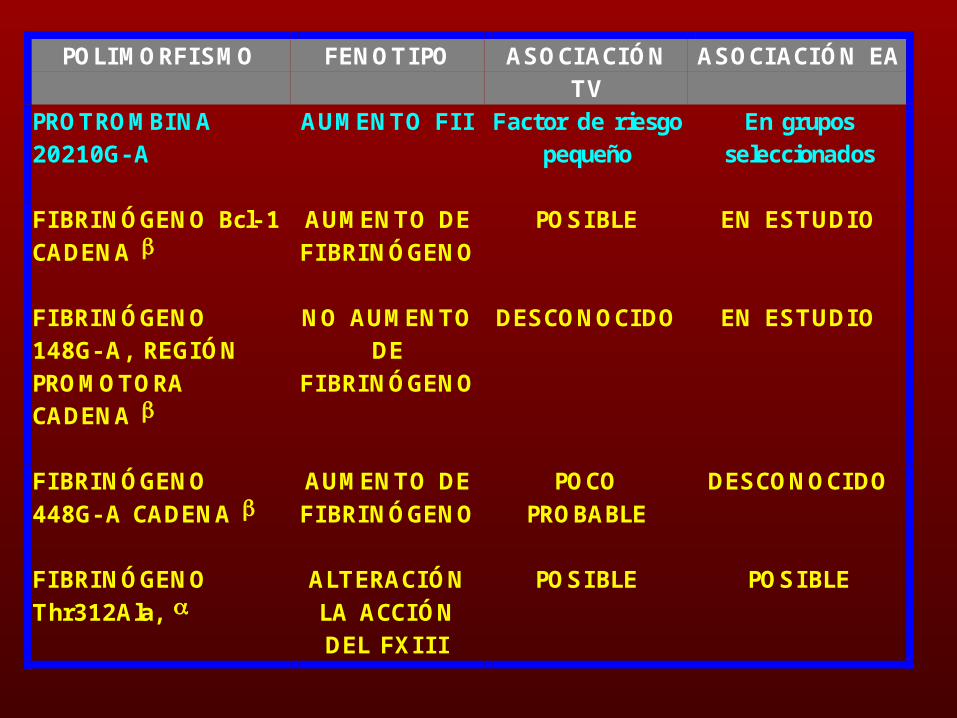
POLIMORFISMO FENOTIPO ASOCIACIÓNTV
ASOCIACIÓN EA
PROTROMBINA20210G- A
AUMENTO FI I Factor de riesgopequeño
En gruposseleccionados
FIBRINÓGENO Bcl- 1CADENA
AUMENTO DEFIBRINÓGENO
POSIBLE EN ESTUDIO
FIBRINÓGENO148G- A, REGIÓNPROMOTORACADENA
NO AUMENTODE
FIBRINÓGENO
DESCONOCIDO EN ESTUDIO
FIBRINÓGENO448G- A CADENA
AUMENTO DEFIBRINÓGENO
POCOPROBABLE
DESCONOCIDO
FIBRINÓGENOThr312Ala,
ALTERACIÓNLA ACCIÓNDEL FXI I I
POSIBLE POSIBLE

