targeted brain delivery of novel nanoparticles via
TRANSCRIPT

TARGETED BRAIN DELIVERY OF NOVEL NANOPARTICLES
VIA THE BLOOD-BRAIN BARRIER THIAMINE TRANSPORTER
by
PAUL RICHARD LOCKMAN, B.S.N.
A DISSERTATION
IN
PHARMACEUTICAL SCIENCES
Submitted to the Graduate Faculty of Texas Tech University Health Sciences Center
in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
Advisory Committee
David D, Allen (Chairperson) Thomas J, Abbruscato
Mansoor A, Khan Reza Mehvar
Joseph D, Roder Quentin R. Smith
Accepted
August, 2003

ACKNOWLEDGEMENTS
I tiiank my mentor and my advisor David D. Allen, R.Ph., Ph.D. for his patience,
uistiiiction, guidance, leadership, and support. This work is the product of hkn giving
time tiirough daily interactions. I also tiiank Quentin R. Smitii, Ph.D. for his critical
review. I also thank tiie remainder of my committee: Thomas J. Abbruscato, Ph.D,
Mansoor Khan, Ph.D., Reza Mehvar, Pharm.D, Ph.D. and Joseph Roder, D.V.M., Ph.D.,
for scientific leadership. Without their effort, tiiis work would not have been completed.
I extend my deepest gratitude to Russell J. Mumper, Ph.D., Joanna M. Koziara,
MS, and Moses Oyewumi, Ph.D., for the nanoparticle preparation, characterization, and
biodistribution studies shown throughout this dissertation. I thank Karen E. Roder BS, for
her technical expertise. I acknowledge the Vascular Biology Research Center at Texas
Tech for critical input. I consider this dissertation a work m partnership with the
aforementioned individuals. I am indebted to the American Federation for Aging
Research: Glen AFAR Research Scholarship Project and an American Foundation for
Pharmaceutical Education pre-doctoral feUowship for financial support.
To my family, I have few words allowed to express my gratitude. Payton and
Karli you give me so much hope and happmess. Mother you set an example of what
someone can be. Laveme and Alfred you constantly provide laughter and joy. John and
Vicki for being awesome parents, grandparents and most importantly friends.
To my wife Melissa, thank you for your unwavering constant love. It is your
support, belief in me, and the promise of our future that has aUowed me to endure.
11

TABLE OF CONTENTS
ACKNOWLEDGEMENTS n
LIST OF TABLES vi
LIST OF FIGURES vu
ABBREVIATIONS ix
CHAPTER
1. INTRODUCTION I
I.l Introduction I
2. NANOPARTICLE TECHNOLOGY FOR DRUG DELIVERY ACROSS THE
BLOOD-BRAIN BARRIER 5
2.1 Introduction 5
2.2 Manufacturing Methods 9
2.3 Nanoparticle Characterization 14
2.4 Mechanisms of Nanoparticle Transport
across the BBB 15
2.5 Nanoparticles Loaded with Analgesics 18
2.6 Nanoparticles Loaded with Chemotherapeutics 20 2.7 Reticuloendothelial System: An Obstacle for CNS Drug
Targeting to the Brain 21 2.8 Current Clinical Use of Nanoparticles 24
2.9 Conclusions 24
2.10 References 26
in

3. IN VIVO AND IN VITRO ASSESSMENT OF BASELINE BLOOD-BRAIN BARRIER PARAMETERS IN THE PRESENCE OF NOVEL NANOPARTICLES 38
3.1 Introduction 38
3.2 Materials and Methods 40
3.3 Results 47
3.4 Discussion 51
3.5 References 56
4. /i\^ 5/71/BLOOD-BRAIN BARRIER TRANSPORT
OF NANOPARTICLES 67
4.1 Introduction 67
4.2 Materials and Methods 69
4.3 Results 75
4.4 Discussion 78
4.5 References 82
5. EVALUATION OF BLOOD-BRAIN BARRIER THIAMINE EFFLUX USING THE IN SITU RAT BRAIN PERFUSION TECHNIQUE 90 5.1 Infroduction 90
5.2 Materials and Methods 92
5.3 Results 97
5.4 Discussion 100
5.5 References 107
IV

6. BRAIN UPTAKE OF THIAMINE-COATED NANOPARTICLES 117
6.1 Introduction 117
6.2 Materials and Methods 119
6.3 Results 128
6.4 Discussion 132
6.5 References 138

LIST OF TABLES
2.1 Ideal properties of polymeric-based nanoparticles for drug delivery across the blood-brain barrier 31
2.2 Examples of nanoparticles used for delivery of drugs across the
blood-brain barrier 32
3.1 Effect of nanoparticles on in vitro blood-brain barrier parameters 59
4.1 Short-term stability of nanoparticles at 4°C 85 4.2 Stability of nanoparticles in various biologically relevant media at 37°C 86
5.1 Capillary localization of [^H]-thiamine and ['" C]-sucrose ti-acer after a 45 s perfusion uptake 110
5.2 Calculated cerebrovascular permeability and saturable kinetic parameters for [^H]-thiamine transport at the blood-bram barrier I l l
5.3 Regional effltix constants for [ H]-thiamine in the absence of vascular thiamine 112
VI

LIST OF FIGURES
2.1 Strategies to increase CNS drug delivery 33
2.2 Various types of drug-loaded nanoparticles for CNS delivery 34
2.3 Pharmacokinetic analysis of drug release from nanoparticle 35
2.4 Mechanisms of drug release from nanoparticle 36
2.5 Mechanisms of nanoparticle CNS entry 37
3.1 Effect of nanoparticles on cerebral perfusion flow 60
3.2 Regional cerebral perfusion flow and vascular integrity in the presence
of nanoparticles 61
3.3 Effect of nanoparticles on vascular volume 62
3.4 Appearance of ['" CJ-sucrose in receiver chamber 63
3.5 Effect of aged nanoparticles on barrier parameters 64
3.6 Choline and thiourea uptake in the presence of nanoparticles 65
3.7 Western blot analysis of blood-brain barrier tight-junction proteins 66
4.1 Entrapment efficiency of [ H]-cetyl alcohol m E72 nanoparticles 87
4.2 Release profile of [ H]-cetyl alcohol from E72 and E78 nanoparticles 88
4.3 Tune course of [^H]-nanoparticle and [''*C]-sucrose uptake uito rat brain 89
5.1 Non-luiear time course of [ H]-thiamine brain uptake 113
5.2 Concentration dependent [ H]-thiamine bram uptake 114
5.3 Time course of [ H]-thiamine washout from brain 115
5.4 Brain efflux [''H]-thiamine constants 116
6.1 Transmission electron micrograph of thiamine-coated nanoparticles 141
Vll

6.2 Tune course of brain uptake for [^H]-nanoparticles and [ H]-thiamine -nanoparticles 142
6.3 Time course of washout from brain for [^H]-nanoparticles and [ H]-thiamine -nanoparticles 143
6.4 [^H]-Thiamine uptake inhibition by nanoparticles 144
6.5 Calculated transfer coefficients for [ H]-nanoparticles and [ H]-thiamme -nanoparticles during 45 to 120 s perfusion time frame 145
6.6 In vivo biodistribution of thiamine-coated and peg-coated nanoparticles 146
Vll l

ABBREVIATIONS
BBB: Blood-brain barrier
CNS: Central Nervous System
NP: Nanoparticle
PA: Cerebrovascular permeability surface-area product
NPE72: Nanoparticle formulation made from Brij72/Tween 80
NPE78: Nanoparticle formulation made from emulsifying wax/Brij78
IX

CHAPTER 1
INTRODUCTION
1.1 Introduction
Nanoparticles (NP) are solid colloidal particles ranging in size from one to 1000
nm that may be utilized as brain drug delivery carriers. NPs may provide significant
advantage to current blood-brain barrier (BBB) penefration sti-ategies by masking drug
permeation Ihniting characteristics. Additionally, NPs may slow drug release in blood
decreasmg peripheral toxicity. This dissertation is focused on tiie development of a novel
NP formtilation that was engineered to target brain.
Previous strategies of brain drug delivery and the advantages of NP technology
are discussed m Chapter 2. Primary methods of NP preparation, characterization and
variable manufacturing factors (i.e., type of polymers and surfactants, NP size, and tiie
drug molecule) are detailed in relation to movement of the colloidal carrier across the
BBB. Currently, reports evaluating NPs for brain dehvery have studied anesthetic and
chemotherapeutic agents. These studies are reviewed for efficacy and mechanisms of
transport. Physiologic factors such as phagocytic activity of the reticuloendothelial
system and protein opsonization may limit the amount of brain delivered drug and
methods to avoid these issues are also discussed.
While NPs have been shown to overcome drug permeation lintuting
characteristics, there are conflicting toxicological data published with regard to BBB
uitegrity and gross mortality. Chapter 3 addresses this issue with two novel NP types

bemg evaluated in vivo using tiie in situ brain perfusion technique and in vitro using
bovine brain microvessel endothelial cells. The results of tiiese initial stiidies show
neitiier NP formulation, demonsh-ate significant differences for cerebral perfusion flow in
vivo. Furthermore, observed in vitro and in vivo data showed no statistical changes m
barrier integrity, membrane permeability, or facilitated choline ti-ansport. Western blot
analyses of occludin and claudin-1 confirmed no protein expression changes with
incubation of eitiier NP. In summary, tiie NP formulations appear to have lack of effect
on primary BBB parameters in established in vitro and in vivo BBB models.
The two novel NP types were tiien evaluated as potential carriers for drugs across
tiie BBB. The NPs were radiolabeled by enti-apment of [•'HJ-cetyl alcohol. Chapter 4
shows entrapment efficiency and release of radiolabel as quality assurance measures. The
brain penetration of both formulations was then measured by the in situ rat brain
perfusion method. For both NP types, statistically significant brain uptake was observed
compared to [ '*C]-sucrose in perfusion time frames of less than 60 s.
Considering: (1) the brain perfusion studies, (2) the lack of toxicity, and (3) the
relative short time of BBB NPs penetration, it appeared the coUoidal carrier primarily
circumvented the BBB by passive permeation. Yet, while brain distribution is critical for
the success of NPs as a delivery system, the abUity of the NP to specifically target brain
should also be considered. The proposed passive permeation of our NP formulations may
hypothetically suggest increased peripheral organ distribution. Thus, to target brain we
incorporated thiamine as a surface ligand on the NP.

Initial kmetic evaluation of tiie BBB thiamine carrier as a NP bram vector is
shovm m chapter five. Thiamine is an essential, positively charged (under physiologic
conditions), water-soluble vitamin requiring tiransport into brain. Evaluation of brain
uptake and efflux of [^H]-thiamine using the in situ rat brain perfusion techiuque was
completed. To confirm [^Hj-tiiiamine brain distiibution was not related to BBB
endotiielial cell uptake, we compared parenchymal and endotiielial cell distribution of
[^H]-thiamine using capillary depletion. The data supported previous literature findings
suggesting BBB thiamine uptake is via a carrier-mediated ti-ansport mechanism, yet
extended the literature by redefining the kinetics with more sensitive methodology. The
influx mechanism and efflux rate were comparable throughout brain regions despite
documented differences in glucose and thiamine metabolism. Based upon BBB thiamine
transport capacity and kinetics the nutrient fransporter may have efficacy as a NP brain
delivery vector.
Thiamine was engineered on the surface of the NP by chemically linking thiamine
to distearoylphosphatidyletholamine via a poly-ethylene glycol spacer (Mw 3350). This
formulation was then radiolabeled. Irutial experiments focused on assessing uptake of
[ H]-NP formulations with and without tiiiamine surface ligands. Biodistribution studies
of both NP formulations were also carried out in Balb/c mice. The results in chapter six
include: (1) effectiveness of microemulsion strategies inNP production, (2) kinetic
modelmg for brain uptake of NP formulations with and without thiamine surface ligands,
and (3) iiutial data suggesting mechaiusms for NP bram entry.

In summary, this dissertation shows the development of a novel NP formulation
that penetrates bram without apparent BBB toxicity. Of significance, the thiamuie-coated
NP associated with the BBB thiamine fransporter and had mcreased brain distribution
after 45 s. We propose the incorporation of a NP thiamine ligand facilitates binding
and/or association with BBB thianune fransporters, which may ultimately aid in brain
targetmg.

CHAPTER 2
NANOPARTICLE TECHNOLOGY FOR DRUG
DELIVERY ACROSS THE BLOOD-
BRAIN BARRIER
2.1 Introduction
Nanoparticles (NPs) are solid colloidal particles ranging in size from one to 1000
nm, consisting of various macromolecules in which tiierapeutic drugs can be adsorbed,
enfrapped or covalently attached. One utility of NPs is to serve as novel drug delivery
carriers to tissues throughout the body. This is accomphshed by masking the membrane
barrier limiting characteristics of the therapeutic drug molecule, as well as to retain drug
stability, with that of the properties of tiie colloidal drug carrier. This disguising of the
drug may allow access across the previously impermeable membrane. Once the NP
reaches the desired tissue, release of the drug may occur by de-sorption, diffiision or
erosion through the NP matrix or polymer wall, or some combination of any or all
mechanisms. Currently, NPs are gaining interest as therapeutic drug carriers across the
blood-braui barrier (BBB).
For drugs to be successfully delivered to theu: target, many factors need to be
considered during manufacture of the NP. Table 2.1 illustrates the requirements for
polymeric based NPs m delivering drug to the cenfral nervous system (CNS). This
chapter will highlight many of the advantages and limitations found in CNS NP
technology.

The BBB represents one of the strictest barriers of m vivo therapeutic drug
delivery. The barrier is defined by restiicted exchange of hydrophilic compounds, small
proteins and charged molecules between tiie plasma and the CNS. The primary
mechanism of regulation centers on the anatomical basis of the BBB. The BBB is
comprised of a contiguous layer of endothelial cells connected by tight junctions that
circumferentially surround tiie entire cell margm at tiie brain capillaries. Similar, but not
identical, junctions exist at the choroid plexus epithelium and arachnoid membrane.
These tight endothelium junctions {zonulae occludens) can be ~ 100 times tighter than
junctions of other capillary endothelitim (1). Thus, the barrier has many of the same
properties of a continuous cell membrane, allowmg lipid soluble molecules transport
across the membrane whereas hydrophilic solutes demonstrate minimal permeation (2).
While the characteristics of the BBB provide a formidable obstacle for drug
therapy m the (3NS, they are not insurmountable. Attempts to overcome the barrier in
vivo have focused on altering barrier integrity or characteristics, or changing the
characteristics of the drug. (Figure 2.1 illusfrates sfrategies utilized to increase CNS drug
delivery). Tight junctions at the BBB have been opened by artificially created osmotic
pressure and the adminisfration of bradykinin analogs such as RMP-7. Junctional operung
of tiie BBB enables paracellular CNS drug delivery across tiie barrier. Specifically,
Rapoport et al. (3) demonstrated that with infracarotid administration of hypertonic
arabinose solutions a fransient (hours) modification in BBB permeability allowed > 20
fold increase in bram concentrations of hydrophihc compounds. Sanovich et al. (4)
demonsfrated increased permeability of tiie BBB to lanthanum by the admmisfration of

tiie bradykinin analog RMP-7. However, opening the barrier by either mechanism allows
CNS entiy of toxins and unwanted molecules, potentially resulting in significant damage
(5).
Since tiiere are risks associated with changing the permeability of tiie BBB,
attempts have been made to modify drugs to more readily cross tiie barrier. Pro-drugs are
an excellent example of such drug manipulation. With this metiiod, tiie original
compound is manipulated to make it more lipid-soluble, providing greater CNS
penefration. While pro-drugs work well, not aU compounds (i.e., neuroti-ophic factors
such as glial derived neurofrophic factor, brain derived neuroti-ophic factor, or nerve
growth factor) may be maiupulated in tiiis way and still maintain tiierapeutic efficacy.
Furthermore, mcreased lipid solubility may significantiy alter pharmacokinetic
parameters such that clearance and half-life become undesfrable as is the case with
chlorambucil derivatives (6). NPs may be superior to both of these techruques since no
manipulation of the barrier or the drug is necessary.
Another alternative for brain drug delivery is utilization of native carriers
expressed at the BBB. Carriers, also known as fransporters, deliver essential hydrophilic
and large compounds across the barrier such as choline, purines, amino acids and
lipoproteins. While physiologically expressed to perform transport of needed nutrients,
they may be used to deliver drugs to the CNS as well. For example, the chemotherapeutic
agent D, L-NAM (for brain tumors) is fransported across the BBB by the large neufral
amino-acid carrier (7). Other drugs also cross the BBB by this carrier, such as baclofen,
melphan, sulfoxime, butiiionine, azaserine, and alpha-methyl DOPA (8). While carriers

are an atfractive means of CNS delivery, the drugs must have carrier-mediated
specificity, tiius limiting tiieir molecular characteristics. NPs may cross the BBB by
passive diffusion or receptor-mediated endocytosis, carrying the drug across the BBB,
witiiout requiring drug molecular specificity. However, if high-affinity ligands for these
fransporters are placed on tiie surface of tiie NP, it may be possible to use NPs as a vector
for brain or other site-directed delivery.
While these fransporters function in the dfrection of influx from blood to brain,
efflux fransporters are also present. These efflux transporters (P-glycoprotein, multi-drug
resistance protein, and others) are likely located at the BBB for detoxification and/or
prevention of nonessential compounds from entering brain. While the natural effect is
beneficial, it is yet another obstacle in delivering drugs to the CNS, as many agents that
readily cross the BBB are subsfrates for efflux fransporters. CNS disposition of a drug
and its metabolites frequently are determined by p-glycoprotein and, furthermore, p-
glycoprotem may function as a defense mechaiusm determining bioavailabiUty and CNS
drug concenfrations (9). Therefore, while non-selective inhibition of efflux fransporters
may lead to a therapeutic benefit, there is risk of CNS or peripheral toxicity.
Direct injection into brain is another approach to cfrcumvent the BBB. This has
been accomplished using different techniques. Some examples of direct CNS drug
delivery include delivering nerve growth factor to an Alzheimer's patient by
infracerebroventricular mfusion into cerebrospmal fluid (10), direct unplantation mto
brain parenchyma of a polymer matiix containing nerve growth factor (11), and
fransplantation of encapsulated cells which secrete nerve growth factor mto forebram

neurons (12). While these techniques can be successful to achieve certain therapeutic
goals, disadvantages exist for dfrect injection, primarily the requfrement of exfremely
mvasive neurosurgery. Limiting tiie potential to freat only gravely ill patients and then
oitiy if tiie affected area is accessible. Furthermore, diffusion of the drug from tiie
injection site may limit efficacy.
While dfrect drug injection into brain may be viable for circumventing the BBB
(considering the limitations described above), there are other strategies as well. For
example, uifracarotid infusion increases the concenfration gradient at the BBB resulting
in increased brain concentrations. However, infracarotid infusions present with
disadvantages of (1) risk of vascular injury and (2) drug streaming with resultant
heterogeneous brain distribution (13).
The use of NPs as colloidal drug carriers may have decided advantage over
previously mentioned approaches to circumvent the BBB. In this chapter, we wiU discuss
NPs in relation to delivery across the BBB, primary methods of preparation, NP
characterization, current research published on NP brain dehvery and the
reticuloendothelial system as an obstacle to CNS delivery.
2.2 Manufacturing Methods
NPs vary in types of polymers, stabiHzers and surfactants used in their
manufacturing. Each excipient added may have a significant affect on brain drug uptake,
drug distiibution and persistence in plasma. When manufacturing NPs as drug carriers in
vivo and in vitro testing should consider tiie factors listed m Table 2.1. Primary

manufactiiring metiiods include: (1) emulsion polymerization, (2) interfacial
polymerization, (3) desolvation evaporation, and (4) solvent deposition (14). Various NP
sti-uctiares may result secondary to each manufacturing metiiod. Furthermore, drug
loading can be accomplished by absorption, adsorption and encapsulation (Figure 2.2).
2.2.1 Emulsion Polymerization
Emulsion polymerization, which characterizes botii radical and aiuoruc
polymerization, is one of the most frequentiy used techniques for NPs production. The
process consists of building a cham of polymers, which acts as a drug carrier, from smgle
monomer units of a given compound. Polymerization occurs spontaneously at room
temperature after initiation by either free radical or ion formation. Triggers for polymer
growth include high-energy radiation, UV tight or hydroxyl ions. Once polymerization is
complete the solution is filtered and neufralized to remove any residual monomers. The
polymers form micelles and droplets (NPs) consisting of approxunately 100 to 10
polymer molecules. The mass of polymers inherent in this type of NP formulation
provides the available space that acts as a carrier for adsorption or absorption of the drug
(Figure 2.2, A and B)(l 5).
Emulsion polymerization has numerous advantages ui NP formulation. It is rapid
compared to other methods, stabilizers and surfactants are generaUy not needed and it is
easily scaled up for large manufacturing requirements (16).
10

Emulsion polymerization can also be accomplished in an organic phase ratiier
tiian an aqueous phase. This process has been adapted for use with polyakyl-
cyanoacrylate NPs.
The primary disadvantages of emulsion polymerization is its requfrement for free-
radicals, radiation or UV light to tiigger polymerization. The necessity of tiiese tiiggers
precludes the incorporation of peptides and proteins during polymerization (Table 2.1).
Hence m order to mamtain the stability of mcorporated proteins or peptides the NPs must
be purified via dialysis and centrifugation to remove residual monomers. Furthermore,
emulsion polymerization also has tiie disadvantage tiiat it requfres large amounts of
orgaiuc solvents and thus creates potential for environmental toxicity (17).
2.2.2 Interfacial Polymerization
Interfacial polymerization is similar to emulsion polymerization in that monomers
are used to create polymers. However, it is mechanistically different. Interfacial
polymerization occurs when an aqueous and organic phase are brought together by
homogenization, emulsification, or micro-fluidization under high-torque mechaiucal
stirring. This precludes the inclusion of peptides/proteins at this step secondary to
mechartical shearing. For example, the creation of polyalkyl-cyanoacrylate nanocapsules
(Figure 2.2, C) was completed when the monomer was dissolved in oil and slowly added
through a small tube to an aqueous phase with constant stirring. The monomer then
spontaneously form 200-300 nm capsules by anionic polymerization. Drug incorporation
11

was accomplished by adding the drug with the monomer in the organic phase. This
encouraged the drug to be enveloped in the matrix of the NP (18).
A subset of interfacial polymerization is the process of adding a solvent mixture
of benzyl benzoate, acetone, and phospholipids to the organic phase containing drug and
monomer. It has been suggested that tiiis process encourages formation of the
nanocapsitie shell between the aqueous phase and the benzyl benzoate drops m the
organic phase (19). One advantage of interfacial polymerization may be the encapsulation
of the drug. Once the drug is encapsulated, it is protected until it reaches the target tissue
and degradation occurs. In the case of CNS delivery, it is desirable to protect or disguise
the drug until it is past the BBB and can be released mto brain.
2.2.3 Denaturation and Desolvation
Macromolecules such as albunun and gelatm can also be used in the production of
NPs. Using such macromolecules capitalizes on the natural affinity between
macromolecule and drug. Two primary processes, oil denahiration and desolvation, are
used to process macromolecules as NPs. Oil emulsion denaturation is where large
macromolecules are frapped m an organic phase by homogenization. Once frapped, tiie
macromolecule is slowly mfroduced to an aqueous phase undergomg constant stirring.
The particles formed by tiie infroduction of tiie two immiscible phases are tiien hardened
by cross-linking witii an aldehyde (20) or heat denahiration (21). Unlike tiie above
preparation metiiods, tiie quantity of tiie macromolecules, temperatiire and emulsification
12

time have little effect on the resultant particle size. The greatest effect seen on this
property is the type of oil used (15).
Macromolecules may also form NPs by "desolvation." Desolvation occurs when
the macromolecule is dissolved in a solvent where macromolecules reside in a swollen,
coiled conformation. The swollen macromolecule is then induced to coil tightly by
changing the environment pH, charge or the use of a desolvating agent such as ethanol.
The macromolecule may then be fixed and hardened by cross-linking to an aldehyde.
Drugs bound to the protein or macromolecule, prior to the cross-linking step, become
enfrapped in the newly-formed particle. The major drawback of this method is that the
quantities of NPs and the drug absorbed are very low compared to other methods (22).
Solid lipid NPs are created by high-pressure homogenization. Solid lipid NPs
share the same benefits of fat emulsions and liposomes while avoiding some of tiie
respective drawbacks. Solid lipid NPs may be sterilized and autoclaved similar to fat
emulsions (23) and possess a soUd matiix which provides a confroUed release avoiding
the burst release seen witii fat emulsions (24).
2.2.4 Nanoparticle Drug Release
Figure 2.3 Ulusfrates pharmacokmetic analysis of tiie release of doxorubicm from
NPs which was characterized by Gupta et al. (25) as a bi-exponential equation:
C,= Ae-"* + Be-P*
13

where C - concentration of drug remaining in the NP at a given tune, A, B = system-
characteristic constants, intercepts of release, a = initial rate constant and p = Secondary
rate constant.
This model suggests there is an initial rapid removal of the drug from tiie NP
possibly related to early loss of drug loosely associated on tiie surface of the NP (Figure
2.4). Once the rapid component of release is complete, tiiere is a slower, much more
confroUed release of drug owing to either NP degradation or diffusion of drug through the
NP matrix or shell. This latter release has been characterized by both zero order and first
order kmetics (25). Zero-order kinetics tend to occur with biodegradable NPs whereas
first order release occurs with non-biodegradable NPs. Regardless of the kinetics of drug
release, it appears release is dependent on NP degradation or erosion of nanocapsule shell
(15).
Drug release is also dependent on the structure of the NP. Drugs may be
incorporated in a solid lipid NP, encapsulated in a NP shell, adsorbed onto the surface or
cross-luiked to the NP. These factors, plus the type and length of the polymer, can have
sigruficant effects on the release of the drug (15).
2.3 Nanoparticle Characterization
The primary characterization of NPs is the size of the newly-formed particle.
Several physicochemical factors may influence NP size. Factors such as pH of tiie
solution used during polymerization, amovmt of initiation tiiggers (heat, radiation etc...)
and concenfration of monomer units may affect NP size m the 100-200 nm range (26,27).
14

However, density, molecular weight, hydrophobicity, surface charge and surface
morphology may also be helpfiil in predicting drug dehvery to specific targets. Kreuter
(15) reviewed tiie primary metiiods of determinmg tiie physical and chemical properties
NPs.
Photon correlation specfroscopy using light scattermg is one of tiie most common
metiiods of sizing. This method relies on Browinian motion tiiat predicts that smaller
particles have increased motion m solution. By illuminatuig the particles witii a laser
beam and analyzing tiie time dependency of the tight changes, the NP can be accurately
sized. Characterizations of molecular weight, density and crystallinity can be
accomplished by gel chromatography, helium compression pycnometry and x-ray
diffraction, respectively (15).
2.4 Mechaiusms of Nanoparticle Transport across the BBB
Transport of NPs across the BBB has been characterized similarly to many of the
known fransport mechanisms described for other drugs (figure 2.5). Passive diffiision at
the BBB occurs when a drug dissolves in the lipid membrane of cerebrovascular
endothelial cells and then released uito the brain. Passive diffusion depends on: (1) the
lipophilicity of the drug, (2) charge, (3) concenfration gradient, (4) molecular weight, and
(5) the degree of protein binding. Passive diffusion is characterized by Pick's law of
diffusion where:
-dC/dt =k(Ci-C2).
15

Transport of drugs across tiie BBB is dependent on carrier proteins at the capillary
endothelial cells. Canier-mediated fransport at the BBB can occur as facilitated fransport
along tiie concentration gradient, active transport (regardless of tiie concenfration
gradient) and endocytosis. Canier-mediated fransport is analyzed witii Michaelis-Menten
saturation kinetics, where:
Rate = Vmax *C + kd. (Km + C)
NPs fransport across tiie BBB has been hypotiiesized to occur by receptor-
mediated endocytosis and/or passive diffusion.
2.4.1 Passive Diffusion
The effect of lipid coating polysaccharide NPs and its effect on fransport across
an in vitro BBB (bovine bram capillary endothelial cells) was evaluated by Fenart et al.
(28). The authors compared uptake of polysaccharide NPs, cross-linked with phosphate
(anioruc) and quaternary ammonium (catioruc) ligands, with and without a surrounding
Upid bilayer. They demonsfrated that when a lipid bilayer contairung dipalmitoyl
phosphatidyl choline and cholesterol coating is applied to the charged NPs, a 3-4-fold
increase in brain uptake was observed. Furthermore, the NP remained mtact as it crossed
the BBB and fransport was not due to altered BBB integrity. They also demonstrated that
albumin, a large protein normally precluded from brain, had a 27-fold increase in uptake
when coated with the same lipid bilayer. However, ui the presence of erythrocytes, a
significant decrease ui fransport was seen, possibly due to an NP-erythrocyte mteraction.
16

The use of NPs for drugs that dononstrate permeation of the BBB by passive
diffiision in die free state may improve die drugs brain distributioD profile. This was most
notably dononstrated widi amitriptyline by Shioeder et aL (29). After anritriptyline was
adsorbed onto polybutjdcyanoaoylate NPs, using die stabilizer dexlran-70,000 and
polysoibate-80 as a surfactant, a 10-fold inaease m brain concentrations was found. The
audiors hypothesized die increase was secondary to an eohaoicemerA of die plasma
concentration resulting in a largo* gradioit at the BBB and thus greater concoitratioas of
die drug ottoing die brain by passive dififiision. Furdionun-e, NP degradation products
may act as adsmption oihancas (30) leading to increased passive diffusion.
2.4.2 Recqjtor-Mediated Endocytosis
The transport medianism of labded polybutylcyanoaoylate NPs coated witii
polysoibate-SO ao-oss die BBB has be«i suggested to be cellular endotiielial endocytosis.
NPs were administered intraarterially and localized by transmission electiron microscopy
and fluorescence microscopy. When die NPs were not coated with surfectants, die
particles remained in die blood vessels (31). Bordiardt et al. (32) confirmed tiiis finding
using ['^CHabeled NPs in brain mio-ovessel endotiielial cells. Furdiennore, iqrtake of
NPs coated widi polysobate-SO was inhibited by die phagocytic inhibitor cytochalsin B
(33).
It has been suggested tiiat apo-E adsorbs onto NPs coated witfi polysorbate 20,40,
60, or 80. A logical conclusion is that polysorbate coated NPs be subject to die same
cndocytotic process low-doisity lipoproteins undergo at the BBB (34).
17

