teorie delle reazioni chimiche - scienze.uniroma2.it · teorie delle reazioni chimiche qualunque...
TRANSCRIPT

1
TEORIE DELLE REAZIONI CHIMICHEQualunque teoria delle reazioni chimiche deve spiegare la dipendenza dalla temperatura delle costanti di equilibrio e delle costanti di velocità.
- Dipendenza degli equilibri dalla temperatura
K = e-ΔGo/RT ΔGo
RT
e poiché ΔGo = -TΔSoΔHo ΔHo
R1T + ΔSo
R
ln K = -
ln K = -
Se ΔHo e ΔSo sono indipendenti dalla temperatura (vero per un intervallo ditemperatura limitato), riportando in grafico ln K in funzione di 1/T, si ha una retta,di pendenza -ΔHo/R
Dalla misura delle costanti di equilibrio a due temperature si può determinareΔHo
se T2 > T11T2
1T1
- > 0ln
K2
K1= ΔHo
R1T2
1T1
-
K2
K1dipende dal segno di ΔHo
K2 K1> cioè se la reazione è endotermicase ΔHo > 0K2 K1< cioè se la reazione è esotermicase ΔHo < 0
- Dipendenza della velocità dalla temperatura
La velocità di una reazione semplice aumenta con l'aumentare della temperatura.un aumento di 10 K provoca un aumento di due o tre volte della costantedi velocità
La relazione quantitativa tra la costante di velocità (k) della reazione e la tempe-ratura assoluta (K) è formulata nell'equazione di Arrhenius (empirica)
ln k = ln A -R
Ea 1T R = 1.987 cal K-1 moli-1 (8.314 J K-1 moli-1)
T in gradi Kelvin
k = A e-Ea/RT A fattore pre-esponenzialeEa energia di attivazione - Ea = RT ln (k/A)
Riportando in grafico log k (o ln k) in funzione di 1/T, si ottiene Ea dallapendenza della rettal'intercetta è log A (o ln A)
Si sottintende che A sia indipendente dalla temperatura

2
In realtà A non è veramente indipendente dalla temperatura
Si può dimostrare che
=/ΔSdove entropia di attivazione=/ΔH entropia di attivazione
k = RTNh
e- ΔG /RT=/= RT
Nhe ΔS /R=/ . e- ΔH /RT=/
RNhlog k
T( ) = log ( ) +=/ΔS
2.303R- ( 2.303R
=/ΔH ) . 1T
A è una costante verso cui converge k (soprattutto se si lavora in unintervallo di temperature relativamente stretto)
=log k T( ) 10.319
=/ΔS+4.573
- (=/ΔH ) . 1
4.573 T
PhSMe + NaIO4 PhS(O)Me + NaIO3
Determinazione di Ea ( ) e di ΔH (------) per la reazione=/

3
Se le concentrazioni sono espresse in M (moli/litro), il tempo in secondi e la temperatura in gradi Kelvin
=/ΔH in cal moli-1
in cal moli-1K-1 = u.e. (unità entropiche) =/ΔS
ΔH =/Ea = + RT
di solito RT è piccolo (~ 0.6 kcal/mole a temp. ambiente)
in condizioni di pseudo-ordine, usando kψ, ΔH è lo stesso (pendenza), ma ΔS no (intercetta). Va perciò determinata usando k e non kψ.
=/
=/
Normalmente i grafici di Arrhenius sono lineari (o almeno non ci sono motivi convincenti per non ritenerli tali)
Grafico di Arrhenius per la conversione sedia-sediadell'anello del cicloesano
La curvatura è un artefatto, introdotto da un errore sistematico della misura
Non improbabile: le costanti di velocità nell'intervallo di temperatura possono variare di ordini di grandezza, richedendo metodi analitici diversi in regioni di temperature diverse.
lavoro sperimentale accuratola curvatura è dovuta a parametri di attivazione che dipendono dalla temperatura
Non tutti accettano questa possibilità
La curvatura è la conseguenza di una reazione complessaPer reazioni reversibili e parallele: kexp = k1 + k2 non si può usare kexp
= RNh
e-ΔS1 /R=/
e ΔS2 /R=/ .ΔH1 /RT=/kexpT ( + e e- ΔH1 /RT=/ )log (k/T) in funzione di (1/T) non è una retta curvatura diagnosticadi situazione complessa
RTNhPer reazioni parallele
k1 k2 =[X]
[Y] =[X][Y] RT
Nh
e-ΔS1 /R=/
e ΔH1 /RT=/
ΔS2 /=/ .e e- ΔH1 /RT=/
In alcune reazioni si osservano grafici di Arrhenius curvi. i possibili motivi sono tre

4
=[X][Y]
log=/ΔS2ΔS1 -=/( )
2.303R- ( =/ΔH2ΔH1 -=/
2.303R ) .T1
=/ΔS2ΔS1 -=/( )=/ΔH2ΔH1 -=/
Riportando in grafico log [X] / [Y] in funzione di - (1/T) si ha una rettapendenza
( ) intercetta
Nel caso di pre-equilibrio kexp = K1 k2
= RNh
(ΔS°1 /Rekexp
Te-ΔS2 /R)=/ (ΔH°1 /RT ΔH2 /RT)=/+ +
Riportando in grafico log ( ) in funzione di (1/T) kexp /T
pendenza variazione complessiva di entalpia=/ΔS2ΔS°1
+( )=/ΔH2ΔH°1
+
( )variazione complessiva di entropiaintercetta
Determinando K1 in funzione della temperatura si determinano ΔH°1 e ΔS°2 dalla variazione complessiva si calcolano ΔH2 e ΔS2
=/ =/
Nel caso di condizioni di stato stazionario kexp = k1k2
k-1 + k2
Sostituendo in k = RTNh
e- ΔG /RT=/= RT
Nhe ΔS /R=/ . e- ΔH /RT=/
e facendo il logaritmo curvatura diagnostica
Se uno dei termini al denominatore può essere trascurato, si hanno semplifica-zioni
k-1 << k2 kexp = k1Se
Se k-1 >> k2 kexp = k1 k2 k-1
= K1k2

5
Grafico di Arrhenius curvo per l'idrolisi del trifluoroacetatodi metile in DMSO
esempioIl legame Co-C della Vitamina B12 subisce omolisi ad opera dell’enzimaribonucleotide trifosfato reduttasi (RTPR)
Questa variazione di pendenza è stata interpre-tata come evidenza di una variazione conforma-zionale dell’enzima ad una forma inattiva al disotto dei 30°C, che perciò richiede isomerizz-azione per avere la forma cataliticamente attiva.
L’isomerizzazione influenza con la suatermodinamica la costante di velocità.
cambiamento di meccanismo
L’analisi di Eyring mette in evidenza che l’entropia di attivazione del processo cata-lizzato dall’enzima è identica a quella della scissione non catalizzata del legami Co-C.
La catalisi viene completamente dal cambiamento di entalpia di attivazione: l’etalpia diattivazione del mccanismo catalizzato è più bassa di 13 kcal/mole, portando ad un aumento della cosante di velocità di 1.6 x 109.

