the chemistry of high and low band gap -conjugated polymers · the chemistry of high and low band...
TRANSCRIPT

The chemistry of high and low band gap -conjugatedpolymersCitation for published version (APA):Mullekom, van, H. A. M. (2000). The chemistry of high and low band gap -conjugated polymers. Eindhoven:Technische Universiteit Eindhoven. https://doi.org/10.6100/IR530045
DOI:10.6100/IR530045
Document status and date:Published: 01/01/2000
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:
www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:
providing details and we will investigate your claim.
Download date: 28. Dec. 2019

?SP�.SPXT^_]d�ZQ�3TRS�LYO�7Zb�-LYO�2L[π�.ZYU`RL_PO�;ZWdXP]^

-7,96�;,20

?SP�.SPXT^_]d�ZQ�3TRS�LYO�7Zb�-LYO�2L[π�.ZYU`RL_PO�;ZWdXP]^
PROEFSCHRIFT
ter verkrijging van de graad van doctor aan de TechnischeUniversiteit Eindhoven, op gezag van de Rector Magnificus,prof.dr. M. Rem, voor een commissie aangewezen door hetCollege voor Promoties in het openbaar te verdedigen opmaandag 24 januari 2000 om 16.00 uur
door
Hubertus Antonius Maria van Mullekom
geboren te Helenaveen

Dit proefschrift is goedgekeurd door de promotoren:
prof.dr. E.W. Meijerenprof.dr. W.J. Feast
Copromotor:dr. J.A.J.M. Vekemans
CIP-DATA LIBRARY TECHNISCHE UNIVERSITEIT EINDHOVEN
Mullekom, H.A.M. van
The chemistry of high and low band gap conjugated polymers / by H.A.M. van Mullekom. -Eindhoven : Technische Universiteit Eindhoven, 2000. - Proefschrift.ISBN 90-386-2791-2NUGI 813Trefwoorden: polymeren; electrische eigenschappen / geleidende polymeren /π-geconjugeerde polymeren; synthese / lichtemitterende diodenSubject headings: polymers; electrical properties / conducting polymers /π-conjugated polymers; synthesis / light-emitting diodes

?LMWP�ZQ�.ZY_PY_^
�� -LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^ �
1.1 Introduction 11.1.1 Conjugated polymers: organic semiconductors 11.1.2 Electrically conducting polymers: Intrinsic vs. extrinsic conductors 31.1.3 Requirements for intrinsic conductors 4
1.2 Minimization of bond length alternation 51.2.1 Introduction 51.2.2 PITN and derivatives 61.2.3 Ladder polymers 81.2.4 Organic analogues to polysulfur nitride 91.2.5 Conclusions 10
1.3 Donor-acceptor systems 111.3.1 Introduction 121.3.2 Acceptor units based on cyano- or nitro substituents 121.3.3 Acceptor units with the electron-deficient atoms close to the backbone 161.3.4 Acceptor units with multiply fused pyrazine and thiadiazole rings 201.3.5 D-A copolymers based on squaraine with very small calculated band gaps 23
1.4 Band gap minimization vs. band gap engineering 251.4.1 Band gap tuning in polymers for Light Emitting Diodes 251.4.2 Influence of mesoscopic ordering on the band gap 28
1.5 Aim and scope of this thesis 29References 30
�� .Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR L[[WTPO _Z L 7Zb -LYO 2L[ � .ZYU`RL_PO ;ZWdXP]
2.1 Introduction 352.2 Design and retrosynthetic analysis 372.3 Synthetic investigations towards the Boc-protected precursor polymer 38
2.3.1 Optimization of the Stille-coupling conditions 392.3.2 Synthesis of the precursor polymer via route A: ABA-B copolymerization 392.3.3 Synthesis of the precursor polymer via route B: A-B homopolymerization 40
2.4 Synthesis and properties of the pyrrole/benzothiadiazole copolymer and its 42model compound2.4.1 Thermal deprotection of Boc-protected precursors 422.4.2 Evidence for hydrogen bonding: NMR and X-ray studies 432.4.3 Band gap and conductivity of the deprotected polymer 46
2.5 Comparison to known thiophene-based analogues 46

2.6 Conclusions 482.7 Experimental section 48References 50
�� -LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_PO π�.ZYU`RL_PO ;ZWdXP]^ �
3.1 Introduction 533.2 Synthesis of the co-oligomers via the Stille coupling methodology 54
3.2.1 Synthesis of pyrrole/2,1,3-benzothiadiazole co-oligomers 543.2.2 Synthesis of pyrrole/quinoxaline co-oligomers 563.2.3 Synthesis of thiophene/2,1,3-benzothiadiazole co-oligomers 57
3.3 Properties of the co-oligomers 583.3.1 UV/Vis spectroscopy 583.3.2 NMR spectroscopy 603.3.3 Cyclic voltammetry 60
3.4 Implications for the band gap engineering of D-A conjugated polymers 623.5 Conclusions 643.6 Experimental Section 65References 69
�� 4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^F���� H_STLOTLeZWP^ "�LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
4.1 Introduction 714.2 Design and retrosynthetic analysis 724.3 Improved synthesis of dithienylbenzobisthiadiazoles 74
4.3.1 Optimization of the dithienylanthracene synthesis 744.3.2 Improved synthesis of dithienylbenzobisthiadiazoles 774.3.3 Copolymerization of dilithiobithiophene with benzobisthiadiazoledione 78
4.4 Copolymerization of dilithiobithiophene with anthraquinone 794.4.1 Introduction 794.4.2 Precursor polymer route towards unsusbstituted poly(dithienylanhtracene)s 794.4.3 Precursor polymer route towards alkoxy-substituted poly- 81(dithienylanhtracene)s
4.5 Conclusions 864.6 Experimental section 87References 90
� , 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT� $�_STPYdWMPYeZMT^_STLOTLeZWP�^ bT_S 0c_]PXPWd 9L]]Zb -LYO 2L[^
5.1 Introduction 935.2 Monomer design 96

5.3 Synthesis and polymerization 975.3.1 Monomer synthesis 975.3.2 Polymerization 98
5.4 Characterization of polymerization products 1005.4.1 UV/Vis/NIR aborption spectroscopy 1025.4.2 MALDI/TOF mass spectrometry 1125.4.3 Acid-doping of poly-7 106
5.5 Polycondensation of other EDOT containing dihydroxycompounds 1085.6 Conclusions 1095.7 Experimental section 110References 112
!� .ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP 8LTY .SLTY QZ] �� -W`P 7TRS_ 0XT__TYR /TZOP^
6.1 Introduction 1156.2 Polymer and monomer design 1176.3 Poly-p-9,10-diphenylanthracene-vinylene PDPAV 119
6.3.1 Synthesis of 9,10-bis(4-formylphenyl)-anthracene monomers 1196.3.2 Synthesis and optical properties of alkoxy-substituted PDPAV 121
6.4 Poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene PDPAFV 1246.4.1 Alternative polymer design 1246.4.2 Synthesis of the fluorene comonomer 1256.4.3 Copolymerization 126
6.5 Conclusions 1286.6 Experimental Section 129References 132
>`XXL]d
>LXPYaL__TYR
.`]]TN`W`X AT_LP
7T^_ ZQ ;`MWTNL_TZY^
/LYVbZZ]O

-7,96 ;,20

.SL[_P]��
-LYO�2L[�.ZY_]ZW�TY�.ZYU`RL_PO�;ZWdXP]^%?ZbL]O^�4Y_]TY^TN�.ZYO`N_Z]^
����4Y_]ZO`N_TZY
Ultimately, conjugated polymers should combine the physical properties of polymers(low specific weight, processibility, solubility, tunable mechanical properties, flexibility etc.)with those of semiconductors to obtain unique and novel materials with numerous excitingapplications. Examples of such applications are large-area, flexible polymer Light EmittingDiodes (pLEDs) that can emit light in virtually any part of the visible spectrum, all-polymericField Effect Transistors (FETs) that give access to high-tech, but low-cost plastic electronicsand polymers with metallic conductivity for the simple fabrication of conductive films, e.g.for electromagnetic shielding or antistatic applications. Besides their attractive materialproperties, the power of conjugated polymers is to be found in the ease of manipulation withtheir chemical structure. This allows the fabrication of materials with tailor-made electronicand/or mechanical properties.1-7
����� .ZYU`RL_PO [ZWdXP]^% Z]RLYTN ^PXTNZYO`N_Z]^
Conjugated polymers are organic semiconductors, that with respect to electronic energylevels hardly differ from inorganic semiconductors. Both have their electrons organized inbands rather than in discrete levels and both have their ground state energy bands eithercompletely filled or completely empty.8,9 The band structure of a conjugated polymeroriginates from the interaction of the π-orbitals of the repeating units throughout the chain.This is exemplified in Figure 1 where the calculated (frontier) energy levels ofoligothiophenes with n = 1 - 4 and of polythiophene are shown as a function of oligomerlength.10 Addition of every new thiophene unit causes hybridization of the energy levelsyielding more and more levels until a point is reached at which there are bands rather thandiscrete levels. Interaction between the π-electrons of neighbouring molecules leads to athree-dimensional band structure.
Analogous to semiconductors, the highest occupied band (which originates from theHOMO of a single thiophene unit) is called the valence band, while the lowest unoccupiedband (originating from the LUMO of a single thiophene unit) is called the conduction band.The difference in energy Eg between these levels is called the band gap. Since π-conjugatedpolymers allow virtually endless manipulation of their chemical structure, control of the bandgap of these semiconductors is a research issue of ongoing interest. This “band gapengineering” may give the polymer its desired electrical and optical properties, and reductionof the band gap to approximately zero is expected to afford an intrinsically conductingpolymer.11-13

.SL[_P] �
�
Sn
3.0
0.0
-3.0
-6.0
-9.0
-12.0
HOMO
LUMO
Valence band
Conduction band
Eg
1 2 3 4 ∞n =
Energy [eV]
Figure 1. Calculated (frontier) energy levels of oligothiophenes with n = 1-4 and of polythiophene, where Eg =band gap; adapted from ref. 10
+ •
+ +
Ox Ox
+ +•
S S
S
S
S
S
S a
S S
S
S
S
S
S c
b
S S
S
S
S
S
S d
e
S
S
S
S
S
S
Sg
i
f
h
•
Ox
Ox
1a
1b
1c
1d
-1 e
-1 e"polaron"
"bipolaron"
"double polaron"
Ox Ox
Ox
Scheme 1. Structural changes in polythiophene upon doping with a suitable oxidant.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�
����� 0WPN_]TNLWWd NZYO`N_TYR [ZWdXP]^% TY_]TY^TN a^� Pc_]TY^TN NZYO`N_Z]^
Since a conjugated polymer is a semiconductor with a finite band gap, conversion into aconductor implies introduction of charges onto the polymer chain which can be accomplishedby various methods. The first method concerns the introduction of charges either by electron-removal (oxidation or p-doping) or -injection (reduction or n-doping). The major part ofconjugated polymers known today is built up of electron-releasing units, making them p-typesemiconductors which can be doped with oxidants like I2, FeCl3 etc. The structural changesthat occur in a conjugated polymer upon oxidation are illustrated in Scheme 1 forpolythiophene.14,15
The removal of one electron from the polythiophene chain (1a) produces a mobilecharge in the form of a radical cation (1b), also called a polaron. The positive charge tends toinduce local atomic displacements ("clothing with phonons"), leading to the polaronicbehaviour. Further oxidation can either convert the polaron into a spinless bipolaron (1c) orintroduce another polaron (1d). In either case, introduction of each positive charge also meansintroduction of a negatively charged counterion (Ox−).
N
N
N
N
H
H
H
H
n
N
N
N
N
H
H
n
-2 H-2 e
N
N
N
N
H
H
n
H
H+ 2 HX
N
N
N
N
H
H
n
H
H
X
2a
2b
2c
2d
insulator
insulator
conductor
conductorX
X X
Scheme 2. Acid doping in polyaniline.

.SL[_P] �
�
The second method consists of acid doping of conjugated polymers having a site thatcan be protonated, which process introduces charges in the main chain. The best knownexample is polyaniline16 (Scheme 2). The neutral leucoemeraldine form 2a can be oxidized tothe emeraldine base 2b without introduction of counterions. However, the emeraldine base 2bonly becomes conductive after treatment with a sufficiently strong acid (HX) whichprotonates the imine nitrogens and, at the same time, introduces a counterion (X-). Theconducting emeraldine 2c can also be represented by the mesomeric structure 2d, in which allphenyl rings are aromatic and radical cations are present on every second nitrogen atom. Thisdegenerate mesomerism is thought to account for a high charge-carrier mobility, and thushigh conductivity, but conformational factors like the crystallinity of polyaniline films alsoplay a crucial role.
In the above two examples, introduction of charge carriers is inevitably accompanied bythe introduction of counter-ions. It is defined here that extrinsically conducting polymers areπ-conjugated polymers that become conductive after doping, i.e. after the introduction ofcharged species that are delocalized along the conjugated main chain (charge carriers),accompanied by the introduction of counter-charged species that are not delocalized alongthe conjugated main chain. It is analogous to interstitial doping in inorganic semiconductors,in which case the counter-charge is also more or less localized on the interstitial species. Thedefinition covers all π-conjugated polymers that are made conductive either by means ofdoping with an oxidizing or reducing agent, or by means of acid doping. Furthermore, “self-doping” polymers like poly(carboxyethylpyrrole)17 and sulfonated polyaniline 3 are notconsidered intrinsically conducting polymers since the negative charge is not delocalizedalong the main chain.
N
N+
N+
N
H H
H Hn
SO3 SO3-
N+
N
N+
N n
SO3SO3 H H
H H
- -
3
-
In contrast, intrinsically conducting polymers are conducting π-conjugated polymerswhich do not need additional doping and are characterized by electrically neutral conjugatedsystems in which some π-electron bands are only partially filled. The character of theirconductivity may range from "hopping" to metallic, depending on the degree of overlap of theπ-orbitals of neighboring molecules.
����� =P\`T]PXPY_^ QZ] TY_]TY^TN NZYO`N_Z]^
To determine the requirements to be met for an intrinsically conducting polymersatisfying the above definition, we have to take a look at metals. These intrinsic conductorsowe their conductivity to the valence band that is only partially filled up to the Fermi-level.To imitate such a partially filled band with a semiconductor, its band gap should be zero or

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
close to zero. Two major approaches towards reduction of the band gap in conjugatedpolymers will be discussed here, guided by some typical examples and in the context ofreaching a zero band gap: i) minimization of bond-length alternation along the main chainand ii) alternation of electron-donor and -acceptors in the main chain.
The first part consists of only a brief discussion about the well-known approachestowards low band gap systems, such as polyisothianaphthenes (PITNs) and ladder polymers,because these approaches are already the subject of several reviews on low band gapsystems.11,18-21 The second part consists of a more elaborate presentation of the variousmolecular designs that are applied for obtaining low band gap conjugated donor-acceptorsystems, including their syntheses. However, many more donor-acceptor conjugated polymersexist, in which the primary goal is not reduction of the band gap but tuning of the band gap,the key issue when certain (non-linear) optical properties are desired. For the latter class ofpolymers other papers may be consulted.22-29
����8TYTXTeL_TZY�ZQ�MZYO�WPYR_S�LW_P]YL_TZY�
����� 4Y_]ZO`N_TZY
The simplest representative of a conjugated polymer is trans-polyacetylene 4 (Figure 2).Its first successful synthesis was described already in 1974,30 however, access topolyacetylene with a well-defined structure and morphology was provided by the “Durham”precursor polymer route.31-34 Polyacetylene would be a metallic conductor if the distancebetween all carbon atoms was identical. In this situation, the double bonds are reallydelocalized along the polymer chain, which situation is depicted in the left-hand side ofFigure 2.35,36 Finding a band gap as the energy difference between a “bonding” ground stateand an “anti-bonding” excited state is impossible due to the equivalence (or degeneracy) −foran infinite chain− of both structures, resulting in a metal-like half-filled band.
anti-bonding
bonding
Eg
n4
Figure 2. Band gap formation by localization of double bonds in trans-polyacetylene 4.

.SL[_P] �
!
However, the equidistant linear chain structure is unstable towards a structuraldeformation of alternating shorter double and longer single bonds, and the result is a finiteband gap Eg. This “bond length alternation” is due to a gain in electronic energy thatovercompensates the loss of "elastic" energy, and is called a Peierls effect.37 Minimizing thebond length alternation along the backbone of a conjugated polymer is an important guidelinein band gap-reduction of which the practical approaches are discussed below.
����� ;4?9 LYO OP]TaL_TaP^
One of the reasons for the high band gap (∼2 eV) of polythiophene 1 is the smallcontribution of the energetically unfavourable quinoid structure to the ground state of thepolymer, resulting in a large single bond character of the thiophene-thiophene linkage andhence a large bond-length alternation. Increasing the double-bond character of the thiophene-thiophene linkage can be accomplished by making the quinoidal structure energetically morefavorable. This is the case in polyisothianaphthene (PITN) 5.38
Upon going from the aromatic to the quinoid state, the loss of aromaticity is thesmallest in 5 because its fused 6-membered ring gains aromaticity. This results in a band gapfor 5 of roughly 1 eV, one full electronvolt lower than that of polythiophene 1.39
Sn S
n
Sn S
n
1
5
aromatic quinoid
Since the discovery of PITN, many papers have appeared on a variety of chemical andelectrochemical syntheses,40-43 as well as on other polymers representing structural variationson the isothianaphthene unit. These include poly(dialkoxyisothianaphthene)s 644
poly(dialkylthienopyrazine)s 7,45 and various fused thienothiophenes 8 - 10.46-48
S
S
S
n
S
S
n
S
NN
n
RR
S
OO
n
RR
S
S
n
R
6 7 8 9 10
An elegant example of applying the above principle for band gap reduction in systemsother than PITN is polyindenofluorene (PIF) 1149 (Scheme 3).

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
"
n
n
Cl
Cl
Cl
Cl
a)
11
Scheme 3. Synthesis of polyindenofluorene 11. Reagents and conditions: a) Co2(CO)8, chlorobenzene.
Although many synthetic routes towards this polymer were investigated, only thedehalogenation condensation of the tetrachloro monomer with various low-valent transitionmetal complexes such as Co2(CO)8 led to high molecular weights (scheme 3). From theabsorption spectra, a band gap of 1.55 eV was estimated, which is quite high compared toPITN. This is explained by the mesomerism in scheme 3. The aromaticity of the centralbenzene ring is lost on going from the structure with a large double bond character to thestructure with a large single-bond character between the units. Moreover, geometrycalculations on model compounds of 11 showed that the torsion angle between theconsecutive aryl units could be as high as 33°.
An approach somewhat different from that of PITN and derivatives was introduced withpolymers 1250-55 and 13.56-61 Unfortunately, polymer 12 with n = m possesses a degenerateground state and, therefore, suffers from the same limitations as polyacetylene 4.
SSn
R R
m XYX
n
m
o
Q
12
13
R = H, Phenyl
X = O, S, N-R, CH=CHY = O, S, SO, SO2, N-R, CH=CH
Q = S, CH=CH,
~~
Though by varying the length and ratio of the quinoidal and aromatic blocks in 12 and13 the band gap can be tuned in between the values for all-quinoidal and all-aromaticconjugated chains, this is not equal to cancelling the bond-length alternation since thereremains always location of double and single bonds. Nevertheless, polymers with the basicstructure of 12 (R = aryl), composed of variable n and m blocks have been prepared, showinglow band gaps. Many monomers containing the basic structure of 13 have also been prepared,yet polymerization of these compounds proved to be difficult.

.SL[_P] �
#
����� 7LOOP] [ZWdXP]^
Another way of cancelling the bond-length alternation is reducing or eliminating thestructural deformations that lead to the localization of alternating double and single bondsalong the conjugated main-chain. This would mean the construction of ladder polymers,11,62-65
of which the best-known example is polyacene 14. This polymer can be regarded as two fusedtrans-polyacetylene chains.
n
n
n
14
Indeed, calculations have shown that polyacene would be a metallic conductor.66
However, due to their difficult synthesis,67 no well-defined examples of these systems areknown. Not all ladder polymers are necessarily zero band gap materials, i.e. polyphenanthrene15, which can be regarded as two fused cis-polyacetylene chains, has a calculated gap of 4-5eV due to the large difference in energy between the aromatic and quinoid structures.18,68,69
n
n
15
Furthermore, conjugated ladder polymers like the poly-p-phenylene derivative 1670-72
exist, which are partially linked with saturated bonds with the purpose of planarization of theconsecutive units. Although these polymers show a reduced band gap compared to their non-planar counterparts such as poly-p-phenylene, it is evident that these systems suffer frombond length alternation as well, and should, therefore, not be classified in the group of “fully”conjugated ladder polymers of type 14 and 15.
16
R
R
R
R
R'
R'
H
H
n

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
$
����� :]RLYTN LYLWZR`P^ _Z [ZWd^`WQ`] YT_]TOP
An elegant way to avoid bond-length alternation is found in the inorganic polymerpolysulfur nitride, (SN)x, the only polymer today which shows metallic conductivity and evensuperconductivity below 0.26 K.73 In the electronic structure of polysulfur nitride, 4mesomers can be recognized in which every second sulfur atom bears a positive charge, andevery second nitrogen atom bears a negative charge (Scheme 4).
S N S Nn
S N S Nn
S N S Nn
S N S Nn
S N S Nn
Scheme 4. Mesomeric structures in polysulfur nitride.
Thus, localization of double bonds in polysulfur nitride is accompanied by localizationof positive and negative charges, which is energetically unfavorable. The ground state ofpolysulfur nitride can, therefore, be regarded as an intermediate of the four mesomers inScheme 4, in which the charges are delocalized along the chain. This means that in theground state, there is no bond-length alternation and, therefore, a zero band gap is expected.In Scheme 4, a neutral structure is drawn for (SN)x in which there is localization of doublebonds, however, in this case every second sulfur atom violates the octet rule.
However, it should also be noted that interconversion between mesomeric structures ismore easy in (SN)x than in “ordinary” conjugated polymers like polyacetylene.Interconversion between the two possible mesomeric structures of polyacetylene requires alldouble bonds of a single PA chain to migrate simultaneously. Although the two mesomericstructures are of the same energy, such a process is expected to have a huge activation energy.From scheme 4, it follows that the migration of double bonds in (SN)x can occur at therepeating unit level, i.e. without migration of double bonds in the rest of the chain. Therefore,this process has a much lower activation energy, and can result in real mesomery, whichimplies a lower band gap.
Unfortunately, (SN)x is not very stable, it is unprocessible and difficult to prepare on alarge scale, which hampers the application of this material. It is therefore worthwhile to see ifthe electronic structure of (SN)x can be mimicked in −processible− conjugated polymers. Thisis exemplified by polymer 17 (Scheme 5).

.SL[_P] �
��
NNS Sn
NNS Sn
H H
"- H2"
NNS Sn
2 Ox
Ox Ox
17
18
19
Scheme 5. Oxidation of polymer 18 towards an organic analogue of polysulfur nitride 17, and possible over-oxidation to the extrinsical conductor 19.
A straightforward route towards polymer 17 implies the oxidation of the known sulfuranalogue of polyaniline 18.74 However, such an oxidation could not be carried outsuccessfully, probably due to the fact that only 1 out of 4 phenyl rings has to be oxidized, aprocess which is very difficult to control. A more likely process to happen is overoxidation tothe extrinsical conductor 19. The similar oxidation of pyrrole-sulfur oligomers towards anoxidation state that is comparable to that of polysulfur nitride or polymer 17 was also foundto be difficult.75
���� .ZYNW`^TZY^
Although, compared to polythiophene, the band gap of PITN and the derivativespresented above are greatly reduced, it would be extremely difficult to reach values down to 0eV. For PITN 5, there is a finite band gap for the fully aromatic structure (large bond lengthalternation due to the 100 % single bond character of the thiophene-thiophene linkage) and afinite band gap for the fully quinoid structure (large bond length alternation due to the 100 %double bond character of the thiophene-thiophene linkage). The minimum band gap is foundat the point where the all-aromatic and all-quinoid contributions to the ground state arebalanced in such a way that the bond-length alternation is canceled39 and the obvious way offinding a polymer which ground state is exactly located at this point is by making chemicalmodifications. Although many structures have been examined theoretically in this respect,76-83
the actual realization of them would be an enormous task.In contrast, ladder-type polyacenes and polysulfurnitride analogues represent polymeric
architectures which are very promising in terms of a vanishingly small band gap, albeit theirsynthetic accessibility is very poor. Fortunately, another approach towards low band gapconjugated polymers exists which combines synthetic accessibility with very low band gaps:the donor-acceptor approach.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
����/ZYZ]�LNNP[_Z]�^d^_PX^
It was shown with PITN 5 that reduction of bond-length alternation by increasing thedouble-bond character between the repeating units of a conjugated polymer, results in adecreased band gap. The driving force for such a process in PITN is the gain in aromaticity ofthe fused benzene ring. The interaction between a strong electron-donor (D) and a strongelectron-acceptor (A) may also give rise to an increased double bond character between theseunits, since they can accommodate the charges that are associated with such a mesomerism(D−A ↔ D+=A−). Hence, a conjugated polymer with an alternating sequence of theappropriate donor- and acceptor-units in the main-chain may have a decreased band gap.
NH2
NH2
S
NS
N
R
R
S
S
S
R
N
NR
R
O
OH OH
O
+
O
O
O
OHOH
S
NS
N
O
O
O
R
Rn
N
N n
O
O
H
H
N
N
O
O
R
Rn
S
NS
N
R
R n
O
O
S
S
SO
O
n
R
N
N
O
O
O
R
Rn
20
21
22
23
24
25
26
27
28
29
30
31
a)
Scheme 6. Preparation of polysquaraines 26 - 29 and polycroconaines 30 and 31. Reagents and conditions: a)mineral acid or strong base, -H2O, alcohol.

.SL[_P] �
��
����� 4Y_]ZO`N_TZY% [ZWd^\`L]LTYP^ LYO [ZWdN]ZNZYLTYP^
The donor-acceptor (D-A) repeating unit strategy was introduced with polymers 26-3184,85 (Scheme 6). The condensation copolymerization of various donor molecules 20-23(R=H or R=Alkyl) with either squaric acid 24 or croconic acid 25 in a higher saturatedalcohol solution with a catalytic amount of either a mineral acid or a strong base undercontinuous removal of water yielded copolymers 26-31. GPC of copolymer 29 with R =heptyl or dodecyl shows molecular weights up to 15 kD (vs. polystyrene). The copolymer 31shows a band gap of 0.45 eV (absorption edge).
The low values were initially explained by the fact that the conjugated main chain ofthese polymers, with their alternation of electron-withdrawing and -releasing units, representsthe one-dimensional analogue of a so-called n-i-p-i semiconductor structure. In thesesemiconductors, the valence- and conduction band are curved by space-charge effects, whichleads to a diminished band gap energy when the spatial alternation of the levels is taken intoaccount. However, calculations have shown that the hybridization of the energy levels of thedonor and the acceptor, particularly the high-lying HOMO of the donor fragment and the low-lying LUMO of the acceptor fragment, yield a D-A monomer with an unusually low HOMO-LUMO separation86,87 (Figure 3). Further hybridization upon chain extension then convergesto the low band gaps.
HOMO
LUMO
Energy
HOMO
LUMO
D AD-A
Figure 3. Hybridization of the energy levels of a donor (D) and acceptor (A) fragment leads to a D-A monomerwith an unusually low HOMO-LUMO energy separation.
For this reason, band gap reduction by means of an alternating donor-acceptor repeatingunit strategy is primarily concerned with the combination of very strong donor and acceptorunits, of which various attempts will be discussed here. In the following part, the variousnarrow band gap donor-acceptor conjugated polymers are subdivided by the nature of theirelectron-accepting unit. For the electron-donating part, thiophene or pyrrole with varioussubstitution patterns often represent the best choice since these are very electron-rich subunitsthat allow numerous chemical transformations.
����� ,NNP[_Z]�`YT_^ ML^PO ZY NdLYZ� Z] YT_]Z�^`M^_T_`PY_^
The most obvious choice for the design of an electron-withdrawing subunit would be anaryl unit substituted with a cyano- or a nitro-group, since the latter two are among the mostwidespread electron withdrawing groups in organic chemistry. By applying the Knoevenagel

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
condensation, of 2-cyanomethylthiophene 32 with thiophene- or furancarboxaldehydes 33a-c,monomer 34a-c could be prepared in good yields88 (Scheme 7).
SCN
X
R
O XS
R
NC
+X
S
R
n
NC
a X = S, R = Hb X = S, R = Mec X = O, R = H
32 33a-c 34a-c 35a-c
a) b)
Scheme 7. Preparation of monomers 34a-c and polymers 35a-c. Reagents and conditions: a) t-BuOK, EtOH, b)electrochemical polymerization, NBu4PF6.
Electrochemical polymerization of these monomers yielded polymers 35a-c, of whichthe polymer 35c was claimed to feature a band gap of 0.6 eV versus 1.5 and 1.4 eV forpolymers 35a and 35b, respectively. However, the electronic absorption spectrum from whichthis low band gap was derived shows a shoulder at high wavelength which may indicateresidual doping and, therefore, obscures the accurate determination of the band gap. This issupported by the physical data of polymers 39a-b89 (Scheme 8).
SCN
RRX
O
O O
XS
NC
O O
RR
+X
S n
NC
O O
R R
a R = H
b R = OO
36a-b 37 38a-b 39a-b
a) b)
Scheme 8. Preparation of monomers 38a-b and polymers 39a-b. Reagents and conditions: a) t-BuOK, EtOH, b)electrochemical polymerization, NBu4ClO4.
Polymers 39a-b were synthesized analogously to polymers 35a-c. Electrochemicaldetermination of the band gap resulted in values of 1.3 for 39a and 1.0 eV for 39b. Since the3,4-ethylenedioxythiophene unit is among the strongest thiophene-based electron-donors, andthe acceptor-unit in 39a-b is unchanged compared to 35a-c, the band gap value of 0.6 eV for35c is doubtful.
Conjugated polymers in which the electron-donating group is pyrrole and/or thiophene,and the electron-accepting group is a cyano-substituted aryl unit, are depicted in scheme 9.90-
92 The polymers were prepared by electrochemical oxidation (acetonitrile/NBu4ClO4) of theircorresponding monomers. The band gaps of polymers 40-43 were estimated at 2.2, 2.7, 1.6and 2.0 eV respectively. Although these values are much lower compared to the identicalpolymers without a cyano-group −except for polymer 41, which suffers from steric hindrancealong the pyrrole-phenyl bond−, the band gaps are still quite high.

.SL[_P] �
��
NN
H
H
CN
n
N
HCN
SN
H CN
n
N
H
SN
H
CN CN
n
NN
H
H
n
CN
40 41
42 43
Scheme 9. Pyrrole-containing polymers 40 - 43.
An example of a conjugated polymer containing an electron-accepting group differingfrom a cyano-substituted aryl unit is depicted in scheme 10.93-95
S S BrBr
NO2O2N
S
NHBocBocHN
S
NHBocBocHN
SnBu3Bu3Sn
SS n
NHBocBocHN
NO2O2N
SS n
NO2O2N
NH2NH2
44
45 46
47
48
a)
b)
c)
d)
e)
Scheme 10. Preparation of polymers 47 and 48. Reagents and conditions: a) Br2; then HNO3/H2SO4, b) Sn,HCl; then Boc2O, Et3N, c) LDA, Bu3SnCl. d) Pd2(dba)3, CuI, AsPh3, THF, e) H+; then Na2CO3.
SS n
N+
O
ON+
O
O
NN
H
H
H
H
SS n
N+
N+
N+
N
O
O O
O
H
H
H
H
SS n
N+
N+
N+
N+
O
O O
O
H
H
H
H
Scheme 11. Rigidification of 48 by donor-acceptor interactions.
Starting with thiophene, successive bromination and nitration gives comonomer 44.Reduction of the nitro-groups and subsequent Boc-protection of the amino-groups givesintermediate 45, which can be converted into the distannyl comonomer 46 by doublelithiation with LDA and subsequent quenching with Bu3SnCl. The Stille copolymerization of44 and 46 then gives the precursor polymer 47, which can be deprotected to the D-A

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�
conjugated copolymer 48 by the action of acid. The solution and solid state optical band gapsfor 48 are 1.4 and 1.1 eV, respectively. It is suggested that this diminished band gaporiginates from rigidification of the conjugated backbone due to the occurrence of mesomericstructures (Scheme 11).
When the band gaps that have been found for the donor-acceptor conjugated polymers(in which a cyano- or nitro-substituted aryl unit is the acceptor) presented above are reviewed,only the value measured for polymer 35c is below 1 eV, a value which is probably due toresidual doping. Since, particularly in the case of scheme 11, very strong electron-donor and -acceptor units are applied, these band gap values fall short of expectations.
To find an explanation for this behavior, we must reconsider the mechanism of band-formation in a conjugated polymer. As mentioned in the introduction, the HOMO and LUMOlevels of the repeating unit disperse into a valence- and conduction band upon chainextension. The degree with which this happens, also called the bandwidth, is represented byWL and WH (Figure 4). The magnitudes of WL and WH are strongly dependent on the degree ofoverlap between the atomic orbitals on the coupling positions of the consecutive aryl units.The maximal values for WL and WH are only reached in the case of an unobstructed overlap.Deviation from this ideal situation can occur when i) steric hindrance forces the consecutivearyl units out of plane, or ii) when the size of the atomic orbitals (AO) at the couplingpositions is diminished. Because in the above examples of donor-acceptor systems stericalhindrance is only present in polymer 41 (scheme 9), the second phenomenon may wellaccount for the disappointing band gaps of polymers 35, 39-43, 48.
HOMO
LUMO
WH
WL
Eg
Monomer Polymer
Figure 4. Dispersion magnitudes WH and WL of the monomer HOMO and LUMO levels, respectively, uponchain-extension.
The comparison of the calculated frontier orbitals of bithiophene 49 with those of(dicyanomethylidenecyclopenta)dithiophene 50 (Figure 5) supports this assumption.96,97 FromFigure 5, it follows that placing intensely electron-withdrawing groups on an aryl unit can,besides lowering the LUMO level, dramatically diminish the size of the AO’s on the couplingpositions (in this case the 2,2’-positions of bithiophene) in the LUMO level. Therefore, themagnitude of WL may be much smaller and, consequently, the band gap may be much largerthan expected in these kinds of polymers.98 This explains why cyano- and nitro-substitutedaryl units are not very efficient in lowering the band gap of donor-acceptor conjugatedpolymers. The cyano- and nitro-group are so strongly electron-withdrawing that in the LUMOof such a unit, the largest orbital density is found outside the main-chain conjugation path.Nevertheless, a band gap of < 1 eV was found for the corresponding polymer of 50, because

.SL[_P] �
�!
the HOMO-LUMO separation in 50 is so narrow (λmax = 576 nm) that it compensates for thesmall LUMO AO coefficients on the 2,2’ positions.
SS SS
NN
HOMO
LUMO
49 50
Figure 5. Calculated frontier orbitals of bithiophene 49 and (dicyanomethylidenecyclopenta)dithiophene 50.
����� ,NNP[_Z]�`YT_^ bT_S _SP PWPN_]ZY�OPQTNTPY_ L_ZX^ NWZ^P _Z _SP NZYU`RL_PO MLNVMZYP
From the above it is clear that finding electron-accepting subunits with large AOcoefficients on the coupling units represents a crucial issue in designing donor-acceptorconjugated polymers with a low band gap. The most obvious approach is selection of an arylunit which bears one or more electronegative atoms in the ring, close to the couplingpositions. The simplest representative of this class is the pyridine ring, which contains anelectron-deficient imine nitrogen.
SBrN
Br
SeBrN
Br
S
NN
Br Br
S
N
n
Se
N
n
S
NN
n
NiLm
NiLm
NiLm
51
52
53
54
55
56
Scheme 12. Synthesis of pyridine-containing polymers 54 - 56.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�"
Via a polycondensation reaction of monomers 51 - 53 using a zerovalent nickelcomplex (NiLm, a mixture of bis(1,5-cyclooctadiene)nickel(0) Ni(COD)2 and a neutralligand, usually bipyridine), polymers 54 - 56 could be prepared in high yields99-101 (Scheme12).
However, the optical data of the polymers are not very encouraging in terms of a lowband gap since the λmax of polymers 54 and 55 is centered around 490 nm, while for polymer56 it is observed around 440 nm. This may be a consequence of the low molecular weightsthat are obtained for these polymers, but the more plausible interpretation is that the pyridineunit acts only as a weak electron-acceptor (E½(red) = -2.15 V vs. mercury pool). Whenincreasing the electronegativity of the acceptor-unit by taking pyrazine instead of pyridine,synthetic problems are encountered because of the difficulty to functionalize (e.g.brominating) and polymerizing pyrazine.102
NS
N NS
N
Br Br
NH2NH2
Br BrBr Br
NN
RR'
NS
N
ArAr
NN
RR'
ArAr
57 58
5960
a)
b)
c)
Scheme 13. Synthesis of quinoxaline and 2,1,3-benzothiadiazole derived building blocks. Reagents andconditions: a) Br2, AcOH, b) Zn, HCl, c) RCOCOR’.
A solution for this problem is fusion of the pyrazine ring onto a unit that can be easilyfunctionalized, e.g. a thiophene or a phenyl ring. The synthetic scheme towards the latterbuilding blocks is depicted in Scheme 13. Bromination of 2,1,3-benzothiadiazole 57(prepared from o-phenylenediamine and SOCl2) gives 4,7-dibromo-2,1,3-benzothiadiazole58103 which can be reduced to dibromo-o-phenylenediamine 59104 by the action of a reducingagent like Zn or SnCl2 in HCl. Reaction of this diamine with a suitable diketone yieldsdibromoquinoxaline 60. This building block serves as the starting-point to construct donor-acceptor co-trimers by means of organometallic aryl-aryl coupling reactions like the Grignardor Stille coupling.105,106 A remarkable spin-off from this reaction scheme is the 2,1,3-benzothiadiazole unit: because it contains two imine nitrogens, it can also serve as anefficient electron-withdrawing unit.
With these precursors, various donor-acceptor polymers containing quinoxaline or2,1,3-benzothiadiazole as the acceptor and thiophene as the donor have been prepared(Scheme 14).

.SL[_P] �
�#
NN
Br Br
RR
S SnMe3Me3SnS
NN
RR
n +
Br Br
NS
N NS
N
SS
NS
N
SSn
NN
Br Br
RR
SS
NN
RR
SSn
NN
RR
61 60 62
58 63 64
60 65 66
a)
b) c)
d) c)
Scheme 14. Synthesis of polymers 62, 64 and 66. Reagents and conditions: a) Pd(PPh3)4, DMF, b) ThSnMe3,Pd(PPh3)2Cl2, THF, c) electrochemical polymerization, d) ThSnMe3, Pd(PPh3)2Cl2, THF.
Polymer 62, where R represents an alkyl or phenyl group, is prepared via the Stillecopolymerization of distannylthiophene 61 with dibromoquinoxaline 60 using a Pd(0)catalyst.99,101,107,108 The Stille coupling, using a Pd(II) species as the catalyst, also gave accessto monomers 63 and 65 (R = CH3 or C6H13) which were electrochemically polymerized topolymers 64 and 66.109,110 The band gaps of polymers 62, 64 and 66 were found to be 1.7, 1.2and 1.4 eV, respectively. If the decrease of the band gap is related to the structure, it followsthat the benzothiadiazole unit is a stronger electron-acceptor than the quinoxaline unit, whichobservation is supported by semi-empirical calculations on the HOMO-LUMO energy levelsof quinoxaline and 2,1,3-benzothiadiazole.110 However, all of these values are still above 1eV, which may be due to the fact that in quinoxaline and 2,1,3-benzothiadiazole the electron-accepting part is again outside the conjugated chain which may adversely affect the LUMOAO-coefficients.
A better performance in the latter field may be expected from acceptor-units in whichthe pyrazine or thiadiazole unit is not fused onto a phenyl ring, but on a thiophene ring e.g.thieno[3,4-b]pyrazine 68 (Scheme 15). In these units, the coupling positions are part of a 5-membered ring and flanked by an electron-donating sulfur atom which is beneficial for thesize of the AO coefficients.111 The synthetic accessibility of such systems is outlined inScheme 15, and starts with dibromodinitrothiophene 44 (See scheme 10). Two routes can befollowed from 44 towards the desired co-oligomers.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�$
S BrBr
NO2O2N
S
NH2NH2
S
NN
RR'
S
NS
N
Br Br S
NS
N
Ar Ar
S ArAr
NO2O2N
S
NH2NH2
ArAr S ArAr
NN
RR'
route A
route B
44 67 68
69
a) b)
c)
d)
e)
f)
g)
d)
Scheme 15. Preparation of thienopyrazine 68 and analogues. Reagents: a) Sn, HCl, b) RCOCOR’, c) NBS; thenPhNSO; then Me3SiCl, d) ArSnMe3, Pd(PPh3)2Cl2, e) Sn, HCl, f) PhNSO; then Me3SiCl, g) RCOCOR’.
Direct reduction of the nitro groups of compound 44 with Sn or SnCl2 in HCl yields3,4-diaminothiophene 67112, under concomitant removal of the bromo groups (Route A).Reaction of this highly unstable compound with a suitable diketone or thionylaniline thenyields thieno[3,4-b]pyrazine 68113 or (dibromo-)thieno[3,4-c]thiadiazole 69.114 Note thatdirect reduction of 44 results in the unstable intermediate 67 which has lost the bromo groups,which is inconvenient when a subsequent arylation step has to be taken. The more convenientroute B circumvents this problem by direct arylation of 44 by means of a Grignard or Stillecross-coupling reaction, followed by reduction of the nitro groups and subsequent ring-closure of the amines towards a thienopyrazine or thienothiadiazole based unit.
Following the above mentioned synthetic strategy, polymers 70 - 73 were prepared viaelectrochemical polymerization of the corresponding monomers. The monomer needed forpolymer 70 was prepared both via route A115 and route B116 in Scheme 15, while themonomers for polymers 71-73 were exclusively prepared via route B.116,117 Unfortunately,only the band gap of polymer 70 was determined: 0.9 eV, the first value below 1 eV for adonor-acceptor conjugated polymer. The absorption maxima of the monomers correspondingto polymers 71, 72, and 73 (529, 712 and 616 nm, respectively) compared to the absorptionmaximum of the monomer for polymer 70 (618 nm) suggest, however, that these arepromising low band gap materials as well. Furthermore, the combination of pyrrole as thedonor and thienothiadiazole as the acceptor gives the most promising values.

.SL[_P] �
��
S S
S
NS
N
n
N N
S
NS
N
n
HH
N N
S n
NNH H
S S
S n
NN
70 71
72 73
An example of a thieno[3,4-b]pyrazine based polymer with a substituted thiophene unitas the donor is the polymer 75118 (Scheme 16), which is electrochemically synthesized frommonomer 74 (prepared using route B in scheme 15) . It is claimed to have a band gap of 0.36eV, a value derived from the solid state absorption spectrum of 75 which shows a λmax = 1430nm (0.86 eV) .
S
NN
S
O O
C6H13H13C6
S
NN
S
O O
C6H13H13C6
n
74 75Scheme 16. Electrochemical preparation of polymer 75 from monomer 74.
Unfortunately, the solid state absorption spectrum of 75 features a shoulder between1.0 and 1.5 eV, the region where the neutral absorption of the analogous polymer 70 is found.This may imply that polymer 75 is not dedoped adequately and the band gap value of 0.36 eVis not reliable. This example, together with that of polymer 35c, illustrates that deriving bandgap values −and intrinsic conductivities− from electropolymerized conjugated polymers isvery intricate and may lead to incorrect values,119 if dedoping is not performed adequately.
����� 0WPN_]ZY�LNNP[_Z]^ bT_S X`W_T[Wd Q`^PO [d]LeTYP LYO _STLOTLeZWP ]TYR^
The electron-withdrawing power of quinoxaline or 2,1,3-benzothiadiazole may befurther increased by fusion of another pyrazine or thiadiazole ring onto the vacant sites of thephenyl ring to yield pyrazinoquinoxaline 76, thiadiazoloquinoxaline 77 andbenzobis(thiadiazole) 78120 (Figure 6). Semi-empirical calculations have shown thatparticularly benzobisthiadiazole 78 is an extremely electron-deficient subunit121 which isattributed to the hypervalent sulfur atom. The occurrence of mesomeric structures is alsoshown in Figure 6.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
NN
N N
NS
N
NS
N
NS
N
N N
HOMO LUMO
NS
N
NS
N
76 77 78
Figure 6. Pyrazinoquinoxaline 76, thiadiazoloquinoxaline 77 and benzobisthiadiazole 78 with approximatefrontier orbitals.
NS
N
BrBr
NO2O2N
NS
N
SS
NO2O2N
NS
N
SS
NH2NH2
SS
NH2NH2
NH2NH2
SS
NN
N N
NS
N
SS
N N
NS
N
SS
NS
N
NS
N
SS
NS
N
n
NS
N
SS
N N
n SS
NN
N N
n
79 80
81 82
83 84 85
86 87 88
a)
b) c)
d) e) f)
g) g) g)
Scheme 17. Synthesis of polymers 86 - 88. Reagents: a) ThSnBu3, Pd(PPh3)2Cl2, b) Fe, AcOH, c) Zn, AcOH, d)PhNSO, Me3SiCl, e) glyoxal. f) diacetyl, g) electrochemical polymerization.
From the approximate HOMO and LUMO AO-coefficients (right hand side in Figure 6)it can be concluded that, though diminished with respect to the HOMO, the LUMO AO-coefficients on the coupling positions are still quite considerable. Together with the

.SL[_P] �
��
observation that donor-acceptor conjugated polymers containing a thiadiazole based acceptorunit give low band gap values, this justifies to consider it as a very promising subunit.
The synthesis of monomers containing one of the units 76 - 78 is outlined in Scheme17. Starting from 5,6-dinitro-4,7-dibromobenzo-2,1,3-thiadiazole 79 (prepared from nitrationof 4,7-dibromobenzo-2,1,3-thiadiazole122), a Stille coupling with 2-trimethylstannylthiophenefurnished the dinitro intermediate 80. Reduction of this compound could be carried out eitherwith iron powder to yield diamine 81, or with zinc powder to yield tetra-amine 82. Thediamine 81 could be reacted with either thionylaniline to yield benzobisthiadiazole containingco-trimer 83, or glyoxal to yield the thiadiazoloquinoxaline containing co-trimer 84. Finally,tetra-amine 82 was reacted twice with diacetyl to yield the pyrazinoquinoxaline containingco-trimer 85. The corresponding polymers 86 - 88 were prepared electrochemically from theirmonomers and showed band gaps of 0.5, 0.7 and 0.9 eV, respectively.109,110,123 With this,polymer 86 enters the band gap region of below 0.5 eV. The logical sequel would besubstitution of the thiophene units in co-trimer 83 by pyrrole units to yield even a lower bandgap. The polymer corresponding to the N-methylpyrrole derivative of 83 has been preparedbut showed an increased band gap of 0.6 eV, presumably due to the steric hindrance of themethyl groups.
Extrapolating the acceptor-design presented before, even better results would beobtained by expansion of the thiophene-based acceptor units like thieno[3,4-b]pyrazine.However, since no free sites are available on thiophene, the only way to expand this acceptoris substitution on the pyrazine ring, preferably with a thiadiazole ring. The monomer 91which contains the thiadiazolo-thienopyrazine subunit has been prepared116 (Scheme 18).
S S
S
NH2NH2
S S
S
NHNH
NS
N
S S
S
NN
NS
N
S S
S
NN
NS
N
n
89 90
92 91
a)
b) c)
d)
Scheme 18. Synthesis of polymer 92. Reagents: a) dimethyl oxaldiimide; then SOCl2, b) NiO2, c) hydroquinone,d) electrochemical polymerization.
Reaction of the diamine 89 (prepared according to Scheme 15) withdimethyloxaldiimide, followed by ring-closure with SOCl2, yielded the thienopiperazinederivative 90. This precursor could be oxidized to the thienopyrazine containing monomer 91

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
by the action of NiO2. Remarkably, this monomer could be reduced back to thethienopiperazine derivative 90 with hydroquinone, which makes this a redox-switchingsystem. The thienopyrazine containing monomer 91 was electrochemically polymerized toyield polymer 92 which featured, after adequate dedoping, a band gap of Eg = 0.3 eV.
As shown before, the band gap of this kind of donor-acceptor polymers with pyrrole asthe electron-donating unit is always lower than that of their thiophene-containing analogues,which would make such a pyrrole-containing polymer the first donor-acceptor conjugatedpolymer to approach the magical value of Eg ~ 0 eV. Based on the synthetic route depicted inScheme 19, the thienopiperazine compound 93 was prepared124 (Scheme 19).
N N
S
NHNH
NS
N
H H
N N
S
NN
NS
N
H H
N N
S
NN
NS
N
n
H H
93 94 95
a)
b)
c)
Scheme 19. Preparation of polymer 95. Reagents: a) NiO2, b) hydroquinone, c) electrochemical polymerization.
Oxidation of this monomer with NiO2 yielded the thienopyrazine containing monomer94 which could not be isolated because it is only stable in solution. However, since theoxidation process was very clean, a solution of pure 94 could be obtained by filtration of thenickel salts. The absorption maximum was found at 1345 nm, an extremely high value forsuch a “small” organic molecule. Electrochemical polymerization of 94 from this solutionyielded polymer 95. Electrochemical determination of the band gap was not possible, sincethere was no potential gap between n- and p-doping peaks. From this fact, it was concludedthat the band gap of polymer 95 is zero. Unfortunately, no optical data were presented tosupport this hypothesis. Furthermore, it was stated that dedoping of this electrochemicallyprepared polymer was not possible due to the zero band gap, which leaves the possibility ofresidual doping open.
���� /−, NZ[ZWdXP]^ ML^PO ZY _SP ^\`L]LTYP `YT_ bT_S aP]d ^XLWW NLWN`WL_PO MLYO RL[^
Reviewing the development in band gap reduction of donor-acceptor conjugatedpolymers, the polysquaraine based polymers with which the approach was introduced are stillamong the lowest band gap polymers known today. Unfortunately, only little work has beendone on polysquaraines containing different donor units, one example being the pyrrolederivatives 96a-d125-129 which are prepared by a simple condensation reaction between theappropriate pyrrole and squaric acid, a reaction known already for a long time130,131 (scheme20).