Multiple mechanisms for NP fransport have been described. While initially tiie
mechanisms seem confradictory, many factors may influence NP BBB penefration. These
include the type of polymers, size of tiie NP, types of surfactants and the drug molecule
itself Further studies should evaluate tiie mechanisms responsible for NP BBB transport,
considering each of these factors.
2.5 Nanoparticles Loaded with Analgesics
Earlier studies in the use of NPs to improve brain drug distribution mvolved
analgesic agents such as dalagrin, kytorphin and the neuromuscular blocking agent
tubocurarine (Table 2.2). The anesthetics were chosen because they exhibit therapeutic
effects when given directly m brain, but with peripheral adminisfration no anesthesia is
seen. These reports suggest the anesthetics do not cross the BBB appreciably from the
plasma ui the time frames evaluated.
Polybutylcyanoacrylate NPs coated with polysorbate-80 have been shown to
deliver the polypeptide dalargin across tiie BBB after IV mjection. When dalagrin was
adsorbed onto tiie polybutylcyanoacrylate NPs coated with polysorbate-80 as a
surfactant, significant and prolonged analgesia took place foUowmg infravenous
adminisfration. After anesthesia was induced, tiie central actmg opiate antagonist
naloxone was administered peripherally causuig a blockade of anestiiesia. This suggests
the dalagrin-induced anestiiesia is mediated by cenfral mechanisms. Furthermore, when
dalagrin is adsorbed onto polybutylcyanoacrylate witiiout tiie surfactant coating no
18

analgesic effect was seen (30, 35). Botii stiidies mdicate that polybutylcyanoacrylate NPs
witii tiie surfactant can aid in the delivery of drugs across tiie BBB.
In addition to dalagrin, stiidies have been conducted to evaluate the analgesic
effects of Kyotorphin loaded NPs (35). It produced cential analgesic effects but only
when tiie particle was stabilized by dextran 70 kDa.
Anestiietics previously demonsfrated to be excluded from brain were shown to
cross tiie BBB by use of NPs (36). These stiidies assessed adsorbed ttibocurarine (a
quaternary ammonium compound that has minimal BBB permeation) onto
polybutylcyanoacrylate particles coated with polysorbate-80. Tubocurarine, when given
mfravenously, is a myoparalytic found in minimal concenfrations in the cerebrospmal
fluid. It does not affect spontaneous and evoked bioelectric activity of the bram when
given infravenously. However, when combined with NPs as described above and
administered peripherally, seizure elecfroencehpalograph patterns were noted m animals.
This study demonsfrates the potential of specific NPs to carry charged cations across the
BBB (36).
NPs have been demonsfrated to fransport charged analgesic agents across the
BBB. Furthermore, these agents produce similar therapeutic effects when given
mfravenously as when admiiustered directiy into the brain. The choice to use analgesic
agents to assess NP brain penefration is simplistic. Transport is determined by induction
of analgesia. However, one must also consider whether tiie proposed analgesic effect is
from tiierapeutic efficacy, simply that of toxicity or a mixture of botii. All of the above
19

stiidies are short-term tenninal stiidies and one must be cautious in extrapolating
application to acute usage in humans.
2.6 Nanoparticles Loaded Witii Chemotiierapeutics
Tumors witiiin brain have provided unique therapeutic chaUenges. Many
chemotiierapeutic drugs are polar molecules tiiat do not readily penefrate the BBB. This
is further complicated by the need to maximize time and exposure concenfration of tiie
chemotiierapeutic agent to cancerous ceUs. However, when tiiese two factors are
maxunized to provide therapeutic efficacy, plasma concenfrations are high resultmg in
significant systemic toxicity. NPs as chemotherapeutic carriers have been studied as a
solution to these issues (Table 2.2).
Doxorubicin, an anthracycline antibiotic, is a chemotherapeutic agent that
intercalates into DNA, resulting in inhibition of DNA synthesis. Doxorubicin is a polar
molecule that does not normally cross the BBB. When doxorubicin was given
infravenously adsorbed on polybutylcyanoacrylate NPs with polysorbate-80 as a
surfactant, CNS doxorubicin concenfrations were therapeutic at ~6mcg/g in the brain
(37). Furthermore, the NP containing doxorubicin administered infravenously to rats with
mtracranially fransplanted glioblastomas led to a cure in -40% of these rats. In confrast,
of the rats that received free doxorubicin, only one survived in seven contiol groups (34,
38).
When the lipophihc anti-cancer drug camphotericin was adsorbed on solid lipid
NPs, the area under the curve and the mean residence time were increased compared to
20

confrol most notably in brain, heart and reticuloendothelial cell containing organs.
Further tiiere was significant protection of the more effective lactone form of the drug
from hydrolysis to tiie carboxalate form. Thus, solid lipid NPs may be a promising
sustained-release and drug targeting system for lipophilic CNS anti-tiimor drugs (39).
The delivery of anti-ttimor drugs by NPs is a promising alternative to surgery and
dfrect injection of drugs m tiie CNS. One significant benefit of ttimor tiierapy witii NPs as
a drug carrier is tiie prolongation of mean residence time ui tiie body. While tius benefit
may mcrease the exposure of tiie tumor to the chemotherapeutic agent, it also prolongs
exposure of the remainder of tiie body to tiie drug, potentially increasmg toxicity.
2.7 Reticuloendotiielial System: An Obstacle for CNS Drug Targetmg
Initially, targeting NPs to brain proved unsuccessful when given infravenously.
Failure of NPs to reach the CNS m appreciable quantity was due to NP uptake by the
reticuloendothelial system (i.e., the mononuclear phagocytic system). The
reticuloendothelial system is a collective group of mononuclear cells originating from
bone marrow that have phagocytic responsibility in removing small foreign particles
from the vascular space. While the cells are foimd throughout the body, a high number of
cells are localized in the liver (Kupffer cells), spleen and bone marrow. The
reticuloendothelial system significantly removes a large portion (up to 80-85%) of NPs
from the vascular space, subsequentiy limiting exposure of NPs at the cerebrovasculature
(40).
21

Sfrategies to overcome reticuloendothelial system uptake mclude external
guidance of magnetically responsive NPs and NP coating with antibodies or hydrophilic
surfactants. Magnetic guidance consists of manufactiiring NPs containing magnetite
(Fe304) and tiie use of an external magnet. Specifically, doxombicin was incorporated
into tiiese "magnetic" NPs (41). When a magnet (3000 gauss) was placed near tiie rat-tail,
a 24-fold increase in tfie area under the curve was observed in comparison to free drug.
This approach has been repeated m NP targeting to the brain. Pulfer and Gallo (42)
mjected magnetic NPs m tiie carotid artery and simultaneously applied an external
magnetic field to the brain area. After sacrificing the animals at 30 minutes and 6 hours,
brain magnetite concenfrations were determined by atomic absorption specfroscopy.
Magnetic brain guidance resulted m mcreased braui concenfrations and decreased non-
target tissue concenfrations. While efficacy of drug delivery has been shown by tius novel
techiuque, it may be impractical for use in human subjects. Issues related to this
technique for human use include duration of magnetic force necessary to exert the effect,
chronic force effects, chronic toxicity of this NP and its metabolites (notably magnetite)
as well as compliance by sick patients.
Another solution to the problem of rapid uptake of NPs by the reticuloendothelial
system is coating with surfactants. Primary surfactants include polaxamine 908 and
polysorbate-80. Poloxamine 908 used as a surfactant on hydrophobic NPs was shown to
reduce reticuloendothelial system uptake m the hver when compared to uncoated drug
(72% versus 19%). There was also a significant reduction of NP uptake by the spleen,
lungs and bone marrow (14). Polysorbate-80 has also been shown to be effective in
22

minimizing NP uptake by tiie reticuloendotiielial system (43). When NPs contaming
bound doxorubicin, were admmistered intravenously, witii and without surfactant,
significant differences were found. The plasma half-life of doxorubicm increased
approximately 4-fold compared to free drug adminisfration. Furthermore, it was noted
tiiat greater concenfrations of the drug were seen m the reticuloendothelial system organs
using uncoated and coated NPs when compared to free drug (37).
A concern regarding polysorbate-80 coated polybutylcyanoacrylate NPs is
toxicity. There is conflicting evidence of lack of toxicity and toxic effects occurring when
this surfactant and polymer is used. Olivier et al. (44) found dalargin adsorbed onto NPs
of polybutylcyanoacrylate coated with polysorbate-80 and administered at 166 mg/kg
resulted in an ~ 30% mortality rate. The surviving mice had significantly decreased
activity, after a short burst of hyperactivity and obvious discomfort. The authors
suggested this toxicity is mediated by rapid esterase biodegradation of the
polybutylcyanoacrylate polymer to toxic compounds. Furthermore, tests in an in vitro
model of the BBB showed mcreased permeability to sucrose, a vascular integrity marker,
suggesting a compromise m the barrier by polybutylcyanoacrylate coated NPs. Kreuter
refutes the interpretation of toxicity suggested by the Olivier study. It was argued that
there was no CNS toxicity since the normal response to dalagrin, an opioid, is a short
hyperactive burst of activity followed by decreased activity. In addition, the BBB
opening was seen in vitro and not in vivo, and multiple other authors have not observed in
vitro BBB opening (35).
23

In summary, phagocytic activity of tiie reticuloendothelial system presents a
major obstacle in delivering NPs to the brain. Some investigators circumvented tiiis
problem by manipulating NP content (magnetite) or adding surfactants (poloxamine and
polysorbate-80). Altiiough all stiidies have shown an increase in bram drug delivery,
tiiere may be a concern regarding tiie toxicity of tiiese novel preparations. Further stiidies
should explore tiie mechanisms of toxicity, LD50's and practicality of use in human
subjects.
2.8 Current Clinical Use of Nanoparticles
Currentiy, tiie only drug marketed using polymeric NPs is the diagnostic agent
Abdoscan® by Nycomed. Abdoscan® is a coUoidal NP contaming crystalline
superparamagnetic fron oxide particles stabilized with low molecular weight dexfran. The
primary use of this novel NP is for diagnostic unaging of spleen and liver tumors.
Abdoscan takes advantage of the NP phagocytosis process occurring in
reticuloendothelial system organs. Phagocytic uptake of colloidal NPs results in increased
magnetic resonance imaging in the organ. Since tumor cells are not capable of
phagocytosis, there is no enhanced unaging in the tumor and a sharp confrast is produced
between healthy and tumor tissue. However, at this time, there are no marketed products
using polymeric NPs for drug delivery across the BBB (45).
2.9 Conclusions
After 30 years of research on polymeric NPs, this delivery system practically does
not exist chnically, yet NPs appear to have significant potential m delivering drugs to
24

brain. It has been demonstrated tiiat NPs can cross tiie barrier intact by passive diffiision
and receptor-mediated endocytosis. Further, site-dfrected bram delivery of NPs may be
possible by tiie use of high affinity NP surface ligands to native BBB fransporters. Once
the NP is in brain, a slow confroUed release of the drug occurs targetmg CNS tissue and
avoiding other organs, which should reduce peripheral or systemic toxicities.
Currentiy, areas of research have focused on analgesic and chemotherapeutic
agents. The use of analgesics is a good choice for basic research determination of NP
crossmg the BBB. However, there is no clinical need to deliver these specific analgesic
agents to brain. The study of NP-loaded chemotherapeutic agents target the therapeutic
problem of cancer in the CNS. The majority of chemotherapeutic agents do not cross tiie
BBB, and ones tiiat do are removed by tiie efflux protein, p-glycoprotem. NPs appear to
mcrease the bram area under the curve of botii doxorubicm and camphotericm. Further
stiidies should consider the evaluation of other CNS diseases, which are limited m
therapeutic freatments by the BBB.
25

2.10 References
1. A.M. Butte, H.C. Jones, and N.J. Abbot. Electiical resistance across tiie blood-brain barrier in anaestiietized rats: a developmental sttidy. J. Physiol, 429: 47-62 (1990).
2. Q.R. Smitii. Advances in Neurology. In: R. Wurtinan, (ed.), Alzheimer's Disease, Raven Press, New York, Vol 51, pp.217-222 (1990).
3. S.I. Rapoport, K. Ohno, W.R. Fredericks and K.D. Pettigrew. Regional cerebrovascular permeability to [ ''Cjsucrose after osmotic opening of the blood-bram barrier. Brain Res. 150(3): 653-657 (1978).
4. E. Sanovich, R.T. Bartus, P.M. Friden, R.L. Dean, H.Q. Le, and M.W. Brightinan. Patiiway across blood-brain barrier opened by tiie bradykinin agonist, RMP-7. Brain Res. 705(1-2): 125-135 (1995).
5. N.H. Greig. Drug delivery to the brain by blood-brain barrier circumvention and drug modification. In: E. A. Neuwelt (ed.). Implications of the Blood-Brain Barrier and its manipulation, Plenum press. New York, pp. 311-367 (1989).
6. N.H. Greig, E.M. Daly, D.J. Sweeney, S.I. Rapoport. Pharmacokinetics of chlorambucil-tertiary butyl ester, a lipophilic chlorambucil derivative that achieves and maintains high concenfrations in brain. Cancer Chemotherapy & Pharmacology. 25(5): 320-325 (1990).
7. Y. Takada, D.T. Vistica, N.H. Greig, D. Purdon, S.I. Rapoport and Q.R. Smitii. Rapid high affinity fransport of a chemotherapeutic amino acid across the blood-brain barrier. Cancer Res. 52(8): 2191-2196(1992).
8. Q.R. Smith. Drug delivery to the brain and the role of carrier mediated transport. In: L.R. Drewes, A.L. Betz (eds.). Frontiers in cerebral vascular biology: Transport and its regulation, Plenum Press, New York, pp. 83-93 (1993).
9. M.F. Fromm. P-glycoprotem: a defense mechanism luniting oral bioavailability and CNS accumulation of drugs. Int. J. Clin. Pharmacol Ther. 38(2): 69-74 (2000).
10. L. Olson, A. Norberg, and H. Von Hoist. Nerve ^ovAh factor affects '^C-nicotme bmding, blood flow, EEG, and verbal episodic memory ui an Alzheuner patient- case report. / . Neural Transm. 4: 79-95 (1992).
11. C.E. Krewson, M.L. Klarman and W.M. Saltzman. Distiibution of nerve growtii factor foUowmg direct delivery to brain mterstitum. Brain Res. 680: 196-206 (1995).
26

12. J.H. Kordower, S.R. Winn, Y.T. Liu, E.J. Mufson, J.R. Sladek Jr, J.P. Hammang, E.E. Baetge and D.F. Emerich. The aged monkey basal forebram: Rescue and sprouting of axotomized basal forebrain neurons after grafts of encapsulated cells secretmg human nerve growtii factor. Proc. Natl Acad. Scl USA. 91(23): 10898-10902 (1994).
13. J.B. Blacklock, D.C. Wright, R.L. Dedrick, R.G. Blasberg, R.J. Lutz, J.L. Doppman and E.H. Oldfield. Drug sfreaming during infra-arterial chemotiierapy. J. Neurosurg. 64(2): 284-91 (1986).
14. J. Kreuter. Nanoparticles and microparticles for drug and vaccme delivery. J. Anat. 189(3): 503-505 (1996).
15. J. Kreuter. Nanoparticles, In: J. Swarbick, J.C. Boylan (eds.). Encyclopedia of Pharmaceutical Technology, Marcel Dekker, New York, pp. 165-190 (1994).
16. J. Kreuter. Large scale production problems and manufacturing of nanoparticles. In: P. Tyle (ed.). Specialized Drug Delivery Systems, Marcel Dekker, New York, pp. 257-266 (1990).
17. G. Birrenbach, P.P. Speiser. Polymerized micelles and thefr use as adjuvants in unmunology. / . Pharm. Scl 65: 1763-1766 (1976).
18. A.L. Khouri, N. Fallouh, L. Roblot-Treupel, H. Fessi, J.P.H. Devissageuet and F. Puissieux. Development of a new process for the manufacture of polyisobutyl-cyanoacrylate nanoparticles. Int. J. Pharm. 28: 125 (1986).
19. H. Fessi, F. Puisiuex, J.P. Devissagauet, N. Ammoury and S. Benita. Nanocapsule formulation by interfacial deposition foUowmg solvent displacement. Int. J. Pharm. 55:R1-R4(1989).
20. J.J. Burger, E. Tomlinson and J.W. Mulder. Incorporation of water-soluble drugs in albumm microspheres. Int. J. Pharm. 23: 333-334 (1985).
21 .1 . ZoUe, F. Hosain and B.A. Rhodes. Preparation of metabolizable radioactive human serum albumm microspheres for studies of the circulation. J. Nucl Med. 11: 73-79 (1970).
22. J.J. Marty, R.C. Oppenheim and P.P. Speiser. Nanoparticles-A new colloidal drug delivery system. Pharm. Acta. Helva. 53: 17-23 (1978).
23. C. Schwarz, W. Mehnert, and J.S. Lucks. Sohd lipid Nanoparticles for confroUed drug dehvery: production, characterization and sterihzation. J. Cont. Rel 30: 83-96 (1994).
27

24. R.H. MuUer, W. Mehnert and J.S.Lucks. Solid lipid Nanoparticles - an altemative colloidal earner for confroUed drug delivery. Eur. J Pharm. Biopharm. 41: 62-69 (1995).
25. P.K. Gupta, C.T. Hung and D.G. Penier. Quantitation of tiie release of doxorubicin from colloidal drug forms using dynamic dialysis. Int. J Pharm. 33: 137-146 (1986).
26. S.J. Douglas, L. Ilium, S.S. Davis and J. Kreuter. Particle size and distiibution of poly(butyl-2-cyanoacrylate) nanoparticles. II. Influence of stabilizers. J. Colloidal Interface Scl 103: 154(1985).
27. U.E. Berg, J. Kreuter, P.P. Speiser, P.P. Influence of the particle size on tiie adjuvant effects of polybutylcyanoacrylate nanoparticles. Pharm. Ind. 48: 75-79 (1986).
28. L. Fenart, A. Casanova, B. Dehouck, C. Duhem, S. Slupek, R. Cecchelli and D. Betbeder. Evaluation of effect of charge and lipid coating on ability of 60 nm Nanoparticles to cross an in vitro model of the blood-brain barrier. J. Pharmacol Exp. Ther. 291(3): 1017-1022 (1999).
29. U. Schroder, P. Sommerfeld, S. Ulrich and B.A. Sabel. Nanoparticle technology for delivery of drugs across the blood-brain barrier. /. Pharm. Scl 87: 1305-1307 (1998).
30. R.N. Alyautidm, D. Gotiier and V. Pefrov. Analgesic activity of the hexapeptide dalagrin adsorbed on the surface of polysorbate-80 coated polybutylcyanoacrylate Nanoparticles. Euro. J. Pharm. Biopharm. 41: 44-48 (1995).
31. J. Kreuter, R. Alyautidin, D.A. Kharkevich and A. A. Ivanov. Passage of peptides through the blood—^brain barrier with coUoidal particles (Nanoparticles). Brain Res. 674(1): 171-174(1995).
32. G. Borchardt, K.L. Audus and F. Shi. Uptake of surfactant-coated poly-methyl-methylacrylate nanoparticles by bovuie brain microvessel endothelial cell monolayers. Int. J. Pharmaceutics. 110: 29-35 (1994).
33. P. Ramge, R.E. Unger, J.B. Olfrogge. Polysorbate-80 coating enhances uptake of polybutylcyanoacrylate (PBCA)-nanoparticles by human, bovine, and murine primary brain capillary endotiielial ceUs. Eur. J. Neuro. 12: 1935-1940 (2000).
34. J. Kreuter. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Del Rev. 47:65-81(2001).
28

35. U. Schroderand B.A. Nanoparticles, a drug carrier system to pass the blood-brain barrier, permit central analgesic effects of i.v. dalagrin injections. Brain Res. 710: 121-124(1996).
36. R.N. Alyautdin, B.E. Tezikov, P. Ramge, D.A. Kharkevich, D.J. Begley and J. Kreuter. Significant entry of tubocurarine into the brain of rats by adsorption to polysorbate-80 coated polybutylcyanoacrylate nanoparticles: an in situ brain perfusion study. J. Microencapsulation. 15 (1): 67-74 (1998).
37. A.E. Gulyaev, S.E. Gelperina, I.N. Skidan, A.S. Anfropov, G.Y. Kivman and J. Kreuter. Significant transport of doxorubicin into the brain with polysorbate-80 coated nanoparticles. Pharm. Res. 16: 1564-1569 (1999).
38. S.E. Gelprina, Z.S. Smimova and A.S. Khalansky. Chemotiierapy of bram ttimors using doxorubicm bound to polysorbate-80coated nanoparticles. Proceedmgs of the 3" worid meeting APV/APGI, Berlui 3/6 April: 441-442 (2000).
39. C.S. Yang, F.L. Lu and Y. Cai. Body distiibution m mice of mfravenously mjected camphotothericin solid lipid nanoparticles and targeting effect on tiie brain. J. Cont. i2e/. 59:299-307(1999).
40. L. Grislain, P. Couvrer and V. Lenaerts. Pharmacokinetics and distribution of a biodegradable drug-carrier. Int. J. Pharm. 15: 333-345 (1983).
41. P.K. Gupta, P.K and C.T. Hung. Targeted delivery of low dose doxorubicin hydrochloride admmistered via magnetic albumin microspheres in rats. / . Microencaps. 7: 85-94 (1990).
42. S.K. Pulfer and J.M. GaUo. Enhanced bram ttimor selectivity of cationic magnetic polysaccharide nucrospheres. J. Drug Target 6: 215-227 (1998).
43 SD Troster U. MuUer and J. Kreuter. Modification of the body distiibution of " poly(metiiyl'metiiyl methylacrylate) nanoparticles by coating with surfactants. Int J
P/iarm. 61:85-100(1990).
44. J.C. Olivier, L. Fenart, R. Chauvet, C. Pariat, R. Cecchelli and W. Couet. Indirect evidence tiiat drug bram targetmg using polysorbate-80 coated polybutylcyanoacrylate nanoparticles is related to toxicity. Pharm. Res. 16(12). 1836-42(1999).
45 R Weissleder, P.F. Hahn, D.D. Stark, P.F. Hahn, J. Marfil, J.F. Gonzalez, S. Sami, • L.E. Todd, and J.T. Ferrucci. The diagnosis of splenic lynjPj^o^^f^^y^J^ ™^f^,S-value of superparamagnetic iron oxide. Am. J Roentgenol 152(1): 175-180 (1989).
29

46. J. Darius, F.P. Meyer, B.A. Sabel and U. Schroeder. Influence of nanoparticles on the brain-to-serum distribution and the metabolism of valproic acid in mice. J. Pharm. Pharmacol 52(9): 1043-1047(2000).
47. A. Fundaro, R. Cavalli, A. Bargoni, D. Vighetto, G.P. Zara and M.R. Gasco. Non-stealth and stealth solid lipid nanoparticles (SLN) carrying doxorubicin: pharmacokinetics and tissue distribution after i.v. adminisfration to rats. Pharmacol Res. 42(4): 337-343 (2000).
30

Table 2.1: Ideal properties of polymeric based nanoparticles for drug delivery across the blood-brain barrier.
Ideal Properties of BBB delivery polymeric-based carriers
Ideal properties of polymeric-based NPs
• Natural or synthetic polymer
• Inexpensive
• Non-toxic
• Biodegradeable^iocompatible
• Non-thrombogenic
• Non-immunogenic
Particle diameter < 100 nanometers
Stable in blood (ie, no opsonization
by proteins)
BBB-targeted (ie, use of cell surface
ligands, receptor mediated
endocytocis)
No activation of neufrophils
No platelet aggregation
Avoidance of the reticuloendothelial
system
Non-uiflanunatory
Prolonged cfrculation time
Scalable and cost effective with
regard to manufacturing process
Amenable to small molecules,
peptides, proteins or nucleic acids.
31
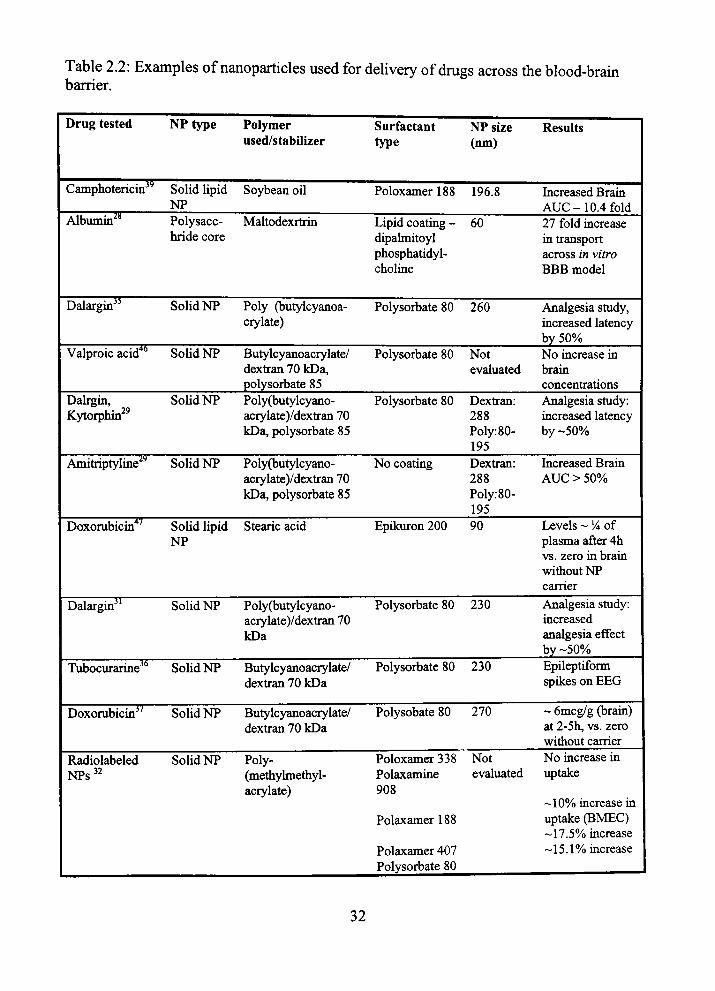
Table 2.2: Examples of nanoparticles used for delivery of drugs across the blood-bram barrier.
Drug tested
Camphotericin'"
Albumin' "
Dalargin'^
Valproic acid'"'
Dalrgin, Kytorphin^'
Amitriptyline^''
Doxorubicin*'
Dalargin""
Tubocurarine'*'
Doxorubicin
Radiolabeled NPs'^
NPtype
Solid lipid NP Polysacc-hride core
Solid NP
Solid NP
SoUdNP
Solid NP
Solid lipid NP
Solid NP
Solid NP
SoUdNP
SoUdNP
Polymer used/stabilizer
Soybean oil
Maltodexrtrin
Poly (butylcyanoa-crylate)
Butylcyanoacrylate/ dextran 70 kDa, polysorbate 85 Poly(butylcyano-acrylate)/dextran 70 kDa, polysorbate 85
Poly(butylcyano-acrylate)/dextran 70 kDa, polysorbate 85
Stearic acid
Poly(butylcyano-acrylate)/dextran 70 kDa
Butylcyanoacrylate/ dextran 70 kDa
Butylcyanoacrylate/ dextran 70 kDa
Poly-(methylmethyl-acrylate)
Surfactant type
Poloxamer 188
Lipid coating -dipalmitoyl phosphatidylcholine
Polysorbate 80
Polysorbate 80
Polysorbate 80
No coating
Epikuron 200
Polysorbate 80
Polysorbate 80
Polysobate 80
Poloxamer 338 Polaxamine 908
Polaxamer 188
Polaxamer 407 Polysorbate 80
NPslze (nm)
196.8
60
260
Not evaluated
Dextran: 288 Poly: 80-195 Dextran: 288 Poly:80-195 90
230
230
270
Not evaluated
Results
Increased Brain AUG-10.4 fold 27 fold increase in transport across in vitro BBB model
Analgesia study, increased latency by 50% No increase in brain concentrations Analgesia study: increased latency by -50%
Increased Brain AUG > 50%
Levels ~ V* of plasma after 4h vs. zero in brain without NP carrier Analgesia study: increased analgesia effect by-50% Epileptiform spikes on EEG
- 6mcg/g (brain) at 2-5h, vs. zero without carrier No increase in uptake
-10% increase in uptake (BMEG) -17.5% increase -15.1% increase
32

Brain Capj^ Endothe
Brain Parenchyma
Pro drug converted to active drug, requires drug modification
Drug may need to be modified to be transported
I Direct injection - invasive
Drug delivered with the addition of other
molecules
Inhibition of efflux transporters, potential toxic accumulation of substrates
Figure 2.1: Sfrategies to increase CNS drug delivery.
33

A. Solid colloidal NP with homogeneous dispersion of drug
B. Solid NP with drug associated on exterior
G.Nanocapsule with drug entrapped
D. Solid colloidal NP with homogeneous dispersion of drug and cell surface ligand
@ Polymeric matrix
* Drug
I—• Cell surface ligand
® SoUdNP
Figure 2.2: Various types of drug-loaded nanoparticles for CNS delivery.
34

A. Initial rapid desorbtion of drug from surface of nanoparticle known as burst effect. May provide initial dose of drug.
B, Slow controlled release of drug. Gharacterized by both first- and zero-order equations. Release is dependent on NP degradation or erosion of nanocapsule shell.
Figure 2.3: Pharmacokinetic analysis of drug release firom nanoparticle.
35

Desorption of drug from polymer surface
^ Degradation ofNP matrix or nanocapsule shell
Drug diffusion through NP matrix
Mechanisms of NP drug release may be associated with one or a combination of any or all proposed mechanisms.
Figure 2.4: Mechanisms of drug release from nanoparticles.
36

Brain Parenchyma
Brain Cap\ Endothel
Concentration gradient driven
Same process as LDL
Manipulation of natural carriers
expressed at luminal endothelium
Figure 2.5: Mechanisms of nanoparticle CNS entry.
37

