texto guÍa de: biofarmacia y farmacocinÉtica...
TRANSCRIPT

TEXTO GUÍA DE:
BIOFARMACIA Y FARMACOCINÉTICA
COMPILADOR:
Percy Alberto Ocampo Rujel

PRÓLOGO DE BIOFARMACIA Y FARMACOCINETICA
Esta asignatura se compone de dos partes íntimamente relacionadas: la Farmacocinética y la Biofarmacia. Ambas disciplinas, junto con la Tecnología Farmacéutica, contribuyen a la consecución de medicamentos eficaces, seguros y estables.
La Farmacocinética General se define como la ciencia que se ocupa del estudio y caracterización de la evolución temporal de los fármacos y de sus metabolitos en el organismo, a través del análisis cinético de las curvas concentración – tiempo, o cantidad – tiempo, obtenidas a partir de muestras de fluidos orgánicos.
La información proporcionada por esta disciplina es sumamente valiosa en clínica ya que permite predecir los niveles que el fármaco va a alcanzar en el organismo, a partir de una determinada forma de dosificación. Esto permite establecer regímenes de dosificación apropiados, y acordes a las características de cada paciente. Por otro lado, la Farmacocinética es indispensable en la evaluación de la biodisponibilidad y por ello, desempeña un papel fundamental en los estudios biofarmacéuticos.
La Biofarmacia por su parte, ha sido definida como la ciencia que estudia la biodisponibilidad de los fármacos en sus formas farmacéuticas, y el modo de alcanzar su óptimo, a través de las interacciones fármaco-forma farmacéutica-sustrato biológico.
Con esta asignatura, se pretende que al finalizar los créditos de la misma el alumno sea capaz de:
1. Conocer los procesos que determinan la entrada de los medicamentos en el organismo, y los factores que afectan su biodisponibilidad.
2. Analizar a través de las curvas de concentración plasmática - tiempo la evolución de los fármacos en el organismo.
3. Interpretar el resultado de las curvas de niveles plasmáticos. 4. Diseñar pautas de individualización posológicas.
Bienvenidos al curso y muchos éxitos.

BIOFARMACIA Y FARMACOCINÉTICA:
PRIMERA UNIDAD, SESION 01.
TEMA 1. IMPORTANCIA DE LOS SABERES PREVIOS PARA ENCARAR CON ÉXITO EL APRENDIZAJE DE LA FARMACOCINETICA
I. DEFINICION Y AMBITOS DE LA FARMACOCINETICA:
1. DEFINICION:
La Farmacocinética es una Rama de la Farmacología que específicamente se encarga del Estudio de:
El análisis matemático de los procesos de Absorción, Distribución, Metabolismo y Excreción (ADME) de las drogas en el organismo. Los factores biológicos, fisiológicos y fisicoquímicos que influencian los procesos de transferencia de las drogas en el organismo, también influyen en la magnitud y extensión de los procesos ADME. En muchos casos, la acción farmacológica y/o toxicológica está muy relacionada con las concentraciones plasmáticas de las drogas. Como consecuencia a través de l estudio de la farmacocinética, el farmacéutico tendrá las capacidades para individualizar la terapéutica de los pacientes.
2. Ubicación y relación de la farmacocinética con otras áreas relacionadas con la terapéutica: ver cuadros anexos.
3. NECESIDADES DE OTRAS AREAS
3.1. BIOLOGIA.
3.2. MATEMATICA I y II
3.3. ANATOMIA Y FISIOLOGIA.
3.4. QUIMICA ORGANICA
3.5 FISICO QUIMICA.
3.6. FISIOPATOLOGIA.
3.7. BIOQUIMICA.
3.8. FARMACOLOGIAS I Y II

3.2 MATEMATICA
LAS MATEMATICAS SON LA BASE PARA EL ENTENDIMIENTO Y ANALISIS DE LOS PROCESOS FARMACOCINETICOS.
Son importantes para explicar situaciones reales que suceden en el organismo cuando los medicamentos se absorven, distribuyen, metabolizan y/o excretan.
Son básicos los conceptos sobre aritmética, álgebra y calculo diferencial e integral.
Premisas básicas:
“Cualquier número multiplicado por cero es igual a cero”
“Cualquier número multiplicado por un número infinito es igual al infinito”
“Cualquier número dividido por cero es indefinido”
“Cualquier número dividido por el infinito es indefinido”
Notación Exponencial:
316000 becomes 3.16 X105
0.00708 becomes 7.08 X10-3
El valor del exponente da el número de lugares los cuales debe moverse el punto decimal, sea a la derecha o izquierda dependiendo del valor positivo o negativo del exponente.
El uso de la notación exponencial será importante para definir procesos y/o cantidades en el orden de las centésimas o milésimas. Para la conversión entre unidades de medida por ej. K a mg o de mg a ug
Cifras Significativas

El manejo de cifras significativas es importante para reducir el tamaño de las cifras que se manejan en los cálculos farmacocinéticas. Se utilizarán solo hasta cuatro cifras significativas.
Manejo de Exponentes
An A: número, n : exponente
Los procesos cinéticos son de naturaleza exponencial, por lo tanto el análisis matemático

incluirá necesariamente el trabajo con exponentes: sumas, restas, multiplicaciones e identidades.
Logaritmos
El logaritmo en base 10 se denomina logaritmo natural
Algunos procesos fisiológicos tienen naturaleza logaritmica, tal como la filtración glomerular o la transferencia de solutos hacia el medio extravascular por filtración celular.
Es una forma de expresar la notación exponencial:
Logaritmo Natural
En vez de expresar el logaritmo en base 10 se utiliza la base natural = e = 2.7182818
Procesos como la filtración glomerular de medicamentos que dependen de la cantidad total de droga que llega al riñón son de naturaleza logarítmica natural. Así mismo el proceso cinético de eliminación por cualquier emuntorio precisa de un análisis Logarítmico Natural.
Conocido como logaritmo hiperbólico o Neperiano

El logaritmo natural y el Neperiano se relacionan de la siguiente manera:
Esto puede ser importante para encontrar las equivalencias de las ecuaciones logarítmicas neperianas con las naturales y facilitar los despejes.
Manejo matemático de Logaritmos:

Logaritmo Negativo:

Dimensiones:
SISTEMA METRICO DECIMAL
El manejo del sistema métrico decimal y sus equivalencias entre unidades y medidas facilitará la resolución de ecuaciones en las cuales será necesario estandarizarlas. pe. Si encontramos un peso orgánico en gramos y una dosis en ug/ml y nos dan una equivalencia de tantos ml de fármaco para tanto peso de paciente.

Tema 2: El Inicio de la Farmacocinética Clínica Tomado de: Paulo Arturo CÁCERES GUIDO. Lat. Am. J. Pharm. 26 (3): 462-7 (2007) INTRODUCCIÓN Luego de la Revolución Científica de media- dos del siglo XVII, donde se produjo la ruptura entre los fundamentos metodológicos y episte- mológicos que procedían de la Grecia clásica, aparecieron las primeras tentativas para elaborar teorías médicas y filosofías novedosas. En aque- llos años William Harvey confirma la circulación de la sangre y la existencia de los capilares (1628), Antonie Van Leeuwenhoek enfoca gló- bulos rojos y espermatozoides entre una miríada de maravillosas observaciones (1673), François Delaboë estudia el papel de la bilis y la saliva y René Antoine Ferchauld de Réaumur’s progresa en el conocimiento de los mecanismos de la di- gestión (1713) 1. El crecimiento del conocimiento científico es lento hasta mitad del siglo XIX, exceptuando al- gunos casos como el de Marie François Xavier Bichat, médico francés que a fines del siglo XVIII refunda la biología, la anatomía y la fisio- logía, despojándola de ideas preconcebidas, im- puestas por siglos (“creaciones brillantes de la imaginación, nacidas en los gabinetes, pero nunca junto al lecho del enfermo”) 1. LOS PRIMEROS TRABAJOS SOBRE DISOLU- CIÓN, ABSORCIÓN Y CONCENTRACIÓN SAN- GUÍNEA
En 1847 Buchanan describe la actividad anestésica del éter en “Physiologic effects of the inhalation of ether”, entendiendo que su acción dependía de la concentración arterial y de la cantidad inhalada 2. En 1862, William Proctor, el llamado Padre de la Farmacia Norteamericana, afirma que “la razón por la cual las píldoras podían atravesar el estómago sin disolverse dependía del estado del paciente, de la composición de la píldora y de la naturaleza de la cubierta”. En 1897, Noyes y Whitney publican en “The Rate of Solution of Solid Substances in Their Own Solution”, las le- yes sobre los factores que afectan a la velocidad de disolución y la ecuación lleva sus nombres 3. Estos autores postulan que el rango de disolu- ción se controla a través de la capa de solución saturada que rodea inmediatamente la superficie de la partícula sólida. Esta teoría es optimizada por Nerst y Brumer en 1904, quienes aplican la ley de difusión de Fick, que relaciona el gra- diente de concentraciones a ambos lados de una membrana, las características de la misma y si el soluto es un ión, el gradiente eléctrico.
En 1902, Hance sugiere que el hecho de que un comprimido contenga la dosis de fármaco apropiada no significa necesariamente que se vaya a producir el efecto terapéutico previsto 3.
Por otro lado, en 1913, Lenor Michaelis y Maude Menten publicarán la ecuación cinética que describe la hidrólisis enzimática de la saca- rosa en fructosa y glucosa, aunque el estudio experimental comienza con Adrian Brown, quien en 1902 ensayó su proceso bioquímico 2.
FISIOLOGÍA, MATEMÁTICA, METABOLISMO Y FÁRMACOCINÉTICA
En 1919, el investigador sueco E. M. Wid- mark expresa en lenguaje matemático la rela- ción entre concentración plasmática y actividad farmacológica. En los años 20 desarrolla ideas sobre el análisis cinético de la eliminación de fármacos y el estudio de dosis múltiples, publi- cando en 1932 una monografía sobre la cinética del metanol y los procesos saturables, con con- ceptos que serían base del desarrollo de la ciné- tica no

lineal 3,4. En 1924, Widmark junto al también sueco J. Tandberg elaboran las ecuaciones de
modelos monocompartimentales, tanto para dosis múlti- ples en bolo como para infusión continua 2 . En marzo de ese año, en su Laboratorio de Fisiolo- gía Aplicada de la Universidad de Yale, Howard Haggard escribe sus hoy clásicos artículos que son parte de la historia contemporánea de la fi- siología y la farmacocinética, analizando los me- canismos de absorción, distribución y elimina- ción del éter etílico, ahondando en los procesos de redistribución en el sistema nervioso central y en su relación cinético-dinámica 5. En 1929, uno de los años más trascendentes en la historia moderna de las ciencias
biomédi- cas, cuando Alexander Fleming comunica el ha- llazgo de la penicilina, nace uno de los concep- tos más importantes en el área de la cinética de drogas, la eliminación o clearance renal, de la mano de Moller, Jolliffe, Smith y Hamilton. En 1931 W. F. Hamilton introduce el concepto de Tiempo Medio de Residencia, lo que involucra tanto a los procesos de liberación-absorción, co- mo a los de disposición (distribución, metabolis- mo y excreción), significando el tiempo medio estadístico que insumen las moléculas adminis- tradas en su pasaje por el organismo 2.
Widmark teoriza, en 1932, sobre la expresión matemática que refleja la eliminación del etanol desde la sangre, definiéndola como de veloci- dad constante. Identifica un proceso farmacoló- gico de eliminación de orden cero, (mecanismo saturable) eliminándose una cantidad constante de fármaco por unidad de tiempo (velocidad de eliminación constante). Esto lo hace en el con- texto de sus trabajos sobre “principios y aplica- ciones de la regulación médico-legal del alco- hol”, en Berlín 2,4.
En la sucesión de hechos que alimentaron el crecimiento de esta rama de la farmacología du- rante las décadas del 20 al 40, aparece la figura del patólogo Rafael Domínguez, prolífico cientí- fico de principios del siglo XX, quien estudió la farmacocinética de sustancias como manitol, xi- losa, galactosa, urea y creatinina. Sobre esta últi- ma, se basó en los trabajos de determinación en orina y sangre que realizaba Otto Folin en la dé- cada de 1910 en su Laboratorio de Bioquímica de la Escuela de Medicina de Harvard 6.
En el Hospital Saint Luke de Cleveland (EE.UU.), Domínguez introduce conceptos fun- damentales de la farmacocinética y puede atri- buírsele la definición de volumen de distribu- ción, así como la formulación matemática de la estimación del rango de absorción de sustancias en función del tiempo 7.
Paralelamente a estos desarrollos, desde la década de 1920, químicos como Eduard Ken- dall, Ortiz de Montellanos y A.Correa comenza- ron estudios que serían la base para que farma- cólogos como Bernard Brodie y Julius Axelrod, entre 1930 a 1950, sentaran, con numerosísimos trabajos en animales y seres humanos, los pila- res del conocimiento respecto del metabolismo de los fármacos. Brodie, considerado por mu- chos como el iniciador de la farmacología clínica y quien se desempeñara durante el período post Segunda Guerra Mundial como jefe del De- partamento de Farmacología Clínica del Instituto Nacional de Salud de los EE.UU., recibe el Pre- mio Lasker en 1967 (un equivalente norteameri- cano del Premio Nobel) por sus extraordinarias contribuciones a esta rama de la ciencia. Cabe mencionar que Axelrod, quien había comenza- do sus estudios sobre metabolismo “in vitro” e “in vivo” hacia mitad del siglo XX con anfetami- nas y catecolaminas, recibe en 1970 el Premio Nobel de Medicina 8.
UNA NUEVA DISCIPLINA Así es como se van sucediendo aportes im- portantes hasta la que hoy día es

considerada la piedra fundacional de la farmacocinética clínica moderna. Esto se da con los trabajos publicados por el biofísico y fisiólogo sueco Torsten Teo- rell, quien en 1937 publica dos artículos, los pri- meros en mostrar un modelo farmacocinético con base fisiológica. La integración con el mo- delo compartimental permite diseñarlos sobre la base de una estructura fisiológica y anatómica real, incorporando aquellos órganos, tejidos y regiones corporales de interés farmacológico y/o toxicológico. Una vez desarrollado un mo- delo adecuado, se pueden extrapolar resultados de una especie animal a otra, cambiando valo- res de parámetros anatómicos y fisiológicos co- rrespondientes a la especie considerada. Teorell demuestra que este modelo reflejaba más fiel- mente la realidad anatómico-fisiológica del or- ganismo. Su objetivo fue “desarrollar relaciones matemáticas generales desde las cuales fuera posible, para propósitos prácticos, describir la cinética de distribución de sustancias en el cuer- po”. Enfatiza sobre los cambios que la absorción podía generar en las concentraciones de sustan- cias en la sangre y en diversos tejidos, por lo que “las conclusiones de este tema podría tener consecuencias prácticas farmacológicas y tera- péuticas” 9.
A pesar de expresarse con suficiente clari- dad, los dos trabajos que Teorell publica en 1937 no son seriamente considerados por la eli- te de la fisiología de la época, siendo olvidados hasta 25 años más tarde, cuando son reevalua- dos por sus aportes. Un primer reconocimiento internacional de sus contribuciones se dio en 1972 en la Conferencia Internacional sobre Far- macología y Farmacocinética que organizó el Centro Internacional Fogerty, del Instituto Na- cional de Salud (EEUU), en Bethesda 2.
Aunque los modelos compartimentales tienen aún muchas aplicaciones, actualmente la te- oría en la que se basan los modelos farmacoci- néticos fisiológicos (Physiologically Based Phar- macokinetic Models) es motivo de múltiples ar- tículos de investigación en diversas disciplinas relacionadas a la medicina, como así también a otras ramas del conocimiento, como la ingenie- ría, especialmente en el área aeronáutica 10.
En términos de absorción, la terminología ha ido evolucionando con el correr de los años. Así, disponibilidad fisiológica es lo que hoy co- nocemos como biodisponibilidad, es decir: la cantidad (o fracción de dosis administrada) y velocidad a la que un fármaco inalterado alcan- za un líquido biológico 11.
La historia específica de este punto, se re- monta a los trabajos que realizan en EE.UU. Da- niel Melnick y Henry Field Jr., en parte gracias a subsidios de la Upjohn Company. En el Depar- tamento de Medicina Interna de la Escuela de Medicina de la Universidad de Michigan deter- minan la estabilidad de la tiamina en orina. Más tarde, ya trasladados a una compañía de investi- gación de alimentos en Long Island, a principios de los 40, junto a Melvin Hochberg y Bernard Oser, Melnick estudia las vitaminas del comple- jo B. Luego, el propio Oser, junto a Melnick y Hochberg y otros colaboradores encaran los pri- meros estudios sistemáticos sobre biodisponibi- lidad en el área de las vitaminas, entendiendo que la normativa de calidad entonces vigente, de las formas de dosificación basada en están- dares de contenido, pureza y potencia es insufi- ciente, e introducen el concepto de “disponibili- dad fisiológica” 12,13.
Hoy la fracción molar terapéuticamente acti- va de la dosis administrada y la velocidad a la que ésta alcanza sus sitios de acción representa la “biodisponibilidad terapéutica”, concepto eminentemente teórico dado que aún no es po- sible verificar experimentalmente el acceso a ca- da uno de los sitios de acción farmacológica. Se apunta, con esta definición, a la calidad total de los medicamentos, incluyendo eficacia y seguri- dad 14.
Surge el campo de investigación con radioi- sótopos cuando Marie y Pierre Curie

descubren el radio y el polonio en 1898, abriéndose de es- ta manera un nuevo campo de investigación. Si bien Pierre Curie estaba desde 1901 contactado con los médicos del Hospital Saint-Louis de Pa- rís en aplicaciones terapéuticas del radio, no fue sino hasta los años 1910-1920 en los que su uso en el dominio médico tomo relevancia 15.
Años después, el biofísico A. K. Solomon investiga con radioisótopos en la Escuela de Me- dicina de Harvard, quien a fines de los 40 publi- ca algunos trabajos utilizando trazadores isotópi- cos. En sus estudios busca “detallar las ecuacio- nes matemáticas que gobiernan las transferen- cias de fluidos de una parte a otra del cuerpo y también la transformación de un compuesto en otro y sus síntesis, mostrando así que muchos de estos principios, que son comunes a la física y la química, también son aplicables a las disci- plinas biológicas”. Sus trabajos incluían el uso de deuterio para observar la síntesis de coleste- rol y el pasaje de sodio del plasma al líquido ce- falorraquídeo 16. Solomon basó parte de su aná- lisis en las ecuaciones que casi un siglo antes (1866) realizara el químico británico Vernon Harcourt, que en la Universidad de Oxford hi- ciera trascendentes aportes al desarrollo de esa área, trabajando junto al matemático William Es- son. Curiosamente, Esson y Charles Lutwidge Dodgson eran potenciales competidores para el puesto de colaborador de Harcourt en sus estu- dios químicos. Finalmente Dodgson se inclina totalmente por su veta artística y, ya con el seu- dónimo de Lewis Carroll, llega a hacerse famoso por obras como “Alicia en el País de la Maravi- llas”. Poco después, los trabajos de Harcourt y Esson servían de base para la recién nacida ci- nética química, luego llamada fisicoquímica 17.
En Europa, por su parte, los avances tampo- co cesaban. Vale destacar a C. Lapp, quien des- de 1948 a 1956, en Francia, trabaja intensamente sobre la cinética de absorción y eliminación de drogas y metabolitos. Ensayando con una im- portante variedad de sustancias busca, en sus escritos esencialmente matemáticos, un acerca- miento a la aplicación clínica de los resultados experimentales 2,4.
Apenas cuatro años después que en 1943 un joven investigador norteamericano descendiente de rusos, Albert Schuarz, junto a un grupo de colaboradores coordinados por el Profesor Sel- man Abraham Waksman, descubriera la estrep- tomicina aislándola de cultivos de Streptomyces griseus, George Boxer y Viola Jelinek comien- zan a estudiar la forma de determinarla química- mente 18.
En los laboratorios de Merck, en New Jersey, Boxer y Jelinek desarrollan un método químico de determinación de estreptomicina en sangre y líquido cefalorraquídeo. Con estos resultados, en 1948, ahondan en el análisis de sus caracte- rísticas cinéticas en administraciones repetidas. Ambos deducen la ecuación que permite el cál- culo de las concentraciones máximas y mínimas del fármaco para el modelo de un compartimen- to, cuando se administran por vía intramuscular dosis múltiples a iguales intervalos de tiempo 19,20. Un año más tarde en EEUU, A. Goldstein pu- blica el primer review específico sobre
proteínas plasmáticas e interacción de drogas en el Phar- macological Reviews. También en 1949 Gaudino trabaja en cinética de inulina, definiendo su vo- lumen extracelular y publicando las ecuaciones que definen un modelo bicompartimental abier- to 2.
En 1949, Drukerei y Kuepfmüeller de la Uni- versidad de Friburg (Alemania) publican una monografía incluyendo conceptos sobre la rela- ción entre la teoría de los receptores y las ecua- ciones farmacocinéticas 2. Aquí irrumpe en esce- na la escuela alemana ya que en dos trabajos presentados en el mismo número de la revista Acta Physiologica et Pharmacologica Neerlandi- ca, De Jongh, Wijnans y simultáneamente Van Gemert y Duyff, plantean ecuaciones matemáti- cas sobre la relación dosis - respuesta farmaco- lógica. Estos trabajos serían los primeros que aplican la modelización farmacocinética/farma-

codinámica, siendo precursores en optimización de regímenes farmacoterapéuticos 2,5. En 1950, De Jongh y Wijnans compararon la duración y acción farmacológica de dosis
altas, bajas intermitentes y continuas en un modelo monocompartimental. Ese mismo año Van Ge- mert and Duyff desarrollaron modelos farmaco- cinéticos-farmacodinámicos comparando distin- tos esquemas de dosis y trabajando también con variables que hoy se definen como farmacoeco- nómicas 3. CONTINUA LA MODERNIZACIÓN
Estos avances en diversos campos, llevan a que en 1953 Friedrich Hartmut Dost (1910-1985) publique Der Blütspiegel: kinetic der konzentra- tionsablaüfe in der krieslanffüssigkeit (De los Niveles Sanguíneos...), afrontando la problemáti- ca del llamado modelo monocompartimental abierto, a la vez que define el término “Phar- makokinetik” 2-4.
Dost era un pediatra con excelente forma- ción matemática, por lo que pudo hacer mano de herramientas de su formación básica para re- dactar adecuadamente este tipo de texto. Es in- teresante destacar que tanto Teorell como Dost utilizaron mucho del tiempo en el que prestaron servicio militar para profundizar sus conoci- mientos matemáticos 4.
En 1968, Dost publica una edición actualizada de su primer libro, al que llamó “Bases de la Farmacocinética” (“Grundlagen der Pharmako- kinetik”), un escrito técnica e históricamente fundamental de la farmacología 2-4. En 1954, Buttler y colaboradores publican, en el Journal of Pharmacology and Experimental Therapeutic, un importante artículo sobre la eliminación, acu- mulación y pautas de dosificación del fenobar- bital 2,4.
A mediados de la década del 50, el Profesor Edward Garrett realiza importantes estudios far- macocinéticos en hombres y animales, como los referentes a antibióticos e hipoglucemiantes y los que evalúan el efecto de esteroides en la disposición del calcio. Estos se profundizarán durante las próximas dos décadas, convirtiendo a E. Garrett en un referente en la etapa de ma- yor complejidad de la farmacocinética, en la dé- cada de 1960 3.
En 1954 se publican dos artículos clásicos de la literatura fisiológica y farmacocinética. S.G. Jokipii y O. Turpeinen escriben “Cinética de la eliminación de glucosa desde la sangre, durante y luego de una infusión intravenosa continua” en The Journal of Clinical Investigation y J. F. Acholar junto a C. F. Code redactan, para Gas- troenterology, “Rango de absorción de agua desde el estómago y el intestino delgado de se- res humanos” 2.
Hacia 1955, Joseph Swintosky, farmacéutico de la Universidad de Wisconsin, quien descolló en numerosos estudios sobre la “física farma- céutica” ó farmacotecnia y que llegara a obtener más de 40 patentes en ese campo, aborda la problemática relacionada a la semivida de elimi- nación y la duración de acción. Swintosky tam- bién ayudó a desarrollar el concepto de biodis- ponibilidad para drogas de liberación prolongada 21.
En 1958 en la Facultad de Farmacia de la Universidad de California, San Francisco, Syd- ney Riegelman inicia, junto a Wilfred Cromwell, sus publicaciones sobre farmacocinética. Por es- tos artículos en el Journal of the American Phar- maceutical Association, titulados en su conjunto “La Cinética de la Absorción Rectal” obtuvo el Ebert Prize en 1959, galardón que se otorga des- de 1873, y es administrado por la American Pharmaceutical Association, siendo el más anti- guo e importante del área farmacéutica en los EE.UU. 22.
Uno de los primeros trabajos que correlacio- nan la respuesta clínica con las concentraciones séricas de una droga, fue publicado en 1958 por P. J. Schiller y F. Buchthal. En el Danish Medical Bulletin muestran su trabajo sobre la acción de la fenitoína en

anormalidades electroencefalo- gráficas de pacientes epilépticos, a la vez que correlaciona las mismas con la dosis, la evolu- ción de su concentración sérica y el tiempo re- querido para alcanzar el estado estacionario 2.
Desde 1960, Ekkehard Krüger-Thiemer, quien trabajó durante muchos años en el Instituto de Investigación de Borstel, Alemania, establece las bases cinéticas para la dosificación de fármacos, publicando varios artículos relacionados con la teoría y aplicación de la farmacocinética a los regímenes de dosificación de sulfonamidas y an- tibióticos 2. REVISIONES, PRIMEROS CONGRESOS Y PERSPECTIVA
En 1961, E. Nelson, quien dos años antes ha- bía publicado, entre otros, un trabajo sobre ab- sorción de tetraciclina, escribe la primera revi- sión sobre Farmacocinética con el título de “Ki- netic of drug absorption, distribution, metabo- lism and excretion” en el Journal of Pharmaceu- tical Science.
El impulso que en estas últimas décadas ha- bía tenido la farmacocinética en Alemania hizo que en 1962 tuviera lugar, en Borstel, el primer simposio que incorpora en su título el término “Farmacocinética”, pudiendo ser considerada como la primera reunión científica internacional de interés en esta disciplina.
Para terminar, podemos afirmar que desde fi- nales de la década de 1960 hasta la actualidad, fundamentalmente las aplicaciones de nuevos descubrimientos en el campo de la química (distintos tipos de cromatografía) y de la infor- mática (nuevos microprocesadores) desencade- narán un sustancial incremento, desde el punto de vista de su aplicabilidad, en la cantidad y ca- lidad de trabajos sobre farmacocinética en las más diversas áreas de la farmacoterapia.
La Farmacocinética comienza así a recono- cerse claramente en forma diferenciada por es- tudiar los procesos cinéticos de los fármacos en los seres vivos y se define hoy como la sucesión de procesos tales como la liberación, absorción, distribución, metabolismo y excreción de los fármacos, estableciendo leyes que rigen estos comportamientos.
La farmacocinética básica encontraría su de- sarrollo más aplicado a la terapéutica de la ma- no de la Farmacocinética Clínica, acercando ele- mentos de Biofarmacia y Farmacotecnia. Esta ra- ma relativamente nueva de la farmacocinética se identifica con la tarea asistencial en el Monito- reo Terapéutico de Drogas (MTD).
El MTD puede definirse como una especiali- dad clínica multidisciplinaria cuyo objetivo es mejorar la calidad de atención del paciente a través del ajuste individualizado de las dosis de drogas para las cuales, la experiencia clínica o los ensayos clínicos han mostrado mejorar la respuesta, ya sea en la población general ó en poblaciones especiales 23.
Aquí concluye la primera etapa de lo que ha sido el nacimiento de un nuevo hijo de la farmacología clásica, la farmacocinética clínica, siendo posible prever que dará aún mucho más al desarrollo de la historia de la farmacoterapia.


1
REFERENCIAS BIBLIOGRÁFICAS 1. Sendrail, M. (1983) “Historia Cultural de la En- fermedad” Ed. Espasa Calpe S.A.,
Madrid Es- paña, págs. 213-44 y 329-60. 2. Wagner, J.G. (1981) Pharmac. Ther. 12: 537-62. 3. Mariño Hernández, E.L.,C. Fernández Lastra & P. Modamio Charles (1998) Farm. Hosp.
22:197-204. 4. Hochhaus, G. (2000) J. Clin. Pharmacol. 40:908-17 5. Haggard, H.W. (1924) J. Biol. Chem. 59: 737-802. 6. Folin, O. (1914) J. Biol. Chem. 17: 469-73; 475-81. 7. Domínguez, R. & E. Pomerene (1934) J. Biol.Chem. 104: 449-71. 8. Axelrod, J. (1970) “Nobel Lecture”, NobelFoundation, Suecia. 9. Paalzow, L.K. (1995) Upsala J. Med. Sci. 100:41-6.
10. Leahy, D.E. (2004) Biosilico 2: 78-84. 11. Obach Vidal, R. & Doménech Berrozpe J. (1997) “Biodisponibilidad” en
“Farmacocinética” Volumen I. “Biofarmacia y Farmacocinética”. Ed. Síntesis. Madrid, págs. 19-41.
12. Melnick, D, & H. Field (1939) J. Biol. Chem.130: 97-107. 13. Melnick, D., M.Hochberg, H.W.Himes & B.L.
Oser (1945) J. Biol. Chem. 160: 1-14. 14. Fagiolino, P. (1999) “Farmacocinética y Tera- péutica”. Ed. Asociación Pro-Fundación
para el Progreso de la Química. Montevideo, págs. 9- 22.
15. Boudia, S. (1997) La Recherche 183: 845-9. 16. Solomon, A.K. (1949) J. Clin. Invest. 28: 1297-
307. 17. Wayne, R.P. (2006) Research. Physical and
Theoretical Chemistry Laboratory. Oxford. 18. Lawrence, F.L. (1997) Fluoride 30: 140. 19. Boxer, G.E. & Jelinek, V.C. (1947) J. Biol.
Chem. 170: 491-500. 20. Boxer G.E, Jelinek V.C., Tompsett R., Dubois R. & Edison A.O. (1948) J. Pharmacol. Exp.
Ther. 92: 226-35. 21. Swintosky, J.V. (1999) Curriculum Vitae. Uni- versity of Kentucky (http://www.uky.edu/
Pharmacy/ps/psfaculty/jvscv.pdf) 22. Benet, L.Z. (1986) Sidney Riegelman. In Me- morian, in University of California History Di-
gital Archives (http://content.cdlib.org/xtf/ view?docId=hb767nb3z6&doc.view=frames&ch unk.id=div00100&toc.depth=1&toc.id=&brand= calisphere).
23. Gross A.S. (1998) Br. J. Clin. Pharmacol. 46: 95-9.

ADMINISTRACIÓN DEL FÁRMACO EN EL ORGANISMO
EN FORMA FARMACÉUTICA
ADADMIMINNIISSTTRARACIÓCIÓNN DEDELL FFÁÁRRMAMACCOO ENEN EELL
ORORGGAANINISMSMOO ENEN FOFORMARMA
Tema 03. DEFINICIONES Y RELACIONES DE LA BIOFARMACIA Y FARMACOCINÉTICA
1. Los fármacos, para producir sus efectos característicos, deben alcanzar concentraciones adecuadas en los sitios donde actúen.
2. as concentraciones logradas, a pesar de que están en función
de la dosis del producto administrado, dependen también de la magnitud y las tasas de absorción, distribución, unión o localización en tejidos, biotransformación y excreción.
3. El efecto producido por un fármaco en el organismo puede
variar de un individuo a otro, ya que está determinado por una compleja serie de eventos, tal como se puede apreciar en la siguiente figura:
ADMINISTRACIÓN DEL FÁRMACO EN EL ORGANISMO
EN FORMA FARMACÉUTICA
FASE BIOFARMACÉUTICA
FASE FARMACOCINÉTICA
FASE FARMACODINÁMICA
• Liberación del p.a.
• Interacción en el sitio de administración
• Absorción • Distribución • Metabolismo • Excreción
ADME
• Interacción fármaco-receptor en el tejido blanco
FÁRMACO DISPONIBLE PARA LA ABSORCIÓN
FÁRMACO DISPONIBLE PARA LA LA ACCIÓN
EFECTO FARMACOLÓGICO
Ariens E. J. Clin Pharmacol Ther 1974; 16: 155

3
BIOFARMACIA 1. Estudio de los factores físicos, químicos y fisicoquímicos de
los medicamentos y las formas farmacéuticas que influencia la acción terapéutica o tóxica de los “productos farmacéuticos” cuando son administrados clínicamente.
2. Estudio de los factores que influyen en la biodisponibilidad de
un fármaco en el hombre o animales, y el uso de esta información para optimizar la forma farmacéutica o el preparado farmacéutico en sus aplicaciones clínicas.
3. Necesario conocer las propiedades fármaco químicas y
biológicas del fármaco en estudio, y las características de la forma farmacéutica soporte.
4. El objetivo es encauzar la liberación del principio activo en
el lugar preciso, para garantizar la correcta absorción.
+
Disolución . . . . . . . . . . . . . . . . .. . . . . ....
FARMACOCINÉTICA 1. Relación matemática que existe entre la dosis del fármaco y la
concentración de éste en un sitio fácilmente accesible del organismo.
2. Ciencia que estudia la cinética de absorción, distribución, metabolismo y excreción de los fármacos en el hombre y los animales. Puede incluir también el curso en el tiempo de la

matemáticos, que se conciben con el afán de interpretar los fenómenos cinéticos, es virtualmente esencial para los estudios farmacocinéticos (Academia de Ciencias Farmacéuticas, Asoc. Farmacéutica de Norteamérica)
3. Estudio del curso en el tiempo de las concentraciones y cantidades de fármacos o metabolitos en los fluidos biológicos, tejidos y excretas, y de la respuesta farmacológica y la construcción de modelos matemáticos apropiados para interpretar dichos datos (Wagner).
4. “Lo que el organismo hace al Fármaco”.
Aplicaciones de la Farmacocinética 1. Industria
a. Diseño de fármacos. b. Biodisponibilidad c. Desarrollo de formulaciones.
2. Clínica a. Aseguramiento del manejo efectivo y seguro de los fármacos
en los pacientes. b. Diseño de regímenes de dosificación. c. Estudio del comportamiento de fármacos en diferentes estado
fisiológicos (pediatría, lactancia, embarazo, geriatría). d. Efecto de estados patológicos en el comportamiento de los
fármacos.
FARMACODINAMIA 1. Ciencia que busca establecer relaciones matemáticas entre las
concentraciones de fármacos en los fluidos biológicos y el efecto producido.

