targeted therapies in epilepsy - canadian league … dravet histamine antagonist bryostatin: kcnt1...
TRANSCRIPT

Targeted Therapies inEpilepsy
Mary B. Connolly
BC Children’s Hospital and UBC

Trials supported by
Biocodex
Novartis
Sage Therapeutics
I will discuss available data on targeted treatments in specific conditions which are off label or not standard therapy.
Disclosures

Objectives
To discuss precision medicine in epilepsy in 2017
To review targeted therapy in neurometabolic disorders, channelopathies, TSC and several specific gene disorders
To review current state of pharmacogenomics in epilepsy
Speculate about how treatment of epileptology will evolve over next few years
3

Epilepsy Challenges in 2017 Seizures remain treatment resistant in 30-40% of
individuals despite new AEDs and other advances.
Surgery is underutilized.
Cognitive and neuropsychiatric co-morbidity high.
Mortality remains high.
Epilepsy genetics shifting from academic pursuit of gene discovery to a clinical discipline based on molecular diagnosis and stratified medicine.
However, in 2017 only a small percentage of patients benefit from precision or targeted therapies

Goals of Epilepsy Treatment
Cure of disease
Control of seizures with absence of side effects
Balance of therapeutic effect and side effects, costs and benefits

Treatment of Epilepsy in 2017
Anti-seizure medications
Dietary therapies
Surgery
Brain Modulation Vagus nerve stimulation, Deep brain stimulation,
Responsive stimulation
Thermocoagulation
Precision treatment guided by genomics
Repurposing of other drugs

Epilepsy in Children: Impact on Learning
Prospective cohort of children with epilepsy diagnosed < 8 years
Early age at seizure onset and pharmaco-resistance resulted in lower IQ
Pharmaco-resistance had most profound impact in 0-3 year group
Berg et al., Neurology 2012

Precision Medicine: Francis Collins
Medicine for most of human history has been one-size-fits-all. But we’re all different, and we’re finding out that the diseases we have lumped together under one label are actually at the molecular level quite distinct.
Precision medicine tries to understand what’s underneath those disease layers and tries not to lump everyone together but think about individual differences.

Essence of Precision Treatment
Is it possible to predict which patients will respond best to a specific treatment
Is there a specific or novel therapy for individual based on genes or pathway involved in causing their epilepsy
Can we predict which patients will develop side effects of treatment
EpiPM Consortium Lancet Neurol 2015; Symonds et al. Current OpinNeurol 2017;

Treatable Neurometabolic Diseases
Rare diseases
Important not to miss diagnosis
Number of treatable metabolic diseases increasing (now ~84)
Newborn screening detects some disorders
Major research interest of team at BCCH (Treatable Intellectual Disability Endeavour -TIDE)
App available to guide physicians with investigations

Inherited Metabolic Epilepsies
Vitamin responsive (B6, PNP, Biotinidase)
Transporter (GLUT1, Cerebral Folate)
Aminoacid and organic disorders (Serine synthesis, Creatine synthesis)
Mitochondrial disorders (KD)
Urea Cycle disorders
Neurotransmitter disorders
Disorders of glucose homeostasis

Pyridoxine Dependent Epilepsy
• Historically diagnosis trial of pyridoxine
• Biomarkers: urine AASA
• Testing for Antiquitin gene mutations
• Can be diagnosed with NGS
• Treatment with pyridoxine alone not effective in all patients