CHAPTER 3
IN VIVO AND IN VITRO ASSESSMENT OF BASELINE
BLOOD-BRAIN BARRIER PARAMETERS IN THE
PRESENCE OF NOVEL NANOPARTICLES
3.1 Infroduction
Brain penefration of tiierapeutic agents is often lunited by tiie blood-brain barrier
(BBB). The BBB is comprised of brain capillary endotiieUal cells connected by tight
junctions {zonulae occludens) that circumferentially surround the cell margin. These tight
junctions may approximate 100 times greater fransendothelial electiical resistance than
junctions of peripheral capillary endothelium (1). Thus, the BBB demonsfrates sunilar
drug permeation restrictions of a continuous cell membrane; i.e., allowmg lipid soluble
molecules fransport across the membrane, whereas compounds that are hydrophilic,
protein bound or are of large molecular weight have little to no permeation (2).
Nanoparticles (NPs) may have utility as drug delivery carriers across the BBB.
These coUoidal particles (size from 1 to 1000 nm) disguise permeation limiting
characteristics of therapeutic molecules with the physical nature of the NP. NPs may
employ numerous combuiations of polymers and surfactants for optunized BBB
penefration. CNS penefration of NPs, loaded with drugs once unpermeable to bram, may
provide therapeutic promise (3-4).
Currentiy NPs manufactured with polybutylcynoacrylate (PBCA) as tiie polymer
and Tween 80 as tiie surfactant have been studied as drug carriers across the BBB (4).
38

However, there are conflictuig data with regard to in vivo toxicity of PBCA-polysorbate
80 NPs. Olivier et al., (5) demonsfrated that dalargm adsorbed onto PBCA-polysorbate
80 NPs resulted in deatii in 3-4 of 10 mice (dose = 166 mg/kg). Furtiiermore, survivmg
animals had significantly decreased activity, after a short burst of hyperactivity and
apparent discomfort. The authors suggest toxicity was mediated by rapid esterase
biodegradation of the PBCA polymer to toxic compounds (6-8). However, Kreuter (4)
refuted the toxicity, since the CNS effects were a normal response to the opioid dalargin.
Tight junctions are mtegral to mauitaming the physiologic role of the BBB (i.e.,
limiting CHSTS entry of toxins). The junctions may be fransientiy opened by artificially
created osmotic pressure as a CNS drug delivery sfrategy (9). However, barrier opemng
poses significant risk for CNS toxin entiy and subsequent damage (10). Alyautdm et al.
(11) mfravenously injecting (12) PBCA tween 80 NPs, demonsfrated an inulm vascular
volume mcrease of 10% after ten minutes and 99% after 45 minutes in tiie presence of
PBCA-polysorbate 80 NPs. While tiie autiior proposed tiiere was not a major opemng of
tiie BBB, it was significant compared to confrols.
In vitro publications of PBCA NP safety have tiie same disparity as tiie apparent
confradictions of m vivo reports. Two previous reports suggest a PBCA-polysorbate 80
NP exposure concenfration of 10 ^g/ml may result m BBMECs tight junction disruption.
(5,13). However, otiier investigators have shown similar concenfrations of PBCA
polysorbate 80 coated NPs have no effect on apical to basal movement of impermeable
markers (4).
39

This chapter addresses changes in BBB parameters issues with regard to two
novel NP foraiulations. These preparations were developed with the rationale that
polymers and surfactants used in manufactiiring should be biocompatible and
biodegradeable. Given tiiis premise, we hypothesize the NP formulations will not
demonsfrate sunilar adverse effects at tiie BBB tiiat are present in vivo and in vitro with
tiie PBCA-polysorbate 80 NPs.
3.2 Materials and Methods
3.2.1 Nanoparticles, Radiochemicals and Antibodies
Emulsifymg wax/Brij 78 NPs (NPE78) and Brij 72/Tween 80 NPs (NPE72) were
prepared from warm oil-in-water microemulsion templates as described elsewhere (14).
Briefly, for the NPE78 formulation 2 mg of emulsifying wax was weighed out into glass
vials and melted at 50-55°C. To the melted oil phase warm Brij 78 solution (100 mM)
was added followed by deionized, filtered (0.2 |im) water to obtain a final volume of
1000 )il and surfactant concenfration of 3 mM. Microemulsion templates formed solid
NPs upon cooling to room temperature. NPE72 were engineered using a similar
procedure, witii 2.3 mM Tween 80 as the surfactant.
The particle size of NPs was measured at 20^0 using a Coulter N4 Plus Sub-
Micron Particle Sizer (Coulter Corporation, Miami, FL). NP suspensions were diluted
with filtered water (1:10) prior to particle sizing and size was measured at 90° tight
scattering for 90 seconds (n=3). For sizing of aged NPE72 and NPE78, tiie NP
suspensions were sealed and stored at 4°C for a period of one week. Prior to particle size
40

measurement aliquots of NPs were allowed to equilibrate to room temperatiire and then
diluted witii filtered water to ensure light scattering intensity within the required range of
tiie instrument (5 x 10'* to 1 x 10 counts per second).
High specific activity [ " CJ-tiuourea (56.0 mCi/mmol) was obtained from
Moravek Biochemicals (Brea, CA., U.S.A). High specific activity [ H]-diazepam (76.0
Ci/mmol), [^H]-choline (79.2 Ci/mmol) and ['" CJ-sucrose (401.0 mCi/mmol) were
obtamed from Perkin Elmer Life Sciences (Boston, MA., U.S.A.). In each experiment,
tiie [ H]-compoimd was dried prior to being dissolved m the buffer, to remove volatile
tritium contammants, mcludmg [ Hj-HjO.
The monoclonal antibodies used were mouse anti-zonulae occuldens (ZO)-l and
rabbit anti-claudin-1 obtained from Zymed Laboratories Inc. (San Fancisco, CA). The
anti- ZO-1 is aimed at the amino acid residues 334-634 of the human recombinant ZO-1
protein. The ZO-1 antibody is specific for ZO-la" and ZO-1 a' isoforms. Claudin-1
recognizes tiie C-terminus of the human/mouse claudin-1 protein. Anti-mouse IgG and
anti-rabbit IgG secondary antibodies were purchased from Sigma (St. Louis, MO).
3.2.2 In Situ Perfusion Procedure
Assessment of m vivo effects of NPs was accomplished by using the in situ rat
brain perfusion technique of Takasato et. al. (15) with modifications (16-17). Briefly,
Male Fischer-344 rats (220-330 g; Charles River Laboratories, Kmgston, NY, n=3-6)
were anesthetized witii sodium pentobarbital (50 mg/kg infraperitoneal). A PE-60
catheter filled with heparinized saUne (100 units/ml) was placed mto tiie left common
41

carotid artery after ligation of tiie left external carotid, occipital and common carotid
arteries. The pterygopalatine artery was left open during tiie experiments (17). Rat body
temperatiire was monitored by rectal probe and maintained at 37°C by a heatmg pad
connected to a feedback device (YSI Indicatmg ControUer, Yellow Springs, OH). The
catiieter was connected to a syringe containing buffered physiologic perfusion fluid
(contammg [in mM]: NaCl 128, NaPOj 2.4, NaHCOj 29.0, KCl 4.2, CaCl 1.5, MgCb
0.9, and D-glucose 9) witii combinations of 0.15 )iCi/nil [ H]-diazepam, 1.0 ^Ci/ml [ H]-
choline, 0.33 ^Ci/nll [ ''C]-sucrose, 0.33 iCi/ml ["*C]-tiiiourea and/or unlabeled NP
formulations. Perfusion fluid was filtered and warmed to 37°C and gassed with 95% air
and 5% CO2. Perfusion fluid was tifrated to a pH of 7.4 witii osmolarity being -290
mOsm. NPs were prepared for perfusion by dilutmg the NP stock concenfration (2
mg/ml) into physiologic buffer to the desured concenfration. The perfusion fluid was
infused into the left carotid artery with an infusion pump for 20-60 seconds at 10
ml/mmute (Harvard Apparatus, South Natick, MA) with a total dose of 200 |j,g NP
delivered (urtiess otherwise specified). Flow was set to maintam a carotid artery pressure
of ~ 120 mm Hg. Rats were decapitated and regional cerebral samples obtauied as
described (15), after removal of the arachnoid membrane and meningeal vessels. The
brain and perfusion fluid samples were then digested overnight at 50°C m 1 ml of IM
piperidme. Ten ml of Fisher Chemical scintillation cocktail (Beckman, FuUerton, CA,
U.S.A.) was added to each vial and the fracer contents assessed by dual-label liquid
scintillation countmg. All studies were approved by tiie Animal Care and Use Committee
42

and were conducted m accordance witii the NIH Guide for tiie Care and Use of
Laboratory Animals.
3.2.3 In Situ Kinetic Analysis
Brain uptake of radiolabeled fracers was determined by calculation of a smgle
time point blood-to-brain fransfer coefficient {K.^J as previously described by Takasato et
al. (15) and Smith (16), from the foUowmg relationship:
Kin = [c,rVvCp,]/(Cp/r) (3.1)
where C j = C . + C ^ is the sum of the amount of fracer remaining m the perfusate in
the blood-brain vessels and the amoimt of fracer that has penefrated uito bram, Vy is brain
vascular volume, defined as a ratio of the vascular marker [ '*C]-sucrose in brain to
perfusion flvud concentration, C ^ is tiie perfusion fluid concenfration of tiie radiolabeled
fracers and T is the net perfusion time with the assumption tiiat uptake is linear.
Apparent cerebrovascular permeability surface-area product (PA) was determmed
using the Crone-Renkin equation (16):
PA =-F In (1 - Ki„/F) (3-2)
where F is tiie cerebral perfusion flow determuied from the uptake of [ H]-diazepam (18).
Regional perfiision flow was used for regional PA determination to account for regional
flow variations.
43

3.2.4 Bovine Brain Microvessel Endothelial Cell Transport Method
Bovine microvessel endothelial cells (BBMEC) were isolated as previously
published (19-21). Briefly, fresh bovine brain was obtamed from a local meat
slaughterhouse and placed in ice-cold buffered essential medium. Meninges and large
surface vessels were carefully removed and discarded. Cerebral gray matter was aspfrated
from the cerebral cortex. To release microvessels from the gray matter a 2.5 h dispase (4
ml of 12.5% dispase sol./50 g gray matter) digestion at 37°C was performed. Tissue
debris was removed by centiifiigation witii 13% dexfran. Removal of pericytes and
asfrocytes was accomphshed by a 4-hour incubation with coUagenase/dispase (3 mg each
in 3 ml of sol /microvessel g). Lastiy, a percoU gradient centiifiigation removed otiier
cellular contaminants.
CeUs were seeded on 12-well Transwell® (Costar, Cambridge, MA) plates (0.4
nm pores) at a density of 50,000 cells/cm , grown to confluency and used on days 10-12.
Culture media was removed prior to fransport experiments and aUowed to equilibrate for
10 minutes in physiologic buffer usuig an oscillatmg-table (122 mM NaCl, 3 mM KCl,
25 mM Na2P04, 1.3 mM K2HPO4, 1.4 mM CaCh, 1.2 mM MgS04,10 mM glucose, 10
mM Hepes; pH~7.4). The basolateral chamber contained 1.5 ml of buffer; apical chamber
contained 0.5 ml, to ensure no change m pressure gradient existed. Transport experiments
were conducted in tiie apical to basal direction for 2 hours witii samplmg tunes of 15, 30,
60, and 120 minutes usmg an oscillating-table for circulation of well content. Buffer was
mamtained at 37°C tiiroughout experimental time frame. Stiidies were completed in tiie
absence (confrol) and presence of two NP fonnulations (200 |ig/ml; we believe a dose
44

significantly higher tiian would be presented to the BBB witii physiologic concenfrations
required in clmical tiierapy), n=6 in all experiments. Study uiitiation began when 0.5 ml
of fransport medium supplemented with test compounds was placed ui the apical
chamber. Apical chamber samplmg was completed (50 jxl, witii replacement) at time zero
for exposure concenfration. Serial 100 |il sampling of the basolateral chamber (with
replacement) occurred at the times listed. At the conclusion, 50 il was sampled from the
apical chamber. Fisher Chemical scintillation cocktail (3 ml) (Beckman, FuUerton, CA.,
U.S.A.) was added to each sample and fracer contents assessed by dual-label liquid
scintillation counting.
3.2.5 /n Vitro Kinetic Analysis
Flux of radiolabeled compounds tiirough BBMECs were determined by
calculation of apparent permeability coefficients (P) as previously described (22) from
the following relationship:
P = Flux/(A*Cdo) (3-3)
where Flux is tiie slope (calculated by linear regression) of pmoles appearing in tiie
receiver chamber versus tune (minutes); A is the area of the membrane m cm; and Cdo is
the donor concenfration at time zero.
3.2.6 Western Blot Analysis
After a 2-hour exposure to NPE72, NPE78 or no freatinent, protein was isolated
from confluent BBMECs seeded on 12-well plates at a density of 50,000 cells/cm on
45

days 10-12. Isolation was completed with the Tri-reagant protocol (Sigma; St Louis,
MO). Briefly, at tiie conclusion of exposure, media was removed and 0.4 ml per 10 /cm
tii-reagant LS was added to each well to lyse cells. Separation of DNA, RNA and protein
was completed witii addition of 0.2 ml of chlorofomi (per ml of tii-reagant). Precipitation
of DNA and protem from tiie mterphase and organic phase was accomplished by addmg
etiianol and isopropanol, respectively. Protein samples were then washed tiiree tunes by a
0.3 M guanidine hydrochloride/95% ethanol solution and centiifiigation 7.5g for five
minutes. Protem was prepared for western blot analysis by pellet dissolution with 1%
SDS and sonication at 65°C. Protem was quantified with a BCA Pierce assay kit (Pierce;
Rockford, IL). After a standard curve was established (r = 0.9847), 20|j.g of proteui from
each group, and molecular weight markers (Amersham Life Science; Buckinghamshire,
UK), were separated using a gradient (4-20%) tris glycine polyacrimde gel (Novex, San
Diego CA.) The protein markers and samples were elecfrophoretically fransferred to
polyvmylidene fluoride membranes (Amersham Life Sciences; Buckinghamshke, UK).
The membrane was mcubated overnight m a blockmg buffer of 5% non-fat dry milk.
After blocking, membranes were washed 3 times with 5% non-fat milk for 20 minutes.
Primary antibodies for ZO-1 and Claudin-1 (1:1,000; 1:500; dUutions respectively) were
incubated for 2 hours at 23°C. Membranes were washed tiiree times witii 5% non-fat miUc
for 20 minutes after which the respective secondary antibodies (1:5,000; 1:5,000;
dilutions respectively) were mcubated for 2 hours. The membranes were washed agam.
Membranes were developed using ECL plus (PerkuiElmer Life Sciences Inc; Boston,
MA). Protein bands and molecular markers were visualized on radiographic film.
46

3.2.7 Statistical Analysis
Data presented are from tiie frontal cerebral cortex for in situ stiidies unless
otherwise specified. Brain PA and permeability coefficients across BBMECs were
evaluated by one-way ANOVA witii a Bonferoni's multiple comparison test. Errors are
reported as standard error of the mean unless otherwise indicated. Differences were
considered statistically significant at/?<0.05. (GraphPad Prism version 2.01 for
Wmdows, GraphPad Software, San Diego, CA USA). Dual labeled scintillation counting
of samples were accomplished with correction for quench, background and efficiency.
Western blot analysis was completed using Scion Image for Windows, version 4.02 Beta,
Scion Corp., USA.
3.3 Results
3.3.1 Effect of NP Types on Cerebral Perfusion Flow
To determine the effect of tiie NPs on cerebral perfusion flow we evaluated brain
permeation of [ H]-diazepam in tiie presence or absence of eitiier NP. [ H]-diazepam
baseline uptake values were determined (4.05 ± 0.33 x 10" ml/s/g). Figure 3.1 shows m
tiie presence of eitiier NP type no significant (p>0.05) alterations in cerebral perfiision
flow were seen at physiologically high NP concenfrations (40 ig/ml; total BBB NP
exposure 200 ig) (Figure 3.1A: NPE72: 3.05 ± 0.68 x 10' ml/s/g; Figure 3.1B: NPE78:
4.10 + 0.18 X 10' ml/s/g). Furtiiermore, lesser concenfrations had no significant impact
on cerebral perfusion flow.
47

Since tiiere is regional variability of cerebral perfiision flow witiun the CNS,
differences in regional BBB [^H]-diazepam uptake were evaluated. Figure 3.2A shows no
overall significant (p>0.05) regional differences in cerebral perfiision flow m the
presence of eitiier NP type. A slight statistical reduction of cerebral perfiision flow was
seen ui tiie parietal cortex (confrol: 6.27 + 0.87 x 10"Ws/g) m tfie presence of NPE78
(4.39 ± 0.40 x 10-2 ml/s/g); p<0.05.
3.3.2 Effect of NP Types on BBB Integrity
To determine if eitiier NP formulation alters tiie integrity of the BBB in vivo,
[ C]-sucrose was incorporated in the perfusion fluid in the presence and absence of
either NP. Figure 3.3 shows no significant changes (p>0.05) for frontal cortex vascular
volume (ml/g) under confrol conditions (1.20 + 0.19 x 10"^nil/g) compared to the
presence of 40 |J.g/nil (total BBB NP exposure 200 \ig) of eitiier NP formulation (Figure
3.3A: NPE72: 0.842 ± 0.146 x 10" ml/g; Figure 3.3B: NPE78: 1.13 ± 0.0012 x 10"
ml/g). Replicated experiments showed subsequent lesser NP concenfrations did not alter
the vascular space measurements from confrol. Regional characterization of the effects of
the NPs on in vivo vascular volume are shown in Figure 3.2B. No significant alterations
in vascular volume were noted in the presence of either NP formulation with tiie
exception of cerebellum volume (1.38 ± 0.17 x 10" ml/g) in tiie presence of NPE72
(0.910 ±0.114x10'^ ml/g).
For in vitro evaluation of BBB tight jimction integrity we evaluated apical to
basal permeation of [ '*C]-sucrose across a BBMEC culttire m tiie presence or absence of
48

eitiier (NPE72; NPE78) fonnulation. Figure 3.4 shows the cumulative appearance of
sucrose picomoles in tiie receiver chamber versus time under confrol and experimental
conditions. For all sampling time points (15, 30, 60 and 120 minutes) no significant
(p>0.05) differences for total ['' C]-sucrose in the receiver chamber between experimental
groups CNPE72; NPE78) were observed. A permeabUity coefficient was calculated for
[•' Cj-sucrose as described. Table 3.1 illusfrates no significant (p>0.05) differences m
[ C]-sucrose permeability coefficients observed m tiie experimental design.
3.3.3 Effect of NP Ostwald Ripening on Cerebral Perfusion Flow and BBB Integrity In Vivo
To assess effects of Ostwald ripening on NPs and the subsequent effects on
cerebral perfusion flow and BBB integrity parameters, the NPs were allowed to age for 1
week at 4°C. [size at day 0 (nm): NPE72 98 ± 2; NPE78 58 ± 1] and at day 7 [size at day
7 (nm): NPE72 206 ± 7; NPE78 95 ± 1]. NP formulations were tiien added to tiie
perfusion fluid in the presence of [''*C]-sucrose and [ H]-diazepam. Figure 3.5A reveals
no significant (p>0.05) alterations m cerebral perfiision flow in the absence (4.05 + 0.33
X 10' ml/s/g) and in tiie presence of 40 |xg/ml (total BBB NP exposure 200 ng) of eitiier
aged NP formulation (NPE72: 2.75 ± 0.45 x 10' ml/s/g); NPE78: 3.42 ± 0.18 x 10"
ml/s/g). Figure 3.5B illusfrates no significant changes (p>0.05) for frontal cortex vascular
volume (ml/g) under confrol (1.20 + 0.19 x lO' ml/g) or in tiie presence of 40 |ig/ml of
eitiier agedNP fonnulation (NPE72: 1.31 ± 0.14 x 10" ml/g; NPE78: 1.24 ± 0.08 x 10"
ml/g).
49

3.3.4 Effect of NP Formulations on BBB Permeation and Facilitated Choline Transport
To ascertain possible effects of NPs on passive permeation at tiie BBB in vivo,
['' CJ-tiiiourea was mcorporated into the perfusion fluid. Figure 3.6A demonsfrates the
passive permeation of ['' CJ-thiourea remains unchanged in vivo (p>0.05) from control
(2.31 +0.18 X 10''* ml/s/g) compared to the presence of either NP formulation (13.33
(Xg/ml) (NP72: 2.42 + 0.64 x 10''* ml/s/g; NP78: 2.06 + 0.59 x 10' nti/s/g). Furtiiermore,
Table 3.1 shows confrol permeability coefficient is not significantly altered in the
presence of either NP formulation in vitro.
Characterization of [ H]-cholme fransport in the presence of the NPs was also
performed to evaluate tiie effects of tiiese NPs on cationic BBB fransport. Figure 3.6B
shows tiie in vivo baseline PA of brain choUne (1.24 ± 0.02 x 10' ml/s/g) in the presence
of eitiier NP formulation (13.33 ig/ml). FacUitated fransport of cholme remains
unchanged (p>0.05) (NPE72:1.50 ± 0.16 x 10' ml/s/g; NPE78:1.35 ± 0.04 x 10"
ml/s/g) during tiie 60-second perfusion. Similar lack of effect was seen in vitro (Table
3.1).
3.3.5 Effect of NPs on BBMECs Tight Junction Protein Expression
To determine if NP exposure resulted m changes of tight junction protem
expression, western blot analyses of ZO-1 and claudin-1 was completed. Figure 3.7
illusfrates protein expression of ZO-1 and claudin-1 m confrol (lanes 1 and 2) and after
exposure to NPE72 (lanes 3 and 4) and NPE78 (lanes 5 and 6). No significant differences
50

were noted between percent control and experimental groups using unaging software
(data not shown).
3.4 Discussion
The results of the studies presented herein demonsfrate that two novel NP
formulations lack adverse effects in vivo and in vitro at the BBB. Parameters evaluated
include: cerebral perfusion flow, integrity, permeability and the facihtated fransport of
choline.
Cerebral perfusion flow has not previously been studied with regard to NP
adminisfration. Yet there are data suggesting microspheres can be used to decrease
cerebral blood flow to the pomt of significant unpedance in the cortical, striatal and
hippocampal regions witii subsequent infarction and necrosis (23). While NPs are an
order of magnitiide smaller tiian microspheres, tiiere is concern NPs may aggregate in
vivo producing larger particles. Therefore, cerebral perfusion flow was determined m tiie
presence of each NP formulation at doses high enough to potentially result in occlusion.
Evaluation of flow was accomplished by calculation of [^H]-diazepam Kin (ml/s/g) and
confrol values calculated agree witii previous results published (15,18) (marker for
cerebral flow given an exfraction of nearly 100%) (15-16). Figure 3.1 and Figure 3.2A
demonsfrates cerebral perfiision flow is not significantiy ahered m the presence of either
NP formulation at doses ranging from 5 ^ig/ml to 40 ^ig/ml witii tiie exception of tiie
parietal cortex in tiie presence of NPE78, where a shght 30% reduction is present.
51

The mechanism of NP brain entiy has not yet been fiiUy elucidated. One
hypotiiesized route is simple paracellular movement after NP mduced BBB tight
junctional opening (4). This is consistent witii reports demonstrating tiiat opening of the
BBB enables paracellular brain entiy of nonnally excluded compounds (9). However,
increasmg barrier permeability also allows brain entry of toxms and unwanted molecules
(10). For tills report, we addressed this potentially toxic route of brain entry by evaluating
[ C]-sucrose (an integrity marker as it does not appreciably cross an intact BBB) (24)
movement in vivo and in vitro m the presence of either NP formulation. Increases of
['"^Cj-sucrose vascular volume would represent mcreased permeability of the BBB, as the
paracellular fransport of [''*C]-sucrose is increased. Figures 3.3 and 3.2B demonsfrates
that in vivo vascular volume, as measured by [''*C]-sucrose, did not significantiy change
in the presence of either NP with the exception of cerebellum volume decrease in the
presence of NPE72.
Olivier et al. (5) measured the flux of [ '*C]-sucrose and [^H]-inulin across
BBMECs (co-cultured with asfrocytes) m the presence of PBCA-polysorbate 80 NPs. At
NP concenfrations of less than 1 |i.g/ml no significant flux aheration of [ '*C]-sucrose or
[^H]-mulin occurred. However, at NP concenfrations between 10-500 p-g/ml; a greater
than 10-fold flux increase occurred witii botii impermeable markers. This data was also
supported by an observed 6.5-fold increase of ["*C]-sucrose across BBMECs at a PBCA-
polysorbate 80 NP concenfration of 10 |Xg/ml (13). However, otiier investigators have
shown PBCA-polysorbate 80 NP concenfrations of 10 and 20 jxg/ml had no significant
52

effect on apical to basal permeation of [ '*C]-sucrose or [ H]-mulin m a BBMECs system
(4).
Based upon previous concenfrations used, and possible therapeutic in vivo NP
concenfrations, we chose to mcubate the BBMECs in the donor chamber with
significantiy higher doses of 200 |xg/ml. Figure 3.4 represents tiie movement of [ ''C]-
sucrose from the donor chamber to the receiver chamber m the presence and absence of
either formitiation. At all times no significant difference in [ " CJ-sucrose accumulations
was observed. Further, the [*'*C]-sucrose confrol permeability coefficient calculated
(Table 3.1) agrees with previously published data (25) and is not statistically altered
experimentally. Given an increased movement of [ '*C]-sucrose from the donor to
receiver chamber would represent decreased integrity of the BBMECs tight junctions
(paracellular fransport of [ ' Cj-sucrose is mcreased), we suggest tiie NP formulations
have little effect on BBMECs tight junction integrity and paracellular permeability m the
time frames evaluated.
The development of coUoidal NPs for large-scale manufactiirmg would partiaUy
depend on NP size stability. In vitro stabUity of NPs m aqueous suspension was tested
over a period of one week at room temperatiire, = 20°C (data not shown) and 4°C. Both
preparations showed superior size stability at 4°C over room or freezing temperattires
remaining well witiun tiie nm requisite for brain delivery. However, given the
approxunate size doublmg we reevaluated cerebral perfiision flow and vascular volume
in vivo witii NPs that have aged for 1 week at 4°C. Figure 3.5 shows 40 ng/ml of aged
NPs did not significantly affect [ H]-diazepam uptake or [ "CJ-sucrose volume.
53

Considering the stability data and lack of parameter effects after 1 week of NP aging,
clinical investigation of the NP products may be further warranted.
Evaluation of BBB permeability was accomplished by analysis of [*'*C]-thiourea
PA. ['"^CJ-Thiourea is considered an in situ perfiision reference solute for low brain
passive permeation, which agrees with standard I.V. injection values (26). The in vivo
[ '*C]-thiourea PA values we obtained are consistent with published data (26). Figure 3.6
illusfrates no significant changes in brain permeability of [ '*C]-tliiourea PA in the
presence of either NP. NP dosing was lowered to accommodate the longer perfusion time
needed to ensure V2 of the slowly penefrating [^''CJ-thiourea had passed into brain (Ctot/Cpf
> 2 X VyCpf/Cpf) (15). Given [ '*C]-tiiiourea is considered a perfusion reference solute it
should behave similarly in vitro. Table 3.1 illusfrates both NP formulations have a lack of
effect on in vitro [''*C]-thiourea permeation.
The BBB is a stiict physical and enzymatic barrier tiiat not only Imiits drug
movement but may lunit passive permeation of essential nutrients. However, requfred
compounds rapidly gam access tiirough BBB transporters. For example, cationic choline
fransport at tiie BBB has been demonsfrated in vivo to be carrier-mediated and sattirable
(27). To evaluate if an interaction between tiie NP constittients (notably tiie nonionic
surfactants Brij 72, Brij 78 and tween 80 used m manufactiiring) and the anionic bmdmg
site (28) of tiie choline fransport protein is present; [^H]-cholme PA values (ml/s/g) were
determined (Figure 3.6) witii confrol values being consistent witii pubhshed values (28).
In tiie presence of eitiier NP, no significant difference for tiie fransport of [^H]-cholme
was observed. Sunilar to tiie [''Cl-ttiiourea experiment NP concenfrations were lowered.
54

Lack of effect on choline transport was also observed in vitro (Table 3.1). This combined
data indicates NPs using nonionic surfactants do not associate appreciably with the
cationic choline fransporter and allow physiologic BBB cholme fransport.
To further explore NP effects on tight junctions western blot analyses were
performed witii purpose to detect the expression of claudin-1 and ZO-1. Claudin-1 is a
major component of tight jvmction proteins given ttiat: (1) it mteracts witii separate
cellular claudin-1 and (2) claudin-1 and -2 are mainly responsible for tight junction sfrand
formation (integral m cell to cell contact). Occludm appears to be an accessory protein
that produces tight junction sfrands to a lesser extent (29). The cytoplamsic, tight junction
accessory proteins, ZO-1, has been shown to play a crucial role of connecting tight
jtmctions to the cytoskeleton. (30). If NPs disrupt tiie tight jimction integrity of
BBMECs, a decreased expression of these two integral proteins may be recognized after
NP exposure. Figure 3.7 illusfrates no apparent change in protein expression after NP
exposure. However, while this data does not account for possible franslocation of the
tight junction proteins into the cytoplasmic domain, considering the absence of [ C]-
sucrose in vitro permeability coefficient changes or in vivo PA changes, it is unlikely
junction integrity is altered in the presence of either NP formulation.
The combined in vivo and in vitro data presented herem support: (1) our
hypothesis the NP formulations appear to have mmimal effect on primary BBB
parameters and (2) tiiese NP formulations should be explored as bram drug delivery
earners.
55

