the role of gut metabolites on regulation of intestinal
TRANSCRIPT

Purdue UniversityPurdue e-Pubs
Open Access Dissertations Theses and Dissertations
January 2015
THE ROLE OF GUT METABOLITES ONREGULATION OF INTESTINAL IMMUNITYMyunghoo KimPurdue University
Follow this and additional works at: https://docs.lib.purdue.edu/open_access_dissertations
This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] foradditional information.
Recommended CitationKim, Myunghoo, "THE ROLE OF GUT METABOLITES ON REGULATION OF INTESTINAL IMMUNITY" (2015). OpenAccess Dissertations. 1121.https://docs.lib.purdue.edu/open_access_dissertations/1121

Graduate School Form 30Updated 1/15/2015
PURDUE UNIVERSITYGRADUATE SCHOOL
Thesis/Dissertation Acceptance
This is to certify that the thesis/dissertation prepared
By
Entitled
For the degree of
Is approved by the final examining committee:
To the best of my knowledge and as understood by the student in the Thesis/Dissertation Agreement, Publication Delay, and Certification Disclaimer (Graduate School Form 32), this thesis/dissertation adheres to the provisions of Purdue University’s “Policy of Integrity in Research” and the use of copyright material.
Approved by Major Professor(s):
Approved by:Head of the Departmental Graduate Program Date
Myunghoo Kim
THE ROLE OF GUT METABOLITES ON REGULATION OF INTESTINAL IMMUNITY
Doctor of Philosophy
Chang H. KimChair
Ramesh Vemulapalli
Arun K. Bhunia
Susan Eicher
Chang H. Kim
Ramesh Vemulapalli 12/3/2015

i
THE ROLE OF GUT METABOLITES ON REGULATION OF INTESTINAL
IMMUNITY
A Dissertation
Submitted to the Faculty
of
Purdue University
by
Myunghoo Kim
In Partial Fulfillment of the
Requirements for the Degree
of
Doctor of Philosophy
December 2015
Purdue University
West Lafayette, Indiana

ii
ACKNOWLEDGEMENTS
First of all, I would like to express my deepest gratitude to my advisor, Dr. Chang Kim.
He always supported me and encouraged me to improve my research skills and perform
cutting edge research. With his guidance, I realized the importance of both creative and
critical thinking abilities in making novel research discoveries.
I also would like to thank my advisory committee members, Dr. Vemulapalli, Dr. Bhunia,
and Dr. Eicher. They gave me helpful advice and valuable suggestions for conducting
and presenting research. My past and current lab colleagues Jeongho Park, Seika
Hashimoto-Hill, Leon Friesen, Youngmin Son, Heejoo Kim, Jinsam Chang, Jeongho
Yoon, Bon-hee Gu, Shankar Thangamani, Benjamine Ulrich, and Yaqing Qie readily
offered me assistance and emotional support during my studies. Finally, I thank my
parents, Joosuk Kim and Sukhyeon Yoon, and my brother Junghoo Kim for their infinite
love and support. And I also appreciate Beobyong Koo and Heebun Kang for their care.
These studies were supported by grants from NIH (R01AI074745, R01DK076616,
1R01AI080769 and 1S10RR028293), Crohn’s and Colitis Foundation of America, and
the National Multiple Sclerosis Society to Chang H. Kim, NIH (R21 AI105620) to
Elizabeth J. Taparowsky and Chang H. Kim, and by NCI (P30 CA023168).

iii
TABLE OF CONTENTS
Page
LIST OF TABLES .............................................................................................................. v
LIST OF FIGURES ........................................................................................................... vi
LIST OF ABBREVIATIONS ............................................................................................. x
ABSTRACT ..................................................................................................................... xiii
CHAPTER 1. LITERATURE REVIEW
1.1 Role of short chain fatty acids on the regulation of intestinal immunity .................. 1
1.1.1 Production of short chain fatty acid ...................................................................... 1
1.1.2 Receptors and transporters of short chain fatty acids ........................................... 2
1.1.3 Function of short chain fatty acid in the regulation of intestinal inflammation.... 3
1.2 Role of retinoic acid on the regulation of immune responses and innate lymphoid
cell migration ............................................................................................................ 6
1.2.1 Function of retinoic acid in the regulation of immune system ............................. 6
1.2.2 Lymphocyte migration program for gut homing .................................................. 8
1.2.3 Innate lymphoid cell subsets ................................................................................. 9
1.2.4 Trafficking receptor expression of ILC subsets .................................................. 10
CHAPTER 2. ROLE OF SHORT CHAIN FATTY ACIDS ON THE REGULATION
OF INTESTINAL IMMUNITY ....................................................................................... 15
2.1 Introduction ............................................................................................................. 15
2.2 Materials and methods ............................................................................................ 17
2.3 Results .................................................................................................................... 23
2.4 Discussion ............................................................................................................. 30
2.5 Summary and future directions ............................................................................... 31

iv
Page
CHAPTER 3. ROLE OF RETINOIC ACID ON THE REGULATION OF INNATE
LYMPHOID CELL SUBSETS' INTESTINAL MIGRATION ....................................... 66
3.1 Introduction ............................................................................................................... 66
3.2 Materials and methods .............................................................................................. 69
3.3 Results. ...................................................................................................................... 75
3.4 Discussion ................................................................................................................. 83
3.5 Summary and future directions ................................................................................. 87
REFERENCES ............................................................................................................... 117
VITA ............................................................................................................................... 129
PUBLICATIONS ............................................................................................................ 131

v
LIST OF TABLES
Table .............................................................................................................................. Page
1.1 Trafficking receptor expression by ILC progenitors, ILC precursors, and mature ILC
subsets in various tissues (Chapter 1) ............................................................................. 14
2.1 Primers used in this study (Chapter 2) ....................................................................... 65

vi
LIST OF FIGURES
Figure ............................................................................................................................. Page
1.1 Roles of SCFAs on the regulation of intestinal immune responses.. .......................... 12
1.2 Retinoic acid effects on the regulation of lymphocyte responses. .............................. 13
2.1 Hypo-inflammatory responses in GPR41-/- and GPR43-/- mice after ethanol-induced
breach of intestinal barrier function. ................................................................................. 34
2.2 Mild histological changes in GPR41-/- and GPR43-/- mice following rectal
administration of ethanol. ................................................................................................. 35
2.3 Reduced neutrophil infiltration in colon tissue following ethanol-induced breach of
intestinal barrier function in GPR41-/- and GPR43-/- mice. ............................................... 36
2.4 Reduced gut permeability and tissue bacteria infiltration in colon tissue following
ethanol-induced breach of intestinal barrier function in GPR41-/- and GPR43-/- mice. .... 37
2.5 Defective induction of inflammation-associated genes in SCFA receptors-deficient
mice following ethanol-induced breach of intestinal barrier function. ............................. 38
2.6 Abnormally low expressions of inflammatory genes in GPR41-/- and GPR43-/- mice
following ethanol-induced breach of intestinal barrier function.. ..................................... 39
2.7 Abnormally low immune responses to TNBS in SCFA receptor-KO mice. .............. 40
2.8 GPR41-/- and GPR43-/- mice were less susceptible to TNBS-induced inflammation . 41
2.9 Abnormally low IL-6 production in GPR41-/- and GPR43-/- mice followed by TNBS
challenge ........................................................................................................................... 42
2.10 Defective cytokine and T cell responses to TNBS in the gut of GPR41-/- and GPR43-
/- mice. ............................................................................................................................... 43
2.11 Defective chemokine expression and neutrophil recruitment in GPR41-/- and GPR43-
/- mice following TNBS administration. ........................................................................... 44
2.12 Increased susceptibility of GPR41-/- and GPR43-/- mice to C. rodentium infection. 45

vii
Figure ............................................................................................................................. Page
2.13 Delayed immune responses of GPR41-/- and GPR43-/- mice to C. rodentium infection.
........................................................................................................................................... 46
2.14 Delayed induction of inflammatory cytokines and chemokines in GPR41-/- and
GPR43-/- mice during C. rodentium infection. .................................................................. 47
2.15 Delayed gut permeability changes of GPR41-/- and GPR43-/- mice to C. rodentium
infection. ........................................................................................................................... 48
2.16 GPR41 and GPR43 do not affect gut permeability in the absence of immunological
challenges ........................................................................................................................ 49
2.17 C2 administration accelerates the immune response to infection………………… 50
2.18 C2 administration promotes chemokine and cytokine expression in colon tissue to C.
rodentium infection. .......................................................................................................... 51
2.19 C2 administration accelerates immune cell responses and gut permeability changes
followed by infection. ..................................................................................................... 52
2.20 No significant changes in commensal bacteria in the gut of GPR41-/- or GPR43-/-
mice ................................................................................................................................. 53
2.21 Differential roles of the SCFA receptors expressed by non-bone marrow versus
marrow-derived cells in promoting the immune response to C. rodentium. .................... 54
2.22 Differential roles of the SCFA receptors expressed by non-bone marrow versus
marrow-derived cells in effector T cell and neutrophil responses to C. rodentium……...55
2.23 GPR41 and GPR43 are highly expressed by intestinal epithelial cells. ................... 56
2.24 Comparison of the roles of SCFA receptors expressed by non-BM versus BM-
derived cells in mediating the inflammatory response to TNBS.. .................................... 57
2.25 SCFA-dependent expression of IL-6 and chemokines by colonic ECs was
suppressed by pertussis toxin. ........................................................................................... 58
2.26 SCFA-dependent expression of inflammatory chemokines and cytokines is
dependent on epithelial GPR41 and GPR43 ..................................................................... 59
2.27 SCFAs activate MEK-ERK pathway in epithelial cells in a GPR41- and GPR43-
dependent manner. ............................................................................................................ 60

viii
Figure ............................................................................................................................. Page
2.28 SCFA-induced expression of the inflammatory cytokine IL-6 is dependent on the
MEK-ERK pathway in epithelial cells. ............................................................................ 61
2.29 Effects of signaling molecule inhibitors on SCFA-dependent expression of
neutrophil-attracting chemokines by epithelial cells.. ...................................................... 62
2.30 Defective SCFA-induced phosphorylation of ERK in SCFA receptor-deficient mice
after infection with C. rodentium.. .................................................................................... 63
2.31 SCFAs induce ATF2 activation in colonic epithelial cells in a GPR41- and GPR43-
dependent manner. ............................................................................................................ 64
3.1 Tissue distribution of ILC subsets. ............................................................................. 88
3.2 Gating strategies for ILC subsets in small intestine. ................................................... 89
3.3 HR expression by total ILCs in various tissues .......................................................... 90
3.4 HR expression by ILC subsets in lymphoid and intestinal tissues ............................. 91
3.5 Tissue-specific homing receptors are required for ILCs population in tissues ........... 92
3.6 Gut HRs required for normal numbers of ILCs in intestines ...................................... 93
3.7 Tissue-specific HRs required for normal population of ILCs .................................... 94
3.8 HR expression by intraepithelial ILC1s and their numbers in gut-HR-deficient mice
.......................................................................................................................................... .95
3.9 Retinoic acid induced HR switch from 2nd LT-HR to gut-HR in ILCs. ..................... 96
3.10 RA induces a HR switch from CCR7 to CCR9 and α4β7 in ILC3s and ILC1s but
not ILC2s. ......................................................................................................................... 97
3.11 RA receptor expression and impact of RAR agonists and antagonists on HR
expression in ILC3s ......................................................................................................... 98
3.12 Impact of RA and Ro41 on chemotaxis of ILC subsets.............................................99
3.13 Mucosal DCs-derived RA induces the expression of gut HRs in ILC3s and ILC1s
......................................................................................................................................... 100
3.14 Vitamin A deficiency reduces expression of gut HR by ILC3s and ILC1s ............ 101
3.15 Decreased ILCs in the intestine of VAN and VAD RAG1-/- mice .......................... 102
3.16 Gating strategies for BM ILC2 progenitors ............................................................ 103
3.17 Expression of gut HRs by BM ILC2Ps in VAN and VAD mice ............................ 104

ix
Figure ............................................................................................................................. Page
3.18 BM ILC2Ps can directly migrate to the intestine .................................................... 105
3.19 Gut HR expression is required for BM ILC2P migration to the intestine .............. 106
3.20 Short-term homing of control versus RA-treated ILC3s to the gut and 2nd LT ...... 107
3.21 RA-induced gut HR expression is required for efficient short-term migration of
ILC3s to the intestine ...................................................................................................... 108
3.22 Gut HRs required for long term population of ILCs ............................................... 109
3.23 VAD RAG1-/- mice are more susceptible to C. rodentium infection ...................... 110
3.24 VAD mice have an impaired ILC3 response in the intestine .................................. 111
3.25 Protective effects of control and RA-treated ILC3s in infected VAD mice ........... 112
3.26 Gut homing receptors are required for optimal ILC3 effector function during C.
rodentium infection ......................................................................................................... 113
3.27 Gut homing receptors are required for optimal ILC3 effector function during C.
rodentium infection ......................................................................................................... 114
3.28 Expression of ILC3 cytokines and antimicrobial peptides following adoptive transfer
of WT or gut HR-deficient ILC3s into VAD mice infected with C. rodentium ............. 115
3.29 Distinctive trafficking receptor switches among ILC subsets ................................ 116

x
LIST OF ABBREVIATIONS
αLP α-lymphoid progenitor
AOM Azoxymethane
ATF2 Activating transcription factor 2
AVNM Ampicilin, Vacomycin, Neomycin, Metronidazole
BM Bone marrow
CCL Chemokine (C-C motif) ligand
CCR CC chemokine receptor
CD Cluster of differentiation
CFU Colony-forming unit
CHILP Common helper-like lymphoid progenitor
CLP Common lymphoid progenitor
CXCL Chemokine (C-X-C motif) ligand
DC Dendritic cell
DSS Dextran sodium sulfate
EC Epithelial cell
ERK Extracellular signal-regulated kinase
FITC Fluorescein isothiocyanate

xi
FoxP3 Forkhead box P3
GC Germinal center
GPR G protein-coupled receptor
HDAC Histone deacetylase
HFD High fiber diet
HR Homing Receptor
Ig Immunoglobulin
IHC Immunohistochemistry
IL Interleukin
Itg Integrin
ILC Innate lymphoid cell
LFD Low fiber diet
LI Large intestine
Lin Lineage
LP Lamina propria
LTi cell Lymphoid tissue inducer cell
MAPK Mitogen-activated protein kinase
MFD Medium fiber diet
MHC Major histocompatibility complex
MLN Mesenteric lymph node
MMP Matrix metalloprotease
mTOR Mammalian target of rapamycin
NK cell Natural killer cell

xii
OVA Ovalbumin
PKC Protein kinase C
PLC Phospholipase C
PP Peyer’s patches
PTX Pertussis toxin
RA Retinoic acid
RALDH Retinaldehyde dehydrogenase
RAR Retinoic acid receptor
RXR Retinoid X receptor
SCFA Short chain fatty acid
SI Small intestine
TNBS 2, 4, 6-trinitrobenzene sulfonic-acid
Treg Regulatory T cell
Th cell T helper cell
VAN Vitamin A normal
VAD Vitamin A deficient

xiii
ABSTRACT
Kim, Myunghoo Ph.D., Purdue University, December 2015. The Role of gut metabolites on regulation of intestinal immunity. Major Professor: Chang H. Kim.
The gastrointestinal tract contains multiple types of immune cells that induce various
immune responses to dietary antigens, commensal bacteria, and pathogens. Maintaining
homeostasis at this mucosal barrier requires stringent regulation of the immune responses,
including mechanisms to convey information about environmental conditions. Dietary
nutrients can be converted into gut metabolites through either host or microbial
enzymatic activities. Accumulating evidence demonstrates that these gut metabolites
have a significant impact on the regulation of intestinal immune responses; however, the
underlying mechanisms by which gut metabolites regulate the intestinal immune system
are incompletely understood. Thus, we have studied the role of gut metabolites,
specifically short chain fatty acids (SCFAs) and retinoic acid (RA), in regulation of
intestinal immune responses. Our research revealed novel pathways that SCFAs and RA
use to contribute to anti-bacteria immunity through modulation of epithelial cells and
innate lymphoid cells (ILCs), respectively.
SCFAs are the major metabolites of dietary fibers and produced from microbial
fermentation in the colon. A mounting body of evidence indicates that SCFAs regulate
cells in the immune system to promote aspects of both immunity and immune tolerance.

xiv
We investigated regulation of the intestinal inflammatory responses by SCFAs and
their receptors, GPR41 and GPR43. The control and SCFA-fed wild type (WT), GPR41-/-,
and GPR43-/- mice were chemically challenged with ethanol and 2, 4, 6-trinitrobenzene
sulfonic-acid (TNBS) or infected with Citrobacter rodentium to gain insights on the roles
of SCFA signaling in inflammatory responses. From these in vivo studies, we reported
that GPR41-/- and GPR43-/- mice had abnormally hypo-inflammatory responses following
chemical challenges. They also had a delayed immune response in clearing C. rodentium
compared to WT mice. SCFA treatment caused intestinal epithelial cells (ECs) to
produce inflammatory mediators both in vitro and in vivo in a GPR41- and GPR43-
dependent manner. These processes were necessary to recruit neutrophils and induce
effector T cells differentiation in the intestine after immune challenges. The underlying
mechanism is that SCFA receptors activated extracellular signal-regulated kinases
(ERK1/2) and p38 mitogen-activated protein kinase (MAPK) signaling pathways in ECs
for production of inflammatory cytokines and chemokines.
Separately, we studied the role of the gut metabolite, RA, in regulating innate
immunity in the intestine. RA is a vitamin A metabolite known to regulate various
immune responses, including development, function, and migration of lymphocytes in the
intestine. However, the roles of RA in innate lymphocyte biology are insufficiently
defined. We identified a novel function of RA in the migration program of ILCs, a class
of newly identified innate lymphocytes to the gut. This regulatory function of RA has a
significant impact on anti-bacterial immunity in the gut. We reported that intestinal ILCs
must undergo a ‘homing receptor (HR) switch’ from a secondary lymphoid tissue
receptor (CCR7) to gut homing receptors (CCR9 and α4β7) to migrate to the intestine.

xv
Mucosal dendritic cells (DCs)-derived RA induced this HR switch, which is required for
effector function of ILCs in the intestine. However, the RA-mediated HR switch occurred
only in ILC1 and ILC3 subsets, but not ILC2s. In contrast, ILC2s acquired gut HRs
during their development in the bone marrow and BM ILC2 precursors could migrate
directly to the intestine. Vitamin A deficient (VAD) mice exhibited higher susceptibility
to C. rodentium infection with decreased ILC3 numbers in the gut compared to mice fed
vitamin A normal diet (VAN). We demonstrated that efficient anti-pathogenic bacterial
immunity in the intestine requires RA-induced gut HRs expression in ILC3s.

1
CHAPTER 1. LITERATURE REVIEW
1.1 Role of short chain fatty acids on the regulation of intestinal immunity
The gastrointestinal tract is exposed to multifarious dietary and commensal
bacterial antigens. The maintenance of immune tolerance to harmless antigens is
necessary for gut health; yet, the capacity to mount efficient immune responses to
pathogens is also crucial. The mechanisms that regulate intestinal immune responses
remain incompletely understood. Commensal bacteria benefit the host through production
of various microbial factors. For example, commensal bacteria metabolize soluble dietary
fibers and produce short chain fatty acids (SCFAs) in the colon. It has been generally
accepted that SCFAs have beneficial impacts on maintenance of gut health. In this
introduction, we will review the production and absorption of SCFAs, as well as
expression of their transporters and receptors by cells, and function of SCFAs in the
regulation of intestinal immune responses.
1.1.1 Production of short chain fatty acids
Host and gut microbial flora are functionally integrated through co-evolution for
mutual benefits. In the gastrointestinal tract commensal bacteria are most densely
populated in the colon, where they exert their effects on the host, largely through
production of various microbial factors. One class of major microbial factors are short c

2
hain fatty acids, which are mainly produced from dietary fibers and non-digestible
carbohydrates through bacterial fermentation. The major species of SCFAs are acetate
(C2), propionate (C3), and butyrate (C4), with relative proportions of C2, C3, C4 in the
colonic lumen being 6:3:1 (1, 2). In the colon tissue, most SCFAs are absorbed into
colonocytes and provide energy for colonocyte biology (3). SCFAs are absorbed into
epithelial cells through SCFA-HCO3 exchange and further transported to distant tissues
including the liver, skeletal muscles, and the pancreas (4-6).
1.1.2 Receptors and transporters of short chain fatty acids
SCFAs can be absorbed into cells through diffusion or transported into cells
through monocarboxylate/solute transporters which expressed by intestinal epithelial
cells (7). The Na+-coupled transporters, SLC16A1 and SLC5A8, mediate SCFA transport
into epithelial cells (8, 9). These transporters are known to be involved in functional roles
of SCFAs such as HDAC inhibition (10, 11). SCFAs can also regulate functional
activities of cell without actually entering it. This is mediated by activating SCFA-
specific receptors, namely surface G-protein-coupled receptors such as GPR41, GPR43,
and GPR109A (Fig. 1.1). SCFAs, especially C3 and C4, can activate GPR41 (also called
free fatty acid receptor 3, FFA3) (12). GPR41 is expressed by many cell types including
intimal epithelial cells, pancreas, spleen, and adipose tissues (12, 13). The expression of
GPR43 (FFA2) was reported that certain immune cells (phagocytes), adipocytes and
epithelial cells (14-16). GPR109A (also called hydroxycarboxylic acid receptor 2, HCA2)
is known to be activated by C4 and niacin as well (17-19). GPR109A is expressed in
adipocytes and colonic epithelial cells (19, 20).