6
TEORIA DELLA COLLISIONEAnalizza la dipendenza della velocità di reazione dalla temperatura sulla base delnumero di collisioni che avvengono nell'unità di tempo.Le collisioni si dividono in due classi
COLLISIONI ELASTICHE: cambiano solo la direzione e la velocità delmovimento delle particelle
COLLISIONI ANELASTICHE (o REATTIVE): cambia in qualche modo lacomposizione chimica delle molecole che collidono
Collisioni fisiche (a) e chimiche (b) delle molecole A e B
Dalla teoria cinetica dei gas si ottiene un'espressione per il numero di collisioni (Z') che avvengono tra due molecole A e B in una unità di volume nel periodo diuna unità di tempo.
nA e nB numero di molecole di massa mA e mB in 1 cm3
μ massa ridotta 1μ
= 1 1+mA mB
molecola cm-3 s-1Z' = 8πRTNμ( )1/2
(rA + rB)2 nAnB
quando nA = nB = 1 cm3 molecola s-1Z = 8πRTNμ
(rA + rB)2
La collisione è possibile solo all'interno di (rA + rB)
dove rA è il raggio di A e rBquello di B

7
per ottenere Z in unità di k, bisogna moltiplicare per il numero di Avogadro edividere per 1000
M-1 s-1Z = 8πRTNμ( 1/2
(rA + rB)2 )N1000
a temperatura ambiente, per molecole con peso molecolare < 100
Z ~ 1011 M-1 s-1
Valore molto maggiore dei k solitamente osservati, dovuto al fatto chenon tutte le collisioni portano a reazione
Perché si abbia reazione, le molecole che collidono devono avere un surplus di energia (energia di attivazione)
Z deve essere moltiplicato per un fattore pre-esponenziale,in accordo con la legge di distribuzione di Boltzmann
k = p Z e-Ea/RT
confrontando con l'equazione di Arrhenius A = pZp <_ 1 per tener conto dell'orientamento
Un test della teoria della collisione consiste nel confrontare i valori osservato ecalcolato del fattore pre-esponenziale A
P =A (osservato)A (calcolato)
Considerando l'estrema semplicità della teoria è notevole che per molte reazioni,in fase gassosa ma anche in soluzione, P sia molto vicino all'unità
EtBr + OH- EtOH 21.4 4.30 3.86 1.11EtO- + MeI EtOH 19.5 2.42 1.93 1.25ClCH2CO2
- + OH- H2O 25.9 4.55 2.86 1.59
C3H6Br2 + I- MeOH 25.1 1.07 1.39 0.77HOCH2CH2Cl + MeI H2O 19.9 25.5 2.78 9.17
4-MeC6H4O- + OH- EtOH 21.2 8.49 1.99 4.27
CH3(CH2)2Cl + I- MeCOMe 20.7 0.085 1.57 0.054
Me2SO4 + CNS- MeOH 17.4 0.19 1.91 0.010
β-C10H7O- + EtI MeOH 21.0 0.10 2.21 0.045
Et3N + EtI PhH 11.2 - - 5.3 x 10-10
py + MeI C3H2Cl4 13.2 - - 2.0 x 10-6
(H2N)2CS EtOH 14.6 - - 1.5 x 10-5
lattosio + H3O+ H2O 24.7 - - 1.17saccarosio + H3O+ H2O 25.8 - - 5.3 x 103
mellibiosio + H3O+ H2O 38.6 - - 1.3 x 109
Tabella. Dati cinetici e valori di P per alcune reazioni in soluzione
Ea, kcal/mole10-11 A
osservato calcolatoP
Reazione Solvente

8
Cause dell'inadeguatezza:- le molecole non sono sferiche- le collisioni possono avere energia non sufficiente- possibili costrizioni steriche- presenza del solvente- possibili contributi entropici molto favorevoli
Svantaggi della teoria della collisionenon fornisce spiegazioni per la barriera di energianon fornisce un metodo per il calcolo dell'energia di attivazione
Vantaggi della teoria della collisione
metodo molto semplice, per definire il comportamento cinetico"normale"
TEORIA DELLO STATO DI TRANSIZIONE
Quando due molecole collidono in una collisione che porta ai prodotti (o una singola molecola esegue i movimenti che provocano la variazione chimica), passano attraverso una configurazione di massima energia potenziale, chiamata STATO DI TRANSIZIONE.
La premessa fondamentale della teoria dello stato di transizione è che i reagentiformino una specie di complesso con una struttura tra lo stato dei reagenti (RS) e lo stato dei prodotti (PS) di una reazione

9
Lo stato di transizione (o struttura di transizione o complesso attivato)si trova lungo la coordinata di reazione (q) e la sua energia rappresentail massimo del profilo di energia
L'assunzione fondamentale della teoria dello stato di transizione e che A e B (RS)sono in equilibrio con il complesso attivato-
L'equilibrio è caratterizzato da una costante di pseudo-equilibrio, K=/
Si assume anche che la velocità complessiva (cioè la la formazione del prodotto)è influenzata anche dalla velocità di decomposizione del complesso attivato, chepuò essere caratterizzata dalla costante di velocità k =/
A + B AB X + YK k =/=/ =/
PEA [AB] = K k [A][B]=/ =/ =/velocità = k [AB ]=/=/
velocità = k2 [A][B]
per la reazione bimolecolare: k2 = K k=/ =/
k =/
AB ha una frequenza vibrazionale (ν) associata al modo di formazione del prodotto lungo la coordinata di reazione
=/
k = κν=/
Se tutte le elongazioni vibrazionali portassero alla decomposizione del complessoattivato verso la formazione del prodotto, k sarebbe numericamente uguale a ν=/
invece
κ = coefficiente di trasmissione 0 < κ < 1_
=/K
La costante di pseudo-equilibrio è legata alle funzioni di partizione (Q) ed alla differenza di energia tra complesso attivato e reagenti allo zero assoluto
=/K =QAB =/QAQB
e - ΔE0 /RT=/
Separando la porzione vibrazionale della funzione di partizione di AB associataalla decomposizione del complesso attivato lungo la coordinata di reazione
=/
si arriva all'espressione
ν non è presentek2 κ = RTNh
=/K'
=/K' è legata all'energia libera di attivazione =/=/ΔG = -RT ln K'

10
=/- ΔG /RT k2 κ = RN e
=/ =/k2 κ = R
N e eΔS /R - ΔH /RT
=/=/ =/
=/ =/
la costante di velocità k2 può essere espressa mediante una delle seguentiformule (equivalenti)
Equazioni di Eyring
Il valore numerico del coefficiente di trasmissione ( ) è incognito e si sceglie 1κ
Le funzioni termodinamiche di attivazione sono la conseguenza della struttura del complesso attivato.
k2 è grande se ΔG e ΔH sono piccole e ΔS è grande
Dai valori numerici sperimentali di ΔH e ΔS si possono avere indicazioni sugli stati di transizione e, di conseguenza, sul meccanismo.
STRUTTURA DEL COMPLESSO ATTIVATO
In casi semplici si possono avere indicazioni sulla simmetria dello stato di transizione
La struttura di un complesso attivato non si può determinare sperimentalmente, perché non ha una vita misurabile. La sua geometria molecolare può essere determinata teoricamente (metodi di ottimizzazione di gradiente quantomeccanico)
Esempio:
In casi più complessi si possono fare ipotesi sulla base del postulato di Hammond
I- + -------------- II1/2- 1/2-
=/
+ I-C IH
HH
CIH
HH
CH
HH
con energia di attivazione piccola, l'energia e la geometria di TS sarà vicina a quella del reagente (TS reagent like) (Fig.a)
con energia di attivazione elevata ed energie simili per reagenti e prodottiTS sarà circa a metàstrada (Fig. b)
con energia di attivazione elevata e reazione endotermica, TS saràsimileal prodotto (TS productlike) (Fig. c)

11
SUPERFICI DI ENERGIA POTENZIALE (PES)
PES: una (iper)superficie di E in funzione delle variabili geometriche che descrivono il sistema reagente:
Reagenti TS … Prodotti
Gli N atomi di una molecola si possono muovere in tre direzioni mutuamente perpendicolari e perciò indipendenti e quindi una molecola ha untotale di 3N gradi di libertà.
la molecola costituisce un'unità
Se gli atomi fossero fissi, gli uni rispetto agli altri, la posizione della molecola rigida nello spazio sarebbe definita dalle tre coordinate cartesiane del suo centro di massa e dai tre angoli rotazionali per indicare il suo orientamento nello spazio
Restano 3N-6 GRADI DI LIBERTA', che sono i movimenti vibrazionali interni degli atomi uno rispetto all'altro (3N-5 se la molecola è lineare).
La vibrazione molecolare totale è complessa, ma con buona appros-simazione si può dividere in 3N-6 modi normali indipendenti
Ciascun modo si può considerare equivalente alla vibrazione di stretchingdi una molecola biatomica
modello oscillatore armonico funzione parabolica
•Per una molecola diatomica, ad es. H–H, l'unica variabile è la distanza rHH. •E = f(r)
Possiamo esprimere l'energia in funzione della distanza interatomica r con la legge di Morse:
PES per una molecola diatomica.
2)( ]1[)( erraeDrE −−−=
In cui l'energia si annulla per r = reper r ∞, E D (energia di dissociazione)per r 0, E aumenta molto rapidamente (repulsione nucleare)

