supplementary material - publish.csiro.au · ammonium molybdate in aqueous sulfuric acid) or...
TRANSCRIPT

10.1071/CH12405_AC CSIRO 2013 Australian Journal of Chemistry 2013, 66(2), 213-217
Supplementary Material
Monoliths for Flow Chemistry: Produced via Visible Light-Initiated
Radical Generation
Farhan R. Bou-Hamdan,a Kathleen Krüger, a Klaus Tauer,a D. Tyler McQuadea,c and
Peter H. Seebergera,b
aMax Planck Institute of Colloids and Interfaces Am Mühlenberg 1, 14476 Potsdam (Germany)
bInstitute of Chemistry and Biochemistry, Freie Universität Berlin Arnimallee 22, 14195
Berlin (Germany)
cDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306 USA.
1.General Information (S2)
2.Procedures and Spectroscopic Data (S4)
3.SEM images and pictures of monoliths (S8)
4.FT-IR Spectra (S17)
5.1H and 13C-NMR Data of All Compounds (S20)

S2
1. General Information
All chemicals were reagent grade and used as supplied except where noted. DCM, 2-Propanol and THF
were HPLC grade. N-methyl-4-aminopyridine was purchased from Alfa Aesar. Irgacure® 819 was
obtained as a gift from BASF, Basel-Switzerland. Hunig’s base, 4,4’-azobis(4-cyanovaleric acid) and
cysteamine hydrochloride was purchased from Sigma Aldrich. Styrene was obtained from Acros
Chemicals and distilled under reduced pressure prior to use. Divinylbenzene was purchased from Alfa
Aesar and passed through a pad of grade III neutral alumina prior to use. FEP tubing (fluorinated ethylene
polymer) was purchased from IDEX Health & Science 1520, natural color, outside diameter (OD) 1/8 in
and inside diameter (ID) 1.55 mm).[1] The term “concentrated under reduced pressure” refers to the
removal of solvents and other volatile material using a rotary evaporator while maintaining a water bath
temperature under 30 °C. The compounds purified over silica gel were further concentrated by the removal
of residual solvent under high vacuum (<0.2 mbar). FEP tubing was selected for its high transmittance and
stability in the UV-vis light range,[2] its flexibility and its high chemical resistance. The temperature in the
polymerization zone during the reaction is estimated to range from 25 to 30 °C, based on temperature
measurements taken around the reaction vial. For safety reasons, the experiments were conducted inside a
fume hood covered with aluminum foil to partially block the intense irradiation of the lamps.
1H NMR spectra were recorded on a Varian 400-MR (400 MHz) spectrometer at ambient temperature. The
proton signal of residual non-deuterated solvent (δ 7.26 ppm for CHCl3) was used as an internal reference
for 1H spectra. Data are reported as follows: chemical shift in parts per million (δ, ppm), multiplicity (s =
singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, qn = quintet and m = multiplet), coupling
constants reported in Hertz (Hz) and integration. 13C spectra were recorded on a Varian VXR-300
spectrometer (at 100 MHz) at ambient temperature. Chemical shifts are reported in parts per million (δ,
ppm). The carbon signal of deuterated solvent (δ 77.16 ppm for CDCl3) was used as an internal reference
for 13C spectra.
Infrared (IR) spectra were recorded as thin films on a Perkin-Elmer 1600 ATR-FTIR spectrophotometer.
GC-MS measurements were performed on a Varian Saturn 2100T GC-MS equipped with a Varian
FactorFour capillary column (Cat. Number: CP8944, VF-5ms, 30 m X 0.25 mm ID, DF 0.25) at the Freie
Universität Berlin, Mass Spectrometry Core Facility. High-resolution mass spectra (HRMS) were recorded

S3
with an Agilent 6210 ESI-TOF mass spectrometer at the Freie Universität Berlin, Mass Spectrometry Core
Facility. 17 W PAR 38 cold white LED lamps, illumination angle 120° were purchased form Conrad (Best-
Nr. 574897 - 62).[3] The intensity spectrum of the LED lamp was recorded with a photonic multichannel
analyzer C10027 (Hamamatsu, Japan). SEM was carried out with a Gemini Leo 1550 microscope. N2
adsorption experiments were conducted at 77K on a Quadrasorb machine from Quantachrome instruments.
Samples were outgassed under reduced pressure overnight at 80 °C. Analytical thin layer chromatography
(TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel.
The TLC plates were visualized with UV light and by staining with Hanessian solution (ceric sulfate and
ammonium molybdate in aqueous sulfuric acid) or anisaldehyde dip. Column chromatography was
perfomed using Kieselgel 60 (230-400 mesh) silica gel.

