supplemental information selective chemical modulation of gene
TRANSCRIPT

Chemistry & Biology, Volume 21
Supplemental Information
Selective Chemical Modulation of Gene Transcription
Favors Oligodendrocyte Lineage Progression
Mar Gacias, Guillermo Gerona-Navarro, Alexander N. Plotnikov, Guangtao Zhang, Lei Zeng, Jasbir Kaur, Gregory Moy, Elena Rusinova, Yoel Rodriguez, Bridget Matikainen, Adam Vincek, Jennifer Joshua, Patrizia Casaccia, and Ming-Ming Zhou

2
Sample Preparation Expression and purification of the recombinant bromodomains of various transcriptional proteins in poly-His tag form were performed using a procedure described previously (Zeng et al., 2008). The protein was purified by affinity chromatography on a nickel-IDA column (Invitrogen), followed by the removal of poly-His tag by thrombin cleavage. Fluorescence Anisotropy Binding Assay Binding affinity of small molecule ligands to various the BrDs of BET proteins and CBP was assessed in a fluorescence anisotropy competition assay using a FITC-labeled MS417 as a probe and an established procedure as described previously (Zhang et al., 2012). Typically, competition assays were carried out with a protein (0.25-1 µM) and the fluorescent probe (80 nM), and increasing concentration of a competing ligand in a sodium phosphate (150 µM) buffer (pH 7.4) in 80 µL. Detection was done after a 1 hour incubation of the fluorescent probe and the protein at room temperature with Safire 2 microplate reader (Tecan). In a competition-binding assay, fluorescent ligand concentration was ≤ 2Kd, and protein concentration was set at which 50-80% of fluorescent ligand is bound. Dissociation constant of a competing ligand was calculated with the correction to Cheng-Prussoff equation introduced by Nicolovska-Coleska and colleagues (Nikolovska-Coleska et al., 2004). Assuming one-site competitive binding model, the equation used to calculate Ki’s from IC50 values recovered from fitting data using Prism:
Ki =I50[ ]
L50[ ]Kd
+P0[ ]Kd
+1!
"#
$
%&
,
where [I50] is the concentration of free inhibitor at 50% inhibition, [L50], the concentration of free labeled ligand at 50% inhibition, and [P0], concentration of free protein at 0% inhibition. Note that Kd for each protein-probe pair is the limit of resolvable Ki in a competition assay. Cell Culture For cell culture experiments, Olineu cell line derived from mouse oligodendrocyte progenitors immortalized with the Neu antigen (kindly provided by Dr. J. Trotter, University of Mainz, Mainz, Germany) were grown at 37°C and 5% CO2 on Poly-D-Lysisne (PDL) coated plates and maintained proliferating in growth medium in ODM medium plus 1% Horse serum as previously described (He et al., 2007). Oli-Neu cells were induced to differentiation in Sato medium (Watkins et al., 2008) T3 at a final concentration of 60 nM (Sigma, T5516). Plasmid Vectors Eukaryotic expression vectors encoding wtBrd2 (pEGFP-C1-Brd2) and mutBrd2 (pEGFP-C1-Brd2 mut) were generous gifts from Dr. D Stanek, Institute of Molecular Genetics of Prague (Hnilicova et al., 2013). RNA Extraction and qPCR Analysis RNA was extracted using Trizol (Invitrogen, #15596-018) and purified with the RNeasy Micro kit (Qiagen, #74004) following the manufacturer’s protocol. 0.5 mg of RNA were reverse transcribed with qScript cDNA Supermix (Quanta, #95048) and qRT-PCR was performed using Perfecta Sybr Fast Mix Rox 1250 (Quanta, #101414-278) at the Mount Sinai Shared Resource Facility (primers listed in Supplemental Table 2). Data were normalized to the internal controls Gapdh or 18S and analyzed using Pfaffl ΔΔCt method. One-Way ANOVA with Dunnett’s multiple comparison test was performed to assess statistical differences between control (DMSO treated) and BrDi treated conditions, with a significance threshold of P<0.05.

3
MTT Assay Cell viability based in mitochondrial respiration was measured with a colorimetric viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Molecular Probes, Eugene, OR). Cells (10000 cells per well of a PDL-coated 96-well plate) were allowed to attach for 24 hours. Stock solutions of compounds (1000 fold concentrated) were prepared in DMSO and then diluted in differentiation media (T3 45 nm, Sigma, T5516) to give a range of final tissue culture concentrations of 0.1 mM to 10 µM for Olinone and 0.01 mM to 0.5 mM for MS417. After 72 h of BrDi treatment, cell viability was examined. Pre-warmed MTT reagent was added to each well (1/10th the volume) to a final concentration of 0.5 mg/mL followed by 5 hours of incubation at 37°C. Following a PBS wash, crystals were solubilized with DMSO and the absorbance of cell supernatants was measured at 540 nm with a background subtraction at 655 nm using a spectrophotometer. Mitochondrial respiration was normalized to that of untreated cells and expressed as percentage of control. One-Way ANOVA with Dunnett’s multiple comparison test was performed to assess statistical differences between control and BrdI treated cells, P<0.05 was considered statistically significant. Chemicals and General Procedures Commercially available reagents and solvents were obtained from Aldrich Chemical Co. (Milwaukee, WI), Fluka Chemical Corp. (Milwaukee, WI), TCI America (Portland, Oregon), Ark Pharm (Livertyville, IL) and Acros Organics USA (Morris Plains, NJ). They were used without any further purification. Reactions were monitored by analytical thin-layer chromatography (TLC) and LC/MS. TLC analysis was performed using Merck silica gel 60 F254 plates. LC/MS analysis was carried out on an Agilent 1100 Series HPLC equipped with a ZORBAX Eclipse XDB-18 analytical column from Agilent (4.6 x 150 mm, 5 µm) and attached to a TOF mass detector equipped with an electrospray ionization source (ESI). A gradient method using H2O/0.1% formic acid (Solvent A) and Acetonitirle/0.1% formic acid (Solvent B) as eluent solvent was implemented with a flow rate of 0.4 mL/min, column temperature at 30oC, UV detection at 210 nm, 254 nm and 280 nm. The gradient method was run in 7 minutes with Solvent A from 90% to 1% and Solvent B from 1% to 99%. Purification was performed using a SP1 purification system (Biotage) with prepacked FLASH silica columns. Chemical Synthetic Procedures The synthesis of the designed compounds was achieved in 4 synthetic steps (Figure S1). Phenylhydrazone 1 was prepared by reaction of phenylhydrazine with commercially available 2,4-piperidinedione in AcOH (10%) under nitrogen atmosphere. The pyrido-indole scaffold was then constructed following the Fisher indole synthesis, by treatment of phenylhydrazone 1 with sulfuric acid (70%) (Rodriguez and Temprano, 1989). The use of other acids such as formic acid, acetic acid, hydrochloric acid or trifluoroacetic acid, which were successfully applied in the Fisher indole synthesis of different substituted indoles, did not produce the expected pyrido-indole ring 2 under different reaction conditions (Barbieri and Grazia, 2006; Gribble, 1994). Next, the 2,3,4,5-tetrahydro-1H-pyrido-[4,3-b]indol-1-one 2 was N-alkylated using lithium bis(trimethylsilyl)amide as a base (Coldham et al., 2007). Other bases such as KOH, NaH or BuLi also led to the correspondent N-alkylated products but with lower yields (Fukuda and Maeda, 1999; Miyamoto et al., 2007). Finally, treatment of N-tert-butoxycarbonyl substituted pyrido-indoles with trifluoroacetic acid and subsequent acetylation with acetyl chloride/propylene oxide afforded the final compounds 4a-d with good yields.