12
Nell'approssimazione armonica (valida per spostamenti piccoli rispetto a re)
La molecola vibra (il legame si stira) attorno a re, e occupa vari livelli vibrazionali
Per una molecola poliatomica si ha una curva di energia potenziale per ciascuno dei 3N-6 modi vibrazionali
L'energia potenziale è caratterizzata da una superficie in uno spazio a 3N-6+1 dimensioni.
L'energia di ciascuna dei 3N-6 modi vibrazionali è quantizzata. Il comportamentoquantico delle vibrazioni è tale, che un modo vibrazionale non può mai perdere tutta la sua energia
Energia del punto zero
Quando due molecole A e B reagiscono, è interessata la superficie del-l'energia potenziale per l'intero processo
Il movimento degli atomi che caratterizza il cambiamento si chiama coordinatadi reazionePRINCIPIO DEL MINIMO MOVIMENTO: saranno favorite quelle reazioni elementariche comportano il minimo cambiamento della posizione degli atomi e dellaconfigurazione elettronica
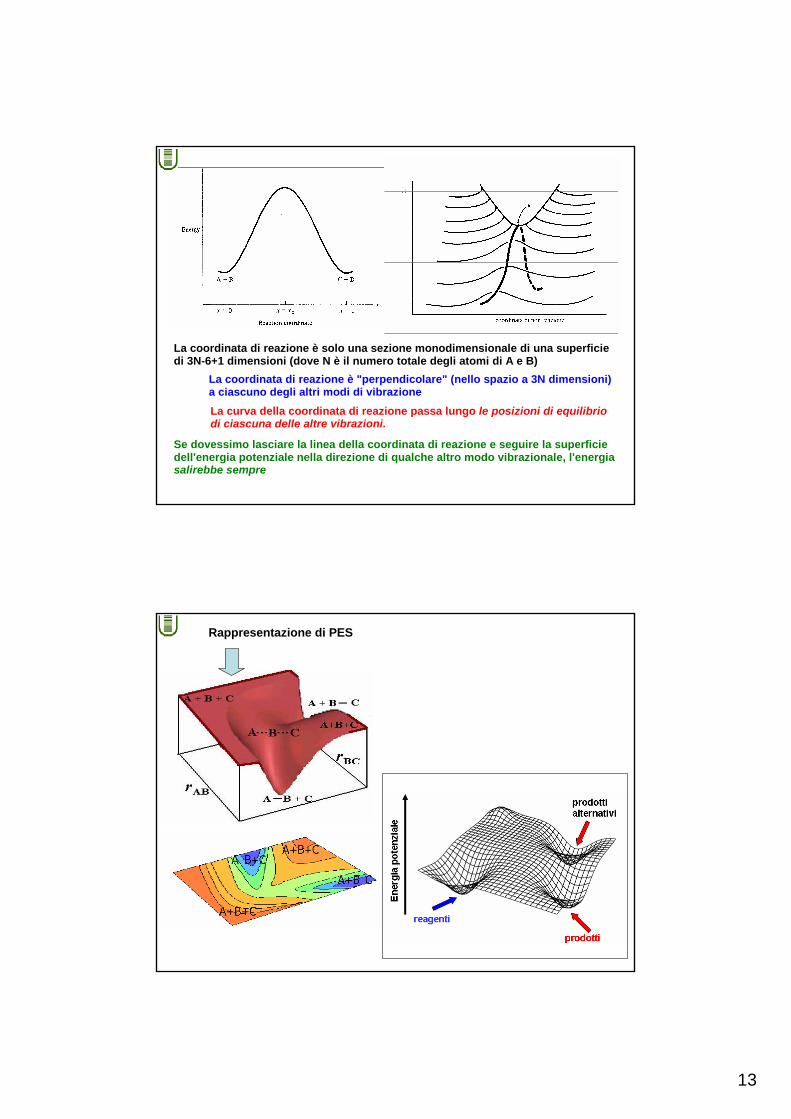
13
La coordinata di reazione è solo una sezione monodimensionale di una superficiedi 3N-6+1 dimensioni (dove N è il numero totale degli atomi di A e B)
La coordinata di reazione è "perpendicolare" (nello spazio a 3N dimensioni)a ciascuno degli altri modi di vibrazioneLa curva della coordinata di reazione passa lungo le posizioni di equilibriodi ciascuna delle altre vibrazioni.
Se dovessimo lasciare la linea della coordinata di reazione e seguire la superficie dell'energia potenziale nella direzione di qualche altro modo vibrazionale, l'energiasalirebbe sempre
Rappresentazione di PES

14
La superficie mostra solo l'energia potenziale.
L'energia totale del sistema molecolare è la somma delle sue energie:vibrazionale, rotazionale, cinetica, potenziale e interna.
Per calcolare la superficie di energia potenziale ci sono tre metodimetodo puramente teorico: usa solo grandezze fisiche fondamentali,come la carica elettricametodo semiempirico: introduce nei calcoli un numero limitato di datisperimentali
metodo empirico: fa largo uso di dati sperimentali
Per una reazione in cui stato iniziale e finale abbiano energia diversa, la superficiedell'energia potenziale non sarà simmetrica
Stato dei reagenti più stabile dello stato dei prodotti
Stato di transizione "repulsivo"o "tardo" (late) o "simile ai prodotti"Il legame AB è molto formato nello stato di transizione
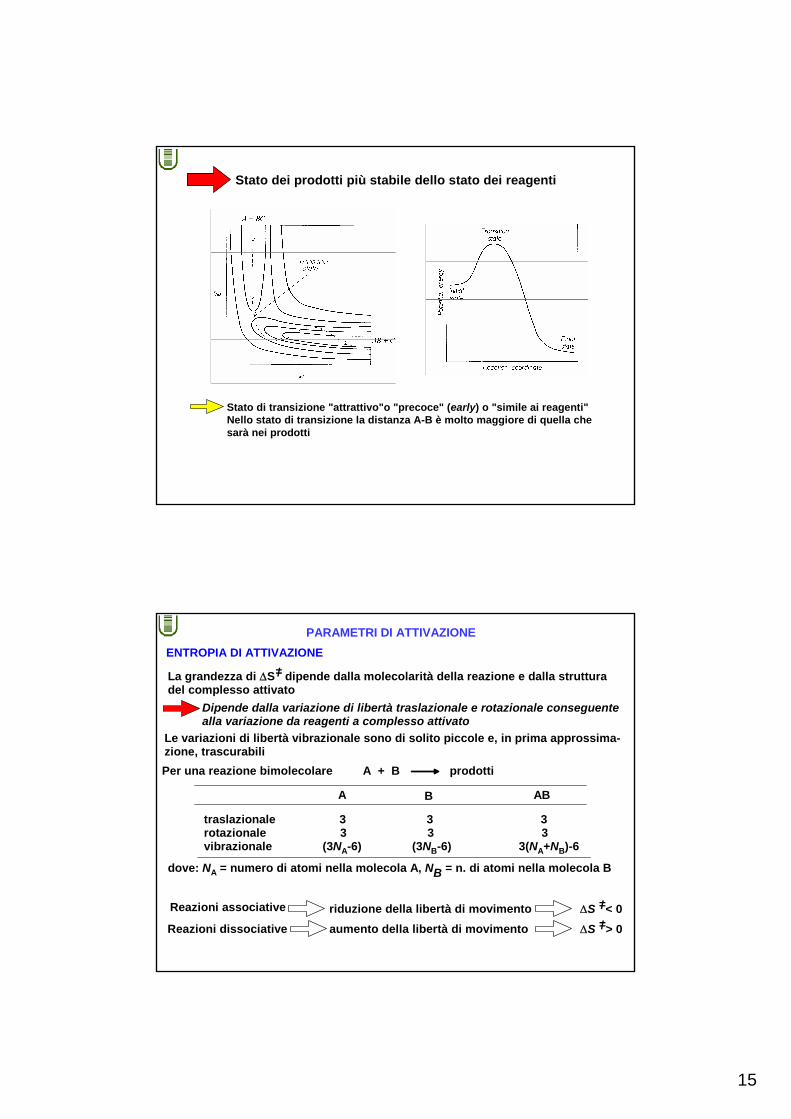
15
Stato dei prodotti più stabile dello stato dei reagenti
Stato di transizione "attrattivo"o "precoce" (early) o "simile ai reagenti"Nello stato di transizione la distanza A-B è molto maggiore di quella che sarà nei prodotti
PARAMETRI DI ATTIVAZIONEENTROPIA DI ATTIVAZIONE
La grandezza di ΔS dipende dalla molecolarità della reazione e dalla strutturadel complesso attivato
=/
Dipende dalla variazione di libertà traslazionale e rotazionale conseguentealla variazione da reagenti a complesso attivato
Le variazioni di libertà vibrazionale sono di solito piccole e, in prima approssima-zione, trascurabili
dove: NA = numero di atomi nella molecola A, NB = n. di atomi nella molecola B
Per una reazione bimolecolare A + B prodotti
traslazionale 3 3 3rotazionale 3 3 3vibrazionale (3NA-6) (3NB-6) 3(NA+NB)-6
A B AB
Reazioni associative riduzione della libertà di movimento =/ΔS < 0Reazioni dissociative aumento della libertà di movimento =/ΔS > 0

