transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism
TRANSCRIPT

Biochimica et Biophysica Acta 1833 (2013) 1527–1541
Contents lists available at SciVerse ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bbamcr
Transferrin iron uptake is stimulated by ascorbate via an intracellularreductive mechanism
Darius J.R. Lane ⁎,1, Sherin Chikhani, Vera Richardson, Des R. Richardson ⁎⁎,1
Iron Metabolism and Chelation Program, Department of Pathology and Bosch Institute, University of Sydney, Sydney, New South Wales 2006, Australia
Abbreviations:BCA, bicinchoninic acid; BPS, bathophenvine serum albumin; CHO, Chinese hamster ovary; CHX, ccytochrome b; DHA, dehydroascorbate; DFO, desferrioxtransporter, isoform 1; Fe2-Tf, diferric transferrin; FTH1, felight chain; GLUT, facilitative glucose transporter; GSH,human umbilical vein endothelial cells; IRE, iron-responsivprotein; LIP, labile iron pool; FeTMPyP, Fe(III)tetrakispentachloride; MnTBAP, Mn(III)tetrakis(4-benzoic acid)Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloSteap3, six transmembrane epithelial antigen of the prosdependent ascorbate transporter; Tf, transferrin; TfR1, traferrin receptor 2; V-ATPase, vacuolar-type H+-ATPase⁎ Corresponding author. Tel.: +61 2 9351 6144.
⁎⁎ Corresponding author. Tel.: +61 2 9036 6548; fax:E-mail addresses: [email protected] (D.J.R.
[email protected] (D.R. Richardson).1 These authors contributed equally as senior authors
0167-4889/$ – see front matter © 2013 Elsevier B.V. Allhttp://dx.doi.org/10.1016/j.bbamcr.2013.02.010
a b s t r a c t
a r t i c l e i n f oArticle history:Received 19 October 2012Received in revised form 14 February 2013Accepted 15 February 2013Available online 26 February 2013
Keywords:TransferrinTransferrin receptorIronIron metabolismAscorbateAscorbic acid
Although ascorbate has long been known to stimulate dietary iron (Fe) absorption and non-transferrin Feuptake, the role of ascorbate in transferrin Fe uptake is unknown. Transferrin is a serum Fe transport protein sup-plying almost all cellular Fe under physiological conditions. We sought to examine ascorbate's role in this pro-cess, particularly as cultured cells are typically ascorbate-deficient. At typical plasma concentrations, ascorbatesignificantly increased 59Fe uptake from transferrin by 1.5–2-fold in a range of cells. Moreover, ascorbateenhanced ferritin expression and increased 59Fe accumulation in ferritin. The lack of effect of cycloheximide orthe cytosolic aconitase inhibitor, oxalomalate, on ascorbate-mediated 59Fe uptake from transferrin indicateincreased ferritin synthesis or cytosolic aconitase activity was not responsible for ascorbate's activity. Experi-ments withmembrane-permeant and -impermeant ascorbate-oxidizing reagents indicate that while extracellu-lar ascorbate is required for stimulation of 59Fe uptake from 59Fe-citrate, only intracellular ascorbate is needed fortransferrin 59Fe uptake. Additionally, experiments with L-ascorbate analogs indicate ascorbate's reducingene-diol moiety is necessary for its stimulatory activity. Importantly, neither N-acetylcysteine nor buthioninesulfoximine, which increase or decrease intracellular glutathione, respectively, affected transferrin-dependent59Fe uptake. Thus, ascorbate's stimulatory effect is not due to a general increase in cellular reducing capacity.Ascorbate also did not affect expression of transferrin receptor 1 or 125I-transferrin cellular flux. However, trans-ferrin receptors, endocytosis, vacuolar-type ATPase activity and endosomal acidification were required forascorbate's stimulatory activity. Therefore, ascorbate is a novel modulator of the classical transferrin Fe uptakepathway, acting via an intracellular reductive mechanism.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
Vitamin C (L-ascorbate) is typically abundant within vertebrate cellsand in extracellular fluids and functions as a one-electron reductant[1,2]. Unlike most animals, a small proportion of vertebrate species (in-cluding humans and other haplorhine primates) are incapable of the syn-thesis of L-ascorbate and develop scurvy if deprived of the vitamin [1].Mammalian cells import ascorbate via: (i) sodium-dependent ascorbate
anthroline disulfonate; BSA, bo-ycloheximide; Dcytb, duodenalamine; DMT1, divalent metalrritin heavy chain; FTL, ferritinreduced glutathione; HUVECs,e element; IRP, iron-regulatory(1-methyl-4-pyridyl)porphyrinporphyrin chloride; MnTMPyP,ride; PHE, o-phenanthroline;tate, isoform 3; SVCT, sodium-nsferrin receptor 1; TfR2, trans-
+61 2 9351 3429.Lane),
.
rights reserved.
transporters (SVCTs) [3]; and/or (ii) as the oxidized form of ascorbate,dehydroascorbate (DHA), by facilitative glucose transporters (GLUTs).In the latter case, DHA is reduced intracellularly to ascorbate byNAD(P)H- and reduced glutathione (GSH)-dependent recycling path-ways [4–7].
Cultured mammalian cells are generally devoid of ascorbate for 3reasons: (i) most mammalian cells, even those from ascorbate-synthesizing species, do not synthesize appreciable levels of ascorbatein culture [1,4,5]; (ii) the vitamin rapidly and irreversibly degradesunder culture conditions [8]; and (iii) cultured cells are typically notsupplementedwith ascorbate [6]. Thus,manyprevious studies performedin vitro to assess basic metabolic processes have lacked a crucial vitaminthat is typically abundant in vivo.
Iron (Fe) is an essential element, being vital for a wide variety ofmetabolic processes [9]. It has long been known that ascorbate defi-ciency leads to anemia and other symptoms typical of scurvy[10–12]. In experimentally-induced scurvy [10,12] and in most clin-ical cases [11,13–15], ascorbate is required to correct this anemia. Addi-tionally, orally administered ascorbate has amarked beneficial effect onhematologic parameters in anemic hemodialysis patients [16]. Howev-er, the mechanisms by which ascorbate elicits these effects are unclear.
Ascorbate enhances dietary non-heme Fe absorption [17,18], whichis typically thought to be themajor role of the vitamin in Femetabolism

1528 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
[17]. Interestingly, the specific release of ascorbate from cells stimulatesFe uptake from low-Mr Fe-citrate complexes [4,19,20]. Such observa-tions suggest that the function of ascorbate in Femetabolism is complex[17].
Once dietary Fe is absorbed and released into the bloodstream,virtually all of the metal is bound to the Fe-transport protein, trans-ferrin (Tf) from which it is distributed to all other tissues throughoutthe body. Strikingly, the role of ascorbate in Tf-bound Fe uptake isunknown. Indeed, although scurvy is associated with a form ofFe-deficiency anemia [10–15], the mechanism responsible has notbeen determined. Tf binds two atoms of Fe(III) to form di-ferricTf (Fe2-Tf), which is delivered to cells after the binding of Fe2-Tf tothe Tf receptor 1 (TfR1) [21,22]. This complex is internalized byreceptor-mediated endocytosis, followed by a vacuolar-type H+-ATPase(V-ATPase)-dependent decrease in endosomal pH that destabilizes thebinding of Fe to Tf [21,23,24]. The Fe(III) must then reduced to Fe(II)prior to its release from the endosome into the cytoplasm via divalentmetal transporter 1 (DMT1; Nramp2) [25] and/or analogous Fe(II) trans-porters such as Zip14 [26]. Once in the cytoplasm, this Fe can enter thelabile Fe pool (LIP) [27], which may consist of Fe-chaperone proteinsthat supply Fe to Fe-containing proteins and/or provide Fe for storagein ferritin [28].
Importantly, a potentially rate-limiting step in the cellular acquisitionof Fe by the Tf-to-cell cycle is the reduction of intra-endosomal Fe, whichdepends on an incompletely understood endosomal ferrireductase activ-ity [29]. Although a candidate ferrireductase in erythroid cells is the ‘sixtransmembrane epithelial antigen of the prostate, isoform 3’ (Steap3),Steap3−/− mice still have 60% of the hemoglobin levels of wild-typemice [30]. This indicates that mammalian cells in vivo possess othermeans of reducing intra-endosomal Fe.
In this study, we examined the role of ascorbate on the cellularuptake of Fe from Fe2-Tf. For the first time, we demonstrate that physi-ological concentrations of ascorbate significantly increase Tf-dependentFe uptake by a range of human cells. Significantly, the mechanism ofascorbate-stimulated Fe uptake from Fe2-Tf is different to that involvedin the ascorbate-stimulated uptake of low-Mr Fe. Indeed, the mecha-nism involved requires: (i) the reductive action of intracellular, butnot extracellular ascorbate; (ii) the expression of functional TfRs 1(or 2), but not changes in cellular Tf uptake/ release or changes inTfR1 expression; and (iii) endocytosis and endosomal acidification.Our data suggest that ascorbate acts via a reductive mechanism involv-ing the enhancement of endosomal ferrireduction.
2. Materials and methods
2.1. Reagents
Unless stated, all chemicals were purchased from Sigma-Aldrich(St. Louis, MO). The following radio-nuclides were purchased fromPerkinElmer (Waltham, MA): [59Fe]Cl3 (Cat. #: NEZ037) and Na125I(Cat. #: NEZ033A). Oxalomalic acid (sodium salt; Cat. #: 13521) andthe following metalloporphyrins were purchased from Cayman Chemi-cal Co. (Ann Arbor, MI, USA): Mn(III)tetrakis(4-benzoic acid) porphyrinchloride (MnTBAP; Cat. #: 75850), Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP; Cat. #: 75852) and Fe(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (FeTMPyP; Cat.#: 75854).
2.2. Cell culture
The human cell lines: melanoma: SK-Mel-28, SK-Mel-2, SK-Mel-5and VMM12; hepatocellular carcinoma: HepG2; neuroepithelioma:SK-N-MC; chronic myelogenous leukemia: K562 (American Type CultureCollection, Manassas, VA); and primary cultures of human umbilicalvein endothelial cells (HUVECs; obtained from Mr. Pat Pisansarakit,Heart Research Institute, Sydney, Australia) were grown as described
[31]. Variant Chinese hamster ovary (CHO-TRVb) cells, which are devoidof endogenous TfRs [32], were grown in MEM supplemented with 10%(v/v) FBS, L-glutamine (2 mM), penicillin (100 U/mL), streptomycin(100 μg/mL), sodium pyruvate (1 mM) and 1× non-essential aminoacids (Gibco; Invitrogen: Mulgrave, VIC, Australia). The CHO-TRVbcells stably transfected to express human TfR1 (TRVb/TfR1) [32], orCHO-TRVb cells transfected with FLAG-tagged human TfR2-α (TRVb/TfR2) [33,34], or the TfR2-α vector (i.e., pcDNA3-FLAG), which impartsneomycin/G418-resistance [33,34] (TRVb/Neo), were grown in themedi-umdescribed above supplementedwith G418 sulfate (200 μg/mL; G418)[33,34]. All CHO-TRVb cell lines were provided by Drs. R. Graham and D.Trinder, Fremantle Hospital, Perth, Western Australia, with the permis-sion of T. McGraw (Weill Cornell Medical College), H. Kawabata (KyotoUniversity, Japan) and H.P. Koeffler (University of California).
2.3. Determination of intracellular ascorbate
Intracellular ascorbate levels were determined either as describedpreviously [4] or by the Tempol-dependent oxidation of ascorbate toDHA followed by condensation of DHA with o-phenylenediamine toform a fluorescent product [35]. Both methods provided identicalresults.
2.4. Determination of intracellular total NADP and NADPH levels
Intracellular total NADP and NADPH levels were determined usinga kit from Abcam (Cambridge, MA).