GLUCOSE TRANSPORTER 1 Deficiency: Variable Clinical Features (SLC2A1)
Neonate
• Main manifestations:– Seizures– Apnea– Abnormal eye
movements
Older children
• Paroxysmal:– Alternating hemiplegia – Intermittent ataxia– Headache, confusion
• Permanent:– Mental retardation– Ataxia– Spasticity
GLUCOSE TRANSPORTER 1 Deficiency
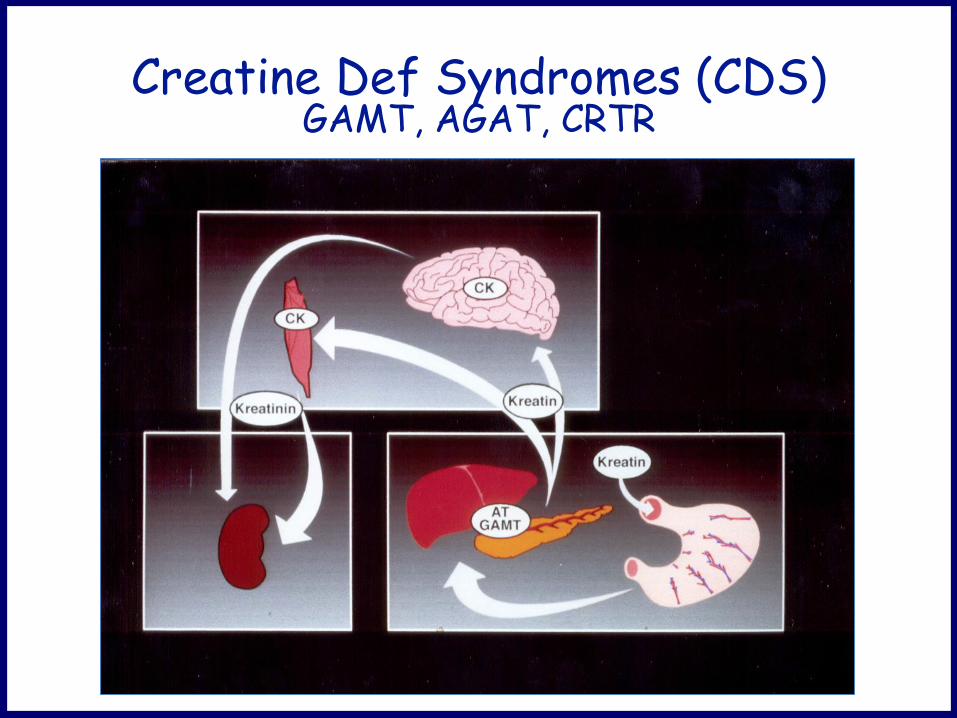
Creatine Def Syndromes (CDS) GAMT, AGAT, CRTR

Sodium Channel Disorders (SCN) In CNS: 4 major SCN family
SCN1A (Nav1.1, 2q24): Dravet syndrome (DS), FS+, FHM3, SUDEP
SCN2A (Nav1.2, 2q24): BFNIS, GEFS+, MFEI, OS, EOEE, DS
SCN3A (Nav1.3, 2q24): Unclear SCN8A (Nav1.6, 12q13): EE
Brunkaus A et al., J Med Genet 2014

Dravet Syndrome
• 1978: 1st described by Charlotte Dravet• Seizure onset: 1st year of life • Hemiclonic or generalized TC seizures • Other seizure types• Normal development prior to seizure onset• Moderate to severe intellectual impairment
Ataxia and crouched gait • Mortality ~ 10%

