studies on the glycosidases in jack bean meal · studies on the glycosidases in jack bean meal ......
TRANSCRIPT

THE JOURNAL cm BIOLOGICAL Camwcrcr Vol. 242, No. 23, Issue of Decembar 10, PP. 54744480, 1967
Prinkd in U.S.A.
Studies on the Glycosidases in Jack Bean Meal
I. ISOLATION AND PROPERTIES OF a-MANNOSIDASE
(Received for publication, June 2, 1967)
Yu-TEH LI
From the Department of Biochemistry, Tulane University Delta Regional Primate Research Center, Covington, Louisiana 70&33
SUMMARY
cY-Mannosidase was purified approximately 500-fold from jack bean meal. This enzyme was able to hydrolyze a-1,6’, a-1, 2’-, and a-1 ,3’-linked oligomannosides, but not phenyl- j%n-mannoside and &1,4’-linked mannobiose. Approxi- mately 5 A of the total mannose present in the yeast mannan was set free by a-mannosidase after prolonged incubation. The fact that no sugars other than mannose were detected in the mannan digests indicates that this enzyme is not a polysaccharidase (endoenzyme) in nature. The enzyme hydrolyzes mannobiose, mannotriose, and mannotetraose derived from yeast mannan.
The K, values obtained were: fi-nitrophenyl-cr-D-mn- noside, 2.5 X low3 M; benzyl-ru-n-mannoside, 3.1 X lOA2 M; methyl-a-D-mannoside, 1.2 X 10-l M. The enzyme was competitively inhibited by mannono-(1 + 4)- and (1 ---t 5)-lactone. With p-nitrophenyl-cu-D-mannoside as sub- strate, Ki values for (1 + 4)- and (1 + 5)-lactone were 1.0 x lo-* M and 1.2 x lo-’ M, respectively.
ar-Mannosidase catalyzes hydrolysis, glycosyl transfer, and synthesis. One of the two disaccharides synthesized from mannose by cu-mannosidase was identified as a-1,6’- linked mannobiose.
cu-Mannosidase (cr-n-mannoside mannohydrolase, EC 3.2.1.24) is known to be widely distributed in plant seeds (1) and animal tissues (2) along with other glycosidases. In contrast to other glycosidases, this enzyme has not been extensively studied. In studies concerned with the enzymes which hydrolyze the carbohydrate portion of glycoproteins, an cr-mannosidase iso- lated from jack bean meal was found to liberate mannose from various glycoprotein preparations (3). The present report describes in detail the isolation and properties of a highly purified cr-mannosidase obtained from jack bean meal. A brief note on the purification of (Y- and &mannosidase from marine gastropods has recently appeared (4).
EXPERIMENTAL PROCEDURE
Materials
The sources for jack bean meal and various substrates, in- cluding chemically synthesized ru-n-mannosides and glyco- protein preparations, have been described earlier (3).
I am grateful to the following individuals for their generous gifts: 0-cY-n-mannopyranosyl-(1 + 2)-0-n-mannopyranose, Drs. C. E. Ballou and Y. C. Lee (5); O-cy-n-mannopyranosyl- (1 -+ 2)-0-cu-n-mannopyranosyl(1 + 2)-0-n-mannopyranose and 0-cu-n-mannopyranosyl-(1 --t 3)-O-cu-n-mannopyranosyl- (1 + 2)-0-or-D-mannopyranosyl-(1 -+ 2)-0-n-mannopyranose, Dr. Y. C. Lee (5); 0-cY-u-mannopyranosyl-(1 -+ 6)-O-n-man- nopyranose, Dr. J. K. N. Jones (6); 0-&n-mannopyranosyl- (1 + 4)-0-n-mannopyranose, Dr. E. T. Reese (7); phenyl$- n-mannoside, Dr. T. Muramatsu (4); mannono-(1 + 4)-lactone and mannono-(1 ---t 5)-lactone, Dr. J. Conchie (8). All other chemicals used were obtained from commercial sources and were of the highest grade. Unless otherwise indicated, all carbo- hydrates referred to in this manuscript are of the D configuration.