S4
2. Experimental Procedures and Spectroscopic Data
Preparation of 6-methacrylamidohexanoic acid (1)
HN
OH
O
OH2N
OH
O , EtNiPr2 (2 equiv.)
DCM:MeOH (10:1), rt, 4 h
O
ON
O
O
To a cooled suspension (0 °C) of caproic acid (1.00 g, 7.62 mmol, 1.50 equiv.) in DCM:MeOH (10:1, 55
mL) was added Hunig’s base (1.97 g, 15.25 mmol, 3.0 equiv.) and N-methacryloxysuccinimide (0.93 g,
5.08 mmol, 1.0 equiv.) and the mixture was allowed to warm up to room temperature and stirred for an
additional 4 h. The reaction mixture was then cooled down to 0oC, acidified by the dropwise addition of
H2SO4 (1M, 15 mL) and extracted with DCM (3X). The combined organic phases were washed with H2O,
brine, dried over Na2SO4 and concentrated to afford 6-methacrylamidohexanoic acid (0.55 g, 54%) as a
colorless oil and was used directly in the following step without any purification. 1H NMR (400 MHz,
CDCl3) 1H NMR (400 MHz, cdcl3) δ 5.84 (bs, 1H), 5.69 – 5.64 (dq, J = 1.0, 1.0 Hz, 1H), 5.34 – 5.31 (dq, J
= 1.5, 1.0 Hz, 1H), 3.32 (td, J = 7.1, 6.0 Hz, 2H), 2.37 (t, J = 7.3 Hz, 2H), 1.96 (dd, J = 1.5, 1.0 Hz, 3H),
1.72 – 1.62 (m, 2H), 1.62 – 1.52 (m, 2H), 1.45 – 1.35 (m, 2H).
Preparation of 2,5-dioxopyrrolidin-1-yl 6-methacrylamidohexanoate (1)
HN
OH
O
O
N-hydroxysuccinimide (1,25 equiv.)
DCC (1.5 equiv.), DCM, rt, 14 h1
HN
O
O
O
N
O
O
To a cooled solution (0 °C) of crude 6-methacrylamidohexanoic acid (0.55 g, 2.74 mmol, 1.00 equiv.) and
N-hydroxysuccinimide (0.40 g, 3.43 mmol, 1.25 equiv.) in DCM (0.1 M, 28 mL) was added DCC (0.85 g,
4.11 mmol, 1.5 equiv.) and the mixture was allowed to warm up to room temperature and stirred at room
temperature for 14 h. The reaction mixture was filtered and concentrated. The residue was dissolved in
minimal DCM and filtered through a cotton pad to remove the residual dicyclohexyl urea product and the
process was repeated a second time. The oily residue was purified by column chromatography (5% then