2.5. Preparation of 59Fe-125I-Transferrin
Human apo-Tf (Sigma-Aldrich) was labeled with [59Fe] or non-radioactive Fe to produce the 59Fe- or Fe-labeled di-ferric proteins,respectively [36]. In some experiments, 59Fe2-Tf or Fe2-Tf were alsolabeled with 125I [36,37].
2.6. Effect of ascorbate on 59Fe2-125I-Tf Uptake by Cells
The effect of ascorbate on cellular 59Fe uptake from 59Fe2-125I-Tf wasexamined using established techniques [36,38]. Briefly, cells were pre-incubated with or without ascorbate (10–500 μM) for 5–60 min/37 °Cbefore the addition of 59Fe2-Tf or 59Fe2\125I-Tf (0.75 μM; [Fe] =1.5 μM) or 59Fe-citrate ([Fe] = 1.5 μM; molar ratio or Fe to citrate =1:100) to the same medium for 3 h/37 °C. In some experiments, DHA(50 μM) was used in place of ascorbate. The cells were then washedfive times on ice with ice-cold PBS to remove non-specifically bound59Fe2-Tf. Internalized 59Fe was determined by standard methods byincubating cells on ice for 30 min/4 °C with the general protease,“Pronase” (1 mg/mL) [23,36]. Cells were then detached followingPronase treatment and centrifuged (16,000 g/4 °C/1 min) in anEppendorf microcentrifuge (Hamburg, Germany). The resulting super-natant represents cell-surface (i.e., Pronase-sensitive; hereinafterreferred to as “cell membrane”) 59Fe and 125I that were released byextracellular proteolysis, while the Pronase-insensitive fraction repre-sents internalized 59Fe and 125I [23,36]. Radioactivity was quantitatedusing a γ-counter (2480 Wizard2, Perkin Elmer, Turku, Finland).
In some experiments, the effect of ascorbate on the affinity ofFe2\125I-Tf binding and the total number of cell-surface Tf-receptorswas assessed by pre-incubating SK-Mel-28 cells with or without ascor-bate (50 μM) for 30 min/37 °C, then cooling the cells to 4 °C and incu-bating them for a further 2 h at 4 °C with increasing concentrations ofFe2\125I-Tf (0.0125–1 μM). Cells were then washed and the amountof specifically bound radioactivity was determined by subtracting theamount of Fe2\125I-Tf bound in the presence of a 200-fold excess ofunlabeled Fe2-Tf. At 4 °C, endocytic internalization of the Fe2-Tf–TfRcomplex is inhibited, but the ligand–receptor interaction is unaffected[36].

1529D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
2.7. Effect of ascorbate on 59Fe and 125I-Tf efflux from cells
The effect of ascorbate on 59Fe and/or 125I-Tf efflux from cellspre-labeled with 59Fe\125I-Tf was examined as previously described[39]. Briefly, cellswere incubatedwith 59Fe\125I-Tf (0.75 μM) for either30 min or 3 h/37 °C. The cells were then placed on ice and washed fivetimes with ice-cold PBS and re-incubated with or without ascorbate(50 μM) for 5–30 min or 3 h/37 °C. At the end of the incubation, theoverlying medium was aspirated into γ-counting tubes and the cellmono-layers solubilized in 2% (w/v) SDS [20]. Levels of 59Fe or 125I-Tfwere determined as above.
2.8. SDS-PAGE and Western blotting
SDS-PAGE (reducing conditions), western blotting and densitome-try were performed as described [40]. The following antibodies wereused to probe for specific proteins. Mouse monoclonal anti-humanTfR1 (clone H68.4) was from Invitrogen (Cat. #: 13-6800). Rabbit poly-clonal anti-human TfR2 was from Abcam (Cat. #: ab83810). Rabbitpolyclonal anti-ferritin heavy chain (FTH1) was from Cell SignalingTechnology (Cat. #: 3998; Danvers, MA). Rabbit polyclonal anti-ferritin light chain (FTL) was from Abcam (Cat. #: ab69090). Rabbitpolyclonal anti-Steap3 was from Abnova (Cat. #: PAB13008). Mousemonoclonal anti-β-actin (clone AC-40; Cat. #: A4700) was fromSigma-Aldrich. The secondary antibodies used (1:5000)were horserad-ish peroxidase-conjugated anti-mouse (Cat. #: A9044) and anti-rabbit(Cat. #: A0545) from Sigma-Aldrich.
Membranes were washed and developed using ECL + Western blotdetection reagent (AmershamBiosciences, Buckinhamshire, UK) and ex-posed to X-ray film. Densitometrywas performed using BioRad QuantityOne software (Hercules, CA). All data were normalized to the loadingcontrol, β-actin. Chemiluminescent immunodetection by X-ray filmyielded a linear signal output within 5% error over (i) the protein loadinglevels employed, and (ii) the exposure time intervals examined. Thiswasconfirmedusing a digital imaging BioRad ChemiDocMP system, showingthat exposures were within the linear range of the film and that chemi-luminescent substrate was not limiting.
2.9. Native PAGE-59Fe-autoradiography
Native PAGE-59Fe-autoradiography was performed using establishedtechniques [41]. Where noted, anti-ferritin “super-shifts” were used toidentify the location of endogenous ferritin by incubating cell extracts(100 μg protein) with 10 μg of rabbit polyclonal anti-horse spleenferritin (Sigma-Aldrich; Cat. #: F6136) or rabbit polyclonal anti-humanferritin (MP Biomedicals; Seven Hills, NSW, Australia; Cat. #: 65077)for 1 h/4 °C. To control for possible non-specific effects of the antibody,an equivalent amount of rabbit polyclonal anti-cyclin D1 (Abcam; Cat.#: ab24249) was used, which had no effect on 59Fe distribution. Thelocation of the low-Mr
59Fe fraction was determined by the incubationof cell extracts with 100 μM of the strong Fe chelator, desferrioxamine(DFO; Novartis, Basel, Switzerland), for 1 h/4 °C [41]. The pre-incubation of cell extracts with DFO binds and reproducibly removeslow-Mr
59Fe [41,42]. Importantly, the DFO-59Fe complex does notappear in the gel due to the net positive charge of the complex at therunning pH, which leads to its migration in the opposite direction to59Fe-ferritin and low-Mr
59Fe.
2.10. Statistical analysis
All statistical analyseswere performed usingGraphPadPrism®v. 5.0(GraphPad Software, California). Means were compared using one-wayANOVA with Bonferroni's post hoc test of significance [4,7,20]. Resultsare expressed as means ± SD (number of experiments) and were con-sidered to be statistically significant when p b 0.05.
3. Results
3.1. The incubation of ascorbate-depleted cells with ascorbate or DHAincreases intracellular ascorbate
Typical human plasma ascorbate concentrations in unsupplementedindividuals are 40–60 μM [43]. Therefore, unless otherwise stated, cellswere incubated with or without ascorbate or ascorbate analogues(50 μM) for 30 min/37 °C before the addition of 59Fe as 59Fe2-Tf to thesame medium for 3 h/37 °C (hereinafter referred to as the ‘standardincubation protocol’). Unless supplemented with the vitamin (seebelow), the cells used in this study were devoid of detectable ascorbate(Table 1, column A). This observation is typical of mammalian cellsgrown under standard culture conditions [4,7,8,19,20].
Importantly, incubation of SK-Mel-28 cells with ascorbate for30 min or 3 h increased intracellular ascorbate fromundetectable levelsto 202–375 pmol/106 cells (Table 1). Incubation of these cells withDHA (50 μM) for 30 min/37 °C, which is rapidly reduced to ascorbateonce imported by cells [6], increased their ascorbate content to534 ± 19 pmol/106 cells (Table 1).
3.2. Ascorbate stimulates cellular 59Fe uptake from 59Fe2-Tf in humancells
In initial studies, we used a wide range of human cells to examinethe effect of ascorbate on 59Fe uptake. Incubation of SK-Mel-28 cells(Fig. 1) with ascorbate significantly (p b 0.001) increased their inter-nalized 59Fe uptake from 59Fe2-Tf by ~2-fold that of cells incubated inthe absence of ascorbate. While there was also a trend towards anincrease in cell-membrane 59Fe uptake, this represented a small propor-tion (b10%) of total 59Fe uptake and was typically not significant(p > 0.05; Fig. 1). Under the same conditions, ascorbate also significant-ly (p b 0.001–0.01) stimulated internalized 59Fe uptake from 59Fe2-Tf inall other human cells examined, including primary HUVECs, and allother human cell lines assessed: SK-Mel-2, SK-Mel-5, VMM12, SK-N-MC, HepG2 and K562 (data not shown). Since SK-Mel-28 cells havebeen well-characterized in terms of their cellular Fe uptake mecha-nisms [36,38,39,44,45], these cells were typically used for all subse-quent characterization experiments.
3.3. Ascorbate-stimulates 59Fe uptake from 59Fe2-Tf in a time- andconcentration-dependent manner and increases the Vmax of
59Fe uptake
The stimulatory effect of ascorbate on 59Fe uptake from Tf contin-ued for at least 24 h (Fig. 2A). Moreover, ascorbate caused a signifi-cant (p b 0.001) increase in 59Fe uptake after only 30 min incubationwith 59Fe2-Tf (Fig. 2B), indicating that stimulation occurred over rela-tively short 59Fe uptake periods. Pre-incubation of SK-Mel-28 cellswith ascorbate from 5 to 60 min/37 °C, followed bywashing to removeextracellular ascorbate,was sufficient in a subsequent 3 h/37 °C incuba-tion to elicit full stimulation of 59Fe uptake from 59Fe2-Tf (Supp. Fig. 1A).Furthermore, ascorbate at concentrations from 10 to 500 μM had nomore stimulatory effect on 59Fe uptake than that observed at 10 μM(Supp. Fig. 1B; 59Fe2-Tf incubation time: 2 h). However, to approximateplasma ascorbate concentrations in healthy humans [43,46], a concen-tration of 50 μM ascorbate was used in all subsequent studies.
We next assessed the dependence of ascorbate-stimulated 59Fe up-take on the concentration of 59Fe2-Tf (0.125–12.5 μM; Fig. 2C). Internal-ized 59Fe uptake was then determined and the results analyzed bynon-linear regression analysis (Fig. 2C). Interestingly, ascorbate signifi-cantly (p b 0.001) increased the Vmax of 59Fe uptake (from 9.2 ± 0.16to 13.0 ± 0.29 × 106 atoms 59Fe/cell/h; please note the different unitsused for the y-axis in Fig. 2C). As typical serum Fe2-Tf concentrationsare 20–30 μM [21], and that ascorbate-stimulated 59Fe uptakeoccurred maximally at a 59Fe2-Tf concentration of 0.75–2 μM

Table 1Intracellular ascorbate levels in SK-Mel-28 cells before and after incubation with ascorbate or DHA at the indicated concentrations and for the indicated times. Results are mean±SD (3 experiments).
Intracellular ascorbate, pmol × (106 cells)−1
Cell line (A) Notreatment
(B) Incubation with ascorbate(50 μM) for 30 min/37 °C
(C) Incubation with ascorbate(50 μM) for 3 h/37 °C
(D) Incubation with DHA(50 μM) for 30 min/37 °C
SK-Mel-28 0 ± 1.0 202 ± 15***(vs A) 375 ± 18***(vs B) 534 ± 19***(vs C)
*** pb0.001.
1530 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
(Fig. 2C), ascorbate-stimulated uptake of Fe from Fe2-Tf is probablyphysiologically relevant.
3.4. Ascorbate and Fe2-Tf increase ferritin levels
We next examined whether ascorbate-stimulated 59Fe uptake corre-lated with alterations TfR1 and ferritin levels. These proteins were exam-ined as their expression levels are post-transcriptionally controlled inan Fe-dependent manner via the iron-regulatory protein (IRP)-iron-responsive element (IRE) system [47,48]. As a consequence, an increasein cytosolic Fe typically decreases TfR1 synthesiswhile it increases ferritinsynthesis [47,49].