Enzyme Assays
Enzyme U&-With p-nitrophenyl-a-n-msnoside as sub- strate, a unit of enzyme was defined as the amount that released 1 pmole of p-nitrophenol per min. Specific activity was ex- pressed as the number of units per mg of protein, as recom- mended by the Report of the Commission on Enzymes of the International Union of Biochemistry (9). a-Mannosidase activity was assayed at 25’, in a Forma-Temp refrigerated and heated bath of Forma Scientific Inc., Marietta, Ohio.
L&ration of p-Nitrophenol-When p-nitrophenyl-a-n-man- noside was used as a substrate, the incubation mixtures con- tained the following components in total volumes of 1 ml: sodium citrate buffer, pH 4.5, 50 pmoles; p-nitrophenyl-a-n- mannoside, 5 pmoles; and 0.1 ml of the properly diluted enzyme solution. Unless otherwise indicated, the mixtures were in- cubated for 5 min. The reaction was terminated by introducing 2 ml of 0.2 M borate buffer, pH 9.8 (10). The liberated p- nitrophenol was quantitatively determined by the use of a Beck- man DU-2 spectrophotometer at its absorption maximum, 466
5474
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from
Issue of December 10, 1967 Y.-T. Li 5475
rnp (e = 1.77 X lo4 M-’ cm-r). @-N-Acetylglucosaminidase and 01- and /I-galactosidases were essentially assayed according to the procedure described for a-mannosidase, except that the substrate was replaced by 1 pmole each of the corresponding p-nitrophenylglycoside.
Detection of Free Mannose-When methyl-a-n-mannoside or beneyl-cr-n-mannoside was used as substrate, the reaction mixtures were essentially similar to that for p-nitrophenyl- cY+mannoside, except t,hat the substrate was replaced by 100 pmoles of methy-a-n-mannoside or 50 pmoles of benzyl-ar-n- mannoside. The reaction was stopped by the addition of 2 ml of Somogyi’s alkaline copper reagent (pH 9.5) (11). The mixture was heated in a boiling water bath for 10 min and then cooled. The amount of cuprous oxide formed was determined by adding Nelson’s arsenomolybdate reagent as described by Somogyi (11). A tube containing bhe same amount of substrate and enzyme was t,reated in the same way as the incubation mixture, but without being incubated, to serve as a blank. It was found that a considerable amount of mannose was lib- erated from methyl-a-n-mannoside or benzyl-a-n-mannoside by heating with Somogyi’s alkaline copper reagent.
Analytical Methods
Protein and Other Determinations-Protein was determined by the method of Lowry et al. (12). Protein concentration of the columneffluent was also estimated spectrophotometrically as pro- posed by Kalckar (13). Carbohydrate content of the column effluent was estimated by the phenol-sulfuric acid method of Dubois et al. (14).
Paper Chromatography-Qualitative identification of mono- saccharides and oligosaccharides in the enzymic digests was made by descending paper chromatography with Whatman No. 1 paper. The solvent systems used were (a) l-butanol-pyridine- 0.1 N HCl (5:3:2) and (b) 1-butanol-ethanol-water (5:1:4, top layer). Reducing sugars were detected by dipping the paper in 2-aminobiphenyl reagent as described by Gordon, Thorn- burg, and Werum (15), and nonreducing sugars by spraying the paper with sodium metaperiodate, potassium permanganate, and benzidine as described by Wolfram and Miller (16).
Purijkation of Enzyme
Ammonium Sulfate Fractionation-Unless otherwise indi- cated, all of the operations were conducted between 0” and 4”. The jack bean meal in 500 g of solutionwasextractedovernight by constant stirring with 2 liters of 33% (NH4)2S04 solution. The mixture was filtered through cheesecloth and centrifuged to ob- tain a clear extract, which was then adjusted to 60% saturation with solid (NH&SO+ The precipitate was collected by cen- trifugation and dissolved in 100 ml of 0.05 M sodium phosphate buffer, pH 7.0.