S5
15% then 25% acetone in DCM) to afford (1) (0.72 g, 89%) as a colorless oil which solidified upon
standing in the freezer. 1H NMR (400 MHz, CDCl3) 1H NMR (400 MHz, cdcl3) δ 5.91 (bs, 1H), 5.71 –
5.64 (dq, J = 1.0, 1.0 Hz, 1H), 5.31 (dq, J = 1.5, 1.0 Hz, 1H), 3.33 (dt, J = 7.0, 6.0 Hz, 2H), 2.84 (d, J = 3.0
Hz, 4H), 2.63 (t, J = 7.2 Hz, 2H), 1.96 (dd, J = 1.5, 1.0 Hz, 3H), 1.79 (dt, J = 14.8, 7.3 Hz, 2H), 1.64 – 1.55
(m, 2H), 1.52 – 1.42 (m, 2H). 13C NMR (100 MHz, cdcl3) δ 169.3, 168.6, 168.6, 140.3, 119.4, 39.4, 31.0,
29.1, 26.0, 25.7, 24.4, 18.9; IR-thin film (ν, cm-1) 3352 (b), 3004, 2942, 2964, 1814, 1780, 1729, 1655,
1607, 1532, 1460, 1433, 1363, 1202, 1073, 1059, 927, 872, 820; HRMS–ESI: M+Na, calc. 319.1270,
meas. 319.1278.
Preparation of N-allyl-N-methyl-4-aminopyridine
a) BuLi (1.1 equiv.), THF, 0 °C, 1 h
b) allylBr (1.2 equiv.), THF, 0 °C, 30 minN
NH
N
N
To a cooled solution (0 °C) of N-methyl-4-aminopyridine (2.0 g, 18.49 mmol, 1.00 equiv.) in THF (0.1 M,
28.0 mL) was added a solution of n-BuLi in hexanes (1.60 M, 12.7 mL, 20.34 mmol, 1.10 equiv.) dropwise
