the purification and properties of formate dehydrogenase ... · vol. 250, no. 17, issue of...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 250, No. 17, Issue of September 10, pp. 6693-6705, 1975
Printed in U.S.A.
The Purification and Properties of Formate Dehydrogenase
and Nitrate Reductase from Escherichia coZi*
HARRY G. ENOCH$ AND ROBERT L. LESTER
From the Department of Biochemistry, College of Medicine,
SUMMARY
The membrane-bound formate dehydrogenase of Esche- richia coli grown anaerobically in the presence of nitrate was solubilized with deoxycholate and purified to near homo- geneity. The purification procedure included ammonium sul- fate fractionation and chromatography on Bio-Gel A-1.5m and DEAE Bio-Gel A in the presence of the nonionic detergent, Triton X-100. This detergent caused a significant decrease in the molecular weight of the soluble formate dehydrogenase complex and allowed the enzyme then to be resolved from other membrane components. Anaerobic conditions were required throughout due to the sensitivity of the enzyme to oxygen inactivation. Formate dehydrogenase was judged to be at least 93 to 99% pure by the following procedures: polyacrylamide gel electrophoresis in the presence of Triton X-100 and sodium dodecyl sulfate, gel filtration, and sedi- mentation velocity studies. The purified enzyme exists as a detergent-protein complex (0.20 & 0.03 g of Triton X-100/g of protein) which has an ~20,~ of 18.1 S and a Stokes radius of 76 A. This corresponds to a molecular weight of 590,000 =t 59,000. The enzyme had an absorbance spectrum of a b- type cytochrome which could be completely reduced by for- mate. The heme content corresponds to an equivalent weight of 154,000 which suggests a tetrameric structure for the enzyme. Formate dehydrogenase was found to contain (in relative molar amounts): 1.0 heme, 0.95 molybdenum, 0.96 selenium, 14 non-heme iron, and 13 acid-labile sulfide. Neither FAD nor FMN could be detected. The enzyme con- tains three polypeptides, designated LY, p, and y, whose molecular weights were estimated by gel electrophoresis in the presence of sodium dodecyl sulfate to be 110,000, 32,000, and 20,000, respectively. After separation of the polypeptides by gel filtration in the presence of sodium dodecyl sulfate LY, p, and y were found in 1: 1.2:0.55 molar ratios. A study of the enzyme obtained from cells grown with [7Se]selenite showed that only the a: polypeptide contained significant amounts of selenium. The enzyme will catalyze the formate-dependent reduction of phenazine methosulfate, dichlorophenolindophenol, methylene blue, nitroblue tetra-
* This investigation was supported by General Research Sup- port Grant RR05374 from the National Institutes of Health.
j Supported by National Institutes of Health Training Grant GM 1026. Portions of this material are taken from the dissertation of H. G. E. submitted to the University of Kentucky. Present address, Department of Biochemistry, University of Connecticut Health Center, Farmington, Conn. OG032.
zolium, benzyl viologen, methyl viologen, ferricyanide, and
(Received for publication, January 29, 1975)
University of Kentucky, Lexington, Kentucky 40506
coenzyme Q6. Cyanide, azide, p-hydroxymercuribenzoate, iodoacetamide, and oxygen inhibit the enzyme.
The procedure which was designed for the purification of formate dehydrogenase also yields a highly purified prepara- tion of nitrate reductase. This nitrate reductase has been shown to contain significant amounts of heme (ENOCH, H. G., AND LESTER, R. L. (1974) Biochem. Biophys. Res. Com- mun. 61, 1234-1241). The enzyme contains three polypeptides with molecular weights of 155,000, 63,000, and 19,000. When measured in the presence of Trition X-100 the Stokes radius of nitrate reductase is 75 A and the SZO+ is 16 S which cor- responds to a molecular wieght of 498,000.
Formate dehydrogenase plays a versatile role in the oxidative metabolism of Escherichia coli. Depending upon the growth conditions formate can provide electrons for the reduction of oxygen to water (formate oxidase), nitrate to nitrite (formate- nitrate reductase), and H+ to hydrogen (formate hydrogenlyase). The activity of the formate dehydrogenase(s) of each of these membrane-associated electron transport systems requires the presence of trace amounts of selenium, molybdenum, and iron in the culture medium (l-3). Previous studies (4) have shown that the absence of selenium and molybdenum in the aerobic culture medium has no effect on the activity of a number of oxidative enzyme systems or on the growth rate and cell yield obtained with a number of different carbon sources. This sug- gests that formate dehydrogenase may be a unique metalloen- zyme in E. coli. Definition of the molecular characteristics of formate dehydrogenase requires highly purified preparations of the enzyme not heretofore obtained. Partially purified prepara- tions have been shown to contain cytochrome b (5, 6), nitrate reductase (5), flavin (5, 6), and selenium (7). Difficulties in purifying the enzyme have been due in part to its oxygen sensi- tivity and its firm association with other membrane components. We undertook to purifv formate dehydrogenase from cells grown anaerobically in the presence of nitrate. These conditions lead to the formation of high levels of formate dehydrogenase, cyto- chrome 6, and nitrate reductase (3, 8, 9) which are thought to be the essential components of the formate-nitrate reductase system (10). We now report a procedure for the preparation of highly purified cytochrome-containing forms of formate de- hydrogenase and nitrate reductase. In addition to being a hemo-
6693
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6694
protein formate dehydrogenase contains significant amounts of selenium, molybdenum, and non-heme iron, but no flavin.
Preliminary accounts of part of this work have been reported
(11, 12).
EXPERIMENTAL PROCEDURI?
Materials
Triton X-100, scintillation grade (Research Products Inter- national), Brij detergents (Atlas Chemical Industries), sodium dodecyl sulfate (Fisher Scientific Co.), and deoxycholic acid (Armour Pharmaceuticals) were used without further purification. Tobacco mosaic virus was a generous gift of Dr. John Shaw. [76Se]Selenite and [l%]bicarbonate were purchased from New England Nuclear. Bio-Gel A-1.5m, Bio-Gel A-15m, and DEAE Bio-Gel A were purchased from Bio-Rad Laboratories; coenzyme Qs, p-hydroxymercuribenzoate, iodoacetamide, N-ethylmalei- mide, n-heptyl hydroxyquinoline-N-oxide, FAD, FMN, phenazine methosulfate, dichlorophenolindophenol, nitroblue tetrazolium, and all proteins (unless otherwise noted) were from Sigma Chem- ical Co.; methyl viologen and benzyl viologen were from Schwarz/ Mann; and methylene blue was from Fisher Scientific Co.
Sedimentation Coeficients
Density gradient centrifugation was performed by the method of Martin and Ames (13). A sample (0.1 ml) was layered on a 12-ml density gradient of sucrose (15 to 50%) in 50 mM Tris-HCl, pH 8. The t,ubes were centrifuged at 35,000 rpm for 16 hours at 4” in a Beckman SW 41 rotor. Each sample included p-galactosidase (kindly provided by A. S. L. Hu, University of Kentucky) as a marker; P-galactosidase, ~20,~ = 16.1 S (14), was assayed with o- nitrophenol p-galactoside (15).
The sedimentation coefficient was also determined in a Beckman model E analytical ultracentrifuge using an An-E rotor. The en- zyme solution (2 ml) containing 0.3 mg of purified formate dehy- drogenase/ml in 0.1% Triton X-100/1 M NaCl/lO mM Tris-HCl, pH 8, was placed in a 30.mm single-sector cell. Centrifugation was conducted at 5” and 42,040 rpm. A single, symmetrical peak was observed which remained unchanged throughout the experiment.
Polyacrylamide Gel Electrophoresis
Triton gel electrophoresis was carried out in 3yo polyacrylamide gels (5 X 65 mm) in 0.1% Triton X-100/0.2 M Tris-HCl, pH 9, at 5”. The electrode buffer was 33 mM Tris-HCl, pH 9. A current of 3 ma/gel was applied for 3 hours. Sodium dodecyl sulfate gel electro- phoresis was carried out in polyacrylamide gels (5 X 65 mm) using the buffer system described by Laemmli (IG, 17). A current of 3 ma/gel was applied until the marker dye reached the bottom of the gel. Gels were stained for protein with Coomassie blue (18). For- mate dehydrogenase activity was located by incubating the gels for 30 min at 30” in a solution (5 ml) containing 250 pmol of for- mate/100 /*g of nitroblue tetrazolium/20 pg of phenazine metho- sulfate/250 pmol of sodium phosphate, pH 7.2. Phenazine metho- sulfate is not required but is highly stimulatory in this reaction. Less than 1 Kg of purified formate dehydrogenase can be detected by this method.
Subunit Molecular Weight by Gel Filtration
A column (0.9 X 92 cm) of Bio-Gel A-1.5m was equilibrated with 0.1% sodium dodecyl sulfate/50 mM sodium phosphate, pH 7.2. Samples in 1% sodium dodecyl sulfate/lyO mercaptoethanol/lOyO glycerol/50 mM sodium phosphate, pH 7.2, were heated at 100” for 1 hour and 0.2 ml was layered on the column. The column was operated at room temperature and 0.8-ml fractions were collected. The distribution coefficient, K,, equals V, - V&‘, - VO (19). The void volume, VO, and the total volume, V, were measured with blue dextran and sucrose, respectively. For measurement of V,, the elution volume, the protein content of each fraction was measured (20). Published values of the molecular weights of stand- ard proteins were used (17, 21).
Separation of Polypeptides of Formate Dehydrogenase by Gel Filtration in Presence of Sodium Dodecyl Sulfate
Purified formate dehydrogenase (1.9 mg) was adsorbed to a column of DEAE-Bio-Gel A, washed with 2 column volumes of 10
mM Tris-HCl, pH 8, to remove the Triton, and then eluted with 1 M NaCl/lO mM Tris-HCl, pH 8. Fractions containing enzyme ac- tivity (all of the activity was recovered) were pooled and am- monium sulfate was added to BOY0 saturation. After centrifugation the precipitate was dissolved in 2% sodium dodecyl sulfate/lyO mercaptoethanol/50 mM sodium phosphate, pH 7.2, and this preparation was heated at 100” for 15 min. Iodoacetamide was added to a final concentration of 0.4 M and the mixture was incu- bated for 4 hours at 30”. The sample then was dialyzed against 1000 volumes of 0.1% sodium dodecyl sulfate/25 mM sodium phos- phate, pH 7.2, for 48 hours at 20”. Solid sucrose was added (2%) and 0.45 ml of this sample (0.79 mg of protein) was layered on a column (0.9 X 175 cm) of Bio-Gel A-1.5m equilibrated with 0.1% sodium dodecyl sulfate/25 mM sodium phosphate, pH 7.2. The column was operated at room temperature with a flow rate of 2.9 ml/hour.