Treatment with Pyridine Acetate Buffer-The enzyme prep- aration obtained above was dialyzed against 0.5 M pyridine acetate buffer, pH 6.0, for 48 hours with several changes of the buffer. The precipitate produced during the dialysis was separated by centrifugation and discarded. The clear dialysate thus obtained was subsequently dialyzed exhaustively against 0.05 M sodium acetate buffer, pH 6.0, containing 0.1 M NaCl. This fraction, designated crude enzyme fraction, could be stored at 4” for several months without appreciable decrease in en- zyme activity.
0 20 40 60 80 100 120 140 160
FRACTION NUMBER
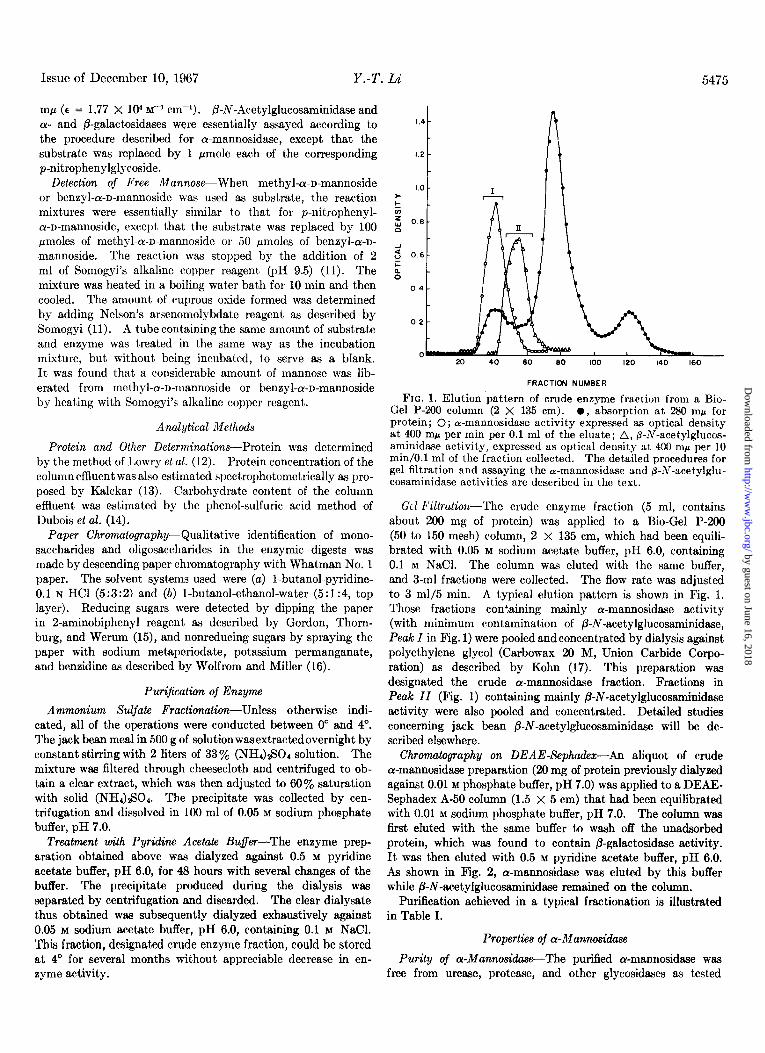
FIG. 1. Elution pattern of crude enzyme fraction from a Bio- Gel P-200 column (2 X 135 cm). l , absorption at 280 rnp for protein; 0; a-mannosidase activity expressed as optical density at 400 rnp per min per 0.1 ml of the eluate; A, a-l\i-acetylglucos- aminidase activity,-expressed as optical density atI 400 niwper 10 min/O.l ml of the fraction collected. The detailed nrocedures for gel filtration and assaying the a-mannosidase and i-N-acetylglu- cosaminidase act,ivities are described in the text.