and the mixture was stirred at 0 °C for 1 h. Allyl bromide (2.7 g, 22.19 mmol, 1.20 equiv.) was then added
and the reaction mixture was stirred for an additional 30 min. Water (50 mL) was added and mixture was
extracted with EtOAc (3X). The combined organic phases were washed with brine, dried over Na2SO4 and
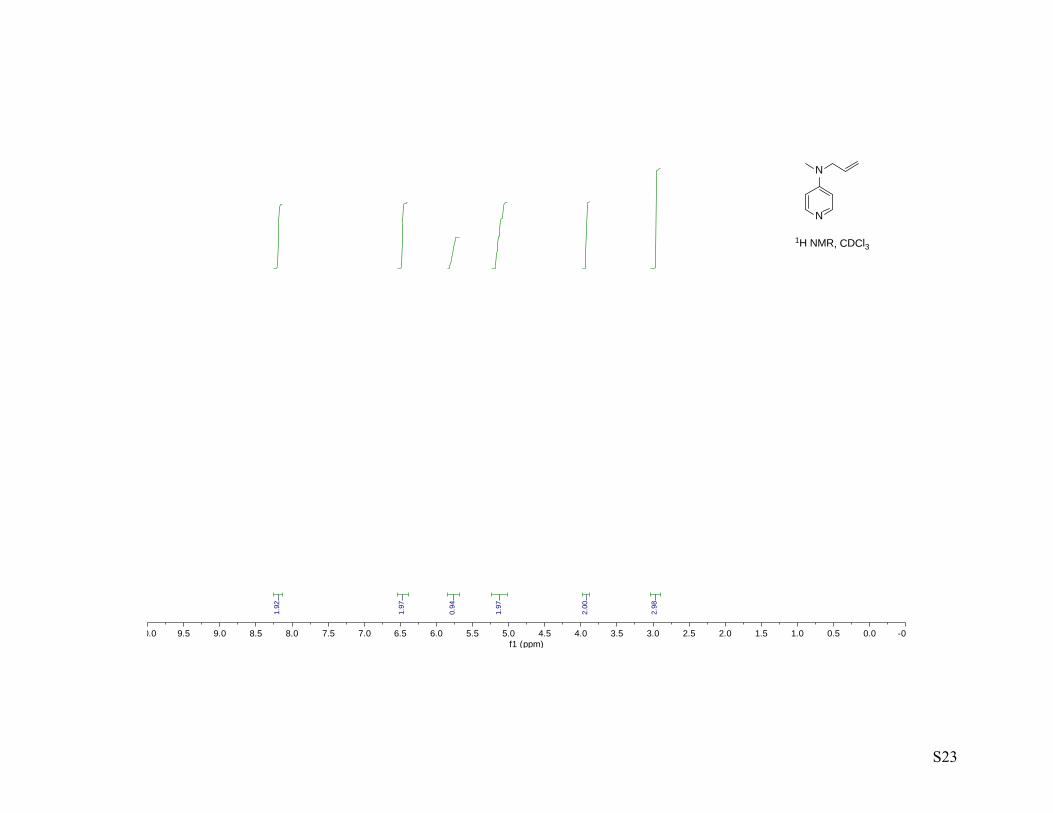
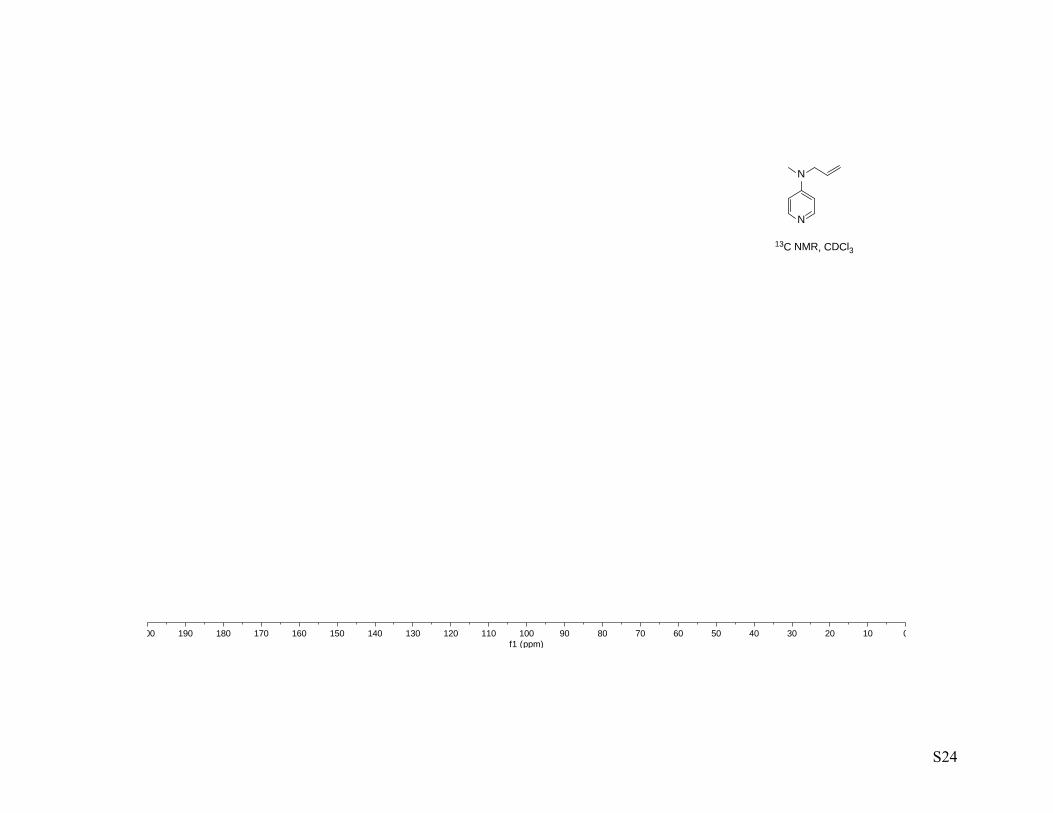
concentrated to afford N-allyl-N-methyl-4-aminopyridine (2.50 g, 91%) as a light yellow oil. 1H NMR (400
MHz, CDCl3) 1H NMR (400 MHz, cdcl3) δ 8.21 – 8.16 (m, 2H), 6.50 – 6.44 (m, 2H), 5.83 – 5.72 (m, 1H),
5.16 (ddt, J = 10.3, 1.5, 1.5 Hz, 1H), 5.10 (ddt, J = 17.2, 1.6, 1.6 Hz, 1H), 3.92 (ddd, J = 4.8, 3.2, 1.6 Hz,
2H), 2.96 (d, J = 1.6 Hz, 3H); 13C NMR (100 MHz, cdcl3) δ 153.7, 150.0, 132.1, 116.7, 106.8, 53.8, 37.3;
IR-thin film (ν, cm-1) 3644 – 2750 (b), 3086, 3033, 3006. 2984, 2910, 1644, 1600, 1541, 1515, 1455, 1435,
1386, 1354, 1228, 1213, 1123, 989, 921, 801; HRMS–ESI: M+H, calc. 149.1079, meas. 149.1074.
Preparation of N-allyl-N-methyl-4-aminopyridinium p-toluenesulfonate
p-TsOH•H2O (1.0 equiv.)
EtOAc, 0 °CN
N
N
N
SO3H
•

