supporting information for pd-catalyzed aerobic...
TRANSCRIPT

S1
Supporting Information for
Pd-Catalyzed Aerobic Oxidative Coupling of Arenes: Evidence for Transmetalation between Two Pd(II)-Aryl Intermediates
Dian Wang, Yusuke Izawa and Shannon S. Stahl*
Department of Chemistry, University of Wisconsin-Madison,
1101 University Avenue, Madison, WI 53706 [email protected]
Table of Contents Page
I. General Considerations S2
II. General Methods for Kinetic Data Acquisition and H/D Exchange Experiments S2
III. Raw Kinetic Data S3
IV. NMR Method to Distinguish between Regioisomers in H/D Exchange Experiments S4
V. Derivation of Reaction Rate Laws S6
VI. Discussion of H/D Exchange Experiments S9
VII. Discussion of KIE Experiments S12

S2
I. General Considerations. 1H NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer. Gas chromatographic analyses were performed on a Shimadzu GC-17A using a RTX-5MS column and referenced to an internal standard (octacosane). All commercial reagents were purchased from Aldrich and used as received unless otherwise noted. Palladium acetate was purchased from Sigma-Aldrich and recrystallized from benzene/HOAc prior to use. 3,3',4,4'-Tetramethylbiphenyl was purchased from Alfa Aesar. II. General Methods for Kinetic Data Acquisition and H/D Exchange Experiments.
Kinetic Data Acquisition. Catalytic aerobic oxidative biaryl coupling reactions were performed using a custom reaction apparatus that enabled up to 48 reactions to be performed simultaneously under a constant pressure of O2 (approx. 1 atm) with controlled temperature and orbital agitation. In a 6 mL vial, palladium acetate (0.02 mmol), 2-fluoropyridine (0.04 mmol), trifluoroacetic acid (0.024 mmol) and acetic acid (2.0 mL) were combined and stirred at room temperature for 30 min. The resulting bright-yellow solution was used as a catalyst stock solution. In a separate 6 mL vial, octacosane (0.06 mmol) and o-xylene (20 mmol, 2.40 mL) were combined and stirred at room temperature for 30 min. The resulting solution was used as a substrate stock solution. Reaction tubes (13 × 100 mm test tubes) were placed in the 48-well parallel reactor mounted on a Glas-Col large capacity mixer. The reactor temperature was set to 80 °C and allowed to equilibrate for 10 min. The catalyst stock solution (400 µL, containing 0.004 mmol Pd) was added, after which the headspace was purged with O2 for 10 min. Injection of the substrate stock solution (480 µL, containing 4 mmol o-xylene) established the t = 0 point. After various time intervals, 100 µL aliquots were removed, quenched/diluted with 900 µL chloroform/pyridine 100:1 solution and analyzed by GC. This procedure also was applied to acquisition of kinetic data for the KIE experiments.
Isotope Exchange Experiments. In a 6 mL vial, palladium acetate (0.02 mmol), 2-fluoropyridine (0.04 mmol), trifluoroacetic acid (0.024 mmol) and acetic acid (2.0 mL) were combined and stirred at room temperature for 30 min. The resulting bright-yellow solution was used as a catalyst stock solution. The reaction tube was placed in a 48-well parallel reactor mounted on a Glas-Col large capacity mixer. The reactor temperature was set to 80 °C and allowed to equilibrate for 10 min, after which the catalyst stock solution (400 µL, containing 0.004 mmol Pd) was added. The headspace was purged with O2 for 10 min and o-xylene-d10 substrate (490 µL) was injected. After 17 h, the reaction mixture was cooled to room temperature and 5 µL of 1,1',2,2'-tetrachloroethane (TCE) was added. A 1H NMR spectrum of the reaction mixture was acquired and the yield of "ArH" was determined with TCE as an internal standard. Into the NMR sample was then added a solution of 0.0025 mmol octacosane in 100 µL CHCl3. The resulting solution was analyzed by GC and the yield of "ArAr" was determined with octacosane as an internal standard. A similar procedure was applied to H/D exchange with o-xylene and DOAc (isotope exchange was determined by 2H NMR spectroscopy with dichloromethane-d2 as internal standard).