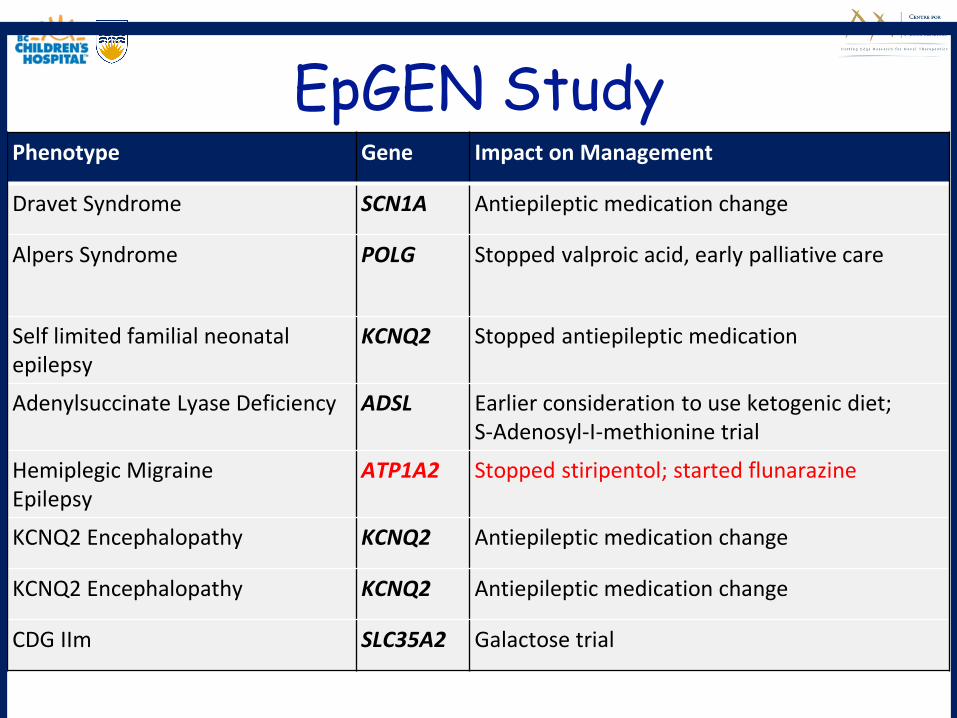
EpGEN StudyPhenotype Gene Impact on Management
Dravet Syndrome SCN1A Antiepileptic medication change
Alpers Syndrome POLG Stopped valproic acid, early palliative care
Self limited familial neonatal epilepsy
KCNQ2 Stopped antiepileptic medication
Adenylsuccinate Lyase Deficiency ADSL Earlier consideration to use ketogenic diet; S-Adenosyl-I-methionine trial
Hemiplegic MigraineEpilepsy
ATP1A2 Stopped stiripentol; started flunarazine
KCNQ2 Encephalopathy KCNQ2 Antiepileptic medication change
KCNQ2 Encephalopathy KCNQ2 Antiepileptic medication change
CDG IIm SLC35A2 Galactose trial

Treatment of Dravet Syndrome
• Seizures treatment resistant in most patients
• Best options: Clobazam, Valproic acid, Topiramate, Stiripentol, Levetiracetam Ketogenic diet
• Avoid: Carbamazepine, Oxcarbazepine, Phenytoin, Lamotrigine, Vigabatrin, Phenobarbital, Rufinamide

Stiripentol
• Modulator of GABA
• Potentiates GABAergic inhibitory post-synaptic currents,maintaining phasic inhibition (by action on α4 GABAA
receptors and tonic inhibition

StiripentolStudy Design N Findings
Dressler et al. 2015
Open label, retrospective
9 All treated with VPA + CLB + STPResponder rate 89%, mean sz reduction 73%
Inoue et al. 2014
Open-label, prospective
24 67% responders (17% sz-free)Significant reduction in sz duration and use of rescue meds
Wirrell et al. 2013
Open-label, retrospective
82 Overall sz frequency reduced in 57-80% depending on co-therapyAll had reduction in frequency of prolonged szs and most had significant reduction in rescue med use and ER visits/hospitalizations
Inoue et al. 2009
Open label, prospective
23 >50% reduction in GTCS in 61% (9% sz free)
Thanh et al. 2002
Open-label, retrospective
46 Seizure frequency decreased by 66% (p<0.001)Reduction in sz duration (p<0.002), status epilepticus (p<0.001)

Fenfluramine
Amphetamine-like agent used as anti-obesity drug
Taken off market due to possible cardiac adverse effects (valve thickening, pulmonary hypertension)
Releases serotonin by disrupting vesicular storage and reversing 5HT transporter function

Fenfluramine
12 pts treated (mean dose 0.34, range 0.12-0.90 mg/kg/d) for mean of 11.4 yrs (range 1-19 yrs)
No pt had received STP, ketogenic diet or bromide
10 (83%) continued on fenfluramine at follow-up
7 (58%) were seizure free for longer than 1 year
Side effects:
Mild, non-progressive heart valve thickening in 2
Decreased appetite in 2
Ceulemans et al. Epilepsia 2012