S6
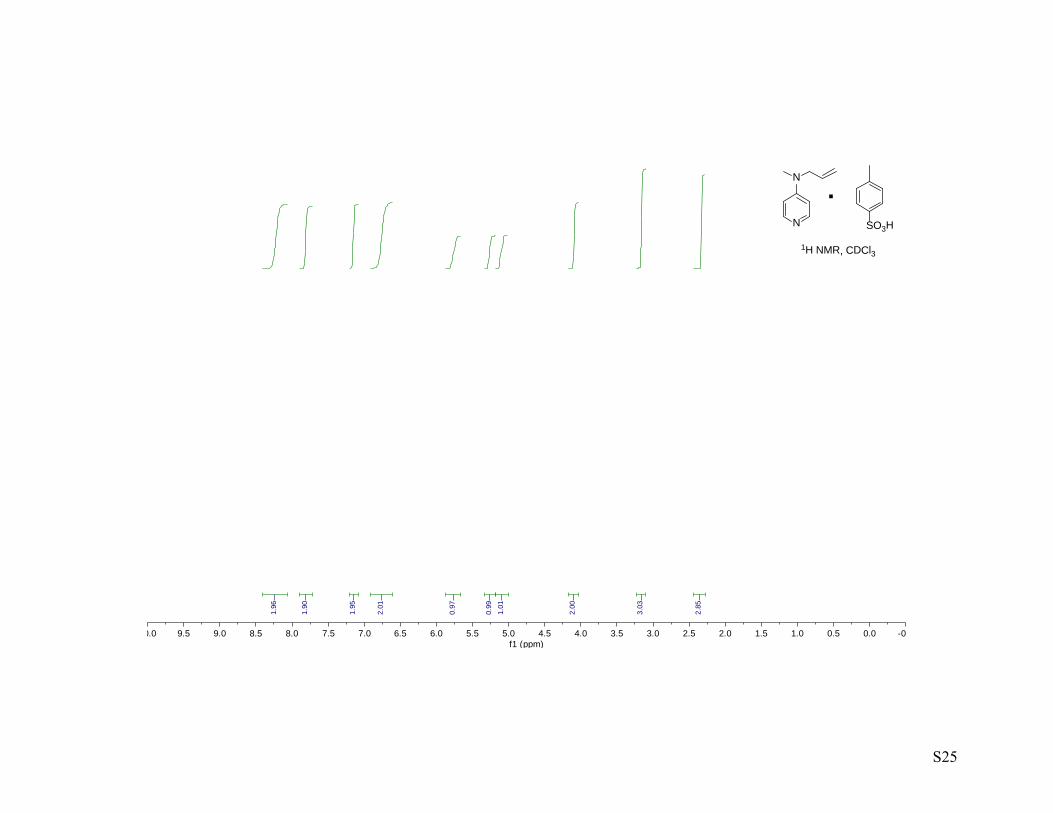
To a cooled solution (0 °C) of N-methyl- N-allyl-4-aminopyridine (1.33 g, 8.97 mmol, 1.00 equiv.) in
EtOAc (15.0 mL) was added a solution of p-TsOH in EtOAc (35.0 mL, 8.97 mmol, 1.00 equiv.) dropwise
and the mixture was stirred at 0 °C for 5 min. The product was collected by filtration and washed with
Et2O to afford N-allyl-N-methyl-4-aminopyridinium p-toluenesulfonate (2.41 g, 84%) as a browm solid. 1H NMR (400 MHz, CDCl3) 1H NMR (400 MHz, cdcl3) δ 8.24 (bs, 2H), 7.86 – 7.79 (m, 2H), 7.16 (m,
2H), 6.76 (bs, 2H), 5.78 (ddt, J = 17.2, 10.3, 4.8 Hz, 1H), 5.28 (ddd, J = 10.4, 1.6, 0.9 Hz, 1H), 5.11 (ddd, J
= 10.4, 1.8, 1.2 Hz, 1H), 4.10 (dt, J = 4.7, 1.8 Hz, 2H), 3.17 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz,
cdcl3) δ 157.4, 142.8, 140.0, 139.9, 129.4, 128.9, 126.2, 118.3, 107.2, 54.8, 38.7, 21.4.
Preparation of N-(3-((2-aminoethyl)thio)propyl)-N-methyl-4-aminopyridine (2)
HSCH2CH2NH2 • HCl (7.0 equiv.)
4,4'-azobis(4-cyanovaleric acid) (0.2 equiv.)H2O, 80 °C, 18 h N
N
N
N
SO3H
•S
NH2
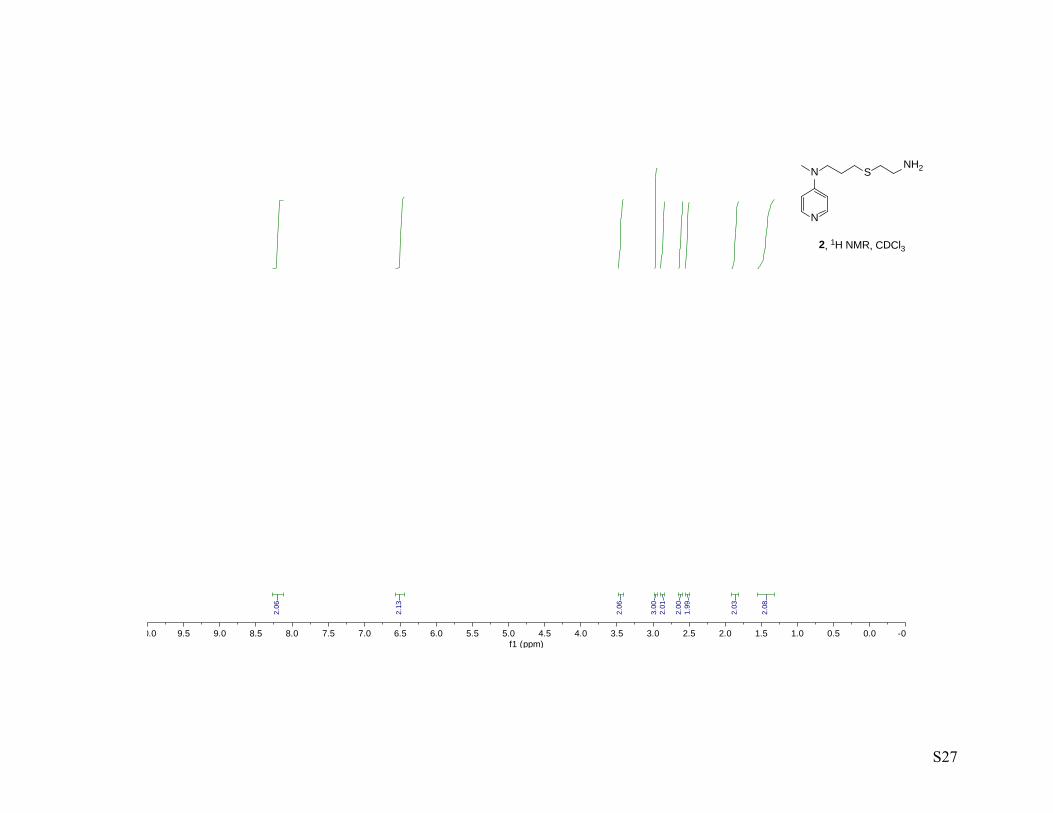
2 N-allyl-N-methyl-4-aminopyridinium p-toluenesulfonate (1.0 g, 3.15 mmol, 1.00 equiv.), cysteamine
hydrochloride (2.5 g, 22.07 mmol, 7.00 equiv.) and 4,4’-azobis(4-cyanovaleric acid) (0.177 g, 0.63 mmol,
0.20 equiv.) were dissolved in degassed water under an atmosphere of argon and the mixture was heated at
80 °C for 18 h. The reaction mixture was then cooled down to 0oC, basified by the dropwise addition of
NaOH (20% w/w, 10 mL) and extracted with EtOAc (4X). The combined organic phases were washed
with NaOH, H2O, brine, dried over Na2SO4 and concentrated to afford N-(3-((2-aminoethyl)thio)propyl)-
N-methyl-4-aminopyridine (2) (0.60 g, 84%) as a light yellow oil. 1H NMR (400 MHz, CDCl3) 1H NMR
(400 MHz, cdcl3) δ8.22 – 8.17 (m, 2H), 6.52 – 6.47 (m, 2H), 3.45 (t, J = 7.3 Hz, 2H), 2.96 (s, 3H), 2.87 (t,
J = 6.4 Hz, 2H), 2.61 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 6.9 Hz, 2H), 1.91 – 1.81 (m, 2H), 1.43 (bs, 2H); 13C
NMR (100 MHz, cdcl3) δ 153.4, 150.1, 106.6, 50.1, 41.2, 37.7, 36.6, 29.2, 26.7; IR-thin film (ν, cm-1) 3352
(b), 3361 (b), 3091, 3038, 2997, 2918, 2861, 1659, 1596, 1540, 1519, 1465, 1437, 1391, 1369, 1279, 1228,
1107, 992, 803; HRMS–ESI: M+H, calc. 226.1378, meas. 226.1380.