S3
III. Raw Kinetic Data. Initial rates of the reactions were defined as the slope of the concentration of biaryl product versus time from t = 0 to ≤ 5% conversion. Figure S1 presents the raw data for Figure 1 in the manuscript.
0
2
4
6
8
10
0 0.5 1 1.5 2 2.5
14.3 mM [Pd]23.9 mM [Pd]49.0 mM [Pd]
[ArA
r] (m
M)
Time (h)
0
0.2
0.4
0.6
0.8
1
0 1 2 3 4 5
1.90 mM [Pd]2.85 mM [Pd]3.80 mM [Pd]4.77 mM [Pd]
[ArA
r] (m
M)
Time (h)
Figure S1. Time courses for the oxidative coupling of o-xylene at varying concentrations of [Pd] [Pd = Pd(OAc)2/2-fluoropyridine/TFAH (1:2:1)]. Reactions conditions: [o-xylene] = 4.5 M (4 mmol, 0.48 mL), HOAc (0.4 mL), O2 (1 atm), 80 °C, [Pd] = 1.90, 2.85, 3.80, 4.77, 14.3, 23.9, 49.0 mM. Figure S2 provides the time courses for the measurement of substrate and solvent KIE values at different [Pd], and represents the raw data for Figure 4 in the manuscript.
0
50
100
150
200
250
0 1 2 3
[Ar-
Ar]
(mM
)
Time (h)
[Pd] = 67.5 mMA
B
C
D0
40
80
120
160
0 1 2 3 4
[Ar-
Ar]
(mM
)
Time (h)
[Pd] = 22.5 mMA
B
CD
0
510
15202530
0 1 2 3 4 5
[Ar-
Ar]
(mM
)
Time (h)
[Pd] = 4.5 mMA
BC D 0
5
10
15
20
25
0 5 10 15 20 25 30
[Ar-
Ar]
(mM
)
Time (h)
[Pd] = 2.7 mMA
BC
D
Figure S2. Time courses for the oxidative biaryl coupling with isotopically-varied substrates and solvents at varying concentrations of [Pd] [Pd = Pd(OAc)2/2-fluoropyridine/TFAH (1:2:1)]. Reactions conditions: [substrate] = 4.5 M (4 mmol, 0.48 mL for o-xylene, 0.49 mL for o-xylene-d10), solvent (0.4 mL), O2 (1 atm), 80 °C, [Pd] = 67.5, 22.5, 4.5, 2.7 mM.
time course(Y, Y' = H/D)
[Pd] (2.7 - 67.5 mM)1 atm O2, 80 oC,Y'OAc,
A. o-xylene/DOAc; B. o-xylene/HOAc;C. o-xylene-d10 /DOAc; D. o-xylene-d10 /HOAc.
Y3C
Y3C
Y Y
Y
Y Y3C
Y3C
Y Y
Y Y
Y Y
CY3
CY3

S4
IV. NMR Method to Distinguish between Regioisomers in H/D Exchange Experiments.
The reaction mixture was cooled to room temperature and 5 µL of 1,1',2,2'-tetrachloroethane (TCE) was added. A portion of the resulting solution (0.050 mL) was mixed with 0.450 mL CDCl3 for NMR analysis. In this solvent system, the peaks in the aromatic region of 1H NMR spectrum are well resolved. The 1H and 13C NMR1 spectra and initial assignments are shown below.
Figure S3. 1H and 13C NMR spectra and initial peak assignments for the reaction mixture of H/D exchange between o-xylene-d10 and HOAc at 0.5 mol % [Pd].
