syntheses and properties of cyclosilazanes and ...szolcsanyi/publications/papers/j. organomet... ·...
TRANSCRIPT

at SciVerse ScienceDirect
Journal of Organometallic Chemistry 732 (2013) 58e91
Contents lists available
Journal of Organometallic Chemistry
journal homepage: www.elsevier .com/locate/ jorganchem
Review
Syntheses and properties of cyclosilazanes and cyclocarbosilazanes
Miroslav Kavala a, Alison Hawkins b, Peter Szolcsányi a,*aDepartment of Organic Chemistry, Slovak University of Technology, Radlinského 9, SK-812 37 Bratislava, SlovakiabChemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, UK
a r t i c l e i n f o
Article history:Received 24 May 2012Received in revised form1 February 2013Accepted 5 February 2013
Keywords:SilazanesSynthesisPropertiesHydrolysisMaterial science
* Corresponding author. Tel.: þ421 2 593 251 62; fE-mail address: [email protected] (P. Szolc
0022-328X/$ e see front matter � 2013 Elsevier B.V.http://dx.doi.org/10.1016/j.jorganchem.2013.02.002
a b s t r a c t
We present a review of synthetic methods and physico-chemical properties of cyclo(carbo)silazanes thatcould form the basis of composite ceramic materials of type Six(Cy)Nz, as well as polymeric materials. Thereview is compiled as a guide for the preparation of such compounds, which have the potential to befurther modified to access novel derivatives. The review is subdivided according to ring size. Each sectionpresents known synthetic methods to obtain various types of cyclo(carbo)silazanes and also highlightstheir possible functionalisation. The review also includes reactions of N-unsubstituted cyclic derivativesand data on the hydrolytic stability of such compounds.
� 2013 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 582. Cyclosilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
2.1. Cyclodisilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 592.2. Cyclotrisilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 662.3. Cyclotetrasilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 742.4. Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
3. Cyclocarbosilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 823.1. Cyclodicarbodisilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 823.2. Cyclotricarbodisilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 883.3. Higher cyclocarbosilazanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89Acknowledgement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
1. Introduction
One of the current trends in the chemistry of materials repre-sents the synthesis of siliconecarbonenitrogen compounds, whichare expected to form the basis of composite ceramic materials oftype SixNy as well as polymeric materials [1]. These compoundspossess suitable material properties, which makes them utilisable
ax: þ421 2 524 953 81.sányi).
All rights reserved.
in various industries such as materials for turbines, heat-resistantmaterials, various coatings and polymer fibres.
We present an overview of physico-chemical properties andsynthetic methods for the preparation of selected types of mono-meric precursors of such materials. The review is intended andcompiled as a synthetic manual for the preparation of such com-pounds, which can be further modified to access novel derivatives.
The review is subdivided by ring size. Each section presentsknown synthetic methods to obtain various types of cyclo-carbosilazanes, and also highlights their possible functionalisation.The review also includes reactions of N-unsubstituted cyclic

Fig. 1. General formula of cyclosilazanes.
R1, R2, R3, R4 = alkyl, aryl, N-alkyl,N-silyl, O-alkyl
12 3 4SiNH
Si
HN
R2R1 R3
R4
Fig. 2. General formula of cyclodisilazanes.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 59
derivatives and important data on the hydrolytic stability ofrespective compounds.
2. Cyclosilazanes
Cyclosilazanes are silicon analogues of saturated carbon het-erocycles e 1,3-diazetidine (n ¼ 1), hexahydrotriazine (n ¼ 2) andtetrazocine (n ¼ 3) e while nitrogen and silicon atoms alternate intheir structure (Fig. 1). This family of compounds has been widelystudied [2], and apart from their properties, synthetic trans-formations towards various functionalised derivatives are of in-terest as well.
Scheme
2.1. Cyclodisilazanes
Cyclodisilazanes form 4-membered rings with the basic struc-ture shown in Fig. 2. The name of this group of compounds has beenderived from the saturated azaheterocycle e 1,3-diazetidine. Car-bon atoms in positions C-2 and C-4 have been replaced by siliconatoms; both carbon and silicon are group 14 elements of the peri-odic table, thus they have the same valence. Substituents arelocated in these positions, thus forming 2,2,4,4-tetrasubstitutedcyclo-2,4-disiladiamines or 2,4-disila-1,3-diazacyclobutanes. Themost common name for this group of compounds is cyclo-2,4-disilazanes. This section concentrates on the synthesis of cyclo-disilazanes with an unsubstituted nitrogen atom, which could becapable of further functionalisation.
2.1.1. Preparation of cyclo-2,4-disilazanesCyclodisilazanes were first mentioned when NMR spectroscopy
and X-ray analysis were in their infancy [3]. Until these twomethods were available, such compounds could not be fully char-acterised or distinguished from the related 6- and 8-memberedisomers. Since cyclo-2,4-disilazanes are sensitive to air moisture,all reactions had to be carried out under a blanket of dry argon ornitrogen.
2.1.1.1. Ammonolysis of chlorosilanes. The cyclosilazane skeletonwas accessed in a step-wise fashion, but only very low yields ofrequired products were obtained [4]. The first step of the sequenceinvolved the reaction of lithiated hexamethyldisilazane 1 withtetrachlorosilane 2which gave trisilazane 3 (Scheme 1). Trisilazane3 further underwent an ammonolysis with a solution of ammoniain benzene which afforded compounds 4 and 5 in 66% combinedyield. Compound 5 was assumed to be the product of a subsequentin situ de-aminative homocoupling of 4. The branched disilazane 5finally underwent thermal condensation under an argon atmo-sphere to yield cyclodisilazane 7 in very low yield (10%). Based on
1.
Scheme 2.
Scheme 3.
Scheme 4.
Scheme 5.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9160

Scheme 6.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 61
reaction monitoring by IR and NMR spectroscopy, the authorsassumed the formation of intermediate 6 (Scheme 1).
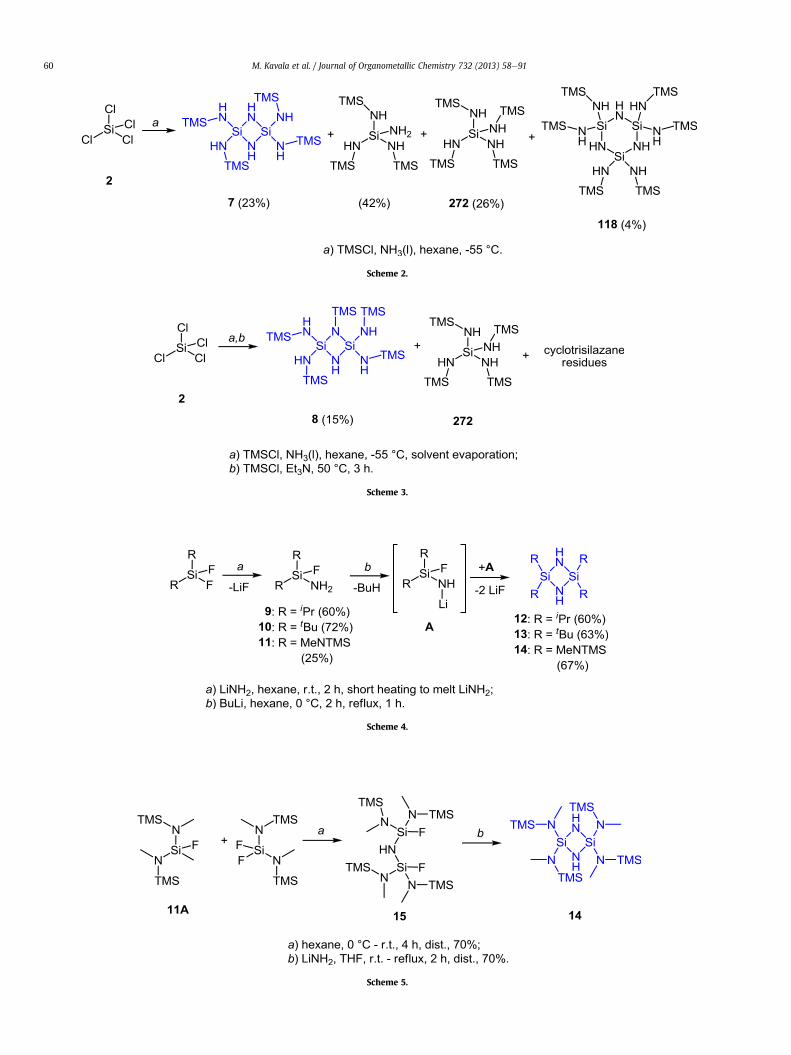
Cyclodisilazane 7 can also be obtained by a one-pot reaction [5]of SiCl4 and TMSCl in the presence of liquid ammonia in 23% yield(Scheme 2). Unfortunately, the formation of cyclodisilazane 7 wasaccompanied by other products including cyclotrisilazane 118 in 4%yield. Isolation of all compounds was non-trivial due to the similarboiling points of the products present in the reaction mixture.
If, however, the solvent was removed after the one-pot trans-formation (2e7), and a fresh portion of TMSCl was added togetherwith triethylamine, the N-monosubstituted derivative 8 wasformed (Scheme 3), together with tetrakis(trimethylsilylamino)silane 272 and cyclotrisilazane residues. However, no yields weregiven for the latter two.
2.1.1.2. Addition of lithium amides to fluorosilanes. The cyclic de-rivatives 7 and 8 are the only compounds prepared by ammonolysisof acyclic chlorosilanes. Generally, the methodology deliveredsubstituted acyclic aminosilanes of type 272; in case of formation ofcyclic compounds only 6- and 8-membered cyclodisilazanes wereformed. In order to obtain 4-membered heterocycles, amines, oftype 9e11, must be converted to harder nucleophiles e lithiumamides [6]. This synthetic methodology employs dialkyldi-fluorosilanes. Since it is extremely difficult to ammonolyse fluo-rosilanes (Si e F bond is significantly stronger than SieN bond:
Scheme
DHSieF ¼ 582 kJ/mol vs. DHSieN ¼ 355 kJ/mol) [7], the first step wassubstitution of a fluoride with lithium amide which formed fluo-rosilanes 9e11 (Scheme 4) [8]. In case of preparation of silane 11,cyclodisilazane 14 was formed in this step in 10% yield. Dia-lkylaminofluorosilanes are isoelectronic with derivatives of silanol(tBu)2SiF(OH), which are known to resist autocondensation withconcomitant release of HF. [9]. Compounds 9e11 are moderatelyacidic and are easily deprotonated by BuLi. Such lithium salts candimerise (A þ A) with concomitant release of LiF unless anotherelectrophile competes in the reaction equilibria. The correspondingsymmetrically substituted cyclosilazanes 12e14 were obtained(Scheme 4).
It has been observed that a solution of lithium derivative 11Awas stable at laboratory temperature and addition of difluoro-diaminosilazane gave disilazane 15 (Scheme 5). When treatedwith lithium amide, cyclodisilazane 14was obtained in 70% yield. Inprinciple, this approach allowed the design of structures whichwere asymmetrically substituted at silicon atoms.
The treatment of lithium derivative 9A with TMSCl gave rise totwo analogues, 16 and 17 (Scheme 6). These were subsequentlydeprotonated by residual 9A which gave disilazanes 18 and 19. Onexposure to BuLi monosubstituted cyclodisilazane 20 was formedin 55% yield.
Interestingly, the strategy shown above could also be used toprepare the tetra-tert-butylcyclodisilaphosphane 22. On exposure
7.

Scheme 8.
Scheme 9.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9162
of difluoro-di-tert-butyl-silane to lithium phosphide, followed byBuLi and subsequent cyclisation, cyclodisilaphosphane 22 could beaccessed via phosphinosilane 21 in 59% overall yield (Scheme 7).
All cyclodisilazanes mentioned thus far were symmetricallysubstituted at silicon. However, when a lithium amide was used tosubstitute the fluorosilane, unsymmetrically substituted cyclo-disilazanes could be prepared [10]. Di-tert-butyldiaminosilazane 23was converted to dilithium salt 23A using two equivalents of BuLi(Scheme 8). On addition of difluoro-dimethylsilane to the dilithiumsalt 23B, cyclotetrasilazane 25 was obtained in 45% yield and
Scheme
dimethyl-di-tert-butylcyclodisilazane 24 was isolated as the minorproduct in 31% yield. If diaminosilane 23 reacted with only oneequivalent of BuLi, a monolithium salt was formed, which thenreacted with dihalosilanes and gave acyclic disilazanes 26 and 27.Although the latter could, in principle, react to form 24 and 25 byreacting with another equivalent of BuLi, this was not observed.
Fluorosubstituted cyclosilazanes constitute another family ofuseful derivatives [11]. In general, they are prepared by the reactionof lithium salts with trifluorosilanes. Exposure of diaminodi-tert-butylsilane 23 to 1 equivalent of BuLi gave monolithium salt 23A.
10.
Scheme 11.
Scheme 12.
Fig. 3. ORTEP diagram of TMEDA-10A obtained by X-ray analysis.
Scheme 13.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 63
Addition of trifluorosilane to lithium salt 23A provided desiredcyclic products 28 and 29 (Scheme 9).
If, however, compounds 23 or 30 were converted to theirrespective dilithium salts 23B and 30B, cyclic derivatives 28, 29, 31and 32 were formed (Scheme 10). According to authors, unlike themonolithium salt, in the case of the dilithium salts, no regenerationof diaminosilanes 23 or 30 was observed.
Scheme 14.