Control and ascorbate-treated SK-Mel-28 cells were incubatedwith non-radioisotopic Fe2-Tf (0.75 μM) for 0–24 h (Fig. 3A). Thelevels of TfR1, FTH1 and FTL were thenmonitored by western blotting.As demonstrated in Fig. 2A, although ascorbate linearly stimulated59Fe uptake over 24 h (Fig. 2A), ascorbate significantly (p b 0.01)down-regulated TfR1 protein by 8 h (Fig. 3A). In contrast, in the ab-sence of ascorbate, significant (p b 0.001) down-regulation of TfR1only occurred by 24 h (Fig. 3A). Thus, TfR1 protein is more acutelydown-regulated by Fe2-Tf in the presence of ascorbate. Conversely,FTH1 and FTL protein levels increased at a significantly (p b 0.001)greater rate over 24 h in ascorbate-treated compared to control cells,with a significant (p b 0.001) increase being evident by 3 h (Fig. 3A,B). Importantly, ascorbate and Fe2-Tf synergistically increasedup-regulation of FTL by 3 h compared to Fe2-Tf or ascorbate alone(Fig. 3B), which was probably due to increased Fe uptake. Similar re-sults were found for FTH1 (data not shown). These results are
Fig. 1. IncubationofhumanmelanomaSK-Mel-28 cellswithascorbate increases internalized59Fe uptake from 59Fe2-Tf. Cells were pre-incubated with or without ascorbate (50 μM) for30 min/37 °C before the addition of 59Fe2-Tf ([Tf] = 0.75 μM; [Fe] = 1.5 μM) to the samemedium for 3 h/37 °C. After this incubation, the cell mono-layers were washed on ice andincubated with Pronase (1 mg/mL) for 30 min/4 °C to obtain the internalized and cell-membrane fractions. Results are mean + SD (3 experiments).
consistent with reports documenting increased de novo synthesis offerritin in cells exposed to a combination of ascorbate and Fe [50–52].
3.5. An increase in ferritin does not explain ascorbate-stimulated 59Feuptake from Tf
The ascorbate/Fe2-Tf-dependent increase in ferritin levels observedhere is probably a consequence, rather than a cause, of ascorbate-stimulated Fe uptake. Indeed, when cells are presented with low-Mr
Fe, ascorbate enhances ferritin expression by an IRP1/cytosolicaconitase “switch” mechanism [51]. Thus, we assessed the effects of:(i) the protein synthesis inhibitor, cycloheximide (20 or 100 μM;CHX) [53], which abrogates global protein synthesis in SK-Mel-28cells [53]; or (ii) the cytosolic aconitase inhibitor, oxalomalate (1 or5 mM) [54], which abolishes aconitase activity of the Fe–sulfur clusterform of IRP1 [54]. Neither compound significantly affected 59Fe uptake(Supp. Fig. 2).
As ascorbate can inhibit ferritin autophagy [55], and ferritin canalso be degraded by the proteasome [56], we examined the effect ofinhibitors of both these pathways on 59Fe uptake (Supp. Fig. 2). Treat-ment of cells with inhibitors of autophagy (3-methyladenine; 5 mM)or the proteasome (MG-132; 10 μM)-conditions previously shown toinhibit the respective degradative pathways [56] – did not affect 59Feuptake (Supp. Fig. 2).
Therefore, our data indicate that ascorbate does not stimulateTf-dependent 59Fe uptake by increasing ferritin levels or by increasingcytosolic aconitase activity.
3.6. Incubation of cells with ascorbate and 59Fe2-Tf increased loading offerritin with 59Fe
To determine whether ascorbate stimulated Fe-loading of ferritin,control and ascorbate-treated SK-Mel-28 cells were incubatedwith 59Fe2-Tf (0.75 μM) for 0.5–24 h (Fig. 4). The location of the ferritinband was determined as described in the Materials and methodssection (Supp. Fig. 3A) [41,53]. While incubating cells with 59Fe2-Tfcaused a time-dependent increase in 59Fe-ferritin, which contained themajority of detectable cellular 59Fe, ascorbate significantly (p b 0.001)increased this by ~2–3 fold at 3, 8 and 24 h (Fig. 4).
Additionally, a putative low-Mr59Fe pool was identified in both
control and ascorbate-treated cell extracts (Fig. 4; see Materials andmethods section), although the latter was always significantly lowerthan the former (data not shown). This pool was verified as “low-Mr”
by its abolishment by a 1 h/4 °C incubation of native cell extracts withDFO (100 μM; Supp. Fig. 3B).
It should be noted that the apparent absence of the 59Fe-Tf bandin the 59Fe autoradiograms presented in Fig. 4 and Supp. Fig. 3 is mostlikely due to their far lower intensity relative to the 59Fe-ferritinand low-Mr
59Fe bands. Indeed, as Tf rapidly donates its Fe to cells,there is only a relatively low steady-state level of 59Fe-Tf in cells. Fur-ther, as 59Fe-Tf does not accumulate in cells, it can be difficult to detectin the presence of 59Fe-ferritin, which does accumulate.

Fig. 2. Characterization of ascorbate stimulated iron uptake from 59Fe2-Tf by SK-Mel-28 cells as a function of: (A, B) incubation timewith 59Fe2-Tf, or (C) 59Fe2-Tf concentration. (A, B) Cellswerepre-incubated with or without ascorbate (50 μM) for 30 min/37 °C before the addition of 59Fe as 59Fe2-Tf ([Tf] = 0.75 μM; [Fe] = 1.5 μM) to the same medium for 1–24 h (A), or 1–60 min(B) at 37 °C. (C) Cells were pre-incubatedwith orwithout ascorbate as in (A, B) before the addition of 59Fe2-Tf ([Tf] = 0.125–12.5 μM; [Fe] = 0.25–25.0 μM) for a further 3 h/37 °C. For (A–C),after incubation with 59Fe2-Tf, cells were washed on ice and then incubated with Pronase (1 mg/mL) for 30 min/4 °C to separate the membrane and internalized fractions. Results aremean + SD (3 experiments).
1531D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
Therefore, the increased amount of Fe from Fe2-Tf reaching thecytoplasm in the presence of ascorbate (i) elevates ferritin expression,probably via an IRP-IRE-regulated increase in de novo ferritin synthesis[51,57]; and (ii) increases ferritin Fe-loading.
3.7. Ascorbate-stimulated 59Fe uptake from 59Fe2-Tf requires intracellularascorbate and not extracellular Fe reduction
Ascorbate-dependent Fe uptake from Fe-citrate requires extracellularascorbate and extracellular ferrireduction [4,19,20]. To determine whetherascorbate-stimulated 59Fe uptake from 59Fe2-Tf occurred by a similarmechanism, control and ascorbate-treated cells were further incubatedwith the cell-impermeant enzyme, ascorbate oxidase (10 U/mL), for a fur-ther 30 min andduring subsequent incubationwith 59Fe2-Tf or 59Fe-citrate(Fig. 5). Ascorbate oxidase,which oxidizes ascorbate toDHA,markedly andsignificantly (p b 0.001) inhibited ascorbate-stimulated 59Fe uptake from59Fe-citrate ([Fe] = 1.5 μM; Fig. 5A), as previously described [4,19,20],but did not significantly affect 59Fe uptake from 59Fe2-Tf ([Fe] = 1.5 μM;Fig. 5B). As ascorbate oxidase only oxidizes extracellular ascorbate[4,7,19], ascorbate-stimulated 59Fe uptake from 59Fe2-Tf does not dependon the continued presence of extracellular ascorbate.
Next, we examined if the extracellular release of 59Fe from 59Fe2-Tfwas required for ascorbate to stimulate 59Fe uptake from this protein,as reported for the activation of Tf-dependent Fe uptake by ferricammonium citrate [58]. Cells were incubated with the membrane-impermeant Fe(II) chelators (200 μM), ferene-S, ferrozine [4,20,59], orbathophenanthroline disulfonate (BPS [45]) for 30 min before additionof 59Fe2-Tf or 59Fe-citrate to the same medium (Fig. 5).
As previously described [4,20], all chelators markedly and signifi-cantly (p b 0.001) inhibited 59Fe uptake from 59Fe-citrate in the pres-ence of ascorbate (Fig. 5A). Importantly, and in contrast to 59Fe uptakefrom 59Fe-citrate, ferene-S, ferrozine and BPS only reduced 59Fe uptakefrom 59Fe2-Tf in the presence of ascorbate by less than 12%, 17% and 36%(p b 0.01), respectively (Fig. 5B). Similarly, co-incubation of cells withthe avid Fe-binding Tf homologue, apo-lactoferrin (4 μM) almostabolished ascorbate-stimulated and basal 59Fe uptake from 59Fe-citrate(Fig. 5A), but only slightly reduced ascorbate-stimulated
59Fe uptake
from 59Fe2-Tf (i.e., by less than 20% of the 59Fe uptake in the presenceof ascorbate; Fig. 5B).
The observation that all chelators caused some inhibition ofascorbate-stimulated 59Fe uptake from 59Fe2-Tf (cf. Fig. 5B) is consis-tent with the finding that Fe can be labilized by co-incubation withboth strong Fe chelators such as BPS and apo-lactoferrin when areductant, such as ascorbate, is present [60].
Therefore, unlike 59Fe uptake from 59Fe citrate, our results suggestthat ascorbate stimulates 59Fe uptake from 59Fe2-Tf by an intracellularmechanism.
3.8. Studies with ascorbate-scavenging metalloporphyrins indicate theneed for intracellular ascorbate in Tf-59Fe uptake
In order to further evaluate the hypothesis that ascorbate-stimulated 59Fe uptake from 59Fe2-Tf requires an intracellular poolof ascorbate, we assessed the ability of the membrane-permeantmetalloporphyrins, MnTMPyP and FeTMPyP, to rapidly catalyze ascor-bate oxidation [61]. These metalloporphyrins catalyze the oxidation

1532 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
of ascorbate at rates 8- and 170-fold, respectively, greater than the rateof ascorbate auto-oxidation at physiological pH [61]. As thesemetalloporphyrins also scavenge superoxide (i.e., MnTMPyP: [62];FeTMPyP: [63]) and peroxynitrite (i.e., MnTMPyP: [64]; FeTMPyP:[65]), the membrane-permeant metalloporphyrin, MnTBAP, whichsimilarly scavenges superoxide [62] and peroxynitrite [61], but is rela-tively unreactive with ascorbate [61], was used to control for thenon-ascorbate oxidizing activities of MnTMyP and FeTMPyP. Allmetalloporphyrins were used at a concentration of 30 μM.
Importantly, when added immediately after pre-incubation of cellswith or without ascorbate, MnTBAP did not affect 59Fe uptake (Fig. 6).However,MnTMPyP partially inhibited (p b 0.001), whereas FeTMPyPalmost abolished (p b 0.001), only ascorbate-stimulated 59Fe uptake(Fig. 6). Notably, although both MnTMPyP and FeTMPyP would haveoxidized both intracellular and extracellular ascorbate, the oxidationof extracellular ascorbate cannot account for the inhibitory action ofthese metalloporphyrins, as ascorbate oxidase, which exclusivelyand rapidly oxidizes extracellular ascorbate [4,7,19], had no significanteffect under the same conditions (Fig. 5B). Thus, these data furthersuggest the involvement of intracellular ascorbate in stimulating59Fe uptake from 59Fe2-Tf.
3.9. The reducing ene-diol moiety is required for ascorbate to stimulate59Fe uptake from 59Fe2-Tf
We next assessed the effects of a range of ascorbate analogues anddegradation products (Fig. 7A) on ascorbate-stimulated 59Fe uptakefrom 59Fe2-Tf (Fig. 7B).