ArHortho + ArHmeta
ArHMe
TCE
HOAc
CHCl3
HOAc C2
C3 C4
CDCl3
HOAc
S5
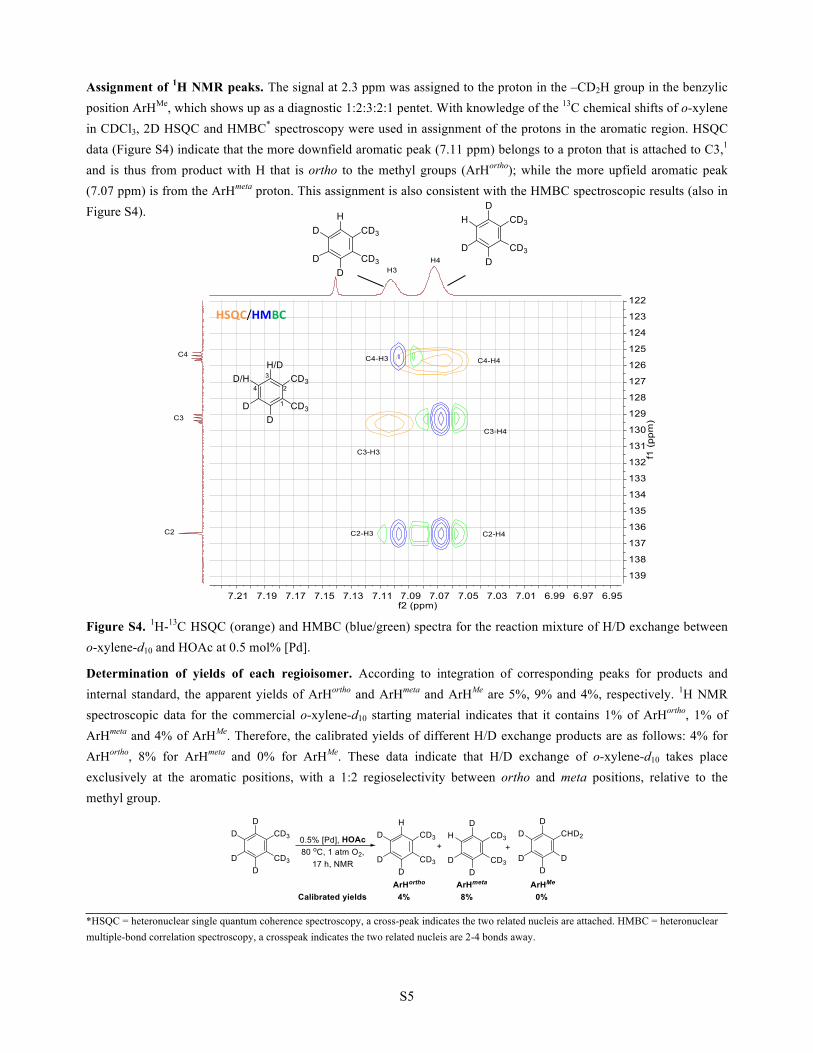
Assignment of 1H NMR peaks. The signal at 2.3 ppm was assigned to the proton in the –CD2H group in the benzylic position ArHMe, which shows up as a diagnostic 1:2:3:2:1 pentet. With knowledge of the 13C chemical shifts of o-xylene in CDCl3, 2D HSQC and HMBC* spectroscopy were used in assignment of the protons in the aromatic region. HSQC data (Figure S4) indicate that the more downfield aromatic peak (7.11 ppm) belongs to a proton that is attached to C3,1 and is thus from product with H that is ortho to the methyl groups (ArHortho); while the more upfield aromatic peak (7.07 ppm) is from the ArHmeta proton. This assignment is also consistent with the HMBC spectroscopic results (also in Figure S4).
Figure S4. 1H-13C HSQC (orange) and HMBC (blue/green) spectra for the reaction mixture of H/D exchange between o-xylene-d10 and HOAc at 0.5 mol% [Pd]. Determination of yields of each regioisomer. According to integration of corresponding peaks for products and internal standard, the apparent yields of ArHortho and ArHmeta and ArHMe are 5%, 9% and 4%, respectively. 1H NMR spectroscopic data for the commercial o-xylene-d10 starting material indicates that it contains 1% of ArHortho, 1% of ArHmeta and 4% of ArHMe. Therefore, the calibrated yields of different H/D exchange products are as follows: 4% for ArHortho, 8% for ArHmeta and 0% for ArHMe. These data indicate that H/D exchange of o-xylene-d10 takes place exclusively at the aromatic positions, with a 1:2 regioselectivity between ortho and meta positions, relative to the methyl group.