5
2. “Lo que el fármaco hace al organismo”: EFECTO FARMACOLÓGICO.
3. El supuesto es que existe una directa relación entre la concentración plasmática del fármaco y la concentración de fármaco en el sitio efector.
El destino en el organismo de un fármaco administrado en una forma farmacéutica se puede apreciar de la siguiente forma:
SITIO DE ACCIÓN EN RECEPTORES
UNIDO LIBRE
RESERVORIO EN TEJIDOS
UNIDO LIBRE
Plasma
ABSORCIÓN FÁRMACO LIBRE
FÁRMACO UNIDO
METABOLITOS
EXCRECIÓN
BIOTRANSFORMACIÓN

FÁFÁRMARMAC
ORGANISMORG
•• ABABSSORCIORCIÓÓNN ••
EFEFECECTOTO FARMFARMACOLÓGICO Ó
ORGANISMO Farmacocinética
• • • •
¾ Introducción del medicamento al organismo (Ingreso, Input) 9 Directo:
ƒ Directamente a la sangre (intravascular,, intravenosa). ƒ Sólo varía la velocidad de ingreso. ƒ Puede ser instantánea (bolus intravenosos) o por infusión
(gota a gota, velocidad constante).
9 Indirecto: ƒ Necesario la absorción para llegar a torrente sanguíneo. ƒ Varía la velocidad y la cantidad de fármaco administrado.


PROCESO LADME: Liberación, absorción, distribución, metabolismo y excreción.
LIBERACIÓN Primer paso que tiene que sufrir un fármaco en el organismo. El proceso finaliza con la disolución del fármaco. Este proceso no existe en el caso de administrar soluciones.
ABSRORCIÓN Proceso mediante el cual las moléculas de fármaco alcanzan la circulación sanguínea. Esto no existe en la administración i.v. Factores fisiológicos implicados en la absorción de fármacos vía oral (tracto gastrointestinal): Tiempo de tránsito (actividad segmental y peristaltismo). Velocidad de vaciado gástrico. Lugar de absorción: estómago, gradiente de absorción en intestino. Área superficial efectiva en los diferentes puntos. Grado de actividad vellosa y vascularidad. Velocidad de flujo sanguíneo hacia el punto de absorción. pH del contenido del lumen. Actividad enzimática, hidrólisis y degradación en el tracto. Concentración de electrolitos en el lumen y en la superficie de la barrera. Tensión superficial e interfacial. Mucus y glucocáliz. Agentes emulsificantes y complejantes (sales biliares). Posición anatómica general y actividad corporal relativa. Temperatura. Integridad de la membrana gastrointestinal. Presión hidrostática e intraluminar.

7
Capacidad reguladora o tampón Tonicidad. Presencia o ausencia de alimentos en el TGI. Ayuno Factores implicados en la absorción parenteral Reacciones locales en el punto de inyección, evidenciados por dolor inmediatamente después o poco después de la inyección, o reacciones tardías con respecto al tiempo de inyección (granuloma de parafina) Los disolventes no acuosos usados en algunas formulaciones pueden ejercer efectos farmacológicos por sí mismos si están en concentraciones suficientemente altas
DISTRIBUCIÓN El fármaco se distribuye en el organismo hasta llegar al equilibrio. El tiempo necesario para que se logre dicho equilibrio es variable para los diferentes fármacos, de modo que su mayor o menor rapidez condicionaría el modelo farmacocinético que se empleará. La distribución es una medida de la extensión de un medicamento de manera ordenada una vez que llega a la circulación. Tejidos altamente prefundidos (75 % débito cardíaco): corazón, pulmones, sistemas hepatoportal, glándulas endocrinas y bajo ciertas condiciones, el cerebro y el sistema espinal. Tejidos escasamente prefundidos (25 % débito cardíaco): músculo, piel, tejido adiposo, médula ósea. Tejidos con perfusión despreciable: huesos, dientes, ligamentos, cartílagos, pelos. Se debe considerar la unión a proteínas que ciertos fármacos presentan, especialmente los de características ácidas o básicas que se unen especialmente a la albúmina y a algunas globulinas.

METABOLISMO Biotransformación metabólica. El órgano fundamental donde ocurre este proceso es el hígado (aumenta la polaridad de las moléculas para solubilizarlas en agua y luego eliminarlas). Reacciones de biotransformación: oxidación, reducción, reacciones de síntesis (Ej.: conjugación con ácido glucorónico) y reacciones de hidrólisis. El metabolismo de medicamentos puede ocurrir en el contenido GI, en la pared GI durante el proceso de absorción y en diversos tejidos del cuerpo, particularmente pulmón y riñones. Algunos fármacos pueden metabolizarse ampliamente en el hígado antes de alcanzar la circulación sistémica. Esto ocurre cuando se administran por vía oral ya que son absorbidos directamente del TGI hacia la circulación portal. El efecto de primer paso puede disminuir sustancialmente la cantidad de fármaco activo que alcanza la circulación sistémica y por lo tanto su biodisponibilidad.
EXCRESIÓN Cuando el medicamento se elimina por la orina o por la bilis sin
ser alterado.
Metabolismo + excreción → eliminación Distribución + eliminación → disposición del fármaco (DISPOSITION)
¾ La orina es la vía fundamental para la eliminación de muchos
medicamentos y metabolismos del organismo. ¾ Los medicamentos unidos a proteínas no son excretados ya que
sólo se filtra la porción no unida de fármaco. ¾ Aclaramiento (clearence o depuración):
9 Mide la capacidad intrínseca que tiene el cuerpo, o los órganos de eliminación (generalmente riñones e hígado) para eliminar un fármaco de la sangre o el plasma.
9 Se expresa en volumen por unidad de tiempo. 9 No es una medida de cantidad de fármaco eliminado, tan
sólo representa el volumen teórico de sangre o plasma que

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
es lavado de fármaco completamente durante un tiempo dado.
9 La cantidad de fármaco eliminado depende de la concentración de fármaco en el plasma, así como de su aclaramiento.

TEMA 04: LA MEMBRANA PLASMÁTICA
La célula está rodeada por una membrana, denominada "membrana plasmática". La membrana delimita el territorio de la célula y controla el contenido químico de la célula. La membrana plasmática representa el límite entre el medio extracelular y el intracelular. Es de gran importancia para los organismos, ya que a su través se transmiten mensajes que permiten a las células realizar numerosas funciones. Es tan fina que no se puede observar con el microscopio óptico, siendo sólo visible con el microscopio electrónico. Presenta las siguientes características:
• Es una estructura continua que rodea a la célula. Por un lado está en contacto con el citoplasma (medio interno) y, por el otro, con el medio extracelular que representa el medio externo.
• Contiene receptores específicos que permiten a la célula interaccionar con mensajeros químicos y emitir la respuesta adecuada.
En la composición química de la membrana entran a formar parte lípidos, proteínas y glúcidos en proporciones aproximadas de 40%, 50% y 10%, respectivamente.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
• Lípidos:
En la membrana de la célula eucariota encontramos tres tipos de lípidos: fosfolípidos, glucolípidos y colesterol. Todos tienen carácter anfipático ; es decir que tienen un doble comportamiento, parte de la molécula es hidrófila y parte de la molécula es hidrófoba por lo que cuando se encuentran en un medio
acuoso se orientan formando una bicapa lipídica
La membrana plasmática no es una estructura estática, sus componentes tienen posibilidades de movimiento, lo que le proporciona una cierta fluidez. Los movimientos que pueden realizar los lípidos son:
o de rotación: es como si girara la molécula en torno a su eje. Es muy frecuente y el responsable en parte de los otros movimientos.
o de difusión lateral: las moléculas se difunden de manera lateral dentro de la misma capa. Es el movimiento más frecuente.
o flip-flop: es el movimiento de la molécula lipídica de una monocapa a la otra gracias a unas enzimas llamadas flipasas. Es el movimiento menos frecuente, por ser energéticamente más desfavorable.
o de flexión: son los movimientos producidos por las colas hidrófobas de los fosfolípidos.
La fluidez es una de las características más importantes de las membranas. Depende de factores como :
o la temperatura, la fluidez aumenta al aumentar la temperatura. o la naturaleza de los lípidos, la presencia de lípidos insaturados y de cadena corta favorecen
el aumento de fluidez; la presencia de colesterol endurece las membranas, reduciendo su fluidez y permeabilidad.
• Proteinas:
Son los componentes de la membrana que desempeñan las funciones específicas (transporte, comunicación, etc). Al igual que en el caso de los lípidos , las proteinas pueden girar alrededor de su eje y muchas de ellas pueden desplazarse lateralmente (difusión lateral) por la membrana. Las proteinas de membrana se clasifican en:
o Proteinas integrales: Están unidas a los lípidos intímamente, suelen atravesar la bicapa lípidica una o varias veces, por esta razón se les llama proteinas de transmembrana.
o Proteinas periféricas: Se localizan a un lado u otro de la bicapa lipídica y están unidas debilmente a las cabezas polares de los lípidos de la membrana u a otras proteinas integrales por enlaces de hidrógeno.
• Glúcidos
Se situan en la superficie externa de las células eucariotas por lo que contribuyen a la asimetría de la membrana. Estos glúcidos son oligosacáridos unidos a los lípidos (glucolípidos), o a las proteinas (glucoproteinas). Esta cubierta de glúcidos representan el carne de identidad de las

células, constituyen la cubierta celular o glucocálix, a la que se atribuyen funciones fundamentales:
o Protege la superficie de las células de posibles lesiones o Confiere viscosidad a las superficies celulares, permitiendo el deslizamiento de células en
movimiento, como , por ejemplo, las sanguineas o Presenta propiedades inmunitarias, por ejemplo los glúcidos del glucocálix de los glóbulos
rojos representan los antígenos propios de los grupos sanguineos del sistema sanguineo ABO.
o Interviene en los fenómenos de reconocimiento celular,particularmente importantes durante el desarrollo embrionario.
o En los procesos de adhesión entre óvulo y espermatozoide.
Con los datos ofrecidos por la microscopía electrónica y los análisis bioquímicos se han elaborado varios modelos de membrana.
En la actualidad el modelo más aceptado es el propuesto por Singer y Nicholson (1972), denominado modelo del mosaico fluido , que presenta las siguientes características:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
• Considera que la membrana es como un mosaico fluido en el que la bicapa lipídica es la red cemetantey las proteinas embebidas en ella, interaccionando unas con otras y con los lípidos. Tanto las proteinas como los lípidos pueden desplazarse lateralmente.
• Los lípidos y las proteinas integrales se hallan dispuestos en mosaico. • Las membranas son estructuras asimétricas en cuanto a la distribución fundamentalmente de los
glúcidos, que sólo se encuentran en la cara externa.
Las funciones de la membrana podrían resumirse en :
1. TRANSPORTE El intercambio de materia entre el interior de la célula y su ambiente externo. Haz clic para ampliar este tema
2. RECONOCIMIENTO Y COMUNICACIÓN Gracias a moléculas situadas en la parte externa de la membrana, que actúan como receptoras de sustancias.
La bicapa lipídica de la membrana actúa como una barrera que separa dos medios acuosos, el medio donde vive la célula y el medio interno celular.
Las células requieren nutrientes del exterior y deben eliminar sustancias de desecho procedentes del metabolismo y mantener su medio interno estable. La membrana presenta una permeabilidad selectiva, ya que permite el paso de pequeñas moléculas, siempre que sean lipófilas, pero regula el paso de moléculas no lipófilas. Los mecanismos de transporte pueden verse en el siguiente esquema:

Transporte de moléculas de baja masa molecular:
1. El transporte pasivo. Es un proceso de difusión de sustancias a través de la membrana. Se produce siempre a favor del gradiente, es decir, de donde hay más hacia el medio donde hay menos. Este tranporte puede darse por:
o Difusión simple . Es el paso de pequeñas moléculas a favor del gradiente; puede realizarse a través de la bicapa lipídica o a través de canales proteícos.
1. Difusión simple a través de la bicapa (1). Así entran moléculas lipídicas como las hormonas esteroideas, anestésicos como el éter y fármacos liposolubles. Y sustancias apolares como el oxígeno y el nitrógeno atmosférico. Algunas

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
moléculas polares de muy pequeño tamaño, como el agua, el CO2, el etanol y la glicerina, también atraviesan la membrana por difusión simple. La difusión del agua recibe el nombre de ósmosis
2. Difusión simple a través de canales (2).Se realiza mediante las denominadas proteínas de canal. Así entran iones como el Na+, K+, Ca2+, Cl-. Las proteínas de canal son proteínas con un orificio o canal interno, cuya apertura está regulada, por ejemplo por ligando, como ocurre con neurotransmisores u hormonas, que se unen a una determinada región, el receptor de la proteína de canal, que sufre una transformación estructural que induce la apertura del canal.
3.
4. o Difusión facilitada (3). Permite el transporte de pequeñas moléculas polares, como los
aminoácidos, monosacáridos, etc, que al no poder atravesar la bicapa lipídica, requieren que proteínas trasmembranosas faciliten su paso. Estas proteínass reciben el nombre de proteínas transportadoras o permeasas que, al unirse a la molécula a transportar sufren un cambio en su estructura que arrastra a dicha molécula hacia el interior de la célula.
2. El transporte activo (4). En este proceso también actúan proteínas de membrana, pero éstas requieren energía, en forma de ATP, para transportar las moléculas al otro lado de la membrana. Se produce cuando el transporte se realiza en contra del gradiente electroquímico. Son ejemplos de transporte activo la bomba de Na/K, y la bomba de Ca.
o La bomba de Na+/K+ Requiere una proteína transmembranosa que bombea Na+ hacia el exterior de la membrana y K+ hacia el interior. Esta proteína actúa contra el gradiente gracias a su actividad como ATP-asa, ya que rompe el ATP para obtener la energía necesaria para el transporte.

Por este mecanismo, se bombea 3 Na+ hacia el exterior y 2 K+ hacia el interior, con la hidrólisis acoplada de ATP. El transporte activo de Na+ y K+ tiene una gran importancia fisiológica. De hecho todas las células animales gastan más del 30% del ATP que producen ( y las células nerviosas más del 70%) para bombear estos iones.
Transporte de moléculas de elevada masa molecular:
Para el transporte de este tipo de moléculas existen tres mecanismos principales: endocitosis, exocitosis y transcitosis. En cualquiera de ellos es fundamental el papel que desempeñan las llamadas vesículas revestidas. Estas vesículas se encuentran rodeadas de filamentos proteicos de clatrina.
1. Endocitosis: Es el proceso por el que la célula capta partículas del medio externo mediante una invaginación de la membrana en la que se engloba la partícula a ingerir. Se produce la estrangulación de la invaginación originándose una vesícula que encierra el material ingerido. Según la naturaleza de las partículas englobadas, se distinguen diversos tipos de endocitosis.
1. Pinocitosis. Implica la ingestión de líquidos y partículas en disolución por pequeñas vesículas revestidas de clatrina.
2. Fagocitosis. Se forman grandes vesículas revestidas o fagosomas que ingieren microorganismos y restos celulares.
3. Endocitosis mediada por un receptor. Es un mecanismo por el que sólo entra la sustancia para la cual existe el correspondiente receptor en la membrana.
Haz clic si quieres ver una fagocitosis animada.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Pinocitosis
Fagocitosis
Endocitosis mediada por receptor
2. Exocitosis. Es el mecanismo por el cual las macromoléculas contenidas en vesículas citoplasmáticas son transportadas desde el interior celular hasta la membrana plasmática, para ser vertidas al medio extracelular. Esto requiere que la membrana de la vesícula y la membrana plasmática se fusionen para que pueda ser vertido el contenido de la vesícula al medio. Mediante este mecanismo, las células son capaces de eliminar sustancias sintetizadas por la célula, o bien sustancias de desecho.

En toda célula existe un equilibrio entre la exocitosis y la endocitosis, para mantener la membrana plasmática y que quede asegurado el mantenimiento del volumen celular.
3. Transcitosis.Es el conjunto de fenómenos que permiten a una sustancia atravesar todo el citoplasma celular desde un polo al otro de la célula. Implica el doble proceso endocitosis-exocitosis. Es propio de células endoteliales que constituyen los capilares sanguineos, transportándose así las sustancias desde el medio sanguineo hasta los tejidos que rodean los capilares.
Aquí tienes dos animaciones donde puedes ver como se deforma la membrana en los procesos de endocitosis y exocitosis

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
igure 3. A schematic model of the different transport mechanisms that most drugs are absorbed with. The
permeability is a key variable in the overall absorption process.

PRIMERA UNIDAD, SESION 2
TEMA 1: FACTORES QUE REGULAN LA ABSORCION DE FARMACOS
La extensión y velocidad con la cual se absorbe un medicamento depende de la eficiencia con la cual se libera de su forma farmacéutica en el sitio de absorción. Es decir el factor limitante de la absorción es el fenómeno biofarmacéutico de liberación.
La fase Biofarmacéutica de liberación consiste en la capacidad que tiene la forma farmacéutica de ceder al medio de disolución las moléculas de PA que contiene; en la extensión y rapidez prevista por la tecnología farmacéutica para la via de administración.
Ver: 1_1. Liberación de medicamentos desde formas farmacéuticas sólidas.
La Liberación de medicamentos depende de varios factores que pueden agruparse teóricamente como:
I) FACTORES FISICO QUIMICOS DE LAS MOLECULAS QUE COMPONEN EL MEDICAMENTO: PA + EXCIPIENTES.
I) FACTORES TECNOLOGICOS Y BIOFARMACEUTICOS. II) FACTORES FISIOPATOLOGICOS DEL PACIENTE.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 2
REVISION:
I) FACTORES FISICO QUIMICOS DE LAS MOLECULAS QUE COMPONEN EL MEDICAMENTO: PA + EXCIPIENTES.
1. PESO y TAMAÑO DE LA MOLECULA.- Moléculas menores de 200 Aº (PM < 500 Daltons) tienen ventajas en la disolución y difusión hacia la membrana e incluso para su difusión Facilitada. Las Moléculas mayores (> 40 hasta 250 Aº tienen menos ventajas) y aquellas de más de 250 Aº tienen que utilizar mecanismo de apoyo para todo el proceso.
El tamaño de las partículas que se desprenden del seno de la ff sólida tienen una influencia importante sobre la velocidad de disolución. Fenómeno de la griseofulvina.
2. COEFICIENTE DE PARTICIÓN. Grado de Liposolubilidad.- Tiene un efecto ambiguo. Mientras que para la Disolución es necesaria una molécula hidrofílica para atravesar la membrana biológica las características lipofílicas son las más apropiadas. Es necesario adicionar a la forma farmacéutica excipientes que les permitan comportarse de esta forma dual y mejorar su biodisponibilidad.
Ver: 1_2. Método Lemke para la determinación de la polaridad de una molécula de medicamento.
3. pKa. INFLUENCIA DEL PH EN LA DISOCIACIÓN Y ESTABILIDAD MOLECULAR.
La absorción es la transferencia de un fármaco desde un sitio de administración hacia la sangre. Los rangos de rapidez y eficacia de la absorción farmacológica dependen de la ruta específica de administración por las diferentes mucosas.
La determinación de la proporción de moléculas ionizadas en el lugar de absorción de luego de la administración de una forma farmacéutica permite predecir la eficacia de la absorción, es decir, la biodisponibilidad de un medicamento.
La ecuación de Henderson - Hasselbalch es útil para determinar esta proporción. Para la translocación del fármaco se requiere que este al liberarse desde su formulación farmacéutica no se disocie al llegar a la membrana celular, osea no se ionice, se mantenga en su carácter estable, no ionizado o liposoluble.
Esta es una característica que debe de ser evaluada en las moléculas de medicamento ácidos y bases débiles.
El efecto del pH en la absorción farmacológica se mide estudiando la ionización de las presentaciones farmacéuticas:
• Los Fármacos Ácidos Débiles [HA]: Liberan un [H+] generando una carga aniónica [A-], para

formar:
AH + H 2 O A - + H3O +
Los Fármacos Alcalinos Débiles [BH+]: Liberan también un [H+]. La forma ionizada de los fármacos base son usualmente cargados, y pierden un protón que produce una base sin carga [B], para formar:
B + H2O BH+ + OH-
El comportamiento de las sustancias farmacéuticas, ácidas o bases débiles depende del pH. El pH de ciertas mucosas, por ejem:
• Cavidad Oral: 5 a 6 pH.
• Mucosa Gástrica: 1 a 3 pH.
• Mucosa Intestinal: 4 a 5 pH.
Estrictamente, el pKa de un compuesto es el logaritmo negativo (-log) de su constante de disociación ácida (Ka). El valor pKa es una ma-nera numérica conveniente para comparar o ex-presar la relativa acidez o basicidad de compues-tos débilmente ionizados en soluciones acuosas o miscibles con solventes hidrofílicos. En el contexto farmacéutico, los términos DISOCIACIÓN ÁCIDA, IONIZACIÓN ÁCIDA Y CONSTANTE DE ACIDEZ, son sinónimos.
Los ÁCIDOS se ionizan para transformar-se en ANIONES cuando ellos pierden uno ó más PROTONES. Las BASES se ionizan cuando ellas ganan uno ó más PROTONES, para transformarse en sus correspondientes ÁCIDOS CONJUGADOS (bases protonadas).
Y Tomando el pK de ciertos fármacos, por ejem:
• Morfina: (Base) 9 pK.
• Azetaminofen: (Ácido) 8 pK.
• Diazepam: (Ácido) 4 pK.
• Aspirina: (Ácido) 3 pK.
Ver: 1_3 Pka de medicamentos.
La ecuación de Henderson-Hasselbalch (frecuentemente mal escrito como Henderson-Hasselbach) fórmula química se utiliza para calcular el pH de una solución buffer, o tampón, a partir del pKa (la constante de disociación del ácido) y de las concentraciones de equilibrio del ácido o base, del ácido o la base conjugada.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 2
donde: S es la sal o especie básica, yA es el ácido o especie ácida En la última ecuación x puede ser a o b indistintamente.
Observaciones:
La ecuación implica el uso de las concentraciones de equilibrio del ácido y su base conjugada. Para el cálculo del pH en soluciones buffer, generalmente se hace una simplificación y se utilizan las concentraciones iniciales del ácido y la sal, por lo tanto se debe tener en cuenta que el valor obtenido es una aproximación y que el error será mayor cuanto mayor sea la diferencia de las concentraciones de equilibrio con las de partida (constante de equilibrio alta). En la misma aproximación, tampoco se considera el aporte del agua, lo cual no es válido para soluciones muy diluidas.
Otro abordaje:
Ejecutando la fórmula de Henderson-Hasselbalch para el ejemplo de Aspirina administrada vía enteral, absorbida en la mucosa gástrica:

Despeje:
3 = 1 + log [AH]/[A][H+]
3 – 1 = log [AH]/[A][H+]
2 = log [AH]/[A][H+]
antilog 2 = [AH]/[A][H+]
100 = [AH]/[A][H+]
100/1= [AH]/[A][H+]
101-100% 100-x%
res x= 99%'
Quiere decir, que la administración enteral de la aspirina, alcanza una absorción casi al 100%, logrando una efctividad de translocación mayor.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 2
Comportamiento derivado de la naturaleza ácida del medicamento y el medio de disolución:
Fármaco pH ácido pH alcalino

& Ph
Fármaco ácido
Predomina la forma no ionizada (liposoluble). Dificulta la disolución, Facilita absorción y dificulta eliminación
Predomina la forma ionizada (hidrosoluble). Facilita la disolución, Dificulta la absorción y facilita eliminación
Fármaco alcalino
Predomina la forma ionizada (hidrosoluble). Facilita la disolución, Dificulta la absorción y facilita eliminación
Predomina la forma no ionizada (liposoluble). Dificulta la disolución. Predomina la absorción y dificulta eliminación
CIDOS Y BASES DEBILES
ÁCIDOS DEBILES BASES DEBILES
DIURÉTICOS: ANTIHISTAMINICOS: NARCÓTICOS:
Teofilina. Difenhidramina. Morfina. Manitol. Dimenhidrinato. Codeina. Aminofilina. Doxilamina. Heroína. Clorotiácido. Tripolididina. Etorfina. Benzotiácida. Clorfeniramina. Nalorfina. Hidroclorotiácida. Bromofeniramina. Naloxona. Furosemida. Tripelenamina. Petidina. Ácido etacrínico. Pirilamina. Fentanil. Metazolamida. Metapirileno Becitramida. Etoxzolamida. ANESTESICOS LOCALES. Metadona. Acetazolamida. Cocaina. COLINERGICOS. SULFAS. Hexilcaina. Betanecol. Sulfametozaxol. Isobucaina. Benzocaina. Carbacol. Sulfaleno. Benoxinato. Metaleolina. Sulfadiacina. Tetracaina. Pirocarpina. Sulfadoxina. Procaina. Neostigmina. Sulfabenzanida Butacaina. Isoflurofato. Sulfaclocina. Cloroprocaina. Fisostigmina. Sulfatroxiazol. Lidocaina. Muscarona. Sulfacitina. Pirrocaina. ANTICOLINERGICOS. Sulfaurea. Dibucaina. Atropina. Sulfisomidina. TRANQUILIZANTES. Escopolamina. Sulfisoxazol diolamina. Meprobanato. Homotropina.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 2
CEFALOSPORINAS. Tibamato. Papavorina. Cefalotina. Clorodiacepóxido. Dimoxilina. Cefacetril Oxacepam. Hiosciamina. Cefazolina. Nitracopam Bromuno de
propantolina. Cefuroxima. Fluracepam Tropicamida. Cloranfenicol. Cefaloxina. Diacepam. Cefradina. Cefoxitina. Cefaloglicina.
OTROS FACTORES FISICOQUÍMICOS QUE AFECTAN LA LIBERACIÓN Y

DISOLUCIÓN:
1. Estructura química. 2. Propiedades de los grupos funcionales. 3. Forma química (p.e. sal, ester, acido, base, etc.). 4. Volatilidad. 5. Refracción de la luz. 6. Polimorfismo.
II) DE LA TECNOLOGIA Y LOS FENOMENOS BIOFARMACEUTICOS.
La fase biofarmacéutica comprende aquellos procesos desde el momento de ser administrado el medicamento, hasta la liberación y disolución del Principio Activo, desde la forma farmacéutica, para dejarlo a disposición del organismo. Esta etapa está determinada por la tecnología de fabricación y el medio de disolución que otorga el epitelio donde se disolverá el medicamento. Por ejemplo la fuerza de compresión de una tableta para que el medicamento se disgregue en el duodeno.
Al ser administrada una F.F vía oral, deben ocurrir una serie de fenómenos hasta alcanzar la disolución o dispersión molecular antes de la difusión hacia la membrana y posterior absorción.
1. DESINTEGRACION. 2. DISPERSION GRANULAR, 3. DISPERSION MOLECULAR,
1. DESINTEGRACIÓN DE LA FORMA FARMACÉUTICA.
Se trata de la fragmentación de la F:F en partes más pequeñas o hasta gránulos al

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 2
hacer contacto con un medio disolvente, generalmente el jugo gástrico o también puede ser en un medio externo al organismo como en las tabletas efervescentes en medio acuoso.
2. DISPERSIÓN GRANULAR, PARTICULAR O DISGREGACION DE LA FORMA FARMACÉUTICA. La disgregación corresponde a la reducción de los gránulos formados en partículas aun más pequeñas.
3. DISPERSIÓN MOLECULAR O DISOLUCION DEL FARMACO. Es el evento en el cual las las partículas del fármaco son de tamaño molecular (soluto) y se encuentran dispersas entre las moléculas del disolvente. Observadas a través del microscopio, las disoluciones aparecen homogéneas y el soluto no puede ser separado por filtración.
Es claro que formas farmacéuticas que ya se encuentran en solución no presentan fase biofarmacéutica.
Los fenómenos de liberación están condicionados por la tecnología utilizada en:

- La obtención del principio activo.- La síntesis y control de calidad del polvo de principio activo sin mezclas racémicas o formas cristalinas insolubles.
- Los procesos de fabricación de la forma farmacéutica final relacionadas con Las Operaciones Unitarias: Secado, Molienda, Tamizado, Mezcla, Compresión. Etc.
- Uso de excipientes como moduladores del proceso de Disolución.
Revisar: 1_4 Aspectos Fisiológicos Y Tecnológicos Relacionados Con La Biodisponibilidad De Medicamentos.
APECTOS BIOFARMACEUTICOS DE LA DISOLUCIÓN.
• En el estómago se disuelven mejor las Bases Débiles • En el intestino se disuelven mejor los Acidos Débiles • Las formas líquidas facilitan el proceso de disolución.
TEORÍAS DE DISOLUCIÓN.
Ver: 1_5 Factores Que Influyen En La Disolución De Medicamentos
- Hace 50 años se reconoció la importancia del proceso de disolución sobre la biodisponibilidad de los F.
- El estudio de este proceso se ha desarrollado desde fines del siglo 19. - 1897. Ecuación de Noyes y Whitney - 1900. Ecuación de Brunner – Tolloczko. - 1904. Ecuación de Nernst - Brunner
• La Ecuación de Nerst y Brunner explican el proceso Biofamacéutico de disolución.
D (Cs – C) A
dW/dt = ------------------------------

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 3
h
dW/dt = W, masa total del soluto disuelto; t, tiempo de disolución.
D = coeficiente de difusión.
A = superficie de intercambio entre la FF y el disolvente
h = espesor de la capa del disolvente
Cs-C = Diferencia de concentraciones entre la capa adyacente a las partículas y el medio de disolución.
FACTORES BIOFARMACÉUTICOS EN EL SITIO DE DISOLUCIÓN:
• Agitación: > agitación > Disolución • Tensión superficial: viscosidad: (n) > n < Disolución • Temperatura: a > tº < n > Disolución • Espesor de la capa disolvente: < h < Disolución.
CLASIFICACIÓN BIOFARMACÉUTICA DE MEDICAMENTOS

VER: 1_6 Clasificación Biofarmaceutica_Implicancias.
III. FACTORES FISIOLÓGICOS Y PATOLOGICOS QUE AFECTAN LA ABSORCION DE FARMACOS.-
3.1. Efecto de la Acidez en los diferentes niveles del tracto GI
pH in the small intestine in healthy humans in the fasted and fed state*
Site Average pH, Average pH, Fasted state fed state
mid-distal 4.9 5.2 duodenum 6.1 5.4 6.3 5.1 jejunum 4.4 - 6.5 5.2 - 6.0 6.6 6.2 ileum 6.5 6.8 - 7.8 6.8 - 8.0 (range) 6.8 - 8.0
from Gray et al. (Pharmacopeial Forum 22; 1943-1945, 1996)
*pH en el intestino humano en estomago vacío y estómago lleno
3.2. Efecto de la velocidad del Vaciamiento Gástrico en la disolución y la absorción.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 3
De manera general el fármaco se absorve mejor luego del estómago, por lo tanto de manera general todos los factores que aceleren el vaciamiento gástrico aumentarán las posibilidades de absorción en el duodeno.
- Contenido Gástrico (CG): a > CG < VG - Dieta sólida: <VG - Grasas: < VG - disminución de pH: ejemplo ceviche, ají, < el VG - Alimentos Calientes < el VG - Alimentos de elevada Osmolaridad: ejm coca cola, sodas, < el VG por aumento de la
viscosidad - Postura Corporal: Erguido > VG; - Yacer sobre el lado Izquierdo < el VG - Estado Mental: Tensión o la Ansiedad: > el VG - Depresión: < el VG - Medicamentos: Los depresores del SNC: Analgésicos narcóticos, - Anticolinérgicos < VG - Sales Biliares: < VG - Metoclopramida: > VG
3.3. Efecto de la motilidad intestinal: - Todo factor que aumenta la motilidad intestinal el Cmax. - De manera general todos los alimentos la motilidad intestinal. - El efecto neto del VG y de la motilidad intestinal es impredecible. - Un aumento exagerado de la de la MI genera un tránsito elevado y menor contacto con la
mucosa. 3.4. Estado de la Mucosa Gastrointestinal.
Contacto con la Mucosa: Son factores negativos la presencia en el intestino de: alimentos, fluidos muy osmolares, formación de complejos tipo quelatos.
Estado de la superficie de la Mucosa.
3.5. Efecto del primer paso. 3.6. Metabolismo en el tubo digestivo por acción de la flora bacteriana. 3.7. Costumbres alimentarias.
- El volumen afecta la motilidad - Las secreciones de las glándulas anexas. - Formación de complejos. - Aumento de secreción de bilis; genera aumento en la secreción de lípidos. - Tetraciclinas con leche, con sales de calcio, con hidroxido de Ca, Mg, Al.
“De manera general los fármacos deberían administrarse con el estómago vacío y + o - 250 ml. De agua”.
3.8. Factores patológicos.
Tiene efectos contradictorios. En general las patologías que afectan la absorción se son aquellas que afectan el vaciamiento gástrico, motilidad intestinal, integridad de la membrana de la mucosa, presencia de flora bacteriana y otros.