Ccl l~‘iltrcliou-The crude enzyme fraction (5 ml, contains about 200 mg of protein) was applied to a Bio-Gel P-200 (50 to 150 mesh) column, 2 x 135 cm, which had been equili- brated with 0.05 M sodium acet.ate buffer, pH 6.0, containing 0.1 M NaCI. The column was eluted with the same buffer, and 3-ml fractions were collected. The flow rate was adjusted to 3 ml/5 min. A typical elution pattern is shown in Fig. 1. Those fractions con:aining mainly cY-mannosidase activity (with minimum contamination of P-N-acetylglucosaminidase, Peak I in Fig. 1) were pooled and concentrated by dialysis against polyethylene glycol (Carbowax 20 M, Union Carbide Corpo- ration) as described by Kohn (17). This preparation was designated the crude ar-mannosidase fraction. Fractions in Peak II (Fig. 1) containing mainly P-N-acetylglucosaminidase activity were also pooled and concentrated. Detailed studies concerning jack bean /3-N-acetylglucosaminidase will be de- scribed elsewhere.
Chromatography on DEAE-Sephadex-An aliquot of crude cr-mannosidase preparation (20 mg of protein previously dialyzed against 0.01 M phosphate buffer, pH 7.0) was applied to a DEAE- Sephadex A-50 column (1.5 x 5 cm) that had been equilibrated with 0.01 M sodium phosphate buffer, pH 7.0. The column was first eluted with the same buffer to wash off the unadsorbed protein, which was found to contain P-galactosidase activity. It was then eluted with 0.5 M pyridine acetate buffer, pH 6.0. As shown in Fig. 2, cr-mannosidase was eluted by this buffer while B-N-acetylglucosaminidase remained on the column.
Purification achieved in a typical fractionation is illustrated in Table I.
Properties of cr-Mannosidase
Purity of cw-Mannosidase-The purified a-mannosidase was free from urease, protease, and other glycosidases as tested
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from
5476 Studies on Glycosidases in Jack Bean Meal. I Vol. 242, No. 23
I.0
0.6
Stability of cr-Mannosiduse-The purified enzyme was un- stable upon lyophilization, freezing, and thawing. When kept at 25’ for 17 hours, the enzyme was stable over the pH range 6.0 to 8.5; however, it became unstable below pH 5.5. When the concentrated preparation (about 20 mg per ml) was prepared in 0.05 M acetate buffer, pH 6.0, and stored at 4’, about 20% decrease in enzyme activity was found after 5 months of storage. Heating the enzyme at 70” for 5 min destroyed more than 50% of its activity. The enzyme was stable at 60” for 5 min.
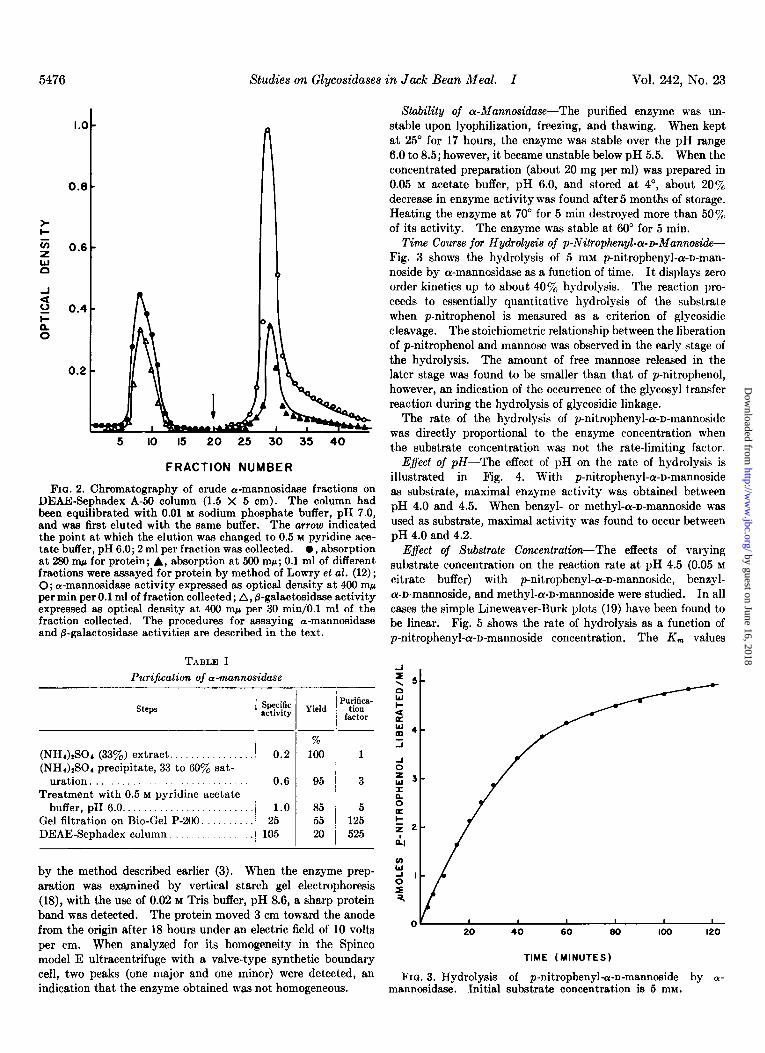