Fenfluramine Studies in DS
Prospective studies
Children on VPA and Clobazam
75% reduction in major seizures
Children on Stiripentol, Clobazam, VPA
Study in Lennox-Gastaut syndrome about
Probable study in adults with DS

KCNQ2 Related Epilepsy
Phenotypes: Benign familial neonatal epilepsy
Epileptic encephalopathy
Encodes potassium voltage-gated channel subfamily KQT member 2
Drugs which act on sodium channels such as carbamazepine and phenytoin often effective
Retigabine shown to be effective in some patients

Adverse Effects of Retigabine
Blue skin discoloration and retinal pigment abnormalities
Urinary retention

KCNT1 Mutations
KCNT1: encodes a weak voltage-dependent and intracellular sodium-activated potassium channel.
Missense mutations:
Autosomal dominant nocturnal frontal lobe epilepsy
De novo gain of function:
Migrating focal epilepsy of infancy

Quinidine in KCNT1 Mutations
Made from Cinchona plant Broad spectrum K channel blocker (KCNT1) Anti arrythmic Patients with malignant migrating epilepsy have
responded to 34-60mg/kg/day Case reports of patients having improvement in
seizures and development with gain of function mutations

Targeted Therapy in Tuberous Sclerosis Complex
cortical tubers cytomegalic neuronsand balloon cells
mTOR pathway

Tuberous Sclerosis Complex (TSC)
90% develop epilepsy
Onset often in 1st year of life
Uncontrolled epilepsy: high risk of autism and intellectual impairment
Advances: Biomarkers of epilepsy and treatment prior to seizure onset
mTOR inhibitors effective for epilepsy

Problems in TSC
How do we predict which patients will do well with surgery?
When to use KD which has mTORinhibition?
What is role of mTOR drugs in epilepsy and how to access them?


mTOR Inhibitors and Epilepsy
TSC mouse models: Rapamycin reversed astrogliosis/neuronal disorganization
Prevent development of seizures Improve learning and attention
Clinical trials: Rapamycin reduced seizure frequency Everolimus Phase 1/2 and EXIST-1 SEGA trials showed
reduction of seizures in some patients EXIST 3 trial: Everolimus effective in reducing seizures

Tubers
Tubers are dynamic entities that evolve over time? Cyst-like cortical tubers progress on MRI Display reactive gliosis on pathology consistent
with a chronic process Elevated mTOR activity in tubers

Exist 3 Trial inTSC366 patients: placebo (n=119), low dose (n=117) or high dose (n=130)
Lee et al, Nature genetics. 2012; D'Gama et al, Annals of Neurology, 2015; French et al, Lancet 2016
>50% sz frequency reduction
15.1% placebo28.2% low dose40% high dose
Median percentage reduction in sz frequency:
14.9% with placebo 29.3% low exposure39.6% high exposure

mTOR Pathway and Other Disorders
Tuberous sclerosis Complex FCD Focal epilepsy without normal imaging Post-traumatic epilepsy Polyhydramnios megalencephaly symptomatic epilepsy
syndrome Neurofibromatoses 1 and 2, Peutz–Jeghers syndrome Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome Proteus syndrome Lhermitte–Duclos syndrome, von Hippel–Lindau
syndrome Huntington’s, Parkinson’s, Schizophrenia, Alzheimer’s Learning and memory

Role of mTOR Inhibitors
Reports of seizure control:– Focal Cortical Dysplasia (esp FCD II)– Polyhydramnios, Megencephaly and Symptomatic
Epilepsy– Hypothalamic Hamartoma

Pathways

Focal Cortical Dysplasia: Genetic Aspects
Somatic and germline mutations Familial or sporadic
GATOR 1 (11%) 1q21.1-q44 duplication affecting AKT3
DEPDC5: 8% of focal epilepsy
+/- focal cortical dysplasia I, IIA, IIB
Seizures from variable foci
NPRL2 and NPRL3: 1–3% of focal epilepsy ;