.SL[_P] �
��
OHOH
OON
R
a: R = Hb: R = C12H25
c: R = C11H22OHd: R = (CH2)3SO3Na
N
O
O R
n +
96
Scheme 20: Polycondensation towards pyrrole - squarene polymers 96a-d.
Unfortunately, the band gaps of polymers 96a-d are in the range of 1 eV, adisappointing value regarding results for the donor-acceptor polymers containing pyrroledescribed earlier. It seems, therefore, that the evolution of the band gap in these polymers isdifferent from what has been described for “neutral” polymers.
Calculations on the polysquaraines of Scheme 6 have shown that their low band gapsoriginate from the narrow HOMO-LUMO energy separation, typical for donor-acceptorfragments, since the calculated band gaps are in good agreement with the experimentalones86,87 (when the hybridization is taken into account that takes place upon chain extension).However, a striking feature for the squaraine-containing polymers discussed in these studies,is that upon hybridization not only the HOMO but also the LUMO shifts upward in energy bysimilar amounts. Based on this, it was predicted that if one would be able to construct apolymer in which, upon chain-extension, the HOMO levels were subject to hybridization,whereas the LUMO level was “pinned” at the originally low value of the fragment, one wouldget an extremely narrow band gap. The “pinning” of the LUMO level may be realized byannihilating the overlap of these orbitals throughout the polymer chain e.g. by making thisinteraction symmetry-forbidden. This is exemplified for polymers 97 - 98 in which thesquaraine units are linked via an amine group, as represented in Figure 7.
NO
O
H
n
Squarene
HOMO
LUMO
pz
Nitrogen
Energy
O
O
n N
H
97
98
Figure 7. “Pinning” of the LUMO level at its originally low value by introducing nitrogen atoms in the main-chain of polysquaraine copolymers.
The interaction between the HOMO of squaraine and the pz orbital of nitrogen results in ahybridization upon chain-extension. However, the interaction between the nitrogen pz orbitaland the LUMO of squaraine is forbidden for symmetry reasons which “pins” the LUMO at itsoriginal level. The calculated band gap of the polymer 97 is 0.5 eV, whereas the calculated

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�
band gap of the polymer 98 is even as low as 0.2 eV. Only very few attempts towards thisclass of systems are known, two of them being the polycarbazole squaraine derivatives 99132
and 100.133 Unfortunately, polymers 99 and 100 were insoluble black powders, which couldhardly be characterized, for which reason no band gap values were reported.
N
n
O
O
N
n
O
O
N
99 100
����-LYO�RL[�XTYTXTeL_TZY�a^��MLYO�RL[�PYRTYPP]TYR
The above examples show that a dramatic decrease in the band gap of conjugatedpolymers can be obtained by the application of two concepts: cancellation of bond-lengthalternation (PITN and derivatives, ladder polymers) and the donor-acceptor repeating unitstrategy. However, many other factors, besides the main-chain chemical constitution, caninfluence the band gap: the nature of (solubilizing) side-chains, the conformation of the main-chain and the occurrence of mesoscopic ordering phenomena (explained in section 1.4.2).Well-known conjugated polymers like poly-p-phenylene vinylene (PPV) and polythiophene(PT) may offer more insight into this matter, since many different substituted PPV and PTderivatives are known,134,135 which are the subject of detailed studies towards conformationalas well as ordering phenomena.136-139 The various effects of these additional factors can beexplained by regarding the band gap engineering of conjugated polymers, that is tuning of theband gap to obtain certain desired (optical) properties.
����� -LYO RL[ _`YTYR TY [ZWdXP]^ QZ] 7TRS_ 0XT__TYR /TZOP^
In the field of polymer light emitting diodes (LEDs),140-142 tuning of the band gap is animportant issue since it determines the emission color. An example is the influence of variousside-chains on the (photo- and electro-) luminescence of substituted polythiophenes 100 - 103(Figure 8).143 The luminescence of this set of polymers covers nearly the whole visiblespectrum.
A trend in Figure 8 is that with increasing size of the solubilizing group, there is a largerdeviation from coplanarity of the subsequent thiophene units. This causes a diminishedextended conjugation and, hence, a hypsochromic shift in the luminescence spectra, while theapplication of a phenyl ring in the solubilizing group enlarges the conjugated systems causinga bathochromic shift in the luminescence spectra. This example shows that control of theconjugation length is a powerful tool for the band gap engineering of conjugated polymers.
Control over the conjugation length can also be achieved by making block copolymers,consisting of conjugated fragments of a discrete length, linked together in a weakly- or non-
.SL[_P] �
�!
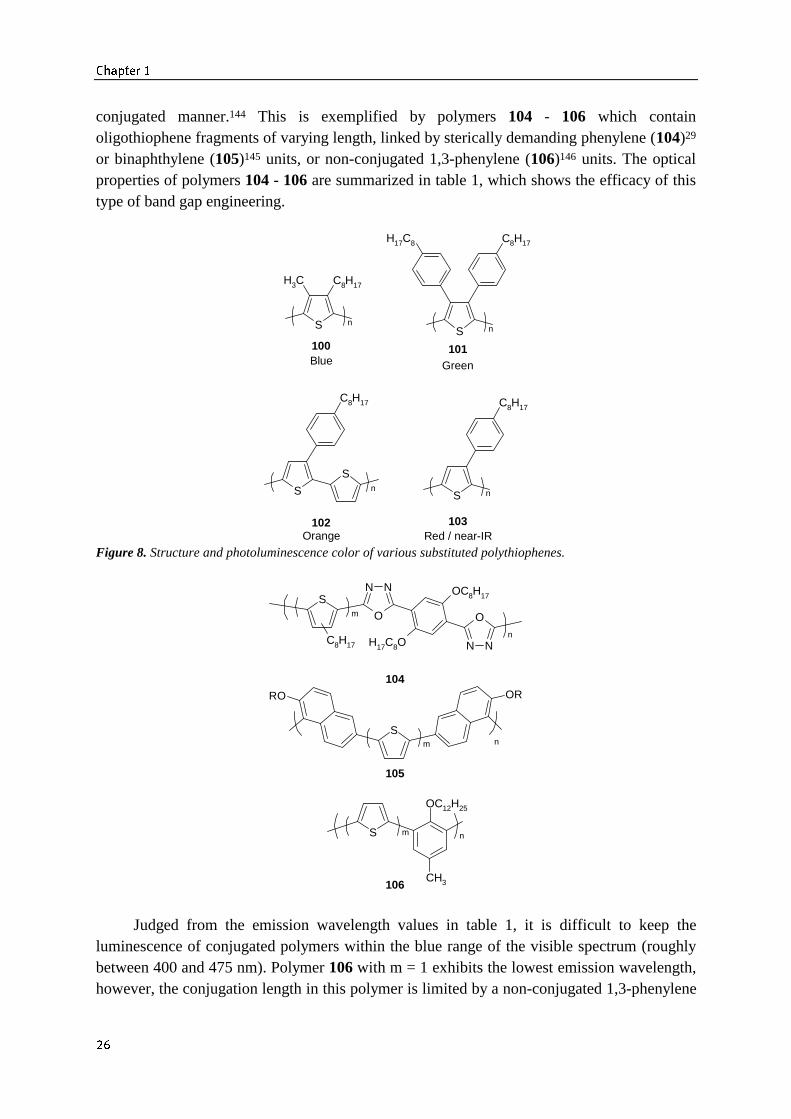
conjugated manner.144 This is exemplified by polymers 104 - 106 which containoligothiophene fragments of varying length, linked by sterically demanding phenylene (104)29
or binaphthylene (105)145 units, or non-conjugated 1,3-phenylene (106)146 units. The opticalproperties of polymers 104 - 106 are summarized in table 1, which shows the efficacy of thistype of band gap engineering.
S n
CH3 C8H17
S n
C8H17H17C8
S n
C8H17
S
S n
C8H17
100 101
102 103
Blue Green
Orange Red / near-IR
Figure 8. Structure and photoluminescence color of various substituted polythiophenes.
NN
O
S
N N
O
n
m
OC8H17
H17C8OC8H17
Sm
n
ORRO
S n
m
OC12H25
CH3
104
105
106
Judged from the emission wavelength values in table 1, it is difficult to keep theluminescence of conjugated polymers within the blue range of the visible spectrum (roughlybetween 400 and 475 nm). Polymer 106 with m = 1 exhibits the lowest emission wavelength,however, the conjugation length in this polymer is limited by a non-conjugated 1,3-phenylene

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�"
“kink” linkage. The disadvantage of reducing the conjugation in this drastic way is a decreasein semiconducting properties.147-149 Therefore, the development of a blue-luminescentconjugated polymer is a difficult enterprise, since two opposite effects must be balancedproperly.150-152
Polymer nr. of thiophene units (m)
Absorptionλmax [nm]
Emissionλmax [nm]
1 420 489104 2 441 530
3 461 579
1 368 475105 2 406 498
4 440 549
1 310 408106 2 390 492
3 406 5264 421 534
Table 1. Optical properties of polymers 104 - 106.
An interesting structural approach towards such systems is the incorporation ofanthracene units in the main chain such as copolymers 107153 and 108.154 The stericallydemanding anthracene unit causes a deviation from coplanarity in the conjugated main-chain,which limits the conjugation length. Furthermore, anthracene derivatives are known for theirhigh photoluminescence quantum yields.155
n
C7H15
C7H15 n
107 108
The interest in derivatives of anthracene is not only due to its fluorescence in or near theblue region of the visible spectrum. Anthracene/thiophene co-oligomers have shown their useas efficient energy-transfer sections in light-harvesting systems.156-158 The unique optical andchemical behavior of anthracene derivatives has also attracted attention in various otherbranches of chemistry.159-164
In conclusion, the band gap of a conjugated polymer can be tuned by limiting theconjugation length to a certain degree, e.g. by the introduction of side-chains or main-chainsubunits with a varying degree of sterical crowding. However, the electronic effects of suchsubstitutions are also of great influence on the band gap, which implies that band gapengineering following this approach may be a matter of trial and error. Band gap engineeringby making block copolymers of a fragment with a discrete conjugation length and a non-

.SL[_P] �
�#
conjugated “spacer” fragment is much more straightforward. This may, however, bedetrimental for its semiconducting properties and/or stability.
����� 4YQW`PYNP ZQ XP^Z^NZ[TN Z]OP]TYR ZY _SP MLYO RL[
The effect of mesoscopic ordering processes, such as the occurrence of crystallinedomains (Figure 9), is of great influence on the physical properties of conjugated polymers, asillustrated by polyaniline: When the emeraldine base of polyaniline 2c/2d (scheme 2) isdoped with strong acids such as p-toluenesulfonic acid, a maximum conductivity of about 5 Scm-1 is obtained.165 However, Smith et al. found that, when spincasting the emeraldine base(doped with camphorsulfonic acid) from m-cresol, it possessed an increased conductivity of200 S cm-1.166 This increase is ascribed to the action of m-cresol, which inducesconformational changes. It has been observed that the camphorsulfonic acid dopedpolyaniline, when spincast from m-cresol, has a higher crystallinity and conductivity than inthe absence of m-cresol.167-169
a) b) c)Figure 9. Schematic drawing of possible mesoscopic ordering phenomena in (conjugated) polymers: a)Amorphous polymer, b) chain-packing in a mesoscopic crystal, c) amorphous and crystalline regions in thesolid phase (e.g. thin films) of the polymer.
The effects of mesoscopic ordering on the band gap of conjugated polymers are lesswell-understood. However, there are apparent effects of such processes on the opticalproperties of conjugated polymers. It is known that upon annealing thin films ofpoly(dialkylfluorene)s, excimer formation occurs, giving rise to a bathochromic shift in theelectroluminescence spectra.170,171 Furthermore, conjugated polymers bearing chiral side-chains can show an aggregation behavior that gives rise to large effects in their circulardichroism (CD) spectra.136-139,172,173
The effect of mesoscopic ordering in low band gap conjugated polymers has not beenstudied in detail yet. The increased conductivity in polyaniline by an increased ordering couldof course also be important for an eventual intrinsical conductor. Additionally, a beneficialeffect of mesoscopic ordering may be found in the stability of low band gap conjugatedpolymers. Conjugated polymers with an extremely low band gap feature a low-lying valenceband, which means that they may be unstable under ambient (air) conditions. Slowing downthe degradation processes by the occurrence of ordering such as chain-packing may help tostabilize these polymers (kinetic stabilization).

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
�$
�� �,TX�LYO�^NZ[P�ZQ�_ST^�_SP^T^
The examples in this introductory chapter show that, in view of band gap minimization,the donor-acceptor repeating unit strategy is presently the most successful approach. Severalexamples of this class of conjugated polymers show band gaps significantly lower than 0.5eV. However, little or no information is accessible about the band gap engineering of thesesystems. Examples are the influence of both donor- and acceptor-unit and the presence ofsecondary interactions like internal hydrogen bonding on the band gap. Moreover, thesynthetic methodologies towards the most promising donor-acceptor conjugated systemsimply inefficient multistep syntheses and need, therefore, drastic improvement. At the sametime, band gap engineering in the reverse direction, i.e. approaches towards high band gapconjugated polymers, often rely on limiting the conjugation length in such a drastic way thatthe main-chain conjugation is lost. The examples of conjugated polymers that feature asufficiently high band gap to exhibit blue luminescence are limited. The aim of this thesis isto prepare conjugated oligomers and polymers that feature both low and high band gaps viaestablished, improved and even novel synthetic methodologies, not only to increase theaccessibility of these systems, but to gain understanding of the various parameters thatinfluence their band gap as well.
The first two following chapters of this thesis deal with the synthesis (via the well-known Pd-catalyzed Stille cross-coupling reaction) and properties of donor-acceptorconjugated oligomers and polymers based on thiophene, pyrrole, quinoxaline and 2,1,3-benzothiadiazole. In chapter 2, the alternating copolymer of pyrrole and 2,1,3-benzothiadiazole is discussed, which represents the first example of a low band gap (1.1 eV)conjugated polymer with a ladder-like structure based on intramolecular hydrogen bonding.Planarization by consequence of the intramolecular hydrogen bond, as proven by studies on amodel compound, is thought to account for the lower band gap of the pyrrole-basedcopolymer compared to the already described analogous thiophene-based copolymers. Inchapter 3, three series of alternating donor-acceptor substituted co-oligomers with differentchain-lengths, consisting of pyrrole or thiophene as the electron-releasing unit andquinoxaline or 2,1,3-benzothiadiazole as the electron-withdrawing unit, are discussed. Thedifferent incremental bathochromic shift of λmax upon chain elongation of the three series ofoligomers compared to homo-oligomers of thiophene and pyrrole is used as a tool in the bandgap engineering of donor-acceptor substituted π-conjugated polymers.
The basis of chapters 4, 5 and 6 is laid by the known synthesis of 9,10-dithienylanthracene. In chapter 4, adaptation of this reaction into an efficient two-stepsynthesis of 4,8-di(thien-2-yl)benzobis[1,2,5]thiadiazoles is presented. Although in principlepossible, this approach did not give access to poly(4,8-dithienylbenzobis[1,2,5]thiadiazole)s.However, the closely related copolymerization of 5,5’-dilithio-2,2’-bithiophene withanthraquinone is introduced as an easy access to well-defined, unsubstituted poly(9,10-dithienylanthracene)s via a soluble, non-conjugated precursor polymer. In chapter 5, a facile,non-oxidative polycondensation towards solution processible, low band gap conjugatedpolymers based on benzobis[1,2,5]thiadiazole and thiophene, featuring band gaps down to 0.3

.SL[_P] �
��
eV, is presented. “Acid-doping” of one of these polymers afforded easy access to thin filmswith a stable conductivity of about 1 S cm-1. The synthetic procedures that were developed inchapter 4 for the preparation of anthracene-containing conjugated systems proved to be usefulin chapter 6. In this chapter, a simple and efficient 3-step procedure towards 9,10-bis(4-formylphenyl)anthracene was developed, which gives facile access to luminescent conjugatedpolymers containing 9,10-diphenylanthracene units in the main-chain, such as the novelalkoxy-substituted poly-p-9,10-diphenylanthracene-vinylene PDPAV and poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene PDPAFV. Studies on model compoundsrevealed some requirements for blue-luminescence in these diphenylanthracene-containingconjugated polymers, an important issue in the design of polymers for blue light-emittingdiodes.
=PQP]PYNP^
(1) MacDiarmid, A. G.; Heeger, A. J. Synth. Met. 1978, 1, 1013.(2) Wegner, G. Angew. Chem. Int. Ed. Engl. 1981, 20, 361.(3) Greene, R. L.; Street, G. B. Science 1984, 226, 651.(4) Bredas, J. L.; Street, G. B. Acc. Chem. Res. 1985, 18, 309.(5) Potember, R. S. Polymer 1987, 28, 574.(6) Stenger-Smith, J. D. Prog. Polym. Sci. 1998, 23, 57.(7) Roncali, J. J. Mater. Chem. 1999, 9, 1875.(8) Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A.; Elsenbaumer, R. L.; Reynolds, J. R., Eds.;
Marcel Dekker: New York, 1998.(9) Organic Conductive Molecules and Polymers; Nalwa, H. S., Ed.; John Wiley & Sons: Chichester,
1997.(10) Salzner, U.; Lagowski, J. B.; Pickup, P. G.; Poirier, P. A. Synth. Met. 1998, 96, 177.(11) Roncali, J. Chem. Rev. 1997, 97, 173.(12) Higgins, S. J. Chem. Soc. Rev. 1997, 26, 247.(13) Bäuerle, P. Adv. Mater. 1993, 5, 879.(14) van Haare, J. A. E. H.; Groenendaal, L.; Peerlings, H. W. I.; Havinga, E. E.; Vekemans, J. A. J. M.;
Janssen, R. A. J.; Meijer, E. W. Chem. Mater. 1995, 7, 1984.(15) Bäuerle, P.; Segelbacher, U.; Maier, A.; Mehring, M. J. Am. Chem. Soc. 1993, 115, 10217.(16) Trivedi, D. C. In Organic Conductive Molecules and Polymers; Nalwa, H. S., Ed.; John Wiley & Sons:
Chichester, 1997; Vol. 2.(17) Wang, R.-S.; Wang, L.-M.; Su, Z.-M.; Fu, Y.-J. Synth. Met. 1995, 69, 511.(18) Tour, J. M. Adv. Mater. 1994, 6, 190.(19) Scherf, U. In Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A., Elsenbaumer, R. L.,
Reynolds, J. R., Eds.; Marcel Dekker: New York, 1998.(20) Pomerantz, M. In Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A., Elsenbaumer, R. L.,
Reynolds, J. R., Eds.; Marcel Dekker: New York, 1998.(21) Kaeriyama, K. In Handbook of Organic Conductive Molecules and Polymers; Nalwa, H. S., Ed.; John
Wiley & Sons: Chichester, 1997; Vol. 2.(22) Demanze, F.; Yassar, A.; Garnier, F. Macromolecules 1996, 29, 4267.(23) Demanze, F.; Yassar, A.; Garnier, F. Synth. Met. 1996, 78, 143.(24) Sotzing, G. A.; Reddinger, J. L.; Reynolds, J. R. Synth. Met. 1997, 84, 199.(25) Reynolds, J. R.; Kumar, A.; Reddinger, J. L.; Sankaran, B.; Sapp, S. A.; Sotzing, G. A. Synth. Met.
1997, 85, 1295.(26) Klärner, G.; Former, C.; Martin, K.; Räder, J.; Müllen, K. Macromolecules 1998, 31, 3571.(27) Francke, V.; Mangel, T.; Müllen, K. Macromolecules 1998, 31, 2447.(28) Zhang, X.; Shetty, A. S.; Jenehke, S. A. Acta Polym. 1998, 49, 52.(29) Yu, W.-L.; Meng, H.; Pei, J.; Lai, Y.-H.; Chua, S.-J.; Huang, W. Chem. Commun. 1998, 1957.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
(30) Ito, T.; Shirakawa, H.; Ikeda, S. J. Polym. Sci., Polym. Chem. 1974, 12, 11.(31) Edwards, J. H.; Feast, W. J. Polymer 1980, 21, 595.(32) Edwards, J. H.; Feast, W. J.; Bott, D. C. Polymer 1984, 25, 395.(33) Feast, W. J.; Parker, D.; Winter, J. N.; Bott, D. C.; Walker, N. S. In Electronic Properties of Polymers
and Related Compounds; Kuzmany, H., Mehring, M., Roth, S., Eds.; Springer: Heidelberg, 1985; Vol.63.
(34) Curran, S.; Stark-Hauser, A.; Roth, S. In Handbook of Organic Conductive Molecules and Polymers;Nalwa, H. S., Ed.; John Wiley & Sons: Chichester, 1997; Vol. 2.
(35) Albright, T. A.; Burdett, J. K.; Whangbo, M.-H. In Orbital Interactions in Chemistry; John Wiley &Sons: New York, 1985.
(36) Tolbert, L. M. Acc. Chem. Res. 1992, 25, 561.(37) Peierls, R. E. Quantum Theory of Solids; Oxford University Press: London, 1956.(38) Wudl, F.; Kobayashi, N.; Heeger, A. J. J. Org. Chem. 1984, 49, 3381.(39) Brédas, J. L.; Heeger, A. J.; Wudl, F. J. Chem. Phys. 1986, 85, 4673.(40) van Asselt, R.; Hoogmartens, I.; Vanderzande, D.; Gelan, J.; Froehling, P. E.; Aussems, M.; Aagaard,
O.; Schellekens, R. Synth. Met. 1995, 74, 65.(41) Chen, S.-A.; Lee, C.-C. Pure & Appl. Chem. 1995, 67, 1983.(42) Paulussen, H.; Vanderzande, D.; Gelan, J. Synth. Met. 1997, 84, 415.(43) Paulussen, H.; Ottenbourgs, B.; Vanderzande, D.; Adriaensens, P.; Gelan, J. Polymer 1997, 38, 5221.(44) Ikenoue, Y.; Wudl, F.; Heeger, A. J. Synth. Met. 1991, 40, 1.(45) Pomerantz, M.; Chaloner-Gill, B.; Harding, L. O.; Tseng, J. J.; Pomerantz, W. J. Synth. Met. 1993, 55-
57, 960.(46) Arbizzani, C.; Catellani, M.; Grazia Cerroni, M.; Mastragostoni, M. Synth. Met. 1997, 84, 249.(47) Pomerantz, M.; Xiaomin, G. Synth. Met. 1997, 84, 243.(48) Inaoka, S.; Collard, D. M. Synth. Met. 1997, 84, 193.(49) Reisch, H.; Wiesler, U.; Scherf, U.; Tuytuylkov, N. Macromolecules 1996, 29, 8204.(50) Braunling, H.; Becker, R.; Blochl, G. Synth. Met. 1991, 41-43, 1539.(51) Braunling, H.; Becker, R.; Blochl, G. Synth. Met. 1993, 55-57, 833.(52) Chen, W.-C.; Jenehke, S. A. Macromolecules 1995, 28, 454.(53) Chen, W.-C.; Jenehke, S. A. Macromolecules 1995, 28, 465.(54) Goto, H.; Akagi, K.; Shirakawa, H. Synth. Met. 1997, 84.(55) Aota, H. A.; Reikan, T.; Matsumoto, A.; Kamachi, M. Chem. Lett. 1997, 527.(56) Hanack, M.; Schmid, U.; Rohrig, U.; Toussaint, J.-M.; Adant, C.; Brédas, J. L. Chem. Ber. 1993, 126,
1487.(57) Hanack, M.; Schmid, U.; Echinger, S.; Teichert, F.; Hieber, J. Synthesis 1993, 634.(58) Echinger, S.; Schmid, U.; Teichert, F.; Hieber, J.; Ritter, H.; Hanack, M. Synth. Met. 1993, 61, 163.(59) Echinger, S.; Schmid, U.; Hieber, J.; Hanack, M. Synth. Met. 1995, 69, 695.(60) Hanack, M.; Dottinger, S. Synth. Met. 1996, 79, 43.(61) Schlick, U.; Teichert, F.; Hanack, M. Synth. Met. 1998, 92, 75.(62) Brédas, J. L.; Baughman, R. H. J. Polymer Sci., Polymer Lett. 1983, 21, 475.(63) Brédas, J. L.; Thémans, B.; André, J. M. J. Chem. Phys. 1983, 78, 6137.(64) Müllen, K.; Scherf, U. Makromol. Chem., Macromol. Symp. 1993, 69, 23.(65) Scherf, U. J. Mater. Chem. 1999, 9, 1853.(66) Kertesz, M. Macromolecules 1995, 28, 1475.(67) Ruud, C. J.; Wang, C.; Baker, G. L. Synth. Met. 1997, 84, 363.(68) Bakhshi, A. K. Mater. Sci. Eng. C3 1995, 249.(69) Yao, Y.; Lamba, J. J. S.; Tour, J. M. J. Am. Chem. Soc. 1998, 120, 2805.(70) Scherf, U.; Müllen, K. Makromol. Chem., Rapid Commun. 1991, 12, 489.(71) Scherf, U.; Müllen, K. Synthesis 1992, 23.(72) Scherf, U.; Müllen, K. Macromolecules 1992, 25, 3546.(73) Banister, A. J.; Gorrell, I. B. Adv. Mater. 1998, 10, 1415.(74) Wang, L. X.; Soczkaguth, T.; Havinga, E. E.; Müllen, K. Angew. Chem. Int. Ed. Engl. 1996, 35, 1495.(75) Groenendaal, L.; Pieterse, K.; Vekemans, J. A. J. M.; Meijer, E. W. Synth. Commun. 1997, 27, 257.(76) Lee, Y. S.; Kertesz, M.; Elsenbaumer, R. L. Chem. Mater. 1990, 2, 526.(77) Kurti, J.; Surjan, P. R.; Kertesz, M. J. Am. Chem. Soc. 1991, 113, 9865.(78) Kurti, J.; Surjan, P. R.; Kertesz, M. Synth. Met. 1992, 49-50, 537.

.SL[_P] �
��
(79) Quattrocchi, C.; Lazzaroni, R.; Brédas, J. L.; Zamboni, R.; Taliani, C. Macromolecules 1993, 26, 1260.(80) Toussaint, J. M.; Brédas, J. L. Macromolecules 1993, 26, 5240.(81) Bakhshi, A. K. J. Mol. Struct. (Theochem) 1996, 361, 259.(82) Hong, S. Y.; Song, J. M. J. Phys. Chem. B 1997, 101, 10248.(83) Hong, S. Y.; Song, J. M. Synth. Met. 1997, 85, 1113.(84) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Polym. Bull. 1992, 29, 119.(85) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Synth. Met. 1993, 55-57, 299.(86) Brocks, G.; Tol, A. J. Phys. Chem. 1996, 100, 1838.(87) Brocks, G.; Tol, A. Synth. Met. 1996, 76, 213.(88) Ho, H. A.; Brisset, H.; Frére, P.; Roncali, J. J. Chem. Soc., Chem. Commun. 1995, 2309.(89) Sotzing, G. A.; Thomas, C. A.; Reynolds, J. R. Macromolecules 1998, 31, 3750.(90) Berlin, A.; Canavesi, A.; Pagani, G.; Schiavon, G.; Zecchin, S.; Zotti, G. Synth. Met. 1997, 84, 451.(91) Zotti, G.; Zecchin, S.; Schiavon, G.; Berlin, A.; Pagani, G.; Borgonovo, M.; Lazzaroni, R. Chem.
Mater. 1997, 9, 2876.(92) Lin, S.-C.; Chen, J.-A.; Liu, M.-H.; Su, Y. O.; Leung, M.-K. J. Org. Chem. 1998, 63, 5059.(93) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1997, 119, 5065.(94) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1998, 120, 5355.(95) Devasagayaraj, A.; Tour, J. M. Macromolecules 1999, 32, 6425.(96) Ferraris, J. P.; Lambert, T. L. J. Chem. Soc., Chem. Commun. 1991, 1268.(97) Toussaint, J. M.; Brédas, J. L. Synth. Met. 1993, 61, 103.(98) Ono, K.; Adachi, A.; Okita, K.; Goto, M.; Yamashita, Y. Chem. Lett. 1998, 545.(99) Yamamoto, T.; Zhou, Z.-h.; Kanbara, T.; Shimura, M.; Kizu, K.; Maruyama, T.; Nakamura, Y.;
Fukuda, T.; Lee, B.-L.; Ooba, N.; Tomaru, S.; Kurihara, T.; Kaino, T.; Kubota, K.; Sasaki, S. J. Am.Chem. Soc. 1996, 118, 10389.
(100) Lee, B.-L.; Yamamoto, T. Macromolecules 1999, 32, 1375.(101) Yamamoto, T. Bull. Chem. Soc. Jpn. 1999, 72, 621.(102) Delnoye, D. A. P.; Sijbesma, R. P.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 1996, 118,
8717.(103) Pilgram, K.; Zupan, M.; Skiles, R. J. Heterocycl. Chem. 1970, 7, 629.(104) Naef, R.; Balli, H. Helv. Chim. Acta 1978, 61, 2958.(105) Stille, J. K. Pure Appl. Chem. 1985, 57, 1771.(106) Stille, J. K. Angew. Chem. 1986, 98, 504.(107) Yamamoto, T.; Kanbara, T.; Ooba, N.; Tomaru, S. Chem. Lett. 1994, 1709.(108) Kanbara, T.; Miyazaki, Y.; Yamamoto, T. J. Pol. Sci. A 1995, 33, 999.(109) Karikomi, M.; Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Am. Chem. Soc. 1995, 117, 6791.(110) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Mater. 1996, 8, 570.(111) Bakhshi, A. K.; Ago, H.; Yoshizawa, K.; Tanaka, K.; Yamabe, T. J. Chem. Phys. 1996, 104, 5528.(112) Outurquin, F.; Paulmier, C. Bull. Soc. Chim. Fr. 1983, II , 153.(113) Outurquin, F.; Paulmier, C. Bull. Soc. Chim. Fr. 1983, II , 159.(114) Tanaka, S.; Tomura, M.; Yamashita, Y. Heterocycles 1994, 37, 693.(115) Tanaka, S.; Yamashita, Y. Synth. Met. 1993, 55-57, 1251.(116) Tanaka, S.; Yamashita, Y. Synth. Met. 1995, 69, 599.(117) Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Chem. Soc., Chem. Commun. 1994, 1585.(118) Akoudad, S.; Roncali, J. Chem. Commun. 1998, 2081.(119) Huang, H.; Pickup, P. G. Chem. Mater. 1998, 10, 2212.(120) Yamashita, Y.; Ono, K.; Tomura, M.; Tanaka, S. Tetrahedron 1997, 53, 10169.(121) Ono, K.; Tanaka, S.; Yamashita, Y. Angew. Chem. 1994, 106, 2030.(122) Uno, T.; Takagi, K.; Tomoeda, M. Chem. Pharm. Bull. 1980, 28, 1909.(123) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Lett. 1996, 63.(124) Tanaka, S.; Yamashita, Y. Synth. Met. 1997, 84, 229.(125) Lynch, D. E.; Geissler, U.; Peterson, I. R.; Floersheimer, M.; Terbrack, R.; Chi, L. F.; Fuchs, H.; Calos,
N. J.; Wood, B.; Kennard, C. H. L.; Langley, G. J. J. Chem. Soc., Perkin Trans. 2 1997, 827.(126) Ajayaghosh, A.; Chenthamarakshan, C. R.; Das, S.; George, M. V. Chem. Mater. 1997, 9, 644.(127) Geissler, U.; Lynch, D. E.; Rohde, N.; Hallensleben, M. L.; Walton, D. J. Synth. Met. 1997, 84, 171.(128) Chenthamarakshan, C. R.; Ajayaghosh, A. Chem. Mater 1998, 10, 1657.(129) Chenthamarakshan, C. R.; Eldo, J.; Ajayaghosh, A. Macromolecules 1999, 32, 251.

-LYO 2L[ .ZY_]ZW TY .ZYU`RL_PO ;ZWdXP]^% ?ZbL]O^ 4Y_]TY^TN .ZYO`N_Z]^
��
(130) Treibs, A.; Jacob, K. Angew. Chem. Int. Ed. Engl. 1965, 4, 694.(131) Treibs, A.; Jacob, K. Justus Liebigs Ann. Chem. 1966, 699, 153.(132) Chen, Y.-Y.; Hall, H. K. Polym. Bull. 1986, 16, 419.(133) Lynch, D. E.; Geissler, U.; Kwiatowski, J.; Whittaker, A. K. Polym. Bull. 1997, 38, 493.(134) Bäuerle, P. In Handbook of Oligo- and Polythiophenes; 1 ed.; Fichou, D., Ed.; Wiley-VCH:
Chichester, 1999.(135) Roncali, J. In Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A., Elsenbaumer, R. L.,
Reynolds, J. R., Eds.; Marcel Dekker: New York, 1997.(136) Bouman, M. M.; Meijer, E. W. Adv. Mater. 1995, 7, 385.(137) Langeveld-Voss, B. M. W.; Janssen, R. A. J.; Christiaans, M. P. T.; Meskers, S.; Dekkers, H. P. J. M.;
Meijer, E. W. J. Am. Chem. Soc. 1996, 118, 4908.(138) Langeveld-Voss, B. M. W.; Peeters, E.; Janssen, R. A. J.; Meijer, E. W. Synth. Met. 1997, 84, 611.(139) Peeters, E.; Christiaans, M. P. T.; Janssen, R. A. J.; Schoo, H. F. M.; Dekkers, H. P. J. M.; Meijer, E.
W. J. Am. Chem. Soc. 1997, 119, 9909-9910.(140) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; Mackey, K.; Friend, R. H.; Burn, P.
L.; Holmes, A. B. Nature 1990, 349, 539.(141) Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem. Int. Ed. Engl. 1998, 37, 402.(142) Segura, J. L. Acta Polym. 1998, 49, 319.(143) Andersson, M. R.; Thomas, O.; Mammo, W.; Svensson, M.; Theander, M.; Inganas, O. J. Mater. Chem.
1999, 9, 1933.(144) Meyers, F.; Heeger, A. J.; Brédas, J. L. J. Chem. Phys. 1992, 97, 2750.(145) Musick, K. Y.; Hu, Q.-S.; Pu, L. Macromolecules 1998, 31, 2933.(146) Kang, B. S.; Seo, M.-L.; Jun, Y. S.; Lee, C. K.; Shin, S. C. Chem. Commun. 1996, 1167.(147) Ahn, T.; Jang, M. S.; Shim, H. K.; Hwang, D.-H.; Zyung, T. Macromolecules 1999, 32, 3279.(148) Cho, H. N.; Kim, D. Y.; Kim, Y. C.; Lee, J. Y.; Kim, C. Y. Adv. Mater. 1997, 9, 326.(149) Cho, H. N.; Kim, J. K.; Kim, D. Y.; Kim, C. Y.; Song, N. W.; Kim, D. Macromolecules 1999, 32,
1476.(150) Grice, A. W.; Tajbakhsh, A.; Burn, P. L.; Bradley, D. C. Adv. Mater. 1997, 9, 1174.(151) Hsieh, B. R.; Yu, Y.; Forsythe, E. W.; Schaaf, G. M.; Feld, W. A. J. Am. Chem. Soc. 1998, 120, 231.(152) Huang, W.; Meng, H.; Yu, W. L.; Gao, J.; Heeger, A. J. Adv. Mater. 1998, 10, 593.(153) Kaeriyama, K.; Tsukahara, Y.; Negoro, S.; Tanigaki, N.; Masuda, H. Synth. Metals 1997, 84, 263.(154) Hodge, P.; Power, G. A.; Rabjohns, M. A. Chem. Commun. 1997, 73.(155) Gershuni, S.; Rabinovitz, M.; Agranat, I.; Derlamn, I. B. J. Phys. Chem. 1980, 84, 517.(156) Vollmer, M. S.; Wurthner, F.; Effenberger, F.; Emele, P.; Meyer, D. U.; Stumpfig, T.; Port, H.; Wolf,
H. C. Chem. Eur. J. 1998, 4, 260.(157) Emele, P.; Meyer, D. U.; Holl, N.; Port, H.; Wolf, H. C.; Wurthner, F.; Bäuerle, P.; Effenberger, F.
Chem. Phys. 1994, 181, 417.(158) Effenberger, F.; Grube, G. Synthesis 1998, 1372.(159) Benshafrut, R.; Rabinovitz, R.; Hoffman, R. E.; Benmergui, N.; Müllen, K.; Iyer, V. S. Eur. J. Org.
Chem. 1999, 12, 37.(160) Mori, Y.; Maeda, K. J. Chem. Soc. Perkin Trans. 2 1996, 113.(161) Mukerjee, A. K.; Margaretha, P.; Agosta, W. C. J. Org. Chem. 1996, 61, 3388.(162) Wong, W. Y.; Wong, W. T. J. Chem. Soc. Dalton Trans. 1996, 1853.(163) Kammermeier, S.; Jones, P. G.; Herges, R. Angew. Chem. 1996, 108, 2834.(164) Becker, H.-D.; Andersson, K. J. Org. Chem. 1983, 48, 4542.(165) Trivedi, D. C.; Dhawan, S. K. Synth. Met. 1993, 58, 309.(166) Cao, Y.; Smith, P.; Heeger, A. J. Synth. Met. 1992, 48, 91.(167) McDiarmid, A. G.; Epstein, A. J. Synth. Met. 1995, 69, 85.(168) Min, Y.; McDiarmid, A. G.; Epstein, A. J. Polymer Preprints 1994, 35, 231.(169) Havinga, E. E.; Bouman, M. M.; Meijer, E. W.; Pomp, A.; Simenon, M. M. J. Synth. Met. 1994, 66, 93.(170) Kreyenschmidt, M.; Klärner, G.; Fuhrer, T.; Ashenhurst, J.; Karg, S.; Chen, W. D.; Lee, V. Y.; Scott, J.
C.; Miller, R. D. Macromolecules 1998, 31, 1099.(171) Klärner, G.; Davey, M. H.; Chen, W.-D.; Campbell Scott, J.; Miller, R. D. Adv. Mater. 1998, 10, 993.(172) Kim, J. K.; Hong, S. I.; Cho, H. N.; Kim, D. Y.; Kim, C. Y. Polymer Bull. 1997, 38, 169.(173) Cornelissen, J. J. L. M.; Peeters, E.; Jansen, R. A. J.; Meijer, E. W. Acta Polym. 1998, 49, 476.

THIS IS A BLANK PAGE (34)

.SL[_P]��
.Z[WLYL]T_d�Md�4Y_]LXZWPN`WL]�3dO]ZRPY�-ZYOTYR�,[[WTPO�_Z�L�7Zb�-LYO2L[�.ZYU`RL_PO�;ZWdXP]
,M^_]LN_% The alternating copolymer of pyrrole and 2,1,3-benzothiadiazole is prepared bythermal conversion of the corresponding N-Boc protected, soluble precursor polymer. Thelatter is obtained from the Pd-catalyzed Stille-polymerization of an AB-type monomer. Itrepresents the first example of a low band gap (1.1 eV) conjugated polymer with a ladder-likestructure based on intramolecular hydrogen bonding. The planarizing power of theintramolecular hydrogen bond between the pyrrole N-H and the 2,1,3-benzothiadiazole iminenitrogen was unambiguously demonstrated by NMR spectroscopic and single crystal X-raystudies on a model compound. Planarization by consequence of the intramolecular hydrogenbonding is thought to account for the lower band gap of the pyrrole-based copolymercompared to the already described analogous thiophene-based copolymers.
����4Y_]ZO`N_TZY
Supramolecular chemistry, often referred to as “chemistry beyond the covalent bond”,1-4
shows the increasing ability of chemists, in their imitation of nature, to apply non-covalentinteractions for the control of the molecular conformation, or even the spontaneous assemblyof molecules into complex structures. In the spectrum of non-covalent interactions that areavailable to the supramolecular chemist, hydrogen bonding occupies a prominent position.5-9
The power of this reversible interaction is illustrated by the class of supramolecular polymersthat possess intermolecular, cooperative multiple hydrogen bonds, which behave like acovalent bond.10-12 Additionally, intramolecular hydrogen bonding shows its use incontrolling molecular conformation, exemplified by a class of rigid-core liquid crystallinemolecules, in which hydrogen bonds force planarization, and hence rigidification, of thearomatic core.13-15
n
H H H
HHHH
H
Figure 1. Steric hindrance between the consecutive units in poly-p-phenylene (PPP).
Planar conformations are also important issues in the field of π-conjugated polymers,since the semiconducting properties of such polymers originate from the π-orbital interactionthroughout the chain. Unfortunately, the repeating unit size and substitution pattern of certainconjugated polymers are such, that there is severe steric hindrance, and deviation from

.SL[_P] �
�!
planarity between the consecutive units. This is best illustrated by poly-p-phenylene16 (Figure1).
The steric hindrance between the ortho-hydrogens of two consecutive units forces themout of plane by 23°. This effect is amplified by the substitution of alkyl or alkoxy substituentson the phenyl rings. The effect of non-coplanarity is evident from the properties of thesepolymers, because the band gap is strongly increased (due to the strongly decreasedbandwidth) and, generally, the conduction properties are decreased upon substitution.17
An obvious solution to this problem is forcing the aryl units back into plane by theintroduction of an additional covalent bond.18,19 This is exemplified in Scheme 1 for poly-p-phenylene derived polymers, in which the consecutive phenyl rings are forced into planeeither by a methylene,20 ethene21,22 or imine bridge,23,24 respectively. However, the syntheticapproaches towards such ladder-type systems are troublesome, since the final ring-closingstep has to be performed on –polydisperse and generally sparingly soluble– polymericmaterials. Furthermore, the solubility of these rigid-rod polymers is generally very small.
n
R
RR'
R'O
O
n
R
R R'H
R' HR' H
n
RR
RR
n
OO RR
OR O R
N
N
N
n
R
R
RNH
NH
NH
n
R O
O R
OR Boc
Boc
Boc
H+
Scheme 1. Three PPP based ladder-polymers together with their polymeric precursors.
The use of hydrogen bonding to induce coplanarity in conjugated polymers wasintroduced by the pyrazine-phenylene based copolymers25 of Figure 2. The advantage of sucha polymer is that a final chemical planarization step is not needed, and that the solubility ofthe polymer can be enhanced by (temporarily) breaking the hydrogen bond (e.g. by adjustingthe pH or the temperature). The polymers of Figure 2 possess a self-assembled ladder-typestructure due to strong hydrogen bonding, a conclusion drawn from the solution 1H-NMRspectra, in which the N-H resonance was found at approx. δ = 12 ppm. Unfortunately, the

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�"
optical data of these polymers cannot be compared to the poly-p-phenylenes since theypossess electron-deficient pyrazine rings, which may give rise to additional charge-transfereffects.
N
N
N H
N
N
NH NH
n
O O
O
R
R
R
R = O-t-Bu or C11H23
Figure 2. A self-assembled ladder-like conjugated polymer.
Maximization of orbital overlap between consecutive monomeric units by planarizationis especially useful when designing low band gap conjugated polymers, since the ultimateband gap is strongly dependent on the extended conjugation. However, no examples areknown of low band gap conjugated ladder polymers. In this chapter, the synthesis andproperties are described of a hydrogen bond containing low band gap conjugated polymerwith a self-assembled ladder-like structure.
����/P^TRY�LYO�=P_]Z^dY_SP_TN�,YLWd^T^
The polymer design is based on the donor-acceptor repeating unit strategy, the mostsuccessful approach towards low band gap conjugated polymers thus far.26,27 Taking intoaccount the structure-property relationships as presented in chapter 1, pyrrole is chosen as theelectron-releasing monomer unit because it is not only a strong electron donor, but a potentialhydrogen bond donor as well. 2,1,3-Benzothiadiazole is chosen as the electron-withdrawingmonomer unit because of its synthetic accessibility and ability to accept hydrogen bonds frompyrrole (due to the presence of imine nitrogens). This design yields target polymer 1, whichalso permits comparison with the already described polymers containing thiophene as theelectron-releasing unit and quinoxaline or 2,1,3-benzothiadiazole as the electron-withdrawingunit, featuring band gaps in the range of 1.2 - 1.6 eV.28-32
N
NS
NH
n
1
N
NS
N
n
OO
2

.SL[_P] �
�#
Due to the expected ladder-like and, hence, rigid-chain properties of polymer 1, itssolubility –and processibility– will be very low. Since substitution of solubilizing groups onthe vacant 3,4-sites of pyrrole or 5,6-positions of 2,1,3-benzothiadiazole will induce sterichindrance and, as a result, deviation from coplanarity, a precursor polymer route will workbest using a removable, solubilizing group. For this purpose, the t-butoxycarbonyl (“Boc”)group at the pyrrole nitrogen is ideal, yielding the precursor polymer 2. Boc-protectedpyrroles can be deprotected easily by heating the neat Boc-protected compound at 200 °C fora short time (typically a few minutes) under evolution of carbon dioxide and isobutene.33
The aryl-aryl coupling of choice for N-Boc protected pyrroles is the Stille coupling34,35
since this reaction has successfully been applied to couple N-Boc protected stannylpyrroleswith a variety of aryl bromides.36-39 Two viable synthetic possibilities exist then towards thedesired precursor polymer 2 (Scheme 2): ABA-B-copolymerization (A) and AB-homopolymerization* (B).
SN N
BrBrA
B2
N
NS
N
Me3Sn Br
Boc
NN
NS
N
SnMe3Me3Sn
Boc Boc
N
NS
N
n
Boc
+
Scheme 2. Retrosynthetic routes towards the precursor polymer 2.
����>dY_SP_TN�TYaP^_TRL_TZY^�_ZbL]O^�_SP�-ZN�[]Z_PN_PO�[]PN`]^Z]�[ZWdXP]
The Pd-catalyzed Stille coupling plays a crucial role in the synthesis of precursorpolymer 2; not only in its (co)polymerization, but also in the monomer preparation. Althoughthe yields in the Stille-coupling of stannylpyrroles with electron-deficient aryl bromides aregenerally good using the solvent system toluene/1 M Na2CO3 and the catalyst Pd0(PPh3)4,36
application of these conditions to the coupling between dibromobenzothiadiazole 340 andstannylpyrrole 441 yielded the Boc-protected co-trimer 5 in only 42% yield (Scheme 3). Sincesuch yields are detrimental for the molecular weights that can be obtained in a correspondingpolymerization, it was decided to optimize the conditions for the Stille coupling of 3 and 4first, by variation in solvent, catalyst and reaction temperature.
* Two additional approaches towards polymer 2 would be an A-B-copolymerization and a BAB-Acopolymerization. However, these approaches imply the usage of 2,5-bis(trimethylstannyl)-N-t-Boc-pyrrole asthe A monomer, a compound that fails to react twice efficiently.41

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�$
SN N
BrBrN SnMe3
Boc
+NN
NS
NBoc Boc
3 4 5
Scheme 3. Synthesis of Boc-protected trimer 5. Standard reagents and conditions: a) Pd(PPh3)4, toluene/1MNa2CO3, reflux 48 h, 42%.
����� :[_TXTeL_TZY ZQ >_TWWP�NZ`[WTYR NZYOT_TZY^
The conditions that are applied in the previously described Stille reactions betweenstannylthiophenes and dibromobenzothiadiazole 3 or dibromoquinoxalines28,32,42 are thesolvent/ catalyst systems THF/PdII(PPh3)2Cl2 , DMF/Pd0(PPh3)4 or DMF/PdII(PPh3)2Cl2 attemperatures between 68 - 80 °C. It was decided to adopt these conditions in the optimizedsynthesis of co-trimer 5. Typically, the reactants 3 and 4 were dissolved in the appropriatesolvent in concentrations of 0.5 - 1.0 mol l−1, after which the solution was blanketed by argon.Catalyst (2 mol% with respect to stannylpyrrole 4) was added and the mixture stirred atelevated temperature for 1 night. The results are summarized in table 1.
Solvent Catalyst Temp. Result (After 1 night)
THF PdII(PPh3)2Cl2 68 °C Low conversionDMF Pd0(PPh3)4 85 °C Low conversionDMF PdII(PPh3)2Cl2 85 °C High conversion/side products
Table 1. Applied solvent/catalyst systems and temperatures, towards the improved synthesis of 5.
The reaction in DMF with PdII(PPh3)2Cl2 as the catalyst gives the highest conversion ofthe stannyl compound 4 after 1 night, however, side products were observed. From the 1H-NMR spectra of the crude reaction mixture, it was concluded that the main side products arethe mono-substituted intermediate, and reaction products in which the Boc group is removed.Presumably, the use of DMF as the solvent at elevated temperatures gives rise to partialremoval of the Boc group in these systems. Though this can be minimized by applying alower temperature, it will also give low conversions since the reaction at 68 °C in THF withthe PdII(PPh3)2Cl2 catalyst proceeds very slowly. Therefore, the reaction was carried out inDMF under catalysis of PdII(PPh3)2Cl2 at the intermediate temperature of 75 °C. After 48 h,complete conversion of the stannyl compound 4 was reached, giving rise to 5 in an isolatedyield of 90 %, unusually high for oligopyrroles prepared via the Stille reaction.36
����� >dY_SP^T^ ZQ _SP []PN`]^Z] [ZWdXP] aTL ]Z`_P ,% ,-,�-�NZ[ZWdXP]TeL_TZY
With the optimized Stille conditions, obtained from the above model reaction, thesynthesis of precursor polymer 2 was attempted via route A (ABA-B-copolymerization). Forthis purpose, co-monomer 6 was prepared first, as outlined in Scheme 4.

.SL[_P] �
��
Br
NS
N
Br
N
NS
N
n
Boc
5
6
2
b)
NN
NS
NBoc Boc
NN
NS
N
SnMe3Me3Sn
Boc Boc
a)
+
3
Scheme 4. Synthesis of co-monomer 6, and attempted synthesis of copolymer 2 via route A. Reagents andconditions: a) LTMP, then SnMe3Cl, THF, -80 °C, 75%, b) PdII(PPh3)2Cl2, DMF, 75 °C.
Double lithiation on the α-sites of the pyrrole 5 with 3 equivalents of Li-tetramethylpiperidide (LTMP) and subsequent quenching of the anion with SnMe3Cl yieldedco-monomer 6 in 75 % yield. Unfortunately, polymerization of co-monomers 3 and 6 wasunsuccessful despite the fact that the optimized solvent/catalyst system DMF/PdII(PPh3)2Cl2was applied. Even the application of other solvent/catalyst/cocatalyst systems, of which theresults are compiled in table 2, did not give rise to polymerization. Typically, co-monomers 3and 6 were dissolved in the appropriate solvent in a 1 : 1 molar ratio and, after blanketingwith argon, 5 mol% of catalyst (in runs 2-4 with Cu2Br2 as a cocatalyst) was added and themixture heated under reflux for 1 night.
Solvent Catalyst Result (after 1 night)
Toluene /1 M Na2CO3 Pd0(PPh3)4 low conversionTHF PdII(PPh3)2Cl2/Cu2Br2 low conversionDioxane PdII(PPh3)2Cl2/Cu2Br2 destannylation of 6p-Xylene PdII(PPh3)2Cl2/Cu2Br2 loss of dibromide 3
Table 2. Solvent/catalyst systems applied in the attempted copolymerization of 3 and 6.
The reactivity of the monomers in toluene/1 M Na2CO3, under catalysis of Pd0(PPh3)4
as well as in THF under catalysis of PdII(PPh3)2Cl2/Cu2Br2 is too low to give polymerization.Unfortunately, the use of the higher boiling solvents dioxane or p-xylene gives rise to loss ofthe co-monomers 6 and 3, respectively. These results illustrate the difficulty of finding asolvent/catalyst combination in which the monomers do not degrade, yet are reactive enoughto polymerize. Therefore, route A was abandoned.
����� >dY_SP^T^ ZQ _SP []PN`]^Z] [ZWdXP] aTL ]Z`_P -% ,-�SZXZ[ZWdXP]TeL_TZY
In view of the difficulty of following route A, route B was chosen as an alternative. Forthis route, however, co-monomer 8 had to be prepared of which the synthesis is outlined inScheme 5.