S7
General procedure for the preparation of the monolith in glass vial
A solution of styrene (60 mol%, 28% v/v), divinylbenzene (40 mol%, 22% v/v), Irgacure® 819 (1 mol%)
and porogen (50% v/v) was loaded into a 15.0 or a 6.6 mm glass vial, and the container was placed
between two LED lamps, placed 20 cm apart, and irradiated for 5 h. The glass vial was removed and the
monolithic structure was extensively washed with MeOH to remove the porogen and any residual non-
polymeric material, resulting in a rigid white monolith. Physical properties of the monoliths were evaluated
by visual and qualitative mechanical inspection, SEM and resistance to flow (back-pressure
measurements).
General procedure for the preparation of the monolith (Omnifit column or FEP tuning)
A solution of styrene (60 mol%, 28% v/v), divinylbenzene (40 mol%, 22% v/v), Irgacure® 819 (1 mol%)
and porogen (50% v/v) was loaded into a 1.55 mm FEP tubing or 10.0 X 6.6 mm Omnifit glass column,
sealed at both ends, and the container was placed between two LED lamps, placed 20 cm apart, and
irradiated for 5 h. The column endings were then replaced with a set of fritted end pieces and extensively
washed with DCM (R2+ unit set to a flow rate of 1.0 mL min−1 for 2 h) to remove the porogen and any
residual non-polymeric material, resulting in a rigid white monolith that completely filled the glass column.
Physical properties of the monoliths were evaluated by visual and qualitative mechanical inspection, SEM
and resistance to flow (back-pressure measurements).