Detergent Studies
The amount of Triton X-100 bound to formate dehydrogenase was determined by a modification of the method described by Helenius and Simons (22) using ion exchange chromatography instead of gel filtration to ensure the separation of Triton X-100 micelles from the detergent-enzyme complex. Purified formate dehydrogenase (1.8 mg) was adsorbed to a column (0.5 X 30 cm) of DEAIS Bio-Gel A equilibrated with 0.1% Triton X-100, 10 mM Tris-HCl, pH 8. After washing with 2 column volumes of the equilibration buffer the formate dehydrogenase was eluted \uith 1 M NaCl in the equilibration buffer. Fractions were assayed for Triton X-100 and protein, and milligrams of bound Triton X-100 per mg of protein were calculated.
The partial specific volume of Triton X-100 was measured at 23” as described by Tanford et al. (23). The viscosity of 0.1% Triton X-100/1 M NaCl/lO mM Tris-HCl, pH 8, was determined in an Ostwald viscometer at 5” using distilled water and acetone as standards.
Stokes Radius
The Stokes radius of each enzyme-Triton X-100 complex was determined by gel filtration according to the method of Tanford et al. (23). A column (0.9 X 140 cm) of Bio-Gel A-1.5m was equili- brated with 0.1% ‘Triton X-100/1 M NaCl/lO mM Tris-HCl, pH 8. A sample (0.4 ml) of purified formate dehydrogenase or nitrate reductase was applied and the column was operated at a flow rate of 5.5 ml/hour (4-5”); 0.9.ml fractions were collected. The column was calibrated in the same manner using standard proteins, omit- ting Triton X-100 from the buffer. The void volume and the total volume were determined using tobacco mosaic virus and sucrose, respectively. Published values of the Stokes radii of standard pro- teins were used (24).
Enzyme Assays Methods
Unless otherwise indicated formate dehydrogenase was meas- ured at 30” by spectrophotometric assay of the anaerobic reduc- tion of dichlorophenolindophenol in the presence of phenazine methosulfate (3). One unit of enzyme activity is defined as that which reduces 1 Mmol of dichlorophenolindophenol/min.
When many fractions were to be assayed, formate dehydrogen- ase was more conveniently measured by the reduction of nitroblue tetrazolium at 30” in open test tubes. The reaction mixture of 1 ml contained enzyme/20 mM formate/O.l M sodium phosphate, pH 7.2. The reaction was initiated by the addition of 20 ~1 of 3.3 mM nitroblue tetrazolium. The reaction was stopped and the reduced dye was solubilized by the addition of 1 ml of a solution containing ethyl acetate/methanol/l N hydrochloric acid (l/1/0.035). The absorbance was measured at 530 nm; an extinction coefficient of 18 mM-1 cm-1 was used for the reduced dye (25). The reaction was linear for at least 5 min or up to an absorbance value of 0.4. A unit of enzyme activity is defined as that which reduces 1 pmol of nitro- blue tetrazolium/min and is equivalent to 0.05 unit assayed with phenazine methosulfate-dichlorophenolindophenol.
Formate-coenzyme Q reductase was assayed anaerobically at 30”. The reaction mixture contained formate dehydrogenase, 1 mM coenzyme Q6, 10 mM formate, 0.09% Triton X-100, and 90 mM Tris-HCl, pH 7. Reduced coenzyme Qs was measured by ferric chloride-dipyridyl (26).
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6695
Nitrate reductase was assayed at 30” by the method of Showe and DeMoss (9). One unit of enzyme activity is defined as that which reduces 1 firno1 of nitrate/min.
Other Assays Methods
Since Triton X-100 interfered with the measurement of protein by the method of Lowry et al. (27), protein was measured by the method of Hirs (20) using bovine serum albumin (Pentex) as a standard.
Heme was estimated as pyridine hemochrome (28). Acid-extract- able and non-extractable FAD and FMN were assayed fluoro- metrically by the method of Wilson and King (29). In the deter- mination of acid nonextractable flavin heat-denatured protein was digested either with 0.5 mg of Pronase (Calbiochem)/mg of protein for 30 min at 60” or with 1 mg of trypsin/mg of protein for 2 hours at 37”. Quinone content was measured after extraction with n-hexane. The extract was taken to dryness and the residue was resuspended in absolute ethanol. The concentration of co- enzyme Q and vitamin KP was determined from the decrease in absorbance at 275 and 268 nm, respectively, after reduction by sodium borohydride (30).
Molybdenum was estimated by the method of Johnson and Ark- ley (31). Samples (1 ml) were mixed with 0.5 ml of perchloric acid and digested at 200” for 2 hours, then 0.1 ml of nitric acid was added and digestion was continued for 2 more hours. Selenium and molybdenum also were determined by neutron activation analysis (General Activation Analysis, Inc., San Diego, Calif.). Non-heme iron was assayed with bathophenanthroline (32) and acid-labile sulfide was assayed with dimethylphenylenediamine (33).
The phospholipid content of formate dehydrogenase was meas- ured by phosphorus analysis (34) of the chloroform/methanol extract (35) of the perchloric acid-precipitated enzyme. Triton X-100 was assayed by the method of Garewal (36) using benzene to extract the detergent-cobaltothiocyanate adduct instead of ethylene dichloride. RNA was measured as hot trichloroacetic acid-soluble pentose by the procedure of Schneider (37) using ribose as a standard. Carbohydrate was measured by the phenol- sulfuric acid method of Ashwell (38). Radioactivity of 7%e and “C were measured by scintillation counting in Triton-toluene counting fluid (39) unless otherwise noted.
Growth Conditions
Escherichia coli (HfrH thi-) was grown anaerobically on a basal anaerobic growth medium (3) containing 0.1 PM sodium selenite, 0.1 FM sodium molybdate, and 1% potassium nitrate and harvested at the end of exponential growth.
Purification of Formate Dehydrogenase and Nitrate Reductase
Step l-Cells were grown in 45-liter batches, rapidly chilled to O-4”, and harvested by centrifugation. The cells were washed twice with 0.9% NaC1/50 mM sodium phosphate, pH 7.2, and the cell paste was stored frozen at -15”. The thawed cell paste from four 45-liter batches (about 500 g wet weight) was suspended in 500 ml of 0.1 M sodium phosphate buffer, pH 7.2 (about 35 mg of pro- tein/ml).
Step %--The cells were disrupted by two passages through a Manton-Gaulin homogenizer at 6,000 p.s.i. All subsequent steps were carried out at O-5” under an atmosphere of NP or Ar unless otherwise noted. Solid ammonium sulfate was slowly added to the homogenate (230 g/liter) and the mixture was stirred for 1 hour. The crude membranes were collected by centrifugation at 15,000 X g for 2 hours and resuspended in 500 ml of 0.1 M Tris-HCl, pH 8. The protein concentration was measured and then 0.1 M Tris-HCl, pH 8, was added to give a final concentration of 4 to 6 mg of pro- tein/ml.
Step S-Sodium deoxycholate (10%) was added to a final concen- tration of 1 mg/mg of protein. Saturated ammonium sulfate in 0.1 M Tris-HCI, pH 8, was then added to give 3Oyc saturation. The suspension was stirred for 3 hours and then the precipitate was removed by centrifugation at 15,000 X g for 3 hours.
Step d--The supernatant was brought to 40y0 saturation with the addition of saturated ammonium sulfate in 0.1 M Tris-HCl, pH 7. After stirring for 1 hour the precipitate was removed by centrifugation at 15,000 X g for 1 hour. The supernatant then was brought to 500/, saturation by adding more saturated ammonium
loo 120 140 160 160
FRACTION NUMBER
FIG. 1. Purification of formate dehydrogenase and nitrate re- ductase by gel filtration on Bio-Gel A-1.5m. Chromatography was performed as described under “Experimental Procedure,” Step 5. A sample of 90 ml (1440 mg of protein) was applied; 9.2-ml fractions were collected. Formate dehydrogenase activity was measured by the nitroblue tetrazolium method. The black bar denotes the frac- tions pooled for Step 6.
sulfate solution. The mixture was stirred for 1 hour and the pre- cipitate was collected by centrifugation at 15,000 X g for 1 hour. The reddish brown precipitate was resuspended in 0.1 M Tris-HCl, pH 7.2, and dialyzed for 24 hours against 20 volumes of this buffer and then for an additional 24 hours against 20 volumes of 0.1 M
Tris-HCl,/l M NaCI, pH 7.2. The dialyzed solution was centrifuged at 15,000 X g for 1 hour and the supernatant was mixed with enough 10% Triton X-100 to give a final concentration of 0.5oj, detergent. All detergent concentrations are expressed as a weight per volume per cent.
Step 6-After 1 hour this solution was layered on a column (5 X 92 cm) of 6% agarose (Bio-Gel A-l .5m) equilibrated with anaerobic 0.5% Triton X-100/1 M NaCl/O.l M Tris-HCI, pH 7.2. The column was eluted with the same solution at a flow rate of 30 ml/hour and fractions were collected in the presence of air (Fig. 1). The rele- vant fractions were assayed and pooled within 3 hours of elution and then dialyzed against two changes each of 15 volumes of 0.1% Triton X-100/10 mrvr Tris-HCl, pH 8, for 20 hours.
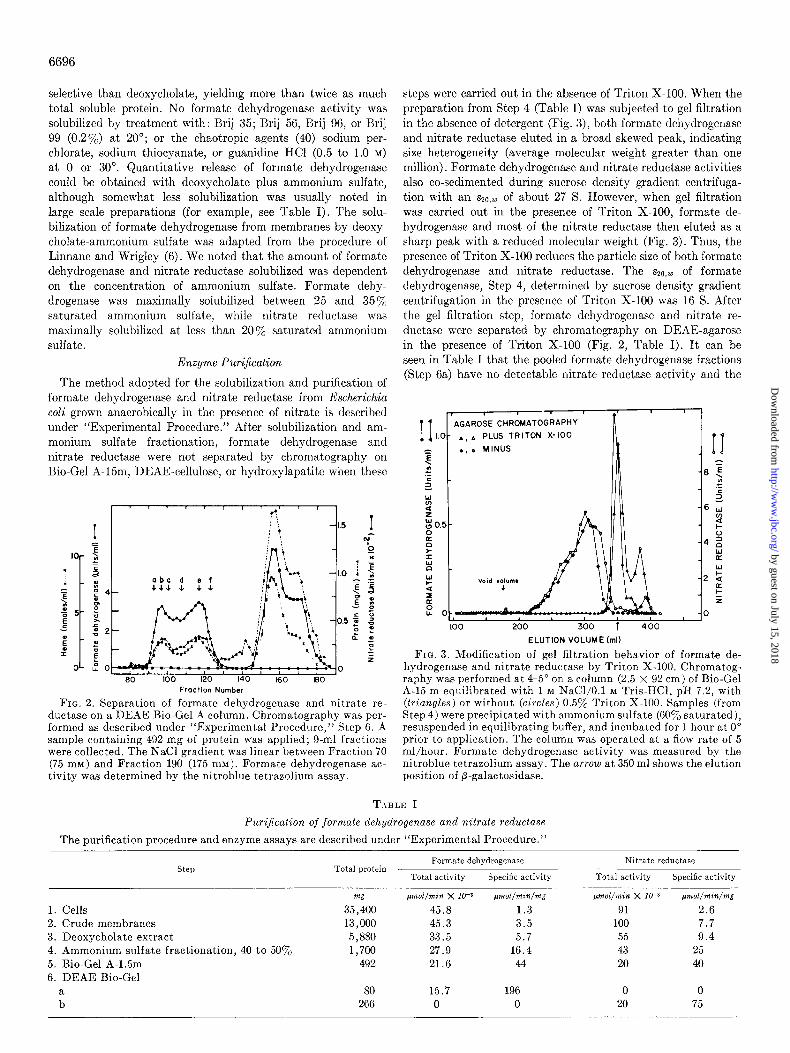
Step B-The dialyzed solution from Step 5 was applied to a col- umn (2.5 X 42 cm) of DISAE-agarose (DEAl;-Bio-Gel A) previ- ously equilibrated anaerobically in the same buffer used for dialy- sis. After washing with 1 column volume of the same buffer, the column was developed with a 2.liter linear gradient from 0 to 0.2 M NaCl in the same Triton-Tris buffer (Fig. 2). Formate dehydrogen- ase and nitrate reductase were separated at this step and both enzymes were stored at 4-5” under an atmosphere of Nz.