- Dependen del grado de severidad y de la concomitancia en la administración de medicamentos.
- Patologías que afectan el pH gástrico: por aumento o por disminución. Hiperacidez, hipo o aclorhidria, toma de med. Bloq H2.
- Edad, a > edad < vaciamiento gástrico. - Migraña, depresión, hipotiroidismo, gastroenteritis, colitis ulcerativa, ulcera gastrica
< el vac. Gástrico. - Anticolinérgicos (atropínicos y muscarínicos), hidróxido de aluminio y la mayoría
de los depresores del sistema nervioso central (narcóticos, hipnóticos, antidepresivos, ansiolíticos, antipsicóticos).
- Alcohol, reduce la velocidad del vaciamiento gástrico. - La gastroenterocolítis, la diarrea crónica inflamatoria y no inflamatoria y el
síndrome de mala absorción ejercen solo efectos menores. - Otros: problemas cardiacos y respiratorios.
III) INTERACCIONES EN LA ABSORCIÓN DE FARMACOS.- Ver: 1_7 Interacciones en la absorción de medicamentos
CUADRO N° INTERACCIONES F-A DURANTE LA ABSORCIÓN
Fármaco Tipo de interacción Recomendación
Anticoagulantes orales Los alimentos ricos en vitamina K (brócoli, coles, coles de Bruselas, espinacas, nabo, lechuga,...) antagonizan su efecto
Mantener una dieta equilibrada sin comer de repente grandes cantidades de estos alimentos
Azitromicina Disminuye la absorción, se reduce la biodisponibilidad un 43%
Separar la ingesta del fármaco de la comida al menos 2 horas
Digoxina Los alimentos ricos en fibra y pectina se unen el fármaco
Tomar el fármaco todos los días a la misma hora en relación con las comidas y no tomarlo con comidas ricas en fibra
Eritromicina Disminuye la absorción de eritromicina base o estearato
Separar la ingesta del fármaco de la comida al menos 2 horas
Fluorquinolonas Disminuye la absorción un 50% porque se forman complejos con cationes divalentes (Fe, Mg, Zn, Ca)
Separar la ingesta del fármaco de la comida al menos 2 horas
Inhibidores de la monoaminooxidasa (fenelcina, isocarboxacida, tranilcipronina)
Crisis hipertensivas si se toman alimentos con alto contenido en tiramina (quesos fermentados, alimentos escabechados, en conservas o ahumados, vino tinto)
Evitar estos alimentos
Levodopa Los aminoácidos inhiben de forma competitiva la absorción
No tomar el fármaco con alimentos ricos en proteínas
Acetaminofén Los alimentos ricos en pectina retrasan la absorción Tomar con el estómago vacío si se tolera
Penicilinas orales Disminución de la absorción Separar la ingesta del fármaco de la comida al menos 2 horas
Teofilina de liberación retardada Las comidas ricas en grasa pueden alterar la velocidad de absorción produciendo concentraciones elevadas de teofilina
No administrar junto con comidas ricas en grasa o tomar 1 hora antes de las comidas
Tetraciclina Los productos lácteos y el hierro disminuyen la absorción de tetraciclina por su efecto quelante
Separar la ingesta del fármaco de la comida al menos 2 horas
Aspirina y antiinflamatorios no esteroideos La comida disminuye la irritación gastrointestinal.
Carbamacepina Los alimentos aumentan la producción de sales biliares con lo que mejoran la disolución y absorción de carbamacepina.
Claritromicina Los alimentos aumentan la absorción en un 50%.
Ciclosporina
La comida aumenta la biodisponibilidad y tiene mejor sabor si se toma con leche. Administrar todos los días igual y monitorizar los niveles plasmáticos de ciclosporina.
Diazepan Los alimentos mejoran la biodisponibilidad, pero se deben separar al menos 1 hora de la leche y los antiácidos.
Eritromicina etilsuccinato Los alimentos pueden incrementar la absorción.
Fenitoína
El retraso del vaciamiento gástrico y el aumento de la secreción biliar mejora la disolución y la absorción, por lo que se debe tomar todos los días a la misma hora en relación con las comidas.
Griseofulvina La comidas ricas en grasa aumentan la absorción.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 3
Itraconazol Los alimentos pueden hacer que la biodisponibilidad llegue al 100%.
Litio El efecto purgante disminuye la absorción por lo que se debe tomar con el estómago lleno.
CUADRO N° INTERACCIONES F – A DURANTE LA ABSORCIÓN


No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 3
UADRO N° INTERACCIONES F-F DURANTE LA ABSORCIÓN

PRIMERA UNIDAD. SESION 4
TEMA 1. FACTORES QUE INFLUYEN EN LA DISTRIBUCIÓN DE MEDICAMENTOS EN FLUIDOS ORGÁNICOS
OBJETIVOS:
- Conocer los distintos mecanismos involucrados en el transporte de fármacos. - Conocer las características esenciales de la unión de fármacos a proteínas circulantes. - Factores de los que depende el paso de fármacos desde plasma a tejidos. Concepto de
flujo regional y barreras tisulares. - Definir, calcular e interpretar desde una perspectiva cinética y clínica el concepto del
Volumen de Distribución. VISION GENERAL DEL PROCESO DE DISTRIBUCIÓN DE FÁRMACOS
La distribución es un proceso farmacocinética en el que tiene lugar el transporte del
fármaco desde su lugar de absorción hasta el órgano diana. No obstante, es importante reseñar que una vez absorbido el fármaco es distribuido no sólo hasta el órgano sobre el que va a actuar, sino que al mismo tiempo llega a otros órganos en donde va a ser eliminado, metabolizado, acumulado.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 3
FACTORES QUE INFLUYEN EN LA DISTRIBUCIÓN
1. Propiedades Físico Químicas del Medicamento. 2. El nivel de fijación a proteinas plasmáticas. 3. La importancia de la fijación a las proteinas tisulares. 4. La perfusión sanguínea tisular.
1.- PROPIEDADES FISICO QUIMICAS DEL MEDICAMENTO
1.- SOLUBILIDAD. Coeficiente de partición.
2.- GRADO DE IONIZACION O pKa. Alteraciones en el Ph del medio pueden afectar la distribución.
3.- PM.- hasta 5000 D fácilmente atraviesan las membranas. De manera general una sustancia con suficiente liposolubilidad y PM de hasta 2000 D pueden atravesar cualquier membrana celular.
4.- TAMAÑO DE LAS MOLECULAS.

2.- FIJACIÓN A PROTEÍNAS PLASMÁTICAS
La mayoría de los fármacos se unen a proteínas plasmáticas para circular en el organismo.
Concepto de tejido indiferente: Tejido que al fijar un fármaco, no genera alteraciones en sus funciones normales, ni tampoco altera las propiedades del fármaco. Esta unión tiene como característica la Reversibilidad, siendo el principal tejido indiferente del organismo las proteínas plasmáticas.
La albúmina (69000) es la principal de estas proteínas vehiculizadoras (la albúmina tiene dos sitios de unión para fármacos, uno para fármacos de carácter ácido, y otro para fármacos de carácter básico).

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 4
Las Globulinas (40000) fijan bases débiles.
Las alfa1 globulinas fijan algunos otros fármacos de carácter básico.
Transcortina fija corticoides, vitamina B12.
La unión de los fármacos a proteínas depende esencialmente de:
1. La concentración de la fracción libre, 2. La constante de asociación, 3. El número de sitios de fijación disponibles por molécula de proteína y 4. La cantidad total de proteínas.
El efecto final de la fijación de los fármacos a las proteinas constituye una cifra estable para cada fármaco. Esta unión es reversible, encontrándose siempre un determinado porcentaje libre y cuando éste se elimina del plasma una nueva cantidad de fármaco se desprende de su unión a proteínas y toma su lugar, de tal modo que las fracciones libre y unida a proteínas permanecen constantes. Ocasionalmente, cuando se sobrepase la capacidad de fijación de la albúmina, aumenta la fracción libre.
Solamente el fármaco libre es activo, puesto que es el único capaz de atravesar barreras y difundir a los tejidos.
En determinadas enfermedades existe un déficit de albúmina circulante, o una menor capacidad de reserva de ésta, por lo que dosis "normales" pueden saturar la capacidad de fijación de las proteínas y elevar la cantidad de fármaco libre. Lo mismo puede ocurrir si se administran fármacos distintos en dosis saturantes con afinidad por el mismo lugar de fijación de la albúmina.
La unión fármaco – proteína plasmática NO ES ESPECIFICA. Esto implica que un fármaco distinto puede desplazar de su unión a las PP al primer fármaco. Esto es importante si el fármaco DESPLAZADOR tiene una Ka por las PP mayor que la Ka del DESPLAZADO. Por otro lado, importa también que la concentración sanguínea del fármaco desplazador sea igual o mayor que la necesaria para tomar todos los sitios activos de las PP.
Ejemplo de sustancias DESPLAZADORAS (alta afinidad, su dosis implica altas concentraciones sanguíneas): Sulfonamidas, Fenilbutazona, Aspirina.
Es necesario tener en cuenta a los fármacos que pueden ser desplazados. Sobre todo aquellos que están unidos a PP en más de un 80%, Esto adquiere importancia cuando el fármaco que va a ser desplazado posee un rango de acción en un estrecho margen terapéutico, o sea, la diferencia

entre las concentraciones sanguíneas en que el fármaco tiene acción útil y aquella en que es tóxica es muy pequeña.
Así, si por alguna razón un fármaco desplaza de su sitio de unión a otro fármaco que posee un estrecho margen de acción, este último puede ver aumentada su concentración sanguínea, y así provocar efectos tóxicos.
Un ejemplo de fármaco de estrecho margen terapéutico es la Warfarina, fármaco ACO de muy estrecho margen terapéutico y que se encuentra unido en un 99% a PP (VER TABLA 01), y por ello con un 1% libre en la sangre, y libre por ello para generar efecto. Basta que el fármaco sea desplazado sólo en un 1% para tener 2% libre, y tener disponible un 100% más de fármaco para ejercer efecto.???
No ocurre lo mismo con la Morfina que tiene un porcentaje de enlace de 35 % y en forma libre de 65%, si otro fármaco desplaza un 5% su unión a la glicoproteína, origina un aumento poco considerable en la fracción libre (70%) y por lo tanto en la concentración disponible para ejercer el efecto.
La importancia de las proteínas plasmáticas en el proceso de distribución, hace necesario que el profesional de Farmacia deba estar atento a la respuesta terapéutica en los siguientes casos:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 4
• Disminución en la síntesis de proteínas (enfermedad hepática) • Incremento en el catabolismo de las proteínas (trauma, cirugía) • Distribución de la albúmina en el espacio extravascular (quemados) • Eliminación excesiva de proteínas (síndrome nefrótico)
3.- FIJACION A PROTEINAS TISULARES.
En los tejidos existen proteinas titulares capaces de fijar los medicamentos. De la relación de afinidades entre las proteinas del plasma y la proteina de los tejidos (KAT) será el valor del Volumen de distribución.
La fijación a las proteinas titulares depende de factores como: Edad, embarazo, la obesidad o enferemedad.

TABLA 01. VALORES DE % DE FIJACION A PROTEINAS Y VOLUMEN DE DISTRIBUCIÓN DE CIERTOS MEDICAMENTOS.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 4
4.- PERFUSION SANGUINEA LOS TEJIDOS:
4.1 FLUJO SANGUÍNEO REGIONAL:
los órganos menos irrigados como pueden ser la piel,el hueso, el tejido graso o el músculo en reposo, reciben menos sangre que otros órganos como el cerebro, el hígado o el riñón. Por ello, el tiempo necesario para alcanzar el equilibrio con el compartimento central es mayor y alternativamente la cantidad de fármaco presente en estos tejidos puede ser inferior. Se exceptúan aquellos casos en los que el fármaco tenga una afinidad especial por un determinado tejido como pueden ser las tetraciclinas (hueso), griseofulvina (piel), o barbitúricos (tejidos ricos en grasa).

SALIDA DEL FÁRMACO DEL INTERIOR VASCULAR:
El fármaco disuelto en sangre se distribuye por todo el organismo, saliendo del compartimento vascular a favor de un gradiente, mediante diferentes mecanismos: a través de los poros, por pinocitosis, o más frecuentemente por difusión pasiva a través de la propia célula endotelial. La velocidad de salida del fármaco al territorio extravascular y de éste al interior celular depende esencialmente de la liposolubilidad del fármaco, si bien en los tejidos inflamados puede estar aumentada.
4.2 COMPARTIMENTOS DE DISTRIBUCION
TABLA 02. COMPARTIMENTOS ACUOSOS DEL ORGANISMO 1.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 4
TABLA 03. COMPARTIMENTOS ACUOSOS DEL ORGANISMO 2.
Una vez que el fármaco se encuentra en plasma, la concentración que alcanza en los diferentes tejidos dependen esencialmente de dos factores: el flujo sanguíneo regional, y la salida del fármaco del interior vascular.

5. - FACTORES QUE MODIFICAN LA DISTRIBUCION 5.1. PROPIEDADES FISICO QUIMICAS
- Liposolubilidad y el PM no se afectan. - Cambios en el pH si afectan.
5.2. ALTERACIONES EN LA FIJACION DE LAS PROTEINAS PLASMATICAS TABLA 04. FACTORES QUE AFECTAN LA FIJACION A PROTEINAS PLASMATICAS.
Proteína Aumento Disminución
Albúmina
Tumor benigno, ejercicio, hipotiroidismo, neurosis, paranoia, psicosis, esquizofrenia.
Edad (neonatos, geriatricos), neumonía bacteriana, quemaduras, cirrosis, fibrosis quística, enfermedad GI, histoplasmosis, lepra, absceso hepático, cáncer, desnutrición severa, mieloma múltiple, síndrome nefrótico, pancreatitis aguda, embarazo, falla renal, cirugía, trauma.
a-glicoproteína
Geriatras, enfermedad celiáca, enf. De Crohn, IAM, falla renal, AR, estrés.
Concentración fetal, síndrome nefrótico, anticonceptivos orales.
Lipoproteínas Diabetes, hipotiroidismo, síndrome nefrótico. Hipertiroidismo, trauma, enfermedad hepática (?).
TABLA 05. Alteración de las proteínas plasmáticas en diferentes estados patológicos
• El principal factor que determina cambios en la fijación de las proteinas plasmáticas son aquellos que modifican la concentración de estas en el plasma.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 4
• El stress que producen diversas condiciones como: infartos, trauma, enfermedades infecciosas ocasiona aumento en la alfa 1 glicoproteina ácida esto origina disminución en la fracción libre del fármaco.
• La disminución de sitios de fijación tiene efecto relativo, tal como sucede cuando existen competidores alostéricos externos o internos. Incluso por metabolitos.
• Dosis altas pueden saturar sitios de fijación.
6. INTERACCIONES DURANTE EL PROCESO DE DISTRIBUCION
TABLA 01. INTERACCIONES DURANTE EL PROCESO DE ABSORCION DE MEDICAMENTOS.

7. BARRERAS BIOLOGICAS: FIG. 01. MEDICAMENTOS QUE ATRAVIESAN LA BARRERA HEMATOENCEFÁLICA PLACENTARIA Y LA LECHE MATERNA. Ver material:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 5
7.1 BARRER
A HEMATOENC
EFÁLICA:
Anatómicament
e, los capila
res que
irrigan el cereb
ro son
distintos de
los del
resto del organismo. En el endotelio apenas hay espacios intercelulares libres, estando todas las células pegadas unas a otras, impidiendo de esta manera la salida a través de poros. Pero, además el 85% de la pared vascular externa está recubierto por terminaciones astrocíticas, que dificultan el paso de fármacos por difusión pasiva. Las condiciones de paso de la BHE pueden alterarse en situaciones tales como una inflamación meníngea, favoreciendo el paso de fármacos al interior cerebral. No obstante, existen áreas cerebrales libres de tales dificultades, como puede ser por ejemplo el área postrema. En los plexos coroideos, en el interior de los ventrículos, se forma el líquido cefaloraquídeo (LCR) donde se considera que la concentración de fármacos es muy semejante a la existente en el espacio extracelular cerebral.

FIGURA 02. LA
BARRERA HEMATOENCEFÁLICA.
Es una separación
funcional entre el cerebro y el resto del organismo; y uno de los componentes de esta barrera se encuentra en los capilares
sanguíneos cerebrales, los cuales son menos permeables, que los del resto del cuerpo, a sustancias que circulan en la sangre. Los pies gliales, extensiones astrocíticas que rodean los vasos capilares, también son parte de la barrera hematoencefálica. A la izquierda se muestra un capilar cerebral y a la derecha un capilar no cerebral. Nótese que la unión entre las células endoteliales de este último muestran aberturas (fenestraciones) que los capilares cerebrales no poseen; éstos, por lo contrario, contienen más mitocondrias, proveedoras de energía de los sistemas de transportación, que acarrean sustancias a uno y a otro lado de la pared capilar.
Alteraciones patológicas de la barrera:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 5
1.- Inflamación de las meninges. 2.- Aumento de la presión craneal. 3.- Isquemia. 4.- Anoxia. 5.- Neoplasias. 6.- Estados convulsivos.
Alteraciones fisiológicas: En el recién nacido no existe barrera la que demora 2 ó 3 semanas en desarrollarse.
7.2 LA BARRERA PLACENTARIA:
Al contrario que a nivel del SNC en la placenta no existen alteraciones morfológicas que dificulten en paso de fármacos al interior del embrión o feto. De hecho, el paso de fármacos al feto depende esencialmente de la liposolubilidad del fármaco y del flujo sanguíneo placentario. Es por ello que casi cualquier fármaco que tome la madre se puede encontrar en tejidos fetales.
El concepto de que la placenta constituye una barrera para los fármacos es falso, el feto esta expuesto en cierto grado, a todos los fármacos administrados a la madre. En general, los fármacos liposolubles (morfina, barbitúricos, anestésicos generales), pasan a través de la placenta por difusión pasiva; la glucosa por difusión facilitada; los iones y aminoácidos por transporte activo; las proteínas (inmunoglobulinas) por pinocitosis; los amonios cuaternarios (relajantes musculares) y las sustancias hidrosolubles de alto peso molecular (mayor de 1000) no atraviesan la barrera placentaria.

Por otra parte, dado que el pH de la sangre fetal es ligeramente inferior al de la sangre materna, tienden a acumularse en el feto los medicamentos de carácter básico.
En general deben evitarse los fármacos durante los 3 primeros meses de embarazo. Ejemplo de sus consecuencias pueden ser el síndrome alcohólico fetal en madres tomadoras crónicas ( corresponde al síndrome de privación alcohólica visto en el feto), teratogénesis, anomalías congénitas, etc.
7.3 HEMATOACUOSA (en el ojo).
FIGURA 03. Barrera Hematoacuosa.
La estructura y el funcionamiento de la barrera hematoacuosa es francamente complejo y en su aspecto morfológico participa tanto un componente epitelial como un componente endotelial. En su estructura es posible diferenciar una parte posterior, que restringe la penetración de solutos a la cámara posterior, y una parte anterior que limita el paso de ciertas sustancias hacia la cámara anterior. Las estructuras anatómicas fundamentales implicadas en la barrera hematoacuosa son el cuerpo ciliar y el iris.
8. ACUMULACION
Cuando la velocidad con que el fármaco es retirado de un tejido específico es mucho menor que la de ingreso, se habla de acumulación (concentración) del fármaco. Los mecanismos de

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 5
acumulación más frecuentemente observados son:
Solubilidad: Si el fármaco es muy liposoluble presenta gran afinidad por el tejido adiposo el cual al ser muy poco perfundido dificulta el regreso del fármaco al torrente circulatorio.
Unión a proteínas específicas: Es el caso de la digoxina, la cual se une a proteínas que únicamente se encuentra en tejido cardiaco. Las fenotiazinas (neurolépticos) se unen a la melanina presente en piel y ojos.
Formación de complejos: Las tetraciclinas reaccionan fácilmente con el calcio formando compuestos insolubles. Por esta razón se acumula en aquellos tejidos ricos en calcio (huesos y dientes)
Activación de sistemas de transporte: La mayoría de fármacos adrenérgicos por su parecido estructural con la adrenalina, activan sistemas de transporte que concentran el fármaco en los órganos blanco.
9. EQUILIBRIO DE REDISTRIBUCION DE MEDICAMENTOS
FIGURA 02: EQUILIBRIO DE DISTRIBUCION
DF-PpF Pp
F Pt F-Pt
EFECTO
D= 100 mgF-Pp (90%) = 90 mg
F (10%) = 10 mg
FF – Pt (90%) = 9.0 mg
F = 1.0 mg
1
2
E ( k = 0.10 h -1)= 10 mg
I
M = 90 mgF-Pp (90%) = 81 mg
F (10%) = 9 mg
F
F – Pt (90%) = 8.1 mg
F = 0.9 mg
1
2
II
E ( k = 0.10 h -1)= 9 mg
D= 81 mgF-Pp (90%) = 72,9 mg
F (10%) = 8,1 mg
F
F – Pt (90%) = 7,29 mg
F = 0,81 mg
1
2
III
E ( k = 0.10 h -1)= 8,1 mg
Modelo bicompartimental
D= dosisF= fármaco libre
Pp = Proteinas plasmática
F-Pp= complejo fármaco – proteina plasmática
F-Pt= complejo fármaco – proteina tisularPt = Proteinas plasmática
E
E= Eliminación
K = constante de eliminación
FIG. 07. PROCESO DE REDISTRIBUCION DE MEDICAMENTOSUna dosis de medicamento se distribuye en el organismo de acuerdo a sus ratios de fijación a las proteinas plasmáticas y tisulares. El proceso de eliminación contribuye a la redistribución del medicamento desde tejidos a la sangre.Pharm. Dr. Percy Ocampo Rujel
M
M = Cantidad de medicamento en el organismo.
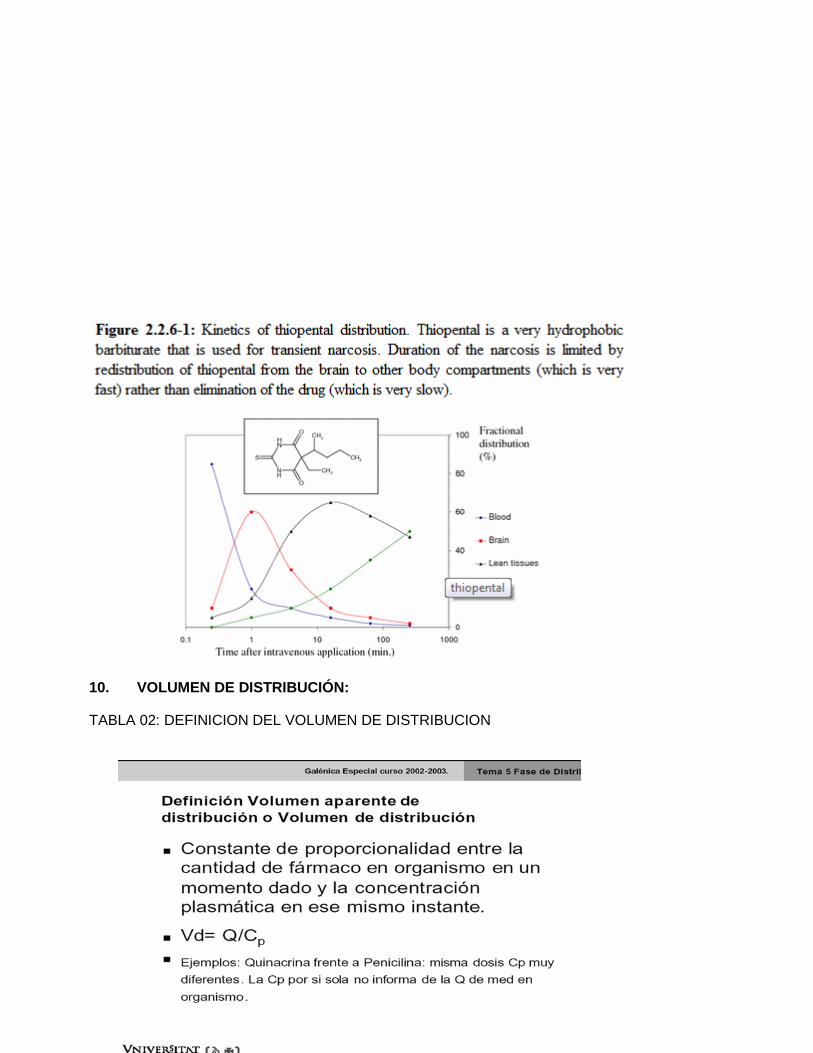
10. VOLUMEN DE DISTRIBUCIÓN:
TABLA 02: DEFINICION DEL VOLUMEN DE DISTRIBUCION

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 5
La unión del fármaco a proteínas y la salida del fármaco del interior vascular condicionan un parámetro que conocemos con el nombre de volumen de distribución (Vd). Se define como Vd el volumen de agua corporal en el que el fármaco se encuentra realmente disuelto. Pero este parámetro de "Vd real" no es fácilmente medible por lo que recurrimos al "Vd aparente" que corresponde a:
DOSIS
Vd = -----------------
Cp
Decimos que este volumen de distribución es aparente porque no refleja con exactitud dónde se encuentra el fármaco (Ver figura 02). Por ejemplo, si la Cp es muy baja el Vd será muy alto, indicándonos que está acumulado en algún tejido; por el contrario, si el fármaco está muy unido a proteínas la Cp será alta y el Vd será bajo.
A pesar de ello nos sirve para conocer la distribución corporal de un fármaco:
Vd 3 litros ----------------- fármaco en plasma
Vd 12 litros ----------------- fármaco en plasma + intersticio
Vd 40 litros ----------------- fármaco en plasma + intersticio + células
El valor clínico que representa conocer el volumen de distribución es limitado.
De manera muy general puede considerarse que si el Vd aumenta a expensas del órgano diana el efecto farmacológico también aumenta, no obstante que la Cp disminuye. Pero si el Vd aumenta debido al depósito de medicamento en el compartimento graso, entonces el efecto farmacológico no se verá incrementado, al contrario.

Es decir cambios en el Vd se reflejarán en la Cp que no forzosamente representan cambios en la respuesta farmacológica.
FIGURA 02: ILUSTRACION DEL EFECTO DEL VOLUMEN DE DISTRIBUCION DENTRO DE LOS TEJIDOS.
Variació
n del Vd
según el
paciente
y sus
característic
as: 1.-La
mujer
embarazada aumenta
su Vd,
el fármaco también se dirige a los nuevos territorios. 2.-Edad: Los niños tienen mayor porcentaje de agua, a diferencia de los ancianos, cuyo porcentaje en grasa es mayor. Ello implica que la liposolubilidad es distinta para los distintas edades con un mismo fármaco. 3.-Patología:Alteraciones de la perfusión sanguínea periférica. 4.-Hipoalbuminemia en fármacos de alta unión a ellos.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 5
5.-Obesidad exagerada. 6.-Acidosis Metabólica: altera la fracción ionizada del fármaco. Deben considerarse estos factores al momento de dar los distintos fármacos.
PRIMERA UNIDAD. SESION 04 TEMA 01: ELIMINACIÓN DE FÁRMACOS: BIOTRANSFORMACION
1. ELIMINACION DE FARMACOS. DEFINICION
En la mayoría de las ocasiones, la finalización del efecto producido por un fármaco se debe, bien a la eliminación desde su lugar de acción hacia el exterior (eliminación propiamente dicha, excresión por ejemplo renal o biliar) o bien porque se altera su estructura (biotransformación o metabolismo de fármacos) y por tanto su capacidad para continuar ejerciendo su efecto.
Las rutas de eliminación mayoritarias del cuerpo para cualquier molécula son la orina y las heces. Menos cantidades pueden ser eliminadas por el sudor, saliva, lágrimas, leche y el aire espirado.
Del fármaco tomado oralmente, parte de él puede no ser absorbido y pasa directamente a través del tracto gastrointestinal y es eliminado con las heces. Como se ve, la vía de administración puede influir también en la vía de excreción. Por ejemplo, puede esperarse que la administración directa en la circulación portal desemboque en una mayor excreción biliar que la administración por vía sistémica.

RAFIC
O NO 01
2. CONSIDERACIONE
S ANA
TÓMICAS. (Ver
Material
Complementario: Anato
mía y fisiología del hígado)
3. DEFINICIÓN DE BIOTRANSFORMACION:
Al conjunto de reacciones bioquímicas que producen modificaciones sobre su estructura química. En términos de metabolismo estas modificaciones pueden producir metabolitos inactivos, metabolitos activos o productos metabólicos con actividad farmacológica distinta a la del fármaco original.
Este proceso se lleva a cabo fundamentalmente en el hígado mediante dos tipos de reacciones que frecuentemente son secuenciales, y que se conocen como reacciones de fase 1 y reacciones de fase 2.
4. METABOLISMO DE PRIMER PASO
Algunos medicamentos son metabolizados con gran eficacia por el hígado a partir de la

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 6
circulación portal, lo que supone la necesidad de administrar dosis mucho mayores cuando se administran por vía oral que por cualquier otra vía.
GRAFICO NO 02: DROGAS QUE SUFREN ALTO/BAJO EFECTO DE PRIMER PASO
5. REACCIONES DE BIOTRANSFORMACION. (Ver Material Complementario: Biotransformación)
5.1. REACCIONES DE FASE 1
Suelen consistir en oxidaciones (hidroxilaciones, N y O-desalquilaciones.) reducciones o hidrólisis. Estas reacciones suelen introducir en la estructura del fármaco un grupo reactivo que lo convierte en más activo quimicamente.
Las reacciones de fase 1 más frecuentes son las oxidaciones, que son catalizadas por un sistema enzimático complejo conocido como sistema de oxigenasas de función mixta, cuyo sistema enzimático más importante es el sistema del citocromo P- 450(CYP), del que existen quizás unas 100 isoenzimas. Las isoenzimas mas importantes para el metabolismo de fármacos en humanos son las CYP 3A4, 2D6 y 2C19.