4
5,6-Dihydro-4-(2-phenylhydrazino)-2(1H)pyridinone (1) Phenylhydrazine (0.880 mL, 8.84 mmol) was added over 5 minutes to a stirred solution of 2,4-piperidinedione (1g, 8.84 mmol) in 10 mL of ethanol under nitrogen atmosphere. After 1h of stirring at room temperature, the resulting suspension was filtered and the solid was then washed with cold water and diethyl ether, to afford 1.60g (82%) of the title compound. 1H NMR (900 MHz, DMSO) δ = 3.16 (m, 2H); 3.25 (m, 2H); 3.33 (m, 2H); 6.71 (bs, 1H, H2); 7.07 (m, 3H); 8.80 (d, 1H, J= 26 Hz). 13C NMR (225 MHz, DMSO) δ= 171.0, 146.8, 142.8, 129.1, 118.8, 112.7, 41.9, 37.2, 26.5. (MS-ESI): m/z calculated for C11H13N3O [M+H]+: 204.11, found 204.11
2,3,4,5-tetrahydro-1H-pyrido-[4,3-b]indol-1-one (2) A solution of 5,6-Dihydro-4-(2-phenylhydrazino)-2(1H)pyridine (1g, 4.93 mmol) was added portionwise to a ice-cold mixture of sulfuric acid (3.5 mL) and water (1.5mL). The reaction progress was monitored by TLC (∼ 3h), then neutralized with ice-cold sodium hydroxide (20mL, 2M) and extracted with EtOAc. The organic phases were combined, washed with brine, dried over sodium sulfate, filtered and evaporated to dryness. Purification by column chromatography, eluting with CH2Cl2/MeOH (15:1)
gave 0.65g (65%) of the indole 2 as a solid. 1H NMR (900 MHz, DMSO) δ = 2.97 (t, 2H, H3, J3-4= 6.6 Hz); 3.47 (t, 2H, H4, J4-3= 6.6 Hz); 7.05 (bs, 1H, H2); 7.11 (m, 2H, H7, H8); 7.40 (d, 1H, H9, J9-
8= 7.2 Hz); 7.89 (d, 1H, H6, J6-7= 7.5 Hz); 11.7 (bs, 1H, H5). 13C NMR (225 MHz, DMSO) δ= 166.3, 144.8, 136.2, 125.7, 121.9, 121.0, 119.9, 111.9, 105.7, 40.4, 22.9. (MS-ESI): m/z calculated for C11H10N2O [M+H]+: 187.10, found 187.08. General procedure for the preparation of N-alkylated-pyridoindoles (3) NaHMDS (0.8 mL, 0.8 mmol of a 1.0 M solution in THF) was added to a solution of 2,3,4,5-tetrahydro-1H-pyrido-[4,3-b]indol-1-one (0.1g, 0.533 mmol) in DMF (5 mL) at -78oC under nitrogen. After 30 min stirring at -78oC, a solution of the correspondent alkylating agent (0.8 mmol) in DMF (1 mL) was added dropwise and the mixture was warmed at room temperature and then heated at 90oC for 12h. Saturated aqueous NaHCO3 was then added to the reaction mixture and the resulting suspension was extracted with EtOAc three times. The combined organic layers were washed with brine and dried with Na2SO4. Filtration and concentration in vacuum afforded the correspondent N-alkyl-pyridoindole (3), which was purified by column chromatography 2,3,4,5-tetrahydro-5-(3’-phthalimidopropyl)- 1H-pyrido-[4,3-b]indol-1-one (208-NPhth)
From compound 2 (0.25g, 1.34 mmol) Alkylating reagent: N-(3-Bromopropyl)phthalimide (0.54 g, 2.01 mmol, 1.5 eq). Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.39g (77%) of the indole 3a as oil. (MS-ESI): m/z calculated for C22H19N3O3 [M+H]+: 374.14, found 374.14.
NH
NH
O
13
4
67
8
9
NH
O
N
NH
34
6
N
NH
O
NO
O