RESULTS
Solubilization studies
Preliminary experiments showed that all of the formate dehydrogenase could be recovered in the membrane-cell en- velope fraction after cell lysis with Iysozyme-EDTA (4). Formate dehydrogenase could be solubilized (i.e. released in a form not sedimented by centrifugation at 100,000 X g for 1 hour) by treatment of the membrane-cell envelope fraction with the following detergents: Triton X-100, Brij 36T, Brij 30 (0.279, or deoxycholate plus ammonium sulfate at 0”. Less than 50% of the original formate dehydrogenase activity was found to be soluble after treatment with Brij 36T or Brij 30 due to incom- plete solubilization and loss of activity. Triton X-100 was less
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
6696
selective than deoxycholate, yielding more than twice as much total soluble protein. No formate dehydrogenase activity was solubilized by treatment with: Brij 35; Brij 56, Brij 96, or Brij 99 (0.2%) at 20’; or the chaotropic agents (40) sodium per- chlorate, sodium thiocyanate, or guanidine HCI (0.5 to 1.0 M)
at 0 or 30”. Quantitative release of formate dehydrogenase could be obtained with deoxycholate plus ammonium sulfate, although somewhat less solubilization was usually noted in large scale preparations (for example, see Table I). The solu- bilization of formate dehydrogenase from membranes by deoxy- cholate-ammonium sulfate was adapted from the procedure of Linnane and Wrigley (6). We noted that the amount of formate dehydrogenase and nitrate reductase solubilized was dependent on the concentration of ammonium sulfate. Formate dehy- drogenase was maximally solubilized between 25 and 35% saturated ammonium sulfate, while nitrate reductase was maximally solubilized at less than 20% saturated ammonium sulfate.
Enzyme Purijication
The method adopted for the solubilization and purification of formate dehydrogenase and nitrate reductase from Escherichia
coli grown anaerobically in the presence of nitrate is described under “Experimental Procedure.” After solubilization and am- monium sulfate fractionation, formate dehydrogenase and nitrate reductase were not separated by chromatography on Bio-Gel A-15m, DEAE-cellulose, or hydroxylapatite when these
Fraction Number
FIG. 2. Separation of formate dehydrogenase and nitrate re- ductase on a DEAE Bio-Gel A column. Chromatography was per- formed as described under “Experimental Procedure,” Step 0. A sample containing 492 mg of protein was applied; g-ml fractions were collected. The NaCl gradient was linear between Fraction 70 (75 mM) and Fraction 190 (175 mM). Formate dehydrogenase ac- tivity was determined by the nitroblue tetrazolium assay.
steps were carried out in the absence of Triton X-100. When the preparation from Step 4 (Table I) was subjected to gel filtration in the absence of detergent (Fig. 3), both formate dehydrogenase and nitrate reductase eluted in a broad skewed peak, indicating size heterogeneity (average molecular weight greater than one million). Formate dehydrogenase and nitrate reductase activities also co-sedimented during sucrose density gradient centrifuga- tion with an szO ,u) of about 27 S. However, when gel filtration was carried out in the presence of Triton X-100, formate de- hydrogenase and most of the nitrate reductase then eluted as a sharp peak with a reduced molecular weight (Fig. 3). Thus, the presence of Triton X-100 reduces the particle size of both formate dehydrogenase and nitrate reductase. The s20,w of formate dehydrogenase, Step 4, determined by sucrose density gradient centrifugation in the presence of Triton X-100 was 16 S. After the gel filtration step, formate dehydrogenase and nitrate re- ductase were separated by chromatography on DEAE-agarose in the presence of Triton X-100 (Fig. 2, Table I). It can be seen in Table I that the pooled formate dehydrogenase fractions (Step 6a) have no detectable nitrate reductase activity and the
ELUTION VOLUME (ml)
FIG. 3. Modification of gel filtration behavior of formate de- hydrogenase and nitrate reductase by Triton X-100. Chromatog- raphy was performed at 4-5” on a column (2.5 X 92 cm) of Bio-Gel A-15 m equilibrated with 1 M NaCl/O.l M Tris-HCl, pH 7.2, with (triangles) or without (circles) 0.5yo Triton X-100. Samples (from Step 4) were precipitated with ammonium sulfate (GOyO saturated), resuspended in equilibrating buffer, and incubated for 1 hour at 0” prior to application. The column was operated at a flow rate of 5 ml/hour. Formate dehydrogenase activity was measured by the nitroblue tetrazolium assay. The arrow at 350 ml shows the elution position of p-galactosidase.
TABLE I
Purijcation of formale dehydrogenase and nitrate reductase
The purification procedure and enzyme assays are described under “Experimental Procedure.”
step Total protein Formate dehydrogenase Nitrate reductase
Total activity Specific activity Total activity Specific activity
1. Cells 2. Crude membranes 3. Deoxycholate extract 4. Ammonium sulfate fractionation, 40 to 5Oc%, 5. Bio-Gel A-1.5m 6. DEAE Bio-Gel
;
35Yoo 13:ooo
5,880 1,700
492
80 15.7 196 0 0 266 0 0 20 75
/.wlol/min x 10-3 &mJl/min/mg @nol/min x 10-a jmol/min/mg
45.8 1.3 91 2.6 45.3 3.5 100 7.7 33.5 5.7 55 9.4 27.9 16.4 43 25 21.6 44 20 40
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6697
pooled nitrate reductase fractions (Step 6b) had no detectable formate dehydrogenase activity.
A 150-fold purification of formate dehydrogenase was ob- tained (Table I) with yields ranging from 34 to 68%. For good recoveries of formate dehydrogenase activity it was essential that the entire purification be carried out in an anaerobic atmosphere. When Steps 1 to 5 (Table I) were carried out anaerobically 100% of the formate dehydrogenase activity was accounted for (including discarded fractions), while in the presence of air, the recovery of formate dehydrogenase at Step 5 varied from 1 to 20%, due to irreversible inactivation of the formate dehydrogenase activity. When Step 6 was performed aerobically 99% of the formate dehydrogenase activity was lost.
Purity of Formate Dehydrogenase
The formate dehydrogenase obtained at the final step (Step 6a) of the purification in most preparations contained a number of aggregated forms of the enzyme. The conditions required to prevent the aggregation of the enzyme have not yet been found. A major problem in the assessment of the purity of formate dehydrogenase was the difficulty in obtaining the enzyme as a single disaggregated species.
Two types of results were obtained at the final step (Step 6) of the purification. During DEAE-agarose chromatography formate dehydrogenase was eluted as a single peak (most prepara- tions, Fig. 4) or as two peaks (Fig. 2) of activity, indicating the presence of multiple forms of the enzymes. We investigated t.he possibility that these two peaks represent different aggregation states of the enzyme. Electrophoresis was performed on Fractions a to f (Fig. 2), and the gels were stained for protein (Fig. 5A). Fractions d to f appeared to contain a single form of the enzyme while Fractions a to c contained a number of different migrating forms of the enzyme. The protein band observed in Fractions d to f was absent in Fractions a and b. With Fractions a and b another protein band was observed which barely penetrated the
0 IO 20 30 40 50 FRACTION NUMBER
FIG. 4. Chromatography of 75%labeled preparation on DEAE- Bio-Gel A. Cells were grown in standard medium (8 liters) supple- mented with lG0 ltCi of 0.1 UM V5Sel selenite. The enzvmes were purified as described under’ “I<xperimental Procedure”” through Step 5 (ammonium sulfate fractionation was between 45 and 58y0). The dialyzed preparat,ion (4.2 mg of protein) was applied to a column (0.9 X 25 cm) of DEAE-Bio-Gel A previously equilibrated with 0.5% Triton X-100/10 mM Tris-HCI, pH 8. The column was developed at 4-5” with a 120-ml linear gradient from 0 to 0.25 M
NaCl in the same buffer (Fractions 1 to 40) and then washed with 1 M NaCl in the same buffer. Fractions of 3 ml were collected in tubes containing 2 ml of 3 M NaCl/O.l M Tris-HCl, pH 7.5. Formate dehydrogenase activity was measured by the nitroblue tetrazolium method.
gels indicating the presence of a very large protein. Similar gels were run on Fractions b and e and stained for formate dehy- drogenase activity (b’ and e’ of Fig. 5A) and bands were observed which corresponded exactly to the protein-stained bands. Sodium dodecyl sulfate gel electrophoresis experiments showed that Fractions a to f did not differ in their polypeptide composition. The same three polypeptides, designated LY, p, and y, are ob- served in each fraction tested (Fig. 5B). This suggests that the different forms of formate dehydrogenase seen after Triton gel electrophoresis represent different aggregation states of the enzyme. The first peak of formate dehydrogenase activity eluted from the DEAE-agarose column (Fig. 2) contains multiple forms of aggregated enzyme, while the second peak of activity appears to contain a single, monodisperse species of the enzyme. The formate dehydrogenase specific activity in both peaks (Fig. 2) is the same (3.2 f 0.1 pmol of nitroblue tetrazolium reduced per min per mg of protein), indicating that the catalytic properties of the enzyme are not greatly affected by aggregation. The heme to protein ratios of both peaks are also the same (7.0 f 0.3 nmol of heme/mg of protein).