También son reacciones de fase 1 alguna reducciones y reacciones hidrolíticas.
5.2. CITOCROMO P-450. El sistema enzimático más importante del metabolismo de fase I es el
citocromo P-450, una superfamilia de enzimas microsomales que catalizan reacciones de oxidación de numerosos fármacos por su capacidad de transferencia de electrones.
Los electrones son aportados por la NADPH-citocromo P-450-reductasa, una flavoproteína que transfiere electrones del NADPH (la forma reducida del fosfato dinu-cleótido de nicotinamida-adenina) al citocromo P-450. Las enzimas del citocromo P-450 están agrupadas en 14 familias de genes de mamífero que comparten secuencias idénticas y 17subfamilias. Se denominan por un símbolo raíz (CYP), seguido de un numeral árabe para la familia, una letra para la subfamilia y otro número árabe para el gen específico.
Las enzimas de las subfamilias 1A, 2B, 2C, 2D y 3A son las más importantes del metabolismo en mamíferos. CYP1A2, CYP2C9, CYP2C19, CYP2D6 y CYP3A4 son los más importantes en el metabolismo humano. La especificidad de las enzimas permite explicar muchas interacciones entre fármacos. En la tabla 298-3 se presentan varios ejemplos de fármacos que interaccionan con enzimas específicas del complejo citocromo P-450 (v. también Interacciones farmacológicas, cap.301). Las diferencias genéticas entre pacientes pueden modificar la respuesta clínica.
5.3. REACCIONES DE FASE 2
Suelen ser reacciones de conjugación, que por regla general, inactivan el fármaco. En términos también generales estas reacciones actúan sobre el grupo reactivo introducido en las reacciones de fase 1, añadiendo un sustituyente más grande, como un glucuronilo, un sulfato o un acetilo que disminuye la liposolublidad y favorece por tanto la eliminación renal o biliar.
La mayoría, aunque no todas, de las reacciones de conjugación del organismo tienen lugar en el hígado.
CUADRO. Rutas de los xenobioticos en los procesos de biotransformación.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 6
5.4. CONJUGACIÓN
La glucuronoconjugación es la reacción de fase II más común, y es la única que ocurre en el sistema enzimático microsomal hepático. Los glucurónidos se secretan por la bilis y se eliminan por la orina. El cloranfenicol, el meprobamato y la morfina son algunos ejemplos de fármacos metabolizados por esta vía.
La conjugación con aminoácidos, como la glutamina y la glicina, produce metabolitos (p. ej., ácido salicilúrico, de la conjugación de ácido salicílico y glicina) fácilmente excretables en la orina, pero que no suelen secretarse por la bilis. La acetilación es la vía metabólica principal de las sulfamidas. La hidralazina, la isoniazida y la procainamida también sufren acetilación. La sulfoconjugación es la reacción entre grupos fenol o alcohol y un sulfato inorgánico, que deriva en parte de aminoácidos que contienen azufre como la cisteína. Los ésteres de sulfato así obtenidos son polares y se excretan rápidamente en la orina. Algunos ejemplos de fármacos que forman sulfatos son: paracetamol, estradiol, metildopa, minoxidil y tiroxina. La metilación es la principal vía metabólica para inactivar algunas catecolaminas. La niacinamida y el tiouracilo también sufren procesos de metilación.
6.- INDUCCIÓN E INHIBICIÓN METABÓLICA

Un gran número de fármacos tienen la propiedad de aumentar o de frenar la capacidad enzimática hepática. Los fármacos y las sustancias inductoras metabólicas actúan incrementando la actividad oxidasa microsomal y la actividad de los sistemas conjugantes, por mecanismos no bien conocidos. Estos efectos, de algunos fármacos, pueden ser importantes en la clínica, al ser responsables de interacciones farmacológicas.
GRAFICO NO 03: INHIBICIÓN Y ACTIVACIÓN METABÓLICA: CONSECUENCIAS.
CUADR
O NO 01:
FACTORES
QUE ALTE
RAN LA ACTIVIDAD ENZIMATICA

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 6
7. CLEARANCE INTRINSECO Y COEFICIENTE DE EXTRACCION HEPATICO La eficiencia del hígado para eliminar fármacos depende de:
1. Cantidad de fármaco que llega al hígado por unidad de tiempo. depende de: -flujo sanguíneo Concentración de fármaco en la sangre 2. Concentración de fármaco libre, no unido a Proteína Plasmática 3. Actividad total del sistema enzimático implicado en la biotransformación, asociado al
clearance intrínseco.
7.1 Coeficiente de Extracción Hepático: (Ver Gráfico No 04)
Para evaluar la capacidad de aclaramiento de un órgano para un determinado fármaco se hace necesario conocer el coeficiente de extracción, que se define según la siguiente ecuación:
Siendo Ca la concentración de entrada y Cv la de salida. Por tanto, el coeficiente de extracción alcanzará valores entre 0 y 1, siendo un parámetro adimensional.
7.2.1 Clearance hepático, Se define como el volumen de sangre depurada de un fármaco por el Hígado por unidad de tiempo. Sus unidades son v. t–1. Para conocer el aclaramiento, deberá multiplicarse el coeficiente de extracción por el flujo sanguíneo de ese órgano concreto.
l aclaramiento total de un fármaco será la suma del aclaramiento de ese fármaco por todas las vías (aclaramiento renal, hepático, etc.). La velocidad de desaparición de un fármaco del organismo será la suma de las velocidades de todos los procesos de eliminación, ya sean mediante metabolización o biotransformación, o a través de las diferentes rutas de excreción (Tabla XII y Figura 5).

TRAFICO NO 04: MECANISMO DE ELIMINACION HEPATICA
7.2. COEFICIENTE DE EXTRACCIÓN HEPÁTICO -BAJO: Diazepam tiene biodisponibilidad alta muy poco se va a
destruir. -INTERMEDIO: Acido acetil salicílico, cloramfenicol. -ELEVADO : Morfina tiene biodisponibilidad baja ya que en una pasada el
hígado lo extrae y lo biotransforma.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 6
CUADRONO 02: COEFICIENTES DE EXTRACCION HEPATICO DE MEDICAMENTOS.
8. MODIFICACIONES EN LA ACTIVIDAD ENZIMATICA
8.1 Limitación de la capacidad metabólica
Para casi cualquier fármaco la velocidad de metabolismo por parte de una enzima o sistema enzimático alcanza un límite máximo (límite de capacidad). A concentraciones terapéuticas por lo general sólo se ocupa una pequeña fracción de los centros activos de la enzima, y la velocidad de metabolismo aumenta de manera proporcional a la

concentración plasmática. Sin embargo, en algunas ocasiones, se encuentra ocupada la mayor parte de los centros activos. En estas circunstancias, la velocidad de metabolismo no aumenta en proporción a la concentración plasmática. El resultado es la limitación de la capacidad metabólica. La difenilhidantoína y el alcohol son dos ejemplos de fármacos con este tipo de metabolismo, lo que explica las diferencias interindividuales en los niveles plasmáticos de difenilhidantoína tras la administración de una dosis diaria de 300 mg.
8.2 Modificaciones debidas a la edad
Los recién nacidos tienen un sistema enzimático microsomal hepático sólo parcialmente desarrollado y, en consecuencia, presentan algunas dificultades para metabolizar muchos fármacos (p. ej., hexobarbital, fenazetina, anfetamina y clorpromazina). La experiencia con el cloranfenicol en recién nacidos muestra claramente las graves consecuencias que se pueden derivar del enlentecimiento de la glucuronoconjugación. Dosis equivalentes en mg/kg de cloranfenicol, bien toleradas por pacientes mayores, pueden provocar una toxicidad grave en los recién nacidos (síndrome del niño gris), asociada a la presencia de niveles plasmáticos elevados de cloranfenicol durante largo tiempo.
A menudo, la capacidad metabólica también se encuentra disminuida en los pacientes ancianos; esta reducción varía en función del fármaco y no es tan grave como en los recién nacidos.
9. OTROS PARÁMETROS FARMACOCINÉTICOS RELACIONADOS CON LA ELIMINACIÓN
9.1 Constante de velocidad de eliminación (ke): por regla general, la eliminación de un fármaco del organismo, mediante biotransformación, excreción o por los dos mecanismos a la vez, es un proceso que sigue una cinética de orden uno, que viene establecido por la siguiente ecuación:
siendo ke la constante de eliminación y Q la cantidad de fármaco remanente. Por tanto, la velocidad de eliminación será proporcional a la cantidad de fármaco remanente en el organismo. Viene expresada en t-1.
9.2 Semivida biológica de eliminación (t1/2): se define como el tiempo necesario para que una concentración plasmática determinada escienda al 50%. Puede calcularse de forma sencilla, conociendo la constante de eliminación. Es un parámetro que orienta sobre el tiempo de permanencia del fármaco en la sangre. Viene expresado en unidades de

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 6
tiempo.
8.3 INTERACCIONES EN LA BIOTRANSFORMACIÓN.


No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 7


No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 7
TEMA 02: ELIMINACIÓN DE FÁRMACOS: EXCRESION
2. EXCRESION DE FÁRMACOS
Proceso por el que un fármaco o un metabolito activo se expulsa del organismo sin modificar más su forma química. En general los fármacos muy polares se eliminan en gran parte sin metabolizar y los muy liposolubles metabolizados.
2. VÍAS DE EXCRESION:
2.1.1 Vía biliar (con recirculación enterohepática)
2.1.2 Leche (de importancia toxicológica para el recién nacido)
2.1.3 Saliva, sudor, piel, pulmones, etc.
2.1.4 Vía renal (la más importante)
El riñón es el principal órgano de excreción y es el responsable de eliminar las sustancias hidrosolubles. El sistema biliar también elimina algunos fármacos y metabolitos. Aunque los fármacos también pueden eliminarse por otras vías (como el intestino, la saliva, el sudor, la leche materna y los pulmones), la contribución global de estas vías suele ser pequeña. La excreción de los anestésicos volátiles a través del aire espirado por los pulmones constituye una excepción. Aunque la eliminación por la leche materna no es demasiado importante para la madre, puede serlo para el lactante.
3. EXCRECIÓN BILIAR
Algunos fármacos sólo se eliminan por esta vía mientras que los otros lo hacen por vía renal y en parte biliar.
MECANISMO: Es un transporte activo desde el hepatocito hasta el polo biliar en contra de un gradiente de concentración. Se conocen 3 sistemas:
• Aniones orgánicos (originales como los conjugados por ejemplo: con el ácido glucurónico). • Cationes orgánicos (compuestos que contienen NH4+) • Otras moléculas: sustancias neutras no ionizables (algunas hormonas y digitálicos) Este mecanismo de transporte puede saturarse cuando hay concentraciones plasmáticas del fármaco elevadas (transporte máximo) y sustancias con propiedades fisicoquímicas similares pueden competir por la excreción a través de esta vía. La excreción biliar es facilitada por diversos factores, como un Peso molecular mayor de 300 g/mol (por lo general, las moléculas más pequeñas se eliminan en cantidades ínfimas), la presencia simultánea de grupos polares y lipófilos y la conjugación (especialmente con ácido glucurónico).

3.1. CIRCULACIÓN ENTEROHEPÁTICA.
Contribuye a aumentar la vida media de los fármacos. Puede alterarse por la presencia de microorganismos y por otros mecanismos. Los fármacos conjugados que se secretan en el intestino también sufren una circulación enterohepática cuando se hidroliza el conjugado y se reabsorbe el fármaco. La excreción biliar es una vía de eliminación del organismo que depende del hecho de que la circulación enterohepática sea incompleta; es decir, cuando no se reabsorbe en el intestino todo el fármaco secretado.
4. ELIMINACIÓN POR LECHE
Es poco importante en cuanto a la cantidad
MECANISMO: Difusión pasiva
FACTORES CONDICIONANTES:
• pK del fármaco (ácido o base débil) • Concentración en plasma y leche • pH de la leche (6-7) • Unión a proteinas de plasma y leche
IMPORTANCIA PRÁCTICA:
• Posibilidad de sensibilización del lactante • Posibilidad de ejercer efectos tóxicos por la inmadurez del lactante
5. ELIMINACION POR SALIVA
Es poco importante en cuanto a la cantidad, puede serlo para monitorizar fármacos.
MECANISMO: Difusión pasiva; pH = 5.8-7.8
Conceptualmente es importante saber que la concentraciones salivares de fármacos son fiel reflejo de la concentración libre de fármaco en plama y en el LCR. Además la obtención de saliva es más fácil que obtención de plasma.
Expecionalmente hay fármacos que se eliminan activamente a saliva y otros que producen toxicidad local.
6. OTROS TIPOS DE EXCRECIONES
Otro tipo de excreción de medicamentos es la excreción sudoral que sigue también un mecanismo de difusión pasiva de la porción no ionizada. Por esta vía se eliminan sustancias tales como el alcohol, la antipirina, la urea y ácidos y bases débiles.
La excreción alveolar afecta a un número de sustancias gaseosas o volátiles a la temperatura del organismo. Sólo es preciso una presión parcial capilar/alveolo positiva para que se produzca su eliminación por difusión pasiva. La intensidad de estos intercambios a nivel de membrana está relacionada con los fenómenos ventilatorios, asegurando la renovación del aire alveolar y la

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 7
irrigación pulmonar. Esta vía de eliminación se utiliza en la práctica médico−legal para las pruebas etílicas.
7. EXCRECIÓN RENAL
7.1 Aspectos Anatómicos Y Fisiológicos. (Revisar Lectura N0 01: ANATOMIA Y FISIOLOGIA RENAL)
7.2 Sistemas de Eliminación Renal: ER = FG + ST – RT
GRAFICO No FACTORES INVOLUCRADOS EN LA ELIMINACION RENAL
7.2.1 Filtración Glomerular
Aproximadamente 1/5 del plasma que llega al glomérulo se filtra a través de los poros del endotelio glomerular, el resto se dirige a los túbulos renales por las arteriolas eferentes. Los fármacos unidos a las proteínas plasmáticas no se filtran; sólo lo hace el fármaco libre. Los principios que rigen la reabsorción tubular de los fármacos son los mismos que en el paso a través de cualquier otra membrana.
Mecanismo: Presión hidrostática

Factores condicionantes:
• Cantidad de sustrato que llega al glomérulo que depende de: • Flujo sanguíneo renal y • La concentración plasmática de la sangre que llega al rinón. • Unión a proteínas plasmáticas. • El número de glomérulos activos. • Tamaño de las partículas (poros de 40 A), PM < de 5000 D
Estos factores pueden relacionarse: FG = VFG x Cf mg/ml (1)
Donde: FG = filtración Glomerular
VFG = Velocidad de filtración glomerular = 130ml/min en ad. Joven.
Cf = Conc. del fármaco libre o no unido a proteinas.
= fp x Cp, donde fp es la fracción del fármaco libre en el plasma ( factor ∞)y Cp es la concentración total del fármaco.
De donde (1) puede quedar: FG = fp x VFG x Cp mg/min
Si FG la suponemos igual a dE/dt. (Cantidad de fármaco eliminado/unidad de tiempo); y por definición:
Y Entonces: ClR = fp x VFG (ml/min)
De tal manera que si fp = 1 entonces y el fármaco solo se filtra por el glomérulo entonces: ClR = VFG (ml/min).
7.2.2 Secreción activa:
En el túbulo proximal existen mecanismos de secreción tubular activa que son importantes para la eliminación de muchos fármacos (p. ej., la penicilina, la mecamilamina y el ácido salicílico). Este proceso requiere energía, así que los inhibidores metabólicos pueden bloquearlo. La capacidad de secreción puede saturarse a concentraciones elevadas y cada sustancia posee su velocidad máxima de secreción característica, denominada transporte máximo.
Los aniones y los cationes disponen de mecanismos de transporte independientes. Normalmente, el sistema secretor de aniones elimina metabolitos conjugados con glicina, sulfatos o ácido glucurónico. Los distintos compuestos aniónicos compiten entre sí por la secreción. Esta competencia puede utilizarse con finalidad terapéutica; por ejemplo, el
dE / dt = ClR x Cp

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 7
probenecid bloquea la secreción normalmente rápida de la penicilina, lo que permite mantener una concentración plasmática de penicilina elevada durante más tiempo. Los cationes orgánicos también compiten entre sí, pero habitualmente no lo hacen con los aniones.
• Existen dos sistemas: uno localizado en el túbulo proximal (el principal) y otro en el túbulo distal.
• Hay un sistema para ácidos y otro para bases. • Mecanismo: Sistema de transporte activo, depende de energía, es saturable y
puede haber competencia por similitud de estructural. • Consideraciones anatómicas: Flujo renal.
Factores condicionantes:
• Cantidad de sustrato que llega al glomérulo que depende de: • Flujo sanguíneo renal y la concentración plasmática de la sangre que llega al
riñón. • La unión del fármaco a las proteínas plasmáticas. • La actividad del sistema de transporte, la que está determinada por la constante de
afinidad del transportador por el fármaco y el número de transportadores disponibles.
7.2.3 Reabsorción pasiva en el túbulo distal y en túbulo proximal.
Los compuestos polares y los iones son incapaces de difundir de nuevo a la circulación y se excretan a no ser que exista algún mecanismo de transporte específico para su reabsorción, como ocurre, por ejemplo, en el caso de la glucosa, el ácido ascórbico y las vitaminas del complejo B.
• Mecanismo: Gradiente de concentración, proceso pasivo. • Factores condicionantes: Características fisicoquímicas del fármaco, pH de orina. • Concentración del fármaco en sangre y orina
8. FACTORES QUE MODIFICAN LA ELIMINACIÓN RENAL
• Aumento de flujo. • Modificación del pH de la orina
8.1. Efecto del pH urinario.
Aunque el filtrado glomerular que llega al túbulo proximal tiene el mismo pH que el plasma, el pH de la orina expulsada oscila entre 4,5 y 8,0, lo cual influye enormemente en la velocidad de excreción de los fármacos. Puesto que las formas no ionizadas de los ácidos y las bases débiles poco polares tienden a reabsorberse fácilmente del filtrado tubular, la acidificación de la orina aumenta la reabsorción de los ácidos débiles (es decir, reduce su excreción) y disminuye la reabsorción de las bases débiles (y, por tanto, se excretan con mayor rapidez). La alcalinización de la orina produce los efectos contrarios.
Estos principios pueden aplicarse a determinados casos de sobredosis para aumentar la eliminación de ácidos o bases débiles. La alcalinización de la orina acelera la eliminación de ácidos débiles como el fenobarbital o la aspirina. Por el contrario, la

acidificación puede acelerar la eliminación urinaria de algunas bases, como la metanfetamina.
El grado con el que los cambios en el pH urinario alteran la eliminación renal total del fármaco depende del grado de contribución de la vía renal en la eliminación total, así como de la polaridad (de la forma no ionizada) y del grado de ionización de la molécula.
• Competición en los sistemas de secreción tubular • Insuficiencia Renal: (Lea el material didactico complementario: Fármacos e nsuficiencia
renal) importante cuando el fármaco se elimina por riñón, habrá que reducir la dosis o alargar los intervalos de dosificación.
CUADRO No EFECTO DEL pH URINARIO PARA LA ACLARACION DE CIERTOS FARMACOS.
9. CALCULO DE LA FUNCION RENAL
Es básico conocer el estado de la filtración glomerular antes de la dosificación de un fármaco. Éste dato se obtiene a partir del análisis del aclaramiento de creatinina. Sin embargo el gran inconveniente radica en que es necesaria una recogida estricta de orina de 24 h. Para obviar este hecho, existen varias fórmulas más simplificadas y normogramas. La más utilizada es la fórmula de Cockfroft y Gault:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 7
Los valores normales de creatinina sérica son: V: 0,6-1,2 mg/dl M: 0,5-1,1 mg/dl
Merece especial atención el caso de pacientes con insuficiencia renal terminal que están sometidos periódicamente a sesiones de hemodiálisis. En dicha situación, hay que tener en cuenta que se van a dar dos procesos cinéticos diferentes, uno que tiene lugar entre dos sesiones de diálisis y que estará condicionado por el grado de insuficiencia renal del paciente; y otro que tiene lugar en el transcurso de las sesiones del hemodiálisis, que estará condicionado por las características del dializador, los flujos de sangre, etc. Por lo tanto será necesario un ajuste de la dosis para cada etapa.
9.1 Aclaramiento Renal
La función renal es normalmente evaluada mediante el grado de capacidad del riñón de excretar desechos solubles, como la urea y el ácido úrico. Generalmente se expresa en términos del aclaramiento renal, aclaramiento renal, que representa el volumen de sangre que es aclarado de medicamento, durante un minuto, por vía renal. Se expresa mediante la siguiente ecuación:
La filtración glomerular se mide mediante la determinación del aclaramiento de inulina (polímero de la fructosa de gran peso molecular), ya que esta sustancia no se fija de modo apreciable a las proteínas plasmáticas y no es reabsorbida ni segregada.
De forma similar, la secreción tubular puede estimarse mediante el aclaramiento del ácido p−aminohipúrico (PAH), que es completamente eliminado por secreción activa, por lo que sirve como medida del flujo plasmático renal.
La glucosa se reabsorbe totalmente, por lo que daría un valor de aclaramiento cero.
Si C1 > 130 ml/min existe secreción activa del fármaco
Si C1 = 130 ml/min, filtración.
Si C1 < 130 ml/min existe reabsorción

9.2 Parámetros Farmacocinéticos.
Los procesos de eliminación de un fármaco pueden expresarse mediante el aclaramiento (Cl), relación existente entre la velocidad de eliminación (Ke) y la concentración en plasma (C), (Cl = Ke/C). Esta relación permanece prácticamente constante para cada fármaco y expresa el volumen de plasma que esdepurado del fármaco por unidad de tiempo. Cuando se considera el proceso de eliminación global de un fármaco se hace referencia al aclaración plasmático (Clp), que es la suma de diferentes aclaramientos (renal, metabólico, biliar, etc.).
El aclaramiento es un parámetro cinético que evalúa los procesos de eliminación, si bien no expresa el tiempo que tarda un fármaco en eliminarse del organismo.
Para ello se re c u r re a otro parámetro, derivado del anterior, denominado semi-vida de eliminación (t1 / 2), que expresa el tiempo necesario para que la concentración plasmática de un fármaco se reduzca a la mitad.
La ecuación que relaciona ambos parámetros es la siguiente: t1 / 2 = 0,693 Vd/Clp, donde Vd es el volumen aparente de distribución y Clp el aclaración plasmático. La tabla 6 muestra los valores de semi-vida de eliminación de diversos medicamentos usuales en la práctica clínica.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 8
`
La insuficiencia renal es el factor responsable de las modificaciones más importantes en la semi-vida de eliminación de los fármacos poco metabolizados que se excretan fundamentalmente a través del riñón. En estos pacientes es necesaria una aceptación de la posología a fin de evitar su acumulación en el organismo, sobre todo en el caso de fármacos potencialmente tóxicos, como los aminoglucósidos y la vancomicina. Habitualmente la posología se ajusta en función de la creatinina sérica o el aclaramiento de creatinina.
10. FACTORES QUE INFLUYEN EN EL ACLARAMIENTO RENAL DE FÁRMACOS.
10.1 Edad
Los mecanismos de secreción tubular renal en niños prematuros e incluso en recién nacidos, no están bien desarrollados y su eficiencia está disminuida. Estudios realizados sobre la eliminación de la inulina sugieren una impermeabilidad parcial de la membrana glomerular o más probablemente una velocidad de flujo sanguíneo menor que el correspondiente al volumen de agua del organismo. Esto adquiere un gran importancia cuando se administran dosis repetidas. La concentración mínima de medicamento justo antes de la administración de cada dosis sucesiva será mayor que la esperada y, por lo tanto, la concentración máxima se puede elevar por encima del máximo permitido, con el consiguiente efecto tóxico.
10.2 Sexo
El aclaramiento renal es aproximadamente el 10 por ciento menor en las mujeres que en los hombres.
10.3 Enfermedad
En enfermedades cardíacas y especialmente en las renales (nefritis, piolonefritis, nefroesclerosis, insuficiencia renal) el funcionalismo renal está muy disminuido. Como consecuencia, los fármacos, o sus productos biotransformados, se acumulan a niveles potencialmente tóxicos. Por tanto, será preciso el ajuste correcto de los regímenes posológicos a la capacidad funcional renal. La aproximación más común para realizar este ajuste se basa en el aclaramiento de la creatinina endógena. Para mantener los niveles de medicamento correctos, se puede disminuir las dosis administrada sin modificar el intervalo de tiempo entre dos dosis sucesivas, o bien

administrar la misma dosis a intervalos más espaciados.
11. INTERACCIONES DE LA EXCRESÓN.
SEGUNDA UNIDAD SEXTA SESIÓN
MODELAJE Y ORDEN FARMACOCINETICO Modificado
de: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide02/capitulo05/01b.html
DEFINICIONES:
La palabra farmacocinética fue empleada por primera vez por Dost(1) en 1953 para describir los procesos de velocidad de cambio de las concentraciones de fármacos en el organismo humano o animal. El propósito de la farmacocinética, como lo establece Wagner,(2) es "el estudio de las velocidades de cambio de la concentración de fármacos y sus metabolitos en los fluidos biológicos, tejidos y excretas, así como también el de la

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 8
respuesta farmacológica y la construcción de modelos adecuados para la interpretación de tales datos".
Los grandes avances alcanzados en los últimos años en esta disciplina han culminado con la, aplicación clínica de los conceptos farmacocinéticos al terreno práctico mediante lo que se denomina la farmacocinética clínica.
La farmacocinética clínica es una disciplina de las ciencias de la salud que tiene por objeto la aplicación de la farmacocinética a la seguridad y el manejo terapéutico efectivo del paciente individual.(3)
La farmacocinética se caracteriza fundamentalmente, entre otros aspectos, por la construcción de modelos que representan un sistema de compartimientos en el organismo y en los cuales se supone que se distribuye el fármaco una vez ingresado a él: Un compartimiento puede ser un grupo de tejidos con características fisiológicas y fisicoquímicas similares, tales como flujo sanguíneo, afinidad por fármacos, etc., que examinaremos más adelante. Después de su introducción al torrente sanguíneo, por un proceso de absorción o bien con una inyección intravenosa, el fármaco se distribuye en estos compartimientos. Este proceso de distribución es, por lo general, rápido y se caracteriza por ser reversible. De este modo, el fármaco presente en la sangre se encuentra en estado de equilibrio con el fármaco en otros líquidos, tejidos u órganos de distribución. Como consecuencia de este equilibrio, los cambios de concentraci6ndel fármaco en la sangre indican cambios de concentración en otros compartimientos. En cambio, la transferencia de fármacos de la sangre a la orina o a otros compartimientos de excreción, así como los procesos de biotransformaci6n de los fármacos en el plasma o tejidos a productos metabólicos, la mayoría de las veces inactivos, suelen ser irreversibles. Esta irreversibilidad da lugar al proceso denominado eliminación, que comprende todos aquellos mecanismos que determinan la eliminación del fármaco desde el organismo ya sea por excreción urinaria, metabolismo o eliminación por otras vías (pulmones, sudor, etc. ).
El conjunto de procesos que intervienen en la distribución del fármaco en el cuerpo puede ser esquematizado de la siguiente manera:
Esta serie de procesos que se suceden tras la administración de un fármaco a un individuo: absorción, distribución, metabolizaci6n y eliminación ha recibido el nombre de sistema ADME. Ritschel, (4) para enfatizar que el proceso de liberación del fármaco, desde un producto medicamentoso que se administra por vía extravascular, es el que determina la absorción y, por lo tanto, los pasos subsiguientes (distribución, metabolismo y eliminación) propone el empleo de la expresión "sistema LADME".

ANÁLISIS COMPARTIMENTAL
La interpretación de los datos experimentales se realiza con la ayuda de modelos basados en compartimientos. La utilización de modelos en farmacocinética supone al organismo dividido en diferentes regiones, unidas entre si, en las cuales el fármaco se distribuye después de su entrada al torrente circulatorio. Si se considera que cada fluido, órgano, tejido o cada célula o grupo de células poseen diferentes características fisicoquímicas y distintos grados de afinidad por los fármacos, podemos imaginar que en realidad el organismo humano o animal consiste en múltiples compartimientos en que, cada uno de ellos, actuaría como un compartimiento individual. Sin embargo, en el organismo animal solamente se tiene acceso a dos fluidos en los cuales es posible investigar la distribución de fármacos: la sangre y la orina. Solo en forma ocasional puede utilizarse otro fluido u órgano al emplear métodos con trazadores radiactivos que se fijan específicamente en determinados tejidos.
El modelo más simple es el llamado modelo abierto de un compartimiento. El término "abierto" se refiere al hecho de que existe un sentido unidireccional de entrada y salida (absorción y eliminación).
El modelo abierto de un compartimiento supone al organismo como un todo homogéneo en el cual se distribuye el fármaco en forma semejante y casi instantánea cuando entra ya sea por un proceso de absorción o bien directamente por medio de una inyección intravenosa. Este tipo de compartimiento estaría formado principalmente por el volumen sanguíneo y los tejidos altamente irrigados, tales como el hígado, los pulmones, los riñones, etc. Este modelo supone también que las velocidades de intercambio entre las diferentes partes del mismo compartimiento, por ejemplo, desde la sangre hacia el hígado, así como el proceso inverso, serían idénticas.
Por otra parte, es posible visualizar también modelos de dos o más compartimientos, representados por tejidos u órganos en los cuales el intercambio es más lento. Estos constituyen los compartimientos periféricos, formados por el tejido adiposo, los tegumentos, los huesos, etc. , tejidos donde el intercambio de fármacos con la sangre no se realiza a la misma velocidad que con los del mismo compartimiento. En otros casos, la sangre engloba, ella misma, dos compartimientos diferentes: la fracción proteica, susceptible de fijar rápidamente el medicamento, y el liquido plasmático.
EL MODELO ABIERTO DE UN COMPARTIMIENTO
El modelo monocompartimental abierto puede representarse por el diagrama siguiente:
En este esquema, el fármaco administrado representa cualquier sitio extravascular de administración. La eliminación constituye el conjunto de procesos mediante los cuales el

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 8
fármaco es retirado del el organismo (excreción urinaria, metabolismo, etc. ); es la constante de velocidad de absorción y K, la constante de velocidad de eliminación o de disposición total.
Según este esquema, el cuerpo se encuentra representado por un compartimiento simple en el cual el fármaco se distribuye uniformemente conforme a un comportamiento cinético particular. Esto implica que el fármaco que entra al organismo desde el sitio de administración, es rápidamente equilibrado en los fluidos de distribución. En otras palabras, la velocidad a la cual el fármaco se distribuye por el cuerpo es lo suficientemente rápida, en relación con su velocidad de eliminación, como para permitir considerar al cuerpo como una solución uniforme del fármaco. Así el paso de éste por el organismo puede representarse adecuadamente por un gráfico de concentración sanguínea en función del tiempo, ya que las muestras sanguíneas representan una solución uniforme.
A pesar de que este modelo es limitado en cuanto al número de 12 fármacos que puede representar adecuadamente, constituye el modelo más sencillo en farmacocinética y posee la ventaja de que sus principios pueden ser aplicados a esquemas más complejos. El modelo básico puede variar respecto al comportamiento cinético, al tipo de administración y al mecanismo de eliminación, por lo cual se consideran por separado los casos en los cuales la entrada del fármaco al torrente circulatorio se realiza mediante una inyección intravenosa rápida, una infusión intravenosa a velocidad constante o a una administración que implique una absorción de primer orden (oral, rectal, intramuscular, etc. ).
El modelo abierto de un compartimiento no representa adecuadamente las verdaderas características de distribución de la mayoría de los fármacos. La distribución instantánea no es posible debido a que los fármacos poseen diferentes grados de afinidad con los tejidos, órganos y fluidos de distribución y, por lo tanto, diferentes velocidades de equilibrio. Los tejidos de gran irrigación, como el hígado, los riñones, los pulmones, etc. logran el estado de equilibrio en forma muy rápida, mientras que los tejidos como el óseo, las grasas, los cartílagos, etc. alcanzan ese estado en forma más lenta, según la solubilidad del fármaco en esos tejidos. Teóricamente, al menos, cada tejido forma un compartimiento individual que tiene relaciones de intercambio con el compartimiento central (sangre) independiente de los otros tejidos.
4. MODELOS MULTICOMPARTIMENTALES
Ka KeE
M
Vd
D = DosisKa = Constante de absorción
M = Cantidad de medicamentos en el Organismo.
Vd = Volumen de Distribución
E = Cantidad de Medicamento

