therapeutic options in inborn errors of metabolism (iem) · pdf filetoxic compounds...
TRANSCRIPT

BeSHG Course 2016
8 January 2016
Center for Inherited Metabolic Diseases
Institut de Pathologie et de Génétique (IPG)
Therapeutic options in Inborn Errors of Metabolism (IEM)
Toxic compounds
Accumulation
Substrate
Deficiency
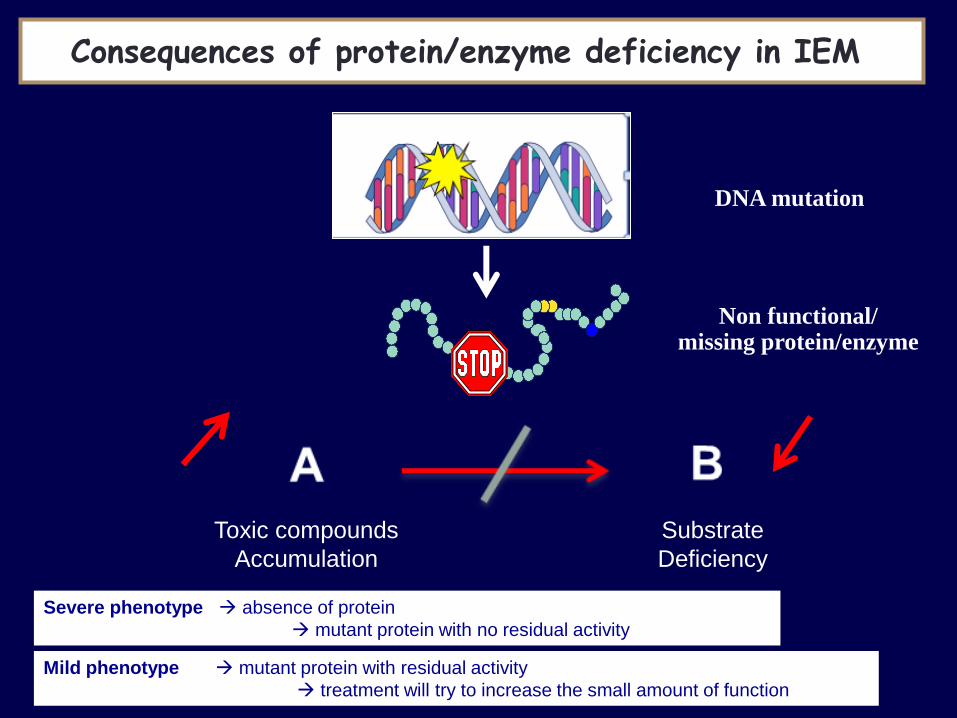
Consequences of protein/enzyme deficiency in IEM
DNA mutation
Non functional/ missing protein/enzyme
Severe phenotype absence of protein
mutant protein with no residual activity
Mild phenotype mutant protein with residual activity
treatment will try to increase the small amount of function

Prenatal symptoms (Echo, RMN)
Neonatal screening
Acute neonatal symptoms
Symptoms after a « free interval »
Specific symptoms (eyes, skin, liver, heart, kidney,..)
Chronic/progressive symptoms (failure to thrive, neurologic
deterioration, ..)
Persistent and without explanation symptoms
Diagnostic circumstances of IEM
For the same enzymatic defect
Neonatal / late onset/ asymtomatic
Groupe 1 Complex Molecules
Groupe 2 Intermediary metabolism : Intoxication type
Groupe 3
Intermediary metabolism : Energy depletion
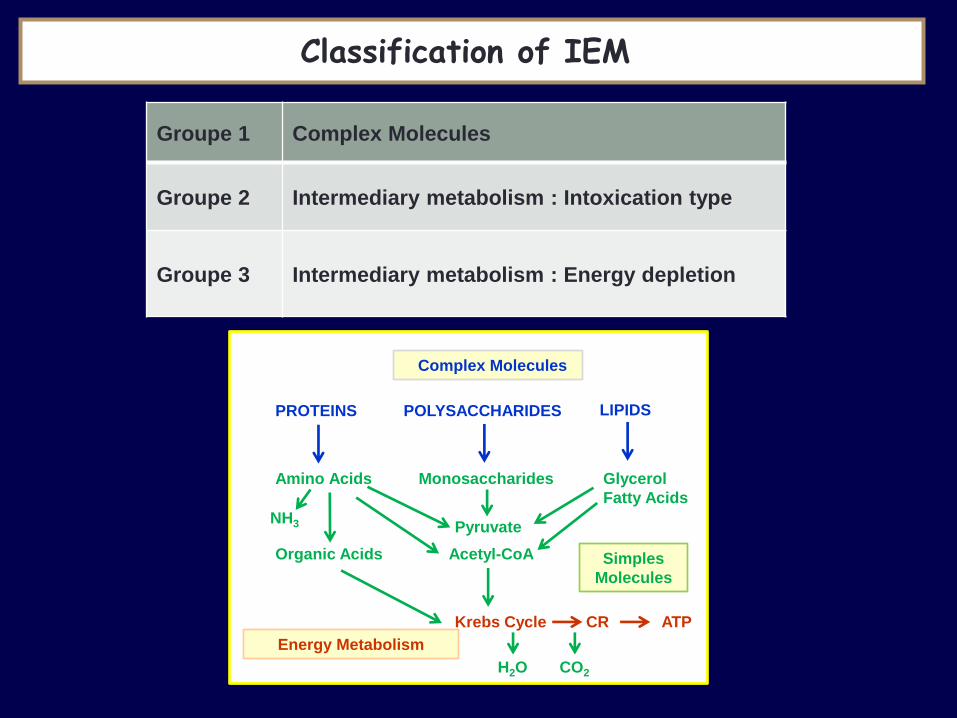
Classification of IEM
Complex Molecules
PROTEINS POLYSACCHARIDES LIPIDS
Amino Acids Monosaccharides Glycerol
Fatty Acids
Organic Acids
Pyruvate
Acetyl-CoA Simples
Molecules
NH3
Krebs Cycle CR ATP
Energy Metabolism
H2O CO2
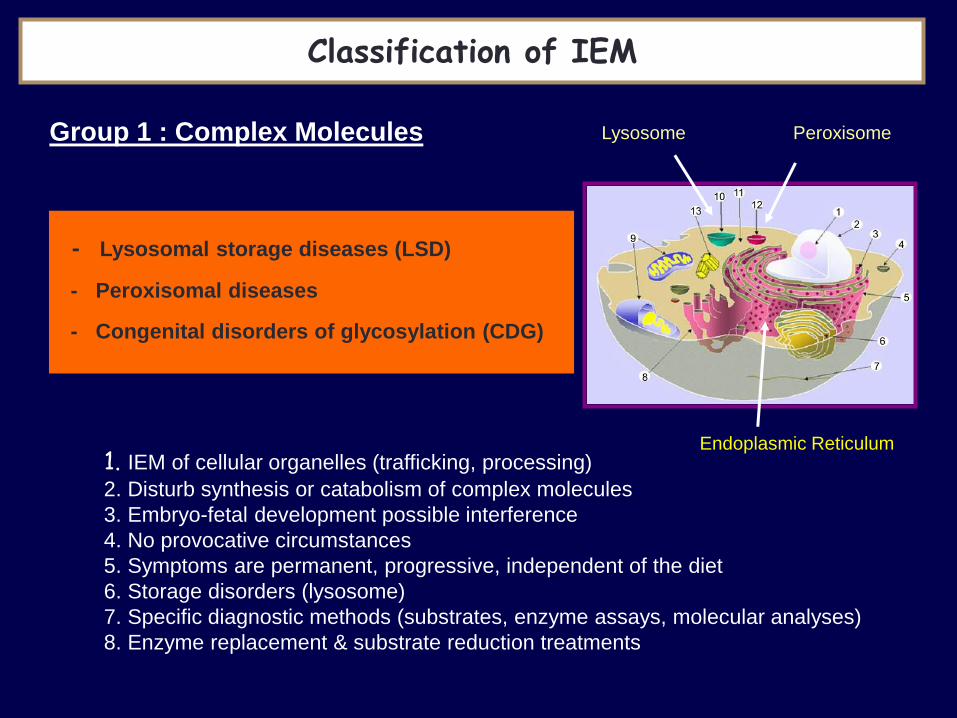
1. IEM of cellular organelles (trafficking, processing)
2. Disturb synthesis or catabolism of complex molecules
3. Embryo-fetal development possible interference
4. No provocative circumstances
5. Symptoms are permanent, progressive, independent of the diet
6. Storage disorders (lysosome)
7. Specific diagnostic methods (substrates, enzyme assays, molecular analyses)
8. Enzyme replacement & substrate reduction treatments
Lysosome Peroxisome
Endoplasmic Reticulum
Classification of IEM
- Lysosomal storage diseases (LSD) - Peroxisomal diseases - Congenital disorders of glycosylation (CDG)
Group 1 : Complex Molecules

(simple molecules) A B
- Aminoacidopathy (PKU, Tyrosinemia, …)
- Urea cycle defect (UCD)
- Organic aciduria (MMA, IVA, PA, …)
- Carbohydrate (galactosemia, hereditary fructose intolerance)
1. Accumulation of toxic compounds
2. No consequence on embryo-fetal development
3. Symptom free interval
4. Clinical signs of intoxication (acute or chronic)
5. Provocative circumstances (catabolism, food intake)
6. Can be late in onset and intermittent
7. Diagnosis based on plasma / urine analyses
8. Most are treatable (extra-corporeal procedures, special diets, « cleaning drugs »)
Group 2 : Intermediary metabolism – Intoxication type
Classification of IEM

- Fatty acids ß-oxidation defects
- Ketone bodies synthesis defects
- Neoglucogenesis defects
- Mitochondrial diseases (Krebs cycle defects and oxidative phosphorylation)
1. Deficiency in energy production or utilization
2. Main target organs (liver, heart, muscle, brain)
3. Embryo-fetal development possible interference
4. Provocative circumstances (catabolism, food)
5. Diagnosis is difficult (function tests, enzymatic assays on various tissues, molecular analyses)
6. Only few are amenable to treatment
Group 3 : Intermediary metabolism – Energy depletion
Classification of IEM
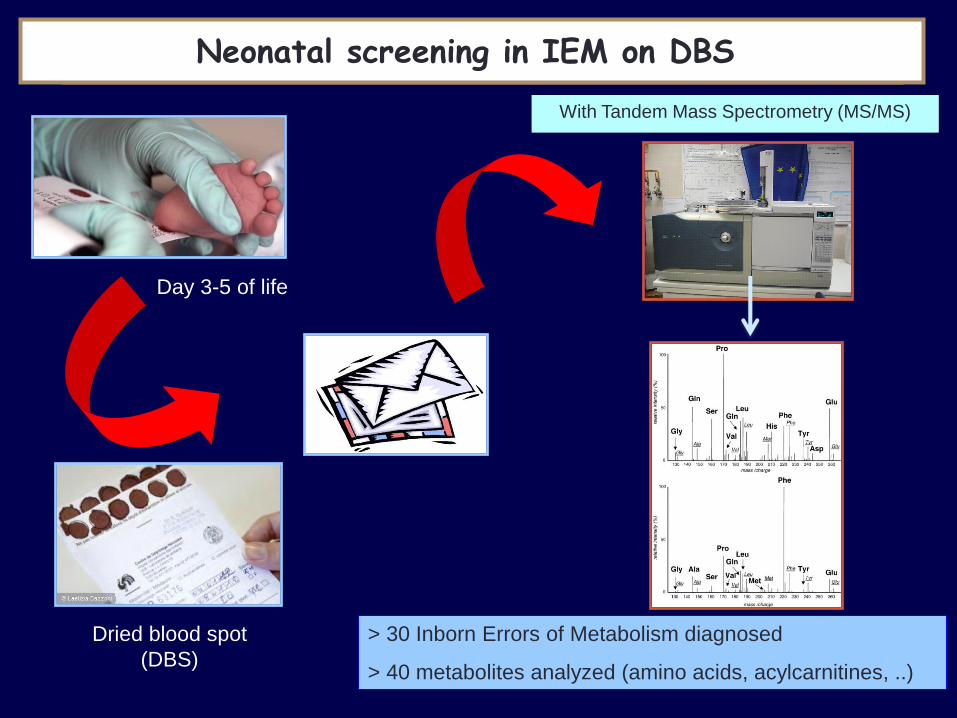
Day 3-5 of life
NEONATAL SCREENING in IEM Neonatal screening in IEM on DBS
Dried blood spot
(DBS)
With Tandem Mass Spectrometry (MS/MS)
> 30 Inborn Errors of Metabolism diagnosed
> 40 metabolites analyzed (amino acids, acylcarnitines, ..)