16
Esempi di reazioni unimolecolari dissociative (in fase gassosa)
2 .ΔS = 17 e.u., 71 kJ mol-1K-1=/
2 ΔS = 9 e.u., 37.6 kJ mol-1K-1=/CH2CH2 CH2
CH2
CH3 CH3 CH3
CH2 CH2
Reazioni unimolecolari che passano attraverso uno stato di transizione ciclicopossono avere entropia di attivazione leggermente negativa (impedimento percerte rotazioni interne)
Esempi:
pirolisi di esteri
Δ
CH2
CH2
OC
CH3O
H.. ..
.
.
. . ..
...... .. ...
.. =/
+
ΔS = -5 e.u., -20.9 kJ mol-1K-1=/
trasposizione di Claisen
CH2
CH2
OH
CH3C OC2H5
OC
CH3O
OH
O
CH2H CHCH2
O
ΔS = -12 e.u., -50.2 kJ mol-1K-1=/
NH2O
Br NH2O
+
+
+ Br-
=/ΔS < 0
=/
=/
=/
Per reazioni bimolecolari soprattutto con stati di transizioneciclici
in fase gassosa:+ ΔS = -19 e.u.
in benzene:+ ΔS = -33 e.u.
ΔS è influenzata da varazione di solvatazione:
=/ΔS < 0=/
=/
Se AB è più solvatato di A + B(ad esempio, gli ioni sono più solvatati delle molecole neutre in solventi polari)
il valore numerico di ΔS dipende dalla polarità del solvente
Esempio: parametri di attivazione determinati in vari solventi per la reazione:

17
Solvente
PhH 31.4 (7.5) - 234 (-56)CHCl3 42.7 (10.2) -192 (-46)MeCOMe 43.9 (10.5) -163 (-39)
SolventeΔH =/
kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)
ΔH =/
kJ mol-1 (kcal mol-1)ΔS =/
J mol-1 K-1(e.u.)
MeOH 49.4 (11.8) -138 (-33)EtOH 55.6 (13.3) -117 (-28)
La formazione del guscio di solvatazione in un solvente apolarerappresenta un apprezzabile aumento di ordine, mentre il solventepolare è già ben ordinato
Bisogna fare attenzione a trarre conclusioni sul meccanismo solo dai valori di entropia di attivazione, a causa dell'effetto del solvente.
L'entropia di attivazione è più alta (> 0) per reazioni unimolecolari e più bassa(< 0) per reazioni bimolecolari.
Reazioni unimolecolari con stati di transizione polare possono avere entropie di attivazione un po' negative, se il solvente è anidro o conpoca acqua
Tabella. Parametri di attivazione per l'idrolisi uni- e bimolecolare di alogenuri alchilici e di esteri (solvente: acqua)
ΔH =/
kJ mol-1 (kcal mol-1)ΔS =/
J mol-1 K-1(e.u.)Composto
Meccanismo
MeCl 106 (25.3) -40.0 (-8.6) SN2MeBr 101 (24.1) -28.0 (-6.7) SN2i-PrCl 104 (24.9) -22.2 (-5.3) SN2i-PrBr 102 (24.4) -5.9 (-1.4) SN2t-BuCl 86 (20.5) 14.2 (3.4) SN1t-BuSMe2 132 (31.6) 65.7 (15.7) SN1MeCO2Me 65 (15.6) -109 (-26.0) AAc2MeCO2Et 65 (15.6) -109 (-26.0) AAc2MeCO2Bu-t 115 (27.5) 58.6 (14.0) AAc1
115 (27.5) 58.6 (14.0) AAc1CO2Me
CH3
CH3 CH3
+