*HSQC = heteronuclear single quantum coherence spectroscopy, a cross-peak indicates the two related nucleis are attached. HMBC = heteronuclear multiple-bond correlation spectroscopy, a crosspeak indicates the two related nucleis are 2-4 bonds away.
HSQC/HMBC

S6
V. Derivation of Reaction Rate Laws 1. Rate laws for the "monometallic" mechanism A simplified "monometallic" mechanism is presented in Scheme S1. Reductive elimination is assumed to be fast relative to the two C–H activation steps.
Scheme S1. Stepwise description for the "monometallic" mechanism
Full rate law derivation: Apply steady-state approximation to PdAr, then:
![𝐏𝐝𝐀𝐫]
!" = k1 [PdL2X2][ArH] - k-1 [HX][PdAr] - k2 [PdAr][ArH] = 0 (S1)
Apply steady-state approximation to PdAr2, then:
![𝐏𝐝𝐀𝐫𝟐]
!" = k2 [PdAr][ArH] - k3 [PdAr2] = 0 (S2)
The observed reaction rate for biaryl product formation is:
rate = ![𝐀𝐫𝐀𝐫]
!" = k3 [PdAr2] (S3)
PdL2X2 is the only observable Pd species present under the reaction conditions (i.e., [PdL2X2] ≈ [Pd]). Eqs S1 and S2 were rearranged in terms of [PdAr] and [PdAr2], respectively:
[PdAr] = !! !" [!"#]
!!! !" !!![!"#] (S4)
[PdAr2] = !!!! !" [!"#]!
!!(!!! !" !!![!"#]) (S5)
rate = !!!! [!"] [!"#]!
!!! !" ! !! !"# (S6)
This expression simplifies under two limiting kinetic scenarios: (1) If the 1st C–H activation is rate-limiting step, k-1 << k2, rate law in Eq S6 simplifies to: rate = k1 [Pd][ArH] = k1obs
[Pd] (S7)
(2) If the 2nd C–H activation is rate-limiting step, k-1 >> k2, rate law in Eq S6 simplifies to:

S7
rate = !!!![!"] [!"#]!
!!! !" = k2obs
[Pd] (S8)
Based on eqs S7 and S8, a first-order [Pd] dependence should be expected for the "monometallic" mechanism. 2. Rate laws for the "bimetallic" mechanism A simplified 'bimetallic' mechanism is described in Scheme S2. The first step is C–H activation between PdL2X2 and arene. The next step is transmetalation between two PdAr intermediate to afford PdAr2. The product-forming reductive elimination step is assumed to be fast relative to C–H activation and transmetalation.
Scheme S2. Stepwise description for the "bimetallic" mechanism
Full rate law derivation:
![𝐏𝐝𝐀𝐫]!"
= k1' [PdL2X2][ArH] - k-1' [HX][PdAr] - 2k2' [PdAr]2 = 0 (S9)
![𝐏𝐝𝐀𝐫𝟐]!"
= k2' [PdAr]2 - k3' [PdAr2] = 0 (S10)
rate = ![𝐀𝐫𝐀𝐫]
!" = k3' [PdAr2] (S11)
PdL2X2 is the only observable Pd species present under the reaction conditions (i.e., [PdL2X2] ≈ [Pd]). Eqs S9 and S10 were rearranged in terms of [PdAr] and [PdAr2], respectively:
[PdAr] = !!!! [!"] [!"#]
!!!! !" ! !!!!" !" !!!!!!!!! !" !"# (S12)
[PdAr2] = !!!!"!!! [!"]! [!"#]!
!!! (!!!!" !" !!!!!! !" !!!!" !" !!!!!!!!! !" !"# !!!!!!!! !" !"# ) (S13)
rate = !!!!"!!! [!"]! [!"#]!
!!!!" [!"]!!!!!! [!"] !!!!" [!"]!!!!!!!!! !" [!"#]!!!!!!!! !" [!"#] (S14)

S8
This expression simplifies under two limiting kinetic scenarios: (1) If C–H activation is rate-limiting step, k-1' << k2', the rate law in eq S14 simplifies to:
rate = !!! [!"] [!"#]
! = k1obs' [Pd] (S15)
(2) If transmetalation is rate-limiting step, k-1 >> k2, the rate law in eq S14 simplifies to:
rate = !!!"!!! !!!!"