• « Consider IEM in parallel with other more common conditions
• Be aware of symptoms that persist and remain unexplained after initial
treatment.
• Don’t confuse a symptom or a syndrome with etiology.
• IEM can present at any age from fetal life to old age.
• Although most IEM are autosomal recessive disorders, majority of
cases appear sporadic.
• Take care first of the patient (emergency treatment), and then of the
family (genetic counselling).
• Initially consider IEM amenable to treatment : »
DON’T MISS A TREATABLE disorder
When to think about IEM ?

Therapeutic approaches to IEM
Pharmacologic enzyme replacement Substrate reduction
Correcting product deficiency
- Replenish depleted product
- Increasing substance supply
- Providing alternate substrate
Transplantation
- Inhibition of enzyme
within pathway :
e.g NTBC in TYR1
- Dietary restriction :
e.g. PKU, galactosemia
Decreasing metabolic toxicity
- Removing toxic metabolite
- Blocking the effect of toxic metabolites
- Co-enzyme treatment
- Enzyme enhancement therapy
Stimulation residual enzyme
A B -
+
- Hematopoietic Stem cell Transfer
- Other organ transplantation
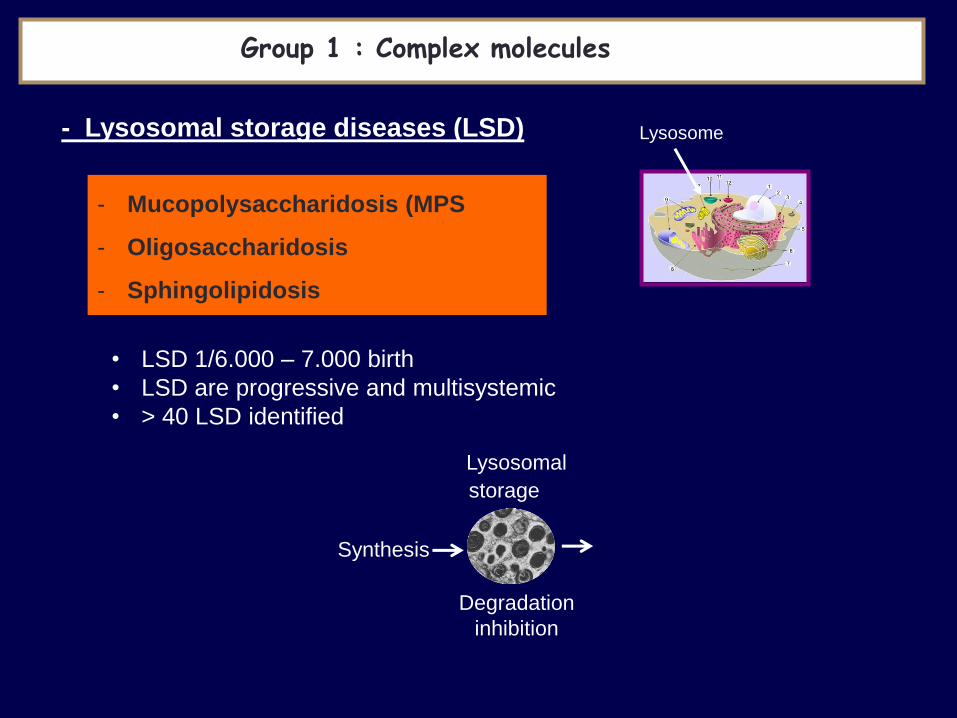
Lysosome
- Mucopolysaccharidosis (MPS
- Oligosaccharidosis
- Sphingolipidosis
Group 1 : Complex molecules
- Lysosomal storage diseases (LSD)
• LSD 1/6.000 – 7.000 birth
• LSD are progressive and multisystemic
• > 40 LSD identified
Synthesis
Degradation
inhibition
Lysosomal
storage
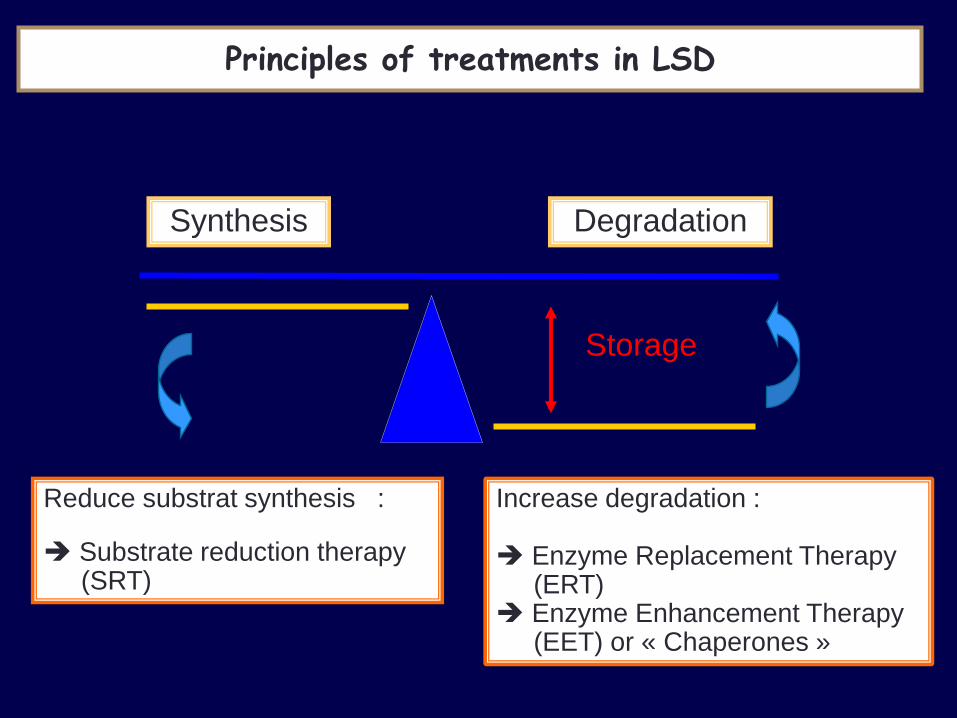
Reduce substrat synthesis :
Substrate reduction therapy (SRT)
Synthesis Degradation
Storage
Principles of treatments in LSD
Increase degradation : Enzyme Replacement Therapy (ERT) Enzyme Enhancement Therapy (EET) or « Chaperones »
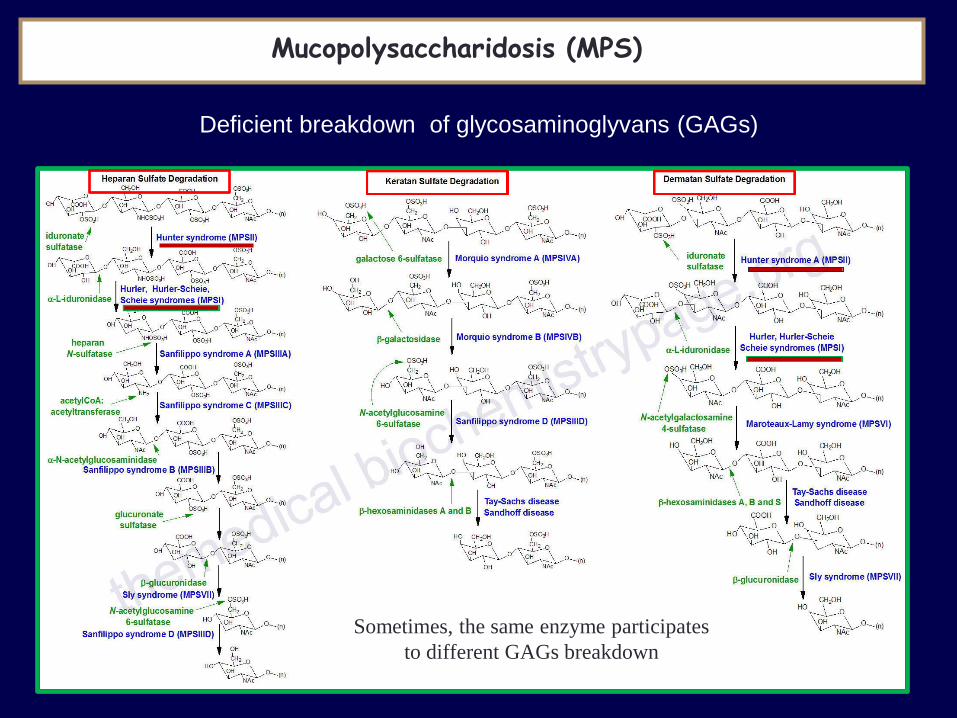
Deficient breakdown of glycosaminoglyvans (GAGs)
Dermatan sulfate degradation Incidence 1/100.000
Mucopolysaccharidosis (MPS)
Sometimes, the same enzyme participates
to different GAGs breakdown

Biochemical Diagnosis in MPS
• The primary diagnostic test for MPS disorders is ALWAYS urine
glycosaminoglycans (GAGs) analysis.
• 10mls urine is required.
• Quantitative analysis. Total GAGs concentration is measured using
Dimethylene Blue (DMB).
• False negative exists mainly in attenuated forms of MPS
Total Glycosaminoglycans
age mg/g creat
<6 mo 135 460
6-12 mo 134 279
1-2 y 81 265
2-4 y 68 188
4-6 y 67 127
6-8 y 50 114
8-10 y 46 103
10-15 y 30 94
15-20 y 13 59
>20 y 13 45

CS
CS : Chondroïtine sulfate
HS : Heparan sulfate
DS : Dermatan sulfate
CS
CS DS
HS
HS
• Qualitative test establishes the identity of the GAGs present
• GAGs are extracted from 2ml urine and precipitated
with Alcian Blue.
• Extract is washed and analysed by 2D electrophoresis.
DS
Biochemical Diagnosis in MPS