3.5 References
1. A.M. Butte, H.C. Jones, and N.J. Abbott. Electiical resistance across the blood-brain barrier in anaesthetized rats: a developmental study, J Physiol 429: 47-62 (1990).
2. Q.R. Smitii. Advances in Neurology. In R. Wurtinan (ed.), Alzheimer's Disease Volume 51, Raven Press, New York, 1990, pp. 217-222.
3. P.R. Lockman, R.J. Mumper, M.A. Khan, and D.D Allen. Nanoparticle technology for drug delivery across tiie blood-brain barrier. Drug Dev Ind Pharm. 28(1): 1-12 (2002).
4. J. Kreuter. Nanoparticulate systems for bram delivery of drugs, Adv Drug Del Rev. 47: 65-81 (2001).
5. J.C. Olivier, L. Fenart, L., R. Chauvet, C. Pariat, R. Cecchelli, and W. Couet. Indirect evidence that drug braui targetmg usuig polysorbate-80 coated polybutylcyanoacrylate nanoparticles is related to toxicity, Pharm Res, 16(12): 1836-42 (1999).
6. C. Lherm, R.H. MuUer, F. Puiseieux, and P. Couvreur. Alkylcyanoacrylate drug carriers: II. Cytotoxicity of cyanoacrylate nanoparticles with different aUcyl chain lengtii, Int J Pharm. 84: 13-22 (1992).
7. R.H. MuUer, C. Lherm, J. Herbort, and P. Couvreur. In vitro model for tiie degradation of alkylcyanoacrylate nanoparticles, 5/omafena/5.11(8): 590-5 (1990).
8. L. Grislain, P. Couvreur, V. Lenaerts, M. Roland, D. Deprez-Decampeneere, and P. Speiser. Pharmacokinetics and distribution of a biodegradable drug carrier, Int J Pharm. 15: 335-345 (1983).
9. S.I. Rapoport, K. Ohno, W.R. Fredericks, and K.D. Pettigrew. Regional cerebrovascular permeability to [14C]-sucrose after osmotic opemng of tiie blood-brain banier. Brain Res. 150(3): 653-657 (1978).
10. N.H. Greig. Drug dehvery to tiie brain by blood-brain banier circumvention and drug modification. In E.A. Neuwelt (ed). Implications of the Blood-Brain Barrier and its manipulation, Plenum Press, New York, 1989, pp. 311-367.
11. R.N. Alyaudtin, A. Reichel, R. Lobenberg, P. Ramge, J. Kreuter, and D.J. Begley. Interaction of poly(butylcyanoacrylate) nanoparticles witii tiie blood-brain bamer in vivo and in vitro, J Drug Target. 9(3): 209-21 (2001).
56

12. U. Bickel. Intravenous Injection/Phannacokinetics. In: W.M. Pardridge, (ed.). Introduction to the Blood Brain Barrier, Cambridge University Press (:ambridge 1998, pp 4-48.
13. S. Stemiger, D. Senker, H. von Briesen, D. Begley and J. Kreuter. The influence of polysorbate 80-coated nanoparticles on bovine brain capUlary endothelial cells in vitro, Proc Int Symp Control Rel Bioact Mater. 26: 789-790 (1999).
14. M.O. Oyevnimi and R.J. Mumper. Gadolinium-loaded nanoparticles engineered from microemulsion templates. Drug Dev Ind Pharm, 28(3): 317-28 (2002).
15. Y. Takasato, S.I. Rapoport, and Q.R. Smitii. An in situ brain perfiision technique to sttidy cerebrovascular transport in the rat. Am J Physiol. 247: 484-493 (1984).
16. Q.R. Smith. Brain perfusion systems for studies of drug uptake and metabolism m tiie cenfral nervous system, Pharm Biotechnol. 8: 285-307 (1996).
17. D.D. AUen, J. Oki, and Q.R. Smith. An update in tiie in situ rat brain perfiision technique: simpler, faster, better, Pharm Res. 14: 337 (1997).
18. S. Momma, M. Aoyagi., S.I Rappaport, and Q.R Smith. Phenylalanine fransport across the blood-brain barrier as studied by the in situ perfusion technique, J Neurochem. 48: 1291-1300(1987).
19. K.L Audus, and R.T. Borchardt. Characterization of an in vitro blood-brain barrier model system for studying drug fransport and metabolism, Pharm Res.3: 81-87 (1986).
20. T.J. Abbruscato, S.A. WiUiams, A. Misicka, A.W. Lipkowski, V.J. Hruby and T.P. Davis TP. Blood-to-cenfral nervous system entry and stabUity of biphalin, a unique double-enkephalin analog, and its halogenated derivatives, J Pharmacol Exp Ther. 276(3): 1049-57 (1996).
21. K.L. Audus and R.T. Borchardt. Bovme braui microvessel endothelial cell monolayers as a model system for the blood-bram barrier, Ann N YAcad Sci. 507: 9-18(1987).
22. J.M. Rose and K.L. Audus. Receptor-mediated angiotensin II franscytosis by brain microvessel endotiiehal cells. Peptides. 19(6): 1023-30 (1998).
23. K. Miyake, S. Takeo, and H. Kaijihara. Sustamed decrease m brain regional blood flow after microsphere embohsm m rats. Stroke. 24(3): 415-20 (1993).
57

24. Q.R Smitii. Quantitation of Blood-brain Banier Permeability. In: E.A. Neuwelt, (ed.). Implications of the Blood-brain barrier and its Manipulation, Volume I, Plenum Press, New York, 1989, pp 85-118.
25. T.J. Abbruscato and T.P. Davis. Combination of hypoxia/aglycemia compromises in vitro blood-brain banier integrity, J Pharmacol Exp Ther. 289: 668-75 (1999).
26. Q.R. Smitii and Y. Takasato. Kinetics of amino acid fransport at tiie blood-brain barrier studied using an in situ brain perfusion technique, Ann N YAcad Sci. 481: 186-201 (1986).
27. D.D. Allen, and Q.R. Smith. Characterization of the blood-brain barrier choline fransporter using tiie in situ rat bram perfusion technique, J Neurochem. 76: 1-11 (2001).
28. P.R Lockman, and D.D AUen. The fransport of choluie: A review. Drug Dev Ind Pharm. 28: 749-771 (2002).
29. M. Furuse, H. Sasaki, K. Fujimoto and S. Tsukita. A single gene product, claudin-1 or -2, reconstitutes tight junction sfrands and recruits occludm in fibroblasts, / Ce//5io/. 143: 391-401 (1998).
30. M.S. Blum, E. Toninelli, J.M. Anderson, M.S. Balda, J. Zhou, L. O'Donnell, R. Pardi, and J.R. Bender. Cytoskeletal reanangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol Heart Circ Physiol 273: H286-H294 (1997).
58

Table 3.1: Effect of nanoparticles on in vitro blood-brain barrier parameters.
Sucrose
Choline
Thiourea
Control
8.6 + 0.4x10"^
6.4 ±0.6x10"*
7.9±0.9xl0' '*
NPE72
9.3 ±0.4x10"^
6.7 ±0.3x10"^
7.6 ±0.3x10"*
NPE78
l l ± l x l O " *
7.6 ±0,6x10"'*
8.6 ±0.6x10''*
BBMEC permeability coefficients for markers of BBB parameters m the presence (200 |j,g /ml) and absence of each NP formulation. Units are cm/min. Data is reported as mean ± SEM (n=6). No significant differences m permeability coefficients were observed in the presence of either NP (p>0.05).
59

0.075 NPE72
o <^ c o 0.050
0) ^
f U
0.025 -
B
0.000
0.075
X
Control 40ug/ml 20ug/ml 10ug/ml
NPE78
X
5ug/nil
o 0.050
3 5* "C > 0 ^ o. E 2
I s
0.025
0.000
X
Control 40ug/ml 20ug/ml 10ug/ml Sug/ml
Nanoparticle concentration
Figure 3.1: Effect of nanoparticles on cerebral perfusion flow.
Frontal cortex [^H]-diazepam (a measurement of cerebral perfusion flow) uptake in the absence (confrol) or presence of either NP formulation at concentrations of 40-5 |ig/ml (total BBB NP exposure 25-200 |Lig). Figure 1 A: represents cerebral perfusion flow in the presence of NPE72. Figure IB: represents cerebral perfusion flow m the presence of NPE78. Data are mean ± SEM (confrol, n=6; NP concenfrations n=3-5). No significant differences were observed in the presence of either NP at the concenfrations evaluated (p>0.05)
60

0.10-1
Regional cerebral perfusion flow in the presence of NPs
Control NPE72 NPE78
Frontal Parietal Occipital Hippocamp Caudate Thalamus Cerebell Pons
0.025
o.ooo
Vascular integrity in the presence of NPs
Confrol NPE72 NPE78
Frontal Parietal Occipital Hippocamp Caudate Thalamus Cerebell Pons
Figure 3.2: Regional cerebral perfusion flow and vascular integrity in the presence of nanoparticles
Evaluation of regional brain cerebral perfusion flow (ml/s/g) (2A) and frontal cortex vascular volume (measured by [*'*C]-sucrose) (ml/g) (2B) in the absence (confrol) and presence (40 ^ig/ml; total BBB NP exposure 200 |xg) of either NP formulation (confrol, n=6; NP experimental groups n=3-4). Data are mean ± SEM. An asterisk (*) indicates a significant difference (p<0.05). Frontal = frontal cortex. Parietal = parietal cortex. Occipital = Occipital cortex, Hippocamp = Hippocampus, Caudate = Caudate/putamen region. Thalamus = thalamus/hypothalamus region, and CerebeU = Cerebellum, Pons = Pons/meduUa region.
61
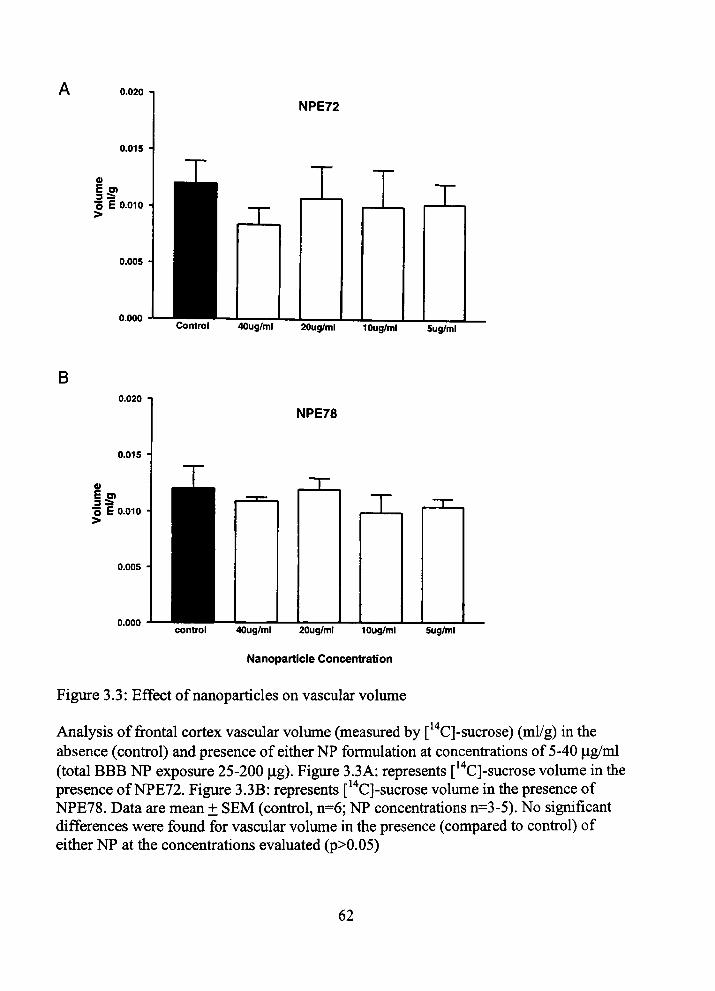
0.020
NPE72
0.000
JL X
Control 40ug/ml 20uc|/ml lOug/ml Sug/ml
B 0,020 1
NPE78
0.000
X
control 40ug/ml 20ug/ml 10ug/ml 5ug/ml
Nanoparticle Concentration
Figure 3.3: Effect of nanoparticles on vascular volume
Analysis of frontal cortex vascular volume (measured by [*'*C]-sucrose) (ml/g) in the absence (confrol) and presence of either NP formulation at concenfrations of 5-40 jig/ml (total BBB NP exposure 25-200 |xg). Figure 3.3A: represents [''*C]-sucrose volume in tiie presence of NPE72. Figure 3.3B: represents [*'*C]-sucrose volume in the presence of NPE78. Data are mean ± SEM (confrol, n=6; NP concenfrations n=3-5). No significant differences were found for vascular volume in the presence (compared to confrol) of either NP at the concenfrations evaluated (p>0.05)
62

2.0
(0 « 1.5 O
E a oT 0) o o 3 0)
o
1.0 •
0.5 •
0.0 25
— 1 —
50 75 I
100
• Control D NPE72 • NPE78
125 — I 150
Minutes
Figure 3.4: Appearance of [*'*C]-sucrose in receiver chamber
Cumulative appearance of [*'*C]-sucrose (picomoles) in the receiver chamber versus time. Data obtained m the absence (confrol) and presence of either NP formulation (200 lig/ml). Data are mean ± SEM; n=6 Slopes are linear regression Imes of [*'*C]-sucrose accumulation. The 95% confidence interval of confrol slope is shown. No significant differences were found for any sampling time point versus confrol in the presence of either NP at any sampling tune (p>0.05).
63
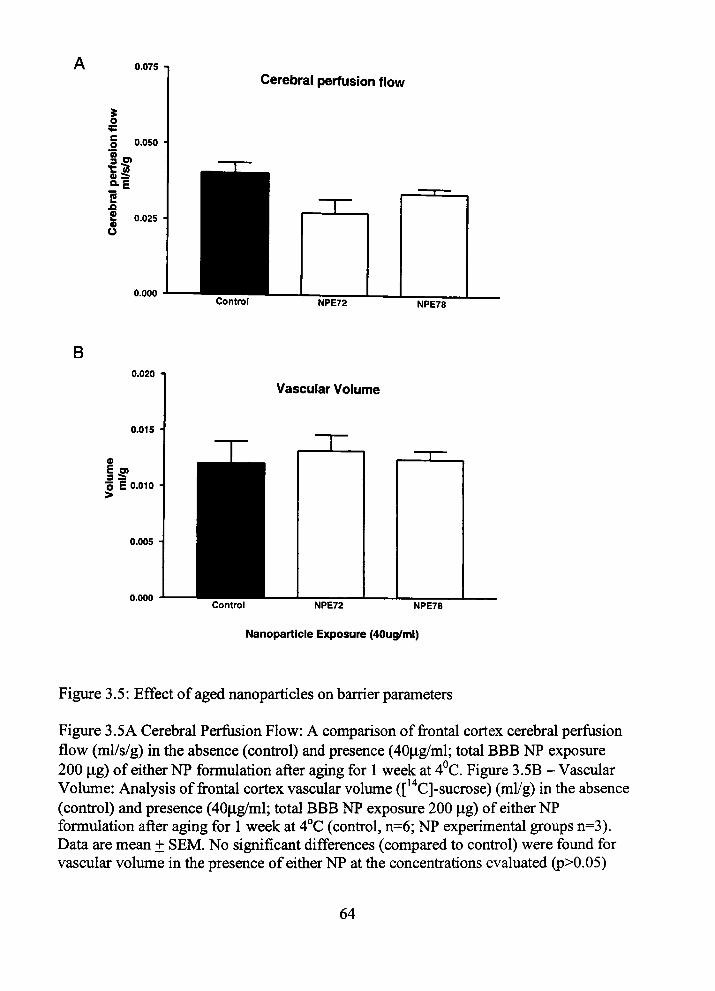
0.075 1
Cerebral perfusion flow
i o
o 3 5) u ^ Q-E 2 9>
0.050
0.025
0.000 Control NPE72 NPE78
B 0.0201
Vascular Volume
0.000 Control NPE72 NPE78
Nanoparticle Exposure (40ug/ml)
Figure 3.5: Effect of aged nanoparticles on barrier parameters
Figure 3.5A Cerebral Perfusion Flow: A comparison of frontal cortex cerebral perfiision flow (ml/s/g) in the absence (confrol) and presence (40|ig/ml; total BBB NP exposure 200 |j,g) of either NP formulation after aging for 1 week at 4°C. Figure 3.5B - Vascular Volume: Analysis of frontal cortex vascular volume ([*'*C]-sucrose) (ml/g) in the absence (confrol) and presence (40|Lig/ml; total BBB NP exposure 200 |ig) of either NP formulation after aging for 1 week at 4°C (confrol, n=6; NP experimental groups n=3). Data are mean ± SEM. No significant differences (compared to confrol) were found for vascular volume in the presence of either NP at the concenfrations evaluated (p>0.05)
64
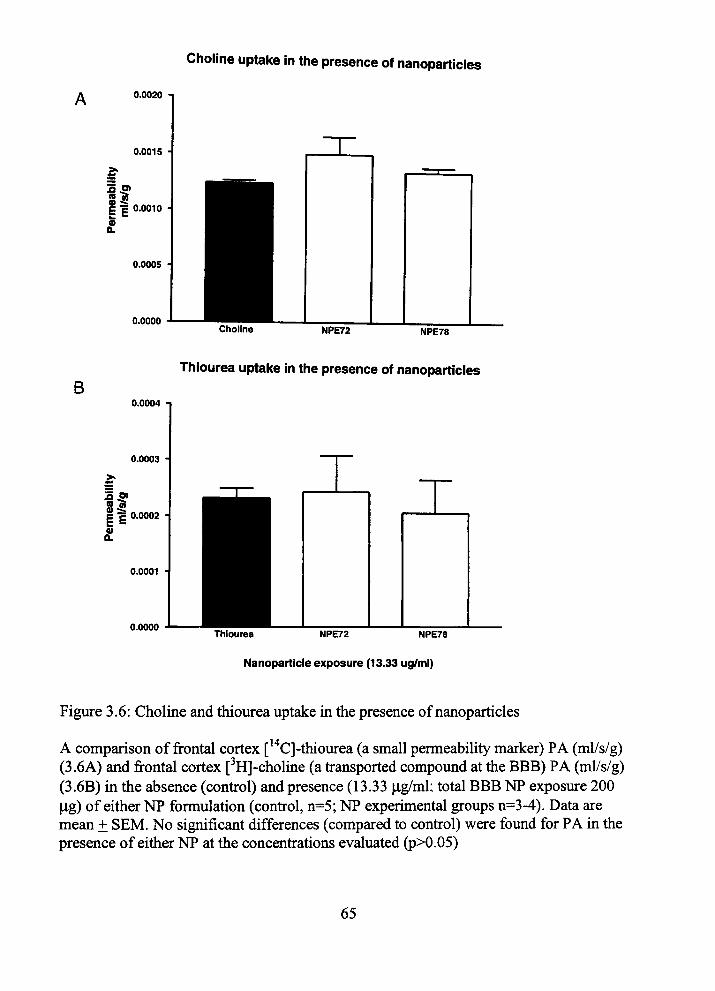
Choline uptake in the presence of nanoparticles
0.0020
0.0015 •
5 » (s a 0) -S: c 1 0.0010
Q.
0.0005 •
0.0000 Choline NPE72 NPE78
B Thiourea uptake in the presence of nanoparticles
0.0004 n
0.0003 •
55»
IE 0) 0.
0.0002 •
0.0001
0.0000 Thiourea NPE72 NPE78
Nanoparticle exposure (13.33 ug/ml)
Figure 3.6: Choline and thiourea uptake in the presence of nanoparticles
A comparison of frontal cortex [''*C]-thiourea (a small permeability marker) PA (ml/s/g) (3.6A) and frontal cortex [ H]-choline (a fransported compound at the BBB) PA (ml/s/g) (3.6B) m the absence (confrol) and presence (13.33 |ig/ml; total BBB NP exposure 200 |4,g) of either NP formulation (confrol, n=5; NP experimental groups n=3-4). Data are mean ± SEM. No significant differences (compared to confrol) were found for PA in the presence of either NP at the concenfrations evaluated (p>0.05)
65
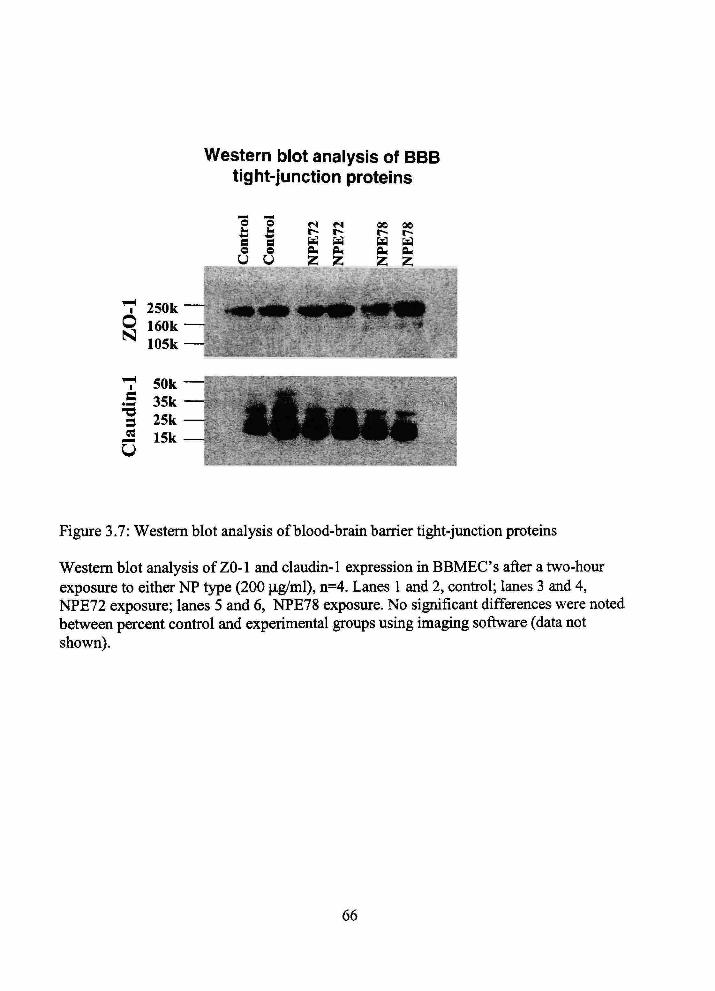
Western blot analysis of BBB tight-junction proteins
o o
a a o o U U
«s t^ ^ CLi
Z
«s t^
m PM z
00 t -txi CM Z.
00 r-a (^ Zi
1 250k
^ 105k
T 50k .S 35k 3 25k
.2 15k
Figure 3.7: Western blot analysis of blood-brain barrier tight-junction proteuis
Western blot analysis of ZO-1 and claudin-1 expression in BBMECs after a two-hour exposure to eitiier NP type (200 |ig/ml), n=4. Lanes 1 and 2, confrol; lanes 3 and 4, NPE72 exposure; lanes 5 and 6, NPE78 exposure. No significant differences were noted between percent confrol and experimental groups using imagmg software (data not shown).
66

CHAPTER 4
/iV5/rt/BLOOD-BRAIN BARRIER TRANSPORT
OF NANOPARTICLES
4.1 Infroduction
The entiy of drug molecules into bram is limited by one of tiie stiictest barriers -
tiie blood-brain banier (BBB). The BBB consists of a continuous layer of endotiielial
cells joined togetiier by tight junctions (zonulae occludens), which severely restiict
paracellular fransport (1). The BBB allows for passive diffiision of small, lipid soluble
molecules, whereas hydrophilic substances or molecules with high molecular weight
have minimal permeation. Movement at tiie BBB is further regulated by effective efflux
fransporters (i.e., multidrug resistance associated protem [MRP], multispecific organic
aiuon fransporter [MOA] and p-glycoprotein [pgp])(2).
There have been multiple attempts to overcome the BBB to deUver cenfral
nervous system (CNS). Artificial opening of the BBB by adminisfration of hyperosmotic
agents or vasoactive molecules (e.g., bradykinin, histamine and serotonin) has been
efficacious in increasing brain distribution of drugs previously impermeable (1). Yet,
while compromising BBB integrity allows paracellular fransport of polar drug molecules
mto brain, there is potential danger of CNS toxicity secondary to concunent entry of
unwelcome molecules. Protems have been included in infracerebroventricular uijections
or unplantation of polymeric devices directly into the brain with pharmacologic benefit
(1,3-4). However, protems have limited regional brain distiibution and tiius, tiie delivery
67

systems must be implanted in close proximity to targeted cells. The necessity of
neuroinvasive surgery limits dkect implantation. Infranasal admmistration has been
successfiil for brain uptake of some molecules into the brain. The olfactory nerve
patiiway allows bypassing tiie BBB; however similar to BBB penefration, uptake by
olfactory neurons depends on lypophilicity and molecular weight of tiie dnig. Further, if
tiie dmg molecule is fransported to tiie CNS by the olfactory epitfielial patiiway it reaches
cerebrospmal fluid (CSF), and still must cross the fimctional CSF-brain banier (5-6). An
altemative sfrategy, tiiat may have less disadvantages tiian the aforementioned
approaches, is tiie use of drug carrier systems such as liposomes (1-2), antibodies (7) or
sohd nanoparticles (NPs) (8,10).
Solid NPs are smaU (1-1000 nm) colloidal particles in which a drug can be
enfrapped or embedded in NP matiix or adsorbed on theu: surface. NPs have been shown
to effectively deliver drugs m a controlled manner (9). Numerous studies have been
performed to demonsfrate the potential use of NPs as drug carriers for brain targeting
(8,10-15). Poly(butylcyanoacrylate) (PBCA) NPs coated witii polysorbate 80 have been
shown to successfully deliver drugs in vivo to the CNS (10). Hexapeptide dalargin, a Leu-
enkephalm analogue with no BBB permeability, adsorbed to the surface of PBCA NPs
caused cenfral analgesia m mice after infravenous adminisfration (12,16). Other drugs
like tubocurarine (11), doxorubicin (13), kytorphin (14), and loperamide (15) were
successfully delivered to the animal brain in vivo. Brain uptake of NPs m these studies
was suggested based on the fact that drugs adsorbed to PBCA NPs caused a resultant
pharmacological effect in the CNS (11-12,14). The brain distribution of drugs dehvered
68

on tiie surface of NPs was also confirmed by quantification of tiie drug itself in brain
tissue (13,17). Stiidies have also demonsfrated uptake of intact NPs in vivo. Troster et al.
mvestigated biodistiibution of [*'*C]-poly(methylmetiiacrylate) NPs coated witii various
surfactants in rats. These authors observed up to a 13-fold increase of radioactivity m tiie
brain witii surfactant-coated NPs over uncoated NPs (18). Kreuter and colleagues
performed fluorescent and election microscopy studies to investigate tiie possibility of
endotiielial cell uptake of PBCA NPs (19). PBCA NPs labeled witii fluorescein
isothiocyanate dexfran were admmistered infravenously to mice. Animals were sacrificed
and brain tissue was analyzed for NP presence. Fluorescence was observed acenfrally in
brain blood vessels and in Purkinje cells of the cerebellum suggesting interaction of
polysorbate 80-coated NPs with the endothelium and subsequent fransport across the
BBB. However, this study was conducted on two animals and no quantification analysis
was performed (19).
The objective of this present study was to assess the brain uptake of two novel
NP-based systems. Emulsifymg wax NPs (E78 NPs) made witii Brij 78 as tiie surfactant
and Brij 72 NPs (E72 NPs) made witii Tween 80 as tiie surfactant were engmeered from
imcroemulsion precursors. NPs were radiolabeled and their fransport across the BBB
was quantified in vivo using a rat brain perfusion method.
4.2 Materials and Methods
Emulsifying wax (E. Wax), polyoxyethylene 20-sorbitan monooleate (Tween 80,
polysorbate 80), polyoxyl 2-stearyl etiier (Brij 72) and DispoDialyzers MWCO 100 kDa
69

were purchased from Spectiiim Chemicals (New Brunswick, NJ). Polyoxyl 20-stearyl
etiier (Brij 78) was obtained from Uniquema (Wilmington, DE). Sephadex G-75,
phosphate buffered saline (PBS) and sodium chloride were purchased from Sigma
Chemicals (St. Louis, MO). l-[^H]-hexadecanol (1 mCi/ml; radiochemical purity greater
tiian 96%) was purchased from Moravek Biochemicals (Brea, CA) and [*'*C]-sucrose
(4.75 mCi/mmol) was obtained from Dupont-New England Nuclear (Boston, MA).
Materials were used as obtained. For all experiments deionized water was filtered
tiirough 0.22 |jm filters (Nalgene International, Rochester, NY).
4.2.1 Preparation of NPs from Microemulsion Precursors
Microemulsion precursors were prepared as reported by Oyewumi and Mumper
(20). Briefly, 2 mg of E. Wax or Brij 72 were weighed out into glass vials. Deionized, 0.2
|im filtered water was added and the mixture was heated to 50-55°C under stirring
conditions to melt the E. Wax or Brij 72. To the milky slurry of E. Wax in water, an
aliquot of 100 mM Brij 78 was added to obtam a fmal volume of 1 ml and final surfactant
concenfration of 3 mM. Microemulsions formed spontaneously after the addition of
surfactant. Warm microemulsion precursors formed sohd E78 NPs upon sunple cooling
to room temperature under stirring conditions. Brij 72 NPs were prepared by tiie same
method with 2.3 mM Tween 80 as final surfactant concenfration. The fmal concenfration
of NPs in all samples was 2 mg/ml.
70

4.2.2 Characterization of NPs
Particle size was measured at 20°C using a Coulter N4 Plus Sub-Micron Particle
Sizer (Coulter Corporation, Miami, FL) at 90° tight scattering for 90 seconds. Prior to
size determination, NP suspensions were diluted with filtered water to ensure light
scattermg intensities were wititin tiie required range of the mstiniment (5 x lO" to 1x10^
counts per second).
Retention of NP size was determined at various temperattires (-20°C, 4°C, 25°C,
and 37°C) and in several different media (10 mM PBS pH 7.4,150 mM sodium chloride,
10% (v/v) fetal bovine serum in 150 mM NaCl, and water). NP suspensions were sealed
and stored at the tested temperatures. Prior to dilution for particle sizing, samples were
left to equilibrate to room temperature. The size stability of NP formulations was also
determined in various media at physiological (37°C) and room temperatures by dilution
of the NPs 1:10 v/v.
4.2.3 NP Radiolabeluig
NPs were radiolabeled by enfrapment of [ H]-hexadecanol (cetyl alcohol).
Radioactive cetyl alcohol in metiianol was pipeted mto glass vials containmg E. Wax or
Brij 72. Vials were left on a hot plate (50°C) to allow complete evaporation of methanol.
After evaporation of metiianol, NPs were formulated as described in above. All
preparations were formulated with theoretical activities of 150 iCi per ml of final
preparation.
71