18
Valori tipici di ΔS# per alcune delle reazioni più comuni
+:
HO
H OEtO
HO H+
:OHH
+O
OH OOH OH
OH
H Me
..
: CH3 IN
:S Cl
O
OEt
Cl
OO
Idrolisi acida degli esteri
-26 eu
Idrolisi acida degli ossaciclopropani
-6 eu
+4.5 eu -17 eu
-31 eu +10 eu +11 eu
Idrolisi acida dell’α-metilglucosideAddizione coniugata
Sostituzione nucleofila Idrolisi del t-BuCl Omolisi di perossidi
ENTALPIA DI ATTIVAZIONE
L'entalpia di attivazione (o energia di attivazione) è la differenza di entalpia (o di energia) tra complesso attivato e reagenti
ΔH =/Il valore numerico di o di Ea è più difficile da prevedere menodiagnostico
Ci si può aspettare un valore più alto per reazioni unimolecolari che per reazionibimolecolari
Esempio:
2
Ea = 258 kJ mol-1
ΔS =/ = + 41.8 J mol-1 K-1
Ea = 96.6 kJ mol-1
ΔS =/ = - 79.5 J mol-1 K-1
=/
in soluzione ΔH =/ è molto influenzato dalla solvatazione
Se la solvatazione stabilizza lo stato di transizione più dei reagenti, ci si aspetta una diminuzione di ΔH
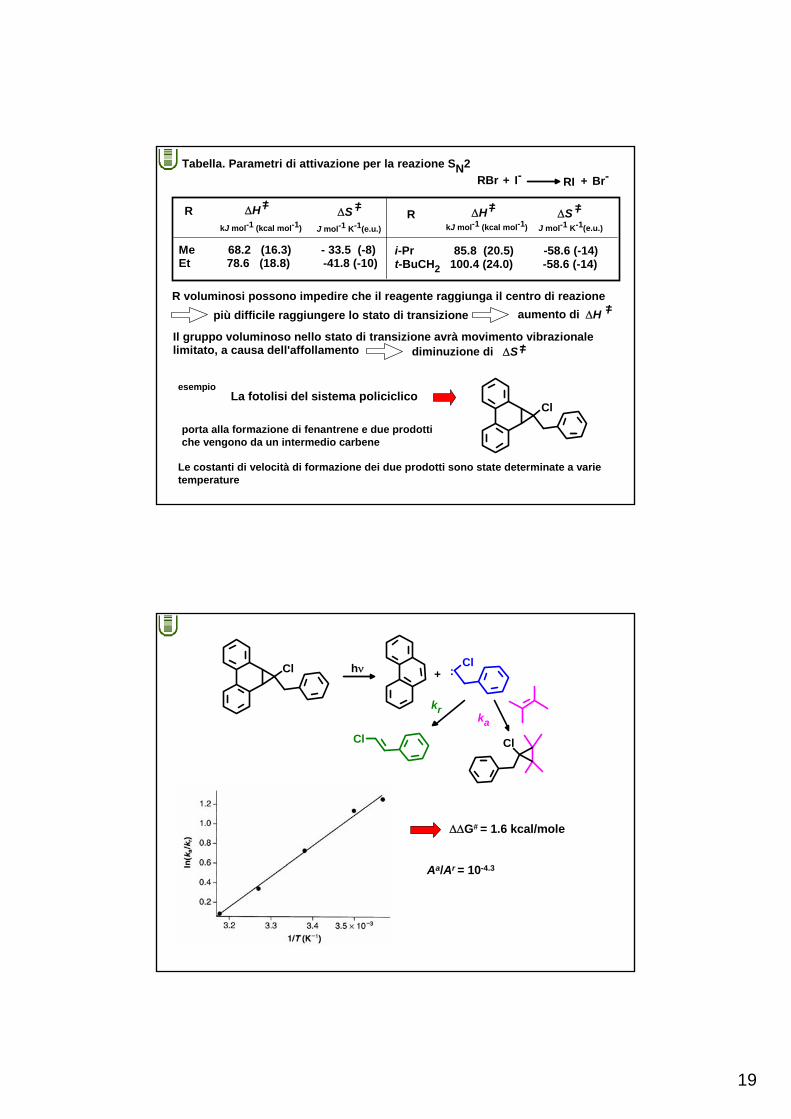
19
+ + Br-Tabella. Parametri di attivazione per la reazione SN2
RBr I- RI
R voluminosi possono impedire che il reagente raggiunga il centro di reazionepiù difficile raggiungere lo stato di transizione aumento di ΔH =/
R
Me 68.2 (16.3) - 33.5 (-8)Et 78.6 (18.8) -41.8 (-10)
ΔH =/
kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)
ΔH =/kJ mol-1 (kcal mol-1)
ΔS =/J mol-1 K-1(e.u.)
i-Pr 85.8 (20.5) -58.6 (-14)t-BuCH2 100.4 (24.0) -58.6 (-14)
R
Il gruppo voluminoso nello stato di transizione avrà movimento vibrazionale limitato, a causa dell'affollamento diminuzione di ΔS =/
esempioLa fotolisi del sistema policiclico
Cl
porta alla formazione di fenantrene e due prodottiche vengono da un intermedio carbene
Le costanti di velocità di formazione dei due prodotti sono state determinate a varietemperature
Cl hν Cl:+
kr
Cl
ka
Cl
ΔΔG# = 1.6 kcal/mole
Aa/Ar = 10-4.3

20
RELAZIONE ISOCINETICA
In alcuni casi i parametri di attivazione per una serie di reazioni possono essere in relazione
ΔS =/ΔH =/ = β + c relazione isocinetica
per una serie di composti che variano solo per il sostituente, fatti reagire nelle stesse condizioni
per una reazione di un solo sistema molecolare, misurata in piùsolventi
Quando la relazione isocinetica è valida, si assume che il meccanismo sia lo stesso in tutta la serie
Quando T = β K, la velocità di ogni termine della serie è la stessa
sostituendo a ΔH il secondo termine della relazione isocinetica:=/
ln k = ln RTNh( ) + -
=/ΔS (T - β)RT
cRT
ln k = ln RTNh( ) +
=/ ΔH TΔS - =/
RT
= 0 a T = β
ln kik = ln RTNh
- cRT
β dimensioni di temperatura assoluta
kik costante di velocità isocinetica(isokinetic)

21
La situazione è illustrata in figura
All'interno della serie, la reazione che è più veloce a T < β, è la più lentaa T > β
ΔH =/Al di sotto della temperatura isocinetica, è più veloce la reazione con piùpiccola; al di sopra della temperatura isocinetica, è più veloce la reazione dellaserie con ΔH più grande.=/
Prendendo due reazione, A e B, di una data serie (due rette della figura) per cuisia valida la relazione isocinetica, con valori diversi di entropia di attivazione, illogaritmo del rapporto delle costanti di velocità è dato da:
kA
kBlog( ) =
(ΔSA - ΔSB )(T-β)=/ =/
RT se ΔSA > ΔSB =/ =/ kA < kB a T < β
kA > kB a T > β
Poiché ΔH e ΔS non si possono determinare molto accuratamente, per stimareβ senza conoscerli esplicitamente
=/ =/
metodo di EXNER
Si prende una serie di composti e si misurano, a due diverse temperature(T1 e T2) due costanti di velocità (k1 e k2) per ciascun termine della serie
log k2 = b log k1 + a
esprimendo ln k1 e ln k2 in funzione dei parametri di attivazione si ha:
log k1 = log RTNh( ) T1ΔS - ΔH =/ =/
RT1
T1T2 (1 - b)
β
=/
+
log k2 = logRTNh( ) T2ΔS - ΔH =/ =/
RT2+
ΔH =(T1- bT2)
=/ΔS -T1T2
(T1- bT2){aR + (b-1)R ln RNh
+ R(b ln T1- lnT2)}=
T1T2 (1 - b)(T1- bT2)

22
occorre determinare i valori di ln k1 e ln k2 per la serie di composti (o di solventi) e determinare b con i minimi quadrati, da b si calcola β usandodue temperature sperimentali
Solvolisi di cloruri di benzoile sostituiti in paracorrelazione di log k a 25°C in funzione di log k a 0°C
Non tutte le reazioni seguono la relazione entalpia-entropiasono pochi gli studi cinetici a temperatura superiore a β
β può assumere valori estremi (0 o ∞ ) quando per una seriecambia solo una delle funzioni termodinamiche, mentre l'altrarimane praticamente costante
poiché la reattività relativa (effetto del sostituente o del solvente) allatemperatura isocinetica si inverte, i risultati ottenuti ad una solatemperatura vanno trattati con molta cautela
La teoria dello stato di transizione è stata molto usata per interpretare le costantidi velocità, ma rimangono dubbi sulla sua validità
nella forma solita non è corretta dal punto di vista quantomeccanico
non spiega l'effetto tunnel
la teoria assume che la coordinata di reazione possa essere separatadagli altri movimenti
calcoli dettagliati per reazioni semplici H. + H2 H2 + H.
hanno dimostrato che l'errore introdotto nella costante di velocità è significativoa temperature fino a 1000K ed è più importante per il movimento di atomi leggeri
sembra che la teoria possa essere inadeguata per processi in cui la coordinata di reazione comporti movimento in più di una dimensione
INADEGUATEZZA DELLO STATO DI TRANSIZIONE

23
DD
DD
DD
NN
DD +
..N2
.
.
NN
DD
DD
DD+
3 : 1
in fase gassosa- N2
Una spiegazione possibile è la formazione di un biradicale, che reagisce in modo sincrono con inversione della configurazione del C da cui sta uscendo N2.
NN
DD
DD
NN
DD ..
Inoltre ci potrebbe essere la formazione di un biradicale al C, in grado di dare i due prodotti in rapporto 1:1
La teoria dello ST potrebbe essere insufficiente
Però i calcoli di dinamica molecolare danno un altro mododi vedere le cose.
Una dettagliata analisi teorica suggerisce che la scissione del secondo legame C-N precede sempre la formazione del legame C-C
DD
.
.è sempre presente
Però una certa frazione della popolazione del biradicale al C si chiude dando il prodotto prevalente, prima di ridistribuire in modo random la sua energia interna
La prevalenza del biciclo “trans” sul “cis” viene dalla traiettoria degli atomi interessati nell’espulsione di N2.
DD
DD
NN
DD ..
N2.
.controllo dinamico della sequenza di reazione
In casi come questo, i modelli strutturali e cinetici semplici possono non essere adeguati.