Biochemical Diagnosis in MPS – GAGs and enzyme deficiency
Genet. Mol. Biol. 1998, vol. 21 n.1, 1678 TLC : thin layer chromatography
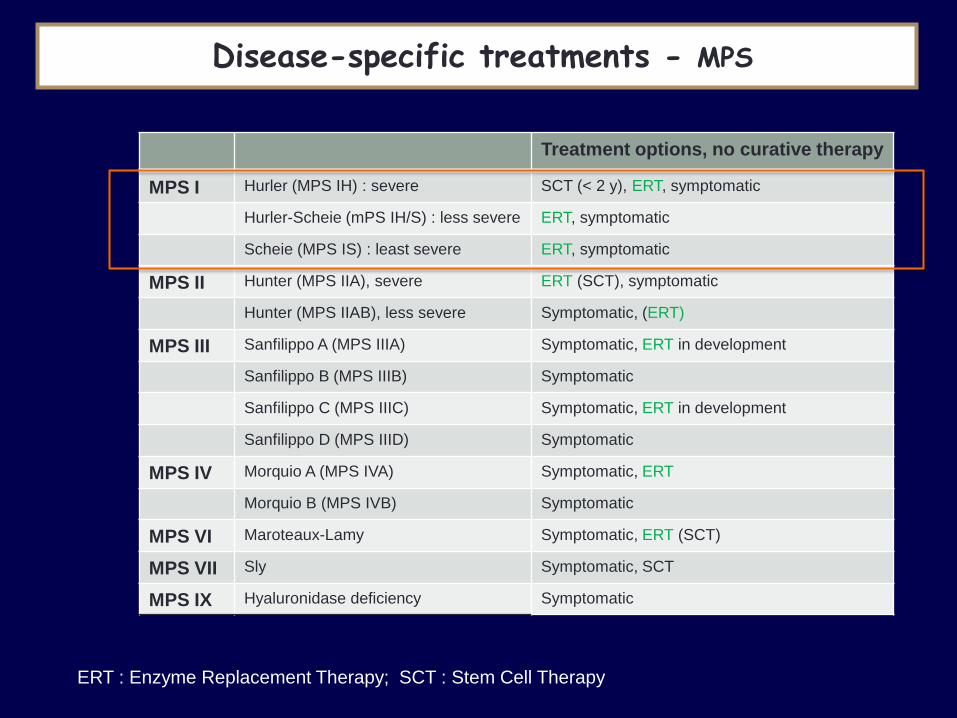
Disease-specific treatments - MPS
Treatment options, no curative therapy
MPS I Hurler (MPS IH) : severe SCT (< 2 y), ERT, symptomatic
Hurler-Scheie (mPS IH/S) : less severe ERT, symptomatic
Scheie (MPS IS) : least severe ERT, symptomatic
MPS II Hunter (MPS IIA), severe ERT (SCT), symptomatic
Hunter (MPS IIAB), less severe Symptomatic, (ERT)
MPS III Sanfilippo A (MPS IIIA) Symptomatic, ERT in development
Sanfilippo B (MPS IIIB) Symptomatic
Sanfilippo C (MPS IIIC) Symptomatic, ERT in development
Sanfilippo D (MPS IIID) Symptomatic
MPS IV Morquio A (MPS IVA) Symptomatic, ERT
Morquio B (MPS IVB) Symptomatic
MPS VI Maroteaux-Lamy Symptomatic, ERT (SCT)
MPS VII Sly Symptomatic, SCT
MPS IX Hyaluronidase deficiency Symptomatic
ERT : Enzyme Replacement Therapy; SCT : Stem Cell Therapy

Age at diagnosis 0.2–7 years 0.2–36 years 2–54 years
Effect on cognition Pronounced mental delay with
loss of acquired skills
No/mild mental delay;
learning disabilities
No
impairment
Mean life expectancy
(untreated) 7 years Approximately
20 years
Adulthood
HURLER HURLER-SCHEIE SCHEIE
SEVERE FORM ATTENUATED FORM
Phenotype distribution* ~65% ~25% ~10%
*based on Moore et al. Orphanet J Rare Dis 2008;3:24 and MPS I Registry data
Treatment according to clinical phenotype in MPS I

*Prediction of disease severity based on clinical
picture, neurodevelopmental testing, genotype and
other relevant information
2 years of age
DQ < 70
MPS I Diagnosis
> 2 years of age
DQ 70 Severe or unknown
phenotype predicted
Attenuated
phenotype predicted*
DQ < 70 DQ 70
ERT (Consider
HSCT
in rare cases)
ERT ERT
Consider
ERT
Consider
HSCT
Treatment algorithm for MPS I
From Muenzer et al. Pediatrics 2009;123:19-29
DQ = Developmental quotient
HSCT = Hematopoietic stem cell transplant
Treatment is adapted to phenotype
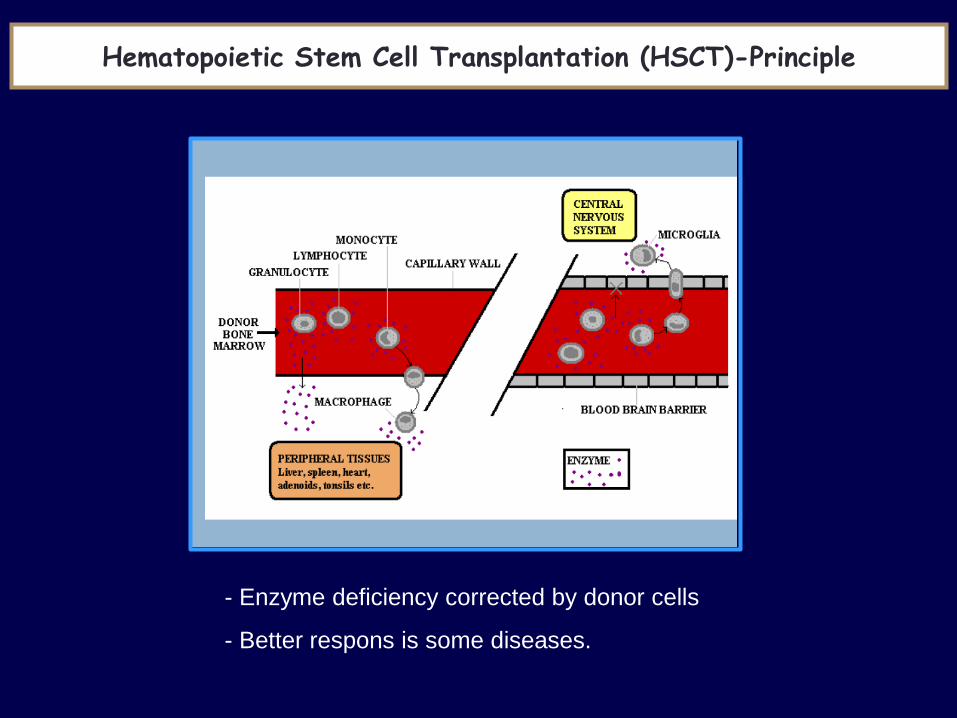
Hematopoietic Stem Cell Transplantation (HSCT)-Principle
- Enzyme deficiency corrected by donor cells
- Better respons is some diseases.

Stem cells are self-renewing cells defined by 2 properties
Stem cell transplantation
1. Ability to proliferate to form the differentiated cell types of a
tissue in vivo
2. Ability to self-renew to form another stem cell
Origin : embryonic, fetal, cord blood, adult
Stem cell transplantation
iPSC cells: Induced-pluripotent stem cells

Stem cell
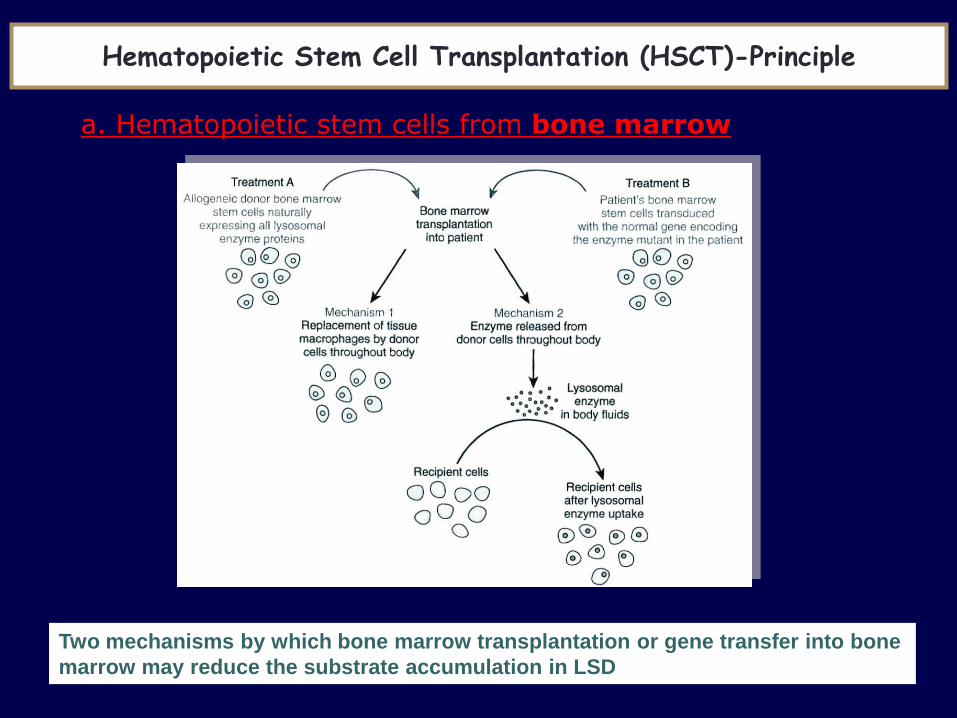
Two mechanisms by which bone marrow transplantation or gene transfer into bone
marrow may reduce the substrate accumulation in LSD
Hematopoietic Stem Cell Transplantation (HSCT)-Principle
a. Hematopoietic stem cells from bone marrow

• Limitations
– Early diagnosis (before irreversible brain damage)
– Matching Donor
– Procedure-related mortality and morbidity
– Variable results on brain and bone
– Do not cure the disease but changes the natural history
BMT - Limitations and Evolution
b. Hematopoietic stem cells from placental cord blood
- Increased tolerance of histoincompatible donor cells
- Reduced risk of graft-versus host disease
- Widely available

HSCT (since 1981) ERT (since 2003)
Patients • Hurler patients <2y who have a
DQ >70
• Due to risks, not recommended
for attenuated forms
• Hurler patients who are not
candidates for HSCT
• Hurler-Scheie and Scheie
patients
Risks • 5-30% mortality
• 60 % complication
• 10-15 % failure
• 40 % risk of mild to moderate
infusion associated reactions
during first 6 months of
treatment
• Very small risk of life-
threatening infusion reactions
Benefits • If performed early enough can
preserve neurocognition
• Prolongs survival
• Some somatic benefits
• No infusion needed
• Some somatic benefits
• For all disease phenotype
• As adjuvant treatment before
HSCT to improve pre-
transplant clinical condition
and engraftment
Limits • Must be done < 2 y • Does not cross BBB
• Weekly infusions for life
Disease-specific treatments in MPS I
BBB : Blood Brain Barrier