4.2.4 Characterization of Radiolabeled NPs ([ H]-NPs)
The enfrapment efficiency of [ H]-cetyl alcohol was detennmed using gel
penneation chromatography (GPC). To obtain GPC elution profiles, 200 il of radioactive
NP suspension was eluted tiu-ough Sephadex G-75 columns (150 x 70 mm) with 10 mM
PBS as tiie mobile phase. NPs were detected by light scattering (CPS) and liquid
scintillation counting (LSC). Additionally, a confrol sample of 300 1 of water spiked
witii 10 |il of [ H]-cetyl alcohol was passed down tiie GPC column and tiie presence of
radioactivity detected by LSC. The enfrapment efficiency (E) was calculated based on the
ratio of radioactivity eluted in the void volume (Pi) and total radioactivity put on tiie
column (Pt) from the relationship:
Pi/Pt = E. (4.1)
The radiolabeled compound release profile was assessed by a dialysis method. A volume
of 300 |il of radiolabeled NP suspension was pipeted into DispoDialyzers (MWCO 100
kDa) and dialyzed against 20 ml of 10 mM PBS for 24 hr at 4°C followed by up to 6 hr at
37°C. At predetermmed time points, 100 |il of sample were withdrawn and radioactivity
was measured by LSC.
4.2.5 Bram Uptake Studies
The uptake of [ H]-NPs into brain was assessed usmg tiie in situ rat brain
perfusion technique of Takasato et al. (21) witii modifications described (22-23).
Perfusions of 15-60 s were used to determme initial brain uptake of the NP formulations.
All studies were approved by Texas Tech University HSC Instihitional Ammal Care and
72

Use Committee and were conducted in accordance witii the NIH Guide for the Care and
Use of Laboratory Anunals. For brain uptake stiidies, tiie radiolabeled NPs were prepared
on tiie day preceding tiie animal experunent. Vials were sealed and shipped at 4°C to
Texas Tech University (Amarillo, TX), where fransport experiments were performed.
4.2.6 In Situ Rat Braui Perfusion
Male Fischer-344 rats (220-330 g; Charles River Laboratories, Kingston, NY)
were anesthetized witii sodium pentobarbital (50 mg/kg mtraperitoneal). A PE-60
catheter filled with heparinized saline (100 units/ml) was placed into the left common
carotid artery after ligation of the left external carotid, occipital and common carotid
arteries. Common carotid artery ligation was accomplished caudal to the catheter
implantation site. The pterygopalatme artery was left open during the experiments (22).
Rat rectal temperature was monitored and maintained at 37°C by a heatmg pad connected
to a feedback device (YSI Indicating ConfroUer, Yellow Sprmgs, OH). The catiieter to
the left common carotid artery was connected to a syrmge containmg buffered
physiologic perfusion fluid (containing [in mM]: NaCl 128, NaPOs 2.4, NaHCOs 29.0,
KCl 4.2, CaCl 1.5, MgCla 0.9, and D-glucose 9) witii 1 iCi/ml [ H]-NP (final NP
concenfration -20 p-g/ml) and 0.3 iCi/ml [*'*C]-sucrose (to detennme vascular volume).
Perfiision fluid was filtered and warmed to 37°C and gassed with 95% O2 and 5% CO2.
The pH and osmolarity of tiiis solution were = 7.35 and 290 mOsm, respectively,
immediately prior to perfiision. The perfiision fluid was mfiised mto tiie left carotid artery
witii an mfiision pump for periods of 15-60 s at 10 ml/mm (Harvard Apparatiis, South
73

Natiick, MA). This perfusion rate was selected to maintain a carotid artery pressure of
-120 mm Hg (21).
Rats were decapitated and cerebral samples obtained as previously described (24).
Briefly, the brain was removed from the skull, and the perfused cerebral hemisphere
dissected on ice after removal of the arachnoid membrane and meningeal vessels. Braui
regions were placed in scintillation vials and weighed. In addition, two 50 |al aliquots of
the perfusion fluid were fransfened to a scintillation vial and weighed. The brain and
perfiision fluid samples were then digested overnight at 50°C m 1 ml of 1 M piperidme.
Ten ml of Fisher Chemical scintillation cocktail (Beckman, FuUerton, CA) was added to
each vial and the fracer contents assessed by dual-label liquid scuitiUation countmg. Dual
labelled scintillation counting of brain and perfusate samples were accomplished with
conection for quench, background and efficiency.
4.2.7 Kinetic Analysis
Concenfrations of NP fracer in brain and perfusion fluid are expressed as dpm/g
brain or dpm/ml perfiision fluid, respectively. [ H]-NP brain uptake was detennmed by
perfiision witii [ H]-NP m separate experiments for 15-60 s periods as described
previously (21, 23). Unidfrectional uptake fransfer constants (Ki„) were calculated from
the following relationship to tiie hnear portion of tiie uptake curve as described (23) from
the equation:
Q*/C* = Ki„T + Vo (4-2)
74

where, Q* is tiie quantity of [ H]-fracer in brain (dpm/g) at the end of perfiision, C* is tiie
perfiision fluid concenfration of [ H]-NP (dpm/ml), T is tiie perfiision time (s) and VQ is
tiie exfrapolated intercept (T = 0 s; "vascular volume" in ml/g). Tracer frapped m the
vascular space was accounted for by the subtraction of [*'*C]-sucrose vascular volume.
Cerebral perfusion flow rate (F) was determined m separate experiments in the presence
botii NP formulations (25).
4.2.8 Statistical Analyses
Data presented are from the frontal cerebral cortex unless otherwise specified.
[ H]-NP brain uptake were fit with linear regression using least squares analysis. For all
data, enors are reported as the standard enor of the mean unless otherwise indicated
(GraphPad Prism Version 3.00 for Windows, GraphPad Software, San Diego, CA).
4.3 Results
Our collaborators, at the University of Kentucky, have recentiy reported on a
novel method to engineer NPs from microemulsion precursors (20, 26-27). Two of tiie
most promismg systems, E. Wax and Brij 72, were chosen for further testing as brain
drug dehvery systems. The novel NPs are composed of biocompatible materials:
emulsifymg wax and Brij 72 as tiie NP matiix materials, and Brij 78 and Tween 80 as
surfactants. Botii NP formulations have particle sizes below 100 nm (20). One of tiie
major requirements for effective NP brain delivery is size, tiius in vitro stability of NPs in
aqueous suspension was tested over a period of one week at room temperature, -20°C
75

(data not shown) and 4°C. Botii NP preparations showed superior stability at 4°C as
compared to 25°C or -20°C. The initial size of E72 NPs stored at 4°C mcreased by 50%
and 88% on day 2 and 4, respectively (Table 4.1). By the end of one week, tiie NPs had
mcreased in size by 100%. E78 NPs were generally more stable tiian tiie E72 NPs. When
stored at 4°C, E78 NPs increased in size by about 60% over the period of one week. The
tendency of tiie NPs to increase in size during storage is not fiiUy understood. It is
possible due to tiie high surface area of NPs m suspension, larger particles grow at tiie
expense of tiie smaller ones m order to reduce tiie overall free energy of the system (i.e.,
Ostwald ripenmg). Table 4.2 shows tiie stability of both E78 NP and E72 NP
formulations m various biologicaUy relevant media at 37°C. Particle size analysis
revealed no significant changes in size at physiological temperature suggesting tiiat both
NP systems remained intact during the tune course of m situ perfusion experiments.
These stability data agree with previous findings (20).
In order to ascertain NP brain uptake, both formulations were radiolabeled with
[ H]-cetyl alcohol. Hydrogen substitution by tritium [ H] atom was made on the first
carbon to avoid the possibility of [ H] hydrogen exchange with water during the time of
experiments. The choice of radioactive compoimd was based on the fact that E. wax is
composed of a mixture of cetostearyl alcohol and polysorbate 60 (4:1 w/w) (28). It was
hypothesized that frace amounts of [ H]-cetyl alcohol (hexadecanol) would be easily
enfrapped in E78 NPs and minimal or no release would be seen. Hexadecanol is
practically insoluble m water. Thus, during the radiolabeling procedure care was taken to
remove all solvent in which cetyl alcohol was supphed. It was also confirmed that tiie
76

labellmg procedure did not affect tiie size of solid NPs (data not shown). The entrapment
efficiency of [ H]-cetyl alcohol was determined by comparing the activity of [ H]-NPs
before and after separation on a gel permeation chromatography column. For both
nanosuspension types, all radioactivity co-eluded witii NPs m the void volume uidicating
high enfrapment efficiency (Figure 4.1). These findings were also confmned by lack of
appearance of a second peak caused by elution of free [ H]-cetyl alcohol. To ensure
stability of [ H]-NPs and association of the label with NPs under conditions which
formulations were ultimately exposed, nanosuspensions were dialyzed against 10 mM
phosphate buffered salme (pH=7.4) for 24 hr at 4°C and 6 hr at 37°C (Figure 4.2). For
E72 NPs, about 2% of the total radioactivity was released after initial dialysis at 4°C. The
amoimt of free radiolabel remained constant throughout the course of experiment. In the
case of E. Wax NPs, a constant level of about 2% radioactivity was detected in dialysis
buffer after 24 hr at 4°C and up to 30 mm at 37°C. After tiiat time, a shght mcrease of
release was noted but reached only 4% after 6 hr at 37°C. Of concern was whetiier
released radioactivity could be due to frace amounts of [ H]-water and/or [ H]-methanol.
The presence of these compounds could create some enor during brain uptake
measurements. It was shown that the steady release of radioactivity from NPs occurs
witiun first 90 min after which it reaches plateau. Additionally, released material passed
down tiie GPC column eluted in tiie same fraction as the free unenfrapped [ H]-cetyl
alcohol standard. It was tiius concluded that tiie radioactivity detected in tiie release
buffer was due to unenfrapped [ H]-cetyl alcohol probably remaming on tiie surface of
tiie NPs.
77

Brain distiibution parameters of [ H]-NPs were evaluated usmg tiie in situ rat
brain perfiision metiiod (21). Uptake of [ H]-NPs (1 Ci/200^g/ml) into bram was
evaluated from 0-60 s. Brain/perfiision fluid ratios (i.e., volume of distiibution or 'space')
were plotted as a function of time and are illusfrated in Figure 4.3. The mtegrity of the
BBB was verified during each experiment by simultaneous vascular volume
measurements using [*''C]-sucrose. [*'*C]-Sucrose vascular volumes in this study ranged
from 0.83 ± 0.2 to 1.2 ± 0.2 x 10" ml/g consistent with an intact BBB during tiie
experimental time frame and previous in situ NP perfusion data (25).
Transfer coefficient values (Kin) across the BBB were calculated from the slope of
[ H]-NP bram accumulation versus tune (Figure 4.3) (23). Calculated Ki„ and PA for
[ H]-NP E72 were 5.66 ± 1.16 x 10" ml/s/g and 6.05 ± 1.37 x 10" ml/s/g, respectively
(r^=0.922) (figure 4.3). A significant difference between [''*C]-sucrose and [ H]-E72 NPs
fransfer coefficients was observed (p<0.001) suggesting braui NP distiibution. The
calculated Ki„ and PA for the [ H]-E78 NPs as shown m Figure 4.3 were 4.11 ± 0.48 x
10' ml/s/g and 4.28 ± 0.68 x 10' ml/s/g, respectively (r =0.973). SunUar to [ H]-E72
NP data, a significant difference between ttie [*'*C]-sucrose and NP formulation fransfer
coefficients was observed (p<0.001).
4.4 Discussion
The data presented herein suggest botii NP fonnulations penefrate the BBB. The
mechanism of NP BBB cncumvention has been suggested m previous literatiire, but not
established. Kreuter (29) argued brain uptake of poty(butylcyanoacrylate) NPs contammg
78

dmg may be due to a number of different mechanisms, such as, (i) fonnation of a dmg
concentration gradient due to NP adhesion to endothelium and subsequent dmg release,
(ii) blocking of p-gfycoprotein (pgp) by tiie surfactant Tween 80 resulting in subsequent
higher brain uptake of dmg, (iii) toxic effects of the NP components on the BBB, (iv)
opening of tiie endotiielium banier due to the presence of surface active agents such as
Tween 80, and/or (v) endocytosis and/or franscytosis of NPs by endotiiehal cells.
Witii regard to NP BBB circumvention evaluated in tius stiidy, tiie uptake of
radiolabeled (dmg-free) NPs was demonstrated in time frames of less tiian 60 s, tiierefore
most likely elimmating mechanisms (i) and (ii) described by Kreuter (29). To evaluate
effects of E. Wax and Brij 72 NPs on BBB toxicity (mechanisms iii and iv), baseline
BBB parameters were screened in the presence of botii types of NP (25). The in situ rat
bram perfusion metiiod was used to investigate tiie mfluence of E78 NPs and E72 NPs on
cerebral perfusion flow, barrier integrity and permeability. Similar studies were
performed in vitro using bovme brain microvessel endothelial cells, an established in
vitro BBB model. The effects of NPs on bovine brain microvessel endothelial ceUs were
tested evaluating barrier integrity, permeability, choline fransport, and tight junctional
protein expression. In vivo and in vitro studies revealed no statistically significant
changes in BBB integrity, permeability or facilitated choline fransport. The presence of
either NP did not lead to any significant differences in cerebral perfusion flow in vivo.
Additionally, Westem blot analysis confirmed that the incubation of NPs with bovine
brain microvessel endothelial cells did not alter occludin and claudm-1 expression. It was
concluded tiiat E78 and E72 NPs have mmunal effect on baselme BBB parameters.
79

tiierefore tiie brain uptake values observed in the present stiidy are not likety to be caused
by toxic effects of NPs on tiie BBB. Further, mechanisms iii) and iv) seem unlikely in the
present stiidies because [*'C]-sucrose vascular volume did not increase m tiie presence of
tiie NPs evaluated (25).
Surface-active agents can compromise membrane integrity and mcrease
penneabihty to dmgs and otiier molecules. Specifically tius mechanism was suggested
for poly(butylcyanoacrylate) tween 80-coated NPs that were successfiil m delivering
dmgs across tiie BBB (29). Similarly, E72 NPs tiiat contamed Tween 80 resuhed in
statistically significant mcrease in brain fransport over E78 NPs (p <0.05). This is in
general agreement witii Kreuter reports (10,19,29). However, in confrast to tiie Kreuter
argument of surfactant induced membrane compromise, we hypothesize the E72 NP does
not penefrate the BBB in tiiis manner based upon the previous BBB integrity data (25).
This argiunent is based upon tiie difference of Tween 80 incorporation during E72 NP
manufacture. Previous reports have Tween 80 adsorbed on the surface of pre-formed
PBCA NPs, whereas in the case of E72 NPs Tween 80 was added during formation of the
microemulsion precursors (which subsequentiy formed the solid NPs), Further, upon
cooling of the microemulsion precursor and during sohd NP formation, the surfactant
may be unmobilized. Thus, we propose with the E72 NPs the hydrophobic taU of Tween
80 may be located m the warm oil droplet with the hydrophilic head group extending into
the extemal aqueous environment. This unmobihzation of the surfactant in the NPs
would likely mmimize, if not elimmate, tiie surface-active properties of the surfactant.
Although this hypothesis was not dfrectiy proven m tiiese present studies, previous
80

reports of coating tiie NPs with hydrophobically modified folate ligands confinned the
phenomena (30-31). Further, the E72 NPs contain only 2.3 mM Tween 80, whereas a 3-
fold higher concenfration was post-coated on tiie surface of the reported PBCA NPs.
Thus, while Tween 80 may be a factor altering BBB permeability in presence of PBCA
NPs, it does not appear to influence BBB integrity in the case of E78 and E72 NP
exposure (25).
Based on the data presented herein and arguments presented above, it appears the
cunent NPs evaluated may penefrate brain via passive permeability, endocytosis and/or
franscytosis. Additional experiments are underway to elucidate the mechanism of
fransport. In situ rat brain perfusion studies with [^H]-NPs will be followed by
subsequent washout steps with perfusion fluid to ensure complete removal of NPs that
may be loosely associated with endothelium.
In summary, two different types of NPs were engineered directly from warm
nucroemulsion precursors. Both NP types were radiolabeled by enfrapment of [^H]-cetyl
alcohol, and tiieir bram uptake evaluated in situ usmg the rat braui perfusion method.
Brain uptake of both NP formulations compared to [*'*C]-sucrose suggests significant
brain penefration. While tiie mechanism of BBB circumvention has not been elucidated,
we have proposed arguments that nanow down possible means. More experiments are
planned to fully understand tiie BBB permeability to these NP formulations.
81

4.7 References
1. W. M. Pardridge. Brain drug targeting, The future of brain drug development. Cambridge University Press, Cambridge, UK, 2001.
2. A. Tsuji. Specific mechanisms for fransporting dmgs mto brain. In D. J. Begley, M.W. Bradbury, J. Kreuter (eds.). The blood-brain barrier and drug delivery to the CNS, Marcel Dekker, New York, 2000, pp. 121-144.
3. C. Krewson, W. M. Saltzman. Transport and elimmation of recombinant human NGF during long-term delivery to tiie brain, 5ram i?e5. 727: 169-181 (1996).
4. C. Krewson, M. L. Klarman, W. M. Saltzman. Distiibution of nerve grov rth factor following durect delivery to brain interstitium. Brain Res. 680: 196-206 (1995).
5. L. Ilium. Transport of dmgs from the nasal cavity to the cenfral nervous system, Eur. J Pharm. Sa. 11: 1-18 (2000).
6. S. Mathison, R. NagiUa, U. B. Kompella. Nasal route for dfrect dehvery of solutes to the cenfral nervous system: fact or fiction?, J.Drug Targ. 5 (6): 415-441 (1998).
7. A. Granholm, D. Albeck, C. Backman, M. Curtis, T. Ebendal, P. Frieden, M. Henry, B. Hoffer, J. Kordower, G. M. Rose, S. Sodersfrom, R.T. Bartus. A non-invasive system for delivering neural growth factors across the blood-brain barrier: a review, Rev. NeuroscL 9: 31-55 (1998).
8. P. R. Lockman, R. J. Mumper, M. A. Khan, D. D. AUen. Nanoparticle technology for dmg delivery across the blood-brain barrier, Drug Dev. Ind. Pharm. 28 (1): 1-12 (2002).
9. K. S. Soppimath, T. M. Ammabhavi, A. R. KuUcami, W. E. Rudzinski. Biodegradable polymeric nanoparticles as dmg delivery devices, J. Controll Rel 70: 1-20 (2001).
10. J. Kreuter. Nanoparticulate systems for brain delivery of dmgs. Adv. Drug Del Rev. Al: 65-81 (2001).
11. R. N. Alyautidm, E. B. Tezikov, P. Ramage, D. A. Kharkevich, D. J. Begly, J. Kreuter. Significant entiy of tubocurarine mto tiie brain of rats by adsorption to polysorbate 80-coated poly(butylcyanoacrylate) nanoparticles: an in situ brain perfiision study, J.Microencap. 15 (1): 67-74 (1998).
12. U. Schroder, B. A. Sabel. Nanoparticles, a dmg carrier system to pass tiie blood-brain barrier, permit cenfral analgesic effects of i.v. dalargm uijections. Brain Res. 710: 121-124(1996).
82

13. A. Gulyaev, S. E. Gelperina, I. N. Skidan, A. S. Antropov, G. Y. Kivman, J. Kreuter. Significant fransport of doxombicin into tiie brain with polysorbate 80 coated nanoparticles, Pharm. Res 16(10): 1564-1569(1999).
14. U. Schroder, P. Sommerfield, S. Uriich, B. Sabel. Nanoparticle technology for delivery of dmgs across the blood-brain banier, J Pharm. Sci. 78 (11): 1305-1307 (1998).
15. R. N. Alyautidm, V. E. Pefrov, K. Langer, A. Berthold, D. A. Kharkievich, J. Kreuter. Delivery of loperamide across tiie blood-brain barrier witii polysorbate 80-coated nanoparticles, Pharm. Res. 14: 325-328 (1997).
16. R. Alyautdin, D. Gotiiier, V. Petrov, D. Kharkevich, J. Kreuter. Analgesic activity of tiie hexapeptide dalargin adsorbed on the surface of polysorbate 80-coated poly(butylcyanoacrylate) nanoparticles, Eur. J. Pharm. Biopharm. 41: 44-48 (1995).
17. S. C. Yang, L. F. Lu, Y. Cai, J. B. Zhu, B. W. Liang, C. Z. Yang. Body distiibution in mice of infravenously mjected camptothecm solid lipid nanoparticles and targeting effect on brain, J. Control Release 59: 299-307 (1999).
18. S. D. Troster, U. MuUer, J. Kreuter. Modification of the body distribution of poly(methyhnethacrylate) nanoparticles in rats by coatmg witii surfactants. Int. J. PAarm. 61:85-100(1990).
19. J. Kreuter, R. N. Alyautdm, D. A. Kharkevich, A. A. Ivanov. Passage of peptides through the blood-brain barrier with coUoidal polymer particles (nanoparticles); Brain Res.eiA: 171-174(1995).
20. M. O. Oyevmmi, R. J. Mumper. Gadolinium loaded nanoparticles engineered from microemulsion templates. Drug Dev. Ind. Pharm. 28 (3): 317-328 (2002).
21. Y. Takasato, S. I. Rapoport, Q. R. Smith. An in situ brain perfusion technique to study cerebrovascular fransport in the rat. Am. J. Physiol. 247: 484-493 (1984).
22. D. D. AUen, J. Oki, Q. R. Smith. An update m tiie in situ rat bram perfiision technique: simpler, faster, better, Pharm. Res. 14: 337 (1997).
23. Q. R. Smitii. Bram perfiision systems for studies of dmg uptake and metabohsm in the cenfral nervous system, Pharm. Biotechnol 8: 285-307 (1996).
24. D. D. Allen, Q. R. Smitii. Characterization of tiie blood-bram barrier cholme fransporter using the in situ rat brain perfusion technique, /. Neurochem. 76:1-11 (2001)
83

25. P. R. Lockman, J. Koziara, K. E. Roder, J. Paulson, T. J. Abbmscato, R. J. Mumper, D. D. Allen. In vivo and in vitro assessment of baseline blood-brain barrier parameters in tiie presence of novel nanoparticles, Pharm. Res. 20:705-713 (2003).
26. M. O. Oyewumi, R. J. Mumper. Engineering Tumor-targeted gadolinium hexanedione nanoparticles for potential application in neufron capture tiierapy, Bioconjugate Chem. 13: 1328-1335 (2002).
27. Z. Cui, R. J. Mumper. Plasmid DNA-Enfrapped Nanoparticles Engmeered from Microemulsion Precursors: In Vitro and In Vivo Evaluation, Bioconjugate Chem. 13: 1319-1327(2002).
28. A. Wade, P. J Weller. Haruibook of pharmaceutical excipients, American Pharmaceutical Association, Washington, DC 1994.
29. J. Kreuter, R. N. Alyautdin. Using nanoparticles to target dmgs to the cenfral nervous system. In D. J. Begley, M. W. Bradbury, J. Kreuter (eds.), The blood-brain barrier and drug delivery to the CNS, Marcel Dekker, New York, 2000, pp. 205-223.
30. M.O. Oyewumi, R.J. Mumper. Engineering tumor-targeted gadolinium hexanedione nanoparticles for potential application in neufron capture therapy. Bioconjugate Chem. 13: 1328-1335(2002).
31. M.O. Oyewumi, R.J. Mumper. Influence of formulation parameters on gadolinium enfrapment and tumor ceU uptake using folate-coated nanoparticles. Inter. J. Pharm. 251:85-97(2003).
84

Table 4.1: Short-term stability of nanoparticles at 4°C.
Nanoparticle size (nm)
Time (days)
0
4 ^^^^^^H^^..^-
NPE78
58 ±1
Ip ; 78±1 ' S 87 ±1
W^-~ 95 + 1
NPE72
98 ±2
•P47±1:BH 184±4 J Of. +-7'?^*^^*^
E. wax nanoparticles (E78 NP) and Brij 72 nanoparticles (E72 NP) particle size was measured immediately after preparation (day 0). Both types of preparations were sealed and stored at 4°C for a period of one week. Prior to nanoparticle size measurement aliquots of samples were allowed to equilibrate to room temperature. Data presented are mean ± SEM (n=3).
85

Table 4.2: Stability of nanoparticles in various biologically relevant media at 37°C
Nanoparticle size (nm)
NP E72 NP E78
Medium
Water
lOmMPBS
ISOmMNaCl
!lO% FBS
Initial time
53 ± 4
55 ± 5
58 ± 6
35 ± 4
1 hour
54 ±8
58 ±6
41 ± 7,
Initial time
82 ± 9
^ffl^^^^^B
73 ±9
53 ± 8
1 hour
78 ± 4
^Z^=F9- - I^B
83 ± 9
57 ± 5
E. Wax and Brij 72 nanoparticles were diluted (1:10 v/v) with water, 10 mM phosphate buffered saline (PBS), 150 mM sodium chloride (NaCl) or 10% (v/v) fetal bovine serum m 150 mM NaCl (FBS). Particle sizes were measured immediately after dilution and after one hour of incubation at 37°C. Data reported are mean ± SEM (n=3).
86

1.5E+06
1.0E406 Q. ^-^ (0 E £ °-.2 "o (0 V O) 3 « = 5.0E+05 .E E at •a
O.OE+00
ZE-(05
0 10 20 30
2.0E+06
"O 1.5E+06 §
o 0)
1.0E+06 ij a
- 5.0E+05 I o u
O.OE+00
0 5 10 15 20 25
GPC Fractions
Figure 4.1: Enfrapment efficiency of [ H]-cetyl alcohol in E72 nanoparticles.
Enfrapment efficiency of [ H]-cetyl alcohol m E72 nanoparticles using gel permeation chromatography (GPC) elution profiles. The GPC fractions contaming cold nanoparticles were detected by laser light scattering (counts per second), [^H]-NP and [^H]-cetyl alcohol were counted by a liquid scintillation counter and expressed as disintegrations per nunute (dpm): (A) counts per second (cps) for cold E72 nanoparticles, (•) dpm for [ H] E72 NP. The inset shows the profile for [^H]-cetyl alcohol alone (•).
87
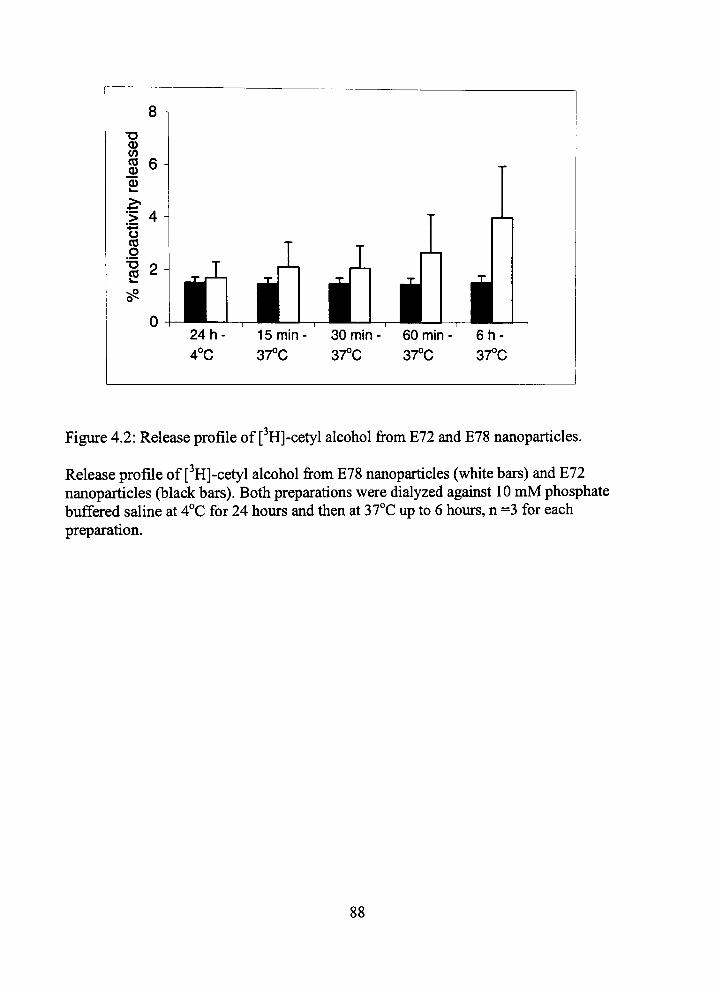
Figure 4.2: Release profile of [ H]-cetyl alcohol from E72 and E78 nanoparticles.
Release profile of [ H]-cetyl alcohol from E78 nanoparticles (white bars) and E72 nanoparticles (black bars). Both preparations were dialyzed agauist 10 mM phosphate buffered saline at 4°C for 24 hours and then at 37°C up to 6 hours, n =3 for each preparation.
88

m o
0.5 ,
0.4
•5'
"5. p 0.2
0.1
0.0
Time (s)
Figure 4.3: Time course of [^H]-nanoparticle and [*'*C]-sucrose uptake mto rat brain
Time course of [ H]-NPs and [*'*C]-sucrose uptake into rat bram during perfusion tunes of 15 to 60 s. A significant difference was noted between [ H]-NP and [''*C]-sucrose distribution parameters (p < 0.001) and between both nanoparticle types (p<0.05).
89

CHAPTER 5
EVALUATION OF BLOOD-BRAIN BARRIER THIAMINE
EFFLUX USING THE IN SITU RAT BRAIN
PERFUSION METHOD
5.1 Infroduction
Thiamme is an essential, water-soluble vitamin required for normal brain glucose
utilization. Dietary thiamine exists and is converted in vivo to three primary states:
thiamine, thiamine monophosphate and thiamme diphosphate (active form). Thiamine
and its monophosphate form are present botii infra and exfracellularly, while the
diphosphate form exists only within the cell. The active diphosphate functions as an
essential enzymatic cofactor for the cytosolic and mitochondrial tricarboxylic acid cycle
enzymes fransketolase, pymvate dehydrogenase and a-ketoglutarate dehydrogenase. The
relationship of thiamine conversion to its active form consists of infracellular thiamine
phosphorylation to the diphosphate form. Thiamine monophosphate must be hydrolyzed
mfracellularly to thiamine before bemg converted to the diphosphate form (1).
Wernicke-Korsakoff syndrome is the preeminent disease process illusfratmg
cenfral nervous system (CNS) thiamine deficiency. This neurologic disorder is seen in a
susceptible population suffering from chronic alcoholism. Characteristics of tiie disease
include memory loss and selective neuronal degeneration (2). The etiology is presumed
to be related to poor nutiition and unpaired intestinal tiiiamme absorption, botii of which
lead to inadequate plasma thiamme levels and subsequent diminished brain distiibution
90