24
GRANDEZZA DELLE QUANTITA' CINETICHE
κTh
~ 1012.8 s-1 a 298 K (κ è la costante di Boltzmann e h quella di Planck)
questo valore dovrebbe rappresentare la velocità attesa per una reazione mono-stadio unimolecolare in fase gassosa, con entalpia ed entropia di attivazione zero
in soluzione, le velocità delle reazioni bimolecolari sono limitate dallavelocità con cui le specie si diffondono tra le molecole di solvente
velocità controllata dalla diffusione
varia con la viscosità del solvente e con la temperaturaper i solventi comuni a temperatura ambiente ~ 1010 M-1s-1
ΔH =/ kJ mol-1 (kcal mol-1)
ΔS =/
J mol-1 K-1(e.u.)κT /h e-ΔH /RT=/
s-1e ΔS /R=/
4.2 (1) 1.15 x 1012 -120 (-30) 2.75 x 10-7
8.4 (2) 2.12 x 1011 -105 (-25) 3.41 x 10-6
21 (5) 1.33 x 109 -84 (-20) 4.23 x 10-5
42 (10) 2.85 x 105 -63 (-15) 5.25 x 10-4
63 (15) 6.10 x 101 -42 (-10) 6.50 x 10-3
84 (20) 1.31 x 10-2 -21 ( -5) 8.07 x 10-2
105 (25) 2.80 x 10-6 0 1126 (30) 6.00 x 10-10 21 (5) 2.14 x 101
146 (35) 1.29 x 10-13 42 (10) 1.54 x 102
167 (40) 2.76 x 10-17 63 (15) 1.91 x 103
La tabella dà un'idea della relazione tra parametri di attivazione e costanti di velocità
ANALISI DI THORNTONApprossimiamo la curva dell'energia potenziale nella regione dello stato di transizione ad una parabola. Supponiamo di fare un piccolo cambiamento della struttura, che renda più difficile procedere verso destra (cambiamentoequivalente ad aumentare l'energia della parte destra della curva più della sinistra)
Si ottiene aggiungendo all'energia in ciascun punto lungo la curva un incremento dΔE°, che aumenta verso destra δΔE° = mx
dove m (in questo caso) è positivo e x è il parametro "coordinata di reazione"
la retta rappresenta la perturbazione δΔE°Se si mette l'origine al vertice della parabola, è facile vedere che il risultato dell'aggiunta della perturbazione alla curva dell'energia potenziale è spostare il suo massimo (e quindi lo stato di transizione) verso destra (curva tratteggiata)
Una perturbazione che renda più difficile il movi-mento da sinistra verso destra, sposta lo TS verso destra lungo la coordinata direazione.Una perturbazione con pendenza negativa, cioèche renda più facile il movimento da sinistra a destra, sposterà la curva (e TS) verso sinistra.
STRUTTURA DELLO STATO DI TRANSIZIONE E DIAGRAMMA TRIDIMENSIONALE DELLA COORDINATA DI REAZIONE

25
Consideriamo come le variazioni strutturali influenzano la posizione dellostato di transizione sulla superficie di energia potenziale rispetto ai gradidi libertà diversi dalla coordinata di reazione.
La curva è una parabola con l'aperura verso l'alto
Se si fa una variazione strutturale che renda più difficile l'allungamento di un legame diverso da quello che si rompe
δΔE° = mz
dove m è positivo e z è una parametro "coordinata non di reazione"
coordinata di vibrazione, z
La curva dell'energia potenzialeperturbata si sposta verso sinistra
Rendere più difficile l'allungamentodel legame ha cambiato la strutturadello stato di transizione in modotale che la distanza di equilibrio dellegame è più corta
Le variazioni di energia dello stato di transizione hanno i seguenti effetti
1. LUNGO LA COORDINATA DI REAZIONE la struttura dello stato di transizione si sposta verso l'estremità che viene innalzata, allon- tanandosi dall'estremità che si abbassa2. PERPENDICOLARMENTE ALLA COORDINATA DI REAZIONE la struttura dello stato di transizione si sposta verso il lato che si abbassa, allontanandosi dal lato che si alza.
Una base strappa un protone da un C ed un gruppo uscente si allontanadal C adiacente
la reazione può seguire meccanismi diversi:
a) Processo sincrono (E2)
B:- + BH + + :X-
stato di transizione C C...........
.....X
B......H
δ-
δ- =/
C CC CX
H

26
b) il protone viene strappato prima dell'uscita del gruppo uscente (E1cb)
B:- + C CX
HBH + C C
X
..-
C CX
..-C C + :X-
c) il gruppo uscente si allontana per primo (E1)
+ :X-
B: + C CH
BH + C C+-
C CX
HC C
H +
Diagrammi di MORE O'FERRALRappresentazione della superficie dell'energia per una reazione E2
Ciascun punto del piano orizzontale rappresenta una particolare combinazione di valori delle coordinate per la scissione del legame C-X e per il trasferimento del protone dal C alla base.L'altezza della superficie al di sopra del piano corrisponde all'energia di quella particolare struttura.
La coordinata di reazione è la proiezione sul piano del cammino ad energia più bassa dai reagenti ai prodotti

27
visto dall'alto:
*p è la proiezione dello stato di transizione in un ipotetico esempio in cui H+ è trasferito a metà e C-X è rotto a metà.
R (reagenti) e P (prodotti) corrispondono a minimi di energia Q (carbanione) e S (carbocatione) corrispondono a massimi di energia
Le frecce indicano la direzione in cui si sposta lo stato di transizionequando:- si alza l'energia di R (o si abbassa quella di P)- si alza l'energia di P (o si abbassa quella di R)- si alza l'energia di Q (o si abbassa quella di S)- si alza l'energia di S (o si abbassa quella di Q)- si alza l'energia lungo RQ (o si abbassa lungo SP)
13241+2 5
Superficie dell'energia e coordinata di reazione per un'ipotetica E2, in cui l'energia dei reagenti è più elevata rispetto all'esempio precedente, mentre l'energia degli altri angoli è invariata
La forma della superficie è variata così da portare la struttura dello stato di transizione più vicino ai reagenti (freccia 1 nella proiezione)
28
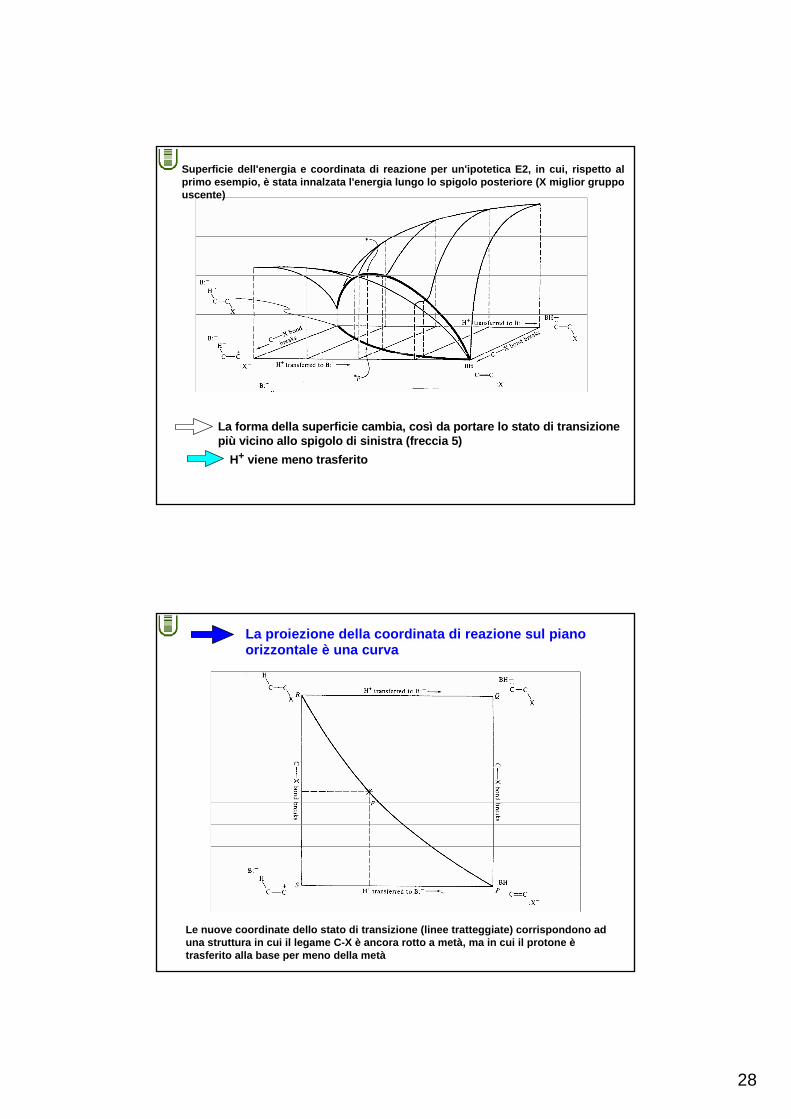
Superficie dell'energia e coordinata di reazione per un'ipotetica E2, in cui, rispetto al primo esempio, è stata innalzata l'energia lungo lo spigolo posteriore (X miglior gruppo uscente)
La forma della superficie cambia, così da portare lo stato di transizionepiù vicino allo spigolo di sinistra (freccia 5)
H+ viene meno trasferito
La proiezione della coordinata di reazione sul pianoorizzontale è una curva
Le nuove coordinate dello stato di transizione (linee tratteggiate) corrispondono ad una struttura in cui il legame C-X è ancora rotto a metà, ma in cui il protone ètrasferito alla base per meno della metà