In other preparations formate dehydrogenase eluted from the DEAE-agarose column in a single peak (Fig. 4). Analysis of these single peaks by Triton gel electrophoresis showed a single form of the enzyme in some preparations, while in other preparations multiple aggregation forms of the enzyme were observed. The cause of aggregation is not known; however, it appears to occur after the gel filtration step and could possibly be initiated under the low ionic strength conditions of Step 6 (Table I). Throughout the remainder of this report the opera- tional term “homogeneous formate dehydrogenase” will be used to denote that enzyme at Step 6a which showed a single band of enzyme activity and protein after Triton gel elec- trophoresis. Although we cannot rigorously exclude that multiple forms of formate dehydrogenase could arise in the course of
I *II ts se <II Y
a b b’ c d e e’ f abcdef
FIG. 5. Polyacrylamide gel electrophoresis of purified formate dehydrogenase in the presence of 0.25y0 Triton X-100 (A) or 0.1% sodium dodecyl sulfate (B). Samples a to f refers to fractions ob- tained after DEAE-agarose chromatography (see Fig. 2). A, samples (40 ~1) were mixed with 10 ~1 of glycerol and layered on a gel. Electrophoresis, protein staining (a, b, c, d, e,f) and formate aehydrogenase activity staining (b’and e’) were performed as described under “Experimental Procedure.” The anode is at the bottom of the gels. B, samples (50 ~1) were mixed with 50 ~1 of 2% sodium dodecyl sulfate/lO% glycerol/50 mM sodium phosphate, pH 7.2, and 2~1 of mercaptoethanol, heated at 100” for 10 min, then 2 ~1 of 0.05y0 bromphenol blue was added before layering the entire sample on a gel (stacking gels and separating gels contained 2.5 and 8% acrylamide, respectively). Electrophoresis and protein staining were performed as described under “Experimental Pro- cedure.” The anode is at the bottom of the gel.
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from
6698
purification via limited deamination or other hydrolytic re- actions, the facile aggregation of isolated enzyme leads us to retain the simple notion that the multiple forms have arisen via aggregation. Aggregation of the homogeneous formate dehy- drogenase occurred when gel electrophoresis was carried out in the absence of Triton X-100 as judged by the appearance of multiple forms of slower migrating enzyme (detected by both protein and enzyme stains, not shown).
Formate dehydrogenase appears to be very nearly pure as judged by Triton gel electrophoresis; however, a small amount of protein ( <l%) was consistently detected as two or three bands near the (Y polypeptide after sodium dodecyl sulfate gel electrophoresis (Fig. 5B). These bands could represent in- completely dissociated polypeptides since they are observed in elevated amounts when the sample is not heated during sodium dodecyl sulfate dissociation. In any case, the enzyme is at least 99% pure if cu, /3, and y are all considered to be subunits of formate dehydrogenase. It can be seen in Fig. 5B that, although each fraction contains the y polypeptide, there was some varia- tion in the density of staining of this polypeptide. This difference could be caused by the difficulty in fixing and staining small proteins in gels (41). It is also possible that the difference could reflect the loss of some of the y polypeptide during purification. On the other hand the y polypeptide could be a tightly associated impurity in formate dehydrogenase amounting to about 7% of the total protein. Sedimentation velocity studies with homo- geneous formate dehydrogenase (see below) did not indicate the presence of any contaminating protein ( <lo%) or multiple forms of the enzyme.
Physical Properties of Formate Dehydrogenase
Subunit Studies-The dissociation of formate dehydrogenase
by sodium dodecyl sulfate (as judged after gel electrophoresis in the presence of this detergent) required incubation at 100”
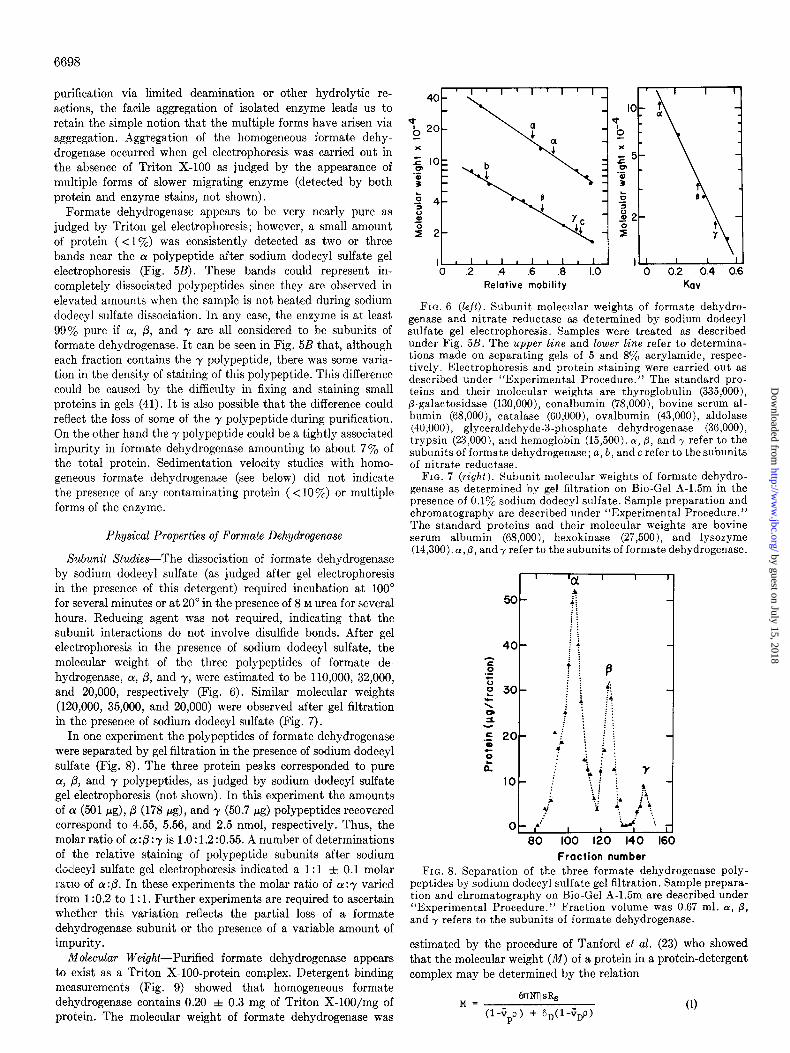
for several minutes or at 20” in the presence of 8 M urea for several hours. Reducing agent was not required, indicating that the subunit interactions do not involve disulfide bonds. After gel electrophoresis in the presence of sodium dodecyl sulfate, the molecular weight of the three polypeptides of formate de- hydrogenase, a, p, and y, were estimated to be 110,000, 32,000, and 20,000, respectively (Fig. 6). Similar molecular weights (120,000, 35,000, and 20,000) were observed after gel filtration in the presence of sodium dodecyl sulfate (Fig. 7).
In one experiment the polypeptides of formate dehydrogenase were separated by gel filtration in the presence of sodium dodecyl sulfate (Fig. 8). The three protein peaks corresponded to pure cu, p, and y polypeptides, as judged by sodium dodecyl sulfate gel electrophoresis (not shown). In this experiment the amounts of a (501 pg), p (178 pg), and y (50.7 c(g) polypeptides recovered correspond to 4.55, 5.56, and 2.5 nmol, respectively. Thus, the molar ratio of a : p : y is 1 .O : 1.2 :0.55. A number of determinations of the relative staining of polypeptide subunits after sodium dodecyl sulfate gel electrophoresis indicated a I : 1 f 0.1 molar ra~lo of a:@. In these experiments the molar ratio of cr :y varied from 1:0.2 to 1: 1. Further experiments are required to ascertain whether this variation reflects the partial loss of a formate dehydrogenase subunit or the presence of a variable amount of impurity.
Molecular Weight-Purified formate dehydrogenase appears to exist as a Triton X-loo-protein complex. Detergent binding
measurements (Fig. 9) showed that homogeneous formate dehydrogenase contains 0.20 f 0.3 mg of Triton X-lOO/mg of protein. The molecular weight of formate dehydrogenase was
‘0.2 Relative mobility Kav
FIG. 6 (left). Subunit molecular weights of formate dehydro- genase and nitrate reductase as determined by sodium dodecyl sulfate gel electrophoresis. Samples were treated as described under Fig. 5B. The upper line and lower line refer to determina- tions made on separating gels of 5 and 8% acrylamide, respec- tively. Electrophoresis and protein staining were carried out as described under “Experimental Procedure.” The standard pro- teins and their molecular weights are thyroglobulin (335,000), p-galactosidase (130,000), conalbumin (78,000); bovine serum al- bumin (68.000). catalase (60.000). ovalbumin (43.000). aldolase (40,000); gly&aldehyde-3‘-phosphate dehydrogenase’ (3G,OOO), trypsin (23,000), and hemoglobin (15,500). 01,p, and y refer to the subunits of formate dehydrogenase; a, b, and c refer to thesubunits of nitrate reductase.
FIG. 7 (right). Subunit molecular weights of formate dehydro- genase as determined by gel filtration on Bio-Gel A-1.5m in the presence of 0.1% sodium dodecyl sulfate. Sample preparation and chromatography are described under “Experimental Procedure.” The standard proteins and their molecular weights are bovine serum albumin (68,000), hexokinase (27,500), and lysozyme (14,300). ~l,p, and? refer to the subunits of formate dehydrogenase.
I ’ ‘cl ’ I I
Fraction number FIG. 8. Separation of the three formate dehydrogenase poly-
peptides by sodium dodecyl sulfate gel filtration. Sample prepara- tion and chromatography on Bio-Gel A-1.5m are described under “Experimental Procedure.” Fraction volume was 0.67 ml. (Y, p, and y refers to the subunits of formate dehydrogenase.
estimated by the procedure of Tanford et al. (23) who showed that the molecular weight (M) of a protein in a protein-detergent complex may be determined by the relation
M= 67Wsb
(l-Gpp) + 6#+)P) (1)
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6699
5
1.6 k 3 I
i 12
;c 1.0 ‘, al
._..” ,*--a._ f
Ot C 0.8 x.i” $ (.I
i : ,,’
p...
0 IO 20 30 40 50 60 70 Fraction number
FIG. 9. Measurement of Triton X-100 binding to formate de- hydrogenase. Chromatography was performed asdescribed under “Experimental Procedure”; O.l-ml fractions were collected. The concentration of unbound detergent (0.89 mg/ml) was taken as the average of Fractions 5 to 40; milligrams of bound Triton X-100 per mg of protein was calculated from the average in Fractions 43 to 49.
0 .2 .4 .6 .8 1.0 Kav
FIG. 10. Stokes radius of formatc dehydrogenase and nitrate reductase as determined by gel filtration. Chromatography on Bio-Gel A-l.5m was performed as described under “Experimental Procedure.” The standard proteins and their Stokes radii are thyroglobulin (81 A), p-galactosidase (68 A), catalase (52 A), alcohol dehydrogenase (45 A), bovine serum albumin (36 A), and cytochrome c (17 A).
where N is Avogadro’s number, 7 the viscosity of the solvent, s the sedimentation coefficient, R, is the Stokes radius of the protein-detergent complex, tip is the partial specific volume of the protein, p is the density of the solvent, 60 is milligrams of bound detergent per mg of protein, and BD is the partial specific volume of the detergent. A value of 0.915 cm3/g was determined for IYD of Triton X-100 in 1 M NaCl. The sedimentation coefficient of homogeneous formate dehydrogenase determined by sedi- mentation velocity at 5” in the presence of 0.1% Triton X-100 and 1 M NaCl was 7.95 S (s*~,~ = 18.1 S). The Stokes radius of the formate dehydrogenase-Triton X-100 complex determined by gel filtration in the presence of 0.1% Triton X-100 and 1 M
NaCl was 76 A (Fig. 10). Only one sharp peak of formate de- hydrogenase activity was observed in these experiments, which indicates that we are looking at a single molecular species of formate dehydrogenase. Assuming a 5, of 0.75 the molecular weight of formate dehydrogenase calculated by Equation 1 is 590,000 f 59,000. The possible error of this determination was large because of the uncertainty of tip. An error of 3% in $, leads to an error of 10% in the molecular weight. The error associated with the determination of R, should be less than 10% (23). The term 6n(l - cop) makes a relatively small contribution to the determination of dl; an error of 10% in this term leads to an error of less than 1% in ,V.