(3). Furtiiennore, inadequate concenfrations of free brain thiamine (and its diphosphate)
are thought to play a role in other neurodegenerative diseases such as progressive
supranuclear palsy, Alzheimer's, Parkinson's and Huntington's diseases (4). The central
link of neurodegnerative disease to tiiiamine deficiency is hypothesized to be the
impairment of thiamuie-dependent enzymes and subsequent reduction in cerebral glucose
utilization (5).
The fransport mechanism of thiamine and thiamine monophosphate into and out
of the cell is critical for thiamine diphosphate concenfrations and has been evaluated
extensively (1). Similarly, fransport of thiamine uito brain from plasma is as fundamental;
given exfracellular brain thiamine concenfrations drive thiamine diphosphate
concenfrations in neuron and glia (4). For thiamine to penefrate the CNS, it must cross
the blood-brain barrier (BBB). The BBB is comprised of a continuous layer of
endothelial cells connected by tight junctions circumferentially sunounding the cell
margin (6). These junctions endow the barrier with properties similar to a cell membrane
with regard to franscellular solute movement. Specifically, lipid soluble molecules diffuse
rapidly across the BBB lipoid endotheUal cell membranes; whereas, hydrophilic
compounds show restricted permeation unless fransported by a carrier exchange
mechanism (7). Because tiiianune is a physiologically charged monovalent cation (8) tiie
BBB attenuates passive brain entiy but aUows brain penefration tiirough a carrier-
mediated fransporter (9,10).
Net thiamine movement mto brain has been reported to be tiie difference between
total influx less proposed, but not demonsfrated, efflux. Influx at tiie BBB is mediated
91

predommantiy by a satiirable carrier-mediated transport mechanism (approximately 90%)
and a nonsaturable mechanism, presumably passive diffusion. A possible efflux
mechanism at the BBB has also been suggested (11) but not fiiUy explored. Thiamme
efflux has been demonsfrated at tiie membranes of neuroblastoma cells (12,13), rat liver
mitochondria (14), E. Coli (15), and Schizosaccharomyces pombe (16).
Considering: (1) we sought to utilize tiiiamme as a NP brain targeting ligand, (2)
cunent literature has implicated thiamme deficiency as an etiology for many significant
neurodegenerative diseases, and (3) cunent BBB kinetic methodologies are more
sensitive than previous methods, we evaluated thiamine kinetics at the BBB, considering
both efflux and influx parameters. Further, the net movement of thiamine into brain is
discussed in its potential relationship to thiamine deficiency-related neurodegenerative
disease.
5.2 Materials and Methods
Uptake of [^Hj-thiamine into brain was assessed using the in situ rat brain
perfiision technique of Takasato et al. (17) with modifications described (7,18). In this
study, short perfusions of 5-135 s were used to determine initial brain uptake. Saturation
kinetics of tiie fransporter were completed at 15 s to mmimize [^H]-thianiine bram efflux.
All studies were approved by tiie Institutional Animal Care and Use Committee and were
conducted in accordance witii the NIH Guide for tiie Care and Use of Laboratory
Animals.
92

5.2.1 Radiochemicals
High specific activity [ H]-thiamine (10 Ci/mmol, >98% purity) was obtained
from American Radiolabeled Chemicals (St. Louis, MO). The [*'*C]-sucrose (4.75
mCi/mmol) was obtained from Dupont-New England Nuclear (Boston, MA). In each
experiment, [ H]-tiiiamine was dried prior to being dissolved m perfusion buffer, to
remove volatile fritium contaminants including [ H]-H20.
5.2.2 Perfusion Procedure
Male Fischer-344 rats (220-330 g; Charles River Laboratories, Kingston, NY)
were anesthetized with sodium pentobarbital (50 mg/kg mfraperitoneal). A PE-60
catheter filled with heparinized saline (100 U/ml) was placed mto the left common
carotid artery after ligation of left extemal carotid, occipital and common carotid arteries.
Common carotid artery ligation was accomplished caudal to the catheter unplantation
site. The pterygopalatine artery was left open during the experiments (18). Rat rectal
temperature was monitored and maintained at 37°C by a heating pad connected to a
feedback device (YSI Indicating ConfroUer, Yellow Springs, OH). The catiieter to tiie left
common carotid artery was connected to a syringe containing buffered physiologic
perfiision fluid (containing [in mM]: NaCl 128, NaPOj 2.4, NaHCOs 29.0, KCl 4.2, CaCl
1.5, MgCl2 0.9, and D-glucose 9) with 0.75 ^Ci/ml [ H]-tiiiamine (final [thiamine] ~ 75
nM) and 0.3 ^Ci/ml [''*C]-sucrose (to determine vascular volume). Perfusion fluid was
filtered and warmed to 37°C and gassed witii 95% O2 and 5% CO2. The pH and
93

osmolarity of tills solution were 7.35 and 290 mOsm, respectively, immediately prior to
perfiision. The perfiision fluid was infiised mto the left carotid artery witii an mfiision
pump for periods of 5-135 s at 10 ml/min (Harvard Apparatiis, Soutii Natick, MA). This
perfiision rate was selected to maintain a carotid artery pressure of ~ 120 mm Hg (17).
Rats were decapitated and cerebral samples obtamed as previously described (19).
Briefly, tiie brain was removed from the skull, and tiie perfiised cerebral hemisphere
dissected on ice after removal of tiie arachnoid membrane and meningeal vessels. Brain
regions were placed in scintillation vials and weighed. In addition, two 50 ^I aliquots of
the perfusion fluid were fransfened to a scintillation vial and weighed. The brain and
perfusion fluid samples were tiien digested overnight at 50°C in 1 ml of 1 M piperidine.
Ten ml of Fisher Chemical scintillation cocktail (Beckman, FuUerton, CA) was added to
each vial and the fracer contents assessed by dual-label liquid scmtillation counting. Dual
labeled scintiUation counting of bram and perfusate samples were accomplished witii
conection for quench, backgroimd and efficiency.
For wash out studies, a 45 s perfusion with [ H]-tiiiamme was completed followed
by a fracer-free buffered physiologic perfusion fluid infusion for 5-60 s. Vascular
conection was accoimted for by subfraction of [*'*C]-sucrose vascular volume. Cerebral
samples were obtained and evaluated similarly as described (19).
To evaluate the distiibution of [ H]-thiamine into brain capillary endothehal ceUs,
capillary depletion was accomplished using the method of Triguero et al. (20). Briefly,
the brain was removed after a 45 s in situ perfiision containing both [ H]-thiamine and
[*'*C]-sucrose. The brain was then homogenized in physiologic buffer (4°C) in a glass
94

homogenizer (6 sfrokes). After homogenization was complete, ice-cold dextran (34%
wt/vol) was added to the homogenate, and fiirther homogenization (4°C) was completed
(4 sfrokes). The homogenate was then cenfrifiiged at 5400 x g for 15 min at 4°C. The
subsequent supematant (bram parenchyma) was tiien carefiiUy separated from tiie pellet
(brain microvasculatiire) and both were prepared for dual label scmtillation counting.
5.2.3 Kinetic Analysis
Concenfrations of fracer in brain and perfusion fluid are expressed as dpm/g brain
or dpm/ml perfiision fluid, respectively. Blood-bram barrier [^H]-tiiiamme fransport mto
brain was determmed by perfusion witii [^H]-tiiiamme (75 nM) for a 5-135 s period as
described previously (7,17). Given an apparent nonlinear uptake partem was observed, a
calculated uptake fransfer constant (Ki„) and a brain efflux rate coefficient (kout) was
estimated from the foUowing relationship as described (7):
Q*/C* = (Ki„/kout)(l - e-"""* ) (5 1)
where, Q* is the quantity of [^H]-fracer m bram (dpm/g) at the end of perfusion, C* is the
perfusion fluid concenfration of [^H]-thiamine (dpm/ml) and T is the perfusion time.
Tracer frapped in the vascular space was accounted for by the subtraction of [*'*C]-
sucrose vascular volume.
Calculation of the effective volume of distribution at steady state is by the
relationship:
Vbr = (Ki„/kout). (5.2)
95

For determination of the satiirable kinetics of [^H]-tiiiamme fransport, a perfiision
tune of 15 s was chosen tiiat allowed an adequate amount of fracer to pass into brain and
mmimized apparent efflux. We then calculated Kii in single pomt uptake experiments m
each bram region from tiie foUowmg relationship as described (7):
K«=[Qtot-VvCpf]/(Cp/D (5.3)
where, Q ^ = Q^^^ + Q^^ represents tiie sum of tiie amount of [^H]-titiamuie remammg in
tiie perfusate m tiie blood-brain vessels and tiie amount of[^H]-tiiiamme that has
penefrated into brain. Cerebral perfusion flow rate (F) was determmed in separate
experiments as previously described (21). Cp is tiie perfiision fluid concenfration of
fracer thiamine and T is the net perfusion time. To ascertain points below the Km, we
decreased perfusion fluid [^H]-tiiiamine concenfration to 37.5 nM by dilution of the
perfusion fluid. This necessitated removal and aggregation of the entire cortical regions
for statistically valid dual labeled scintUlation countmg.
Kin values were converted to apparent cerebrovascular permeability-surface area
products (PA) using the Crone-Renkin equation (7):
PA = -Fhi(l-Kto/F) (5.4)
where F is tiie cerebral perfusion flow determined from the uptake of [^H]-diazepam (21).
Regional perfusion flow was used for regional PA determination to account for regional
flow variations. In all mstances, PA differed by < 2% from Kin, because F exceeded Kin
by > 40 fold.
96

Concenfration dependent [^H]-tiiiamine brain uptake was evaluated as a smgle
saturable and non-saturable process where:
PA = [Vn^/(K„ + Cpf)] +KD. (5.5)
5.2.4 Statistical Analysis
Data presented are from the frontal cerebral cortex unless otherwise specified.
[ H]-thianune brain uptake and PA reduction over time were fit with non-linear
regression using least squares analysis. Washout regression lines were calculated with
least squares Imear regression. One-way ANOVA analysis followed by a Bonferoni's
multiple comparison test were used for evaluation of regional brain uptake and efflux of
[^H]-tiiiamine. For all data, enors are reported as standard enor of the mean unless
otherwise indicated. Differences were considered statistically significant a priori at the
p<0.05 level. (GraphPad Prism version 3.00 for Wmdows, GraphPad Software, San
Diego, CA USA).
5.3 Resuhs
5.3.1 [^H]-Thiamine Brain Uptake
Figure 5.1 illusfrates tiie time course of [^H]-tiiiamme (75 nM) bram uptake
measured using the in situ rat bram perfusion method. The bram/perfiision fluid
concenfration ratio of [^H]-tiiiamme mcreased non-linearly over 5-135 s in tiie absence of
unlabeled tiuamine. Brain/perfusion fluid ratios (i.e., volume of distiibution or 'space')
were fit to equation (1) (r^ = 0.978) (Figure 5.1). The calculated Kin and estimated kout for
97

[^H]-thiamme uptake were 1.05 ± 0.16 x 10" ml/s/g and 2.68 ± 0.58 x 10" s'',
respectively. The calculated brain distiibution volume at steady state (Kin/kout) equaled
3.92 ± 0.28 ml/g x 10" . Of significance, [ H]-tiiiamine bram distiibution is significantiy
mfluenced by a considerable efflux rate.
To ascertain if [ H]-tiiiamine brain distiibution seen m figure 5.1 is overestimated
secondary to capillary endothelial association, we evaluated cellular localization of [ H]-
thiamine and [*''C]-sucrose (Table 5.1) using capillary depletion (20). Rats were perfused
witii botii fracers for 45 s. We observed a significant (p <0.05) brain parenchyma
(Dhomogenate) accumulation of [ H]-thiamine (compared to [*'*C]-sucrose) and a non
significant difference of the fracers associating with the pellet (Dpeiiet).
5.3.2 Transport Kinetics for [ H]-Thiamme
To evaluate saturable fransport kinetics a perfusion tune of 15 s was chosen and
evaluated as single time brain uptake experiments with data being fit to equation 5 (r =
0.948) (7,17). Saturable kinetic parameters were evaluated with the addition of unlabeled
thiamine ([^H]-thiamine concenfration ~ 75 nM) to the perfusion fluid at concenfrations
of 25 to 10,000 nM. Figure 5.2 shows dose-dependent reduction of ['H]-thiamine PA in
the presence of unlabeled thiamine for the frontal cortical region. Saturable kinetic
parameters observed were: Km = 96 ± 6 nM, Vmax = 8.7 ± 1.7 pmol/min/g, and IQ = 0.42
± 0.04 X lO"'' ml/s/g. To evaluate points 75 nM tiie perfusate concenfration was decreased
to 37.5 nM by fracer dilution. Given the subsequent decreased fracer penefrating brain it
was necessary to evaluate tiie entire cortical region as a smgle region. The PA obtamed in
98

tills experiment (data not shown) was consistent witii the saturable kinetic data shown m
Figure 5.2. Table 5.2 hsts tiie estimated satiirable fransport parameters in various brain
regions. Significant differences for PA were seen in the occipital cortex, hippocampus
and tiie caudate putamen region, whereas minimal regional differences were noted for Km
and Vmax.
5.3.3 Efflux of [ H]-Thiamine at the BBB
Since we observed non-linear brain uptake of [ H]-thiamine over time, we
calculated brain efflux constants using a washout method. Briefly, we evaluated the fracer
brain/perfusion ratios after 45 s loading of [ H]-thianiine with a following "wash" of
thiamine free saline for periods of 5 - 60 s. Figure 5.3 shows the brain perfusion ratio in
the frontal cortex dropped significantly over a 60 s period (0 s: 2.38 ± 0.27 nti/g x 10';
60 s: 1.04 ± 0.15 X 10" ml/g). The efflux constant can be calculated by either first- or
zero-order kinetics. The best fit was based upon zero-order kinetics where tiie linear
regressed slope was equal to -2.20 ± 0.24 x 10"'* ml/g. However, in order to make
comparison to the mitial calculated uptake kout rate we assumed first-order bram
elmunation. We then convert the Imear regressed slope to a rate constant where: kout =
hi2/ti/2 (tiie efflux rate is 1.30 ± 0.17 x 10" s'*). The latter rate is tiien comparable to tiie
rate determined in Figure 5.1. Table 5.3 shows tiie efflux rate by tiie washout method is
regionally homogeneous. The calculated efflux constant (kout) for each brain region was
not significantiy different among regions (p > 0.05).
99

To evaluate if efflux could be stimulated, such as a trans-stimulation mechanism,
subsequent washout experiments were completed with unlabeled thiamme (300 nM)
present in tiie wash. The brain/perfiision ratio of [^H]-tiiiamine at 15 s was significantiy
reduced when 300 nM was added (thiamme free saline: 2.15 ± 0.04 ml/g x 10'^ 300 nM
thiamine 1.12 ± 0.02 ml/g x 10" ). Figure 5.4 shows tiie calculated efflux constants m tiie
presence of varying vascular concentrations of thiamine. The efflux rate constant m the
presence of 75 nM thiamine (Figure 5.1; estunated during uptake usmg equation 1) was ~
2-fold higher than in the absence of thiamine and approximately half that of the presence
of 300 nM thiamine (Figure 5.4). This data suggested that the presence of vascular
thiamuie stimulated brain efflux. Regional examination of efflux constants at 300 nM
were not significant from frontal cortical values (data not shown).
5.4 Discussion
The results of the studies presented herein confirm previous reports that thiamine
influx at the BBB is via a carrier-mediated fransport mechanism, yet provide a new
estimation of saturation kmetics. The unportance of this report is two-fold. First, net
thiamine brain accumulation is significantiy uifluenced by a rapid efflux mechanism tiiat
may be frans-stimulated. Second, tiie observation of homogeneous influx and efflux of
thiamine at the BBB may explain regional depletion of thiamme dependent enzymes m
neurodegenerative disease Imked to thiamme deficiency.
Initial experiments to detennine brain [^H]-tiiianime distiibution during perfiision
tune frames revealed 135 s distiibution volumes to be consistent witii previous work (9).
100

However, m confrast to earher work tiiat suggested linear tiiiamine uptake m the initial
period, we observed a non-linear [^H]-thiamine brain uptake distribution pattern. The
disparity is most explained by methodological considerations. In situ brain perfiisions can
accurately detect brain distiibution of a high specific activity radiolabeled compound at 5
s (17). The non-lmear uptake suggested rapid uptake and subsequent efflux of [^H]-
tiiiamine from brain (Figure 5.1). Similar non-lmear uptake profiles are seen witii dmgs
having significant brain efflux such as cyclosporin A, vmcristme, theophyUine and
iodoantipyrine (22). Previous thiamine brain distribution literature has suggested an
efflux mechanism may be present (11), but tiiis mechanism has not been explored in
detail.
To confirm initial [^H]-thiamine distribution was not overestimated secondary to
BBB endothelial cell accumulation and/or association, we compared [^H]-thiamine brain
parenchyma and cell distribution after a 45 s perfusion. Table 5.1 shows significant [^H]-
thiamine brain parenchyma distiibution (homogenate distribution compared to [*'*C]-
sucrose) and a non-significant difference of the fracers associating with the pellet (BBB
endothelial cells). This data suggests [ H]-thiamine brain accumulation is related to
penefration and unlikely to be related to significant endothelial cell thiamine uptake.
In order to evaluate saturable fransport kmetics using in situ rat perfusions vdth
significant efflux, it is necessary to perform smgle time uptake experiments with and
without unlabeled compound for inhibition (7). Given the non-lmear tune dependent
[^H]-tiiiamine uptake, we chose a perfusion tune based upon two factors: (1) it was long
enough for [^H]-thiamine detection, and (2) it was short enough to mmimize apparent
101

efflux. Thus, a perfiision time of 15 s was chosen given [ H]-thiamine brain uptake was
approximatefy linear at this time. Figure 5.2 shows tiiat addition of unlabeled thiamme
(25 to 10,000 nM) to the perfiision fluid resulted in a dose-dependent reduction of PA,
consistent witii a satiirable canier-mediated process. Data points below tiie Km were
evaluated by decreasing perfiisate fracer concentrations and evaluation of tiie entue
cortex as a single region. The PA obtained from tius approach (data not shown) was
consisent witii tiie satiirable kinetic data shown m Figure 5.2.
Considering tfie canier kmetics (Table 5.2), tiie observed Vmax has somewhat
large enors, which may be attributable to using [ H]-tiiiamine levels (75 nM) close to tiie
calculated Km value. Higher specific activity [ H]-tiiiamine (not commercially available)
would aUow better characterization of tiie satiiration kinetics (i.e., tiie pomts below Km)
and provide less enor. However, Vmax estimates obtained in this report (1.4 to 14.4
pmol/min/g) are consistent witii previous estunates reported at tiie BBB (16.5 to 18.6
pmol/mm/g) (9) and m a neuroblastoma cell tine (12).
The apparent Km values (~75-130 nM) obtained are less than previous apparent
Km estimates of 0.61, 0.78, 2.5 and 4.98 |JM (9-11,23). The cunent values may be more
physiologically consistent with baseluie plasma thiamine levels that range from ~ 6 to
240 nM (24-27).
Differences amongst the kinetic values may be related to the experimental tune
frames and tiiiamine concenfrations. The data herein were completed in time frames from
5-135 s with high specific activity [ Hj-thiamine (concenfrations ~ 75 nM with no
contribution from plasma thiamine); whereas previous studies used uiitial time frames of
102

1-2 minutes, low specific activity [*'*C]-tiiiamine (concenfrations ~ 280 nM) (11) m
addition to baseline plasma thiamine concentrations that may cause an overestimation in
Km values. Otiier differences may be from metiiodology, not simultaneously considermg
brain vascular space or not accounting for tiiiamine erytiirocyte accumulation (28). The in
situ rat perfusion method using high specific activity compounds accounts for the
described limitations (7).
To investigate and acciu-ately account for the reported BBB thiamine efflux
mechaiusm (11), we perfused the brain with [ H]-thiamine for 45 s followed by a fracer
and thiamine free perfusion buffer for 5-60 s. Significant efflux from the brain was
observed over the one-min period in the frontal cortical region (Figure 5.3). The
calculated [ H]-thiamine efflux constant (assuming first-order elimination) confirmed the
efflux observed during initial uptake experiments. Examination of regional [ H]-thiamine
efflux is shown in Table 5.3 and the estimated efflux constant for each brain region was
not significantly different.
To determme if additional tiiiamme could stimulate efflux, we perfused the brain
with [^H]-thiamine for 45 s, but witii 300 nM of tiiianune added to tiie wash buffer
(vascular thiamme concenfration). Thiamine included m the wash at a concenfration of
300 nM resuhed in a significant uicrease in [ Hj-tiuamme efflux from bram at 15 s. This
data is compared to tiie efflux constant calculated m tiie presence of 75 nM (during initial
uptake experiment) in Figure 5.4. The calculated efflux constants showed a significant
difference m tiie presence of varymg concenfrations of vascular tiiiamuie. This evidence
of efflux stimulation suggests a frans-stimulation or self-exchange mechanism may be
103

present for thiamine fransport at tiie BBB. A similar mechanism of increased thiamine
efflux was found in a neuroblastoma thiamine homeostasis stiidy (12). In this stiidy,
neuroblastoma cells loaded with radiolabeled thiamine had significantly increased effiux
in tiie presence of increasing amounts of extemal tiuamme. The autiior suggested tiie
high-affinity tiiiamme influx carrier is a self-exchange mechanism tiiat is sensitive to
extemal thiamine and intracellular thiamine diphosphate concenfrations.
Explanation as to the physiologic rationale for thiamine efflux from brain has
not been elucidated; however, we propose two possibilities. Ffrst, it is possible the
presence of high levels of plasma thiamine quickly saturate the mfracellular environment
of the BBB with free thiamine (to be converted to thiamine diphosphate), which in tum
stimulates the BBB efflux mechanism. Second, Bettendorf (12) proposed thiamine efflux
from neuroblastoma cells is simply related to the storage of excessive thiamine. This
rationale is based on previous work (29) demonsfrating enormous doses of thiamme
resulted hi only small mcreased brain concenfrations but significantly augmented
thiamine concenfrations in liver and erythrocytes. The hypothesis is the latter two
compartments store thiamine for periods of deficiency, allowing remaining tissues to
efflux excessive thiamine for storage. Whetiier this mechaiusm is by self-exchange or by
an alternate pathway wanants furtiier evaluation. Future work may also explore the
possible asymmetric contiibution of the luminal and ablummal membranes in eitiier
influx or efflux of thiamine movement at the BBB.
Of major significance in tius report is [ H]-tiiiamme bram uptake and efflux is to a
large extent regionally homogeneous. A hallmark characteristic of tiiiamme deficiency
104

induced Wemicke-Korsakoff syndrome is tiie selective neuronal cell degeneration and/or
deatii (2). Specifically, tiie vulnerable brain regions mclude: tiialamic nuclei, inferior
colliculus, inferior olivary, mammillary bodies and tiie lateral vestibular. Regions where
tiiis is not observed include tiie caudate nucleus, frontal-parietal cortex and tiie
hippocampus (30). This regional vulnerability has been positively conelated to tfie rate of
tiiiamine neuronal uptake and metabolism in the specific brain regions. For example,
Rindi et al. (31) demonsfrated tiiat tiie cerebellum region has the greatest turnover and
mflux rate of thiamine, while the cerebral cortical region was tiie lowest. Given the
regional differences in bram thiamine utilization, we initially hypotiiesized there would
be significant regional differences in BBB tfuanune fransport. However, the
characterization of regional PA, influx kinetics and efflux rates described herem
demonsfrate thiamine homeostasis at the BBB is essentially consistent throughout brain
regions. This unifomuty, compared to regional thiamine utilization rates, may suggest
vulnerable brain regions are depleted of thiamine (at a greater rate than non-vulnerable
regions) imder conditions of low brain delivery, i.e., low plasma levels secondary to
chronic alcoholism.
A cunent hypothesis with regard to neurodegenerative disease Imked to thiamine
deficiency is regional depletion (under conditions of low plasma levels) leads to regional
production impairment of mitochondrial tricarboxylic acid cycle thiamme- dependent
enzymes. Subsequentiy, regional enzymatic decrease dunuiishes cellular cerebral glucose
metabolism in areas of high glucose utilization (vuhierable brain regions) ultimately
resulting in selective neuronal degeneration (4). This argument can be associated witii tiie
105

data presented in tiiis report in two ways: (1) regionally homogenous BBB tiuamine
mflux and efflux constants despite reports of regionally significant differences between
tiuamine/glucose brain utilization rates and (2) the suggestion of rapid brain thiamuie
depletion as evidenced by significant BBB tiiiamine efflux.
Also of significance in tiiis report is the comparison of endothelial cell association
ui perfusion experiments between thiamine and choline; both physiologically charged
cations. [ H]-choline non-specifically binds to BBB endothelia during perfusion
distribution experiments, as seen by post-perfiision washouts resultuig m an immediate
reduction in the brain/perfusion ratio (18). This suggests cationic molecules may
nonspecifically bmd to BBB endothelia. However, tiie time course of [^H]-thianune
during our washout study (Figure 5.3) revealed an essentially linear decrease of the
brain/perfiision ratio, suggestive of braui efflux with minimal endothehal disassociation.
In summary, this research confirms previous literature suggestuig BBB tiiiamine
uptake is via a carrier-mediated fransport mechanism, yet extends the literature by
redefining the BBB kinetics with a more sensitive methodology. With regard to BBB
thiamine homeostasis, accumulation was significantly influenced by a considerable efflux
rate. Fiuther evaluation of tiie efflux rates demonsfrated efflux stunulation m the presence
of mcreased vascular tiiiamine concenfrations. The observation of regionally homogenous
tiuamme homoeostasis at tiie BBB may be of significant relevance to neurodegenerative
disease linked to thiamine deficiency.
106

5.6 References
1. O.K. Singleton, Martin P.R. Molecular mechanisms of tiuamine utilization, Curr Mol Mei/. 1: 197-207(2001).
2. M. Victor, R.D. Adams, Collins G.H. The Wemicke-Korsakoff syndrome. A clinical and pathological stiidy of 245 patients, 82 witii post-mortem exammations, Contemp. Neurol Ser. 7: 1-206 (1971).
3. A.D. Thomson. Mechanisms of vitamm deficiency in chronic alcohol misusers and tiie development of tiie Wemicke-Korsakoff syndrome. Alcohol Alcohol Suppl 35 Suppl: 2-7 (2000).
4. G.E. Gibson, H. Zhang. Interactions of oxidative sfress with tiiiamme homeostasis promote neurodegeneration, Neurochem. Int 40: 493-504 (2002).
5. A.M. Hakim, H.M. Pappius. The effect of thiamine deficiency on local cerebral glucose utilization, ^«n. Neurol 9: 334-9 (1981).
6. A.M. Butt A.M., H.C. Jones, N.J. Abbott. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study, J. Physiol 429: 47-62 (1990).
7. Q.R. Smith. Brain perfusion systems for studies of dmg uptake and metabolism in the cenfral nervous system, Pharm. Biotechnol 8: 285-307 (1996).
8. T. Komai, H. Shindo. Stmctural specificities for the active fransport system of thiamme in rat small intestme, J. Nutr. Scl Vitaminol (Tokyo) 20: 179-87 (1974).
9. J. Greenwood, E.R. Love, O.E. Pratt. Kinetics of thiamine fransport across the blood-bram barrier m tiie rat, J. Physiol 327: 95-103 (1982).
10. C. Patrini, C. Reggiani, U. Laforenza, G. Rindi. Blood-brain fransport of thiamme monophosphate m the rat: a kinetic study in vivo, J. Neurochem. 50: 90-3 (1988).
11. J. Greenwood, P.J. Luthert, O.E. Pratt, P.L. Lantos. Transport of thiamin across the blood-brain barrier of the rat in the absence of aerobic metabolism. Brain Res. 399: 148-51 (1986).
12. L. Bettendorff Thiamine homeostasis ui neuroblastoma ceUs. Neurochem. Int. 26: 295-302 (1995).
107

13. L. Bettendorff, P. Wins. Mechanism of thiamine transport in neuroblastoma cells. Inhibition of a high affinity carrier by sodium channel activators and dependence of tiiiamine uptake on membrane potential and infracellular ATP, /. Biol Chem. 269: 14379-85(1994).
14. M. Barile, S. Passarella, E. Quagliariello. Thiamine pyrophosphate uptake mto isolated rat liver mitochondria. Arch. Biochem. Biophys. 280: 352-7 (1990).
15. T. Nishimune, M. Tomita, Z.Yoshii, R. Hayashi. Counterflow efflux of thiamm m Escherichia coli, Biochim. Biophys. Acta. 649: 419-26 (1981).
16. K. Hfrose K, R. Chumnantana, T. Nakashima, M. Ashiuchi, T.Yagi. Efflux system for pyridoxine in Schizosaccharomyces pombe, Biosci. Biotechnol Biochem. 64: 2675-9 (2000).
17. Y. Takasato, S.I. Rapoport, Q.R. Smith. An in situ brain perfusion technique to study cerebrovascular fransport m the rat. Am. J. Physiol. 247: 484-493 (1984).
18. D.D. Allen, J. Oki, Q.R. Smith. An update in the in situ rat brain perfusion technique: simpler, faster, better, Pharm. Res. 14 suppl: 337 (1997).
19. D.D. Allen, Q.R. Smith. Characterization of the blood-brain barrier choline fransporter using the in situ rat brain perfusion technique, J. Neurochem. 16: 1-11 (2001).
20. D. Triguero, J. Buciak, W.M. Pardridge. Capillary depletion method for quantification of blood-brain barrier fransport of circulatmg peptides and plasma protems, J. Neurochem. 54: 1882-8 (1990).
21. S. Momma, M. Aoyagi, S.I. Rapoport, Q.R. Smith. Phenylalanme fransport across the blood-brain barrier as studied by the in situ perfusion technique, / Neurochem. 48: 1291-1300(1987).
22. H. Murakami, H. Takanaga, H. Matsuo, H. Ohtani, Y. Sawada. Comparison of blood-brain barrier permeability in mice and rats using in situ brain perfusion technique. Am. J Physiol Heart Circ. Physiol 279: H1022-8 (2000).
23. C. Reggiani, C. Patrini, G. Rindi. Nervous tissue tiiiamine metabolism in vivo. I. Transport of thiamine and tiiiamine monophosphate from plasma to different brain regions of the rat. Brain Res. 293: 319-27 (1984).
24. W. Weber, H. Kewitz. Detemunation of thiamme in human plasma and its pharmacokinetics, Eur J. Clin. Pharmacol 28: 213-9 (1985).
108