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
��
SN N
BrBr
BrN
NS
N
Me3Sn
Boc
N SnMe3
Boc
+ BrN
NS
NBoc
N
NS
N
n
Boc
a)
b)
c)
3 4 7
82
Scheme 5. Synthesis of compound 7, monomer 8 and copolymer 2. Reagents and conditions: a) Pd(PPh)3,toluene/1M Na2CO3, reflux 24 h, 47%, b) LTMP, then SnMe3Cl, -100 °C, 62%, c) PdII(PPh3)2Cl2, DMF, 85 °C,168 h.
Application of the standard conditions toluene/1M Na2CO3 with the Pd0(PPh3)4 catalystin the reaction of dibromobenzothiadiazole 3 with stannylpyrrole 4, yielded the bromocompound 7 in 47 % yield. Lithiation at the α-position of pyrrole with LTMP, followed byquenching with SnMe3Cl yielded monomer 8 in 62 % yield which could be purified bycrystallization from n-hexane. The polymerization of 8 in DMF under catalysis ofPdII(PPh3)2Cl2 at 75 °C was monitored by size exclusion chromatography (SEC), of which thetraces, normalized to the monomer peak (rightmost large peak) after 48, 96 and 168 hours aredisplayed in Figure 3.
0 5 10 15 time (min)
168 hrs
96 hrs
24 hrs
Monomer
n = 4 3 2
Det
ecto
r ou
tput
(A
.U.)
Figure 3. SEC traces of the polymerization of 9 after 24, 96 and 168 h, normalized to monomer peak (Column:PL500Å (0.1-40 kD), eluent: CHCl3, flow rate: 1 ml min-1, UV detection at 254 nm).
From this picture it follows that oligomerization has taken place. The peaks for n = 2, 3,4… can be clearly distinguished, if it is assumed that the peak for the oligomer with n = 2 isnext to the monomer peak. After 7 days, no significant further increase of the molecularweight was observed and hence, the reaction was worked up. The higher molecular weightfraction of the prepolymer was separated by precipitation in diethyl ether in only 13 % yield,

.SL[_P] �
��
due to the large amount of oligomeric material present in the reaction mixture. From the SECtraces of Figure 3, it follows that the molecular weights that can be attained in thepolymerization of 8 are not high, since oligomers with n = 2, 3, 4…. can be distinguished.The SEC traces of the high-molecular weight fraction that was obtained with precipitationtogether with that of the crude polymerization mixture are shown in Figure 4.
0 5 10 15 time (min)
Monomer
23
45
6n =
Det
ecto
r ou
tput
(A
.U.)
Figure 4. SEC traces of polymer 2. Top: crude polymerization mixture. Bottom: high molecular weight fraction,obtained from precipitation of the crude mixture in diethyl ether (Column: PL500Å (0.1-40 kD), eluent: CHCl3,flow rate: 1 ml min-1, UV detection at 254 nm).
Calibration of the SEC curve with polystyrene standards is ineffectual for these kinds ofrigid-rod conjugated polymers, since the relation between hydrodynamic volume andmolecular weight is completely different. However, if it is assumed again that the peak nextto the monomer peak in the SEC trace of the crude polymerization mixture is the one with n =2, the top of the SEC trace for the precipitated precursor polymer 2 is located at about 8repeating units, i.e. ~16 aromatic units linked together (Mp ∼ 2.4 kg mol-1). Of course, noaccurate determination of Mn and Mw can be made since the width of the MWD is not known.
In general, the yields obtained in the Stille coupling of pyrrole-containing oligomersdrop dramatically with increasing substrate size.43 Therefore, it is reasonable to assume that ina Stille polymerization, the reactivity of the growing chain decreases with increasing chainlength. This is an explanation for the moderate molecular weights that are obtained in ourpolymerization, which is supported by the fact that the Stille polymerization towards otherconjugated polymers did not give very high molecular weights either.25,32
����>dY_SP^T^�LYO�[]Z[P]_TP^�ZQ�_SP�[d]]ZWP�MPYeZ_STLOTLeZWP�NZ[ZWdXP]�LYO�T_^�XZOPWNZX[Z`YO
����� ?SP]XLW OP[]Z_PN_TZY ZQ -ZN�[]Z_PN_PO []PN`]^Z]^
Polymer 1 can conveniently be prepared by thermal deprotection of the purifiedpolymer 2, as depicted in Scheme 6. In the same way, model compound 9 can be prepared bythermal deprotection of its precursor 5, of which the preparation is described in section 2.3.1.

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
��
N
NS
N
n
Boc
5
2
NN
NS
NBoc Boc
NS
N
Nn
H
NS
N
NN
H H
a)
b)
1
9
Scheme 6. Preparation of polymer 1 and its model compound 9. Conditions: a) Heat film at 200 °C, 15 min. b)Heat solid at 200 °C, 30 min, 96 %.
Since polymer 1 is expected to be an insoluble and intractable material, processing wasperformed with the Boc-protected precursor polymer 2 by spincasting a thin film of 2 on glassfrom a CHCl3 solution. Heating the film of 2 at 200 °C for 15 minutes caused quantitativecleavage of the Boc groups (confirmed by IR spectroscopy) and a color change from brown toblue. This film of 1 was then used for optical characterization and doping studies. Similarly,heating neat 5 at 200 °C for 30 minutes yielded the co-trimer 9 in 96 % yield.
����� 0aTOPYNP QZ] SdO]ZRPY MZYOTYR% 98= LYO C�]Ld ^_`OTP^ ZY _SP XZOPW NZX[Z`YO
An indication for the presence of a hydrogen bond between the pyrrole N-H and theimine nitrogen of the benzothiadiazole unit is found in the 1H-NMR spectrum of co-trimer 9,since the pyrrole N-H absorption (δN-H) is found as a sharp singlet at 10.9 ppm (CDCl3),typical for a N-H with an additional hydrogen bond. However, such a high value is notabnormal for pyrroles, e.g. 2,5-diphenylpyrrole43 features a δN-H = 10.2 ppm in THF-d8. Tojudge the strength of an intramolecular hydrogen bond by NMR more quantitatively, thechange in the position of δN-H as a function of concentration and temperature wasinvestigated. When the 1H-NMR spectra of co-trimer 9 were recorded in CDCl3 atconcentrations of 5.0 mg ml-1, 9.4 mg ml-1 and 21.3 mg ml-1, the δN-H remained at 10.9 ppm.Furthermore, when a sample of 9 in toluene-d8 was measured at 25 °C and at 100 °C, the δN-H
shifted from 10.46 ppm to 10.33 ppm, which corresponds to a ∆δN-H/∆T = 1.73⋅10-3 ppm K-1.These values are typical, together with the observed independence of δN-H on concentration,44
for the presence of a strong intramolecular hydrogen bond. The temperature dependence ∆δN-
H/∆T = 1.73⋅10-3 ppm K-1 is even smaller than the best value of ∆δN-H/∆T = 2.8⋅10-3 ppm K-1
found in acylated bipyridines with a strong intramolecular hydrogen bond.45 Therefore, it issuggested that in solution, a strong hydrogen bond is present between the pyrrole N-H and theimine nitrogen of the benzothiadiazole unit of compound 9.
Finally, the occurrence of solid-state hydrogen bonds in a molecule can be deducedfrom its X-ray structure. Crystals of X-ray quality of co-trimer 9 were obtained bycrystallization at -18 °C in CH3CN. The crystallographic data46 are collected in Table 3. Table4 summarizes the typical bond distances and angles, as well as the inter- and intramolecular

.SL[_P] �
��
interaction found in the crystal. The bond distances in table 4 nicely show the localizedcharacter of the double bonds in the 2,1,3-benzothiadiazole unit between C(4) - C(5) and N(3)- C(9). This “o-quinoidal” character, which is also found in isothianaphthene and derivatives,is thought to contribute to the diminishing of the band gap in the corresponding polymer.47,48
In contrast, the double bonds in pyrrole show a more delocalized character, resulting inapproximately equal bond distances. The deviation from coplanarity of the pyrrole and 2,1,3-benzothiadiazole ring is in the range of -6.8° − 8.5°. The nearly coplanar conformation of 9 isillustrated in Figure 5. Remarkably, compound 9 also features an N-H⋅⋅⋅⋅S hydrogen bondwith the sulfur atom of a neighboring molecule, thus forming a dimer. In the crystal (Figure5b), these dimers are packed perpendicularly.
Data 9
formula C14H10N4Srecryst. solvent CH3CNformula weight 266.33color, habit purple needleslattice type monoclinicspace group P21/c
cell dimensionsa (Å) 12.202 (2)b (Å) 5.1015 (10)c (Å) 19.686 (3)α (deg) 90β (deg) 106.023 (10)γ (deg) 90
V (Å3) 1177.8Z 4Dc (g/cm3) 1.5019 (5)F(000) 552µ (cm-1) 2.6R1 0.069wR2 0.167
Table 3. Crystallographic details for compound 9: data collection and refinement parameters.
NS
N
NN
H H
9
10
114
9
3
2
51213
148
6
716
17
1
15
Benzothiadiazolebond lengths
Distance (Å) Pyrrole bondslengths
Distance (Å)
atoms C(5) C(4) 1.38 C(11) C(12) 1.38C(4) C(9) 1.42 C(12) C(13) 1.39C(9) N(3) 1.36 C(13) C(14) 1.37C(9) C(8) 1.43 C(14) N(10) 1.37
N(10) C(11) 1.37
Table 4. Bond distances, angles and hydrogen bonds, found in the crystal of 9 (Continued on next page).

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�
Pyrrole-benzoth. bond lengths Distance (Å)
atoms C(4) C(11) 1.45C(7) C(16) 1.45
Pyrrole-benzothiadiazole torsion angles Angle
atoms C(8) C(7) C(16) N(15) 8.0°C(9) C(4) C(11) N(10) -6.8°C(6) C(7) C(16) N(15) -171.5 °C(5) C(4) C(11) N(10) 171.7°
H-donor H H-acceptr. D-A dist. (A) D−H−A angle
intra N(10) H(10) N(3) 2.81 115°inter N(10) H(10) S(2) 3.58 162°
Table 4 (Continued).
a)
b)Figure 5. PLUTON Representations of the crystal structures of compound 5. a) Dimer formation by N-H⋅⋅⋅Shydrogen bonds. b) Perpendicular packing of the dimers in the crystal.

.SL[_P] �
�!
����� -LYO RL[ LYO NZYO`N_TaT_d ZQ _SP OP[]Z_PN_PO [ZWdXP]
The optical absorption spectrum of the neutral deprotected polymer 1 as a thin film onglass is displayed in Figure 6. The absorption maximum of polymer 1 is situated at λmax = 704nm (1.76 eV), while the band gap, derived from the onset of absorbance (indicated by thedashed line in Figure 6) is estimated at 1.1 eV. In the high wavelength tail of the absorptionpeak, there is a small shoulder which may indicate unintentional doping, e.g. by oxygen fromair.
~1.1 eV
1.76 eV
0
0.1
0.2
0.3
0.4
0.5
0 1 2 3 4energy [eV]
AB
S
Figure 6. Absorption spectrum of polymer 2 (film on glass).
On exposure of a film of polymer 1 to iodine vapor, the initially blue film turns greenafter a few minutes. This color change is reflected in the UV/Vis/NIR absorption spectrum ofthe doped polymer by the occurrence of a very broad band, centered at λmax = 1960 nm. Theneutral peak at λ = 704 nm is also present, which indicates incomplete doping. Prolongedexposure to iodine vapor did not increase the doping level, probably due to the limitedpermeability of iodine vapor through the whole film. The conductivity of the doped samples,determined with the four-probe method, was in the order of 1 S cm-1, but decreased uponstanding.
�� �.ZX[L]T^ZY�_Z�VYZbY�_STZ[SPYP�ML^PO�LYLWZR`P^
The structures of the already described thiophene and quinoxaline or 2,1,3-benzothiadiazole containing copolymers 10 - 12 are depicted in Figure 7, together with theirreported (optical) band gaps. Polymer 1 with Eg = 1.1 eV (thin film) possesses a band gap thatis lower than any of the polymers 10 - 12, even with its low degree of polymerization. Ofcourse, the more electron-releasing character of pyrrole compared to thiophene is one of thereasons for the lower band gap of 1. However, in polymers 11 and 12, the electron-releasingunit is a bithiophene species which evidently is also a strong electron-releasing unit, in view

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�"
of the 0.2 eV decrease in band gap between 10 and 11. Therefore, it is suggested that theadditional, planarizing hydrogen bond interaction that is present in polymer 1 also contributesto a diminishing of the band gap.
NN
RR
n
S
NS
N
SSn SS
n
NN
RR
10 11 12
Eg = 1.6 eV (R = n-hexyl) 1.4 eV (R = n-methyl) 1.2 eV(solution) (thin film) (thin film)
ref. 32 28,29 28,29
Figure 7. Structures and reported band gaps of polymers 10 - 12
Evidence for the significance of the hydrogen bond as a planarizing force was found inthe reported X-ray crystal structure of the monomer of polymer 11, 5,8-dithien-2-ylquinoxaline 12 (R = CH3).28 Although this crystal structure shows coplanarity of thethiophene and quinoxaline rings, a structural disorder is present, associated with a 180°rotation around the thiophene-quinoxaline bond, schematically represented in Figure 8. Thisdisorder was also found for other thiophene-based co-trimers of this type.
It is likely that the observed coplanarity of 12 is a consequence of crystal-packing,which means that in solution the thiophene rings have a considerable freedom to rotatearound the thiophene-quinoxaline bond. As a consequence, there is a realistic possibility ofnon-coplanarity of the consecutive units in amorphous thin films of the correspondingpolymer 11. In the crystal structure of model compound 9, no disorder of any kind wasobserved. Furthermore, the hydrogen bond in 9 was shown to be operative as a stronglyplanarizing force even in solution.
N N
X
YY
X
Y = 20% S + 80% C
12
X = 80% S + 20%C
Figure 8. Schematic representation of the disorder, found in the crystal structure of 12 (ref. 28)
With the above observations in mind, it is concluded that the use of hydrogen bondingin these systems is a valid approach towards planarization of the conjugated backbone, andeven a prerequisite if polymers with a vanishingly small band gap are pursued.

.SL[_P] �
�#
��!�.ZYNW`^TZY^
Polymer 1 is the first example of a low band gap conjugated polymer with a self-assembled ladder-like structure based on intramolecular hydrogen bonding. The preparationvia the Stille polymerization of a Boc-protected precursor monomer, followed by spincastingof the soluble precursor polymer on a suitable substrate and subsequent cleavage of the Bocgroups by heating, gives access to stable, transparent thin films of 1, featuring an absorptionmaximum of 704 nm and an estimated band gap of 1.1 eV. Significant planarizing power wasascribed to the intramolecular hydrogen bond, based on NMR spectroscopic and single crystalX-ray studies on model compound 9. Comparison of the physical data of polymer 1 and itsmodel compound 9 with analogous, thiophene based polymers and model compoundsdemonstrated the importance of planarization in this type of low band gap conjugatedpolymers.
Of course, band gap reduction is not only concerned with planarization of theconsecutive monomer units. The alternating donor-acceptor repeating unit strategy that isapplied in this chapter, implies the combination of the proper donor and acceptor unitsHowever, the various parameters influencing the band gap in these systems, such as differentcombinations of donor- and acceptor units and the evolution of the band gap with increasingchain length, are not known. Therefore, it was decided to synthesize a series of well-definedoligomers with different electron-donating and -accepting units and varying chain lengths tostudy these parameters in detail, as will be described in chapter 3.
��"�0c[P]TXPY_LW�>PN_TZY
2PYP]LW _PNSYT\`P^� All solvents and reagents were reagent grade and used as received. THF was distilled overNa/benzophenone prior to use. DMF was stored over BaO. For column chromatography, Merck silica gel 60(particle size 0.063-0.200 mm) or Merck aluminum oxide 90 (neutral; activity I deactivated with 7 wt % ofwater) were used. Melting points are uncorrected and determined using a Büchi melting point apparatus (afterDr. Tottoli). NMR spectra were recorded on a Bruker AM-400 spectrometer at frequencies of 400.1 and 100.6MHz for 1H and 13C nuclei, respectively, or on a Varian Gemini spectrometer at frequencies of 300.1 and 75.0MHz for 1H and 13C nuclei, respectively. Tetramethylsilane (TMS) was used as an internal standard for 1H NMRand CDCl3 or DMSO-d6 for 13C NMR. UV/Vis spectra were recorded on a Perkin-Elmer Lambda 3B UV/Vis orLambda 900 UV/Vis/NIR spectrometer. Infrared (FT-IR) spectra were recorded on a Perkin-Elmer 1605 FT-IRspectrophotometer with wavenumbers between 4400 and 450 cm-1. Elemental analyses were performed on aPerkin Elmer 2400 Series II CHN Analyzer. GC/MS measurements were performed on a Shimadzu GCMS-QP5000. Size Exclusion Chromatography was performed on a Shimadzu LC-10AT system in combination with aPolymer Laboratories PL500Å SEC column (0.1 - 40 kD) with CHCl3 as the eluent at a flow rate of 1 ml min-1
and UV detection at 254 nm. 4,7-Dibromo-2,1,3-benzothiadiazole40 3 and 2-trimethylstannyl-N-t-butoxycarbonylpyrrole43 4 were prepared according to literature procedures.
��"�-T^��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��������MPYeZ_STLOTLeZWP F H�� In a 250 mL flask, N-t-butoxycarbonyl-2-trimethylstannyl-pyrrole 4 (4.5 g, 14 mmol) and 4-bromo-2,1,3-benzothiadiazole 3 (2.0 g, 6.8 mmol) weredissolved in a mixture of toluene and 1 M Na2CO3 (1:1, 100 mL). This mixture was deaerated and brought underan argon atmosphere. Then, tetrakis(triphenylphosphine)palladium(0) (2 mol%) was added and the resultingmixture was heated under reflux for 48 h. Subsequently, the reaction mixture was allowed to cool to roomtemperature, and the organic layer was separated from the aqueous layer. The water layer was extracted threetimes with ether, and the combined organic layers were dried over MgSO4, filtered and evaporated to give crude5 (3.53 g). Column chromatography over Al2O3 with hexane : dichloromethane 5:1 yielded pure 5 (1.32 g, 2.83

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�$
mmol, 41.6 %) as a fluorescent orange solid. 1H-NMR (400 MHz, CDCl3) δ: 7.58 (s, 2H, H btd), 7.49 (dd, J =3.3 and 1.8 Hz, 2H, H-2 pyr), 6.41 (dd, J = 3.3 and 1.8 Hz, 2H, H-4 pyr), 6.34 (t, J = 3.3 Hz, 2H, H-3 pyr), 1.19(s, 9H, Me Boc). 13C-NMR (100 MHz, CDCl3) δ: 154.8, 149.1, 130.2, 127.4, 125.9, 123.4, 115.6, 110.8, 83.4,27.4. UV/Vis (CHCl3): λmax = 401 nm. IR (KBr): ν = 2976, 1743, 1318, 1138, 846-400 cm-1. Anal. calcd. forC24H26N4O4S: C, 61.78; H, 5.61; N, 12.00. Found: C, 61.17; H, 6.05; N, 11.82.
��"�-T^�� �_]TXP_SdW^_LYYdW�9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��������MPYeZ_STLOTLeZWP F!H �� In an oven-dried50 mL flask, a solution of 2,2,6,6-tetramethylpiperidine (TMP, 0.252 g, 1.61 mmol) in dry THF (25 mL) wascooled to -80 °C and subsequently treated with a 1.35 M solution of n-butyllithium in hexane (1.2 mL, 1.6mmol). This mixture was stirred for 15 min at -80 °C, warmed to room temperature, stirred for another 15 minand recooled to -80 °C. A solution of 5 (0.25 g, 0.54 mmol) in dry THF (5 mL) was added and the reactionmixture was stirred at -80 °C for 60 min, warmed to room temperature, stirred for another 15 min and recooledto -80 °C. The reaction mixture was then quenched with a solution of SnMe3Cl (0.32 g, 1.6 mmol) in THF (5mL) and subsequently warmed to room temperature. The THF was evaporated and the residue was dissolved inether/water. The layers were separated and the water layer was extracted with ether. The combined organiclayers were washed with a 0.01 M HCl solution, dried over MgSO4 and evaporated to give the crude distannylcompound 6 as a dark oil. This oil was dissolved in CH2Cl2 and filtered over Al2O3 (CH2Cl2) which, afterevaporation, gave the pure distannyl compound 6 (0.31 g, 0.39 mmol, 73 %) as smelly orange solid. 1H-NMR(400 MHz, CDCl3) δ: 7.52 (s, 2H, H btd), 6.50 (d, J = 3.2 Hz, 2H, H-4 pyr), 6.46 (d, J = 3.0 Hz, 2H, H-3 pyr),0.85 (s, 9H, Me Boc), 0.39 (s, Sn sat.), 0.32 (s, 9H, Me-Sn), 0.25 (s, Sn sat.). 13C-NMR (100 MHz, CDCl3) δ:154.7, 150.1, 139.1, 133.3, 128.4, 127.2, 121.4, 116.7, 83.0, 27.0, -7.43.
��-]ZXZ�"��9�_�M`_ZcdNL]MZYdW[d]]ZW���dW��������MPYeZ_STLOTLeZWP F"H �� This compound was prepared, asdescribed for compound 5, by means of a Stille coupling between N-t-butoxycarbonyl-2-trimethylstannyl-pyrrole4 (2.3 g, 7.0 mmol) and 4-bromo-2,1,3-benzothiadiazole 3 (2.0 g, 6.8 mmol) in a mixture of 25 ml toluene and25 ml 1 M Na2CO3; reaction time 24 h. Column chromatography of the crude reaction workup product (3.57 g)over alumina (300 g) with hexane/CH2Cl2 = 6:1 yielded pure 8 as a fluorescent yellow solid (1.22 g, 3.2 mmol,47%). 1H-NMR (400 MHz, CDCl3) δ: 7.84 (d, J = 7.5 Hz, 1H, H-5 btd), 7.49 (dd, J = 3.3 and 1.7 Hz, 1H, H-2pyr), 7.43 (d, J =7.4 Hz, 1H, H-6 btd), 6.40 (dd, J = 3.3 and 1.8 Hz, 1H, H-4 pyr), 6.33 (t, J = 3.3 Hz, 1H, H-3pyr), 1.19 (s, 9H, H-Boc). 13C-NMR (100 MHz, CDCl3) δ: 154.1, 153.0, 148.8, 131.9, 129.2, 127.9, 123.6,115.9, 113.1, 110.9, 83.5, 27.4. UV/Vis (CHCl3): λmax = 388 nm. IR (KBr): ν = 2979, 1737, 1323, 1158, 846-489 cm-1. Anal. calcd. for C15H14N3SO2Br: C, 47.38; H, 3.71; N, 11.05. Found: C, 47.59; H, 3.88; N, 11.45.
��-]ZXZ�"�� �_]TXP_SdW^_LYYdW�9�_�M`_ZcdNL]MZYdW[d]]ZW���dW��������MPYeZ_STLOTLeZWP F#H �� In an oven-dried 50 ml flask, TMP (0.31 g, 2.0 mmol) was dissolved in 20 ml dry THF. This solution was cooled to -80 °Cand, subsequently, a 1.6 M solution of n-BuLi in hexane (1.25 ml, 2.0 mmol) was added. This mixture wasstirred for 15 min at -80 °C, warmed to room temperature, stirred for another 15 min and recooled to -100 °C. Asolution of 8 (0.50 g, 1.3 mmol) in 5 ml dry THF was added and the reaction mixture was stirred at -100 °C for30 min, after which the reaction was quenched with a solution of SnMe3Cl (0.42 g, 2.1 mmol) in 5 ml dry THFand allowed to warm to room temperature. THF was evaporated and the residue dissolved in ether/water. Thelayers were separated and the aqueous layer was extracted with ether. The combined organic layers were washedwith a 0.01 M HCl solution, dried over MgSO4 and evaporated to give the crude monomer 9 as a yellow/greenfluorescent solid (0.44 g, 0.81 mmol, 62 %) which could be recrystallized from hexane. 1H-NMR (400 MHz,CDCl3) δ: 7.85 (d, J = 7.5 Hz, 1H, H-5 btd), 7.39 (d, J =7.5 Hz, 1H, H-6 btd), 6.48 (m, 2H, H-3,4 pyr), 0.86 (s,9H, H-Boc), 0.41 (s, Sn sat.), 0.32 (s, 9H, Me-Sn), 0.22 (s, Sn sat.). 13C-NMR (100 MHz, CDCl3) δ: 155.1,153.7, 151.4, 140.2, 132.6, 129.9, 128.2, 122.0, 117.8, 113.2, 83.7, 27.6, -6.3. UV/Vis (CHCl3): λmax = 403 nm.IR (KBr): ν = 2978, 1726, 1324, 1154, 847-526 cm-1. Anal. calcd. for C18H22N3SO2BrSn: C, 39.81; H, 4.08; N,7.74. Found: C, 40.14; H, 4.31; N, 8.00.
��"�-T^��[d]]ZW���dW��������MPYeZ_STLOTLeZWP F$H�� Compound 5 (0.196 g, 0.43 mmol) was put in a 10 mL flaskand heated on an oil bath at 200 °C. Evolution of CO2 and isobutene and a rapid color change of the solid wereobserved. After 30 min the flask was allowed to cool to room temperature, in which pure 7 (0.1084 g, 0.407mmol, 95.5 %) was found as a deep purple solid. M.p. > 200 °C. 1H-NMR (400 MHz, CDCl3) δ: 10.9 (s, 1H, N-H), 7.84 (s, 1H, H btd), 7.03 (m, 1H, H-3 pyr), 6.88 (m, 1H, H-4 pyr), 6.37 (m, 1H, H-5 pyr). 13C-NMR (100MHz, CDCl3) δ: 152.5, 129.5, 123.2, 121.4, 120.0, 110.0, 107.0. UV/Vis (CHCl3): λmax = 532 nm. IR (KBr): ν

.SL[_P] �
�
= 3415, 1481, 1113, 888-450 cm-1. GC/MS: M+. 265.95 (100%). Anal. calcd. for C14H10N4S: C, 63.61; H, 3.05;N, 21.20. Found: C, 63.92; H, 3.47; N, 21.18.
,-, � , NZ[ZWdXP]TeL_TZY L__PX[_^� _d[TNLW PcLX[WP% A mixture of the distannyl co-monomer 6 (0.15 g, 0.189mmol), dibromo co-monomer 3 (0.0557 g, 0.189 mmol) and K2CO3 (0.052 g, 0.38 mmol) in distilled THF (10ml), dioxane (10 ml) or xylene (5 ml) was blanketed by argon using a single-stage vacuum pump. PdII(PPh3)2Cl2catalyst (4 mol%, 5.3 mg, 7.6 µmol) and Cu2Br2 (2.7 mg, 9.0 µmol) were added and the mixture heated underreflux for 1 night. However, no product formation was observed.
��"�-T^��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��������MPYeZ_STLOTLeZWP F H� ]PLN_TZY TY /81�� In a 25 mL flask, N-t-butoxycarbonyl-2-trimethylstannyl-pyrrole 4 (0.39 g, 1.17 mmol) and 4-bromo-2,1,3-benzothiadiazole 3 (0.15g, 0.50 mmol) were dissolved in 10 ml dry DMF. The solution was degassed at room temperature with a single-stage vacuum pump and blanketed with dry argon. This degassing cycle was repeated 3 times. Pd(PPh3)2Cl2 (8.5mg, 12 µmol) was added and the solution was blanketed again by Argon following the above procedure. Thereaction mixture was stirred at 75 °C for 2 nights, after which the solution was cooled to room temperature andmixed with ether. The organic layer was washed well with icecold water to remove the DMF, dried over MgSO4
and evaporated to give crude 5 as an orange oil. This oil was subjected to column chromatography over Al2O3
with hexane : dichloromethane 5:1 to yield pure 5 (1.32 g, 2.83 mmol, 41.6 %) as a fluorescent orange solid.(Analytical data: vide supra)
;ZWd��9�_�M`_ZcdNL]MZYdW[d]]ZW���dW��NZ�������MPYeZ_STLOTLeZWP F�H�� In a dry 25 ml flask, monomer 9 (0.33g, 0.6 mmol) was dissolved in 10 ml dry DMF. The solution was degassed at room temperature with a single-stage vacuum pump and blanketed with dry Argon. This degassing cycle was repeated 3 times. Pd(PPh3)2Cl2 (8.5mg, 12 µmol) was added and the solution was blanketed again by argon following the above procedure. Thereaction mixture was then stirred at 75 °C for 7 days, after which the reaction mixture was poured in Et2O. Theprecipitate (75 mg) was filtered off, dissolved in CHCl3 and filtered over silica with CHCl3 and THF,respectively. The THF fraction was concentrated and reprecipitated in Et2O to yield the precursor polymer 2 as adark solid (19.1 mg, ~13 %). 1H-NMR (400 MHz, CDCl3) δ: 7.8 (broad s, H-btd), 6.6 (broad s, H-pyr), 0.8(broad s, Boc). 13C-NMR (100 MHz, CDCl3) δ: 154.2, 149.1, 132.4, 128.1, 127.5, 114.2, 83.4, 27.0. UV/Vis(CHCl3): λmax = 434 nm. IR (KBr): ν = 2976, 1749, 1306, 1136, 872-731 cm-1.
;ZWd��[d]]ZW���dW��NZ�������MPYeZ_STLOTLeZWP F�H�� A film of the precursor polymer 2 on glass (spincast fromCHCl3 solution) was heated at 200 °C for 15 min, during which time a rapid color change from reddish brown todeep blue was observed. UV (solid film): λmax = 704 nm. IR (KBr): ν = 3347, 1569, 1474, 1338, 1239, 1119,1048, 873, 772 cm-1.
=PQP]PYNP^
(1) Lehn, J.-M. Makromol. Chem. Makromol. Symp. 1993, 69, 1.(2) Lehn, J.-M. Angew. Chem. Int. Ed. Engl. 1988, 27, 89.(3) Lehn, J.-M. Angew. Chem. Int. Ed. Engl. 1990, 29, 1304.(4) Fouquey, C.; Lehn, J.-M.; Levelut, A.-M. Adv. Mater. 1990, 2, 254.(5) Bohanon, T. M.; Denzinger, S.; Fink, R.; Paulus, W.; Ringsdorf, H.; Weck, M. Angew. Chem. Int. Ed.
Engl. 1995, 34, 58.(6) Grotzfeld, M.; Branda, N.; Rebek Jr., J. Science 1996, 271, 487.(7) Hamilton, A. D.; Pant, N.; Muhldorf, A. V. Pure & Appl. Chem. 1988, 60, 533.(8) Lange, R. F. M.; Meijer, E. W. Macromolecules 1995, 28, 782.(9) Mathias, J. P.; Simanek, E. E.; Whitesides, G. M. J. Am. Chem. Soc. 1994, 116.(10) Beijer, F. H.; Sijbesma, R. P.; Kooijman, H.; Spek, A. L.; Meijer, E. W. J. Am. Chem. Soc. 1998, 120,
6761.(11) Beijer, F. H.; Kooijman, H.; Spek, A. L.; Sijbesma, R. P.; Meijer, E. W. Angew. Chem. Int. Ed. Engl.
1998, 37, 75.(12) Sijbesma, R. P.; Beijer, F. H.; Brunsveld, L.; Folmer, B. J. B.; Hirschberg, J.; Lange, R. F. M.; Lowe, J.
K. L.; Meijer, E. W. Science 1997, 278, 1601.(13) Palmans, A. R. A.; Vekemans, J. A. J. M.; Meijer, E. W. Recl. Trav. Chim. Pays-Bas 1995, 114, 277.

.Z[WLYL]T_d Md 4Y_]LXZWPN`WL] 3dO]ZRPY -ZYOTYR ,[[WTPO _Z L 7Zb -LYO 2L[ .ZYU`RL_PO ;ZWdXP]
�
(14) Palmans, A. R. A.; Vekemans, J. A. J. M.; Havinga, E. E.; Meijer, E. W. Angew. Chem. Int. Ed. Engl.1997, 36, 2648.
(15) Palmans, A. R. A.; Vekemans, J. A. J. M.; Hikmet, R. A.; Fischer, H.; Meijer, E. W. Adv. Mater. 1998,10, 873.
(16) Tour, J. M. Adv. Mater. 1994, 6, 190.(17) Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A.; Elsenbaumer, R. L.; Reynolds, J. R., Eds.;
Marcel Dekker: New York, 1998.(18) Scherf, U. In Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A., Elsenbaumer, R. L.,
Reynolds, J. R., Eds.; Marcel Dekker: New York, 1998.(19) Scherf, U. J. Mater. Chem. 1999, 9, 1853.(20) Scherf, U.; Müllen, K. Makromol. Chem. Rapid Commun. 1991, 12, 489.(21) Chmil, K.; Scherf, U. Acta Polym. 1997, 48, 208.(22) Chmil, K.; Scherf, U. Makromol. Chem. Rapid Commun. 1993, 14, 217.(23) Tour, J. M.; Lamba, J. J. S. J. Am. Chem. Soc. 1993, 115, 4935.(24) Tour, J. M.; Lamba, J. J. S. J. Am. Chem. Soc. 1994, 116, 11723.(25) Delnoye, D. A. P.; Sijbesma, R. P.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 1996, 118,
8717.(26) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Polym. Bull. 1992, 29, 119.(27) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Synth. Met. 1993, 55-57, 299.(28) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Mater. 1996, 8, 570.(29) Karikomi, M.; Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Am. Chem. Soc. 1995, 117, 6791.(30) Lee, B.-L.; Yamamoto, T. Macromolecules 1999, 32, 1375-1382.(31) Yamamoto, T.; Zhou, Z.-h.; Kanbara, T.; Shimura, M.; Kizu, K.; Maruyama, T.; Nakamura, Y.;
Fukuda, T.; Lee, B.-L.; Ooba, N.; Tomaru, S.; Kurihara, T.; Kaino, T.; Kubota, K.; Sasaki, S. J. Am.Chem. Soc. 1996, 118, 10389-10399.
(32) Kanbara, T.; Miyazaki, Y.; Yamamoto, T. J. Pol. Sci. A 1995, 33, 999.(33) Rawal, V. H.; Cava, M. P. Tetrahedron Lett. 1985, 26, 61.(34) Stille, J. K. Pure Appl. Chem. 1985, 57, 1771.(35) Stille, J. K. Angew. Chem. 1986, 98, 504.(36) Groenendaal, L.; Peerlings, H. W. I.; Havinga, E. E.; Vekemans, J. A. J. M.; Meijer, E. W. Synth. Met.
1995, 69, 467.(37) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G. Synthesis 1991, 613.(38) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G. Synth. Met. 1992, 51, 299.(39) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G.; Zotti, G.; Zerbi, G. Synth. Met. 1993, 55,
1096.(40) Pilgram, K.; Zupan, M.; Skiles, R. J. Heterocycl. Chem. 1970, 7, 629.(41) Martina, S. Ph D Thesis 1992, University of Mainz, Mainz.(42) Tamao, K.; Shigeki, O.; Yamaguchi, S. Chem. Commun. 1996, 1873.(43) Groenendaal, L., Ph D Thesis 1996, Eindhoven University of Technology, Eindhoven.(44) Hamuro, Y.; Geib, S. J.; Hamilton, A. D. Angew. Chem. 1994, 106, 465.(45) Palmans, A. R. A., Ph D Thesis 1998, Eindhoven University of Technology, Eindhoven.(46) Spek, A. L. Acta Cryst. 1990, A46, C34.(47) Ferraris, J. P.; Bravo, A.; Kim, W.; Hrncir, D. C. J. Chem. Soc., Chem. Commun. 1994, 991.(48) Kurti, J.; Surjan, P. R.; Kertesz, M. J. Am. Chem. Soc. 1991, 113, 9865.

THIS IS A BLANK PAGE (52)

.SL[_P]��
-LYO�2L[�0YRTYPP]TYR�ZQ�/ZYZ]�,NNP[_Z]>`M^_T_`_PO�π�.ZYU`RL_PO�;ZWdXP]^
,M^_]LN_% Three series of alternating donor-acceptor substituted co-oligomers (with variouschain lengths), consisting of pyrrole or thiophene as the electron-releasing unit andquinoxaline or 2,1,3-benzothiadiazole as the electron-withdrawing unit, have been preparedby application of the Pd-catalyzed Stille coupling methodology. The trimethylstannyl-group isalways located at the electron-rich unit, while the bromo substituent is always located at theelectron-deficient unit. Furthermore, the Boc protecting group is used in the synthesis of thepyrrole containing oligomers. The incremental bathochromic shift of λmax upon chainelongation of the three series of oligomers is less than that of the homo-oligomers ofthiophene and pyrrole, which finds its origin in a diminished dispersion (hybridization) of theLUMO level upon chain elongation. The latter conclusion was drawn after comparing theoxidation and reduction behavior of the thiophene/benzothiadiazole oligomers with that ofthiophene oligomers. The incremental bathochromic shift shows similarity for all three seriesof oligomers, which is used as a tool in the band gap engineering of donor-acceptorsubstituted π-conjugated polymers.
����4Y_]ZO`N_TZY
One of the most successful approaches towards low band gap conjugated polymerspresented in chapter 1, implies the application of an alternating sequence of donor-acceptorunits in the π-conjugated polymer chain.1-5 Indeed, a narrow band gap can be obtained bystarting from a monomer which already has a narrow HOMO-LUMO energy separation. Thestructural variations that lead to a narrowing of the band gap in these systems are not wellunderstood though, because many polymers have been prepared that satisfy the donor-acceptor strategy, but still show a rather high band gap.6-9
Consequently, it is also unknown whether the evolution of the band gap upon chainelongation is comparable to that of “ordinary” π-conjugated polymers, like polythiophene andpolypyrrole. The question is relevant not only for the understanding of band gap engineering,but also with respect to theoretical considerations, that have revealed the unique electronicproperties of the donor-acceptor type systems.10,11
The growing ability of chemists to prepare and handle large conjugated moleculararchitectures,12-19 has led to many studies on the dependence of chain length and absorptionmaximum in conjugated oligomers of increasing size.20-28 However, alternating donor-acceptor conjugated oligomers have not yet been studied systematically.

.SL[_P] �
�
In this chapter, the synthesis of three series of donor-acceptor oligomers 1-11 (Figure 1)is described as well as the investigations concerning the dependence of the absorptionmaximum on chain-length, leading to the formulation of some consequences for the design oflow band gap conjugated donor-acceptor polymers.
11
NS
N
SSS S
NS
NNS
N
SS SS
10
NSN
S
S
98
NS
N
SS
7
NS
N
S
NN
NH
N
NNH
6
N
NN
N
HH
54
N
N N H
NS
N
N
H
N
H
NS
N
N S
N
3
NS
N
N
H
N
H
2
NS
N
N
H
1
Figure 1. Donor-acceptor oligomers investigated in this study.
The design relies on oligomeric equivalents of not only the pyrrole/2,1,3-benzothiadiazole containing copolymer described in chapter 2, but their thiophene andquinoxaline containing counterparts as well. Again, the synthetic approach towards thesesystems is based on the Stille coupling.
����>dY_SP^T^�ZQ�_SP�NZ�ZWTRZXP]^�aTL�_SP�>_TWWP�NZ`[WTYR�XP_SZOZWZRd
����� >dY_SP^T^ ZQ [d]]ZWP�������MPYeZ_STLOTLeZWP NZ�ZWTRZXP]^
The synthesis of oligomers 1-3 is outlined in Scheme 1. The Stille coupling29,30 givesaccess to the appropriate oligomers. The combination of the solvent system toluene/1 MNa2CO3 and the catalyst Pd0(PPh3)4 31-34 is used here in the synthesis of pyrrole containingoligomers 1-6. Thus, the bromide 1235 was reacted with N-t-Boc-2-trimethylstannylpyrrole1336 in a boiling two-phase system of toluene and 1 M Na2CO3 (1:1) under catalysis of

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
Pd0(PPh3)4 for 48 hours, to give the Boc-protected precursor 1,Boc in 78% yield. Thisprecursor was deprotected by shortly heating the solid at 200 °C to give 1.37
g)
f)
e)
d)
c)
b)
a)
13
N
Boc
Me3Sn
12
NS
N
Br
NS
N
N
Boc
1,Boc
2,Boc
NS
N
NN
BocBoc
NS
N
BrBr
14
N
SN
N N
NS
N
NS
NBoc Boc
3,BocN
SN
N
BocMe3Sn
15
1 2 3
Scheme 1: Synthesis of pyrrole/2,1,3-benzothiadiazole co-oligomers. Reagents and conditions: a) Pd(PPh3)4,toluene, 1M Na2CO3, reflux 48 h, 78 %, b) Pd(PPh3)4, toluene, 1M Na2CO3, reflux 48 h, 42 %, c) heat, 200 °C,30 min., 95 %, d) heat, 200 °C, 30 min., 96 %, e) heat, 0.1 mm Hg, 200 °C, 30 min., 100 %, f) Pd(PPh3)4,toluene, 1M Na2CO3, reflux 72 h, 11 %, g) LTMP, then SnMe3Cl, THF, -80 °C, 57 %.
Analogously, 2,Boc (See chapter 2) was synthesized in 42% yield from the dibromide14 and trimethylstannylpyrrole 13, and subsequently deprotected to afford 2. The synthesis of3 requires the intermediate stannyl-compound 15 which was prepared in 57% yield from1,Boc by deprotonation with Li-tetramethylpiperidide (LTMP) in THF at -80 °C, andsubsequent quenching with SnMe3Cl. Compound 15 was then reacted under the standardconditions with dibromide 14 to give 3,Boc in 11 % yield, which upon deprotection gave 3. Ageneral trend in the Stille couplings used here, is that with increasing oligomer size thereaction yields drop, which is presumably due to the more difficult ligand exchange withlarger molecules.38

.SL[_P] �
!
����� >dY_SP^T^ ZQ [d]]ZWP�\`TYZcLWTYP NZ�ZWTRZXP]^
The synthesis of oligomers 4-6 is outlined in Scheme 2. The Pd0(PPh3)4 catalyzedStille-reactions are again performed in the boiling two-phase system toluene/1M Na2CO3
(1:1).
h)
g)f)e)
d)c)
a)
b)
N
BocMe3Sn
N N
Br
NN
16
N
Boc
Me3Sn
13 17
Br
NN
Br
N
Boc NN
4,Boc
18
N
Boc NN
Br
5,Boc
19
6,Boc
N NN
N
NN
Boc
BocN
NN
N
Boc Boc
654
Scheme 2: Synthesis of pyrrole/quinoxaline co-oligomers. Reagents and conditions: a) Pd(PPh3)4, toluene, 1MNa2CO3, reflux 72 h, 23 %, b) Pd(PPh3)4, toluene, 1M Na2CO3, reflux 72 h, 36 %, c) Pd(PPh3)4, toluene, 1MNa2CO3, reflux 72 h, 75 %, d) Pd(PPh3)4, toluene, 1M Na2CO3, reflux 72 h, 85 %, e) heat, 200 °C, 30 min., 95%, f) heat, 200 °C, 30 min., 95 %, g) heat, 0.1 mm Hg, 200 °C, 30 min., 90 %, h) LTMP, then SnMe3Cl, THF, -80 °C, 32 %.
Thus, the bromide 1639,40 was reacted with 13 to give 4,Boc in 36% yield. Thermaldeprotection of this compound then gave 4. The synthesis of co-trimer 5 differed from that ofits benzothiadiazole analogue 2. In view of the longer reaction times needed to complete theStille coupling involving bromoquinoxalines compared to that involvingbromobenzothiadiazoles (and hence the greater probability of by-product formation), theintermediate compound 18 was isolated (23% yield) and thereafter reacted again with anadditional equivalent of 13 to give the desired 5,Boc in 75 % yield. Thermal deprotection thengave 5. For the synthesis of co-tetramer 6, the trimethylstannyl compound 19 was preparedfrom 4,Boc first —analogous to the synthesis of 15 from 1,Boc— in 32 % yield. Subsequentreaction with 18 gave 6,Boc in 85 % yield and thermal deprotection then gave crude 6.

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
"
During this deprotection, an unidentified by-product was formed, hence 6 had to be purifiedby precipitation from THF in hexane.
As mentioned before, the Stille coupling of bromoquinoxalines withtrimethylstannylpyrroles proceeds more slowly than that of bromobenzothiadiazoles. Eitherthe lower electron-withdrawing power or the more sterically demanding 6-membered rings ofquinoxaline lead to less activation of this substrate compared to benzothiadiazole. In thisview, it is strange that the yields in the synthesis of 5,Boc and 6,Boc are quite high, since thebromide here is expected to be even less activated. A lower yield would also be expected inview of the oligomer size. It seems, therefore, that the introduction of the first substituent ismore difficult than the introduction of the second one.
����� >dY_SP^T^ ZQ _STZ[SPYP�MPYeZ_STLOTLeZWP NZ�ZWTRZXP]^
The Stille coupling of 2-tributylstannylthiophene with 4,7-dibromo-2,1,3-benzothiadiazole has previously been described,41,42 employing THF as the solvent andPdII(PPh3)2Cl2 as the catalyst.
f)
e)
d)c)
b)
a)
NS
N
Br
12
SMe3Sn
20
7
SSnMe3
S
21
9
14
NS
N
BrBr
8
22
NS
N
S SMe3Sn
11
10
Scheme 3. Synthesis of thiophene/2,1,3-benzothiadiazole co-oligomers. Reagents and conditions: a)Pd(PPh3)2Cl2, DMF, 75 °C, 1 h, 89 %, b) Pd(PPh3)2Cl2, DMF, 75 °C, 1.5 h, 56 %, c) Pd(PPh3)2Cl2, DMF, 75°C, 1 h, 75 %, d) LTMP, then SnMe3Cl, THF, -80 °C, 99 %, e) air, Pd(PPh3)2Cl2, toluene, reflux, 18 h, 15 %, f)Pd(PPh3)2Cl2, DMF, 75 °C, 2 h, 14 %.

.SL[_P] �
#
However, analogous to what is described in chapter 2, it was found that reaction of 2-trimethylstannylthiophene with 4,7-dibromo-2,1,3-benzothiadiazole in dry DMF at 75 °Cwith the PdII(PPh3)2Cl2 catalyst gave a cleaner reaction in comparable yields. The syntheticScheme is outlined in Scheme 3. Thus, 7 was synthesized in 89% yield frombromobenzothiadiazole 12 and trimethylstannylthiophene 20 while 8 was synthesized from 2equivalents of 20 and dibromobenzothiadiazole 14 in 56% yield. To investigate the effect ofbithiophene as the electron-releasing unit, co-trimer 9 and co-tetramer 10 were synthesizedfrom 2-trimethylstannylbithiophene 21 and (di)bromobenzothiadiazoles 12 and 14, in 75%and 15% yield, respectively. Especially notable is the low yield of 10, probably connectedwith the size of the oligomer and its low solubility. Finally, co-hexamer 11 was synthesizedby a PdII(PPh3)2Cl2 catalyzed oxidative coupling of the trimethylstannyl compound 22 in thepresence of air. This reaction is often encountered as a side reaction during a Stille couplingwhen the reaction mixture is not adequately deaerated. Other examples in which this homo-coupling is utilized are known.43
Thus, co-trimer 8 was monostannylated to 22 in 99% yield, in the same way ascompounds 1,Boc and 4,Boc, with LTMP and SnMe3Cl in THF at -80 °C. Subsequently,stannyl compound 22 was heated under reflux in toluene in the presence of air andPdII(PPh3)2Cl2 to give 11 in 15% yield. The ESI-MS spectrum showed minor amounts ofmethyl and dimethyl substituted derivatives of 11, the origin of which is not yet clarified.Compound 11 is insoluble in most organic solvents, and is slightly soluble in solvents likeCHCl3 and DMSO.
����;]Z[P]_TP^�ZQ�_SP�NZ�ZWTRZXP]^
����� @A�AT^ ^[PN_]Z^NZ[d
The donor-acceptor character of the oligomers is manifested in the UV/Vis spectra(Table 1).
Class Compound λmax [nm] Emax [eV]
1 442 2.81“P-B” 2 532 2.33
3 599 2.07
4 421 2.95“P-Q” 5 502 2.47
6 535 2.32
7 390 3.188 447 2.77
“T-B” 9 429 2.8910 502 2.4711 521 2.38
Table 1. Optical properties of co-oligomers 1 - 11.
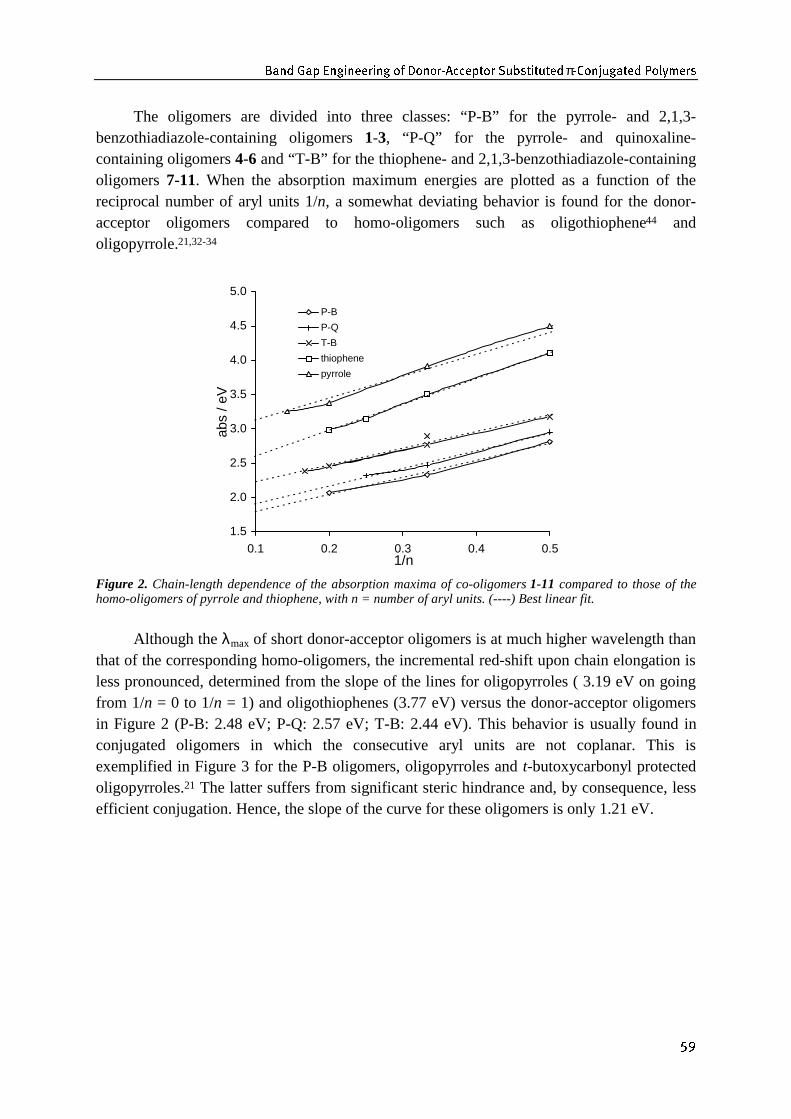
-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
$
The oligomers are divided into three classes: “P-B” for the pyrrole- and 2,1,3-benzothiadiazole-containing oligomers 1-3, “P-Q” for the pyrrole- and quinoxaline-containing oligomers 4-6 and “T-B” for the thiophene- and 2,1,3-benzothiadiazole-containingoligomers 7-11. When the absorption maximum energies are plotted as a function of thereciprocal number of aryl units 1/n, a somewhat deviating behavior is found for the donor-acceptor oligomers compared to homo-oligomers such as oligothiophene44 andoligopyrrole.21,32-34
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0.1 0.2 0.3 0.4 0.51/n
abs
/ eV
P-B
P-Q
T-B
thiophene
pyrrole
Figure 2. Chain-length dependence of the absorption maxima of co-oligomers 1-11 compared to those of thehomo-oligomers of pyrrole and thiophene, with n = number of aryl units. (----) Best linear fit.
Although the λmax of short donor-acceptor oligomers is at much higher wavelength thanthat of the corresponding homo-oligomers, the incremental red-shift upon chain elongation isless pronounced, determined from the slope of the lines for oligopyrroles ( 3.19 eV on goingfrom 1/n = 0 to 1/n = 1) and oligothiophenes (3.77 eV) versus the donor-acceptor oligomersin Figure 2 (P-B: 2.48 eV; P-Q: 2.57 eV; T-B: 2.44 eV). This behavior is usually found inconjugated oligomers in which the consecutive aryl units are not coplanar. This isexemplified in Figure 3 for the P-B oligomers, oligopyrroles and t-butoxycarbonyl protectedoligopyrroles.21 The latter suffers from significant steric hindrance and, by consequence, lessefficient conjugation. Hence, the slope of the curve for these oligomers is only 1.21 eV.