Time Course for Hydrolysis of p-Nitrophenyl-a-~Mannoside- Fig. 3 shows the hydrolysis of 5 mM p-nitrophenyl-or-D-man- noside by ar-mannosidase as a function of time. It displays zero order kinetics up to about 40% hydrolysis. The reaction pro- ceeds to essentially quantitative hydrolysis of the substrate when p-nitrophenol is measured as a criterion of glycosidic cleavage. The stoichiometric relationship between the liberation of p-nitrophenol and mannose was observed in the early stage of the hydrolysis. The amount of free mannose released in the later stage was found to be smaller than that of p-nitrophenol, however, an indication of the occurrence of the glycosyl transfer reaction during the hydrolysis of glycosidic linkage.
5 IO I5 20 25 30 35 40
FRACTION NUMBER
FIG. 2. Chromatography of crude a-mannosidase fractions on DEAE-Sephadex A-50 column (1.5 X 5 cm). The column had been equilibrated with 0.01 M sodium phosphate buffer, pH 7.0, and w&s first eluted with the same buffer. The arrow indicated the point at which the elution was changed to 0.5 M pyridine ace- tate buffer, pH 6.0; 2 ml per fraction was collected. 0, absorption at 280 ~QJ for protein; A, absorption at 500 rnp; 0.1 ml of different fractions were assayed for protein by method of Lowry et al. (12); 0; a-mannosidase activity expressed as optical density at 400 ~QJ per min per 0.1 ml of fraction collected; A, @-galactosidase activity expressed as optical density at 400 rnp per 30 min/O.l ml of the fraction collected. The procedures for assaying a-mannosidase and &galactosidase activities are described in the text.
TABLE I Purification of a-mannosidase
steps Yield
(NH&Sot (33%) extract .............. (NH&SO, precipitate, 33 to 60% sat-
uration ............................. Treatment with 0.5 M pyridine acetate
buffer, pH 6.0. ...................... Gel filtration on Bio-Gel P-200. ....... DEAE-Sephadex column. .............
0.2
0.6
1.0 25
105
% 100
95
85 55
20
Purifica- tion
factor
1
3
5 125
525
by the method described earlier (3). When the enzyme prep- aration was exmined by vertical starch gel electrophoresis (18), with the use of 0.02 M Tris buffer, pH 8.6, a sharp protein band w&s detected. The protein moved 3 cm toward the anode from the origin after 18 hours under an electric field of 10 volts per cm. When analyzed for its homogeneity in the Spinco model E ultracentrifuge with a valve-type synthetic boundary cell, two peaks (one major and one minor) were detected, an indication that the enzyme obtained was not homogeneous.
The rate of the hydrolysis of p-nitrophenyl-cY-D-mannoside was directly proportional to the enzyme concentration when the substrate concentration was not the rate-limiting factor.