Scheme 15.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9164
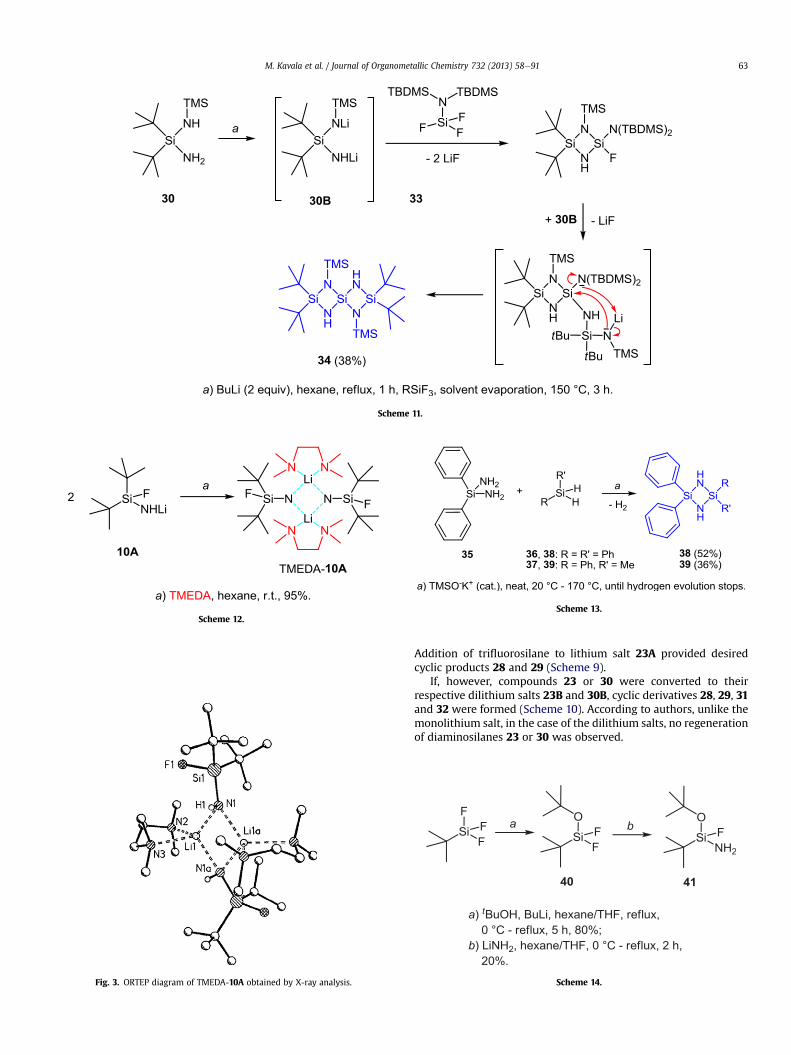
Exploitation of this methodology also allowed the preparationof spiro-dicyclosilazanes 34 (Scheme 11) [11]. After formation ofdilithium salt 30B, addition of silyl protected aminofluorosilane 33to 30B gave spirocycle 34 in 38% yield. The mechanism is outlinedin Scheme 11 and the structure of 34 was confirmed by singlecrystal X-ray diffraction studies.
As previously mentioned, lithium derivatives of aminosilanes9Ae11A are stable crystalline compounds when kept under anargon or nitrogen atmosphere. and at laboratory temperature. Atthe same time, they are nearly insoluble in non-polar organic sol-vents [12]. However, their complete dissolution can be achieved bythe addition of tetramethylenediamine (TMEDA). The dissolution isfacilitated by the formation of dimeric complex TMEDA-10A(Scheme 12), which was stable enough to be isolated by solventevaporation and crystallisation at 0 �C. The structure was eluci-dated by single crystal X-ray diffraction studies (Fig. 3).
2.1.1.3. Dehydrogenative coupling. In the reactions of silazanes withorganosilanes, the formation of N-substituted 4-membered ringswas observed (Scheme 8) [13]. An interesting idea was to employ
Table 1Physical data of cyclo-2,4-disilazanes.
Comp. Melting point Boiling point
7 [4] e 153e155 �C (1.5 Torr)8 [4] e 100e120 �C (0.06 Torr)12 [6a] e 55 �C (0.01 Torr)13 [6a] 93 �C e
14 [6c] e 105 �C (0.04 Torr)20 [6c] e 80 �C (0.008 Torr)22 [6c] 114 �C 95 �C (0.008 Torr)24 [10] e 48 �C (0.04 Torr)28 [11] e 96 �C (0.008 Torr)29 [11] e 130 �C (0.008 Torr)31 [11] e 116 �C (0.008 Torr)32 [11] e 124 �C (0.008 Torr)34 [11] 174 �C e
38 [14] 206 �C e
39 [14] 65 �C e
44 [15] 105 �C e
silanolate base TMSOeKþ as a catalyst [14]. When diphenyldiami-nosilane 35 was employed with catalytic TMSOeKþ and eitherdiorganosilane 36 or 37, cyclodisilazane derivatives 38 and 39wereobtained in 52% and 36% yield respectively, with concomitantrelease of molecular H2 (Scheme 13).
2.1.1.4. Nucleophilic substitution and ammonolysis of halogen silanes.In Sections 2.1.1.1 and 2.1.1.2, two methods for the synthesis ofcyclodisilazane derivatives were presented: an ammonolysis ofchlorosilanes and the substitution of fluorosilanes with lithiumamides. There is a third methodology that combines both ap-proaches and uses tert-butyl trihalogensilanes as the starting ma-terial [15]. Thus, the reaction of tert-butyltrifluorosilanes withlithium butanolate furnished difluorosilanol 40 in good yield(Scheme 14). This substrate is an analogue of difluorosilanes 9e10(Section 2.1.1.2), but the subsequent reaction with lithium amideto the silazane derivative 41 proceeded in only 20% yield. Thereason for such an outcome might have been the bulkiness of tert-butyl substituent(s) at silicon, which could have prevented theaccess of the nucleophile.
On exposure of tert-butyltrichlorosilane to lithium tert-buta-nolate, dichlorosiloxane 42 was isolated in 85% yield (Scheme 15).Chlorosiloxane 42 was subsequently attacked by ammonia, whichgave diaminosiloxazane 43 in moderate 40% yield. Deprotonationwith 1 equivalent of BuLi producedmonolithium salt 43A, which byreaction with 40 afforded cyclodisiloxazane derivative 44, accom-panied by the formation of acyclic disiloxazane 45. According toNMR analysis, Z- and E-isomers were formed in a 3 : 8 (Z:E) ratioand the latter was the major product. The stereochemistry of the
Scheme 16.

Scheme 17.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 65
major (E) isomer was determined by single crystal X-ray diffractionanalysis (Table 1).
2.1.2. Reactions of cyclo-2,4-disilazanesThere are two known types of reactions of cyclodisilazanes
described in the literature. Owing to their unsubstituted aminogroup, the first of the transformations is N-functionalisation(Scheme 17). The second transformation is the substitution ofsilicon-bound halogens and related transformations (Scheme 16).
2.1.2.1. N-functionalisation. Unsubstituted amino groups of cyclo-disilazanes are weak nucleophiles and cannot perform the SieXsubstitution. Therefore much stronger nucleophiles e metal am-ides emust be used, obtained by deprotonation of the amine with
Table 2N-functionalisation of cyclodisilazanes.
Comp. R R1 R2 R3
46 [8] iPr iPr SiMe2F H47 [8] iPr iPr SiMe2F SiMe48 [6c] iPr iPr SiMe3 H49 [6c] iPr iPr Si(iPr)2F H50 [6c] iPr iPr SiMePhF H51 [6c] iPr iPr Si(MeNTMS)2F H52 [6c] iPr iPr SiMe3 SiMe53 [6c] iPr iPr SiF3 SiMe54 [6c] iPr iPr SiPhF2 SiMe55 [17] iPr iPr Si(N(TMS)2)F2 H56 [17] iPr iPr SiMe2F SiF357 [17] iPr iPr Si(N(TMS)2)F2 Si(N(58 [17] iPr iPr Si(iPr)2F Si(N(59 [17] iPr iPr Si(NMeTMS)2F Si(N(60 [17] iPr iPr BF(N(TMS)2) SiMe61 [10] tBu Me SiMe2F H62 [10] tBu Me SiMe2F SiMe63 [18] tBu iPr SitBuF2 H64 [18] tBu iPr SitBuF2 SiPh65 [11] tBu R1 ¼ F
R10 ¼ tBuSitBuMe2 SiMe
66 [11] tBu R1 ¼ OMeR10 ¼ tBu
SitBuMe2 H
67 [11] tBu R1 ¼ OMeR10 ¼ tBu
SitBuMe2 SiMe
a strong base, such as butyllithium. Whilst a single equivalent ofBuLi led to a monolithium salt, 2 equivalents of BuLi gave adilithium salt in a regioselective fashion (Scheme 17), i.e. 1equivalent of BuLi does not give 0.5 equivalents of the dilithiumsalt but rather 1 equivalent of the monolithium salt. Substitutionsat silicon proceed via an SN2 mechanism: the addition of thelithium salt first forms a pentasubstituted silicon anion, whichsubsequently loses X� to form LiX and the substituted product(Scheme 16) [11]. Silicon compounds which undergo substitutionreactions always do so via an SN2 mechanism, and not by an SN1mechanism [16]. The reason is that unlike carbon, silicon hasavailable d-orbitals and thus SN2eSi pathway is extremely rapid.Compounds described in the literature, prepared by this method,are given in Table 2.
Method Yield Boiling or melting point
b 71% 52 �C (0.01 Torr)2F a 40% 202 �C
b 55% 80 �C (0.008 Torr)b 60% 118 �C (0.008 Torr)b 75% 112 �C (0.008 Torr)b 40% 150 �C (0.008 Torr)
3 a 40% 303 �C3 b 80% 136 �C3 b 65% 128 �C (0.008 Torr)
b 60% 121 �C (0.008 Torr)b 16% 89 �C (0.008 Torr)
TMS)2)F2 a 49% 158 �CTMS)2)F2 b 42% 142 �C (0.008 Torr)TMS)2)F2 b 41% 158 �C (0.008 Torr)3 b 52% 118 �C (0.008 Torr)
b 70% 48 �C (0.008 Torr)2F a 71% 119 �C (0.008 Torr)
b 10% 80 �C (0.008 Torr)F2 b 50% 98 �C3 b 45% 132 �C (0.008 Torr)
a 85% 121 �C (0.008 Torr)
2F b 8% 114 �C (0.008 Torr)
Scheme 18.
Scheme 19.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9166
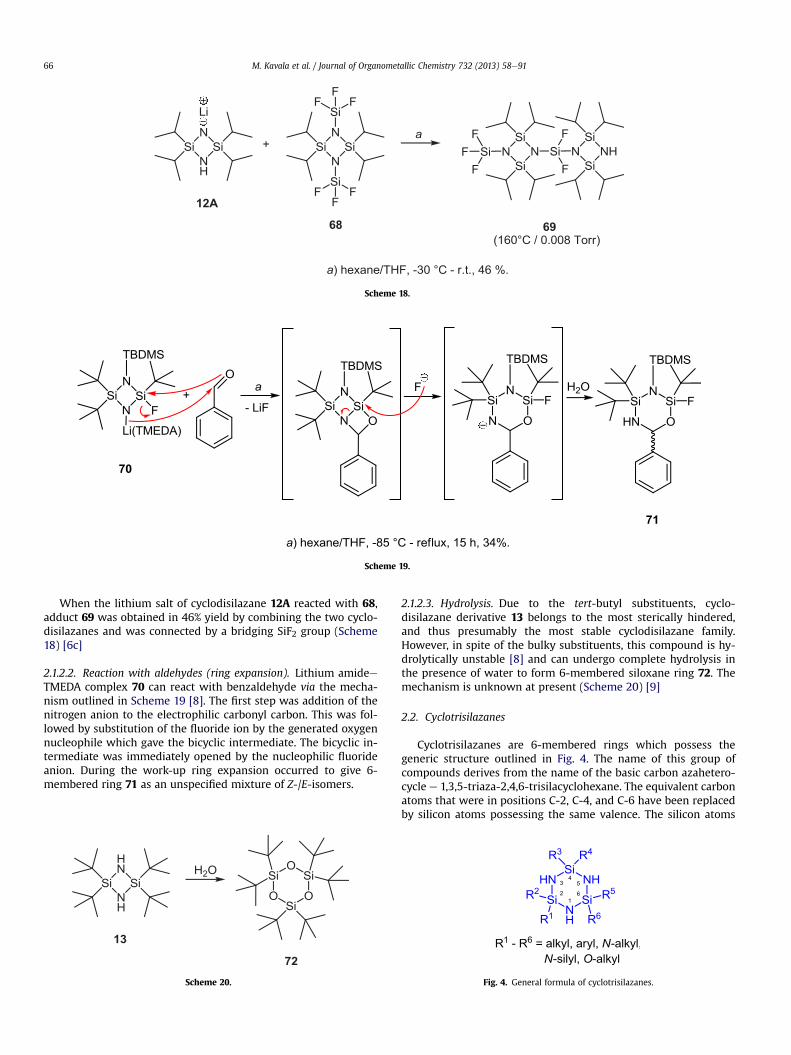
When the lithium salt of cyclodisilazane 12A reacted with 68,adduct 69 was obtained in 46% yield by combining the two cyclo-disilazanes and was connected by a bridging SiF2 group (Scheme18) [6c]
2.1.2.2. Reaction with aldehydes (ring expansion). Lithium amideeTMEDA complex 70 can react with benzaldehyde via the mecha-nism outlined in Scheme 19 [8]. The first step was addition of thenitrogen anion to the electrophilic carbonyl carbon. This was fol-lowed by substitution of the fluoride ion by the generated oxygennucleophile which gave the bicyclic intermediate. The bicyclic in-termediate was immediately opened by the nucleophilic fluorideanion. During the work-up ring expansion occurred to give 6-membered ring 71 as an unspecified mixture of Z-/E-isomers.
Scheme 20.
2.1.2.3. Hydrolysis. Due to the tert-butyl substituents, cyclo-disilazane derivative 13 belongs to the most sterically hindered,and thus presumably the most stable cyclodisilazane family.However, in spite of the bulky substituents, this compound is hy-drolytically unstable [8] and can undergo complete hydrolysis inthe presence of water to form 6-membered siloxane ring 72. Themechanism is unknown at present (Scheme 20) [9]
2.2. Cyclotrisilazanes
Cyclotrisilazanes are 6-membered rings which possess thegeneric structure outlined in Fig. 4. The name of this group ofcompounds derives from the name of the basic carbon azahetero-cycle e 1,3,5-triaza-2,4,6-trisilacyclohexane. The equivalent carbonatoms that were in positions C-2, C-4, and C-6 have been replacedby silicon atoms possessing the same valence. The silicon atoms
Fig. 4. General formula of cyclotrisilazanes.