Fig. 3. Ascorbate potentiates Fe-dependent ferritin expression, but this is not the cause of ascorbor without ascorbate (50 μM) for 30 min and then Fe2-Tf (0.75 μM) added to this media and t(i.e., transferrin receptor 1 (TfR1), ferritin heavy chain (FTH1), ferritin light chain (FTL) and thesitometry of the indicated bands is also shown. (B) The observed increase in FTL expression afteascorbate (50 μM) and Fe2-Tf (0.75 μM). Western blots (A and B) are typical results from 3 ex
The effect of a change in side-chain stereoisomerism was assessedwith the L-ascorbate epimer, D-isoascorbate (Fig. 7A). Interestingly,D-isoascorbate can substitute for L-ascorbate in several biologically-relevant, enzyme-catalyzed reactions and may be imported by cells,albeit at a lower rate than L-ascorbate [3,66,67]. Interestingly, the stim-ulation of 59Fe uptake from 59Fe2-Tf by D-isoascorbatewas equivalent tothat of L-ascorbate in both cell lines, indicating that stimulation of 59Feuptake by ascorbate is not stereo-specific (Fig. 7B).
The influence of changing side chain hydrophobicity was assessedwith L-ascorbate-6-palmitate (Fig. 7A), a highly lipophilic, palmitoylatedL-ascorbate derivative [68], and 5,6-isopropylidene-L-ascorbate, whichis lipophilic and a potential substrate for SVCTs [67]. Like L-ascorbate,these derivatives have potent reducing activity due to the 2,3-ene-diolmoiety (Fig. 7A) [66,69]. The stimulation of Tf-dependent 59Fe uptakeby L-ascorbate-6-palmitate was very similar to that provided by 5,6-isopropylidene-L-ascorbate, and in both cases the stimulation was onlymarginally lower than that with L-ascorbate (Fig. 7B). Thus, stimulationof 59Fe uptake does not appear to be sensitive to side chain structureand/or hydrophobicity.
We then assessed the effect of modifications to the electron-dense2,3-ene-diolmoiety (Fig. 7A), which, togetherwith the conjugated struc-ture of the five-membered lactone ring, is essential for the one-electronreducing activity of ascorbate [70]. The importance of this group wasassessed using the two L-ascorbate derivatives, L-ascorbate-2-phosphateand L-ascorbate-2-sulfate (Fig. 7A), both of which lack reducing activity.
While dephosphorylation of L-ascorbate-2-phosphate by phospha-tases leads to the slow release of L-ascorbate that can be accumulatedby cells [71], the corresponding sulfate group in L-ascorbate-2-sulfateis not cleavable bymammalian cells, and correspondingly, thismolecule
ate-stimulated 59Fe uptake from transferrin. (A) SK-Mel-28 cells were pre-incubatedwithhe cells then incubated for 0, 3, 8 or 24 h/37 °C. The expression of the indicated proteinsloading control, β-actin) was thenmonitored bywestern blotting. (lower panels) The den-r a 3 h/37 °C incubation of SK-Mel-28 cells in (A) was dependent on incubation with bothperiments, while all graphical data are mean + SD (3 experiments).

Fig. 5. Ascorbate-stimulated 59Fe uptake from 59Fe-citrate (A), but not 59Fe2-Tf (B), requires extracellular ascorbate and extracellular labilization of 59Fe. SK-Mel-28 cells werepre-incubated with either ascorbate (50 μM) or control medium alone without ascorbate and then either vehicle medium, ascorbate oxidase (10 U/mL), ferene-S (200 μM),ferrozine (200 μM), apo-lactoferrin (4 μM) or bathophenanthroline disulfonate (BPS; 200 μM) were added, and the cells incubated for a further 30 min/37 °C incubation. Then,59Fe was added to the same medium as either 59Fe-citrate ([Fe] = 1.5 μM; molar ratio of Fe to citrate = 1:100) (A), or 59Fe2-Tf ([Tf] = 0.75 μM; [Fe] = 1.5 μM) (B), and thecells were incubated for a further 3 h/37 °C. After this incubation, cells were washed on ice and then incubated with Pronase (1 mg/mL) for 30 min/4 °C to obtain the internalizedand cell-membrane fractions. Results shown are mean + SD (3 experiments).
Fig. 4. Ascorbate-stimulated 59Fe uptake from Fe2-Tf leads to increased loading of ferritin with 59Fe. (top panel) SK-Mel-28 cells were pre-incubated with/without ascorbate (50 μM) for30 min/37 °C and then 59Fe2-Tf (0.75 μM) was added and the cells incubated for a further 0.5, 3, 8 or 24 h/37 °C incubation. At each time point, cells were harvested and cytoplasmicextracts were obtained under native conditions at 4 °C and native PAGE 59Fe autoradiography performed. (lower panel) The densitometry of the 59Fe-ferritin and low-Mr
59Fe bands isalso shown, respectively. The native PAGE results are typical results from 3 experiments, while the densitometry in (lower panel) is mean ± SD (3 experiments).
1533D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541

Fig. 6. Cell-permeant ascorbate-scavenging metalloporphyrins further suggest the in-volvement of intracellular ascorbate in the stimulation of Tf-dependent 59Fe uptake.SK-Mel-28 cells were pre-incubated with control medium or ascorbate (50 μM) for30 min/37 °C. Vehicle medium (MEM + 0.1% DMSO), MnTBAP, MnTMPyP or FeTMPyP(all at a final concentration of 30 μM) were then added to the same medium, and thecells incubated for an additional 30 min/37 °C. Then, 59Fe2-Tf (0.75 μM) was added andthe cells incubated for a further 3 h/37 °C. After this incubation, cells were washed onice and incubated with Pronase (1 mg/mL) for 30 min/4 °C to obtain the internalizedand cell-membrane fractions. Results shown represent internalized 59Fe uptake and aremean + SD (3 experiments).
1534 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
is not a source of cellular ascorbate [72]. We observed that although thestimulation of cellular 59Fe uptake from 59Fe2-Tf by ascorbate-2-phosphate was significantly (p b 0.001) less than L-ascorbate, it wasstill significantly (p b 0.01) greater than the ascorbate-free control(Fig. 7B). In contrast, L-ascorbate-2-sulfate provided no stimulation(Fig. 7B). Thus, the 2,3-ene-diol moiety is crucial for stimulation ofTf-dependent 59Fe uptake.
It is important to note that as L-ascorbate-2-phosphate andL-ascorbate-2-sulfate may not be substrates for SVCTs [67], these an-alogues may not readily be taken up by cells. Hence, the requirementfor an intact ene-diol in stimulating 59Fe uptake reflects a require-ment for either (i) the reducing activity of ascorbate; and/or (ii) theSVCT-dependent uptake of ascorbate.
Under standard cell culture conditions, ascorbate is rapidly oxidized toits two-electron oxidized form, DHA [8]. As expected on the basis of arequirement for intracellular ascorbate in stimulating Tf-dependent 59Feuptake, pre-treatment of cells with DHA, which increased intracellularascorbate (Table 1), stimulated 59Fe uptake equivalently to that ofL-ascorbate (Fig. 7B).
As DHA itself is an unstable molecule that undergoes rapid andirreversible delactonization under physiological and cell culture condi-tions [1], we examined the following commonDHAdegradation productsfor their ability to stimulate Tf-dependent 59Fe uptake: L-(+)-erythrulose,L-(+)-threonate, L-threose and oxalate [1] (Fig. 7A,B). None of these deg-radation products had any effect on Tf-dependent 59Fe uptake, whichdemonstrates a requirement for the intact vitamin.
3.10. Ascorbate-stimulated 59Fe uptake from Tf is not due to a generalincrease in cellular reducing capacity
To examine whether a general increase in cellular reducing capacitycould account for ascorbate's effect on 59Fe uptake, SK-Mel-28 cellswere pre-incubated with the following for 24 h/37 °C prior to imple-mentation of the standard incubation protocol: (i) control medium;(ii) the membrane-permeant GSH precursor, N-acetylcysteine (NAC;5 mM) [73], which increases cell GSH in SK-Mel-28 cells [74]; or(iii) theγ-glutamylcysteine synthetase inhibitor, buthionine sulfoximine
(BSO; 10 μM), which markedly attenuates GSH levels in these cells[75,76]. The results (Fig. 7C) show that modulating cellular GSH had nosignificant effect on 59Fe uptake (Fig. 7C). Thus, the stimulatory effectof ascorbate on 59Fe uptake cannot be attributed to a general increasein cellular reducing capacity.
Next, we assessed whether ascorbate specifically caused an in-crease in cellular NADPH levels (Fig. 7D). This was important to assessas NADPH is thought to be the major electron donor for theendosomal ferrireductase, Steap3, which contributes to Fe uptakefrom Tf in some cells [30] (see also Section 3.11). Interestingly, whileascorbate had no effect on total NADP levels (i.e., NADP+ + NADPH),incubation with ascorbate significantly (p b 0.001) lowered NADPHlevels (Fig. 7D). The latter is probably due to the requirementfor NADPH in maintaining intracellular ascorbate [1]. There-fore, the ability of ascorbate to stimulate Tf-dependent Fe up-take cannot be explained by an increase in cellular NADPHlevels.
3.11. Ascorbate-stimulated 59Fe uptake from 59Fe2-Tf is not caused byeither changes in levels of TfR1/2 or Steap3, or changes in cellular 125I-Tfuptake
Next, we assessed whether the stimulation of Tf-dependent 59Feuptake by ascorbate was due to alterations in Tf binding to cells, Tfuptake and/or Tf cycling. First, using the standard incubation proto-col, we confirmed that ascorbate, with or without Fe2-Tf, had no effecton TfR1 expression within 3 h, despite there being a marked increasein 59Fe uptake in the presence of ascorbate over this period (Fig. 8A;cf. TfR1 expression in Fig. 3A). Additionally, when cells were incubat-ed with ascorbate and Fe2-Tf as described in Fig. 3A (see Section 3.4),there was no significant change in TfR2 or Steap3 protein levelsover 0–24 h compared to the relevant controls (Supp. Fig. 4). Thus,at least in SK-Mel-28 cells, ascorbate does not appear to stimulateTf-dependent 59Fe uptake by modulating the levels of these proteins.It should be noted that although there was some trend towards adecrease in TfR2 levels in the presence of both ascorbate and Fe2-Tf,this was not found to be significant (Supp. Fig. 4). This is consistentwith the observation that unlike TfR1, TfR2 expression does not ap-pear to be Fe-regulated [77]. Moreover, although TfR2 protein canbe stabilized by the addition of Fe2-Tf in cells of hepatic origin[78,79], we did not observe this effect in SK-Mel-28 cells (Supp. Fig.4). This may be due to the following observations: (i) the stabilizationof TfR2 by Fe2-Tf appears to be specific to cells of hepatic origin[78,79] (e.g., endogenous TfR2 is not regulated by Fe2-Tf in K562cells [79]); and (ii) while our experiments employed a relativelylow Fe2-Tf concentration of 0.75 μM, the stabilization of TfR2 byFe2-Tf in HepG2 cells was found to be most pronounced at markedlyhigher Fe2-Tf concentrations (i.e., 2.5–25 μM) [78].
Next, we assessed if ascorbate affected the number and/or affinity ofTf-binding sites at the cell surface (see Materials and methods section).The apparent dissociation constants and total number of cell-surfaceTf-binding sites were determined using non-linear regression analysis(Fig. 8B). Our data clearly demonstrate that ascorbate did not signifi-cantly affect the affinity of Fe2\125I-Tf-binding, or the total number ofspecific cell-surface Tf-binding sites [control = 35,800 ± 1500/cell(n = 6); ascorbate-treated = 37,100 ± 1800/cell (n = 6)].