Composition of Formate Dehydrogenase
Selenium-When formate dehydrogenase was purified from E. coli grown in the presence of 0.1 PM [7%e]selenite the enzyme
4
? 03 2
h* 0
$1 t
0 !
FIG. 11. Sodium dodecyl sulfate gel electrophoresis of ‘&Se- labeled formate dehydrogenase. The sample, 50 ~1 of Fraction 14 (Fig. 4), was treated as described under Fig. 5B and the sample was layered on a gel (stacking gel and separating gel contained 2.5 and 8% acrylamide, respectively). Electrophoresis and protein staining were performed as described under “Experimental Pro- cedure.” Protein stain was estimated by a densitometer tracing (..... ) and the gel was then sliced and ?%e radioactivity was determined using a Nuclear-Chicago y-counter with a Picker Cliniscaler. 01,p, and y refer to the subunits of formate dehydro- genase.
activity which was eluted from the DEAE-agarose column was coincident with 75Se radioactivity and protein (Fig. 4). From the specific radioactivity of selenium in the growth medium (114 cpm/pmol of selenium) and in formate dehydrogenase (496 cpm/pg of protein) the selenium content of formate de- hydrogenase was estimated to be 4.4 nmol of selenium/mg of protein. Nitrat,e raductase from the same preparation contained less than 0.1 nmol of selenium/mg of protein, indicating that little selenium is nonspecifically incorporated into protein (for example, as the selenium analogs of the sulfur-containing amino acids) under these conditions. Somewhat higher levels of selenium (6.2 nmol/mg of protein or 0.95 mol/mol of heme) were de- tected by neutron activation analysis in another preparation of formate dehydrogenase.
The 75Se of formate dehydrogenase was not removed by dialysis at pH 7 or by precipitation with 10% trichloroacetic acid or 95% acetone. Jenkins (42) has shown that selenite can be nonenzymatically incorporated into protein. He suggested that the selenium was covalently bound between the sulfurs of half-cystine residues and showed that this form of selenium could be removed by dialysis of the selenoprotein at pH 11.5 or by treatment with 0.5 M mercaptoethanol, 50 mM sodium sulfite, or 5 mM KCN at 30” for 30 min. None of these treat- ments resulted in the release of ‘%e from formate dehydrogenase suggesting that the enzyme may not contain selenium as seleno- trisulfide. 7Se could be released (trichloroacetic acid-soluble) by treatment of formate dehydrogenase with 3% H202 for 1 hour at 30”.
75%labeled formate dehydrogenase (Step 6) was dissociated in sodium dodecyl sulfate and subjected to gel electrophoresis in the presence of this detergent. The densitometer tracing of the Coomassie blue-stained gel and the 75Se radioactivity of the sliced gel fractions are shown in Fig. 11. It can be seen that the bulk of the radioactivity was present in the o( polypeptide. Insignificant amounts of radioactive selenium were associated with the p and y polypeptides.
Heme-After DEAE-agarose chromatography the formate dehydrogenase-containing fractions were colored (reddish brown) and were found to contain heme. The heme content was coincident with formate dehydrogenase activity and protein (Fig. 2). When the homogeneous enzyme was subjected to gel filtration (Fig. 12), the heme content (based on the absorbance
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6700
TARLE II
Composition of formale dehydroqenase
Details of the analytical procedures are given under “Experi- mental Procedure” or in the text (for [76Se] selenium analysis).
I L ,’
2 3 60- - 0.6 L 5 ii
2 z *- - 0.4
H -
11 I I t ,
25 30 36 40
FRACTION NUMBER
FIG. 12. Rechromatography of purified formate dehydrogenase on Bio Gel A-15m. A sample of Step 6 formate dehydrogenase (4 mg of protein in 0.5 ml) was layered on a column (1 X 143 cm) of Bio-Gel A-15m eqllilibrated with 0.1% Triton X-100/1 M NaCI/ 0.1 M Tris-HCl, pH 7.4. The column was developed with the same buffer at a flow rate of 1.8 ml/hour (4-5”); 2.0-ml fractions were collected.
1.4- “Ml ’ ’ l’~~~l~~~~l~~~‘l”‘l”“~‘_
Formote Dehydrogemse -
- ox8dued wreduced
0.6 - 0 e :: 6 0.6- 2 .a -
04-
02-
Or,,,,,,,,,,,,,~~l~~~~1~~~~l~~~~l~~~~l,~~~r- 3clo 400 500 600
Wavelength (nm)
FIG. 13. The oxidized and reduced absorption spectra of for- mate dehydrogenase. Purified formate dehydrogenase was ad- sorbed to a DEAE-agarose column, washed with 10 volumes of 10 mM Tris-HCl, pH 8, to remove Triton X-100, and then eluted with 0.2 M NaCl/lO mM Tris-HCI, pH 8. The spectra were recorded at 20” under anaerobic conditions in a cuvette of l-cm path length. The sample contained O.G3 mg of protein/ml. The reduced spec- trum was obtained 5 min after the addition of 50 mM formate and was identical (visible portion) to that obtained by the addition of solid sodium dithionite.
of oxidized enzyme at 414 nm) was again coincident with formate dehydrogenase activity and protein and a constant ratio of heme (A 414 .,) to protein (milligrams per ml) was observed, 5.8 f 0.4. The specific activity of formate dehydrogenase was nearly constant (107 f 13 pmol/min/mg of protein) in Fractions 31 to 34 (Fig. 12) which represent the bulk of the total activity (90%). The decreased specific activity in the later fractions could be due to the presence of some inactive formate dehy- drogenase or other impurity.
The absorbance spectra of oxidized and reduced formate dehydrogenase are shown in Fig. 13. The spectra were obtained after the removal of Triton X-100 because this detergent ab-
Component nmol/mg of protein Molar ratio
Heme F.5 1 Molybdenum 6.2a 0.95 Molybdenum 4.8” 0.74 Selenium 4.4c 0.68 Selenium 6.2* 0.95 Non-heme iron 89 14 Acid-labile sulfide 82 13 Coenzyme Qd <0.3 <0.05 Vitamin Ei@ <0.3 <0.05 Flavin <O.l <0.02 Phospholipid <2 <0.3
Q Determined by chemical assay. * Determined by neutron activation analysis. c Determined by T&Se specific radioactivity. d Determination was made on detergent-free enzyme. The puri-
fied formate dehydrogenase was adsorbed to a l)EAE-Bio-Gel A column, washed with 10 volumes of 10 mM Tris-HCl, pH 8, and then eluted with 0.2 M NaCl/lO mM Tris-HCl, pH 8.
sorbs strongly in the ultraviolet region (E:z = 24). The oxidized and reduced absorbance maxima are those of a b-type cyto- chrome and are essentially the same as those previously found in partially purified preparations of formate dehydrogenase (5, 6). The cytochrome of formate dehydrogenase became com- pletely reduced in the presence of formate (i.e. compared to dithionite reduction). Under anaerobic conditions this reduction was rapid: half of the cytochrome was reduced immediately (<2 s) and 95% reduction was obtained in less than 1 min at 20”. The heme content of purified formate dehydrogenase is 6.5 nmol/mg of protein. This is 4.2-fold higher than that reported for partially purified formate dehydrogenase obtained from aerobically grown E. coli (6).
Other Components-Chemical analysis showed that homo- geneous formate dehydrogenase contains molybdenum, non- heme iron, and acid-labile sulfide (Table II). No acid-releasable flavin was found in our purified preparations of formate de- hydrogenase and bound flavin was not detected after proteolytic digestion of the enzyme. The fluorescence of added FAD and FINN were unchanged after being carried through the same analyses in the presence of formate dehydrogenase, indicating that our lack of flavin detection was not due to quenching of the fluorescence (e.g. by heme) or reduction of flavin by the enzyme. Formate dehydrogenase has long been considered to be a flavin- containing enzyme (43) since flavin was detected in partially purified preparations of this enzyme (5, 6). Itagaki et al. (5), however, did not report the amount of flavin in their preparation and Linnane and Wrigley (6) reported the presence of only 0.35 mol of flavin/mol of heme in their preparation. It would now appear that the E. coli formate dehydrogenase is not a flavo- protein.
Quinone was not detected in our preparation of formate dehydrogenase (Table II). The removal of Triton X-100 (which interfered with the quinone measurement) had no effect on the reduction of cytochrome b by formate.
The purified formate dehydrogenase (Step 6a) contains less than 0.2y0 phospholipid and less than 0.4y0 RNA. Total sugar analysis by the phenol-sulfuric acid method revealed the presence
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6701
of 2% carbohydrate (as glucose equivalents). We have not yet attempted to verify this result by identification of specific sugar residues. The composition of formate dehydrogenase is sum- marized in Table II.
Catalytic Characteristics of Purified Formate Dehydrogenase
The highest specific activity of formate dehydrogenase which we have observed was 220 prnol of phenazine methosulfate-di- chlorophenolindophenol reduced/min/mg of protein, yielding a turnover number of 33,800 mol/min/mol of heme. The K, for formate was 0.12 mM. It does not seem relevant to compare the specific activity of this enzyme with less pure preparations of others due to differences in assay procedures and to the fact that activity is rapidly lost in the presence of oxygen (see below). We have observed homogeneous preparations of formate de- hydrogenase having specific act.ivities with phenazine methosul- fate-dichlorophenolindophenol as low as 20 pmol/min/mg of pro- tein due to less than rigorous exclusion of oxygen at Step 5 or Step 6 of the purification.
Accepfor Specificity-Of the electron acceptors tested formate dehydrogenase showed the highest activity with phenazine methosulfate plus dichlorophenolindophenol (Table III). Di- chlorophenolindophenol alone is a good acceptor for the enzyme. Itagaki et al. (5) reported that partially purified formate dehydro- genase was inactive with dichlorophenolindophenol. Although the basis for this difference is not clear it should be noted that the cytochrome b of their preparation was not reduced by formate. The particulate and the soluble, homogeneous formate dehydro- genase have similar relative activities with dichlorophenolindo- phenol and nitroblue tetrazolium (Table III), indicating that cer- tain catalytic properties of the enzyme have been maintained.
TABLE III
Electron acceptor specijicity of formate dehydrogenase
All reactions were carried out anaerobically at 30”. The reduc- tion of electron acceptors (except coenzyme Q6) was monitored spectrophotometrically. The reaction mixtures (4 ml) contained formate dehydrogenase, acceptor, 20 mM formate, and 0.15 M
sodium phosphate, pH i.2. Coenzyme Qs reduction was measured as described under “Experimental Procedure.” All activities are based on a two-electron transfer. PMS, phenazine methosulfa.te; DCI, dichlorophenolindophenol.