POLIMORFISMO FENOTIPO ASOCIACIÓNTV
ASOCIACIÓN EA
FACTOR VI IArg355Gln
DISMINUCIÓNFVI I
DESCONOCIDO POSIBLEMENTEPROTECTOR
FACTOR VI I H7H7 DISMINUCIÓNFVI I
DESCONOCIDO POSIBLEMENTEPROTECTOR
FACTOR VI I G73A DISMINUCIÓNFVI I
DESCONOCIDO POSIBLEMENTEPROTECTOR
FACTOR XI I ISUBUNIDAD AVal34Leu
ACTIVIDADINCREMENTADA
POSIBLEMENTEPROTECTOR
POSIBLEMENTEPROTECTOR
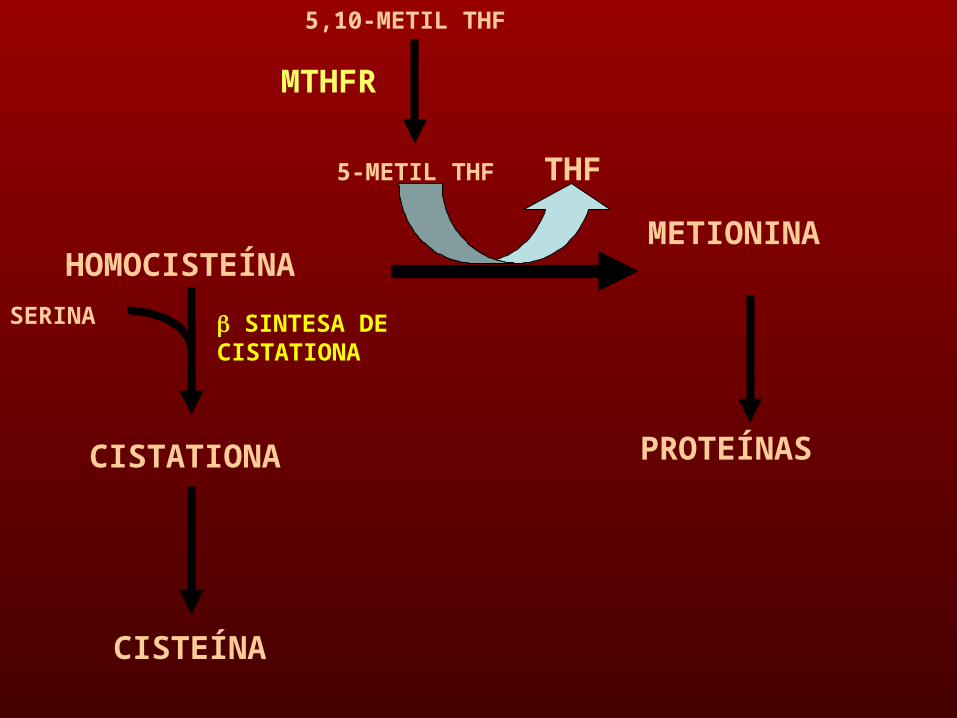
METIONINA
SERINA
HOMOCISTEÍNA
CISTATIONA
SINTESA DE CISTATIONA
CISTEÍNA
5,10-METIL THF
5-METIL THF THF
MTHFR
PROTEÍNAS
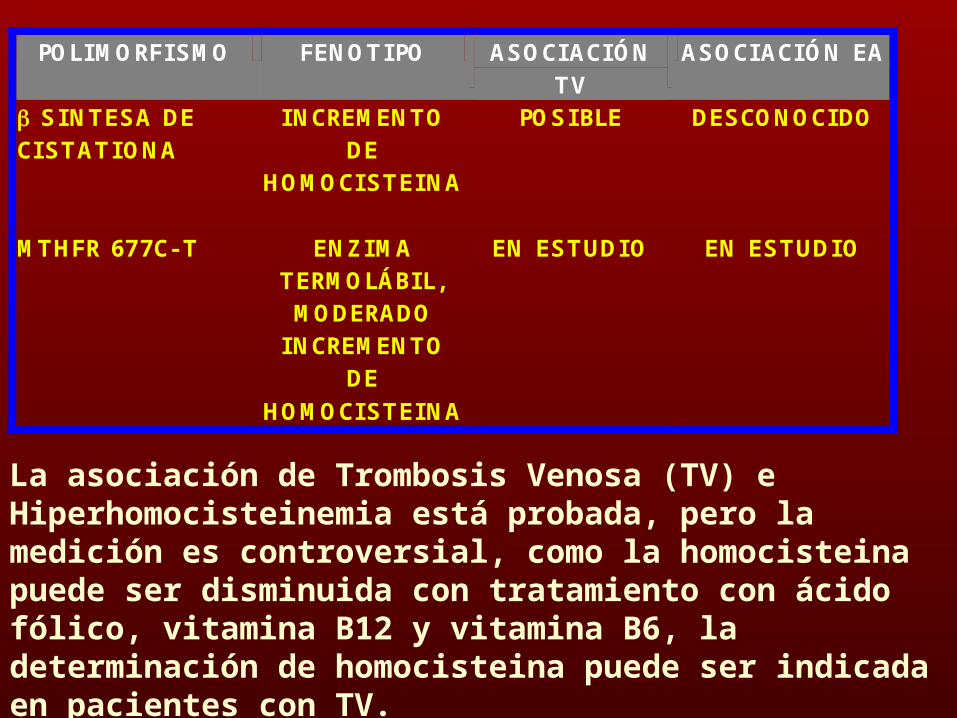
POLIMORFISMO FENOTIPO ASOCIACIÓN TV
ASOCIACIÓN EA
SINTESA DE CISTATIONA
INCREMENTO DE
HOMOCISTEINA
POSIBLE DESCONOCIDO
MTHFR 677C- T
ENZIMA TERMOLÁBIL, MODERADO
INCREMENTO DE
HOMOCISTEINA
EN ESTUDIO EN ESTUDIO
La asociación de Trombosis Venosa (TV) e Hiperhomocisteinemia está probada, pero la medición es controversial, como la homocisteina puede ser disminuida con tratamiento con ácido fólico, vitamina B12 y vitamina B6, la determinación de homocisteina puede ser indicada en pacientes con TV.