.SL[_P] �
!�
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0.1 0.2 0.3 0.4 0.51/n
abs
/ eV
P-B
N-Boc-pyrrole
pyrrole
Nn
Boc
Nn
H
N n
H
NS
N
/2
Figure 3. Chain-length dependence of the absorption maxima of compounds 1-3 (P-B series) compared to thoseof the homo-oligomers of pyrrole and N-Boc-pyrrole. n = number of aryl units.
����� 98= >[PN_]Z^NZ[d
The P-B as well as the P-Q oligomers are expected to be planar by virtue of hydrogenbonding, as depicted in Figure 1 by the dashed lines. In the proton NMR spectra ofcompounds 1-6 (CDCl3), the pyrrole N-H signal is found at low field (Table 2) which is anindication for such a hydrogen bond.
Compound δN-H in CDCl3 [ppm]
1 10.92 10.93 12.14 11.95 11.96 14.2/11.9
Table 2 Position of the pyrrole N-H absorption in the 1H -NMR spectrum (CDCl3) of compounds 1 - 6.
As shown in chapter 2, this signal showed no concentration dependence and a verysmall temperature dependence, which is an additional indication for strong intramolecularhydrogen bonding.45-47 Therefore, the smaller slope of the donor-acceptor oligomers in Figure2 is considered not to be caused by conformational factors but rather by electronic factors.
����� .dNWTN aZW_LXXP_]d
The bathochromic shift in the absorption maximum upon extension of a conjugated π-system originates from the dispersion of the HOMO and LUMO levels of the monomericunits into new bands until, in the case of conjugated polymers, a broad valence- andconduction band have emerged, from which these polymers lend their semiconductingproperties. The less pronounced bathochromic shift for the donor-acceptor oligomers should,

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
!�
therefore, be caused by a diminished dispersion of the HOMO and/or LUMO level(s) uponextension of these systems. To check this hypothesis, the cyclic Voltammograms ofcompounds 7, 9 and 10 were measured in a 0.1 M Bu4NPF6 solution in CH2Cl2 versus astandard calomel electrode (SCE), of which the data are summarized in table 3.
Compound Oxidation E01 [V] Reduction E0
1 [V]
7 1.62 -1.609 1.30 -1.4010 0.98 -1.32
Table 3. Cyclic voltammetry data for compounds 7, 9 and 10 vs. SCE in 0.1 M Bu4NPF6 in CH2Cl2.
Meerholz et al.48 have investigated the oxidation- and reduction potentials (vs.Ag/AgCl) of oligothiophenes versus the inverse chain length 1/n, where they determined thefollowing fits:
Thiophenes:
reduction E01(N) = −1.95 1/n − 1.42 (1)
oxidation E01(N) = +1.80 1/n + 0.35 (2)
In Figure 4, equations 1 and 2 are plotted together with the oxidation- and reduction data ofthe T-B oligomers.
-3
-2
-1
0
1
2
0.1 0.2 0.3 0.4 0.5 0.6
1/n
Pot
entia
l / V
Figure 4. Chain-length dependence of oxidation- and reduction potentials for oligothiophenes () vs.Ag/AgCl (eqs. 1 and 2) and for T-B oligomer oxidation (----××----) and reduction (----∗----) vs. SCE. n = Numberof aryl units.
Linear regression of the data for the T-B oligomers gives the following values:
T-B oligomers:
reduction E01(N) = −0.95 1/n − 1.11 (3)
oxidation E01(N) = +2.11 1/n + 0.57 (4)

.SL[_P] �
!�
Whereas the dispersion of the HOMO level for the T-B oligomers upon chainelongation is comparable to that of oligothiophenes (as concluded from the slopes ofequations 2 and 4), the dispersion of the LUMO level for the T-B oligomers is significantlylower (eqs. 1 and 3). The smaller slope for the donor-acceptor oligomers in Figure 2 istherefore mainly caused by the smaller dispersion of the LUMO level upon chain elongationcompared to homo-oligomers such as oligothiophenes and oligopyrroles.
����4X[WTNL_TZY^�QZ]�_SP�MLYO�RL[�PYRTYPP]TYR�ZQ�/�,�NZYU`RL_PO�[ZWdXP]^
The degree of dispersion of the HOMO and LUMO levels depends on the size of theAO coefficients on the coupling positions of the monomers. Preliminary semi-empiricalPMO/MNDO calculations49 on bithiophene and T-B oligomer 7 reveal that the LUMO AOcoefficients of the latter are indeed smaller, as was expected on the basis of the CV results. Inthe LUMO, the largest electron-density is found in the electron-deficient part of the donor-acceptor systems. When this part is located outside the polymer backbone, the dispersion ofthe LUMO level is diminished.
Therefore, it must be kept in mind that, when designing low band gap donor-acceptorconjugated polymers of this type, the initial reduction of the energy separation betweenHOMO and LUMO levels cannot be scaled to homopolymers such as polythiophene, toprevent a too optimistic estimation of the band gap. Preferentially, the acceptor unit musthave its electron-accepting part incorporated in the conjugated backbone.
When Figure 2 is reconsidered, it is remarkable that the slope for all three donor-acceptor oligomers is more or less equal at about 2.5 eV when going from 1/n = 0 to 1/n = 1.If it is assumed that this slope is found for all donor-acceptor oligomers, it gives us aquantitative guideline to design donor-acceptor conjugated polymers with the desired(optical) properties. This is exemplified in Figure 5, where the “low band gap area”, forexample, is supposed to start with polymers that have an absorption maximum of < 1 eV. Ofcourse, a few limitations hold:
1) The prediction is only valid for alternating donor-acceptor conjugated copolymers thatresemble the oligomers 1 - 11 and are completely planar.
2) Figure 5 is based on absorption maxima, not on band gaps. The “intrinsic conductors”area in the graph corresponds to polymers with an absorption maximum of 0 eV. Sincethe band gap is always lower in energy than the absorption maximum, this area may startalready at higher energy.
3) No experimental data are available to check the assumption that in the low-energy regionof Figure 5 the slope of the curve is still 2.5 eV.
4) For large numbers of n, it has been found that the linear relationship between Emax and1/n does not hold anymore.50 Beyond a certain number of repeat units (referred to as the“effective conjugation length” nECL), the absorption maximum is no longer shifted tohigher wavelengths, indicating that some kind of saturation is reached. To account forthis effect, the dotted lines beneath the “low band gap” and “intrinsic conductors” areas

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
!�
are introduced in Figure 5. An effective conjugation length nECL = 10 is assumed, whichmay seem rather low for these completely planar systems, yet it serves merely to indicatethe boundary case since a conjugation length of 10 repeat units is the minimum forsterically non-hindered conjugated polymers.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
0 0.1 0.2 0.3 0.4 0.5
1/n
ener
gy /
eV
P-B
low band-gap area
intrinsic conductors area
Figure 5. Application of the universal slope of 2.5 eV on low band gap donor-acceptor copolymers (for whichEabs(n→∞) = 1 eV is taken) and -possibly intrinsically conducting- zero band gap donor-acceptor copolymers(for which Egap(n→∞) = 0 is estimated by Eabs(n→∞) = 0).
With the aid of Figure 5, and bearing the many limitations in mind, it can now bepredicted that when a donor-acceptor conjugated copolymer is pursued with an absorptionmaximum of 1 eV (upper edge of the “low band gap area” in fig. 5), the corresponding D-Adimer must show a λmax of >550 nm (>620 nm with nECL = 10). A polymer with ahypothetical absorption maximum of 0 eV, must have a corresponding D-A dimer with anabsorption maximum of > 990 nm (>1240 nm with nECL = 10).
S
N N
NS
N
NN
H H
23
In a recent report of Tanaka et al., the donor-acceptor co-trimer 23 is described, thatexhibits an absorption maximum of 1345 nm.51 This value approaches the value of 1488 nmthat is predicted by the “intrinsic conductors” line in Figure 5 for a trimer, and, taken intoaccount the many assumptions, it may indeed be a monomer candidate for an intrinsicallyconducting polymer.

.SL[_P] �
!�
�� �.ZYNW`^TZY^
Various “push-pull” conjugated oligomers with varying chain length, consisting ofpyrrole or thiophene as the electron-rich subunit and 2,1,3-benzothiadiazole or quinoxaline asthe electron-deficient subunit, can be synthesized in moderate yields by means of the Stillecoupling. The incremental bathochromic shift in the absorption spectra upon chain elongationof the investigated oligomers —typically ~ 2.5 eV when going from 1/n = 0 to 1/n = 1— isnot as large as in homopolymers such as polythiophene (3.77 eV) and polypyrrole (3.19 eV).Cyclic voltammetry measurements show that this is mainly due to the diminished dispersionof the LUMO level upon chain elongation, which is supported by semi-empirical calculationsthat predict small LUMO AO coefficients at the coupling positions of the D-A monomers.
The incremental bathochromic shift appears to be ~ 2.5 eV for all three different classesof donor-acceptor oligomers. Based on this equality, predictions can be made —if someassumptions are taken into account— to which requirements the monomers must meet toyield low band gap or even intrinsically conducting conjugated polymers. For a polymer withan absorption maximum below 1 eV, the corresponding D-A co-dimer must show anabsorption maximum > 550 nm, whereas a hypothetical polymer with an absorptionmaximum of 0 eV requires a co-dimer with an absorption maximum of at least 990 nm. Withthese guidelines, it was verified that the recently published oligomer 23 indeed is a monomercandidate for an intrinsically conducting polymer.
However, compound 23 is unstable, probably due to its narrow HOMO-LUMO energyseparation and, consequently, ease of being oxidized and/or reduced. Therefore, compound 23is difficult to prepare. An interesting alternative offers a series of benzobisthiadiazolecontaining co-oligomers which are stable under ambient conditions and also display a narrowHOMO-LUMO energy separation. The co-trimer 24, for example, shows an absorptionmaximum of 700 nm while the corresponding polymer possesses an absorption maximum ofEmax = 1 eV and a band gap of Eg = 0.5 eV.41,42
NS
N
NS
N
S
S
24
The oligomers and polymers of this type satisfy the general rule for donor-acceptor π-conjugated systems −the incremental bathochromic shift of 2.5 eV− introduced above. Thepredicted value of Ep = 0.95 eV, based on the absorption maximum of 24, corresponds withthe reported experimental absorption maximum of Emax = 1 eV. Rational design byintroducing structural variations in oligomers with the basic structure of 24 may lead tooligomeric and polymeric systems with even smaller band gaps. Unfortunately, the synthesis

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
!
of these oligomers implies an inefficient multi-step procedure52 for which an alternative two-step synthesis was developed. These results will be presented in the next chapter.
��!�0c[P]TXPY_LW�^PN_TZY
2PYP]LW _PNSYT\`P^� Electrospray-MS (ESI/MS) measurements were performed on a Perkin-Elmer/Sciex API300 mass spectrometer. Cyclic voltammetry was performed in CH2Cl2/Bu4NPF6 (0.1 mol l-1) at 295 K, scan rate100 mV s-1, potential vs. SCE calibrated using Fc/Fc+ (0.470 V). For other general techniques the reader isreferred to section 2.5.
���9�_�-`_ZcdNL]MZYdW�[d]]ZW���dW��������MPYeZ_STLOTLeZWP F��-ZNH�� In a 50 ml flask, N-t-butoxycarbonyl-2-trimethylstannyl-pyrrole 13 (1.04 g, 3.10 mmol) and 4-bromo-2,1,3-benzothiadiazole 12 (0.65 g, 3.00 mmol)were dissolved in a mixture of toluene and 1 M Na2CO3 (1:1, 20 ml). This mixture was deaerated and broughtunder an argon atmosphere. Then, tetrakis(triphenylphosphine)palladium(0) (2 mol%) was added and theresulting mixture was heated under reflux for 48 h. Subsequently, the reaction mixture was allowed to cool toroom temperature, and the organic layer was separated from the aqueous layer. The water layer was extractedthree times with ether, and the combined organic layers were dried with MgSO4, filtered and evaporated to givecrude 1,Boc as a brown oil (1.25 g). This oil was subjected to column chromatography on Al2O3 (hexane :dichloromethane = 5:1 as the eluent) to give pure 1,Boc (0.49 g, 0.00163 mol, 77.8 %) as a fluorescent green-yellow solid. M.p. 71 °C; dec. 200 °C. (1H-NMR (400 MHz, CDCl3) δ: 7.95 (dd, J = 8.7 and 1.1 Hz, 1H, H-7btd), 7.60 (dd, J = 8.7 and 6.8 Hz, 1H, H-6 btd), 7.54 (dd, J = 6.8 and 1.1 Hz, 1H, H-5 btd), 7.50 (m, 1H, H-5pyr), 6.40 (dd, J = 3.3 and 1.77 Hz, 1H, H-3 pyr), 6.33 (m, 1H, H-4 pyr), 1.10 (s, 9H, Me Boc). 13C-NMR (100MHz, CDCl3) δ: 154.7, 154.5, 149.0, 130.0, 129.4, 128.5, 127.5, 123.4, 120.5, 115.6, 110.7, 83.18, 27.29.UV/Vis (CHCl3): λmax = 369 nm. IR (KBr): ν = 2975, 1737, 1312, 1149, 848-448 cm-1. Anal. calcd. forC15H15N3O2S: C, 59.78; H, 5.02; N, 13.94. Found: C, 60.13; H, 5.09; N, 13.97.
���;d]]ZW���dW��������MPYeZ_STLOTLeZWP F�H�� ��-ZN (6 mg, 0.2 mmol) was put in a 10 ml flask and heated on anoil bath at 200 °C. Evolution of CO2 and isobutene and a rapid color change of the solid were observed. After 30min the flask was allowed to cool to room temperature, in which pure 1 (4 mg, 0.19 mmol, 95 %) was found as adark yellow solid. M.p. > 200 °C. 1H-NMR (400 MHz, CDCl3) δ: 10.9 (s, 1H, N-H), 7.78 (dd, J = 7.2 and 0.7Hz, 1H, H-7 btd), 7.74 (dd, J = 8.8 and 0.7 Hz, 1H, H-5 btd), 7.55 (dd, J = 8.8 and 7.2 Hz, 1H, H-6 btd), 7.02(m, 1H, H-5 pyr), 6.89 (m, 1H, H-3 pyr), 6.35 (m, 1H, H-4 pyr). 13C-NMR (100 MHz, CDCl3) δ: 155.5, 151.5,130.2, 129.2, 124.8, 121.9, 120.3, 117.7, 109.9, 107.5. UV/Vis (CHCl3): λmax = 442 nm. IR (KBr): ν = 3395,1481, 1091, 880-450 cm-1. MS (70 eV, EI) m/z (%): 200.85 (100) [M+. -H]. Anal. calcd. for C10H7N3S: C,59.68; H, 3.51; N, 20.88; Found: C, 59.13; H, 3.63; N, 20.61.
��"�-T^��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��������MPYeZ_STLOTLeZWP F��-ZNH�� This compound was prepared, asdescribed for 1,Boc, by a Stille coupling between dibromo-2,1,3-benzothiadiazole 14 (2.0 g, 6.8 mmol) and N-Boc-2-trimethylstannylpyrrole 13 (4.5 g, 14 mmol) in a mixture of toluene and 1M Na2CO3 (1:1, 100 ml),reaction time 48 h. Column chromatography of the crude reaction workup product (3.53 g) over Al2O3 withhexane : dichloromethane 5:1 yielded pure 2,Boc (1.32 g, 2.83 mmol, 41.6 %) as a fluorescent orange solid. 1H-NMR (400 MHz, CDCl3) δ: 7.58 (s, 2H, H btd), 7.49 (dd, J = 3.3 and 1.8 Hz, 2H, H-5 pyr), 6.41 (dd, J = 3.3and 1.8 Hz, 2H, H-3 pyr), 6.34 (t, J = 3.3 Hz, 2H, H-4 pyr), 1.19 (s, 9H, Me Boc). 13C-NMR (100 MHz, CDCl3)δ: 154.8, 149.1, 130.2, 127.4, 125.9, 123.4, 115.6, 110.8, 83.4, 27.4. UV/Vis (CHCl3): λmax = 401 nm. IR(KBr): ν = 2976, 1743, 1318, 1138, 846-400 cm-1. Anal. calcd. for C24H26N4O4S: C, 61.78; H, 5.61; N, 12.00.Found: C, 61.17; H, 6.05; N, 11.82.
��"�-T^��[d]]ZW���dW��������MPYeZ_STLOTLeZWP F�H�� This compound was prepared, as described for 1, from2,Boc (0.196 g, 0.43 mmol) to give 2 (0.1084 g, 0.407 mmol, 95.5 %) as a deep purple solid.M.p. > 200 °C. 1H-NMR (400 MHz, CDCl3) δ: 10.9 (s, 1H, N-H), 7.84 (s, 1H, H btd), 7.03 (m, 1H, H-5 pyr),6.88 (m, 1H, H-3 pyr), 6.37 (m, 1H, H-4 pyr). 13C-NMR (100 MHz, CDCl3) δ: 152.5, 129.5, 123.2, 121.4,120.0, 110.0, 107.0. UV/Vis (CHCl3): λmax = 532 nm. IR (KBr): ν = 3415, 1481, 1113, 888-450 cm-1. GC/MS:M+. 265.95 (100%). Anal. calcd. for C14H10N4S: C, 63.61; H, 3.05; N, 21.20. Found: C, 63.92; H, 3.47; N,21.18

.SL[_P] �
!!
���9�_�-`_ZcdNL]MZYdW���_]TXP_SdW^_LYYdW�[d]]ZW� �dW��������MPYeZ_STLOTLeZWP F� H�� In a 50 ml flask, asolution of 2,2,6,6-tetramethylpiperidine (TMP, 0.1 g, 0.7 mmol) in dry THF (20 ml) was cooled to -80 °C andsubsequently treated with a 1.6 M solution of n-butyllithium in hexane (0.44 ml, 0.7 mmol). This mixture wasstirred for 15 min at -80 °C, warmed to room temperature, stirred for another 15 min and recooled to -80 °C. Asolution of 1,Boc (0.191 g, 0.63 mmol) in THF (5 ml) was added and the reaction mixture was stirred at -80 °Cfor 30 min. The reaction mixture was then quenched with a solution of SnMe3Cl (0.14 g, 0.7 mmol) in THF (5ml) and subsequently warmed to room temperature. The THF was evaporated and the residue was dissolved inether/water. The layers were separated and the water layer was extracted with ether. The combined organiclayers were washed with brine, dried over MgSO4 and evaporated to give the crude stannyl compound 15 as adark oil. This oil was dissolved in hexane and filtered over Al2O3, which, after evaporation, gave the purestannyl compound 15 (0.169 g, 0.36 mmol, 57 %) as a fluorescent yellow/green oil. 1H-NMR (400 MHz, CDCl3)δ: 7.93 (dd, J = 8.8 and 1.1 Hz, 1H, H-7 btd), 7.59 (dd, J = 8.8 and 6.7 Hz, 1H, H-6 btd), 7.49 (dd, J = 6.7 and1.1 Hz, 1H, H-5 btd), 6.49 (d, J = 3.2 Hz, 1H, H-3 pyr), 6.46 (d, J = 3.1 Hz, 1H, H-4 pyr), 0.79 (s, 9H, MeBoc), 0.32 (s, 9H, SnMe3). 13C-NMR (100 MHz, CDCl3) δ: 154.9, 154.7, 151.0, 139.0, 133.2, 129.7, 129.4,127.2, 121.3, 120.2, 116.8, 82.75, 26.93, -8.0. Anal. calcd. for C18H23N3O2SnS: C, 46.58; H, 4.99; N, 9.05.Found: C, 46.90; H, 5.31; N, 8.66.
��"�-T^�F ��������MPYeZ_STLOTLeZW���dW��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dWH�������MPYeZ_STLOTLeZWP F��-ZNH��
This compound was prepared, as described for 1,Boc, by a Stille coupling between 15 (0.169 g, 0.36 mmol) and4,7-dibromo-2,1,3-benzothiadiazole 14 (0.051 g, 0.17 mmol) in a mixture of toluene and 1M Na2CO3 (1:1, 10ml), reaction time 72 h. The crude reaction product (0.22 g) was subjected to column chromatography overAl2O3 with hexane:dichloromethane = 3:1 as the eluent to give pure 3,Boc (30 mg, 0.031 mmol, 11.3 %) as afluorescent yellow oil. 1H-NMR (400 MHz, CDCl3) δ: 8.00 (dd, J = 8.7 and 1.4 Hz, 2H, H-7 btd’), 7.79 (s, 2H,H-5 btd), 7.72 (dd, J = 6.9 and 1.4 Hz, 2H, H-5 btd’), 7.67 (dd, J = 8.6 and 6.8 Hz, 2H, H-6 btd’), 6.55 (d, J =3.4 Hz, 2H, pyr), 6.52 (d, J = 3.5 Hz, 2H, pyr), 0.74 (s, 9H, Me Boc). 13C-NMR (100 MHz, CDCl3) δ: 154.8,154.4, 154.2, 149.1, 132.4, 132.3, 129.6, 128.5, 128.1, 127.5, 120.7, 114.3, 114.1, 83.26, 26.94. UV/Vis(CHCl3): λmax = 412 nm. IR (KBr): ν = 2976, 1751, 1304, 1146, 872-400 cm-1. Anal. calcd. for C36H30N8O4S3:C, 58.84; H, 4.11; N, 15.24. Found: C, 58.79; H, 3.96; N, 15.25.
��"�-T^�F ��������MPYeZ_STLOTLeZW���dW��[d]]ZW���dWH�������MPYeZ_STLOTLeZWP F�H�� This compound wasprepared as described for 1, however, the deprotection was performed under vacuum for 45 min. In this way, theBoc protected precursor 3,Boc (22.9 mg, 0.031 mmol) was deprotected to yield 3 (16.5 mg, 0.031 mmol, 100%)as a deep blue powder. M.p. > 200 °C. 1H-NMR (400 MHz, CDCl3) δ: 12.1 (s, 2H, N-H), 7.94 (s, 2H, H btd),7.89 (dd, J = 8.0 and 0.8 Hz, 2H, H-7 btd’), 7.83 (dd, J = 8.8 and 0.9 Hz, 2H, H-5 btd’), 7.66 (dd, J = 8.8 and8.1 Hz, 2H, H-6 btd’), 7.1 (m, 2H, H-3,4 pyr). 13C-NMR (100 MHz, CDCl3) δ: 130.1, 123.5, 122.3, 118.2,110.0, 109.6 (owing to the poor solubility of this compound in CDCl3, only the peaks corresponding to carbonnuclei bearing a proton could be detected). UV/Vis (CHCl3): λmax = 599 nm. IR (KBr): ν = 3308 (broad), 1475,1120, 875-450 cm-1. ES/MS: m/z: 533.9 [M+ +H]. Anal. calcd. for C26H14N8S3: C, 58.41; H, 2.64; N, 20.96.Found: C, 58.26; H, 2.70; N, 20.34.
��9�_�-`_ZcdNL]MZYdW�[d]]ZWP���dW��\`TYZcLWTYP F��-ZNH�� This compound was prepared, as described for1,Boc, by a Stille coupling between 5-bromoquinoxaline 16 (0.30 g, 1.44 mmol) and 13 (0.57 g, 1.7 mmol) in amixture of toluene and 1M Na2CO3 (1:1, 20 ml) reaction time 72 h. Chromatography of the crude reactionproduct (0.8 g) on Al2O3 with hexane:dichloromethane = 3:1 as the eluent gave 4,Boc as a fluorescent yellowsolid. 1H-NMR (400 MHz, CDCl3) δ: 8.81 (dd, J = 4.9 and 1.8 Hz, 2H, H-2,3 qui), 8.10 (t, J = 5.1 Hz, 1H, H-7qui), 7.77 (dd, J = 4.7 and 0.4 Hz, 2H, H-6,8 qui), 7.52 (d, J = 2.8 Hz, 1H, H-5 pyr), 6.35 (d, J = 2.8 Hz, 2H, H-3,4 pyr), 1.02 (s, 9H, Me Boc). 13C-NMR (100 MHz, CDCl3) δ: 149.1, 144.5, 144.3, 142.7, 142.5, 134.8, 130.6,129.8, 129.5, 129.1, 122.7, 115.2, 110.6, 82.72, 27.21. Anal. calcd. for C17H17N3O2: C, 69.14; H, 5.80; N,14.23. Found: C, 69.13; H, 5.89; N, 14.09.
��;d]]ZW���dW��\`TYZcLWTYP F�H�� This compound was prepared, as described for 1, from 4,Boc (0.15 g, 0.51mmol) to give 4 (0.11 g, 0.41 mmol, 95.2 %) as a dark yellow solid. 1H-NMR (300 MHz, CDCl3) δ: 11.98 (s,1H, N-H), 8.89 (d, J = 1.7 Hz, 1H, H-2,3 qui), 8.83 (d, J = 1.8 Hz, 1H, H-2,3 qui), 8.15 (dd, J = 7.5 and 1.3 Hz,1H, H-8 qui), 7.89 (dd, J = 8.4 and 1.3 Hz, 1H, H-6 qui), 7.78 (t, J = 3.8 Hz, 1H, H-7 qui), 7.03 (m, 1H, H-5pyr), 6.93 (m, 1H, H-3 pyr), 6.35 (m, 1H, H-4 pyr). 13C-NMR (75 MHz, CDCl3) δ: 144.7, 143.8, 142.6, 139.4,

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
!"
129.8, 130.5, 126.3, 125.3, 119.9, 100.3, 100.0. UV/Vis (CHCl3): λmax = 421 nm. IR (KBr): ν = 3332, 1493,1465, 1088, 760, 734 cm-1. MS (70 eV, EI): m/z (%): 195 (100) [M+ -H].
�-]ZXZ�#��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��\`TYZcLWTYP F�#H�� This compound was prepared, as described for1,Boc, by a Stille coupling between 5,8-dibromoquinoxaline 17 (1.40 g, 4.7 mmol) and N-t-butoxycarbonyl-2-trimethylstannylpyrrole 13 (1.7 g, 5.2 mmol) in a mixture of toluene and 1 M Na2CO3 (1:1, 40 ml); reaction time72 h. Chromatography of the crude reaction product (1.0 g) over Al2O3 with hexane:dichloromethane = 2:1 asthe eluent gave 18 as a fluorescent yellow solid (0.40 g, 1.08 mmol, 23 %). 1H-NMR (300 MHz, CDCl3) δ: 8.84(d, J = 1.8 Hz, 1H, H-2,3 qui), 8.75 (d, J = 1.7 Hz, 1H, H-2,3 qui), 8.01 (d, J = 7.8 Hz, 1H, H-6,7 qui), 7.56 (d,J = 7.7 Hz, 1H, H-6,7 qui), 7.41 (m, 1H, H-5 pyr), 6.26 (m, 2H, H-3,4 pyr), 1.00 (s, 9H, Me Boc). 13C-NMR (75MHz, CDCl3) δ: 148.9, 146.0, 144.9, 144.8, 143.3, 134.7, 133.6, 133.0, 129.9, 123.3, 122.9, 115.4, 110.7,83.00, 27.25.
�#�-T^��9�_�M`_ZcdNL]MZYdW�[d]]ZW���dW��\`TYZcLWTYP F �-ZNH�� This compound was prepared, as described for1,Boc, by means of a Stille coupling between 18 (0.10 g, 0.27 mmol) and 13 (0.10 g, 0.22 mmol) in a mixture oftoluene and 1M Na2CO3 (1:1, 20 ml), reaction time 72 h. Chromatography of the crude reaction product (0.6 g)over Al2O3 with hexane:dichloromethane = 1:1 as the eluent gave 5,Boc as a fluorescent yellow oil (0.17 g, 0.17mmol, 75 %). 1H-NMR (300 MHz, CDCl3) δ: 8.78 (s, 2H, H-2,3 qui), 7.80 (s, 2H, H-5,6 qui), 7.52 (m, 2H, H-5pyr), 6.38 (m, 4H, H-3,4 pyr), 1.12 (Me Boc). 13C-NMR (75 MHz, CDCl3) δ: 149.8, 144.1, 142.2, 136.4, 131.8,129.7, 123.4, 115.7, 111.8, 27.9.
�#�-T^��[d]]ZW���dW��\`TYZcLWTYP F H�� This compound was prepared, as described for 1, from 5,Boc (0.10 g,0.20 mmol) to give 5 as a dark red solid (0.047 g, 0.18 mmol, 95%). 1H-NMR (300 MHz, CDCl3) δ: 11.9 (s, 2H,N-H), 8.87 (s, 2H, H-2,3 qui), 8.12 (s, 2H, H-6,7 qui), 7.01 (m, 2H, H-5 pyr), 6.90 (m, 2H, H-3 pyr), 6.35 (m,2H, H-4 pyr). 13C-NMR (75 MHz, CDCl3) δ: 142.4, 142.3, 130.6, 126.5, 125.9, 119.6, 109.3, 107.6. UV/Vis(CHCl3): λmax = 505 nm. IR (KBr): ν = 3362, 1469, 1107, 1083, 794, 725 cm-1. ESI/MS: m/z: 261.2 [M+ +H].
��9�_�-`_ZcdNL]MZYdW���_]TXP_SdW^_LYYdW�[d]]ZW� �dW��\`TYZcLWTYP F�$H�� This compound was prepared, asdescribed for 15, in dry THF (20 ml) with TMP (0.11 g, 0.75 mmol), n-BuLi (0.47 ml of a 1.6 M solution inhexane, 0.75 mmol), 4,Boc (0.20 g, 0.68 mmol) and SnMe3Cl (0.11 g, 0.75 mmol). The crude reaction workupproduct was subjected to column chromatography over Al2O3 with hexane : dichloromethane = 2:1 as the eluentto give 19 as a yellow oil (0.10 g, 0.22 mmol, 32 %) which was used in the next step without further purification.The proton spectrum of 19 is qualitatively equal to that of 4,Boc, except for the absence of the α-proton ofpyrrole and the appearance of the SnMe3 peak at 0.3 ppm.
���#��9�_�-`_ZcdNL]MZYdW�[d]]ZW���dW��\`TYZcLWTY� �dW�� ��\`TYZcLWTY� �dW��9�_�M`_ZcdNL]MZYdW�[d]]ZWP
F!�-ZNH�� This compound was prepared, as described for 1,Boc, by a Stille coupling between 18 (0.09 g, 0.24mmol) and 19 (0.10 g, 0.22 mmol) in a mixture of toluene and 1 M Na2CO3 (1:1, 20 ml); reaction time 72 h.Chromatography of the crude reaction product (0.8 g) over Al2O3 with hexane:dichloromethane = 1:1 as theeluent gave 6,Boc as a fluorescent yellow solid (0.12 g, 0.20 mmol, 85 %). 1H-NMR (400 MHz, CDCl3) δ: 8.81(d, J = 1.8 Hz, 1H, H-2 qui’), 8.79 (d, J = 1.8 Hz, 1H, H-3 qui’), 8.74 (d, J = 1.8 Hz, 1H, H-2 qui), 8.71 (d, J =1.8 Hz, 1H, H-3 qui), 8.13 (dd, J = 8.5 and 1.4 Hz, 1H, H-8 qui’), 8.00 (m, 2H, H-6,7 qui’), 7.85 (m, 2H, H-6,7qui), 7.51 (m, 1H, H-5 pyr’), 6.49 (s, 2H, H-3,4 pyr), 6.37 (m, 2H, H-3,4 pyr’), 1.10 (Me Boc), 0.59 (Me Boc’).13C-NMR (100 MHz, CDCl3) δ: 149.1, 149.0, 142.3, 142.2, 142.0, 141.9, 141.4, 141.3, 141.2, 141.2, 131.2,131.1, 131.0, 130.9, 130.8, 130.5, 130.1, 130.0, 129.9, 129.8, 129.7, 122.0, 112.2, 111.5, 110, 81.3, 81.0, 28.8,28.5.
���#��;d]]ZW���dW��\`TYZcLWTY� �dW�� ��\`TYZcLWTY� �dW��[d]]ZWP F!H�� This compound was prepared, as describedfor 1, from 5,Boc (0.10 g, 0.17 mmol) to give crude 5 as a purple solid. This solid was dissolved in THF andprecipitated in hexane to give pure 5 as purple needles (0.058 g, 0.15 mmol, 90 %). 1H-NMR (400 MHz, CDCl3)δ: 14.2 (s, 1H, N-H pyr), 11.9 (s, 1H, N-H pyr’), 9.01 (d, J = 1.7 Hz, 1H, H-3 qui), 8.98 (d, J = 1.7 Hz, 1H, H-1qui), 8.92 (d, J = 1.7 Hz, 1H, H-3 qui’), 8.89 (d, J = 1.6 Hz, 1H, H-2 qui’), 8.16 (m, 3H, H-6,7 qui & H-8 qui’),7.90 (dd, J = 8.3 and 1.1 Hz, 1H, H-6 qui’), 7.78 (t, J = 7.9 Hz, 1H, H-7 qui’), 7.00 (m, 4H, H-pyr’ & H-4 pyr),6.37 (m, 1H, H-4 pyr’). 13C-NMR (100 MHz, CDCl3) δ: 144.7, 143.8, 142.7, 142.5, 142.3, 139.9, 132.2, 131.8,130.7, 130.4, 130.0, 126.6, 126.4, 126.1, 125.8, 125.5, 119.7, 109.7, 109.4, 109.3, 107.8. UV/Vis (CHCl3): λmax

.SL[_P] �
!#
= 535 nm. IR (KBr): ν = 2959 (very broad), 1692, 1657, 1589, 1529, 1255, 771.8 cm-1. ESI/MS: m/z: 389.2 [M+
+H].
���?STPY���dW��������MPYeZ_STLOTLeZWP F"H�� 4-Bromo-2,1,3-benzothiadiazole 12 (1.00 g, 4.63 mmol) and 2-trimethylstannylthiophene 20 (1.14 g, 4.63 mmol) were dissolved in dry DMF (25 ml). The solution wasdegassed by evacuation of the flask with a single-stage vacuum pump (until effervescence of air ceased) andsubsequent introduction of dry argon gas. After repeating this cycle three times, Pd(II)(PPh3)2Cl2 catalyst (0.03g, 0.043 mmol) was added and the reaction mixture stirred at 75 °C for 60 min. The resulting orange solutionwas diluted with ether and extracted five times with ice-water to remove DMF. The ether layer was dried withMgSO4 and evaporated to give crude 7 (1.06 g), which was purified by sublimation of the solid in a kugelrohrapparatus at approx. 100 °C to give 0.90 g 7 (4.1 mmol, 89%) as a yellow-green solid. M.p. 46 °C. 1H-NMR(400 MHz, CDCl3, 300 MHz): δ: 8.11 (dd, J = 3.8 and 1.3 Hz, 1H, H-5 th), 7.9 (dd, J = 8.8 and 1.1 Hz, 1H, H-7btd), 7.85 (dd, J = 7.1 and 1.1 Hz, 1H, H-5 btd), 7.6 (dd, J = 7.2 and 8.8 Hz, 1H, H-6 btd), 7.5 (dd, J = 5.0 and1.1 Hz, 1H, H-3 th), 7.21 (dd, J = 3.8 and 5.0 Hz, 1H, H-4 th). 13C-NMR (100 MHz, CDCl3): δ: 155.4, 152.1,139.2, 132.1, 129.5, 127.9, 127.7, 126.7, 125.4, 120.0. UV/Vis (CHCl3) λmax = 390 nm. IR (KBr): ν: 1589,1541, 1484, 1427, 1210, 1166, 1046, 852, 820, 804, 753, 688, 504 cm-1. Anal. calcd. for C10H6N2S2: C, 55.02;H, 2.77; N, 12.83. Found: C, 55.46; H, 2.75; N, 12.66.
-T^���"��_STPY���dW��������MPYeZ_STLOTLeZWP F#H�� This compound was prepared as described for 7, from 4,7-dibromo-2,1,3-benzothiadiazole 14 (1.13 g, 3.84 mmol) and 2-trimethylstannylthiophene 20 (1.90 g, 7.69 mmol)in dry DMF (25 ml) with Pd(II)(PPh3)2Cl2 catalyst (0.108 g); reaction time 90 min. The crude reaction workupproduct was crystallized from CHCl3/hexane after treatment with norit to give 8 as highly fluorescent orangeneedles (0.64 g, 2.13 mmol, 55.5 %). M.p. 118 °C. 1H-NMR (400 MHz, CDCl3): δ: 8.07 (dd, J = 3.8 and 1.2Hz, 2H, H-5 th), 7.80 (s, 2H, H-5,6 btd), 7.42 (dd, J = 5.1 and 1.1 Hz, 2H, H-3 th), 7.18 (dd, J = 3.8 and 5.1 Hz,2H, H-4 th). 13C-NMR (100 MHz, CDCl3): δ [ppm]: 152.6, 139.3, 128.0, 126.0, 127.5, 126.8, 125.8. UV/Vis(CHCl3) λmax = 447 nm. IR (KBr): ν: 1526, 1481, 1422, 1379, 1216, 1073, 1042, 881, 825, 710, 700, 690, 508cm-1. Anal. calcd. for C14H8N2S3: C, 55.97; H, 2.68; N, 9.32. Found: C, 55.63; H, 2.56; N, 9.10.
������u�-T_STPY� �dW��������MPYeZ_STLOTLeZWP F$H�� This compound was prepared as described for 7, from 4-bromo-2,1,3-benzothiadiazole 12 (0.50 g, 2.32 mmol) and 2-thieno-5-trimethylstannylthiophene 21 (0.76 g, 2.32mol) in dry DMF (15 ml) with Pd(II)(PPh3)2Cl2 catalyst (0.032 g); reaction time 60 min. The crude reactionworkup product was chromatographed on silica gel with a gradient of dichloromethane/hexane (1:5 → 1:1) aseluent to give, after evaporation of the solvent, 9 as fluorescent orange plates (0.52 g, 1.73 mol, 74.8 %). M.p.156 °C. 1H-NMR (300 MHz, CDCl3): δ: 8.04 (dd, J = 3.9 and 1.1 Hz, 1H, H-5 th’), 7.9 (dd, J = 8.8 and 1.1 Hz,1H, H-7 btd), 7.83 (dd, J = 7.1 and 1.1 Hz, 1H, H-5 btd), 7.63 (dd, J = 7.1 and 8.6 Hz, 1H, H-6 btd), 7.3 (m,3H, H-3 th’ H-3,4 th), 7.07 (dd, J = 3.9 and 4.9 Hz, 1H, H-4 th’). 13C-NMR (75 MHz, CDCl3): δ: 155.1, 152.1,139.0, 138.5, 137.4, 129.6, 128.5, 128.0, 125.0, 124.6, 124.5, 124.1, 120.0. UV/Vis (CHCl3) λmax = 429 nm. IR(KBr): ν: 1527, 1479, 1445, 1039, 828, 800, 745, 715, 524, 491 cm-1. Anal. calcd. for C14H8N2S3: C, 55.97; H,2.68; N, 9.32. Found: C, 56.09; H, 2.74; N, 9.23.
-T^���"�����u�MT_STPY� �dW��������MPYeZ_STLOTLeZWP F��H�� This compound was prepared from 4,7-dibromo-2,1,3-benzothiadiazole 12 (0.22 g, 0.75 mmol) and 2-thienyl-5-trimethylstannylthiophene 21 (0.50 g, 1.5 mmol)in dry DMF (15 ml) with Pd(II)(PPh3)2Cl2 catalyst (0.042 g), reaction time 120 min. The crude reaction workupproduct was crystallized from CHCl3 to give 10 as lustrous, copper-like plates (50 mg, 0.108 mmol, 14.4 %).M.p. 188 °C. 1H-NMR (400 MHz, DMSO-d6): δ: 8.17 (s, 2H, H-5,6 btd), 8.15 (dd, 3.9 and 1.1 Hz, 2H, H-5 th’),7.59 (dd, J = 5.1 and 1.1 Hz, 2H, H-3 th’), 7.47 (m, 4H, H-3,4 th), 7.16 (dd, J = 3.6 and 5.1 Hz, 2H, H-4 th’).UV/Vis (CHCl3) λmax = 505 nm. IR (KBr): ν: 1480, 1226, 1064, 840, 796, 698, 683, 527 cm-1. Anal. calcd. forC22H12N2S5: C, 56.86; H, 2.60; N, 6.03. Found: C, 56.90; H, 2.60; N, 5.69.
"��?STPY���dW�������_]TXP_SdW^_LYYdW_STPY� �dW��������MPYeZ_STLOTLeZWP F��H�� 2,2,6,6-Tetramethyl-piperidine(TMP, 0.265 g, 1.7 mmol) was dissolved in dry THF (25 ml) under an argon atmosphere. The solution wascooled to -78 °C on a dry ice/acetone bath and n-butyllithium (1.06 ml of a 1.6 M solution in hexane, 1.7 mmol)was added rapidly. The resulting solution was allowed to warm to and kept at room temperature for 10 min, andsubsequently recooled to -78 °C. At this temperature, a solution of 8 (0.40 g, 1.3 mmol) in dry THF (5 ml) wasadded dropwise. The resulting deeply coloured solution was kept at -78 °C for 30 min., at which temperature asolution of SnMe3Cl (0.338 g, 1.7 mmol) in dry THF (5 ml) was added. The reaction mixture was then allowed

-LYO 2L[ 0YRTYPP]TYR ZQ /ZYZ]�,NNP[_Z] >`M^_T_`_POπ�.ZYU`RL_PO ;ZWdXP]^
!$
to warm to room temperature, and mixed with ether. The organic phase was extracted three times with dilutehydrochloric acid to remove TMP, dried over MgSO4 and evaporated to give a brown oil. This oil was filteredover aluminum oxide using hexane as the eluent. Evaporation of the orange coloured hexane filtrate gave 22 asfluorescent orange plates (0.55 g, 1.28 mmol, 99 %). 1H-NMR (400 MHz, CDCl3): δ: 8.17 (dd, J = 3.5 and 2.0Hz, 1H, H-5 th’), 8.10 (m, 1H, H-4 th’), 7.84 (s, 2H, H-5,6 btd), 7.44 (m, 1H, H-4 th), 7.29 (dd, J = 3.6 and 1.5Hz, 1H, H-3 th’), 7.20 (m, 1H, H-3 th), 0.43 (s, 9H, H-SnMe3).13C-NMR (100 MHz, CDCl3): δ: 152.6, 145.0,140.2, 139.3, 136.1, 128.5, 128.4, 128.0, 127.5, 127.3, 126.8, 126.6, 125.7, -8.17. Anal. calcd. forC17H16N2S3Sn: C, 44.07; H, 3.48; N, 6.04. Found: C, 43.67; H, 3.84; N, 5.45.
-T^� � u��"�_STPY���dW�������MPYeZ_STLOTLeZW���dW�����u�MT_STZ[SPYP F��H�� To a solution of trimethylstannylcompound 22 (0.12 g, 0.28 mmol) in 25 ml of toluene was added 0.02 g of Pd(II)PPh3Cl2 catalyst, withoutexclusion of air. The reaction mixture was then refluxed for 18 h, allowed to cool down to room temperature andthe black precipitate filtered off. The precipitate (ca. 0.07 g) was subjected to soxhlet extraction with hexane andchloroform. The intensely red coloured chloroform fraction was evaporated to give 0.06 g of a black powder.This powder was crystallized from chloroform at -20 °C to give 11 as a black powder (12.5 mg, 0.021 mmol,14.8 %). M.p. > 300 °C. UV/Vis (CHCl3) λmax = 521 nm. IR (KBr): ν: 1479, 1434, 1205, 1046, 881.2, 829.0,796.1, 695.8, 513.2 cm-1. ES/MS m/z = 598.1 (612.1, 626.2) amu. Anal. calcd. for C28H14N4S6: C, 56.16; H,2.36; N, 9.36. Found: C, 55.59; H, 2.55; N, 8.88.
=PQP]PYNP^
(1) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Polym. Bull. 1992, 29, 119.(2) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Synth. Met. 1993, 55-57, 299.(3) Tanaka, S.; Yamashita, Y. Synth. Met. 1993, 55-57, 1251.(4) Tanaka, S.; Yamashita, Y. Synth. Met. 1995, 69, 599.(5) Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Chem. Soc., Chem. Commun. 1994, 1585.(6) Brockmann, T. W.; Tour, J. M. J. Am. Chem. Soc. 1994, 116.(7) Brockmann, T. M.; Tour, J. M. J. Am. Chem. Soc. 1995, 117, 4437.(8) Zotti, G.; Zecchin, S.; Schiavon, G.; Berlin, A.; Pagani, G.; Canavesi, A.; Casalbore-Miceli, G. Synth.
Met. 1996, 78, 51.(9) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1997, 119, 5065.(10) Bakhshi, A. K.; Ago, H.; Yoshizawa, K.; Tanaka, K.; Yamabe, T. J. Chem. Phys. 1996, 104, 5528.(11) Bakhshi, A. K.; Ladik, J. Indian J. Chem. Sec. A 1997, 36, 1.(12) Albers, W. M.; Canters, G. W.; Reedijk, J. Tetrahedron 1995, 51, 3895.(13) Bauerle, P.; Wurthner, F.; Gotz, G.; Effenberger, F. Synthesis 1993, 1099.(14) Effenberger, F.; Wurthner, F.; Steybe, F. J. Org. Chem. 1995, 60, 2082.(15) Horne, J. C.; Blanchard, G. J.; LeGoff, E. J. Am. Chem. Soc. 1995, 117, 9551.(16) Muller, U.; Baumgarten, M. J. Am. Chem. Soc. 1995, 117, 5840.(17) Nakayama, J.; Dong, H.; Sawada, K.; Ishii, A.; Kumakura, S. Tetrahedron 1996, 52, 471.(18) ten Hoeve, W.; Wynberg, H.; Havinga, E. E.; Meijer, E. W. J. Am. Chem. Soc. 1991, 113, 5887.(19) Wei, Y.; Yang, Y.; Yeh, J.-M. Chem. Mater. 1996, 8, 2659-2666.(20) Bauerle, P.; Fischer, T.; Bidlingmeier, B.; Stabel, A.; Rabe, J. P. Angew. Chem. Int. Ed. Engl. 1995, 34,
303.(21) Groenendaal, L.; Peerlings, H. W. I.; van Dongen, J. L. J.; Havinga, E. E.; Vekemans, J. A. J. M.;
Meijer, E. W. Macromolecules 1995, 28, 116.(22) Jestin, I.; Frere, P.; Mercier, N.; Levillain, E.; Stievenard, D.; Roncali, J. J. Am. Chem. Soc. 1998, 120,
8150.(23) Kesslen, E. C.; Euler, W. B. Tetrahedron Lett. 1995, 36, 4725.(24) Klaerner, G.; Former, C.; Yan, X.; Richert, R.; Müllen, K. Adv. Mater. 1996, 8, 932.(25) Maddux, T.; Li, W.; Luping, Y. J. Am. Chem. Soc. 1997, 119, 844.(26) Pearson, D. L.; Schumm, J. S.; Tour, J. M. Macromolecules 1994, 27, 2348.(27) Pushkara Rao, V.; Jen, A. K.-Y.; Wong, K. Y.; Drost, K. J. Tetrahedron Lett. 1993, 34, 1747.(28) Yu, Y.; Gunic, E.; Zinger, B.; Miller, L. L. J. Am. Chem. Soc. 1996, 118, 1013.(29) Stille, J. K. Pure Appl. Chem. 1985, 57, 1771.(30) Stille, J. K. Angew. Chem. 1986, 98, 504.

.SL[_P] �
"�
(31) Groenendaal, L.; Peerlings, H. W. I.; Havinga, E. E.; Vekemans, J. A. J. M.; Meijer, E. W. Synth. Met.1995, 69, 467.
(32) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G. Synthesis 1991, 613.(33) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G. Synth. Met. 1992, 51, 299.(34) Martina, S.; Enkelmann, V.; Schluter, A.-D.; Wegner, G.; Zotti, G.; Zerbi, G. Synth. Met. 1993, 55,
1096.(35) Pilgram, K.; Zupan, M.; Skiles, R. J. Heterocycl. Chem. 1970, 7, 629.(36) Groenendaal, L.; van Loo, M. E.; Vekemans, J. A. J. M.; Meijer, E. W. Synth. Commun. 1995, 25,
1589.(37) Rawal, V. H.; Cava, M. P. Tetrahedron Lett. 1985, 26, 61.(38) Farina, V.; Krishnan, B. J. Am. Chem. Soc. 1991, 113, 9585.(39) Kanbara, T.; Yamamoto, T. Macromolecules 1993, 26, 3464.(40) Naef, R.; Balli, H. Helv. Chim. Acta 1978, 61, 2958.(41) Karikomi, M.; Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Am. Chem. Soc. 1995, 117, 6791.(42) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Mater. 1996, 8, 570.(43) Tamao, K.; Shigeki, O.; Yamaguchi, S. Chem. Commun. 1996, 1873.(44) Havinga, E. E.; Rotte, I.; Meijer, E. W.; ten Hoeve, W.; Wynberg, H. Synth. Met. 1991, 41-43, 473.(45) Hamuro, Y.; Geib, S. J.; Hamilton, A. D. Angew. Chem. 1994, 106, 465.(46) Palmans, A. R. A.; Vekemans, J. A. J. M.; Meijer, E. W. Recl. Trav. Chim. Pays-Bas 1995, 114, 277.(47) Palmans, A. R. A.; Vekemans, J. A. J. M.; Fischer, H.; Hikmet, R. A.; Meijer, E. W. Chem. Eur. J.
1997, 3, 300.(48) Meerholz, K.; Heinze, J. Electrochim. Acta 1996, 41, 1839.(49) Pomerantz, M.; Cardona, R.; Rooney, P. Macromolecules 1989, 22, 304.(50) Meier, H.; Stalmach, U.; Kolshorn, H. Acta Polym. 1997, 48, 379.(51) Tanaka, S.; Yamashita, Y. Synth. Met. 1997, 84, 229.(52) Yamashita, Y.; Ono, K.; Tomura, M.; Tanaka, S. Tetrahedron 1997, 53, 10169.