25. L. Bettendorff, C. Grandfils, C. De Rycker, E. Schoffeniels. Determmation of thiamine and its phosphate esters in human blood semm at femtomole levels, J. Chromatogr. 382: 297-302 (1986).
26. C. Patrini, E. Pemcca, C. Reggiani, G. Rindi. Effects of phenytoui on tiie in vivo kinetics of thiamine and its phosphoesters in rat nervous tissues, Brain Res. 628: 179-86 (1993).
27. CM. Tallaksen, T. Bohmer, J. Karlsen, H. BeU. Determination of tiiiamin and its phosphate esters in human blood, plasma, and urine. Methods Enzymol 219: 61-1A (1997).
28. D. Casu-ola, C. Patrini, G. Fenari, G. Rindi. Thiamm fransport by human erythrocytes and ghosts, J Membr. Biol 118: 11-8 (1990).
29. H. Sanemori, T. Kawasaki. Thiamme tiiphosphate metabohsm and its turnover in tfie rat liver, Experientia. 38: 1044-5 (1982).
30. K. Todd, R.F. Butterworth. Mechanisms of selective neuronal cell deatfi due to tiuamine deficiency,^nn. N.Y. Acad Scl 893: 404-11 (1999).
31. G. Rindi, C. Patiini, V. Comincioli, C. Reggiani. Thiamine content and tiimover rates of some rat nervous regions, using labeled tiiiamine as a fracer. Brain Res. 181: 369-80 (1980).
109

Table 5.1: Capillary localization of [ H]-thiamuie and [*'*C]-sucrose fracer after a 45 s perfusion uptake
Marker Total percent of tracer associated with pellet
[ H]-thianaine
[ C]-sucrose
0.4 ±0.1%
0.8 + 0.2
Cellular localization of [ H]-thiamme and [*'*C]-sucrose using capillary depletion. Values reported are for 45 s perfiisions of [ H]-thianune and [ '*C]-sucrose at fracer concenfrations. Values shown are mean ± SEM, n=3-5.
110

Table 5.2 Calculated cerebrovascular permeability and saturable kinetic parameters for [^H]-thiamine fransport at the blood-bram barrier.
Brain Region
Frontal cortex
Parietal cort^H
Occipital cortex
PA Km (nM)
1.06 ±0.17 96 ±6
'max
pmol/min/g
8.7 ±1.7
• p 4 i 0 . 0 5 84 + 4 3.6±0:9
0.83 ±0.21* 97 ±8 3.1 ±0.6
Kd (ml/s/g X 10 )
0.42 ± 0.04
oleWM^
0,39 ±0.03
Hippocampu§
Caudate/ Putamen Thalamus/ M Hypothalamus
Cerebellum
M&^^^.^J^ZiJ^^^^wJ:'^'^ - 4-9 ^ _ -31 ± 0,06*
0.78 ±0.10* 75 ± 4 1.4 ±0.6 0.39 ±0.03
f ^+O. lO 8 6 ± 4 ^ ^ B 5 , 2 ± 1 . 3 0.31 ±0 .04^
0.89 + 0.08 89 + 8
Pons/MeduUa 0.85 + 0.02 86 + 7
5.0 ±1.9 0.28 ±0.05*
3.8 + 0:8 ^0.26 ±0.04*
Regional cerebrovascular PA (ml/s/g) x 10^ calculated Vmax, Km and Kd values for blood-brain barrier [^H]-tiiianune transport mto bram. Values are reported for 15 s perfiisions of [^H]-tiuamme at fracer concenfrations (75nM). *Differs significantiy from mean value for frontal cortex (p< 0.05) Values are mean ± SEM, n=3-5.
I l l

Table 5.3: Regional efflux constants for [ H]-thiamine in the absence of vascular thiamine.
Brain Region
Frontal cortex
Parietal cortex ^^Kj^
Occipital cortex
' Hippocampus " •
Caudate/Putamen
Kout
s"* X 10
1.28±0.17
H K . 5 3 ± 0 . 4 0 ' .
1.36 ±0.65
W^^^f^^'
1.25 + 0.61
Thalamus/Hypotiialamus ; 1.46 ± 0.15
Cerebellimi 1.15 + 0.15
Pons/MeduUa ^1.45 + 0.39
Calculated regional efflux constants in the presence of 0 nM thiamine. Efflux constants were evaluated by a 45 s loading of [^H]-ttuamme witii a following "wash" of thiamine and fracer free saline for periods of 5 - 60 s. The efflux constant is calculated by linear regression where kout = ln2/tV2. The calculated efflux constant (kout) for each brain region was not significantly different from tiie frontal cortex (p > 0.05).
112

Table 5.3: Regional efflux constants for [ H]-thiamine in the absence of vascular thiamine.
Braui Region
Frontal cortex
Parietal cortex , ^ ^ ^ ^ H
Occipital cortex
Hippocampus
Caudate/Putamen
Thalamus/Hypothalamus
Cerebellum
Pons/MeduUa '-'^^^^^
Kout
s'* X 10
1.28 ±0.17
B;.53±0.40
1.36 ±0.65
1.2^i:b:2d^
1.25 ±0,61
,;L46 + A.i5j
1.15±0.15
R.45±0.39
Calculated regional efflux constants in the presence of 0 nM thiamine. Efflux constants were evaluated by a 45 s loading of [^H]-tiiianiine witii a following "wash" of thiamine and fracer free saline for periods of 5 - 60 s. The efflux constant is calculated by linear regression where kout = ln2/t'/2. The calculated efflux constant (kout) for each braui region was not significantly different from the frontal cortex (p > 0.05).
112

to
a>
Q.
GO
0.05-1
0.04-
0.03-
0.02
0.01-
0.00
Kin = 1.05 ± 0.16 X10''' ml/s/g
kout = 2 . 6 8 * 0.58 X 10"^ S"
— I 150
Time (s)
Figure 5.1 Non-linear time course of [ H]-thiamine brain uptake
Non-linear time course of [ H]-thiamine (75 nM) bram uptake during physiologic salme perfusion. The fine represents the least squares fit of equation (1). Data are mean ± SEM for frontal cortex; n=3-5 for all points.
113
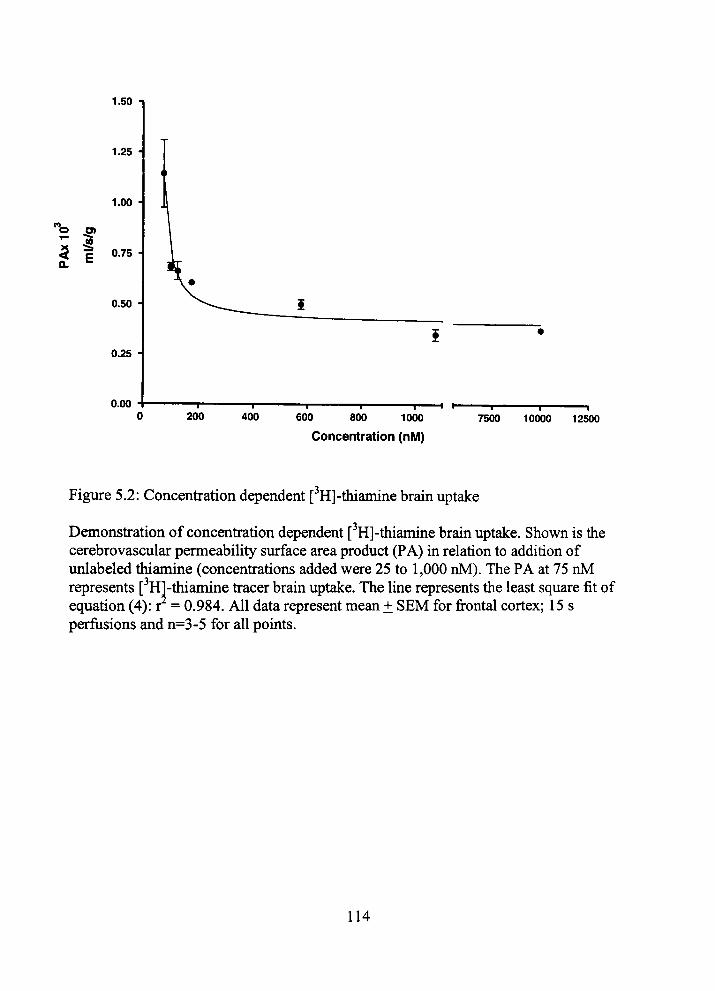
1.50 n
1.25 •
1.00 •
< p 0.75 H
a. =
0.50
0.25
0.00 200
I 400 600 800 1000
Concentration (nM) 7500
— I 1 10000 12500
Figure 5.2: Concenfration dependent [^H]-tiiiamine brain uptake
Demonsfration of concenfration dependent [^H]-thiamine brain uptake. Shown is the cerebrovascular permeability surface area product (PA) in relation to addition of unlabeled thiamine (concentrations added were 25 to 1,000 nM). The PA at 75 nM represents [^Hl-thiamine fracer brain uptake. The Ime represents the least square fit of equation (4): r = 0.984. All data represent mean ± SEM for frontal cortex; 15 s perfiisions and n=3-5 for all points.
114
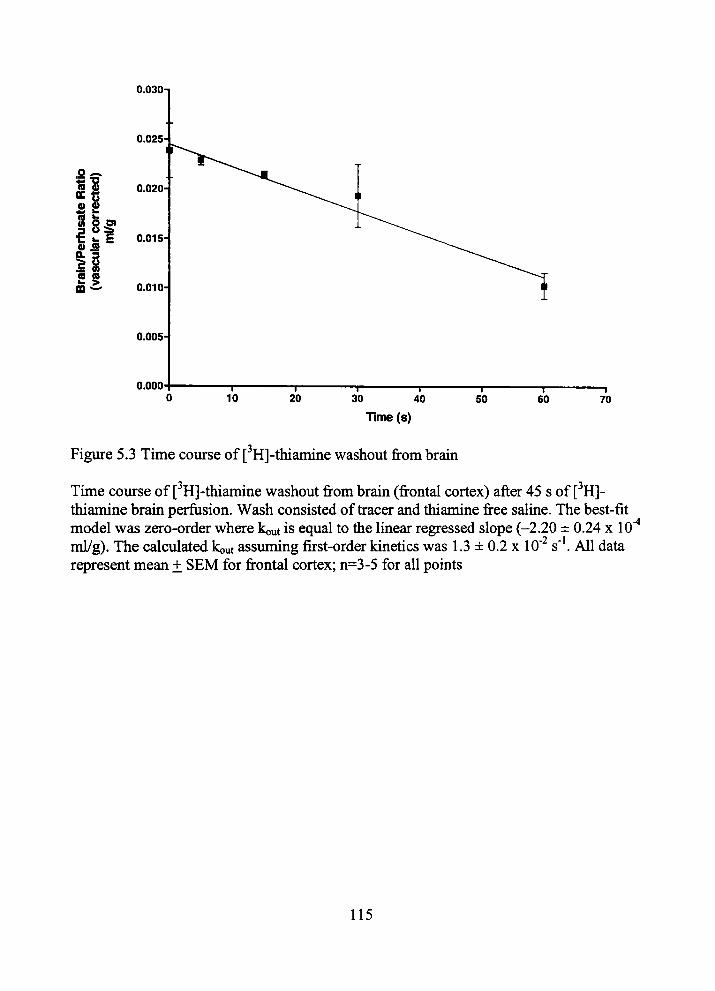
0.030-1
0.025
o ^ 15^ « s *< k
rfus
a rc
or
ml/g
Q) IS CL 3 I S CB CO k >
0.020
0.015
0.010
0.005-
0.000
Time(s)
Figure 5.3 Time course of [ H]-thiamine washout from brain
Tune coiurse of [ H]-thiaimne washout from brain (frontal cortex) after 45 s of [ H]-thiamine brain perfusion. Wash consisted of fracer and thiamine free saline. The best-fit model was zero-order where kout is equal to the Imear regressed slope (-2.20 ± 0.24 x 10''* ml/g). The calculated kout assuming first-order kinetics was 1.3 ± 0.2 x lO' s'*. All data represent mean ± SEM for frontal cortex; n=3-5 for all points
115

6.0
4.5
(0 CM
o
s 3.0
1.5
0.0 75 300
Vascular thiamine concentration (nM)
Figure 5.4: Bram efflux [ H]-thiamine constants
Brain efflux [^H]-thiainine constants at vascular thiamine concenfrations of 0, 75 and 300 nM. Efflux constants were determined at 15 s. An (*) mdicates (p<0.05) and (**) mdicates (p<0.01) when the brain perfusion ratio is compared to 0 nM concenfration. Data are mean ± SEM for frontal cortex; n=3-5 for all data pomts.
116

CHAPTER 6
BRAIN UPTAKE OF THIAMINE-COATED
NANOPARTICLES
6.1 Infroduction
Effective bram dmg delivery is limited by tiie blood-bram barrier (BBB). This
mterface between plasma and brain consists of both brain microvascular endotiiehum and
tiie sunoundmg glia. The BBB significantiy restiicts water-soluble, charged and high
molecular weight tiierapeutics to the vascular space while allowing brain parenchyma
penefration of small and/or lipohillic molecules (1). Mechanisms of permeability
regulation mclude: (1) microvascular endotiielial tight junctions (2), (2) enzymatic
regulation (3), and (3) active bram efflux (4). While multiple sfrategies have been
employed to circumvent the BBB, an emerging approach is the use of nanoparticles
(NPs). NPs are defined as colloidal particles less than 1000 nm m size. The colloid
combinations can be adjusted to allow ahnost any therapeutic dmg to be adsorbed or
enfrapped. This method effectively disguises the membrane barrier luniting
characteristics of the dmg molecule, and protects it from enzymatic degradation. Once
the NP has mvaded the brain parenchyma, therapeutic dmg release from the colloidal
carrier may occur by de-sorption, diffusion through tiie NP matiix or polymer wall and or
NP erosion (5, 6).
Indeed, NP technology has been shown to improve bram distiibution of dmgs
previously impermeable, i.e., dalagrin, tubociu-arine and doxombicin (7-9). While
117

improved brain delivery has been demonstrated, the mechanism of NP BBB
circumvention is still theorized (6). Potential metiiods of NP brain penefration include
paracellular movement (after endotiielial cell tight junction compromise), sunple passive
diffusion, fransport and or endocytocis (5, 6).
We have recently shown significant brain uptake of a novel NP fonnulation,
comprised of emulsifying wax and Brij 78 (10), usmg the in situ rat bram perfiision
technique (10, 11). It was demonsfrated brain uptake of NPs in vivo and in vitro occuned
without any effect on significant baseline BBB parameters, i.e. cerebral perfusion flow,
barrier mtegrity and permeability (12). Considering: (1) tiie bram perfusion stiidies, (2)
tiie lack of toxicity and (3) tiie relative short time (<60 s) for the NPs to penefrate tfie
BBB, it appeared that the colloidal carrier systems studied primarily penefrated the BBB
by passive permeation.
While brain distribution is critical for the success of NPs as a delivery system, the
ability of the NPs to specifically target brain should also be considered. The proposed
passive permeation of the NPs may also lead to increased peripheral organ distribution.
Thus, to specifically target brain, a thiamine Ugand was incorporated on the surface of the
NPs. The targeting of NPs is not witiiout precedent. Various types of NP targeting ligands
have been employed includmg antibodies, peptides and vitamins (13,14). Thiamme is a
water-soluble micronutrient that is essential for normal cell function, growth and
development. The consideration of tiiiamme as a cell specific ligand for targeted delivery
can be rationalized smce all eukaryotic cells have a specified fransport mechanism for
tiiiamme. hi tiiis regard, our coUaborators at tiie University of Kentiicky previously
118

stiidied the effectiveness of using tiie tiiiamine ligand m tiimor-targeting (15). It was
observed that thiamine-coated gadolinium NPs had specific association with human
breast cancer cells that expressed the thiamine fransporters THTRl and THTR2 (15).
Similarly, given the rich number of nutrient transporters at the BBB, the tiiiamine
ligand should bind to the BBB tiiiamine fransporter, subsequentiy increasing tiie number
of NPs at tiie BBB interface. Furtiier, based upon BBB tiiiamme fransport capacity and
kinetics (16,17), this nutrient fransporter has been suggested as a brain dmg delivery
vector (18). We hypothesize that the thiamme-coated NPs (thiamine-NP) will specifically
favor brain distribution by either (1) a facilitated fransport mechanism or (2) increased
passive diffusion secondary to increased concenfration gradient of NPs at the BBB
mterface (due to association with the thiamme fransporter). In the present study,
thiamine-coated NPs (intended for brain delivery) were enguieered from microemulsion
precursors. The brain uptake and distribution of NPs with and with out tiiiamme as a
bram targeting ligand were also investigated.
6.2 Materials and Methods
6.2.1 Materials
Emulsifying wax (E. Wax) and DispoDialyzers MWCO 100 kDa were purchased
from Spectirum Chemicals (New Bnmswick, NJ). Polyoxyl 20-stearyl etiier (Brij 78) was
obtained from Uniquema (Wilmington, DE). Distearoylphosphatidylethanolamine
(DSPE)-PEG-NHS (Mw 3350) was purchased from Shearwater Polymers (HunsviUe,
AL). Thiamme hydrochloride was purchased from Aldrich Chemicals (Milwaukee, WI).
119

Sephadex G-75, Sepharose CL-4B, potassium ferricyanide, phosphate buffered saline
(PBS), fetal bovine semm and sodium chloride were purchased from Sigma Chemicals
(St. Louis, MO). Mice (female; BALB/c) were purchased from Harlan (Indianapolis, IN).
6.2.2 Radiochemicals
High specific activity [^H]-thiamine (10 Ci/mmol, >98% purity) was obtained
from American Radiolabeled Chemicals (St. Louis, MO). [*'*C]-Sucrose (4.75
mCi/mmol) was obtained from Dupont-New England Nuclear (Boston, MA). 1-[^H]-
Hexadecanol (1 mCi/ml; radiochemical purity > 96%) was purchased from Moravek
Biochemicals (Brea, CA). For all experiments, deionized water was filtered through 0.22
fim filters (Nalgene International, Rochester, NY). Indium-111 chloride (2 mCi) in 0.05
M HCL was purchased from Perkui Elmer (Boston, MA). In each perfusion experiment,
[^H]-thiamine was dried prior to being dissolved in perfusion buffer, to remove volatile
tritium contaminants including [ H]-H20.
6.2.3 Preparation of Nanoparticles from Microemulsion Precursors
Solid NPs were prepared from oil-in-water microemulsion precursors as described
previously (19). Briefly, 2 mg of emulsifymg wax (oil phase) was accurately weighed,
placed into a glass vial and melted on a hotplate. To the mehed matiix at 55°C, polyoxyl
20-stearyl ether (Brij 78; final concenfration of 3 mM) was added under magnetic
stining. Water was added to make a final volume of 1 ml. The fonnation of oil-in-water
microemulsion was verified by tiie clarity of tiie mixhire and by photon conelation
120

specfroscopy (PCS) using an N4 Plus Submicron Particle Sizer at 55''C. Sohd NPs were
obtamed by simple cooling the warni microemulsion to room temperatiire in one vessel.
The cured NPs (E78 NPs) were characterized based on size, size disfribution and
morphology.
6.2.4. Preparation of Thiamine-Coated Nanoparticles
A tiiiamine ligand was syntiiesized by chemically Imking tiiianune to DSPE via a
PEG spacer (Mw 3350) as reported earlier (15). Usmg a stock aqueous solution of
tiuamine ligand (tiiiamine-PEG-DSPE), 0.2% w/w tiiiamme ligand was added to cured
NP suspensions at 25°C. The mixtiire was gentiy stured for 4 h at 25°C. The efficiency of
thiamuie attachment/adsorption was assessed by gel permeation chromatography (GPC)
elution profiles usmg a Sepharose CL-4B column. Briefly, 80 |xL of thiamine-coated NP
suspensions were passed down the Sepharose CL-4B column (1.5 cm x 8 cm) using
deionized water (0.22 |j,m filtered) as the mobile phase. The elution of thiamuie-coated
NPs and free thiamine ligand in all GPC fractions was detected by laser light scattering
counts per second (CPS) and thiochrome assay. The GPC elution profiles of confrol NPs
(witiiout thiamine) and free thiamuie ligand were obtained to serve as references. Based
on the GPC elution profiles, tiie efficiency of tiiiamine hgand coatmg was calculated as
tiie percentage of the ratio of tiie area under tfuamine-coated NP profiles to tiie area under
the total elution profiles. Calculation of the concentration of tiiiamine and total number of
tiuamine molecules used in coating NPs was based on coating efficiency data.
121

6.2.5 Characterization of Thiamme-Coated Nanoparticles
Photon conelation spectroscopy (PCS). The particle size of thiamme-coated NPs
was detennined using an N4 Plus Sub-Micron Particle Sizer at 20°C by scattering light at
angle 90° for 180 s (Beckman Couher Corporation, Miami, FL). Prior to particle size
measurements, tiie NPs were diluted (1:10 v/v) witii water, to ensure tiiat tfie light
scattering signal as indicated by the particle counts per second was witfun tiie sensitivity
range of the instrument.
6.2.6 Characterization of Thiamme-Coated Nanoparticles
Gel permeation chromatography (GPC). To obtam the GPC elution profiles of
NPs, 80 \i[ of NP suspensions was passed down a Sephadex G-75 column (1.5 cm x 8
cm) using deionized water (0.22 \im filtered) as tiie mobile phase. The elution of
thiamine-coated NPs was detected by laser light scattering counts per second and
thiochrome assay using fluorescence specfroscopy (Hitachi Model F-2000). The
thiochrome assay (15) involved oxidizmg thiamine in each GPC fraction to thiochrome
and subsequentiy measuring fluorescence intensity at 365 nm (excitation) and 445 nm
(emission).
6.2.7 Characterization of Thiamme-Coated Nanoparticles
Transmission election microscopy (TEM). The size and morphology of NPs were
observed using a Jeol Elecfron Microscope m tiie hnagmg Facility Unit of the University
of Kentiicky. A carbon-coated 200-mesh copper specunen grid was glow-discharged for
1.5 min. One drop of NP suspension was deposited on tiie grid and allowed to stand for
122

1.5 min after which any excess fluid was removed witfi filter paper. The grid was later
stamed with 1 drop of 1% uranyl acetate (0.22 ^m fihered) for 30 s and any excess staui
removed. The grids were aUowed to dry for an additional 10 mm before exammation
under the elecfron microscope.
6.2.8 Nanoparticle Radiolabeling
To allow for easy detection m tiie brain perfusion studies, the NP preparation
method was modified as described earlier (10) to mclude a radiofracer in both the confrol
NPs ([^H]-NPs) and tiie tiiiamme coated NPs ([^H]-thiamme-NPs). Briefly, a frace
amount of [ H]-hexadecanol (cetyl alcohol) was added to emulsifying wax prior to the
formation of the microemulsion precursors at 55°C. Nanoparticles containing enfrapped
[^H]-hexadecanol were cured from warm microemulsions as described above. The
enfrapment efficiency of the radiofracer was ~100%. AU final preparations had
theoretical activities of 150 |j-Ci per ml.
6.2.9 Perfusion Procedure
Initial experiments were focused on assessuig uptake of [^H]-tiuamme-NPs and
[^H]-NPs into bram. The results obtamed for [ H]-NP were consistent witii earlier
observations by Koziara et al. (10). Briefly, uptake was evaluated using tiie in situ rat
brain perfiision technique of Takasato et al. (11) witii modifications described (20,21).
Perfiisions of 5 - 120 s were used to detennine initial bram uptake of tiie NPs. All stiidies
123

were approved by tiie Institiitional Animal Care and Use Committee and were conducted
m accordance with tiie NIH Guide for tiie Care and Use of Laboratory Animals.
Male Fischer-344 rats (220-330 g; Charles River Laboratories, Kingston, NY)
were anestiietized witii sodium pentobarbital (50 mg/kg intraperitoneal). A PE-60
catiieter filled witii heparinized salme (100 units/ml) was placed into the left common
carotid artery after ligation of the left extemal carotid, occipital and common carotid
arteries. Common carotid artery ligation was accomplished caudal to tiie catfieter
implantation site. The pterygopalatine artery was left open during tfie experiments (21).
Rat rectal temperature was monitored and maintamed at 37°C by a heating pad connected
to a feedback device (YSI Indicating ConfroUer, Yellow Springs, OH). The catiieter to
the left common carotid artery was connected to a syringe contaming buffered
physiologic perfusion fluid (contaming [in mM]: NaCl 128, NaPOs 2.4, NaHCOs 29.0,
KCl 4.2, CaCl 1.5, MgClz 0.9, and D-glucose 9) with 1 fiCi/ml [^H]-NPs or [^H]-
thiamine-NPs (final NP concenfration -20 |ig/ml) and 0.3 fiCi/ml [''*C]-sucrose (to
determine vascular volume). Perfiision fluid was filtered and warmed to 37°C and gassed
witii 95% O2 and 5% CO2. The pH and osmolarity of tiiis solution were ~ 7.35 and 290
mOsm, respectively, immediately prior to perfiision. The perfiision fluid was infused into
the left carotid artery with an infusion pump for periods of 5-120 s at 10 ml/min (Harvard
Apparatus, South Natick, MA). This perfiision rate was selected to maintam a carotid
artery pressure of-120 mm Hg (11).
Rats were decapitated and cerebral samples obtamed as previously described (21).
Briefly, tiie bram was removed from the skull, and tiie perfiised cerebral hemisphere
124

dissected on ice after removal of tfie arachnoid membrane and meningeal vessels. Bram
regions were placed in scintillation vials and weighed. In addition, two 50-jil aliquots of
tiie perfiision fluid were fransfened to a scmtillation vial and weighed. The brain and
perfiision fluid samples were then digested overnight at 50°C in 1 ml of 1 M piperidine.
Ten ml of Fisher Chemical scmtillation cocktail (Beckman, FuUerton, CA) was added to
each vial and the fracer contents assessed by dual-label hquid scintillation countmg. Dual
labeled scintillation counting of brain and perfiisate samples were accomplished with
conection for quench, background and efficiency.
6.2.10 Kinetic Analysis
Concenfrations of NP fracer in brain and perfusion fluid are expressed as dpm/g
brain or dpm/ml perfusion fluid, respectively. Blood-bram banier [ H]-NP brain uptake
was determuied by perfusion with [^H]-thiamine-NPs and [^H]-NPs m separate
experunents for 5 - 120 s periods as described previously (10,11,20). Given the
apparent linear uptake of the [ H]-NPs in the time frames evaluated a unidfrectional
uptake fransfer constant (Kin) was calculated from the following relationship to the linear
portion of the uptake curve as described (20):
Q*/C* = KinT + Vo (6.1)
where, Q* is the quantity of [^H]-fracer m brain (dpm/g) at tfie end of perfiision, C* is the
perfusion fluid concenfration of [^H]-NP (dpm/ml), T is tfie perfusion time (s) and VQ is
tfie exfrapolated mtercept (T = 0 s; "vascular volume" m ml/g). Tracer trapped in tfie
vascular space was accounted for by the subfraction of [*'*C]-sucrose vascular volume.
125

Given the [^H]-tiuamine-NPs had an apparent nonlmear mitial bram uptake
pattern, a calculated uptake fransfer constant (Ki„) and a brain efflux rate coefficient (ko.)
was estimated from tiie following relationship as described (20):
Q*/C* = (Kin/kout)(l-e-'^°"''^) (6.2)
where Q* is tiie quantity of [^H]-fracer in brain (dpm/g) at the end of perfiision, C* is the
perfiision fluid concenfration of [^H]-tfiiamme (dpm/ml) and T is tfie perfiision time.
Tracer frapped in tiie vascular space was accounted for by tfie subfraction of [*'*C]-
sucrose vascular volume.
For detemunation of ['H]-tiuamine BBB uptake, we calculated Kin by single-pomt
uptake experiments at 15 s in from tiie foUowmg relationship as described (20):
Kin=[Q,,-V^Cp,]/(Cp/T) (63)
where, Q ^ = Q^^ + Q^ ^ tfie sum of the amount of tfiiamme remaining m tiie perfiisate m
tiie blood-brain vessels and tiie amount of tiiiamine tfiat has penefrated mto brain.
Cerebral perfiision flow rate (F) was determined in separate experiments (12). C ^ is the
perfusion fluid concenfration of fracer thiamme and T is tiie net perfusion time. Kin values
were tiien converted to apparent cerebrovascular permeabihty-surface area products (PA)
using the Crone-Renkin equation (20),
PA = -Fln(l-Kin/F). (6.4)
6.2.11 In Vivo Biodistribution Studies in Mice
Biodistribution studies of thiamine-coated NPs were carried out in BALB/c mice
(female; 16 g weight) after a 7-day acclimation period at the University of Kentucky. The
126

stiidies were approved by the University of Kentiicky Anunal Care and Use Committee in
accordance witii tiie NIH Guide for the Care and Use of laboratory ammals. The two
types of modifications that were carried out on thiamine-coated NP preparations used m
the biodistiibution experiments mclude: (1) addition of DSPE-PEG (Mw 3000; m 3%
w/w concenfration) to emulsifying wax during the preparation of microemulsion
precursors, and (2) enfrapment of a frace quantity of indium-111 acetylacetonate (['**In]-
AcAc) in NPs. [*^'ln]-Iabeled NPs allow fast and effective detection of NPs in various
tissues using a gamma counter. After entrapment of the [^^*In]-label, thiamine-coated
NPs were prepared as described earlier. The two types of NPs used ui the biodistribution
experiments were: (1) thiamine-coated NPs, and (2) confrol NPs (PEG-coated) that were
not coated with the thiamine ligand. Prior to admmisfration to mice, all NP preparations
were diluted (1:1) with 0.9% sodium chloride solution (0.2 |jm filtered). Each mouse was
anestiietized and then mjected (by tail vein adminisfration) with a dose of 0.5 g/kg of
either thiamme-coated or PEG-coated NPs. At 2 or 6 hr post-injection, tiie mice were
sacrificed. The biodistiibution of tiie [***In] radioactivity in tissues such as flie blood,
liver, lungs, kidneys, heart, spleen and brain (obtained from each mouse) was measured
by a Cobra II auto gamma counter (Parkard Bioscience Company, Meriden, CT). The
residual radioactivity m tiie tail of each mouse was also measured to enable exact
quantification of the injected dose. The total blood volume of a mouse was assumed to be
7.5% (v/w) of tiie total mouse (22, 23).
127