3
Upon binding of SCFAs to G-protein receptors, the cytoplasmic region of the
receptors are coupled to G-protein family Gαi/o or Gαq, which regulate the adenylate
cyclase pathway and the phospholipase C (PLC) pathway, respectively (21, 22). Binding
of GPCR by ligands induces activation of mitogen-activated protein kinases (MAPKs)
(p38 and ERK), protein kinase C (PKC), which leads to activates key cellular processes
such as proliferation, motility, and regulation of gene expression (e.g. expression of
cytokines and chemokines) (23).
1.1.3 Function of short chain fatty acid in the regulation of intestinal inflammation
It has been generally accepted that dietary fiber intake contributes the
maintenance of gut health. Numerous sources indicate that dietary fiber or SCFA
administration regulates intestinal inflammation in both humans and mice. These
beneficial effects of SCFAs are mainly mediated by regulation of certain immune cell
types, such as intestinal epithelial cells, T cells, and phagocytes. In this section, we will
discuss functional roles of SCFAs on the regulation of intestinal immune responses
through specific immune cells types. We will review impacts of SCFAs on epithelial cells,
T-cells, and phagocytes.
Intestinal epithelial cells are not simply a physical barrier in the intestine. Intestinal
epithelial cells can be considered immune cells, given that they play significant roles in
regulation of intestinal immune responses through recognition of antigens and production
of various immune modulators. SCFAs can be internalized by intestinal epithelial cells or
activate the SCFA receptors on intestinal epithelial cells. Therefore, the biological
functions of these cells can be readily affected by the concentrations of SCFAs in the

4
intestine. Among SCFAs, C4 (and to a lesser extent C2 and C3) can be readily utilized by
colonocytes as an energy source (3). C4 regulates survival, apoptosis, proliferation, and
differentiation of colonocytes through the control of the cell cycle (24, 25). The
underlying mechanism is C4-mediated hyperacetylation of histones, resulting in
enhanced accessibility of the transcriptional machinery for gene expression, which
regulate cellular responses (26, 27). For example, C4-mediated HDAC inhibition is
known to regulate the cell cycle inhibitor, p21Waf1/Cip1, and the pro-apoptotic protein,
BAK, in transformed cells (28, 29).The most important role of intestinal epithelial cells is
providing a barrier between luminal antigens and intestinal tissues. The separation
between luminal contents and tissues can be mediated by physical and molecular means.
The energy that SCFAs provide to epithelial cells contributes to maintenance of an intact
epithelium, required for the physical barrier. Anti-microbial peptides and mucins protect
the gastrointestinal mucosa against the invasion of pathogenic bacteria and gut
microbiota (30-32). SCFAs regulate various antimicrobial peptides and mucins
production by epithelial cells. C4 treatment enhances MUC2 expression and cathelicidin
(LL-37) in epithelial cell lines (33, 34).
Intestinal epithelial cells produce cytokines and chemokines upon recognition of
danger signals such as commensal bacteria, pathogens, and allergens. A mounting body
of evidence reports that SCFAs regulate production of various regulatory molecules by
epithelial cells. SCFAs are known to suppress expression of inflammatory mediators,
including IL-8, MCP-1, CXCL-1, and CXCL-10, from epithelial cells (35-37). Some
reports indicate that SCFA receptors are involved in the production of cytokines by
epithelial cells. For example, GPR109a-/- or GPR43-/- colonic epithelial cells were

5
defective in producing IL-18 in response to C4 (38). GPR43 regulates inflammasome
activation for IL-18 production in epithelial cells (39).
T cells are one of the key players in the regulation of intestinal inflammation. There
are some indications that SCFAs directly and indirectly regulate T-cell responses,
including their differentiation and cytokine production in the gut. C4 inhibits lymphocyte
proliferation with reduced IL-2 production (40, 41). SCFAs enhance IFN-γ and IL-10
production by cultured lymphocytes in certain condition (42). Recently, it has been
reported that SCFAs induce Treg generation, resulting in improved suppression of
intestinal inflammation (43). The underlying mechanism is that GPR43-dependent
HDAC inhibition by SCFAs induces FoxP3 expression in colonic T cells (44, 45).
Another mechanism of SCFA-mediated T cell regulation is through control of mTOR
activity (46). It has been reported that SCFAs increase mTOR activity (phosphorylation
of S6K protein), which contributes to Th1 and Th17 cell differentiation and IL-10
expression. The same study also reported that SCFAs induce hyper-acetylation of S6K in
T cells, leading to increased mTOR activity (46). Collectively, SCFAs regulate T-cell
differentiation and function through genomic hyperacetylation and metabolic changes
(Fig. 1.1).
Another potential target of SCFAs is phagocytes, including neutrophils,
macrophages, and dendritic cells. A mounting body of evidence reported that SCFAs
modulate activities of phagocytes in a SCFA-receptor-dependent or -independent manner.
SCFAs regulate migration of neutrophils migration into inflamed tissue sites in a GPR43-
dependent manner (15, 22). It has been reported that MAPKs and PKC signaling
mediated chemotaxis of neutrophils in response to chemoattractants. The recruitment of

6
neutrophils by SCFAs, through GPR43, contributes to the regulation of intestinal
inflammation and immune responses. Also, SCFAs regulate the production of TNF-α,
CINC-2αβ, and NO by neutrophils (47, 48). SCFAs can also affect production of
inflammatory mediators by macrophages. Generally, SCFAs promote production of anti-
inflammatory mediators but suppress pro-inflammatory mediators from macrophages.
For instance, C4 elevated IL-10 production but suppressed the production of TNF-α, IL-
1β, IL-6, and nitric oxide by macrophages (49). GPR109A is known to regulate IL-18
production by colonic macrophages (38). SCFAs also affect dendritic cells, with the most
well-reported function being control of development and maturation (50-52). Generally,
SCFAs inhibit development and maturation of DCs. SCFAs suppressed DC development
and maturation from BM progenitors in vitro (11). GPR109A activation regulates the
phenotype of colonic macrophages and DCs, which in turn affects the generation of Treg
cells (38).
1.2 Role of retinoic acid on the regulation of immune responses and innate
lymphoid cell migration
1.2.1 Function of retinoic acid in the regulation of immune system
Vitamin A deficiency is widely believed to lead to immune deficiencies in the
body. Retinoic acid, a major vitamin A metabolite, regulates broad aspects of immune
responses (53). Dietary vitamin A is absorbed in the gut tissue and transported into the
blood circulation to enter the liver (54, 55). Upon entering liver cells, retinol or retinal
can be converted into retinoic acid (RA) by retinaldehyde dehydrogenase (RALDH) (56,

7
57). RA can bind retinoic acid receptors (RAR) and retinoid X receptor (RXR) in the
cytoplasm (57, 58). All-trans RA binds to RXR via heterodimers with RAR to regulate
variety of gene expression. RA production is not limited to the liver; in gut-associated
secondary lymphoid tissue, mucosal DCs process vitamin A and produce RA, effectively
regulating local immune cell activity (59). RA has significant impacts on the
differentiation, activation, and migration of lymphocytes (Fig. 1.2). RA promotes plasma
cell differentiation and IgA expression in mucosal tissues (60-62). This is implicated in
anti-bacterial immunity in the intestine. RA also regulates T-cell differentiation and
activation. It has been reported that RA induces Treg generation in the intestine, which is
implicated in the immune tolerance in the intestine (63, 64). RA also regulates effector T
cell differentiation. Low dose of RA enhances Th17 cell differentiation (65). RA
suppresses Th1 cell differentiation in vitro (66, 67). RA is also required for T-cell
activation upon antigen priming. RARα deficiency induced an impaired Th1 response to
T. gondii infection (68). RA as a gut tissue factor regulates gut tissue tropism of adaptive
lymphocytes by upregulation of gut homing receptors (HR). Mucosal DC-derived RA
induces the upregulation of gut homing receptors, CCR9 and α4β7, in T and B
lymphocytes (69, 70). Vitamin A deficiency reduced gut-HR expression by lymphocytes
and decreased numbers of lymphocytes in the intestine (71). Recently, it has been
reported that RA also regulates the development, expansion, and function of innate
lymphocytes. RA promotes IL-22 expression by γδT-cells and type 3 innate lymphoid
cells (ILC3s) through RARα directly binding to promoter regions of the IL-22 gene (72).
Vitamin A status affects the balance of ILC subsets in the intestine. Spencer et al.,
reported that RA expands ILC3 but suppresses ILC2 in the gut (73). This is implicated in

8
anti-bacteria and anti-parasite immunity in mucosal tissues. RA also has a significant role
in the development of ILC3s. RA-RAR signaling is important for lymphoid tissue
inducer cell (LTi cell) development in the gut tissue of the fetus (74). This is the one of
the factors that emphasizes the importance of maternal vitamin A on the immunity of
their offspring. Collectively, RA has significant impacts on the regulation of innate and
adaptive immunity in mucosal tissues.
1.2.2 Lymphocyte migration program for gut homing
Lymphocytes circulate in the bloodstream and can migrate to certain tissue sites
in respond to specific signals such as chemokines (75, 76). Lymphocytes express tissue-
specific chemokine receptors and integrins for directing their migration. The interactions
between chemokine receptors/integrins and chemokines/adhesion molecules guide
lymphocytes to migrate to certain tissues sites. The gut homing of lymphocytes is the
most well established migration program. CCR6, CCR9, CCR10, CD103, and α4β7 are
known as gut homing receptors (HRs) and these receptors play significant roles in
migration and localization of lymphocytes to intestinal tissues (70, 71, 77-81). Beyond
gut HR expression, lymphocytes undergo more sophisticated migration programs to
migrate to the intestine. The naive T cells can migrate to secondary lymphoid tissues by
utilizing CCR7 and CD62L (70, 71). In gut-associated secondary lymphoid tissues such
as the mesenteric lymph nodes (MLN) and Peyer’s patches (PP), T cells undergo a
homing receptor switch to change preferential migration from secondary lymphoid
tissues to intestinal tissue. RA, as an intestinal tissue factor, can induce this homing
receptor switch in lymphocytes. Upon antigen priming, RA suppresses expression of the

9
secondary lymphoid tissue receptors, CCR7 and CD62L, but induces expression of gut
HRs, CCR9 and α4β7. Unlike T cells, innate lymphocytes are not thought to have
sophisticated migration programs. Instead, they usually upregulate trafficking receptors
in response to inflammatory signals and then migrate to sites of inflammation (82).
1.2.3 Innate lymphoid cell (ILC) subsets
ILCs are a recently identified family of heterogeneous innate immune
lymphocytes. ILCs play important roles in host defense against pathogens and tissue
inflammation (83, 84). Interestingly, ILCs do not express antigen-specific receptors, but
they share many functional phenotypes with CD4+ T-cells (84, 85). For example, ILCs
can produce the cytokines IFN-γ, IL-5, IL-9, IL-13, IL-17A, and IL-22 that are typically
known as signature cytokines for CD4+ T-cell subsets. ILCs also can express common
CD4+ T-cell transcription factors, including T-bet, GATA-3, and RORγt, which are
required for their development and functional activity (86). ILCs are subdivided into
three groups, based largely on the expression of signature transcription factors and
effector cytokines. ILC1s require the transcription factor T-bet (Tbx21) and produce IFN-
γ, and as such resemble Th1 cells. ILC1s can induce chronic inflammation in the tissue
and promote anti-intracellular bacteria immunity (87). ILC2s, which produce IL-5 and
IL-13, require RORα, GATA-3, and Tcf7 for development in a similar manner as Th2
cells (88). ILC2s are involved in the regulation of metabolic homeostasis, anti-parasite
immunity, and allergic diseases (88, 89). ILC2s produce cytokines in response to IL-25
and IL-33 secreted by epithelial cells and are known to initiate a Th2 response in the lung.
ILC3s require RORγt and GATA-3 for development and IL-17A and IL-22 production,

10
which mediates gut tissue homeostasis and induces anti-extracellular bacteria immunity
(90, 91). The functional transcriptional phenotype of ILC3s is similar to that of Th17
cells. Interestingly, ILCs regulate T-cell responses in mucosal tissues in an antigen-
dependent manner (92). Like professional antigen-presenting cells, ILC2s and ILC3s
express MHCII to present antigens (90, 93). MHCII-dependent antigen presentation by
ILCs is critical for the regulation of T-cell homeostasis in the lung and intestine.
1.2.4 Trafficking receptor expression of ILC subsets
Recent reports revealed that homing receptor expression is required for ILC
development, migration, and effector function. In this section, we reviewed tissue
trafficking receptor expression by ILCs during development and maturation. Table 1.1
shows homing receptor (HR) expression by ILC progenitors, ILC precursors, and mature
ILCs. ILC progenitors have a broad migration potential for peripheral tissues. BM
common lymphoid progenitors (CLPs) express CCR7. ILC progenitors such as
CXCR6+ α-lymphoid progenitor (αLP), and Id2+NFIL+ common helper-like lymphoid
progenitor (CHILP), Id2+PLZF+ ILCP express α4β7 (87, 94, 95). This is intriguing
because these receptors are implicated in immune cell migration and cell-cell interaction
in various peripheral tissues (96). Among ILC precursors in the BM, ILC2Ps express
integrin α4β7 and CCR9 (88, 97).
ILCs express tissue-specific but largely subset-independent HRs, and this
acquisition of HRs is thought to occur during their functional maturation in the periphery.
Mouse skin ILC2s express CD103 (Itg-αE) and CXCR6 (98). CD103 mediates
lymphocyte interaction with epithelial cells through E-cadherin. On the other hand, the

11
ILC2s in the small intestinal-LP express CCR4, CCR8, CCR9, CXCR4, and CXCR6 (86).
Thus, the HR profile of gut ILC2s is largely different from that of skin ILC2s, but many
HRs are shared with Th2 cells. On the other hand, ILC3s express numerous HRs
reminiscent of Th17 cells. The migration of ILC3s to lymph nodes is regulated by CCR7
(99). LTi ILC3s migrate from the small intestine to the draining mesenteric lymph nodes
in a CCR7-dependent manner. Gut ILC3s express CCR9 and α4β7 (97). In addition to
CCR9 and α4β7, NKp46+ ILC3s express CXCR6 to migrate local site of small intestine
in respond to CXCL16, which produced by CX3CR1+ myeloid cells (100). CD4+ LTi
ILC3s express CCR6 and are localized to cryptopatches and isolated lymphoid follicles in
the gut (101, 102). ILC3s in the small intestine additionally express CCR1, CXCR3, and
CXCR4, which may promote ILC3 localization and interaction with other cell types for
effector function (86, 103).
ILC1s share many HRs with ILC3s. CCR9 and α4β7 are required for optimal
population and migration of ILC1s to the intestine. They also express CXCR3, CXCR4,
CCR5, and CXCR6. CCR5 appears to be preferentially expressed by ILC1s among ILCs
(86). The intestinal intraepithelial compartment is enriched with CD103-expressing
human ILC1 (104, 105). Overall, NK cells and non-NK ILC1s share many trafficking
receptors. Both NK cells and ILC1s in the small intestinal lamina propria express CCR5,
CCR9, CXCR3, CXCR4, and CXCR6 (86). NK cells in the spleen and liver variably
express CX3CR1, CCR5, CXCR3, and CXCR4. Some NK-cell subsets migrate to lymph
nodes and promote Th1 responses, a process dependent on CCR7 or CXCR3 (106, 107).
CXCR4 is important for NK cell retention in the bone marrow, and the sphingosine-1-
phosphate receptor S1P5 promotes NK egress from the bone marrow (108, 109).

12
Figure 1.1 Roles of SCFAs on the regulation of intestinal immune responses.
Dietary fibers are converted into SCFAs by bacterial fermentation in the intestine. SCFAs
regulate immune cell activities through two different pathways. First, SCFAs activate
GPCR (GPR41, GPR43, and GPR109A) to induce intracellular signaling pathway to
produce inflammatory mediators. Second, SCFAs transport into cells and regulate
cellular metabolism and gene expression by non-GPCR regulation.

13
Figure 1.2 Retinoic acid effects on the regulation of lymphocyte responses.
Vitamin A is converted into retinoic acid (RA) through CD103+ mucosal dendritic cells.
RA is known to regulate gut homing and differentiation of adaptive lymphocytes. RA
induces gut homing receptors by adaptive lymphocytes and promotes regulatory T-cells
and plasma cell differentiation in T- and B- lymphocyte, respectively.

14
Table 1.1 Trafficking receptor expression by ILC progenitors, ILC precursors, and
mature ILC subsets in various tissues.
ILC subsets such as NK, ILC1, ILC2, and ILC3 can be made from a group of progenitor
cells such as CLP, αLP, CHILP, and ILCP in the bone marrow or from fetal liver
progenitors. The HRs expressed by human ILCs are enclosed in parentheses (Except for
ILC progenitors) to distinguish them from the HRs expressed by mouse ILCs. Peripheral
ILC subsets express a group of HRs, which can be different depending on tissue sites and
ILC subsets. ND, not determined; Ref, References.
Progenitors Precursors Mature ILCs Bone marrow (or fetal liver)
ILC subsets
Spleen Lymph nodes
Intestines Skin Lungs
CLP (CCR7) αLP (α4β7, CXCR6) CHILP (α4β7) ILCP (α4β7)
NKP CXCR4, S1P5
NK CCR5 CXCR3,4 CX3CR1
CCR7, CXCR3
Itg-α1,α2,α4 CCR5,7,9 CXCR3,4,6
(CLA, CCR5,8 CXCR3)
CCR2,4 CX3CR1
ILC1P ND ILC1 CCR7 α4β7,
CCR7,9
(Itg-aE), Itg-α1, α4β7 CCR5,9 (CXCR3,4,6)
(CXCR3) ND
ILC2P CCR9, α4β7 ILC2 CCR9
α4β7 CCR9 α4β7
CCR4,6,8,9 CXCR4, (CXCR5, 6 CRTH2)
CLA,CD103 CCR4,6,10, (CXCR5,6, CRTH2)
ND
ILC3P CCR6, α4β7 ILC3
CCR6, CCR7 CXCR5
α4β7 CCR6,7,9
α4β7 CCR1,6, 9 CXCR3-6
ND ND
Ref. (87, 94, 95, 110)
(88, 94, 97, 101, 109) Ref.
(86, 97, 111, 112)
(97, 99, 106, 107)
(86-88, 97, 100, 104, 105, 113, 114)
(98, 114-117)
(118, 119)

15
CHAPTER 2. ROLE OF SHORT CHAIN FATTY ACIDS ON THE REGULATION OF INTESTINAL IMMUNITY
2.1 Introduction
The mucosal surface of the intestinal tract provides a major entry point for food
antigens, commensal bacterial products, and pathogens (15). The gut immune system
normally maintains immune tolerance to the harmless antigens but can mount effective
immune responses upon infection by pathogens. The mechanism(s) that control the
balanced immunity and immune tolerance in the intestine, however, remain incompletely
understood. The intestine is densely populated with commensal bacteria which
metabolize undigested carbohydrate fibers in the colon (120, 121). Evidence is available
that the gut commensal bacteria produce factors that profoundly affect the immune
system (122).
Short chain fatty acids (SCFAs) are the major metabolites of dietary fibers and
produced from microbial fermentation in the colon (123). SCFAs are volatile fatty acids
with 1 to 6 carbon atoms and include acetate (C2), propionate (C3), and butyrate (C4) as
the major SCFAs. Intestinal epithelial cells play significant roles in regulation of
intestinal immune responses through recognition of antigens and production of various
immune modulators. And the biological functions of these cells can be readily affected by
the concentrations of SCFAs in the intestine. SCFAs, exemplified by relatively more e

16
xtensively studied butyrate, have regulatory effects on various aspects of
epithelial cell biology (25). SCFAs serve as an energy source for epithelial cells and
regulate intestinal epithelial cell proliferation, differentiation, and apoptosis (124, 125).
SCFAs are transported into cells through monocarboxylate transporters and solute
transporters (126). Also, SCFAs activate cells through GPR41 and GPR43. GPR41 and
GPR43 are expressed highly by gut epithelial cells (127). GPR41 and GPR43 are
expressed also by adipocytes and implicated in regulation of leptin expression and energy
balance (13, 128). GPR43 is expressed by phagocytes and controls phagocyte functions
and tissue inflammation (21, 129). The functions of SCFAs in regulation of inflammatory
responses and anti-bacterial immunity in the intestine are poorly understood. Also, roles
of SCFA receptors, GPR41 and GPR43 on the regulation of inflammatory responses in
the intestine are still unknown.
Here, we investigated the functions of SCFAs and their receptors in regulation of an
acute immune response following chemical or pathogenic bacteria infection-induced
inflammatory immune responses. We report that GPR41 and GPR43 expression in
intestinal epithelial cells are required for induction of effective inflammatory immune
responses. SCFAs and their receptors promote acute inflammatory responses in the
intestine for efficient protection of the host from pathogens. Overall, we provide
evidences that SCFAs have significant impacts on regulation of anti-bacterial immunity
and inflammation in the intestine.

17
2.2 Materials and methods
Animals
All experiments with animals in this study were approved by the Purdue Animal
Care and Use Committee. CD45.2 and CD45.1 C57BL/6 mice were from Harlan and
Jackson Laboratory, respectively. GPR43-/- C57BL/6 mice were from Deltagen (San
Mateo, CA), and GPR41-/- C57BL/6 mice were described previously (13). All mice were
maintained at Purdue for at least 2 years before use. Mice were maintained on a regular
rodent chow (Harlan 2018S Global 18% Protein Rodent Diet). Mice were weaned at 3
weeks of age.
In vivo studies
For induction of intestinal inflammation with ethanol and TNBS, mice were fasted
overnight and received an intrarectal administration of 100 µl of 50% ethanol using a
round-tip needle. Intestinal inflammation with TNBS was induced as described
previously (137). Mice were sacrificed one day after ethanol challenge or three days after
the TNBS challenge. For pathogenic bacterial infection, mice were infected with C.
rodentium (DBS100). For infection of C2-administered mice, mice were fed with C2
(sodium acetate, 200 mM, pH 7.5) in drinking water for 4 weeks and infected with C.
rodentium (1010 CFU/mouse). When indicated, mice were injected i.p. with antibodies
(50 µg/injection for each antibody) to neutralize IL-6 (MP5-20F3 on day 0 and 2) and
deplete neutrophils (RB6-8C5 on day -2 and 0). Weight change and stool consistency
were monitored during the experimental period. Mice were monitored daily for changes

18
in body weight, signs of illness, and stool scores. Stool consistency scores were measured
as described previously (23). Briefly, stool scores were measured as follows: 0, normal
stool; 1, soft stool; 2, diarrhea; 3, diarrhea and anal bleeding. Mice challenged by TNBS
or C. rodentium were monitored daily for changes in body weight and stool scores.
Infected mice were monitored for stool C. rodentium burden.
Scoring of the intestinal inflammation
Histological changes in the distal colon following rectal administration with 50%
ethanol with or without ethanol and TNBS (Sigma-Aldrich, St. Louis, MO). Colon tissues
in paraffin were cut into 6 μm-thick sections and stained with hematoxylin and eosin
(H&E). The sections were semiquantitatively graded on a 0-4 scale based on pathological
features, including the extent of leukocyte infiltration, crypt elongation/abscess, bowel
wall thickening, mucosa hyperplasia, and ulceration. 0, no evidence of inflammation; 1,
low level of leukocyte infiltration in <10% high power fields (hpf), no structural changes
observed; 2, moderate leukocyte infiltration in 10–25% hpf, crypt elongation, and bowel
wall thickening; 3, high level of leukocyte infiltration in 25–50% hpf, bowel wall
thickening and mucosa hyperplasia; and 4, severe leukocyte infiltration in >50% hpf,
crypt elongation, bowel wall thickening, mucosa hyperplasia, and ulceration.
Assessment of C. rodentium load
For enumeration of fecal C. rodentium, fecal pellets were collected, weighed, and
homogenized in sterile PBS. The fecal homogenates were serially diluted and plated in
duplicate on MacConkey agar plates and incubated aerobically for 24 h at 37 oC.

19
Bacterial colony forming units (CFU) were calculated per g of fecal pellet. For C.
rodentium in the spleen and liver, organ homogenates were plated on MacConkey agar.
Generation of bone marrow chimeras
Congenic mouse strains with allergic variants with CD45 (CD45.1 and CD45.2)
were used for chimeric study. Host mice were exposed to 500 cGy of ionizing radiation
twice and were reconstituted with 10 million donor bone marrow cells by tail vein
injection. The mice were kept for 8 weeks for hematopoietic reconstitution and were
sensitized and administered with TNBS, or infected with C. rodentium. The marrow
reconstitution efficiency was greater than 95% (based on spleen).
Confocal microscopy
Frozen and paraffin-embedded tissue sections were prepared at 6-8 μm
thicknesses. Colon sections were stained respectively with rabbit polyclonal antibodies to
ZO-1 (Invitrogen) and Phospho-p44/42 MAPK (Erk1/2) (Cell signaling technology). The
slides were further stained with fluorescently labeled polyclonal anti-rabbit IgG
antibodies (Invitrogen). Tissue sections were stained with anti-Gr-1 (RB6-8C5) and
Hoechst 33342 (Invitrogen) when indicated. The fluorescence images were collected with
a SP5 II laser scanning microscope system (Leica). Colonic paraffin-embedded sections
were stained with H&E. Sections were semi-quantitatively graded on a 0-4 scale based on
pathological features.