Scheme 21.
Scheme 22.
Scheme 23.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 67
tend to be substituted, thus forming the 2,2,4,4,6,6-substituted1,3,5-triaza-2,4,6-trisilacyclohexanes and2,2,4,4,6,6-substitutedcyclotrisilazanes. The most common name for this group of com-pounds is cyclo-2,4,6-trisilazanes. This section concentrates on thesynthesis of cyclotrisilazanes with an unsubstituted nitrogen atom,thus allowing for further functionalisation.
Table 3Physical data of cyclotrisilazanes.
Comp. R1 R2 Method
73 [19] Me Me a
75 [19] Et Et a76 [23] Me Et b77 [23] Me nBu b78 [23] Me nHex b79 [23] Me nOct b80 [23] Me nNon b81 [23] Et nOct b82 [25] Ph Ph b83 [28] Me Ph b84 [29] iPrO iPrO b85 [30] nBuO nBuO b86 [31] H Et b87 [32] H iPr b88 [33] nBu nBu b89 [34] Me iPr b90 [34] Me tBuO b91 [34] Ph iPr b92 [24] Me iBu b93 [33] Me cycHex b94 [35] Me Ph(CH2)2 b95 [35] Me (CH2)3CN b96 [35] Et Ph(CH2)2 b97 [35] Me CH2CH(Me) (Ph) b98 [35] Et CH2CH(Me) (Ph) b99 [35] Me CH2CH(Me) (CO2Me) b100 [35] Et CH2CH(Me) (CO2Me) b101 [36] Me CH¼CH2 b
2.2.1. Preparation of cyclotrisilazanes2.2.1.1. Ammonolysis of chlorosilanes
2.2.1.1.1. Ammonolysis of dichlorosilanes. The first report con-cerning the preparation of cyclotrisilazanes employed a substitu-tion reaction of dimethylchlorosilane with ammonia, a processgenerally called ammonolysis [19e21]. The first synthetically usefulmethod described in detail was published in 1948. Thus, dime-thylchlorosilane, which was dissolved in liquid ammonia, wasconverted to hexamethylcyclotrisilazane (HMCTS) 73 in 45% yield(Scheme 21). The process also generated the octamethylcyclote-trasilazane (OMCTS) 74 as a side product (25%) as well as otherminor products (silazanes, polymeric silazanes etc.). When gaseousammonia was used, benzene served as the solvent. The authorsclaimed that under such conditions, the combined yield of 73 and74 was only slightly higher [22], but a lower 73 vs. 74 ratio wasobserved compared to using liquid ammonia, but the exact detailswere not disclosed.
The chemistry outlined in Scheme 22 highlights the mostfrequently used and cited synthetic approach to cyclotrisilazanes. Alarge group of symmetrically substituted cyclotrisilazane de-rivatives have been prepared by this method; an overview is givenin Table 3.
Yield Melting point Boiling point
42% �10 �C 188 �C (760 Torr)111e112 �C (85 Torr)
85% �41 �C 150 �C (10 Torr)75% e 91e93 �C (3 Torr)73% e 126e128 �C (1 Torr)68% e 171e173 �C (1 Torr)60% e 185e189 �C (1 Torr)53% e 233e234 �C (1 Torr) [24]53% e 224e228 �C (1 Torr)79% [26] 213 �C decomp. at 369 �C [27]43% 115e116 �C 218e220 �C (1 Torr)33% e 147 �C (4 Torr)50% e 245e250 �C (12 Torr)7% e 62 �C (0.5 Torr)20% e 87 �C (1 Torr)e e 193 �C (2 Torr)65% e 94e96 �C (1 Torr)64% e 109e110 �C (1 Torr)39% e 227e230 �C (1 Torr)45% e 131e132 �C (1 Torr)43% e 192e195 �C (1 Torr)57% e 263e265 �C (3 Torr)42% e 256e260 �C (1.5 Torr)63% e 270e275 �C (2.6 Torr)48% e 245 �C (1.5 Torr)46% e 360 �C (2.5 Torr)29% e 184e187 �C (0.2 Torr)30% e 252e255 �C (3 Torr)34% e 100 �C (7 Torr)

Table 4Preparation of vinyl cyclotrisilazanes.
Comp. Dichlorosilanes Products bp
R1 R2 R3 R4 R1 R2 R3 R4 R5 R6
102 [36] Me Me Me CH¼CH2 Me Me Me CH¼CH2 Me CH¼CH2 68e70 �C (3 Torr)103 [36] Me Me Me CH¼CH2 Me Me Me CH¼CH2 Me Me 54e56 �C (3 Torr)104 [36] Et Et Me CH¼CH2 Et Et Me CH¼CH2 Me CH¼CH2 84e86 �C (1 Torr)105 [36] Et Et Me CH¼CH2 Et Et Me CH¼CH2 Et Me 105e107 �C (1 Torr)
Scheme 24.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9168
The ammonolysis of dichlorosilazanes (Scheme 22), which led tosymmetrically substituted cyclotrisilazane derivatives 73e101, wasbased on the reaction of dichlorosilanes with ammonia. Anotherpossible approach to synthesise cyclotrisilazanes utilised thetransformation of a mixture of two different dichlorosilanesreacting with ammonia. Gaseous ammonia was bubbled through abenzene solution of dichlorosilanes at laboratory temperature andunsymmetrically substituted compounds 102e105 were obtained(Scheme 23). (Scheme 23) [36]. This method has often been usedfor the preparation of vinyl derivatives (Table 4). Compounds 102/103 and 104/105 were isolated as mixtures of separable productsfrom a single reaction. Although not discussed by the authors, dueto the low yields obtained (12e15%), the methodology was syn-thetically unfeasible.
2.2.1.1.2. Aminolysis of dichlorosilanes. Derivatives of cyclo-trisilazanes can also be prepared by treating diaminosilazanes withdichlorosilanes [37]. The transformation proceeded in diethyl etherin the presence of Et3N (to neutralise HCl) at low temperaturesbefore heating to reflux in toluene (Scheme 24). This syntheticmethod was used to prepare aryl-substituted compounds 106e109which were isolated in low to moderate yields (Tables 5 and 6).
2.2.1.1.3. Aminolysis of dichlorodisilazanes. The reaction of dia-minosilazanes with acyclic dichlorodisilazanes gives cyclo-trisilazanes (Scheme 25, compounds 110, 111) [37]. The reactionwas catalysed by Et3N and takes place in Et2O at low temperatures(Scheme 25, Table 7). This method has been used to prepare onlytwo derivatives, 110 and/or 111 in rather low yields (37% and 39%respectively), however, the methodology certainly possesses prac-tical synthetic potential.
2.2.1.1.4. Simultaneous ammonolysis of dichlorosilanes with tri-chlorosilanes. Ammonolysis of two silanes, which have different
Table 5Preparation of aryl cyclotrisilazanes.
Comp. Diaminosilane Dichlorosilane Product Yield (%)
R1 R2 R3 R4 R1 R2 R3 R4
106 [37] Ph Ph Me CH¼CH2 Ph Ph Me CH¼CH2 47107 [37] Ph Me Me CH¼CH2 Ph Me Me CH¼CH2 42108 [37] Ph Ph Me H Ph Ph Me H 44109 [37] Ph Me Me H Ph Me Me H 48
substitution patterns, afforded a mixture of various cyclosilazanes(Scheme 26) [24]. The major product was 6-membered cyclo-trisilazane 112 (22%) and 8-membered cyclotetrasilazane derivative113 (15%). As well as the isolation of compounds 112 and 113, twobicyclic skeletons 114 and 115were formed, albeit in very low yields(9% and 11% respectively). No further reactions were attempted onthese derivatives; the bulky substituents attached to the siliconblocked the amino moiety. The selectivity of the reaction was poorand therefore, the methodology was clearly unsuitable for thepreparation of cyclotrisilazanes. However, cyclotrisilazane 112 wasaccessible by the ammonolysis of iso-butylmethyldichlorosilane(Section 2.2.1.1.1) in 45% yield (Table 8).
2.2.1.1.5. Ammonolysis of tetrachlorosilane. When a cooledethereal solution of tetrachlorosilane was treated with gaseousammonia, hexachlorodisilazane 116 was isolated as the majorproduct, by acidic cleavage of polysilazanes that are produced(Scheme 27) [38,39]. However, the minor product, 117 (mp 164 �C,sublimes at 70e80 �C/1 Torr), is of more synthetic interest. Cyclo-trisilazane 117 is a reactive intermediate: substitution of the chlo-rine atoms allowed for further derivatisation of the silicon atom.Although the ORTEP diagram was not disclosed, X-ray analysis of117 showed that the 6-membered ring adopted a planar configu-ration [40]. Moreover, the SieN bond in this compound is only1.68 �A long, which is the shortest observed SieN bond reported inthe literature. The bond length thus lies between the standardsingle SieN bond length (1.80�A) and the double bond Si]N length(1.56 �A) which indicates rather polar SieN bonds in 117.
In Section 2.1.1.1 (Scheme 2), ammonolysis of tetrachlorosilanein the presence of TMSCl was described. Side product 118 wasisolated in only 4% yield (mp 94e100 �C, Scheme 28). Thus, bothmethods described clearly indicated that ammonolysis of tetra-chlorosilanewas unsuitable for the preparation of cyclotrisilazanes.
2.2.1.2. Substitution of trichlorosilanes with metal amides2.2.1.2.1. Substitution with sodium amide. Substituted cyclo-
silazanes can also be obtained by the reaction of phenylchlorosilanewith metallic sodium in liquid ammonia (Scheme 29) [41].Although the authors designated the reaction as ammonolysis, itwas in fact a nucleophilic substitution of halogens by the NH2
�
anion, since the first step of the reactionwas the in situ formation ofsodium amide. Once formed, the amide attacked the trichlorosilanederivative to form the cyclotrisilazane 119 (m.p. 64e68 �C) in 70%yield. This is the most effective synthetic approach to cyclo-trisilazanes as far as yield is concerned.
Table 6Physical data of aryl cyclotrisilazanes.
Comp. Melting point Boiling point
106 [37] 112 �C 230e238 �C (1 Torr)107 [37] e 175e180 �C (1 Torr)108 [37] 121e123 �C 240e245 �C (2 Torr)109 [37] e 168e172 �C (1 Torr)