Enzyme replacement therapies in LSD
Disease ERT
Gaucher type 1 Cerezyme® (Imiglucerase)
VIPRIV® (Velaglucerase α)
Elelyso® (Taliglucerase α)
CHO cells
Human cells
Carrot cells
Fabry disease Replagal® (agalsidase α)
Fabrazyme® (agalsidase β)
Human cells
CHO cells
MPS I (H,HS,S) Aldurazyme® CHO cells
MPS II Elaprase® CHO cells
MPS VI Naglazyme® CHO cells
Pompe disease Myozyme® CHO cells
CHO cells : Chinese Hamster Ovary cells
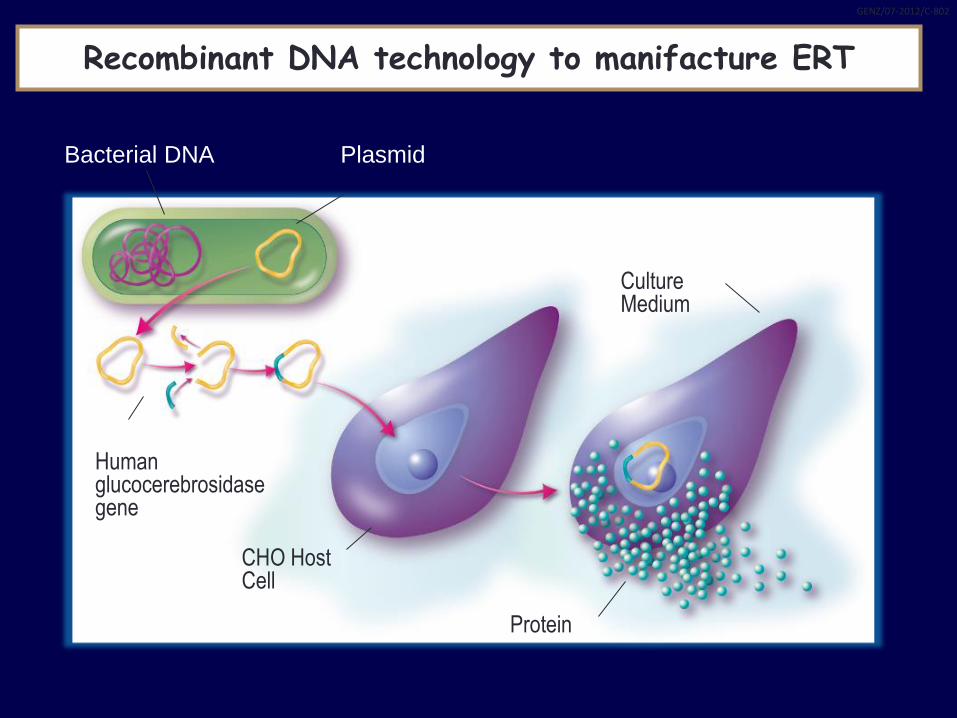
Bacterial DNA Plasmid
Culture Medium
Protein
CHO Host Cell
Human glucocerebrosidase gene
GENZ/07-2012/C-802
Recombinant DNA technology to manifacture ERT
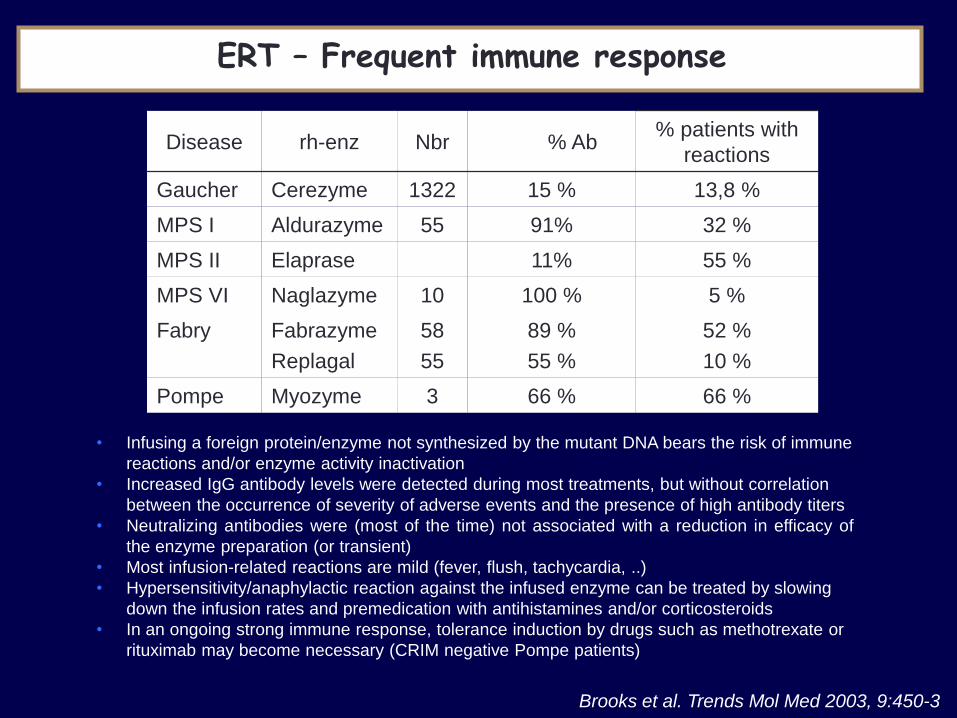
Disease rh-enz Nbr % Ab % patients with
reactions
Gaucher Cerezyme 1322 15 % 13,8 %
MPS I Aldurazyme 55 91% 32 %
MPS II Elaprase 11% 55 %
MPS VI Naglazyme 10 100 % 5 %
Fabry Fabrazyme
Replagal
58
55
89 %
55 %
52 %
10 %
Pompe Myozyme 3 66 % 66 %
Brooks et al. Trends Mol Med 2003, 9:450-3
ERT – Frequent immune response
• Infusing a foreign protein/enzyme not synthesized by the mutant DNA bears the risk of immune
reactions and/or enzyme activity inactivation
• Increased IgG antibody levels were detected during most treatments, but without correlation
between the occurrence of severity of adverse events and the presence of high antibody titers
• Neutralizing antibodies were (most of the time) not associated with a reduction in efficacy of
the enzyme preparation (or transient)
• Most infusion-related reactions are mild (fever, flush, tachycardia, ..)
• Hypersensitivity/anaphylactic reaction against the infused enzyme can be treated by slowing
down the infusion rates and premedication with antihistamines and/or corticosteroids
• In an ongoing strong immune response, tolerance induction by drugs such as methotrexate or
rituximab may become necessary (CRIM negative Pompe patients)

SRT are efficient if there is persistant residual
degradation activity
Synthesis Degradation
Storage SRT
• Possible application on glycosphingolipids synthesis
• Application with Gaucher disease
Substrate Reduction Therapy (SRT) in LSD - principle

Glycosphingolipids Storage Diseases
Glycosphingolipids
storage Gaucher
cell
Glucocerebrosidase
Heterogeneous group of diseases

Charrow J et al., Arch Intern Med 2000;160:2835.
Heterogeneity in clinical
presentation
Clinical diagnosis at every age
Pathologic fracture (15%)
Osteonecrosis (25%)
Osteopenia (42%)
Anemia (64%)
Thrombocytopenia (56%)
Hepatomegaly (79%)
Splenomegaly (87%)
Bone pain (63%)
Bone crisis (33%)
Joint collapse (8%)
General symptoms:
• Fatigue
• Easy bruising/bleeding
• Menorraghia
• Decreased appetite
• Abdominal pain
• Growth retardation
• Slow pubertal development
Bone marrow infiltration
(40%)
Interstitial Pulmonary
fibrosis
Erlenmeyer flask
deformity (46%)
Multisystemic symptoms in Gaucher Disease

Phenotypic continuum in Gaucher Disease
Non-neuronopathic GD
~ 95 %
Asymptomatic
Skeletal disease
Visceral disease
2e neurologic
involvement
Parkinsonian
Manifestations
Eye movement
disorder
Hydrocephalus,
cardiac valve
calcifications
Myoclonic
epilepsy
Progressive
neurological
degeneration
Congenital
icthyosis
Hydrops
fetalis
Neurological manifestations Type 1
Type 3 Type 2
Neuronopathic GD
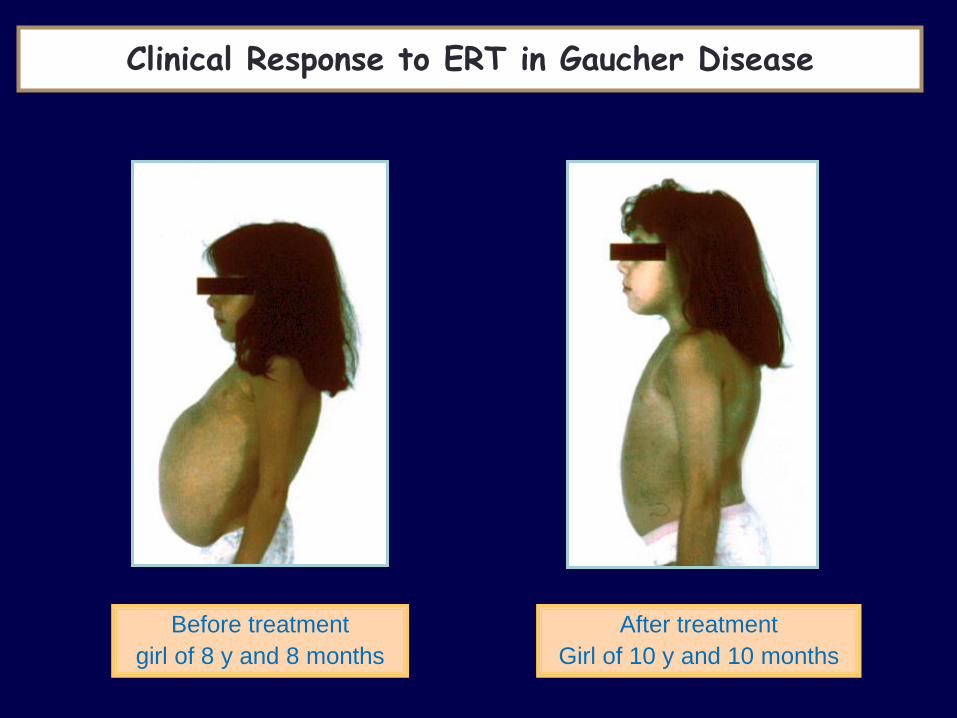
Before treatment
girl of 8 y and 8 months
After treatment
Girl of 10 y and 10 months
Clinical Response to ERT in Gaucher Disease

n=420 n=458
Liver Volume Spleen Volume
Andersson et al, Pediatrics 2008;122(6):1182-1190S
n=768
n=771
Platelet Count Hemoglobin Level
Visceral organ and
hematologic responses to
ERT treatment in children
Clinical Response to ERT in Gaucher Disease

No brain access - tried in Gaucher type II (neurologic form)
Immune response
Limited results on bone
Limitations of ERT
Intravenous infusion therapy
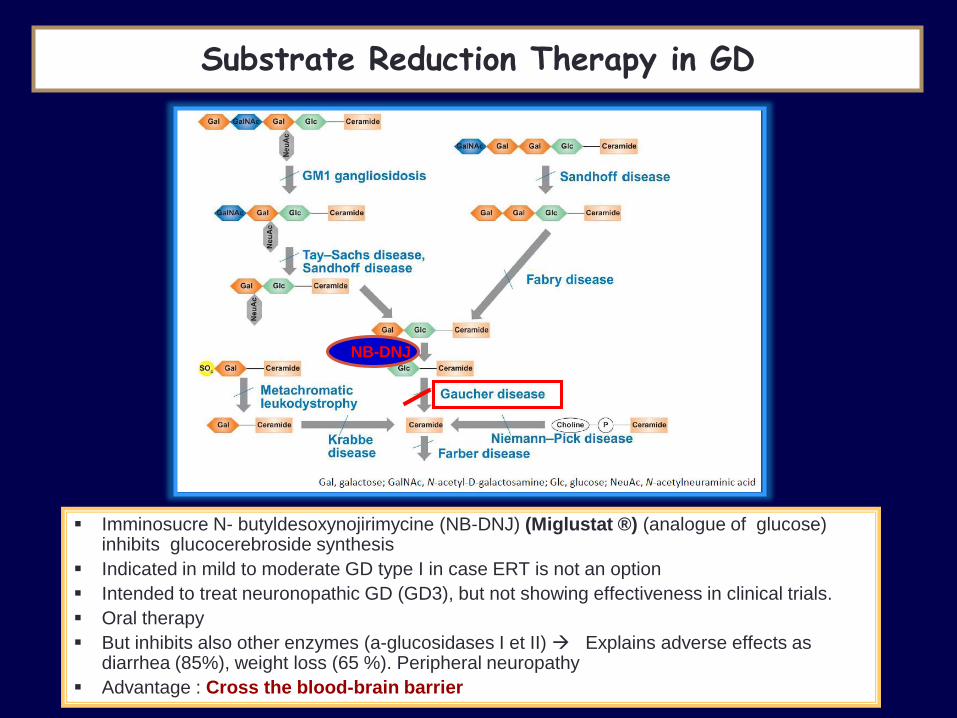
NB-DNJ
Substrate Reduction Therapy in GD
Imminosucre N- butyldesoxynojirimycine (NB-DNJ) (Miglustat ®) (analogue of glucose) inhibits glucocerebroside synthesis
Indicated in mild to moderate GD type I in case ERT is not an option
Intended to treat neuronopathic GD (GD3), but not showing effectiveness in clinical trials.
Oral therapy
But inhibits also other enzymes (a-glucosidases I et II) Explains adverse effects as diarrhea (85%), weight loss (65 %). Peripheral neuropathy
Advantage : Cross the blood-brain barrier
NB-DNJ