20
Gut permeability
Mice were fasted overnight, then received an intragastrical injection of
fluorescein isothiocyanate (FITC)-conjugated dextran (0.2 mg/g of body weight; average
MW 3,000-5,000; Sigma-Aldrich) using a round-tip feeding needle. The mice were
sacrificed 3 hours later, and the FITC-dextran concentration in the plasma was
determined with a fluorescent microplate reader (excitation at 485 nm and emission at
535 nm; Biotek Synergy HT).
In vitro culture of colonic epithelial cells
Lamina propria (LP) cells and colonic ECs (purity > 95% based on CD45- cells) were
isolated as described previously (138). For expression of GPR41 and GPR43, colonic EC
(CD326+ cells) were isolated by Miltenyi magnetic beads. Colonic ECs (2×105 cells/well)
were cultured for 6 days, and C2 (10 mM) or C3 (1 mM) was added during this culture
period. These cells were further activated with LPS (Sigma-Aldrich; 1 µg/ml) or a
commensal bacterial antigen (CBA, 20 µg/ml) for 24 h. Alternatively, colonic ECs,
established for 3 days, were treated with kinase inhibitors (PD98059, U0126, SB203580,
and SP600125; Enzo Life Sciences, Inc., Farmingdale, NY) and SCFAs for 3 days.
Concentrations of indicated cytokines in serum and conditioned medium were measured
in triplicates with a Luminex assay (Luminex Corporation, Austin, TX) or an ELISA
method.

21
Flow cytometry
Flow cytometry was performed as described previously (138). For neutrophils, tissue
cells were stained with antibodies to Gr-1 (RB6-8C5) and 7/4 (Ly-6B.2; Serotec). For
intracellular staining for IL-17 and IFN-γ, T cells were stained first for surface antigens
and then activated in RPMI 1640 (10% FBS) with PMA (50 ng/ml; Sigma Aldrich),
ionomycin (1 μM; Sigma Aldrich) and monensin (2 mM; Sigma Aldrich) for 4 h. Then,
cells were fixed, permeabilized and stained with antibodies to mIL-17A (TC11-18H10.1)
or mIFN-γ (XMG1.2). For FoxP3+ cells, cells were stained with anti-mFoxP3 (FJK-16s)
according to the manufacturer's protocol (eBioscience). Stained cells were analyzed with
FACS Canto II (BD Bioscience). For phosphorylation of ERK and ATF2 in ECs, cells
were fixed and permeabilized with perm III buffer (BD Bioscience) and stained with
antibodies to Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) or Phospho-ATF-2
(Thr71) (Cell signaling). Donkey anti-rabbit IgG-FITC (BioLegend) was used as the
secondary antibody. Spleen and intestinal lamina propria cells were examined.
Microarray and quantitative, real time PCR
Colon tissues were frozen in liquid nitrogen and ground using mortar and pestle.
The array data were analyzed as described and deposited at:
http://www.ncbi.nlm.nih.gov/geo (GSE36569). qPCR was performed as previously
described. For bacterial 16S Ribosomal RNA Gene Analysis, total DNA was isolated
from colon tissues and fecal pellets using QIAamp DNA Stool Mini Kit (Qiagen,
Valencia, CA). For total eubacteria in tissues, a pair of primers was used to amplify the
16S ribosomal RNA (rRNA) gene sequence conserved in all commensal bacteria. The

22
PCR was performed using Maxima SYBR Green/ROX qPCR Master Mix (Thermo
Scientific, Logan, UT). Tissue-associated bacteria levels based on the Ct values of the
amplified conserved 16S rRNA gene sequence were normalized with the signal of
amplified mouse genomic (β-actin) gene. The abundance of specific bacterial groups was
measured with group-specific 16S rRNA gene primers (synthesized by Integrated DNA
Technologies). The signals of group-specific 16S rRNA gene were normalized with that
of the total eubacteria 16S rRNA gene. Primers information is described in Table 2.1.
Statistical analysis
Student’s t-test, Mann-Whitney test, and One-Way Analysis Of Variance
(ANOVA) were used to determine significance of the differences between groups. p
values < or = 0.05 were considered significant.

23
2.3 Results
GPR41 and GPR43 are required for normal level of inflammatory response
following ethanol-induced breach of the gut barrier
To gain general idea about immunological function of SCFA receptors, GPR41
and GPR43 in the induction of inflammatory responses, we injected ethanol into WT,
GPR41-/-, and GPR43-/- mice to transiently breached the gut barrier function. The ethanol
challenge induced decrease in the weight of WT mice. However, the GPR41- and
GPR43-deficient mice showed mild response in weight change to ethanol challenge (Fig.
2.1A). Also, WT mice had shorter and thicker colon tissue in gross examination than both
KO mice (Fig. 2.1B). In line with less inflammatory responses in SCFA receptors
deficiency, both KO mice showed reduced leukocyte infiltration in the mucosa and
submucosa, thus they have reduced histology scores (Fig. 2.2). The ethanol treatment
induced infiltration of neutrophils within the colon tissue. In this regard, infiltration of
neutrophils in the colonic lamina propria was abnormally low in the SCFA-receptor KO
mice compared to WT mice (Fig. 2.3A). Immunohistochemistry (IHC) revealed that only
WT mice had significantly increased Gr-1+ cells in colon tissue following ethanol
challenge (Fig. 2.3B). Inflammation induces elevation of gut permeability and this leads
to facilitated antigen recognition by antigen presenting cells, resulting in further
inflammation. We investigated gut permeability changes during inflammation. IHC
revealed that GPR41-/- and GPR43-/- mice maintain ZO-1 expression following the
ethanol treatment but WT mice lost ZO-1 expression in colon tissue (Fig. 2.3B). We
further determined gut permeability by FITC-dextran leakage. Compared to WT mice,

24
the GPR41 and GPR43 KO mice showed mild gut permeability changes (Fig. 2.4A). In
line with smaller gut permeability change in GPR41-/- and GPR43-/- mice, they had
smaller increases in tissue bacteria compared to WT mice (Fig. 2.4B). We performed
microarray for examination of inflammatory responses-related genes expression regulated
by SCFA receptors. Interestingly, the patterns of gene expression in colon tissues of
GPR41- and GPR43-deficient mice are similar (Fig. 2.5A). Many genes, including
chemokines, inflammatory cytokines, and metalloproteinases induced in WT mice were
not induced in the two SCFA-receptor KO mice (Fig. 2.5A, B). These findings also were
confirmed by qPCR (Fig. 2.6). Results indicate that SCFA receptors, GPR41 and GPR43,
positively regulate inflammatory gene expression after immune challenge.
GPR41 and GPR43 are necessary for an efficient induction of inflammatory
response to TNBS
We further examined the inflammatory responses to TNBS in WT, GPR41, and
GPR43 KO mice. Like ethanol treatment, TNBS treatment decreased BW in WT mice by
20% but, the same challenge induced a smaller level of BW loss in GPR41 and GPR43
KO mice. Only WT, but not the KO mice, had increased stool scores on day 2 (Fig. 2.7A
and B). Colon length of WT mice was significantly shorter than those of both KO mice
(Fig. 2.7C). Also, the distal colon of both KO mice strains had less leukocyte infiltration,
mucosa hyperplasia, and tissue damage after TNBS challenge (Fig. 2.8). Notably, IL-6
levels are lower in both SCFA receptor-deficient mice compared to those of WT mice
(Fig. 2.9). Induction of Th1 (CD4+IFN-γ+ cells)-associated genes (IL-12, IFN-γ, and T-
bet) was defective in both GPR41-/- and GPR43-/- mice (Fig. 2.10A). In line with this, the

25
frequency of Th1 cells (CD4+IFN-γ+) was decreased in the small intestine, colon, and
MLN of the KO mice (Fig. 2.10B). Moreover, induction of neutrophil responses
including chemoattractants, CXCL1, CXCL2, and CCL2 expression in the colon was
abnormally low in GPR41-/- and GPR43-/- mice (Fig. 2.11A and B). These results indicate
that SCFA receptors play positive roles in mounting the acute inflammatory response to
TNBS.
GPR41- and GPR43-deficient mice display delayed pathogen clearance
From abnormal induction of inflammatory responses in GPR41- and GPR43-
deficient conditions, we assumed that GPR41 and GPR43 play a significant role in
control pathogenic bacteria in the intestine. We utilized a C. rodentium infection model to
determine the role of SCFA receptors in mounting immune responses to bacterial
pathogens. As expected, GPR41-/- and GPR43-/- mice were more susceptible to infection
than WT mice based on weight changes and stool consistency scores (Fig. 2.12A and B).
Both KO mice showed a delayed clearance of C. rodentium from the intestinal tissue in
late stage of infection (Fig. 2.12C). Both KO mice had more detectable bacteria in
peripheral organs such as liver and spleen than WT mice, indicating increased
translocation of the pathogenic bacteria (Fig. 2.12C). Both KO mice had low responses in
neutrophil recruitment, and frequencies of effector T cells in the colon (Fig. 2.13A and B).
We found that the expression kinetics of inflammatory cytokines were delayed in both
KO mice by 1-2 weeks (Fig. 2.14A and B). Regarding the gut permeability, GPR41 and
GPR43 KO mice showed a delayed kinetics in the change of gut permeability and
bacterial infiltration in the colon tissue (Fig. 2.15A and B). However, there are no

26
differences on gut permeability and tight junction protein (ZO-1) expression between WT
and SCFA receptor-deficient mice (Fig. 2.16A and B). In addition, C2 administration had
no impacts on the gut permeability in WT mice. These results suggest that differences in
gut permeability are mainly mediated by different levels of inflammation after immune
challenge.
Acetate (C2) administration facilitates anti-pathogenic bacterial immunity
in the intestine
Next, we investigated the role of SCFAs on the anti-pathogenic bacterial
immunity in the gut. We administered C2 (100 mM) to mice and examined immune
responses and bacterial clearance in the gut. In infection studies, C2-treated mice were
more resistant to infection based on BW changes and stool consistency scores compared
to control mice (Fig. 2.17A). Also, C2-fed mice had lower intestinal C. rodentium loads
(Fig. 2.17B). Regarding the immune responses to infection, C2-fed mice showed
accelerated kinetics of inflammatory cytokine and chemokine expression (Fig. 2.18A and
B). In line with this, C2 administration increased recruitment of neutrophils and Th17
(CD4+IL-17A+) cells in the cecum (Fig. 2.19A and B). C2 administration also facilitated
a change in gut permeability (Fig. 2.19C). Thus, C2 administration accelerated the overall
immune response during the infection. To demonstrate the importance of facilitated
immune responses by SCFA administration, we neutralized or depleted IL-6 and
neutrophils in animals fed control or C2 water during infection. As expected, antibody
injection delayed immune responses, including of IL-6-Th17 axis and CXCL1/2-
neutrophil (Gr-1+ cells) axis and increased susceptibility to the C. rodentium infection

27
(Fig. 2.18A and B). This indicates the important roles of IL-6 and neutrophils in
induction of the anti-pathogenic bacterial immunity (Fig. 2.19A and B). Notably, the C2
effects on immunity were abolished by the antibody treatments. We also tested if
antibody injection affects gut permeability during infection. As expected, antibody
injection delayed increase of gut permeability at an early time points (Fig. 2.19C). At
later time points, however, a greater change in gut permeability occurred in the antibody-
injected animals (Fig. 2.19C). We also examined the levels of seven bacterial groups in
the gut of GPR41-/- and GPR43-/- mice to determine whether SCFA receptor deficiency
may affect the gut microbiota in the gut. There was no significant change in the gut
microbiota profile (Fig. 2.20).
Identification of potential immune cell types involved in SCFA receptor-mediated
anti-bacterial immune responses
To determine the relative contribution of SCFA receptors expressed by marrow
versus non-marrow-derived cells in immune responses, we prepared bone marrow-
chimeras with WT recipients and GPR41-/- or GPR43-/- marrow donors or vice versa.
Clinical symptom and immune responses in chimera were examined following C.
rodentium infection (Fig. 2.21). Host mice (WT) reconstituted with WT or KO marrow
showed comparable clinical symptoms and bacterial clearance. In contrast, the KO host,
reconstituted with WT marrow, lost weight significantly, and higher stool scores and
bacteria load (Fig. 2.21A-C). In line with the delayed clearance of C. rodentium,
frequencies of Th17 (CD4+IL-17A+) cells and neutrophils were lower in the colon of both
SCFA receptor-KO hosts compared to other mice (Fig. 2.22A and B). From BM chimera

28
experiments, we concluded that expression of SCFA receptors by non-hematopoietic
cells play significant roles in anti-bacterial immunity compared to those by hematopoietic
cells. We also confirmed this finding in the TNBS-induced colitis model (Fig. 2.23). In
line with BM chimera studies, both GPR41 and GPR43 mRNAs were highly expressed
by CD326+ (EpCAM) epithelial cells (Fig. 2.24).
SCFAs-receptors axis activates production of chemokines and cytokines in colonic
epithelial cells in a MEK-ERK-dependent manner
High expression of SCFAs receptors in epithelial cells and the BM chimera study
promoted us to investigated role of SCFAs and their receptors on the regulation of
immunological function of intestinal epithelial cells (ECs). First, we compared
production of cytokines and chemokines between control and SCFA-treated cultured
primary epithelial cells. The expression of IL-6, CXCL1, and CXCL10 in colonic
epithelial cell cultures was increased by C2 or C3 treatment. This induction was
completely abolished when cells were pretreated with pertussis toxin (PTX), which is an
inhibitor of G0/i-coupled receptors such as GPR41 and GPR43 (Fig. 2.25). The ECs
isolated from GPR41-/- or GPR43-/- mice were largely unresponsive to C2 or C3 in
expression of the cytokines and chemokines beyond basal levels (Fig. 2.26A and B).
There results suggest that GPR41 and GPR43 are involved in SCFA-mediated
inflammatory molecules production by ECs. We studied the intracellular pathways
important for SCFA-dependent expression of inflammatory mediators. As the
intracellular mechanism, we identified that SCFAs activate ERK signaling pathway in
colonic ECs (Fig. 2.27). And GPR41- and GPR43-deficient ECs are largely unresponsive

29
in ERK activation to SCFA treatment (Fig. 2.27), suggesting that ERK activation is
induced by the SCFA signals through GPR41 and GPR43. We also confirmed that ERK
inhibitors PD98059 (a MEK1 inhibitor) and U0126 (a MEK1/2 inhibitor) suppressed
ERK activation in ECs (Fig. 2.27). In addition, these inhibitors suppressed SCFA-
induced IL-6, CXCL1, and CXCL2 expression (Fig. 2.28 and Fig. 2.29). This ERK
activation was suppressed by two inhibitors of ERK: PD98059 (a MEK1 inhibitor) and
U0126 (a MEK1/2 inhibitor) (Fig. 2.28). These inhibitors also suppressed the expression
of IL-6, CXCL1 and CXCL2 induced in response to C2 or C3 (Fig. 2.29). Also, p38
MAPK is implicated in the SCFA receptor signaling in a breast cancer cell line.
SB203508 (a MAPK inhibitor) was less effective than the ERK inhibitors but still
suppressive at a high concentration However, a c-Jun N-terminal kinases/JNK inhibitor,
SP600125, was ineffective (Fig. 2.28 and Fig. 2.29). IHC revealed that in vivo
administration of C2 increased activation of ERK in colons of WT, but not GPR41 or
GPR43 KO mice after C. rodentium infection (Fig. 2.30). We also examined downstream
molecules activation of ERK. Activating transcription factor 2 (ATF2) is involved in
expression of inflammatory cytokines and chemokines. SCFAs induce transcription
factor ATF2 in WT cells, but not both KO ECs (Fig. 2.31).

30
2.4 Discussion
We investigated the roles of SCFAs and their receptors in the regulation of
intestinal immunity. Defective immune responses including changes in gut permeability,
expression of inflammatory mediators, leukocyte infiltration, and tissue inflammation
were detected in the mice deficient in GPR41 or GPR43 following immune challenge.
Also, expression of GPR41 and GPR43 by ECs is required for mounting effective
inflammatory responses including effector T-cell responses and neutrophil accumulation
in inflamed tissue. Our results establish important roles of SCFA signaling in the
induction of immune responses through activation of epithelial cells.
SCFAs are highly enriched in the colon tissue, and their receptors, are highly
expressed by intestinal ECs. It has been reported that butyrate can decrease the tight
junction permeability (139, 140). Unexpectedly, ethanol challenge induces a transient
increase in mucosal permeability in the intestine and triggers gut bacterial entrance into
the lamina propria area in WT mice. However, GPR41-/- or GPR43-/- mice had
significantly milder changes in gut permeability compared to WT mice. Moreover,
expression of the tight junction protein ZO-1 was better maintained in the SCFA
receptor-deficient mice than WT mice following the ethanol treatment. But, tight junction
protein expression is comparable in WT and both KO mice without immune challenge.
These results suggest that activation of the SCFA receptors functions to increase the gut
epithelial permeability, with a probably outcome of mounting an effective immune
response after a challenge.

31
Neutrophils accumulate in the intestine during inflammatory responses, and
chemoattractants including CXCL1 and CXCL2 are major players in recruiting
leukocytes (141). Mixed conclusions regarding the roles of SCFAs in regulation of gut
inflammation have been reported. SCFA administration increased gut expression of
inflammatory cytokines in some studies (142). Others reported that SCFAs, especially
butyrate, have an anti-inflammatory role in gut inflammation (143). Recently,
contradictory results on the impact of GPR43 on DSS-induced colitis have been reported
(21, 22). Thus, the function of SCFAs and GPR43 in regulation of inflammation in the
intestine remains inconclusive. Our results demonstrated that SCFA receptor-deficient
mice displayed hypo-inflammation following the ethanol or TNBS treatment. In line with
the positive roles of SCFA receptors in mediating inflammation, induction of major
chemokines, such as CXCL1 and CXCL2, and inflammatory cytokines, such as IL-1β,
IL-6, and IL-12, in the colon was significantly lower in GPR41-/- and GPR43-/- mice
compared to WT mice. Collectively, SCFAs receptors play a positive role in induction of
inflammatory responses.
With C. rodentium infection model, we examined anti-bacterial immunity in
SCFA-treated mice or SCFA receptor-deficient mice. Interestingly, we found that both
KO mice showed delayed clearance of pathogenic bacteria in the intestine. In line with
this, both KO mice failed to induce the acute inflammatory response post infection.
Acetate (C2) administration accelerated anti-bacterial immunity based on clearance of the
pathogen and induction of inflammatory molecules such as cytokines and chemokines.
These results suggest that SCFA-receptor signaling has a significant role in anti-bacterial
immunity in the intestine.

32
SCFA receptors are highly expressed by epithelial cells. Thus, these receptors are
more likely play a significant role in regulation of intestinal immune response through
epithelial cells. The chimera studies indicate that the GPR41 and GPR43 expressed by the
non-marrow-derived cells have significantly larger impacts than marrow-derived cells in
mediating tissue inflammation, mounting a T-cell response, and generating neutrophil
accumulation in the TNBS-induced inflammation model and C. rodentium infection
model. This result is also supported by our in vitro study with intestinal ECs that
indicates that SCFA receptors are required for SCFA-mediating inflammatory mediator
production by colonic ECs. These data suggests the SCFA-receptor axis in epithelial cells
has a significant impact on effective induction of anti-bacterial immunity.
As an intracellular mechanism, we found that activation of MEK-ERK and p38
MAPK pathways and the ATF2 transcription factor are required for normal expression of
inflammatory cytokines and chemokines by ECs in response to SCFAs. In summary, gut
microbiota-produced SCFAs activate SCFA receptors on colonic ECs, which leads to
conditions ECs to mount effective inflammatory responses.

33
2.5 Summary and future directions
We identified SCFA have positive function on induction of inflammatory responses
followed by chemical or pathogen challenges. SCFAs activate GPR41 and GPR43 on
intestinal epithelial cells to induce production of inflammatory mediators. Underlying
mechanism is that SCFA-receptors axis activate ERK signaling to induce gene
expressions of inflammatory mediators. SCFA-receptor-mediated inflammatory
responses have implicated in anti-bacterial immunity in the intestine. The efficient anti-
bacterial immunity contributes the maintenance of gut homeostasis in the steady state.
Therefore there is a possibility that SCFAs potentially have long term effects on the
regulation of immune homeostasis through multiple immune cell types. Thus, effects of
dietary fibers or SCFAs on the regulation of inflammation-associated tumorigenesis need
to be studied. And impacts of SCFAs on the regulation of other immune cells such as B
cells, which are important for anti-bacterial immunity and inflammation are need to be
further investigated.

34
Figure 2.1 Hypo-inflammatory responses in GPR41-/- and GPR43-/- mice after
ethanol-induced breach of intestinal barrier function.
Clinical symptom of WT, GPR41-, and GPR43-deficient mice following ethanol
challenge. Weight change (A) and gross appearance, and colon length (B) in mice
following the ethanol treatment. Mice were treated by intra-rectal injection of 50%
ethanol. All of the data were obtained at 24 h after ethanol treatment. Pooled data from
three independent experiments are shown. Significant differences (p<0.05) from
untreated WT mice* or ethanol-treated WT mice.**

35
Figure 2.2 Mild histological changes in GPR41-/- and GPR43-/- mice following rectal
administration of ethanol.
Histological changes in the distal colon of WT, GPR41-/-, and GPR43-/- mice following
rectal administration of 50% ethanol. Colon tissues in paraffin were stained with
hematoxylin and eosin (H&E). The sections were graded on a 0-4 scale based on
pathological features, including the extent of leukocyte infiltration, crypt
elongation/abscess, bowel wall thickening, mucosa hyperplasia, and ulceration. 0, no
evidence of inflammation; 1, low level of leukocyte infiltration in <10% high power
fields (hpf), no structural changes observed; 2, moderate leukocyte infiltration in 10–25%
hpf, crypt elongation, and bowel wall thickening; 3, high level of leukocyte infiltration in
25–50% hpf, bowel wall thickening and mucosa hyperplasia; and 4, severe leukocyte
infiltration in >50% hpf, crypt elongation, bowel wall thickening, mucosa hyperplasia,
and ulceration. *Significant differences from WT mice.

36
Figure 2.3 Reduced neutrophil infiltration in colon tissue following ethanol-induced
breach of intestinal barrier function in GPR41-/- and GPR43-/- mice.
(A) Neutrophil infiltration in the colon following the ethanol treatment. Neutrophil
frequency (%) among colonic lamina propria cells is shown. Neutrophils were stained by
anti-Gr-1 and anti-7/4 antibody and data were acquired by flow cytometry. (B)
Expression of ZO-1 and GR-1 in the colon during the inflammation was examined by
IHC. For immunofluorescence staining, 6 μm-thick frozen and paraffin colon sections
were stained respectively with rabbit polyclonal antibodies to ZO-1 and anti-Gr-1 and
Hoechst 33342.

37
Figure 2.4 Reduced gut permeability and tissue bacteria infiltration in colon tissue
following ethanol-induced breach of intestinal barrier function in GPR41-/- and
GPR43-/- mice.
(A) The gut barrier function was determined based on FITC-dextran leakage into the
blood following the ethanol treatment (n=10-12/group). For measurement of gut
permeability, mice fasted overnight, received intragastrically with fluorescein
isothiocyanate (FITC)-conjugated dextran. The mice were sacrificed 3 h later, and the
FITC-dextran concentration in the plasma was determined with a fluorescent microplate
reader. (B) Levels of colon tissue-associated commensal bacteria following the ethanol
treatment (n=10-13/group). Bacterial 16S rRNA gene levels in the colon tissue were
examined by qPCR for a conserved region of the gene. All of the data were obtained at
24 h after ETOH treatment. Pooled data from three independent experiments are shown.
Significant differences (p<0.05) from untreated WT mice* or ethanol-treated WT
mice.**

38
Figure 2.5 Defective induction of inflammation-associated genes in SCFA receptors-
deficient mice following ethanol-induced breach of intestinal barrier function.
(A) Genes differentially expressed in the colon tissues of WT versus SCFA-receptor KO
mice following the ethanol treatment. Microarray data are shown in a multi-plot format
showing gene expression differences between WT and KO mice. (B) Treeviews of
selected chemokine and cytokine genes up- or down-regulated following the ethanol
treatment.