EL MODELO ABIERTO DE DOS COMPARTIMIENTOS
Uno de los modelos más comunes, adaptable a la mayoría de los fármacos, es el modelo de dos compartimientos en el cual los tejidos del cuerpo se clasifican en dos categorías: los que se equilibran instantáneamente o casi instantáneamente y los que requieren algún tiempo para lograr el equilibrio. Un modelo de esta naturaleza puede ser esquematizado de la siguiente manera:
La utilización de este modelo supone que:
- el organismo está constituido por dos compartimientos: uno Q, llamado compartimiento central, y otro P denominado periférico o tisular.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 8
- la cantidad de fármaco A llega al compartimiento central a una velocidad caracterizada por la constante y en seguida se distribuye en el compartimiento periférico de manera reversible y de acuerdo con las constantes de velocidad y que caracterizan el paso del fármaco desde el compartimiento central hasta el periférico y desde éste hasta el central, respectivamente.
- la eliminación del fármaco se realiza a partir del compartimiento central, proceso definido por la constante de velocidad de primer orden .
En este modelo, el fármaco llega al compartimiento central donde se distribuye con rapidez y luego pasa al compartimiento periférico. La velocidad de distribución en este último compartimiento es función de varios factores, tales como la velocidad de circulación sanguínea, el grado de irrigación y el coeficiente de partición del fármaco entre los dos compartimientos.
5. FARMACOCINETICA NO LINEAL
La linealidad farmacocinética puede ser definida como una "proporcionalidad directa de las velocidades de transferencia entre los diferentes compartimientos y las concentraciones o diferencias de concentraciones".(42)
En farmacocinética, un sistema lineal tiene una consecuencia importante: el área bajo la curva de concentración sanguínea en función del tiempo, después de una administración intravenosa, es función directa de la dosis administrada. Es decir, si la dosis de un fármaco produce un valor determinado de área bajo la curva, una dosis doble o triple producirá un valor doble o triple, respectivamente, del área bajo la curva (Fig. 28 a).
DKa Ke
EM
Vd
MODELO BICOMPARTIMENTAL
1
2
K12 K21
D = DosisKa = Constante de absorciónM = Cantidad de medicamentos
en el Organismo.Vd = Volumen de Distribución
Ke = Constante de eliminación
CINETICA DE PRIMER ORDEN
Después de la administración I.V. de un fármaco existe un período, la fase de distribución, en el que las concentraciones plasmáticas totales decaen muy rápidamente

Numerosos fármacos, en ciertas condiciones experimentales, no conservan esta linealidad, es decir,. los parámetros farmacocinéticos presentan desviaciones cuando se administran diferentes dosis y el proceso cinético está regido por la magnitud de la dosis. La no linealidad se reconoce, justamente, por las desviaciones farmacocinéticas que pueden producirse. Además del área bajo la curva también pueden alterarse la constante de velocidad de eliminación y la vida media biológica del fármaco, como consecuencia de desviaciones en la distribución del fármaco.
Respecto a la mayoría de los fármacos, las desviaciones de la linealidad de sus relaciones dosis-concentración se consideran despreciables en el rango de dosis empleado en terapéutica y se supone que la distribución del fármaco se realiza en forma uniforme y más rápida que la velocidad de eliminación, correspondiendo esta última a un proceso cinético de primer orden.
También suelen observarse desviaciones de la linealidad en la cinética de absorción, ocasionadas posiblemente por una baja solubilidad del fármaco en los fluidos gastrointestinales, por una baja cinética de disolución o, si la absorción se realiza mediante un transporte activo, por una saturación de este proceso (Fig. 28 b).
El reconocimiento de la no linealidad farmacocinética puede hacerse administrando dos dosis diferentes de un fármaco a un mismo individuo en dos ocasiones distintas. La curva de concentración plasmática, en ambos casos, puede diferir notablemente, sobre todo en la pendiente de la fase de eliminación. Por otra parte, las áreas bajo la curva de concentración sanguínea en función del tiempo, al ser medidas difieren de lo esperado. En general, cuando no existe linealidad farmacocinética, la concentración plasmática máxima del fármaco puede ser menor o mayor que lo esperado al aumentar la dosis.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 8
La alteración de la linealidad puede manifestarse en diferentes procesos: distribución, eliminación o absorción.
a) Distribución. Las desviaciones en la fase de distribución pueden ser causadas por multicompartimentalización de los fluidos biológicos y tejidos y, sobre todo, por la unión a proteínas. Muchos fármacos, especialmente ácidos o básicos, se unen a las proteínas del plasma, de preferencia a las albúminas y algunas veces a las globulinas.
La interacción entre un fármaco libre, , con los sitios libres de la proteína, P, para formar el complejo fármaco-proteína, D-P, obedece a la ley de acción de masas, como se indica en la ecuación siguiente:
en que es la concentración del fármaco unido a la proteína; es la concentración de fármaco libre y P es la concentración de proteína.
Por lo general se mencionan tres formas lineales de la ecuación [5. 2]:(43)
Ecuación de Scatchard:

Como el grado de unión no es constante, sino que depende de la concentración del fármaco en el plasma, la unión a proteínas que, como se mencionó antes, puede ser descrita por la ley de acción de masas, es de gran importancia a causa de la relación no lineal de dosis-concentración. La no linealidad es más pronunciada en aquellos fármacos que presentan una alta capacidad de unión a proteínas. Mientras más pequeña. es la dosis, las curvas de concentración en el agua plasmática en función del tiempo tienden a ser paralelas en un gráfico semilogarítmico. Estas curvas poseen, además, otra propiedad interesante: la pendiente de todas las curvas con idéntica constante de disociación e idéntico número total de lugares de unión es igual para una cierta concentración. Esto significa que cada conjunto de curvas, con idénticos valores de k1 y P, es congruente y superponible. De esto puede concluirse que en los fármacos que exhiben una apreciable capacidad de unión a proteínas la vida media biológica tiene diferentes valores para diferentes concentraciones en el plasma.
b) Eliminación. La desaparición de un fármaco de los fluidos del cuerpo puede ocurrir principalmente por dos mecanismos: biotransformación (metabolismo) y/o excreción a través de diversos órganos, como los riñones, los pulmones, la piel, etc.
Desde el punto de vista farmacocinético, la constante de velocidad de eliminación comprende la suma de las constantes de velocidad de biotransformación y de excrecíón. Por otra parte, en los modelos farmacocinéticos, la cantidad de fármaco dentro del tracto gastrointestinal es considerada como si estuviera fuera del organismo y, por lo tanto, el fármaco eliminado por las heces, sin que haya sido absorbido, no se considera como eliminado. Esto no se aplica a aquellos fármacos que se pierden durante el ciclo enterohepático, el cual puede o no incluir biotransformación del fármaco.
Durante la fase de metabolización, son frecuentes las desviaciones de la linealidad cinética. La linealidad se cumple solo si la concentración de enzima es alta en relación.con la concentración del fármaco, observándose una cinética de primer orden. Si la concentración de fármaco es alta, se observa la saturación de la enzima y el cambio a una cinética de orden cero, mientras que las concentraciones intermedias dan origen a cinéticas dependientes de la dosis y regidas por la ecuación de Michaelis-Menten.(44)
Los procesos metabólicos se describen, a menudo, como procesos irreversibles de primer orden. Esto puede ser considerado como una aproximación en los modelos farmacocinéticos, ya que las reacciones enzimáticas, en las cuales en muchos casos interviene la biotransformación, pueden ser reversibles. Por ejemplo, la acetilación de las sulfamidas es un proceso reversible, como lo han descrito numerosos investigadores. La ecuación de Michaelis-Menten ha sido empleada para describir las reacciones metabólicas causadas por la saturabilidad de estos procesos, pero es preciso tomar en cuenta que esta ecuación supone la irreversibilidad de la reacción. Esta suposición puede adquirir validez si la reacción inversa es mucho más lenta que la reacción metabólica y si el metabolito no es removido de los fluidos del organismo.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
La ecuación de Michaelis-Menten se expresa:
donde dC/dt representa la velocidad de cambio de concentración del fármaco a tiempo t; C es la concentración del fármaco a tiempo t; es la velocidad máxima teórica del proceso y es la constante de Michaelis, que es equivalente a la concentración cuando la velocidad de la reacción es la mitad del máximo.
Generalmente, es mucho más grande que C; luego, la ecuación [5. 8] se reduce a:
Es por este motivo que, a bajas concentraciones, la cinética de Michaelis-Menten se considera como una cinética de primer orden.
En aquellos casos en que C , la ecuación [5. 8] se reduce a:
En tales condiciones, la velocidad es independiente de la concentración de fármaco, de manera que el proceso se realiza a una velocidad constante. La cinética de biotransformaci6n del etanol y de los salicilatos ha sido descrita a base de esta última ecuación.(46)
En los procesos no lineales podemos calcular los parámetros de la ecuación [5. 8] si se toma el valor recíproco de ella y se expresa -dC/dt como V:
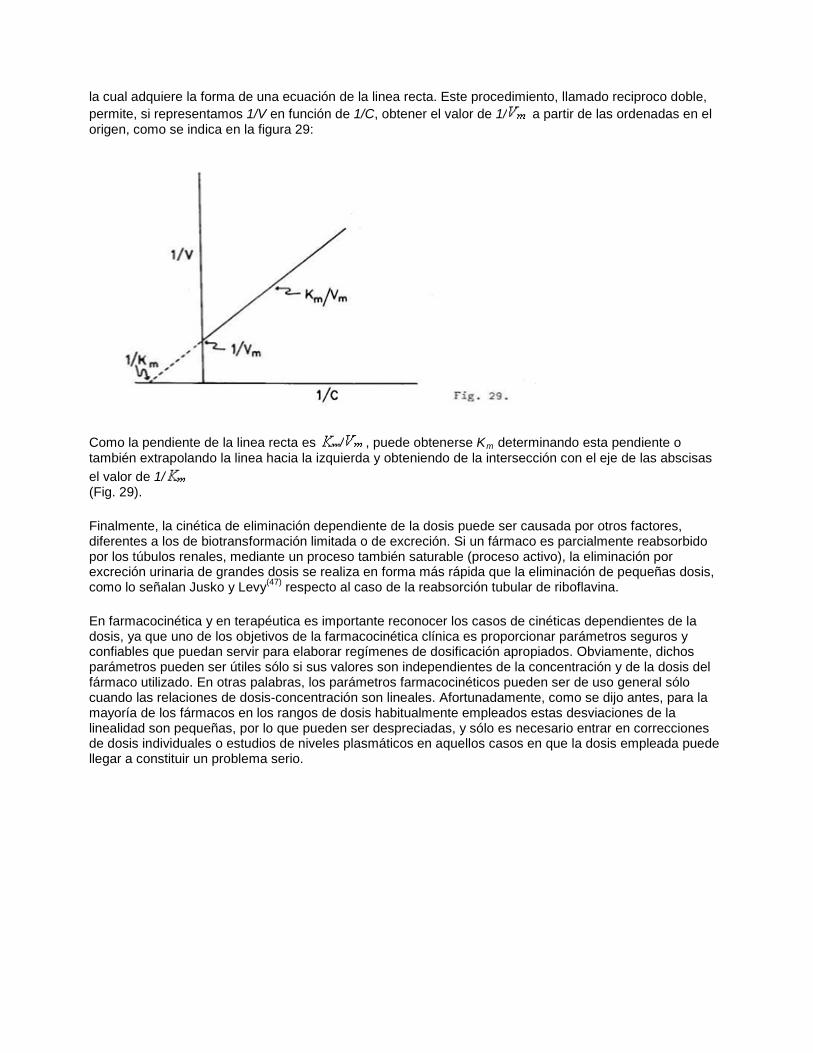
la cual adquiere la forma de una ecuación de la linea recta. Este procedimiento, llamado reciproco doble, permite, si representamos 1/V en función de 1/C, obtener el valor de 1/ a partir de las ordenadas en el origen, como se indica en la figura 29:
Como la pendiente de la linea recta es / , puede obtenerse Km determinando esta pendiente o también extrapolando la linea hacia la izquierda y obteniendo de la intersección con el eje de las abscisas el valor de 1/ (Fig. 29).
Finalmente, la cinética de eliminación dependiente de la dosis puede ser causada por otros factores, diferentes a los de biotransformación limitada o de excreción. Si un fármaco es parcialmente reabsorbido por los túbulos renales, mediante un proceso también saturable (proceso activo), la eliminación por excreción urinaria de grandes dosis se realiza en forma más rápida que la eliminación de pequeñas dosis, como lo señalan Jusko y Levy(47) respecto al caso de la reabsorción tubular de riboflavina.
En farmacocinética y en terapéutica es importante reconocer los casos de cinéticas dependientes de la dosis, ya que uno de los objetivos de la farmacocinética clínica es proporcionar parámetros seguros y confiables que puedan servir para elaborar regímenes de dosificación apropiados. Obviamente, dichos parámetros pueden ser útiles sólo si sus valores son independientes de la concentración y de la dosis del fármaco utilizado. En otras palabras, los parámetros farmacocinéticos pueden ser de uso general sólo cuando las relaciones de dosis-concentración son lineales. Afortunadamente, como se dijo antes, para la mayoría de los fármacos en los rangos de dosis habitualmente empleados estas desviaciones de la linealidad son pequeñas, por lo que pueden ser despreciadas, y sólo es necesario entrar en correcciones de dosis individuales o estudios de niveles plasmáticos en aquellos casos en que la dosis empleada puede llegar a constituir un problema serio.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
SÉTIMA SESIÓN CALCULO DE PARAMETROS FARMACOCINETICOS LUEGO DE UNA
ADMINISTRACION INTRAVENOSA RAPIDA. UTILIZANDO DATOS DE Cp.
Referencia: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado
desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
A. A partir de Datos de concentración sanguínea.
El esquema más sencillo es el que representa una eliminación conforme a una cinética de primer orden luego de una inyección intravenosa del fármaco.
Como la administración puede considerarse instantánea y la distribución en el organismo es rápida, la farmacocinética puede describirse como un simple proceso de transferencia entre dos compartimentos, tal cual se presenta en el esquema siguiente:
donde Q representa la cantidad de fármaco en el organismo considerado como un solo compartimento con un volumen Vd; E es la cantidad total de fármaco eliminado por diferentes vías. Además, según este esquema, el fármaco se elimina en forma inalterada, es decir, no ha experimentado modificaciones por biotransformación (metabolismo) a su paso por el organismo.
La velocidad de eliminación del fármaco desde el organismo corresponde a una cinética de primer orden, ya que es función de la concentración del medicamento en el volumen total del compartimento. La ecuación que describe el proceso de cambio de la cantidad de fármaco en el compartimento sanguíneo en función del tiempo es:
dQ = -KQ ---------------------------------------------------------------------------------------[2.15] dt
en que dQ/dt es la velocidad de cambio de la cantidad de fármaco en el organismo y el signo negativo en el segundo miembro de la ecuación indica que la cantidad decrece con el tiempo. La integración de la ecuación [2.15] entre los límites de tiempo cero y t conduce a:
Q = Q0 e-kt-------------------------------------------------------------------------------------[2.16]
donde Q0 representa la cantidad inicial de fármaco en el organismo (dosis administrada) y Q, la cantidad de

fármaco a un tiempo t. Esta ecuación también puede expresarse en forma logarítmica como:
log Q = log Q0 - Kt ----------------------------------------------------------------------------[2.17] ......................2,303
Es difícil determinar las cantidades de fármaco, simbolizado por Q y Q0, si no se conoce exactamente el volumen de fluido de distribución del individuo. Por otra parte, lo que se mide en la sangre es la concentración de fármaco. Aquí es donde la noción de volumen aparente de distribución encuentra su utilidad práctica, ya que permite expresar concentraciones en cantidades y viceversa:
C = Q/Vd ---------------------------------------------------------------------------------[2.18]
Q = C Vd ------------------------------------------------------------------------------[2.19]
Por lo tanto:
log C = log C0 -_ Kt ----------------------------------------------------------------[2.20] ................... . 2.303
De este modo, al elaborar un gráfico del logaritmo de la concentración plasmática en función del tiempo, se obtiene una recta cuya pendiente es –K/2,303, con una ordenada en el origen igual al logaritmo de ta concentración a tiempo cero, como se indica en la figura 2.4.
Fig. 2.4. Cálculo de la constante de velocidad de eliminación en gráfico semilogarítmico de concentración plasmática en función del tiempo.
En consecuencia, como se conoce la dosis inyectada, puede calcularse el volumen de distribución aparente del fármaco, de acuerdo a la ecuación [2.1].
Otra manera de calcular K consiste en emplear el concepto de tiempo medio de eliminación del fármaco, el cual puede ser obtenido en forma gráfica a partir de la figura 2.4.
En consecuencia:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
[2.21]
Problema 2.2 Calcular la constante de velocidad de eliminación, el volumen de distribución aparente y la vida media de eliminación de un fármaco que, al ser inyectado en una dosis de 200 mg por vía intravenosa, se distribuye de acuerdo a un modelo monocompartimental. Los datos de concentración plasmática en función del tiempo son:
t (hr) C (mg/L) 0,25
0,5
0,75
1,0
1,5
2,0
3,0
4,0
6,0
19,3
18,6
17,9
17,2
16,0
14,8
12,7
10,9
8,1
Si se realiza un gráfico de C versus t, se verá que la eliminación del fármaco es exponencial. Luego, otro gráfico de log C versus t será lineal y la pendiente (m) de la recta es igual a -K/2,303. En este caso,, la pendiente es igual a -0,065 por lo que K = m x 2,303 = -0,15hr-1.
Al prolongar la recta hacia la ordenada, el intercepto nos da el valor de log C0 = 2,995 y antilog n = 20 mg /ml.
Como Vd = Dosis ------------------ C0
Vd = 200 mg = 10 Litros -----20 mg/L
t1/2 = 0,693 = 4,6 horas --------- 0,15

OCTAVA SESIÓN CALCULO DE PARAMETROS FARMACOCINETICOS LUEGO DE UNA ADMINISTRACION
INTRAVENOSA RAPIDA. UTILIZANDO DATOS DE CONCENTRACIÓN URINARIA.
Tomado desde: Referencia: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado
desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
En los estudios farmacocinéticos, es corriente la determinación de parámetros de la cantidad acumulativa de fármacos que se excretan por la orina durante un tiempo adecuado para que todo el fármaco se elimine por esta vía. En la aplicación de este tipo de métodos deben satisfacerse varios requisitos:
a) el fármaco debe excretarse por el riñón por lo menos en un 10% en forma inalterada, (no metabolizada) con el fin de asegurar la precisión del método.
b) salvo excepciones, debe recolectarse la orina hasta que todo el fármaco no metabolizado o sus metabolitos sean excretados, sin perder ninguna fracción. Se considera apropiado, para fines prácticos, recolectar la orina por lo menos hasta 7 vidas medias de eliminación donde, teóricamente, se ha eliminado el 99,2 % de la dosis administrada.
c) se supone que la velocidad de excreción urinaria del fármaco corresponde a un proceso cinético de primer orden.
Si se considera el modelo representado por el esquema, que simboliza la eliminación de un fármaco que no ha experimentado biotransformación, la velocidad con que éste se elimina queda expresada por la ecuación diferencial:
dE = KQ ---------------------------------------------------------------[2.22] dt
donde dE/dt representa la velocidad de excreción instantánea del fármaco en estudio. Si el valor de Q, expresado por la ecuación [2.16], se substituye en la ecuación [2.22] se obtiene:
dE = KQ0 e-kt -----------------------------------------------------------------------------------[2.23] dt
La integración de la ecuación [2.23] conduce a:
E = Q0 (1 - e-kt) --------------------------------------------------------------------------------[2.24]

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
En esta ecuación, 1 - e-kt representa la fracción acumulativa de fármaco excretado por la orina. Por ejemplo, si se considera un período de eliminación igual al tiempo medio de eliminación (t1/2), la fracción de fármaco eliminado sería de 0,5, es decir, se ha eliminado el 50%. En 2 t1/2, se elimina el 75%, y así sucesivamente, para luego de 7 t1/2 encontrar que la fracción eliminada es 0,992, o sea, se ha eliminado el 99,2% del fármaco en forma inalterada.
En términos prácticos, como veremos más adelante, para un período superior a 7 vidas medias de eliminación se considera que el fármaco se ha eliminado totalmente, o que se ha llegado a la excreción a tiempo infinito.
La ecuación 2.24 reordenada queda como
1 - E – e-kt ----------------------------------------------------------------------------------------[2.25] --- Q0
y expresada en forma logarítmica:
log (1 – E) = - Kt _ ------------------------------------------------------------------------------[2.26] ---------- Q0 --2,303
E, representa la cantidad acumulativa del fármaco no metabolizado excretado por la orina hasta un tiempo t. La relación E/Q0 es la fracción de la dosis excretada, por lo que 1- E/Q0 es la fracción que queda por excretarse y que puede ser expresada como porcentaje no excretado al ser multiplicada por 100. De la ecuación (2.26) se deduce que el gráfico que representa el logaritmo de fracción de la dosis no excretada (o el log del porcentaje no excretado), en función del tiempo, es una recta cuya pendiente es igual a -K/2,303, como se indica en la figura 2.5.
Fig. 2.5. Gráfico semilogarítmico del porcentaje de la dosis de fármaco no excretado en función del tiempo, para calcular la constante de velocidad de eliminación total.
La mayoría de los fármacos se excretan parcial o totalmente metabolizados. En estos casos podemos considerar un esquema como el siguiente:

que indica que el fármaco, además de ser excretado por la orina en forma inalterada, experimenta también una transformación metabólica por un proceso cinético de primer orden, descrito por la constante de velocidad km Los metabolitos en este sistema se eliminan también por vía urinaria.
En este esquema, ME es el metabolito excretado por la orina; M, el metabolito en el volumen de distribución y km es la constante de velocidad de excreción urinaria del metabolito. Luego, la constante de velocidad de eliminación total del proceso es:
K= ke + km ---------------------------------------------------------------------------------[2.27]
donde ke es la constante de velocidad de excreción urinaria del fármaco no metabolizado.
Si se considera primero la cantidad del fármaco que se excreta inalterado por la orina, E, la velocidad de cambio del mismo puede expresarse por la ecuación diferencial siguiente:
dE =ke Q ---------------------------------------------------------------------------------- [2.28] dt
la que por integración conduce a:
E = Ke Q0 (1 – e-Kt) -------------------------------------------------------------------[2.29] ----K
Si se recolecta la orina durante un tiempo prolongado, e-Kt adquiere un valor muy pequeño, de modo que para t = ∞ esta forma exponencial toma un valor nulo. Así, la ecuación [2.29] queda:
E = k0 Q0 -------------------------------------------------------------------------------[2.30] --------- K
en la cual E es la cantidad de fármaco inalterado que se excreta por la orina a tiempo infinito. La ecuación [2.30] puede expresarse también como:
E = ke ------------------------------------------------------------------------------------[2.31] Q0 -----K
donde ke/K(o E /Q0) corresponde a la fracción de la dosis excretada sin metabolizar por la orina.
Se puede concluir pues, que el proceso mediante el cual el fármaco desaparece del torrente sanguíneo está regido por dos constantes de velocidad y que la fracción que se elimina sin modificación es igual a la constante de velocidad de excreción de la porción no modificada dividida por la suma de las dos constantes de eliminación, ke y Km. Si estas dos últimas constantes fueran exactamente iguales, la cantidad eliminada sin metabolizar sería igual al 50% de la dosis.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 9
En la ecuación [2.29], al substituir keQ0/K por E (ecuación [2.30]), se obtiene:
E = E (1 – e-Kt) -----------------------------------------------------------------------------------------[2.32]
y, por lo tanto:
1 - E = e-Kt [2.33] ----E
que en forma logarítmica queda como:
log ( 1 - E ) = - Kt _----------------------------------------------------------------------------------------[2.34] -----------E -- 2,303
o bien, luego del reordenamiento de la ecuación [2.33]:
log (E - E) = log ke Q0 - _Kt_ -----------------------------------------------------------------------[2.35] ------------------------- - K --------2,303
Por consiguiente, al representar gráficamente el logaritmo de la diferencia entre la cantidad de fármaco excretado a tiempo infinito y la cantidad excretada a cada intervalo en función del tiempo de cada recolección, se obtiene una línea recta en la cual la pendiente es -K/2,303, de donde puede calcularse el valor de la constante de eliminación total, K. Como la ordenada en el origen o intercepto de este gráfico viene dada por log keQ0/K, de su valor, conociendo K y la dosis inyectada Q0, se puede calcular ke, la constante de excreción del fármaco sin metabolizar. Finalmente, por la ecuación [2.27] podremos obtener el valor de km, la constante de velocidad de metabolización.
La necesidad de conocer exactamente el valor de E , o sea, la cantidad, total de fármaco eliminado por la orina, hace imprescindible que la recolección de ésta se realice hasta que la excreción sea completa. Por este motivo, el método no puede aplicarse si se pierde una muestra de orina o no se llega a la excreción total, ya que la cantidad recolectada no representa el verdadero valor de la cantidad excretada. Como lo habíamos señalado anteriormente, en términos prácticos se considera como excresión total, lo excretado después de transcurridas siete vidas medias de eliminación del producto.
Otro método aplicable al estudio de parámetros farmacocinéticos, aprovechando la excreción urinaria de fármacos, es el que recurre al cálculo de la velocidad de excreción (9). La ecuación [2.22] expresa la velocidad instantánea de excreción urinaria de un fármaco. Esta velocidad no puede ser medida, pero para intervalos cortos de tiempo, dE/dt puede estimarse como aproximadamente igual a E/ t, donde E es el cambio de E en un intervalo de tiempo t:
[2.36]
Ecuación que en forma lógica pasa a:
[2.37]
E/ t representa la velocidad promedio de excreción, la cual no puede ser medida, pero se supone que alcanza su valor más aproximado en la mitad del intervalo de tiempo de recolección de las muestras de orina. Por este motivo, la forma correcta de obtener los parámetros farmacocinéticos por este método consiste en representar gráficamente el logaritmo de ∆E/∆t versus el tiempo intermedio de los intervalos de recolección de las muestras. examinadas, (tm), como se indica en la figura 2.6:

Fig. 2.6. Gráfico semilogarítmico de la velocidad de excreción urinaria de un fármaco no metabolizado en función del tiempo intermedio de la recolección de muestras de orina.
Este método puede ocasionar errores en la determinación de las constantes si los períodos de recolección urinaria son muy prolongados, pero si éstos no son superiores a la vida media biológica del fármaco, el error no excede de un 2%. Evidentemente, los problemas surgen cuando las vidas medias biológicas son muy cortas, ya que a veces es imposible conseguir una recolección urinaria en forma tan seguida. Sin embargo, este método presenta la ventaja de que no es necesario llegar hasta la eliminación total del fármaco.
Tal como se aprecia en la figura 2.6, la pendiente de la recta obtenida es igual -K/2,303, lo que permite calcular la constante de disposición total de fármaco. Como la ordenada en el origen da el valor de keQ0, se puede obtener fácilmente el valor de ke y después el de km, aplicando la ecuación [2.27]
La velocidad de excreción urinaria también permite medir la depuración renal de un fármaco que se elimina sin experimentar metabolización, ya que:
[2.38]
En donde Cm es la concentración plasmática medida en la mitad del intervalo de recolección de muestras de orina. Luego, si se elabora un gráfico de la velocidad de excreción urinaria en varios intervalos, en función de Cm, se tiene que la depuración renal corresponde a la pendiente de la línea recta obtenida, como se ilustra en la figura 2.7.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Fig. 2.7 Relación entre velocidad de excreción urinaria de un fármaco no metabolizado y la concentración plasmática medida en la mitad del intervalo de recolección de muestras de orina.
Como puede apreciarse, la velocidad de excreción urinaria es proporcional a la concentración plasmática medida en la mitad del intervalo de recolección de las muestras de orina, por lo que la depuración renal puede obtenerse dividiendo la velocidad de excreción urinaria en un determinado intervalo, por la concentración plasmática correspondiente al tiempo intermedio de dicho intervalo.
NOVENA SESIÓN
CÁLCULO DE PARÁMETROS FARMACOCINETICOS AL ADMINISTRAR UNA INYECCIÓN INTRAVENOSA CONTINUA (INFUSIÓN)
Editado por: Percy Alberto Ocampo Rujel
Referencia: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
1. A Partir de Datos de Concentración Sanguínea
En aquellos casos en que la velocidad de entrada del fármaco al torrente sanguíneo se realiza de acuerdo con un proceso cinético de orden cero, como es el caso de una administración continua a velocidad constante (infusión intravenosa) y cuando la velocidad de eliminación desde el sistema es de tipo exponencial, la cantidad de fármaco tiende a acumularse hasta alcanzar un estado de equilibrio estacionario. Este equilibrio significa que la cantidad de fármaco en el sistema ha aumentado hasta un valor máximo, que se mantiene constante, lográndose un nivel constante o estacionario ("plateau"). En este estado estacionario, la velocidad de eliminación se iguala con la velocidad de entrada del fármaco al volumen de distribución.

El estado estacionario puede ser alterado si se modifica la velocidad a la cual se administra la infusión y por cambio de la constante de velocidad de eliminación o ambos parámetros. Cualquiera de éstos cambios da lugar a un nuevo estado estacionario.
Si se aplican estos principios a una administración intravenosa a velocidad constante, se tendrá un modelo como el esquematizado a continuación:
La ecuación diferencial que describe el proceso de cambio de Q en función del tiempo es:
Como la velocidad de eliminación se expresa por la concentración del fármaco en el compartimiento sanguíneo más que por la cantidad total de fármaco que contenga, la ecuación [2.44] se puede expresar en función de la concentración dividiendo ambos términos por el volumen de distribución:
Esta ecuación demuestra que con una infusión constante se debe alcanzar el "nivel estacionario" at infinito, ya que el término (1- ) para este tiempo se reduce a la unidad. En consecuencia:
donde y representan la cantidad y la concentración del fármaco en el estado estacionario, respectivamente.
Si en la ecuación [2.45] se reemplaza el valor de de la ecuación [2.47] se obtiene:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
ecuación en la cual C/ es la fracción del estado estacionario que se alcanza a un tiempo t. De esta manera, es posible calcular el tiempo en que se logra una fracción determinada del estado estacionario si se administra una infusión intravenosa. Si se representa el tiempo en función de vidas medias de eliminación, se obtendrá un gráfico como el de la figura 9.
La ecuación [2.49] puede expresarse también como:
Fig. 9. Acumulación de fármaco en el organismo durante la administración continua, a velocidad constante.
por lo que en el caso de una infusión intravenosa, una vez alcanzado el estado de equilibrio estacionario, al representar gráficamente la fracción que falta para alcanzar este estado, expresado en forma logarítmica en función del tiempo, se puede obtener la constante de velocidad de eliminación a partir de la pendiente de la recta que se origina, como lo indica la figura 10:
Todas estas ecuaciones tienen una aplicación práctica muy importante, ya que si se conoce Vd y K o la vida media biológica de un fármaco, puede calcularse el valor del nivel estacionario que se alcanzará con una velocidad de infusión conocida. También puede calcularse la velocidad de infusión para mantener un nivel terapéutico del fármaco dentro de una fracción del estado estacionario. Por ejemplo, si mediante ínfusion intravenosa se administra a un paciente lidocaina hasta alcanzar un nivel terapéutico efectivo de 3 mg/l de sangre y se desea mantener este nivel constante durante un tiempo determinado, puede calcularse

la velocidad de administración (ko) sabiendo que la vida media biológica de este fármaco es de 1, 7 h (K = 0,408 ) y que Vd es de alrededor de 120 1 en una persona de 70 Kg. Luego:
Q = Vd Q = 3 mg/1 x 120 1 = 360 mg
= QK = 0,408 x 360 mg = 147 mg/h o 2, 45 mg/min.
De modo que en este paciente, si se le administra una infusión a una velocidad de 2, 45 mg por minuto de lidocaina, al cabo de 10 vidas medias biológicas se habrá logrado el estado estacionario y éste se mantendrá constante durante todo el tiempo que dure la infusión. Si se quisiera lograr este nivel de 3 mg/1 en forma inmediata, se debe inyectar una dosis de 360 mg del fármaco por vía intravenosa rápida y mantener el nivel sanguíneo mediante una infusión a una velocidad de 2, 45 mg/min, lo cual resulta más práctico.
La ecuación [2.45] es útil también para calcular el volumen de distribución aparente de fármacos administrados mediante infusión intravenosa, conociendo K, ko y la concentración plasmática que se va alcanzando a diversos tiempos de infusión, ya que el gráfico de C versus (1 – ) es una recta cuya pendiente es ko/VdK, como se indica en la figura 11:
Al suspenderse la infusión, la concentración plasmática desciende exponencialmente, tal como sucede tras una inyección intravenosa instantánea. La ecuación que describe el proceso de eliminación es similar a la ecuación [2.6], solo que la concentración a tiempo inicial , pasa a ser :
ecuaciones en las cuales es el tiempo que transcurre desde la suspensión de la infusión. Si ésta se suspende antes de alcanzar el estado estacionario, la ecuación que describe la variación de C respecto al tiempo es:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
De manera que al suspender la infusión, dentro del estado estacionario o antes de alcanzar éste, la evolución sanguínea se realiza en forma exponencial y la constante de velocidad de eliminación puede obtenerse de la recta de la eliminación de primer orden del gráfico de concentración versus tiempo.
TERCERA UNIDAD UNDÉCIMA SESIÓN
TEMA 1: CÁLCULO DE PARÁMETROS FARMACOCINETICOS DE UNA ADMINSITRACIÓN EXTRAVASCULAR. METODO DE WAGNER Y NELSON

Editado de : Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado
desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
2. Método de Wagner y Nelson
Uno de los métodos que, indiscutiblemente, ha alcanzado mayor difusión por su fácil aplicación es el método ideado por Wagner y Nelson (17.18) para calcular la constante de velocidad de absorción y la cantidad de fármaco absorbida después de su administración oral.
Si se considera que la cantidad total de un fármaco absorbida a un tiempo t es igual a lo que se tiene en el cuerpo más la que se ha eliminado por metabolismo, excreción urinaria u otros mecanismos, se tiene que:
A = Q +E
Luego, la variación de A se encuentra definida por:
área bajo la curva de absorción hasta el tiempo t. Cuando la absorción termina, adquiere un valor constante puesto que no queda más fármaco para ser absorbido y, por lo tanto, el valor máximo de la función es:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Si se representan los valores de en función de t, se obtiene una curva que adopta una forma asintótica en el momento en que termina el proceso de absorción, como lo indica en la figura 15.
La ecuación [2. 62] expresa la desaparición del fármaco desde el tracto gastrointestinal. En ella AD representa la cantidad inicial de fármaco en el tracto que puede ser absorbida o que está disponible para la absorción y que corresponde a la cantidad absorbida a tiempo infinito, esto es:
por lo que, efectuando los reemplazos respectivos en la ecuación [2. 85], se obtiene:
Luego, al representar el logaritmo de la cantidad de fármaco que queda por absorberse en función del tiempo, se obtiene una linea recta con una pendiente igual a , 303, de donde es posible calcular la constante de velocidad de absorción de primer orden, según puede concluirse de la figura 16.