We then compared the effect of ascorbate on the relative distribu-tions of 59Fe and 125I-Tf when SK-Mel-28 cells were co-incubated withdoubly-labeled 59Fe2\125I-Tf (0.75 μM). While pre-incubation of cellswith ascorbate followed by incubation with 59Fe2\125I-Tf (0.75 μM)resulted in significantly (p b 0.001) more 59Fe uptake into the internal-ized compartment (Fig. 8C), ascorbate did not significantly affect 125I-Tfuptake into either the internalized or cell-membrane compartment(Fig. 8D). Therefore, ascorbate-stimulated 59Fe uptake from 59Fe2-Tf isnot due to up-regulation of TfRs or increased Tf uptake.

Fig. 7. The reducing ene-diol moiety of ascorbate is necessary for stimulation of 59Fe uptake from 59Fe2-Tf (A,B) and the effect of ascorbate is not due to a general increase in cellularreducing capacity (C,D). (A) Line drawings of the structures of the ascorbate analogues and degradation products assessed. (B) In these studies, SK-Mel-28 cells were pre-incubatedwith or without ascorbate or its analogues/degradation products (50 μM) for 30 min/37 °C before the addition of 59Fe2-Tf ([Tf] = 0.75 μM; [Fe] = 1.5 μM) to the same medium fora further 3 h/37 °C. (C) Alternatively, SK-Mel-28 cells were treated with either control medium, N-acetylcysteine (NAC; 5 mM) or buthionine sulfoximine (BSO; 10 μM) for 24 h/37 °C, following which cells were washed and re-incubated in fresh medium with/without ascorbate (50 μM) for 30 min, followed by the addition of 59Fe2-Tf as in (B) to the samemedium. In both (B) and (C), following incubation with 59Fe2-Tf, cells were washed on ice and incubated with Pronase (1 mg/mL) for 30 min/4 °C to obtain the internalized andcell-membrane fractions. (D) SK-Mel-28 cells were incubated with/without ascorbate (50 μM) as in (B), then Fe2-Tf (0.75 μM) or vehicle were added to the same medium for afurther 3 h/37 °C. Cells were harvested and total NADP (NADP-total) and NADPH levels were determined. Results are mean + SD (3 experiments).
1535D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
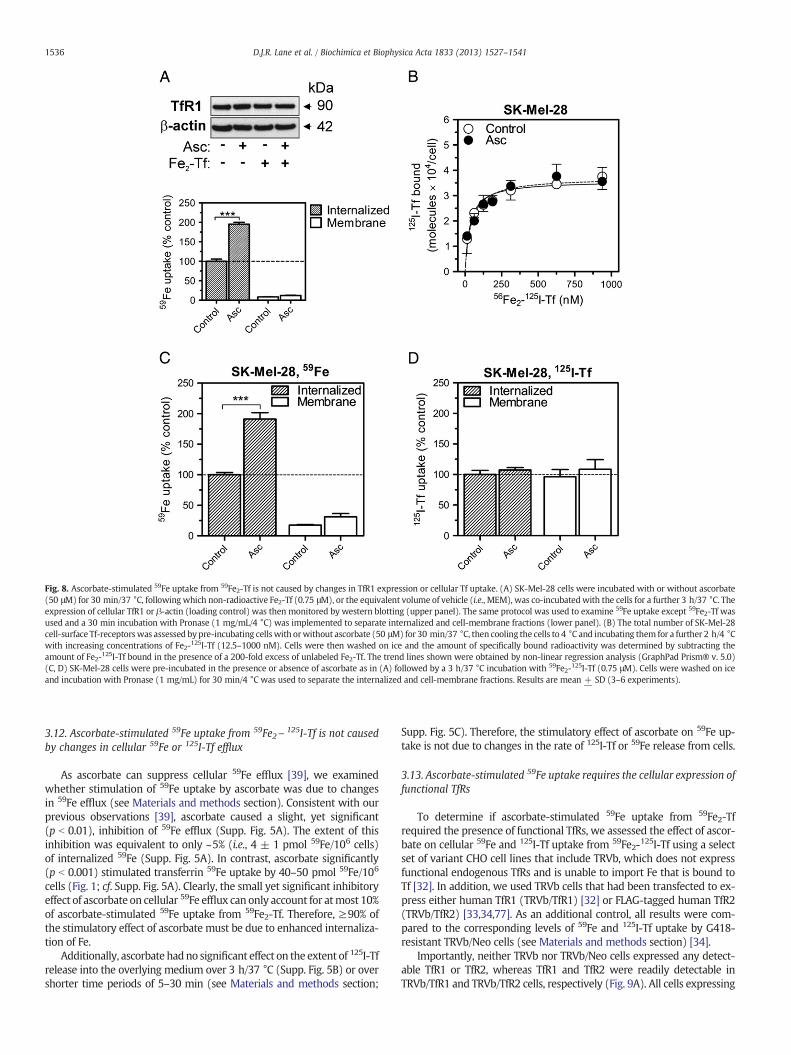
Fig. 8. Ascorbate-stimulated 59Fe uptake from 59Fe2-Tf is not caused by changes in TfR1 expression or cellular Tf uptake. (A) SK-Mel-28 cells were incubated with or without ascorbate(50 μM) for 30 min/37 °C, following which non-radioactive Fe2-Tf (0.75 μM), or the equivalent volume of vehicle (i.e., MEM), was co-incubatedwith the cells for a further 3 h/37 °C. Theexpression of cellular TfR1 or β-actin (loading control) was then monitored by western blotting (upper panel). The same protocol was used to examine 59Fe uptake except 59Fe2-Tf wasused and a 30 min incubation with Pronase (1 mg/mL/4 °C) was implemented to separate internalized and cell-membrane fractions (lower panel). (B) The total number of SK-Mel-28cell-surface Tf-receptorswas assessed by pre-incubating cellswith orwithout ascorbate (50 μM) for 30 min/37 °C, then cooling the cells to 4 °C and incubating them for a further 2 h/4 °Cwith increasing concentrations of Fe2-125I-Tf (12.5–1000 nM). Cells were then washed on ice and the amount of specifically bound radioactivity was determined by subtracting theamount of Fe2-125I-Tf bound in the presence of a 200-fold excess of unlabeled Fe2-Tf. The trend lines shown were obtained by non-linear regression analysis (GraphPad Prism® v. 5.0)(C, D) SK-Mel-28 cells were pre-incubated in the presence or absence of ascorbate as in (A) followed by a 3 h/37 °C incubation with 59Fe2-125I-Tf (0.75 μM). Cells were washed on iceand incubation with Pronase (1 mg/mL) for 30 min/4 °C was used to separate the internalized and cell-membrane fractions. Results are mean + SD (3–6 experiments).
1536 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
3.12. Ascorbate-stimulated 59Fe uptake from 59Fe2\125I-Tf is not caused
by changes in cellular 59Fe or 125I-Tf efflux
As ascorbate can suppress cellular 59Fe efflux [39], we examinedwhether stimulation of 59Fe uptake by ascorbate was due to changesin 59Fe efflux (see Materials and methods section). Consistent with ourprevious observations [39], ascorbate caused a slight, yet significant(p b 0.01), inhibition of 59Fe efflux (Supp. Fig. 5A). The extent of thisinhibition was equivalent to only ~5% (i.e., 4 ± 1 pmol 59Fe/106 cells)of internalized 59Fe (Supp. Fig. 5A). In contrast, ascorbate significantly(p b 0.001) stimulated transferrin 59Fe uptake by 40–50 pmol 59Fe/106
cells (Fig. 1; cf. Supp. Fig. 5A). Clearly, the small yet significant inhibitoryeffect of ascorbate on cellular 59Fe efflux can only account for atmost 10%of ascorbate-stimulated 59Fe uptake from 59Fe2-Tf. Therefore, ≥90% ofthe stimulatory effect of ascorbate must be due to enhanced internaliza-tion of Fe.
Additionally, ascorbate hadno significant effect on the extent of 125I-Tfrelease into the overlying medium over 3 h/37 °C (Supp. Fig. 5B) or overshorter time periods of 5–30 min (see Materials and methods section;
Supp. Fig. 5C). Therefore, the stimulatory effect of ascorbate on 59Fe up-take is not due to changes in the rate of 125I-Tf or 59Fe release from cells.
3.13. Ascorbate-stimulated 59Fe uptake requires the cellular expression offunctional TfRs
To determine if ascorbate-stimulated 59Fe uptake from 59Fe2-Tfrequired the presence of functional TfRs, we assessed the effect of ascor-bate on cellular 59Fe and 125I-Tf uptake from 59Fe2-125I-Tf using a selectset of variant CHO cell lines that include TRVb, which does not expressfunctional endogenous TfRs and is unable to import Fe that is bound toTf [32]. In addition, we used TRVb cells that had been transfected to ex-press either human TfR1 (TRVb/TfR1) [32] or FLAG-tagged human TfR2(TRVb/TfR2) [33,34,77]. As an additional control, all results were com-pared to the corresponding levels of 59Fe and 125I-Tf uptake by G418-resistant TRVb/Neo cells (see Materials and methods section) [34].
Importantly, neither TRVb nor TRVb/Neo cells expressed any detect-able TfR1 or TfR2, whereas TfR1 and TfR2 were readily detectable inTRVb/TfR1 and TRVb/TfR2 cells, respectively (Fig. 9A). All cells expressing

Fig. 9. Ascorbate-stimulated 59Fe uptake requires the expression of functional TfRs.(A) Whole-cell lysates were obtained and the solubilized proteins separated by SDS-PAGEand the expression of TfR1, TfR2 or β-actin (loading control) was assessed by westernblotting for the following variant Chinese Hamster Ovary (CHO) cells: TRVb (variant CHOcells devoid of endogenous TfRs; TRVb), or G418/neomycin-resistant TRVb cells transfectedwith either pcDNA3-FLAG (TRVb/Neo), human TfR1 (TRVb/TfR1) or FLAG-tagged humanTfR2 (TRVb/TfR2). (B,C) Cells were pre-incubated with/without ascorbate (50 μM) for30 min/37 °C before the addition of vehicle medium, ascorbate oxidase (Asc Ox; 10 U/mL)or FeTMPyP (30 μM) to the same medium for a further 30 min/37 °C. Then, 59Fe2\125I-Tf(0.75 μM) was added to the same medium and cells were incubated for a further3 h/37 °C. After this incubation, cells were washed on ice and then incubated withPronase (1 mg/mL) for 30 min/4 °C to obtain the internalized and cell-membranefractions. Results shown are mean + SD (3 experiments).
1537D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
TfRs (i.e., TRVb/TfR1 and TRVb/TfR2) accumulated significantly moreinternalized 59Fe and 125I-Tf from 59Fe2\125I-Tf (0.75 μM) than TRVb orTRVb/Neo cells (Fig. 9B,C). Indeed, the latter two cell lines accumulated
negligible levels of 59Fe under these conditions (Fig. 9B). Moreover,these cells also accumulated very low levels of 125I-Tf compared to theTfR-expressing cells (Fig. 9C). The TRVb/TfR1 cells accumulated thehighest levels of internalized 59Fe and 125I-Tf (Fig. 9B,C), which is consis-tent with the apparent primary function of TfR1 in Tf-dependent Feuptake [80]. Interestingly, TRVb/TfR2 cells accumulated substantiallylower levels of both radiolabels, albeit at levelsmarkedly and significantly(p b 0.01) higher than that of the TRVb or TRVb/Neo cells (Fig. 9B,C).Although the major function of TfR2 appears to be as a regulator of theexpression of the hormone of Fe metabolism, hepcidin [49], TfR2 mayalso play some role in Tf-dependent Fe uptake [34]. Indeed, the heterolo-gous expression of human TfR2 in TRVb cells has previously been shownto promote Tf-dependent 59Fe uptake in an analogous manner to TfR1under certain conditions [34].