5
2,3,4,5-tetrahydro-5-(4’-phthalimidobutyl)- 1H-pyrido-[4,3-b]indol-1-one (308-NPhth) From compound 2 (0.25g, 1.34 mmol) Alkylating reagent: N-(4-Bromopropyl)phthalimide (0.57 g, 2.01 mmol, 1.5 eq). Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.40g (77%) of the indole 3b as foam. (MS-ESI): m/z calculated for C23H21N3O3 [M+H]+: 388.16, found 388.21.
2,3,4,5-tetrahydro-5-(6’-phthalimidopentyl)- 1H-pyrido-[4,3-b]indol-1-one (408-NPhth)
From compound 2 (0.25g, 1.34 mmol) Alkylating reagent: N-(5-Bromopentyl)phthalimide (0.59 g, 2.01 mmol, 1.5 eq). Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.39g (73%) of the indole 3c as foam.
2,3,4,5-tetrahydro-5-(6’-phthalimidohexyl)- 1H-pyrido-[4,3-b]indol-1-one (508-NPhth) From compound 2 (0.25g, 1.34 mmol) Alkylating reagent: N-(6-Bromohexyl)phthalimide (0.62 g, 2.01 mmol, 1.5 eq). Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.37g (66%) of the indole 3d as a foam.
Deprotection of the Phthalimide Group from the N-Alkylated-Pyridoindoles and Subsequent Acetylation of the Free Amino Group. (1) Deprotection - 0.35g (8.32 mmol) of the pyridoindol were dissolved in 25 mL of acetonitrile. Next, 0.5 mL of a solution of hydrazine hydrate 51% in water were added and the solution was refluxed at 80oC for 1h. The reaction was followed by LC/MS analysis until the total deprotection of the phthalimide group (if needed, an extra 5 eq of hydrazine hydrate are added after 1h to
N
NH
NO
O
O
N
NH
O
NHO
N
NH
O
NO
O

6
complete the deprotection). The deprotected indol is isolated by either filtration, or precipitation as a hydrochloride salt (after addition of HCl to the reaction solution), and used in the following step without further purification. (2) Acetylation - The compound obtained in the previous step was dissolved in CH3CN/MeOH (50:50). Next, pyridine (1.1 eq) and acetic anhydride (1.1 eq) were added to the solution and the mixture was reacted under microwave irradiation for 5 min at 80oC. Next, the solvent was evaporated to dryness. The resulting oil was finally purified by column chromatography. 2,3,4,5-tetrahydro-5-(3’-acetamidopropyl)-1H-pyrido-[4,3-b]indol-1-one (4a)
From 3a (0.35g, 0.93 mmol) Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.165 g (72%) of the acetyl-substituted indole 4a as a solid.1H NMR (900 MHz, DMSO)δ = 1.86 (s, 3H, CH3), 2.08 (m, 2H, H2’), 2.99 (m, 2H, H3’), 3.08 (t, 2H, H3, J3-4= 6.2 Hz); 3.48 (t, 2H, H4, J4-3= 6.2 Hz); 4.19 (t, 2H, H1’, J1’-2’ = 5.1 Hz), 6.73 (bs, 1H, NH); 7.14 (m, 2H, H7, H8); 7.54 (d, 1H, H9, J9-8= 7.2 Hz); 7.90 (d, 1H, H6, J6-7= 7.3 Hz). 7.99 (bs, 1H, NH). 13C NMR (225 MHz, DMSO) δ= 169.4, 166.0, 145.5, 136.6, 125.5, 122.0, 121.3, 120.2, 110.5, 105.4, 43.1, 40.1, 39.9, 28.6, 27.2, 21.7 (MS-ESI): m/z calculated for C16H19N3O2 [M+H]+: 286.15, found 286.16.
2,3,4,5-tetrahydro-5-(4’-acetamidobutyl)-1H-pyrido-[4,3-b]indol-1-one (4b) From 3b (0.35g, 0.90 mmol) Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.21g (78%) of the acetyl-substituted indole 4b as a solid. 1H NMR (900 MHz, DMSO) δ = 1.47 (m, 2H, H2’), 1.72 (s, 3H, CH3), 3.01 (m, 6H, H3’, H4’, H3); 3.48 (t, 2H, H4, J4-3= 6.3 Hz); 4.18 (t, 2H, H1’, J1’-2’ = 5.1 Hz), 6.73 (bs, 1H, NH); 7.15 (m, 2H, H7, H8); 7.52 (d, 1H, H9, J9-8= 8.1 Hz); 7.83 (bs, 1H, NH), 7.91 (d, 1H, H6, J6-7= 7.6 Hz). 13C NMR (225 MHz, DMSO) δ= 166.1, 156.1, 145.5, 136.7, 125.5, 121.9, 120.2, 110.6, 111.9, 105.3, 43.1, 40.0, 38.3, 27.4, 26.9, 22.9, 21.7. (MS-ESI): m/z calculated for C17H21N3O2 [M+H]+: 300.16, found 300.16.
2,3,4,5-tetrahydro-5-(6’-acetamidopentyl)-1H-pyrido-[4,3-b]indol-1-one (4c)
From 3c (0.35g, 0.87 mmol) Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.20g (74%) of the acetyl-substituted indole 4c as solid. 1H NMR (600 MHz, DMSO) δ = 1.26 (m, 2H, H4’), 1.39 (m, 2H, H3’), 1.68 (m, 2H, H2’), 1.77 (s, 3H, CH3), 2.99 (m, 4H, H5’, H3); 3.47 (t, 2H, H4, J4-3= 6.2 Hz); 4.15 (t, 2H, H1’, J1’-2’ = 5.4 Hz), 7.11 (bs, 1H, NH); 7.16 (m, 2H, H7, H8); 7.51 (d, 1H, H9, J9-8= 7.1 Hz); 7.77 (bs, 1H, NH); 7.91 (d, 1H, H6, J6-7= 7.3 Hz).
N
NH
O
HN
O
13
4
67
8
9
1'
2'
3'
N
NH
O
NH
13
4
67
8
9
1'
2'
3'
O
4'
N
NH
O
NH
1
3
4
6
7
8 9
1'2'
3'
4'
5'
O