Substrate reduction therapy in GD
Eliglustat ® , new SRT
Approved for GD type I (FDA 2004, EMEA 2015 approved)
First line oral therapy in mild to moderate GD type 1
Effects : reduction of liver and spleen volume, increase of hemoglobin and platelet count
Effects on bone (more rapid than with ERT)
Adverse effects (≥10%): arthralgia, headache, nausea, fatigue, back pain, pain in extremities.
Contraindications: Dosage adapted to metaboliser profil (poor (PM)/intermediate (IM)/extensive (EM).
Electrocardiographic changes and potential cardiac arrhythmias. Not recommended in patients with pre-existing cardiac disease, long QT syndrome, and concomitant use of class Ia and class III antiarrhythmics.
Limits : Does not Cross the blood-brain barrier

Normal « misfolding »
chaperones specific of active site
Enzyme Enhancement therapy or pharmacological « charperone »

• Novel treatment concept in development for Fabry, Pompe, Gaucher disease
• Competitive inhibitor of the enzyme at subinhibitory concentrations can act as a
chemical chaperone leading to correct conformation of the mutant enzyme
Advantage :
- Orally administered
- Better biodistribution profile in comparison with recombinant enzyme
- Binds to amenable mutant forms of enzyme and increases trafficking
to lysosomes
- Chaperons are not immunogenic
Disadvantage :
- Treatment effect is restricted to patients with amenable mutations.
- Only 10–15% Pompe patients are amenable to chaperons, 30-50%
of currently diagnosed Fabry patients are believed to have mutations
amenable to treatment (under evaluation)
Enzyme Enhancement therapy or pharmacological « charperone »

- Aminoacidopathy (PKU, tyrosinemia, …)
- Urea cycle defect (UCD)
- Organic aciduria (MMA, IVA, PA, …)
- Carbohydrate (galactosemia, hereditary fructose intolerance)
Group 2 : Intermediary metabolism – Intoxication type
Classification of IEM
- Fatty acids ß-oxidation defects
- Neoglucogenesis defects
- Ketone bodies synthesis defects
- Mitochondrial diseases (oxydative phosphorylation and Krebs cycle defects)
Group 3 : Intermediary metabolism – Energy depletion

Neurological
Deterioration
PKU
MSUD
MMA
PA, IVA
UCD
MCD
Predominant
Seizures
Pyridoxine
Pyridoxal P
Folinic acid
MCD
3PGD
GLUT-1
Jaundice
Liver failure
Galactosemia
Fructosemia
TYR-1
CDG-1b
Bile acids
LCHAD
Cardiac
FAO
Persistent
Hypoglycemia
PHHI
FAO
Glycogenosis
Hormones
Emergency treatment must be undertaken
in parallel with investigations
First think of treatable disorders
IEM (Intoxication, Energy depletion)

Neurological
Deterioration
PKU
MSUD
MMA
PA, IVA
UCD
MCD
Predominant
Seizures
Pyridoxine
Pyridoxal P
Folinic acid
MCD
3PGD
GLUT-1
Jaundice
Liver failure
Galactosemia
Fructosemia
TYR-1
CDG-1b
Bile acids
LCHAD
Cardiac
FAO
Persistent
Hypoglycemia
PHHI
FAO
Glycogenosis
Hormones
First think of treatable disorders
Anabolism
Epuration
Special diets
Glucose
Vitamins
Amino Acids
Special diet
Avoid fasting
L-carnitine
Special diet
IEM (Intoxication, Energy depletion)
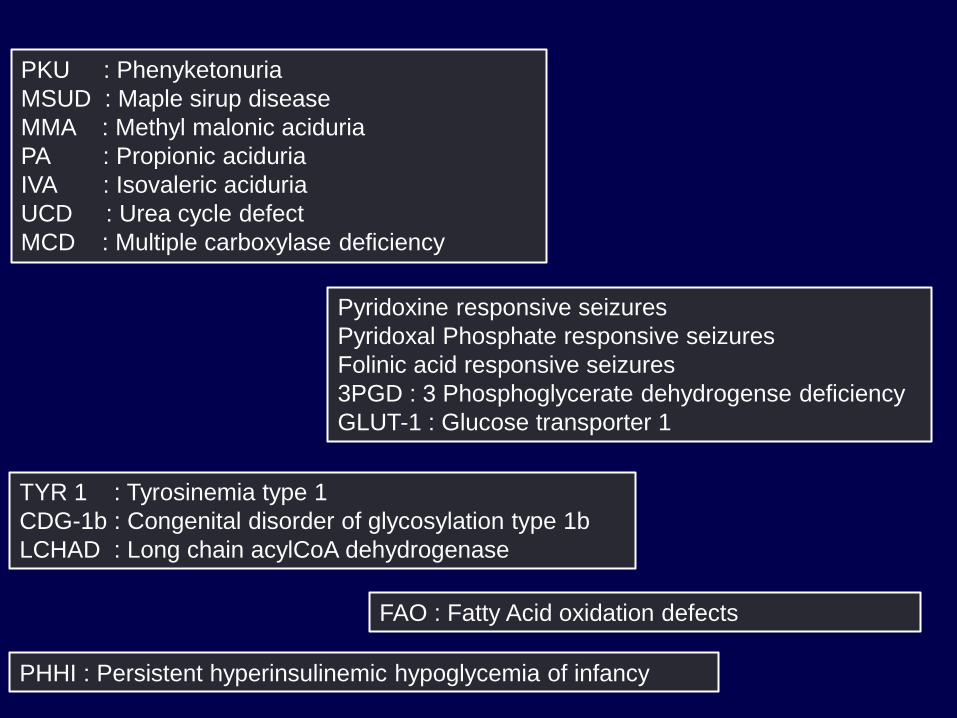
PKU : Phenyketonuria
MSUD : Maple sirup disease
MMA : Methyl malonic aciduria
PA : Propionic aciduria
IVA : Isovaleric aciduria
UCD : Urea cycle defect
MCD : Multiple carboxylase deficiency
Pyridoxine responsive seizures
Pyridoxal Phosphate responsive seizures
Folinic acid responsive seizures
3PGD : 3 Phosphoglycerate dehydrogense deficiency
GLUT-1 : Glucose transporter 1
TYR 1 : Tyrosinemia type 1
CDG-1b : Congenital disorder of glycosylation type 1b
LCHAD : Long chain acylCoA dehydrogenase
FAO : Fatty Acid oxidation defects
PHHI : Persistent hyperinsulinemic hypoglycemia of infancy

Disease specific treatments – Neurologic deterioration
PKU : Phenylketonuria
• Autosomal recessive disease
• 1 / 10 000 birth
• PAH gene (chromosome 12)
• PAH - tetrameric structure
• Classic PKU is caused by a complete or near-complete
deficiency of phenylalanine hydroxylase activity (PAH) in liver.
• PAH deficiency results in intolerance to the dietary intake of the
essential amino acid phenylalanine and produces a spectrum of
disorders

Phenylalanine Hydroxylase (PAH) system
BH4, a natural
cofactor of PAH

Dried blood spot on filter paper :
Guthrie card bacterial inhibition assay
R Guthrie in early 1960s
PKU = first metabolic disease detected through Neonatal Screening - increased PHE levels

0
5
10
15
20
25
30
35
40
45
50
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
1 - 3
60 - 180
> 20
>1200
10 - 20
600 - 1200
3 - 10
180 - 600
Normal Hyperphenylalaninemia atypic PKU Classical PKU
mg/dl
µmol/l
PHE
PHE plasmatic levels and PKU classification
PAH activity PHE level Daily PHE
tolerance
Classical PKU :
0-1 % > 20 mg/dl 200 – 350 mg
Atypical PKU :
1-3 % 10 - 20 mg/dl
350 - 850 mg
Non-PKU
Hyperphenylalaninemia
3 - 5 % PHE 3 - 10 > 850 mg

Mild to severe mental retardation
Neurologic symptoms
• Microcephaly
• Gait instability, tremor
• Epilepsy
• Autistic behavior
• Auto and hetero aggressivity
Eczema
Decreased skin and hair pigmentation
(Blond hair, blue eyes)
Structural brain changes and white matter
abnormalities
Musty body odor (typical)
Untreated classical PKU

Horst Bickel (1953)
Phenylketonuria = Low-phenylalanine diet
Lancet, 1953; 2, 812-813
First dietetic treatment for an IEM
Control of natural protein intake according to PHE tolerance
Avoidance of high protein food
(milk, dairy products, meat, fish, chicken, eggs, beans and nuts,...)
Phenylalanine-free formula (amino acids mixture with vitamins and
oligoelements)
Low protein food (manufactured hypoproteic bread, pasta, biscuits, ...)
No control of protein-free food

0 9 15 11
0
6
12
20
Adult
France
15
Germany
5
GB
4
8
Phe mg/dl
USA
5
2
10 18 (y)
22
10
International recommendations for PHE control according to age and country – no universal consensus

• Enzyme is synthesized but activity is
null or decreased
• PKU as a model of « misfolding » +++
http://www.bh4.org/biopku.html
PAH gene – Importance of missense mutations
• BH4 = Natural cofactor of aromatic
amino acid hydroxylases
• Sapropterin (6R-BH4) synthetic form
of tetrahydrobiopterin
• Stabilization of the active tetramer
forms of the mutant protein
• Protection from inactivation
• Acts as a « chemical chaperone »,
preventing misfolding
• Orphan drug (FDA and EMEA)
~ 500 mutations worldwide

Responder
Different responses to oral BH4 loading test (20mg/kg)
• About 70 % of mild PKU patients proved to be responder
• About 10 % of classical PKU patient respond to BH4
• In PKU patients responsive to BH4, oral treatment could be used in addition to a
restrictive low-phenylalanine diet and might even replace the diet in some instances.
• Limited adverse effects : upper airway tract infection, headache, vomiting, abdominal pain,
diarhhea, fever, back pain.
• Limits : palatability, compliance
hours