Ascorbate significantly (p b 0.001) stimulated the uptake of 59Fe, butnot 125I-Tf, from 59Fe-125I-Tf exclusively in TRVb/TfR1 and TRVb/TfR2cells (Fig. 9B,C). Notably, and consistent with our previous data (Figs. 5and 6), the ascorbate-enhanced 59Fe uptake in TfR1-expressing cellswas insensitive to ascorbate oxidase, yet sensitive to the membrane-permeant ascorbate-oxidizing metalloporphyrin, FeTMPyP (Fig. 9B).
Further, ascorbate had no effect on 59Fe or 125I-Tf uptake by cellsnot expressing TfRs (i.e., TRVb or TRVb/Neo cells; Fig. 9B,C). Theobservation that ascorbate also enhanced Tf-dependent 59Fe uptakein TRVb/TfR2 cells indicates TfR2 is sufficient for ascorbate to stimu-late Tf-dependent 59Fe uptake from Tf. Therefore, the expressionof functional cellular TfRs is necessary for ascorbate-stimulatedTf-dependent 59Fe uptake to occur.
It should be noted here that, although the human TfR1 cDNA (viz.,“pCD-TR1”) used by McGraw et al. [32] to create CHO-TRVb/TfR1 cellscontains IREs in the 3′-untranslated region [81], the expression ofhuman TfR1 by these cells was not affected by ascorbate over the 3 hinterval, as shown by the equivalent 125I-Tf uptake by control andascorbate-treated cells (Fig. 9C). Thus, a change in human TfR1expression cannot account for the stimulatory effect of ascorbateon Tf-dependent 59Fe uptake in CHO-TRVb/TfR1 cells.
3.14. Ascorbate-stimulated Tf-dependent 59Fe uptake requires endocytosisand intravesicular acidification
As the major route of Tf-dependent Fe uptake occurs by thereceptor-mediated endocytic internalization of the Fe2-Tf complex[21,22], this process is likely to be required for the stimulatory effectof ascorbate on 59Fe uptake to occur.
First, to examine the involvement of endocytosis in Tf-dependent 59Feuptake by control and ascorbate-treated cells, we assessed the effect ofthe general inhibitor of endocytosis, phenylglyoxal [45,82]. Phenylglyoxal(5 mM) inhibits both 59Fe and 59I-Tf uptake from the endocytic internal-ization of 59Fe2-Tf by SK-Mel-28 cells [45]. In the present study, afterincubation of SK-Mel-28 cells with or without ascorbate, phenylglyoxal(5 mM) [45] was added to the same medium for a further 30 min/37 °Cbefore addition of 59Fe2-Tf. In these experiments, phenylglyoxal reducedboth control and ascorbate-stimulated 59Fe uptake from 59Fe2-Tf to~5% of the vehicle-treated cells (Fig. 10). These data suggest endocytosisof the TfR1-59Fe2-Tf complex is required for ascorbate-stimulatedTf-dependent 59Fe uptake.
Next, we assessed the effect on Tf-dependent 59Fe uptake ofthe lysosomotropic agents, chloroquine and NH4Cl [83]. Thesechemicals cause alkalinization of normally acidified intracellular vesi-cles (e.g., endosomes and lysosomes [83]) [84]. As endosomal acidifica-tion is required for the release of Fe from Fe2-Tf within the endosome[21,84,85], these agents inhibit Tf-dependent Fe uptake [21,45,84].The results presented in Fig. 10 clearly show that both chloroquine(0.5 mM) and NH4Cl (5 mM) markedly and significantly (p b 0.001)inhibited Tf-dependent 59Fe uptake in the presence or absence ofascorbate.

Fig. 10. Ascorbate-stimulated Tf-dependent 59Fe uptake requires endocytosis and intra-vesicular acidification. SK-Mel-28 cells were pre-incubated with/without ascorbate(50 μM) for 30 min/37 °C, then either: chloroquine (0.5 mM), NH4Cl (5 mM), bafilomycinA1 (170 nM) or phenylglyoxal (5 mM) were added and the incubation continued for30 min/37 °C. Then 59Fe2-Tf (0.75 μM)was added to the samemedium and the cells incu-bated for a further 3 h/37 °C. After this incubation, the cellmono-layerswerewashed on iceand then incubated with Pronase (1 mg/mL) for 30 min/4 °C to separate the internalizedand cell-membrane fractions. Results shown represent internalized 59Fe uptake and aremean + SD (3 experiments).
1538 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
In mammals, a bafilomycin A1-sensitive V-ATPase acidifies endo-somes and lysosomes at the expense of ATP [86]. The inhibition ofV-ATPase by bafilomycin A1 (150–250 nM) also causes endosomal al-kalinization that inhibits Tf-dependent 59Fe uptake [87,88]. Similarly tothe lysosomotropic agents, bafilomycin A1 (170 nM) significantly(p b 0.001) inhibited Tf-dependent 59Fe uptake (Fig. 10). Interestingly,in the presence of chloroquine, NH4Cl and bafilomycin A1, ascorbate stillstimulated 59Fe uptake by approximately 1.5–2-fold of the respectivecontrol value (Fig. 10). Thus, intravesicular alkalinization does not total-ly abolish ascorbate-stimulated 59Fe uptake from 59Fe2-Tf, which maybe due to residual 59Fe uptake under these conditions.
4. Discussion
4.1. A novel role for ascorbate in enhancing Fe uptake from Fe-Tf
For thefirst time,we have demonstrated that ascorbatemarkedly en-hances Fe uptake from the major plasma Fe donor, Fe-Tf, in human cells.This is an important finding, as ascorbate is a ubiquitous and normallyabundant cellular reductant in vivo, yet it is absent under standard cellculture conditions [8]. Significantly, typical physiological plasmaascorbate concentrations enhanced 59Fe uptake by up to 1.5–2-foldfrom 59Fe2-Tf, which was accompanied by a corresponding increasein cellular ferritin expression and ferritin-59Fe loading. Although ascor-bate can increase ferritin either by promoting de novo synthesis [51] orby inhibiting ferritin autophagy [55], we demonstrated that none ofthese processes were responsible for ascorbate's ability to stimulateTf-dependent 59Fe uptake. Indeed, these results support the generalnotion that ferritin does not contribute to Tf-dependent Fe uptakeper se, although it serves as the major Fe storage depot for nascentlyinternalized Fe.
4.2. The molecular mechanism involved in ascorbate-stimulated Feuptake from Fe2-Tf
To further dissect the ascorbate's mechanism-of-action, we firstadopted a comparative approach to determine key differences betweenthe ascorbate-stimulated uptake from Fe-citrate and Fe2-Tf. We deter-mined that, unlike 59Fe-citrate [4,19,20], the uptake of 59Fe from 59Fe2-Tf
was largely independent of: (i) the reductive action of extracellular ascor-bate; and (ii) the extracellular labilization of Fe from Fe2-Tf. In fact, the re-sults of our experiments with membrane-impermeant (e.g., ascorbateoxidase) andmembrane-permeant (e.g., FeTMPyP) ascorbate-oxidizing re-agents strongly suggest that ascorbate acts intracellularly to enhanceTf-dependent Fe uptake.
Consistent with these findings, and with the fact that most biolog-ical activities of ascorbate are mediated by the molecule's reducingactivity [1,70], our results demonstrated that the reducing ene-diolmoiety of ascorbate was required for stimulation of Fe uptake. Impor-tantly, we also showed that this was not due to a general increase incellular reducing capacity in the presence of ascorbate (Fig. 7C), or toa specific increase in NADPH. In fact, ascorbate decreased cellularNADPH (Fig. 7D). Taken together, our results suggest that ascorbateenhances Tf-dependent 59Fe uptake in a manner dependent on thismolecule.
Intriguingly, while ascorbate did not affect the cellular flux of Tf,another important finding was that the expression of human TfRs 1or 2 in CHO-TRVb cells was necessary for the stimulatory effect ofascorbate (Fig. 9B). We also showed that this stimulation by ascorbatewas probably mediated by intracellular ascorbate and was not theresult of a change in TfR1 or 2 expression. Our observation that ascor-bate stimulated Fe uptake in a TfR2-dependentmanner lends some sup-port to the notion that, in addition to acting as a regulator of hepcidinproduction and systemic Fe homeostasis [89], TfR2 may contribute tothe cellular acquisition of Fe from Fe-Tf [34,90]. However, any directrole for TfR2 in Tf-dependent Fe uptake is likely to be minor in compar-ison to TfR1 [91,92].
Finally, using an array of well-characterized inhibitors of endocy-tosis or endosome acidification, we demonstrated that endocytosisand intracellular vesicle acidification were important for the mecha-nism by which ascorbate enhances Tf-dependent Fe uptake(Fig. 10). Specifically, endocytosis was necessary for stimulation tooccur, while blockade of intracellular vesicle acidification inhibitedboth control and ascorbate-stimulated 59Fe uptake similarly, al-though the relative stimulation provided by ascorbate was not affect-ed (Fig. 10). Together, these results demonstrate that: (i) endocytosisis essential for ascorbate to stimulate Tf-dependent Fe uptake; and(ii) intracellular vesicle acidification facilitates both control andascorbate-stimulated Fe uptake from Fe2-Tf equally. These observa-tions show that ascorbate acts to enhance Tf-dependent Fe uptakedownstream of an endocytosis event, which is further enhanced by in-tracellular vesicle acidification.
Taken together, our results indicate that ascorbate acts intracellu-larly via a reductive mechanism to enhance Tf-dependent Fe deliveryof Fe to cells, which occurs subsequent to the TfR-dependent endocy-tosis of Fe2-Tf complexes (Fig. 11). Considering the mechanisticinsights provided herein, and the observation by others that ascorbatepromotes mobilization of 59Fe from isolated Tf-containing endosomes[29,93], we propose a model for ascorbate's activity in which ascorbateenhances intra-endosomal ferrireduction and, consequently, Fe mobili-zation from the Tf-containing endosome (Fig. 11). As suggested by stud-ies with isolated endosomes containing Tf, this could occur by directuptake of ascorbate into the endosomal vesicle followed by chemicalferrireduction [94], and/or by provision of reducing equivalents fromascorbate to an endosomal ferrireductase [29,93]. As the only confirmedferrireductase of the Tf-to-cell Fe uptake cycle is Steap3, which appearsto be most active in this role in erythroid cells [30], the possibility thatascorbate stimulates Steap3 activity, either directly or indirectly, shouldnow be examined. However, our findings that ascorbate did notincrease cellular NADPH (Fig. 7D), a putative electron source for Steap3'sferrireductase activity [30], or increase Steap3 expression (Supp. Fig. 4),suggest that ascorbate does not act via Steap3 to enhance Tf-dependentFe uptake. Therefore, the possibility that other as-yet-to-be-identifiedendosomal ferrireductases may utilize ascorbate as an electron sourcemust also be considered.

Fig. 11. Model for the action of ascorbate in stimulating cellular iron (Fe) uptake from transferrin (Tf). (A) Under control conditions, Fe2-Tf binds to cell surface Tf receptors (TfR) and isinternalized by endocytosis. The endosomes that form become acidified via the proton-pumping vacuolar-type H+-ATPase (V-ATPase). Acidification leads to release of Fe from Tf, whileFe-free Tf (apo-Tf) remains bound to the TfR. The labilized intra-endosomal Fe3+ is then reduced to Fe2+, probably by a reductase and transported across the endosomalmembrane by thedivalent metal transporter 1 (DMT1) and/or Zip14. The apo-Tf–TfR complex returns to the cell surface, where apo-Tf dissociates at neutral pH. After transport by DMT1/Zip14, Fe initiallyenters the labile Fe pool (LIP) and can be stored in ferritin. (B) In contrast, in the presence of ascorbate, the vitamin enhances: (i) Tf-dependent Fe uptake; (ii) ferritin expression; and(iii) Fe deposition in ferritin. Importantly, the ascorbate-dependent increase in the LIP probably causes increased ferritin synthesis. Our results indicate that ascorbate acts intracellularlyvia a reductive mechanism that probably (as indicated by dotted lines) either involves direct import of ascorbate into the Tf cycle endosome and direct or chemical ferrireduction and/orprovision of electrons to an endosomal ferrireductase to enhance Tf-dependent Fe delivery of Fe to cells.