.SL[_P]��
4X[]ZaPO�>dY_SP_TN�;]ZNPO`]P^�?ZbL]O^�/T_STPYdW�MPYeZMT^F���� H_STLOTLeZWP^�LYO�;ZWd�OT_STPYdWLY_S]LNPYP�^
,M^_]LN_%�Based on the optimization of the known synthesis of 9,10-dithienylanthracene, atwo-step synthesis of 4,8-di(thien-2-yl)benzobis[1,2,5]thiadiazole is presented. This synthesisreplaces the original, inefficient five-step procedure. Although the copolymerization of 5,5’-dilithio-2,2’-bithiophene with benzobis[1,2,5]thiadiazole-4,8-dione did not give access topoly(4,8-dithienylbenzobis[1,2,5]thiadiazole)s, the corresponding copolymerization withanthraquinone was successful. This copolymerization represents an easy access to well-defined, unsubstituted poly(9,10-dithienylanthracene)s via a soluble, non-conjugatedprecursor polymer.
����4Y_]ZO`N_TZY
The structure-property relationship between the magnitude of the HOMO-LUMOenergy separation and the size of the oligomer was investigated in chapter 3 for variousdonor-acceptor systems. It followed that the design of the acceptor unit is crucial for theultimate band gap of the donor-acceptor polymer. Among the most powerful and versatileacceptor systems is the benzobis[1,2,5]thiadiazole unit 1. It has proven its efficiency inelectron acceptors like 2 for various molecular organic conductors1 as well as in donor-acceptor conjugated polymers2,3 like 3.
NS
N
NS
N
NS
N
NS
N
R R
CN CN
CNCN
NS
N
NS
N
S
S
n
1 2 3
Unfortunately, the preparation of co-oligomers containing thiophene andbenzobis[1,2,5]thiadiazole implies an inefficient multistep synthesis4, which is outlined inScheme 1. Due to the electron-withdrawing properties of the thiadiazole ring, introduction ofthe two nitro groups in step b) is very inefficient5 and is accompanied by the formation of alarge amount of by-products, which have to be separated by column-chromatography. Thisprevents the multigram-scale synthesis of the benzobis[1,2,5]thiadiazole containingmonomers. Moreover, the electrochemical preparation of polymer 3, although it providesfacile and quick access to a conjugated polymer, suffers from a number of drawbacks: i) thepolymerization process is known to give rise to a certain percentage (depending on the

.SL[_P] �
"�
monomer) of mislinkages in the backbone which diminishes the effective conjugation length,ii) the polymer is only available as a thin film on a conducting substrate (usually ITO/glass)and iii) the polymer is obtained in its doped state6. The latter fact is a severe drawback for lowband gap conjugated polymers, since the low-energy side of their absorption spectrum isseriously affected by the doping level and may give rise to inaccurate determination of thephysical properties, such as the band gap and intrinsic conductivity.
NS
N NS
N
BrBr
NS
N
BrBr
NO2O2N
NS
N
S
S
NO2O2N
NS
N
NS
N
S
S
a) b)
c)
d)e)
NS
N
S
S
NH2NH2
Scheme 1. Published synthesis of 4,7-dithienylbenzobis[1,2,5]thiadiazole: Reagents and conditions: a) Br2,AcOH, 90%, b) H2SO4/HNO3, 8 %, c) Bu3SnTh, Pd(PPh3)2Cl2, THF, 47 %, d) Fe, AcOH, 58%, e) N-sulfinylaniline, Me3SiCl, pyridine, 82%.
Therefore, the chemistry involving co-monomers and -polymers of thiophene andbenzobis[1,2,5]thiadiazole units needs significant improvement. It is not only desirable todevelop an alternative procedure that replaces the highly inefficient synthesis of Scheme 1,also a chemical polymerization route towards polymers with the basic structure of 3, yieldingneutral (undoped), processible low band gap conjugated polymers would be highlyappreciated.
����/P^TRY�LYO�=P_]Z^dY_SP_TN�,YLWd^T^
The opportunity for an alternative synthesis towards systems containing thebenzo[1,2,5]bisthiadiazole unit is provided by the established two-step synthesis of 9,10-di(thien-2-yl)anthracene 6 (Scheme 2). The reaction of anthraquinone, 4 with a large excessof 2-thienylmagnesium iodide, followed by aqueous workup, yields the dihydroxyintermediate 5 as a mixture of syn and anti diastereomers which can be reduced withKI/NaHSO3 to yield the dithienylanthracene 6 in good yield7. Adopting this approach forbenzobisthiadiazole analogues implies the usage of benzobis[1,2,5]thiadiazole-4,8-dione, 7, acompound previously described8,9 (Scheme 3). Furthermore, 2-thienylmagnesium iodide maybe substituted by 2-thienyllithium10 since the Grignard reaction with anthraquinone requires a
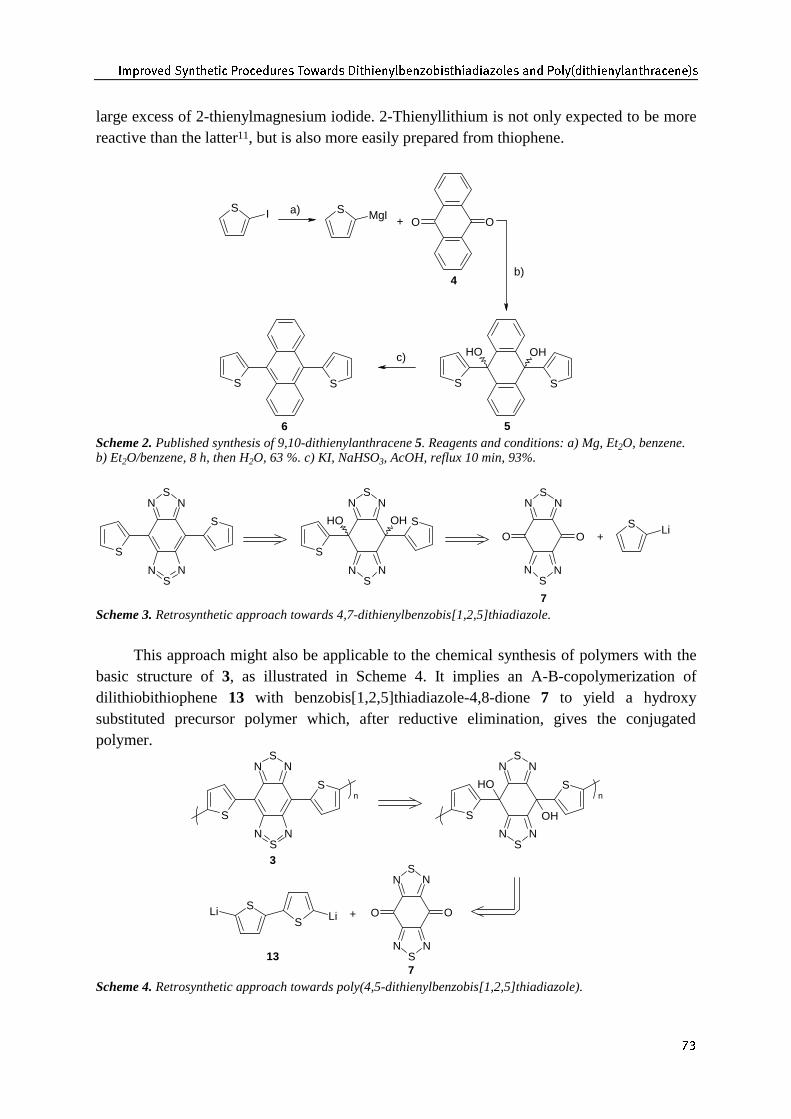
4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
"�
large excess of 2-thienylmagnesium iodide. 2-Thienyllithium is not only expected to be morereactive than the latter11, but is also more easily prepared from thiophene.
S IOO+
S S
OH OH
S S
4
56
S MgIa)
b)
c)
Scheme 2. Published synthesis of 9,10-dithienylanthracene 5. Reagents and conditions: a) Mg, Et2O, benzene.b) Et2O/benzene, 8 h, then H2O, 63 %. c) KI, NaHSO3, AcOH, reflux 10 min, 93%.
NS
N
NS
N
S
S
NS
N
NS
N
S
S
OHOH
NS
N
NS
N
OOS Li
+
7
Scheme 3. Retrosynthetic approach towards 4,7-dithienylbenzobis[1,2,5]thiadiazole.
This approach might also be applicable to the chemical synthesis of polymers with thebasic structure of 3, as illustrated in Scheme 4. It implies an A-B-copolymerization ofdilithiobithiophene 13 with benzobis[1,2,5]thiadiazole-4,8-dione 7 to yield a hydroxysubstituted precursor polymer which, after reductive elimination, gives the conjugatedpolymer.
NS
N
NS
N
S
Sn
O O
NS
N
NS
N
S
SLi Li
NS
N
NS
N
S
Sn OH
OH
+
3
713
Scheme 4. Retrosynthetic approach towards poly(4,5-dithienylbenzobis[1,2,5]thiadiazole).

.SL[_P] �
"�
In this chapter, the alternative synthesis of 4,8-dithienobenzobis[1,2,5]thiadiazolesfollowing the synthetic strategy of Scheme 3 is described, as well as the attempts towards analternative synthesis of polymers with the basic structure of 3.
����4X[]ZaPO�^dY_SP^T^�ZQ�OT_STPYdWMPYeZMT^F���� H_STLOTLeZWP^
The improved synthesis of dithienylbenzobis[1,2,5]thiadiazoles, following theapproach of Scheme 3, is preceded here by the optimization of the dithienylanthracenesynthesis. This model reaction should furnish the appropriate reaction conditions for thebenzobis[1,2,5]thiadiazole based oligomeric and polymeric systems.
����� :[_TXTeL_TZY ZQ _SP OT_STPYdWLY_S]LNPYP ^dY_SP^T^
Although the reaction of thienyllithium derivatives with anthrone is known10, the use of2-thienyllithium in the synthesis of the dithienylanthracene 6 has not been reported yet(Scheme 5).
O OS Li
+
4
5,anti
S
SOH
OH SS
OHOH
5,syn
S a)
b)
Scheme 5. Synthesis of diastereomers 5. Reagents and conditions: a) n-BuLi, THF, -80 °C. b) THF, r.t., thenH2O, quant.
Addition of anthraquinone 4 to a solution of 2-thienyllithium (prepared from theaddition of an n-BuLi solution to a solution of thiophene in dry THF) at -80 °C followed byaqueous workup yielded dihydroxy intermediate 5 as a mixture of syn and anti diastereomers,also observed for the analogous reaction with thienylmagnesium iodide7, in quantitative yield.Crystallization of this mixture from CHCl3 yielded one isomer in its pure form. Regarding theincreased polarity of the syn isomer and, hence, its diminished solubility in chloroform (arelatively apolar solvent) the pure isomer is assumed to be the syn-isomer. Furthermore, theother isomer could be obtained quantitatively from equilibration towards one isomer bytreatment of the crude diastereomeric mixture with aqueous HCl. This process is visualized inScheme 6.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
"
S
OH S
OH
S
OH OH
S
S
S
OH+
HO
H
5,syn
5,anti
+ H+
- H2O
Scheme 6. Reaction of the diastereomeric mixture 5 with aqueous acid.
Treating a mixture of 5,syn and 5,anti in CH3COOH with a few drops of aqueous HCl(37%) causes protonation of the alcohol group, followed by the release of water, leaving apositive charge on the tertiary carbon. The use of aqueous HCl implies the presence of a largeexcess of water which immediately attacks the carbocation to give back either of the isomers,depending on the side at which water attacks. The anti isomer is expected to be formedpreferentially, since it is likely to be the thermodynamically most stable isomer: it lacks adipole moment and its thiophene units are further apart, giving rise to diminished stericcrowding.
The described reductions of analogues of dihydroanthracene 5, using powerful reducingagents like SnIICl2 12 or NaHSO3/KI in boiling acetic acid,7 are not useful since the reactionconditions have to be applicable to 2,1,3-thiadiazole containing heterocycles, which aresensitive towards reduction.13 Therefore, an alternative procedure was investigated usingmineral acids. It is known that hydroiodic acid reduces diols of type 5 within a few minutes atambient temperature,14-16 via the proposed reaction sequence of Scheme 7.
Ar
OH
Ar
OH
RR
RR
Ar
I
Ar
I
RR
RR
RR
RR
ArAra) b)
Scheme 7. Reductive elimination using HI. a) + 2 HI, −2 H2O. b) −I2
Protonation of the alcohols, followed by the attack of iodide ions yields a diiodointermediate, which is unstable17 and eliminates I2 −via a mechanism that has not beenclarified yet− to give the aromatized compound. Indeed, compound 5 was reduced in this wayto the anthracene derivative 6. However, since HI is only available as an aqueous solution, alarge excess is necessary to guarantee sufficient protonation of the hydroxy groups in 5. Dueto the reducing power of HI, this will make the procedure again unsuitable for the reductionof 2,1,3-thiadiazole-containing analogues.
To provide possible alternatives, the action of the mineral acids HCl, HBr and HI oncompound 5 was investigated (Scheme 8).

.SL[_P] �
"!
S S
OH OH
SS XSS+
5 6 8
a)
Scheme 8. Reaction of dihydroxycompound 5 with mineral acids. a) + 2 HX, -2 H2O, -X2
A suspension of compound 5 in AcOH was treated with gaseous HCl, a solution of HBrin AcOH and a solution of HI in H2O, respectively. Not only the unsubstituteddithienylanthracene 6 but also the monohalogenated derivative 8 (confirmed by elementalanalysis) were, depending on the acid, formed in different ratios. Halogenation is known tooccur during similar reductive eliminations with HCl.18-20 The results (based on 1H-NMRintegrals) are summarized in table 1.
Acid Time/temp Ratio6 8
HCl 5 min./r.t. 0 % 100 % (X = Cl)HBr 30 min./r.t. 76 %* 24 %* (X = Br)HI 15 min./reflux 100 % 0 % (X = I)
* EstimatedTable 1. Product distribution after treatment of dihydroxycompound 5 with various mineral acids.
It was concluded that halogenation occurs at the 5-position (or α-position) of thiopheneby comparing the coupling constants of the thiophene protons in the 1H-NMR of 8. Thecommon coupling constants between the protons at the 3, 4 and 5 positions that are found in2-substituted thiophenes (see the experimental data of chapter 3) are depicted in Figure 1.
SR H
HHJ ~ 5.0 Hz
J ~ 3.5 Hz
J ~ 1.0 Hz1
2
43
5
Figure 1. Common 1H-NMR coupling constants between the protons at the 3, 4 and 5 positions in 2-substitutedthiophenes.
Only substitution at the 5-position can result in a pair of doublets with J ~ 3.5 Hz. Thisis the case in the 1H-NMR of 8, where a pair of doublets is found (7.09 and 6.94 ppm) with acoupling of J = 3.6 Hz. It is remarkable that only monohalogenated products are produced,which excludes the possibility that halogenation is a result of the action of the Cl2, Br2 or I2that is formed upon elimination. The latter process would inevitably yield dihalogenatedspecies, besides mono- and non-halogenated species. Presumably, the elimination of the

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
""
dihalo intermediate (e.g. the diiodo intermediate of Scheme 7) is catalyzed by halide ions (X−)according to Scheme 9.
Ar Ar
X X
RR
RR
X
RR
RR
ArAr + X2 + X
Scheme 9. Suggested catalysis of the reductive elimination by halide ions.
The mechanism of Scheme 9 implies that the capability of X− to catalyse the reaction is
proportional to its nucleophilicity. Because thiophene itself is nucleophilic, it is also able to“catalyse” the reaction according to Scheme 9, probably via an intramolecular process to yieldthe monohalogenated species. The increase in nucleophilicity in the series Cl−, Br− and I−
explains the different ratios between 6 and 8. Cl− is only a weak nucleophile compared tothiophene, resulting in exclusive formation of the monochlorinated thiophene species. Thenucleophilicity of Br− is more comparable to that of thiophene, which implies competitionbetween Br− and thiophene, resulting in a mixture of monobrominated and unsubstitutedthiophene species. I− is far more nucleophilic than thiophene and, therefore, governs thereaction, resulting in non-iodinated thiophene species. It should be remarked that thepercentage of 8,X=Br is not completely accurate due to overlap of signals in 1H-NMR, and,due to its high boiling point, the impossibility of using gas chromatography to quantify theresult.
The above observations imply that pure HCl and HBr cannot be applied in the reductiveelimination of 5, if unsubstituted 6 is to be obtained. Therefore, the halogenation step must becircumvented by the addition of iodide ions. Indeed, when a mixture of thedihydroxycompound 5 and 5 equivalents of NaI in acetic acid was treated with a 33 % HBrsolution in acetic acid (approx. 5 eq.) at room temperature, the anthracene derivative 6 wasformed within 5 minutes in 99 % yield. The use of hydrobromic acid is the most convenient,since a 33% solution in acetic acid is commercially available.
����� 4X[]ZaPO ^dY_SP^T^ ZQ OT_STPYdWMPYeZMT^F���� H_STLOTLeZWP^
Adopting the conditions described for the synthesis of 5, the reaction between 2-thienyllithium derivatives and benzobis[1,2,5]thiadiazol-4,8-dione 7 was performed (Scheme10).
SR Li+
7
O O
NS
N
NS
N
S
S
NS
N
NS
N
OH
OHR
R
R = HR = n-C6H13
9: R = H10: R = n-C6H13
SR
R = HR = n-C6H13
a) b)
Scheme 10. Synthesis of dihydroxycompounds 9 and 10. Reagents and conditions: a) n-BuLi, THF, -80 °C, b)THF, -80 to -40 °C, 15 - 180 min, then AcOH, 59 - 64 %.

.SL[_P] �
"#
Addition of dione 7 to a solution of 2-thienyllithium or 2-(5-hexylthienyl)lithium inTHF at −80 °C, followed by quenching with AcOH and aqueous workup affordeddihydroxycompounds 9 and 10 in 59 and 64 % yield, respectively. The use of AcOH wasbeneficial since quenching with water generates OH− ions, which can attack the thiadiazolerings. A remarkable observation is that, in contrast to anthraquinone, only one isomer isobtained in this reaction, the structure of which has not been identified.
Applying the reaction conditions, used for the reduction of dihydroxyanthracene 5, todihydroxycompounds 9 and 10 yields benzobis[1,2,5]thiadiazole compounds 11 and 12,respectively (Scheme 11).
S
S
NS
N
NS
N
OH
OHR
R
9: R = H10: R = n-C6H13
S
S
NS
N
NS
NR
R
11: R = H12: R = n-C6H13
a)
Scheme 11. Synthesis of aromatized compounds 11 and 12. Reagents and conditions: a) NaI/HBr, AcOH, r.t.,10 - 30 min, 67 - 53 %.
Addition of a 33 % HBr solution in AcOH to a mixture of 9 or 10 and NaI (5 eq.) inAcOH gave, after 10 to 30 minutes of stirring and aqueous workup, the compounds 11 and 12in 67 and 53 % yield, respectively. The reported purification of 11 by sublimation at 300 °Cin vacuo3 did not work properly, which resulted in dissatisfying microanalytical analyses.NMR, UV and mass spectral data, however, are in full agreement with the structure of 11.
The above procedure implies a drastic improvement of the synthesis of compounds withthe basic structure of 11. Compounds 11 and 12 show strongly red-shifted absorption maximaof λmax = 700 nm and λmax = 756 nm, respectively, which is due to their strong donor-acceptorinteraction. Increasing the donor-strength of the thiophene unit by substitution of electron-donating hexyl groups on the 5-positions, results in a bathochromic shift of 56 nm (0.13 eV).The effect of replacing thiophene with even stronger electron-donating units such as pyrroleor alkyloxy-substituted thiophenes, is therefore an exciting challenge in order to obtainmonomers which will give rise to polymers with a narrowing energy separation.
����� .Z[ZWdXP]TeL_TZY ZQ OTWT_STZMT_STZ[SPYP bT_S MPYeZMT^F���� H_STLOTLeZWPOTZYP
The copolymerization of benzobis[1,2,5]thiadiazol-4,8-dione 7 and dilithio-bithiophene 13 according to Scheme 4, was attempted by the addition of the quinone 7 to asolution of 13, in THF at a temperature of −80°C, followed by prolonged stirring at roomtemperature. Unfortunately, the reaction was completely unsuccessful because in every run,insoluble black tars were obtained which could not be characterized, thus preventingdetermination of the exact reason of failure. When the yields, obtained in the reaction ofthienyllithium with benzobisthiadiazolone 7 and with anthraquinones are compared, it can beconcluded that the first reaction is far from quantitative. Presumably, this is obstructing the

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
"$
copolymerization process. Regarding the quantitative yields that are obtained in the reactionof thienyllithium with anthraquinone, this would be a more appropriate unit for thecopolymerization with dilithiobithiophene, the attempts of which are described in section 4.4.
Fortunately, the alternative synthesis of dithienylbenzobis[1,2,5]thiadiazoles furnishedanother possibility of preparing polymers with the basic structure of 3. It was found thattreatment of dihydroxycompounds of type 9 with formic acid gave rise to a polycondensationreaction of which the detailed investigations are described in chapter 5. Furthermore, theinsight that was gained by the addition of thienyllithium derivatives to anthraquinone couldbe extended to other aryllithium derivatives. This opened the way to conjugated polymerscontaining the diphenylanthracene moiety, the results of which are described in chapter 6.
����.Z[ZWdXP]TeL_TZY�ZQ�OTWT_STZMT_STZ[SPYP�bT_S�LY_S]L\`TYZYP
����� 4Y_]ZO`N_TZY
Substitution of anthraquinone for benzobis[1,2,5]thiadiazol-4,8-dione in the attemptedcopolymerization with 2,2’-dilithiobithiophene should give access to poly(dithienyl-anthracene)s. Although the resulting conjugated polymers will not feature low band gaps,they are still very attractive, because there is an increasing interest in anthracene containingoligomers and polymers, due to their unique optical and chemical behavior, in variousbranches of chemistry.21-26 The fluorescence of anthracene derivatives in or near the blueregion of the visible spectrum, has initiated many attempts to incorporate anthracene-basedunits in the main-chain27-31 or side chain32,33 of conjugated polymers, with the aim to preparea blue-emissive material for light-emitting diodes (LEDs). Oligomers containing thiopheneand anthracene show their use as efficient energy-transfer sections in light-harvestingsystems.34-36
The synthetic methods that are applied to incorporate the anthracene unit in theexamples described above, often make use of an organometallic coupling to a 9,10-dihaloanthracene. In the case of conjugated polymers, such an approach requires thesubstitution of solubilizing alkyl- or alkoxygroups on the main-chain, which may beundesirable. Therefore, the anthracene-based precursor polymer system may, analogously toScheme 4, provide an interesting alternative for the preparation of unsubstituted conjugatedpolymers containing the anthracene moiety. However, the substitution of alkyl- oralkoxygroups remains possible by making the appropriate modifications on monomers 13 or4.
����� ;]PN`]^Z] [ZWdXP] ]Z`_P _ZbL]O^ `Y^`M^_T_`_PO [ZWd�OT_STPYdWLY_S]LNPYP�^
The ability of copolymerizing aryldilithium salts with substituted quinones wasinvestigated by the reaction of dilithiobithiophene 13 with anthraquinone 4 (Scheme 12). Theacid-base reaction between 2 equivalents of n-BuLi and 2,2’-bithiophene in dry THF resultedin the dilithiobithiophene 13. Addition of 1 equivalent of solid anthraquinone 4 to thereaction mixture at -80 °C yielded, after stirring at -80 °C for 30 min. and subsequently at r.t.

.SL[_P] �
#�
for 180 min, followed by aqueous workup, the crude precursor polymer 14. The crudeprecursor polymer was dissolved in hot THF, precipitated in acetone and filtered to give ahigher molecular weight fraction in 27% yield. Evaporation of the acetone filtrate gave theresidual lower molecular weight fraction in 67% yield (total 94%). Size exclusionchromatography (SEC) in THF gave an indication of the molecular weights for the lower andhigher molecular weight fractions at Mw = 1.4 kD and 2.6 kD (vs. polystyrene), respectively.
S
S
n
OHOHO OS
SLiLi
4
13
14
+S
S a) b)
Scheme 12. Synthesis of precursor polymer 14. Reagents and conditions: a) n-BuLi, THF, -80 °C, b) THF, -80°C, 30 min, r.t., 180 min., then H2O, 94%.
Because the precursor polymer 14 is not conjugated, it lacks rigid-rod character and isstill moderately soluble in polar solvents like THF and N-methylpyrrolidone. However, theconjugated rigid-rod polymer that is produced in the reduction step will be insoluble becauseit lacks solubilizing groups. Therefore, the precursor polymer must be processed prior to thereductive elimination. Since it has been shown that the reductive elimination can beperformed by the action of HBr, exposing a film of the precursor polymer to HBr vapourshould produce the conjugated analogue 15. The occurrence of competitive halogenation dueto the use of pure HBr, as observed in the dihydroxy compound 6, is expected to be negligibledue to the low number of free α-thiophene sites present in the polymer.
300 400 500 600 700 800
Wavelength [nm]
270
272418
412328 580A
bsor
ptio
n [A
.U.]
a)
c)
b)500 600 700 800
500 600 700 800
Figure 2. Thin film UV spectra (stacked) of a) the untreated precursor polymer 14; b) directly after treatment of14 with HBr vapour; c) after heating of the HBr-treated film of 14 at 100 °C.
A solution of the higher molecular weight fraction of precursor polymer 14 in THF wasused for spincasting a transparent film on glass. This film was put in a container that wassubsequently evacuated and refilled with HBr gas, which caused the film to colorize from

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
#�
transparent to blue. Subsequent heating of the film at 100 °C for 1 minute resulted in a color-change from blue to yellow. The changes in the absorption spectra in the various stages of thereductive elimination are depicted in Figure 2. The precursor polymer absorption maximumof λmax = 328 nm corresponds with the bithiophene absorption. The structural changes uponexposure of precursor polymer 14 to HBr vapour are depicted in Scheme 13. Protonation ofthe alcohols by HBr introduces charges on the polymer. Because the bithiophene unit is astrong electron-donor, charge delocalization effects may occur, which give rise to the highwavelength absorption at λ = 580 nm. The anthracene absorption at λ = 272 nm indicates thatthe reductive elimination process is taking place. This fairly slow process is promoted byshortly heating the film, giving rise to the conjugated polymer 15 with λmax = 412 nm and thecharacteristic anthracene absorption at λ = 270 nm.
S
S
n
OHOH
S
S
n
OHBr
S
S
n
colorless blue yellow14 15
a) b)+
_
Scheme 13. Suggested structural changes upon treatment of polymer 14 with HBr vapour. Reagents andconditions: a) HBr(g), −H2O, b) HBr(g), −H2O, then heat, −Br2.
In the UV/Vis absorption spectrum of the conjugated polymer 15, the bithiopheneabsorption at 328 nm is still present. Although it is not excluded that this absorption ispresent in the conjugated polymer too35 due to the expected twist along the anthracene-thiophene bond, it can also indicate incomplete elimination. In the IR spectrum of 15 (film onKBr) the –O-H stretch (present in the IR spectrum of the precursor polymer 14) at 3306 cm-1
is greatly reduced, which is an indication for reduction. However, due to the insolubility ofthe conjugated polymer, the effectiveness of the elimination process could not be investigatedin further detail.
����� ;]PN`]^Z] [ZWdXP] ]Z`_P _ZbL]O^ LWVZcd�^`M^_T_`_PO [ZWd�OT_STPYdWLY_S]LNPYP�^
In order to gain more insight into the effectiveness of the copolymerization ofdilithiobithiophene 13 with anthraquinone and the subsequent reduction step, it was decidedto prepare soluble analogues of these polymers, by substitution of an alkoxy chain on theanthracene unit. This replacement of anthraquinone by dialkoxyanthraquinone for thesynthesis of more processible and soluble polymers is depicted in Scheme 14. Thecopolymerization with dilithiobithiophene 13 was performed analogously to thecopolymerization of the unsubstituted anthraquinone 4: addition of the quinone 16 (preparedfrom 2,6-dihydroxyanthraquinone and tetradecylbromide in DMF in 79 % yield37) to asolution of 13 in THF, followed by aqueous workup. Precipitation in hexane, followed bySoxhlet extraction with hexane and THF, subsequently, yielded the precursor polymer 17 in45% yield.

.SL[_P] �
#�
S
S
n
OHOH
C14H29O
OC14H29
O O
C14H29O
OC14H29
S
SLiLi
13
+S
S a) b)
16 17
Scheme 14. Synthesis of precursor polymer 17. Reagents and conditions: a) n-BuLi, THF, -80 °C, b) THF, -80°C, 30 min, r.t., 180 min. then H2O, 45 %.
From the SEC traces of the precursor polymer 17, a molecular weight of Mw = 4.5 kD(vs. polystyrene) followed. Since the precursor polymer 17 is soluble in solvents like CHCl3,it was possible to perform NMR spectroscopy. To obtain reference material, the reactionbetween thienyllithium and quinone 16 was performed as well (Scheme 15).
O O
C14H29O
OC14H29
S Li+
16
18,anti
S
SOH
OH
OC14H29
C14H29O
SS
OHOH
OC14H29
C14H29O
18,syn
S a)
b)
Scheme 15: Synthesis of dihydroxycompounds 18. Reagents and conditions: a) n-BuLi, THF, -80 °C, b) THF, -80 °C to r.t., quant.
In a procedure completely analogous to the preparation of 5, compound 18 wasobtained quantitatively as a mixture of syn and anti diastereomers. The 13C-NMR spectrum ofthis mixture shows a close similarity to the spectrum of the crude precursor polymer 17(Figure 3), which means that the syn/anti isomerism is also present along the polymer mainchain.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
#�
Figure 3: 100 MHz 13C-NMR spectrum (aromatic part) of diastereomers 18,syn and 18,anti (top) and polymer17 (bottom).
From the 13C-NMR spectra in Figure 3, it can be deduced that the copolymerization ofdilithiobithiophene with anthraquinone yields well-defined polymers, consistent with theproposed structures of 14 and 17. The broadness of the 13C-NMR peaks may be aconsequence of the relatively low molecular weights that can be obtained in this reaction.
Reduction of the precursor polymer 17 can now be performed in solution, since theconjugated polymer 19 will be soluble (Scheme 14).
S
S
n
OHOH
C14H29O
OC14H29
C14H29O
S
S
n
OC14H29
17 19
a)
Scheme 16. Reductive elimination of precursor polymer 17. Reagents and conditions: a) 2.5 eq 33% HBr, THF,30 min.
Treating a solution of the precursor polymer 17 in THF with 2.5 eq. of a 33% HBrsolution in AcOH causes strong coloration of the solution. Pouring the polymerizationmixture in methanol results in precipitation of the crude polymer 19 as a yellow solid. TheSEC trace of the crude polymer is depicted in Figure 4, whose shape reveals the presence ofoligomers. If it is assumed that these oligomers have the general structure depicted in the topleft corner of Figure 4 (i.e. polymer 19 with thiophene endgroups), and the rightmost peakoriginates from the n = 1 oligomer (it has been checked that this is not bithiophene noranthraquinone), the average degree of polymerization (DP) that can be reached is 7, i.e. amolecular weight of 5.5 kD. The molecular weight at the peak top of n = 7 vs. polystyrenestandards is 10.9 kD, which means an overestimation with a factor ~2. Fractionation of thepolydisperse, crude polymer mixture could be achieved by Soxhlet extraction using hexane,chloroform, THF and toluene, subsequently during 18 hours (for each solvent). The SECtraces of the various fractions are depicted in Figure 5.

.SL[_P] �
#�
0 5 10 15 time (min)
dete
ctor
out
put [
A.U
.]
1
2
3
456n = 7
S
S
n
S
S
C14H27O
OC14H27
Figure 4. SEC trace and proposed structure of polymer 19 (Column: PL500Å (0.1-40 kD), eluent: CHCl3, flowrate: 1 ml min-1, UV detection at 254 nm).
0 5 10 15 time [min]
dete
ctor
out
put [
A.U
.]
16267
13947
a)
b)
c)
d)
e)
Figure 5. SEC traces of Soxhlet-fractions of polymer 19. a) crude polymer; b) hexane fraction; c) chloroformfraction; d) THF fraction; e) toluene fraction. The Mw at the peak top vs. polystyrene standards is displayed forthe latter two traces (Column: PL500Å (0.1-40 kD), eluent: CHCl3, flow rate: 1 ml min-1, UV detection at 254nm).
From Figure 5, it follows that Soxhlet extraction with hexane and chloroform mainlyseparates out the oligomers with 1 ≤ n ≤ 6. The high molecular weight fractions, for which themolecular weights (vs. polystyrene, which value may be overestimated by a factor ~2, asshown above) at the peak top are displayed, are obtained by extraction with THF and/ortoluene. The weight fraction of polymer that is extracted by the various solvents is: hexane:40%, CHCl3: 27 % and THF/toluene: 33%. This means that the yield of high-molecularweight material in this polymerization is 33 % at maximum.
The most obvious reason for the low molecular weights obtained is non-stoichiometryin the copolymerization of co-monomers 13 and 16. It is assumed that dilithiobithiophene 13is prepared from bithiophene by a quantitative reaction with exactly 2 equivalents of n-BuLi.However, due to inevitable volumetric errors, deterioration and hence change in molarity ofthe n-BuLi solution and non-completeness of the acid-base reaction, some mono-lithiatedbithiophene may be present. This is then responsible for the diminished molecular weight.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
#
Addition of more than 2 equivalents of n-BuLi is not allowed since it also reacts withanthraquinone. An attempt has been made to work completely equimolarly by preparingdilithiobithiophene in diethyl ether, followed by filtration and weighing of the dilithium saltwith Schlenck techniques. However, the salt seemed prone to deterioration (change in color)in its solid form at elevated temperatures.
The 13C-NMR spectrum of the high-molecular weight fraction of polymer 19 showedno quaternary C-OH peaks and, therefore, reduction is complete. When the absorptionspectrum of a spincast film on glass of the solution-reduced polymer 19 is compared to thespectrum of a spincast film of the HBr vapour-reduced precursor polymer 17, they arequalitatively equal (Figure 6).
300 400 5000.0
0.5
1.0
1.5
2.0
2.5
a)
b)
430
272
AB
S [a
.u.]
nm
500
Figure 6. a) Solid-state (film on glass) UV/Vis spectra of a) a solution-reduced film of 19, stacked on top of b) afilm of the precursor polymer 17 after treatment with HBr(g) , and subsequent heating at 100 °C for 1 min.
Thus, the reductive elimination of spincast films of precursor polymers 14 and 17 bythe action of HBr-vapor is an efficient procedure, which makes the process a viable approachtowards poly(dithienylanthracene)s with or without solubilizing groups.
300 400 500 600 700 800
0
100
200
300
400502
433
276
Flu
ores
cenc
e [a
.u.]
nm
Figure 7. Fluorescence spectra of polymer 19 in CHCl3. () Excitation spectrum (emission at 502 nm).(⋅⋅⋅⋅⋅⋅⋅⋅⋅) Emission spectrum (excitation at 275 nm).

.SL[_P] �
#!
The anthracene moiety in polymers 15 and 19 may give rise to interesting fluorescencephenomena. For this reason, the fluorescence of polymer 19 was investigated in chloroformsolutions and in the solid state. Indeed, polymer 19 exhibits green fluorescence in CHCl3
solutions, which is exemplified by the excitation- and emission spectra (Figure 7).Unfortunately, the fluorescence of polymer 19 in the solid state (spincast film) is stronglyquenched.
�� �.ZYNW`^TZY^
The study towards optimized reaction conditions for the model reaction between 2-thienyllithium and anthraquinone and the subsequent reductive elimination has yielded anefficient, alternative two step synthesis of 4,8-di(thien-2-yl)benzobis[1,2,5]thiadiazoles 11and 12 that can replace the original inefficient five-step synthesis. Particularly the crucial stepin this synthesis, reductive elimination of a dihydroxy intermediate, needed optimizationsince the conditions used in the reported procedures for analogous compounds do not allowthiadiazole rings in the reactant. The copolymerization of 5,5’-dilithio-2,2’-bithiophene 13with benzobis[1,2,5]thiadiazole-4,8-dione 7 was unsuccessful, probably due to the non-quantitative addition of thienyllithium to the quinone. Insolubility of the products obtained inthis reaction concealed the reasons for failure. In conclusion, this route is not suitable as anon-electrochemical approach towards dithienobenzobis[1,2,5]thiadiazole-based conjugatedlow band gap polymers.
However, the successful copolymerization of dilithiobithiophene with anthraquinonesproved to be a very effective approach towards well-defined thiophene-anthracenecopolymers. The copolymerization of 5,5’-dilithio-2,2’-bithiophene 13 with anthraquinone 4gave access to a non-conjugated, soluble precursor polymer 14. This precursor polymer 14could be spincast from THF solution, and the film could be converted into the conjugatedpolymer poly(9,10-dithienylanthracene) 15 by the action of HBr vapour, as demonstrated byUV spectroscopy. Soluble alkoxy-substituted analogues 17 and 19 showed a degree ofpolymerization up to DP = 7, (Mn = 5.5 kD). These fairly low molecular weights are hard toimprove due to the requirement of working exactly equimolarly to obtain high molecularweights in a copolymerization. The reductive elimination of the precursor polymer 17 showedto be quantitative both in solution (as proven by the 13C-NMR of the soluble alkoxyderivative 19), and in the solid state (by comparison of the UV spectra of solution- and film-reduced polymers). Polymer 19 exhibited green fluorescence in CHCl3 solutions, however, inthe solid state this fluorescence was strongly quenched. The use of other aryldilithium saltsand fused quinones in this polymerization has not been exploited yet, the attempts of whichmay open up a completely new approach to a wide variety conjugated polymers.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
#"
��!�0c[P]TXPY_LW�>PN_TZY
2PYP]LW _PNSYT\`P^� Size Exclusion Chromatography (SEC) for precursor polymer 14 was performed on aWaters GPC system in THF. SEC for precursor polymer 17 and polymer 19 was performed in CHCl3 on aShimadzu LC10 system with a Polymer Laboratories PL500Å column (0.1 - 40 kD), flow rate = 1 ml/min, UVdetection at 254 nm. Anthraquinone 4 (Fluka, p.a. quality) was used as received. Benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione 7 8,9 and 2,2’-bithiophene38 were prepared according to literature procedures .For other general techniques, the reader is referred to chapter 2.
$����-T^�_STPY���dW��$����OTSdO]ZLY_S]LNPYP�$����OTZW F H�� A solution of thiophene (2.78 g, 0.033 mol) inanhydrous THF (100 ml) was cooled, under an inert atmosphere of Argon, to -78 °C. To this solution was addeddropwise a 1.6 M solution of n-BuLi in hexane (22.5 ml, 0.036 mol) over 10 min. The resulting mixture wasstirred at -78 °C for 15 min, allowed to warm to room temperature, stirred for 30 min at this temperature andcooled again to -78 °C. Solid anthraquinone 4 (3.12 g, 0.015 mol) was then added and the reaction mixtureallowed to warm to room temperature. After complete disappearance of the solid anthraquinone, the resultingreddish solution was quenched with water. Ether (300 ml) was added and the layers were separated. The organicphase was washed thoroughly with water, dried over MgSO4 and evaporated to afford a white powder, whichcould be crystallized from a mixture of acetic acid and water to give 5.3 g of 5, being a mixture of syn and antidiastereomers. One of these isomers could be isolated by crystallization of the mixture from CHCl3 giving thefollowing spectral data: 1H-NMR (400 MHz, CDCl3): δ: 7.63 (m, 4H, H1,4-anth), 7.35 (m, 4H, H2,3 anth), 7.25(dd, J = 5.1 and 1.2 Hz, 2H, H5 th), 6.91 (dd, J = 3.6 and 5.1 Hz, 2H, H4 th), 6.74 (dd, J = 3.6 and 1.2 Hz, 2H,H3 th), 2.80 (s, 2H, OH). 13C-NMR (100 MHz, CDCl3): δ: 152.3 (C2 th), 140.4 (C4a,9a anth), 128.7 (C2,3anth), 127.4 (C6,7 anth), 126.6, 125.9, 125.4 (C3,4,5 th), 73.5 (C9,10 anth). IR (KBr): ν: 3324 (O-H), 1702,1482 - 450 cm-1. Anal. calcd. for C22H16O2S2 (376.4990): C, 70.18; H, 4.28. Found: C, 69.80; H, 4.32.To a suspension of 5,syn and 5,anti (0.10 g) in CH3COOH (50 ml) were added a few drops of concentrated HCl(37% in H2O). After stirring this mixture for 2 h at room temperature, it was poured into a saturated NaHCO3
solution (250 ml). The aqueous phase was extracted with ether, dried over MgSO4 and evaporated to give 0.10 gof a white solid, being the second isomer. 1H-NMR (400 MHz, CDCl3): δ: 7.95 (m, 4H, H1,4 anth), 7.46 (m, 4H,H2,3 anth), 6.95 (dd, J = 5.1 and 1.2 Hz, 2H, H5 th), 6.37 (dd, J = 5.1 and 3.7 Hz, 2H, H4 th), 5.84 (dd, J = 3.7and 1.2 Hz, 2H, H3 th), 2.93 (broad s, 2H, -OH). 13C-NMR (100 MHz, CDCl3): δ: 149.5 (C2 th), 140.1 (C1a,4aanth), 128.3 (C2,3 anth), 126.6, 125.9 (C3,4 th), 125.6 (C1,4 anth; C5 th) 72.1 (C9-10 anth). Anal. calcd. forC22H16O2S2 (376.4990): C, 70.18; H, 4.28. Found: C, 69.96; H, 4.46.
=PLN_TZY ZQ _SP OTL^_P]PZXP]TN XTc_`]P �^dY LYO �LY_T bT_S XTYP]LW LNTO^�
BT_S 3.W% $�� �NSWZ]Z_STPY���dW������_STPY���dW�LY_S]LNPYP F#�C(.WH�� Through a stirred suspension of 5 (0.25g, 0.664 mmol) in anhydrous acetic acid (10 ml) was passed a stream of dry HCl gas for 5 min. The reactionmixture was then stirred for 30 min under an atmosphere of Ar. The HCl and acetic acid were subsequentlyremoved in vacuo to leave 8,X=Cl as a yellow solid (0.250 g, 0.663 mmol, 99.9 %). m.p. 174 °C. 1H-NMR (400MHz, CDCl3): δ: 7.92 (m, 2H, H anth), 7.87 (m, 2H, H anth), 7.58 (dd, J = 5.1 and 1.1 Hz, 1H, H5 th), 7.40 (m,4H, H anth), 7.27 (dd, J = 5.1 and 3.4 Hz, 1H, H4 th), 7.18 (dd, J = 3.4 and 1.1 Hz, 1H, H3 th), 7.09 (d, J = 3.6Hz, 1H, H3 thCl), 6.94 (d, J = 3.6 Hz, 1H, H4 thCl). 13C-NMR (100 MHz, CDCl3): δ: 138.6 (C2 thCl), 137.8(C2 th), 131.4 (C3 th), 130.9 (C9 anth), 130.8 (C10 anth), 129.5, 128.9 (C4,5 th), 128.8 (C-Cl thCl) 127.1-125.7(C anth, th, thCl). IR (KBr): ν: 1436-511 cm-1. Anal. calcd. for C22H13S2Cl (376.9297): C, 70.10; H, 3.48.Found: C, 69.82; H, 3.47.BT_S 3-]% To a stirred suspension of 5 (0.11 g, 0.29 mmol) in anhydrous acetic acid (1 ml) was added a 33%solution of HBr in acetic acid (0.055 ml, 0.320 mmol). The reaction mixture was stirred for 30 min under anatmosphere of Ar. The HBr and acetic acid were subsequently removed in vacuo to leave a yellow solid (0.12 g),being a mixture of unsubstituted 6 and bromo substituted 8,X=Br.BT_S 34% $����MT^�_STPY���dW�LY_S]LNPYP F!H�� To a stirred suspension of 5 (0.25 g, 0.66 mmol) in anhydrousacetic acid (1 ml) was added a 55% solution of HI in water (d = 1.7, 0.3 ml, 4.0 mmol). The mixture was heatedat reflux for 15 min, allowed to cool to room temperature and poured into water. Ether was added and the layersseparated. The organic layer was washed with water, 0.1 M NaOH and a 10 % NaS2O8 solution, respecitvely,dried over MgSO4 and evaporated to give a yellow solid which could be crystallized from toluene/i-PrOH,giving 6 as yellow crystals (0.22 g, acetic acid (0.055 ml, 0.64 mmol, 96.8 %). 1H-NMR (400 MHz, CDCl3): δ:7.86 (m, 4H, H anth), 7.59 (dd, J = 5.1 and 1.1 Hz, 2H, H5 th), 7.37 (m, 4H, H anth), 7.28 (dd, J = 3.5 and 5.1Hz, 2H, H4 th), 7.19 (dd, J = 3.4 and 1.3 Hz, 2H, H3 th). 13C-NMR (100 MHz, CDCl3): δ: 138.9 (C2 th), 131.4

.SL[_P] �
##
(C4a, 9a anth), 130.2 (C9, 10 anth), 129.5 (C3 th), 127.1, 126.8 (C4, 5 th), 126.6, 125.6 (C2,3 anth). UV/Vis(CHCl3) λ = 263.0, 360.2, 379.1, 399.9 nm. IR (KBr): ν: 1436-511 cm-1. Anal. calcd. for C22H14S2 (342.4846):C, 77.15; H, 4.12. Found: C, 77.03; H, 4.29.BT_S 9L4�34% $����MT^�_STPY���dW�LY_S]LNPYP F!H�� To a stirred suspension of 5 (0.25 g, 0.664 mmol) inanhydrous acetic acid (10 ml) was added NaI (0.5 g, 3.32 mmol). To this mixture was added dropwise a 33%solution of HBr in acetic acid (about 5.6 M, 0.6 ml, 3.3 mmol). The resulting brown reaction mixture was stirredfor 5 min and subsequently poured into water (200 ml). Just enough Na2S2O5 was added to decolorize theaqueous mixture. The yellowish precipitate was filtered, washed thoroughly with water and dried under vacuumto give 6 (0.225 g, 0.657 mmol, 99 %) with the identical spectral and microanalytical properties as in thereduction with pure HI.
��#�-T^�_STPY���dW����#�OTSdO]ZMPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP���#�OTZW F$H�� A solution ofthiophene (0.394 g, 4.68 mmol) in anhydrous THF (75 ml) was cooled, under an inert argon atmosphere, to -78°C. To this solution was added dropwise a 1.6 M solution of n-BuLi in hexane (3.0 ml) over 10 min. Theresulting mixture was stirred at -78 °C for 15 min, allowed to warm to room temperature, stirred for 30 min atthis temperature and cooled again to -78 °C. Solid benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione 7 (0.50 g,2.23 mmol) was added at such a rate as to keep the internal temperature below -60 °C. After complete addition,the purple reaction mixture was allowed to warm to -40 °C, stirred at this temperature for 15 min, and cooledagain to -78 °C. A solution of acetic acid in anhydrous THF (50 %, 2 ml) was added at this temperature andsubsequently the solution was warmed to room temperature. Ethyl acetate (150 ml) and water (100 ml) wereadded, the layers were separated and the organic layer was washed thoroughly with water and then dried overMgSO4. After evaporation of the solvent crude 9 remained, which was crystallized from acetic acid/water to give9 as colorless prisms (0.52 g, 1.32 mmol, 59.2 %). m.p. 185 °C (tr), 228 °C. 1H-NMR (400 MHz, DMSO-d6): δ:7.66 (s, 2H, OH), 7.45 (dd, J = 5.1 and 1.1 Hz, 2H, H5 th), 6.77 (dd, J = 5.1 and 3.7 Hz, 2H, H4 th), 6.31 (dd, J= 3.6 and 1.1 Hz, 2H, H3 th). 13C-NMR (100 MHz, DMSO-d6): δ: 161.6 (C=N), 147.4 (C2 th), 127.1, 126.3,125.5 (C3,4,5 th), 71.5 (C-OH). IR (KBr): ν: 3403, 3282, 1431-510 cm-1. Anal. calcd. for C14H8N4S4O2
(392.5066): C, 42.84; H, 2.05; N, 14.27. Found: C, 42.80; H, 2.06; N, 14.03.
��#�-T^� �SPcdW_STPY���dW����#�OTSdO]ZMPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP���#�OTZW F��H�� A solution offreshly distilled 2-hexylthiophene (0.394 g, 2.34 mmol) in anhydrous THF (40 ml) was cooled, under an inertargon atmosphere, to -78 °C. To this solution was added dropwise a 1.6 M solution of n-BuLi in hexane (1.46ml) over 10 min. The resulting mixture was stirred at -78 °C for 30 min, allowed to warm to room temperature,stirred for 15 min at this temperature and cooled again to -78 °C. Solid benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione 7 (0.25 g, 1.12 mmol) was added at such a rate as to keep the internal temperature below -60 °C. Aftercomplete addition, the reaction mixture was stirred at -78 °C for 3 h. A solution of acetic acid in anhydrous THF(50 %, 1 ml) was added at this temperature and subsequently the solution was warmed to room temperature.Ethyl acetate (100 ml) and water (100 ml) were added, the layers were separated and the organic layer waswashed well with water and then dried over MgSO4. After evaporation of the solvent crude 10 remained, whichwas subjected to column chromatography over silica gel with EtOAc/heptane = 1:2 as the eluent to yield pure 10(0.40 g, 0.71 mmol, 63.7 %). 1H-NMR (400 MHz, CDCl3): δ: 6.33 (m, 2H, H4 th), 6.10 (d, J = 3.6 Hz, 2H, H3th), 4.44 (s, 2H, OH), 2.63 (t, J = 7.4 Hz, 4H, th-CH2-), 1.51 (m, 4H, th-CH2CH2-), 1.26 (m, 12H, hex), 0.85 (t, J= 7.0 Hz, 6H, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 160.2 (C=N), 148.8 (C2 th), 142.1 (C5 th), 126.4, 123.5(C3,4 th), 72.5 (C-OH), 31.8, 31.4, 31.3, 30.0, 22.4, 14.0 (C hexyl). IR (KBr): ν: 3430, 2957, 2925, 2851, 1461-522 cm-1. Anal. calcd. for C26H32N4S4O2 (560.8282): C, 55.68; H, 5.75; N, 9.99. Found: C, 55.41; H, 5.85; N,9.93.
��#�-T^�_STPY���dW�MPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP F��H��To a stirred suspension of 9 (0.196 g, 0.50mmol) in anhydrous acetic acid (5 ml) was added NaI (0.37 g, 2.5 mmol). To this mixture was added dropwise a33% solution of HBr in acetic acid (about 5.6 M, 0.44 ml, 2.5 mmol). The resulting brown/green reactionmixture was stirred for 15 min and subsequently poured into water (200 ml). The precipitate was filtered off,washed well with water and methanol and dried to give 11 as a black/blue solid (0.12 g, 0.33 mmol, 66.0 %). 1H-NMR (400 MHz, CDCl3): δ: 9.01 (dd, J = 4.0 and 1.0 Hz, 2H, H3 th), 7.71 (dd, J = 5.1 and 1.0 Hz, 2H, H5 th),7.35 (dd, J = 4.0 and 5.1 Hz, 2H, H4 th). 13C-NMR (100 MHz, CDCl3): δ: 151.3 (C4a,8a bbt), 137.6 (C2 th),132.7, 130.8, 127.9 (C3,4,5 th), 113.9 (C4,8 bbt). GC/MS m/z (amu): 358. Anal. calcd. for C14H6N4S4
(358.4922): C, 46.91; H, 1.69; N, 15.63. Found: C, 45.22; H, 2.04; N, 14.31.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
#$
��#�-T^� �SPcdW_STPY���dW�MPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP F��H�� To a stirred suspension of 10 (0.50g, 0.892 mmol) in anhydrous acetic acid (10 ml) was added NaI (0.4 g, 2.67 mmol). To this mixture was addeddropwise a 33% solution of HBr in acetic acid (about 5.6 M, 0.5 ml, 2.8 mmol). The resulting brown reactionmixture was stirred for 15 min and subsequently poured into water (200 ml). The precipitate was filtered off andwashed thoroughly with water and then methanol. The blueish/green solid was taken up in a minimal amount ofCH2Cl2 and precipitated in MeOH. The precipitate was filtered, dried and recrystallized from heptane to give 12as a blue powder (0.25 g, 0.475 mmol, 53.3 %). 1H-NMR (400 MHz, CDCl3): δ: 8.65 (d, J = 3.8 Hz, 2H, H3 th),6.92 (d, J = 3.8 Hz, 2H, H4 th), 2.92 (t, J = 7.7 Hz, 4H, th-CH2-), 1.79 (m, 4H, th-CH2CH2-), 1.45-1.33 (m, 12H,hex), 0.91 (t, J = 7.1 Hz, 6H, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 151.9 (C2 th), 150.7 (C=N), 135.4 (C5th), 132.7, 125.4 (C3,4 th), 113.0 (C4,8 bbt), 31.6, 31.5, 30.4, 30.0, 22.6, 14.1 (C hexyl). UV/Vis (CHCl3): λmax
= 756 nm. IR (KBr): ν: 2955, 2924, 2852 1450-516 cm-1. Anal. calcd. for C26H30N4S4 (526.8138): C, 59.28; H,5.74; N, 10.64. Found: C, 58.83; H, 5.75; N, 10.27.
.Z[ZWdXP] F��H�� To a solution of 2,2’-bithiophene (0.75 g, 4.51 mmol) in anhydrous THF (100 ml) at -80 °Cunder an inert atmosphere of argon, was added a solution of n-BuLi in hexane (1.6 M, 5.64 ml, 9.02 mmol). Theresulting mixture was stirred at -80 °C for 30 min, warmed to room temperature, stirred for 15 min, and recooledto -80 °C. Solid anthraquinone (0.939 g, 4.51 mmol) was added and the reaction mixture stirred at -80 °C for 30min, allowed to warm to room temperature and stirred for another 3 hours. The reaction mixture was then pouredin water (1000 ml) under vigorous stirring, and the pH of the waterlayer was brought between 6 and 7 byaddition of a HCl solution. The white precipitate was filtered, washed well with water and dried to give the crudeprecursor polymer 14 as a white powder (1.6 g). This powder was dissolved in hot THF (about 100 ml) andslowly poured into cold acetone (1000 ml). The precipitate was filtered, giving the higher molecular weightfraction of the precursor polymer 14 as a white powder (0.46 g, 27 %) of Mn = 2171 D, Mw = 2638 D, polydisp.= 1.2 (vs. polystyrene). UV (film) λmax = 328 nm. IR (film on KBr): ν: 3306, 2960, 2872, 1654, 1600, 1447-753cm-1. The acetone filtrate was evaporated to give the lower molecular weight fraction of the precursor polymer14 as a white powder (1.14 g, 67 %) of Mn = 1220 D, Mw = 1424 D, polydisp. = 1.2 (vs. polystyrene).
.Z[ZWdXP] F� H� � A glass plate (~ 1 x 1 cm), coated with a spincast film (from THF) of the precursor polymer14 was put in a 100 ml flask fitted with an air in/outlet. The flask was evacuated (12 torr) and refilled with dryHBr gas, which caused the film of 14 to change from colorless to blue. After 1 minute, the glass plate was takenout of the flask and put on a hotplate at 100 °C for 1 minute, resulting in a yellow transparent film of polymer15. UV (film): λ = 270, 328 nm. IR (KBr): ν: 3064, 2957, 1670, 1600, 1438-766 cm-1.
��!�/T�Y�_P_]LOPNdWZcdLY_S]L\`TYZYP F�!H�� This compound was prepared according to literature procedures37
from 2,6-dihydroxyanthraquinone (18.0 g, 0.075 mol), K2CO3 (72 g, 0.52 mol) and tetradecyl bromide (83 g, 0.3mol) in DMF (250 ml) yielding, after crystallization from AcOH, 16 as a fluffy greenish powder. (37.7 g, 0.060mol, 79.4 %). 1H-NMR (400 MHz, CDCl3): δ: 8.21 (d, J = 8.6 Hz, 2H, H4), 7.70 (sd, J = 2.6 Hz, 2H, H1), 7.21(dd, J = 8.6 and 2.6 Hz, 2H, H2), 4.14 (t, J = 6.5 Hz, 4H, -OCH2), 1.84 (m, 4H, -OCH2CH2), 1.49, 1.45, 1.26(44 H), 0.88 (t, J= 6.9 Hz, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 182.3 (C=O), 164.0, 135.8, 129.6, 126.9,120.9, 110.5, 68.8, 31.9, 29.7 - 29.3, 29.0, 25.9, 22.7, 14.1.
.Z[ZWdXP] F�"H�� To a solution of 2,2’-bithiophene (0.532 g, 3.20 mmol) in anhydrous THF (100 ml) at -80 °Cunder an inert atmosphere of argon, was added a solution of n-BuLi in hexane (1.6 M, 4.00 ml, 6.40 mmol). Theresulting mixture was stirred at -80 °C for 30 min, warmed to room temperature, stirred for 15 min, after whichsolid 2,6-di-n-tetradecyloxy-anthraquinone 16 (2.025 g, 3.20 mmol) was added at r.t. The reaction mixtureimmediately turned brown. After stirring this mixture for 2 hours, the color had changed from brown to yellow.At this point, the reaction mixture was quenched with AcOH and subsequently mixed with CH2Cl2 (~200 ml).The resulting organic layer was washed well with water and a 1 M NaHCO3 solution (to remove the excessAcOH), dried over MgSO4 and concentrated on a rotary evaporator to a volume of about 50 ml. Precipitation inhexane followed by filtration (G4 glass filter) and drying afforded the precursor polymer as a white solid, whichwas subjected to soxhletted extraction with hexane (18 h) and THF, respectively. The THF fraction wasevaporated to give the precursor polymer 17 as a white solid (1.11 g, 43.4 %) of Mw = 4475 D, Mn = 4202 D,Polydisp. = 1.06 (vs. polystyrene). 1H-NMR (400 MHz, CDCl3): δ: 7.9 - 5.5 (aromatic protons) 3.9 (-OCH2-),1.7 (-OCH2CH2-), 1.3 (-CH2-), 0.9 (-CH3). 13C-NMR (100 MHz, CDCl3): δ: 160, 151, 148, 142, 137, 132, 129 -121, 115, 112, 110 (aromatic C), 73.2 (C-OH), 72.0 (C-OH), 68 (-OCH2-), 31.9 - 13.6. Anal. calcd. for(C50H70O4S2)n ((799.24)n): C, 75.14; H, 8.82. Found: C, 74.53; H, 8.74.