Scheme 25.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 69
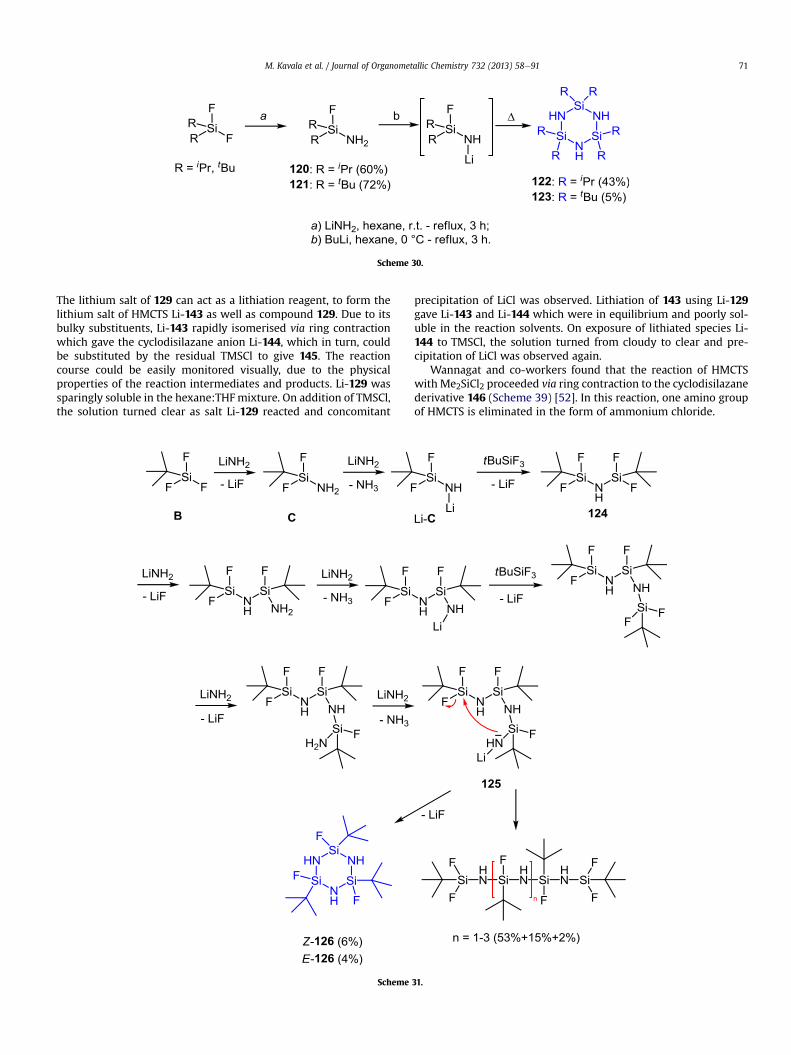
2.2.1.2.2. Substitution with lithium amide. Lithium amide is apowerful nucleophilic reagent, capable of converting di-iso-pro-pyldifluorosilane and di-tert-butyldifluorosilane to distillable sila-zanes 120 and 121 (Scheme 30) [6a,8]. Their reaction with BuLifurnished lithium salts, which in the absence of other nucleophilesafforded, along with other major cyclodisilazanes (analogous tothose in Scheme 31), cyclotrisilazanes 122 and 123 [6b]. The tert-butyl derivative 123 is the bulkiest known cyclotrisilazane thatcannot be prepared from di-tert-butyldiaminosilane by directammonolysis [33]; substitution with lithium amide followed bylithiation and cyclisation is at present, the only method available toaccess tert-butyl derivative 123, whichwas isolated in only 5% yield.
Starting from a trifluorosilane derivative, 2,4,6-trifluorocyclo-trisilazane derivatives can be prepared [41]. The reaction mecha-nism for the preparation of trifluorosilazane 126 has been postulatedand is outlined in Scheme 31.The primary product of the reactionbetween tert-butyltrifluorosilane and lithium amide was believed tobe too reactive to be isolated; when lithium amide was used, tert-butyl aminosilane C was immediately converted to the lithium saltLieC. The salt reacted with tert-butyltrifluorosilane which gave dis-ilazane 124. Two further cycles of the first two steps proceeded,which led eventually to 125. The loss of LiF gave the desired cyclo-silazane derivative 126, albeit in only 10% yield (Z:E 3:2 ratio). Themajor products isolated from the reaction mixture were open-chainpolysilazanes.
2.2.1.2.3. Autocondensation by acid. The first step of the syn-thesis involved the aminolysis of tetrachlorosilane using an etherealsolution of dimethylamine, which gave the tris(dimethylamido)chlorosilane (Scheme 32) [42].Subsequent conversion to tris(di-methylamido)silylamine 127 proceeded after the addition of liquidammonia. The following autocondensation, catalysed by tri-fluromethanesulfonic acid, gave the cyclotrisilazane derivative 128in moderate yield (61%). The structure of 128 was elucidated bysingle crystal X-ray diffraction (Fig. 5). The Si3N3 6-membered ringwas found to be nearly planar: the mean deviation of silicon andnitrogen atoms fromplanaritywas amere 0.105�A. Each silicon atomwas surrounded by two exocyclic and two endocyclic nitrogenatoms, thus a tetrahedral SiN4 structure in each subunit was created(Fig. 5).
2.2.1.2.4. Contraction of octamethylcyclotetrasilazane. Octamethyl-cyclotetrasilazane (OMCTS) is capable of ring-contraction to hexam-ethylcyclotrisilazane (HMCTS) in an acid-catalysed and high-
Table 7Physical data of vinyl cyclotrisilazanes.
Comp. Yield Melting point Boiling point
110 [37] 39% 38e40 �C 170e172 �C (1 Torr)111 [37] 37% e 106e110 �C (1 Torr)
temperature process [43]. When OMCTS was heated in the presenceof a Lewis acid (AlCl3) or weakly acidic ammonium sulphate, it couldbe converted to HMCTS 73 in almost quantitative yields (Scheme 33).Since both compounds are already commercially available, thismethodology was not explored further in the literature.
2.2.2. Reactions of cyclotrisilazanesUnless otherwise stated, all reactions described below pro-
ceeded in deoxygenated solvents under a blanket of dry argon ornitrogen.
2.2.2.1. N-functionalisation. The reactions of (lithiated) cyclo-trisilazanes led to cleavage of the SieN bond and hence, to theformation of acyclic compounds [21]. In contrast, reactions be-tween lithiated HMCTS 73 and fluorosilanes led to the formation ofstable substitution products [44]. Lithium salts were easily gener-ated when BuLi was employed. This deprotonation proceededselectively, i.e. 1 equivalent of BuLi gave a monolithium salt with noformation of dilithium salt; another equivalent of BuLi was required(Scheme 34). The reaction of lithiated HMCTS with tBuSiF3 gavetBuSiF2-substituted 6-membered ring 129 [45]; a similar reactionwith two equivalents of tBuSiF3 afforded the di-tBuSiF2-substitutedcyclotrisilazane 130.
The TMS-substituted HMTCS 131 could be converted to themono- and dilithium salts when BuLi was used, and was dependanton the amount of base employed as observed in Scheme 34. Themonolithium salt reacted with fluorosilane derivatives which gavecompounds 132e136 (Scheme 35) [46,47]. Further derivatisation of133 and 135 could lead to the tris-N,N0,N00-substituted cyclo-trisilazanes 137 and 138 [48]. The reactions of the dilithium de-rivative with dimethylfluorosilane or phenyltrifluorosilane led tothe corresponding disubstituted products 139 and 140 in goodyields (85% and 80% respectively).
2.2.2.2. Contractions of lithiated hexamethylcyclotrisilazanes.Detailed studies have shown that depending on reaction temper-ature, the cyclotrisilazane anion (particularly the disubstituted one)is in equilibriumwith the 4-membered ring anion (Scheme 36). Theequilibrium is further affected by the stereoelectronic properties ofthe substituents as well as by the Lewis character of the respectivereagent (acidity vs. basicity) [49]
In general, the following rules apply:
1. Thermal effects: At higher temperature, the spatial contactbetween the Si-atom and the N-anion within ring improves.Thus, the ring contraction becomes more feasible.
2. Stereoelectronic effects: Donor substituents that enhance thenitrogen basicity (possessing either þI or þM effect) favour ringcontraction, while electron-accepting substituents (with �I or

Scheme 26.
Table 8Physical data of isobutyl cyclotrisilazanes.
Comp. Yield (%) Boiling point
112 [24] 22 131e132 �C (1 Torr)113 [24] 15 141e142 �C (1 Torr)114 [24] 9 164e165 �C (1 Torr)115 [24] 11 224e226 �C (1 Torr)
Scheme 28.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9170
�M effect) disfavour it. Large substituents at nitrogen enforce asmaller SieNeSi angle within the ring as well as shortening ofthe SieN distance around the ring, thus favouring ringcontraction.
3. Properties of electrophile: If the electrophile is a Lewis acid,basic character of the ring decreases, thus creating a situationin which either the 6-membered ring tends to be retained, orthe 4-membered ring expanded.
Klingebiel and co-workers found that isomerisation could becontrolled by temperature [50]. Thus, in the reaction of lithiated129 with tBuSiF3 and Me2SiF2 at �50 �C, a substitution occurredwhich gave compounds 130 and 141, and no change in ring size wasobserved. However, the analogous reactionwith tBuSiF3 carried outat laboratory temperature induced a ring contraction and formedthe isomeric cyclodisilazane 140 (Scheme 37).
Scheme 27.
Compounds 129, 130, 141 and 142 can react with BuLi to givestable lithiated or dilithiated derivatives. If Li-129 reacted withTMSCl in an equimolar ratio, the substitution would make thesecond amino group of HMTCS 143 (Scheme 38) more basic [47].
Scheme 29.
Scheme 30.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 71
The lithium salt of 129 can act as a lithiation reagent, to form thelithium salt of HMCTS Li-143 as well as compound 129. Due to itsbulky substituents, Li-143 rapidly isomerised via ring contractionwhich gave the cyclodisilazane anion Li-144, which in turn, couldbe substituted by the residual TMSCl to give 145. The reactioncourse could be easily monitored visually, due to the physicalproperties of the reaction intermediates and products. Li-129 wassparingly soluble in the hexane:THF mixture. On addition of TMSCl,the solution turned clear as salt Li-129 reacted and concomitant
Scheme
precipitation of LiCl was observed. Lithiation of 143 using Li-129gave Li-143 and Li-144 which were in equilibrium and poorly sol-uble in the reaction solvents. On exposure of lithiated species Li-144 to TMSCl, the solution turned from cloudy to clear and pre-cipitation of LiCl was observed again.
Wannagat and co-workers found that the reaction of HMCTSwith Me2SiCl2 proceeded via ring contraction to the cyclodisilazanederivative 146 (Scheme 39) [52]. In this reaction, one amino groupof HMCTS is eliminated in the form of ammonium chloride.
31.
Scheme 32.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9172
2.2.2.3. Acidic hydrolysis. Andrianov and Kotrelev found thatcyclotrisilazanes readily hydrolyse in acidic media [23]. Experi-ments were carried out with compounds 77, 78 and 80 in boilingbenzene containing sulphuric acid (Scheme 40). After 24 h underthese conditions, the starting cyclotrisilazanes were converted tosilanols in 53%, 43% and 36% yield respectively. The conversion wasdetermined from the amount of the created ammonium salt.
2.2.2.4. Reactions in basic conditions. As mentioned in Section2.2.2.1, a strong base (BuLi) can deprotonate the free amino group toform a relatively stable salt. The use of a catalytic amount of otherstrong bases (KOH, EtONa) led to the formation of unstable in-termediates, which in turn, could further attack another moleculeof HMCTS 73 (Scheme 41) [23]. However, only the first steps of atentative mechanism was postulated. The mechanism of an anal-ogous process of the degradation of 76e78 has not been described,although the corresponding products 147e149 were analysed inconsiderable detail (Scheme 42).
2.2.2.4.1. Hexamethylcyclotrisilazane as a dehydrating agent.HMCTS 73 is considered an effective dehydrating agent [53]. Itsreaction with benzamide 150, acetamide 151, or substituted N-substituted amides 152,153 at 180e200 �C gave rise to benzonitrile
Fig. 5. ORTEP diagram of 128 obtained by single crystal X-ray structural analysis.
and acetonitrile respectively in low to excellent yields (27e95%)(Scheme 43). The HMCTS hydrolysed to form complex siloxanes.As an advantage over the classical dehydration and/or reductionagents (acidic: SOCl2, P2O5, POCl3; basic: NaBH4, LiAlH4), facileisolation of products was observed. On the other hand, high reac-tion temperatures (200 �C) were a drawback. The authors claimedthat the given yields were not optimised, since the reactions pro-ceeded with incomplete conversion and were stopped prematurely(due to the prolonged heating at 200 �C). All preparations of nitriles150e153 described proceeded neat without solvent.
Similarly, the reaction of HMCTS 73 with syn-benzaldoxime 154led to the formation of benzonitrile (Scheme 44). The analogy be-tween compounds 150 and 154 is obvious from the tautomericforms of 150 (Scheme 45). The authors also suggested amechanismfor this transformation (Scheme 46).
2.2.2.4.2. HMCTS as a silylation agent. HMCTS 73 has beenwidely employed in organic synthesis as a reagent used to intro-duce a silyl protecting group, and therefore could be used forderivatisation of samples in gas-chromatography analysis [54].Silicon-containing functional groups often replace an acidichydrogen on heteroatoms (eOH, eNHR). Silylated derivatives ofvarious alcohols, carboxylic acids, phenols, amino acids, and aminesare mostly more volatile than the parent non-silanized derivativesdue to the absence of hydrogen bonds. The compounds with sily-lated heteroatoms are usually more stable under a variety of con-ditions and also facilitate a chemoselective course of reaction.Silanes are often used for the protection of bifunctional molecules,such as primary amines or vicinal diols. Such a process has beentermed cyclosilylation (Scheme 47155e156) [55]. HMCTS 73 can beseen as a protecting group for the hydroxyl group or, that 73 wassubjected to alcoholysis [26]
Scheme 33.
Scheme 34.
NH
NLiSi
Si SiN
Si
NH
Si
Si SiN
SiNSiR'
RF
NH
NHSi
Si Si
NSi
NLi
NLiSi
Si SiN
Si Si
Si Si
NSi
NSiR
R´F
N
SiR R´F
131 132: R = R' = F (85%)133: R = R' = Me (80%)
139: R1 = R2 = Me (85%)140: R1 = F, R2 = Ph (80%)
a) BuLi (1 equiv), hexane, -78 °C - r.t., RR'SiF2 (1 equiv), 20 h;b) BuLi (2 equiv), hexane, r.t., R1R2SiF2 (2 equiv), 16 h.
a
b
a N
Si
Si SiN
SiNSiR'
RF
SiR'''R''
F137: R = R' = Me
R'' = F, R''' = Ph (65%)134: R = F, R' = tBu (30%)135: R = R' = iBu (90%)136: R = H, R' = F (15%)
138: R = R' = iBuR'' = Me, R''' = Ph (90%)
RR'SiF2
Scheme 35.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 73