MSUD : Maple Sirup Disease
MMA : Methylmalonic Aciduria
PA : Propionic Aciduria
IVA : Isovaleric Aciduria
Disease specific treatments – Neurologic deterioration
Acute and chronic
« intoxication »
• Metabolic decompensation precipitated by :
prolonged fasting, protein overload, infection
• Ketoacidosis
• Accumulation of abnormal organic acids or
carboxylic acids (GC/MS) in urine
• Abnormal Amino Acid chromatography (MSUD)
• Abnormal acylcarnitine profile (on paper filter)
• Neonatal form : metabolic encephalopathy : lethargy, feeding problems, dehydratation, truncal
hypotonia/limb hypertonia, myoclonic jerks, neurovegetative dysregulation cerebral oedema,
coma, multi-organ failure; unusual odor (maple sirup-like odor of urine)
• Chronic intermittent form (up to aduldhood) : recurrent episodes of ketoacidotic coma,
lethargy and ataxia, focal neurologic signs, Reye syndrome
• Chronic progressive form : failure to thrive, chronic vomiting, anorexia, osteoporosis,
hypotonia, psychomotor delay, recurrent infections (particularly candida)
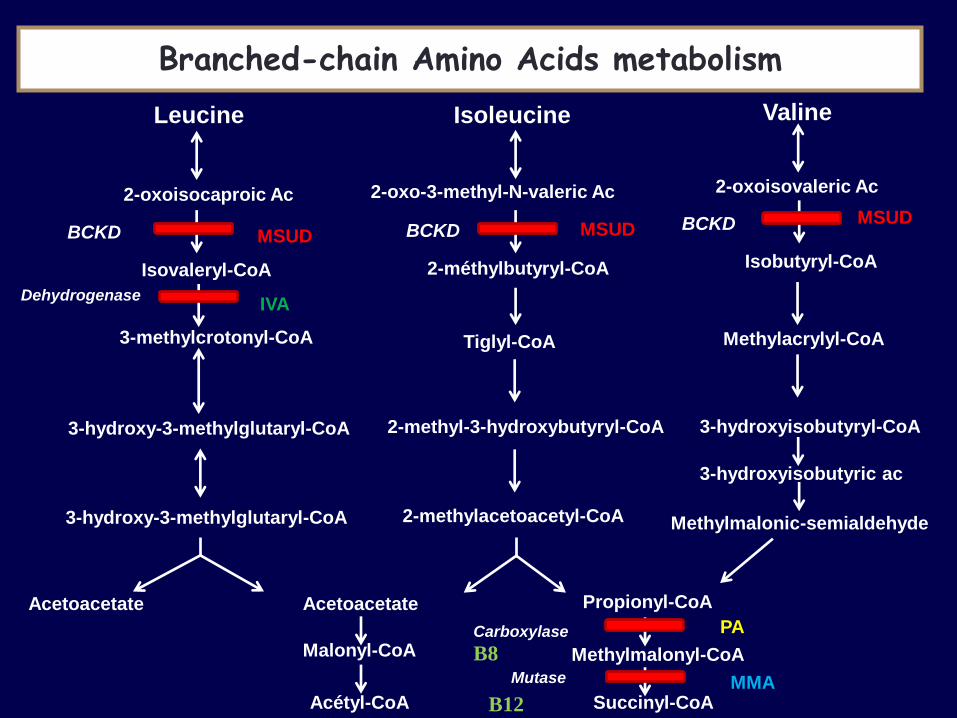
Leucine Isoleucine Valine
2-oxoisocaproic Ac 2-oxo-3-methyl-N-valeric Ac 2-oxoisovaleric Ac
Isovaleryl-CoA 2-méthylbutyryl-CoA Isobutyryl-CoA
3-hydroxy-3-methylglutaryl-CoA
Tiglyl-CoA Methylacrylyl-CoA
2-methyl-3-hydroxybutyryl-CoA 3-hydroxyisobutyryl-CoA
3-hydroxy-3-methylglutaryl-CoA 2-methylacetoacetyl-CoA
3-hydroxyisobutyric ac
Methylmalonic-semialdehyde
Acetoacetate Acetoacetate
Malonyl-CoA
Acétyl-CoA
Propionyl-CoA
Methylmalonyl-CoA
Succinyl-CoA
3-methylcrotonyl-CoA
BCKD BCKD BCKD MSUD MSUD
MSUD
IVA
PA
MMA
Dehydrogenase
Carboxylase
Mutase
Branched-chain Amino Acids metabolism
B12
B8
MSUD : Maple Sirup Disease
MMA : Methylmalonic Aciduria
PA : Propionic Aciduria
IVA : Isovaleric Aciduria
Acute treatment : - Interrupt catabolic state with high dose glucose IV
- Counteract acidosis
- Stop protein intake
- Remove toxins – depending on the disease and lab findings : increase diuresis,
dialysis, haemo(dia)filtration
- Consider specific treatment (L-Carnitine, B12, B8,..)
Long term treatment :
- Diet – protein restriction (according to safe values, beware of protein deficiency in
case of overtreatment)
- Supplementation of unaffected amino acids mixture
- Supplementations of minerals, vitamins or cofactors
- L-Carnitine in all disorders that cause intramitochondrial accumulation of CoA esters
Disease specific treatments – Neurologic deterioration
Acute and chronic intoxication
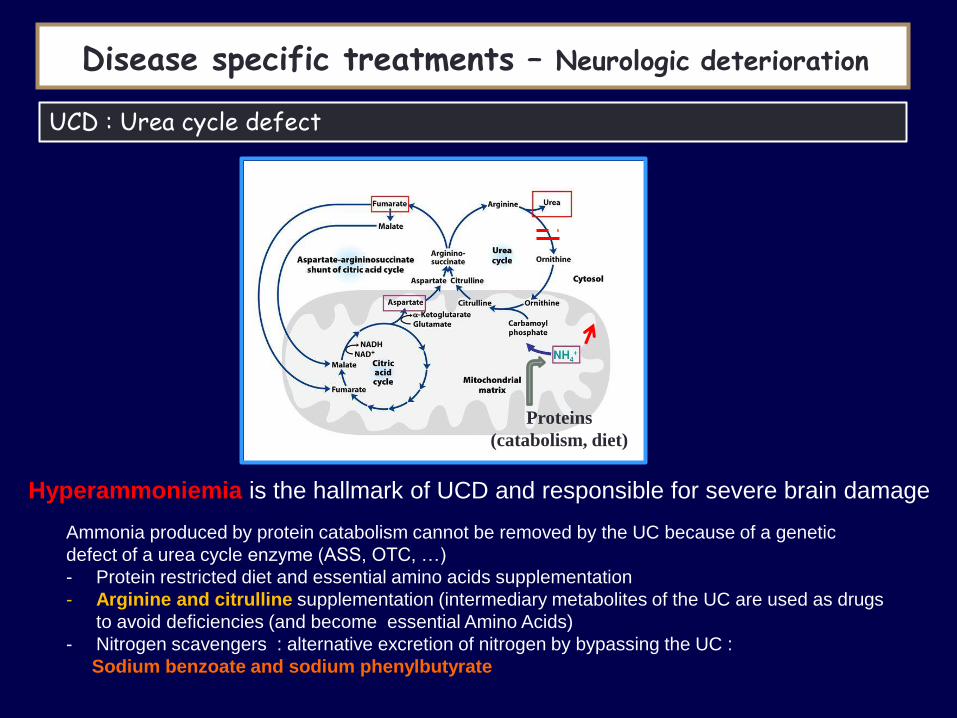
Hyperammoniemia is the hallmark of UCD and responsible for severe brain damage
UCD : Urea cycle defect
Disease specific treatments
Proteins
(catabolism, diet)
Ammonia produced by protein catabolism cannot be removed by the UC because of a genetic
defect of a urea cycle enzyme (ASS, OTC, …)
- Protein restricted diet and essential amino acids supplementation
- Arginine and citrulline supplementation (intermediary metabolites of the UC are used as drugs
to avoid deficiencies (and become essential Amino Acids)
- Nitrogen scavengers : alternative excretion of nitrogen by bypassing the UC :
Sodium benzoate and sodium phenylbutyrate
Disease specific treatments – Neurologic deterioration
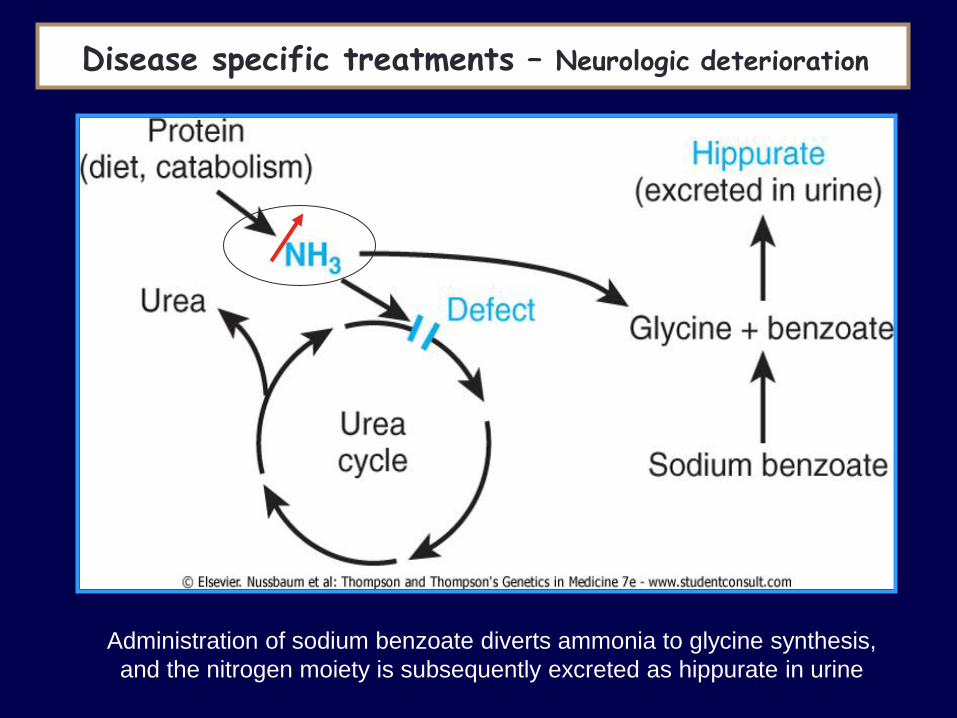
Administration of sodium benzoate diverts ammonia to glycine synthesis,
and the nitrogen moiety is subsequently excreted as hippurate in urine
Disease specific treatments Disease specific treatments – Neurologic deterioration
TREATMENT :
- Biotinidase deficiency : Biotin 5-10 mg/day (oral)
- HLCS : Biotin 10-20 (-40)mg/day
MCD : Multiple Carboxylase Deficiency (Biotinidase, Holocarboxylase synthetase)
Biotin
cycle Biotinidase deficiency must be ruled out in every
child with unexplained neurologic symptoms even
in absence of cutaneous or laboratory symptoms
Improvement of most symptoms with treatment
Vitaminotherapy
Disease specific treatments – Neurologic deterioration

Holocarboxylase
synthetase (HCLC)
Biotinidase
First symptoms First days of life 1w till 10 y
Hypotonia, coma,
Seizures, hypothermia
Hypotonia, ataxia,
Seizures
Cutaneous symptoms Alopecia, skin rash Alopecia, scaly perioral/facial rash
Periorificial eczema
pigmentation deficit (loss of hair color)
Complications Deafness and optic atrophy
Periventricular leucodystrophy
Or thalami abnormalities (MRI)
Spinal cord and progressive spastic paresis
Intellectual disability and developmental delay
Recurrent viral or fungal infections < immune
dysfunction
Severe metabolic acidosis Metabolic ketoacidosis
Organic aciduria : propionylglycine,
tiglylglycine, methylcitrate, 3-
hydroxypropionique, 3-methylcrotonylglycine, 3-
hydroxyisovalérique
Enzyme activity
Fibroblasts, lymphocytes DBS or plasma
MCD : Multiple Carboxylase Deficiency (Biotinidase, Holocarboxylase synthetase)
Disease specific treatments – Neurologic deterioration Disease specific treatments – Neurologic deterioration

Neurological
Deterioration
PKU
MSUD
MMA
PA, IVA
UCD
MCD
Predominant
Seizures
Pyridoxine
Pyridoxal P
Folinic acid
MCD
3PGD
GLUT-1
Jaundice
Liver failure
Galactosemia
Fructosemia
TYR-1
CDG-1b
Bile acids
LCHAD
Cardiac
FAO
Persistent
Hypoglycemia
PHHI
FAO
Glycogenosis
Hormones
First think of treatable disorders
Anabolism
Epuration
Special diets
Glucose
Vitamins
Amino Acids
Special diet
Avoid fasting
L-carnitine
Special diet