39
Figure 2.6 Abnormally low expressions of inflammatory genes in GPR41-/- and
GPR43-/- mice following ethanol-induced breach of intestinal barrier function.
qPCR analysis of selected genes for distal colon tissues. All of the data were obtained
from colon tissues from WT, GPR41-/-, and GPR43-/- mice at 24 h after ethanol treatment.
Pooled data from three independent experiments are shown. Significant differences
(p<0.05) from untreated WT mice* or ethanol-treated WT mice.**

40
Figure 2.7 Abnormally low immune responses to TNBS in SCFA receptors-KO mice.
Weight change (A), stool consistency scores (B), colon length (C) following the TNBS
treatment. Mice were sacrificed on 3 day post TNBS injection. Stool consistency scores
were measured as follows: 0, normal stool; 1, soft stool; 2, diarrhea; 3, diarrhea and anal
bleeding. Weight change and stool scores were monitored during experimental period.
Mice were sacrificed three days after the TNBS rectal administration and measured colon
length. Combined (n=12-30/group) or representative data from 4 independent
experiments are shown. Significant differences (p<0.05) from untreated WT mice* or
TNBS-treated WT mice.**

41
Figure 2.8 GPR41-/- and GPR43-/- mice were less susceptible to TNBS-induced
inflammation.
Histological changes in the distal colon following rectal administration of TNBS. Colon
tissues in paraffin were stained with H&E. The sections were graded on a 0-4 scale based
on pathological features, including the extent of leukocyte infiltration, crypt
elongation/abscess, bowel wall thickening, mucosa hyperplasia, and ulceration. 0, no
evidence of inflammation; 1, low level of leukocyte infiltration in <10% high power
fields (hpf), no structural changes observed; 2, moderate leukocyte infiltration in 10–25%
hpf, crypt elongation, and bowel wall thickening; 3, high level of leukocyte infiltration in
25–50% hpf, bowel wall thickening and mucosa hyperplasia; and 4, severe leukocyte
infiltration in >50% hpf, crypt elongation, bowel wall thickening, mucosa hyperplasia,
and ulceration. *Significant differences from WT mice.

42
Figure 2.9 Abnormally low IL-6 production in GPR41-/- and GPR43-/- mice followed
by TNBS challenge.
Levels of IL-6 in the blood or conditioned media of colonic ECs prepared from TNBS-
treated mice. The concentration of IL-6 was examined by ELISA. Mice were sacrificed
three days after the TNBS rectal administration. Blood was collected for measurement of
serum IL-6 concentration. Colonic epithelial cells were isolated from WT, GPR41-/-, and
GPR43-/- mic and cultured in the presence of LPS for stimulation. Conditioned media of
colonic ECs were harvested for ELISA assay. Significant differences (p<0.05) from
untreated WT mice* or TNBS-treated WT mice.**

43
Figure 2.10 Defective cytokine and T cell responses to TNBS in the gut of GPR41-/-
and GPR43-/- mice.
(A) Expression of signature genes for Th1, Th17 and Tregs in the distal colon. qPCR was
performed for indicated genes. (B) Frequencies of Th1 (CD4+IFN-γ+ cells), Th17
(CD4+IL-17A+ cells), and Tregs (CD4+FoxP3+ cells), in the intestinal LP and MLN. Flow
cytometry was performed for examination of T-cell subset frequencies. Percentages of
CD4+ T-cells are shown. All of the data were obtained on day 3 following the TNBS
treatment. Pooled data obtained from 5 independent experiments are shown. Significant
differences from the untreated wild type (WT) mice* or TNBS-treated WT mice.**

44
Figure 2.11 Defective chemokine expression and neutrophil recruitment in GPR41-/-
and GPR43-/- mice following TNBS administration.
(A) Infiltration of phagocytes in the large intestinal lamina propria. Frequencies of Gr-1+
cells were examined by flow cytometry. (B) Expression of chemokines in the distal colon.
qPCR was performed. Values were normalized to β-actin. All of the data were obtained
on day 3 following TNBS treatment. Combined (n=12-30/group) or representative data
from 4 independent experiments are shown. Significant differences (p<0.05) from
untreated WT mice* or TNBS-treated WT mice.**
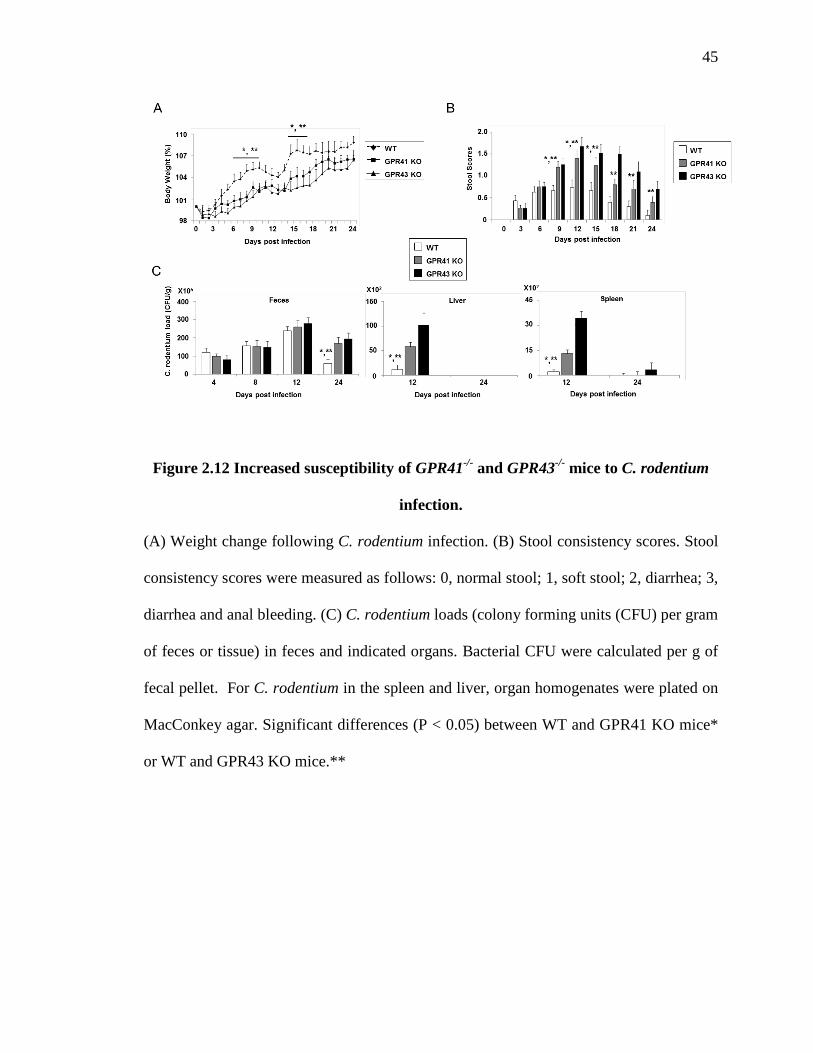
45
Figure 2.12 Increased susceptibility of GPR41-/- and GPR43-/- mice to C. rodentium
infection.
(A) Weight change following C. rodentium infection. (B) Stool consistency scores. Stool
consistency scores were measured as follows: 0, normal stool; 1, soft stool; 2, diarrhea; 3,
diarrhea and anal bleeding. (C) C. rodentium loads (colony forming units (CFU) per gram
of feces or tissue) in feces and indicated organs. Bacterial CFU were calculated per g of
fecal pellet. For C. rodentium in the spleen and liver, organ homogenates were plated on
MacConkey agar. Significant differences (P < 0.05) between WT and GPR41 KO mice*
or WT and GPR43 KO mice.**

46
Figure 2.13 Delayed immune responses of GPR41-/- and GPR43-/- mice to
C. rodentium infection.
(A) Neutrophil infiltration in the colon during infection. Gr-1+ cells were examined. Mice
were sacrificed at indicated time pointes and colonic neutrophil frequencies were
examined by flow cytometry. (B) Th1 (CD4+IFN-γ+ cells) and Th17 (CD4+IL-17A+ cells)
frequencies in the colon at 2 weeks post infection. Percentages of CD4+ T-cells are
shown. Flow cytometry was performed for panel A and B. *Significant differences
between indicated groups or from infected WT mice

47
Figure 2.14 Delayed induction of inflammatory cytokines and chemokines in
GPR41-/- and GPR43-/- mice during C. rodentium infection.
(A) Expression of pro-inflammatory cytokines in the cecum (n=13-15/group). (B)
Expression of CXCL1 and CXCL2 in the cecum (n=10-12/group). WT, GPR41-/-, and
GPR43-/- mice were sacrificed for examination of kinetics of inflammatory mediators
expression in cecum tissues. qPCR was performed. Combined data of 3 independent
experiments are shown. Significant differences (P < 0.05) between WT and GPR41 KO
mice* or WT and GPR43 KO mice.**

48
Figure 2.15 Delayed gut permeability changes of GPR41-/- and GPR43-/- mice to
C. rodentium infection.
(A) Gut permeability change and (B) tissue-associated eubacteria following infection.
For measurement of gut permeability, mice fasted overnight, received intragastrically
with fluorescein isothiocyanate (FITC)-conjugated dextran. And mice were sacrificed 3 h
later. Tissue-associated eubacteria levels based on the Ct values of the amplified
conserved 16S rRNA gene sequence were normalized with the signal of the amplified
mouse genomic β-actin gene in each sample. Pooled data obtained from 3 independent
experiments are shown (4-5/group). Significant differences (P < 0.05) between WT and
GPR41 KO mice* or WT and GPR43 KO mice.**
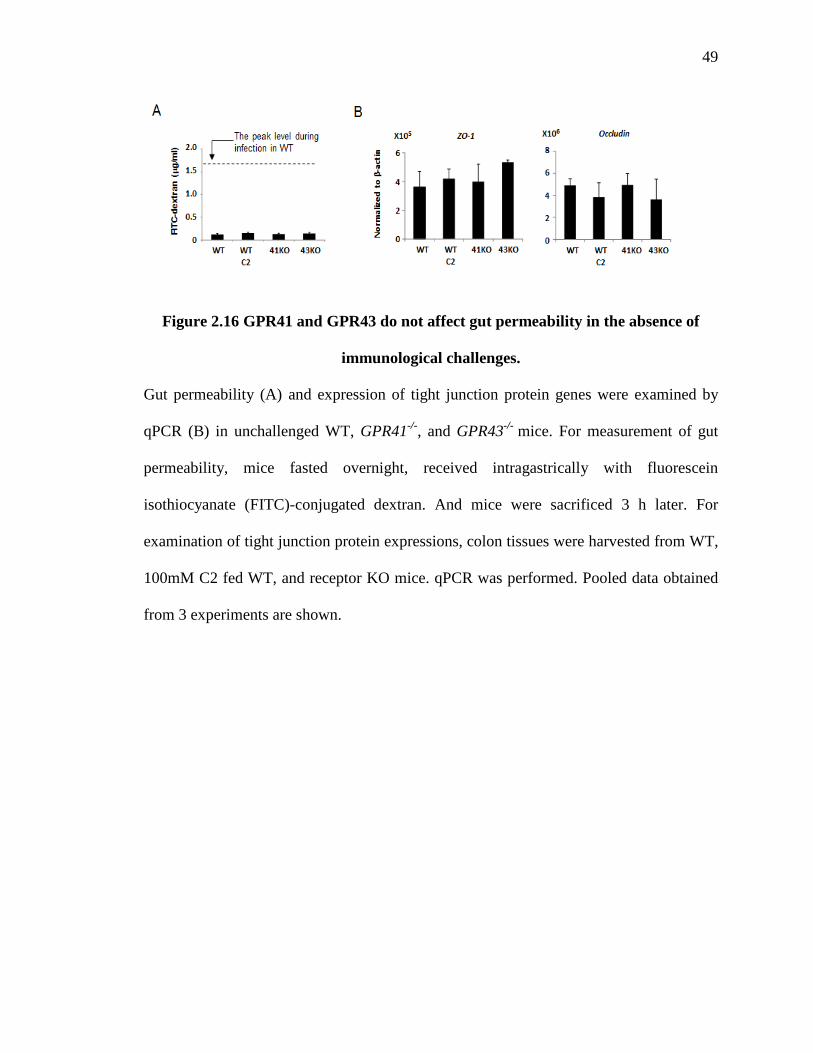
49
Figure 2.16 GPR41 and GPR43 do not affect gut permeability in the absence of
immunological challenges.
Gut permeability (A) and expression of tight junction protein genes were examined by
qPCR (B) in unchallenged WT, GPR41-/-, and GPR43-/- mice. For measurement of gut
permeability, mice fasted overnight, received intragastrically with fluorescein
isothiocyanate (FITC)-conjugated dextran. And mice were sacrificed 3 h later. For
examination of tight junction protein expressions, colon tissues were harvested from WT,
100mM C2 fed WT, and receptor KO mice. qPCR was performed. Pooled data obtained
from 3 experiments are shown.

50
Figure 2.17 C2 administration accelerates the immune response to infection.
(A) Weight change and stool consistency scores of WT and C2-fed mice following C.
rodentium infection. Stool consistency scores were measured as follows: 0, normal stool;
1, soft stool; 2, diarrhea; 3, diarrhea and anal bleeding. (B) Fecal C. rodentium load.
Pooled data obtained from 3 independent experiments are shown (n=10-15/group). WT
and C2-fed mice were infected with C. rodentium. When indicated, mice were injected
i.p. with antibodies to neutralize IL-6 (on day 0 and 2) and deplete neutrophils (on day -2
and 0). Significant differences (P < 0.05) between WT and WT-C2 * or WT and WT-Ab-
Ly6G/IL6**.

51
Figure 2.18 C2 administration promotes chemokine and cytokine expression in
colon tissue to C. rodentium infection.
Expression of neutrophil chemoattractants, CXCL1 and CXCL2 (A) and expression of IL-
6 (B). Pooled data obtained from 3 independent experiments are shown (n=10-15/group.
WT and 100mM C2-fed mice were infected by C. rodentium. When indicated, mice were
injected i.p. with antibodies to neutralize IL-6 (on day 0 and 2) and deplete neutrophils
(on day -2 and 0).Significant differences (P < 0.05) between WT and WT-C2 * or WT
and WT-Ab-Ly6G/IL6**.

52
Figure 2.19 C2 administration accelerates immune cell responses and gut
permeability changes followed by infection.
(A) Neutrophils (Gr-1+) in colon. (B) Th17 (CD4+IL-17A+) cells in the colon. Relative
values are shown by normalized values to untreated mice. (C) Gut permeability change.
Some mice were injected twice with antibodies to neutralize IL-6 (day 0 and 2) and
deplete neutrophils (day -2 and 0). WT and C2-fed mice were infected by C. rodentium.
When indicated, mice were injected i.p. with antibodies to neutralize IL-6 (on day 0 and 2)
and deplete neutrophils (on day -2 and 0). Pooled data obtained from 3 independent
experiments are shown (n=10-15/group). Significant differences (P < 0.05) between WT
and WT-C2 * or WT and WT-Ab-Ly6G/IL6**.

53
Figure 2.20 No significant changes in commensal bacteria in the gut of
GPR41-/- or GPR43-/- mice.
The levels of major bacterial groups in the gut were analyzed by qPCR examination of
the 16S rRNA gene isolated from the stool of wild-type, GPR41-/-, and GPR43-/- (n = 8-9
for each group) mice using the primers listed in Table 2.1. The copy numbers of 16S
rRNA genes for specific bacterial groups were normalized by that of the total eubacteria.
Abundance of the commensal bacterial groups is shown in relative to WT levels.

54
Figure 2.21 Differential roles of the SCFA receptors expressed by non-bone marrow
versus marrow-derived cells in promoting the immune response to C. rodentium.
(A) Weight change following C. rodentium infection in bone marrow chimera mice. (B)
Stool consistency scores. Stool consistency scores were measured as follows: 0, normal
stool; 1, soft stool; 2, diarrhea; 3, diarrhea and anal bleeding. (C) Fecal C. rodentium load.
For generating chimeras, irradiated recipient mice were reconstituted with donor marrow
cells. The mice were kept for 8 weeks for hematopoietic reconstitution and were
sensitized and orally infected with C. rodentium. Significant differences from WT → WT
mice*.

55
Figure 2.22 Differential roles of the SCFA receptors expressed by non-bone marrow
versus marrow-derived cells in effector T cell and neutrophil responses to C.
rodentium.
(A) Th17 (CD4+IL-17A+) cells in the colon. (B) Infiltration of the colon by neutrophils
(Gr-1+). Chimera mice were infected at 8 weeks after the bone marrow-transfer
(donorhost). Flow cytometry was performed for panel A and B. Pooled data (8-10
mice/group) obtained from 2-3 independent experiments are shown. Significant
differences from WTWT mice* or between indicated groups.**

56
Figure 2.23 GPR41 and GPR43 are highly expressed by intestinal epithelial cells.
Expression of GPR41 and GPR43 mRNA by Gr-1+ neutrophils, CD326+ ECs, CD19+ B
cells, and CD4+ T cells was examined by qPCR. Both CD19+ B cells and CD4+ T cells
were isolated from spleen. CD326+ ECs were isolated from colon. Gr-1+ neutrophils were
isolated from BM. WT mice were used for cell isolations.

57
Figure 2.24 Comparison of the roles of SCFA receptors expressed by non-BM
versus BM-derived cells in mediating the inflammatory response to TNBS.
(A) Weight change in marrow-transferred mice (n=10-23/group). (B) Stool consistency
scores. Stool consistency scores were measured as follows: 0, normal stool; 1, soft stool;
2, diarrhea; 3, diarrhea and anal bleeding. (C) Gross appearance and length of the colon.
(D) Infiltration of the colon by neutrophils (Gr-1+ cells). Frequency of neutrophils in
untreated-WT mice is shown. (E) Th1 (CD4+IFN-γ+) frequency in the colon. All of the
data were obtained on day 3 following TNBS treatment. Pooled data obtained from three
independent experiments are shown. Significant differences from WTWT mice* or
between indicated groups.**

58
Figure 2.25 SCFA-dependent expression of IL-6 and chemokines by colonic ECs
was suppressed by pertussis toxin.
Expression of IL-6, CXCL1, and CXCL10 by ECs in response to SCFAs and bacterial
products (LPS or commensal bacterial extract) was examined with qPCR. ECs,
established for 3 days, were treated with pertussis toxin (PTX, FL; 0.1 μg/ml) for 1 h
before treatment with the SCFAs for 6 days. The cells were then treated with LPS (1
μg/ml) or commensal bacterial extract (20 μg/ml) for 24 h. Pooled data from three
independent experiments (n=3) are shown. *Significant differences from the untreated
WT control groups.

59
Figure 2.26 SCFA-dependent expression of inflammatory chemokines and cytokines
is dependent on epithelial GPR41 and GPR43.
(A) Production of inflammatory cytokines by WT, GPR41-/-, and GPR43-/- colonic ECs
pre-treated with SCFAs and LPS. (B) Expression of chemokines by colonic ECs in
response to SCFAs and LPS. Colonic ECs were cultured for 6 days, and C2 (10 mM) or
C3 (1 mM) was added during this culture period. These cells were further activated with
lipopolysaccharide (LPS; 1 µg/ml) or a commensal bacterial extract (20 µg/ml) for 24
h*Significant differences from the untreated WT control groups.
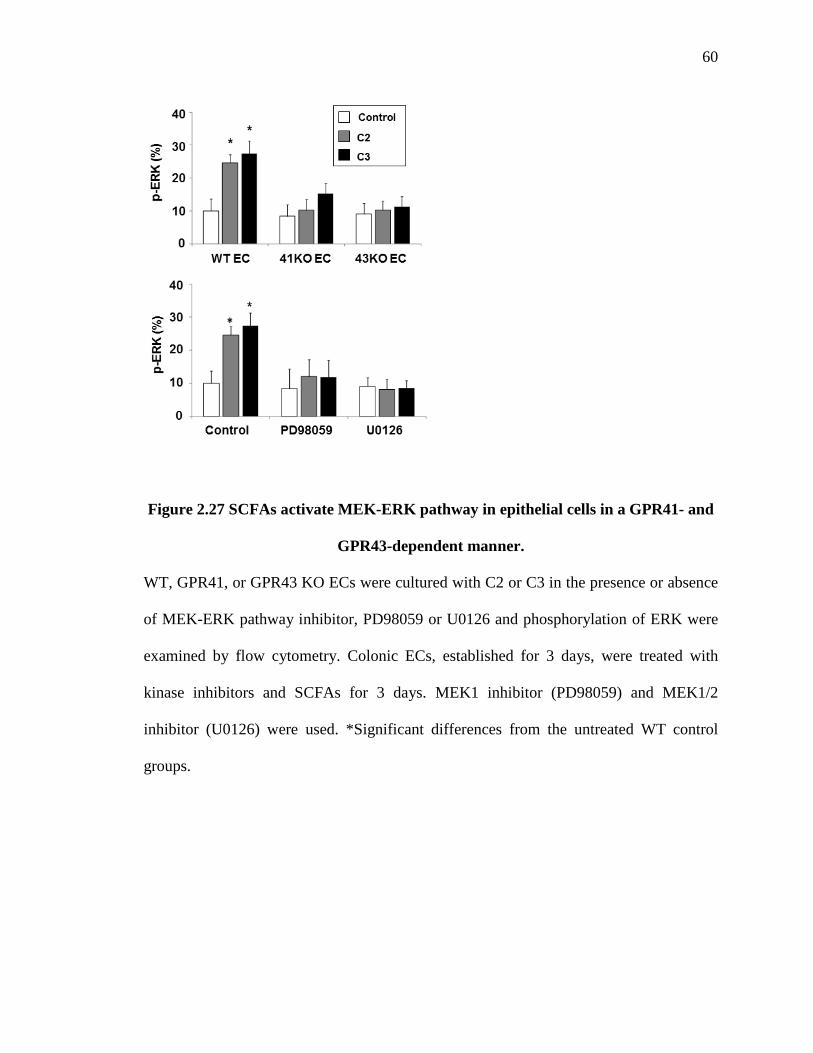
60
Figure 2.27 SCFAs activate MEK-ERK pathway in epithelial cells in a GPR41- and
GPR43-dependent manner.
WT, GPR41, or GPR43 KO ECs were cultured with C2 or C3 in the presence or absence
of MEK-ERK pathway inhibitor, PD98059 or U0126 and phosphorylation of ERK were
examined by flow cytometry. Colonic ECs, established for 3 days, were treated with
kinase inhibitors and SCFAs for 3 days. MEK1 inhibitor (PD98059) and MEK1/2
inhibitor (U0126) were used. *Significant differences from the untreated WT control
groups.

61
Figure 2.28 SCFA-induced expression of the inflammatory cytokine IL-6 is
dependent on the MEK-ERK pathway in epithelial cells.
Effects of signaling molecule inhibitors on SCFA-dependent expression of IL-6 were
examined. Colonic ECs, established for 3 days, were treated with kinase inhibitors and
SCFAs for 3 days. qPCR was performed for examination of IL-6 expression. MAPK
inhibitor (SB203508), c-Jun N-terminal kinases/JNK inhibitor (SP600125), MEK1
inhibitor (PD98059), and MEK1/2 inhibitor (U0126) were used. Significant differences
(P < 0.05) between control and inhibitor-treated groups for C2* or C3**.