.SL[_P] �
$�
��!�/T�Y�_P_]LOPNdWZcd�$����MT^�_STPY���dW��$����OTSdO]ZcdOTSdO]ZLY_S]LNPYP F�#H�� This compound wasprepared, as described for 5, from thiophene (0.25 g, 3.0 mmol), n-BuLi (1.85 ml of a 1.6 M solution in hexane,3.0 mmol) and 2,6-tetradecyloxyanthraquinone 16 (0.90 g, 1.4 mmol) in dry THF (50 ml) to give 18 as a mixtureof syn and anti diastereomers (1.11 g, 1.39 mmol, 98.2 %). 1H-NMR (400 MHz, CDCl3): δ: 7.81 (d, J = 8.6 Hz,2H, H4 anth), 7.48 (d, J = 8.7 Hz, 2H, H4 anth), 7.44 (sd, J = 2.5 Hz, 2H, H1 anth), 7.22 (dd, J = 5.1 and 1.2Hz, 2H, H3 anth), 7.10 (sd, J = 2.6 Hz, 2H, H1 anth), 6.95 (dd, J = 5.1 and 1.3 Hz, 2H, H3 anth), 6.93 - 6.84(6H, thiophene signals), 6.75 (dd, J = 3.6 and 1.3 Hz, 2H, H3 th), 6.38 (dd, J = 5.1 and 3.6 Hz, 2H, H4 th), 5.88(dd, J = 3.6 and 1.4 Hz, 2H, H5 th), 4.00 (t, J = 6.6 Hz, 4H, -OCH2), 3.89 (t, J = 6.7 Hz, 4H, -OCH2), 1.81 (m,4H, -OCH2CH2), 1.72 (m, 4H, OCH2CH2), 1.47 - 1.26 (44 H), 0.88 (m, 6H, -CH3). 13C-NMR (100 MHz,CDCl3): δ: 159.1, 158.9, 152.7, 150.0, 142.2, 141.9, 132.6, 132.4, 128.9, 127.1, 126.5, 126.2, 125.6, 125.5,125.1, 115.3, 115.1, 112.3, 110.7, 73.4, 72.0, 68.3, 68.0, 31.9, 29.7 - 29.1, 26.0, 25.9, 25.5, 22.7, 20.6, 14.1.Anal. calcd. for C50H72O4S2 (801.248): C, 74.95; H, 9.06. Found: C, 75.01; H, 9.31.
.Z[ZWdXP] F�$H�� To a solution of the precursor polymer 17 (2.0 g, 5.0 mmol -OH groups) in THF (50 ml) wasadded a solution of HBr in AcOH (33%, 1.25 ml, ~6.25 mmol). The reaction mixture immediately turned green,and became yellow after a few min. Stirring was continued for 30 min after which the reaction mixture wasslowly poured, under vigorous stirring, in methanol (1000 ml). The precipitate was filtered, washed well withmethanol and dried to give 1.9 g of crude 19 as a yellow powder. Separation of the lower molecular weightoligomers was performed by soxhlett extraction with hexane and chloroform for 18 h, respectively, giving 1.33 gof low-molecular weight material. Continued soxhlett extraction with THF and toluene for 18 h, respectively,yielded 0.64 g (33 %) of high-molecular weight material of 19 with an approximate moleculare weight of Mw =14 - 16 kD (vs. polystyrene). 1H-NMR (400 MHz, CDCl3): δ: 7.94 (br d, J = 9.0 Hz, 2H, H4 anth), 7.50 (br s,2H, H1 anth), 7.25, 7.20, 7.18 (br peaks, 6H, H th/H3 anth), 4.03 (br s, 4H, -OCH2-), 1.83 (br s, 4H, -OCH2CH2-), 1.50 - 1.24 (br peaks, 44H), 0.87 (br s, 4H, -OCH3). 13C-NMR (100 MHz, CDCl3): δ: 156.3, 138.6,131.2, 130.2, 129.0, 128.5, 128.2, 127.9, 127.6, 123.9, 120.9 (aromatic peaks), 68.0 (-OCH2-), 31.9, 29.7 - 29.1,26.2, 22.7, 14.1. Anal. calcd. for (C50H68O2S2)n ((765.2178)n): C, 78.48; H, 8.96. Found: C, 76.27; H, 7.58.
=PQP]PYNP^
(1) Yamashita, Y.; Suzuki, T.; Mukai, T. J. Chem. Soc. Chem. Commun. 1987, 1184.(2) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Mater. 1996, 8, 570.(3) Karikomi, M.; Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Am. Chem. Soc. 1995, 117, 6791.(4) Yamashita, Y.; Ono, K.; Tomura, M.; Tanaka, S. Tetrahedron 1997, 53, 10169.(5) Uno, T.; Takagi, K.; Tomoeda, M. Chem. Pharm. Bull. 1980, 28, 1909.(6) Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A.; Elsenbaumer, R. L.; Reynolds, J. R., Eds.;
Marcel Dekker: New York, 1998.(7) Etienne, A. Bull. Chim. Soc. Fr. 1947, 634.(8) Winkelmann, E. Tetrahedron 1969, 25, 2427.(9) Neidlein, R.; Tran-Viet, D.; Gieren, A.; Kokkindidis, M.; Wilckens, R. Chem. Ber. 1982, 115, 2898.(10) Wurthner, F.; Vollmer, M. S.; Effenberger, F.; Emele, P.; Meyer, D. U.; Port, H.; Wolf, H. C. J. Am.
Chem. Soc. 1995, 117, 8090.(11) Duveen, D.; Willemart, A. J. Chem. Soc. 1939, 116.(12) Skowronski, R.; Chodkiewicz, W.; Cadiot, P. Bull. Chim. Soc. Fr. 1967, 4235.(13) Naef, R.; Balli, H. Helv. Chim. Acta 1978, 61, 2958.(14) Rio, G. Ann. Chem. 1954, 9, 182.(15) Clar, E.; Kelly, W.; Wright, J. W. J. Chem. Soc. 1954, 76, 1108.(16) Dodge, J. A.; Chamberlin, R. A. Tetrahedron Lett. 1988, 29, 1359.(17) Ingold, C. K.; Marshall, P. G. J. Chem. Soc. 1926, 3080.(18) Etienne, A.; Salmon, J. Bull. Chim. Soc. Fr. 1954, 1127.(19) Bergmann, E.; Blum-Bergmann, O. J. Am. Chem. Soc. 1937, 59, 1439.(20) Alonso, F.; Barba, I.; Yus, M. Tetrahedron 1990, 46, 2069.(21) Benshafrut, R.; Rabinovitz, R.; Hoffman, R. E.; Benmergui, N.; Müllen, K.; Iyer, V. S. Eur. J. Org.
Chem. 1999, 12, 37.(22) Mori, Y.; Maeda, K. J. Chem. Soc. Perkin Trans. 2 1996, 113.

4X[]ZaPO >dY_SP_TN ;]ZNPO`]P^ ?ZbL]O^ /T_STPYdWMPYeZMT^_STLOTLeZWP^ LYO ;ZWd�OT_STPYdWLY_S]LNPYP�^
$�
(23) Mukerjee, A. K.; Margaretha, P.; Agosta, W. C. J. Org. Chem. 1996, 61, 3388.(24) Wong, W. Y.; Wong, W. T. J. Chem. Soc. Dalton Trans. 1996, 1853.(25) Kammermeier, S.; Jones, P. G.; Herges, R. Angew. Chem. 1996, 108, 2834.(26) Becker, H.-D.; Andersson, K. J. Org. Chem. 1983, 48, 4542.(27) Yunhi, K.; Kwon, S.; Dongsik, Y.; Rubner, M. F.; Wrighton, M. S. Chem. Mater. 1997, 9, 2699.(28) Hirohata, M.; Tada, K.; Kawai, T.; Onoda, M.; Yoshino, K. Synth. Met. 1997, 85, 1273.(29) Kaeriyama, K.; Tsukahara, Y.; Negoro, S.; Tanigaki, N.; Masuda, H. Synth. Met. 1997, 84, 263.(30) Klarner, G.; Davey, M. H.; Chen, W. D.; Scott, J. C.; Miller, R. D. Adv. Mater. 1998, 10, 993.(31) Hodge, P.; Power, G. A.; Rabjohns, M. A. Chem. Commun. 1997, 73.(32) Bouche, C. M.; Berdague, P.; Facoetti, H.; Robin, P.; Lebarny, P.; Schott, M. Synt. Met. 1996, 81, 191.(33) Chung, S. J.; Kim, K. K.; Jin, J. I. Polymer 1999, 40, 1943.(34) Vollmer, M. S.; Wurthner, F.; Effenberger, F.; Emele, P.; Meyer, D. U.; Stumpfig, T.; Port, H.; Wolf,
H. C. Chem. Eur. J. 1998, 3, 260.(35) Emele, P.; Meyer, D. U.; Holl, N.; Port, H.; Wolf, H. C.; Wurthner, F.; Bauerle, P.; Effenberger, F.
Chem. Phys. 1994, 181, 417.(36) Effenberger, F.; Grube, G. Synthesis 1998, 3, 1372.(37) Fages, F.; Desvergne, J.-P.; Bouas-Laurent, H. Bull. Soc. Chim. Fr. 1985, 959.(38) Kagan, J.; Arora, J.; Sudershan, K. Heterocycles 1983, 20, 1937.

THIS IS A BLANK PAGE (92)

.SL[_P]�
,�9ZY�ZcTOL_TaP�;ZWdNZYOPY^L_TZY�?ZbL]O^>ZW`_TZY�;]ZNP^^TMWP�;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLeZWP�^
bT_S�0c_]PXPWd�9L]]Zb�-LYO�2L[^�
,M^_]LN_% A facile, non-oxidative polycondensation towards solution processible, low bandgap conjugated polymers based on benzobis[1,2,5]thiadiazole and thiophene is presented.The polymer containing benzobis[1,2,5]thiadiazole and EDOT units in the main-chainfeatures a band gap of 0.3 eV, which value is the lowest known for non-oxidatively prepared,processible conjugated polymers. Furthermore, this polymer is prone to acid-doping byvarious organic acids, giving thin films with a stable conductivity of 1 S cm-1
���4Y_]ZO`N_TZY
The preparation of a conjugated polymer by means of chemical or electrochemicaloxidation of a suitable monomer, implies acquirement of the polymer in the doped state dueto the excess of oxidant. Therefore, the final step in such a synthesis involves dedoping,which must be carried out accurately when the neutral polymer is to be obtained. This isespecially important in low band gap polymers, since the band gap, their primary physicalproperty, can only be derived correctly when the polymer is in its neutral state. Unfortunately,all conjugated polymers with a reported band gap of Eg < 0.5 eV are preparedelectrochemically1-3 which is also a disadvantage in terms of processibility, characterizationand applicability. Preferably, low band gap conjugated polymers should be synthesized via anon-oxidative polymerization method, as applied to other π-conjugated polymer. Examplesare a polycondensation,4-7 dehalogenation8-10 or a cross-coupling reaction like the Stille-coupling.11-13 However, the reported band gaps of the polymers prepared in this way allfeature values above 0.5 eV, which is still far from the ultimate goal of ~0 eV.
NS
N
NS
N
S
S
n
O O
NS
N
NS
N
S
SLi Li
NS
N
NS
N
S
S
n
OH
OH
Scheme 1. Attempted non-oxidative approach towards poly(dithienylbenzobis[1,2,5]thiadiazole)s, described inchapter 4.
In view of the low band gaps that are obtained with thiophene/benzobis-[1,2,5]thiadiazole based conjugated polymers,14-16 an improved synthesis of their monomers

.SL[_P]
$�
was developed (chapter 4). Although in principle possible, this did not provide a non-oxidative route towards such conjugated polymers (Scheme 1). The successful polymerizationcould only be carried out with an anthracene-based model system.
An alternative for a non-oxidative preparation of such polymers is inspired by thehistorical reaction of thiophene with isatin,17 leading to the indophenin dye. (Scheme 2). Themechanism of this reaction was suggested in 1939 by Steinkopf and Hanske.18 Reaction ofisatin 1 with thiophene yields, upon treatment with sulphuric acid, the hydroxy compound 2.Compound 2 loses water, under the influence of acid, to yield a reactive intermediate thatreacts with 2 at the α-positions, resulting, after the loss of water and H+, in the indophenin 3.
N OH
OS
S
NH
O
OH
N S
O
H
NN S
S
O
O
H H
+ H+
+ H+
- H2O
- H+
- H2O
1 2
3
+
NS
O
HSNH
O
H
H
OH
2
- H+
+
+
Scheme 2. Reaction of thiophene with isatin 1, under the influence of sulphuric acid, to yield the indophenindye 3.
Although some earlier attempts have been made to adopt the indophenin structure inconjugated polymers,19,20 the more rational adaptation of the indophenin-mechanism wasintroduced with the acid-induced polymerization of dihydroxycompound 421-23 (Scheme 3).
SSSS
OH
H OH
H
SSSS
OH H
H
SSSS
H OHSS
SSOH H
H
H
+H+
-H2O
4
5
+ 4- H+, -H2O
+
Scheme 3. Polycondensation of the dihydroxyoligothiophene 4 under the influence of acid.

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
$
Analogous to Scheme 2, protonation of the dihydroxycompound 4, followed by the lossof water, leads to a reactive intermediate. Reaction of this intermediate with 4 via the α-positions, results, after the loss of water and a proton, in dihydroxycompound 5. Thiscompound can then enter the reaction cycle again, thus giving rise to polymerization. Thecrucial step in this polymerization is the transfer of the carbocation, formed upon protonationand subsequent loss of water in the first step, from the secondary carbon atom to thethiophene unit. The reactive species in this polymerization, the positively charged thiopheneunit, shows a close similarity to that in the oxidative FeCl3 polymerization of thiophene.24
This implies that the use of thiophenes with vacant β-positions in the reaction of Scheme 3can give rise to “mislinkages”. Furthermore, the possibility exists here that the carbocation istransferred to one of the “wrong” central thiophene units, with the possibility of side-reactions. The observation that the products, obtained from polycondensation of monomer 4,are insoluble and intractable powders, may be explained by these facts.
Reconsidering the dihydroxycompound 6, an intermediate in the alternative synthesis ofdithienobenzobis[1,2,5]thiadiazoles (chapter 4), it appears to be an appropriate monomer fora polymerization process analogous to that depicted in Scheme 3 (Scheme 4).
NS
N
NS
N
S
S
OH
OHN
SN
NS
N
S
S OH
NS
N
NS
N
S
S OHN
SN
NS
N
S
S
OH
6
+ H+
- H2O
+ 6- H+, - H2O
NS
N
NS
N
S
S
n
poly-6
+
Scheme 4. Polycondensation of dihydroxybenzobis[1,2,5]thiadiazole 6 under the influence of acid, analogousto Scheme 3.
Protonation of one of the hydroxygroups in compound 6 and subsequent loss of watermay lead to a reactive thiophene species which can react similarly with another molecule 6. Itis essential that the acid, upon protonation of the hydroxy groups, generates a non-nucleophilic counter-ion, because otherwise reductive elimination will take place (chapter 4).A major difference in reactivity of 6 compared to 4 is expected because the crucial step,transfer of the carbocation onto thiophene, will be much faster in the case of 6, due to thestrong electron-accepting properties of the benzobisthiadiazole unit.

.SL[_P]
$!
���8ZYZXP]�OP^TRY
From the mechanism of Scheme 4, some requirements for the monomers can bedefined, to ensure a successful polymerization. Since the reactive species in thispolymerization shows a close similarity to that of the oxidative FeCl3 polymerization ofthiophene, the β-positions on the thiophene units in monomers of type 6 should be blocked toavoid mislinkages. With the expectation in mind that the unsubstituted polymers will beinsoluble materials, solubilizing groups are the blocking groups of choice.
Furthermore, for the crucial charge-transfer step to be as fast as possible, the electron-donating properties of the thiophene unit should be maximized. This also increases the donor-acceptor interaction in the polymer, thus leading to a diminished band gap. Combination ofthis fact with the desired blocking of the β sites, suggests the placement of (electron donating)alkyl- or alkoxy-groups on thiophene. It should be emphasized, however, that the substitutionof alkyl- or alkoxy groups may lead to steric hindrance, and hence deviation from coplanarityof the thiophene and central benzobisthiadiazole unit. This is probably unfavorable for therate of the charge-transfer step as well. Translating the above requirements into structures hasled to the following monomer design:
NS
N
NS
N
S
S OH
OH
NS
N
NS
N
S
S OH
OHOO
OO
NS
N
NS
N
S
S OH
OHC6H13O
OC6H13
OC6H13
OC6H13
NS
N
NS
N
S
S OH
OHH13C6
C6H13
6
7
8
9
Monomer 6, described in chapter 4, serves as a reference compound. Monomer 7contains the strong electron-donor 3,4-ethylenedioxythiophene,25,26 while monomer 8contains the solubilizing 3,4-di-n-hexyloxythienyl units. Finally, monomer 9 only has asolubilizing group at the 4-position which may limit the deviation from coplanarity of thethiophene units. In this chapter, the synthesis of compounds 7 - 9 and polymerization ofcompounds 6 - 9 is described, as well as the characterization of the polymerization products.†
* The polymerization was discovered during the attempted reductive elimination of compound 7. Application ofthe standard conditions for this reaction (addition of HBr to a solution of 7 and NaI in AcOH), led to aninsoluble black precipitate. In an attempt to use formic acid at elevated temperatures as the reducing agent, itwas noticed that upon addition of 7 to formic acid, strong coloration and an increase in viscosity occurred.

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
$"
���>dY_SP^T^�LYO�[ZWdXP]TeL_TZY
���� 8ZYZXP] >dY_SP^T^
The synthetic approach towards monomers 7 - 9 is identical to the synthesis ofmonomer 6, described in chapter 4: Reaction of the appropriate thienyllithium withbenzobis[1,2,5]thiadiazole-4,8-dione (Scheme 5).
S
R2R1
+ O O
NS
N
NS
N
S
S
NS
N
NS
N
OH
OH
R2
R2R 1
R 1S
R2R1
Lia) b)
7: R1 = R2 = -OCH2CH2O-8: R1 = R2 = -O-n-C6H13
9: R1 = -n-C6H13; R2 = H
(6: R1 = R2 = H)
Scheme 5. Synthesis of monomers 7 - 9. Reagents and conditions: a) n-BuLi, THF, -78 °C (30 min) → r.t. (30min); b) THF, -78 °C (1 h) → -15 °C (1 h) → r.t. (1 - 18 h), AcOH, 17 - 51 %.
Deprotonation of 3,4-ethylenedioxythiophene (“EDOT”) with n-BuLi in THF, followedby the addition of benzobis[1,2,5]thiadiazole-4,8-dione, initially yielded thedihydroxycompound 7 in only 19 % yield. Two reasons for this low yield can be given.Firstly, the reaction produces the hydroxycompound 7 as a mixture of syn and anti isomers,and isolation by crystallization yielded only one isomer. Secondly, EDOT forms a complexwith the product 7 in an EDOT : 7 ratio of 1 : 1. This is probably due to the electron-richnature of EDOT and the electron-deficient nature of the benzobis[1,2,5]thiadiazole moiety,giving rise to the formation of charge-transfer complexes. Efficient separation of EDOT and 7by crystallization required a relatively large amount of solvent (to prevent cocrystallization ofEDOT and 7), which is disadvantageous for the yield. However, an improved isolationprocedure consisted in Soxhlet extraction of the crude mixture of EDOT and 7 with hexaneand EtOAc, subsequently. Because the hydroxycompound 7 is insoluble in hexane, EDOT isextracted by hexane, after which extraction of the remaining solid with EtOAc yields theEDOT-free hydroxycompound 7 in, after crystallization, 51% yield. Crystallization of 7 fromacetone resulted in the incorporation of 2 equivalents of acetone in the crystal.
Similarly, deprotonation of 3,4-di-n-hexyloxythiophene with n-BuLi in THF, followedby the addition of benzobis[1,2,5]thiadiazole-4,8-dione, yielded, after crystallization, thedihydroxycompound 8 in 41% yield. Although only one isomer (syn or anti, not identified) isformed in this reaction, the formation of a complex between 3,4-di-n-hexyloxythiophene andthe product 8, which were separated in the crystallization step, was observed here too.
Finally, monomer 9 was prepared in 16% yield by deprotonation of 3-n-hexylthiophenewith n-BuLi in THF, followed by the addition of benzobis[1,2,5]thiadiazole-4,8-dione. Onlyone syn/anti isomer was obtained, however, since both the 2 and 5 positions of 3-n-hexylthiophene are prone to lithiation, a mixture of products was obtained from whicholigomer 9 (in slight excess) could be isolated by crystallization from heptane/CH2Cl2. A

.SL[_P]
$#
better yield in this reaction (based on benzobis[1,2,5]thiadiazole-4,8-dione) can be obtainedwhen 4 equivalents of lithiated 3-n-hexylthiophene are used. Benzobis[1,2,5]thiadiazole-4,8-dione reacts preferentially with 5-lithio-3-n-hexylthiophene because of the smaller sterichindrance, which results in about 80% conversion (based on 1H-NMR) into the desiredcompound 9.
���� ;ZWdXP]TeL_TZY
The polymerization of compounds 6 - 9, according to Scheme 4, requires an acid thatgenerates a non-nucleophilic counterion upon dissociation. It was found that formic acid isstrong enough to protonate the hydroxygroups in 6 - 9 efficiently. Dissolving the parenthydroxycompound 6 in pure formic acid led to an intense green coloration and a very fastincrease of the viscosity. After a few minutes, insoluble material was deposited, whichobstructs the processing and characterization. It was found that polymerization of 6 in a 1 : 1mixture of CHCl3/HCOOH afforded a homogeneous reaction mixture after overnight stirring.Precipitation of this solution in water or methanol afforded poly-6 as a black, insolublepowder. However, processing the homogeneous CHCl3/HCOOH mixture into thin films ispossible.
Dissolving 7 in formic acid at room temperature also resulted in strong coloration ofthe solution and a gradual increase of viscosity, however, no deposition of insoluble materialwas observed. In Figure 1, the increase in viscosity, relative to pure formic acid, as a functionof time is displayed when 7 is dissolved in formic acid (4.13 mg ml-1) at t = 0.
NS
N
NS
N
S
S OH
OHOO
OO
0 500 1000 1500 20000
2
4
6
8
10
12
14
Rel
ativ
e vi
scos
ity
Reaction time [min]
Figure 1. Viscosity increase as a function of time when 4.13 mg ml-1 of 7 is dissolved in formic acid at t = 0
A tenfold increase in viscosity is observed after approximately 24 hours, which is astrong indication for polymerization. Precipitation in water or methanol, or evaporation of theformic acid polymerization mixture afforded poly-7 as a black, insoluble material. It isremarkable that neat poly-7 cannot be redissolved in formic acid, although the polymerizationmixture remains a homogeneous solution. Therefore, poly-7 must also be processed from itspolymerization mixture in formic acid.

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
$$
Due to the low solubility of the tetrahexyloxycompound 8 and dihexylcompound 9 inHCOOH at room temperature, no polymerization was observed under these conditions,although coloration occurred when such mixtures were heated to reflux temperature. A milderprocedure consisted of dissolving compounds 8 or 9 in CHCl3, followed by the addition of anequal volume of CF3COOH or HCOOH. In this way, the addition of HCOOH to a solution ofdihexylcompound 9 in CHCl3 gave rise to rapid coloration already at room temperature. Bycontrast, a solution of tetrahexyloxycompound 8 in CHCl3 had to be heated, after treatmentwith HCOOH or CF3COOH, at an elevated temperature of 50 °C for a longer time to ensurepolymerization. This effect is probably due to the hexyloxy substituent adjacent to thebenzobisthiadiazole unit in 8, which gives rise to steric hindrance and deviation fromcoplanarity. Therefore, the crucial charge-transfer step is slower in 8 and heating is necessaryto accelerate the polymerization rate. Surprisingly, neat poly-9 is an insoluble material,indicating that only one hexyl substituent per thiophene unit is not sufficient to guaranteesolubility. In contrast, poly-8 is soluble by virtue of its 2 hexyloxy substituents per thiopheneunit. In Figure 2, the SEC traces of the polymerization of tetrahexyloxycompound 8 in a 1:1mixture of CHCl3/HCOOH at 50 °C after 1, 2, 3 and 5 nights reaction time are displayed,normalized to the monomer peak.
0 5 10 15 time (min)
dete
ctor
out
put [
A.U
.]
5 nights
3 nights
2 nights1 nights
monomer
Figure 2. SEC traces of the polymerization of 8 in HCOOH/CHCl3 at 50 °C after 1, 2, 3 and 5 nights (Column:PL1E4Å (1 - 400 kD), Eluent: CHCl3, Flow rate: 1 ml min-1, UV detection at 254 nm).
The leftmost, broad peak at high Mw is ascribed to the aggregation of the polymer, sinceit is strongly dependent on the sample concentration and not on reaction time. The rightmostpeak at low Mw increases upon reaction time, thus indicating a low-molecular weight by-product, which was not identified. It should be noted that this peak is present with UVdetection at 254 nm, but not with Vis-detection at 700 nm, thus excluding the possibility thatthis compound is a conjugated benzobisthiadiazole species. After 5 days, the monomer peakhas almost disappeared and a peak corresponding to higher molecular weight material (Mp = 7kD vs. polystyrene) has appeared. A large amount of lower molecular weight material ispresent too, which is another indication for the more difficult polymerization of 8 comparedto 6, 7 and 9. The crude material from the polymerization of 8 could be subjected to

.SL[_P]
���
preparative SEC using CH2Cl2 as the eluent to yield a higher molecular weight fraction in 45% yield with an Mw = 11.6 kD, Mn = 7.3 kD, polydisp. = 1.6 (vs. polystyrene).
���.SL]LN_P]TeL_TZY�ZQ�[ZWdXP]TeL_TZY�[]ZO`N_^
���� @A�AT^�94= LM^Z][_TZY ^[PN_]Z^NZ[d
Stable, greenish/transparent thin films of poly-6-9 could be directly spincasted fromtheir HCOOH or CHCl3/HCOOH polymerization mixtures (after at least 12 hours ofpolymerization) onto glass. The UV/Vis/NIR absorption spectra of these polymer thin filmsare depicted in Figure 3.
0 1 2 3 40.0
0.2
0.4
0.6
1.63 eV
Abs
orpt
ion
[a.u
.]
Energy [eV]
0 1 2 3 40.0
0.2
0.4
0.6 1.16 eV0.66 eV
Abs
orpt
ion
[a.u
.]
Energy [e.v.]
0 1 2 3 40
2
4
6
1.52 eV
1.29 eV
Abs
orpt
ion
[a.u
.]
Energy [eV]0 1 2 3 4
0.0
0.2
0.4
0.6
0.8
1.01.32 eV
Abs
orpt
ion
[a.u
.]
Energy [eV]
a b
c d
Figure 3. Absorption spectra of thin films of poly-6-9, spincasted from their polymerization mixtures; a) poly-6,b) (⋅⋅⋅⋅⋅⋅⋅) poly-7 immediately after spincasting, () = after heating the film at 200°C for 15 min.; c) poly-8,() = Purified high-molecular weight fraction, (⋅⋅⋅⋅⋅⋅⋅⋅) = Crude polymerization mixture; d) poly-9.
The absorption maximum of poly-6 (Emax = 1.65 eV, Figure 3a), spincast from theCHCl3/HCOOH polymerization mixture, differs from the value of the already reported,electrochemically prepared identical polymer27 (Emax = 1.0 eV). This could be either due tothe presence of a large amount of oligomers, and/or the occurrence of mislinkages in poly-6.
The absorption spectrum of poly-7 (Figure 3b, dashed line), immediately afterspincasting from the HCOOH polymerization mixture, features a low-energy peak at Emax =0.66 eV (1879 nm), a typical value for doped conjugated polymers. Moreover, this film showsan electrical conductivity of about 1 S cm-1, immediately after spincasting, which valuedecreases upon standing. It is suggested that the polymer is doped by the action of formic

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
���
acid. “Acid-doping” is best known from polyaniline, where the protonation of the iminenitrogens of the emeraldine base by strong acids leads to a highly conductive polymer.28 Sincepoly-7 also features imine nitrogens, formic acid may act as a dopant. Indeed, heating of thespincasted film of poly-7 at 200 °C for 15 minutes resulted in evaporation of formic acid,accompanied by the disappearance of electrical conductivity, and a shift of the absorptionmaximum towards 1.16 eV (Figure 3b, solid line), a value that is in agreement with polymersof this type.27 Surprisingly, only poly-7 is prone to acid-doping, which is studied in moredetail in section 5.4.3.
The absorption spectrum of the crude poly-8, spincast from the CHCl3/HCOOHpolymerization mixture, shows its maximum at Emax = 1.52 eV (Figure 3c, dashed line),however, the purified high-molecular weight fraction of poly-8 with Mw = 11 kD (vs.polystyrene) shows a red-shifted maximum of Emax = 1.29 eV (solid line in Figure 3d) whichis more in line with the expected value for these kinds of polymers. Therefore, the increasedabsorption maximum energy of poly-6 is probably also due to the presence of oligomericmaterial. An indication is the fact that the polymerization of 6 in HCOOH/CHCl3 remainshomogeneous, in contrast to the polymerization in pure HCOOH, where a very quick increaseof viscosity and deposition of insoluble material was observed. It seems that polymerizationin pure HCOOH produces higher molecular weight material than polymerization in a mixtureof CHCl3/HCOOH.
The absorption spectrum of poly-9, spincast from the CHCl3/HCOOH polymerizationmixture (Figure 3d), features a peak with an absorption maximum of 1.32 eV and a smallshoulder at higher energy, which is again assigned to oligomeric material. Indeed, evaporationof the polymerization mixture of poly-9, and subjecting the remaining solid to Soxhletextraction with CHCl3, yielded a small amount of oligomeric material. The SEC trace of these“CHCl3 extractables” is displayed in Figure 4.
0 5 10 15 time (min)
2555
2068
1409
1001
Det
ecto
r ou
tput
(a.
u.)
Figure 4. SEC trace of CHCl3 extractables of poly-9 (Column: PL500Å (0.1 kD - 40 kD), eluent: CHCl3, Flowrate = 1 ml min-1, UV detection at 254 nm).
The latter result indicates that the use of CHCl3/HCOOH mixtures is not beneficial forthe molecular weight obtained in this polymerization. Perhaps, the apolar character of CHCl3
causes a diminished stabilization of the charged, reactive intermediate.

.SL[_P]
���
The absorption maxima and band gaps, determined from the onset of absorbance, ofpoly-6-9 are summarized in table 1
Polymer Absorption max. [eV] Band gap† [eV]
poly-6 1.65‡ (751 nm) -‡
poly-7 1.16 (1069 nm) 0.3poly-8 1.29# (961 nm) 0.9#
poly-9 1.32 (939 nm) 0.9
Table 1. Absorption maxima and band gaps for poly-6-9. † From onset of absorbance. ‡ Not accurate due topresence of oligomers. # High-molecular weight fraction.
The band gap of poly-7 is centered about 0.3 eV. This is among the lowest valuesknown today for conjugated polymers, and certainly the lowest for solution-processible, non-oxidatively prepared polymers. The higher absorption maximum energy and band gap for thehigh molecular weight fraction of poly-8 compared to poly-7, is explained by the increasedsteric crowding in poly-8 due to the hexyloxy substituents. Additionally, the more electron-donating power of EDOT compared to dialkoxythiophenes results in a stronger donor-acceptor interaction and a lower band gap in poly-7. The absorption maximum and band gapenergy of poly-8 and poly-9 are approximately equal. The increased absorption maximumenergy of poly-6 is due to the low molecular weights accompanying the polymerization of 6in HCOOH/CHCl3.
���� 8,7/4�?:1 8L^^ ^[PN_]ZXP_]d
The use of Matrix Assisted Laser Desorption Ionization (MALDI) in combinationwith Time Of Flight (TOF) mass spectrometry is a growing tool in polymer characterization.However, application of this technique for the characterization of conjugated polymers isscarce.29,30
The use of the α-cyano-4-hydroxycinnamic acid matrix proved to be successful in theionization of poly-6, poly-7 and poly-9. Figure 5 shows the TOF mass spectrum of poly-6,measured in reflectron mode, which allows for a resolution of m/∆m = 5000 i.e. resolution atthe isotope level for masses <5000. Based on its mass spectrum, structure 10 is proposed forpoly-6.
NS
N
S
NS
N
S
NS
N
S
NS
N
SOH
NS
N
S
NS
N
SOH
n
10
The endgroups suggested for polymer 10 are based on the appearance of triplets for n= 1, 2, 3, ….. (exemplified by the peak for n =2 in the insert of Figure 5), with a massseparation of approximately 17 amu. Due to the proposed fragmentation during ionization,

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
���
the labile C−OH bond is broken, resulting in three species with respectively two, one and no -OH groups. The triplets cannot be due to multiple charged species, since the isotope patternof all peaks show a mass separation of 1 amu. The structure of 10 corresponds with theexpected one, based on the mechanism of Scheme 4.
1400 1420 1440 1460 1480 15000
1000
2000
3000
4000
5000
1457
1424
1440
n = 2
Cou
nts
Mass [m/z]
1000 2000 3000 40000
1000
2000
3000
4000
5000
8765
4
3
2n = 1
Cou
nts
Mass [m/z]
1000 2000 3000 40000
1000
2000
3000
4000
5000
8765
4
3
2n = 1
Cou
nts
Mass [m/z]
Figure 5. MALDI-TOF mass spectrum of poly-6.
The presence of oligomers of 10 with n = 1 to 8, i.e. 9 to 30 aryl units, can bedetected. These values indicate that poly-6 is indeed of low-molecular weight, as alreadydeduced from its UV/Vis/NIR absorption spectrum.
The MALDI-TOF spectrum of poly-7 shows a complex character (Figure 6). Similarto poly-6, peaks could be detected corresponding to the structure 11 with n = 1 to 7 and itsanalogues with one and two hydroxygroups eliminated.
1000 2000 3000 4000 50000
1000
2000
3000
Cou
nts
Mass [m/z]
1200 1400 1600 1800 20000
1000
2000
3000
Cou
nts
Mass [m/z]
n = 1
2
3
4
5
67
n = 2
n = 2-2 EDOT
n = 2- EDOT
n = 1
n = 1- EDOT
Figure 6. MALDI-TOF mass spectrum of poly-7
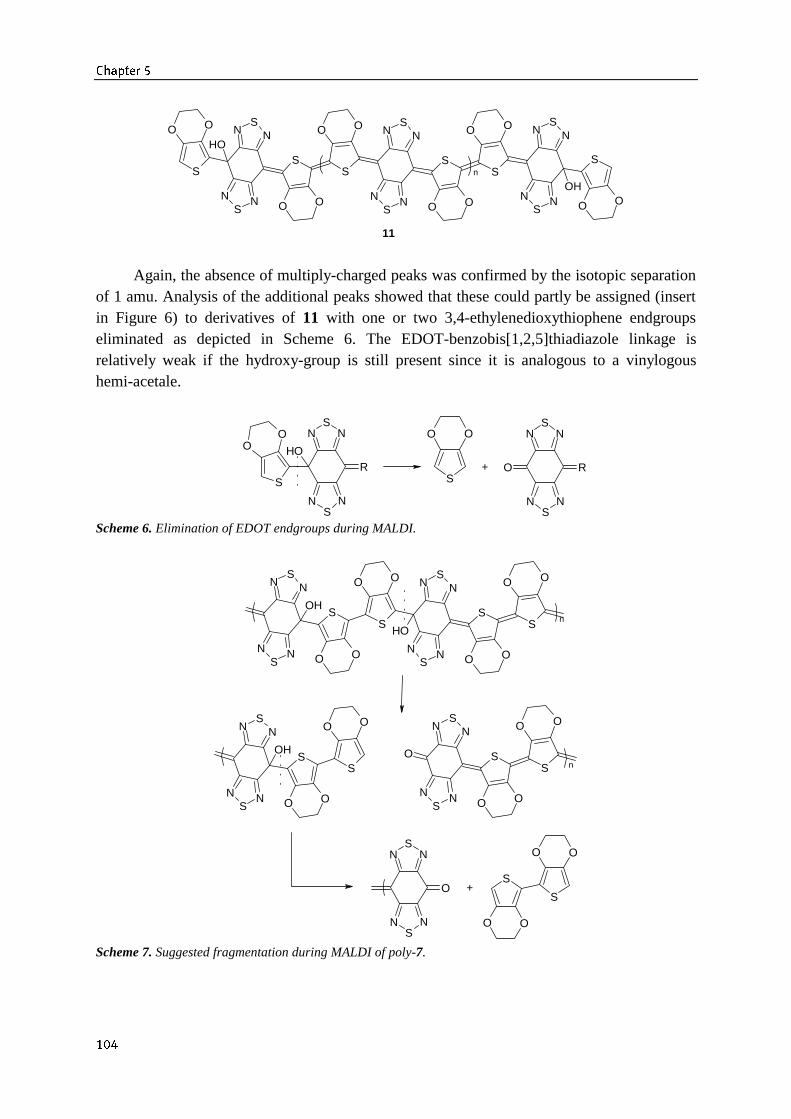
.SL[_P]
���
NS
N
S
NS
N
S
NS
N
S
NS
N
SOH
NS
N
S
NS
N
SOH
n
OO
O O
OO
O O
OO
O O
11
Again, the absence of multiply-charged peaks was confirmed by the isotopic separationof 1 amu. Analysis of the additional peaks showed that these could partly be assigned (insertin Figure 6) to derivatives of 11 with one or two 3,4-ethylenedioxythiophene endgroupseliminated as depicted in Scheme 6. The EDOT-benzobis[1,2,5]thiadiazole linkage isrelatively weak if the hydroxy-group is still present since it is analogous to a vinylogoushemi-acetale.
NS
N
RS
NS
N
OH
OO
NS
N
R
NS
N
OS
OO
+
Scheme 6. Elimination of EDOT endgroups during MALDI.
NS
N
S
NS
N
S
NS
N
NS
N
SS
O O
OO
O O
OO
OHn OH
NS
N
S
NS
N
S
NS
N
NS
N
SS
O O
OO
O O
OO
n OOH
S
S
O O
OO
NS
N
NS
N
O +
Scheme 7. Suggested fragmentation during MALDI of poly-7.

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
��
Analysis of the residual peaks suggested that main-chain fragments were present.Although this type of fragmentation is highly unlikely during the relatively mild MALDI,“weak spots”, giving rise to chain fracture can be assigned, assumed that poly-7 contains acertain percentages of hydroxy groups in the main chain (Scheme 7).
The occurrence of hydroxy groups in the main-chain may be rationalized by the lowoxidation potential of EDOT.25 Since the polymerization is not performed under inertatmosphere, a certain percentage of the monomer units can give rise to oxidative (e.g. by air)homo-coupling, yielding the proposed fragment in the main-chain. Moreover, since fragments11 contain terminal EDOT units, such an oxidative homo-coupling may occur duringprocessing of the polymer. In conclusion, the following fragmentation processes can bedistinguished during MALDI of poly-7:
1) Cleavage of 1 and 2 hydroxygroups in structure 11.2) Cleavage of a terminal EDOT unit from 11, according to Scheme 63) Fracture of the main-chain according to Scheme 7, and subsequent cleavage of
hydroxygroups and EDOT units from the fragmentation products.Calculation of all possible fragmentation products that are produced in either of the
above three processes for various chain lengths yields a calculated mass-spectrum, which issuperimposable to the experimental spectrum. This is exemplified in Figure 7 for massesbetween 1200 and 2700 amu.
1200 1400 1600 1800 2000 2200 2400 2600
0
1000
2000
3000
Cou
nts
Mass [m/z]
Figure 7. Experimental (top) and calculated (bottom) MALDI mass-spectrum of poly-7, taking into account allpossible fragmentation processes.
Though not completely equal (the measured small peaks at 1970 amu and 2440 amu donot occur in the simulation), the calculated mass-spectrum is in good accordance with theexperimental one, which affirms the above mentioned processes. The occurrence of main-chain fracture during MALDI implies that no accurate estimation of the molecular weight ofpoly-7 can be made on the basis of its MALDI-TOF mass spectrum. The presence ofsaturated segments (Scheme 7) in the main-chain of poly-7 is not in accordance with itsaccurate UV/Vis/NIR absorption spectrum and low band gap. It is likely that during the

.SL[_P]
��!
heating step, elimination of the hydroxygroups via a homolytic bond scission occurs, possiblyby the aid of formic acid. Similar processes are known from terphenyl-based systems.31
The MALDI-TOF spectrum of poly-9 is qualitatively equal to that of poly-6 (Figure 8),including the “triplet” pattern of peaks due to cleavage of hydroxygroups. Hence, structure 12is suggested for poly-9.
12
NS
N
S
NS
N
S
NS
N
S
NS
N
SOH
NS
N
S
NS
N
SOH
n
H13C6
C6H13
H13C6
C6H13
H13C6
C6H13
1000 2000 3000 4000 50000
2000
4000
6000
654
3
2n = 1
Cou
nts
Mass [m/z]Figure 8. MALDI-TOF mass spectrum of poly-9
In the mass spectrum depicted in Figure 8, oligomers of 12 with n = 1 - 6 can bedistinguished. However, the observation that the CHCl3 extractable oligomeric fraction ofpoly-9 (section 5.4.2 and Figure 4) is only very small, is not reflected in its mass spectrum. Inthis light, it should be emphasized that MALDI-TOF can discriminate longer chains byselective desorption of oligomers.32,33 In support of this is the fact that no MALDI-TOF massspectrum could be obtained from the high-molecular weight fraction of poly-8.
���� ,NTO�OZ[TYR ZQ [ZWd�"
Freshly spincast films of poly-7 exhibit an electrical conductivity of ~1 S cm-1. Due toevaporation of HCOOH, this value gradually decreases upon standing. Interestingly, poly-7can be redoped by exposure of a neutral film to the vapour of acids such as HBr ortrifluoroacetic acid. However, the use of such volatile acids always results in slow dedoping,which can be circumvented by the use of a high-boiling acid. Polyaniline is the best-knownexample of a conjugated polymer that becomes conductive by acid-doping,34 typically by (±)-camphorsulphonic acid35 or p-toluenesulphonic acid.36,37 Indeed, addition of either (±)-camphorsulphonic acid (CSA) or p-toluenesulphonic acid (PTSA, 1 equivalent per monomer

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
��"
unit) to a HCOOH solution of poly-7 and subsequent spincasting yielded films with a stableconductivity of about 1 S cm-1. Heating of such films did not lead to dedoping and decrease inconductivity.
The influence of the amount of acid on the doping level of poly-7 was investigated byUV/Vis/NIR spectroscopy. Thin films were spincasted from three solutions of poly-7 inHCOOH with 0.5, 1 and 2 equivalents of p-toluenesulphonic acid per monomer unit,respectively, and heated shortly at 200°C. The UV/Vis/NIR absorption spectra are depicted inFigure 9.
1 2 3 4
ABS[a.u.]
eVFigure 9. UV/Vis/NIR absorption spectra of thin films of poly-7: (——) 2 eq. PTSA, (-----) 1 eq. PTSA. (········),0.5 eq. PTSA.
Addition of 0.5 eq. of PTSA results in a shift of the absorption maximum to Emax = 0.75eV. However, a shoulder is still present at ~1.2 eV, corresponding to the neutral polymer (seealso Figure 3c), which is an indication for incomplete doping. Addition of 1 eq. of PTSAresulted in a shift of the absorption maximum to Emax = 0.65 eV. The addition of 2equivalents of PTSA did not result in a further shift of the absorption maximum, though theshoulder at 1.2 eV was more reduced. Surprisingly, all films were of comparable conductivityin the range of 1 S cm-1. Unfortunately, addition of strong acids such as PTSA and CSA to asolution of poly-7 in HCOOH resulted in partial aggregation/precipitation of the polymer.This is detrimental for the film-forming properties of poly-7, as reflected in the opaquecharacter of films containing the PTSA or CSA dopant. It is suspected that the aggregation isdue to the low pKa of CSA and PTSA (pKa < 1), since HCOOH (pKa = 3.75) solutions ofpoly-7 remain homogeneous. The use of salicylic acid (b.p. = 211 °C, pKa = 2.97) did notgive rise to aggregation when added to a solution of poly-7 in HCOOH. Spincasting fromsuch solutions yielded homogeneous thin films featuring a stable conductivity between 1 and10 S cm-1. The proposed mechanism that accounts for the conductivity of poly-7 upon acid-doping is depicted in Scheme 8. The electrical conductivity of conjugated polymers is basedon the existence of charge carriers in the main-chain upon doping (chapter 1). Protonation ofimine nitrogens in poly-7 can create a cation in the polymer main chain. A prerequisite for themechanism of Scheme 8 to work is the ability of the EDOT unit, by virtue of its electron-richcharacter, to donate an electron to the benzobisthiadiazole unit. This explains why poly-6,

.SL[_P]
��#
poly-8 and poly-9 are not prone to acid-doping, since the electron-donating power of thethiophene unit is too small in these polymers.
S
OO
NS
N
S
NS
N+O
On
HX
S
OO
NS
N
S+
NS
NO
On
H
X
S
OO
NS
N
S
NS
+ NO
On
HX
Scheme 8. Proposed mechanism of acid-doping in poly-7. Protonation of imine nitrogens (left) can lead tocharged species in the main-chain.
� �;ZWdNZYOPY^L_TZY�ZQ�Z_SP]�0/:?�NZY_LTYTYR�OTSdO]ZcdNZX[Z`YO^
A crucial step in the polycondensation of compounds 6 - 9 is, according to Scheme 4transfer of the carbocation from the benzobis[1,2,5]thiadiazole unit onto thiophene. This stepis assumed to occur by virtue of the electron-withdrawing character of thebenzobis[1,2,5]thiadiazole unit and the electron-releasing character of the (substituted)thiophene. Furthermore, these two units should be coplanar to secure efficient electron-transfer. The importance of these two requirements is exemplified by behavior of theanthracene-based compound 13.
S
SOH
OH
OO
OC14H29
H29C14O
OO
13
Compound 13 was synthesized in high yield from EDOT and 2,6-di-n-tetradecyloxyanthraquinone, in a procedure analogous to the preparation of 9,10-dithienylanthracenes, described in chapter 4. Dissolving 13 in a mixture of CHCl3/HCOOHdoes not give rise to polymerization, but to the formation of a relatively stable carbocation.Hence, coplanarity and/or the presence of an electron-acceptor in the monomer is indeedimportant for the polycondensation according to Scheme 4 to occur.
An example of a monomer, differing from monomers 6 - 9, yet containing an electron-deficient unit and, therefore, potentially prone to the polycondensation depicted in Scheme 4is the tetrachlorocompound 14, of which the synthesis is described in Scheme 9. Addition ofchloranil to a solution of lithiated EDOT in THF yields, after workup with a NH4Cl solution,the crude dihydroxycompound 14 as a mixture of syn and anti diastereomers, complexed withEDOT, an effect previously observed in the synthesis of compounds 7 and 8. Crystallization

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
��$
from THF furnished pure 14 in 36 % yield. Dissolving compound 14 in pure HCOOH ledonly to coloration after heating to reflux temperature. Although a slight increase in viscositywas observed after reflux for 3 days, the UV/Vis/NIR absorption spectrum of a spincastedfilm of this solution is indecisive (Figure 10).
S
O O
+ O O
ClCl
Cl Cl
S
SOH
OH
ClCl
Cl Cl
OO
OO
S Li
O O
a) b)
14
Scheme 9. Preparation of compound 14. Reagents and conditions: a) n-BuLi, THF, -78 °C (30 min) → r.t. (30min); b) THF, -78 °C → r.t. (1 h), NH4Cl, 36 %.
0 1 2 3 4 50.00
0.05
0.10
0.15
0.20
0.25
0.30
Abs
orpt
ion
[a.u
.]
eVFigure 10. UV/Vis/NIR thin film absorption spectrum of poly-11.
The absorption maximum at Emax = 5 eV (248 nm) does not correspond to what wouldbe expected for poly-14. Only a very small signal around 2 eV is encountered that may be dueto poly-14. It is, therefore, unlikely that compound 14 can be polymerized according toScheme 4. Presumably, the four chlorine atoms do not decrease the electron-deficiencysufficiently to guarantee efficient charge transfer. Furthermore, the bulkiness of the chloro-substituents must not be underestimated. A better performance may be expected from thetetrafluoro analogue, since the fluoro atoms are not only more electronegative, but alsosmaller. Compounds 13 and 14 illustrate the importance of monomer design in the synthesisof conjugated polymers according to Scheme 4. The successful monomer should contain astrong electron acceptor, and may not suffer from steric crowding that causes deviation fromcoplanarity.
�!�.ZYNW`^TZY^
The acid-mediated polycondensation of dihydroxy-monomers 6 - 9 represents a facileand non-oxidative route towards solution processible, low band gap conjugated polymersbased on benzobis[1,2,5]thiadiazole and thiophene. The polymer containing