29

30
CoOLO
CoL
H
H
LL L
L
PO O
O OMe-
X
CoOLO
CoL
H
LL L
L
PO O
O OMe-
X
PO O
OMe
CoOLO
CoL
H
LL L
L
-
XO-
PO O
OMeO
-
CoOLO
CoL
H
LL L
L
H
-
- H+..
PO O
O OMe-
XX
O-
PO O
OMeOH
-
HO:
+
esempio: uso dei diagrammi di More O’Ferrall in catalisi
La scissione catalitica dei diesteri dell’acido fosforico è molto studiata (modello della scissione di DNA e RNA).
Tra i migliori catalizzatori: complessi metallici dinucleari
Reazione catalizzata:
Reazione non catalizzata:
Sperimentale: reazione non catalizzata ca. 37% rottura gruppo uscente
reazione catalizzata ca. 77% rottura gruppo uscente
Il Nu migliore dovrebbe attaccare il P senza bisogno di uscita del gruppo uscenteAnalisi con diagramma di More O’Ferral
R
Sistemi con due anelli a 4 termini: si alzal’energia dello spigolo
P
Lungo la coordinata di reazione lo ST simuove verso lo spigolo in alto a destra; lungo la coordinata non di reazione simuove verso lo spigolo in basso a destra
La risultante mantieneuguale y, aumenta x

31
REAZIONI SINCRONE E REAZIONI IN PIU' STADI
Quando nello sviluppo di uno stato di transizione è coinvolto più di un processo, per specificare il grado con cui questo processo è avvenuto c'è bisogno di più di una variabile del progresso.
considerando una reazione generaleY- + G-X Y-G + X-
dove G rappresenta la struttura centrale che resta sostanzialmente invariata, Y- è il gruppo che attacca e X- quello che esce.
Se il processo è sincrono Y......G......Xδ- δ- =/
basta la coordinata di reazione
La reazione può avvenire in più stadi:
Come si colloca lo stato di transizione?
G X G+ + X- ; G+ + Y- G Y I+ Y- + G X G XY( )- G Y + X- I-
Nel meccanismo sincrono puro, per la descrizione dello stato di transizione nonserve considerare intermedi anionici o cationici, ma sono possibili deviazioni daquesta situazione ideale e perciò serve un modo per misurare quanto carattereanionico o cationico è "mescolato" allo stato di transizione
tipo di reazioneDescrizione dello stato
R I+ I- P
Y- R-X Y-R+X- Y-R-X+ Y-R X-
B- H-R-R-X B- H-R-R+X- BH -R-R-X BH R=R X-
Y- RCOX Y-RCO+X- RC(O-)XY RCOY X-
Y+ Ar-X (Y-Ar-X)+ Y+Ar-X+ Y-Ar X+
Y- Ar-X Y-Ar+X- (Y-Ar-X)- Y-Ar X-
Sostituzione nucleofila alifatica
Sostituzione nucleofila acilica
β-Eliminazione
Sostituzione elettrofila aromatica
Sostituzione nucleofila aromatica
Diagrammi RPI (reagent-intermediate-product)

32
METODO DI Pross e Shaik (o DEL LEGAME DI VALENZA)
Le teorie MO e VB sono teoricamente equivalenti, dato che entrambeportano alla stessa funzione d'onda.
il metodo di Pross e Shaik consiste nell'esaminare gli stati elettronici dei legami di valenza di reagenti e prodotti e di connetterli su un diagramma dell'energia infunzione della coordinata di reazione, chiamato diagramma di correlazione distato.
Consideriamo la rottura del legame polare R-X (con X più elettronegativodi R)
In prima approssimazione lo stato fondamentale ed il primo stato eccitato di RXsi possono rappresentare:
R-Xstato fondamentale = R. .X
R-Xprimo stato eccitato = R+ :X-
Variazione delle due configurazioni con la distanza R-X in FASE GASSOSA(Figura a sinistra):
Una descrizione molto migliore di questi due stati si ottiene mescolando partedella configurazione ad energia più elevata a quella più bassa (in modo legante)e parte di quella ad energia più bassa con quella più alta (in modo antilegante)
R-Xstato fondamentale = R. .X + (R+ :X-)λ
R-Xprimo stato eccitato = R+ :X- + (R. .X)λ
L'energia di questi stati corretti varia dando la linea tratteggiata (figura a sinistra)

33
All'aumentare della distanza tra R e X, la regione dello spazio in cui c'èun'elevata probabilità di trovare il secondo elettrone su X ha unasovrapposizione MOLTO PICCOLA con la regione dello spazio in cui c'è un'elevata probabilità di trovare l'elettrone dispari su R
In soluzione (figura di destra) la configurazione R+:X- è fortementestabilizzata per solvatazione
primo stato eccitato = R. .X configurazione delle particelle dissociate: stato fondamentale = R+ :X-
Se non ci fosse nessuna interazione di configurazione, le curve delle dueconfigurazioni si incrocerebbero
nella realtà, questo incrocio "nelle intenzioni" non avviene
Man mano che il legame R-X è stirato, l'energia della configurazione fondamentaleaumenta, fino a che diventa quasi uguale all'energia della configurazione del primostato eccitato
Man mano che le energie delle due configurazioni diventano quasi uguali,aumenta l'interazione tra loro.

34
avoided crossing (incrocio evitato)l'aumento dell'interazione diminuisce l'energia della curva più bassa ed aumentaquella dell'energia più alta, nella zona dell'incrocio atteso e l'incrocio viene evitato
Il risultato è che le energie dello stato elettronico seguono le curvetratteggiate e la reazione allo stato fondamentale in soluzione dà unacoppia ionica
Effetto di un cambiamento delle condizioni di reazione sullo stato di transizione?
La diminuzione di energia dello stato fondamentale del prodotto (a) o l'aumento di energia dello stato fondamentale del reagente fanno sì che l'incrocio evitato (cioè lo stato di transizione) avvenga prima lungo la coordinata di reazione.
Nel modello di Pross e Shaik anche le energie del primo stato eccitato direagenti e prodotti influisce sulla posizione dello stato di transizione, in-fluenzando la pendenza della curva
Effetto, sulla posizione dello stato di transizione per la rottura di R-X, dell'aumento di energia del primo stato eccitato dei reagenti (curva tratteggiata)
il modello di Pross e Shaik rende molto semplice che cosa è che provocala barriera di energia
Si forma una barriera, perché i reagenti devono aprire i loro gusci di valenza(stirare o rompere legami) e perciò aumentare di energia, fino a che incontranouno stato elettronico decrescente, nel quale possono passare.