F16. Cálculo de la constante de velocidad de absorción de primer orden, de acuerdo con el método de Wagner y Nelson.
a) Cálculo del área bajo la curva (ABC)
La ecuación de Wagner y Nelson está basada en el cálculo de las áreas bajo la curva de absorción, es decir, aquella área delimitada por el gráfico obtenida al representar la concentración plasmática en función del tiempo. En los estudios de biodisponibilidad es importante determinar exactamente estas áreas ya que, en virtud de la ley de Dost, (19) "la relación del área bajo la curva deconcentración sanguínea en función del tiempo, luego de una administración oral, y la que se obtiene después de una inyección intravenosa de la misma dosis del fármaco es una medida de la absorción del fármaco administrado".
Para la determinación del área bajo la curva pueden utilizarse varios métodos:
i) Los niveles sanguíneos o plasmáticos se representan en función del tiempo en papel milimétrico. Las curvas se cortan y se pesan en balanza analítica.
ii) Se determina mediante un planímetro, el área de las curvas dibujadas en papel milimétrico.
iii) El método de la "regla trapezoidal": la curva se divide en secciones que se aproximan a trapecios en su forma (Fig. 17) y se calcula el área de cada una de ellas mediante la fórmula siguiente:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Fig. 17. Cálculo del área bajo la curva por el método de los trapecios.
En el caso dé la figura 17, el área bajo la curva hasta tiempo seria:
El área bajo la curva se expresa mediante la concentración indicada en la ordenada multiplicada por el tiempo. En la figura 17 se ha expresado la concentración plasmática en mg/1 y el tiempo en horas, por lo que la dimensión del área bajo la curva será de mg/l X h.
Para una correcta interpretación de la cantidad absorbida, el ABC debe calcularse hasta tiempo infinito, ya que la fase de eliminación puede representar un valor importante del área total, especialmente en aquellos fármacos que poseen vidas medias de eliminación muy largas. Por lo general, los experimentos se detienen a tiempos anteriores a la desaparición total del fármaco de la sangre, por lo cual es preciso calcular el área desde el tiempo final del experimento hasta t = oo. Para realizar el cálculo hay que conocer el valor de la constante de velocidad de eliminación del fármaco. En el caso de una inyección intravenosa, el área bajo la curva se obtiene de la integración de la ecuación que describe la evolución de concentración plasmática en función del tiempo:
Esto significa que el área total bajo la curva, en una administración intravenosa rápida, se determina dividiendo la concentración inicial por la constante de velocidad de eliminación. Del

mismo modo, al dividir cualquier valor de concentración plasmática en la fase de eliminación, tras una administración oral, se obtiene el ABC desde ese punto hasta infinito. Por lo general, se utiliza el último punto experimental de la curva, , de modo que el área a es:
b) Aplicación de la ecuación de Wagner y Nelson
La ecuación de Wagner y Nelson puede aplicarse a los datos sobre concentración sanguínea de un fármaco administrado por cualquier vía extravascular. En el ejemplo presentado a continuación se han tomado los valores de concentración plasmática de salicilatos totales después de la administración rectal de aspirina a conejos. Los datos tabulados conforme a la ecuaci6nde Wagner y Nelson se consignan en la Tabla 1.
Tabla I (a) Tiempo (h)
(b)
( g /ml)
(c) (ABC) ( g/ml) x h
(d) K(ABC)
( g/ml)
0, 25 4, 497 0, 562 0, 053 4, 550 0, 50 8, 267 2, 158 0, 202 8, 469 1, 0 9, 705 6, 651 0, 623 10, 328 2, 0 10, 568 16, 788 1, 157 12, 139 3, 0 9, 693 26, 919 2, 520 12, 213 4, 0 8, 778 36, 155 3, 384 12, 162 5, 0 7, 991 44, 539 4, 169 12, 160 7, 0 6, 635 59, 165 5,538 12, 173
En esta tabla, la columna (b) representa la concentración plasmática medida a los tiempos indicados en (a); la columna (c) agrupa las áreas bajo la curva en forma acumulativa, calculadas por el método de los trapecios; y la columna (e) contiene la cantidad absorbida (b + d).
De acuerdo con los valores representados en la Tabla I, se puede concluir que a las dos horas el fármaco se absorbió totalmente o, por lo menos, ya no se absorbe más. El promedio de las 5 últimas horas para es de 12, 169. Sin embargo, la manera correcta de obtener el valor de la cantidad total absorbida es recurriendo al método de los mínimos cuadrados de los valores asintoticos, con el cual se consigue un valor de 12, 164. Si se representan los valores logarítmicos de ( ) versus tiempo, resulta una recta como se indica en la figura 18, de cuya pendiente se obtiene el valor de la constante de velocidad de absorción del fármaco en estudio.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
El área bajo la curva desde tiempo inicial hasta 7 h está dada por la columna (c): 59, 165 g / ml X h. El área desde 7 h hasta tiempo infinito se obtiene dividiendo 6, 635 g / ml, la concentración plasmática a las 7 h, por la constante de velocidad de eliminación calculada de la recta exponencial obtenida a partir de las 3 h en un gráfico de log versus t, cuyo valor es de 0, 0936
:
De modo que el área total bajo la curva es de 130, 051 g / ml x h.

TERCERA UNIDAD UNDÉCIMA SESIÓN:
TEMA 2. CÁLCULO DE PARÁMETROS FARMACOCINETICOS DE UNA ADMINSITRACIÓN
EXTRAVASCULAR. METODO DE RESIDUALES
Tomado de: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado
desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
Cuando se administra un fármaco por vía oral u otra vía que no sea la intravenosa, cabe suponer que se realiza una transferencia de un compartimento a otro por un proceso cinético de primer orden. La secuencia más simple de los procesos que ello entraña puede representarse por el esquema siguiente:
en el cual A representa el fármaco que está siendo absorbido desde el tracto gastrointestinal; Q el fármaco que aparece en la sangre; ka la constante de velocidad de absorción de primer orden; E y K tienen el mismo significado mencionado en los modelos anteriores.
La velocidad con que desaparece el fármaco desde el tracto gastrointestinal puede ser descrita mediante una expresión de primer orden:
Esto significa que la dosis administrada disminuye con el tiempo, en tanto que en la sangre la concentración va aumentando hasta llegar a un máximo para luego comenzar a decrecer en forma exponencial cuando toda la dosis es absorbida. En este caso, el paso desde el tracto gastrointestinal a la sangre es irreversible, por lo que para t=infinito, el valor en el primero es nulo ya que la transferencia es virtualmente completa.
La ecuación [2.78] integrada toma la forma:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
[2.79] o bien, en su forma logarítmica:
[2.80]
en las cuales At es la cantidad de fármaco que queda en el tracto gastrointestinal a tiempo t y A0 sería la cantidad inicialmente administrada, es decir, la dosis si ésta hubiera sido totalmente absorbida.
Estas ecuaciones expresan la disminución de A en función del tiempo y es muy difícil emplearlas para determinar la constante de velocidad de absorción ka y la cantidad que va quedando, por la imposibilidad de medir ésta en las condiciones normales de trabajo. Sólo es factible de emplearse en casos de administración de compuestos con trazadores radiactivos o en trabajos con animales, por ejemplo ratas, donde puede exponerse el estómago o el intestino, introducir una solución e ir evaluando la cantidad remanente en función del tiempo.
La variación del fármaco en el compartimento sanguíneo es más compleja, pues representa el balance entre lo que ingresa y lo que se elimina. Es decir, por un lado va llegando fármaco al torrente circulatorio, pero por otro lado va siendo eliminado por diferentes mecanismos. Sin embargo, la sangre tiene la ventaja de constituir un compartimento al cual se puede tener acceso: se toman muestras sanguíneas y se evalúa la cantidad de fármaco en cualquier momento del experimento.
La variación de la cantidad de fármaco en la sangre está dada por la ecuación siguiente:
[2.81]
Estos dos procesos, el de absorción y el de eliminación del fármaco, corresponden a procesos cinéticos de primer orden.
Al substituir en la ecuación [2.81] el valor de A dado en la ecuación [2.79], se obtiene:
[2.82]
La integración de esta ecuación conduce a:
[2.83]
Si se dividen ambos términos por el volumen de distribución aparente, se obtiene esta misma ecuación expresada en términos de concentración. Como A0 corresponde a la dosis administrada, es preciso tomar en cuenta que no toda ella puede ser absorbida, como se ha explicado anteriormente. Por ello, es más apropiado emplear el término "fracción de la dosis absorbida", FD. En una administración oral, esta fracción adquiere valores inferiores a 1, por las razones antes anotadas.
Conforme a estas consideraciones, la ecuación [2.83] puede expresarse de la manera siguiente:

[2.84]
La mayoría de las veces ka posee un valor que es de mayor magnitud que K, por lo que a través del tiempo, e-k
at adquiere un valor nulo y la ecuación [2.84] se reduce a:
[2.85]
Ecuación que puede expresarse en forma logarítmica como:
[2.86]
Esta última ecuación expresa la evolución de la concentración plasmática respecto al tiempo una vez que la absorción cesa y el único proceso de velocidad involucrado es el de eliminación, en tanto que la ecuación [2.84] representa todo el proceso de absorción y de eliminación. Esto se encuentra representado en la figura 2.13.
Fig. 2.13 Evolución de la concentración sanguínea de una fármaco que se introduce al organismo por vía extravascular por un proceso cinético de primer orden.
Si a la ecuación [2.75] se le resta la ecuación [2.84], se obtiene la concentración residual CR;
[2.87]
o bien:
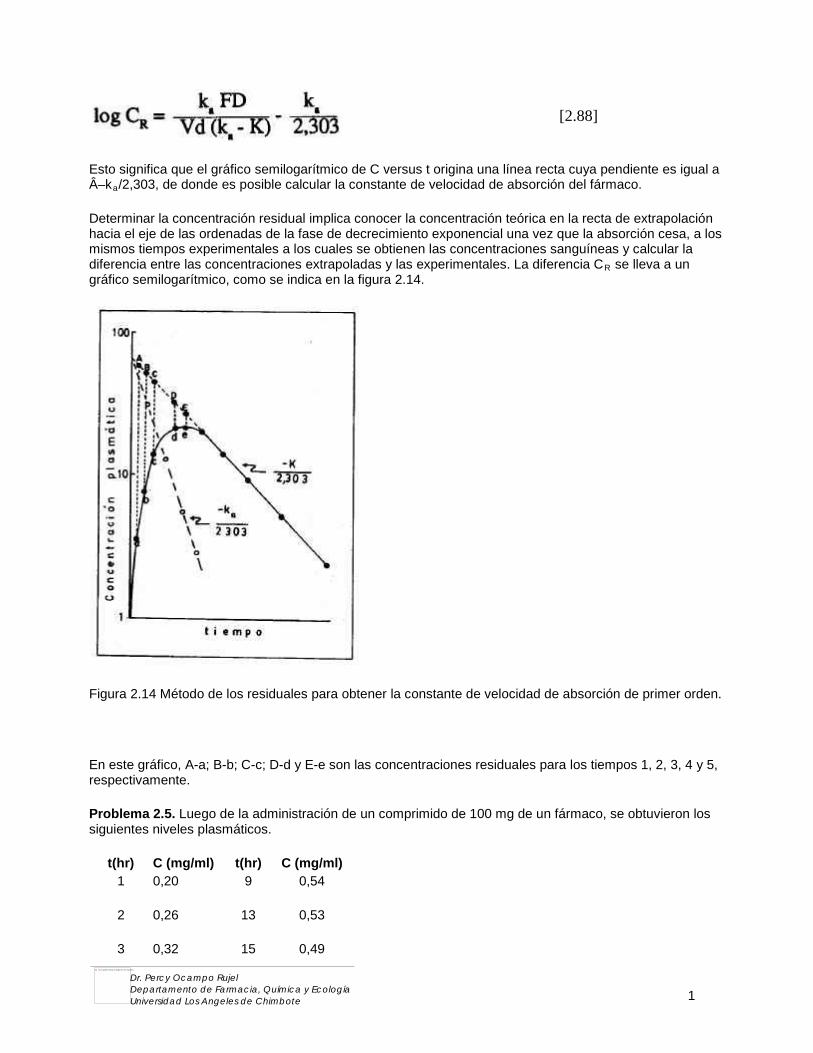
No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
[2.88]
Esto significa que el gráfico semilogarítmico de C versus t origina una línea recta cuya pendiente es igual a –ka/2,303, de donde es posible calcular la constante de velocidad de absorción del fármaco.
Determinar la concentración residual implica conocer la concentración teórica en la recta de extrapolación hacia el eje de las ordenadas de la fase de decrecimiento exponencial una vez que la absorción cesa, a los mismos tiempos experimentales a los cuales se obtienen las concentraciones sanguíneas y calcular la diferencia entre las concentraciones extrapoladas y las experimentales. La diferencia CR se lleva a un gráfico semilogarítmico, como se indica en la figura 2.14.
Figura 2.14 Método de los residuales para obtener la constante de velocidad de absorción de primer orden.
En este gráfico, A-a; B-b; C-c; D-d y E-e son las concentraciones residuales para los tiempos 1, 2, 3, 4 y 5, respectivamente.
Problema 2.5. Luego de la administración de un comprimido de 100 mg de un fármaco, se obtuvieron los siguientes niveles plasmáticos.
t(hr) C (mg/ml) t(hr) C (mg/ml) 1
2
3
0,20
0,26
0,32
9
13
15
0,54
0,53
0,49

5
7
0,42
0,48
18
22
25
0,40
0,30
0,21
Calcular la constante de velocidad de eliminación, la vida media de eliminación y la constante de velocidad de absorción.
Respuesta: La constante de disposición total (o de velocidad de eliminación) puede determinarse de la fase de eliminación exponencial, por ejemplo desde las 15 a las 25 horas.
Esto da un K = 0,083 hr-1
luego, t1/2 = 0693 = 8,35 hr ------------------ 0,083
En el gráfico de log C versus t, encontramos que los valores extrapolados son:
t(hr) C(mg/ml) Cextrap.(mg/ml) CR 1
2
3
5
7
0,2
0,20
0,32
0,42
0,48
1,61
1,48
1,36
1,16
0,97
1,41
1,22
1,04
0,74
0,49
Un gráfico de log CR versus t da una pendiente
m = 0,0764
luego ka = 0,0764 x 2,303 = 0,176 hr-1
Las ecuaciones [2.76] y [2.78] poseen el mismo intercepto. Sin embargo, cuando se administran formas farmacéuticas sólidas, como comprimidos, grageas o cápsulas, estos valores no suelen coincidir debido a una cesión retardada del fármaco y, por lo tanto, de su absorción, ya que se necesita cierto tiempo para que la absorción comience. Este tiempo de latencia, t0, puede determinarse sí sé conoce ka y K, a partir de la ecuación siguiente:
[2.89]
donde A es el intercepto de la ecuación [2.88] y B la de la ecuación [2.86].

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
En los casos donde se aprecia un tiempo de latencia, la ecuación que describe el proceso de absorción y eliminación de un fármaco administrado por vía oral o por cualquier otra vía que implique una absorción de primer orden es:
[2.90]
para t>t0
Si se conociera la fracción de la dosis absorbida se podría calcular el volumen de distribución del fármaco a partir del valor del intercepto (n), de la ecuación [2.86], ya que:
[2.91]
De la integración de la ecuación [2.74] de t=0 a t=infinito, se obtiene:
[2.92]
en la cual ∫0∞ Cdt es el área bajo la curva de concentración sanguínea o plasmática de t=0 a t=infinito.
Por lo tanto, el Vd es igual a:
[2.93]
Desafortunadamente, suele ser difícil determinar la fracción absorbida a meros que se comparen las áreas bajo la curva, luego de administrar una inyección intravenosa del fármaco. Por este motivo, en el mejor de los casos sólo se consiguen aproximaciones del volumen de distribución cuando se administra un fármaco por vía oral y se supone una absorción completa o casi completa como sucede en el caso de administrar una solución del fármaco.
Como lo habíamos expresado anteriormente ka, la mayoría de las veces es de mayor magnitud que K. En algunas oportunidades esto no se cumple y K es mayor que ka, lo cual da origen a un modelo llamado "flip-flop". Cuando esto ocurre, en un gráfico de concentración versus tiempo, la fase terminal representa la velocidad de absorción, en tanto que a través del método de los residuales se obtiene la constante de velocidad de disposición total del proceso. Esto lo vemos ejemplificado en la figura 2.15.

Fig. 2.15. Modelo "flip-flop".
Este tipo de molo puede ser identificado al administrar una dosis intravenosa y una oral. La constante de la fase terminal para la administración intravenosa no coincide con la obtenida de la fase terminal de la administración oral, pero sí es coincidente con la constante obtenida de acuerdo a los residuales.
TERCERA UNIDAD DUODECIMA SESIÓN
MODELO BICOMPARTIMENTAL
CÁLCULO DE PARÁMETROS FARMACOCINETICOS AL ADMINISTRAR UNA INYECCIÓN INTRAVENOSA RAPIDA (BOLUS)
Referencia: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
1. Análisis de la concentración de fármaco en la sangre.
Uno de los modelos más comunes, adaptable a la mayoría de los fármacos, es, el modelo de dos compartimentos. En este modelo, se ha supuesto que el fármaco se distribuye en dos compartimentos. Uno es conocido como compartimento central, representado por la sangre, el agua extracelular y los tejidos altamente irrigados como el corazón, los pulmones, los riñones, etc.; en este compartimento el fármaco se distribuye rápidamente. El segundo compartimento, conocido como compartimento periférico o tisular, contiene los tejidos que se equilibran más lentamente con el fármaco. Este modelo supone, además, una eliminación desde el compartimento central, como se representa en el esquema 5.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
En este modelo el fármaco, a consecuencia de una inyección intravenosa rápida, llega al compartimento central donde se distribuye con rapidez y luego. pasa al compartimiento periférico. La velocidad de distribución en este último compartimento es función de varios factores, tales como la velocidad de circulación sanguínea, el gran de irrigación y el coeficiente de reparto del fármaco entre los dos compartimientos.
Si se representa el logaritmo de la concentración plasmática en función del tiempo, se obtiene una curva de tipo biexponencial como lo muestra la figura 2.24.
Fig. 2.24. Gráfico del logaritmo de la concentración plasmática en función del tiempo de una fármaco que se distribuye según un modelo de dos compartimentos, luego de una dosis i.v.
La primera parte de la curva corresponde a la fase de distribución. Luego de un tiempo el fármaco logra el equilibrio entre el compartimento central y los tejidos menos irrigados y cuando este equilibrio se logra, la pérdida del fármaco desde el compartimento central se realiza de acuerda a un proceso cinético de primer orden, debido a todos los procesos de eliminación desde el cuerpo. Este proceso, más lento, corresponde a la fase de eliminación propiamente tal.
Esta curva biexponencial queda definida por la ecuación:
C = Ae -αt + Be -ßt -------------------------------------------------------[2.185]

Si esta curva se resuelve por el método de los residuales, se tiene qUe A y B son los respectivos interceptos de las rectas logarítmicas de distribución y de eliminación.
Las constantes α y β son constantes de la fase de distribución y de eliminación, respectivamente. Los inteceptos A y B son, realmente, constantes híbridas, donde se encuentran involucradas otras constantes y pueden expresarse por las ecuaciones siguientes:
-[2.186]
-[2.187]
donde Vc es volumen de distribución del compartimento central, k10, k12 y k21, son microconstantes que relacionan la cantidad de fármaco por unidad de tiempo entre los dos compartimentos. Los valores de estas microconstantes no pueden ser determinadas por medida directa puesto que no puede medirse la concentración del fármaco en el compartimento periférico en forma directa.
Los valores de A, B, α y β, pueden deducirse a partir de los gráficos correspondientes a los resultados experimentales. Las constantes k10, k12 y k21 están definidas por las siguientes relaciones:
Como se ha señalado, los valores de A, B, y pueden deducirse de la curva biexponencial representada por la figura 2.25:
a) , de la pendiente del tramo lineal del gráfico de concentración plasmática en función del tiempo, ya que al alcanzarse el equilibrio de distribución, la ecuación [2.175] adopta la forma siguiente:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
C = B e - t -------------------------------------------------------------------------------[2.196]
lo cual significa que la expresión semilogarítmica de las concentraciones plasmáticas en función del tiempo corresponde a un gráfico cuyo tramo terminal es una línea recta con pendiente - / 2,303.
b) B, de la intersección de la recta obtenida anteriormente en la fase de eliminación, con el eje de las ordenadas, de acuerdo a la ecuación [2.196].
c) la diferencia entre las ecuaciones,[2.185] y [2.196] proporciona los valores de concentración residual correspondiente a los tiempos anteriores al equilibrio entre los compartimentos:
CR = A e - t --------------------------------------------------------------------------------------[2.197]
por lo que un gráfico del logaritmo de la concentración residual (CR) en función de t origina una recta cuya pendiente es - /2,303.
d) de la intersección de la recta anterior en el eje de las ordenadas, se obtiene A.
La constante β, que algunos autores mencionan como la constante de velocidad lenta del proceso, no es la constante de velocidad de eliminación como es corriente encontrarla en algunos trabajos. Según Riegelman y colaboradores (23) es la constante de velocidad de disposición, ya que implica los procesos de distribución y eliminación. Para ilustrar la relación entre la verdadera constante de velocidad de eliminación kl0 y β, basta tomar la ecuación [2.192] y expresarla en la forma siguiente:
k10 ---= --C0 ----------------------------------------------------------------[2.198] A/ +-- B/
La constante describe la desaparición del fármaco desde el compartimento central por excreción o por metabolismo o por distribución a otro compartimento. En cambio la constante k1o describe la eliminación del fármaco desde el compartimento central. Por este motivo, la vida media de eliminación o vida media biológica en un modelo de dos compartimentos es igual a 0,693/ β.
El valor de la relación A/ es, a menudo despreciable en relación con la magnitud de B/ y la ecuación [2.198]puede reducirse a la expresión:
-[2.199]
La relación CO/ β varía de un fármaco a otro, pero a menudo se sitúa entre valores de 1,5 a 2,5 . Sin embargo, en fármacos que se distribuyen en gran proporción fuera del compartimento central, suelen encontrarse constantes de proporcionalidad mucho más grandes. Por ejemplo, la digoxina posee un k10 por lo menos 15 a 40 veces más grande que (24).
Nivel de fármaco en el compartimento periférico
La ecuación diferencial que describe la velocidad de cambio de la cantidad de fármaco en el compartimento periférico puede representarse por:

[2.200] ecuación que por integración lleva a:
[2.201]
que describe la evolución de la cantidad de fármaco en el compartimento periférico después de una administración i.v.. De acuerdo con esta ecuación, existe una fase rápida descrita por la constante α qué corresponde a la entrada del fármaco al compartimento periférico. Como, por definición α >>β, el término e -αt tiende a cero y la ecuación se reduce a:
[2.202]
luego, la pendiente de la fase terminal en un gráfico de log P en función de t es igual a - /2,303. Esto quiere decir que en la fase de postdistribución la cantidad de fármaco en el plasma y en el compartimento periférico declinan en forma paralela.
2. A partir de dates de excreción urinaria.
Si se considera el esquema 5, la constante de eliminación desde el compartimento central, k10, es la suma de las constantes individuales de velocidad que. caracterizan otros procesos paralelos de eliminación. La velocidad de excreción del fármaco intacto por la orina puede representarse por la ecuación:
[2.203]
donde ke es la constante de velocidad de 9ccreción urinaria del fármaco intacto o no metabolizado. Como el valor de Q puede deducirse de las ecuaciones [2.185], [2.1861] y [2.187], resulta:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Las ecuaciones (2.203) y (2.205) expresan la velocidad de excreción urinaria del fármaco no metabolizado. El análisis de esta expresión revela que la velocidad de excreción aumenta, alcanza un máximo y luego disminuye exponencialmente. Esto último sucede cuando a un tiempo finito, e - t tiende a un valor nulo, ya que >> . La figura 2.25 representa esta situación e indica que puede calcularse de la fase de decrecimiento exponencial lograda una vez que se alcanza el equilibrio de distribución.
Fig. 2.25. Velocidad de excreción urinaria en función del tiempo medio entre los intervalos de recolección de muestras de orina en un modelo de dos compartimentos.
La constante a puede obtenerse aplicando la técnica de los residuales como se indica también en la figura 2.25. De esta misma figura se obtienen las ordenadas en el origen A' y B', cuya suma conduce a:
[2.209]

y finalmente a:
[2.209]
o bien:
[2.209]
de modo que, como se conoce la dosis inyectada, Q0, se puede calcular la contante de velocidad de excreción urinaria del fármaco no metabolizado, k. Las restantes constantes también pueden calcularse con los datos obtenidos de la figura 2.25, ya que:
[2.212]
y k12 se obtiene de acuerdo con la ecuación [2.194]
Otro método aplicable a la farmacocinética de fármacos excretados por la orina en forma no metabolizada es el método de la cantidad que queda por ser eliminada, similar al descrito en el modelo de un compartimento. La ecuación [2.176] por integración conduce a:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Tenemos así, una curva biexponencial representada por la figura 2.26 que expresa la cantidad de fármaco que queda por eliminarse en función del tiempo. La fase de distribución queda indicada por la primera parte de la curva y luego, cuando e- t alcanza un valor nulo, origina una recta cuya pendiente es - /2,303, de donde puede obtenerse el valor de .
Fig. 2.26 Gráfico semilogarítmico de la cantidad de fármaco no metabolizado que permanece sin excretarse en función del tiempo después de una administración i.v., en un modulo de dos compartimentos.
Si se aplica el método de los residuales, se obtendrá el valor de y A", mientras que B" se obtendrá de la extrapolación de la recta de la fase de postdistribución.
La suma de A" y B" da el valor de de E
Las constantes k12 y k21 se obtienen de las ecuaciones (2.194) y (2.195), respectivamente.
2.2.3.1.3 CALCULO DE LA CONSTANTE DE VELOCIDAD DE ABSORCION EN UN MODELO DE DOS COMPARTIMENTOS.
El modelo que representa esta situación farmacocinética es esencialmente el indicado por el esquema

siguiente:
La constante de absorción en la mayoría de los fármacos administrados en forma fácil de absorber es mayor que la velocidad de eliminación y, como por definición >> , en algún momento los términos e-k t y e- t se aproximan a cero y la ecuación [2.232] se reduce a:
C = A2 e- t -------------------------------------------------------------------------[2.236]
De ahí que el gráfico del logaritmo de la concentración plasmática en función del tiempo represente una curva triexponencial cuyo segmento terminal es rectilíneo. Esta curva queda expresada en [2.236], de donde puede obtenerse el valor de , ya que la pendiente de esta recta es – /2,303 y el valor de A2 se obtiene de la ordenada en el origen. Por el método de los residuales es posible obtener A1, A3 y ka . Sin embargo, para muchos fármacos, ka, tiene una magnitud similar a α, de modo que la fase de distribución no se observa después de una administración oral y las curvas de LogC versus t suelen aparentar ser bioexponenciales; es decir, estas curvas dan la impresión de que el fármaco se absorbe conforme a un modelo de dos compartimentos. Por este motivo, en algunos casos es imprescindible recurrir a la inyección intravenosa para visualizar la fase a ya que ésta no puede manifestarse por el método de los residuales debido a que se encuentra enmascarada por k;
De la integración de la ecuación [2.202] entre los límites de tiempo t=O y t= obtiene el área total bajo la curva de concentración plasmática versus tiempo:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
A. METODO DE LOO Y RIEGELMAN
Es una extensión del método de Wagner y Nelson (11) visto para un modelo monocompartimental. Tal como en aquel método, no existen limitaciones en cuanto al orden del proceso de absorción. Sin embargo, este método requiere que el fármaco sea administrado previamente en forma intravenosa con el objeto de determinar las constantes de intercambio.
La ecuación de Loo y Riegelmann (29) se expresa corrientemente por:
donde Ct y Cp representan las concentraciones en el comportamiento central y el comportamiento periférico, respectivamente. CP se evalúa a base de la ecuación diferencial que representa la velocidad de intercambio con el comportamiento periférico:
Como k12 y k21 pueden determinarse a partir de los datos de concentración plasmática,
si ésta se expresa en función del tiempo, la solución de la ecuación [2.241] se obtiene suponiendo que entre dos intervalos de tiempo, t n y t n-1, la curva de nivel plasmático es aproximadamente recta y, por lo tanto, es posible sustituir C por Ctn basándose en la ecuación de la línea recta:

t=t luego el área hasta infinito dividiendo la última concentración obtenida por B, Finalmente, se estima el logaritmo del porcentaje de fármaco no absorbido, el cual se lleva a un gráfico en función del tiempo calculándose la constante de absorción desde la pendiente de la recta obtenida.
En el ejemplo dado en las tablas 2.4 y 2.5, se ilustra el caso de un producto hipotético, administrado en una dosis de 500 mg y en el cual una inyección intravenosa originó las siguientes constantes:
K10 = 0,15 hr -1 ; k12 = 0,28 hr -1 y k2l = 0,33 hr -1
La tabla 2.4 indica la serie de cálculos realizados a partir de los datos sanguíneos de la columna 2, a los tiempo de la columna 1, para obtener la concentración en el compartimento periféricos para cada tiempo (Cp)tn.
Luego, el método prosigue de la misma manera que el método de Wagner y Nelson, calculando las áreas bajo la curva para cada tiempo, multiplicándolas por k10 y sumándole Ct, y (CP)tn para cada tiempo como se ejemplifica en la tabla 2.5.
El método de Loo y Riegelmann se puede usar para fármacos que presentan una farmacocinética lineal en un modelo multicompartimental en el cual se supone que la eliminación tiene lugar exclusivamente desde el compartimento central. Si bien tal suposición puede ser correcta para la mayoría de los fármacos, en otros, esta aproximación no puede ser la adecuada. Además, este método está basado en la suposición de que los parámetros farmacocinéticos son los mismos durante una administración intravenosa y una oral, lo cual constituye una restricción en el sentido de que para aplicar el método de Loo y Riegelman es preciso contar con los parámetros obtenidos por vía intravenosa. Por otra parte, la ecuación de Loo y Riegelman proporciona solamente valores aproximados de At/Vd, ya que supone un segmento lineal entre dos concentraciones y tiempo ( C y t).
B. METODO DE WAGNER PARA DOS COMPARTIMENTOS
Wagner (30), en un intento por subsanar esta restricción, desarrolló un método que permitiría aplicar el método de Wagner y Nelson. Posteriormente (31), desarrolló ecuaciones de absorción exactas para los casos en que la disposición de los fármacos es descrita por uno, dos o tres términos exponenciales y de

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
aplicabilidad más simple que el método de Loo y Riegelman, aún cuando para los modelos multicompartimentales es igualmente necesario contar con los datos farmacocinéticos obtenidos por una administración intravenosa.
La ecuación para un modelo bicompartimental es:
Tabla 2.4. Ejemplo de cálculo de la concentración de fármaco en el compartimento periférico.
Tabla 2.5. Cálculo de la Fracción no absorbida.
Tabla 2.6. Ejemplo de la aplicación del Método de Wagner para un modelo de dos compartimentos.
El tercer término de la ecuación [2.244] es una expresión exacta para la obtención de (Cp)tn y reemplaza a la ecuación [2.241]. Cabe hacer notar que la segunda integral lleva el término k21t con signo positivo.
En la tabla 2.6 se realiza un ejercicio aplicando esta ecuación modificada de Wagner.
La última columna de la tabla 2.6 representa la fracción no absorbida al tiempo de la primera columna. El logaritmo de la fracción no absorbida versus tiempo origina una recta con pendiente negativa de donde puede obtenerse la constante de velocidad de absorción, ka, que en este ejemplo es de 0,458 hr-1.
C. METODOS DE CONVOLUCION Y DECONVOLUCION.
Los estudios de biodisponibilidad, especialmente, determinan la velocidad de entrada del fármaco al organismo por un proceso de absorción, la mayoría de las veces, de primer orden.
Aparte de los métodos mencionados anteriormente, se han descrito numerosos métodos para evaluar la absorción como un factor de entrada ("input"), los cuales pueden catalogarse como métodos modelo dependientes y modelo independientes.
Los métodos modelo dependientes son los que hemos analizado hasta ahora, los cuales se ajustan a un modelo específico para la absorción. Por ej. absorción de orden cero o de primer orden y modelos mono o multicompartimentales. Estos métodos han sido objeto de críticas frecuentes ya que no permiten obtener estimaciones confiables de las cinéticas de entrada o de disposición debido a que suponen procesos simples, a veces, lejos de la realidad.
Los modelos independientes presentan una mayor flexibilidad puesto que no se ajustan a modelo alguno ni a tipo de cinética en particular y se basan en un análisis matemático de sistemas lineales. Esta última característica los hace ser más complejos ya que es necesario un manejo matemático complicado y los métodos de análisis de datos son altamente sensibles al error que presentan los datos.
Si tenemos las funciones siguientes : P, Q y G, que representan respectivamente las funciones de "entrada", de "salida" y la respuesta característica del sistema, y suponemos que Q(t)=0 para t<0, las tres funciones están relacionadas por la integral de convolución:

Los métodos de deconvolución para resolver la ecuación [2.245] constituyen una herramienta para determinar la liberación y absorción de fármacos, en los casos que exista una administración intravenosa o una dosis administrada por una vía extravascular (corrientemente oral).
El término "entrada" al cual nos hemos referido, indica la velocidad a la cual una substancia entra al sistema en un punto particular. La entrada puede provenir de una fuente externa (forma farmacéutica) o de un proceso metabólico "in vivo". El término "respuesta" está abierto a un amplio rango de interpretaciones, dependiendo de la propiedad particular del sistema interesado. Una posible respuesta es la concentración sanguínea o plasmática de una substancia o un metabolito de esa substancia, la velocidad de excreción urinaria de una substancia o de un metabolito de esa substancia, o bien, puede tratarse de una respuesta farmacológica.
El problema es estimar la función P(t) cuando se dispone de los datos experimentales G(t) y Q(t). Este procedimiento es el que se denomina corrientemente como "deconvolución numérica".
Aún cuando G(t) y Q(t) son conocidos analíticamente, la integral definida por la ecuación {2.245] no puede ser resuelta, excepto en ciertos casos, para P(t), a través de las transformadas de Laplace:
q(s) = p(s) g(s) -----------------------------------------------------------------------------------------------------------[2.246]
cuya solución es: p(s) = y(s) -----------------------------------------------------------------------------------------------------------------[2.247] -- - - - g(s)
donde p(s), q(s) y g(s) son las transformadas de Laplace para P(t) y G(t), respectivamente.
Vaughan y Dennis (32) han empleado una función de entrada tipo "escalera", en un conjunto de pulsos rectangulares de duración Pj – Pj-1 y de intensidad Ij, comenzando cada uno a t = pj-1.
La deconvolución permite calcular la entrada al sistema (porcentaje de absorción) y la convolución, operación inversa, permite determinar la función respuesta a partir de una función de entrada (por ej. el porcentaje disuelto "in vitro").
Por otra parte, Cutler (33-35) ha propuesto una deconvolución numérica mediante los cuadrados menores, que se caracteriza por emplear un operador lineal (L) conocido, sobre la función de entrada para obtener la función de salida.
Si tenemos una unidad de dosis de una substancia de interés, situada en un punto particular a tiempo t=0, G(t) es la respuesta resultante, la cual se denomina "unidad de respuesta al impulso". Si desearnos calcular la velocidad de absorción , el impulso "unidad de dosis" requerido para la aplicación de la ecuación [2.245] es la dosis introducida a la circulación sanguínea.
La aplicación farmacocinética de P(t) y G(t) requiere una especial atención. En la práctica la entrada es, generalmente, especificada de antemano y la unidad impulso respuesta es determinada de la respuesta a una entrada conocida. El cálculo se simplifica cuando P(t) se efectúa a una velocidad constante.
Süverkrüp y cols. (36) han comparado algunos métodos de deconvolución, como el método trapezoidal, el de balance de material y el de los cuadrados menores Trabajos de esta naturaleza han realizado también Chan y Gibaldi (37).