Relative formate dehydrogenase
Acceptor Acceptor concentration
activity Wavelength
HOltlO- Partic- geneous ulate eIlZyRX ellZyIIle”
PMS + DC1 DC1 Ferricyanide Nitroblue tetra-
zolium Methyl viologen Benzyl viologen Methylene blue Coenzyme Q6 FAD FMN NAD+ NADP+
?nM n?n
0.24 + 0.087 600 1006 100’ 0.087 600 23 25 2.0 420 20 0
0.066 530 15 15 1.0 600 0.2 1.0 600 2.4 0.04 650 30 48 1.0 40 0.072 450 0 0.1 450 0 1.0 340 0 0 1.0 340 0 0
a Step 2 enzyme (Table I). b Specific activity was 142 pmol/min/mg of protein. c Specific activity was 5.2 pmol/min/mg of protein.
The lack of ferricyanide reductase activity in membranes suggests that the preparation may consist of oriented vesicles which are not accessible to bulky anions. The activity with methylene blue and viologen dyes was similar to that previously reported (5). The purified E. coli enzyme could not use NAD+ or NADP+ as electron acceptors (Table III). Neither FAD nor FMN were active as acceptors (Table III) and neither were stimulatory in the assay of formate dehydrogenase with phenazine metho- sulfate and dichlorophenolindophenol. Quinone has been impli- cated as an essential component of the formate-nitrate reductase in E. coli (44). Coenzyme Q was shown to be an acceptor for a particulate preparation of formate dehydrogenase; however, the specific activity was only I y0 of that with methylene blue (44). We found coenzyme Q6 to be a very effective electron acceptor for the purified formate dehydrogenase (Table III).
Inhibitors-The over-all pattern of sensitivity of purified formate dehydrogenase toward a number of inhibitors was similar to that previously reported for partially purified prepara- tions of the enzyme (5,6), in that the activity measured by the re- duction of dichlorophenolindophenol with phenazine methosulfate was very sensitive to inhibition by cyanide and azide and was partially inhibited by iodoacetamide, p-hydroxymercuribenzoate, and n-heptyl hydroxyquinoline-N-oxide (Table IV). Only 9 y0 inhibition of formate dehydrogenase was observed when formate (20 mM) when present in the incubation of the enzyme with iodoacetamide. Each of these inhibitors had significantly less effect on the activity measured with other electron acceptors. It is particularly noteworthy that the activity measured with methylene blue was not inhibited by cyanide. Itagaki et al. (5) reported that partially purified formate dehydrogenase was inhibited 71% by 5 mM cyanide when the enzyme activity was assayed manometrically with methylene blue. However, others (45, 46) have shown that the activity of particulate formate dehydrogenase of E. coli measured with methylene blue was not inhibited by 1 mM cyanide, a level which caused complete in- hibition of the formate oxidase activity of the preparation. The
TABLE IV
Effect of inhibitors on various formate dehydrogenase acticities
The reactions were carried out as described in Table III. Puri- fied formate dehydrogenase, Step 6a (Table I), and inhibitor were incubated anaerobically for 5 min at 30” in 0.15 M sodium phos- phate, pH 7.2, and then the reaction was initiated by the addition of electron acceptor and formate (20 mM). The same conditions apply for oxygen inhibition except the incubation and reaction were conducted in the presence of air. PMS, phenazine metho- sulfate; DCI, dichlorophenolindophenol.
Addition Relative formate dehvdromnase activitv
PMS + DC1
DCI Me;ben Ferri- Nitroblue cyanide tetrazolium
None 100 NaCN, 5 mM 0.5 NaNa, 5 rnM 9.9 Iodoacetamide, 5 mM 55 N-Ethylmaleimide, A
rnM 99
n-Heptyl hydroxy- quinoline-AT-oxide, 0.1 rnM 60
Oxygen (air), 0.25 rnM
p-Hydroxymercuri- benzoate, 0.1 mM 41
100 100 100 100 7 105 30 27
61 52 51 35 90
98
102 124 109
61 50 34
87 84
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6702
results obtained here suggest that the enzyme may contain a number of different catalytic sites for interaction with these artificial electron acceptors.
The enzyme activity measured with dichlorophenolindophenol, ferricyanide, and nitroblue tetrazolium was inhibited by oxygen (Table IV). This inhibition may be related to the oxygen in- activation of the enzyme. It should be noted, however, that the inhibition is immediate, requiring no prior incubat.ion with oxygen, while the time course of inactivation is much slower. This suggests that oxygen may compete with other acceptors.
Only the activity with phenazine methosulfate-dichlorophenol- indophenol was inhibited by n-heptyl hydrosyquinoline-N-oside, which is a potent inhibitor of formate-nitrate reduction in the particulate fraction of E. coli (47) and also in the system re- constructed from purified formate dehydrogenase, nitrate re- ductase, and coenzyme Qs (12). The level of this inhibitor used in the experiment in Table IV causes a much greater inhibition of formate-nitrate reduction (loo%, Ref. 48) than formate- phenazine methosulfate-dichlorophenolindophenol reduction. We also found that formate-coenzgme Qs reduction is not affected at the same level of inhibitor.
Stability-Formate dehydrogenase activity is lost when the enzyme is exposed to oxygen. This instability is enhanced in low ionic strength buffers, detergents, at temperatures above O”, and at pH greater than 7. The enzyme was remarkably stable in air in the presence of 1 M NaCl (in 10 mM Tris-HCl, pH 8) with less than 5% loss of activity after 24 hours (0”). The same incubation in the absence of NaCl resulted in a 90% loss of activity. The purified enzyme (Step 6a) could be stored under Nz at O-4” for up to 9 months with no loss of activity. The enzyme was unstable to freezing at all stages of purity.
Formate dehydrogenase is extremely sensitive to inactivation by oxygen in the presence of formate. Purified formate de- hydrogenase, Step 6a (0.15 mg of protein/ml) was completely inactivated by aerobic incubation in 75 mM sodium phosphate, pH 7, for 1 hour at 20” in the presence of 50 mM formate, while only 40% of the activity was lost when the same incubation was carried out in the absence of formate. Ko activity was lost when the same incubation was carried out anaerobically even in the presence of formate. Similar results were observed by Gale (46) for a particulate preparation of formate-methylene blue reductase. He showed that the inactivation process was not due to peroxide formation. He further reported that the activity could be recovered after anaerobiosis was restored. When our purified enzyme was inactivated in the presence of formate no activity could be recovered by anaerobic incubation for 3 hours. Furthermore, the inactivated enzyme was inactive with dichlorophenolindophenol, methylene blue, nitroblue tetrazolium, and ferricganide as electron acceptors and the heme was no longer reducible with formate.
Purified formate dehydrogenase (0.15 mg of protein/ml) could be heated to 60” at pH 8.5 (in 0.75 M Tris-HCl) anaerobically for 15 min with no loss of activity. After heating under the same conditions aerobically, no activity was recovered after 5 min. The anaerobically heated enzyme displayed the same rela- tive activities with phenazine methosulfate-dichlorophenolindo- phenol, dichlorophenolindophenol, methylene blue, nitroblue tetrazolium, and ferricyanide as unheated enzyme, and the heme was still reducible by formate.
Reversibility of Reactions Catalyzed by Formate Dehydrogenase- If formate dehydrogenase (E) catalyzes the reversible oxidation of formate (F) in the absence of an electron acceptor, yielding COn and reduced enzyme (EHJ, F + E +-+ EH2 + Con, then
it should be possible to demonstrate a CO*-formate exchange reaction. Step 5 enzyme (3.7) units was incubated anaerobically at 30” in 1 ml of 1 mM [14C]bicarbonate (180,000 cpm)/O.l mM formate/ mM sodium phosphate, pH 7.2, for 5 to 15 min. The reaction was stopped by the addition of 0.1 ml of 1 M sodium [12C]bicarbonate. After the addition of 0.1 ml of 5 M sodium formate buffer, pH 3, bicarbonate was removed by bubbling with Nz for 5 min. The radioactivity of a 0.4.ml aliquot then was determined by scintillation counting. No detectable counts were found in formate; this was equivalent to less than 0.01% of the rate measured by dichlorophenolindophenol reduction with phenazine methosulfate, indicating that formate oxidation is not reversible under these conditions. The same results were observed when the reaction was carried out in 50 mM sodium acetate buffer, pH 4.9, or 0.1 M Tris-HCl, pH 8.9.
Formate dehydrogenase could not catalyze the reduction of CO2 in the presence of reduced methyl viologen. The oxidation- reduction potential at pH 7 is -0.42 volts for COz/formate and -0.44 for oxidized/reduced methyl viologen. A reaction mixture of 4 ml containing 0.3 mM methyl viologen, 1 mM
EDTA, and 0.15 M sodium phosphate, pH 7.2, was made an- aerobic by bubbling with argon and then sodium dithionite was added to give a final concentration of 0.27 mM reduced methyl viologen. The non-enzymatic rate of oxidation of methyl viologen was monitored spectrophotometrically at 600 nm (less than 2 nmol/min). No change in the rate was observed following the addition of purified formate dehydrogenase, Step 6 (15 pg) and sodium bicarbonate (40 pmol). The rate was less than 0.01 y. of formate-phenazine methosulfate-dichlorophenolindophenol re- duction.
Properties of Purified Nitrate Reducfase
Our procedure for the purification of formate dehydrogenase also yielded a highly purified preparation of nitrate reductase. As shown in Table I nitrate reductase was purified 29-fold over whole cells with a 22% yield. In other preparations yields as high as 72% were obtained. The yields of nitrate reductase were variable and sometimes low because the purification procedure was adapted for maximal enrichment of formate dehydrogenase and each purification step was designed on that basis without regard to nitrate reductase. Only one-half of the nitrate reductase was solubilized in Step 3 (Table I) ; however, if 10% saturated ammonium sulfate was used, essentially all of the nitrate reductase could be solubilized along with 20 to 40% of the formate dehydrogenase.
The nitrate reductase activity which was eluted from DEAE- agarose at Step 6 showed a peak with a shoulder (Fig. 2). Frac- tions 152 to 174 (Fig. 2) had a constant specific activity (75 & 3 pmol of nitrite produced/min/mg of protein). All nitrate re- ductase fractions were colored (reddish brown) and contained heme (Fig. 2) ; however, the heme to protein ratio is higher in the peak than in the shoulder. We have previously reported that this suggests that two forms of the enzyme exist which differ in their heme content (12). These two forms of nitrate reductase which are partially resolved by DEAE-agarose chromatography may be completely separated by gel electrophoresis in the presence of Triton X-100; the forms enriched in the peak (nitrate reductase I) and shoulder (nitrat.e reductase II) fractions have been estimated to contain 6.7 and 3.2 nmol of heme/mg of protein, respectively (12).
The purified preparation of nitrate reductase, Step 6b, con- tains three types of polypeptides (12) whose molecular weights were estimated by sodium dodecyl sulfate gel electrophoresis
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

to be 155,000, 63,000, and 19,000 (Fig. 6). There is some evidence that the heme may be associated with the 19,000.dalton poly- peptide (12).