1539D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
In conclusion, our novel finding that ascorbate stimulatesTf-dependent Fe uptake is significant for the following reasons: (i) thema-jority of in vitro studies on Tf-dependent Fe uptake have been withperformed with ascorbate-depleted cells; and (ii) severe ascorbate-deficiency in humans (i.e., scurvy), as well as experimentally-inducedascorbate deficiency in guinea pigs, causes an “anemia of scurvy”[11,12]. The observation that ascorbate enhances Fe uptake from Fe2-Tfin all human cells examined suggests that the many previous studieson cellular Fe metabolism and Tf-dependent Fe uptake may haveoverlooked the involvement of this important reductant. Moreover, theability of ascorbate to enhance Fe uptake from Fe2-Tf, which is thoughtto be the major donor of Fe to the erythropoietic compartment [22],could explain the metabolic defect that contributes to ascorbate-deficiency-induced anemia.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamcr.2013.02.010.
Acknowledgements
D.J.R.L thanks the Cancer Institute New South Wales for an EarlyCareer Fellowship [10/ECF/2-18] and the National Health and MedicalResearch Council (NHMRC) of Australia for an Early Career PostdoctoralFellowship [1013810]. D.R.R. thanks the NHMRC for a Senior PrincipalResearch Fellowship and Project Grants.
References
[1] C.L. Linster, E. Van Schaftingen, Vitamin C. Biosynthesis, recycling and degradationin mammals, FEBS J. 274 (2007) 1–22.
[2] J. Mandl, A. Szarka, G. Banhegyi, Vitamin C: update on physiology and pharmacology,Br. J. Pharmacol. 157 (2009) 1097–1110.
[3] H. Tsukaguchi, T. Tokui, B. Mackenzie, U.V. Berger, X.Z. Chen, Y. Wang, R.F. Brubaker,M.A. Hediger, A family of mammalian Na+-dependent L-ascorbic acid transporters,Nature 399 (1999) 70–75.
[4] D.J.R. Lane, A. Lawen, Non-transferrin iron reduction and uptake are regulated bytransmembrane ascorbate cycling inK562 cells, J. Biol. Chem. 283 (2008) 12701–12708.
[5] D.J.R. Lane, A. Lawen, Transplasma membrane electron transport comes in twoflavors, Biofactors 34 (2009) 191–200.
[6] D.J.R. Lane, A. Lawen, Ascorbate and plasma membrane electron transport—enzymesvs efflux, Free Radic. Biol. Med. 47 (2009) 485–495.
[7] D.J.R. Lane, S.R. Robinson, H. Czerwinska, A. Lawen, A role for Na+/H+ exchangersand intracellular pH in regulating vitamin C-driven electron transport across theplasma membrane, Biochem. J. 428 (2010) 191–200.
[8] H. Frikke-Schmidt, J. Lykkesfeldt, Keeping the intracellular vitamin C at a physiologi-cally relevant level in endothelial cell culture, Anal. Biochem. 397 (2010) 135–137.
[9] L.L. Dunn, Y.S. Rahmanto, D.R. Richardson, Iron uptake and metabolism in thenew millennium, Trends Cell Biol. 17 (2007) 93–100.
[10] A. Gosiewska, F. Mahmoodian, B. Peterkofsky, Gene expression of iron-relatedproteins during iron deficiency caused by scurvy in guinea pigs, Arch. Biochem.Biophys. 325 (1996) 295–303.
[11] E.V. Cox, M.J. Meynell, B.E. Northam, W.T. Cooke, The anaemia of scurvy, Am. J.Med. 42 (1967) 220–227.
[12] S.R. Mettier, W.B. Chew, The anemia of scurvy. Effect of vitamin C diet on bloodformation in experimental scurvy of guinea pigs, J. Exp. Med. 55 (1932) 971–980.
[13] N.G. Clark, N.F. Sheard, J.F. Kelleher, Treatment of iron-deficiency anemia complicatedby scurvy and folic acid deficiency, Nutr. Rev. 50 (1992) 134–137.
[14] E. Cacciola, U. Consoli, R. Giustolisi, Ascorbic acid deficiency may be a cause ofrefractoriness to iron-therapy in the treatment of iron-deficiency anemia,Haematologica 79 (1994) 96–97.
[15] E. Butensky, P. Harmatz, B. Lubin, Nutritional anemias, in: C. Duggan, J.B. Watkins,W.A. Walker (Eds.), Nurition in pediatrics: basic science, clinical applications, BCDecker Inc., Hamilton, Ontario, Canada, 2008, pp. 701–711.
[16] W.D. Sirover, A.A. Siddiqui, R.L. Benz, Beneficial hematologic effects of daily oralascorbic acid therapy in ESRDpatients with anemia and abnormal iron homeostasis:a preliminary study, Ren. Fail. 30 (2008) 884–889.
[17] B.D. Atanassova, K.N. Tzatchev, Ascorbic acid—important for iron metabolism,Folia Med. (Plovdiv) 50 (2008) 11–16.
[18] L. Hallberg, M. Brune, L. Rossander, Effect of ascorbic acid on iron absorption fromdifferent types of meals. Studies with ascorbic acid-rich foods and syntheticascorbic acid given in different amounts with different meals, Hum. Nutr. Appl. Nutr.140A (1986) 97–113.

1540 D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
[19] J.M. May, Z.-c. Qu, S. Mendiratta, Role of ascorbic acid in transferrin-independentreduction and uptake of iron by U-937 cells, Biochem. Pharmacol. 57 (1999)1275–1282.
[20] D.J.R. Lane, S.R. Robinson, H. Czerwinska, G.M. Bishop, A. Lawen, Two routes ofiron accumulation in astrocytes: ascorbate-dependent ferrous iron uptake viathe divalent metal transporter (DMT1) plus an independent route for ferriciron, Biochem. J. 432 (2010) 123–132.
[21] E.H. Morgan, Transferrin, biochemistry, physiology and clinical significance, Mol.Aspects Med. 4 (1981) 1–123.
[22] D.R. Richardson, D.J.R. Lane, E.M. Becker, M.L. Huang, M. Whitnall, Y.S. Rahmanto,A.D. Sheftel, P. Ponka, Mitochondrial iron trafficking and the integration of ironmetabolism between the mitochondrion and cytosol, Proc. Natl. Acad. Sci. U.S.A.107 (2010) 10775–10782.
[23] B.J. Iacopetta, E.H. Morgan, The kinetics of transferrin endocytosis and iron uptakefrom transferrin in rabbit reticulocytes, J. Biol. Chem. 258 (1983) 9108–9115.
[24] R.S. Ohgami, D.R. Campagna, A. McDonald, M.D. Fleming, The Steap proteins aremetalloreductases, Blood 108 (2006) 1388–1394.
[25] H. Gunshin, B. Mackenzie, U.V. Berger, Y. Gunshin, M.F. Romero, W.F. Boron, S.Nussberger, J.L. Gollan, M.A. Hediger, Cloning and characterization of a mammalianproton-coupled metal-ion transporter, Nature 388 (1997) 482–488.
[26] N. Zhao, J. Gao, C.A. Enns, M.D. Knutson, ZRT/IRT-like protein 14 (ZIP14) promotesthe cellular assimilation of iron from transferrin, J. Biol. Chem. 285 (2010)32141–33250.
[27] O. Kakhlon, Z.I. Cabantchik, The labile iron pool: characterization, measurement,and participation in cellular processes, Free Radic. Biol. Med. 33 (2002) 1037–1046.
[28] C.C. Philpott, Coming into view: eukaryotic iron chaperones and intracellular irondelivery, J. Biol. Chem. 287 (2012) 13518–13523.
[29] M.-T. Nunez, V. Gaete, J.A. Watkins, J. Glass, Mobilization of iron from endocyticvesicles. The effects of acidification and reduction, J. Biol. Chem. 265 (1990) 6688–6692.
[30] R.S. Ohgami, D.R. Campagna, E.L. Greer, B. Antiochos, A. McDonald, J. Chen, J.J.Sharp, Y. Fujiwara, J.E. Barker, M.D. Fleming, Identification of a ferrireductaserequired for efficient transferrin-dependent iron uptake in erythroid cells, Nat.Genet. 37 (2005) 1264–1269.
[31] F. Saletta, Y. Suryo Rahmanto, E. Noulsri, D.R. Richardson, Iron chelator-mediatedalterations in gene expression: identification of novel iron-regulated moleculesthat are molecular targets of hypoxia-inducible factor-1α and p53, Mol. Pharmacol.77 (2010) 443–458.
[32] T.E. McGraw, L. Greenfield, F.R. Maxfield, Functional expression of the humantransferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenoustransferrin receptor, J. Cell Biol. 105 (1987) 207–214.
[33] H. Kawabata, R. Yang, T. Hirama, P.T. Vuong, S. Kawano, A.F. Gombart, H.P.Koeffler, Molecular cloning of transferrin receptor 2. A newmember of the transferrinreceptor-like family, J. Biol. Chem. 274 (1999) 20826–20832.
[34] R.M. Graham, G.M. Reutens, C.E. Herbison, R.D. Delima, A.C. Chua, J.K. Olynyk, D.Trinder, Transferrin receptor 2 mediates uptake of transferrin-bound andnon-transferrin-bound iron, J. Hepatol. 48 (2008) 327–334.
[35] J.M. Vislisel, F.Q. Schafer, G.R. Buettner, A simple and sensitive assay for ascorbateusing a plate reader, Anal. Biochem. 365 (2007) 31–39.
[36] D.R. Richardson, E. Baker, The uptake of iron and transferrin by the human malignantmelanoma cell, Biochim. Biophys. Acta 1053 (1990) 1–12.
[37] A.S. McFarlane, Efficient trace-labelling of proteins with iodine, Nature 182 (1958) 53.[38] D. Richardson, E. Baker, Twomechanisms of iron uptake from transferrin bymelanoma
cells. The effect of desferrioxamine and ferric ammonium citrate, J. Biol. Chem.267 (1992) 13972–13979.
[39] D.R. Richardson, Role of ceruloplasmin and ascorbate in cellular iron release,J. Lab. Clin. Med. 134 (1999) 454–465.
[40] J. Gao, D.R. Richardson, The potential of iron chelators of the pyridoxalisonicotinoylhydrazone class as effectiveantiproliferative agents, IV: Themechanismsinvolved in inhibiting cell-cycle progression, Blood 98 (2001) 842–850.
[41] D.R. Richardson, P. Ponka, D. Vyoral, Distribution of iron in reticulocytes afterinhibition of heme synthesis with succinylacetone: examination of the intermediatesinvolved in iron metabolism, Blood 87 (1996) 3477–3488.
[42] D.R. Richardson, K. Milnes, The potential of iron chelators of the pyridoxalisonicotinoyl hydrazone class as effective antiproliferative agents II: the mechanismof action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone, Blood 89 (1997) 3025–3038.
[43] R.M. Evans, L. Currie, A. Campbell, The distribution of ascorbic acid between variouscellular components of blood, in normal individuals, and its relation to the plasmaconcentration, Br. J. Nutr. 47 (1982) 473–482.
[44] D. Richardson, E. Baker, The uptake of inorganic iron complexes by humanmelanomacells, Biochim. Biophys. Acta 1093 (1991) 20–28.
[45] D.R. Richardson, E. Baker, Two saturablemechanisms of iron uptake from transferrin inhumanmelanoma cells: the effect of transferrin concentration, chelators, and meta-bolic probes on transferrin and iron uptake, J. Cell. Physiol. 161 (1994) 160–168.