LOS ESTUDIOS MOLECULARES DEL ADN O ARN SON PRUEBAS DIRECTAS BASADAS EN LA AMPLIFICACIÓN O DETECCIÓN DIRECTA DE LAS SECUENCIAS DEL GEN ADN/ARN QUE PORTAN LA MUTACIÓN.
LOS SISTEMAS MÁS UTILIZADOS DE PCR SON:
DIGESTIÓN CON ENZIMAS DE RESTRICCIÓN (MnlI-FACTOR V LEIDEN Y HindIII -G20210A.
AMPLIFICACIÓN ALELO ESPECÍFICAS.
HIBRIDIZACIÓN ALELO ESPECÍFICA.
DETECCIÓN FLUORESCENTE DE PRODUCTO DE PCR CON SONDA ALELO ESPECÍFICA (1 ETAPA).

PCR Y DIGESTIÓN CON ENZIMAS DE RESTRICCIÓN MNL1- FACTOR V LEIDEN)
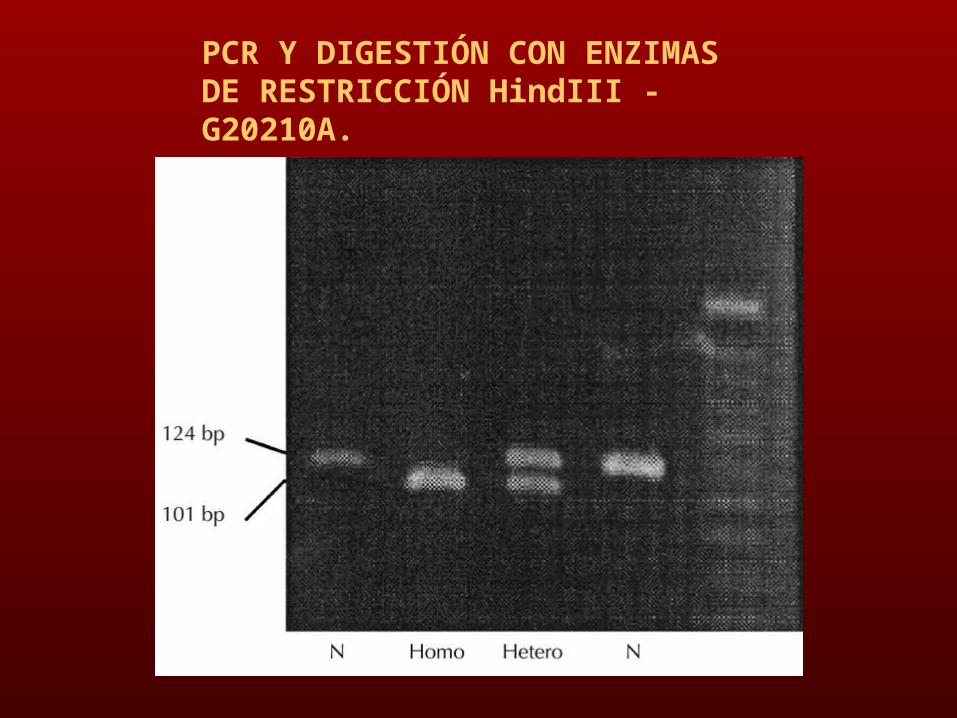
PCR Y DIGESTIÓN CON ENZIMAS DE RESTRICCIÓN HindIII - G20210A.