7
2,3,4,5-tetrahydro-5-(6’-acetamidohexyl)-1H-pyrido-[4,3-b]indol-1-one (4d) From 3d (0.35g, 0.84 mmol) Column chromatography: Eluted with CH2Cl2/MeOH (15:1) to give 0.21g (78%) of the acetyl-substituted indole 4d as solid. 1H NMR (900 MHz, DMSO) δ = 1.23 (m, 4H, H3’, H4’), 1.38 (m, 2H, H2’), 1.78 (s, 3H, CH3), 2.99 (m, 6H, H5’, H6’, H3); 3.48 (t, 2H, H4, J4-3= 6.3 Hz); 4.20 (t, 2H, H1’, J1’-
2’ = 5.2 Hz), 7.2 (bs, 1H, NH); 7.42 (m, 2H, H7, H8); 7.79 (d, 1H, H9, J9-8= 7.3 Hz); 8.05 (d, 1H, H6, J6-7= 7.4 Hz), 9.3 (bs, 1H, NH). 13C NMR (225 MHz, DMSO) δ= 166.0, 156.2, 145.4, 136.2, 125.4, 121.9, 121.1, 120.2, 110.7, 105.3, 43.2, 40.0, 38.7, 29.8, 29.7, 26.3, 22.7, 22.4, 21.7 (MS-ESI): m/z calculated for C19H25N3O2 [M+H]+: 328.19, found 328.20.
(E)-N-(4-cyanophenyl)-4-((4-hydroxy-3,5-dimethylphenyl)diazenyl)benzenesulfon-amide (MS611) MS611 was synthesized using a procedure we recently reported (Zhang et al., 2013). A 50 mL round-bottomed flask was charged with 4-amino-N-(4-cyanophenyl)benzene- sulfonamide (255.0 mg, 0.93 mmol) and concentrated HCl (552 mg, 468 µL, 5.60 mmol). The mixture was dissolved in a MeOH/ACN mixture (3 mL/3mL). The solution was cooled to 0 °C and stirred for 15 min. Isoamyl nitrite (109.3 mg, 125 µL, 0.93 mmol) was added drop wise under argon over 10 min. The solution was stirred at 0 °C for 45 min. Meanwhile, to another 50 mL round-bottomed flask were added 2,6-dimethylphenol 57.0 mg, 0.47 mmol) and potassium carbonate (322 mg, 2.33 mmol). To this mixture were added methanol (1.0 mL) and DI H2O (8.0 mL). The solution was deoxygenated for 15 min by argon. The resultant solution was cooled to 0 °C. The previously prepared amber color diazonium ion was added drop wise under argon over 15 min. At the end of the addition, the pH of the solution was maintained between 8–10. The solution was allowed to stir at 0°C for 1 h and then quenched with 1 N HCl to reach pH 1. The precipitate was filtered under vacuum and provided the title compound as a fine orange powder (145 mg, 77%). 1H NMR (600 MHz, MeOD-d4) δ 8.50 (d, J = 1.9 Hz, 1H), 8.13 (d, J = 8.6 Hz, 2H), 7.95 (dd, J = 8.7, 2.2 Hz, 1H), 7.93 (d, J = 8.6 Hz, 2H), 7.61 (s, 2H), 7.23 (d, J = 8.8 Hz, 1H), 2.29 (s, 6H). HRMS (m/z) calculated for C20H18N5O3S+ [M+H]+, 408.1130; found 408.1131. Purity >99%, tR = 4.5 min.
SN NHO
O
CN
NHO
3,5-dimethoxyaniline HCl salt (1) A solution of 3,5-dimethoxyaniline (1.99 g, 0.13 mol) in diethyl ether (50 mL) in a 250 mL 3-necked round bottom flask was cooled to 0 °C. HCl gas was bubbled through the solution over 15 min. After stirring at 15 min at 10°C, the mixture was filtered, washed with isopropylacetate (5 mL), and dried overnight on high vacuum at room temperature to give the hydrochloride (1.99 g, 80%), as a white solid.
N
NH
O
13
4
67
8
9
1'
2'
3'
NH
4'
O
5'6'