S8
A. 1-Dodecanol, 15.0 mm glass vial, 2 X 17 W LED lamps:
Picture
SEM images

S9
B. 1-Dodecanol:THF, 15.0 mm glass vial, 2 X 17 W LED lamps:
SEM images

S10
BET measurements: Surface area = 197 m2g-1
0 20 40 60 80 100 120
0,000
0,002
0,004
0,006
0,008
0,010
0,012
0,014
0,016
0,018
dV(r
) (cc
/A/g
)
Half pore width (A)

S11
C. THF, 15.0 cm glass vial, 2 X 17 W LED lamps:
SEM images

S12
D. i-Propanol, 6.6 mm glass vial, 2 X 17 W LED lamps:
SEM images

S13
E. 1-Dodecanol, 1.55 mm FEP tubing, 2 X 17 W LED lamps:
SEM images

S14
F. 1-Dodecanol, 1.55 mm FEP tubing, Hannovia 450 W medium pressure Hg lamp:
G. 1-Dodecanol, 6.6 mm glass vial, two standard fluorescent tubes (Osram L 18W, light
color 840, lumilux, cool white):
Procedure for the preparation of the DMAP-functionalized monolith in an Omnifit column
A solution of styrene (50 mol%, 20% v/v), divinylbenzene (40 mol%, 20% v/v), N-hydroxysuccinimidyl
ester 1 (10 mol%), Irgacure® 819 (1 mol%) and i-PrOH (50% v/v) was loaded into a 10.0 X 6.6 mm
Omnifit glass column, sealed at both ends, and the container was placed between two LED lamps, placed
20 cm apart, and irradiated for 5 h. The column endings were then replaced with a set of fritted end pieces
and extensively washed with DCM (R2+ unit set to a flow rate of 1.0 mL min−1 for 2 h) to remove the
porogen and any residual non-polymeric material, resulting in a rigid white monolith that completely filled
the glass column. The NHS-monolith was then treated with a solution of 2 (3 equiv. relative to NHS) and
Hunig’s base (3 equiv.) in DCM at a flow rate of 500 µL/min for 16 hours at room temperature using a
closed loop to minimize the amount of 2 needed to maximally functionalize the column. The monolith was
then washed with DCM for 2 hours at a flow rate of 1 mL/min and directly used in acylation studies.