[!"]! [!"#]! [!"]!
= k2obs' [Pd]2 (S16)
Rate laws (S15) and (S16) are consistent with the observed 2nd/1st order [Pd] dependence.

S9
VI. Discussion of H/D Exchange Experiments. H/D exchange and biaryl product formation arise, respectively, from the protonolysis and forward reaction of the PdAr intermediate (Scheme S3). Scheme S3. Proposed pathways for H/D exchange and product formation in experiments between arene ArY and solvent Y'OAc for the "monometallic" and "bimetallic" mechanisms.
Formation of ArY' (H/D exchange) in Monometallic and Bimetallic Mechanisms: d[ArY']/dt = k-1[PdAr][Y'X] (or k-1'[PdAr][Y'X]) (S17) Formation of ArAr product: "Monometallic" mechanism: d[ArAr]/dt = k2[PdAr][ArY] (S18) "Bimetallic" mechanism: d[ArAr]/dt = k2'[PdAr]2 (S19) The expressions for [PdAr] were derived above for the two different mechanisms (eqs S4 and S12). The modified expressions below (eqs S4' and S12') are identical, but incorporate the more generic use of " Y " and " Y' " to reflect the mixture of H and D in the substrate and solvent:
"Monometallic" mechanism: [PdAr] = !! !" [!"#]
!!! !!! !!![!"#] (S4')
"Bimetallic" mechanism: [PdAr] = !!!! [!"] [!"#]
!!!! !!! ! !!!!" !!! !!!!!!!!! !" !"# (S12')
1. H/D exchange between o-xylene-d10 and HOAc. The H/D exchange between o-xylene-d10 and HOAc reveals that H/D exchange is first-order in [Pd], but product formation is second-order in [Pd] (Figure 3B). The "bimetallic" mechanism rationalizes these results as follows: In this reaction, the steady state concentration of PdAr intermediate is low due to the slow activation of o-xylene-d10 substrate (k1' is small). As a result, transmetalation between two molecules of PdAr is the rate-limiting step over the entire [Pd] range in the experiment. Since k1' is small, in eq S12', 8k1'k2' << (k-1')2 and the equation simplifies to:
[PdAr] = !!! !" [!"#] !!!! !!!
(S20)
The expression for [PdAr] in eq S20 can be substituted into eqs S17 and S19 to obtain rate laws in terms of [Pd]: d[ArY']/dt = k1' [ArY][Pd] (S21)
+ LnPdX2+ Y'X, k-1
- YX, k1LnPdArX Pd0Ln + ArAr
Product Formation
(L = 2Fpy; X = TFA, OAc; Y, Y' = H, D)
Protonolysis
LnPdAr2k2, + ArY
PdAr
-YX
"Monometallic":
"Bimetallic":
- YX, k1'
+ Y'X, k-1'- LnPdX2
LnPdAr2k2'
+ LnPdX2 LnPdArX
+ LnPdX2 LnPdAr'X
ArYPdAr
- YX, k1'
+ Y'X, k-1'
Protonolysis
Pd0Ln + ArAr
Product FormationArY'
ArYArY'
ArY
ArY'

S10
d[ArAr]/dt = !!!"!!! [!"#]!
!!!!" [!!!]![Pd]2 (S22)
Eqs S21 and S22 are consistent with the H/D exchange results in Figure 3B, which show that H/D exchange is first-order in [Pd] and product formation is second-order in [Pd]. Inconsistency with the "monometallic" mechanism. The expression for [PdAr] in eq S4' can be substituted into eqs S17 and S18 to obtain rate laws in terms of [Pd] for the monometallic mechanism:
d[ArY']/dt = !!!!! !" !"# [!!!] !!! !!! !!![!"#]
(S23)
d[ArAr]/dt = !!!! !" !"# !