8
4,6-dimethoxyisatin (2) A mixture of the hydrochloride above (1, 1.99 g, 0.01 mol) and oxalyl chloride (22.6 mL, 0.26 mol) in a 100 mL round-bottomed flask equipped with a reflux condenser was heated to 170 °C for 2 h with stirring. The solution mixture changed from a brownish suspension to a transparent solution and then some yellowish green precipitate started to appear. The oxalyl chloride was distilled from the reaction mixture. The flask was cooled to 0°C and methanol (4 mL) was added. The reaction mixture was heated to reflux (85 °C) for 1 h, filtered while hot, and washed with methanol (5 mL) to give the 4,6-dimethoxyisatin (1.49 g, 67%) as a yellow-green solid. 1H NMR (DMSO-d6) δ 10.92 (s, 1H), 6.17 (d, J = 1.5, 1H), 6.02 (d, J = 1.5, 1H), 3.88 (s, 3H), 3.86 (s, 3H). HRMS calculated for C10H10NO4
+ [M+H]+ 208.0610; found 208.0711. Purity >99%, tR = 3.6 min. 2-amino-4,6-dimethoxybenzoic acid (3) To a heated solution (70 °C) of the isatin (2, 1.49 g, 7.2 mmol) in aqueous NaOH (40%, 14.1 mL) was added H2O2 (35%, 3.75 mL) slowly over 10 min. After the addition of each portion of H2O2, the internal reaction temperature (initially 64 °C.) increased (to a maximum temp of 80 °C). After the addition was complete, the foaming reaction mixture was then stirred for an additional 2 h at 70 °C. The mixture was allowed to stir overnight while cooling to room temperature. The mixture was heated to 80 °C. Additional H2O2 (0.7 mL) was added, and the mixture was stirred at 80 °C for a further 2 h until the reaction was complete. After cooling to 10°C, aqueous Na2S2O3 (13.70 mL, saturated) was added. The mixture was brought to pH 8 with HCl (37%, 14.5 mL) and pH 6 with acetic acid (glacial, 2.7 mL), without allowing the reaction mixture to warm to greater than 20 °C. Filtration of the reaction mixture and washing with water (30 mL) gave the expected amino acid as a tan solid (0.76 g, 61%). 1H NMR (DMSO-d6) δ 6.85 (br s, 1H), 6.00 (s, 1H), 5.86 (s, 1H), 3.83 (s, 3H), 3.78 (s, 3H). HRMS calculated for C9H12NO4 [M+H]+ 198.0766; found 198.0844. Purity >99%, tR = 4.3 min. 2-amino-4,6-dimethoxybenzamide (A) To a solution of 3 (0.76 g, 3.85 mmol) in anhydrous THF (36 mL) was added EDCI (0.77 g, 4.23 mmol), HOBt (0.57 g, 4.91 mol), and NMM (0.47 mL), and the mixture was allowed to stir at room temperature for 3 h. Aqueous NH3 (10.8 mL, 30%) was added, and the mixture was stirred at room temperature for 16 h and immediate precipitate was observed. Water (14 mL) was added, and the mixture was extracted with DCM (2 × 2.5 mL). The combined extracts were washed with water (2 × 5.0 mL). Concentration, formation of a slurry with ether (15 mL), filtration, and drying under high vacuum afforded A (0.29 g, 44%) as a brown solid. 1H NMR (CDCl3) δ 7.69 (s, 1H), 6.41 (br s, 2H), 5.80 (d, J = 2.7, 1H), 5.78 (d, J = 2.7, 1H), 5.36 (s, 1H), 3.87 (s, 3H), 3.78 (s, 3H). HRMS calculated for C9H13N2O3 [M+H]+ 197.0926; found 197.1004. Purity >99%, tR = 3.9 min. 4-(2-hydroxyethoxy)-3,5-dimethylbenzaldehyde (B) 4-hydroxy-3,5-dimethylbenzalde- hyde (0.70 g, 4.66 mmol), K2CO3 (97 mg, 0.7 mmol) and DMF (1.4 mL) were mixed and stirred at 110 °C under nitrogen. Ethylene carbonate (0.456 g, 5.1 mmol) in DMF (485 mL) was added to the mixture over 10 min. The reaction mixture was stirred at 110 °C for 12 hours. The reaction mixture was cooled to 25 °C and water (50 mL) was added followed by a mixture of dichloromethane and heptane (v/v=3:2, 50 mL). The mixture was agitated for 30 minutes. The organic layer was isolated and the aqueous layer was back extracted with a mixture of dichloromethane and heptane (v/v=3:2, 50 mL). Very heavy emulsion was developed. Eventually the emulsion was separated by centrifuge at 4000 rpm for 10 min. The combined organic layers were washed with aqueous sodium hydroxide (3.0 M, 25 mL), followed by three washes with water (3 × 50 mL), and dried over sodium sulfate. Dichloromethane was removed by distillation, keeping the temperature below 40 °C. The mixture was dried under vacuum until constant weight to afford intermediate B (384 mg, 43%) as a dark liquid. 1H NMR (DMSO-d6) δ 9.82 (s, 1H), 7.54 (s, 2H), 4.96 (s, 1H), 3.85 (m, 2H),

9
3.74 (m, 2H), 2.29 (s, 6H). HRMS calculated for C9H15O3 [M+H]+ 195.1021, found 197.1164. Purity >99%, tR = 4.2 min. 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)-5,7-dimethoxyquinazolin-4(3H)-one (C) (RVX-208, or MS765) Aldehyde B (106.0 mg, 0.55 mmol)), N,N-dimethylacetamide (DMA, 2.0 mL), and intermediate C (100.7 mg, 0.51 mmol) were combined and p-toluenesulfonic acid monohydrate (10.4 mg, 55 mmol) and 1/3 of the required sodium bisulfite (44.1 mg, 0.42 mmol) were added. The mixture was heated to 115 °C and stirred for 100 min before the second 1/3 of the required sodium bisulfite (44.1 mg, 0.42 mmol) was added. The remaining sodium bisulfite (44.1 mg, 0.42 mmol) was added after another 100 min. The reaction mixture was stirred at 115 °C until the reaction was complete. The reaction mixture was cooled to 25 °C and added to water (20 mL). The mixture was stirred at 20 °C for 6 h to complete the crystallization. The crude material was isolated by filtration, washed with water (4 mL) and dried under vacuum. The crude material was dissolved in DMA (2 mL) at 80 °C. The solution was cooled to 60 °C and heptane (10 mL) was slowly added over a period of 1 h, maintaining a temperature above 35°C. The solution was cooled to 35 °C and stirred at 35 °C for 1 h. The solid was isolated by filtration, washed with heptane (1 mL) and dried under vacuum. The dry solid was added to MeOH (3 mL) and purified either by recrystallization or by HPLC. The pure final product C appeared as a pale yellow powder (180 mg, 90%). 1H NMR (DMSO-d6) δ 11.81 (s, 1H), 7.88 (s, 2H), 6.72 (d, J = 2.3 Hz, 1H), 6.50 (d, J = 2.3 Hz, 1H), 4.89 (t, J = 5.5 Hz, 1H), 3.87 (s, 2H), 3.83 (s, 6H), 3.71 (q, J = 4.9 Hz, 2H), 2.29 (s, 6H). HRMS calculated for C20H23N2O5 [M+H]+ 371.160; found 371.163. Purity >99%, tR = 4.5 min.