.SL[_P]
���
benzobis[1,2,5]thiadiazole and EDOT units in the main-chain features a band gap of 0.3 eV,which is, to our knowledge, the lowest value known for non-oxidatively prepared, processibleconjugated polymers.
Polymerization of monomers 6 - 9 in HCOOH or CHCl3/HCOOH mixtures wasdemonstrated to occur unambiguously on the basis of viscosity and SEC measurements. Theuse of a mixture of CHCl3/HCOOH gave rise to lower molecular weight material, asexpressed in the UV/Vis/NIR absorption spectra, probably due to the less polar character ofthese mixtures. MALDI-TOF mass spectrometry on poly-6, poly-7 and poly-9 showed thatthese polymers are well-defined and possess the structure that is expected from thepolycondensation mechanism described in Scheme 4, including the correct endgroups. Poly-7was shown to contain some saturated fragments in the main-chain due to partial oxidativedimerization of EDOT units. However, no effect of these saturations was found in theUV/Vis/NIR absorption spectra of poly-7, which is either due to the small amount ofsaturations present, or to the possibility of their elimination during the heating step of poly-7.Only molecular weights up to 4 - 5 kD could be detected in the MALDI-TOF mass spectra,probably due to discrimination of higher molecular weight chains by the selective desorptionof oligomers.
Poly-7 is prone to undergo acid-doping with various organic acids. The use of volatileacids resulted in slow dedoping and loss of conductivity in films of poly-7, whereas the use ofhigh-boiling acids gave films with a stable conductivity in time. Addition of sulphonic aciddopants with a low pKa to solutions of poly-7 in HCOOH gave rise to aggregation, and hencepoor film-forming properties. Doping with salicylic acid, an acid with a high boiling pointand a moderate pKa, resulted in good films of poly-7 with a stable conductivity in the order of~ 1 S cm-1.
The attempted polycondensation of compounds whose structure is not related to 6,confirmed two important design criteria for successful monomers: the presence in themonomer of a strong electron acceptor unit, and the absence of steric crowding. Obviously,this relates to the crucial charge-transfer step in the polycondensation mechanism of Scheme4. With these criteria in mind, we are currently designing novel monomer architectures,supposed to give access to solution processible, intrinsically conducting conjugated polymers.
�"�0c[P]TXPY_LW�>PN_TZY
2PYP]LW _PNSYT\`P^% The preparation of compound 6 is described in chapter 4. Benzobisthiadiazole-4,8-dionewas prepared according to ref. 38. 3,4-Ethylenedioxythiophene was obtained from Bayer Polymerforschung(Krefeld). 3,4-di-n-hexyloxythiophene and 3-hexylthiophene were prepared according to ref. 39. The preparationof 2,6-di-n-tetradecyloxyanthraquinone is described in chapter 4. Spincasting was performed on a HeadwayResearch spincoater. Conductivity measurements were performed at Bayer Polymerforschung (Krefeld) usingthe two-point probe method. Viscosity measurements were performed using an Schott Geräte Ubelohdeviscometer. SEC was performed in CHCl3 on a Shimadzu LC10 system with a Polymer Laboratories PL500 (0.1- 40 kD) or PL1E4Å (1 - 400 kD) column, flow rate = 1 ml/min, UV detection at 254 nm. MALDI-TOF massspectra were recorded using the following procedure: To a solution of the polymer in HCOOH orCHCl3/HCOOH (20 µl, c = 0.3 mg/ml) was added a solution of α-cyano-4-hydroxycinnamic acid in THF (10 µl,c = 20 mg/ml). After being intimately mixed, 0.3 µl of the resulting mixture was brought on a sample plate andallowed to evaporate. The resulting spots were then analyzed using a Perseptive Biosystems Voyager DE-Pro

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
���
MALDI-TOF mass spectrometer (Accelerating voltage: 25000 V; Grid voltage: 77.000 %; Guide wire voltage:0.050 %; Delay: 400 ms; Low mass gate: 1000 amu). For further general procedures the reader is referred tochapter 2.
��#�/TSdO]Zcd���#�OT�����P_SdWPYPOTZcd_STPY���dW�MPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP �"��� To asolution of 3,4-ethylenedioxythiophene (0.761 g, 5.35 mol) in dry THF (50 ml) at -80 °C under an argonatmosphere was added a 1.6 M solution of n-butyllithium in hexane (3.21 ml, 5.13 mmol). The solution wasstirred at -80 °C for 30 min, allowed to warm to room temperature, stirred at this temperature for 30 min. andrecooled to -80 °C. Solid benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione (0.50 g, 2.23 mmol) was added atthis temperature and the resulting mixture was stirred at -80 °C (1 h), -15 °C (1 h) and r.t. (1 h), respectively,after which the reaction was quenched with AcOH (2 ml). After the addition of EtOAc to the reaction mixture,the resulting organic layer was washed thoroughly with water, dried over MgSO4 and evaporated to give amixture of crude 7 and unreacted 3,4-ethylenedioxythiophene, which was taken up in CH2Cl2 (20 ml) andprecipitated in hexane (250 ml). Filtration yielded the crude 7 (contaminated with a small amount of 3,4-ethylenedioxythiophene) as a pale brown powder (1.0 g). Crystallization from acetone afforded 7 as brownplates, in which 2 molecules of acetone per molecule of 7 were cocrystallized (0.26 g, 0.42 mmol, 19 %). 1H-NMR (400 MHz, acetone-d6): δ: 6.44 (s, 2H, H5 th), 3.96 (m, 4H, -OCH2-), 3.76 (m, 4H, -OCH2-). 13C-NMR(100 MHz, CDCl3): δ: 163.0 (C=N), 142.9 (C3 th), 138.3 (C4 th), 122.9 (C2 th), 99.4 (C5 th), 71.5 (C-OH),66.2, 65.8 (-OCH2CH2O-). IR (KBr): ν: 3392 (OH), 3101, 1705, 1690, 1495, 1438 - 800 cm-1. Anal. calcd. forC18H12N4S4O6 + 2 C3H6O (508.5794 + 116.1594): C, 46.14; H, 3.87; N, 8.97. Found: C, 45.90; H, 3.62; N,8.94.An improved workup procedure consists in subjecting the crude 7, obtained from the precipitation step, toSoxhlet extraction with hexane (to remove the final traces of 3,4-ethylenedioxythiophene) and EtOAc (to isolate7). Evaporation of the EtOAc fraction, followed by crystallization of the resulting solid from EtOAc yields thepure 7 in 51% yield.
��#�/TSdO]Zcd���#�OT�����OT�Y�SPcdWZcd_STPY���dW�MPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP �#��� Thiscompound was prepared, as described for 7, from 3,4-di-n-hexyloxythiophene (2.0 g, 7.03 mmol), an 1.6 M n-BuLi solution in hexane (4.29 ml, 6.86 mmol) and benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione (0.733 g,3.27 mmol), with this difference that, before quenching with AcOH, the reaction mixture was stirred at roomtemperature for 18 h. Workup consisted in extraction with EtOAc/water and crystallization of the crude reactionproduct from heptane to yield 8 as a pale brown solid (1.06 g, 1.33 mmol, 40.9 %). Further purification waspossible by recrystallization from isopropanol. 1H-NMR (300 MHz, CDCl3): δ: 6.21 (s, 2H, H5 th), 4.40 (s, 2H,OH), 3.87 (t, J = 6.6 Hz, 4H, -OCH2-), 3.41 (t, J = 6.9 Hz, 4H, -OCH2-), 1.8 - 0.9 (44H, hexyl). 13C-NMR (75MHz, CDCl3): δ: 160.9 (C=N), 149.5 (C3 th), 140.8 (C4 th), 127.9 (C2 th), 95.8 (C5 th), 72.6 (C-OH), 70.4,69.8 (-OCH2-), 31.4, 31.3, 29.0, 25.7, 25.0, 22.5, 14.0 (hexyl). IR (KBr): ν: 3430 (OH), 3105, 2955, 2930,2858, 1496 - 727 (1496, 1468, 1377, 1173, 997 822) cm-1. Anal. calcd. for C38H54O6N4S4 (791.1312): C, 57.69;H, 6.88; N, 7.08. Found: C, 57.87; H, 6.56; N, 6.94.
��#�/TSdO]Zcd���#�OT���Y�SPcdW_STPY���dW�MPYeZF����N%�� �NuH�MT^F���� H_STLOTLeZWP �$��� This compound wasprepared, as described for 9, from 3-hexylthiophene (0.79 g, 4.69 mmol), a 1.6 M solution of n-BuLi in hexane(2.93 ml, 4.68 mmol) and benzo[1,2-c:4,5-c’]-bis[1,2,5]thiadiazole-4,8-dione (0.50 g, 2.23 mmol). The crudereaction workup product was crystallized from heptane/CH2Cl2 to yield pure 9 as a pale brown solid (0.21 g,0.37 mmol, 16.8 %). 1H-NMR (400 MHz, CDCl3): δ: 6.82 (d, J = 1.5 Hz, 2H, H5 th), 6.20 (d, J = 1.5 Hz, 2H,H3 th), 4.10 (s, 2H, OH), 2.32 (t, J = 7.9 Hz, 4H, th-CH2-), 1.34 - 1.21 (m, 16H, -CH2-), 0.90 (t, J = 7.2 Hz, 6H,-CH3). 13C-NMR (100 MHz, CDCl3): δ: 161.1 (C=N), 145.8, 144.1 (C2,4 th), 128.9, 123.3 (C3,5 th), 78.2 (C-OH), 33.2, 32.8, 31.6, 30.3, 24.0, 15.4 (hexyl). IR (KBr): ν: 3326 (OH), 2954, 2925, 2853, 1548 - 520 cm-1.Anal. calcd. for C26H32O2N4S4 (560.8282): C, 55.68; H, 5,75; N, 9.99. Found: C, 55.49; H, 5.54; N, 9.81.
$����OT�����P_SdWPYPOTZcd_STPY���dW����!�OT�Y�_P_]LOPNdWZcd�$����OTSdO]ZcdOTSdO]ZLY_SLNPYP F��H�� Thiscompound was prepared following the procedure for the reactions of thienyllithium with anthraquinone asdescribed in chapter 4 from 3,4-ethylenedioxythiophene (0.75 g, 5.3 mmol), n-BuLi (3.3 ml of a 1.6 M solutionin hexane, 5.3 mmol) and 2,6-tetradecyloxyanthraquinone (1.59 g, 1.4 mmol) in dry THF (50 ml) to give crude13 (2.3 g, 98%). Crystallization from pentane afforded pure 13 as a white powder. (0.65 g, 0.71 mmol, 27%). 7.1H-NMR (400 MHz, CDCl3): δ: 7.42 (d, J = 8.8 Hz, 2H, H3 anth), 7.00 (d, J = 2.6 Hz, 2H, H7 anth), 6.82 (dd, J= 8.8 and 2.7 Hz, 2H, H1 anth), 6.29 (s, 2H, H5 th), 3.75 - 4.00 (m, 10H, -OCH2CH2O- + OH), 1.72 (m, 4H, -

.SL[_P]
���
OCH2C13), 1.62 - 1.21 (m, 48H), 0.88 (t, J = 6.8 Hz, 6H, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 158.9, 141.5,140.4, 135.0, 131.1, 129.3, 125.3, 115.8, 111.9, 96.6, 71.0, 67.9, 64.3, 64.1, 31.9, 29.7, 29.6, 29.4, 29.3, 29.2,26.0, 22.7, 14.1. IR (ATR): ν: 3467 (OH), 2918, 2850, 1612, 1492, 1437, 1362 - 676 cm-1. Anal. calcd. forC54H76O8S2: C, 70.71; H, 8.35. Found: C, 70.47; H, 8.24.
����/T�����P_SdWPYPOTZcd_STPY���dW������OTSdO]Zcd����� �!�_P_]LNSWZ]ZOTSdO]ZMPYePYP F��H��To a solutionof 3,4-ethylenedioxythiophene (1.0 g, 7.0 mmol) in dry THF (50 ml) at -80 °C under an argon atmosphere wasadded a 1.6 M solution of n-Butyllithium in hexane (4.4 ml, 7.0 mmol). The solution was stirred at -80 °C for 30min., allowed to warm to room temperature, stirred at this temperature for 30 min. and recooled to -80 °C.Chloranil (0.72 g, 2.9 mmol) was added at this temperature, after which the reaction mixture was allowed towarm to room temperature. After 1 h, the reaction was quenched with a saturated solution of NH4Cl. Ether wasadded and the layers were separated. The organic layer was washed thoroughly with water, dried over MgSO4
and evaporated. The residue was dissolved in CHCl3 and precipitated in hexane, giving 1.3 g of crude 14 as amixture of syn/anti isomers, complexed with 3,4-ethylenedioxythiophene. Crystallization from THF affordedwhite crystals of pure 14 (0.56 g, 1.06 mmol, 36 %) as a mixture of syn and anti isomers. 1H-NMR (400 MHz,CDCl3): δ: 6.58 (s, 2H, H5 th), 6.45 (s, 2H, H5 th), 4.12 (m, 8H, -OCH2CH2O-), 3.17 (s, 2H, -OH), 3.16 (s, 2H,-OH). IR (ATR): ν: 3457 (OH), 1628, 1491, 1435, 1367 - 675 cm-1. Anal. calcd for C18H12Cl4O6S2: C, 40.77; H,2.28. Found: C, 41.01, H, 2.41.
;ZWd�!�[ZWd�$� _d[TNLW [ZWdXP]TeL_TZY Pc[P]TXPY_% Monomer 6/9 (25 mg) was dissolved in 5 ml CHCl3.Formic acid (5 ml) was added under stirring, and the resulting green solution stirred at room temperature for atleast 12 h. The resulting solution of poly-6/poly-9 can then be used for e.g. spincasting.
;ZWd�"� RPYP]LW []ZNPO`]P: Finely powdered 7 (25 mg) was added to formic acid (10 ml) and stirred at roomtemperature for at least 12 h. The resulting solution of poly-7 in formic acid can then be used for e.g.spincasting. The viscosity of the solution continues to increase upon standing, to reach a gel-state within a fewdays, depending on the concentration. A sample of the polymerization mixture was evaporated to dryness andkept under vacuum for 72 h, giving the following microanalytical analysis: Anal. calcd. for (C18H8O4N4S4)n: C,45.75; H, 1.71; N, 11.86. Found: C, 43.92; H, 1.73; N, 11.10.
;ZWd�#�� Monomer 8 (210 mg) was dissolved in CHCl3 (5 ml), after which formic acid (5 ml) was added understirring. After heating the resulting reaction mixture at 50 °C for 5 days, it was evaporated to dryness. Theresidue was taken up in CHCl3, and precipitated in hexane. The black precipitate was filtered and dried to give~200 mg of crude poly-8, which was subjected to preparative SEC over a BioBeads column using CH2Cl2 as theeluent, to give 90 mg (44.7 %) of the high-molecular weight fraction of poly-8. Mw = 11.6 kD, Mn = 7.3 kD,Polydisp. = 1.6 (vs. polystyrene). 13C-NMR (100 MHz, CDCl3): δ: 154.4, 153.7, 150.7, 73-76 (-OCH2-), 32.7 -33.3, 30.3 - 31.6, 26.8 - 27.0, 23.8 - 24.0, 15.3 (-OCH2C5H11).
=PQP]PYNP^
(1) Akoudad, S.; Roncali, J. Chem. Commun. 1998, 2081.(2) Roncali, J. Chem. Rev. 1997, 97, 173.(3) Huang, H.; Pickup, P. G. Chem. Mater. 1998, 10, 2212.(4) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Polym. Bull. 1992, 29, 119.(5) Havinga, E. E.; ten Hoeve, W.; Wynberg, H. Synth. Met. 1993, 55-57, 299.(6) Chen, S.-A.; Lee, C.-C. Polymer 1996, 37, 518(7) Chen, S.-A.; Lee, C.-C. Pure & Appl. Chem. 1995, 67, 1983.(8) Reisch, H.; Wiesler, U.; Scherf, U.; Tuytuylkov, N. Macromolecules 1996, 29, 8204.(9) Lee, B.-L.; Yamamoto, T. Macromolecules 1999, 32, 1375.(10) Yamamoto, T.; Zhou, Z.-h.; Kanbara, T.; Shimura, M.; Kizu, K.; Maruyama, T.; Nakamura, Y.;
Fukuda, T.; Lee, B.-L.; Ooba, N.; Tomaru, S.; Kurihara, T.; Kaino, T.; Kubota, K.; Sasaki, S. J. Am.Chem. Soc. 1996, 118, 10389.
(11) Kanbara, T.; Miyazaki, Y.; Yamamoto, T. J. Pol. Sci. A 1995, 33, 999.(12) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1997, 119, 5065.(13) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1998, 120, 5355.(14) Tanaka, S.; Yamashita, Y. Synth. Met. 1993, 55-57, 1251.

, 9ZY�ZcTOL_TaP ;ZWdNZYOPY^L_TZY ?ZbL]O^ >ZW`_TZY ;]ZNP^^TMWP ;ZWd�OT_STPYdWMPYeZMT^F���� H_STLOTLm
���
(15) Tanaka, S.; Yamashita, Y. Synth. Met. 1995, 69, 599.(16) Kitamura, C.; Tanaka, S.; Yamashita, Y. J. Chem. Soc., Chem. Commun. 1994, 1585.(17) von Baeyer, A. Ber. 1879, 12, 1311.(18) Steinkopf, W.; Hanske, W. Liebigs Ann. Chem. 1939, 541, 238.(19) Martinez, F.; Naarmann, H. Synth. Met. 1990, 39, 195.(20) Martinez, F.; Naarmann, H. Angew. Makromol. Chem. 1990, 178, 1.(21) Braunling, H.; Becker, R.; Blochl, G. Synth. Met. 1991, 41-43, 1539.(22) Braunling, H.; Becker, R.; Blochl, G. Synth. Met. 1993, 55-57, 833.(23) Chen, W.-C.; Jenekhe, S. A. Macromolecules 1995, 28, 465.(24) McCullough, R. D.; Ewbank, P. C. In Handbook of Conducting Polymers; 2 ed.; Skotheim, T. A.,
Elsenbaumer, R. L., Reynolds, J. R., Eds.; John Wiley & Sons: Chichester, 1998.(25) Yamamoto, T.; Abla, M. Synth. Met. 1999, 100, 237.(26) Jonas, F.; Krafft, W.; Muys, B. Macromol. Symp. 1994, 169.(27) Kitamura, C.; Tanaka, S.; Yamashita, Y. Chem. Mater. 1996, 8, 570.(28) Trivedi, D. C. In Organic Conductive Molecules and Polymers; Nalwa, H. S., Ed.; John Wiley & Sons:
Chichester, 1997; Vol. 2.(29) Liu, J.; McCullough, R. D. Polymer Preprints 1999, 40, 695.(30) Rader, H. J.; Spickermann, J.; Krevenschmidt, M.; Mullen, K. Macromol. Chem. Phys. 1996, 197,
3285.(31) Morrow, G. W.; Schwind, B. Synth. Commun. 1995, 25, 269.(32) Lehrle, R. S.; Sarson, D. S. Int. J. Polymer Degradation and Stability 1996, 51, 197.(33) Axelsson, J.; Scrivener, E.; Haddleton, D. M.; Derrick, P. J. Macromolecules 1996, 29, 8875.(34) Surville, R.; Josefowicz, M.; Yu, L. T.; Perichon, J.; Buvet, R. Electrochim. Acta 1968, 13, 1451.(35) Cao, Y.; Smith, P.; Heeger, A. J. Synth. Met. 1992, 48, 91.(36) Trivedi, D. C.; Dhawan, S. K. Synth. Met. 1993, 58, 309.(37) Dhawan, S. K.; Trivedi, D. C. Polym. Int. 1992, 25, 55.(38) Neidlein, R.; Tran-Viet, D.; Gieren, A.; Kokkindidis, M.; Wilckens, R. Chem. Ber. 1982, 115, 2898.(39) Langeveld-Voss, B. M. W. Ph D Thesis 1999, Eindhoven University of Technology, Eindhoven.

THIS IS A BLANK PAGE (114)

.SL[_P]�!
.ZYU`RL_PO�;ZWdXP]^�bT_S�$����/T[SPYdWLY_S]LNPYP�@YT_^TY�_SP�8LTY�.SLTY�QZ]�-W`P�7TRS_�0XT__TYR�/TZOP^
,M^_]LN_% A simple and efficient 3-step procedure towards 9,10-bis(4-formylphenyl)anthracene was developed, which compound gave facile access to blueluminescent conjugated polymers containing 9,10-diphenylanthracene units in the mainchain. The novel alkoxy-substituted poly-p-9,10-diphenylanthracene-vinylene PDPAV wasprepared, and the thin film emission maximum was located at 480 nm, implying greenemission. By comparing the fluorescence spectra of an unsubstituted and an alkoxy-substituted model compound, the unexpected red-shift in the emission spectrum was ascribedto the presence of alkoxygroups on the anthracene nucleus of 9,10-diphenylanthracene. Aconjugated copolymer consisting of an unsubstituted 9,10-diphenylanthracene and a 9,9-dialkylfluorene unit, connected by vinylene linkages (poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene PDPAFV), exhibited intense blue fluorescence with an emissionmaximum at λem = 455 nm, which makes PDPAFV a promising conjugated polymer for itsuse in blue light-emitting diodes.
!���4Y_]ZO`N_TZY
Charge injection in a semiconductor under the influence of an electric field, followedby radiative recombination of these charges, called electroluminescence (EL), is the workingprinciple of a Light Emitting Diode (LED). Presently, the active materials used in commercialLEDs are inorganic semiconductors. However, it was shown with an anthracene single crystalthat molecular organic materials can also be prone to electroluminescence.1 Selection of theappropriate organic molecules, based on their fluorescence properties and ability to transportcharges, gave rise to a variety of LEDs emitting in different parts of the visible spectrum.2
The only property in which these organic materials were inferior to their inorganiccounterparts is the long-term structural stability.
Some π-conjugated polymers are, due to the unique combination of theirsemiconducting and luminescent character, also capable to yield EL. This was shown for thefirst time with a LED based on poly-p-phenylene vinylene (PPV).3 Their ease of processingand tuneable bandgap (= tuneable emission colour) make conjugated polymers very attractivefor their use in LEDs, which is reflected in the immense research effort performed in this areaover the past decade.4,5
The ongoing search for the ideal active material in a stable blue light-emittingpolymer LED illustrates the importance of “band gap engineering” in the opposite direction ofwhich was discussed in the preceding chapters, since blue light emission (around 450 nm) ofa conjugated polymer requires a large bandgap of about 2.7 - 3.0 eV. Indeed, the first blue

.SL[_P] !
��!
light-emitting polymer LEDs (with low stability, however) were made from the large-bandgappolymer poly-p-phenylene (PPP).6
The present approaches towards high-bandgap systems for blue-light emitting diodesconsist of limiting the conjugation length by the introduction of 1,3-phenylene “kink”linkages,7-9 silicon sp3 linkages,10-14 or the use of nonconjugated polymers with a blueluminescent chromophore in the side chain15,16 or the main-chain.17-19 The disadvantages ofreducing the conjugation in these drastic ways is a major decrease in semiconductingproperties, which for instance results in the need for high onset voltages.
Although the use of a fully conjugated polymer may result in enhancedsemiconducting properties, keeping the photo- and electroluminescence of these polymerswithin the blue part of the visible spectrum (roughly between 400 and 500 nm) is difficult dueto their extended conjugation and, hence, decreased bandgaps.20-22 The incorporation of unitsin the backbone which exhibit efficient blue photoluminescence, but allow for a conjugationthrough the backbone like diphenylanthracene 1 and fluorene 2 , is therefore very interesting.
1 2
HH
The most extensively studied polymers are the poly-2,7-fluorenylenes, owing to theirthermal and chemical stability, high fluorescence quantum yields and ease of preparation andfunctionalization.7,8,23-26 However, it was noticed that poly-2,7-(9,9-dialkylfluorenylene)sshow a strong tendency to form excimers due to aggregation in the solid state, leading to ared-shift in the emission bands.27,28 This effect could be circumvented by the incorporation ofsterically demanding anthracene units in the main-chain.29
Although it is known that non-conjugated polymers bearing a 9,10-diphenylanthraceneunit in the main-18 or side-chain16 perform very well in blue light-emitting LEDs, theincorporation of this unit in conjugated polymers is less widespread. Examples are polymer 3,where the phenylanthracene side-chain in combination with the PPV backbone gives rise to abroad-wavelength emission,30 and polymers 431 and 5,32 copolymers of anthracene andbenzene of which, unfortunately, no EL data were provided.
n
C7H15
C7H15
n
n
3
4 5

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
��"
The facile access to 9,10-diarylanthracenes, presented in chapter 4 of this thesis, may bea fruitful approach towards monomers for conjugated polymers containing the 9,10-diphenylanthracene moiety.
!���;ZWdXP]�LYO�XZYZXP]�OP^TRY
The basic structure of the desired polymers can be described as 9,10-diphenylanthracene units connected via a unit which does not interrupt the main-chainconjugation (Figure 1). However, this π-conjugated unit may not extend the conjugation toomuch as to keep the luminescence of the polymer within the blue range of the visiblespectrum.
n π-conjugated
unit
Figure 1. Desired structure of 9,10-diphenylanthracene containing conjugated polymers.
Connecting the consecutive diphenylanthracene units by double bonds yields thesimplest representative 6 of such a polymer. The emission maxima of the stilbene (351 nm)33
and diphenylanthracene (431 nm)34 fragments in cyclohexane (Figure 2) are displayed aswell. Although the fluorescence in cyclohexane is not necessarily equal to the fluorescence inthe (thin film) solid state, these values are promising in terms of polymer 6 exhibiting blueluminescence.
n
6
Diphenylanthraceneemission max. = 431 nm(cyclohexane)
Stilbeneemission max. = 351 nm(cyclohexane)
Figure 2. Poly-p-9,10-diphenylanthracene-vinylene PDPAV 6, and emission maxima in cyclohexane of somestructural fragments.
Furthermore, the synthesis of polymer 6 can rely on the multitude of syntheticapproaches towards PPV and derived polymers that are presently known.5 Since the reductive

.SL[_P] !
��#
polymerization of aromatic dialdehydes, using Zn/TiCl4, easily provides poly-arylene-vinylenes,35-37 this reaction was selected for the synthesis of polymer 6, which implies thesynthesis of dialdehydes of type 7 (Scheme 1).
n
6
O
O
7Scheme 1. Retrosynthetic approach towards polymer 6.
The solubility of polymer 6 may be expected to be very low, which must becircumvented by substitution of alkyl- or alkoxygroups on the conjugated backbone. Theintroduction of solublizing groups can be performed either on the benzene or on theanthracene nucleus, depending on the synthetic accessibility. Unfortunately, no syntheticapproaches towards dialdehyde 7 are presently known. Therefore, the addition of aryllithiumderivatives to anthraquinone (chapter 4) is investigated as a viable synthetic route towards 7(Scheme 2), prior to the preparation of monomers carrying solubilizing groups.
O
O
7
BrBr
BrBr
OH
OHO OBrBr +
8
9
Scheme 2. Retrosynthetic approach towards dialdehyde 7.
The synthesis of aromatic aldehydes from their bromides using BuLi/DMF is wellestablished.38 Although the synthesis of the dibromide 8 via dihydroxycompound 9 isknown39,40 it implies a very inefficient procedure based on Grignard reagents. The latter maybe replaced by aryllithium derivatives, to yield reactions analogous to those introduced inchapter 4. In this chapter, the synthesis of dialdehyde 7 is presented, as well as thehomopolymerization of an alkoxysubstituted analogue and copolymerization of 7 with a

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
��$
fluorenyl derivative to yield conjugated polymers containing the 9,10-diphenylanthracene unitin the main-chain.
!���;ZWd�[�$����OT[SPYdWLY_S]LNPYP�aTYdWPYP�;/;,A
!���� >dY_SP^T^ ZQ $����MT^���QZ]XdW[SPYdW��LY_S]LNPYP XZYZXP]^
The synthesis of dialdehyde 7, which is outlined in Scheme 3, follows the procedurethat was developed in chapter 4 for dithienylanthracenes.
BrBr LiBr OO+
BrBr
OH
OHBrBr
O
O
a) b)
c)
d)
98
7
Scheme 3. Synthesis of dialdehyde 7. Reagents and conditions: a) n-BuLi, THF, 45 min. -80 °C, b) THF, -80 °Cto r.t., 60 min. AcOH, 79%, c) HBr, AcOH, r.t., 60 min., 65 - 90 %, d) n-BuLi, Et2O, r.t. 60 min., then DMF, 0°C, 15 min; then 1 M HCl, 99 %.
Addition of an equimolar amount of n-BuLi to a solution of p-dibromobenzene in THFat -80 °C resulted in a monolithiated species, which reacts with half an equivalent ofanthraquinone to give, after quenching with AcOH, the dihydroxycompound 9 in 79% yield.Reduction of compound 9 following the procedure described in chapter 4 using NaI/HBr inAcOH yielded the dibromocompound 8 in 65% yield. However, an improved procedureconsisted of treating a suspension of 9 in AcOH with HBr, without the addition of NaI, togive 8 in 90% yield. It was found that the use of NaI is not necessary in the reduction of 9,because competitive mono-halogenation, as observed for the thiophene analogues, did nottake place. This is presumably due to the reduced reactivity of halogenated phenyl derivativescompared to thiophene. Lithiation of dibromocompound 8 at -80 °C in THF wasunsuccessful. However, lithiation at room temperature in Et2O, followed by quenching with
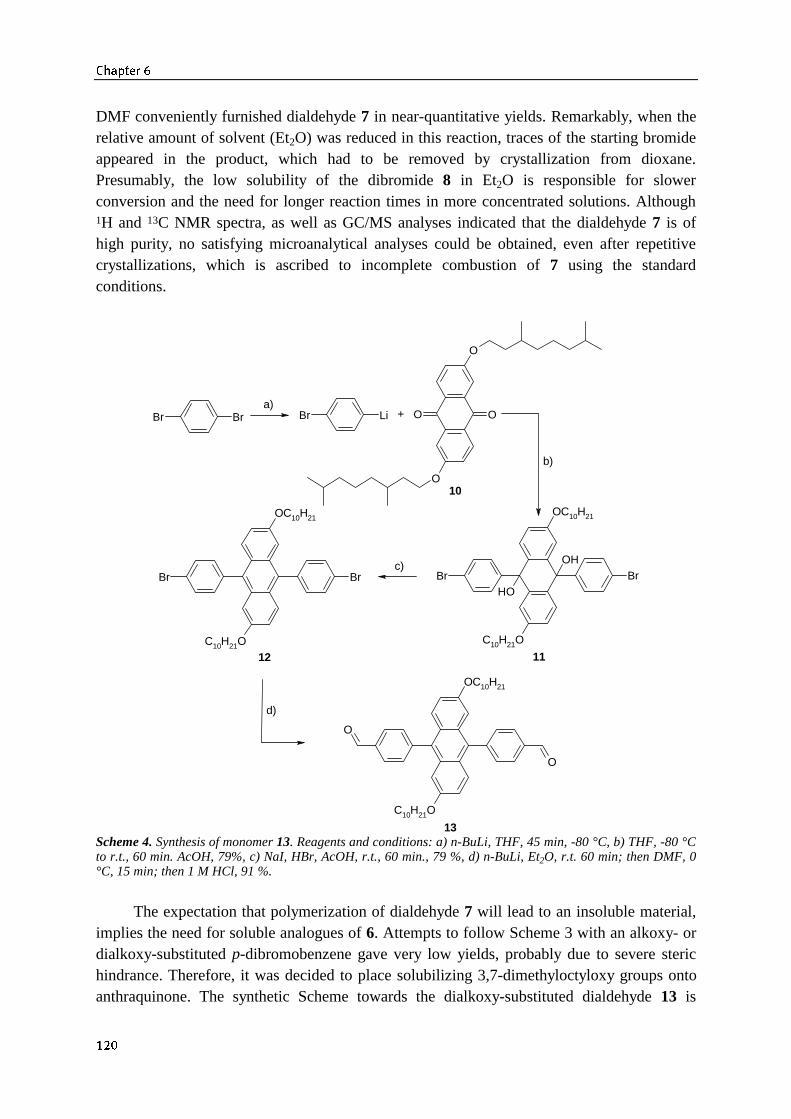
.SL[_P] !
���
DMF conveniently furnished dialdehyde 7 in near-quantitative yields. Remarkably, when therelative amount of solvent (Et2O) was reduced in this reaction, traces of the starting bromideappeared in the product, which had to be removed by crystallization from dioxane.Presumably, the low solubility of the dibromide 8 in Et2O is responsible for slowerconversion and the need for longer reaction times in more concentrated solutions. Although1H and 13C NMR spectra, as well as GC/MS analyses indicated that the dialdehyde 7 is ofhigh purity, no satisfying microanalytical analyses could be obtained, even after repetitivecrystallizations, which is ascribed to incomplete combustion of 7 using the standardconditions.
BrBr LiBr OO
O
O
+
BrBrOH
OH
OC10H21
C10H21O
BrBr
OC10H21
C10H21O
O
O
OC10H21
C10H21O
b)
c)
1112
13
10
a)
d)
Scheme 4. Synthesis of monomer 13. Reagents and conditions: a) n-BuLi, THF, 45 min, -80 °C, b) THF, -80 °Cto r.t., 60 min. AcOH, 79%, c) NaI, HBr, AcOH, r.t., 60 min., 79 %, d) n-BuLi, Et2O, r.t. 60 min; then DMF, 0°C, 15 min; then 1 M HCl, 91 %.
The expectation that polymerization of dialdehyde 7 will lead to an insoluble material,implies the need for soluble analogues of 6. Attempts to follow Scheme 3 with an alkoxy- ordialkoxy-substituted p-dibromobenzene gave very low yields, probably due to severe sterichindrance. Therefore, it was decided to place solubilizing 3,7-dimethyloctyloxy groups ontoanthraquinone. The synthetic Scheme towards the dialkoxy-substituted dialdehyde 13 is

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
���
outlined in Scheme 4. Thus, dialkoxylation of anthraquinone was accomplished by aWilliamson-type reaction of 2,6-dihydroxyanthraquinone with 3,4-dimethyloctyl tosylate toyield the dialkoxyanthraquinone 10 in 56 % yield. The subsequent steps in Scheme 4 via thedihydroxycompound 11 and dibromocompound 12 towards the dialdehyde 13 are entirelyanalogous to the synthesis of the unsubstituted aldehyde 7 in Scheme 3, with comparableyields.
!���� >dY_SP^T^ LYO Z[_TNLW []Z[P]_TP^ ZQ LWVZcd ^`M^_T_`_PO ;/;,A
The monomeric dialdehyde 13 was polymerized using a TiCl4/Zn induced reductivecoupling35-37 (Scheme 5).
O
O
C10H21O
OC10H21
13
C10H21O
OC10H21
n
14
a)
Scheme 5. Synthesis of polymer PDPAV 14. Reagents and conditions: a) Zn/TiCl4, THF, reflux, 16 h, 42%.
McMurry-coupling of the monomeric aldehyde 13 by a suspension of Zn/TiCl4 in THFduring 16 h at reflux temperature, gave the crude polymer 14. Intensive washing of thepolymer in a soxhlet-extractor with methanol and acetone, subsequently, followed byextraction with THF and precipitation in heptane, gave the pure polymer 14 in 42 % yield.The molecular weight of this purified fraction, determined by SEC in CHCl3, is Mw = 13.9kD, Mn = 7.8 kD, D = 1.8 (vs. polystyrene). 1H and 13C-NMR spectra correspond with theproposed structure. Its solubility in CHCl3 and THF is good, and spincasting polymer 14 fromCHCl3 or THF solutions yields yellow transparent thin films of good quality. The optical(absorption and fluorescence spectra) characteristics of polymer 14 are summarized in Figure3.
The absorption spectrum of polymer 14 shows maxima at λmax = 420, 400, 353, 320 and271 nm. The intense absorption at 271 nm is characteristic for aromatic compoundscontaining anthracene. The absorption spectra in solution and the −thin film− solid state areidentical, and the thin film excitation spectrum shows close similarity with the absorptionspectra. The thin film emission spectrum shows a maximum at λmax,em = 480 nm, with a longtail up to 700 nm. This implies that the emission colour of polymer 14 is perceived by thehuman eye as green. This was not expected on the basis of the emission maxima of the twomain-chain fragments of polymer 6 (stilbene and 9,10-diphenylanthracene, Figure 2). Twopossible reasons for the increased emission wavelength can be given: i) The extendedconjugation in polymer 14 causes a decrease in bandgap and, hence, a redshift of theabsorption and emission maximum or ii) the alkoxygroups on polymer 14 cause, due to their

.SL[_P] !
���
electron-releasing character, an increase of the absorption/emission wavelength and/or Stokesshift.
300 400 500 600 700
336 418
400
270480 nm
Flu
ores
cenc
e / A
BS
[a.u
.]
Wavelength [nm]Figure 3. Absorption and fluorescence spectra of polymer 14. (): excitation spectrum (thin film, emissionat λ = 500 nm). (------) emission spectrum (thin film, excitation at λ = 418 nm). (⋅⋅⋅⋅⋅⋅⋅⋅⋅): absorption spectrum(thin film). (-⋅-⋅-⋅-⋅-⋅): absorption spectrum (chloroform solution).
Because the comparison of monomers 7 and 13 is intricate due to the presence ofelectron-withdrawing aldehyde groups, model compounds 15 and 16 were prepared (Scheme6) to investigate the reason for the unexpected increase in emission wavelength of polymer14.
O
O
R
R
R
R
7 : R = H13 : R = OC10H21
15 : R = H16 : R = OC10H21
P
O
OEtEtO+ a)
Scheme 6. Synthesis of model compounds 15 and 16. Reagents and conditions. a) t-BuOK, DMF, r.t. 2 h, 81-78%.

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
���
The Wittig-Horner reaction between diethyl benzylphosphonate and aldehydes 7 and 13gave model compounds 15 and 16 in 81 and 78 %, respectively. The solid state (powder)fluorescence spectra of 15 and 16 are depicted in Figures 4a and 4b, respectively.
300 400 500 600 700
391
457 nm
Wavelength [nm]
Flu
ores
cenc
e [a
.u.]
a)
300 400 500 600 700
492 nm390
453
Wavelength [nm]
Flu
ores
cens
e [a
.u.]
b)Figure 4. a) Solid state (powder) fluorescence spectra of 15. () Excitation spectrum (emission at 457 nm),(------) emission spectrum (excitation at 365 nm). b) Solid state (powder) fluorescence spectra of 16. ()excitation spectrum (emission at 492 nm), (------) emission spectrum (excitation at 390 nm).
From the fluorescence spectra of compounds 15 and 16 it follows that the presence ofalkoxy-substituents on the anthracene nucleus causes the appearance of an additional peak inthe excitation spectra at λ = 453 nm and an increase of the emission maximum wavelengthfrom λmax,em = 457 (blue) to λmax,em = 492 nm (green). Therefore, the increased emissionwavelength (λ = 499 nm) of polymer 14 is due to the alkoxy substituents on the anthracenenucleus. If soluble conjugated polymers are to be obtained with the 9,10-diphenylanthracenemoiety in the main-chain, substitution of the anthracene nucleus with alkoxygroups isundesirable, of which the consequences are evaluated in the next section.

.SL[_P] !
���
!���;ZWd�[�OT[SPYdWLY_S]LNPYP�QW`Z]PYdWPYP�aTYdWPYP^�;/;,1A
!���� ,W_P]YL_TaP [ZWdXP] OP^TRY
The difficulty of following the synthesis depicted in Scheme 3 in case of alkoxy-substituted phenyls, together with the undesired 2,6-alkoxy-substitution of the anthracenenucleus, implies that incorporation of unsubstituted 9,10-diphenylanthracene units in themain-chain must be considered for obtaining these types of blue luminescent conjugatedpolymers. As a consequence, the polymer-design shown in Figure 2 has to be evaluated again:a requirement for the connecting conjugated unit to be added to those already discussed(section 6.2), is the ability of this unit to carry solubilizing groups.
As mentioned in the introduction (section 6.1), the fluorene unit 2 is very appropriate inthis light, since it is an efficient blue luminescent species41 and easily functionalized withalkyl substituents at the 9,9-positions without dramatically altering the fluorescence.42 Thus,the copolymer depicted in Figure 5 is suggested as a candidate for a 9,10-diphenylanthracenecontaining, soluble blue luminescent polymer.
2,7-distyrylfluoreneemission max = 427 nmR = n-propyl(cyclohexane)
9,10-diphenylanthraceneemission max. = 431 nm(cyclohexane)
RR
n
RR
9,10-bis(4-styrylphenyl)anthracene 15emission max = 457 nm(neat)
Figure 5. Poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene (PDPAFV), and emission maxima of somefragments.
Again, the expected blue luminescence of this polymer is based on the comparison ofthe fluorescence data of some main chain fragments: 2,7-Distyrylfluorene,43
diphenylanthracene,34 and 9,10-bis(4-styrylphenyl)anthracene (this chapter), possessingemission maxima (Figure 5) within the blue range of the visible spectrum. Retrosyntheticanalysis shows that the aldehyde 7 can also be used in the synthesis of the fluorene-containingpolymer (Scheme 7). The synthesis of the dialkylfluorenyl bismethylphosphonate of Scheme7 has been described previously.43 Furthermore, the copolymerization of such co-monomers

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
��
with various aromatic aldehydes towards blue luminescent conjugated polymers is alsoknown.8,44 However, incorporation of the 9,10-diphenylanthracene unit in this way is notknown, due to the previous unavailability of dialdehyde 7.
RR
n
O
O
7
P
O
P
O
R R
OEtOEt
EtOOEt
Scheme 7. Retrosynthetic analysis of poly-p-9,10-diphenylanthracene-fluorenylene-vinylene.
!���� >dY_SP^T^ ZQ _SP QW`Z]PYP NZ�XZYZXP]
The synthesis of the fluorene co-monomers of type 19 has been described,43, and isused here with some minor adaptations (Scheme 8). Again, the 3,7-dimethyloctyl group wasselected as the solubilizing group. Application of this group in its racemic form is expected tobe beneficial for solubilizing the polymer chain, because of diminished regularity and chain-packing possibilities. This may partly compensate for the relatively small fraction ofsolubilizing groups in the polymer.
P
O
P
O
H21C10 C10H21
OEtOEt
EtOOEt
Cl
C10H21H21C10
BrBr
a)
b)
c)
+
1718
19
Scheme 8. Synthesis of fluorene co-monomer 19. Reagents and conditions: a) t-BuOK, NaI, DMF, r.t., 72 h,82%, b) paraformaldehyde, NaBr, H2SO4 (conc.), AcOH, 80°C, 24 h, 85%, c) (EtO)3P, 140 °C, 24 h, 71%.

.SL[_P] !
��!
The NaI-catalysed reaction of fluorene with 3,7-dimethyloctylchloride in the presenceof potassium-t-butoxide gave the dialkylfluorene 17 in 82% yield, as a mixture of thediastereomers. Bromomethylation of 17 using a mixture of paraformaldehyde/NaBr andH2SO4 at 80 °C furnished the bisbromomethylcompound 18 in 85% yield. Conversion intothe bisphosphonate 19 was accomplished in 71% yield by heating the bisbromomethylcompound 18 in a large excess of (EtO)3P. The fluorene compounds 17 - 19 are colourlessviscous oils.
!���� .Z[ZWdXP]TeL_TZY
The Wittig-Horner copolymerization of aromatic dialdehydes and dialkylfluorenebisphosphonates has been described using NaOEt or K-O-t-Bu in EtOH,9,28, however, lowyields and quite low molecular weights (around 5 kD) were obtained under these conditions.The solubility of copolymer 20 (Scheme 9) in EtOH is expected to be rather low, which maymean premature precipitation and diminished molecular weights. Therefore, THF wasregarded as a more appropriate solvent for the copolymerization.
O
O
P
O
P
O
H21C10 C10H21
OEtOEt
EtOOEt
197
H21C10 C10H21
n
20
a)
+
Scheme 9. Synthesis of copolymer 20. Reagents and conditions: a) K-O-t-Bu, THF, r.t., 4 h, then PhCHO, 1 h,then AcOH, 73 %.
Treatment of a solution of a 1 : 1 mixture of co-monomers 7 and 19 with K-O-t-Bu inTHF at r.t. resulted in the formation of copolymer 20. To ensure complete removal of thephosphonate endgroups, an excess of benzaldehyde was added at the end of thepolymerization. Quenching with AcOH and subsequent workup then afforded the crude 20,which could be purified by precipitation in acetone to give 20 in 73 % yield. The molecularweight of this purified fraction, determined by SEC in CHCl3, is Mw = 22.4 kD; Mn = 6.1 kD;D = 3.6 (vs. polystyrene). 1H and 13C-NMR spectra as well as microanalytical analysescorrespond to the suggested structure of 20. In Figure 5, the aromatic part of the 13C-NMRspectrum of polymer 20 is depicted which shows 15 of the expected 16 signals.

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
��"
150 140 130 120
Figure 6. Aromatic part of the 125 MHz 13C-NMR spectrum of polymer 20.
Polymer 20 is sparingly soluble in chloroform and toluene, and moderately soluble inTHF and cyclopentanone. Stable yellow transparent films of polymer 20 could be cast fromTHF/toluene solutions onto glass. Figure 7 shows the absorption, excitation and emissionspectra of polymer 20 as thin films on glass.
300 400 500 600 700
381
387455 nm
Flu
ores
cenc
e / A
BS
[a.u
.]
Wavelength [nm]
Figure 7. Solid-state (thin film on glass) fluorescence and absorption spectra of polymer 20. () Excitationspectrum (emission at 455 nm). (⋅⋅⋅⋅⋅⋅⋅⋅⋅) Emission spectrum (excitation at 385 nm). (-⋅⋅-⋅⋅-) Absorption spectrum.
The films show an intense, sky blue fluorescence with an emission maximum of 455nm. The absorption and emission spectra show only a small overlap, which diminishes theluminescence self-absorption. The emission maximum of 455 nm is close to that of modelcompound 15, a structural fragment of polymer 20 (Figure 5), which may indicate that thisfragment is governing the luminescence.
Annealing a film of 20 at 100 °C for a longer time (typically a few hours) in air did notdramatically influence the luminescence color, which is an indication that excimer formation,as observed for poly(dialkylfluorenylene)s, is negligible. Probably, the diphenylanthraceneunit prevents the aggregation of the polymer chains, an effect that was also observed forcopolymers of dialkylfluorene and anthracene.29 However, a decrease in fluorescenceintensity after annealing for a long time (24 h) was observed. Furthermore, exposing films of

.SL[_P] !
��#
polymer 20 to intense UV light (365 nm) in air for a longer time (24 h) causes a red-shift inthe luminescence. The degradation processes in polyfluorenes under the influence of UV lighthave been studied in detail45 and are ascribed to the formation of fluorenone derivativeswhich quench the luminescence. Due to the presence of 9,9-dialkylfluorenylene units in 20,formation of fluorenone derivatives is unlikely. Because in polymer 20 a diphenylanthraceneunit is present, it should not be excluded that degradation of this unit can occur as well, sinceit has long been known that photo-oxidation processes easily occur in diarylanthracenes whenthey are exposed to UV light in air.46,47 Nevertheless, it was found that polyfluorenes, andespecially fluorene copolymers with anthracene, have an increased stability compared toPPV-type polymers.45
The electroluminescence of polymer 20 is currently under investigation.
!� �.ZYNW`^TZY^
Starting from 1,4-dibromobenzene and anthraquinone, an efficient 3-step proceduretowards 9,10-di(4-formylphenyl)anthracene 7 was developed. The pivotal role of thismolecule in the synthesis of conjugated polymers incorporating 9,10-diphenylanthraceneunits in the main chain was demonstrated by the synthesis of two types of luminescentpolymers.
Reductive polymerization of the alkoxy substituted dialdehyde 13 yielded the novelpoly-p-9,10-diphenylanthracene-vinylene PDPAV 14. Based on the known blue-fluorescentproperties of the −unsubstituted− main-chain fragments of this polymer, PDPAV 14 wasexpected to exhibit intense blue fluorescence as well. However, the (broad) emissionmaximum of the alkoxy-substituted PDPAV 14 was located at 480 nm, giving greenemission. By comparing the fluorescence spectra of an unsubstituted and an alkoxy-substituted model compound, the bathochromic shift in the emission spectrum was ascribedto the location of alkoxygroups on the anthracene nucleus of 9,10-diphenylanthracene.Therefore, an important design criterion for blue luminescent 9,10-diphenylanthracene-containing conjugated polymers is that substitution of solubilizing alkoxygroups on theanthracene nucleus is undesirable. Together with the failure to synthesize solublediphenylanthracene derivatives with alkoxygroups on the phenylrings, this implies that blueluminescence in these polymers is only secured when the 9,10-diphenylanthracene unitsremains unsubstituted.
Therefore, a conjugated copolymer was designed consisting of an unsubstituted 9,10-diphenylanthracene and a 9,9-dialkylfluorene unit, connected by vinylene linkages. It wasverified that substitution of alkyl groups on fluorene does not result in a bathochromic shift ofthe luminescence into the green region of the visible spectrum. Indeed, the poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene PDPAFV 20, obtained via a Wittig-Hornercopolymerization of dialdehyde 7 with bisphosphonate 19, exhibited intense bluefluorescence with an emission maximum at λem = 455 nm. Degradation of PDPAFV 20,which causes undesired changes in the fluorescence spectra, only occurred after heating the

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
��$
film at 100 °C for a long time in air, or exhibition to intense UV-light in air. The aboveproperties make PDPAFV 20 a promising conjugated polymer for the application in bluelight-emitting diodes, the investigation of which is now underway.
!�!�0c[P]TXPY_LW�>PN_TZY
2PYP]LW []ZNPO`]P^% For general procedures, the reader is referred to section 2.5
$����-T^����M]ZXZ[SPYdW��$����OTSdO]ZcdOTSdO]ZLY_S]LNPYP F$H�� Under an inert atmosphere of argon, 1,4-dibromobenzene (7.50 g, 0.032 mol) was dissolved in dry THF (100 ml) and the resulting solution cooled to -80°C. A 1.6 M solution of n-BuLi in hexane (18.2 ml, 0.029 mol) was added dropwise at this temperature, afterwhich the solution was stirred at -80 °C for 45 min. Solid anthraquinone (3.31 g, 0.016 mol) was added in oneportion at -80 °C and the reaction mixture allowed to warm to room temperature. Stirring was continued for 60min. at this temperature, after which acetic acid (10 ml) was added. The reaction mixture was concentrated on arotary evaporator and the residue taken up in ethanol (100 ml). Precipitation of this solution in water (300 ml),followed by filtration and drying, yielded crude 9 as a pale brown powder. Crystallization from CHCl3/THFyielded pure 9 as white crystals (6.53 g, 0.0125 mol, 78.7 %). 1H-NMR (300 MHz, DMSO-d6): δ: 7.82 (m, 4H,H anth), 7.44 (m, 4H, H anth), 7.15 (d, J = 8.8 Hz, 4H, H3,5 ph), 6.87 (d, J = 8.8 Hz, 4H, H2,6 ph), 5.3 (broad s,2H, -OH). 13C-NMR (75 MHz, DMSO-d6): δ: 145.6 (C1 ph), 141.3 (C4a anth), 130.4, 129.7, 127.7, 127.3 (C-Hanth + C-H ph), 120.3 (C-Br), 73.9 (C-OH). IR (ATR): ν: 3457, 1486, 1393, 1326, 1154, 1073, 1008, 885, 818,763 cm-1. Anal. calcd for C26H18Br2O2 (522.24): C, 59.80; H, 3.47. Found: C, 61.04; H, 3.70.
$����-T^����M]ZXZ[SPYdW��LY_S]LNPYP F#H�� ;]ZNPO`]P �� To a suspension of 9 (6.5 g, 0.012 mol) and NaI (9.3g, 0.062 mol) in acetic acid (75 ml) at room temperature was slowly added a 33% solution of HBr in AcOH (11ml). After complete addition, the reaction mixture was stirred at room temperature for another 60 min. andsubsequently poured into 1000 ml water. The suspension was treated with an aqueous solution of Na2S2O5 untilcolorless, filtered, washed thoroughly with water and dried. The resulting cream-coloured product wasrecrystallized from THF/Hexane to give pure 8 as white crystals (3.78 g, 7.74 mmol, 64.5 %). 1H-NMR (300MHz, CDCl3): δ: 7.77 (d, J = 8.5 Hz, 4H, H2 ph), 7.68 (m, 4H, H1 anth), 7.38 (d + m, 8H, H3 ph + H2 anth).13C-NMR (75 MHz, CDCl3): δ: 137.9, 136.1 (C1 ph + C9,10 anth), 133.1, 131.8 (C-H ph), 129.8 (C4a anth),126.8, 125.6 (C-H anth), 121.9 (C-Br). IR (ATR): ν: 1487, 1389, 1098, 1068, 1009, 940, 813, 767 cm-1. Anal.calcd. for C26H16Br2 (488.22): C, 63.96; H, 3.30. Found: C, 63.98; H, 3.29.;]ZNPO`]P �% To a stirred suspension of 9 (21.6 g, 0.04 mol) in AcOH (200 ml) at room temperature was slowlyadded a 33% solution of HBr in AcOH (37.5 ml). After complete addition, the reaction mixture was stirred atroom temperature for another 60 min. The suspension was filtered and the residue washed with AcOH until thewashings were colourless. The filtrate was collected and dried to give 8 as a cream-coloured solid (17.5 g, 0.036mol, 89.6 %) with the identical analytical data as above.
$����-T^����QZ]XdWNL]MZcLWOPSdOP��LY_S]LNPYP F"H�� A suspension of 8 (1.0 g, 2.05 mmol) in dry Et2O (100ml) was cooled, under an inert atmosphere of argon, to -10 °C. A 1.6 M solution of n-BuLi in hexane (3.2 ml,5.12 mmol) was added dropwise at this temperature and the resulting yellow suspension allowed to warm toroom temperature. After stirring at this temperature for 60 min., the reaction mixture was cooled to 0 °C andquenched with dry DMF (1 ml). The resulting suspension was allowed to warm to room temperature and stirredfor 15 min. A solution of HCl (1 M, 100 ml) was added to the reaction mixture, and the Et2O evaporated on arotary evaporator. The resulting aqueous suspension was filtered and the residue thoroughly washed with waterand dried to give 7 as a yellowish solid (0.78 g, 2.01 mmol, 98.5 %) which could be crystallized from dioxane.1H-NMR (300 MHz, CDCl3): δ: 10.24 (s, 2H, -CHO), 8.18 (d, J = 6.3 Hz, 4H, H ph), 7.71 (d, J = 6.4 Hz, 4H, Hph), 7.64 (m, 4H, H2 anth), 7.40 (m, 4H, H1 anth). 13C-NMR (75 MHz, CDCl3): δ: 192.0 (-CHO), 145.8 (C1ph), 136.3, 135.9 (C4 ph + C9,10 anth), 132.2, 130.0 (C-H ph), 129.5 (C4a anth), 126.7, 125.8 (C-H anth). IR(ATR): ν: 1694, 1603, 1372, 1302, 1207, 1171, 1010, 941, 818, 768, 671 cm-1 , Anal. calcd. for C28H18O2: C,87.03; H, 4.69. Found: C, 85.60; H, 4.72. GC/MS (EI): 386 (M+⋅, 100%), 357 (M+ - CHO), 326 (M+ -2 CHO),252 (M+ -2 CHO, - Ph).