E$ecf of pH-The effect of pH on the rate of hydrolysis is illustrated in Fig. 4. With p-nitrophenyl-a-n-mannoside as substrate, maximal enzyme activity was obtained between pH 4.0 and 4.5. When benzyl- or methyl-Lu-n-mannoside was used as substrate, maximal activity was found to occur between pH 4.0 and 4.2.
Effect of Substrate Concentration-The effects of varying substrate concentration on the reaction rate at pH 4.5 (0.05 M citrate buffer) with p-nitrophenyl-a-n-mannoside, benzyl- a-n-mannoside, and methyl-a-n-mannoside were studied. In all cases the simple Lineweaver-Burk plots (19) have been found to be linear. Fig. 5 shows the rate of hydrolysis as a function of p-nitrophenyl-cr-n-mannoside concentration. The K, values
TIME (MINUTES)
FIQ. 3. Hydrolysis of p-nitrophenyle-n-mannoside by O- mannosidase. Initial substrate concentration is 5 mM.
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of December 10, 1967 Y.-T, La’ 5477
' ' ' 3 4 5 6 3 4 5 6 3 4 5 6
PH
FIG. 4. Effect of pH on a-mannosidasc activity. A, p-nitro- phenyla-n-mannoside; B, bcnzyl-cr-n-mennoside; C, methyl-a- n-mannoside. Assays were performed in 0.05 M citrate buffer. For other assay conditions see text.
obtained from the plots were p-nitrophenyl-cu-n-mrtnnoside, 2.5 X 10-a M; benzyl-cr-n-mannoside, 3.1 X lo+ M; methyl- Lu-n-mannoside, 1.2 X 10-l M.
Inhibition Studies-Among various metal ions tested, Ag+ and Hg* were found to be potent inhibitors for a-mannosidase. p-Chloromercuribenzoate did not inhibit the cY-mannosidase activity. The enzyme was also strongly inhibited by mannono- (1 + 4)- and -(l -+ 5)-lactone, the latter being much the more potent inhibitor. Both (1 + 4)- and (1 + 5)-la&one acted as competitive inhibitors as determined by the Lineweaver-Burk plot (Fig. 6). With p-nitrophenyl-cY-n-mannoside, Ki values for (1 -P 4)- and (1 + 5)-lactone were estimated to be 1.0 X 10m2 M
and 1.2 X lop4 M, respectively. Substrate Specificity-In addition to hydrolyzing methyl-,
benzyl-, and p-nitrophenyl-ol-n-mannosides, as described in the previous sections, cY-mannosidase also liberated mannose from various natural cr-mannosides, which include O-cu-n-manno- pyranosyl- (1 -+ 2)0-n-mannopyranose (M-2), O-CY-D-
mannopyranosyl-(1 --f 6)-0-n-mannopyranose (M-2’), O- cu-n-mannopyranosyl-(1 + 2)-0-cr-n-mannopyranosyl-(1 -+ 2)-O-n-mannopyranose (M-3), 0-cr-n-mannopyranosyl-(1 + 3)- 0-Lu-n-mannopyranosyl-(1 --+ 2)-0-cY-n-mannopyranosyl-(1 + 2)-O-n-mannopyranose (M-4), and mannosyl-rhamnose obtained from Salmonella typhimurium 0 antigen? Fig. 7 is the chro- matogram indicating the hydrolysis of mannotriose (M-3) by cY-mannosidase; mannobiose (M-2) was detected as an inter- mediate of the hydrolytic product. When mannotetraose (M4) was incubated with cr-mannosidase, both mannotriose (M-3) and mannobiose (M-2) were detected as the intermediates of the hydrolytic products. Exhaustive digestion of yeast mannan with a-mannosidase resulted in the liberation of about 5% of the total mannose in this polysaccharide. When the enzymic digests of yeast mannan were examined at various time intervals by paper chromatography, only mannose was found to have been liberated.
cr-Mannosidase was not able to hydrolyze phenyl-&D-man- noside and 0-@-n-mannopyranosyl-(1 -+ 4)-0-n-mannopyranose.