10
REFERENCES: Barbieri, V., and Grazia, M.F. (2006). Microwave-assisted one-pot synthesis of substituted tetrahydrocarbazole and 8,9,10,11-tetrahydro-7H-pyrido[a]carbazoles. Tetrahedron Letters 47, 8289-8292.
Coldham, I., Dobson, B.C., Fletcher, S.R., and Franklin, A.I. (2007). Intramolecular Diploar Cycloaddition Reactions to Give Substituted Indoles - A Formal Synthesis of Deethylibophyllidine. Eur J Org Chem, 2676-2686.
Fukuda, T., and Maeda, K. (1999). Directed C-7 lithiation of 1-(2,2-diethylbutanoyl)indoles. Tetrahedron 55, 9151-9162.
Gribble, G.W. (1994). Recent Developments in Indole Ring Synthesis-Methodology and Applications. Contemp Org Synth 1, 145-172.
He, Y., Dupree, J., Wang, J., Sandoval, J., Li, J., Liu, H., Shi, Y., Nave, K.A., and Casaccia-Bonnefil, P. (2007). The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 55, 217-230.
Hnilicova, J., Hozeifi, S., Stejskalova, E., Duskova, E., Poser, I., Humpolickova, J., Hof, M., and Stanek, D. (2013). The C-terminal domain of Brd2 is important for chromatin interaction and regulation of transcription and alternative splicing. Mol Biol Cell 24, 3557-3568.
Miyamoto, H., Okawa, Y., Nakazaki, A., and Kobayashi, S. (2007). Total synthesis of (±)-debromoflustramine B and E and (±)-debromoflustramide B based on one-pot intramolecular Ullmann coupling and Claisen rearrangement. Tetrahedron Letters 48, 1805-1808.
Nikolovska-Coleska, Z., Wang, R., Fang, X., Pan, H., Tomita, Y., Li, P., Roller, P.P., Krajewski, K., Saito, N.G., Stuckey, J.A., et al. (2004). Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem 332, 261-273.
Rodriguez, J.-G., and Temprano, F.J. (1989). Synthesis of 4-(N,N- dimethylaminoethyl)-1,2,3,4-tetrahydrocarbazole: molecular structure and reactivity of the 1,2-dihydrocarbazol-4(3H)-one and derivatives. J Chem Soc Perkins Trans I, 2117-2122.
Watkins, J., Basu, S., and Bogenhagen, D.F. (2008). A quantitative proteomic analysis of mitochondrial participation in p19 cell neuronal differentiation. J Proteome Res 7, 328-338.
Zeng, L., Zhang, Q., Gerona-Navarro, G., Moshkina, N., and Zhou, M.M. (2008). Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure 16, 643-652.
Zhang, G., Liu, R., Zhong, Y., Plotnikov, A.N., Zhang, W., Zeng, L., Rusinova, E., Gerona-Nevarro, G., Moshkina, N., Joshua, J., et al. (2012). Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem 287, 28840-28851.
Zhang, G., Plotnikov, A.N., Rusinova, E., Shen, T., Morohashi, K., Joshua, J., Zeng, L., Mujtaba, S., Ohlmeyer, M., and Zhou, M.M. (2013). Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem 56, 9251-9264.
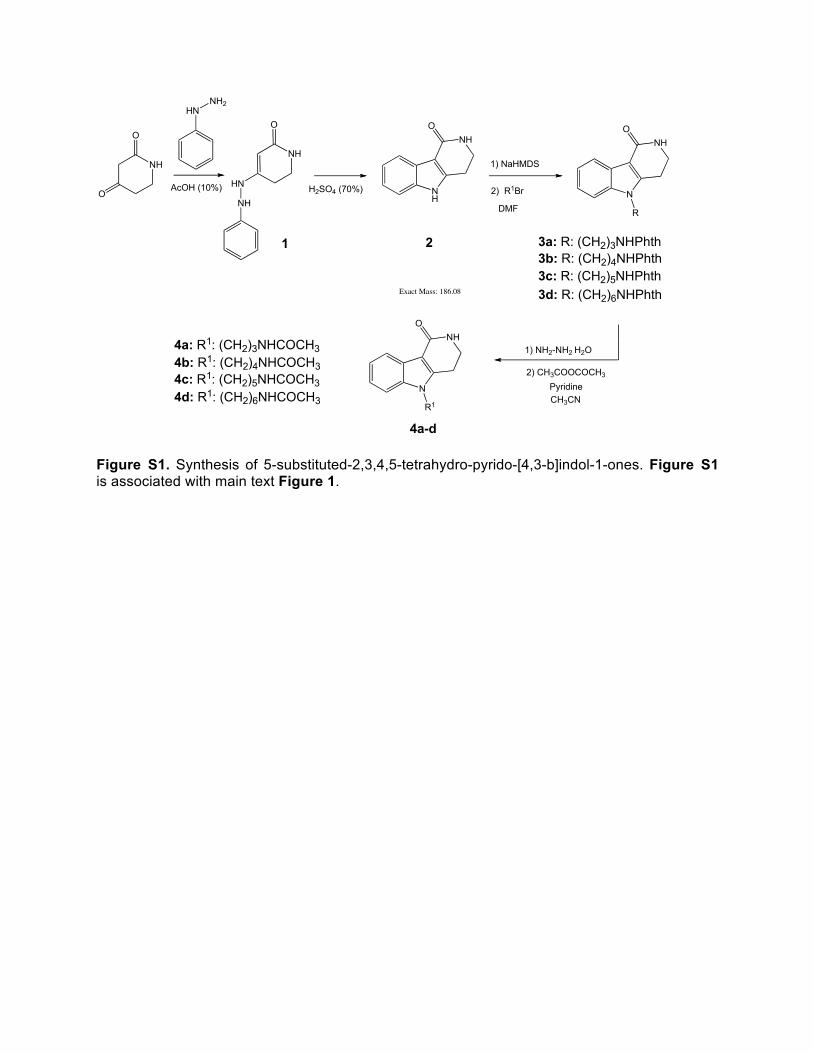
Figure S1. Synthesis of 5-substituted-2,3,4,5-tetrahydro-pyrido-[4,3-b]indol-1-ones. Figure S1 is associated with main text Figure 1.
HNNH2
NH
O
O
NH
O
HN
NH
AcOH (10%) NH
NHO
H2SO4 (70%) N
NHO
R
1) NaHMDS
1) NH2-NH2 H2O
2) CH3COOCOCH3
1 2 3a: R: (CH2)3NHPhth3b: R: (CH2)4NHPhth3c: R: (CH2)5NHPhth
Exact Mass: 186.08
N
NHO
R1
2) R1Br
DMF
CH3CN
4a-d
4a: R1: (CH2)3NHCOCH34b: R1: (CH2)4NHCOCH34c: R1: (CH2)5NHCOCH34d: R1: (CH2)6NHCOCH3
3d: R: (CH2)6NHPhth
Pyridine