62
Figure 2.29 Effects of signaling molecule inhibitors on SCFA-dependent expression
of neutrophil-attracting chemokines by epithelial cells.
ECs, established for 3 days, were treated with inhibitors and SCFAs for 6 days. The cells
were then treated with LPS (1 μg/ml) for 24 h before qPCR analysis of CXCL1 (A) and
CXCL2 (B). MAPK inhibitor (SB203508), c-Jun N-terminal kinases/JNK inhibitor
(SP600125), MEK1 inhibitor (PD98059), and MEK1/2 inhibitor (U0126) Pooled data
obtained from three independent experiments (n=3) are shown. *Significant differences
from control groups (no inhibitor treatment).

63
Figure 2.30 Defective SCFA-induced phosphorylation of ERK in SCFA receptor-
deficient mice after infection with C. rodentium.
ERK expressions by colon tissues of WT, GPR41-/-, and GPR43-/- mice at 5 days after
infection with C. rodentium were examined by IHC. WT, GPR41-/-, and GPR43-/- mice
fed regular water or C2 (100 mM) water before initiation of infection study. For
immunofluorescence staining, frozen colon sections were stained respectively with
Phospho-p44/42 MAPK (Erk1/2).

64
Figure 2.31 SCFAs induce ATF2 activation in colonic epithelial cells in a GPR41-
and GPR43-dependent manner.
Phosphorylation of ATF2 in WT, GPR41-/-, and GPR43-/- ECs were examined by flow
cytometry. Colonic ECs, established for 3 days, were treated with kinase inhibitors and
SCFAs for 3 days. Pooled data obtained from three independent experiments (n=3) are
shown. Significant differences (p<0.05) from C2/C3-untreated*

65
Table 2.1 Primers used in this study (Chapter 2)
Quantitative real time PCR (qPCR) for mouse genes was performed. The abundance of
specific bacterial groups was measured with group-specific 16S rRNA gene primers. The
signals of group-specific 16S rRNA gene were normalized with that of the total eubacteria
16S rRNA gene in each sample. *Gene accession numbers.
Gene (mouse) Forward Reverse References* IL17A GAC TCT CCA CCG CAATG CGG GTC TCT GTT TAG GCT NM_010552.3
RoRγt AGC CAC TGC ATT CCC AGT TTC T TGA AAG CCG CTT GGC AAA CT NM_011281.3 IFNγ AGA CAA TCA GGC CAT CAG CA CGA ATC AGC AGC GAC TCC TTT NM_008337.4 T-bet ATG CCG CTG AAT TGG AAG GT ACC TCG CAG AAA GCC ATG AAG T NM_019507.2
FoxP3 CAG CTG CCT ACA GTG CCC CTA G CAT TTG CCA GCA GTG GGT AG NM_054039.2 IL-6 CTG GTC TTC TGG AGT ACC ATA GC TGC CGA GTA GAT CTC AAA GTG
AC NM_031168.2
IL-12 CTA GGA TCG GAC CCT GCA GG CTA GGA TCG GAC CCT GCA GG NM_008354.2 CCL2 GTT GGC TCA GCC AGA TGC A AGC CTA CTC ATT GGG ATC ATC
TTG NM_000882.3
CCL5 TCA CCA TCA TCC TCA CTG C TGA ACC CAC TTC TTC TCT GG NM_013653.3
CXCL1 CGT CGC TTC TCT GTG CA TGG GGA GAC CTT TTA GCA NM_008176.3 CXCL2 AAC CAC CGA GCT ACA GG CCT TTC GAG GTC AGT TAG C NM_009140.2 CXCL10 GGT CTG AGT GGG ACT CAA GG TTT GGC TAA ACG CTT TCA TT NM_021274.2
CXCL13 GCA GAA TGA GGC TCA GCA CAG AGA GAA TGA GTG ACC TCG AAC NM_018866.2 MMP9 TGT ACA CAG GCA AGA CCG TG CTC CAG CAT CCA GTG CAT CG NM_013599.3 MMP10 CCT TCA GAC TTA GAT GCT GCC GTA CTA CTA GTC GTG TCG TCG NM_019471.3
IL1β GTA CAA GGA GAA CAA AGC AAC CTG TGC CTA AGG TAC CAC TTC NM_008361.4
Ffar3 CTG GCT GTT GTG ACG CTT CGG TGA CAA ATT CAG AAC TCT NM_001033316.3 Ffar2 CTT CTT GCA GCC ACA CTG CT CAC TCC TTG GAT GGC TCT TC NM_001168511.1 Bacterial 16S rRNA gene Forward Reverse References Eubacteria ACT CCT ACG GGA GGC AGC AGT ATT ACC GCG GCT GCT GGC (145) Enterobacteriaceae GTG CCA GCA GCC GCG GTA A GCC TCA AGG GCA CAA CCT CCA
AG (146)
Bacteroides GGT TCT GAG AGG AGG TCC C GCT GCC TCC CGT AGG AGT (147)
Mouse intestinal bacteroides
CCA GCA GCC GCG GTA ATA GCG ATT CCG CAT ACT TCT C (148)
Clostridium coccoides ACT CCT ACG GGA GGC AGC GCT TCT TAG TCA GGT ACC GTC AT
(149)
Clostridium perfringens CGC ATA ACG TTG AAA GAT GG CCT TGG TAG GCC GTT ACC C (150) Segmented filamentous bacteria
GAC GCT GAG GCA TGA GAG CAT GAC GGC ACG GAT TGT TAT TCA (151)
Bacillus GCG GCG TGC CTA ATA CAT GC CTT CAT CAC TCA CGC GGC GT (152)

66
CHAPTER 3. ROLE OF RETINOIC ACID ON THE REGULATION OF INNATE LYMPHOID CELL SUBSETS’ INTESTINAL MIGRATION
3.1 Introduction
Innate lymphoid cells (ILCs) do not have antigen receptors but play immune
regulatory functions in a manner somewhat reminiscent of T cells (153). ILCs play
important roles in host defense against pathogens and can mediate tissue inflammation
(73, 105, 154). Three groups of ILCs with distinct characteristics in development and
effector function have been identified. Group 1 ILCs (ILC1), including NK cells, are
induced by the transcription factor T-bet and produce IFN-γ (155). The development of
group 2 ILCs (ILC2), which produce IL-5 and IL-13, require ROR-α, Gata3, and Tcf7
for development (156). Group 3 ILCs (ILC3) require RORγt and Gata3 for development
and are composed of IL-17/IL-22-producing LTi cells, IL-17/IL-22/IFN-γ-producing
ILCs, and IL-22-producing natural cytotoxicity triggering receptor (NCR)+ ILCs (91,
157). The importance of these ILCs in mounting innate immunity and regulating
inflammatory responses in the intestine and other mucosal tissues is now firmly
established.
Migration of lymphocytes is regulated by chemokines and integrins (75, 158).
Naïve T cells utilize CCR7 and CD62L to migrate to secondary lymphoid tissues. Upon
antigen priming, T cells undergo trafficking receptor switches. Gut-homing T cells down-

67
regulate CCR7 and CD62L but up-regulate a chemokine receptor, CCR9, and an integrin,
α4β7 (70, 71). CCR9+α4β7+ cells migrate to the small intestine, while CCR9-α4β7+ cells
are likely to migrate into the colon. Acquisition of these receptors occurs in gut-
associated secondary lymphoid tissues such as mesenteric lymph node (MLN) and
Peyer’s patches (PP). CCL25, a chemokine expressed in the small intestine (SI), activates
CCR9 for adhesion triggering and chemotaxis (159). α4β7 binds MAdCAM-1, which is
widely expressed on gut endothelial cells (160). It is generally accepted that innate
immune cells such as NK cells, do not undergo a sophisticated trafficking receptor switch
for specific migration into a tissue site. All ILC subsets are highly enriched in the
intestine compared to other tissue sites. However, the mechanism for ILC enrichment and
population in the intestine has been unclear.
Recently, it has been reported that RA regulates the development, expansion, and
effector function of ILCs. RA-RAR signaling is important for lymphoid tissue inducer
cell (LTi cell) development in the gut tissue of the fetus (74). RA expands ILC3s but
suppresses ILC2s in in the gut (73). In addition, RA promotes IL-22 expression by ILC3s
through RARα (72). However, role of RA on ILC migration program have not been
investigated, yet.
We hypothesized that ILC migration to the intestine is not random but is regulated
specifically by certain trafficking receptors. We investigated migration programs of ILC
subsets and role of intestinal tissue factor, RA on the regulation of homing receptor
expression in ILC subsets. We report here that retinoic acid induce homing receptor
switches from lymphoid tissue receptor to gut homing receptor in ILC1 and ILC3 in the
periphery. However, ILC2 acquire gut homing receptors expression in the bone marrow.

68
Hence, ILC subsets have conserved and diverse migration program for gut homing and
gut metabolite, RA have significant impact on ILC migration program in subset-specific
manner.

69
3.2 Materials & methods
Animals
All animal experiments in this study were approved by the Purdue Animal Care
and Use Committee. All strains were kept at Purdue for at least 1 year. C57BL/6 mice
were from Harlan. CD45.1 congenic (B6.SJL-Ptprca Pepcb/BoyJ), CD90.1 congenic
(B6.PL-Thy1a/CyJ), Rag1-/- (B6.129s7-Rag1tm1Mom/J), Ccr7-/- (B6.129P2(C)-Ccr7tm1Rfor/J),
and Itgβ7-/- (C57BL/6-Itgb7tm1Cgn/J) mice were purchased from the Jackson Laboratory.
Ccr9-/- mice were obtained from described previously (161). VAD and VAN Rag1-/- or
C57BL/6 mice were generated by feeding 0 and 2,500 IU/kg of retinyl acetate containing
rodent diets (Harlan Teklad diets, Indianapolis, IN). Special diets were given to mice
following birth and after weaning until they were 11-12 weeks of age.
Identification of mature ILC subsets and bone marrow ILC progenitors
Cells from intestinal lamina propria, PP, spleen, MLN, and blood were prepared
as previously described. For ILC1s and ILC3s, cells were first stained with antibodies to
following the common lineage markers: CD3ε (clone 145-2C11), CD5 (53-7.3), CD8α
(53-6.7), CD19 (6D5), B220 (RA3-6B2), CD11b (M1/70), CD11c (N418), Ter119 (Ter-
119), F4/80 (BM8), Gr-1 (RB6-8C5), TCRβ (H57-597), TCRγδ (GL3), CD49b (DX5),
and FcεR1α (MAR-1) and other antigens (CD45.2/104, CD90.2/53-2.1, CD90.1/OX-7,
and/or CD127/A7R34), and then fixed and permeabilized for further staining for
intracellular antigens (T-bet/eBio-4B10, GATA-3/TWAJ, RORγt/AFKJS-9 and/or
Eomes/Dan11mag). For ILC2s, cells were stained with antibodies to the common lineage

70
markers mentioned above and NK1.1/PK136, CD90.2, CD90.1, CD127, Sca-1/D7,
KLRG-1/2F1, T1/ST2 (DIH9), and GATA-3. Intraepithelial cells were stained with
antibodies to the common lineage markers, NKp46 (29A1.4), and NK1.1. All antibodies
were from BioLegend or eBioscience. Gating strategies for mature ILC subsets are
described in Fig. 2.1. Isolated BM cells were stained with antibodies to CD127, Sca-1,
CD25, KLRG-1 and BM lineage markers (CD3ε, CD5, CD8α, B220, CD11b, Gr-1, Ter-
119, TCRγδ, and NK1.1). BM ILC2P and relatively more mature KLRG-1+ ILC2 cells
were respectively identified as Lin-CD127+Sca-1hiCD25+KLRG-1- and Lin-CD127+Sca-
1hiCD25+KLRG-1+ cells. The expression of homing receptors by ILCs and ILC2P was
determined using antibodies to CCR7 (4B12), CCR9 (242503), α4β7 (DATK32), and
CD103 (2E7).
Cell isolation and culture
Lineage negative cells (Lin-) were isolated from the spleen by depleting cells
expressing CD3ε, CD5, CD19, B220, CD11b, CD11c, Ter119, F4/80, and Gr-1
(splenocyte depletion markers) using biotin-labeled antibodies and anti-biotin beads
(Miltenyi Biotec). ILC3s were isolated by FACS (Lin-NK1.1-KLRG-1-CD90hiCD127+)
or MACS (Lin-NK1.1-KLRG-1-CD90hi). BM Lin- cells were prepared by depleting cells
expressing the BM lineage markers and Ly-6B.2 (7/4) by MACS and used for the BM
ILC2P in vivo short homing study. Lin- cells were cultured for 4 days in complete RPMI
1640 medium (10% FBS) with ILC subset-specific condition. (ILC3: Cecal bacterial
antigen (CBA, 10 µg/ml), IL-7 (20 ng/ml) and IL-23 (20 ng/ml); ILC1: CBA, IL-7 and
IL-15 (20 ng/ml); ILC2: IL-7). When indicated, RA (10 nM, all-trans RA), Ro41-5253

71
(500 nM, a RARα antagonist), AM580 (10 nM, RARα agonist), AC55649 (10 nM, a
RARβ2 agonist), or LE540 (500 nM, a pan-RAR antagonist), were added to culture.
These chemicals were from Sigma-Aldrich or Enzo Life Sciences. For DC and ILC co-
culture, CD11c+ cells were isolated from spleen and MLN with CD11c microbeads
(Miltenyi Biotec) (purity > 90%). 5 × 104 spleen and MLN-DCs, pulsed with CBA (10
µg/ml) for 3 h, were co-cultured with 5 × 105 spleen Lin- cells in complete RPMI medium
containing 10% FBS (non-heat inactivated) for 4 days. Cells were harvested and stained
for HRs and ILC-associated antigens and transcription factors.
Chemotaxis assay
Spleen Lin- cells (5×105/well), cultured in the presence or absence of RA (10 nM)
or Ro41-5253 (500 nM) in ILC subset-specific culture conditions for 4 days, were loaded
in the upper chamber of Transwell inserts (Corning) and allowed to migrate to the lower
chamber containing murine CCL25 (3 μg/ml, R&D Systems) for 3 h. The migrated cells
in the lower chambers were harvested, stained with antibodies to identify ILC1s, ILC2s,
and ILC3s, and counted by a time-based flow cytometry.
In vivo migration studies
For short-term migration of ILCs, cultured CD45.2+CD90.1+ WT and
CD45.2+CD90.2+ HR-deficient spleen Lin- cells (10-15M cells) were co-injected i.v. into
CD45.1 mice. Host mice were euthanized 20 hours later, and indicated organs were
harvested. The numbers of injected ILC3s migrated into each organ or in the blood were
determined by flow cytometry. The relative homing index for HR-deficient ILCs was

72
calculated as below: Homing index of homing receptor-deficient ILC for organs A=
[CD90.2+ HR-deficient- ILC in organ A] / [CD90.1+ WT ILC in organ A] ÷ [CD90.2+
HR-deficient ILC in injected cells] / [CD90.1+ WT ILC in injected cells]. Similar
experiments were performed for control versus RA-treated ILCs. For migration of BM
ILC2Ps, CD45.2+ Lin- BM cells, isolated from WT or HR-deficient BM (~35 M
cells/mouse), were singly injected i.v. into CD45.1 mice. The host mice were sacrificed
44 hours later, and indicated organs were harvested and examined for numbers of
transferred ILC2 lineage cells in each organ. Migration efficiency of HR-deficient
ILC2Ps to an organ was determined based on the normalized absolute numbers of
transferred ILC2 lineage cells in each organ that were divided by the total numbers of the
transferred ILC2 lineage cells in indicated organs.
For long-term population of BM-derived ILC subsets, a 1:1 mixture of BM cells (5×106
each) from WT mice (CD45.2+CD90.1+) and HR-deficient mice (CD45.2+CD90.2+) were
co-injected i.v. into irradiated (2×500 cGy with a 3 hour interval) host mice (CD45.1)
(The reconstitution efficiency: higher than 95% based on spleen). After 8 weeks, mice
were sacrificed for examination of ILC subset frequencies and numbers.
Confocal microscopy
Ileum tissues were frozen in Tissue-Tek OCT Compound (Sakura), and 9-μm
tissue sections were made using a cryostat (Leica). The sections were fixed in cold
acetone, and stained with fluorochrome-conjugated antibodies to pan-cytokeratin (Clone
AE1/AE3), CD90.2, and CD3 (Clone 17A2) or CD4. The images were collected with a
SP5 II (Leica) confocal microscope.

73
Infection with C. rodentium and ILC3-enriched cell transfer
VAN and VAD RAG1-/- mice were infected with C. rodentium (DBS100, 109
CFU/mouse) and their weight changes (%), stool consistency, and bacterial burden were
monitored. For ILC3 transfer into those mice, VAD mice were injected i.v. with WT
control ILC3s, WT RA-treated ILC3s, or gut HR-deficient RA-treated ILC3s two times
on day -2 and day 2 post-infection. Mice were sacrificed on day 9 post-infection.
qPCR
qPCR was performed as described before (162) to detect mRNA expression for
Il22, Il17a and anti-microbial peptide genes in SI and proximal colon tissues. The
primers information used for qPCR as followed.
β-actin: Forward: AGAAGAGCTACGAGCTGCCTGAC Reverse: TACTCCTGCTT
GCTGATCCACAT; IL17a: Forward: GAC TCT CCA CCG CAATG Reverse: CGG
GTC TCT GTT TAG GCT; IL22: Forward: GGC CAG CCT TGC AGA TAA CAA
Reverse: TCA GAG ACA TAA ACA GCA GGT CC; RegIIIβ: Forward: GAG GCC TGG
AGG ACA CCT C Reverse: GTA TCC CGT TGA AGT GGA GTG T ; RegIIIγ: Forward:
ATG GCT CCT ATT GCT ATG CC Reverse: RegIIIγ-R GGA ACT TAA ACG TCT GTA
TCC C

74
Statistical analysis
Student’s t-test (paired. Two-tailed) was used to determine significance of the differences
between two groups. p values < or = 0.05 were considered significant. Repeated
measures ANOVA was used to determine differences between two sets of weight change
data (SAS, version 9.2, SAS Institute Inc. Cary, NC). All error bars indicate standard
error of the mean (SEM).

75
3.3 Results
ILC subset distribution in various tissues
ILC subsets are widely distributed in various tissues but most densely populate
mucosal tissues, where their innate immune functions can be effectively utilized. For
example, ILCs in the intestines and lungs play significant roles in the regulation of innate
immunity against pathogenic microbes. ILCs are also detected in peripheral tissues and
regulate host metabolism. ILC subsets differently populate various tissues, which may
indicate their functional relevance to the immunological regulation of certain tissues. We
examine ILC subset (NK cells, ILC1, ILC2, and ILC3) in different tissues sites. Figure
3.1 shows the proportions of ILC subsets in each tissue site. NK cells (CD3-CD19-
CD127-NKp46+NK1.1+ or intestinal NK cells: CD3-CD19-CD127-RORγt-NKp46+NK1.1+
cells) are mostly present in secondary lymphoid tissues rather than intestinal tissue.
Among the total population of ILCs, NK cells comprise ~95% in the spleen and MLN
(Fig. 3.1). NK cells were also detected in lung and fat at modest levels. Non-NK ILC1s
are broadly distributed in various tissues, but they are a minor population compared to
NK cells and other ILC subsets. ILC2s (Lin-CD90+CD127+RORγt-GATA3+ cells) were
detected in the most of the analyzed tissues. It is noteworthy that ILC2s make up the
largest ILC population in the colonic lamina propria, skin, and fat tissue. In fat tissue and
skin, ILC2s comprised ~45% and ~90% of total ILCs, respectively (Fig. 3.1). This may
related to their regulatory functions such as allergic responses and fat metabolism. ILC3s
are enriched in the intestine, especially in the small intestinal lamina propria (Fig 3.1). In

76
summary, the distinct distribution of ILC subsets suggests specific roles of ILC subsets in
different tissue sites.
ILCs express homing receptors in tissue- and subset-specific manner
First we attempted to detect ILCs in different tissues. We established gating
strategy to detect total ILCs (Lin-CD90+CD127+) and ILC subsets (ILC1: Lin-
CD127+CD90+Eomes-RORγt-T-bet+, ILC2: Lin-NK1.1- CD127+CD90+Sca-1+KLRG-
1+GATA-3+), and ILC3: T-bet- or T-bet+ Lin-CD127+CD90+RORγt+GATA-3- (Fig. 3.2).
With these gating strategies, we examined homing receptors (HR) expression by ILCs in
various tissues to determine whether ILCs express HR in a tissue dependent manner.
Interestingly, ILCs display distinct HR expression pattern between secondary lymphoid
and intestinal tissues (Fig. 3.3). For example, CCR7 was highly expressed by in splenic
and mesenteric lymph node (MLN)-ILCs, whereas CCR9 and α4β7 was highly expressed
by the intestinal ILCs (Fig. 3.3). These data indicate that ILCs express HRs in a tissue-
specific manner. Next, we also examine HR expression in ILC subsets to determine
whether HR expression by ILCs is in a subset-specific manner. CCR7 is highly expressed
by splenic and MLN-ILC3s and MLN-ILC1s (Fig. 3.4). However, ILC2s did not express
CCR7 in either secondary lymphoid or intestinal tissues (Fig. 3.4). Many CCR9+ or
α4β7+ ILC1s and ILC3s were detected in the small intestine and colon. CCR9 expression
was uniformly high on ILC2s from all organs, but α4β7 expression was reduced in
intestinal ILC2s (Fig. 3.4). In general, ILC3s resemble ILC1s in terms of HR expression
profiles but are different than ILC2s. These results suggest that ILC subsets have shared
and distinctive HR expression profiles.