Vitamin responsive neonatal seizures :
- Pyridoxine (vit B6) -responsive seizures - Antiquitin (ALDH7A1) mutation - Pyridoxal Phosphate (PLP) -responsive seizures - PNPO (pyridox(am)ine 5-phosphate-oxidase) deficiency - Folinic acid -reponsive seizures (identical to pyridoxine-dependant epilepsy)
P5P (B6 active form) is cofactor of
> 100 transamination and decarboxylation
reactions in various pathways (serotonin and
dopamine synthesis, GABA synthesis, alanine,
glycine, threonine metabolism
Disease specific treatments – Seizures
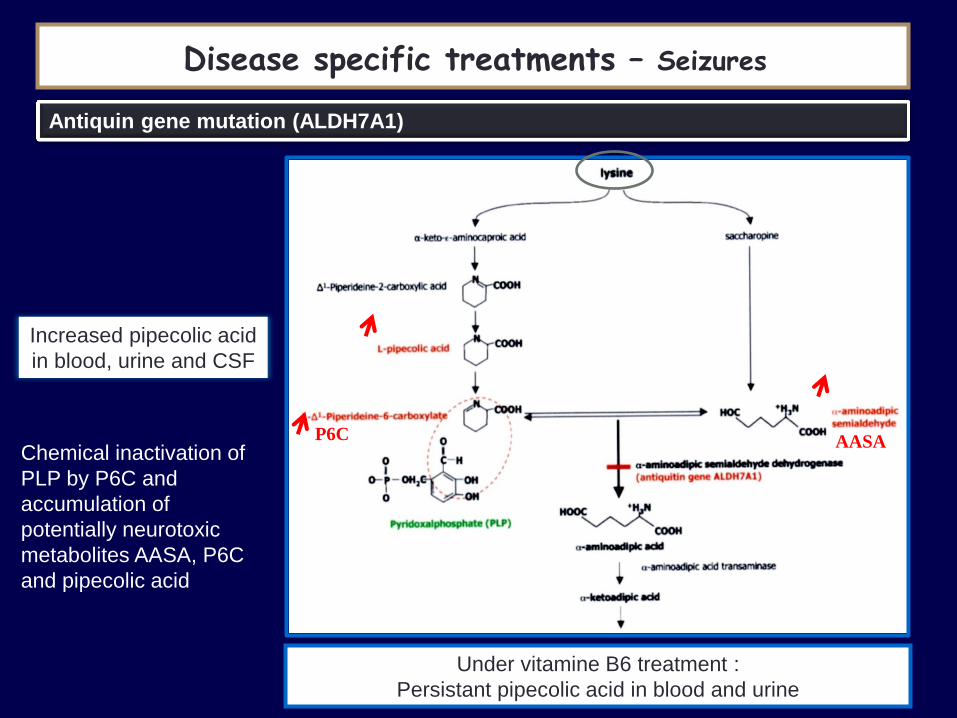
Antiquin gene mutation (ALDH7A1)
Increased pipecolic acid
in blood, urine and CSF
Under vitamine B6 treatment :
Persistant pipecolic acid in blood and urine
Chemical inactivation of
PLP by P6C and
accumulation of
potentially neurotoxic
metabolites AASA, P6C
and pipecolic acid
Disease specific treatments – Seizures
AASA P6C

PNPO deficiency
Clinical :
• Refractory neonatal seizures not responsive to pyridoxine but to
pyridoxal phosphate
• Microcephaly
• Prematurity
• Hypotonia
Diagnosis : CSF : Alanine, threonine, glycine
urine : vanillactic acid
Gene : PNPO
Therapy : pyridoxal phosphate 30 mg/kg/day oral in 3 doses
Disease specific treatments – Seizures

Disease specific treatments – Seizures
Pyridoxine 30 mg/kg/J
Pyridoxal-phosphate 10-50 mg/kg/j
Acide folinique 10 mg/j
Biotine 10-50 mg/j
In case of neonatal seizure suspected to be an IEM
A vitaminotherapy is never contraindicated

Dimethylglycine Sarcosine
Glycine
Serine
NH3 + CO2
Serine
Glycine
Methyl THF
3-P-glycerate Glucose P-serine P-OHpyruvate
Mitochondria
Methyl THF
Methyl THF
3PGDH
3PSP
3-PGDH (phosphoglycerate dehydrogenase) deficiency or
Serine synthesis deficiency
Disease specific treatments – Seizures

Severe congenital microcephaly ++
Epileptic encephalopathy
Psychomotor delay
Spastic tetraparesis
Cataracte (sometimes)
Growth retardation
Hypogonadism
3-PGDH (phosphoglycerate dehydrogenase) deficiency or serine synthesis def
Serine/glycine given with different treatment dosages
but a favorable responses were observed :
- Major reduction in seizure frequency
- Some patients became seizure free
- Increased white matter volume
- Progress of psychomotor development in patients,
diagnosed early, and treated with a high dose of L-
serine
- Prenatal treatment and normal head
circumference at birth. Normal neurologic
development at 12 y
De Koning et al, 2013
Disease specific treatments – Seizures
Low serine in CSF and borderline in
plasma (in fasted state)
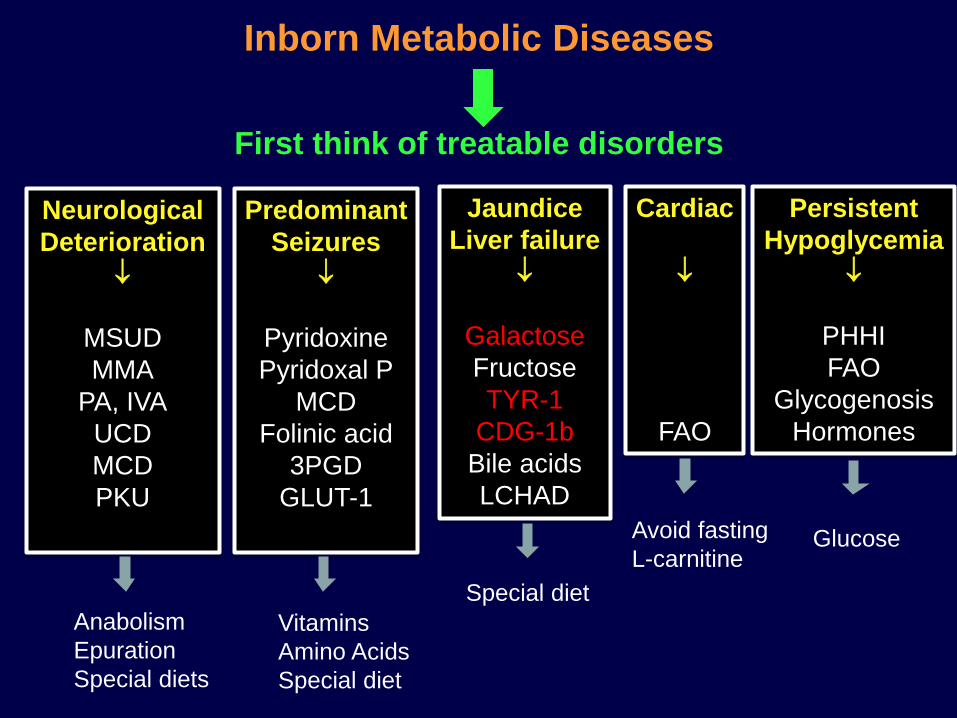
Neurological
Deterioration
MSUD
MMA
PA, IVA
UCD
MCD
PKU
Predominant
Seizures
Pyridoxine
Pyridoxal P
MCD
Folinic acid
3PGD
GLUT-1
Jaundice
Liver failure
Galactose
Fructose
TYR-1
CDG-1b
Bile acids
LCHAD
Cardiac
FAO
Persistent
Hypoglycemia
PHHI
FAO
Glycogenosis
Hormones
Inborn Metabolic Diseases
First think of treatable disorders
Anabolism
Epuration
Special diets
Glucose
Vitamins
Amino Acids
Special diet
Avoid fasting
L-carnitine
Special diet
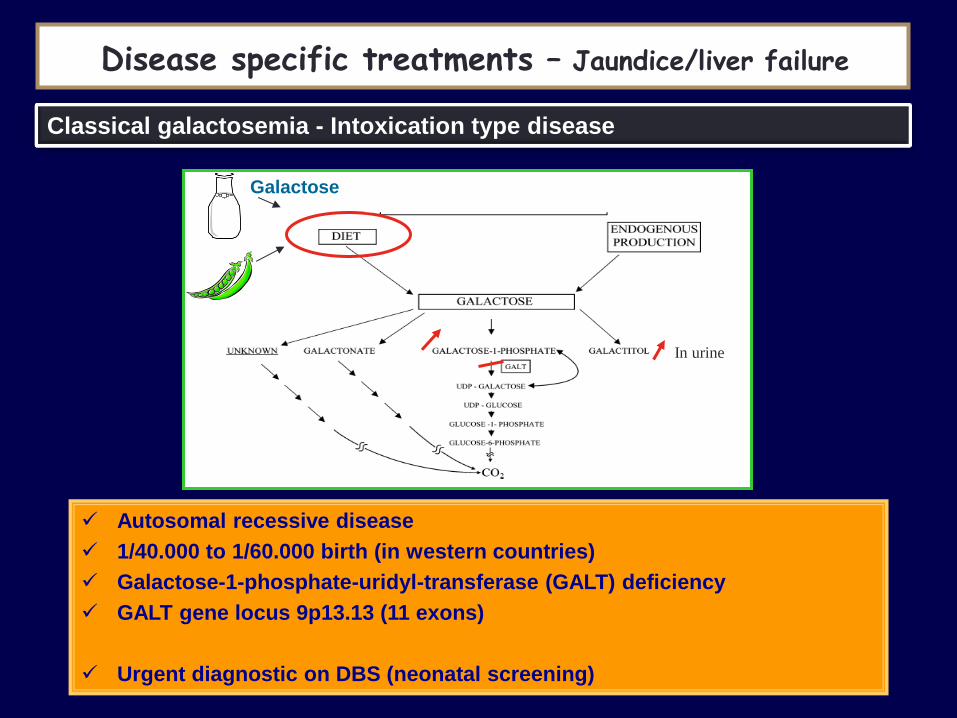
Galactose
Disease specific treatments – Jaundice/liver failure
Classical galactosemia - Intoxication type disease
Autosomal recessive disease
1/40.000 to 1/60.000 birth (in western countries)
Galactose-1-phosphate-uridyl-transferase (GALT) deficiency
GALT gene locus 9p13.13 (11 exons)
Urgent diagnostic on DBS (neonatal screening)
In urine

First symptoms, first weeks of life
Gastrointestinal problems, feeding difficulties, failure to thrive, lethargy
Hepatomegaly
Severe hepatic insufficiency (jaundice, bleeding tendency, hypoglycemia) and death if not promptly treated
E.Coli infection frequent
Diagnosis :
– Galactose, GALT activity on DBS
– Total red blood cell (RBC) Gal-1-P concentration
– Galactitol on urine
Urgent treatment : removal of LACTOSE and GALACTOSE from diet
(soya or lactose free formula- Olac®) rapid recovery
Disease specific treatments – Jaundice/liver failure
Classical galactosemia – Intoxication type disease

Disease specific treatments – Jaundice/liver failure
Classical galactosemia and consequences
Mild growth retardation
Delayed speech development
Verbal dyspraxia
Difficulties with spatial orientation
Decreased concentration ability
Reading difficulties
Abnormal brain white matter
In women, in general puberty is normal but may be delayed
- Premature ovarian insufficiency (81%)
Despite galactose restricted diet started soon after birth Despite compliance to diet
Despite Gal-1-P concentration within normal range
Risk: cataract
Resolves under galactose restricted diet, prognosis is good
Theorical risk at adolescence when diet is released
At 2 y : 80% patients IQ > 80
At 12 y : 80 % patients IQ < 80