!!! !!! !!![!"#] (S24)
Eqs S23 and S24 shows that both H/D exchange and product formation exhibit a first-order dependence on [Pd], which is inconsistent with the observed 2nd-order [Pd] dependence for product formation (Figure 3B). 2. H/D Exchange between o-xylene and AcOD. Results. The H/D exchange between o-xylene and HOAc was performed at 0.1 mol% [Pd] under the catalytic conditions. The reaction affords 4.3% of D incorporation into the substrate (both aromatic positions; o- vs. m- selectivity could not be determined) and 0.8% of biaryl product. The D incorporation was characterized by 2H NMR spectroscopy with dichloromethane-d2 as the internal standard (Figure S5). The biaryl product was quantified by GC with octacosane as internal standard.
Figure S5. 2H NMR spectrum of the reaction mixture of H/D exchange between o-xylene and CH3COOD at 0.1% [Pd] with the corresponding peak assignments. Peaks between 2–3 ppm may arise from the residual deuterium in o-xylene starting material and CH3COOD solvent, as suggested by control experiments in the absence of [Pd]. Reaction conditions: CH3COOD (0.4 mL), O2 (1 atm), 80 °C, [o-xylene] = 4.5 M (4 mmol), [Pd] = 4.5 mM, 17 h.
DOAc
ArD
CD2Cl2

S11
This experiment was further carried out at different [Pd], and the results reveal that product formation is first-order in [Pd] whereas H/D exchange shows curvature, indicating less than first-order in [Pd] (Figure S6).
0
2
4
6
8
10
12
14
16
0
1
2
3
4
5
0 5 10 15 20 25
H/D
Exc
hang
e (%
, ArD
)Product Form
ation (%, A
rAr)
[Pd] (mM)
Product Formation
H/D Exchange
Figure S6. H/D exchange experiments at varying [Pd] and the dependences of H/D exchange and product formation on [Pd]. Reaction conditions: CH3COOD (0.4 mL), O2 (1 atm), 80 °C, [o-xylene] = 4.5 M (4 mmol), [Pd] = 1.35–22.5 mM, 17 h. H/D exchange: 2H NMR yield. Product Formation: GC yield. Discussion. In this reaction, protonolysis of the PdAr intermediate in DOAc is comparatively slow (i.e., k-1' is small), and C–H activation is effectively irreversible and rate-limiting. For the bimetallic mechanism, since k-1' is small, (k-1')2 << 8k1'k2' and eq S12' simplifies to the following: [PdAr] = (1/2)1/2(k1'/k2')1/2 [ArY]1/2[Pd]1/2 (S25) This expression for [PdAr] can be substituted into eqs S17 and S19 to obtain rate laws in terms of [Pd]: d[ArY']/dt =(1/2)1/2 k-1' (k1'/k2')1/2 [ArY]1/2[Y'X] [Pd]1/2 (S26) d[ArAr]/dt = (1/2)k1'[ArY] [Pd] (S27) Eqs S26 and S27 account for less-than-first-order dependence on [Pd] for H/D exchange and first-order dependence on [Pd] for product formation (Figure S6).

S12
VII. Discussion of KIE Experiments. The oxidative biaryl coupling reaction with isotopically-varied substrates and solvents were performed at different [Pd] (Figure S2). The results (Figure 4) reveal that when [Pd] increases from 2.7 mM to 67.5 mM, (1) Substrate KIE drops from 25 to 18 when HOAc is the solvent; (2) Substrate KIE drops from 12 to 7.9 when DOAc is the solvent; (3) (1/Solvent KIE) drops from 11 to 3.1 when o-xylene is the substrate; (4) (1/Solvent KIE) drops from 23 to 7 when o-xylene-d10 is the substrate. These results are consistent with the "bimetallic" mechanism and inconsistent with the "monometallic" mechanism, as elaborated below. 1. KIE analysis showing consistency with the "bimetallic" mechanism. The rate law for the "bimetallic" mechanism in eq S14 contains isotopically-sensitive terms k1' and k-1'.
rate = !!!!"!!! [!"]! [!"#]!