.SL[_P] !
���
��!�-T^����"�OTXP_SdWZN_dWZcd�LY_S]L\`TYZYP F��H�� 2,6-Dihydroxyanthraquinone (2.75 g, 11.4 mmol), 3,7-dimethyloctyl tosylate (7.15 g, 22.9 mmol) and NBu4Cl (0.25) were dissolved in dry MEK (50 ml) under an inertatmosphere of argon. Under stirring, K2CO3 (9.5 g, 68.7 mmol) was added and the resulting reaction mixturerefluxed under argon for 12 h. After cooling to r.t., the inorganic salts were removed by filtration and the filtrateevaporated. The residue was taken up in Et2O, filtered over a short column of silica with Et2O and the eluateevaporated. The remaining solid was crystallized from hexane to give 10 as yellow crystals (3.3 g, 6.33 mmol,55.6 %). 1H-NMR (300 MHz, CDCl3): δ: 8.22 (d, J = 8.7 Hz, 2H, H4 anth), 7.70 (d, J = 2.6 Hz, 2H, H1 anth),7.21 (dd, J = 8.6 and 2.6 Hz, 2H, H3 anth), 4.17 (m, 4H, -OCH2-), 1.9 - 1.1 (m, H alkyl), 0.98 (d, J = 6.5 Hz,6H, -CH3), 0.88 (d, J = 6.5 Hz, 12H, -CH3). 13C-NMR (75 MHz, CDCl3): δ: 182.8 (C=O), 164.6 (C-O-Alkyl)136.4 (C3a anth), 130.2 (C-H anth), 127.5 (C4a anth), 121.5 (C-H anth), 111.1 (C-H anth), 67.8 (-OCH2-), 39.8,37.8, 37.8, 36.5, 30.4, 28.5, 25.2, 23.3, 23.2, 20.2 (C alk). Anal calcd. for C34H48O4 (520.75): C, 78.42; H, 9.29.Found: C, 78.49; H, 9.59.
$����-T^���M]ZXZ[SPYdW����!�MT^����"�OTXP_SdWZN_dWZcd��$����OTSdO]ZcdOTSdO]ZLY_S]LNPYP F��H�� Prepa-red as described for 9 from 1,4-dibromobenzene (1.70 g, 7.2 mmol), a 1.6 M solution of n-BuLi in hexane (3.2ml, 5.12 mmol) and 10 (1.50 g, 2.9 mmol) in dry THF (50 ml) to give 2.3 g of 11, which was used in the nextstep without further purification. 1H-NMR (400 MHz, CDCl3): δ: 7.45 (d, J = 8.7 Hz, 2H, H4 anth), 7.18 (d, J =8.6 Hz, 4H, H3 ph), 7.04 (d, J= 2.6 Hz, 2H, H1 anth), 6.92 (d, J = 8.7 Hz, 4H, H2 ph), 6.84 (dd, J = 8.7 and 2.6Hz, 2H, H3 anth), 3.70 (m, 4H, -OCH2-), 2.06 (s, 2H, -OH), 1.1 - 1.9 (m, 20H, alk), 0.91 (d, J = 6.5 Hz, 6H, -CH3), 0.86 (d, J = 6.5 Hz, 12H, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 158.9 (C-OCH2-), 144.5, 142.0, 132.6(C4a,3a anth + C1 ph), 130.7, 129.0 (C-H ph), 128.7 (C4 anth), 121.1 (C-Br), 115.0 (C3 anth), 112.3 (C1 anth),74.2 (C-OH), 66.4 (-OCH2-), 39.2, 37.3, 36.1, 29.8, 28.0, 24.6, 22.7, 22.6, 19.6 (alk).
$����-T^���M]ZXZ[SPYdW����!�MT^����"�OTXP_SdWZN_dWZcd�LY_S]LNPYP F��H�� Prepared as described for 8following procedure 1 from 11 (2.30 g, 2.76 mmol), NaI (2.55, 17.0 mmol) and a 33 % solution of HBr inAcOH (3 ml) to give, after crystallization from i-PrOH/CH2Cl2 pure 12 (1.70 g, 2.18 mmol, 79.1 %) as yellowcrystals. 1H-NMR (300 MHz, CDCl3): δ: 7.74 (d, J = 8.3 Hz, 4H, H2 ph), 7.48 (d, J = 9.5 Hz, 2H, H4 anth),7.35 (d, J = 8.3 Hz, 4H, H3 ph), 7.03 (dd, J = 9.5 and 2.5 Hz, 4H, H3 anth), 6.78 (d, J = 2.5 Hz, 2H, H1 anth),4.88 (m, 4H, -OCH2-), 1.9 - 1.1 (m, 20H, alk), 0.88 (m, 18H, -CH3). Anal. calcd. for C46H56Br2O2 (800.76): C,69.00; H, 7.05. Found: C, 69.50; H, 7.18.
$����-T^����QZ]XdW[SPYdW����!�MT^����"�OTXP_SdWZN_dWZcd�LY_S]LNPYP F��H�� Prepared as described for 7 from12 (1.30 g, 1.62 mmol), a 1.6 M solution of n-BuLi in hexane (2.54 ml, 4.06 mmol) and DMF (±1 ml) in dryether (100 ml) to give, after crystallization from i-PrOH pure 13 as yellow needles (1.03 g, 1.47 mmol, 91.0 %).1H-NMR (400 MHz, CDCl3): δ: 10.2 (s, 2H, -CHO), 8.14 (d, J = 8.3 Hz, 4H, H3 ph), 7.66 (d, J = 8.2 Hz, 4H,H2 ph), 7.45 (d, J = 9.5 Hz, 2H, H4 anth), 7.05 (dd, J = 9.5 and 2.5 Hz, 2H, H3 anth), 6.73 (d, J = 2.4 Hz, H1anth), 3.83 (m, 4H, -OCH2-), 1.8 - 1.1 (m, 20H, alk), 0.85 (m, 18H, -CH3). 13C-NMR (100 MHz, CDCl3): δ:192.0 (CHO), 155.8 (C-OAlk), 146.3, 135.6, 134.0 (C1,4 ph + C9a anth), 132.0 (C-H ph), 130.0 (C-H ph),129.0, 127.6, 126.3 (C anth), 120.6, 103.3 (C-H anth), 66.0 (-OCH2-), 39.2, 37.2, 35.8, 29.7, 27.9, 24.6, 22.7,22.6, 19.5 (C alk).IR (ATR): ν: 2925, 1703, 1626, 1603, 1462, 1383, 1287, 1205, 1120, 1018, 808 cm-1. Anal.calcd. for C48H58O4: C, 82.48; H, 8.36. Found: C, 82.78; H, 8.65.
;ZWd�[�$����OT[SPYdW���!�MT^����"�OTXP_SdWZN_dWZcd�LY_S]LNPYP�aTYdWPYP ;/;,A F��H�� The reductivepolymerization was carried out following the method of Cooke and Wagener.36 A suspension of zinc dust (281mg, 4.30 mmol) in dry THF (100 ml) was cooled to -10 °C under an inert atmosphere of argon. TiCl4 (0.24 ml,2.15 mmol) was added dropwise at this temperature and the resulting mixture was heated at reflux for 45 min toobtain a black solution. After cooling to r.t., a solution of 13 (250 mg, 0.358 mmol) in dry THF (5 ml) wasadded dropwise and the resulting polymerization mixture was heated under reflux for 16 h. After cooling thepolymerization mixture to r.t., 100 ml of a 10 % K2CO3 solution was added and the resulting mixture stirred for30 min, during which time the color of the mixture changed to bright yellow. The yellow solid which hadseparated was filtered, washed with MeOH and, subsequently, subjected to Soxhlet-extraction with methanol,acetone and THF (24 h each), respectively. The THF fraction was evaporated, taken up in CHCl3 andprecipitated in heptane to give PDPAV 14 as a yellow amorphous solid (100 mg, 0.15 mmol, 41.9 %). 1H-NMR(400 MHz, CDCl3): δ: 6.5 - 8.0 (aromatic/vinylic protons), 3.5 - 4.0 (-OCH2-), 0.5 - 2.0 (alkoxy protons). 13C-NMR (100 MHz, CDCl3): δ: 155.5 (C-OAlk), 139.5, 139.0 (C1,4 ph), 135.5 (C9,10 anth), 131.7 - 126.7 (ph +anth + vinyl), 119.9 (C3 anth), 103.9 (C1 anth). IR (ATR): ν: 2925, 1610, 1464, 1274, 1211, 1047, 1020, 817

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
���
cm-1. GPC (CHCl3): Mw = 13.9 kD, Mn = 7.8, D = 1.8 (vs. polystyrene). Anal. calcd. for (C48H58O2)n (666.98)n:C, 86.44; H, 8.76. Found: C, 82.41; H, 8.98.
$����-T^���_]LY^�^_d]dW[SPYdW�LY_S]LNPYP F� H�� To a solution of diethyl benzylphosphonate (479 mg, 2.10mmol) in dry DMF (25 ml) under an inert atmosphere of argon at r.t. was added K-O-t-Bu (245 mg, 2.10 mmol).This mixture was stirred until a clear solution was obtained. Subsequently, a suspension of 7 (270 mg, 0.70mmol) in dry DMF (10 ml) was added dropwise, and the mixture stirred at r.t. for 2 h. The reaction mixture waspoured in water (400 ml) and neutralized with 1 N HCl. The precipitate was filtered, washed with water anddried. Crystallization from toluene yielded the pure 15 as cream-coloured crystals (302 mg, 0.57 mmol, 81.4 %).1H-NMR (300 MHz, CDCl3): δ: 7.78 (m, 8H, styryl), 7.61 (d, J = 8.0 Hz, 4H, H ph), 7.50 (d, J = 8.1 Hz, 4H, Hph), 7.46 - 7.28 (m, 14H, styryl + anth). IR (ATR): ν: 3023, 1595, 492, 1445, 1391, 1107 - 670 cm-1. Anal.calcd. for C42H30 (534.70): C, 94.35; H, 5.66. Found: C, 93.74; H, 5.47.
$����-T^���_]LY^�^_d]dW[SPYdW����!�MT^����"�OTXP_SdWZN_dWZcd��LY_S]LNPYP F�!H�� This compound wasprepared as described for 15 from diethyl benzylphosphonate (245 mg, 1.0 mmol), K-O-t-Bu (112 mg, 3.0mmol) and 13 (250 mg, 0.36 mmol). After pouring the reaction mixture in water and neutralization, the waterlayer was extracted with Et2O (2 times). The combined organic layers were thoroughly washed with water, driedover MgSO4 and evaporated to give a yellow solid, which was crystallized from i-PrOH to afford the pure 16 asyellow needles (0.22 g, 0.28 mmol, 77.8 %). 1H-NMR (300 MHz, CDCl3): δ: 7.77 (d, J = 8.3 Hz, 4H, H ph),7.60 (m, 6H, styryl + anth + vinyl), 7.48 (d, J = 8.2 Hz, H ph), 7.43 (t, J = 7.5 Hz, 4H, styryl), 7.30 (m, 6H,styryl + anth + vinyl), 7.03 (dd, J = 2.6 and 9.5 Hz, 2H, H3 anth), 6.91 (d, J = 2.4 Hz, 2H, H1 anth) 3.90 (m, 4H,-OCH2-), 1.8 - 1.1 (m, 20H, alk), 0.86 (t, J = 7.3 Hz, 18H, -CH3). 13C-NMR (100 MHz, CDCl3): δ: 155.5 (C-OAlk), 138.4, 137.3, 131.6, 129.5, 128.9, 128.8, 128.5, 128.1, 127.7, 126.9, 126.7, 126.6, 120.1 (aromatic +vinylic C), 103.8 (C4 anth), 66.0 (-OCH2-), 39.2, 37.3, 35.9, 29.7, 27.9, 24.6, 22.7, 22.6, 19.6. IR (ATR): ν:3027, 2925, 1625, 1457, 1382, 1210, 1121 - 689. Anal. calcd. for C58H66O2 (795.16): C, 87.61; H, 8.37. Found:C, 87.68; H, 8.47.
$�$�-T^����"�OTXP_SdWZN_dW��QW`Z]PYP F�"H�� The alkylation was carried out by an adaptation of the procedure ofKelley et al.43 To a solution of KO-t-Bu (2.02 g, 0.018 mol) in dry DMF (25 ml) at r.t. was added fluorene (1.0g, 6.00 mmol). The reaction mixture turned deeply orange, after which 1-chloro-3,7-dimethyloctane (2.65 g,0.015 mol) was added dropwise (exothermic reaction). After the addition of NaI (100 mg), the reaction mixturewas stirred at r.t. for 3 days (blue-coloration) and subsequently poured into water. The aqueous layer wasextracted with Et2O three times, and the combined organic layers washed thoroughly with water. After dryingwith MgSO4 and evaporation of the solvent, crude 17 remained as a mixture with the alkyl chloride, which wasdistilled off in a kugelrohr apparatus. The distillation residue was subjected to filtration over a short column ofsilica with hexane. Evaporation of the hexane filtrate gave the pure 17 (2.2 g, 4.92 mmol, 82.1 %) as acolourless, viscous oil. 1H-NMR (300 MHz, CDCl3): δ: 7.74 (d, J = 7.5 Hz, 2H, H4,5), 7.35 (m, 6H,H1,2,3,6,7,8), 2.00 (m, 4H, -CH2-), 1.6 - 0.4 (38H, alk). 13C-NMR (75 MHz, CDCl3): δ: 150.7, 141.3 (C1a, 4a),127.1, 126.7, 122.9, 119.7 (C1,2,3,4), 54.9 (C9), 39.3, 37.7, 36.7, 33.0, 30.5, 28.0, 24.7, 22.8, 22.7, 19.6 (alkyl).IR (ATR): ν: 2925, 1449, 1365, 734 cm-1. Anal. calcd for C33H50 (446.77): C, 88.72; H, 11.28. Found: C, 89.14;H, 11.64.
��"�-T^��M]ZXZXP_SdW��$�$�MT^����"�OTXP_SdWZN_dW��QW`Z]PYP F�#H�� A mixture of 17 (1.30 g, 2.90 mmol),NaBr (2.10 g, 20 mmol) and 90 % paraformaldehyde (1.45 g, 44 mmol) in AcOH (25 ml) was heated to 80 °C,after which concentrated H2SO4 (1.6 ml) was slowly added. The resulting reaction mixture was stirred at 80 °Cfor 24 h., cooled to r.t. and poured into water. The aqueous layer was extracted with Et2O, after which thecombined organic layers were washed with aqueous Na2CO3 and water, dried over MgSO4 and evaporated togive the crude 18 as a viscous oil. This oil was subjected to filtration over alumina (deactivated with 7 % w/wwater) with hexane. Evaporation of the hexane filtrate the pure 18 as a colourless oil (1.56 g, 2.47 mmol, 85.2%). 1H-NMR (300 MHz, CDCl3): δ: 7.65 (d, 8.1 Hz, 2H, H4,5), 7.38 (m, 6H, H1,2,3,6,7,8), 4.62 (s, 4H, -CH2Br), 2.00 (m, 4H, -CH2-), 1.5 - 0.3 (38H, alk). 13C-NMR (75 MHz, CDCl3): δ: 151.7, 141.0 (C1a,4a), 137.1,128.0, 123.8, 120.1 (C1,2,3,4), 55.1 (C9), 39.3, 37.4, 36.7, 34.5, 32.9, 31.7, 30.5, 28.0, 24.7, 22.8, 19.6 (alkyl +CH2Br). IR (ATR): ν: 2925, 1466, 1365, 1217, 819, 738, 673. Anal. calcd for C35H52Br2 (632.61): C, 66.45; H,8.29. Found: C, 66.69; H, 8.47.

.SL[_P] !
���
��"�-T^��OTP_SdW[SZ^[SZYZXP_SdW��$�$�MT^����"�OTXP_SdWZN_dW��QW`Z]PYP F�$H�� A solution of 18 (1.56 g, 2.47mmol) in (EtO)3P (10 ml) was heated at 140 °C for 24 h. The excess (EtO)3P was removed by vacuumdistillation to give 19 as a colourless oil. (1.31 g, 1.75 mmol, 71 %). 1H-NMR (400 MHz, CDCl3): δ: 7.59 (d, J= 7.6 Hz, 2H, H4,5), 7.25 (m, 6H, H1,2,3,6,7,8), 3.97 (m, 8H, POCH2CH3), 3.21 (d, J = 21.6 Hz, -CH2PO(OEt)2), 1.95 (m, 4H, -CH2-), 0.3 - 1.5 (50H, alkyl + POCH2CH3). 13C-NMR (100 MHz, CDCl3): δ:151.1, 139.8 (C1a,4a), 130.4, 128.7, 124.3, 119.8 (C1,2,3,4), 62.4 (-CH2PO(OEt)2), 55.1 (C9), 39.6, 38.2, 37.2,35.2, 33.8, 33.4, 31.3, 28.3, 25.1, 23.0, 19.8, 16.9 (alkyl + OEt). IR (ATR): ν: 2926, 1469, 1251, 1025, 958, 849cm-1. Anal. calcd. for C43H72O6P2 (747.00): C, 69.14; H, 9,72. Found: C, 69.24; H, 10.04.
;ZWd�[�$����OT[SPYdWLY_S]LNPYP���"�MT^�$����"�OTXP_SdWZN_dW��QW`Z]PYdWPYP�aTYdWPYP ;/;,1A F��H�� To amixture of bisphosphonate 19 (8.95 g, 0.0120 mol) and dialdehyde 7 (4.34 g, 0.0120 mol) in anhydrous THF(900 ml) at r.t. under an inert atmosphere of argon, was quickly added, under vigorous stirring, a solution of K-O-t-Bu (5.4 g, 0.048 mol) in anhydrous THF (100 ml). The reaction mixture immediately turned red/purple, andstirring was continued for 1 h, after which an extra 2 equivalents of K-O-t-Bu (2.7 g, 0.024 mol) in THF (50 ml)were added. After stirring this mixture at r.t. for 3 h, benzaldehyde (3 ml) was added, and the mixture stirred foran additional 90 min. The copolymerization was quenched by the addition of AcOH (20 ml), and the resultingyellow mixture was slowly poured in a vigorously stirred mixture of MeOH (3000 ml) and water (1000 ml). Theyellow precipitate was filtered, washed thoroughly with MeOH and dried to give the crude PDPAFV 20 as ayellow powder. This powder was taken up in THF (500 ml) and reprecipitated in acetone (3500 ml). Theprecipitate was collected by filtration to give 7.27 g of PDPAFV 20 as a yellow powder (7.27 g, 73 %). 1H-NMR(400 MHz, CDCl3): δ: 7.84, 7.62, 7.56, 7.41 (aromatic H), 1.47 - 0.87 (aliphatic H). 13C-NMR (125 MHz,CDCl3): δ: 151.9, 141.1, 138.5, 137.2, 136.6, 132.0, 130.2, 127.9, 127.3, 126.7, 125.4, 121.1, 120.3 (aromaticC), 55.1 (C9 fl), 39.5, 36.9, 33.2, 28.2, 24.9, 23.0, 22.9, 19.9 (aliphatic C). Anal. calcd. for (C63H68)n: C, 91.69;H, 8.31. Found: C, 86.49; H, 8.24.
=PQP]PYNP^
(1) Helfrich, W.; Schneider, W. G. Phys. Rev. Lett. 1965, 14, 229.(2) Tang, C. W.; VanSlyke, S. A. Appl. Phys. Lett. 1987, 51, 913.(3) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; Mackey, K.; Friend, R. H.; Burn, P.
L.; Holmes, A. B. Nature 1990, 349, 539.(4) Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem. Int. Ed. Engl. 1998, 37, 402.(5) Segura, J. L. Acta Polym. 1998, 49, 319.(6) Grem, G.; Leditzky, G.; Ullrich, B.; Leising, G. Adv. Mater. 1992, 4, 36.(7) Cho, H. N.; Kim, J. K.; Kim, D. Y.; Kim, C. Y.; Song, N. W.; Kim, D. Macromolecules 1999, 32,
1476.(8) Cho, H. N.; Kim, D. Y.; Kim, Y. C.; Lee, J. Y.; Kim, C. Y. Adv. Mater. 1997, 9, 326.(9) Ahn, T.; Jang, M. S.; Shim, H. K.; Hwang, D.-H.; Zyung, T. Macromolecules 1999, 32, 3279.(10) Satoh, S.; Suzuki, H.; Kimata, Y.; Kuriyama, A. Synth. Met. 1996, 79, 97.(11) Cimrova, V.; Neher, D.; Remmers, M.; Kminek, I. Adv. Mater. 1998, 10, 676.(12) Kim, K. D.; Park, J. S.; Kim, H. K.; Lee, T. B.; No, K. T. 1998 1998, 31, 7267.(13) Garten, F.; Cacialli, F.; Hilberer, A.; Esselink, F. J.; van Dam, Y.; Schlatmann, A. R.; Friend, R. H.;
Klapwijk, T. M.; Hadziioannou, G. Adv. Mater. 1997, 9, 127.(14) Garten, F.; Hilberer, A.; Cacialli, F.; Esselink, F. J.; van Dam, Y.; Schlatmann, A. R.; Friend, R. H.;
Klapwijk, T. M.; Hadziioannou, G. Synth. Met. 1997, 85, 1253.(15) Bouché, C.-M.; Berdagué, P.; Facoetti, H.; Robin; LeBarny, P.; Schott, M. Synth. Met. 1996, 81, 191.(16) Boyd, T. J.; Geerts, Y.; Lee, J.-K.; Fogg, D. E.; Lavoie, G. G.; Schrock, R. R.; Rubner, M. F.
Macromolecules 1997, 30, 3553.(17) Cumming, W.; Gaudiana, R. A.; Hutchinson, K.; Kolb, E.; Ingwall; Mehta, P.; Minns, R. A.; Petersen,
C. P.; Waldman, D. J.M.S.-Pure Appl. Chem. 1996, A33, 1301.(18) Kim, Y.; Kwon, S.; Yoo, D.; Rubner, M. F.; Wrighton, M. S. Chem. Mater. 1997, 9, 2699.(19) Pyo, S. M.; Kim, S. I.; Shin, T. J.; Park, H. K.; Ree, M.; Park, K. H.; Kang, J. S. Macromolecules 1998,
31, 4777.(20) Hsieh, B. R.; Yu, Y.; Forsythe, E. W.; Schaaf, G. M.; Feld, W. A. J. Am. Chem. Soc. 1998, 120, 231.(21) Huang, W.; Meng, H.; Yu, W. L.; Gao, J.; Heeger, A. J. Adv. Mater. 1998, 10, 593.

.ZYU`RL_PO ;ZWdXP]^ bT_S $����/T[SPYdWLY_S]LNPYP @YT_^ TY _SP XLTY NSLTY QZ] -W`P 7TRS_�0XT__TYR /TZOP^
���
(22) Grice, A. W.; Tajbakhsh, A.; Burn, P. L.; Bradley, D. C. Adv. Mater. 1997, 9, 1174.(23) Pei, Q.; Yang, Y. J. Am. Chem. Soc. 1996, 118, 7416.(24) Ranger, M.; Leclerc, M. Chem. Commun. 1997, 1597.(25) Ranger, M.; Rondeau, D.; Leclerc, M. Macromolecules 1997, 30, 7686.(26) Woo, E. P.; Inbasekaran, M.; Shiang, W. R.; Roof, G. R. WO Patent 1997, 97/05184.(27) Kreyenschmidt, M.; Klaerner, G.; Fuhrer, T.; Ashenhurst, J.; Karg, S.; Chen, W. D.; Lee, V. Y.; Scott,
J. C.; Miller, R. D. Macromolecules 1998, 31.(28) Kim, J. K.; Hong, S. I.; Cho, H. N.; Kim, D. Y.; Kim, C. Y. Polymer Bull. 1997, 38, 169.(29) Klaerner, G.; Davey, M. H.; Chen, W.-D.; Campbell Scott, J.; Miller, R. D. Adv. Mater. 1998, 10, 993.(30) Chung, S.-J.; Jin, J.-I.; Lee, C.-H.; Lee, C.-E. Adv. Mater. 1998, 10, 984.(31) Kaeriyama, K.; Tsukahara, Y.; Negoro, S.; Tanigaki, N.; Masuda, H. Synth. Metals 1997, 84, 263.(32) Hodge, P.; Power, G. A.; Rabjohns, M. A. Chem. Commun. 1997, 73.(33) Kaupp, G.; Stark, M. Chem. Ber. 1981, 114, 2217.(34) Gershuni, S.; Rabinovitz, M.; Agranat, I.; Derlamn, I. B. J. Phys. Chem. 1980, 84, 517.(35) Cornelissen, J. J. L. M.; Peeters, E.; Jansen, R. A. J.; Meijer, E. W. Acta Polymerica 1998, 49, 476.(36) Cooke, A. W.; Wagener, K. B. Macromolecules 1991, 26, 2704.(37) Iwatsuki, S.; Kubo, M.; Ito, Y. Chem. Lett. 1993, 1085.(38) Feringa, B. L.; Hulst, R.; Rikers, R.; Brandsma, L. Synthesis 1988, 316.(39) Ingold; Marshall J. Chem. Soc. 1926, 3086.(40) Dufraisse; Morgoulis-Molho Bull. Soc. Chim. Fr. 1940, 7, 928.(41) McGowan, W. M.; Hilinski, E. F. J. Am. Chem. Soc. 1995, 117, 9019.(42) Davydov, S. N.; Rodionov, A. N.; Shigorin, D. N.; Syutkina, O. P.; Krasnova, T. L. Russ. J. Phys.
Chem. 1981, 55, 444.(43) Kelley, C. J.; Ghiorghis, A.; Kauffman, J. M. J. Chem. Res. Miniprint 1997, 12, 2701.(44) Kim, C. Y.; Cho, H. N.; Kim, D. Y.; Kim, Y. C.; Lee, J. Y.; Kim, J. K. US Patent 1998, 55,807,974.(45) Bliznyuk, V. N.; Carter, S. A.; Scott, J. C.; Klaerner, G.; Miller, R. D.; Miller, D. C. Macromolecules
1999, 32, 361.(46) Etienne, A. Bull. Chim. Soc. Fr. 1947, 634.(47) Duveen, D.; Willemart, A. J. Chem. Soc. 1939, 116.

THIS IS A BLANK PAGE (134)

>`XXL]d
Conjugated polymers combine the optical and electronic properties of semiconductorswith the attractive material properties (such as processibility) of polymers to obtain uniqueand novel materials. They feature numerous exciting applications such as large-area, flexiblepolymer light emitting diodes (pLEDs) that can emit light in virtually any part of the visiblespectrum, and all-polymeric field effect transistors (FETs) that give access to high-tech,though low-cost plastic electronics. The ease of manipulation with the chemical structure of apolymer allows the fabrication of functional materials with tailor-made electronic and/ormechanical properties. This thesis describes the synthesis and characterization of conjugatedpolymers and oligomeric (short-chain) systems consisting of alternating electron-releasingand electron-withdrawing aryl units along the main-chain. These “donor-acceptor” (DA)conjugated polymers represent the most successful organic analogues of low band gapsemiconductors so far, and are considered a move into the direction of polymers exhibitingmetallic conductivity without the need for additional doping, i.e. intrinsically conductingpolymers. Surprisingly, it was found that the chemistry towards these low band gap systems isalso applicable to the preparation of high band gap conjugated polymers which, for example,find their use in blue light-emitting diodes.
Because the semiconducting properties of conjugated polymers originate from the π-orbital overlap of the repeat units throughout the chain, planarization of these units isimportant to guarantee efficient overlap. The alternating copolymer of pyrrole and 2,1,3-benzothiadiazole represents the first example of a low band gap (1.1 eV) conjugated polymerwith a ladder-like structure based on intramolecular hydrogen bonding. It is prepared bythermal conversion of the corresponding N-Boc protected, soluble precursor polymer. Thelatter is obtained from the Pd-catalyzed Stille-polymerization of an AB-type monomer. Theplanarizing power of the intramolecular hydrogen bond between the pyrrole N-H and the2,1,3-benzothiadiazole imine nitrogen was unambiguously demonstrated by NMRspectroscopic and single crystal X-ray studies on a model compound. Planarization as aconsequence of intramolecular hydrogen bonding is suggested to account for the lower bandgap of the pyrrole-based copolymer compared to the already described thiophene analogues.
The structural variations that lead to a narrowing of the band gap in donor-acceptorconjugated systems are not very well understood. It was unknown, for example, whether theevolution of the band gap upon chain elongation of these systems is comparable to that of“ordinary” π-conjugated polymers like polythiophene and polypyrrole. Therefore, three seriesof alternating donor-acceptor substituted co-oligomers (with various chain lengths),consisting of pyrrole or thiophene as the electron-releasing unit and quinoxaline or 2,1,3-benzothiadiazole as the electron-withdrawing unit were prepared, by application of the Pd-catalyzed Stille coupling methodology. The incremental bathochromic shift of λmax uponchain elongation of the three series of oligomers is less than that of the homo-oligomers ofthiophene and pyrrole, which finds its origin in a diminished dispersion (hybridization) of the

LUMO level upon chain elongation. The latter conclusion was drawn after comparing theoxidation and reduction behavior of the thiophene/benzothiadiazole oligomers with that ofthiophene oligomers. The incremental bathochromic shift is similar for all three series ofoligomers. This fact could be used as a tool in the band-gap engineering of donor-acceptorsubstituted π-conjugated polymers. Extrapolation of these results indicated that thiophene-benzobis[1,2,5]thiadiazole based co-oligomers are among the most promising donor-acceptorsystems.
The preparation of thiophene-benzobis[1,2,5]thiadiazole based co-oligomers implies aninefficient multistep synthesis. Therefore, a two-step synthesis of 4,8-di(thien-2-yl)benzobis[1,2,5]thiadiazoles was developed, that can replace the original, inefficient five-step procedure. Moreover, a closely related copolymerization reaction gave easy access towell-defined poly(9,10-dithienylanthracene)s via a non-conjugated precursor polymer.
Because the copolymerization strategy towards poly(9,10-dithienylanthracene)s couldnot be adapted to yield poly(4,8-dithienylbenzobis[1,2,5]thiadiazole)s, an alternativepolymerization for these systems was developed. It represents a facile, non-oxidativepolycondensation towards solution processible, low band gap conjugated polymers. Thepolymer containing benzobis[1,2,5]thiadiazole and 3,4-ethylenedioxythiophene (“EDOT”)units in the main-chain features a band gap of 0.3 eV, which is the lowest known value fornon-oxidatively prepared, processible conjugated polymers. Furthermore, this polymer isprone to acid-doping by various organic acids, giving thin films with a stable conductivity of1 S cm-1
Finally, minor adaptations in the chemistry that is described in earlier chapters, led to asimple and efficient 3-step procedure towards 9,10-bis(4-formylphenyl)anthracene. Thiscompound gave facile access to blue luminescent conjugated polymers containing 9,10-diphenylanthracene units in the main chain. The novel alkoxy-substituted poly-p-9,10-diphenylanthracene-vinylene (PDPAV) was prepared, and the thin film emission maximumwas located at 480 nm, implying green emission. By comparing the fluorescence spectra of anunsubstituted and an alkoxy-substituted model compound, the red-shift in the emissionspectrum was ascribed to the presence of alkoxygroups on the anthracene nucleus of 9,10-diphenylanthracene. A conjugated copolymer consisting of an unsubstituted 9,10-diphenylanthracene and a 9,9-dialkylfluorene unit, connected by vinylene linkages (poly-p-9,10-diphenylanthracene-2,7-fluorenylene-vinylene, PDPAFV), exhibited intense bluefluorescence with an emission maximum at λem = 455 nm. This makes PDPAFV a promisingconjugated polymer for blue light-emitting diodes.

>LXPYaL__TYR
Geconjugeerde polymeren combineren de optische en elektronische eigenschappenvan halfgeleiders met de aantrekkelijke eigenschappen van polymeren (bijv. verwerkbaarheid,flexibiliteit), hetgeen unieke nieuwe materialen met tal van bijzondere eigenschappenoplevert. Voorbeelden zijn flexibele polymere licht emitterende diodes (PLEDs) met eengroot oppervlak, die licht van vrijwel elke kleur kunnen uitzenden, en polymere transistoren(FETs), die goedkope “plastic electronica” mogelijk maken. Het gemak waar men dechemische structuur van een polymeer mee kan manipuleren, maakt het mogelijk omfunctionele materialen met op maat gesneden electronische- of materiaaleigenschappen terealiseren. In dit proefschrift wordt de synthese en karakterisering van geconjugeerdepolymeren en oligomere (hoog- en laagmoleculaire) modelsystemen beschreven, die eenalternerende reeks van elektronen-stuwende (-donerende) en elektronen-zuigende (-accepterende) eenheden bezitten. Deze “donor-acceptor” geconjugeerde polymerenvertegenwoordigen tot dusver de meest succesvolle organische analoga van lage band gaphalfgeleiders, en worden gezien als een stap in de richting van polymeren die de elektrischestroom geleiden zonder additionele doping, ofwel intrinsieke (metallische) geleiders.Verrassend genoeg bleken de chemische reacties die in dit proefschrift werden ontwikkeldvoor lage band gap geconjugeerde polymeren, ook toepasbaar te zijn voor hoge bandgapgeconjugeerde polymeren. Laatstgenoemde polymeren vinden hun toepassing als delichtgevende, actieve laag in blauw licht emitterende diodes.
Omdat de haflgeleidende eigenschappen van een geconjugeerd polymeer het resultaatzijn van de orbitaaloverlap tussen de repeterende eenheden, is het belangrijk dat dezeeenheden in één vlak liggen (coplanair zijn). Het copolymeer van pyrrole en benzothiadiazoolis het eerste voorbeeld van een lage band gap geconjugeerd polymeer, waarin de repeterendeeenheden in hetzelfde vlak worden gehouden d.m.v. intramoleculaire waterstofbruggen. Hetwordt gemaakt uit het N-Boc beschermde, oplosbare precursor polymeer, dat op zijn beurtwordt verkregen uit een Pd-gekatalyseerde Stille-polymerisatie van een AB-type monomeer.Het vermogen van de intramoleculaire waterstofbrug om de repeterende eenheden coplanairte krijgen werd aangetoond aan de hand van NMR-metingen en kristalstructuuropheldering(röntgendiffractie) aan een modelverbinding. Het bereiken van coplanariteit d.m.v. internewaterstofbrugvorming wordt gezien als een belangrijke reden voor de lagere bandgap van ditpyrool-gebaseerde polymeer in vergelijking met bekende thiofeen-analoga.
De structurele parameters die leiden tot een verlaging van de band gap in donor-acceptor geconjugeerde systemen zijn nog niet geheel doorgrond. Het is bijvoorbeeld nietbekend of de verandering van de band gap in deze systemen vergelijkbaar is met klassiekegeconjugeerde polymeren zoals polypyrrool en polythiofeen. Om hier meer licht op teschijnen zijn drie reeksen van donor-acceptor gesubstitueerde co-oligomeren metverschillende lengtes gesynthetiseerd d.m.v. de Pd-gekatalyseerde Stille-koppeling. Dewaargenomen bathochrome verschuiving van het absorptiemaximum bij toenemende

ketenlengte is geringer dan die van homo-oligomeren van pyrrool of thiofeen. Dit wordttoegeschreven aan een geringere dispersie van de LUMO band bij ketenverlenging van dezesystemen. De bathochrome verschuivingen als functie van toenemende ketenlengte zijnonderling nagenoeg gelijk voor de drie reeksen co-oligomeren. Dit fenomeen kan gebruiktworden als een hulpmiddel bij de “band-gap engineering” van D-A gesubstitueerdegeconjugeerde polymeren. Hieruit bleek o.a. dat co-oligomeren op basis vanbenzobis[1,2,5]thiadiazool en thiofeen veelbelovend zijn als het gaat om een lage bandgap.
Helaas konden co-oligomeren op basis van benzobis[1,2,5]thiadiazool en thiofeen totnu toe alleen via een inefficiënte meerstaps-synthese worden bereid. Daarom werd eentweestapssynthese van 4,8-di(thien-2-yl)benzobis[1,2,5]thiadiazole ontwikkeld, diebovengenoemde inefficiënte synthese kan vervangen. Bovendien werd via een nauw verwantecopolymerisatie gemakkelijk toegang verkregen tot goed gedefinieerde poly(9,10-dithienylanhtracen)en, via niet-geconjugeerde precursor polymeren.
Omdat laatsgenoemde copolymerisatie niet kon worden aangepast voor poly(4,8-dithienylbenzobis[1,2,5]thiadiazole)n, werd een alternatieve polymerisatie voor dezesystemen ontwikkeld. Deze komt neer op een eenvoudige, niet-oxidatieve polycondensatiedie toegang verschaft tot uit oplossing verwerkbare lage bandgap polymeren. Het polymeergebaseerd op benzobis[1,2,5]thiadiazole en 3,4-ethyleendioxythiofeen (“EDOT”) heeft eenbandgap van 0.3 eV, één van de laagste waarden tot nog toe voor niet-oxidatief bereide,verwerkbare geconjugeerde polymeren. Bovendien kan het polymeer zeer eenvoudig wordengedoopt met allerlei anorganische zuren, wat elektrisch geleidende dunne films oplevert meteen stabiele geleidbaarheid van ongeveer 1 S cm-1.
Als laatste werd een eenvoudige synthese voor 9,10-bis(4-formylfenyl)anthraceenontwikkeld, op basis van de chemie die in eerdere hoofdstukken staat beschreven. Dezeverbinding bood eenvoudige toegang tot blauw fluorescerende polymeren met 9,10-difenylanthraceen-eenheden in de hoofdketen. Het polymeer poly-p-9,10-difenylanthraceen-vinyleen (PDPAV) werd gesynthetiseerd, waarvan het emissie-maximum (dunne film) wasgelocaliseerd rond de 480 nm, wat groene emissie betekent. Vergelijking van defluorescentiespectra van een ongesubstitueerde en een alkoxy-gesubstitueerdemodelverbinding leerde dat de roodverschuiving in de fluorescentiemaxima kan wordentoegeschreven aan de aanwezigheid van alkoxygroepen op de anthraceenkern. Eengeconjugeerd polymeer gebaseerd op een ongesubstitueerde difenylanthraceen- en een 9,9-dialkylfluoreen-eenheid, verbonden door vinyleen fragmenten (poly-p-9,10-difenylanthraceen-2,7-fluorenyleen-vinyleen PDPAFV) gaf een intens blauwe fluorescentie,met een emissie maximum bij 455 nm, wat betekent dat PDPAFV een veelbelovend polymeeris om toegepast te worden in blauw lichtgevende diodes.

.`]]TN`W`X�AT_LP
Robert van Mullekom werd geboren op 12 oktober 1971 te Helenaveen. Na een VWOopleiding aan het College Asten-Someren te Asten werd in 1990 begonnen aan de studieScheikundige Technologie aan de Technische Universiteit Eindhoven, alwaar hetpropaedeutisch examen werd behaald in 1991. Het afsluitend examen werd in oktober 1995met goed gevolg afgelegd, middels een afstudeerstage bij de vakgroep Organische Chemie(prof.dr. E.W. Meijer). Vanaf november 1995 tot en met december 1999 was de schrijver indienst van de Technische Universiteit Eindhoven als AIO in dezelfde vakgroep, onder leidingvan prof.dr. E.W. Meijer en dr. J.A.J.M. Vekemans. De belangrijkste resultaten van ditonderzoek staan beschreven in dit proefschrift. Vanaf maart 2000 zal de schrijver als post-docwerkzaam zijn in de groep van prof.dr. J.S. Siegel aan de universiteit van Californië, SanDiego (USA) op het gebied van HIV-integrase inhibitoren.
Robert van Mullekom was born in Helenaveen, the Netherlands on October 12th, 1971.In 1990 he obtained his VWO (pre-university education) degree at the “College Asten-Someren” in Asten, the Netherlands. He continued with the study of Chemical Engineering atthe Eindhoven University of Technology in that same year, and graduated in October 1995.His graduation work was conducted at the laboratory of Organic Chemistry under thesupervision of prof.dr. E.W. Meijer. From November, 1995 up to and including December,1999 the author has been working for the Eindhoven University of Technology as a Ph Dstudent in the same group, under the supervision of prof.dr. E.W. Meijer and dr. J.A.J.M.Vekemans. The most important results of the investigations are described in this thesis.Starting March 2000, the author will work as a post-doc in the group of prof.dr. J.S. Siegel atthe University of California, San Diego (USA) in the area of HIV-integrase inhibitors.

7T^_�ZQ�;`MWTNL_TZY^
H.A.M van Mullekom, J.A.J.M. Vekemans and E.W. Meijer: “Alternating Copolymer ofPyrrole and 2,1,3-Benzothiadiazole”, Chem. Commun. 1996, 2163-2164.
J.A.J.M. Vekemans, L. Groenendaal, A.R.A. Palmans, D.A.P. Delnoye, H.A.M.vanMullekom and E.W. Meijer: “Coplanarity by Hydrogen Bonding in Well-definedOligoheterocycles”, Bull. Soc. Chim. Belg. 1996, 105, 659-674
H.A.M. van Mullekom, J.A.J.M. Vekemans and E.W. Meijer: “Band Gap Engineering ofDonor-Acceptor Substituted Conjugated Polymers”, Proceedings of the 4th EuropeanConference on Molecular Electronics, 1997, 50-54
H.A.M. van Mullekom, J.A.J.M. Vekemans and E.W. Meijer: “Band Gap Engineering ofDonor-Acceptor Substituted Conjugated Polymers”, Chem. Eur. J. 1998, 4, 1235-1243
H.A.M. van Mullekom, J.A.J.M. Vekemans and E.W. Meijer: “Band Gap Engineering ofDonor-Acceptor Conjugated Polymers”, Polymer Prep. 1998, 39, 1002-1003
H.A.M. van Mullekom, J.A.J.M. Vekemans, E.W. Meijer and L. Groenendaal “NeueThiophene und Verfahren zu ihrer Polymerisation”, German Patent Application Le 33 850,1999 (Bayer AG, Leverkusen, Germany).
H.A.M. van Mullekom, J.A.J.M. Vekemans and E.W. Meijer: “Non-oxidativePolycondensation Towards Solution-Processible Poly(dithienylbenzobis[1,2,5]thiadiazole)swith Extremely Narrow Band Gaps.”, J. Am. Chem. Soc., Submitted.

/LYVbZZ]O
Vanzelfsprekend wil ik mijn proefschrift beëindigen met hen te bedanken, zonder wieik alle hier beschreven onderzoekingen niet had kunnen uitvoeren. Het vertrouwen datprof.dr. Bert Meijer mij de afgelopen vier jaren heeft geschonken is voor mijn ontwikkelingals jonge wetenschapper en mens van doorslaggevend belang geweest, en zal het altijdblijven. Voor de vrijheid die hij mij bood in het doen van onderzoek, en het bijsturen wanneerdit nodig was ben ik hem dankbaar. Dit geldt ook voor mijn dagelijkse begeleider, dr. JefVekemans, die vaak de juiste suggesties op cruciale punten in het onderzoek wist aan tedragen. Zijn indrukwekkende kennis van de organische chemie, gecombineerd met debereidheid deze, op welk tijdstip van de dag dan ook, met groot enthousiasme los te laten opeen probleem, maken van hem de ideale begeleider. Problemen van meer fysische aardwerden vaak verhelderd door discussies met dr.ir. René Janssen en dr. Edsko Havinga.Laatstgenoemden waren ook altijd bereid tot het kritisch nakijken van een manuscript, inwelk opzicht ik ook prof.dr. Bert Hulshof en prof.dr. James Feast wil bedanken. De kennis enkunde van ing. Joost van Dongen op het gebied van de analytische chemie, in combinatie metzijn gezonde wantrouwen in de geclaimde zuiverheid van een verbinding, maakten een zovolledig mogelijke karakterisering van de door mij gesynthetiseerde preparaten mogelijk.Tevens gaat mijn dank uit naar de onderzoeksschool polymeren PTN voor het financieren vandit onderzoek, en NWO voor het beschikbaar stellen van een SIR-reisbeurs.
Verder wil ik nog bedanken de studenten René Scholtes en Arjen Funhoff voor huninzet tijdens hun respectievelijke research- en afstudeerstages, drs. Joke Apperloo voor devele CV metingen, ir. Martin Struijk voor kamergenootschap en CV metingen, ing. Richardvan Someren en ing. Michel Fransen voor het opschalen van diverse syntheses, dr. HuubKooijman (Universiteit Utrecht) voor het maken van de kristalstrukturen in hoofdstuk 2, dr.ir.L. Groenendaal (Bayer AG, Uerdingen) voor geleidingsmetingen en het beschikbaar stellenvan EDOT, dr. Herman Schoo en Kornel Hoekers (Beiden TNO Industrie, Eindhoven), voorhet maken van LEDjes, ir. Bas de Waal en ing. Jolanda Spiering voor het beschikbaar stellenvan diverse gesubstitueerde thiofenen, Ingrid vd Boomen, Hans Damen, Henk Eding, Hannyvd Lee en Hanneke Veldhoen, zonder wie de vakgroep snel tot stilstand zou komen, entenslotte eenieder die hier onvermeld is gebleven door onoplettendheid mijnerzijds.
Ik eindig hier met mijn ouders te bedanken, die mij altijd hebben gestimuleerd om tegaan studeren, en natuurlijk mijn vrouw Pien.

THIS IS A BLANK PAGE

STELLINGEN
behorende bij het proefschrift
The Chemistry of High and Low Band Gap π-Conjugated Polymers
door Hubertus A.M. van Mullekom
1. Het verkondigen van een “very low band gap” of een “extremely narrow band gap” bij elektrochemisch gesynthetiseerde π-geconjugeerde polymeren dient, i.v.m. eventuele achtergebleven doping, zeer omzichtig te gebeuren.
Huang, H.; Pickup, P.G. Chem. Mater. 1998, 10, 2212. Akoudad, S.; Roncali, J. Chem. Commun., 1998, 2081.
2. De door Yamashita et al. beschreven synthese van dibromobenzo[1,2-c:4,5-c’]bis[1,2,5]-
thiadiazool is moeilijk reproduceerbaar. Ono, K.; Tanaka, S.; Yamashita, Y. Angew.Chem., 1994, 106, 2030. Yamashita, Y.; Ono, K.; Tomura, M.;Tanaka, S. Tetrahedron, 1997, 53, 10169.
3. Het hieronder afgebeelde geconjugeerde polymeer poly(4,8-bis(pyrrol-2-yl)benzo[1,2-
c:4,5-c’]bis[1,2,5]thiadiazole) zal een intrinsieke elektrische geleider zijn.
NS
N
NS
N
N
N
H
Hn
4. Het als een noviteit publiceren van een reeds lang bekend concept is in het licht van een
volledig nieuw doel te billijken, kwalijker wordt het wanneer dit nieuwe doel een verkapte versie is van het oorspronkelijke doel.
Zhang, Q.T.; Tour, J.M. J. Am. Chem. Soc. 1997, 119, 5065. Zhang, Q.T.; Tour, J.M. J. Am. Chem. Soc. 1998, 120, 5355.
5. Het “afvlakken” van de curve λmax = f(n) voor hogere waarden van n, aanvoeren als een indicatie voor het niet lineair zijn van de curve Emax = f(1/n), getuigt van een slechte kennis van middelbare school wiskunde.
λmax is het absorptiemaximum, als golflengte, van een bepaald π-geconjugeerd oligomeer, en n zijn lengte; Emax is hetzelfde absorptiemaximum, nu als energie. Stalmach, U.; Kolshorn, H.; Brehm, I; Meier, H. Liebigs Ann. 1996, 1449.

6. π-Geconjugeerde polymeren kunnen pas dan volwaardige “organische halfgeleiders” genoemd worden, wanneer zij, vergelijkbaar met anorganische halfgeleiders, anisotrope eigenschappen vertonen.
Sirringhaus, H.; Brown, P.J.; Friend, R.H.; Nielsen, M.M.; Bechaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.; de Leeuw, D. Nature 1999, 401, 685.
7. Een gezonde hekel aan kolomchromatografie siert de synthetisch organisch chemicus. 8. Monsterintroductie wordt met een autosampler niet automatisch simpeler. 9. Elementanalyse is en blijft waardevol, omdat het als één van de weinige analytische
technieken iets zegt over het volledige monster. 10. In de discussie over de lange termijn oplossingen voor het fileprobleem vergeet men vaak
dat de olievoorraden in de wereld eindig zijn. 11. Een wielrenner die zijn krachten kent en verdeelt, hoeft zelfs op de flanken van de
Galibier nauwelijks uit het zadel.