The Stokes radius of nitrate reductase was estimated to be 75 A when determined in the presence of Triton X-100 (Fig. 10). The s20,w of nitrate reductase b was estimated to be 16 S by sucrose density gradient centrifugation in the presence of Triton X-100. The molecular weight of nitrate reductase was approximated using Equation 1 and ignoring any contribution made by bound detergent (i.e. omitting the sn(l - finp) term). Using a 0, of 0.725 cm3/g (49), the molecular weight was esti- mated to be 498,000 f 10%.
step enzyme specific activity was increased 515% while 7SSe specific activity increased only 17%). Thus, the significance of selenium in this preparation is not clear. Shum and Murphy (7) obtained a partially purified preparation of formate dehydro- genase from E. coli grown in the presence of [7%e]selenite and showed that enzyme activity cosedimented with 7%e during sucrose density gradient centrifugation. The selenium content of the enzyme from E. coli (7) and C. thermoaceticum (56) was not determined.
DISCUSSION
The purified formate dehydrogenase appears to exist as a globular’ protein-detergent complex free of phospholipid. Our results indicate that the enzyme is composed of three types of polypeptide chains and has the subunit structure (y4fi4y2. The molecular weight of this structure is 608,000 f 60,000 based on the molecular weights of the polypeptides. This is in good agree- ment with the value calculated from the heme content (molecu- lar weight = 615,000 based on 4 hemes/mol) and the value based on the sedimentation coefficient and Stokes radius (molecu- lar weight = 590,000). However, in view of the possible varia- tion in the amount of the y polypeptide it seems prudent to consider alternative structures. If some of the y polypeptide is lost in the course of purification, the actual structure may be (Y&I (molecular weight = 648,000) or, if y represents a tightly associated impurity (of about 7%), a structure of c& (molecular weight = 568,000) is possible.
We have shown that selenium incorporation into formate dehydrogenase is highly specific. The specific radioactivity of nitrate reductase was less than 3% of that of 7?%labeled formate dehydrogenase obtained from the same preparation. The data of Table II indicate that the enzyme may contain equimolar amounts of selenium, molybdenum and heme (4 mol of each/m01 of enzyme).
The selenium was shown to be associated with a polypeptide of molecular weight 110,000 and would appear to be covalently bound since rather drastic conditions were required to remove it. The enzyme does not contain acid-labile selenide, nullifying any possible role of selenium as a replacement of sulfide in the non-heme iron centers. It is not yet known if selenium partici- pates in the enzyme catalysis. The selenoenzymes, glutathione peroxidase (63), glycine reductase (52j, and formate dehydro- genase (this work) are all inhibited by iodoacetamide. It is pos- sible that a selenol rather than a thiol group is modified by the alkylating agent which could indicate a functional role of sele- nium in these enzymes.
This work has directly established that E. coli formate de- hydrogenase is a selenoenzyme. Selenium was recently shown to be a component of glutathione peroxidase of erythrocytes (50, 51) and a protein (“Protein A”) of the glycine reductase system of Clostridium sticklandii (52). Glutathione peroxidase (molecular weight = 84,000) contains 4 mol of selenium/m01 of enzyme. “Protein A” (molecular weight = 12,000) contains 1 mol of selenium/mol of protein (53). A selenium-containing protein has been isolated from lamb muscle (54, 55) and identified as a hemoprotein (molecular weight = 10,000).
No flavin was found in purified formate dehydrogenase and the lack of involvement of flavin in the reaction is further indi- cated by the inability of flavin to act as an electron acceptor or to stimulate the reduction of dichlorophenolindophenol and phenazine methosulfate. The soluble NAD+-dependent formate dehydrogenase of Pseudomonas oxalaticus has been reported to contain 1 mol of FMN as well as 5 to 8 mol of iron and 7 to 8 mol of labile-sulfide/300,000 g of protein (64).
In 1954, Pinsent (1) observed that the formation of formate dehydrogenase activity in E. coli required the presence of trace amounts of selenium, molybdenum, and iron in the culture medium. This was the first enzyme for which a selenium require- ment had been demonstrated. Similar requirements for the formation of formate dehydrogenase activity in a number of bacteria (56-58) suggest that selenium may be a general con- stituent of all formate dehydrogenases. Earlier work on selenium- containing preparations of formate dehydrogenase was carried out with preparations of unestablished purity or of low activity (7, 56) and could therefore not defmitively establish selenium as a component of the enzyme. Further uncertainty results from the fact that selenium can be randomly incorporated into pro- tein as selenocystine (59-61) and as selenotrisulfide (42, 62). Andreesen and Ljungdahl (56) showed that a selenium-contain- ing formate dehydrogenase can be obtained from Clostridium thermoaceficum grown in the presence of [7%e]selenite. However, the 75Se specific activity (counts per min per mg of protein) remained nearly constant during five purification steps (in one
1 The minimum possible radius of a globular particle, Rmin = [3M(o, + 6~8~)/4rrN]~‘~. Ratios of R,/R,i, up to about 1.25 define globular particles (23). R,/R,i, for formate dehydrogenase is 1.26 , __ ~.. --..
Relatively few enzymes have been reported to contain molyb- denum as a constituent. These are the xanthine oxidase (65), aldehyde oxidase (66), and sulfite oxidase (67) of mammalian tissues, xanthine dehydrogenase (68), and nitrogenase (69, 70) of bacteria, and the nitrate reductase of plants, fungi, algae, and bacteria (71, 72). With the identification of formate dehydro- genase as a molybdoprotein it appears that of the six known molybdenum-containing enzymes two may be found together in the membrane-bound complex which catalyzes the reduction of nitrate by formate (i.e. formate dehydrogenase and nitrate reductase). This fact explains the pleiotropy of some chlorate- resistant mutants of E. coli (i.e. ChlD mutants) which are thought to have a defective molybdenum processing system (73). Neither formate dehydrogenase nor nitrate reductase activities can be detected in the ChlD mutant unless high levels of molybdate (0.1 mM) are provided in the growth medium. Formate dehydro- genase and nitrate reductase are inhibited by cyanide which may indicate a functional role of molybdenum in these enzymes since cyanide has been shown to bind stoichiometrically to the molybdenum of molybdoenzymes (74) ; b-type cytochromes do not typically bind cyanide. The oxidized and reduced cytochrome b spectra of formate dehydrogenase and nitrate reductase are indistinguishable on the basis of their absorbance maxima2 (12). The heme of nitrate reductase would appear to be in a more labile configuration since heating the enzyme at pH 8.5 for 5
2 The absorbance maxima of reduced nitrate reductase reported (using LIZ = 608,000). in Ref. 12should be corrected to: 559,531, and 428nm.
6703
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6704
min results in a complete loss of the cytochrome spectrum and heme (12), while formate dehydrogenase after the same treat- ment retained all of its heme in a form which was still reducible by formate. Nitrate reductase (12) and formate dehydrogenase (this work) both have complex subunit structures apparently composed of three nonidentical polypeptide chains. The enzymes do not share a common polypeptide.
Formate dehydrogenase has widespread occurrence in animals, plants, yeast, and many bacteria (75) ; however, highly purified enzyme has been obtained from only two sources: P. oxalaticus (64) and C. thermoaceticum (76). The soluble enzyme of plants (77) and animals (78) use NADf as the physiological electron acceptor, while the soluble bacterial enzymes have been shown to be specific for NAD+ (64), NADP+ (76), or ferredoxin (79, 80). The particulate enzyme of Desulfovibirio vulgaris utilizes a c-type cytochrome as the natural acceptor (81).
In E. coli under conditions of nitrate respiration, nitrate is the ultimate acceptor of electrons from formate. Electron flow from formate to nitrate is probably mediated by a complex chain of electron carriers which are present in the two purified protein complexes, formate dehydrogenase and nitrate reductase, plus a lipid carrier (quinone) :
formate quinone nitrate HCOOH + dehydrogenase ____) reductase __) NO;
(Se,Mo,Fe,heme) (heme,Mo,Fe)
A functional role for these carriers and the intramolecular se- quence of electron flow remains to be shown for both of these enzymes. A number of lines of evidence indicate a role for the heme in this pathway. Maximal levels of formate dehydrogenase, cytochrome b, and nitrate reductase in anaerobically grown cells require the addition of molybdenum (3) and nitrate (3, 8) to the culture medium which implies a coordinate regulation of these components. Formate causes the reduction of cytochrome b in whole cells (48, 82), membrane preparations (83), and the re- constructed formate-nitrate reductase system (12). The reduced cytochrome b is reoxidized by the addition of excess nitrate in each case. Rapid kinetic studies would be helpful in determining if the rate of cytochrome b reduction is compatible with the rate of the over-all reaction (formate-nitrate reduction). Formate- nitrate reductase appears to require a quinone cofactor in addi- tion to the protein-bound electron carriers.
Itagaki (44) has reported that formate-nitrate reductase ac- tivity is lost in quinone-depleted membrane particles and may be partially restored by the addition of coenzyme Q (vitamin Kz was 38% as effective on a mole per mole basis). It was re- cently shown (12) that coenzyme Q6 stimulated formate-nitrate reductase activity 33.fold in a system containing only purified formate dehydrogenase and nitrate reductase. The site(s) of quinone action remains to be established.
The key components of the formate-nitrate reductase system of E. coli, formate dehydrogenase and nitrate reductase, are associated with the cell membrane where they may exist as a discrete complex. Evidence for the existence of such a complex was presented by Itagaki et al. (5) who obtained a soluble prepa- ration of formate dehydrogenase, cytochrome b, and nitrate re- ductase with each enriched IO-fold over membranes. Our evi- dence suggests that formate dehydrogenase and nitrate reductase may be tightly associated after their release from the membrane. During the purification of formate dehydrogenase we found that the pooled agarose eluates (Table 1) contains little protein that is not associated with either formate dehydrogenase or nitrate reductase (see Fig. 4). Formate dehydrogenase and nitrate re-
ductase were not separated on the basis of size or charge by a number of purification steps carried out in the absence of deter- gent. It is possible that the association of these enzymes in vitro is fortuitous and does not reflect the nature of the in vivo inter- action. It is not clear that formate dehydrogenase and nitrate reductase must interact in the membrane since electron transfer between the enzymes could be facilitated by a mobile quinone carrier.
Experiments with lysozyme-EDTA (4) indicated that about 20% of the cellular protein of E. coli is associated with the cell envelope fraction. Using the purification factors obtained from Table I, one can calculate that formate dehydrogenase and ni- trate reductase represent 3.3 and 17% of the protein, respec- tively, of the cell envelope. Both these enzymes are located on the cytoplasmic membrane which makes up about one-third of the protein of the cell envelope (84). Thus, 10% of the protein of the cytoplasmic membrane is present as formate dehydrogenase and 52% of the protein is present as nitrate reductaseP The large amount of protein of this organelle associated with the formate-nitrate reductase system indicates the importance of this system under anaerobic growth conditions (in the presence of nitrate). Other donors can provide electrons for nitrate re- duction in E. coli, for example, NADH, succinate, and lactate (82, 85) and these membrane-bound dehydrogenases may also form complexes with nitrate reductase. However, formate ap- pears to be the major electron donor for nitrate reduction during anaerobic growth in the presence of nitrate (3, 48, 85).