77
Tissue-specific HR expression required to populate ILCs in certain tissue sites
Since ILCs express HRs in a tissue dependent manner, we investigated whether
HR is functionally important for ILCs populating specific tissue sites. To do that, we
examined ILCs in mice deficient in the tissue-specific HRs: CCR7, CCR9, and Itgβ7.
Total ILCs were significantly decreased in the MLN of Ccr7-/- mice (Fig. 3.5). ILC
numbers were decreased specifically in the SI of Ccr9-/- mice (Fig. 3.5). ILCs were
decreased in both the small intestine (SI) and colon of Itgβ7-/- mice. In contrast, ILCs
were increased in the spleen and MLN of Ccr9-/- or Itgβ7-/- mice (Fig. 3.5). We also
confirmed that total ILC numbers in the SI were decreased in CCR9- or Itg-β7-deficient
mice by confocal microscopy (Fig. 3.7). More specifically, we examined ILC subset
frequencies and numbers in those HR-deficient mice. CCR9 deficiency affected ILC2
numbers in the small intestine (Fig. 3.6). In contrast, ILC1s and ILC3s were decreased in
the small intestine and colon of Ccr9-/- mice and Itgβ7-/- mice, respectively. ILC1s and
ILC3s were decreased in the MLN of Ccr7-/- mice. CCR9 is highly expressed by
intraepithelial (IEL)-ILC1 (Lin-NKp46+NK1.1+ cells) (Fig 3.8). In line with this, IEL-
ILC1 numbers were moderately decreased in the small intestine of CCR9-deficient mice
(Fig 3.8). A few IEL-ILC1s expressed α4β7 and CD103 (Fig. 3.8).
RA induces HR switch from secondary lymphoid tissue-HR to gut-HR in ILC1s and
ILC3s
We investigated the tissue-specific regulatory factors affecting gut-HR expression
in ILCs. RA is known as gut-tissue-specific factor because RA promotes the induction of

78
gut-HR in adaptive immune cells. Thus, we examined if RA induces the expression of
gut-HR in ILCs. RA induced CCR9 and α4β7 expression on ILCs while reducing CCR7
expression (Fig. 3.9A). Co-staining of CCR7 and gut homing receptors revealed that RA
induces a switch from CCR7+CCR9-α4β7- to CCR7-CCR9+ and/or α4β7+ expression in
total ILCs (Fig. 3.9B). It has been reported that RA affects the balance of ILC subsets.
Therefore, there is the possibility that RA induced an altered balance of ILC subsets
rather than expression of gut-HR. To rule out this possibility, we tested for a HR switch
in each ILC subset. RA induced HR switches were observed in ILC1s and ILC3s but not
in ILC2s (Fig. 3.10).
Among RA receptors, the transcription factors RARα is the most highly
expressed by ILCs (Fig. 3.11A). In line with this, AM580, an RARα agonist, efficiently
induced gut HRs in ILC3s (Fig. 3.11B). LE540 (a pan-RAR antagonist) and Ro41-5253
(an RARα antagonist) further decreased the basal expression levels of CCR9 and α4β7
on cultured ILC3s (Fig. 3.11B). The chemotactic activity of ILC1s/ILC3s to CCL25 was
increased by RA treatment, but was suppressed by Ro41-5253 (Fig. 3.12). ILC2s
migrated to CCL25 as efficiently as RA-treated ILC1s and ILC3s. Dendritic cells (DCs)
express retinal aldehyde dehydrogenases (RALDHs), which convert retinaldehyde to RA;
therefore mucosal DCs are a major cellular source for RA in the intestine. When ILC3s
were cocultured with MLN-DC in RA-free medium, MLN-DCs induced gut-HRs in
ILC3s and ILC1s (Fig. 3.13A and B). Interestingly, MLN-DCs had higher capacity to
induce HR switch in ILC1s and ILC3s compared to Spleen-DCs. RARα antagonist Ro41-
5253 or an RALDH inhibitor, DEAB, effectively abolished the HR switch in ILC1s and

79
ILC3s. These results imply that mucosal DCs-derived RA induces a HR switch in
intestinal tissue-associated lymphoid tissue such as the MLN.
Vitamin A status affects HR expression by ILC1s and ILC3s, but not ILC2s
From previous research, we found that RA is important for a HR switch in ILC1s
and ILC3s in vitro. We further investigated the role of RA on HR expression by ILC
subsets in vivo. We generated vitamin A normal (VAN) and vitamin A deficient (VAD)
RAG1-/- mice to examine the HR phenotype of ILCs. As expected, VAD mice had
reduced gut-HR expression in ILC1s and ILC3s of the small intestine (Fig. 3.14).
However, CCR7 expression was only slightly increased compared to their counterparts in
VAN mice (Fig. 3.14). However, vitamin A status did not affect the HR phenotype of
ILC2s in vivo (Fig. 3.14). We also found that total ILC numbers were decreased in the
intestines of VAD mice by flow cytometry and confocal microscopy (Fig. 3.15A and B).
BM ILC2 progenitors express gut HRs and directly migrate to the intestine
Since ILC2s broadly express gut-HR in secondary lymphoid and intestinal tissues.
We assumed ILC2Ps acquire gut-HR expression in early stages unlike ILC1s and ILC3s.
We could detect ILC2Ps in the BM by gating on Lin-CD45+Sca1highCD25+KLRG1+ or –
cells (Fig. 3.16). Interestingly, those cells express the gut-HRs, CCR9 and α4β7, more
highly compared to other Lin- cells (Fig. 3.17). Additionally, we tested whether
expression of gut-HR by BM ILC2Ps is influence by vitamin A status. Gut HR
expression by BM ILC2Ps was not affected in vitamin A deficiency (Fig. 3.17). Then, we
tested whether BM-ILC2Ps directly migrate to the intestine with those gut-HRs. We

80
transferred CD45.2 BM cells into CD45.1 congenic mice for a short-term migration assay.
Surprisingly, up to 20-30% ILC2Ps were detected in intestinal tissues (Fig. 32.18). We
also determined if gut-HRs affect ILC2Ps homing properties by a short term migration
study. Results indicated that CCR9 is necessary for normal migration of ILC2Ps to the
small intestine specifically, and Itg-β7 was required for migration of ILC2Ps to the MLN,
small intestine, and colon. These results indicate that acquisition of gut HRs expression in
the BM guides ILC2 migration to the intestine.
Importance of RA-induced gut HR for ILC3 migration to the intestine
We next asked if RA-induced CCR9 and α4β7 expression is important for
migration of ILC3s to the intestine. We performed a short term migration study using
CD45.2+ CD90.1+ RA-treated ILC3s and CD45.2+ CD90.2+ control ILC3s that were co-
injected into CD45.1+ CD90.2+ mice (Fig. 3.20). The RA-treated ILC3s were highly
efficient in migrating to the small intestine and colon compared to control ILC3s (Fig.
3.20). We also performed a short term migration study with HR-deficient ILC3s (Fig.
3.20). We prepared RA-treated ILC3s (WT, Ccr9-/-, and Itgβ7-/-) or control ILC3s (WT
and Ccr7-/- ILC3). Ccr9-/- was required for RA-treated ILC3 migration to the small
intestine (Fig. 3.20). Results showed that Itgβ7-/- RA-treated ILC3s and Ccr7-/- ILC3s
were not efficient in migrating to the intestine and MLN, respectively (Fig. 3.21). To see
the long term effect of HRs of ILCs in migrating to lymphoid tissues and intestine, mixed
BM chimeras were generated by mixed CD45.2 WT and HR-deficient BM cells
transplanted into lethally-irradiated CD45.1 mice. Ccr7-/- ILC3s and Ccr9-/- ILC3s were
inefficient in populating the MLN and small intestine, respectively (Fig. 3.22). Itgβ7-/-

81
ILC3s failed to normally populate the intestine (Fig. 3.22). Ccr9-/- ILC2s failed to
populate the small intestine, and Itg-β7 deficiency slightly decreased the ILC2 population
in the colon. These results indicate that the distinct expression of tissue-specific homing
receptors expression determines the population of ILC subsets in the intestine and
lymphoid tissues.
RA-treated ILC3s complete with their HRs can effectively restore defective mucosal
immunity in vitamin A deficient hosts
VAD RAG1-/- mice are more susceptible to C. rodentium infection than VAN
RAG1-/- mice and are deficient in ILC3s in the intestine (Fig. 3.23). These VAD mice had
reduced IL-17 and IL-22 expression and antimicrobial peptide expression in intestinal
tissues (Fig. 3.24A and B). We tested if RA-induced gut HRs are important for ILC3-
mediated anti-bacteria immunity against pathogenic bacteria. Only RA-treated ILC3
transfer showed a protective effect in infected VAD RAG1-/- mice (Fig. 3.25). However, it
is already reported that RA promotes ILC3 expansion and IL-22 expression, which could
explain the protective function of ILC3s in VAD mice. To rule out this possibility, we
performed the transfer of WT versus Ccr9-/- or Itgβ7-/- ILC3s into VAD mice (Fig. 3.26).
These gut-HR-deficient ILC3s showed similar expansion and IL-22 expression induced
by RA in cell preparation (not shown). Compared to Ccr9-/- and Itgβ7-/- ILC3s, WT
ILC3s showed more effectiveness in protecting VAD mice (Fig. 3.26). In line with this,
WT ILC3 transfer efficiently increased ILC3 numbers in intestines compared to Ccr9-/-
and Itgβ7-/- ILC3s (Fig. 3.27). In addition, the transfer of RA-treated ILC3s restored the
expression of cytokines (IL-17a and IL-22) and anti-microbial peptides (RegIIIβ and

82
RegIIIγ) in the intestine of VAD RAG1-/- mice in a largely CCR9- or Itgβ7-dependent
manner.

83
3.4 Discussion
Innate lymphoid cells (ILCs) are highly enriched in the intestine, and their
importance in innate immunity has been well established. Recently, it has been reported
that RA regulates innate lymphoid cell biology, including development, expansion, and
effector function in the intestine. However, it is unclear how ILCs populate the intestine
and if RA regulates their migration. Therefore, we focused our research on how ILCs
migrate to the intestine and whether RA as a major intestine-specific cue controls ILC
trafficking behavior. Our study has established the migration behavior of ILCs to the
intestine. We identified that RA play a significant role in regulating ILC migration for
population of and effector function in the intestine.
We demonstrated that ILCs utilize gut-homing receptors, CCR9 and α4β7 to
migrate into the intestine. α4β7 acts as an adhesion molecule to bind MadCAM-1, which
is expressed broadly on intestinal endothelial cells (163, 164). α4β7 is required for
migration to both SI and colon. The role of α4β7 for ILC migration into the whole
intestine is well supported by our data that ILC1 and ILC3 in both SI and colon express
α4β7. The CCR9-specific chemokine, CCL25, is specifically expressed in the small
intestine (165). SI ILCs most highly express CCR9. Our data indicates that many of ILCs
in the MLN also express CCR9, but CCR9-deficient mice have normal levels of ILCs in
MLN. This result suggested the potential that the MLN may be a generating site for gut-
homing ILCs. MLN ILC3s were most affected in CCR7-/- mice, whereas intestinal ILC3s
were affected in CCR9-/- or Itg-β7-/- mice. In a short-term migration experiment, CCR9-/-

84
ILC3s were defective in migration only to SI, whereas Itg-β7-/- ILC3 were defective in
migration to both colon and SI. CCR7-/- ILC3s were not efficient in migrating to the
MLN. Thus, these results indicate that ILC3s utilize tissue-specific homing receptors to
selectively migrate to different tissues.
Interestingly, ILC1s and ILC3s can undergo sophisticated trafficking receptor
switches to change their tissue tropism. Spleen ILCs do not express CCR9 or α4β7 at
significant frequencies. However, dendritic cells isolated from MLN change the
trafficking receptor expression phenotype of spleen ILC1s and ILC3s through production
of RA. Thus, these ILCs have the potential to undergo trafficking receptor switches in a
manner highly similar to gut-homing T and B cells. We demonstrated that RA is a key
regulatory metabolite in up-regulating the expression of gut homing receptors on ILCs.
Indeed, RA activates RARs, which function to induce the transcription of CCR9 and Itg-
α4 genes. We found that pan-RAR antagonist (LE540) or RARα antagonists (Ro41) can
down-regulate the expression of gut-homing receptors on ILCs. This fact supports the
idea that ILCs utilize RARα, for the induction of the gut-homing receptors. A question of
interest is if all ILC subsets are regulated by RA. It has been documented that ILC2s are
expanded in VAD mice whereas ILC3s are expanded by RA. These data indicate an
apparent role of RA in regulation of ILC proliferation and survival in a subset-specific
manner. Regarding homing receptor expression, ILC subsets differ in expression of
CCR9 and α4β7. ILC1s and ILC3s highly expressed the two gut HRs only in the
intestine, but the number of ILC2s expressing α4β7 were lower than ILC1s and ILC3s in
the intestine. Moreover, ILC1s and ILC3s express CCR7 specifically in the MLN, but

85
ILC2s do not express CCR7 in both secondary lymphoid and intestinal tissue. In VAD
mice, we also found correlating discrepancies in ILC subsets. Vitamin A deficiency
affects HR expression patterns in ILC1s and ILC3s but not in ILC2s. These results
suggest that ILC2s have different migration programs from ILC1s and ILC3s. In fact,
BM ILC2Ps express CCR9 and α4β7 in bone marrow. And BM ILC2Ps can directly
migrate to intestine, largely independent of gut-homing receptor expression. Unlike
ILC1s and ILC3s, ILC2s do not undergo a HR switch in peripheral tissue, but rather they
acquire gut HR expression during an early development stage.
ILC3 deficiencies induce an impaired IL-22-antimicrobial peptide axis, leading to
defective intestinal immunity (166). In this regard, the decreased immunity to C.
rodentium infection in VAD mice is, in part, due to reduced numbers of ILC3s in the
intestine. We demonstrated the possibility that the homing behavior of ILC3s can be
changed by RA ex vivo to efficiently induce migration to the intestine and restore
mucosal immunity in VAD mice. Importantly, without the expression of gut HRs, RA-
treated ILC3s were not able to provide effective immunity to mucosal pathogens,
underlying the importance of the migration ability in determining the effector function of
ILCs.
The results provided novel insights into the migration program of ILCs to the
intestine. ILC1s and ILC3s can reprogram their trafficking receptor expression to migrate
into the intestine, a process regulated by RA. This indicates that certain innate immune
cells possess sophisticated migration programs in a manner similar to adaptive immune
cells. For ILC2s however, the expression of gut HRs occurs not in the periphery but early
in the bone marrow at the ILC2P stage. Thus, our findings indicate the gut metabolite,

86
RA, regulates the gut migration program of certain ILC subsets, with implications for
anti-bacterial immunity in the intestine.

87
3.5 Summary and future directions
We reported that the vitamin A metabolite retinoic acid, produced by intestinal
dendritic cells, is required for homing receptor switch from lymphoid tissue receptor,
CCR7 to gut homing receptors, CCR9 and α4β7 by ILC1s and ILC3s. ILC3s deficient in
expression of CCR9 and α4β7 were defective in not only their migration but also effector
function in the intestine. In contrast, ILC2s acquire gut homing receptors in a largely RA-
independent manner during their development in the bone marrow and can migrate
directly to the intestine. Thus, distinct programs regulate the migration of ILC subsets to
the intestine for regulation of innate immunity. Beyond the gut homing of ILCs, ILC
migration program to other tissue sites such as lung, skin, and fat need to be investigated
in the future.

88
Figure 3.1 Tissue distribution of ILC subsets.
Average percent frequencies of NK, ILC1, ILC2, and ILC3 in total ILCs in indicated
tissues are shown. NK cells were identified based on CD3-CD19-CD127-NKp46+NK1.1+
(most tissues), CD3-CD19-RORγt-NKp46+NK1.1+ (intestinal LP). ILC1 were identified
by Lin-RORγt-CD127+NKp46+NK1.1+. ILC2 were identified by Lin-
CD127+CD90+KLRG1+GATA3+ (most tissues), Lin-CD90+CD25+GATA3+ (spleen and
skin), or Lin-CD127+CD90+T1/ST2+GATA3+ (lung and Fat) phenotype. ILC3 were
identified by Lin-GATA3-CD127+CD90+RORγt+ phenotype. MLN, mesenteric lymph
node; LP, lamina propria.

89
Figure 3.2 Gating strategies for ILC subsets in small intestine.
Cells were isolated from the small intestine of C57BL/6 mice and stained with antibodies
to lineage-specific antigens and indicated ILC-associated antigens. ILC1 (Lin-
CD127+CD90+Eomes-RORγt-T-bet+), ILC2 (Lin-NK1.1- CD127+CD90+Sca-1+KLRG-
1+GATA-3+), and ILC3 (T-bet- or T-bet+ Lin-CD127+CD90+RORγt+GATA-3-) subsets
were identified as shown.
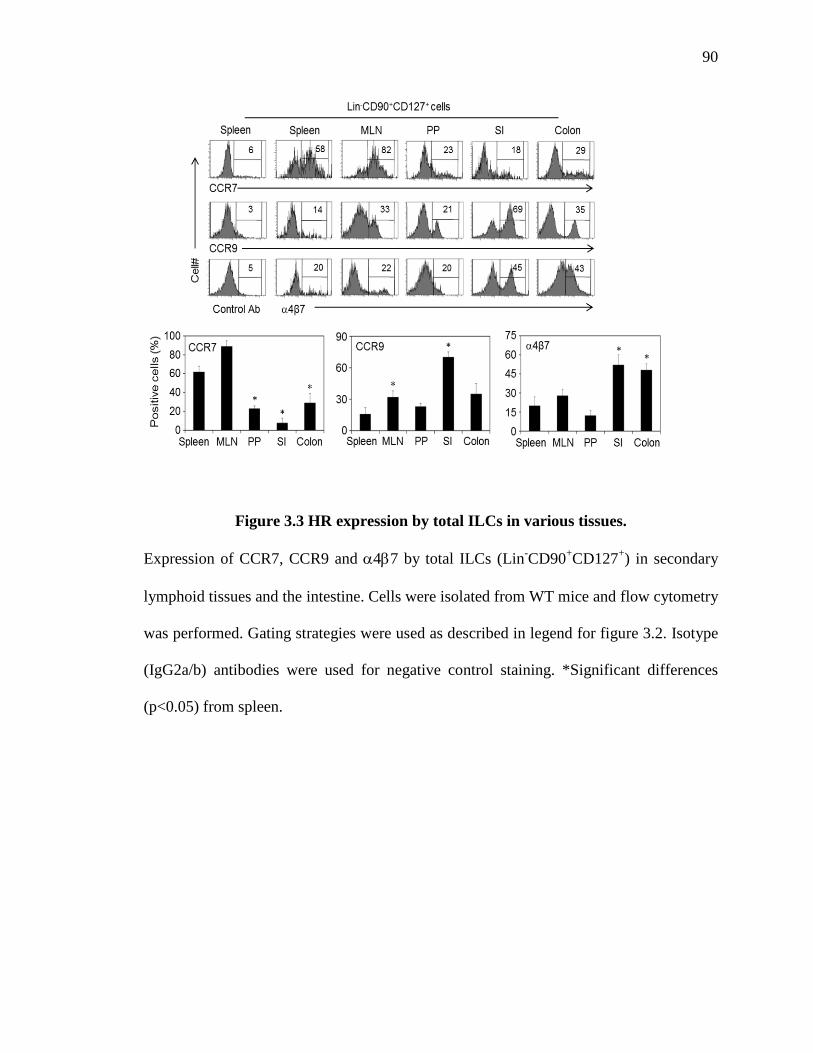
90
Figure 3.3 HR expression by total ILCs in various tissues.
Expression of CCR7, CCR9 and α4β7 by total ILCs (Lin-CD90+CD127+) in secondary
lymphoid tissues and the intestine. Cells were isolated from WT mice and flow cytometry
was performed. Gating strategies were used as described in legend for figure 3.2. Isotype
(IgG2a/b) antibodies were used for negative control staining. *Significant differences
(p<0.05) from spleen.

91
Figure 3.4 HR expression by ILC subsets in lymphoid and intestinal tissues.
Expression of homing receptors by ILC subsets in the spleen, MLN, SI, and colon. Data
from RAG1-/- or C57BL/6 mice were essentially identical and combined (n=15). Gating
strategies for ILC subsets are shown in Fig. 2.1. *Significant differences (p<0.05) from
ILC2. **The spleen T-bet+ ILC3 population was very small (~0.1% of Lin-
CD127+CD90+ cells) and not shown.

92
Figure 3.5 Tissue-specific homing receptors are required for ILCs population in
tissues.
Frequencies of Lin-CD90+CD127+ total ILCs in lymphoid tissues and the intestine of WT,
Ccr7-/-, Ccr9-/- or Itgβ7-/- mice. Dot plots show Lin- cells. Pooled data obtained from at
least 3 different experiments (n=15) are shown. *Significant differences (p<0.05) from
WT.

93
Figure 3.6 Gut HRs required for normal numbers of ILCs in intestines.
CD3-CD90+ ILCs in the small intestine of WT, CCR9-/- or Itgβ7-/- mice were imaged by
laser scanning confocal microscopy. Frozen sections were stained by CD3, CD90, and
pan-cytokeratin. Average numbers of CD3-CD90+ ILCs per villus and section are shown
in the graph. *Significant differences (p<0.05) from WT.

94
Figure 3.7 Tissue-specific HRs required for normal population of ILCs.
Frequencies of ILC subsets (ILC1, ILC2, and ILC3) in lymphoid tissues and the intestine
of WT, Ccr7-/-, Ccr9-/- or Itgβ7-/- mice. Relative values of ILC numbers in HR-deficient
mice to those of WT are showing. Pooled data obtained from at least 3 different
experiments (n=15) are shown. *Significant differences (p<0.05) from WT.

95
Figure 3.8 HR expression by intraepithelial ILC1s and their numbers in gut-HR-
deficient mice.
(A) Expression of CCR9, α4β7, and CD103 by intraepithelial ILC1s (Lin-
NKp46+NK1.1+ cells) and γδ T cells in the small intestine. (B) Numbers of intraepithelial
ILC1s in the small intestine and colon of WT, CCR9-/-, and Itg-β7-/- mice. *Significant
differences between indicated pairs or from WT (n=4).

96
Figure 3.9 Retinoic acid induced HR switch from 2nd LT-HR to gut-HR in ILCs.
(A) Induction of gut homing receptors and down-regulation of CCR7 on ILCs by RA. (B)
Induction of a homing receptor switch from CCR7 to CCR9 and α4β7 in cultured ILCs
by RA. Expression of HRs on Lin-CD90+CD127+ ILCs was assessed after the culture of
spleen Lin- cells for 4 days with RA or Ro41-5253 (an RARα antagonist). For panel B,
cultured cells were co-stained by CCR7, and CCR9 and α4β7.

97
Figure 3.10 RA induces a HR switch from CCR7 to CCR9 and α4β7 in ILC3s and
ILC1s but not ILC2s.
HR expression in ILC1s, ILC2s, and ILC3s cultured with RA or Ro41. ILC subset-
specific culture conditions were used. (ILC3: Cecal bacterial antigen (CBA, 10 µg/ml),
IL-7 (20 ng/ml) and IL-23 (20 ng/ml); ILC1: CBA, IL-7 and IL-15 (20 ng/ml); ILC2: IL-
7). *Significant differences (p<0.05) from controls.

98
Figure 3.11 RA receptor expression and impact of RAR agonists and antagonists on
HR expression in ILC3s.
(A) RA receptors expression was examined in total ILC and CD4+ T-cells by qPCR. ILCs
(Lin-CD90+ cells) and CD4+ T-cells were isolated from WT-spleen. (B) Splenic ILCs
were cultured in an ILC3 culture condition in the presence of indicated RAR agonists and
antagonists. Cultured cells were stained for HR and ILC3 markers. Pooled data obtained
from at least 3 different experiments are shown. *Significant differences (p<0.05) from
controls.
B
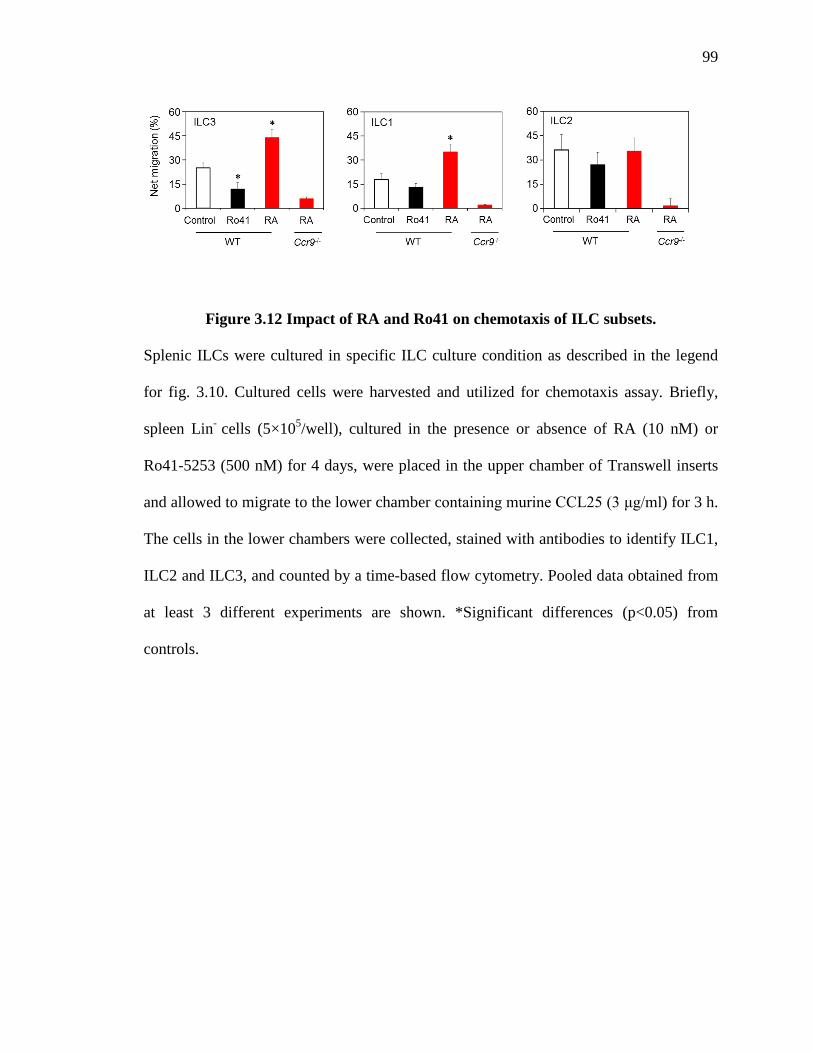
99
Figure 3.12 Impact of RA and Ro41 on chemotaxis of ILC subsets.
Splenic ILCs were cultured in specific ILC culture condition as described in the legend
for fig. 3.10. Cultured cells were harvested and utilized for chemotaxis assay. Briefly,
spleen Lin- cells (5×105/well), cultured in the presence or absence of RA (10 nM) or
Ro41-5253 (500 nM) for 4 days, were placed in the upper chamber of Transwell inserts
and allowed to migrate to the lower chamber containing murine CCL25 (3 μg/ml) for 3 h.
The cells in the lower chambers were collected, stained with antibodies to identify ILC1,
ILC2 and ILC3, and counted by a time-based flow cytometry. Pooled data obtained from
at least 3 different experiments are shown. *Significant differences (p<0.05) from
controls.