Scheme 36.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9174
2.3. Cyclotetrasilazanes
Cyclotetrasilazanes are eight-membered rings with the basicstructure shown in Fig. 6. Their systematic name is derived from1,3,5,7-tetraaza-2,4,6,8-tetrasilacyclooctane. Ring positions 2, 4, 6and 8 are occupied by four-valent silicon atoms which are analo-gous to carbon as both elements belong to group 14. The entiresection focuses on the synthesis of cyclotetrasilazanes withunsubstituted nitrogen atoms e compounds which are able toundergo further functionalisation.
2.3.1. Preparation of cyclotetrasilazanesSynthetic methods for the preparation of cyclotetrasilazanes
often follow those used for cyclotrisilazanes (Section 2.2.1). Actu-ally, in the majority of cases, the eight-membered cyclo-tetrasilazanes are merely side products during the synthesis of six-membered cyclotrisilazanes.
2.3.1.1. Ammonolysis of chlorosilanes2.3.1.1.1. Ammonolysis of dichlorosilanes. The historically first,
and even now most frequently used method for the preparation ofcyclotetrasilazanes is ammonolysis [19e21]. Brewer and Habercarried out ammonolysis of dichlorodimethylsilane using liquidammonia at �78 �C. Although the major product was HMCTS 73,the octamethylcyclotetrasilazane OMCTS 74 could be isolated in25% yield (Scheme 21). Ammonolysis using benzene solution ofgaseous ammonia gave a slightly higher combined yield of 73 and74, as well as a higher ratio of tetramer 74 to trimer 73 compared to
Scheme
when the reaction was performed in neat liquid ammonia [22].However, the authors did not disclose any data on yields and/orreaction conditions, probably because they originally planned tosynthesise the cyclotrisilazane 73. Presently OMTCS 74 iscommercially available.
The concept presented in Scheme 48 represents the most oftenused route to cyclotetrasilazanes. It is used to synthesise symmet-rically substituted cyclotetrasilazane derivatives, of which anoverview and yields are given in Table 9. The yields given are thehighest obtained for the respective compound.
Zachernyuk’s group successfully used this method for thepreparation of cyclotetrasilazane 172 [58,59] where the siliconatoms were substituted by 6-membered siloxane rings (Scheme49). In this case again, cyclotrisilazane 173 was formed as a majorproduct.
2.3.1.1.2. Ammonolysis of mixtures of dichlorosilanes. The actionof ammonia in benzene on chlorosilazane derivatives resulted inthe condensation to various cyclosilazane structures (Section2.2.1.1.1, Scheme 23). Replacement of iso-butyltrichlorosilane bynonyltrichlorosilane in this reaction furnished cyclotetrasilazane174 as the major product in 30% yield and 8-membered ringcyclotetrasilazane 113 was isolated as a minor product in 12% yield(Scheme 50).
Methylvinyldichlorosilane reacted with ammonia to givecyclotetrasilazane 175 as the minor product in 12% yield (Scheme51). The major product of the reaction was the 6-membered vinylderivative 110whichwas isolated in 37% yield. The authors assertedthat the main reason for the observed low yields was the formationof polymeric material of unknown structure with a "glassy"appearance. In the case of the identical reaction in the presence ofdimethyl-dichlorosilane, the reaction gave rise to the cyclo-tetrasilazane derivative 177, carrying two reactive vinyl groups aswell as the 6-membered derivatives 178 and 179. The authorsenvisioned further functionalisation of the vinyl group, althoughsuch reactions are as yet unreported in the literature.
2.3.1.1.3. Aminolysis of dichlorodisilazanes. An effective methodof cyclotetrasilazane preparation is the reaction of 1,3-diaminodisilazanes with 1,3-dichlorodisilazanes (Schemes 52 and
37.

Scheme 38.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 75
53). Compared with the previously mentioned methods, averageyields of 50% could be achieved which was a significant improve-ment on yields obtained by other approaches (Tables 10 and 11).
2.3.1.2. Substitution of chlorosilanes with metal amides2.3.1.2.1. Substitution with lithium amide. Attempts to prepare
cyclotrisilazanes by the substitution with lithium amide (see alsoSection 2.2.1.2.2, Scheme 30) led to the formation of analogousoctasubstituted cyclotetrasilazanes 190 and 191, albeit in traceyields (Scheme 54) [60,61]. An interesting feature of derivative 190was an even more pronounced ring planarisation than in OMCTS74. This was believed to be due to the bulkiness of the tert-butylsubstituents in 190. In the case of dilithium salt 192, on exposure toMe2SiCl2 cyclotetrasilazane 193 was isolated in 45% yield.
2.3.1.2.2. Substitution with sodium amide. The reaction ofmethyl- or ethyltrichlorosilane with sodium amide prepared in situgave rise to interesting cage structures 194 and 195 (Scheme55) [41,62].Analysis by single crystal X-ray diffraction allowed theelucidation of the conformations of polycyclic cyclotetrasilazanes194 and 195. As depicted in Fig. 7194 and 195 consist of two fusedcyclotetrasilazanes, connected by two bridging amino groups, andcan also be viewed as two cyclotrisilazanes bridged by three aminogroups.
Scheme 39.
2.3.2. Reactions of cyclotetrasilazanesEight-membered cyclotetrasilazanes are moisture sensitive,
which was observed with the analogous six-membered cyclo-silazanes. All reactions have to be carried out under a blanket of dryargon or nitrogen.
2.3.2.1. N-functionalisation. Free amino groups of cyclo-tetrasilazanes are readily deprotonated by BuLi and the nucleo-philic lithium amides that form are capable of substituting silicon(and boron) halides. Thus, deprotonation of 74 furnished a mono-lithium salt that readily reacted with tetrafluorosilane (Scheme56) [48]. However, the product of monosubstitution 196 was iso-lated in only 10% yield. Such a substitution increased the basicity ofthe remaining amino groups, so that the free amine could bedeprotonated sufficiently by the primarily formed monolithiumsalt which then underwent subsequent substitutions. The authorsindicated that the enhanced basicity maywell be the reason for lowyields of themonosubstituted cyclotetrasilazane. Other alternativescan be found in Section 2.3.2.4.
The nucleophilic lithium amides can attack organoboranes aswell as organosilanes. The reaction gave acceptable yields, and
Scheme 40.
Scheme 41.
Scheme 42.
R
O
NHNSiNH
SiNH
Si
RN+ + +3 NHR'R''
a
150: R = Ph, R' = R'' = H151: R = Me, R' = R'' = H152: R = Ph, R' = H, R'' = Me153: R = R' = Me, R'' = Ph
R = Ph (95% from 150)(43% from 152)
R = Me (85% from 151)(27% from 153)
3 3R''
R'
(OSiMe2)n
a) 110-200 °C, 48-100 h.
Scheme 43.
Scheme 44.
Scheme 45.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9176
interestingly, it proceeded via a regioselective disubstitution; themajor products observed were 1,5-disubstituted rings 197e200,not the equivalent 1,3-disubstituted compounds (Schemes 57 and58) [63]
2.3.2.2. Alcoholysis and aminolysis of cyclotetrasilazanes.Alcohols as protic solvents will cause alcoholysis of cyclosilazaneswhich is why all reactions have been carried out in non-polaraprotic solvents. For example, cyclotetrasilazane 74 was readily
Scheme 46.
Scheme 47.
Fig. 6. General formula of cyclotetrasilazanes. Scheme 48.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 77
Table 9Preparation and physical data of cyclotetrasilazanes.
Comp. R1 R2 Yield (%) Melting point Boiling point
74 [19] Me Me 25 97 �C 134e135 �C (35 Torr)228 �C (755 Torr)
159 [19] Et Et e 16 �C 192 �C (8 Torr)160 [56] Me Ph e 202e204 �C e
161 [56] Bn Bn e 165e165.5 �C 126e128 �C (1 Torr)162 [29] iPrO iPrO e e 191 �C (2 Torr)163 [23] Me Et 21 e 136e139 �C (3 Torr)164 [23] Me nBu 22 e 162e165 �C (T1 Torr)165 [23] Me nHex 25 e 220e224 �C (1 Torr)166 [23] PhO PhO 50 129 �C e
167 [57] Ph EtO 11 e 289e292 �C (1 Torr)168 [35] Et (CH2)3CN 61 e 295e300 �C (2 Torr)169 [31] H Me 25 e 102 �C (0.5 Torr)170 [31] H Et 50 e 54 �C (1 Torr)172 [58] OeSiMe2eOeSiMe2eO 35% 172 �C
Scheme 49.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9178
opened by various alcohols to disilazane diols 201e203 underacidic catalysis (Scheme 59) [26]
In a similar manner, cyclotetrasilazane 74 can react with triol204 to form cyclic siloxane 205 in 70% yield (Scheme 60) [64]
In this context, amines (mostly anilines) behave analogously toalcohols. The reaction of aniline with OMCTS led to the isolation ofdianiline derivatives of dimethylsilane 206e210 in 64e98% yield(Scheme 61) [65]
2.3.2.3. Reactions with acids. The reactions with protic partnerssuch as alcohols and weakly basic amines, demonstrates the
Scheme
essential lability of OMCTS. However, this is in marked contrast tothe reactions of 74reactions with HX. On exposure to strong acid-s,the 8-membered ring is opened, to form haloderivatives of dis-ilazanes 211e213 (Scheme 62) [26,66,67]. The first step is likely toinvolve protonation of the amino group, followed by nucleophilicsubstitution of a Si-atom with a halide anion. Finally, the ring isopened with the formation of NH4X and hygroscopic hal-odisilazanes 211e213.
2.3.2.4. Ring contractions. The lithium salts of cyclotetrasilazanesobey the same rules as lithium salts of 4- and 6-membered cycles
50.

Scheme 51.
178-185
a
a) Et3N, Et2O, -70 °C - reflux, 4 h.
HN
SiHN Si
NH
SiNHSi
R3
R2R1
SiNH2
HN
Si
R3H2N
SiR2Cl
HN
SiR1
Cl +
R1 ,R2 , R3 - see Table 10
Scheme 52.
186-189
a
a) Et3N, Et2O, -70 °C - ref lux, 4 h.
HN
SiHN Si
NH
SiNHSi R5
R4
R2R3R1
SiNH2
HN
SiR4
R5H2N
Si
R2
R3
Cl
HN
Si
R1Cl +
R1 ,R2 , R3 , R4 ,R5 - see Table 11
Scheme 53.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 79