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
TERCERA UNIDAD DECIMOTERCERA SESIÓN
EL MODELO ABIERTO DE DOS COMPARTIMENTOS
Referencia: Aquiles Arancibia y col. Fundamentos de Farmacia Clínica. Universidad de Chile. Biblioteca Digital de la Universidad de Chile. Tomado desde: http://mazinger.sisib.uchile.cl/repositorio/lb/ciencias_quimicas_y_farmaceuticas/cide01/cap2/2-2-1-1.html
2.2.3.1.1. CALCULO DE PARAMETROS A PARTIR DE UNA INYECCION INTRAVENOSA RAPIDA
1. Análisis de la concentración de fármaco en la sangre.
Uno de los modelos más comunes, adaptable a la mayoría de los fármacos, es, el modelo de dos compartimentos. En este modelo, se ha supuesto que el fármaco se distribuye en dos compartimentos. Uno es conocido como compartimento central, representado por la sangre, el agua extracelular y los tejidos altamente irrigados como el corazón, los pulmones, los riñones, etc.; en este compartimento el fármaco se distribuye rápidamente. El segundo compartimento, conocido como compartimento periférico o tisular, contiene los tejidos que se equilibran más lentamente con el fármaco. Este modelo supone, además, una eliminación desde el compartimento central, como se representa en el esquema 5.
En este modelo el fármaco, a consecuencia de una inyección intravenosa rápida, llega al compartimento central donde se distribuye con rapidez y luego. pasa al compartimiento periférico. La velocidad de distribución en este último compartimento es función de varios factores, tales como la velocidad de circulación sanguínea, el gran de irrigación y el coeficiente de reparto del fármaco entre los dos compartimientos.
Si se representa el logaritmo de la concentración plasmática en fondón t3d tiempo, se obtiene una curva de tipo biexponencial como lo muestra la figura 2.24.

Fig. 2.24. Gráfico del logaritmo de la concentración plasmática en función del tiempo de una fármaco que se distribuye según un modelo de dos compartimentos, luego de una dosis i.v.
La primera parte de la curva corresponde a la fase de distribución. Luego de un tiempo el fármaco logra el equilibrio entre el compartimento central y los tejidos menos irrigados y cuando este equilibrio se logra, la pérdida del fármaco desde el compartimento central se realiza de acuerda a un proceso cinético de primer orden, debido a todos los procesos de eliminación desde el cuerpo. Este proceso, más lento, corresponde a la fase de eliminación propiamente tal.
Esta curva biexponencial queda definida por la ecuación:
C = Ae -αt + Be -ßt -------------------------------------------------------[2.185]
Si esta curva se resuelve por el método de los residuales, se tiene qUe A y B son los respectivos interceptos de las rectas logarítmicas de distribución y de eliminación.
Las constantes α y β son constantes de la fase de distribución y de eliminación, respectivamente. Los inteceptos A y B son, realmente, constantes híbridas, donde se encuentran involucradas otras constantes y pueden expresarse por las ecuaciones siguientes:
-[2.186]
-[2.187]
donde Vc es volumen de distribución del compartimento central, k10, k12 y k21, son microconstantes que relacionan la cantidad de fármaco por unidad de tiempo entre los dos compartimentos. Los valores de estas microconstantes no pueden ser determinadas por medida directa puesto que no puede medirse la concentración del fármaco en el compartimento periférico en forma directa.
Los valores de A, B, α y β, pueden deducirse a partir de los gráficos correspondientes a los resultados experimentales. Las constantes k10, k12 y k21 están definidas por las siguientes relaciones:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Como se ha señalado, los valores de A, B, y pueden deducirse de la curva biexponencial representada por la figura 2.25:
a) , de la pendiente del tramo lineal del gráfico de concentración plasmática en función del tiempo, ya que al alcanzarse el equilibrio de distribución, la ecuación [2.175] adopta la forma siguiente:
C = B e - t -------------------------------------------------------------------------------[2.196]
lo cual significa que la expresión semilogarítmica de las concentraciones plasmáticas en función del tiempo corresponde a un gráfico cuyo tramo terminal es una línea recta con pendiente - / 2,303.
b) B, de la intersección de la recta obtenida anteriormente en la fase de eliminación, con el eje de las ordenadas, de acuerdo a la ecuación [2.196].
c) la diferencia entre las ecuaciones,[2.185] y [2.196] proporciona los valores de concentración residual correspondiente a los tiempos anteriores al equilibrio entre los compartimentos:
CR = A e - t --------------------------------------------------------------------------------------[2.197]
por lo que un gráfico del logaritmo de la concentración residual (CR) en función de t origina una recta cuya pendiente es - /2,303.
d) de la intersección de la recta anterior en el eje de las ordenadas, se obtiene A.
La constante β, que algunos autores mencionan como la constante de velocidad lenta del proceso, no es la constante de velocidad de eliminación como es corriente encontrarla en algunos trabajos. Según Riegelman y colaboradores (23) es la constante de velocidad de disposición, ya que implica los procesos de distribución y eliminación. Para ilustrar la relación entre la verdadera constante de velocidad de

eliminación kl0 y β, basta tomar la ecuación [2.192] y expresarla en la forma siguiente:
k10 ---= - -C0 ----------------------------------------------------------------------------------------------[2.198] A/ α +-- B/
La constante B describe la desaparición del fármaco desde el compartimento central por excreción o por metabolismo o por distribución a otro compartimento. En cambio la constante k1o describe la eliminación del fármaco desde el compartimento central. Por este motivo, la vida media de eliminación o vida media biológica en un modelo de dos compartimentos es igual a 0,693/ β.
El valor de la relación A/ es, a menudo despreciable en relación con la magnitud de B/ y la ecuación [2.198]puede reducirse a la expresión:
-[2.199]
La relación CO/ β varía de un fármaco a otro, pero a menudo se sitúa entre valores de 1,5 a 2,5 . Sin embargo, en fármacos que se distribuyen en gran proporción fuera del compartimento central, suelen encontrarse constantes de proporcionalidad mucho más grandes. Por ejemplo, la digoxina posee un k10 por lo menos 15 a 40 veces más grande que (24).
Nivel de fármaco en el compartimento periférico
La ecuación diferencial que describe la velocidad de cambio de la cantidad de fármaco en el compartimento periférico puede representarse por:
[2.200] ecuación que por integración lleva a:
[2.201]
que describe la evolución de la cantidad de fármaco en el compartimento periférico después de una administración i.v.. De acuerdo con esta ecuación, existe una fase rápida descrita por la constante α qué corresponde a la entrada del fármaco al compartimento periférico. Como, por definición α >>β, el término e -αt tiende a cero y la ecuación se reduce a:
[2.202]
luego, la pendiente de la fase terminal en un gráfico de log P en función de t es igual a - /2,303. Esto quiere decir que en la fase de postdistribución la cantidad de fármaco en el plasma y en el compartimento periférico declinan en forma paralela.
2. A partir de dates de excreción urinaria.
Si se considera el esquema 5, la constante de eliminación desde el compartimento central, k10, es la suma de las constantes individuales de velocidad que. caracterizan otros procesos paralelos de eliminación. La velocidad de excreción del fármaco intacto por la orina puede representarse por la ecuación:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
[2.203]
donde ke es la constante de velocidad de 9ccreción urinaria del fármaco intacto o no metabolizado. Como el valor de Q puede deducirse de las ecuaciones [2.185], [2.1861] y [2.187], resulta:
Las ecuaciones (2.203) y (2.205) expresan la velocidad de excreción urinaria del fármaco no metabolizado. El análisis de esta expresión revela que la velocidad de excreción aumenta, alcanza un máximo y luego disminuye exponencialmente. Esto último sucede cuando a un tiempo finito, e - t tiende a un valor nulo, ya que >> . La figura 2.25 representa esta situación e indica que puede calcularse de la fase de decrecimiento exponencial lograda una vez que se alcanza el equilibrio de distribución.

Fig. 2.25. Velocidad de excreción urinaria en función del tiempo medio entre los intervalos de recolección de muestras de orina en un modelo de dos compartimentos.
La constante a puede obtenerse aplicando la técnica de los residuales como se indica también en la figura 2.25. De esta misma figura se obtienen las ordenadas en el origen A' y B', cuya suma conduce a:
[2.209]
y finalmente a:
[2.209]
o bien:
[2.209]
de modo que, como se conoce la dosis inyectada, Q0, se puede calcular la contante de velocidad de excreción urinaria del fármaco no metabolizado, k. Las restantes constantes también pueden calcularse con los datos obtenidos de la figura 2.25, ya que:
[2.212]
y k12 se obtiene de acuerdo con la ecuación [2.194]
Otro método aplicable a la farmacocinética de fármacos excretados por la orina en forma no metabolizada es el método de la cantidad que queda por ser eliminada, similar al descrito en el modelo de un compartimento. La ecuación [2.176] por integración conduce a:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Tenemos así, una curva biexponencial representada por la figura 2.26 que expresa la cantidad de fármaco que queda por eliminarse en función del tiempo. La fase de distribución queda indicada por la primera parte de la curva y luego, cuando e- t alcanza un valor nulo, origina una recta cuya pendiente es - /2,303, de donde puede obtenerse el valor de .
Fig. 2.26 Gráfico semilogarítmico de la cantidad de fármaco no metabolizado que permanece sin excretarse en función del tiempo después de una administración i.v., en un modulo de dos compartimentos.
Si se aplica el método de los residuales, se obtendrá el valor de y A", mientras que B" se obtendrá de la extrapolación de la recta de la fase de postdistribución.
La suma de A" y B" da el valor de de E

Las constantes k12 y k21 se obtienen de las ecuaciones (2.194) y (2.195), respectivamente.
DECIMO CUARTA SESIÓN
BASES FARMACOCINÉTICAS PARA EL DISEÑO DE UNA PAUTA TERAPÉUTICA 1. CONCEPTO DE PAUTA TERAPÉUTICA:
Una vez conocidos los parámetros farmacocinéticos (fundamentalmente volumen de distribución y vida media) estamos en condiciones de diseñar una pauta terapéutica (dosis e intervalo de administración) que nos permita alcanzar una concentración plasmática en equilibrio estacionario dentro de un rango terapéutico adecuado:

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
2. RANGO TERAPÉUTICO
Es aquel intervalo de concentraciones plasmáticas comprendido entre la concentración mínima eficaz (aquélla por encima de la cual se inicien los efectos terapéuticos) y la concentración mínima tóxica (aquélla por encima de la cual empiezan a observarse efectos tóxicos).
3. ACUMULACIÓN DE FÁRMACOS:
Los fármacos son habitualmente prescritos para tomarlos en dosis fijas y a intervalos también fijos. Por ej.: 100 mg diarios o 25 mg, 3 veces al día. En asociación con este tipo de administración intermitente, los niveles en el organismo fluctúan y se van acumulando, hasta que en un determinado momento alcanzan equilibrio estacionario (e.e.). se define como equilibrio estacionario aquella concentración en la que el organismo absorbe y elimina la misma concentración en cada intervalo entre dosis.
El equilibrio estacionario es independiente de la dosis administrada y se alcanza siempre y para todos los fármacos tras un periodo de administración de 5 veces la vida media.
Durante el equilibrio estacionario existen oscilaciones (en administración oral) que dependen de la velocidad de eliminación y del intervalo entre dosis, que se calculan:

4. DOSIS DE ATAQUE Y DOSIS DE MANTENIMIENTO.
La dosis administrada de un fármaco está habitualmente calculada para que su concentración en equilibrio estacionario coincida con el rango terapéutico. Por lo tanto, durante el tiempo que tarda en llegar el fármaco hasta el equilibrio estacionario estará en rango infraterapéutico, por lo que habrá que administrar una dosis de ataque superior a la de mantenimiento con objeto de llegar desde el primer momento al rango adecuado.
La relación entre la dosis de ataque y la dosis de mantenimiento va a depender del intervalo de administración y de la vida media. Como norma general se puede calcular mediante la siguiente fórmula:
Donde P es igual al intervalo de administración dividido entre la vida media (cuando ambos coinciden P = 1).
5. DEBEMOS DISTINGUIR 3 GRANDES GRUPOS DE FÁRMACOS:
- Aquéllos cuya vida media oscila entre 4 y 24 h
- Aquéllos cuya vida media es inferior a 4 h
- Aquéllos cuya vida media es superior a 24 h

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
En los fármacos de vida media entre 4 y 24 horas intentaremos hacer coincidir la vida media con el intervalo de administración (P = 1) y daremos una dosis de ataque si hace falta.
Los intervalos pueden ser mayores que la vida media si el rango terapéutico lo permite.
En el grupo de fármacos de vida media corta la pauta anterior es a menudo impracticable, sobre todo si su índice terapéutico es pequeño. Cuando estas dos circunstancias concurren tenemos que recurrir a administrar el fármaco mediante una infusión continua, como ocurre por ej. con la heparina. Si el índice terapéutico es grande, hecho que sólo ocurre con algunos antibióticos, podemos recurrir a dar dosis mucho mayores que las requeridas.
En los fármacos con vida media larga (superior a 24 horas) la administración debe ser diaria para evitar fallos en el cumplimiento terapéutico, siendo necesario con frecuencia dar una dosis de ataque para alcanzar pronto los niveles terapéuticos.
6. SERIE DE DOSIS INDIVIDUALES.


No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
6. ACUMULACIÓN DE FÁRMACOS EN EL ORGANISMO
El término acumulación, según una definición cualitativa, es: " la cantidad de fármaco en el organismo, después de la administración repetida de una dosis, es más alta que luego de la administración de una sola dosis". De acuerdo con esta definición, sólo es posible la acumulación después de una administración repetida de un fármaco.
Wagner (23) expresa la acumulación mediante el "índice de acumulación", :

Como se ha señalado antes, en un régimen de dosificación en el cual se administra la mitad de la dosis inicial a intervalos iguales a la vida media biológica del fármaco, se obtendrá un nivel estacionario. En este caso, = 1, 443, de modo que cualquier régimen de dosificación en el cual sea mayor que esta cifra, producirá una acumulación del fármaco en el organismo.
El índice de acumulación permite conocer la extensión de la acumulación de un fármaco conforme a un régimen propuesto o la magnitud del cambio si se varía el régimen. Por ejemplo, si se administra un fármaco que tiene una vida media biológica de 24 h (K = 0, 0289 ) y se repite la dosis cada 24 h, el es 1, 443. En cambio, si se repite la dosis cada 8 h, el es 4, 32, lo cual significa que la acumulación se triplica con este último régimen.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
ANEXO
EJERCICIOS PROPUESTOS
Instrucciones generales 1.- Uso del papel semilogarítmico El papel semilogarítmico representa una herramienta de uso muy simple para identificar modelos compartimentales. Usted también puede identificar los modelos utilizando un programa Excel. Sin embargo, el uso del papel no requiere de un computador, por ello, en todos los libros de Farmacocinética se encuentran gráficos hechos con este papel. Este papel presenta una distribución lineal en el eje de las abscisas y una logarítmica (en base 10 es el papel del que Ud. dispone) en el eje de las ordenadas. El papel presenta los datos experimentales (en forma visual) en el punto que ocuparía si Ud. hubiese aplicado logaritmo al número, pero en vez de poner el valor del logaritmo, pone el valor original. Ej: si Ud. necesita el log10, en vez de poner 1 en el eje, pone 10 (el valor original) pero la distribución de la escala la hace aparecer visualmente como si hubiese aplicado logaritmo. Si se fija en la ordenada, los números están ubicados en ciclos de 10. La convención que debe seguir es respetar un ciclo de 10 de diferencia entre uno y otro. Ud. deci de cual es el valor de inicio y de ahí en adelante tiene que asignarle a cada ciclo una potencia de 10 superior al anterior. Ej: 1-10-100 o 0.1-1-10 o 10-100-1000 Mediante este sistema, se hace más fácil trabajar con los números, ya que se usan las concentraciones originales y se evita trabajar con números de a lo menos 3 cifras significativas, como sería si Ud. quisiera trabajar con los logaritmos.
2.- En los siguientes problemas se utilizarán las abreviaturas que se indican:
Cpl o C = concentración plasmática t= tiempo K= constante de velocidad de eliminación t1/2= vida media Cl= clearance ABC= área bajo la curva de concentración plasmática versus tiempo Vd= volumen de distribución ku= constante de velocidad de excreción urinaria CEE o CSS= concentración en estado estacionario o en steady state
3.- Usted puede complementar los ejercicios de esta guía con los que aparecen en los siguientes libros: - Farmacocinética Básica e Aplicada. Silvia Storpirtis, María Nella Gai, Daniel Rossi de Campo s, José Eduardo Gonçalves. Ed. Guanabara Koogan, Rio de Janeiro, 2011. - Applied Biopharmaceutics & Pharmacokinetics. Leon Shargel, Susanna Wu-Pong, Andrew B.C.Yu. Fifth Edition. Mc Graw Hill. 2005. - DiPiro J, Blouin R, Pruemer J, Spruill W. Concepts in clinical pharmacokinetics. A self- instructional course. American Society of Health System Pharmacists Inc. 1996.

PROBLEMAS.
TEMA: modelo de 1 compartimento, bolus iv, datos plasmáticos. Objetivos: 1 (parcial), 2-4, 5(parcial),8,13
1.- Identifique si los siguientes fármacos siguen una cinética lineal. ¿Lo hacen en todo el rango de dosis, en ninguno, o en una parte de él?
FÁRMACO
LACIDIPINO ACIDO ACETILSALICÍLICO FENITOÍNA Dosis (mg) ABC (ugh/l) Dosis/día Cmax(ug/ml) Dosis/día CEE
(mg) (mg) 2 4,5 100 0,15 300 5,2 4 9,2 200 0,32 350 8 6 14,15 500 0,8 400 15 8 24,25 3000 8 425 20
2.- En un fármaco que sigue un modelo farmacocinético lineal, monocompartimental y que es administrado mediante un bolus intravenoso, ¿cuánto tiempo se requiere para eliminar el 99% de la dosis, cuando ésta es de 100 mg y cuando es de 200mg?
3.- Un fármaco tiene un Cl de 5 mL/min y un Vd de 15 litros. Su unión a proteínas es de 10%. ¿Cuál es su vida media?
4.- Para dos fármacos con las siguientes características: Metoprolol: Vd = 300 L Unión a proteínas plasmáticas: 13% Fenilbutazona: Vd = 17 L Unión a proteínas plasmáticas: 99%
Analice el impacto de la unión a proteínas plasmáticas sobre el volumen de distribución

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
5.- La tabla siguiente resume los datos de concentración plasmática después de administrar un bolus iv (37,5 mg) de un fármaco:
t(h)
0,8
2,0
4,0
5,0
7,0
10,0
Cpl(mg/L) 2,1 1,2 0,58 0,32 0,13 0,041
a.- Grafique estos datos en papel semilogarítmico y estime en forma gráfica la t1/2, K, Vd, Cl y ABC.
b.- Calcule t1/2, K, Cl, Vd y ABC.
c.- Escriba el modelo que describe el comportamiento de este fármaco.
d.- Si se administra el doble de la dosis, analice qué ocurre con las Cpl, ABC, Vd, K, Cl y t1/2.
e.- Si este fármaco se elimina en forma inalterada por el riñón en un 80%, analice qué pasa con la vida media y el ABC si el paciente experimenta una pérdida de 60% de su función renal.
f.- Este fármaco presenta un 75% de unión a albúmina. ¿Cuáles parámetros farmacocinéticos se alterarían si disminuye la concentración de albúmina plasmática?

6.- Se administró una dosis de 250 mg de un principio activo a un voluntario adulto de sexo masculino de 63 kg de peso. Se tomaron muestras de sangre a los tiempos indicados obteniéndose los siguientes valores de concentración plasmática del antibiótico. La administración se efectuó mediante un "bolus" intravenoso:
Tiempo (horas)
Cpl (ug/mL)
0,5
9,3
1,0
5,6
1,5
3,8
2,0
2,5
3,0
1,4
4,0 0,6
5,0 0,3
a) Graficar los valores de concentración plasmática versus tiempo en papel semilogarítmico. b) Identifique el modelo que mejor describe el comportamiento del fármaco y calcule todos los parámetros farmacocinéticos. R: K = 0,755 h-1 t1/2 = 0,92 h Vd = 20,3 L Cl = 15,3 L/h
ABC = 16,31 ug h/mL
7.- A un paciente se le administra una inyección intravenosa que contiene 250 mg de un fármaco. Se recolectan muestras de sangre cada hora y son analizadas obteniéndose los siguientes valores:
Tiempo (horas)
Cpl (mg/L)
1 5,32
2 4,75
3 4,20
5 3,30
7 2,67
Calcular: - La constante de velocidad de eliminación - El volumen de distribución aparente
K = 0,116 h-1 Vd = 41,9 L Co = 0,60 mg/100 mL Cl = 4,86 L/h

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
8.- A un individuo de 70 kg de peso se le administra una dosis intravenosa de 250 mg de ampicilina anhidra, obteniéndose los siguientes niveles plasmáticos.
Tiempo (horas)
Cpl (mg/L)
0,25
2,231
0,5
1,932
0,75
1,671
1,0
1,450
1,5
1,083
2,0 0,812
2,5 0,608
3,0 0,456
4,0 0,256
Calcular la constante de velocidad de eliminación, la vida media de eliminación, el volumen aparente de distribución, la depuración total y al área bajo la curva hasta tiempo infinito.
R : K= 0,577 h-1 t1/2 = 1,2 h Vd = 97 L Cl = 56 L/h ABC = 4,46 mg h/L
9.- Se inyecta por vía intravenosa 500 mg de un fármaco cuya vida media biológica es de 7 horas y cuyo comportamiento farmacocinético puede describirse de acuerdo a un modelo monocompartimental. Calcular la cantidad de fármaco presente en el organismo 18 horas después de la inyección.
R: 84 mg
10.- Para un medicamento que presenta una t1/2 de 4 h y un Vd de 25 L (en un
individuo de 70 Kg), calcular la dosis requerida para que al ser administrado por vía i.v. rápida alcance una concentración de 2,4 mg/L a las 6 h después de su administración. R = 170 mg
11.- La aminofilina presenta una variabilidad interindividual alta, de forma tal que la t1/2 que aparece en la literatura sólo puede servir como referencia. A un paciente se le inyecta por vía i.v. rápida una dosis de aminofilina que genera inmediatamente después de la dosis una Cpl de teofilina de 20 ug/mL. Al cabo de 6 horas la Cpl es de 5 ug/mL. Calcule la vida media de teofilina en este paciente. R = 3 h
TEMA: modelo de 1 compartimento, bolus iv, excreción urinaria.
Objetivos: 1(parcial), 2-4, 5 (parcial),8,13
12.- Al estudiar la cinética de disposición de una sulfa, después de administrar 1 g en forma de inyección intravenosa rápida, se obtuvieron los siguientes datos:
a.- De concentración plasmática
t(h)
1
2
4
6
8
Cpl(mg/L) 58 51 42,5 36 29
b.- De excreción urinaria del fármaco inalterado
Intervalo(h)
0-3
3-6
6-9
9-12
12-15
15-24
24-48
Xu(mg) 240 180 96 69,6 55 101 48
Calcule los parámetros farmacocinéticos a partir de los 2 líquidos biológicos y compárelos.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
13- Luego de la administración de 250 mg de un fármaco por vía intravenosa, se obtuvieron los siguientes valores de excreción urinaria:
Tiempo (horas)
Farmaco inalterado excretado en orina (mg)
0,5 10,0
1,0 19,4
2,0 36,3
3,0 51,0
4,0 63,8
6,0 84,7
8,0 100,5
12,0 121,6
16,0 133,7
24,0 144,6
48,0 149,8
72,0 150,0
Identifique qué tratamiento(s) de datos puede utilizar y calcule: - Vida media y constante de velocidad de eliminación del fármaco. - Fracción de la dosis que se excreta en forma inalterada. - Constante de velocidad de excreción renal
R: K = 0,14 h-1 t!/2 = 5 h fu = 0,62 ku = 0,086 h-1

14.- Se inyecta por vía intravenosa 250 mg de un fármaco cuyo volumen de distribución es de 10 litros. Se recolecta orina en diversos períodos de tiempo obteniéndose los siguientes resultados.
Calcular:
Periodo de recolección (horas)
Concentración de fármaco en orina (mg/L)
Volumen de orina (L)
0 - 1 24,70 0,35
1 - 2 14,48 0,53
2 - 4 8,97 1,25
4 - 6 12,17 0,78
6 - 8 8,70 0,85
8 - 12 5,95 1,75
12 - 24 5,31 2,25
24 - 36 0,00 2,25
6,7 h
= 29%
- La constante de velocidad de eliminación - La constante de velocidad de metabolización - La constante de velocidad de excreción renal - La vida media biológica - El clearance renal - La vida media biológica en caso de anuria - La vida media biológica en caso de una función renal disminuída en un 50% - La concentración plasmática a las 3 horas después de la inyección El porcentaje de biotransformación y el de fármaco no metabolizado en individuos normales. R: K = 0,12 h-1 km = 0,086 h-1 ku = 0,035 h-1
t1/2 = 5,7 h Clr= 0,35 L/h t1/2 (anuria)= 8 h t1/2 (renal al 50%)=
Cpl 3 horas = 17,3 mg/L % biotransformación = 71% % no metabolizado
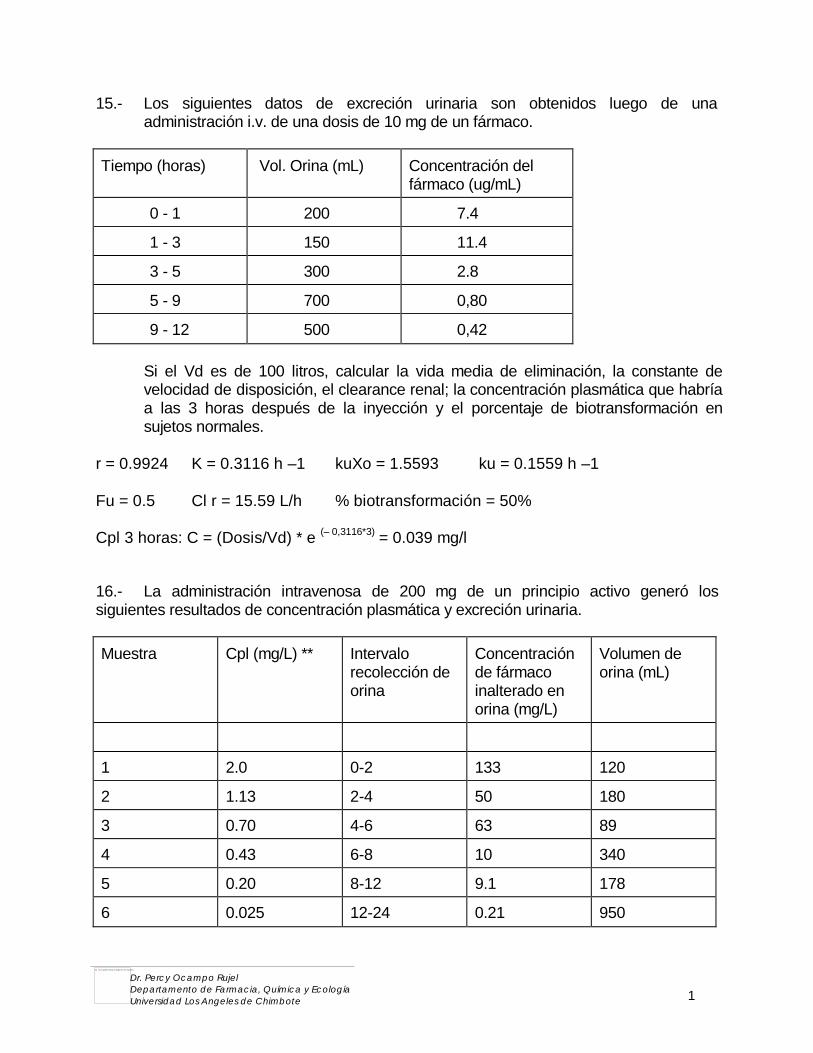
No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
15.- Los siguientes datos de excreción urinaria son obtenidos luego de una administración i.v. de una dosis de 10 mg de un fármaco.
Tiempo (horas)
Vol. Orina (mL)
Concentración del fármaco (ug/mL)
0 - 1 200 7.4
1 - 3 150 11.4
3 - 5 300 2.8
5 - 9 700 0,80
9 - 12 500 0,42
Si el Vd es de 100 litros, calcular la vida media de eliminación, la constante de velocidad de disposición, el clearance renal; la concentración plasmática que habría a las 3 horas después de la inyección y el porcentaje de biotransformación en sujetos normales.
r = 0.9924 K = 0.3116 h –1 kuXo = 1.5593 ku = 0.1559 h –1
Fu = 0.5 Cl r = 15.59 L/h % biotransformación = 50%
Cpl 3 horas: C = (Dosis/Vd) * e (– 0,3116*3) = 0.039 mg/l
16.- La administración intravenosa de 200 mg de un principio activo generó los siguientes resultados de concentración plasmática y excreción urinaria.
Muestra
Cpl (mg/L) **
Intervalo recolección de orina
Concentración de fármaco inalterado en orina (mg/L)
Volumen de orina (mL)
1 2.0 0-2 133 120
2 1.13 2-4 50 180
3 0.70 4-6 63 89
4 0.43 6-8 10 340
5 0.20 8-12 9.1 178
6 0.025 12-24 0.21 950

**: muestra obtenida en la mitad del intervalo de recolección de orina
a.- Defina si el comportamiento de este principio activo se ajusta a algún modelo farmacocinético particular. b.- Obtenga los parámetros farmacocinéticos correspondientes. c.- Compare y discuta los parámetros que se pueden obtener tanto de los datos de concentración plasmática como de los de excreción urinaria.
TEMA: modelo de 1 compartimento, infusión iv.
Objetivos: 1,2,3,6,8,13
17 .- Se administra una infusión intravenosa de 200 mg/L a una velocidad de 2 mg/min de un fármaco que tiene una vida media de 16 horas y un volumen de distribución de 40 litros. Calcular el tiempo que se necesita para alcanzar el 70% del estado estacionario.
R= 27,8 horas
18.- Un fármaco se administra por infusión intravenosa a una velocidad de 38 mg/h, obteniéndose un estado estacionario de 5 mg/L. Al suspender la infusión las concentraciones sanguíneas son:
Tiempo (horas)
Cpl (ug/mL)
2 3,4
4 2,2
6 1,5
8 1,0
Calcular: la t1/2, K, Vd y Cl - ¿Cuál sería la concentración esperada 2 horas después del término de la infusión? - Si la velocidad de la infusión aumenta a 50 mg/h, calcular el nuevo valor del
estado estacionario. - ¿Cuál sería la dosis de carga necesaria para alcanzar inmediatamente una
concentración de 5 mg/L y cuál sería la velocidad de infusión necesaria para mantener esta Cpl? R: t1/2 = 3,6 h K = 0,19 h-1

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
19.- A un paciente se le inyecta un fármaco por infusión intravenosa a una velocidad de 1,3 g/hr. Los niveles plasmáticos del fármaco, alcanzados son los siguientes:
t (horas)
C (ug/mL)
5 17,4
10 25,4
15 29,6
20 31,5
Si el tiempo medio de eliminación de este fármaco es de 4,6 horas ¿cuál será el clearance total y el volumen de distribución aparente?. R: Al graficar C versus (1-e -Kt) de la pendiente se obtiene Css = 33,5 ug/mL
K = 0,151 h-1 Vd = 256,6 L Cl = 38,75 L/h
20.- En procedimientos quirúrgicos prolongados, se administra succinilcolina por infusión intravenosa para lograr la relajación muscular. La dosis inicial habitual es 20 mg seguida por una infusión continua de 0,5 mg/min. La infusión debe ajustarse individualmente por la gran variación interindividual en el metabolismo. Estimar la vida madia de eliminación de la succinilcolina en pacientes que requieren 0,5 mg/min y 5,0 mg/min para mantener 20 mg en el organismo. R: 1,5 h-1 y 15 h-1
21.- Calcular los valores de Vd, K, t1/2 y Cl a partir de los datos de la tabla, obtenidos después de una infusión iv a una velocidad de 50 mg/h por 7,5 horas.
t(h)
0
2
4
6
7,5
9
12
15
Cpl (mg/L)
0 3.4 5.4 6.5 7.0 4.6 2.0 0.9
Antes de hacer cualquier cálculo, analice mirando los datos disponibles si al suspender la infusión existiría la posibilidad de haber alcanzado el estado estacionario.