100
Figure 3.13 Mucosal DCs-derived RA induces the expression of gut HRs
in ILC3s and ILC1s.
(A) Mucosal DCs induce a homing receptor switch in ILC3s. (B) Mucosal DCs induce
the homing receptor switch in ILC1s. CD11c+ DCs, freshly isolated from spleen or MLN,
and spleen Lin- cells were co-cultured (ratio=1:10, 4 days) for 4 days in the presence of
Ro41-5253 or DEAB (an RALDH inhibitor). Pooled data obtained from at least 3
different experiments (n=3) are shown. *Significant differences (p<0.05) from controls.

101
Figure 3.14 Vitamin A deficiency reduces expression of gut HR by ILC3s and ILC1s.
Expression of HRs by ILC subsets in the small intestine of VAN and VAD mice. Mice
were fed with vitamin A deficient (VAD) or normal (VAN) diets for 11-12 weeks before
examination of ILC subsets. SI-LP cells were isolated and stained by antibodies for HRs
and ILC subset markers. Flow cytometry was performed. Pooled data obtained from at
least 3 different experiments (n=8-12) are shown. *Significant differences (p<0.05) from
VAN.

102
Figure 3.15 Decreased ILCs in the intestine of VAN and VAD RAG1-/- mice.
(A) Confocal imaging of ILCs Average numbers of CD90+ ILCs per each villus and
section are shown in the graph. IHC was performed to detect total ILCs in the intestine.
(B) Total ILCs (Lin-CD127+CD90+ cells) numbers in VAN and VAD mice. Flow
cytometry was performed. Mice were fed with vitamin A deficient (VAD) or normal
(VAN) diets for 11-12 weeks before examination of ILC subsets. Pooled data obtained
from at least 3 different experiments (n=12) are shown. *Significant differences (p<0.05)
from VAN.
A
B

103
Figure 3.16 Gating strategies for BM ILC2 progenitors.
BM cells were stained with antibodies to the “BM lineage markers” markers (CD3ε, CD5,
CD8α, B220, CD11b, Gr-1, Ter-119, TCRγδ, and NK1.1), CD127, Sca-1, CD25, CD45.2,
and KLRG-1. BM ILC2Ps were identified as Lin-CD127+CD45.2+Sca-1highCD25+KLRG-
1- or +.

104
Figure 3.17 Expression of gut HRs by BM ILC2Ps in VAN and VAD mice.
Gut-HR expressions by BM ILC2P of VAN and VAD mice were examined by flow
cytometry. Mice were fed with vitamin A deficient (VAD) or normal (VAN) diets for 11-
12 weeks before examination of ILC2P. KLRG1 staining was used to differentiate mature
and immature ILC2 lineage cells in BM. Pooled data obtained from 3-7 different
experiments are shown.

105
Figure 3.18 BM ILC2Ps can directly migrate to the intestine.
(A) Tissue distribution of BM KLRG-1- and KLRG-1+ ILC2 lineage cells 44h after cell
transfer (i.v.). Lin- Ly-6B.2- BM cells were injected i.v. into host mice and sacrificed 44
hours later. (B) Representative dot plots for migrated BM-derived cells. The “Lin”
mixture refers to the “BM lineage markers.” For migration of BM ILC2P, CD45.2+ Lin-
BM cells, isolated from WT mice were singly injected i.v. into CD45.1 mice. The host
mice were sacrificed 44h later, and indicated organs were harvested and examined by
flow cytometry for numbers of transferred ILC2 lineage cells in each organ. Pooled data
obtained from 3-7 different experiments (n=3-10) are shown.
A

106
Figure 3.19 Gut HR expression is required for BM ILC2P migration to the intestine.
Short term migration of WT versus gut HR-deficient BM ILC2P. Lin- Ly-6B.2- BM cells
were injected i.v. into host mice and sacrificed 44 hours later. Migration efficiency of
HR-deficient ILC2P to an organ was determined based on the normalized absolute
numbers of transferred ILC2P in each organ that were divided by the total numbers of the
transferred ILC2P in indicated organs.*Significant differences (p<0.05) from WT. Pooled
data obtained from 3-7 different experiments (n=3-10) are shown.

107
Figure 3.20 Short-term homing of control versus RA-treated ILC3s to the gut and
2nd LT.
Spleen cells, depleted of cells expressing the “splenocyte depletion markers” mentioned
in the Materials and Methods section, were cultured for 4 days in an ILC3 culture
condition (commensal bacteria extract, IL-7, and IL-23) for 4 days. Cultured cells were
utilized for short term (20 hr) migration study as described in material & method. Pooled
data obtained from 2-3 different experiments (n=7-10) are shown. *Significant
differences (p<0.05) from control or between WT and homing receptor-deficient ILCs.

108
Figure 3.21 RA-induced gut HR expression is required for efficient short-term
migration of ILC3s to the intestine.
Short-term homing of WT versus homing receptor-deficient ILC3. Spleen cells, depleted
of cells expressing the “splenocyte depletion markers” mentioned in the Materials and
Methods section, were cultured for 4 days in an ILC3 culture condition (commensal
bacteria extract, IL-7, and IL-23) for 4 days. The cells, except Ccr7-/- and matching WT
cells, were treated with RA for the homing experiment. Short term migration studies were
performed as described in material & method.*Significant differences (p<0.05) from
control or between WT and homing receptor-deficient ILCs.

109
Figure 3.22 Gut HRs required for long term population of ILCs.
A long-term competitive population study of WT and homing receptor-deficient ILC
subsets following mixed (1:1) BM transfer from CD45.2+CD90.1+ WT and
CD45.2+CD90.2+ homing receptor-deficient mice. The ILC subsets were examined in the
indicated organs 8 weeks after the mixed BM transfer. Pooled data obtained from 2-3
different experiments (n=7-10) are shown. *Significant differences (p<0.05) from control
or between WT and homing receptor-deficient ILCs.

110
Figure 3.23 VAD RAG1-/- mice are more susceptible to C. rodentium infection.
Mice were fed with vitamin A deficient (VAD) or normal (VAN) diets for 11-12 weeks
before examination of anti-bacterial immunity. VAN and VAD RAG1-/- mice were
infected with C. rondentium. Infected mice were monitored for weight change (%), stool
consistency, and bacterial load in feces and peripheral tissues. Stool consistency scores
were measured as follows: 0, normal stool; 1, soft stool; 2, mild diarrhea; 3, severe
diarrhea; 4, anal bleeding. *Significant differences (p<0.05) from VAN mice.
Bac
teria
l bur
den
(CFU
/g, f
eces
)
Bac
teria
l bur
den
(CFU
/g, o
rgan
)

111
Figure 3.24 VAD mice have an impaired ILC3 response in the intestine.
Reduced cytokines and antimicrobial peptide expression (A), and intestinal ILC3
numbers (B) in VAD mice with or without infection. Mice were fed with vitamin A
deficient (VAD) or normal (VAN) diets for 11-12 weeks before examination of ILC3
responses. Infected mice were euthanized at 6 days post infection. qPCR was performed
for examination of expression of cytokines and antimicrobial peptides in intestinal tissues.
Flow cytometry was performed for examination of absolute numbers of ILC3 (Lin-
CD127+CD90+RORγt+ cells).
A
B

112
Figure 3.25 Protective effects of control and RA-treated ILC3s in infected VAD
mice.
Purified ILCs (Lin-NK1.1-KLRG-1-CD90high) were cultured with RA (10 nM) in an ILC3
culture condition (commensal bacteria extract, IL-7, and IL-23) for 4 days for the cell
transfer. Control and RA-treated ILC3s were transferred into VAD mice and infected
with C. rodentium. Weight change and C. rodentium load were monitored. Pooled data
obtained from 2 different experiments (n=7-8) are shown. *Significant differences from
the control (none) groups.

113
Figure 3.26 Gut homing receptors are required for optimal ILC3 effector function
during C. rodentium infection.
Protective effects of WT versus gut homing receptor-deficient RA-ILCs on C. rodentium
infection. Purified ILCs from WT, Ccr9-/-, or Itgβ7-/- mice, were cultured with RA (10
nM) in an ILC3 culture condition for 4 days before the cell transfer. Weight change (A),
stool scores (B), and pathogen burden (C) were determined. Stool consistency scores
were measured as follows: 0, normal stool; 1, soft stool; 2, diarrhea; 3, diarrhea and anal
bleeding. Pooled data obtained from 2 different experiments (n=7-8) are shown.
*Significant differences between VAN and VAD or from the control (none) groups.

114
Figure 3.27 Tissue distributions of ILC3s following adoptive transfer of WT or gut-
HR-deficient ILC3s into VAD mice infected with C. rodentium.
(A) ILC3 numbers in various tissues of infected VAD mice after ILC3 enriched cell
transfer. (B) Representative dot plots for ILC3 in the colon. Lin-CD127+ cells are shown.
VAD mice were injected i.v. with WT RA-ILC3 or HR-deficient RA-ILC3 two times on
day -2 and 2 post-infection with C. rodentium. After infection, mice were monitored for
anti-bacterial immunity. Mice were sacrificed on day 9 post-infection. Pooled data
obtained from 2 different experiments (n=7-8) are shown. *Significant differences from
the control (none) groups.
B
A

115
Figure 3.28 Expression of ILC3 cytokines and antimicrobial peptides following
adoptive transfer of WT or gut HR-deficient ILC3s into VAD mice infected with
C. rodentium.
VAD mice were injected i.v. with WT RA-ILC3 or HR-deficient RA-ILC3 two times on
day -2 and 2 post-infection with C. rodentium. Mice were sacrificed on day 9 post-
infection. Colon tissues were harvested after infection. qPCR was performed. Pooled data
obtained from 2 different experiments are shown. *Significant differences between VAN
and VAD or from the control (none) groups.
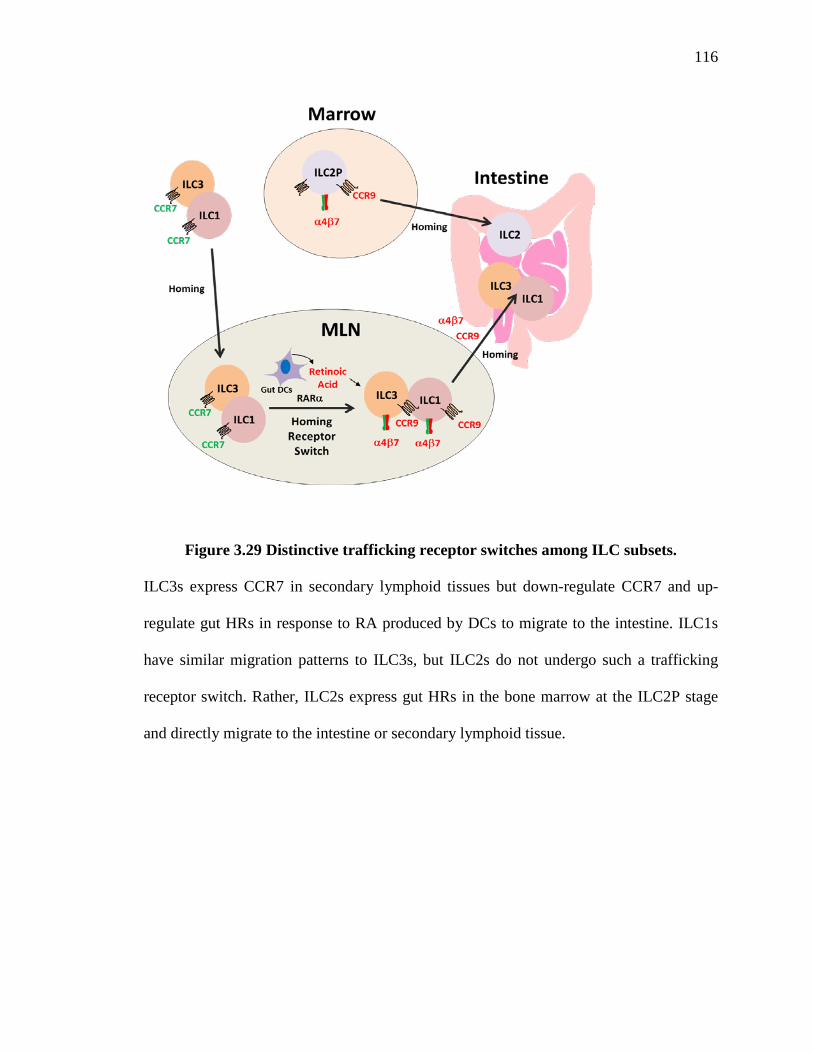
116
Figure 3.29 Distinctive trafficking receptor switches among ILC subsets.
ILC3s express CCR7 in secondary lymphoid tissues but down-regulate CCR7 and up-
regulate gut HRs in response to RA produced by DCs to migrate to the intestine. ILC1s
have similar migration patterns to ILC3s, but ILC2s do not undergo such a trafficking
receptor switch. Rather, ILC2s express gut HRs in the bone marrow at the ILC2P stage
and directly migrate to the intestine or secondary lymphoid tissue.

116
REFERENCES

117
REFERENCES
1. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3(10):858-76. 2. Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proceedings of the Nutrition Society. 2003;62(01):67-72. 3. McNeil N. The contribution of the large intestine to energy supplies in man. The American journal of clinical nutrition. 1984;39(2):338-42. 4. Hadjiagapiou C, Schmidt L, Dudeja PK, Layden TJ, Ramaswamy K. Mechanism (s) of butyrate transport in Caco-2 cells: role of monocarboxylate transporter 1. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2000;279(4):G775-G80. 5. Alrefai W, Tyagi S, Gill R, Saksena S, Hadjiagapiou C, Mansour F, et al. Regulation of butyrate uptake in Caco-2 cells by phorbol 12-myristate 13-acetate. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2004;286(2):G197-G203. 6. Ritzhaupt A, Ellis A, Hosie KB, Shirazi‐Beechey SP. The characterization of butyrate transport across pig and human colonic luminal membrane. The Journal of physiology. 1998;507(3):819-30. 7. Binder HJ. Role of colonic short-chain fatty acid transport in diarrhea. Annual Review of Physiology. 2010;72:297-313. 8. Gopal E, Miyauchi S, Martin PM, Ananth S, Roon P, Smith SB, et al. Transport of nicotinate and structurally related compounds by human SMCT1 (SLC5A8) and its relevance to drug transport in the mammalian intestinal tract. Pharmaceutical research. 2007;24(3):575-84. 9. Coady MJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. The human tumour suppressor gene SLC5A8 expresses a Na+–monocarboxylate cotransporter. The Journal of physiology. 2004;557(3):719-31. 10. Miyauchi S, Gopal E, Fei Y-J, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. Journal of Biological Chemistry. 2004;279(14):13293-6. 11. Singh N, Thangaraju M, Prasad PD, Martin PM, Lambert NA, Boettger T, et al. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. Journal of Biological Chemistry. 2010;285(36):27601-8. 12. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. Journal of Biological Chemistry. 2003;278(13):11312-9.

118
13. Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(4):1045-50. 14. Karaki S-i, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell and tissue research. 2006;324(3):353-60. 15. Le Poul E, Loison C, Struyf S, Springael J-Y, Lannoy V, Decobecq M-E, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. Journal of Biological Chemistry. 2003;278(28):25481-9. 16. Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA 2 R, expressed on leukocytes and activated by short-chain fatty acids. Biochemical and biophysical research communications. 2003;303(4):1047-52. 17. Offermanns S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends in pharmacological sciences. 2006;27(7):384-90. 18. Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, et al. Molecular identification of high and low affinity receptors for nicotinic acid. Journal of Biological Chemistry. 2003;278(11):9869-74. 19. Taggart AK, Kero J, Gan X, Cai T-Q, Cheng K, Ippolito M, et al. (D)-β-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. Journal of Biological Chemistry. 2005;280(29):26649-52. 20. Cresci GA, Thangaraju M, Mellinger JD, Liu K, Ganapathy V. Colonic gene expression in conventional and germ-free mice with a focus on the butyrate receptor GPR109A and the butyrate transporter SLC5A8. Journal of Gastrointestinal Surgery. 2010;14(3):449-61. 21. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282-6. 22. Vinolo M, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly Y, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One. 2011;6(6):e21205-e. 23. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145(2):396-406. e10. 24. Mortensen FV, Langkilde NC, Joergensen JC, Hessov I. Short-chain fatty acids stimulate mucosal cell proliferation in the closed human rectum after Hartmann’s procedure. International journal of colorectal disease. 1999;14(3):150-4. 25. Hague A, Singh B, Paraskeva C. Butyrate acts as a survival factor for colonic epithelial cells: further fuel for the in vivo versus in vitro debate. Gastroenterology. 1997;112(3):1036-40.

119
26. Gibson PR, Rosella O, Wilson AJ, Mariadason JM, Rickard K, Byron K, et al. Colonic epithelial cell activation and the paradoxical effects of butyrate. Carcinogenesis. 1999;20(4):539-44. 27. Daly K, Shirazi-Beechey SP. Microarray analysis of butyrate regulated genes in colonic epithelial cells. DNA and cell biology. 2006;25(1):49-62. 28. Chirakkal H, Leech S, Brookes K, Prais A, Waby J, Corfe B. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene. 2006;25(54):7192-200. 29. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. The Journal of nutrition. 2002;132(5):1012-7. 30. Shen L, Turner JR. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. American journal of physiology-gastrointestinal and liver physiology. 2006;290(4):G577-G82. 31. McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, et al. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. Journal of Clinical Investigation. 2007;117(8):2313. 32. Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422(6931):522-6. 33. Willemsen L, Koetsier M, Van Deventer S, Van Tol E. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E1 and E2 production by intestinal myofibroblasts. Gut. 2003;52(10):1442-7. 34. Barcelo A, Claustre J, Moro F, Chayvialle J, Cuber J, Plaisancie P. Mucin secretion is modulated by luminal factors in the isolated vascularly perfused rat colon. Gut. 2000;46(2):218-24. 35. Blais M, Seidman EG, Asselin C. Dual Effect of Butyrate on IL-1 β-Mediated Intestinal Epithelial Cell Inflammatory Response. DNA and cell biology. 2007;26(3):133-47. 36. Leung CH, Lam W, Ma DL, Gullen EA, Cheng YC. Butyrate mediates nucleotide‐binding and oligomerisation domain (NOD) 2‐dependent mucosal immune responses against peptidoglycan. European journal of immunology. 2009;39(12):3529-37. 37. Fusunyan RD, Quinn JJ, Fujimoto M, MacDermott RP, Sanderson IR. Butyrate switches the pattern of chemokine secretion by intestinal epithelial cells through histone acetylation. Molecular medicine. 1999;5(9):631. 38. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128-39. 39. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nature communications. 2015;6. 40. Nancey S, Bienvenu J, Coffin B, Andre F, Descos L, Flourié B. Butyrate strongly inhibits in vitro stimulated release of cytokines in blood. Digestive diseases and sciences. 2002;47(4):921-8.

120
41. Cavaglieri CR, Nishiyama A, Fernandes LC, Curi R, Miles EA, Calder PC. Differential effects of short-chain fatty acids on proliferation and production of pro-and anti-inflammatory cytokines by cultured lymphocytes. Life sciences. 2003;73(13):1683-90. 42. Kurita-Ochiai T, Fukushima K, Ochiai K. Volatile fatty acids, metabolic by-products of periodontopathic bacteria, inhibit lymphocyte proliferation and cytokine production. Journal of dental research. 1995;74(7):1367-73. 43. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446-50. 44. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569-73. 45. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, Liu H, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451-5. 46. Park J, Kim M, Kang S, Jannasch A, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal immunology. 2015;8(1):80-93. 47. Eftimiadi C, Buzzi E, Tonetti M, Buffa P, Buffa D, Van Steenbergen M, et al. Short-chain fatty acids produced by anaerobic bacteria alter the physiological responses of human neutrophils to chemotactic peptide. Journal of Infection. 1987;14(1):43-53. 48. Carretta M, Conejeros I, Hidalgo M, Burgos R. Propionate induces the release of granules from bovine neutrophils. Journal of dairy science. 2013;96(4):2507-20. 49. Park J-S, Lee E-J, Lee J-C, Kim W-K, Kim H-S. Anti-inflammatory effects of short chain fatty acids in IFN-γ-stimulated RAW 264.7 murine macrophage cells: Involvement of NF-κB and ERK signaling pathways. International immunopharmacology. 2007;7(1):70-7. 50. Wang B, Morinobu A, Horiuchi M, Liu J, Kumagai S. Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cellular immunology. 2008;253(1):54-8. 51. Nascimento CR, Freire-de-Lima CG, de Oliveira AdS, Rumjanek FD, Rumjanek VM. The short chain fatty acid sodium butyrate regulates the induction of CD1a in developing dendritic cells. Immunobiology. 2011;216(3):275-84. 52. Berndt BE, Zhang M, Owyang SY, Cole TS, Wang TW, Luther J, et al. Butyrate increases IL-23 production by stimulated dendritic cells. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2012;303(12):G1384-G92. 53. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13-22. 54. Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. Journal of neurobiology. 2006;66(7):606-30. 55. Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315(5813):820-5.

121
56. Mic FA, Molotkov A, Benbrook DM, Duester G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proceedings of the National Academy of Sciences. 2003;100(12):7135-40. 57. Chambon P. A decade of molecular biology of retinoic acid receptors. The FASEB Journal. 1996;10(9):940-54. 58. Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129(4):723-33. 59. Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4+ T cells activated in cutaneous or mucosal lymphoid tissues. The Journal of experimental medicine. 2002;195(1):135-41. 60. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303(5664):1662-5. 61. Mora JR, Iwata M, Eksteen B, Song S-Y, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314(5802):1157-60. 62. Fagarasan S, Kinoshita K, Muramatsu M, Ikuta K, Honjo T. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 2001;413(6856):639-43. 63. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun C-M, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β–and retinoic acid–dependent mechanism. The Journal of experimental medicine. 2007;204(8):1757-64. 64. Sun C-M, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. The Journal of experimental medicine. 2007;204(8):1775-85. 65. Uematsu S, Fujimoto K, Jang MH, Yang B-G, Jung Y-J, Nishiyama M, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nature immunology. 2008;9(7):769-76. 66. Cantorna MT, Nashold FE, Chun TY, Hayes CE. Vitamin A down-regulation of IFN-gamma synthesis in cloned mouse Th1 lymphocytes depends on the CD28 costimulatory pathway. The Journal of Immunology. 1996;156(8):2674-9. 67. Stephensen CB, Rasooly R, Jiang X, Ceddia MA, Weaver CT, Chandraratna RA, et al. Vitamin A enhances in vitro Th2 development via retinoid X receptor pathway. The Journal of Immunology. 2002;168(9):4495-503. 68. Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, et al. Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity. 2011;34(3):435-47. 69. Johansson-Lindbom B, Svensson M, Wurbel M-A, Malissen B, Márquez G, Agace W. Selective Generation of Gut Tropic T Cells in Gut-associated Lymphoid Tissue (GALT) Requirement for GALT Dendritic Cells and Adjuvant. The Journal of experimental medicine. 2003;198(6):963-9. 70. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song S-Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527-38.