0 10 20 30 40
0 0.5 1.0 1.5 2.0
0
-1.0
-2.0
-3.0
-4.0
0
-2.0
-6.0
-8.0
-10.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
-4.0
A
4b / Olinone 4a 4c
4d 5 6
Compd. n Kd (µM)
!H (kcal/mol)
T!S (kcal/mol)
!G (kcal/mol)
4a 0.96 ± 0.02 52.6 ± 1.3 -4.19 ± 0.15 1.35 -5.54
4b / Olinone 0.98 ± 0.03 3.4 ± 0.2 -9.21 ± 0.27 -2.04 -7.17
4c 1.05 ± 0.04 47.2 ± 2.2 -7.11 ± 0.12 -1.50 -5.61
4f 1.03 ± 0.02 21.3 ± 0.4 -6.29 ± 0.09 -0.23 -6.06
5 0.99 ± 0.04 89.1 ± 3.3 -3.99 ± 0.24 1.25 -5.25
6 0.98 ± 0.03 76.8 ± 1.7 -2.99 ± 0.38 -1.32 -5.31
2.5 3.0 3.5
0 10 20 30 40
0 1.0 2.0
0
-1.0
-2.0
-3.0
0
-1.0
-2.0
-3.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
3.0 4.0 5.0
0 10 20 30 40
0 1.0 2.0
0
-0.5
-1.0
-1.5
0
-1.0
-2.0
-3.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
3.0 4.0 5.0
-2.0
0 10 20 30 40
0 1.0
0
-0.5
-1.0
-2.0
0
-2.0
-4.0
-6.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
2.0 3.0 4.0
-2.5
-1.5
0 10 20 30 40
0 1.0
0
-0.5
-1.0
-2.0
0
-1.0
-2.0
-4.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
2.0 3.0 4.0
-2.5
-3.0
-1.5
0 10 20 30 40
0 1.0
0
-0.5
-1.0
0
-1.0
-2.0
-3.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
2.0 3.0 5.0
-2.0
-1.5
4.0
4a (CH2)3NHCOCH3
Comp. R1
4b (CH2)4NHCOCH34c (CH2)5NHCOCH34d (CH2)6NHCOCH35 (CH2)4NH26 H
Figure S2. Structure-guided design of olinone as a selective BrD inhibitor for the BrD1 of BET proteins. (A) Upper panel, Table listing thermodynamic parameters of olinone and its analogs binding to BRD4-BrD1. Lower panel, isothermal titration calorimetry measurements of olinone and its analogs to BRD4-BrD1. Figure S2A is associated with main text Figure 1.

B
123
125
117
121
15N
(ppm
)
115
7.78.18.58.99.39.71H (ppm)
7.3
113
119
6.9
111
123
125
117
121
15N
(ppm
)
115
7.78.18.58.99.39.71H (ppm)
7.3
113
119
6.9
111
BRD4-BrD1 free vs. + MS417 (1:1)
123
125
117
12115N
(ppm
)
115
7.88.28.69.09.49.81H (ppm)
7.4
113
119
7.0
111
123
125
117
12115N
(ppm
)
115
1H (ppm)
113
119
111
BRD4-BrD2 free vs. + MS417 (1:1)
7.88.28.69.09.49.8 7.4 7.0
BRD4-BrD1 free vs. + Olinone (1:1) BRD4-BrD2 free vs. + Olinone (1:1)
15N
(ppm
)
1H (ppm)
123
125
117
121
115
113
119
111
7.88.28.69.09.49.8 7.4 7.0
CBP free vs. + Olinone (1:1)
15N
(ppm
)
1H (ppm)
123
125
117
121
115
113
119
111
7.88.28.69.09.49.8 7.4 7.0
PCAF free vs. + Olinone (1:1)
Figure S2. Structure-guided design of olinone as a selective BrD inhibitor for the BrD1 of BET proteins.(B) Selectivity of BrDis olinone and MS417 binding to different bromodomains, as assessed by 2D 1H-15N HSQC spectra of a given bromodomain in the free form (black) and in the presence of olinone (red) or MS417 (red). Protein:ligand molar ratio was kept at 1:1. Figure S2B is associated with main text Figure 1.

C
Figure S2. Structure-guided design of olinone as a selective BrD inhibitor for the BrD1 of BET proteins.(C) Selectivity of olinone binding to different bromodomains, as assessed by isothermal titration calorimetry measurement. Table listing thermodynamic parameters of olinone binding to the various bromodomains that represent different subgroups of the human BrD family. Figure S2C is associated with main text Figure 1.
BrDs n Kd (µM)
!H (kcal/mol)
T!S (kcal/mol)
!G (kcal/mol)
BRD4-BrD1 0.97 ± 0.03 3.3 -6.62 ± 0.03 0.93 -7.55
BRD4-BrD2 - > 300 - - -
BRD3-BrD1 0.99 ± 0.03 3.7 -5.98 ± 0.25 -0.36 -6.34
BRD3-BrD2 - > 300 - - -
BRD2-BrD1 0.99 ± 0.01 8.6 -8.13 ± 0.15 1.56 -6.57
BRD2-BrD2 - > 300 - - -
CBP 1.07 ± 0.08 33 -2.18 ± 0.30 3.81 -5.99
PHIP-BrD2 0.98 ± 0.03 103.4 -7.53 ± 0.33 -3.61 -3.61
BRD7 - N.B. - - -
PCAF - N.B. - - -
ASH1L - N.B. - - -
TAF1L - N.B. - - -
SMARCA4 - N.B. - - -
BAZ1B - N.B. - - -
BAZ2B - N.B. - - -
ATAD2 - N.B. - - -
BPTF - N.B. - - -
TRIM24 - N.B. - - -
0 10 20 30 40 50
0 0.5 1.0 1.5 2.0
0
-0.5
-1.0
-1.5
-2.0
0
-2.0
-6.0
-8.0
-10.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
-4.0
0 10 20 30 40 50
0 0.5 1.0 1.5 2.0
0
-0.5
-1.0
-1.5
-2.0
0
-2.0
-6.0
-8.0
-10.0
Time (min)
Molar Ratio
μca
l/sec
Inje
ctan
t (kc
al/m
ol)
-4.0
CBP BrD PHIP-BrD2