Disease specific treatments – Jaundice/liver failure
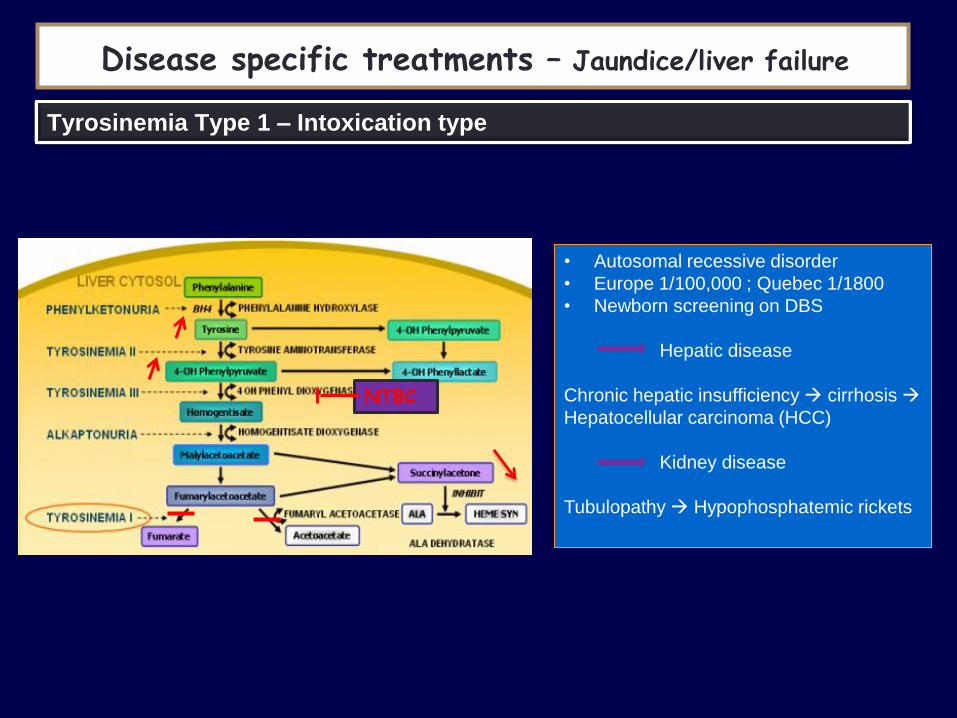
Tyrosinemia Type 1 – Intoxication type
• Autosomal recessive disorder
• Europe 1/100,000 ; Quebec 1/1800
• Newborn screening on DBS
Hepatic disease
Chronic hepatic insufficiency cirrhosis
Hepatocellular carcinoma (HCC)
Kidney disease
Tubulopathy Hypophosphatemic rickets
Disease specific treatments – Jaundice/liver failure
Tyrosinemia Type 1 – Intoxication type
• Autosomal recessive disorder
• Europe 1/100,000 ; Quebec 1/1800
• Newborn screening on DBS
Hepatic disease
Chronic hepatic insufficiency cirrhosis
Hepatocellular carcinoma (HCC)
Kidney disease
Tubulopathy Hypophosphatemic rickets
NTBC

Disease specific treatments – Jaundice/liver failure
Tyrosinemia Type 1 – basis of treatment
1. Dietetic : hypoproteic diet (1964) and Tyrosine/PHE free amino acid
mixture supplementation
At short term improves hepatic symptoms,
decrudescence of tubulopathy
BUT … at long term liver failure, HCC are not prevented
2. Orthotopic Liver transplantation (1976) cure hepatic and neurologic
symptoms (5-10 % mortality) – now reserved to patient with acute liver
failure and fail to respond to NTBC and patients suspected with HCC
3. Kidney and liver transplant
4. Hematin (in case of porphyric crisis before transplant)
5. NTBC (1991) – a « pharmacologic inhibitor » that inhibits Tyrosine
degradation at an early step to prevent production of toxic compounds
normalisation of hepatic function
Risk of HCC very low if NTBC started < 6 months

Disease specific treatments – Jaundice/liver failure
Congenital Disorder of glycosylation 1b
PMI :
Phosphomannose
-Isomerase

Disease specific treatments – Jaundice/liver failure
Congenital Disorder of glycosylation 1b
500
20
3
0
<10
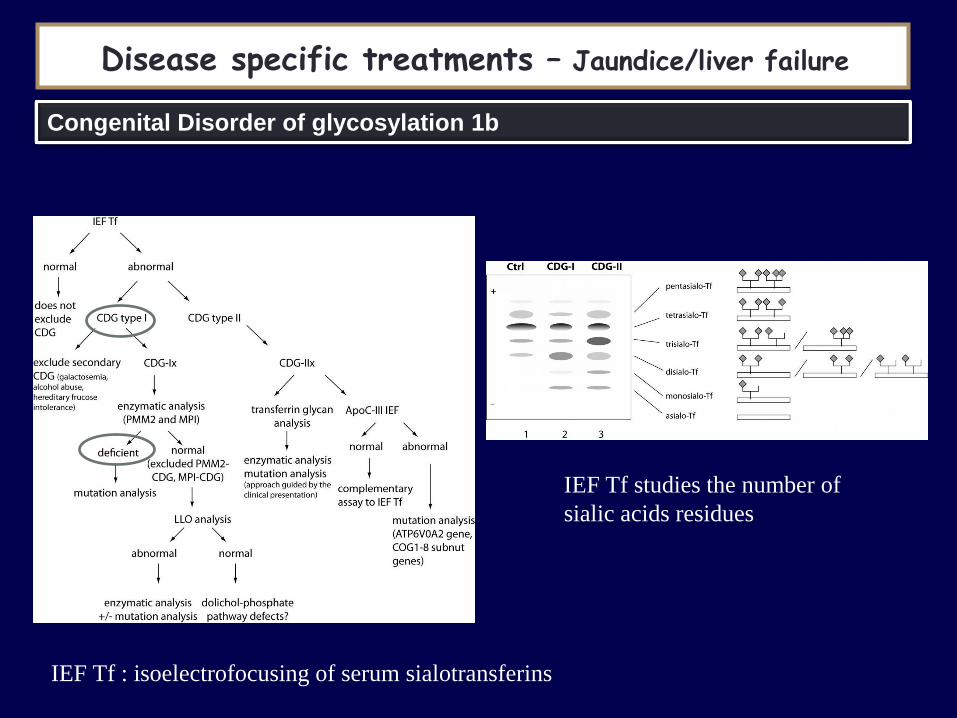
Disease specific treatments – Jaundice/liver failure
Congenital Disorder of glycosylation 1b
IEF Tf : isoelectrofocusing of serum sialotransferins
IEF Tf studies the number of
sialic acids residues

Disease specific treatments – Jaundice/liver failure
Congenital Disorder of glycosylation 1b
• Mannose 1g/kg/day divided in 5 doses
– Clinical recovery (liver, digestive)
– ATIII normalisation
– IEF Tf pattern improves after several months
• Only one treatable CDG syndrome
• Digestive and liver symptoms – diarrhea, exsudative enteropathy (hypoalbuminemia)
– hepatomegaly, cytolysis, cirrhosis, chronic liver disease
• hypoglycemia (hyperinsulinism)
• Kidney : tubulopathy, proteinuria
• Ig deficit (infections)
• Bleeding/thrombosis (XI, ATIII, prot C, prot S)
• No neurologic symptoms
• Mild asymptomatic forms

Fatty Acids B-Oxidation Defects – Energy depletion type
Disease specific treatments – Cardiomyopathy
Cardiac arrhythmia
Acute or chronic cardiomyopathy
(LVH or dilated)
Mucle pain,
Rhabdomyolysis
Hypoketotic hypoglycemia

Energy
depletion
- Carnitine transporteur
- CPT1, translocase, CPT2
- VLCAD (Very long Chain AcylCoA
dehydrogenase),
- LCHAD (Long Chain AcylCoA
dehydrogenase ,
- MCAD (Medium Chain acylCoA
dehydrogenase),
- SCAD (short chain acylCoA
dehydrogenase), SCHAD
- ETF (electrons transfert) LCHAD
MCAD
SCHAD
Cardiomyopathy and conductive defects

Cardiomyopathy and conductive defects
• Simultaneous determination of Free fatty acids and Ketones (3-
Hydroxybutyrate) is essential for a rapid diagnosis in case of
Hypoglycemia
• Acylcarnitine profile is usually diagnostic
(plasma, filter paper)
• Organic acids analysis (dicarboxylic acids from
ω oxidation) and serum carnitine may be helpful
• Enzyme studies (leukocytes, fibroblasts)
• Molecular studies confirm diagnosis (common mutation in MCAD and
LCHAD)
• Challenge tests (fasting test, oil challenge) are indicated only in
selected, exceptional cases and should be carried out in specialised
metabolic centers (danger of acute cardiotoxicity and others)
C8

Loïc (4/11/87)
11 y : dyspnea after efforts and vomiting
• Personal history : BW 3400 g, Height 51,5 cm; HC 37 cm.
• A 10 1/2 ans : Frequent vomiting. Hypoglycemia ++
• A 10 3/4 ans : After a running competition, dizziness and transient amaurosis and vomiting.
Since then, feels tired and have muscle weakness after walking (3 km)
Regular physical activity otherwise
• Clinical Exam :
W 30 kg (P10-P25), H 135 cm (P10-P25), TA 90/50 mmHg.
Systolic murmur 2/6, Liver enlarged (3-4 cm)
Normal Peripheral pulse
Scapular muscle weakness
Case report
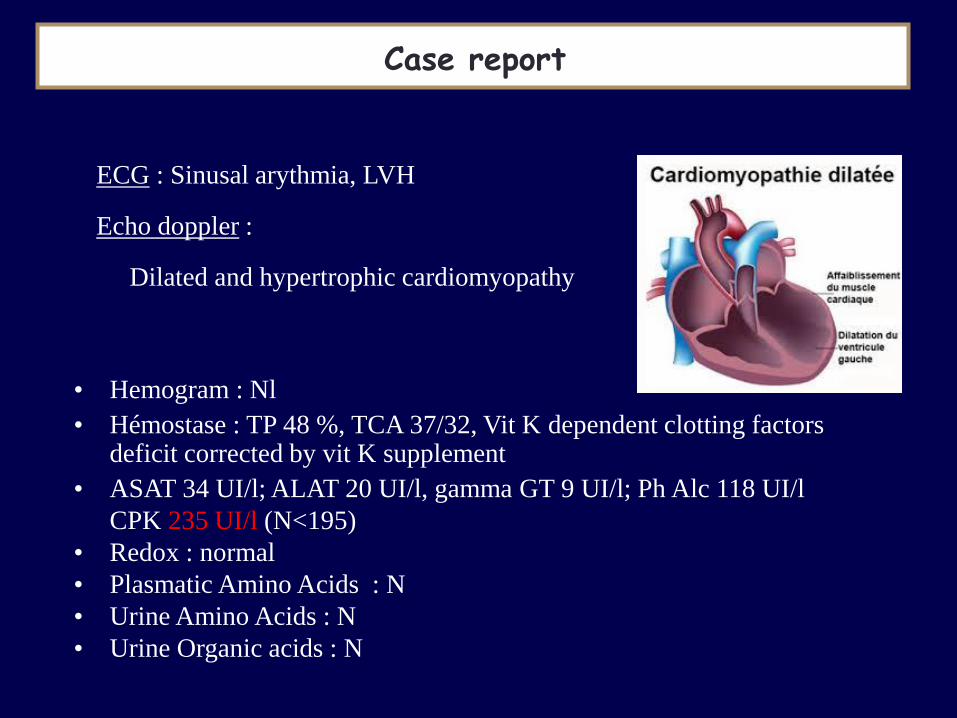
ECG : Sinusal arythmia, LVH Echo doppler : Dilated and hypertrophic cardiomyopathy
• Hemogram : Nl
• Hémostase : TP 48 %, TCA 37/32, Vit K dependent clotting factors deficit corrected by vit K supplement
• ASAT 34 UI/l; ALAT 20 UI/l, gamma GT 9 UI/l; Ph Alc 118 UI/l
CPK 235 UI/l (N<195)
• Redox : normal
• Plasmatic Amino Acids : N
• Urine Amino Acids : N
• Urine Organic acids : N
Case report

• Plasmatic Free Carnitine 4 uMol/l ( N 22-64)
• Plasmatic Total Carnitine 12 uMol/l ( N 34-77)
• Plasmatic Esterified Carnitine 14 uMol/l (low)
• Urine Free Carnitine 20.9 umol/mmol creat
• Urine Totale Carnitine 42.2 umol/mmol creat
• FA Oxidation on lymphocytes (Dr Brivet, Bicêtre) :
Long chain FA oxidation defect corrected after incubatiton with L-Carnitine
• Impaired Skin Fibroblast Carnitine Uptake
Patient control Normal values (n=10)
Total Uptake
(pmol/min/mg prot
0,60 1,60 1,55 - 2,90
Specific uptake
(pmol/min/mg prot)
0,0 1,20 1,19 - 2,40
Case report

• At 12 y : walk 12 km without being tired
• At 13 y : normalisation of left ventricular function. FE 65 %.
• Slight Left ventricular hypertrophy (10-11 mm).
• Stop treatment (digoxine, IEC)
• Increase of muscular mass
• Weight gain
Treatment : L-Carnitine 100 mg/kg/j/day
Case report