S15
General procedure for the continuous flow acylation of 1-phenylethanol.
A Vapourtec R series flow reactor system[4] was set up. With the pump set to flow at the required flow rate,
a solution of 1-phenylethanol (0.5 mmol, 1 equiv.), Ac2O (0.75 mmol, 70 µL, 1.5 equiv.), DIPEA (0.75
mmol, 130 µL, 1.5 equiv.) in DCM (0.25 M) was loaded into the reactor via a 2 mL injection loop. The
system was configured to collect 5 mL of the product solution in a single vessel containing MeOH (2.5
mmol, 100 µL, 5 equiv.). An aliquot of the product solution was diluted 100-fold and analyzed by GC-MS.
The concentrations of 1-phenylethanol and 1-phenethylacetate were calculated using the GC calibration
curves shown below. Background acylation is below detection limits.
GC-MS calibration curves
A. 1-Phenylethanol: retention time = 6.60 min, Area = 3.06 * 108 (Concentration), R2 = 0.994
B. 1- -phenethylacetate: retention time = 7.64 min, Area = 1.37 * 108 (Concentration), R2 =
0.998
0,0000 0,0005 0,0010 0,0015 0,00200
50000100000150000200000250000300000350000400000450000500000550000600000650000700000
1-Phenylethanol 1-Phenethylacetate
Area
Concentration (M)

S16
Catalysis Results
Run Flow Rate
(mL/min)
Residence
Time (min)
5 mol% DMAP 10 mol% DMAP Calculated Conversion (%)
Area SM Area Product Area SM Area Product 5 mol% 10 mol%
1 2.4 0.5 154473 56259 111135 89806 14 27 2 1.2 1 144968 99961 77605 135843 24 44 3 0.6 2 94630 148585 46371 182172 41 64 4 0.3 4 57546 147725 22042 255783 53 84 5 0.15 8 37243 212843 1000 327442 72 99
0 1 2 3 4 5 6 7 8 90
10
20
30
40
50
60
70
80
90
100
(5 mol%) (10 mol%)
Conv
ersio
n (%
)
Residence Time (min)

S17
1. IR Spectra
4000 3500 3000 2500 2000 1500 100080
85
90
95
Tran
smitta
nce (
%)
Wavenumber (cm-1)
IR Spectrum of the Unfunctionalized PS Monolith

S18
4000 3500 3000 2500 2000 1500 100090
92
94
96
98Tr
ansm
ittan
ce (%
)
Wavenumber (cm-1)
IR Spectrum of the NHS-functionalized PS Monolith

S19
4000 3500 3000 2500 2000 1500 100086
88
90
92
94
Tran
smitt
ance
(%)
Wavenumber (cm-1)
IR Spectrum of the DMAP-functionalized PS Monolith

S20
2. 1H and 13C-NMR Data
-00.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.50.0f1 (ppm)
2.19
4.19
3.08
2.00
1.96
0.95
0.97
0.91
1H NMR, CDCl3
HN
OH
O
O

S21
-00.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.50.0f1 (ppm)
2.06
3.16
2.07
3.02
2.00
4.12
1.97
1.05
0.98
0.89
1, 1H NMR, CDCl3
HN
O
O
O
N
O
O

S22
010203040506070809010011012013014015016017018019000f1 (ppm)
1, 13C NMR, CDCl3
HN
O
O
O
N
O
O
S23
-00.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.50.0f1 (ppm)
2.98
2.00
1.97
0.94
1.97
1.92
1H NMR, CDCl3
N
N
S24
010203040506070809010011012013014015016017018019000f1 (ppm)
13C NMR, CDCl3
N
N
S25
-00.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.50.0f1 (ppm)
2.85
3.03
2.00
1.01
0.99
0.97
2.01
1.95
1.90
1.96
1H NMR, CDCl3
N
N
·SO3H

S26
010203040506070809010011012013014015016017018019000f1 (ppm)
13C NMR, CDCl3
N
N
·SO3H
S27
-00.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.50.0f1 (ppm)
2.08
2.03
1.99
2.00
2.01
3.00
2.06
2.13
2.06
2, 1H NMR, CDCl3
N
N SNH2

S28
010203040506070809010011012013014015016017018019000f1 (ppm)
2, 13C NMR, CDCl3
N
N SNH2

S29
[1] http://www.idex-hs.com
[2] (a) A. M. S. Galante, O. L. Galante and L. L. Campos, Nucl. Instrum. Meth. A, 2010, 619, 177; (b) B. D. A. Hook, W. Dohle, P. R.
Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558.
[3] http://www.conrad.de
[4] http://www.vapourtec.co.uk/