D
Figure S2. Structure-guided design of olinone as a selective BrD inhibitor for the BrD1 of BET proteins. (D) Binding affinity of various small molecule inhibitors to the BrDs of BET proteins and CBP, as determined in a fluorescence anisotropy binding assay using a FITC-labeled MS417 as a probe, in which a small molecule BrD inhibitor’s ability to compete off FITC-MS417 binding to the given BrD was evaluated as a function of ligand concentration. Ki was calculated from IC50 as described in detail in Experimental Methods. The errors in IC50 values recovered from fitting competition curves reflect 95% confidence intervals. Figure S2D is associated with main text Figure 6.
Protein Olinone MS417 MS611 MS765 (RVX208)
Ki (μM) Ki (μM) Ki (μM) Ki (μM) BRD4-BrD1 17.2 <0.042 0.41 3.0
BRD4-BrD2 No Binding <0.039 41.3 0.33
BRD4-BrD1/2 9.5 <0.017 0.55 0.82
BRD4-BrD1/2-N140A No Binding 0.023 4.99 0.83
BRD4-BrD1/2-N433A 13.2 <0.012 0.48 2.03
BRD3-BrD1 27.5 <0.11 3.39 18.0
BRD3-BrD2 No Binding <0.12 3.68 0.61
BRD2-BrD1 39.5 <0.15 1.17 18.9
BRD2-BrD2 No Binding <0.13 5.58 0.76
CBP BrD >100 18.0 44.0 31.6

Figure S3. Dose dependent effects of BrD inhibitors on mOPC differentiation. (A) Experimental timeline. (B) Dose response of MS417 and Olinone treatment during mOPC differentiation. Primary cultured mouse OPC were treated with MS417 and Olinone at different doses (MS417: 0.01 and 0.05 μM; Olinone: 1 and 5 μM) for 72 hours. The graph represents the qRT-PCR results for the expression levels of differentiation markers. Results were normalized to Gapdh, with relative mRNA levels in DMSO treated control cells set to 1 MS417 inhibitory effect is doses dependent and 24 hours of treatment are enough to induce myelin gene repression. (both doses of MS417 and Olinone 1 μM , n=3; One-Way ANOVA with Dunnett’s post test,* P<0.05; ** P<0.01; ***P<0.001. Olinone 5 μM, n=1). Figure S3 is associated with main text Figure 2.
A
C
B

Figure S4. The synthetic scheme for compound C (also known as RVX-208, or MS765) was adapted from literature (Hansen et al., 2009). Figure S4 is associated with main text Figure 6. Reference: Hansen, H.C., Chopade, S.P., Citineni, J.R., Short, R.P., and Yiannikouros, G.P. (2009). Methods of Preparing Quinazolinone Derivatives, W.I.P.O.I. Bureau, ed. (USA), pp. 1-36.
NH2
OO
NH2
OO
.HClHCl (g) O
OCl Cl
MeOH
HN
O
O
O
O
H2O2
NaOH OO
NH2
O
OH
OO
NH2
O
NH2NH3 H2O
1 2
3
O
HO O
OO+ O
O OH
H
N
NH
O
O
O
O OH
PTSA, NaHSO3
A
A
B
C
.

Figure S5. Selective bromodomain inhibition affects the progression of primary OPCs towards a fully differen-tiated phenotype. Representative confocal images of mOPCs treated for 72h with BrDi (Olinone at 1 μM, Compound 5 at 1 μM, MS417 at 0.05 μM, MS566 at 0.05 μM, MS611 and MS765/RVX-208 at 0.5 μM) in differentiation medium (DM) and stained with MBP antibody (grey) and DAPI (Blue). Scale bar: 100 μm. (n=3 with 7-8 sections per condition). Figure S5 is associated with main text Figure 6.

Table S1. Data Collection and Refinement Statistics for the Crystal Structure of the BRD4-BrD1/Olinone complexa.
Table S1 is related to main text Figure 1
BRD4-BrD1/Olinone PDB Code Data collection
4QB3
Space group P212121 Cell dimension a, b, c (Å) 36.94, 44.12, 78.44 α, β, γ (°) 90.0, 90.0, 90.0 Resolution (Å) 10.0-0.94 (0.96-0.94) Measured reflections 649,857 Unique reflections 83,348 Rmerge (%) 6.4 (80.1)b I / σ 39.8 (3.0) Completeness (%) 99.5 (100) Redundancy 7.8 (7.6) Refinement Resolution (Å) 10.0 - 0.94 No. reflections 79,108 Rwork / Rfree (%) 13.6 / 15.4 No. atoms Protein 1,076 Ligand 22 Water 295 B-factors Protein 8.9 Ligand 6.9 Water 29.4 R.m.s. deviations Bond length (Å) 0.028 Bond angles (°) 2.47 Ramachandran plot % residues
Favored 99.2 Additional allowed 0.8 Generously allowed 0 Disallowed 0
aSee Methods section for exact experimental conditions. bValues in parentheses are for highest-resolution shell.