DEPDC5 and GATOR1 Mutations in Focal Epilepsy
» GATOR1: 21%

mTOR Inhibitors in Post-traumatic Epilepsy
Changes in brain involve mTORC1 activation Axon sprouting, increased synthesis and
phosphorylation of proteins, cell migration, and neurogenesis
TBI mouse model studies with rapamycin: GABAergic inhibition reduce mossy fiber sprouting and excitation of
dentate granule cells Decreased seizure frequency

Pharmacogenomics in Epilepsy
• How genetic factors affect response to treatment –efficacy and adverse drug reactions
• Genetic categories of variation:• Drug pharmacokinetics, drug pharmacodynamics, epilepsy
genes, ? Other
• Impact of NGS on pharmacogenomics in epilepsy and precision medicine yet to come• Collaborative projects: EpiPGX, CPNDS (The Canadian
Pharmacogenomics Network for Drug Safety)

Influence of genetic factors on response and adverse reactions to AEDs: established evidence
Mediator Genetic Factor Effect Recommendations
Pharmacokinetics & Pharmacodynamics
Variation in CYP2C9 (*2/*3)
Risk of developing concentration-dependent neurotoxicity from phenytoin
Pre-treatment genetic testing not routineMonitor clinically and drug levels
Epilepsy Genes Mutations in SLC2A1
Bi-allelic mutations in ALDH7A1
GLUT-1 deficiency
B6-dependent epilepsy
CSF/genetic testingketogenic diet
AASA/genetic testingPyridoxine or pyridoxal 5’ phosphate
Other HLA-B*15:02
HLA-B*13:01
SJS and TEN induced carbamazepine/ other aromatic AEDS in patients from Han Chinese, other South Asia groups
Increased risk carbazmazepine-induced hypersensitivity reactions-European, Japanese, all ancestries
Pre-treatment testing groups at risk (Level A)a
If positive-use alternative as 1st line
As above except all ancestries (Level B)a
S Balestrini & S Sisodiya Neuroscience Letters 2017. aAmstutz U et al Epilepsia 2014.
rash

Subject 033• 9 month presentation focal status-left arm
• Development normal
• Family history febrile seizures in father, paternal aunt and paternal great uncle
• Failed 5 anti-seizure medications
• MRI, neurometabolic, microarray negative

033 – Genetic Findings 4 weeks
Compound heterozygous mutations in POLG (each inherited by a parent) likely pathogenic ALPERS syndrome
POLG chr15:89866657 C/G rs113994097 C/TDDD NM_001126131 exon13 c.G2243C p.W748S
POLG chr15:89871929 C/G novel C/DDDD NM_001126131 exon5 c.G1157C p.R386P

033 – Genetic findings ~ 4 weeks
• The proband is a compound heterozygous for two POLGmutations – likely pathogenic– ALPERS syndrome
-> immediate impact on clinical care!?
POLG chr15:89866657 C/G rs113994097 C/TDDD NM_001126131 exon13 c.G2243C p.W748S
POLG chr15:89871929 C/G novel C/DDDD NM_001126131 exon5 c.G1157C p.R386P
Immediate clinical impact (valproic acid)


Potential Treatments
Clemizole: Dravet histamine antagonist
Bryostatin: KCNT1 Macrolide lactone: inhibit K current
Silencing microRNAs: sz & neuroprotective results regulate post-transcriptional expression of
protein coding mRNA, dendritic spine morphology
SAGE 217: Dravet, Rett syndrome

The Future
• Early diagnostic NGS in early onset epilepsy or epilepsy of unknown cause
• Drugs targeted towards genetic defect or molecular pathway or to bypass the pathway disturbed be genetic variant
• Functional test systems measuring single gene targets and multicomponent networks will be required
• Pharmacogenomics essential to reduce side effects in both patients and embryos at risk
• Potential for further novel or repurposed therapies akin to "Personalized Oncogenomic Program”
• Malformations of brain development and prevention of post traumatic epilepsy: ? Use mTOR inhibitors in the future
Rosenow et al. Epil Behav 2017;Bauer et al. Epil Behav 2017; Lindhout Lancet Neurol 2015;