!!!!" [!"]!!!!!! [!"] !!!!" [!"]!!!!!!!!! !" [!"#]!!!!!!!! !" [!"#] (S14)
The rate constant k1' corresponds to C–H activation of the arene substrate; k-1' corresponds to protonolysis of the Pd aryl intermediate by the solvent (HOAc or DOAc). Both k1H'/k1D' and k-1H'/k-1D' are expected to have intrinsic values of ~3–5. The substrate and solvent KIEs arise from the following initial rate measurements: Substrate KIE = rate (o-xylene) / rate (o-xylene-d10) (S28) 1/Solvent KIE = rate (DOAc) / rate (HOAc) (S29) KIEs under limiting kinetic scenarios. Scenario A: If C–H activation is rate-limiting step, k-1'2 << k1'k2'[Pd], the rate law in eq S14 simplifies to:
rate = !!! [!"] [!"#]
! (S15)
Reaction with o-xylene as substrate:
rate (o-xylene) = !!!! [!"] [!"#]
! (S30)
Reaction with o-xylene-d10 as substrate:
rate (o-xylene-d10) = !!!! [!"] [!"#]
! (S31)
Since there is no k-1' term in eq S15, rate (DOAc) = rate (HOAc) (S32) In this scenario, eqs S28–S32 will lead to the following isotope effects: Substrate KIE = k1H'/k1D' ~ 3–5. (S33) 1/Solvent KIE = 1. (S34) Scenario B: If transmetalation is rate-limiting step, k-1'2 >> k1'k2'[Pd], the rate law in eq S14 simplifies to:
rate = !!!"!!! !!!!"
[!"]! [!"#]! [!"]!
(S16)

S13
Reaction with o-xylene as substrate:
rate (o-xylene) = !!!!" !!! !!!!"
[!"]! [!"#]! [!"]!
(S35)
Reaction with o-xylene-d10 as substrate:
rate (o-xylene-d10) = !!!!" !!! !!!!"
[!"]! [!"#]! [!"]!
(S36)
Reaction with HOAc as solvent:
rate (HOAc) = !!!"!!! !!!!!"
[!"]! [!"#]! [!"]!
(S37)
Reaction with DOAc as solvent:
rate (DOAc) = !!!"!!! !!!!!"
[!"]! [!"#]! [!"]!
(S38)
In this scenario, eqs S28, S35, and S36 will lead to the following substrate KIE:
Substrate KIE = !!"!" !!!!" =
𝑘1H′
𝑘1D′
!
~ 9–25. (S39)
and eqs S29, S37, and S38 will lead to the following solvent KIE:
Solvent KIE = !!!"!" !!!!!" ~ 0.040–0.11 (S40)
1/Solvent KIE = !!!"!" !!!!!" =
𝑘−1H′
𝑘−1D′
!
~ 9–25 (S41)
The derived KIE values (eqs S33, S34, S39, and S41) are summarized in Table S1.
Table S1. Theoretical KIE values under limiting kinetic scenarios of the "bimetallic" mechanism Scenario A Scenario B
Rate-limiting step C–H activation transmetalation Requirement for rate constants k-1'2 << k1'k2'[Pd] k-1'2 >> k1'k2'[Pd]
Theoretical Substrate KIE 3—5 9—25 Theoretical (1/Solvent KIE) 1 9—25
The actual reaction conditions exhibit kinetic behavior that is between Scenarios A and B. The relative value of k-1'2 and k1'k2'[Pd] will determine the ratio of contribution from Scenario A vs. Scenario B to the reaction. Based on Table S1, the observed KIE values and their change as a function of [Pd], substrate and solvent can be rationalized as follow. Magnitude of KIE values. Table S1 and the analysis above predicts a substrate KIE between 3 and 25, and a (1/Solvent KIE) between 1 and 25. These predictions are consistent with the measured values in Figure 4. Dependence of KIE on [Pd]. When [Pd] increases, the term k1'k2'[Pd] increases and the reaction becomes closer to the rate-limiting C–H activation scenario A, which leads to smaller substrate KIE and (1/Solvent KIE) values. The experimental results show that both Substrate KIE and (1/Solvent KIE) decrease when [Pd] increases. Dependence of Substrate KIE on solvent. The measured KIE in HOAc is larger than in DOAc. This result is due to k-1'2 being larger for HOAc than DOAc, and, therefore, the reaction is closer to Scenario B, which leads to larger

S14
substrate KIE value. Dependence of Solvent KIE on substrate. The measured (1/Solvent KIE) with o-xylene is smaller than with o-xylene-d10. This result is due to that the k1' value is larger for o-xylene than o-xylene-d10. Therefore, for reactions with o-xylene, the k1'k2'[Pd] term is larger and the reaction is closer to Scenario A, which leads to smaller (1/Solvent) value. 2. Inconsistency with the "monometallic" mechanism. The rate law for the "monometallic" mechanism in eq S6 contains isotopically-sensitive terms k1, k-1 and k2.