The availability of purified formate dehydrogenase and nitrate reductase and the knowledge of their composition will now per- mit experiments designed to study the regulation of synthesis of subunits and their assembly into a functional membrane complex.
REFERENCES
5.
6.
7. 8.
9.
10. 11. 12.
13.
14. 15.
1G. 17.
18.
19.
20.
PINSENT, J. (1954) Biochem. J. 67, lo-16 FUICUYAMA, T., AND ORDAL, E. J. (1965) J. Bacterial 90, 673-
680 LESTER, R. L., AND DnMoss, J. A. (1971) J. Bacferiol. 106,
1006-1014 EXOCH, H. G., lj~~ LESTER, R. L. (1972) J. Bacterial. 110, 103%
1040 IT.~G.~KI. E.. FUJIT~. T.. .~RTD S.~TO. It. (1962) J. Biochem. 62.
131-14i ’ ’ ’ I
LINNANE, A. W., .IND WRIGLEY, C. W. (1963) Biochim. Biophys. Acta 77, 408-418
SHUM, A. C., .\ND MURPHY, J. (1972) J. Bacterial. 110, 447-449 WIMPNNNY, J. W. T., AND COLE, J. -4. (1967) Biochim. Biophys.
Acta 148, 233-242 SHOWE, M. K., AND DxMoss, J. A. (1968) J. Bacterial. 96,
1305- 1313 TANIGUCHI, 8. (1961) 2. Allg. Mikrobiol. 1, 341-375 ENOCH, H. G., AND LESTIEIL, It. L. (1974) Fed. Proc. 33, 1577 ENOCH, II. G., AND LESTER, It. L. (1974) Biochem. Biophys.
Kes. Commun. 61, 1234- 1241 MARTIN, R. G., AND AMES, B. N. (1961) J. Biol. Chcm. 236,
1372-1379 SUND, H., AND WEINER, K. (1963) Biochem. 2. 337, 24-34 HISSTIN, S., FEINGOLD, D. S., AND SCHRBMM, M. (1955) Methods
Enzymol. 1, 231-257 LAICMMLI, U. K. (1970) iVature 227, 680-685 L.\E:MMLI, U. K., .&ND F.\vRE, M. (1973) J. Mol. Biol. 80, 575-
599 WE:~ER, K., AND OSDORN, M. (1969) J. Biol. Chem. 244, 4406-
4412 LAURENT, T. C., AND KILL.~NDER, J. (1964) J. Chromatogr. 14,
317-330 HIRS, C. H. W. (1967) Methods Enzymol. 11, 325-329
3 MacGregor et al. (49) reported that 15y0 of the protein in the cytoplasmic membrane was nitrate reductase.
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

6705
21. KLOTZ, I., AND D.~RNALL, D. (1969) Science 166, 127-128 22. HELENIUS, A., AND SIMONS, K. (1972) J. Biol. Chem. 247,
3656-3661 23. TANFORD, C., NOZ~KI, Y. REYNOLDS, J. A., AND M:~KINO, S.
(1974) Biochemistry 13, 2369-2376 24. ACKERS G. K., (1964) Biochemistry 3, 723-730 25. LESTER, R. L., AND SMITH, A. L. (1961) Biochim. Biophys. Acta
47, 475-496 26. LESTER, R. L., H~TEFI, Y., WIDMER, C., AND CR,~NF,, F. L.
(1959) Biochim. Biovhus. Acta 33. 169-185 27. L~~RY:O. H.. ROSEB~O;GH, N. J.,F,lnn, A. L., AND RANDALL.
R. J.‘(1951)‘J. Biol. Chem: 193, i65-27i 28. RIESKE. J. S. (1967) Methods Enzumol. 10.488-493 29. WILSON; D. F:, AND KING, T. E”. (1964) J. Biol. Chem. 239,
2683-2690 30. BRODIE, A. F. (1963) Methods Enzymol. 6, 295-308 31. JOHNSON, C. M., AND ARKLEY, T. H. (1954) Anal. Chem. 26,
572-574 32. DOEG, K. A., AND ZIEGLER, D. M. (1962) Arch. Biochem. Bio-
phys. 97, 37-40 33. FOGO, J. K., AND POPOU-SICY, M. (1949) Anal. Chem. 21.732-734 34. B.~RL~TT, G. It. (1959) J. Biol. Chem. 234,466-468 35. BLIGH, E. G., AND DYER, W. J. (1959) Can. J. Biochem. Physiol
37, 911-917 36. G~RE~~L, H. S. (1973) Anal. Biochem. 64, 319-324 37. SCHNEIDER. W. C. (1957) Methods Enzumol. 3.680-684 38. ASHWELL, 6. (1966) Methods Enzymoi. 8, 85-95 39. TALWALKAR, It. T., AND LESTER, R. L. (1973) Biochim. Bio-
phys. Acta 306, 412-421 40. HATEFI, Y., AND HANSTEIN, W. G. (1969) Proc. Natl. Acad.
Sci. 0. S: A. 62, 1129-1136 41. HOUSTON. L. L. (1971) Anal. Biochem. 44. 81-88 42. JENI~INS, ‘K. J. (i968)‘Can. J. Biochem. 46, 1417- 1425 43. WHITE, k., H.~NDL&, P., AND SMITH, E. L. (1964) Principles
of Biochemistru. D. 345. McGraw-Hill. New York 44. ITAGAICI, E. (1964j 3. Bidchem. 66, 432-445 45. COOIC, R. P., HALDANE, J. B. S., AND MAPSON, L. W. (1931)
Biochem. J. 26, 534-550 46. G.~LE, E. P. (1939) Biochem. J. 33, 1012-1027 47. IID.%, K., AND TANIGUCHI, S. (1959) J. Biochem. 46, 1041-1055 48. RUIZ-HERRERA, J., AND DEMOSS, J. A. (1969) J. Bacterial. 99,
720-729 49. MACGREGOR, C. H., SCHNAITMAN, C. A., NORM~YNSELL, D. E.,
AND HODGINS, M. G. (1974) J. Biol. Chem. 249, 5321-5327 50. OH, S.-H., GANTHF:R, H. E., .~ND HOEKSTRA, W. G. (1974)
Biochemistry 13, 1825-1829 51. FLOH~., L., G~~NZLER, W. A., AND SCHOCK, H. H. (1973) FEBS
Lett. 32, 132-134 52. TURNER, D. C., .~ND ST.IDTMAN, T. C. (1973) Arch. Biochem.
Biophys. 164, 366-381 53. ST.~DTMAN, T. (1974) Fed. Proc. 33, 1291 54. PEDER~ON, N. D., WH.INGER, P. D., WESWIG, P. H., AND
MUTH, 0. H. (1972) Bioinorg. Chem. 2, 33-45 55. WH.INGER, P. D., PEDERSON, N. D., WESWIG, P. H. (1973)
Biochem. Biophys. Res. Commun. 63, 1031-1035
56. ANDREESEN, 5. R., AND LJUNGDAHL, L. G. (1973) J. Bacterial. 116, 867-873
57. ANDREESEN, J. It., EL GH~ZZ.~WI, E., .IND GOTTSCH~LK, G. (1974) Arch. Mikrobiol. 96. 103-118
58. ST:\DT&N, T. C. (1974) Science 183, 915-922 59. WI~ISS, K. F., AYIZES, J. C., AND KRAFT, A. A. (1965) J. Bac-
teriol. 90, 857-862 60. Tuvrs, T., .%ND WILLIAMS, H. H. (1961) J. Biol. Chem. 236,
597-601 61. BLAU, M. (1961) Biochim. Biophys. Acta 49, 389-390 62. GANTHER, H. E., ‘ZND CORCOR~N, C. (1969) Biochemistry 8,
2557-2563 63. FLOH~, L., AND G~~NZLER, W. A. (1974) in Glutathicne, p. 132
(FlohB. L.. Benohr. H. Ch.. Sies. H.. Waller. H. D.. and Wended, A:, eds), Academic &ess,‘Ne& York ’ ’
64. H~~PNxI~, T., ,~ND TR~~UTWEIN, A. (1972) 2. Naturforsch. 27, 1075-1076
65. BRAY, R. C. (1963) in The Enzymes (Boyer, P. D., Lardy, H., and Myrblick, K., eds) Vol. VII, p. 533, Academic Press, New York
66. R.~J~GoP.~L.~N, K. V., AND HANDLER, P. (1968) in Biological Oxidation (Singer, T. P., ed) p. 301, Interscience Publishers, New York
67. COHEN, H. J., FRIDOVICH, I., AND R~J.~GoP.~L~N, K. V. (1971) J. Biol. Chem. 246, 374-382
68. SMITH, S. T., R‘~J,YGOPALAN, K. V., AND HANDLER, P. (1967) J. Biol. Chem. 242, 4108-4117
69. MORTE:NSON, L. E. (1966) Biochim. Biophys. Acta 127, 18-25 70. BULEN, W. A., ASD LECOMTE, J. R. (1966) Proc. Natl. Acad.
Sci. U. S. A. 66,979-986 71. BEEVERS. L.. AND H.IGEMAN. R. H. (19701 Annu. Rev. Plant
Physioi. 26, 495-522 ’ ’ ’ 72. Papers, W. J. (1973) Bacterial. Rev. 37, 405-452 73. GLASER, J. H., AND D~Moss, J. A. (1971) J. Bacterial. 108,
854-860 74. COUGHL.IN, M. P., RAJ~GOP~L.~N, K. V., AND HANDLER, P.
(1969) J. Biol. Chem. 244, 2658-2663 75. ENOCH, H. G. (1975) Ph.D. thesis, University of Kentucky 76. ANDREESEN, J. R., A&D LJUNGDBHL, L. G. (1974) J. Bacterial.
120, 6-14 77. DAVISON, D. (1951) Biochem. J. 49,520-526 78. ~~~~~~~~~ M. B., AND VENNESLAND, B. (1950) J. Biol. Chem.
186, 667-682 79. BRILL, W. J., WOLIN, E. A., AND WOLFF:, R. S. (1964) Science
144, 297-298 80. THAUER, R. K., FUCHS, G., AND JUNGERMANN, K. (1974) J.
Bacterial. 118, 758-760 81. YAGI, T. (1969) J. Biochem. 66, 473-478 82. COLE, J. A., AND WIMPENNY, J. W. (1968) Bicchim. Biophys.
Acta 162, 39-48 83. SATO, R., AND Ecanl1, F. (1949) Bull. Chem. Sot. Japan 22, 137 84. SCHNAITMAN, C. A. (1971) J. Bacterial. 108, 545-552 85. TANIGUCHI, S., AND ITAGAKI, E. (1960) Biochim. Biophys.
Acta 44, 263-279
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from

H G Enoch and R L Lesterfrom Escherichia coli.
The purification and properties of formate dehydrogenase and nitrate reductase
1975, 250:6693-6705.J. Biol. Chem.
http://www.jbc.org/content/250/17/6693Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/250/17/6693.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on July 15, 2018http://w
ww
.jbc.org/D
ownloaded from