[46] S.C. Rumsey, M. Levine, Absorption, transport and disposition of ascorbic acid inhumans, J. Nutr. Biochem. 9 (1998) 116–130.
[47] M.U. Muckenthaler, B. Galy, M.W. Hentze, Systemic iron homeostasis and theiron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network,Annu. Rev. Nutr. 28 (2008) 197–213.
[48] A. Lawen, D.J.R. Lane, Mammalian iron homeostasis in health and disease: uptake,storage, transport, and molecular mechanisms of action, Antioxid. Redox Signal.(2013), http://dx.doi.org/10.1089/ars.2011.4271, (Epub ahead of print).
[49] M.W. Hentze, M.U. Muckenthaler, B. Galy, C. Camaschella, Two to tango: regulationof mammalian iron metabolism, Cell 142 (2010) 24–38.
[50] K.R. Bridges, K.E. Hoffman, The effects of ascorbic acid on the intracellular metabolismof iron and ferritin, J. Biol. Chem. 261 (1986) 14273–14277.
[51] I. Toth, K.R. Bridges, Ascorbic acid enhances ferritin mRNA translation by anIRP/aconitase switch, J. Biol. Chem. 270 (1995) 19540–19544.
[52] M. Goralska, J. Harned, L.N. Fleisher, M.C. McGahan, The effect of ascorbic acid andferric ammonium citrate on iron uptake and storage in lens epithelial cells, Exp.Eye Res. 66 (1998) 687–697.
[53] J.C. Kwok, D.R. Richardson, Examination of the mechanism(s) involved indoxorubicin-mediated iron accumulation in ferritin: studies using metabolicinhibitors, protein synthesis inhibitors, and lysosomotropic agents, Mol. Pharmacol.65 (2004) 181–195.
[54] M. Festa, A. Colonna, C. Pietropaolo, A. Ruffo, Oxalomalate, a competitive inhibitorof aconitase, modulates the RNA-binding activity of iron-regulatory proteins,Biochem. J. 348 (Pt. 2) (2000) 315–320.
[55] K.R. Bridges, Ascorbic acid inhibits lysosomal autophagy of ferritin, J. Biol. Chem.262 (1987) 14773–14778.
[56] I. De Domenico, D.M. Ward, J. Kaplan, Specific iron chelators determine the routeof ferritin degradation, Blood 114 (2009) 4546–4551.
[57] I. Toth, J.T. Rogers, J.A. McPhee, S.M. Elliott, S.L. Abramson, K.R. Bridges, Ascorbicacid enhances iron-induced ferritin translation in human leukemia and hepatomacells, J. Biol. Chem. 270 (1995) 2846–2852.
[58] D.R. Richardson, P. Ponka, Identification of a mechanism of iron uptake by cellswhich is stimulated by hydroxyl radicals generated via the iron-catalysedHaber–Weiss reaction, Biochim. Biophys. Acta 1269 (1995) 105–114.
[59] D.J.R. Lane, A. Lawen, A highly sensitive colorimetric microplate ferrocyanideassay applied to ascorbate-stimulated transplasmamembrane ferricyanide reductionand mitochondrial succinate oxidation, Anal. Biochem. 373 (2008) 287–295.
[60] E.H. Morgan, Studies on the mechanism of iron release from transferrin, Biochim.Biophys. Acta 580 (1979) 312–326.
[61] J.P. Crow, Manganese and iron porphyrins catalyze peroxynitrite decompositionand simultaneously increase nitration and oxidant yield: implications for theiruse as peroxynitrite scavengers in vivo, Arch. Biochem. Biophys. 371 (1999)41–52.
[62] K.M. Faulkner, S.I. Liochev, I. Fridovich, Stable Mn(III) porphyrins mimic superox-ide dismutase in vitro and substitute for it in vivo, J. Biol. Chem. 269 (1994)23471–23476.
[63] Y. Ilan, J. Rabani, I. Fridovich, R.F. Pasternack, Superoxide dismuting activity of aniron porphyrin, Inorg. Nucl. Chem. Lett. 17 (1981) 93–96.
[64] J. Lee, J.A. Hunt, J.T. Groves, Rapid decomposition of peroxynitrite by manganeseporphyrin-antioxidant redox couples, Bioorg.Med. Chem. Lett. 7 (1997) 2913–2918.
[65] M.P. Jensen, D.P. Riley, Peroxynitrite decomposition activity of iron porphyrincomplexes, Inorg. Chem. 41 (2002) 4788–4797.
[66] E. Flashman, S.L. Davies, K.K. Yeoh, C.J. Schofield, Investigating the dependence ofthe hypoxia-inducible factor hydroxylases (factor inhibitingHIF andprolyl hydroxylasedomain 2) on ascorbate and other reducing agents, Biochem. J. 427 (2010) 135–142.
[67] S.C. Rumsey, R.W. Welch, H.M. Garraffo, P. Ge, S.F. Lu, A.T. Crossman, K.L. Kirk, M.Levine, Specificity of ascorbate analogs for ascorbate transport. Synthesis and detectionof [125I]6-deoxy-6-iodo-L-ascorbic acid and characterization of its ascorbate-specifictransport properties, J. Biol. Chem. 274 (1999) 23215–23222.
[68] J.M. May, Z.C. Qu, C.E. Cobb, Accessibility and reactivity of ascorbate 6-palmitatebound to erythrocyte membranes, Free Radic. Biol. Med. 21 (1996) 471–480.
[69] J.M. May, Z.-c. Qu, R.R. Whitesell, Ascorbate is the major electron donor for atransmembrane oxidoreductase of human erythrocytes, Biochim. Biophys. Acta1238 (1995) 127–136.
[70] H. Asard, Ascorbate, in: R. Banerjee (Ed.), Redox Biochemistry, Wiley, Hoboken,2007, pp. 22–27.
[71] M. Fujiwara, N. Nagao, K. Monden, M. Misumi, K. Kageyama, K. Yamamoto, N.Miwa, Enhanced protection against peroxidation-induced mortality of aorticendothelial cells by ascorbic acid-2-O-phosphate abundantly accumulated inthe cell as the dephosphorylated form, Free Radic. Res. 27 (1997) 97–104.
[72] L.J. Machlin, F. Garcia, W. Kuenzig, C.B. Richter, H.E. Spiegel, M. Brin, Lack ofantiscorbutic activity of ascorbate 2-sulfate in the rhesus monkey, Am. J. Clin. Nutr.29 (1976) 825–831.
[73] I.A. Cotgreave, N-acetylcysteine: pharmacological considerations and experimentaland clinical applications, Adv. Pharmacol. 38 (1997) 205–227.
[74] Y. Yu, D.R. Richardson, Cellular iron depletion stimulates the JNK and p38 MAPKsignaling transduction pathways, dissociation of ASK1-thioredoxin, and activationof ASK1, J. Biol. Chem. 286 (2011) 15413–15427.
[75] S.C. McNeely, A.C. Belshoff, B.F. Taylor, T.W. Fan, M.J. McCabe Jr., A.R. Pinhas, J.C.States, Sensitivity to sodium arsenite in human melanoma cells depends uponsusceptibility to arsenite-induced mitotic arrest, Toxicol. Appl. Pharmacol. 229(2008) 252–261.
[76] J.C. Kwok, D.R. Richardson, Anthracyclines induce accumulation of iron in ferritinin myocardial and neoplastic cells: inhibition of the ferritin iron mobilizationpathway, Mol. Pharmacol. 63 (2003) 849–861.
[77] H. Kawabata, R.S. Germain, P.T. Vuong, T. Nakamaki, J.W. Said, H.P. Koeffler,Transferrin receptor 2-α supports cell growth both in iron-chelated culturedcells and in vivo, J. Biol. Chem. 275 (2000) 16618–16625.
[78] M.B. Johnson, C.A. Enns, Diferric transferrin regulates transferrin receptor 2 proteinstability, Blood 104 (2004) 4287–4293.
[79] A. Robb, M. Wessling-Resnick, Regulation of transferrin receptor 2 protein levelsby transferrin, Blood 104 (2004) 4294–4299.
[80] P. Aisen, Transferrin receptor 1, Int. J. Biochem. Cell Biol. 36 (2004) 2137–2143.[81] E.W. Mullner, L.C. Kuhn, A stem-loop in the 3′ untranslated region mediates
iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm,Cell 53 (1988) 815–825.
[82] M.L. van Schaik, R.S. Weening, D. Roos, Phenylglyoxal is not a selective inhibitorof phagocytosis, J. Cell Sci. 38 (1979) 331–343.

1541D.J.R. Lane et al. / Biochimica et Biophysica Acta 1833 (2013) 1527–1541
[83] O.A. Weisz, Acidification and protein traffic, Int. Rev. Cytol. 226 (2003)259–319.
[84] L.M. Sorokin, E.H. Morgan, G.C. Yeoh, Transferrin endocytosis and iron uptake indeveloping myogenic cells in culture: effects of microtubular and metabolic in-hibitors, sulphydryl reagents and lysosomotrophic agents, J. Cell. Physiol. 137(1988) 483–489.
[85] B. Mackenzie, M.A. Hediger, SLC11 family of H+-coupled metal-ion transportersNRAMP1 and DMT1, Pflugers Arch. 447 (2004) 571–579.
[86] T. Yoshimori, A. Yamamoto, Y. Moriyama, M. Futai, Y. Tashiro, Bafilomycin A1,a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification andprotein degradation in lysosomes of cultured cells, J. Biol. Chem. 266 (1991)17707–17712.
[87] E.J. Bowman, A. Siebers, K. Altendorf, Bafilomycins: a class of inhibitors of membraneATPases frommicroorganisms, animal cells, and plant cells, Proc. Natl. Acad. Sci. U.S.A.85 (1988) 7972–7976.
[88] L.S. Johnson, K.W. Dunn, B. Pytowski, T.E. McGraw, Endosome acidificationand receptor trafficking: bafilomycin A1 slows receptor externalization by amechanism involving the receptor's internalization motif, Mol. Biol. Cell 4 (1993)1251–1266.
[89] J. Gao, J. Chen, M. Kramer, H. Tsukamoto, A.S. Zhang, C.A. Enns, Interaction of thehereditary hemochromatosis protein HFE with transferrin receptor 2 is requiredfor transferrin-induced hepcidin expression, Cell Metab. 9 (2009) 217–227.
[90] M.B. Johnson, J. Chen, N. Murchison, F.A. Green, C.A. Enns, Transferrin receptor 2:evidence for ligand-induced stabilization and redirection to a recycling pathway,Mol. Biol. Cell 18 (2007) 743–754.
[91] C.E. Herbison, K. Thorstensen, A.C. Chua, R.M. Graham, P. Leedman, J.K. Olynyk, D.Trinder, The role of transferrin receptor 1 and 2 in transferrin-bound iron uptakein human hepatoma cells, Am. J. Physiol. Cell Physiol. 297 (2009) C1567–C1575.
[92] A.C. Chua, R.D. Delima, E.H. Morgan, C.E. Herbison, J.E. Tirnitz-Parker, R.M.Graham, R.E. Fleming, R.S. Britton, B.R. Bacon, J.K. Olynyk, D. Trinder, Iron uptakefrom plasma transferrin by a transferrin receptor 2 mutant mouse model ofhaemochromatosis, J. Hepatol. 52 (2010) 425–431.
[93] J.A. Watkins, J.D. Altazan, P. Elder, C.-Y. Li, M.-T. Nunez, X.-X. Cui, J. Glass, Kineticcharacterization of reductant dependent processes of iron mobilization fromendocytic vesicles, Biochemistry 31 (1992) 5820–5830.
[94] A. Escobar, V. Gaete, M.T. Nunez, Effect of ascorbate in the reduction oftransferrin-associated iron in endocytic vesicles, J. Bioenerg. Biomembr. 24 (1992)227–233.