122
71. Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. The Journal of Immunology. 2007;179(6):3724-33. 72. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. The Journal of experimental medicine. 2013;210(6):1117-24. 73. Spencer S, Wilhelm C, Yang Q, Hall J, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. 2014;343(6169):432-7. 74. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508(7494):123-7. 75. Agace WW. T-cell recruitment to the intestinal mucosa. Trends in immunology. 2008;29(11):514-22. 76. Gorfu G, Rivera-Nieves J, Ley K. Role of β7 integrins in intestinal lymphocyte homing and retention. Current molecular medicine. 2009;9(7):836. 77. Pan J, Kunkel EJ, Gosslar U, Lazarus N, Langdon P, Broadwell K, et al. Cutting edge: a novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. The Journal of Immunology. 2000;165(6):2943-9. 78. Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. Journal of Clinical Investigation. 2003;111(7):1001. 79. Agace WW, Higgins JM, Sadasivan B, Brenner MB, Parker CM. T-lymphocyte–epithelial-cell interactions: integrin α E (CD103) β 7, LEEP-CAM and chemokines. Current opinion in cell biology. 2000;12(5):563-8. 80. Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Förster R, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. The Journal of experimental medicine. 2005;202(8):1063-73. 81. Farstad IN, Halstensen TS, Kvale D, Fausa O, Brandtzaeg P. Topographic distribution of homing receptors on B and T cells in human gut-associated lymphoid tissue: relation of L-selectin and integrin alpha 4 beta 7 to naive and memory phenotypes. The American journal of pathology. 1997;150(1):187. 82. Morris M, Ley K. Trafficking of natural killer cells. Current molecular medicine. 2004;4(4):431-8. 83. Cortez VS, Robinette ML, Colonna M. Innate lymphoid cells: new insights into function and development. Current opinion in immunology. 2015;32:71-7. 84. Wojno EDT, Artis D. Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell host & microbe. 2012;12(4):445-57. 85. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41(3):354-65. 86. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nature immunology. 2015.

123
87. Klose CS, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157(2):340-56. 88. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37(4):634-48. 89. Wolterink RGK, Serafini N, Van Nimwegen M, Vosshenrich CA, De Bruijn MJ, Pereira DF, et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proceedings of the National Academy of Sciences. 2013;110(25):10240-5. 90. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343(6178):1249288. 91. Serafini N, Wolterink RGK, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, et al. Gata3 drives development of RORγt+ group 3 innate lymphoid cells. The Journal of experimental medicine. 2014;211(2):199-208. 92. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498(7452):113-7. 93. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283-95. 94. Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, et al. Notch signaling is necessary for adult, but not fetal, development of ROR [gamma] t+ innate lymphoid cells. Nature immunology. 2011;12(10):949-58. 95. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508(7496):397-401. 96. Kraal G, Schornagel K, Streeter PR, Holzmann B, Butcher EC. Expression of the mucosal vascular addressin, MAdCAM-1, on sinus-lining cells in the spleen. The American journal of pathology. 1995;147(3):763. 97. Kim MH, Taparowsky EJ, Kim CH. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity. 2015;43(1):107-19. 98. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nature immunology. 2013;14(6):564-73. 99. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nature communications. 2015;6. 100. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld J-C, Frankel G, et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity. 2014;41(5):776-88. 101. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORγt+ innate lymphoid cells. Science. 2010;330(6004):665-9.

124
102. Lügering A, Ross M, Sieker M, Heidemann J, Williams I, Domschke W, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clinical & Experimental Immunology. 2010;160(3):440-9. 103. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-ROR [ggr] t+ innate lymphoid cells. Nature. 2013;494(7436):261-5. 104. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12-and IL-15-responsive IFN-γ-producing cells. Immunity. 2013;38(4):769-81. 105. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature immunology. 2013;14(3):221-9. 106. Martín-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nature immunology. 2004;5(12):1260-5. 107. Mailliard RB, Alber SM, Shen H, Watkins SC, Kirkwood JM, Herberman RB, et al. IL-18–induced CD83+ CCR7+ NK helper cells. The journal of experimental medicine. 2005;202(7):941-53. 108. Beider K, Nagler A, Wald O, Franitza S, Dagan-Berger M, Wald H, et al. Involvement of CXCR4 and IL-2 in the homing and retention of human NK and NK T cells to the bone marrow and spleen of NOD/SCID mice. Blood. 2003;102(6):1951-8. 109. Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, et al. T-bet–dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. The Journal of experimental medicine. 2009;206(11):2469-81. 110. Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood. 2010;115(10):1897-905. 111. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nature immunology. 2014;15(4):354-64. 112. Kim MY, Rossi S, Withers D, McConnell F, Toellner KM, Gaspal F, et al. Heterogeneity of lymphoid tissue inducer cell populations present in embryonic and adult mouse lymphoid tissues. Immunology. 2008;124(2):166-74. 113. Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORγt on the development of NKp46+ cell populations in gut and skin. Nature immunology. 2009;10(1):75-82. 114. Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25-and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nature immunology. 2011;12(11):1055-62. 115. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. The Journal of experimental medicine. 2013;210(13):2939-50.

125
116. Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A. CD56brightCD16–NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. European journal of immunology. 2006;36(1):118-28. 117. Ebert LM, Meuter S, Moser B. Homing and function of human skin γδ T cells and NK cells: relevance for tumor surveillance. The Journal of Immunology. 2006;176(7):4331-6. 118. Yu YRA, Fong AM, Combadiere C, Gao JL, Murphy PM, Patel DD. Defective antitumor responses in CX3CR1‐deficient mice. International journal of cancer. 2007;121(2):316-22. 119. van Helden MJ, Zaiss DM, Sijts AJ. CCR2 defines a distinct population of NK cells and mediates their migration during influenza virus infection in mice. 2012. 120. Ventura M, Turroni F, Canchaya C, Vaughan EE, O'Toole PW, Sinderen Dv. Microbial diversity in the human intestine and novel insights from metagenomics. 2009. 121. Hamer HM, De Preter V, Windey K, Verbeke K. Functional analysis of colonic bacterial metabolism: relevant to health? American Journal of Physiology-Gastrointestinal and Liver Physiology. 2012;302(1):G1-G9. 122. Jarchum I, Pamer EG. Regulation of innate and adaptive immunity by the commensal microbiota. Current opinion in immunology. 2011;23(3):353-60. 123. Macia L, Thorburn AN, Binge LC, Marino E, Rogers KE, Maslowski KM, et al. Microbial influences on epithelial integrity and immune function as a basis for inflammatory diseases. Immunological reviews. 2012;245(1):164-76. 124. Hague A, Elder DJ, Hicks DJ, Paraskeva C. Apoptosis in colorectal tumour cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. International Journal of Cancer. 1995;60(3):400-6. 125. Sakata T, Von Engelhardt W. Stimulatory effect of short chain fatty acids on the epithelial cell proliferation in rat large intestine. Comparative Biochemistry and Physiology Part A: Physiology. 1983;74(2):459-62. 126. Thwaites DT, Anderson CM. H+‐coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Experimental physiology. 2007;92(4):603-19. 127. Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki S, Kuwahara A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol. 2008;59(Suppl 2):251-62. 128. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proceedings of the National Academy of Sciences. 2008;105(43):16767-72. 129. Sina C, Gavrilova O, Förster M, Till A, Derer S, Hildebrand F, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. The Journal of Immunology. 2009;183(11):7514-22. 130. Alexander KL, Targan SR, Elson CO. Microbiota activation and regulation of innate and adaptive immunity. Immunological reviews. 2014;260(1):206-20. 131. Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nature immunology. 2011;12(1):5-9.

126
132. Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288(5474):2222-6. 133. Lim BO, Yamada K, Nonaka M, Kuramoto Y, Hung P, Sugano M. Dietary fibers modulate indices of intestinal immune function in rats. The Journal of nutrition. 1997;127(5):663-7. 134. Schley P, Field C. The immune-enhancing effects of dietary fibres and prebiotics. British Journal of Nutrition. 2002;87(S2):S221-S30. 135. Nguyen KA, Cao Y, Chen JR, Townsend Jr CM, Ko TC. Dietary fiber enhances a tumor suppressor signaling pathway in the gut. Annals of surgery. 2006;243(5):619. 136. Ishikawa H, Akedo I, Otani T, Suzuki T, Nakamura T, Takeyama I, et al. Randomized trial of dietary fiber and Lactobacillus casei administration for prevention of colorectal tumors. International Journal of Cancer. 2005;116(5):762-7. 137. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nature protocols. 2007;2(3):541-6. 138. Kang SG, Park J, Cho JY, Ulrich B, Kim CH. Complementary roles of retinoic acid and TGF-β1 in coordinated expression of mucosal integrins by T cells. Mucosal immunology. 2011;4(1):66-82. 139. Peng L, He Z, Chen W, Holzman IR, Lin J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatric research. 2007;61(1):37-41. 140. Wang H-B, Wang P-Y, Wang X, Wan Y-L, Liu Y-C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Digestive diseases and sciences. 2012;57(12):3126-35. 141. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121(24):4930-7. 142. Lührs H, Gerke T, Müller J, Melcher R, Schauber J, Boxberger F, et al. Butyrate inhibits NF-κB activation in lamina propria macrophages of patients with ulcerative colitis. Scandinavian journal of gastroenterology. 2002;37(4):458-66. 143. Säemann MD, Böhmig GA, Österreicher CH, Burtscher H, Parolini O, Diakos C, et al. Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. The FASEB Journal. 2000;14(15):2380-2. 144. Tlaskalová-Hogenová H, Štěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cellular & molecular immunology. 2011;8(2):110-20. 145. Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Applied and environmental microbiology. 1990;56(6):1919-25. 146. Goodfellow M, Stackebrandt E. Nucleic acid techniques in bacterial systematics: J. Wiley; 1991.

127
147. Doré J, Sghir A, Hannequart-Gramet G, Corthier G, Pochart P. Design and evaluation of a 16S rRNA-targeted oligonucleotide probe for specific detection and quantitation of human faecal Bacteroides populations. Systematic and Applied Microbiology. 1998;21(1):65-71. 148. Salzman NH, de Jong H, Paterson Y, Harmsen HJ, Welling GW, Bos NA. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology. 2002;148(11):3651-60. 149. Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Applied and Environmental Microbiology. 1998;64(9):3336-45. 150. Wise MG, Siragusa GR. Quantitative detection of Clostridium perfringens in the broiler fowl gastrointestinal tract by real-time PCR. Applied and environmental microbiology. 2005;71(7):3911-6. 151. Snel J, Heinen P, Blok H, Carman R, Duncan A, Allen P, et al. Comparison of 16S rRNA sequences of segmented filamentous bacteria isolated from mice, rats, and chickens and proposal of “Candidatus Arthromitus”. International journal of systematic bacteriology. 1995;45(4):780-2. 152. De Clerck E, Van Mol K, Jannes G, Rossau R, De Vos P. Design of a 5′ exonuclease‐based real‐time PCR assay for simultaneous detection of Bacillus licheniformis, members of the ‘B. cereus group’and B. fumarioli in gelatine. Letters in applied microbiology. 2004;39(1):109-15. 153. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual review of immunology. 2012;30:647-75. 154. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature immunology. 2011;12(1):21-7. 155. Maloy KJ, Uhlig HH. ILC1 populations join the border patrol. Immunity. 2013;38(4):630-2. 156. Zhou L. Striking similarity: GATA-3 regulates ILC2 and Th2 cells. Immunity. 2012;37(4):589-91. 157. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nature Reviews Immunology. 2013;13(2):145-9. 158. Sheridan BS, Lefrançois L. Regional and mucosal memory T cells. Nature immunology. 2011;12(6):485-91. 159. Kunkel EJ, Campbell JJ, Haraldsen G, Pan J, Boisvert J, Roberts AI, et al. Lymphocyte Cc Chemokine Receptor 9 and Epithelial Thymus-Expressed Chemokine (Teck) Expression Distinguish the Small Intestinal Immune Compartment Epithelial Expression of Tissue-Specific Chemokines as an Organizing Principle in Regional Immunity. The Journal of experimental medicine. 2000;192(5):761-8. 160. Walsh G, Symon F, Lazarovits A, Wardlaw A. Integrin α4β7 mediates human eosinophil interaction with MAdCAM‐1, VCAM‐1 and fibronectin. Immunology. 1996;89(1):112-9. 161. Uehara S, Grinberg A, Farber JM, Love PE. A role for CCR9 in T lymphocyte development and migration. The Journal of Immunology. 2002;168(6):2811-9.

128
162. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145(2):396-406 e1-10. 163. Shyjan AM, Bertagnolli M, Kenney CJ, Briskin MJ. Human mucosal addressin cell adhesion molecule-1 (MAdCAM-1) demonstrates structural and functional similarities to the alpha 4 beta 7-integrin binding domains of murine MAdCAM-1, but extreme divergence of mucin-like sequences. The Journal of Immunology. 1996;156(8):2851-7. 164. Rott LS, Rosé JR, Bass D, Williams MB, Greenberg HB, Butcher EC. Expression of mucosal homing receptor alpha4beta7 by circulating CD4+ cells with memory for intestinal rotavirus. Journal of Clinical Investigation. 1997;100(5):1204. 165. Amersi FF, Terando AM, Goto Y, Scolyer RA, Thompson JF, Tran AN, et al. Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clinical cancer research. 2008;14(3):638-45. 166. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nature immunology. 2011;12(5):383-90.

128
VITA

129
VITA
Myunghoo Kim
Education 2010-2015 Ph.D. in Microbiology and Immunology, Veterinary Medicine
Purdue University, West Lafayette, USA (Advisor: Chang H. Kim)
2009-2010 Research assistance
Seoul National University
2007-2009 M.S. in Agricultural Biotechnology (Ruminant Nutrition and Microbiology)
Seoul National University, Seoul, Korea (Advisor: Jong-Kyu Ha)
2000-2007 B.S. in Agricultural Biotechnology
Seoul National University, Seoul, Korea
AWARDS
[1] PVM Graduate Student Research Award. 2015.
[2] AAI Trainee Poster Award. 2014.
Myung H. Kim, Seung G. Kang, Jeong H. Park, Masashi Yanagisawa, and Chang H. Kim The commensal bacteria metabolites short chain fatty acids positively regulate epithelial innate immune
responses in the gut.
[3] 7th Joint symposium Best oral Presentation Award. 2009.
Myung H. Kim, S. Y. Lee, I. H. Kwon, J. Y. Ko, S. H. Yang and J. K. Ha. Effects of administration of
fermented SBM and hydrolyzed-yeast on growth performance, diarrhea and immuno-physiological
characteristics in calves with weaning challenge. Korea-Japan-China Joint Ruminant Symposium, Korea.

130
[4] AAAP Excellent Poster Presentation Award at animal health. 2008.
Myung H. Kim, Santi upadhaya, J. K. Ha. Selection and identification of bacteria having Ochratoxin A
degrading ability by using the whole rumen fluid and bacterial isolates of ruminants and intestinal fluid of
non-ruminant and its bacterial isolates. Asian-Australasian Association of Animal Production Societies
Animal Science Congress Proceedings, Vietnam.
[5] AAAP Excellent Poster Presentation Award at animal health. 2008.
Myung H. Kim, J. Y. Ko, C. H. Yun, J. K. Ha. Effect of administration of immune enhancer and AO
treated SBM on immune response and growth performance of neonatal calves. Asian-Australasian
Association of Animal Production Societies Animal Science Congress Proceedings, Vietnam.
Oral Presentation
Presentation of recipient of 2015 PVM Graduate Student Research Award, 2015, 28th Annual Phi Zeta
Research Day, USA
CIS talk, 2014, Purdue Cancer center, USA
Korean Ruminant symposium, 2010, Korea
Korea-Japan-China Joint Ruminant Symposium, 2009, Korea.
Asian-Australasian Association of Animal Production Societies Animal Science Congress, 2008, Vietnam
Korea-Japan-China Joint Ruminant Symposium, 2007, Korea.
Poster Presentation
Phi-zeta presentation, 2015
Phi-zeta presentation, 2014
AAI Annual meeting, 2014
SIGMA XI poster presentation competition, 2014
Cancer research retreat, 2014
Cancer research retreat, 2013
GATROENTEROLOGY SYMPOSIUM, 2013
Phi-zeta presentation, 2013
Phi-zeta presentation, 2012
Autumn Immunology Conference, 2012
AAAP Poster Presentation Award at animal, 2008
6th Korea-Japan-China Joint Ruminant Symposium, 2007

130
PUBLICATIONS

131
PUBLICATIONS
[1] Myung H. Kim, Elizabeth J. Taparowsky, and Chang H. Kim. Retinoic acid differentially regulates the
migration of innate lymphoid cell subsets to the gut. 2015. 43, 1–13. July. Immunity.
[2] Shankar Thangamani, Myung H. Kim,Youngmin Son, Xinxin Huang, Heejoo Kim, Jee H. Lee,
Jungyoon Cho, Benjamin Ulrich, Hal E. Broxmeyer, and Chang H. Kim. Progesterone directly up-regulates
vitamin D receptor gene expression for efficient regulation of T cells by calcitriol, 2015, Cutting Edge (JI).
(Co-first author).
[3] Jeong H. Park, Myung H. Kim, Seung G. Kang, AH. Jannasch, B. Cooper, J Patterson, CH. Kim.
Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases
and regulation of the mTOR-S6K pathway. 2014. Jun 11. doi: 10.1038/mi.2014.44. Mucosal Immunology.
[4] Rukhsana Jabeen, Ritobrata Goswami, Olufolakemi Awe, Aishwarya Kulkarni, Evelyn T. Nguyen,
Andrea Attenasio, Daniel Walsh, Matthew R. Olson, Myung H. Kim, Robert S. Tepper, Jie Sun, Chang H.
Kim, Elizabeth J. Taparowsky, Baohua Zhou, and Mark H. Kaplan. Th9 cell development requires a
BATF-regulated transcriptional network. 2013. doi:10.1172/JCI69489. Journal of Clinical Investigation.
[5] Myung H. Kim, Seung G. Kang, Jeong H. Park, Masashi Yanagisawa, and Chang H. Kim. Short chain
fatty acids regulate anti-bacterial inflammatory responses and clearance of pathogenic bacteria through
epithelial GPR41 and GPR43. 2013. 145:396-406. Gastroenterology.
[6] Chuanwu Wang, Shankar Thangamani, Myung H. Kim, Bon-Hee Gu, Jee H. Lee, Elizabeth J.
Taparowsky, and Chang H. Kim. BATF is required for normal expression of gut-homing receptors by T
helper cells in response to retinoic acid. 2013. Vol. 210 No. 3. Journal of Experimental Medicine.
[7] Jinsam Chang, Shankar Thangamani, Myung H. Kim, Benjamin Ulrich, Sidney M. Morris Jr. and
Chang H. Kim. Retinoic acid promotes the development of Arg1-expressing dendritic cells for the
regulation of T-cell differentiation. 2013. 43: 967–978. European Journal of Immunology.
[8] J. K. Seo, Myung H. Kim, J. Y. Yang, H. J. Kim, C. H. Lee, K. H. Kim and Jong K. Ha. Effects of
Synchronicity of Carbohydrate and Protein Degradation on Rumen Fermentation Characteristics and
Microbial Protein Synthesis. 2013. Vol. 26, No. 3:358-365. Asian-Australasian Journal of Animal Science.
[9] Myung H. Kim, C.H. Yun, C.H. Lee, J.K. Ha. The effects of fermented soybean meal on growth
performance, diarrhea, stress- related serum proteins, and immune cells in Holstein calves after weaning.
2012. 95: 5203–5212. Journal of Dairy Science.

132
[10] In Hyuk Kwon, Myung H. Kim, Cheol-Heui Yun, Jong Yeol Go, Chan Ho Lee, Hyun June Lee,
Wisut Phipek, Jong K. Ha. Effects of Fermented Soybean Meal on Immune Response of Weaned Calves
with Experimentally Induced Lipopolysaccharide Challenge. 2011. Vol. 24, No. 7:957-964. Asian-
Australasian Journal of Animal Science.
[11] Santi Devi Upadhaya, Liu Yang, Ja Kyeom Seo, Myung H. Kim, Chang-kyu Lee, Chan Ho Lee, Jong
K. Ha. Effect of Feed Types on Ochratoxin A Disappearance in Goat Rumen Fluid. 2011. Vol. 24, No. 2:
198-205. Asian-Australasian Journal of Animal Science.
[12] Myung H. Kim, C. H. Yun, K. L. Kim, J. Y. Ko, J. Y Song and Jong K. Ha. Changes of
immunoglobulins and lymphocyte subpopulations in Holstein calves challenged with Escherichia coli
lipopolysaccharide. Vol. 24, No. 5: 696-706. 2011. Asian-Australasian Journal of Animal Science.
[13] Myung H. Kim, J. K. Seo. C. H. Yun, S. J. Kang, J. Y. Ko and J. K. Ha. Effects of hydrolyzed yeast
supplementation in calf starter on the immune response of neonatal calves experimentally-challenged with
microbes. 2011. 5:6, 953–960. Animal.
[14] Myung H. Kim, Ji-Young Yang, Santi Devi Upadhaya, Hyun-Jun Lee, Cheol-Heui Yun, Jong K. Ha.
The stress of weaning influences serum concentrations of acute-phase proteins, iron-binding proteins,
inflammatory cytokines, cortisol, and leukocyte subsets in Holstein calves. 2011. 12(2), 151-157. Journal
of Veterinary Science.
[15] Ja K. Seo, Seon W. Kim, Myung H. Kim, Santi D. Upadhaya, Dong K. Kam, Jong K. Ha. Direct-fed
Microbials for Ruminant Animals. 2010. Vol. 23, No. 12: 1657-1667. Asian-Australasian Journal of
Animal Science.
[16] Myung H. Kim, K. J. Jo, H. S. Kim and J. K. Ha. Effects of fermented SBM on growth performance,
diarrhea incidence and immune-response of neonatal calves. 2010. 81, 475–481. Animal Science Journal.
[17] Myung H. Kim, C. H. Yun. J. Y. Ko, J. S. Kang, S. J. Kang, W. S. Lee, J. H. Kim and J. K. Ha.
Change of immunophysiological characteristics in neonatal calves experimentally challenged with mixture
of bacteria and virus. 2009. 92:5534–5543. Journal of Dairy Science.
REVIEW
Chang H. Kim, Jeong H. Park, and Myung H. Kim. Regulation of T cells and inflammation by gut
microbiota-derived short chain fatty acids. Immune Network. 2015.
Chang H. Kim, Seika Hashimoto-Hill, and Myung H. Kim. Migration and tissue tropism of innate
lymphoid cells. Trend in Immunology. (In press)

133
BOOK CHAPTER
Myung. H Kim and Chang H. Kim. Short Chain Fatty Acids in Regulation of the Immune System and
Tissue Inflammation in the Intestine. Ed. Cong-Jun Li. In Butyrate: Food Sources, Functions and Health
Benefits. Nova Science Publishers. 2014.