Table 10Preparation and physical data of cyclotetrasilazanes.
Comp. R1 R2 R3 Yield Melting pointa Boiling point
178 [37] CH¼CH2 CH¼CH2 Ph 40% 80e81 �C 210e215 �C (1 Torr)179 [60] Me Me Me 37% e 101e105 �C (1 Torr)180 [60] CH¼CH2 CH¼CH2 Me 45% e 148e152 �C (1 Torr)181 [60] Me Me Ph 53% 94 �C 193e196 �C (1 Torr)182 [60] Me Ph Me 43% 119e120 �C
104e105 �C194e198 �C (1 Torr)
183 [60] CH¼CH2 Ph CH¼CH2 38% e 191e198 �C (1 Torr)184 [60] Ph Me Ph 47% e 228e231 �C (1 Torr)185 [60] Ph Ph Ph 60% 83e85 �C
154e155 �C204e206 �C [56]
256e263 �C (1 Torr)
a Different mp corresponds to various Z-/E-isomers.
Table 11Preparation and physical data of cyclotetrasilazanes.
Comp. R1 R2 R3 R4 R5 Yield Melting point Boiling point
186 [60] Me Ph Ph Me Me 7% 165.5e166 �C e
187 [60] Me Me Me Me Me 34% 79.5 �C 179e182 �C (1 Torr)188 [60] CH¼CH2 CH¼CH2 Me Me Me 25% e 196e205 �C (1 Torr)189 [60] Me Me Me Ph Ph 43% 164.5e165 �C e
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9180
(Section 2.2.2.2). However, it is not yet clear whether the ringcontraction from 8-membered to 4-membered ring proceedsdirectly, or by a 6-membered ring intermediate (Scheme 63). The 6-membered ring was, as mentioned earlier, in equilibrium with theanion of the 4-membered ring (cf. Scheme 36).
In order to preserve the 8-membered ring, it was imperativethat all reactions with the OMTCS anion were carried out at lowtemperatures (�78 �C). Reactions at higher temperatures inevitablyled to ring-contraction.
The capability of the 8-membered ring to contract to a 6-membered ring [43] can be utilised in the targeted synthesis ofcyclotrisilazanes, as mentioned in Section 2.2.1.2.4. Heating OMCTS74 in the presence of an acidic catalyst, either AlCl3 or ammoniumsulphate, gave rise to HMCTS 73 in nearly quantitative yields(Scheme 64).
Scheme 55.
2.4. Summary
Cyclosilazanes have been studied for more than 60 years, mak-ing them a well known group of compounds. They are unstablewhen exposed to air, undergo a gradual hydrolysis bymoisture, andhave to be stored and manipulated in non-aqueous media andunder a blanket of dry argon or nitrogen.
Scheme
� The 4-membered cyclosilazanes can be accessed directly byheating lithium salts of halosilazanes; they are generallyinaccessible by ammonolysis.
� On the contrary, 6-membered cyclosilazanes are not preparedby thermal reactions of halosilazane salts, but arise as majorproducts of ammonolysis of chlorosilane derivatives.
54.
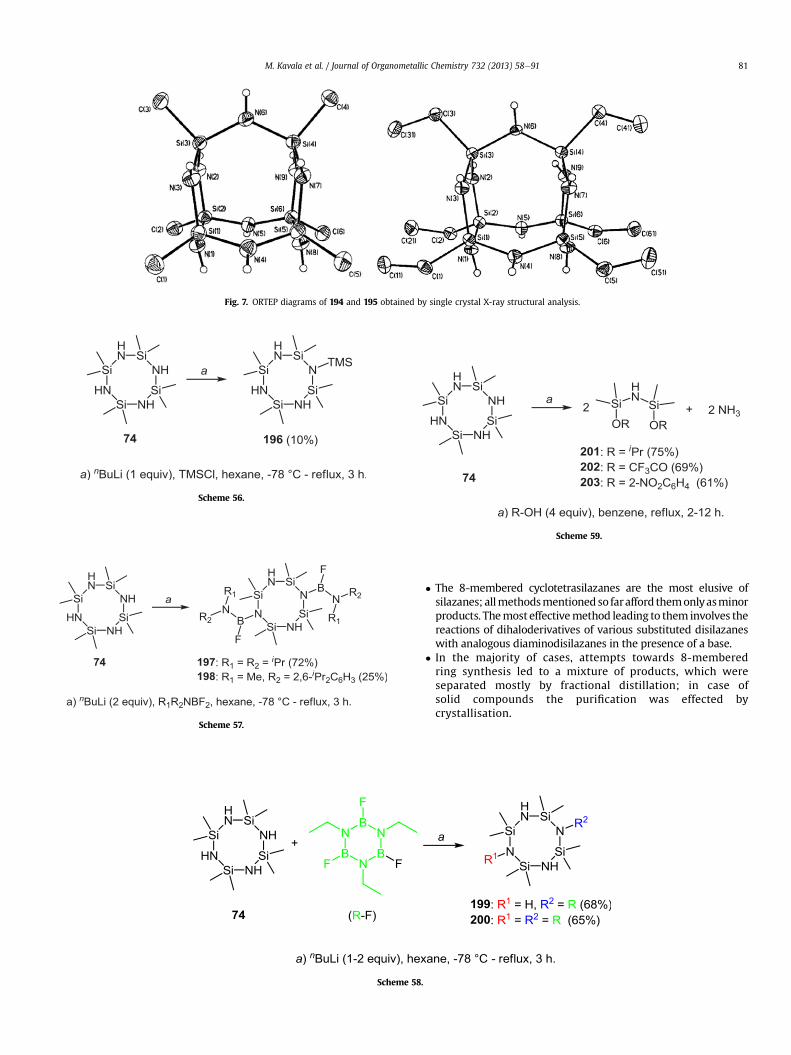
Fig. 7. ORTEP diagrams of 194 and 195 obtained by single crystal X-ray structural analysis.
Scheme 56.
Scheme 57.
Scheme
Scheme 59.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 81
� The 8-membered cyclotetrasilazanes are the most elusive ofsilazanes; allmethodsmentionedso farafford themonlyasminorproducts. Themost effectivemethod leading to them involves thereactions of dihaloderivatives of various substituted disilazaneswith analogous diaminodisilazanes in the presence of a base.
� In the majority of cases, attempts towards 8-memberedring synthesis led to a mixture of products, which wereseparated mostly by fractional distillation; in case ofsolid compounds the purification was effected bycrystallisation.
58.

Scheme 60.
Scheme 61.
Scheme 62.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9182
3. Cyclocarbosilazanes
3.1. Cyclodicarbodisilazanes
Cyclodicarbodisilazanes are 5-membered heterocyclic ringswith the generic structure shown in Fig. 8. The name of this groupof compounds derives from the saturated azaheterocycle,
Scheme
pyrrolidine. Carbon atoms in positions C-2 and C-5 have beenreplaced by silicon atoms from the same group of the periodic table.Typically, substituents are found in these positions, thus 2,5-substitutions would afford 2,2,5,5-substituted 2,5-disilapyrrolidines, or 2,2,5,5-substituted 2,5-disila-1-azacyclopentanes. In this section, we deal mainly with dis-ilapyrrolidines with unsubstituted nitrogen atoms.
63.

Scheme 64.
Fig. 8. General formula of cyclodicarbosilazanes.
Table 12Physical data of cyclotetrasilazanes.
Comp. Melting point Boiling point
218 150 �C e
219 e 141 �C (760 Torr)92 �C (185 Torr)
220 e 98e99 �C (3 Torr)221 e 135 �C (3 Torr)
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 83
3.1.1. Preparation of 2,5-disilapyrrolidinesCyclocarbodisilazanes, which are silicon analogues of carbon
compounds, have been researched mainly for differences in prop-erties and reactivity as a direct result of this atom exchange. Allsyntheses reported in the literature are based on a single type ofreaction.
3.1.1.1. Ammonolysis of acyclic dichlorosilanes. The first reportedsynthetic methodology for the preparation of 2,5-disila-1-azacyclopentanes was the ammonolysis of acyclic 1,2-bis(chlorodiphenylsilyl)ethane 214, which led to 2,5-disilapyrrolidine 218 [68]. Other substitution derivatives 219e221were accessed by this method from dichloridisilanes 215e217(Scheme 65, Table 12). Whilst however, Haberland [69] reportedthe preparation of cyclic products 218 and 219 in considerableexperimental detail, Andrianov [70] did not fully report his syn-thetic procedure so we can only assume he employed the sameconditions as Haberland. This may be the reason why the subse-quently published synthetic papers [71,72] refer to and quote onlyHaberland’s work. However, none of the authors indicate therelative configuration of chiral compound 221,.
2,2,5,5-Tetramethyl-2,5-disila-1-azacyclopentane 219 has sincebecome commercially available and as a result, its synthesis nolonger appears in the literature.
Scheme
Interestingly, Andrianov and co-workers tested 2,5-disilapyrrolidine 219 for its bioactivity [73]. They found it to beantibacterial and suppressed the growth of cultures of Gram-negative bacteria Escherichia coli., as well as Gram-positive bacte-ria Staphylococcus aureus.
3.1.1.2. Radical cyclisation. An interesting approach to access the2,5-disilapiperidine skeleton utilised a radical-initiated intermo-lecular hydrosilylation, followed by an in situ cyclisation. Seyferthand co-workers [74,75] carried out a transformation of 1,1,3,3-tetramethyl-1,3-divinylsilazane 222 using 20 equivalents of trie-thylsilane 223; an excess was necessary in order to obtain higheryields of the products. In the presence of a catalytic amount ofradical initiator e di-tert-butylperoxide (DTBP), cyclic products 224and 225 were obtained in 43% overall yield (Scheme 66). The pro-posed mechanism involved a radical process, in which the initiallyformed silyl radical added to olefin 222. The addition was followedby a non-selective 5-exo vs. 6-endo cyclisation, which gave anapproximately equimolar mixture of disilapiperidines 224 and 225(Scheme 67). However, this transformation suffers from a numberof drawbacks; apart from low regioselectivity of cyclisation, bothproducts 224 and 225 are virtually inseparable from each other.
3.1.2. Reactions of 2,5-disilapyrrolidinesSubstituted 2,5-disilapyrrolidines are moisture and acid sensi-
tive, and are easily hydrolysed to silanols, following the generalpattern observed in most cyclo(carbo)silazanes. Therefore, the re-actions have to be carried out in a dry atmosphere and usingdeoxygenated solvents.
3.1.2.1. Acidic hydrolysis. In acidic aqueous media, cyclo-carbodisilazanes 226 and 227 undergo a complete hydrolysis to the5-membered cyclocarbodisiloxane 228, or even down to acyclicdisilanol 229 (Scheme 68). The process is accompanied by release ofthe corresponding primary amine [71]
In addition to mineral and haloacids, hydrolysis of 2,5-disilapyrrolidines can also be initiated by acidic phenols. The re-action of 219 with 2,2-diphenylpropane 232 in dioxane led to theopening of the silazane ring [68]. Under the given reaction
65.
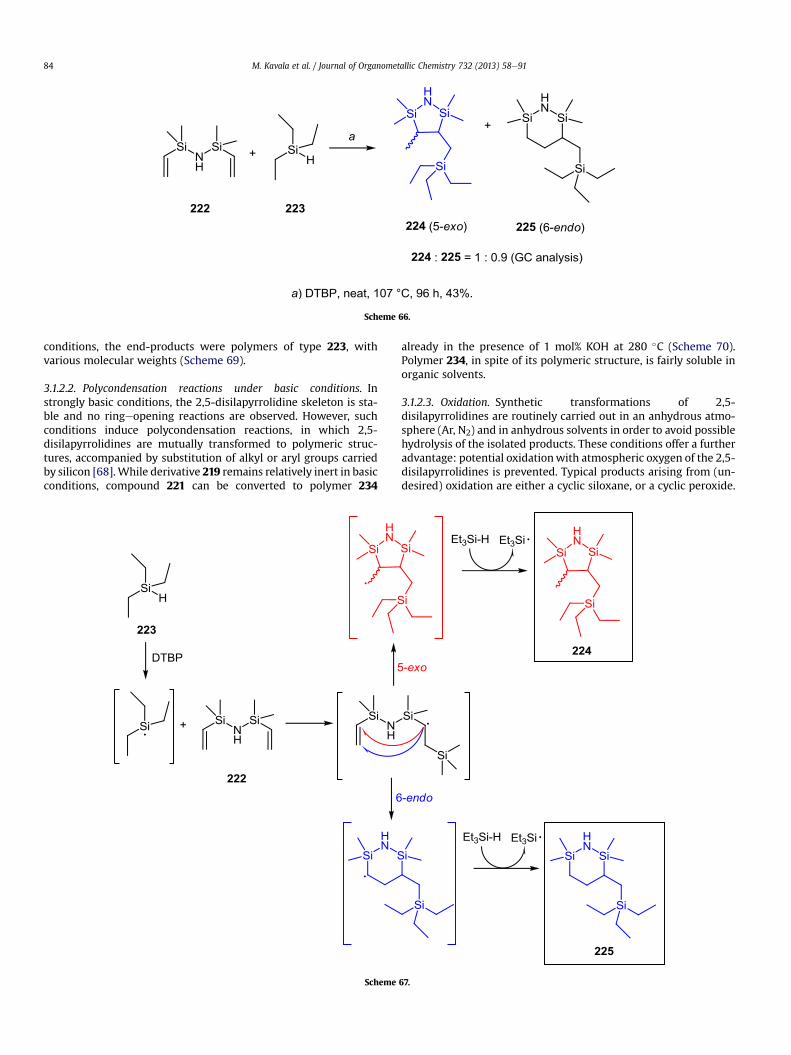
Scheme 66.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9184
conditions, the end-products were polymers of type 223, withvarious molecular weights (Scheme 69).
3.1.2.2. Polycondensation reactions under basic conditions. Instrongly basic conditions, the 2,5-disilapyrrolidine skeleton is sta-ble and no ringeopening reactions are observed. However, suchconditions induce polycondensation reactions, in which 2,5-disilapyrrolidines are mutually transformed to polymeric struc-tures, accompanied by substitution of alkyl or aryl groups carriedby silicon [68].While derivative 219 remains relatively inert in basicconditions, compound 221 can be converted to polymer 234
Scheme
already in the presence of 1 mol% KOH at 280 �C (Scheme 70).Polymer 234, in spite of its polymeric structure, is fairly soluble inorganic solvents.
3.1.2.3. Oxidation. Synthetic transformations of 2,5-disilapyrrolidines are routinely carried out in an anhydrous atmo-sphere (Ar, N2) and in anhydrous solvents in order to avoid possiblehydrolysis of the isolated products. These conditions offer a furtheradvantage: potential oxidationwith atmospheric oxygen of the 2,5-disilapyrrolidines is prevented. Typical products arising from (un-desired) oxidation are either a cyclic siloxane, or a cyclic peroxide.
67.