DEFICIENCIA DE AT 0.02 1.9-4.3
5.0
DEFICIENCIA DE PROTEÍNA C 0.2 3.2-4.8
6.5
DEFICIENCIA DE PROTEÍNA S 1.3 2.3-4.3
2.4
RESISTENCIA PROTEÍNA C ACTIVADA
4.8 (0.05)
18.8-40
6.6
PROTROMBINA G20210A 2.7 (0.06)
7.1-16
2.8
HIPERHOMOCISTEINEMIA 5 10 2.5
FACTOR DE RIESGO
PREVALENCIA EN
POBLACIÓN SANA
PREVALENCIA EN
POBLACIÓN CON TEV
RR TEV

Objetivos de los estudios de trombofilias.
• Conocer la etiología e informar al paciente (una condición trombofílica en un paciente con trombosis no implica conocer la causa, sino un factor que contribuyó al evento).
• Decidir sobre el tratamiento. Duración, Intensidad, Detención.
• Actuar sobre futuras situaciones de riesgo del paciente.
• Acciones sobre familiares.

Niveles de Evidencias.
• Nivel 1: Recomendación basada en 1 o más estudios prospectivos bien diseñados.
• Nivel 2: Estudios retrospectivos o estudios anecdóticos múltiples con consenso.
• Nivel 3: Estudios anecdóticos aislados y consenso de expertos.

Selección de pruebas. Nivel de Evidencia Selección de pruebas. Nivel de Evidencia y propósitos clínicos.y propósitos clínicos.
AF AT PC PS RPCA FVL
Etiología. 2 2 2 2 2 2
Tto. 2 3 3 3 *** ***
Paciente *** 3 3 3 *** ***
Familiares n/a 3 3 3 *** 3
***Datos insuficientes, n/a no aplicable.

Recomendaciones.
• Factor V Leiden (RPCA), Proteína C, Proteína S, AT y G20210A. Pacientes con TVP (jóvenes o con historia familiar). Nivel 2 con algunos nivel 1.
• ACA y AL: Pacientes con ETV idiopática o asociada con Enfermedades autoinmunes o en ausencia de historia familar. Nivel 2 con algunos nivel 1.

PACIENTES DE QUE DEBEN SER EVALUADOS PARA TROMBOFILIAS CONGÉNITAS. Evidencia Nivel 2.
•HISTORIA DE TROMBOSIS RECURRENTE.
•TROMBOSIS EN LA JUVENTUD (ANTES DE LOS 50 AÑOS).
•TROMBOSIS NO PROVOCADA A CUALQUIER EDAD.
•TROMBOSIS NEONATAL FULMINANTE.
•TROMBOSIS EN SITIOS INUSUALES (CEREBRAL, MESENTÉRICA, PORTAL, HEPÁTICA ,RENAL).
•TROMBOSIS ASOCIADA AL EMBARAZO O PUERPERIO, USO DE CONTRACEPTIVOS ORALES Y TERAPIA HORMONAL SUSTITUTIVA..
•TROMBOSIS ASOCIADA A CIRUGÍA U OTRAS PROVOCACIONES.

Recomendaciones.
No realizar estudios funcionales o antigénicos durante la fase aguda, incluyendo VTE.
No realizar estudios funcionales o antigénicos mientras el paciente está anticoagulado.
Confirmar resultados anormales
Confirmar pruebas anormales en parientes de primer grado.
Evidencias nivel 2 y 3.

BIBLIOGRAFIA:•Seigsohn U, Lubetsky A. Genetic Susceptibility to Venoous Thrombosisi. N. England J Med, Vol 344, No 19, 2001, 1222-1331.
•Kottke-Marchant K. Genetic Polymorphism Assciated with Venous and Arterial Thrombosis. Arch Pathol Lab Med, Vol 126, 2002, 295-304.
•Endler G, Mannhalter C. Polymorfism in coagulation factor genes and their impact on arterial and venous thrombosis, Clinica Chimica Acta, Vol 330, 2003, 31-55.
•Van cott EM, Laposata M, Prins MH. Laboratory Evaluation of Hipercoagulability with Venous or Arterial Thrombosisi. Arch Pathol Lab Med, Vol 126, 2002, 1281-1295.
•College of American Patologist Consensus conference XXXVI: Diagnostic Issue in Thrombophilia. Arch Pathol Lab med, Vol 126, 2002.

DIAGNÓSTICO DE TROMBOFILIAS.
Septiembre 2004