6.2.12 Statistical Analysis
Brain tissue data presented are from the frontal cerebral cortex unless otherwise
specified. [^H]-tiiiamuie-NP and [ H]-NP brain uptake over tune were fit with non-lmear
regression and linear regression, respectively, using least squares analysis. One-way
analysis of variance followed by a Bonferoni's multiple comparison test were used to
evaluate the significance of [^H]-thiamine brain uptake inhibition in tiie presence of tiie
NP formulations. For all data, enors are reported as standard enor of tiie mean, unless
otiierwise mdicated. Differences were considered statistically significant a/^rion at tiie
p<0.05 level. (GraphPad Prism version 3.00 for Windows, GraphPad Software, San
Diego, CA).
6.3 Results
6.3.1 Preparation and Characterization of Thiamine-Coated Nanoparticles
Microemulsions (oU-in-water) were used as precursors to obtain solid NPs. NPs
were cured from microemulsions by sunple cooling m one vessel. Emulsifymg wax was
used as tiie mafrix (oU phase) and Brij 78 as tfie surfactant. The choice and tfie
concenfration of tiie components of tfie microemulsion was based on earher stiidies (19).
To obtain tiuamme-coated NPs, a tiiiamine ligand was syntiiesized using tiie procedure
earlier described (15) and added to NP suspensions at 25''C. The tiuamme hgand
contained a DSPE group and a PEG spacer. Tlie attachment of thiamme Ugand to NPs
was confimied by gel pemieation chromatography. The elution of free thiamine
(unattached to NPs) and thiamine ligand attached to NPs was detected by tiiiochrome
128

assay and laser light scattering. The NP coating efficiency was calculated from GPC
elution profiles. Based on the thiochrome assay, the NPs (2 mg/ml) contained 10 ^M of
thiamine. Thiamine-coated NPs were characterized by laser light scattering, gel
permeation chromatography and fransmission elecfron microscopy (TEM). The TEM
micrograph of tiiiamine-coated NPs is shown ui Figure 6.1, indicatmg NPs size of ~ 100
nm. Additional measurements by laser light scattering showed tiiat tiie mean diameter of
thiamme-coated NPs was 67 ± 8 nm witfi a polydispersity index of 0.2. The NPs were
stable when incubated with PBS (pH 7.4) at 37°C for 60 min (data not shown).
6.3.2 Nanoparticle Braui Uptake
Initial bram distiibution parameters of [ H]-NPs were evaluated using tiie rat
bram perfiision metiiod (11). ['H]-NP and [ H]-tiiiamine-NP bram uptake (1
^Ci/200|Xg/ml) were evaluated from 0-120 s. In all experiments BBB integrity was
verified witii concunent [i'C]-sucrose vascular volume measurements. [*'C]-Sucrose
vascular volumes ranged from 0.76 ± 0.13 to 1.1 ± 0.2 x 10" ml/g, consistent with
previous in situ NP brain perfiision data demonsfrating an intact BBB (12).
Brain/perfiision fluid ratios (i.e., volume of disfribution or 'space') were plotted as
a fimction of time during tiie initial 45 s period and shown in Figure 6.2. Given tiiat tiie
brain uptake of tiie [ H]-NPs was apparently linear in tius time frame, a unidkectional
fransfer coefficient (Kin) was calculated (3.37 ± 0.24 x 10^ ml/s/g) accordmg to equation
1. In confrast to tfie [ H]-NP linear uptake, tiie [ H]-tiiiamine-NP demonsfrated non-lmear
uptake during tiie mitial 45 s uptake time. The [3H]-tiiiamine-NP uptake data were fit to
129

equation 2, for a calculated Ki„ and kout of 7.11 ± 1.96 x 10' ml/s/g and 5.55 ± 2.40 x 10'
s"^ respectively.
To confirm tiie [^H]-tiiiamine-NP efflux rate seen in tfie initial experiments
washout studies were completed (16,21). Briefly, tiie method consists of evaluating tiie
fracer brain/perfiision ratios after: (1) a 45 s loading of either [ H]-NP or [^H]-thiamme-
NP and (2) an unmediate subsequent "wash" of NP/fracer free salme for periods of 15 -
30 s. Figure 6.3 shows brain perfusion ratios had no significant reduction during the 30 s
fracer free wash period. Furtiier, the efflux constant, based upon the luiear regression of
tiie slope, for either fonnulation ([^H]-NP: - 5.0 ± 0.8 x lO"" s' ; [^H]-tiiianune-NP: 1.6 ±
0.8 X 10^ s'*) was not significant from a slope of zero.
To determine if tiie NP thianune hgand is associatmg with tiie tiiiamine BBB
fransporter, we calculated (equations 3 and 4) a PA for [^H]-tiiiamine usmg single time
point uptake experiments for 15 s witii results shown in Figure 6.4. The calculated confrol
[^H]-thiamme PA (11.6 ± 0.7 x 10"'* ml/s/g) and tiie approximate 20% mhibition (9.4 ±
0.6 X 10''* ml/s/g) with 100 nM unlabeled thiamme agree witii recentiy published in situ
rat bram perfiision data for [^H]-tiiiamine (16). Consistent witii tiie above data, tiie
presence of [^H]-tiuamme-NPs (total NP concenfration of 20 ig/ml witii thiamme hgand
concenfration of approximately 100 nM) resulted m mhibition (8.9 ± 0.2 x lO"* ml/s/g)
similar to tiie presence of 100 nM tiiiamme. The uncoated NP had no apparent inhibition
of [^H]-tiiiamme BBB fransport.
To evaluate if [^H]-tiiiamine-NPs are accessmg brain via tiie BBB tiuamine
fransporter, we incorporated unlabeled tiiiamme mto tiie perfiision fluid at concenfrations
130

(50 |iM) tiiat would completely inhibit BBB tiiiamine transport, and subsequentiy
decrease [^H]-thiamine-NP brain distiibution (16). Figure 6.2 (empty cfrcles and inset)
shows brain distiibution volume of [^H]-tiiiamine-NP with 50 iM of tiuamme
incorporated into the perfusion fluid. The 15 s bram distiibution volume was not
significantiy different from tiie value obtamed in tiie absence of tiuamme (0.13 ± 0.08
ml/g; 0.091 ± 0.008 ml/g). However, in confrast to tiie expected decrease of brain
distiibution at 45 s, we observed a significant increase (p<0.05) of [^H]-tfiianune-NP
brain distiibution m tiie presence of 50 ^M thiamine (0.12 ± 0.02 ml/g; 0.17 ± 0.02 ml/g).
Subsequent perfusion experunents to 120 s were completed to determine extended
time frame brain distiibution. In confrast to the non-linear and linear uptake patterns seen
in early perfusion experiments, both the [^H]-NP and the [^H]-thiamine-NP had a linear
brain uptake pattern from 45 to 120 s. The calculated Kin (equation 6.1) during this time
period for each NP is shown in figure 6.5. The [^H]-thiamine-NPs had a significantiy
(p<0.05) increased Kin (9.78 ± 1.06 x 10' ml/s/g) compared to tfie uncoated NPs (7.00 ±
0.29x10'^ ml/s/g).
6.3.3 In Vivo Biodistribution Studies in Mice
The results of the biodistribution experiments in mice (BALB/c) are shown in
Figures 6.6A and 6.B. Each mouse was injected witii 0.5 g/kg of NP suspensions m 0.9%
sodium chloride. The results shovra in Figure 6.6A indicate tfiat tfiiamme coating did not
have a significant effect on final organ biodistribution (p>0.05). The two NPs had long
cu-culating properties (Figure 6.6A). Specifically, tiie tfiiamine-coated NPs had amounts
131

of radioactivity in circulation at 2 and 6 hr of 79% injected dose (ID) and 65% ID,
respectively. The cumulative radioactivity disfributed to reticuloendotfiehal system
tissues (liver and spleen) were 13% ID and 22% ID at 2 and 6 hr, respectivdy. NP
stabihty was demonstrated by the low accumulated radioactivity m tfie lungs (Figure
6.6A). The amounts of radioactivity disfributed to tfie brain for tiie two types of NPs are
shown m Figure 6.6B. In botfi NPs very low levels of radioactivity were observed m
otiier tissues such as tiie lungs, kidneys and heart. For both tiuamine-coated and PEG-
coated NPs, tfie average bram radioactivity levels for tiiiamme-coated NPs at 2 and 6 hr
were slightiy less tfian 0.5% ID and did not differ significantiy (p>0.05) (figure 6.6B)
6.4 Discussion
The results of tiie stiidies presented herem demonsfrated: (1) the effectiveness of
microemulsion sfrategies for NP production, (2) kmetic modelmg for bram uptake of NPs
v ith and without thiamme surface ligands, and (3) mitial data suggestuig mechanisms for
NP brain entry. Comparison of the brain distribution of the NPs demonsfrated that the
[ H]-thiamine-NPs associated with the BBB thiamine fransporter and had increased brain
distribution during the uptake period of 45 -120 s. This data suggests the thiamine ligand
on the NPs may facilitate binding and/or association with BBB thiamine fransporters. The
relevance of this report is two-fold; first, to our knowledge there is no otfier work that has
specifically targeted NPs to brain in this manner, and second we have provided a highly
sensitive kinetic analysis of NP movement at the BBB.
132

The NPs used in this stiidy were prepared using oU-in-water microemulsions as
precursors. The effectiveness of tiie microemulsion sfrategy is in agreement witii earlier
stiidies (19). The application of microemulsions as templates for NP production offers
numerous advantages, demonstrated in tiiis report, such as: (1) simphstic production of
NPs ranging approximately 100 nm in diameter, (2) ease of microemulsion incorporation
of hydrophobic compounds m tiie oil droplets (facilitatmg enfrapment in cured NPs), and
(3) inclusion of site-specific ligands in the NP preparations.
As shown in the TEM micrograph (Figure 6.1), NP size averaged -100 nm. The
frend observed from laser tight scattering also confumed thiamme-coated NPs were -100
nm with unimodal size distribution. Prior to brain perfusion studies, the stability of the
NPs in relevant media was demonsfrated (data not shown). Based on previous studies
using liposomes and other macromolecules, we considered that the NP size of 100 nm as
suitable for ceU targetmg and confroUed release. The preparation technique also
demonsfrated significant versatility in that, for both the perfusion studies and
biodistribution studies, different radiofracers were included m the NPs and enfrapped at
-100%. efficiency (data not shovm) simUar to previously published data (10).
The synthesized thiamme ligand has both DSPE groups and a PEG spacer. The
DSPE hydrophobic groups were engineered to act as anchors and uifluence the
insertion/adsorption of tfie tfuamine hgand to tfie NP. The PEG spacer was included to
facilitate flexibility and cell recognition of ligand on NP surface. Thiamine-coated NPs
were obtained by adding 0.2% w/w thiamine hgand to NP suspensions at 25°C. This NP
133

formulation has been shown in vitro to positively associate with human breast cancer
cells expressing tiie thiamine fransporters THTRl and THTR2 (15).
To determine if tiie tiuamine-coated NPs would sunUarly associate witii BBB
thiamine fransporters we completed bram uptake evaluation for both the [ H]-NPs and
[^H]-thiamine-NPs using tiie in situ rat bram perfusion technique (11) (Figure 6.2). The
calculated fransfer coefficient (Kin) based upon tiie initial 45 s linear uptake for tiie [ H]-
NPs is consistent with previously published in situ NP perfusion data (10). However, in
confrast to the previous report, the [ H]-thiamme-NPs had an mitial brain distribution
pattern that was non-linear in nature suggesting efflux or delayed bram penefration.
Of interest, the placement of thiamine ligands on the NP created a brain
distiibution configuration consistent with [ H]-thianiine brain uptake and efflux during in
situ brain perfusions of less tiian 120 s (16). Therefore, after evaluation of mitial NP brain
distiibution we chose to evaluate [ H]-tiiiamuie-NP and [ H]-NP efflux (Figure 6.3)
Specifically, tiie [ H]-tiuamine-NP complex confrasted [ H]-tiiiamine BBB movement, in
that there was no significant efflux during the washout study. However, sunilar to [ H]-
thiamine BBB movement there was no apparent BBB endotiiehal ceU disassociation as
seen by post-perflision washouts resultmg in an immediate reduction ui tiie
brain/perfiision ratio (16, 21).
Given tfie lack of efflux, we hypothesized tiie non-luiear [ H]-tiiiamine-NP uptake
pattern was secondary to: (1) facilitated but delayed fransport mto bram via tiie BBB
tiuamme fransporter witiiout subsequent efflux, and/or (2) tiie ligand-NP complex was
associating witii tiie carrier creatmg a subsequent delay in passive brain penneation. To
134

detennine if a substrate is being fransported across the BBB, experiments should be
completed to show substrate transporter association and saturabihty (20). Therefore, we
performed [ H]-thiamuie uptake experiments at 15 s with and witiiout the presence of
both NPs and unlabeled thiamine. The experimental tune frame was based on previous
[ H]-thiamine BBB kinetic modelmg (i.e., the time was long enough for [^H]-thianune
brain detection yet was short enough to minimize apparent efflux) (16). Figure 6.4 shows
addition of unlabeled thiamine (100 nM) to the perfusion fluid resulted in a reduction of
PA consistent with previously published results (16). Of major significance the uncoated
NPs did not inhibit BBB [^H]-thiamine fransport yet the thiamine-NPs did (thiamme
concenfration - 100 nm). The abihty of the latter NP type to inhibit [^H]-tfiianime BBB
uptake sfrongly suggests NP complex association withthiamine BBB fransporter proteins.
To determine if [^H]-thiamme-NP is accessing brain via tiie BBB tiiiamine
fransporter, we evaluated saturabihty by mcorporation of unlabeled tiiiamine (50 |aM)
into the perfusion fluid. If fransport occurs tiiis concenfration will completely inhibit
BBB [^H]-thiamme-NP bram distiibution via the respective carrier (16). hi confrast to the
expected findmg (i.e., mhibiting bram penefration of [^H]-tiiiamme-NP with unlabeled
tiuamine). Figure 6.2 (mset) shows the presence of unlabeled tiiiamuie resulted m a
significant mcrease of [^H]-tiuanime-NP bram distiibution. This data unphes addition of
tiiiamine ligands to tiie NP does not result m BBB penefration via facilitated thiamine
BBB fransport but ratiier, tiie ligand-NP complex is associatmg with tiie tiuamine BBB
fransporter creating a delay in passive BBB penneation.
135

After detennining BBB thiamine fransporter association witii [^H]-thiamme-NP,
we hypotiiesized tiiere may be accumulation of tiie thiamine-coated NPs at tiie BBB.
Potentially, NP BBB accumulation may lead to improved brain disfribution by mcreasing
tiie concenfration gradient. To assess tiiis hypotiiesis, we extended perfiision tune frames
to 60, 75, 90 and 120 s. In confrast to the non-linear and Imear brain uptake pattern seen
in tiie mitial 45 s brain distiibution time frame, both NPs had apparent Imear bram
penetration during extended perfusions. Figure 6.5 shows the uptake fransfer coefficient
(Kin) for [ H]-tiiianiine-NPs is significantiy greater tiian [^H]-NPs (perfusion tune frame
- 45 to 120 s). This data further supports tiie hypothesis of tiie tiiiamine ligand associating
witii the thiamine BBB fransporter unproves overall NP brain distiibution.
To assess the improved NP brain uptake we completed biodistribution studies.
The NPs were modified to contain a DSPE-PEG spacer based upon: (1) stabilization of
the cured NPs in cuculation (24) and (2) PEG-coated macromolecules display specific
affinity for braui endothelial cells (25). As shown ui Figure 6.6A, both thiamuie and
PEG-coated (confrol) NPs had long circulating properties most likely due to PEG
molecules preventing or at least minimizmg reticuloendothelial NP recognition. It is also
noteworthy there was an apparent lack of NP aggregation (leading to rapid limg
deposition) as demonsfrated by the low levels of radioactivity observed in the lungs
(Figure 6.6A).
For both thiamine-coated and PEG-coated NPs, low levels of radioactivity were
distributed to the brain at tiie tune pomts studied. In tiie biodistiibution studies (Figure
6.6B), thiamine ligand coatmg on NPs did not have a significant effect on bram
136

radioactivity levels (in comparison to control) possibly due to a number of factors
including: (1) addition of 0.2% w/w of tiiiamine ligand may result in low and insufficient
density of tiiiamine ligand coatmg on NPs in vivo, (2) bram distiibution at extended time
pomts (beyond 6 hr) were not stiidied, (3) normal blood thiamine levels m mice may
competitively inhibit brain uptake of tiiiamme-coated NPs, (4) the thiamine NP complex
may interact with erythrocyte thiamine transporters, and (5) in vivo shear flow and
dilution effects could dinunish the amount of thiamine-coated NP association with BBB.
In summary, the results presented herein indicate the addition of a thiamine ligand
to the NPs causes association with the BBB thiamine transporter. This association may
create an accumulation of NPs at the BBB, which ultimately increases brain distribution
during perfiision time frames. Of major importance, the long circulating property of both
NPs mcreases their potential utility in site-specific and confroUed dmg delivery.
137

6.5 Reference.s
' 755^8^2002)^^* ^""^ "^^ ^^"^ '^^"^^'^ ^"^ *^ ^'^'"- *^ ''^''"^^' '^''^^' ^"''^"- ^ ( >"
2. M.W. Brightinan. Morphology of blood-brain interfaces, Exp Eye Res. 25 (Suppl): 1-25
3. WM. Pardridge. Bram dmg targeting and gene technologies, Jpn. J Pharmacol 87(2):
4. T. Terasaki. Development of bram efflux index (BEI) method and its application to tiie blood-bram banier efflux fransport stiidy, m: W.M. Pardridge (Ed), Introduction to the Blood-Brain Barrier, Cambridge University Press, New York, pp. 24-31 (1998).
5. P. R. Lockman, R. J. Mumper, M. A. Khan, D. D. Allen. Nanoparticle technology for dmg delivery across flie blood-brain barrier, Drug Dev. Ind. Pharm. 28(1): 1-12 (2002).
6. J. Kreuter, R. N. Alyautdin. Using nanoparticles to target dmgs to tiie cenfral nervous system. In D. J. Begley, M. W. Bradbury, J. Kreuter (eds.). The blood-brain barrier and drug delivery to the CNS, Marcel Dekker, New York, pp. 205-223 (2000).
7. A. Gulyaev, S. E. Gelperina, I. N. Skidan, A. S. Anfropov, G. Y. Kivman, J. Kreuter. Significant fransport of doxombicm into tiie brain with polysorbate 80 coated nanoparticles. Pharm. Res. 16(10): 1564-1569(1999).
8. U. Schroder, B. A. Sabel. Nanoparticles, a dmg carrier system to pass tfie blood-brain barrier, permit cenfral analgesic effects of i.v. dalargm uijections. Brain Res. 710(1-2): 121-124(1996).
9. R.N. Alyautidin, E.B. Tezikov, P.D. Ramage, A. Kharkevich, D.J. Begly, J. Kreuter. Significant entry of tubocurarine into the bram of rats by adsorption to polysorbate 80-coated poly(butylcyanoacrylate) nanoparticles: an in situ brain perfusion study, J. Microencap. 15(1): 67-74 (1998).
10. J. Koziara, P.R. Lockman, D.D. Allen, R.J. Mumper. In situ blood-bram barrier fransport of nanoparticles, Pharm. Res. Submitted (2003).
11. Y. Takasato, S.I. Rapoport, Q.R. Smith. An in situ brain perfiision technique to study cerebrovascular fransport in tiie rat. Am. J. Physiol 247: 484-493 (1984).
12. P.R. Lockman, J. Koziara, K.E. Roder, J. Paulson, T.J. Abbmscato, R.J. Mumper, D.D. Allen. In vivo and in vitro assessment of baselme blood-brain barrier parameters in the presence of novel nanoparticles, Pharm. Res. 20(5): 705-713 (2003).
138

13. S.P Vyas, A. Singh, V. Sihorkar. Ligand-receptor mediated dmg delivery: an emergmg paradigm in cellular dmg targeting, Crit. Rev. Ther. Drug Carrier Syst 189(1)- 1-76 (2001). ^'
14. S. Wang, P.S Low. Folate-mediated targeting of antineoplastic dmgs, unaging agents and nucleic acids to cancer ceUs, J. Control Rel 53(1-3): 39-48 (1998).
15. M.O. Oyewumi, S. Liu, J.A. Moscow, R. J. Mumper. Specific association of thiamine-coated gadolmium nanoparticles with human breast cancer cells expressing thiamine fransporters, Bioconjugate Chem. 14(2): 404-411 (2003).
16. P.R. Lockman, R.J. Mumper, D.D. Allen. Evaluation of blood-brain barrier tiiiamine efflux using tiie in situ rat brain perfusion method, J. Neurochem. In press (2003).
17. J. Greenwood, E.R. Love, O.E. Pratt. Kinetics of thiamine fransport across the blood-bram barrier in tiie rat, / . Physiol 327: 95-103 (1982).
18. Q.R. Smitii, Dmg delivery to tiie bram and the role of carrier mediated fransport, in: L.R. Drewes, A.L. Betz (Eds.), Frontiers in Cerebral Vascular Biology: Transport and its Regulation. Plenum Press, New York, pp. 83-93 (1993).
19. M. O. Oyewoiini, R. J. Mumper. Gadolmium loaded nanoparticles engineered from microemulsion templates, Z>rMg Dev. Ind. Pharm. 28(3): 317-328 (2002).
20. Q.R. Smith. Brain perfusion systems for studies of dmg uptake and metabolism in the cenfral nervous system, Pharm. Biotechnol 8: 285-307 (1996).
21. D.D. Allen, Q.R. Smith. Characterization of the blood-brain barrier choline fransporter using the in situ rat bram perfiision technique, J. Neurochem. 76(4): 1032-1041 (2001).
22. B. Davies, T. Morris. Physiological parameters in laboratoty animals and humans, Pharm. Res. 10(7): 1093-1095 (1993).
23. V.C.F. Mosquieira, P. Legrand, J. Morgart, M. Vert, E. Mysiakme, R. Gref, J. Devissaguet, G. Barrat. Biodistribution of long-circulating PEG-grafted nano-capsules m mice: effects of Peg chain length and density, Pharm. Res. 18(10): 4111-4119 (2001).
24. M.O.Oyevnimi, R.J. Mumper. Tumor-targeted delivery of folate-coated gadolinium nanoparticles m athymic nude mice. Proceedmgs of Annual Conference of ConfroUed Release Society, Glasgow-Scotland. Absfract Accepted (2003).
139

25.1. Brigger, J. Morizet, G. Auber, H. Chacun, M. Tenier-Lamcombe, P. Couvreur, G. Vassal. Poly(ethylene gycol)-coated hexadecylcyanozcrylate nanosperes display a combined effect for brain tumor targeting, J. Pharmacol Exp. Ther. 303(3): 928-936 (2002).
140
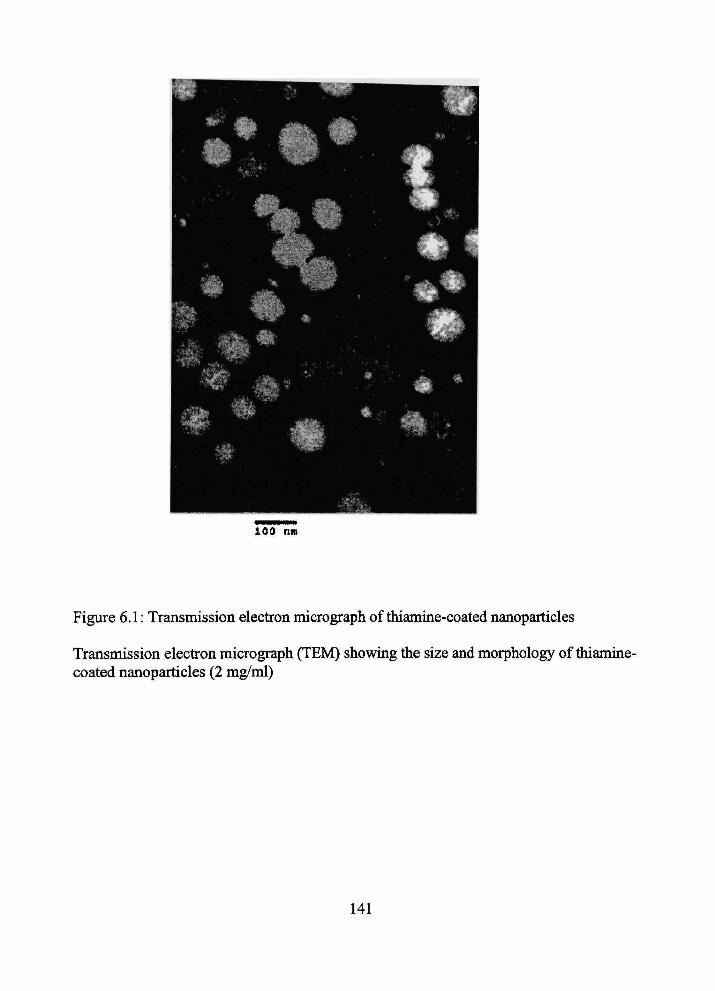
100 nm
Figure 6.1: Transmission elecfron micrograph of thiamine-coated nanoparticles
Transmission elecfron micrograph (TEM) showing the size and morphology of thiamine-coated nanoparticles (2 mg/ml)
141

0.25-1
° [ Hl-thiamine-NP with 50 nM thiamine present 1 — 30 40 50
•nme(s)
Figure 6.2 Tune course of bram uptake for [^H]-nanoparticles and [^H]-tfiianime -nanoparticles.
The tune course of brain uptake for tfie [•'H]-NP and [^H]-thiamine-NP formulations during perfusion times of 5 to 45 s. The open circles represent the bram distribution voliune of the [^H]-thiamine-NP in the presence of 50 |iM thiamine. Inset: The brain distribution volume of [^H]-tliiamme-NP with 50 |iM of thiamine incorporated into the perfiision fluid. Data are for frontal cerebral cortex and represent the mean ± SEM; n=3-5. Similar patterns were observed for other brain regions; data not shown. All values were conected for vascular volume using concunent [''*C]-sucrose distribution measurements.
142

0.2S-I
0.20:
.9 n
S 0.15H (B
•C E
1 0.10-w ID
0.05-
0.00-10
— I — 15
rH]-NP
[^H]-thlamine-NP
20 —r-25 30 35
Washout Time (s)
Figure 6.3 Time course of washout from brain for ['H]-nanoparticles and [^H]-thiamine -nanoparticles.
•y T
Time coiu-se of [ H]-NP and [ H]-thiamme-NP washout from brain (frontal cortex) after 45 s of brain perfusion uptake. Wash consisted of tracer free saline. The Imear regression of both slopes were not significant from zero (p>0.05) indicatmg little to no efflux. All data represent mean ± SEM for frontal cortex; n=3-5 for all points.
143

X
« c
1 (S
t
15.0
12.5
10.0
7,5
5,0 •
2.5
0.0 Control Thiamine Uncoated-NP Thiamine-NP
Figure 6.4: [ H]-Thiamine uptake inhibition by nanoparticles
[^H]-Thiamine brain uptake in the presence of unlabeled thiamine (100 nM), the uncoated NP formulation (20 jig/ml; total NP dose - 200 ig) and the tfiiamine coated NP formulation (NP concenfration was 20 mg/ml; thiamine ligand concenfration -100 nM). A * indicates differs significantly (p<0.05) from confrol. Data are for frontal cerebral cortex and represent the mean ± SEM; n=3-5.
144

c f
0.0125-1
0.0100
0.0075-
0.0050-
0.0025
0.0000
Uptake time 45 -120 s
Figiu-e 6.5 Calculated fransfer coefficients for [^H]-nanoparticles and ['H]-thiamine-nanoparticles during 45 to 120 s perfusion time frame.
Calculated transfer coefficients for both NP formulations during the 45 to 120 s perfiision time frame. A * indicates differs significantiy (p<0.05) from confrol. Data are for frontal cerebral cortex and represent the mean ± SEM; n=3-5.
145

A) nTHNP(6hr)
• THNP(2 hr)
lCTRNP(6hr)
1CTRNP(2 hr)
Blood Liver Lungs Kidneys Heart Spleen
B) 0.8 -
0.7 -
g 0-6 o D 0.5
•|« 0,3
S? 0.2
0.1
0 -
THNP CTRNP
Nanoparticle Fbrmulations
Figure 6.6: In vivo biodistribution of thiamuie coated and peg coated nanoparticles
/111 A) In vivo biodistiibution studies of ( In-labeled) thiamine-coated nanparticles (THNP) and PEG-coated nanoparticles (CTRNP) m mice (Balb/c). Biodistribution of radioactivity was carried out at 2 and 6 hr-post injection of nanoparticles (dose: 0.5g/ kg mouse weight). The mice were sacrificed and the amounts of radioactivity in each tissue was measured by a gamma counter. B) The amounts of radioactivity distributed to the brain of mice (Balb/c) at 2 hr (white bars) and 6 hr post-injection of (** In-labeled) tiiiamme-coated nanparticles (THNP) and PEG-coated nanoparticles (CTRNP). Data represents the mean ± SD (n= 5-7 mice).
146