Scheme 69.
Scheme 70.
Scheme 68.
Scheme 71.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 85

Scheme 72.
Scheme 73.
Fig. 9. General formula of cyclotricarbosilazanes.
Scheme
Table 13Physical data of 2,6-disilapiperidines.
Comp. Melting point Boiling point
245 e 158e160 �C (760 Torr)55e56 �C (21 Torr)
246 35e35.5 �C 145e146 �C (1 Torr)
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9186
In the reaction of 2,5-disilapyrrolidine 219 with a urea/hydrogenperoxide complex in dry dichloromethane, an oxidation takesplace, and disilacyclohexane 235was isolated in 68% yield [76]. Theminor side products of the transformation are disiloxane 228 and apolymer 236 (Scheme 71).
3.1.2.4. Hydroboration. The ease of hydrolysis and/or oxidation of2,5-disilapyrrolidines indicates that the SieN bond is rather labile(DH ¼ 355 kJ/mol). Hydroboration of the SieN bond in derivative219 using either diborane 237 or 238 gave silaborazanes 239 and240 (Scheme 72) [77]. Analysis of the 13C NMR spectra indicates arotation barrier around the BeN bond in compound 240 (in CDCl3at r.t.), hence the presence of two conformers 240 and 240’ in a 1:1ratio.
74.
Scheme 75.
Scheme 76.
Scheme 77.
Scheme 78.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 87

Scheme 79.
Scheme 80.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9188
3.1.2.5. N-functionalisation of 2,5-disilapyrrolidines. The 2,2,5,5-tetramethyl-2,6-disilapyrrolidine 219 possesses a relatively acidicamino group, which can be deprotonated by strong, non-nucleophilic bases (BuLi, metal hydrides). Deprotonation gave sta-ble metal amides, which demonstrated their dual character byacting as both bases and nucleophiles; they can therefore be used ina Gabriel-like synthesis of primary amines [71]. The preparation ofN-substituted derivatives of cyclodisilapyrrolidines 241 and 242may serve as an example of such functionalisation, albeit in syn-thetically unattractive yields (Scheme 73).
3.2. Cyclotricarbodisilazanes
Cyclotricarbodisilazanes are six-membered cyclic compoundswith the basic skeleton outlined in Fig. 9. Their nomenclature wasderived from the generic name of the saturated azaheterocycle,piperidine. Carbon atoms in positions C-2 and C-6 in piperidinehave been replaced by two silicon atoms from the same group ascarbon in the periodic table to give cyclotricarbodisilazanes. Thesesubstituted silicon derivatives can either be referred to as 2,2,6,6-substituted 2,6-disilapiperidine (in the case of this review) or2,2,6,6-substituted 2,6-disila-1-azacyclohexane. In this section, weshall deal mainly with synthetic methods leading to dis-ilapiperidines with an unsubstituted nitrogen atom.
3.2.1. Preparation of 2,6-disilapiperidinesAs it has been mentioned in Section 3.1.1 cyclocarbodisilazanes
have been scarcely mentioned in the literature. However, in recentyears there has been a revival of interest not only in their effectivesynthesis, but also in the exploration of their useful properties andapplications in novel materials.
3.2.1.1. Ammonolysis of acyclic dichlorosilanes. As mentioned inprevious sections, the first and still the most frequently methodused to generate an SieN bondwas the cyclisation ammonolysis ofdichlorosilanes. The preparation of two types of 2,6-disilapiperidines 245 and 246, have been described in the litera-ture, which are both based on the reaction between ammonia andcorresponding a,u-bis-(diorganochlorosilyl)-alkanes 243 and 244(Scheme 74). While Andrianov [70] gave neither experimental
Table 14Preparation of N-substituted 2,6-disilapiperidines.
Comp. RX Yield
264 EtBr 50%265 nPrBr 61%266 nBuBr 72%267 nOctBr 89%268 BnCl 85%269 peCH3C6H4CH2Cl 85%
details of the preparation of 245 or 246 (Table 13), nor the relativeconfiguration of the chiral derivative 246, the synthesis of 2,6-disilapiperidine 245 has been described in detail by Hosomi [71].The preparation of key dichlorosilane 243 (now commerciallyavailable) employed allyltrimethylsilane and methyldichlorosilaneas starting materials. The cyclisation ammonolysis of 243 itself waseffected by bubbling gaseous ammonia through an ethereal solu-tion of 243 at 0 �Cwhich gave target 2,6-disilapiperidine 245 in 67%yield.
3.2.1.2. Radical cyclisation. Another known method which can leadto the 2,6-disilapiperidine skeleton, is a radical cyclisation reaction.So far, there is only one known synthesis of 2,6-disilapiperidinesusing such methodology [74,75]. It is apparent from the keytransformation that a C-substituted product should be isolated, in asimilar manner to the reactivity observed in Section 3.1.1.2 (Scheme75). The corresponding 5-exo (224) and 6-endo (225) cyclisationproducts were formed in an approximately 1:1 ratio (224 dr 1:0.9).Themechanism is outlined in Scheme 67 (in Section 3.1.1.2). In viewof the fact that the regioselectivity is low for this reaction, and thatthe products are inseparable from each other, the method has un-doubtedly questionable synthetic value as far as preparation of thedisilapiperidine skeleton is concerned.
3.2.2. Reactions of 2,6-disilapiperidinesJust like other cyclosilazanes, 2,6-disilapiperidines are relatively
sensitive to water and acidic media and can undergo hydrolysis tosilanols. Consequently, their synthesis and subsequent trans-formations have to be carried out under a blanket of dry argon ornitrogen.
3.2.2.1. Acidic hydrolysis. Cyclotricarbodisilazanes are unstable inacidic media and readily undergo hydrolysis. A typical example wasshown by the acidic hydrolysis ofN-alkyl derivatives 247 and 248 tothe corresponding amines 249 and 250 respectively. Thus, thecyclocarbodisilazane ring was converted to cyclocarbodisiloxane
Scheme 81.

Table 15Physical data of eight-membered cyclocarbosilazanes.
Comp. Melting point Boiling point
268 31.5e33 �C 86e88 �C (1 Torr)269 133e134 �C e
270 118 �C Sublimation 80 �C (“under high vacuum”)271 29 �C 142 �C (“under high vacuum”)
Scheme 82.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 89
251, which underwent subsequent hydrolysis to acyclic disilanol252 (Scheme 76) [71]
Opening of the 2,6-disilapiperidine ring can be easily performedwhenweak acids such as phenols are employed [70]. In the reactionof tetramethyl-substituted disilapiperidine 245 with 2,2-diphenolylpropane 253 in 1,4-dioxane, the cyclic skeleton of 245was cleaved and polymer 245 was formed (Scheme 77). The au-thors asserted that the opening of the 2,6-disilapiperidine ring viaSieN bond cleavage was to a great extent, controlled by electrondensity at the nitrogen atom which in turn is dependent on thecharacter of substituents at silicon, not by the ring size of thecarbosilazane.
3.2.2.2. Polycondensation reactions under basic conditions.Polycondensation reactions represent transformations in basicmedia which led to polymerisation of 2,6-disilapiperidines. Thereaction does not involve opening of the ring, however, it didinvolve the substitution of alkyl or aryl substituents at silicon [70].When there were dimethyl substituents on the silicon atom (245),the silicon remained inert to nucleophilic attack. However, in thecase of dimethyl-diphenyl-substituted derivative 246 a nucleo-philic attack became possible: in the presence of 1 mol% KOH at280 �C, 246 polymerised to form compound 255 (Scheme 78).Combustion analysis data and absence of characteristic NH bands inthe IR spectra indicated the formation of macromolecule 255.Remarkably, such polymers are, according to the authors, highlysoluble in organic solvents.
Scheme
3.2.2.3. N-functionalisation of 2,6-disilapiperidines. Compared toHMDS 262, 2,2,6,6-tetramethyl-2,6-disilapiperidine 245 has analmost planar structure with a sterically non-congested nitrogenatom [71]. Cyclocarbosilazane 245 can be considered to be ananalogue of phthalimide which is used in the Gabriel synthesis ofprimary amines. Thus, in principle, it could be possible to preparevarious N-functionalised derivatives 245, since the potassium saltK-245 exhibited significant nucleophilicity towards carbon elec-trophiles. Potassium amide (K-245), easily accessible by deproto-nation of 245 by potassium hydride [78], was thermally stable inTHF and reacted with alkyl chlorides or alkyl bromides at reflux, togive the corresponding N-alkylated 2,6-disilapiperidines 256e261(Scheme 79). Organohalides 267e269 gave better yields than bro-mides 264e266 (Table 14).
In order to demonstrate the significant degree of planarisationand enhanced nucleophilicity of K-245 over acyclic silyl analogues,a reaction between potassium salt of hexamethyldisilazane K-262and the requisite alkyl bromidewas carried out (Scheme 80). HMDSgave lower yields of compounds 263e265 presumably due to thedecreased nucleophilicity and increased steric hindrance.
3.3. Higher cyclocarbosilazanes
This group includes cyclocarbosilazanes possessing larger than8-membered rings, as well as more than one ring and bridgedstructures.
3.3.1. Synthesis of higher cyclocarbosilazanesIn contrast to other organosilicon compounds, there is
comparatively little information on synthesis and physico-chemicalproperties of cyclocarbosilazanes. Larger cyclocarbosilazane ringsare even less well known, and are mostly formed as side and/orunexpected products of various chemical transformations. It isevident that their preparation and synthetic utility has not beenstudied in much detail. However, as reaction side products, thesecompounds have been characterised by spectral and other analyt-ical methods.
3.3.1.1. Eight-membered cyclocarbosilazanes. In their efforts tosynthesise 2,4-disila-azetidine derivative 267 by ammonolysis of1,1-bis(dimethylchlorosilyl)methane 266, the authors succeeded inpreparing 8-membered cyclocarbosilazane 268 as the majorproduct (Scheme 81) [70]. It was a colourless crystalline solid(Table 15) which allowed its effective separation from other com-pounds and facile purification. Compound 268 is the only highercyclocarbosilazane known in the literature that is accessed in asynthetically utilisable yield. All other described derivatives wereformed only as minor products in low, or even trace amounts.
The ammonolysis of 1,2-bis(dimethylchlorosilyl)ethane 216 byliquid ammonia in diethyl ether at low temperatures gave 2,5-
83.