22.- La teofilina es un medicamento que habitualmente requiere monitorización debido a que presenta una gran variabilidad interindividual en su comportamiento farmacocinético y un estrecho margen terapéutico (10-20 mg/l). Su comportamiento farmacocinético en algunos pacientes puede describirse como monocompartimental, en cambio, en otros pacientes se describe mejor de acuerdo a un modelo bicompartimental.
Según la literatura, en promedio la vida media es de 5 horas y su volumen de distribución es de 0.45 l/kg. Utilizando estos datos, se calculó una dosis de carga de 550 mg de aminofilina administrada en forma de bolus i.v. para un sujeto de 70 kg de peso. Después de la inyección se tomaron muestras de sangre las que fueron analizadas para teofilina, encontrándose los siguientes valores de concentración plasmática (Cpl)
Tiempo (h) Cpl (mg/l)
0.5 14.2 1 13.5 3 9.0 6 5.8
a.- Determine los parámetros farmacocinéticos que caracterizan el comportamiento de la teofilina para este sujeto en particular. Calcular la dosis de carga que se necesitaría para alcanzar una Cpl de 14 mg/l y una velocidad de infusión que le permita mantener esta concentración. b.- Por razones de seguridad para el paciente, el médico opina que sería mejor administrar una infusión i.v rápida seguida de una más lenta, en vez de la combinación (bolus i.v + infusión). Calcule la velocidad de infusión necesaria para alcanzar la Cpl adecuada en 30 minutos y la velocidad de infusión necesaria para mantener esta Cpl. c.- Si se enfrentara a la siguiente situación en clínica: un paciente con su función de eliminación disminuida ¿esperaría que el valor de Cpl en estado estacionario fuera el mismo si mantiene las condiciones de administración que en a y b? ¿El tiempo necesario para alcanzar el estado estacionario sería el mismo?
R: a.- Los parámetros farmacocinéticos para este sujeto son: Vd = 28,4 L K = 0,1665 h-1 t1/2 = 4,2 h Cl = 4,73 L/h ABC= 93 mg h/L La dosis de carga es de 400 mg de teofilina o de 500 mg de aminofilina y una velocidad de infusión de 66,2 mg/h o 1,1 mg/min de teofilina (1,4 mg/min de aminofilina). b.- 13,8 mg/min durante 30 minutos y luego cambiar a 1,1 mg/min

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
TEMA: modelo de 1 compartimento, administración extravascular.
Objetivos: 1-4, 7,8,13 23.- Cuando se administró una dosis oral de 100 mg de un fármaco nuevo a un grupo de voluntarios se obtuvieron los siguientes datos plasmáticos promedios.
t (horas)
Clp (ug/mL)
0 0
1 1,88
2 2,06
3 2,27
5 2,65
7 2,87
9 2,46
12 2,08
18 1,33
24 0,813
36 0,297
48 0,107
a) ¿Cuál es el modelo compartimental que mejor define el comportamiento de este fármaco? b) ¿Cuál es el tiempo medio de eliminación del fármaco en este grupo de voluntarios? c) ¿Cuál es la velocidad de absorción del fármaco?.
R: a.- 1 compartimento b.- 8,25 h c.- 0,306 h-1

24.- Al administrar una dosis de 500 mg de un fármaco por vía oral se encuentra que la ecuación que describe la concentración plasmática en función del tiempo es:
C = 28,5 (e 0.123t - e -0.438t)
¿Cuál es el tmáx, la Cmáx, la vida media de eliminación y el volumen de distribución de este fármaco?. Suponga una absorción completa.
R: tmax= 4h Cmax=12,5 mg/L t1/2= 5,6 h Vd= 24,4 L
25.- A un paciente se le administra un comprimido de 250 mg de un fármaco y se determina la concentración sanguínea a diversos tiempos posteriores a la administración obteniéndose los siguientes resultados:
t (horas)
Cpl (ug/mL)
0,25 1,56
0,5 2,66
1 3,91
1,5 4,41
2 4,51
3 4,20
4 3,68
5 3,17
6 2,71
7 2,31
Calcular:
a) La constante de velocidad de eliminación total del fármaco. b) La constante de velocidad de absorción c) El volumen de distribución, suponiendo una biodisponibilidad del 100%
R: K= 0,155 h-1 ka= 1,36 h-1 Vd = 41,6 L
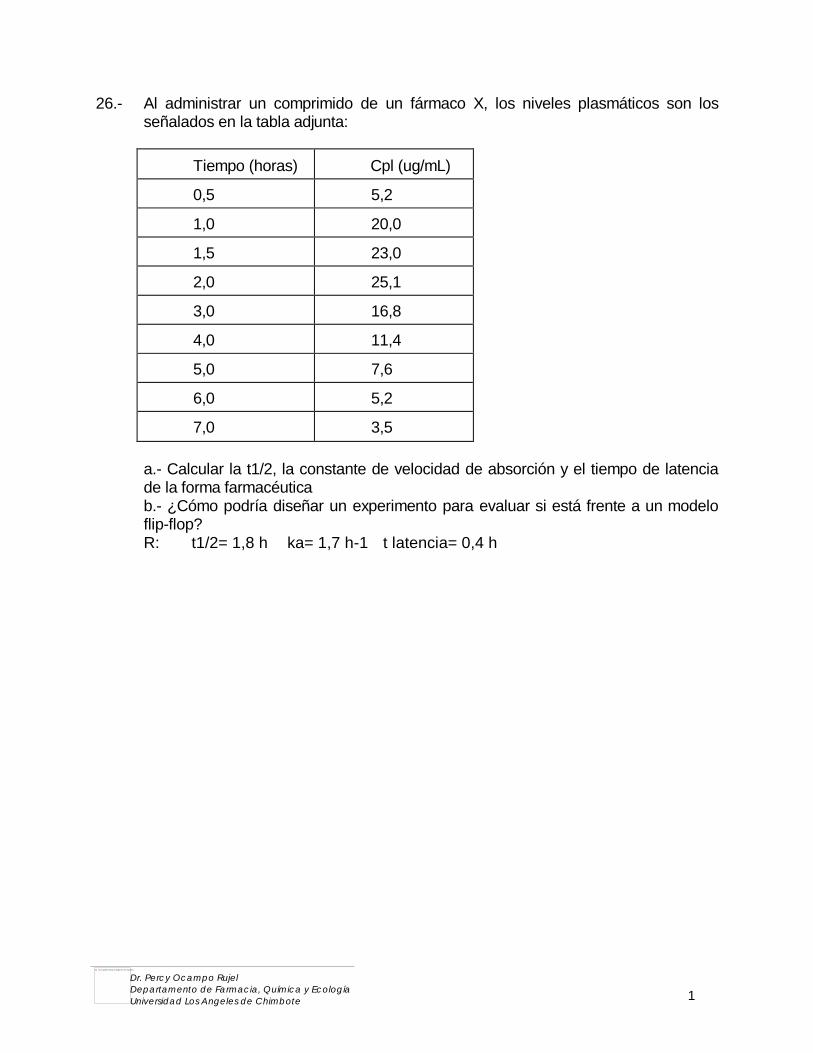
No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
26.- Al administrar un comprimido de un fármaco X, los niveles plasmáticos son los señalados en la tabla adjunta:
Tiempo (horas)
Cpl (ug/mL)
0,5 5,2
1,0 20,0
1,5 23,0
2,0 25,1
3,0 16,8
4,0 11,4
5,0 7,6
6,0 5,2
7,0 3,5
a.- Calcular la t1/2, la constante de velocidad de absorción y el tiempo de latencia de la forma farmacéutica b.- ¿Cómo podría diseñar un experimento para evaluar si está frente a un modelo flip-flop? R: t1/2= 1,8 h ka= 1,7 h-1 t latencia= 0,4 h

27.- La administración iv de un bolus de 100 mg generó los siguientes resultados:
Tiempo (horas)
Cpl (ug/mL)
0,25 3,75
0,5 2,8
1,0 1,75
1,5 1,1
2 0,55 La administración de la misma dosis oral en forma de comprimido generó los siguientes resultados:
Tiempo (horas)
Cpl (ug/mL)
0,5 0,57
1,0 0,88
1,5 1,03
2,0 1,07
2,5 1,04
3,0 0,98
4,0 0,82
6,0 0,50
8,0 0,29
12,0 0,09 a.- Calcule las constantes de velocidad involucradas b.- ¿Cree usted que se halla frente a un modelo flip-flop? c.- ¿Tiene tiempo de latencia el comprimido?
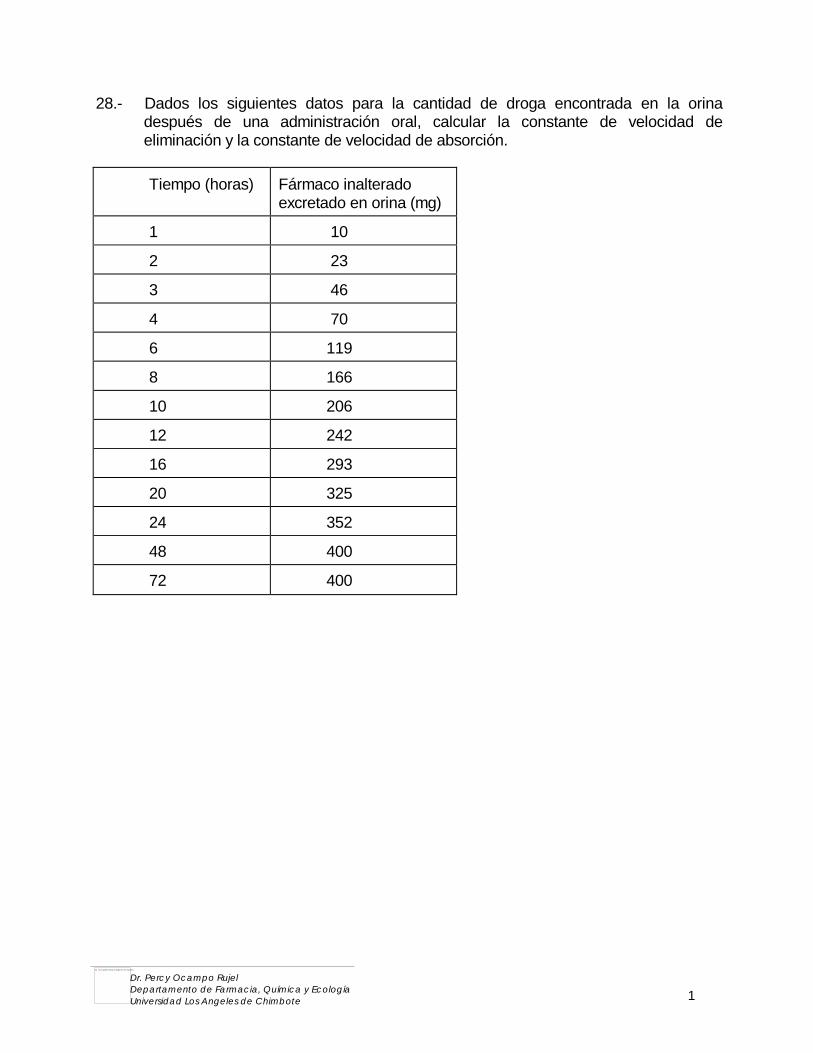
No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
28.- Dados los siguientes datos para la cantidad de droga encontrada en la orina después de una administración oral, calcular la constante de velocidad de eliminación y la constante de velocidad de absorción.
Tiempo (horas)
Fármaco inalterado excretado en orina (mg)
1 10
2 23
3 46
4 70
6 119
8 166
10 206
12 242
16 293
20 325
24 352
48 400
72 400

TEMA: modelo de 1 compartimento, administración en dosis múltiple.
Objetivos: 13, 14,15,16 29.- Un fármaco se administra de acuerdo a un régimen de dosificación múltiple por vía
intravenosa cada 8 horas. Las concentraciones plasmáticas analizadas en el mismo individuo luego de una dosis única de 100 mg en una experiencia anterior fueron las siguientes:
Tiempo (horas)
Concentración (mg/L)
1 1,222
2 1,004
3 0,810
4 0,671
6 0,447
En este nuevo tratamiento de dosis repetidas de 100 mg, calcular: a) La concentración promedio en el estado de equilibrio b) Las fluctuaciones máximas y mínimas de la concentración dentro del estado estacionario.
30.- La warfarina tiene una vida media de eliminación de 54 horas. En un régimen de
administración de dosis múltiple por vía intravenosa se administran 5 mg cada 24 horas. Compare la acumulación de warfarina en relación a un régimen de administración de 48 horas. R: 3,78 y 2,2
31.- Si un fármaco con una vida media de 12 h se administra cada 6 horas y se obtiene
una Cpl máxima en el estado estacionario de 10 mg/L, ¿cuál sería la Cpl máxima que se alcanzó después de administrar la quinta dosis? R: 8,2 mg/L
32.- Una dosis de 100 mg de un fármaco X es admnistrado a dos pacientes cada 8 horas. ¿Cuál paciente (A ó B) alcanzará mayores Cpl en el estado estacionario?
Paciente K (1/h) Vd (L) A 0.2 10 B 0.4 20
R: el paciente A

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
33.- Una dosis de 50 mg de un fármaco X es administrado cada 8 horas hasta que se
alcanza el estado estacionario. El ABC obtenida en dicho estado es de 16 mg h/L. ¿Cuál es la Cpl promedio en el intervalo de dosificación y cuál será el valor del clearance? R: 2 mg/L y 3,125 L/h
34.- Un médico ordena un tratamiento con un antibiótico en un esquema de 200 mg cada 8 horas mediante inyecciones repetitivas de bolos intravenosos. El volumen de distribución aparente descrito para el antibiótico es de 0.2 L/kg y tiene una vida media de eliminación de seis horas. Los estudios clínicos, microbiológicos y toxicológicos indican que la mínima concentración sérica efectiva es de 2 ug/ml y la mínima concentración sérica tóxica es de 16 ug/ml. Este régimen de dosificación es prescrito para un paciente adulto de 80 Kg. de peso y cuyo clearence de creatinina es de 122 milílitros por minuto. De acuerdo a sus resultados comente si el esquema propuesto por el médico es apropiado o no lo es. Si no lo es, exprese su punto de vista y sugiera Ud. un régimen de dosificación alternativo para el paciente con los fundamentos correspondientes.
35.- La vancomicina presenta las siguientes características: - se administra por vía intravenosa en forma de infusiones cortas de 2 horas de
duración, diluidas en suero, para evitar la aparición de una reacción adversa llamada “Síndrome del hombre rojo”
- su vida media es de 6 horas (función renal normal) - su volumen de distribución es de 0,47 L/Kg - su margen terapéutico oscila entre 5 y 40 ug/mL - se elimina en forma inalterada por la orina en un 95%, por filtración glomerular - se presenta en frascos ampolla de dosis de 1000 mg
Para un paciente hospitalizado varón de 65 Kg de peso con una función renal normal: a.- El tratamiento estándar es de 1 gramo cada 12 horas.¿Le parece adecuado? b.- Si el paciente experimentara una brusca disminución de su función renal a un 50% ¿qué dosificación propondría como alternativa a la del punto c? c.- En pacientes grandes quemados, al tercer día comienza la denominada fase hipermetabólica, que se caracteriza por un aumento del Cl de creatinina de un 100%. ¿Qué tendría que hacer con el tratamiento para que sea eficaz?

36.- Ceftriaxona es una cefalosporina que presenta una vida media más larga que otras de la misma familia como una de sus características más relevantes, permitiendo su administración una vez al día. En la farmacia se cuenta con frascos-ampolla de 1000 mg para uso intravenoso. Los parámetros farmacocinéticos promedio son: Vida media: 8 horas Vd: 0.16 L/Kg Cl: 0.24 mL/min/Kg Este antibiótico se administra de forma tal que la dosis del frasco ampolla ingresa al organismo mediante una infusión corta de 1 hora. ¿Qué opina respecto de la acumulación de este fármaco? Considerando un paciente de peso 70 Kg que presenta un cuadro infeccioso por neumococo y que el MIC para neumococo es de 2 ug/ml, evalúe cuánto tiempo permanece la Cpl por sobre la MIC. En el periodo más crítico de la enfermedad es fundamental que la Cpl del antibiótico permanezca la mayor parte del tiempo sobre la MIC. De acuerdo con este criterio ¿sería suficiente una administración diaria?
Tema: modelo de 2 compartimentos
Objetivos: 1(parcial), 9 al 13 37.- A un individuo de 70 kg de peso se le administró una inyección i.v. de un fármaco. Las concentraciones de éste en la sangre obtenidas a diferentes tiempos fueron:
t (horas)
C (ug/mL)
0,17 36,2
0,33 34,0
0,50 27,0
0,67 23,0
1,0 20,8
1,5 17,8
2,0 16,5
3,0 13,9
4,0 12,0
6,0 8,7
7,0 7,7
18,0 3,2
23,0 2,4

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
Establecer el modelo que describe la evolución de la concentración plasmática en función del tiempo.
38.- A un niño de 4 años de edad y 16,4 Kg de peso, que presenta un asma crónica severa, se le administra una dosis intravenosa de aminofilina equivalente a 3,2 mg/Kg de teofilina. La medición de los niveles plasmáticos (Cpl) de teofilina se presenta en la tabla:
tiempo(horas)
0.25
0.5
1
2
3
4
6
8
10
Cpl(ug/mL) 10.62 9.15 7.42 5.78 4.77 3.98 2.78 1.94 1.35
a.- Escriba la ecuación que describe el modelo farmacocinético para teofilina.
b.- Calcule los parámetros farmacocinéticos pertinentes.
c.- ¿Esperaría usted que la ecuación farmacocinética establecida describa el comportamiento de la teofilina en otro niño de la misma edad y peso? Fundamente su opinión.
39.- Luego de la administración intravenosa de 500 mg de amoxicilina a un sujeto adulto, se obtuvieron las siguientes concentraciones plasmáticas:
t(min)
Cpl(ug/mL)
5 42.3
10 38.3
15 30.3
20 23.4
30 18.6
45 13.8
60 11.2
90 7.7
120 5.0
180 2.8
240 1.1
300 0.6

Al administrar la misma dosis en forma oral al mismo sujeto se obtuvieron las siguientes concentraciones plasmáticas:
t(min)
30
45
60
75
90
120
180
240
300
360
Cpl (ug/mL)
1.2 6.7 10.2 12.1 12.6 11.1 5.1 2.5 1.1 0.6
¿A qué tipo de modelos compartimentales se ajustan los datos obtenidos? ¿Qué explicación tiene el comportamiento de la administración oral?
Calcule: α y ß ABC i.v y oral t1/2 α y ß Cl k10,k12 y k21 ka
Los volúmenes de distribución que caracterizan al modelo La Biodisponibilidad
R: β= 0.01229 min-1 B= 23.15 mg/L α= 0.0957 min-1 A= 40.41 mg/L V1= 7.9 L ABC = 2305.9 mg min/L Vd área = 17.64 L Cl= 218 ml/min K10= 0.0276 min-1 k12= 0.03779 min-1 k21= 0.0426 min-1
BD forma oral= 80% (ABC oral= 1834.7)

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
40.- Las siguientes concentraciones plasmáticas se obtuvieron después de administrar por vía oral dos comprimidos de 300 mg de carbonato de litio.
t (h)
Cpl (ug/mL)
0.08 0.035
0.16 0.05
0.25 0.22
0.33 0.71
0.5 1.43
0.75 2.89
1 3.57
1.5 3.96
2 3.84
3 3.24
4 2.72
5 2.38
6 2.16
8 1.81
12 1.30
18 1.10
24 0.91 Calcular las constantes que caracterizan al modelo
R: beta= 0.0297 h-1 alfa= 0.296 h-1 ka= 2.741 h-1

Tema: Biodisponibilidad y perfiles de absorción
41.- Los siguientes datos se obtuvieron luego de la administración de dos formas farmacéuticas al mismo sujeto en dos ocasiones diferentes. El sujeto era un hombre de 38 años y 78 kg de peso.
Vía i.v. oral
Dosis 150 mg 300 mg
Forma farmacéutica Solución comprimido
Tiempo(h) Cpl(ug/mL) Cpl(ug/mL)
0 15 0
1 13,5 2
2 12,3 2.6
3 11 3.2
5 8,8 4.2
7 7,3 4.8
9 5,8 5.4
12 3,8 5.3
15 3,1 4.9
18 2,3 4
22 1,5 3
25 1,1 2.1
a) Haga una estimación de la biodisponibilidad absoluta del preparado oral. b) Ejecute un tratamiento de los datos que le permita obtener una apreciación del perfil
de absorción del preparado oral y calcule la constante de velocidad de absorción. Perfil de absorción: t % absorbido 1 20 2 27,6 3 35,7 5 51,2 ka= 0,1575 h-1 7 64,1 9 78 13 94

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
42.- Tratar de acuerdo al método de Wagner y Nelson los siguientes datos de concentración plasmática que se obtuvieron al administrar 60 mg de un medicamento.
Tiempo (h) Cpl (mg/l)
0 0.5 3.58 1 5.37
1.5 6.33 2 6.75
2.5 6.89 3 6.83
3.5 6.68 4 6.43 5 5.9 6 5.38 8 4.39
10 3.58 12 2.9 14 2.36
a) Obtener gráficos de porcentaje de fármaco absorbido y porcentaje de fármaco no absorbido versus tiempo.
b) Estimar la constante de velocidad de absorción. R: t % absorbido 0,5 41,16 1 63,79 1,5 77,91 ka = 1,02 h-1
2 86,38 2,5 91,87 3 95,13

Tema: teoría estadística de los momentos Objetivos: 17 43.- Al administrar un fármaco por vía i.v se obtuvieron los siguientes datos de
concentraciones plasmáticas versus tiempo.
t(h)
Cpl(mg/L)
0 10
1 9.7
2 9.42
4 8.87
5 8.35
8 7.87
10 7.41
14 6.57
18 5.83
24 4.87
36 3.40
48 2.37
60 1.67
72 1.15
Calcular el tiempo medio de residencia (TMR)

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
44.- Se diseñó en nuestro laboratorio una forma farmacéutica de Nifedipino de 20 mg de liberación controlada, en base a un coprecipitado con PEG. Se llevaron a cabo estudios "in vitro" e "in vivo" de la formulación definitiva. Es estudio "in vivo" consistió en una evaluación de la biodisponibilidad comparativa con el producto innovador del mercado. El estudio se realizó en forma cruzada, utilizando el esquema del cuadrado latino de 2x2, sobre una población de ocho voluntarios, formada por dos grupos de cuatro sujetos, los que ingirieron en forma alterada el comprimido diseñado y el producto innovador, hasta alcanzar el estado estacionario. Cada grupo de voluntarios fue mantenido administrándosele un comprimido cada 12 horas por tres días, para alcanzar la concentración de estado estable (Css). Los resultados de concentración plasmática en el intervalo de dosificación fueron los siguientes:
Cpl (ug/mL)
Tiempo (horas)
Compr. diseñado
Producto innovador
0,0 0 0
0,5 17,81 20,87
1,0 23,10 30,68
1,5 30,10 33,74
2,0 31,57 31,46
3,0 34,46 28,43
4,0 32,33 23,93
6,0 27,56 20,49
8,0 22,12 16,92
10,0 17,96 14,51
12,0 15,14 11,70
Calcule el tiempo medio de residencia (TMR) para ambos productos y comente los resultados. Evalúe su biodisponibilidad e identifique a qué tipo de biodisponibilidad corresponde.

45.- Se administraron 500 mg de un fármaco por vía oral, en dos ocasiones diferentes. Una vez en forma de solución y la otra en una forma farmacéutica sólida. Los siguientes son los valores de concentración plasmática versus tiempo para ambas situaciones.
Cpl (ug/mL)
Tiempo (horas)
Solución
F.f. sólida
0,25 22,94
0,50 31,35 3,89
1,75 32,32
1,00 32,38 8,89
1,50 28,58 13,98
2,00 25,11 18,44
2,50 22,40
3,00 20,82 26,01
3,50 25,94
4,00 18,47 23,64
5,00 16,85 19,97
6,00 15,53 17,78
8,00 13,29 14,97
10,00 11,40 12,82
12,00 9,78 10,98
14,00 8,39 9,43
24,00 3,89 4,38
28,00 2,87 3,22
32,00 2,11 2,37
36,00 1,55 1,74
48,00 0,62 0,69
a) Calcule los TMR para ambas situaciones. b) Calcule el tiempo medio de Disolución (TMD) para la forma farmacéutica
sólida c) Comente sus resultados.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
46.- Los estudios de bioequivalencia deben proporcionar algún tipo de medida de la cantidad efectivamente absorbida desde la forma farmacéutica y de la velocidad de absorción. Los datos experimentales que se presentan a continuación corresponden a los obtenidos de un voluntario al cual se administró un comprimido de 250 mg de un fármaco y se determinaron las concentraciones plasmáticas a diversos tiempos posteriores a la administración obteniéndose los siguientes resultados:
t (horas)
Cpl (ug/mL)
0,25 1,56
0,5 2,66
1 3,91
1,5 4,41
2 4,51
3 4,20
4 3,68
5 3,17
6 2,71
7 2,31
El objetivo de los cálculos que se solicitan es obtener parámetros farmacocinéticos que sean de utilidad en un estudio de bioequivalencia.
Calcular:
a) La constante de velocidad de eliminación total del fármaco. b) La constante de velocidad de absorción c) El ABC total d) El tiempo máximo y la Cmax reales e) Las concentraciones que alcanzaría si se administra en dosis múltiple con un
intervalo de dosificación de 6 horas. Establezca a qué tiempo se produciría el máximo de concentración con esta administración y la Cmax que alcanzaría.. R: K = 0,1554 h-1 ka= 1,287 h-1 ABC= 38,7 ug h/mL tmax= 1,41 h

Tema: disolución
Objetivos: 27, 28 49.- El estudio de una cinética de disolución de comprimidos experimentales de paracetamol dio los siguientes resultados de porcentaje disuelto en función del tiempo:
t (min)
% disuelto
5 34,9
10 57,7
15 72,5
20 82,1
30 92,4
Calcular la constante de velocidad de disolución y el t50% y t80% de disolución.
50.- Un comprimido de Glibenclamida se somete al ensayo de cinética de disolución en un medio solvente determinado, obteniéndose los siguientes resultados:
t (min)
% disuelto
10 40,7
20 54,2
30 64,9
45 76,4
Calcular la constante de velocidad de disolución y el t 85%
51.-Al determinar la cantidad disuelta de un comprimido de ácido acetilsalicílico de 500 mg se encontró que a los 15 minutos se habían disuelto 425 mg. Suponiendo que el proceso de disolución es de primer orden y que el comprimido no presenta tiempo de latencia, calcular:
a) La constante de velocidad de disolución. b) La cantidad de fármaco que había disuelta a los 5 minutos de iniciado el ensayo.

No se puede mostrar la imagen en este momento.
Dr. Percy Ocampo Rujel Departamento de Farmacia, Química y Ecología Universidad Los Angeles de Chimbote 1
52.- Se realizó un estudio de cinética de disolución de comprimidos de Cimetidina de 200 mg. Los siguientes son los datos de cantidad de fármaco disuelto (mg) en la alícuota extraída (5 ml) versus el tiempo de muestreo. El volumen del medio de disolución es de 900 mL.
Tiempo (min)
Cantidad en alicuota (mg)
2 0,496
4 0,770
6 0,951
8 1,048
10 1,100
12 1,135
14 1,148
15 1,152
16 1,155
18 1,158
∞ 1,162
a) ¿Cuál es el orden cinético del proceso de disolución? b) Calcule la constante de velocidad de disolución (Kd), la vida media de disolución (t50%), el porcentaje de fármaco disuelto a los 30 min y el tiempo para que se disuelva el 90% del fármaco (t90%) c)¿Qué podría deducir a partir de sus resultados experimentales en relación con la dosificación del comprimido?. d) ¿Presenta tiempo de latencia esta forma farmacéutica? Comente.

53.- Al comparar 4 productos del mercado, que contienen 300 mg de un principio activo, con un producto de referencia mediante una cinética de disolución, hecha según las condiciones experimentales descritas para ese principio activo en la USP, se obtuvieron los siguientes resultados:
Producto Tiempo (minutos)
5 10 15 30 45 60 75 % disuelto sobre lo declarado
Referencia 16 30 40 67 80 87 89 Producto 1 8 18 28 51 71 88 92 Producto 2 14 25 36 69 84 89 93 Producto 3 18 32 43 78 86 93 94 Producto 4 30 50 78 89 91 93 95
a.- Establezca cuál o cuáles productos pueden considerarse similares al producto de referencia. b.- Calcule la Eficiencia de la disolución para los productos en ensayo