rate = !!!! [!"] [!"#]!
!!! !" ! !! !"# (S6)
k1 and k2 are determined by the substrate and k-1 is determined by the solvent. The values k1H/k1D, k2H/k2D, and k-1H/k-1D are expected to have intrinsic values of ~ 3–5. The substrate and solvent KIEs arise from the following initial rate measurements: Substrate KIE = rate (o-xylene) / rate (o-xylene-d10) (S28) 1/Solvent KIE = rate (DOAc) / rate (HOAc) (S29) Scenario A: If the first C–H activation is rate-limiting step, k-1 << k2, rate law (eq S6) simplifies to: rate = k1 [Pd][ArH] (S7) Similar to Scenario A in the "bimetallic" mechanism, the following isotope effects are expected: Substrate KIE = 3–5; (S42) 1/Solvent KIE = 1. (S43)
Scenario B: If the second C–H activation is rate-limiting step, k-1 >>k2, rate law (eq S6) simplifies to:
rate = !!!![!"] [!"#]!
!!! !" (S8)
Reaction with o-xylene as substrate:
rate (o-xylene) = !!!!!! !!!
[!"] [!"#]! [!"]
(S44)
Reaction with o-xylene-d10 as substrate:
rate (o-xylene-d10) = !!!!!! !!!
[!"][!"#]! [!"]
(S45)
Reaction with HOAc as solvent:
rate (HOAc) = !!!! !!!!
[!"] [!"#]! [!"]
(S46)
Reaction with DOAc as solvent:
rate (DOAc) = !!!! !!!!
[!"] [!"#]! [!"]
(S47)
In this scenario, eqs S28, S44, and S45 will lead to the following substrate KIE:
Substrate KIE = !!!!!! !!!!!!
=(!!! !!!
)(!!! !!!
) = 9–25 (S48)
and eqs S29, S46, and S47 will lead to the following solvent KIE:

S15
1/Solvent KIE = !!!! !!!!
= 3–5 (S49)
The derived KIE values (eqs S42, S43, S48 and S49) are summarized in Table S2.
Table S2. Theoretical KIE values under limiting kinetic scenarios of the "monometallic" mechanism Scenario A Scenario B
Rate-limiting step 1st C–H activation 2nd C–H activation Requirement for rate constants k-1 << k2 k-1 >> k2
Theoretical Substrate KIE 3—5 9—25 Theoretical (1/Solvent KIE) 1 3—5
The theoretical KIE values of the "monometallic" mechanism are inconsistent with experimental values. Specifically, (1) according to Table S2, (1/Solvent KIE) has a theoretical range of 1–5, which cannot explain a number of very large (1/Solvent KIE) values observed experimentally (e.g., 23, 13, 11 and 8.5; see Figure 4B in the manuscript). (2) according to Table S2, the contribution from each limiting scenario to the behavior of the real reaction is dependent on the relative value between k-1 and k2 and should be independent of [Pd] and k1. Therefore, the observed Substrate KIE and (1/Solvent KIE) values should be independent of [Pd] and the nature of substrate. This is inconsistent with the observed significant change of KIE as a function of [Pd] and the difference in 1/Solvent KIE between o-xylene and o-xylene-d10 as substrate. References. (1) For 13C NMR chemical shift of o-xylene, see: Spectral Database for Organic Compounds (SDBS); 13C NMR spectrum; SDBS No.: 1028; CDS-00-332; http://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi (accessed March 28, 2014).