35
Applicando il modello ad una reazione SN
Si sviluppa una descrizione VB scrivendo una combinazione linearedi un insieme ragionevole di configurazioni
Y:- + R-X R-Y + X:-
lo stato iniziale è dominato dalla configurazione R e lo stato finale da P,anche se ciascuno stato comprende contributi da tutte le configurazioni
L'energia della configurazione di R aumenta, perché è energeticamente sfavorevole mantenere la configurazione dello stato iniziale man mano che si procede lungo la coordinata di reazioneL'energia dello stato P diminuisce, msan mano che ci si avvicina allostato finaleNel punto in cui queste configurazioni si incontrano, si ha un "incrocioevitato" ed il sistema in reazione segue il cammino ad energia più bassa.
Y:- R. .X R, simile ai reagentiY. . R :X- P, simile ai prodotti
Y:- R+ :X- I+, simile al carbocationeY:- R:- X+ I-, simile al carbanione
Una variazione strutturale che stabilizzi P rispetto a R abbassa il profilodi energia di P, dando uno stato di transizione anticipato (comportamentosecondo Hammond)

36
Supponendo che la configurazione I+ non contribuisca in modo significativo agli stati iniziale e finale, ma sia significativa nella zona di mezzo della coordinata di reazione:

37
TEORIA DELLA VELOCITA' DI MarcusLa teoria di Marcus considera che la barriera di energia della reazione siacostituita da due contributi
una parte termodinamica, da ΔG°una parte cinetica intrinseca, che è la barriera che esisterebbese reagenti e prodotti avessero la stessa energia libera
Considerando la reazione:HO- + H3CBr HOCH3 + Br-
Marcus descrive la coordinata di reazione per questo processo come due parabole che si intersecano
una che rappresenta la superficie dell'energia potenzialeper lo stiramento del legame C-Br nei reagentiuna che rappresenta la superficie dell'energia potenzialeper lo stiramento del legame C-OH nel prodotto
La forma e la collocazione di queste parabole sono determinate dalla variazionecomplessiva di energia libera e dalla barriera intrinseca.
2. Si fanno intersecare due parabole per la reazione simmetrica
Br- + H3CBr BrCH3 + Br-
La reazione è termoneutra e le parabole si fanno incrociare in modo da intersecarsi ad una altezza che corrisponde all'energia libera di attivazione sperimentale
Per determinare la barriera intrinseca:1. Si fanno intersecare due parabole per la reazione simmetrica
HO- + H3COH HOCH3 + HO-
La reazione è termoneutra e le parabole si fannoincrociare in modo da intersecarsi ad una altezzache corrisponde all'energia libera di attivazione sperimentale
ΔG = 174.9 kJ/mole = 41.8 kcal/mole=/
ΔG = 99.2 kcal/mole = 23.7 kJ/mole =/

38
3. La teoria di Marcus definisce la barriera intrinseca disegnando una nuova coppia di parabole, la cui intersezione avviene ad una altezza che corrisponde alla media delle barriere delle due reazioni simmetriche
ΔG = 137.2 kcal/mole = 32.8 kJ/mole =/
4. La barriera per la reazione vera viene costruita spostando le parabole mediate verticalmente, così da ottenere il ΔG° osservato sperimentalmente
ΔG = 137.2 kcal/mole = 32.8 kJ/mole =/
Il nuovo punto di intersezione è dato dalla formula di Marcus:
dalla geometria analiticaΔG= =/intr
+ 12
ΔG° +(ΔG°)2
16 ΔG =/intr
=/ΔG
ΔG=/intr energia di attivazione intrinseca (media delle barriere di attivazione
delle due reazioni simmetriche)
ΔG=/intr
= 12
=/ΔG( XX+ =/ΔG
YY )=/ΔG calcolato=/ΔG sperimentale 95.0 kJ/mole =22.71 kcal/mole
92.5 kJ/mole =22.1 kcal/mole
(ΔG°)2
16 ΔG=/intr
di solito è piccolo e trascurabile (tutte le volte che ΔG° < ΔGo confrontabile)
=/
ΔG= =/intr
+ 12
ΔG°=/ΔG
Dalla geometria del modello di Marcus si può derivare la posizione dello statodi transizione lungo la coordinata di reazione, come il valore x del punto diintersezione delle parabole =/
8 ΔG =/intr
= +12
ΔG°x
Rendere ΔG° più negativo anticipa lo stato di transizione; rendere ΔG°più positivo posticipa lo stato di transizione

39
Per quanto riguarda l'influenza di variazioni della barriera di attivazione intrinseca:
Per una reazione esotermica (ΔG° < 0)
abbassare ΔG=/intr lasciando invariato ΔG° ANTICIPA LO STATO DI
TRANSIZIONE
Per una reazione endotermica (ΔG° > 0)
abbassare ΔGintr lasciando invariato ΔG° RITARDA LO STATO DITRANSIZIONE
Accordo con il postulato di Hammond

40
Dando a ciascuna curva del modello di Marcus un peso decrescente con l'aumentare della distanza dal suo minimo e sommandoli si ottiene una parabola con laconcavità verso il basso, analoga al modello di Thornton, che risponde allo stesso modo alle perturbazioni (linea continua nella figura)
Non sono molti i sistemi per cui si possa definire facilmente la reazione di “autoscambio”. La teoria di Marcus non è direttamente applicabile a molte reazioni complesse.
ST
Se la reazione diventa piùesoergonica, diventa più veloce
ad un certo punto non si ha barriera
Rendendo ancora piùesoergonica la reazione, la barriera si reintroduce
Regione invertitadi Marcus
Energie libere sempre più negative rallentano la velocità

41
L’impatto maggiore della teoria di Marcus si è avuto nello studio ditrasferimento di elettrone (in chimica ed in biologia)
elettrochimica, reazioni redox, reazioni enzimatiche, fotosintesi
I trasferimenti di elettrone avvengono da un DONATORE (D) ad un ACCETTORE (A), con una velocità molto maggiore (10-16) delle vibrazioni nucleari (10-13): perciò I nuclei non cambiano posizione
L’energia degli orbitali di donatore ed accettore deve essere la stessa prima del trasferimento.
I livelli di energia degli orbitali del donatore e dell’accettore sono in continuo flussodovuto ai movimenti nucleari interni ed ai movimenti del solvente
Per il trasferimento, il donatore e l’accettore devonoraggiungere contemporaneamente particolari geometriee solvatazioni che diano livelli di energia in accordo.
L’energia necessaria per portare alla stessa energiagli orbitali di donatore ed accettore si chiama energiadi riorganizzazione, che costiruisce la barriera per iltrasferimento di elettrone.
ΔG =/ = λ4
1 +(ΔG°)2
λ
λ = energia di riorganizzazione
- .
- .
accettore
accettore
k
λ = λi + λo λi = energia di riorganizzazione interna, misura le differenze dienergia dovute a variazioni di lunghezze ed angoli di legame
λo = misura l’energia coinvolta nella riorganizzazione del gusciodi solvatazione necessaria per il trasferimento di elettrone
Conferma della “regione invertita di Marcus”
Il trasferimento di elettrone avviene attraverso un sistema rigido steroidico
Accettore e donatore vengono mantenuti alla stessa distanza, ma cambiandoaccettore, cambia il suo potenziale di riduzione
L’anione rdicale del bifenile è stato generato per pulse radiolisi

42
La velocità di reazione aumenta e poi diminuisce, anche se le reazionisono sempre più esoergoniche