Fig. 10. ORTEP diagram of 271 obtained by single crystal X-ray structural analysis.
M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e9190
disilapyrrolidine 219 (see Section 3.1.1.1). When the reaction wascarried out in toluene, the major product was a barely identified“glassy” polymer. However, an NH-bridged 8-membered ring 269was isolated as a minor product in 18% yield (Scheme 82) [79,80]
3.3.1.2. Nine- and higher-membered cyclocarbosilazanes.Hermann and co-workers [72] performed an ammonolysis ofacyclic dichlorodisilane 216 in an attempt to synthesise 2,5-cyclodicarbodisilazane 219 (see Section 3.1.1.1). As well asisolating the major target compound 2,2,5,5-tetramethyl-2,5-disilapyrrolidine 219 in 48% yield, the reaction also gave two mi-nor products: the 10-membered tetrasiladiazacyclodecane deriva-tive 270 in 3% yield as well as the 15-membered derivative 271 in4% yield. The distillation residue after their separation containednon-volatile polymer 272 in 24% yield (Scheme 83). The authorsfound that analysis by single crystal X-ray diffraction of the minorproducts 270 and 271 revealed a high degree of planarity in thestructures (Fig. 10Table 15).
3.3.2. Reactions of higher cyclocarbosilazanesAll 8- and/or higher-membered cyclocarbosilazanes obtained
have been isolated either as unexpected products (268), or as sideproducts (269e271). The reason for this finding lies in the fact thatno further transformations of these derivatives have been re-ported. The authors of the respective papers merely mention thefact that the compounds are moisture-sensitive which makesthem liable to undergo hydrolysis to form the corresponding si-loxanes and silanols in a similar manner to cyclodicarbo- andcyclotricarbodisilazanes (Sections 3.1.2.1 and 3.2.2.1).
Acknowledgement
We are gratuful to Dr. Peter Zálupský for his help with themanuscript preparation. We thank Prof. Franti�sek Pova�zanec forhelpful discussions. This work was supported by the Science andTechnology Assistance Agency under contract No. APVV-0014-11.
References
[1] (a) E. Kroke, Y.-L. Li, Ch. Konestchny, E. Lecomte, C. Fasel, R. Riedel, Mater. Sci.Eng. 26 (2000) 97;(b) B. Jaschke, U. Klingebiel, R. Riedel, N. Doslik, R. Gadow, Appl. Organometal.Chem. 14 (2000) 671;(c) Ch. Duriez, E. Framery, T. Bérangére, T. Pascaline, P. Miele, M. Vaultier,B. Bonnetot, J. Organomet. Chem. 657 (2002) 107;(d) N. Helmold, L. Verena, U. Klingebiel, S. Schmatz, in: N. Auner, J. Weis (Eds.),Organosilicon Chemistry V: From Molecules to Materials, Wiley-VCH, Wein-heim, 2003, p. 261;(e) R.M. Laine, A. Sellinger, Si-containing ceramic precursors, in: Z. Rappoport,
Y. Apeloig (Eds.), The Chemistry of Organic Silicon Compounds, Vol. 2, JohnWiley & Sons, Chichester, 2003.
[2] (a) P. Neugebauer, B. Jaschke, U. Klingebiel, Recent developments in thechemistry of compounds with siliconenitrogen bonds, in: Z. Rappoport,Y. Apeloig (Eds.), The Chemistry of Organic Silicon Compounds, Vol. 3, JohnWiley & Sons, Chichester, 2003;(b) U. Klingebiel, N. Helmold, S. Schmatz, Adv. Organomet. Chem. 54 (2006) 1;(c) P.Neugebauer, B.Jaschke, U.Klingebiel, Recent Developments in theChemistry of Compounds with SiliconeNitrogen Bonds in Patai’s Chemistry ofFunctional Groups, 2009; (d) D.A. Armitage, R.J.P. Corriu, T.C. Kendrick,B. Parbhoo, T.D. Tilley, J.W. White, J.C. Young, Organosilicon Nitrogen Com-pounds in the SiliconeHeteroatom Bond (Chapter 11), John Wiley & Sons,Chichester, 2010.
[3] A. Stock, K. Somieski, Ber 54 (1921) 1921.[4] K.A. Andrianov, G.V. Kotrelev, V.V. Kazakova, I.E. Rogov, Russ. Chem. Bull. 24
(1975) 2489.[5] K.A. Andrianov, M.M. Ilín, V.N. Talanov, I.I. Zhursakovskaya, Russ. Chem. Bull.
25 (1976) 2432.[6] (a) U. Klingebiel, N. Vater, Angew. Chem. 94 (1982) 870;
(b) U. Klingebiel, N. Vater, Chem. Ber 116 (1983) 3277;(c) U. Kliebisch, U. Klingebiel, N. Vater, Chem. Ber 118 (1985) 4561.
[7] http://chemed.chem.wisc.edu/chempaths/GenChem-Textbook/Bond-Enthalpies-718.html.
[8] U. Klingebiel, N. Vater, Angew. Chem. Suppl. (1982) 1865.[9] U. Klingebiel, Angew. Chem. 93 (1981) 696. Angew. Chem. Int. Ed. Engl. 20
(1981) 678.[10] H.-J. Rakebrandt, U. Klingebiel, M. Noltemeyer, U. Wenzel, D. Mootz,
J. Organomet. Chem. 524 (1996) 237.[11] C. Reiche, U. Klingebiel, M. Noltemeyer, Z. Naturforsch 58b (2003), 939.[12] T.Kottke, U.Klingebiel, M.Noltemeyer, U.Pieper, S.Walter, D.Stalke, Chem. Ber.
124 (1991) 1941.[13] W. Fink, Helv. Chim. Acta 47 (1964) 498.[14] K.A. Andrianov, Zh.S. Syrtsova, V.M. Kopylov, T.K. Augustinene, Chem. Het-
erocycl. Comp. (1972) 679.[15] S. Dielkus, D. Grosskopf, R. Herbst-Irmer, U. Klingebiel, Z. Naturforsch. 50b
(1995) 844.[16] (a) S.E. Thomas, Organic Synthesis. The Roles of Boron and Silicon, Oxford
University Press, Oxford, 1991. p. 48;(b) P. Wothers, N. Greeves, S. Warren, J. Clayden, Organic Chemistry, OxfordUniversity Press, New York, 2001. p. 1288.
[17] E. Egert, U. Kliebisch, U. Klingebiel, D. Schmidt, Z. Naturforsch. 42b (1987) 23.[18] D. Grosskopf, U. Klingebiel, Z. Anorg. Allg. Chem. 691 (1993) 1857.[19] S.D. Brewer, Ch.P. Haber, J. Am. Chem. Soc. 70 (1948) 3888.[20] U. Wannagat, Angew. Chem. 71 (1959) 574. Fortschr. Chem. Forsch. 9 (1967)
102.[21] W. Fink, Angew. Chem. 73 (1961) 736. Angew. Chem. 78 (1966) 803.[22] R.C. Osthoff, S.W. Kantor, Inorg. Synth. 5 (1957) 62.[23] K.A. Andrianov, G.V. Kotrelev, J. Organomet. Chem. 7 (1967) 217.[24] K.A. Andrianov, G.V. Kotrelev, Chem. Heterocycl. Comp. 5 (1969) 585.[25] E. Larrson, L. Bjellerup, J. Am. Chem. Soc. 75 (1953) 995.[26] U. Wannagat, E. Bogusch, P. Geymayer, Monatsh. Chem. 95 (1964) 801.[27] W. Fink, Helv. Chim. Acta 52 (1969) 1841.[28] K. Hizawa, E. Nojimoto, Kogyo Kagaku Zasshi 59 (1956) 1445. Chem. Abstracts
53 (1959) 4176.[29] R. Schwarz, F. Weigel, Z. Anorg Allg. Chem. 268 (1952) 291.[30] E. Larsson, Acta Chem. Scan 8 (1954) 1084.[31] E.A. Semenova, D.Y. Zhinkin, K.A. Andrianov, Russ. Chem. Bull. (1962) 246.[32] K. Ruehlmann, G. Tuchtenhagen, Z. Chem. 5 (1965) 107.[33] L.H. Sommer, L.J. Tyler, J. Am. Chem. Soc. 76 (1954) 1030.[34] K.A. Andrianov, J. Gen. Chem. USSR (Engl. Transl.) 42 (1972) 854.

M. Kavala et al. / Journal of Organometallic Chemistry 732 (2013) 58e91 91
[35] E.P. Lebedev, V.O. Reikhsfeld, J. Gen. Chem. USSR (Engl. Transl.) 40 (1970)1070, 1084.
[36] K.A. Andrianov, I. Khayduk, L.M. Khananashvili, Russ. Chem. Bull. (1963) 860.[37] A.A. Zhdanov, K.A. Andrianov, T.V. Astapova, B.D. Lavrukhin, Russ. Chem. Bull.
(1978) 1858.[38] U. Wannagat, P. Schmidt, M. Schulze, Angew. Chem. 79 (1967) 409.[39] U. Wannagat, H. Moretto, P. Schmidt, M. Schulze, Z. Anorg. Allg. Chem. 381
(1971) 288.[40] A. Zinnius, PhD Thesis, Technische Universität Braunschweig, Germany, 1970.[41] C. Ackerhans, B. Räke, R. Krätzner, P. Müller, H.W. Roesky, I. Usón, Eur. J. Inorg.
Chem. (2000) 827.[42] R. Rovai, Ch.W. Lehmann, J. Bradley, Angew. Chem. Int. Ed. 38 (1999) 2036.[43] T. Shinohara, A. Yokoo, M. Kudo, M. Iwabuchi, K. Matsumura, U.S. Patent
5466847A, 1995.[44] U. Klingebiel, D. Enterling, A. Meller, Chem. Berichte. 110 (1977) 1277.[45] D. Enterling, U. Klingebiel, A. Meller, Z. Naturforsch. 33b (1978) 527.[46] M. Hesse, U. Klingebiel, J. Organomet. Chem. C1 (1981) 221.[47] U. Klingebiel, L. Skoda, A. Meller, Z. Anorg. Allg. Chem. 467 (1980) 131.[48] U. Klingebiel, D. Enterling, L. Skoda, A.Meller, J. Organomet. Chem. 135 (1977) 167.[49] U. Klingebiel, M. Noltemeyer, H.-J. Rakebrandt, Z. Anorg. Allg. Chem. 623
(1997) 281.[50] E. Egert, U. Kliebisch, U. Klingebiel, D. Schmid, Z. Anorg. Allg. Chem. 548
(1987) 89.[52] U. Wannagat, E. Bogush, P. Geymayer, Monatsh. Chem. 102 (1971) 1825.[53] W.E. Dennis, J. Org. Chem. 35 (1970) 3253.[54] L. Birkofer, O. Stuhl, J. Organomet. Chem. C1 (1979) 164.[55] L. Birkofer, O. Stuhl, J. Organomet. Chem. C16 (1979) 177.[56] M. Wada, T. Suda, R. Okawara, J. Organomet. Chem. 65 (1974) 335.[57] K.A. Andrianov, G.A. Kotrelev, B.D. Lavrukhin, L.N. Orobinskaya, Russ. Chem.
Bull. 10 (1967) 2179.[58] A.B. Zachernyuk, V.B. Isaev, B.B. Lavrukhin, A.A. Zhdanov, J. Gen. Chem. USSR
(Engl. Trans.) 60 (1990) 2107.[59] A.B. Zachernyuk, A.A. Zhdanov, V.B. Isaev, Yu.E. Ovchinnikov, V.E. Shklover,
Yu.T. Struchkov, Dokl. Chem. 295 (1987) 338.[60] K.A. Andrianov, A.A. Zhdanov, B.A. Astapov, B.D. Lavrukhin, T.V. Strelkova,
T.V. Astapova, Russ. Chem. Bull. 28 (1979) 2373.[61] T. Kottke, U. Klingebiel, M. Noltemeyer, U. Piper, S. Waler, D. Stalke, Chem. Ber.
124 (1991) 1941.[62] B. Räke, H.W. Roesky, I. Usón, P. Müller, Angew. Chem. Int. Ed. 37 (1998) 1432.[63] B. Jaschke, N. Helmond, I. Müller, T. Pape, M. Noltemeyer, R. Herbst-Irmer,
U. Klingebiel, Z. Anorg. Allg. Chem. 628 (2002) 2071.[64] J.P. Pometan, C. Dumas, J.P. Fourtilian, P. Curtois, B. Maillard, Eur. J. Med. Chem.
16 (1981) 425.[65] B. Dejak, J. Kulpinski, Z. Lasocki, S. Piechucki, B. Pol, Acad. Sci-chem. 35 (1988)
121.[66] U. Wannagat, E. Bogusch, P. Geymayer, Monatsh. Chem. 99 (1968) 1198.[67] U. Wannagat, E. Bogusch, Monatsh. Chem. 102 (1971) 1806.[68] S.W. Jarvie, D. Lewis, J. Chem. Soc. (1963) 4758.[69] R.H. Baney, G.G. Haberland, J. Organomet. Chem. 5 (1966) 320.[70] K.A. Andrianov, G.V. Kotrelev, A.M. Kononov, I.M. Prudnik, Dokl. Akad. Nauk
SSSR Ser. Khim. 216 (1974) 1041.[71] A. Hosomi, S. Kohra, Y. Tominaga, M. Inaba, H. Sakurai, Chem. Pharm. Bull. 36
(1988) 2342.[72] W.A. Herrmann, F. Dyckhoff, E. Herdtweck, Chem. Ber. 125 (1992) 2651.[73] K.A. Andrianov, Y.S. Nabokov, I.M. Prudnik, A.M. Kolonov, G.V. Kotrelev,
Pharm. Chem. J. 10 (1976) 745.
[74] D. Seyferth, H. Friedrich, S.W. Krska, Z. Naturforsch. Chem. Sci. 49 (1994) 1818.[75] S.W. Krska, Cyclisation and cyclopolymerization of silicon-containing dienes,
Ph.D. thesis, MIT, Cambridge, Massachusetts, USA, 1997.[76] W. Adam, R. Albert, Tetrahedron Lett. 33 (1992) 8015.[77] B. Wrackmeyer, B. Schwarze, W. Milius, J. Organomet. Chem. 489 (1995) 201.[78] C.A. Brown, Synthesis (1974) 427.[79] K.A. Andrianov, G.V. Kotrelev, I.M. Prudnik, A.A. Lukovnikov, Bull. Acad. Sci.
USSR, Div. Chem. Sci. (English Translation) 22 (1973) 1822.[80] G. Motz, T. Schmalz, G. Glatz, R. Kempe, B. Wrackmeyer, W. Milius, Z. Anorg.
Allg. Chem. 636 (2010) 1056.
Miroslav Kavala obtained his Bc. (2007) andMSc. (2009)degrees at the Slovak University of Technology (STU) inBratislava, Slovakia. He spent one semester at LincolnMemorial University in Harrogate, USA (2005) and wastrained in Pharma Discovery Research at F. Hoffmann-LaRoche, Ltd. In Basel, Switzerland (2008e2009). Under thesupervision of Dr. Peter Szolcsányi, he is currently, fin-ishing his PhD. project dealing with stable organicradicals.
AlisonHawkinswas born in Oxford in 1987. She receivedher MChem in 2009 from Durham University whichincluded a year working at UCB Celltech synthesisingpeptidomimetic libraries. She completed her DPhil. underthe supervision of Prof. Darren Dixon at Oxford Universitywhere sheworked on the total synthesis ofmanzamine A.
Peter Szolcsányi obtained hisMSc. and PhD. degrees at theDept. of Organic Chemistry, Slovak University of Technol-ogy (STU) in Bratislava, Slovakia. During his postgraduatestudies he spent study stayswith Prof. S. G. Davies inOxford(asymmetric synthesis of aplysillamide B, 1995e1996), andwith Prof. S. V. Ley in Cambridge (first total synthesis oferythroskyrine, 1998). He then pursued the postdoctoralstay with Prof. Andrea Vasella at ETH Zürich (2000e2002)working on the total synthesis of guanofosfocin analogues.In 2003, he accepted the position of Deputy Head ofChemistry at Biorex R&D Ltd. in Veszprém, Hungary. A yearlater, he eventually began his independent career as anAssistant Professor at his alma mater. In January 2009, hewas promoted to the rank of Associate Professor.