Liberatim of Mannose from Various Glycoproteins by a- Mannosfdose-Jack bean cr-mannosidase was able to liberate mannose from ovalbumin, ovalbumin-glycopeptide, ovomucoid, and orosomucoid, its described previously (3). It also liberated
1 D. C. H. Bray, personal communication, 1966.
$ x I02
5 10 IS
P-NITROPHENYL-d-0-MANNOSIDE (M X IO’)(S)
FIG. 5. Effect of p-nitrophenyla-n-mannoside concentration on reaction rate. For details concerning assay conditions, see the text.
2 4 6 6 IO
i x IO3
FIG. 6. Inhibition of a-mannosidase activity by mannono-(1 + 4)- and (1 + 5)-lactone ss assayed by the method of Lineweaver and Burk (19). p-Nitrophenyl-or-n-mannoside was used as a substrate.
mannose from glycopeptides isolated from the deoxycholate- soluble fraction of rat liver microsomes.2 The enzyme liberated mannose from the glycoproteins or glycopeptides only when the terminal cu-mannosidic linkage was available. The rate of the
*Y.-T. Li, S. C. Li, and M. R. Shetlar, manuscript in preparation.
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from

5478 Studies on Glycoddases in Jack Bean Meal. I Vol. 242, No. 23
FIQ. 7. Hydrolysis of Oa-r+mannopyranosyl-(1 ---f 2)-0-a-D- mannopyranosyl-(1 -+ P)-0-D-mannopyranose by wmannosidase. Mannotriose (2 mg) was incubated with 50 units of enzyme in 0.05 M citrate buffer, pH 4.5. Final volume of the incubation mixture was 0.1 ml. At various time intervals, 10 ~1 of the incubation mixture were spotted directly to Whatman No. 1 paper. 8, standard; M, mannose; M-d, Oa-n-mannopyranesyl-(1 + 2)-0- D-mannopyranose.
liberation of mannose units from various glycoproteins varied from case to case, since the reaction rate was greatly affected by the polypeptide moiety and the length and the sequential arrangement of the oligosaccharide chain in the glycoprotein. It is, therefore, essential to check the extent of hydrolysis several times during the course of incubation. The liberation of man- nose from glycoprotein was usually much slower than that of glycopeptide derived from the same glycoprotein. In general, 25 units of the enzyme liberated 90% or mom of the terminal mannose from 1 ml of glycoprotein or glycopeptide solution containing 0.02 to 0.05 M of terminal cu-mannosidic units in 20 hours at 25“.
Glycosyltransjemse Activity of or-Mannosi&e-Glycosidases are known to catalyze transfer as well as hydrolytic reactions (20, 21). The mannosyl transfer reaction catalyzed by LY- marmosidaze was verified by the identification of new oligo- saccharides formed in the enzymic digests of p-nitrophenyl- or methyl-cu-n-mannopyranoside. These digests were found to contain disaccharides corresponding to O-cu-n-mannopyranosyl- (1 + 2)-0-n-mannopyranose and O-ar-n-mannopyranosyl- (1 + 6)-O-n-mannopyranoside, as detected by paper chromatog- raphy. Since the amount of the oligosaccharides produced was so small, no attempt was made to further verify the structure of these two disaccharides. The occurrence of mannosyl transfer reaction catalyzed by or-mannosidase ww further observed following the addition of methanol to the reaction mixture. The production of free mannose was greatly retarded when methanol (2 M) was present in the reaction mixture, but there was no appreciable change in the rate of the liberation of
p-nitrophenol. This result indicates that the mannosyl moiety was transferred to methanol. When this reaction mixture was subjected to paper chromatography, a nonreducing sugar with Rp identical with methyl-cr-n-mannoside was detected.
Synthetic Properties of a-Mannotidase-Besides hydrolysis and glycosyl transfer, glycosidases were also known to catalyze the condensation of monosaccharide to .form disaccharides and oligosaccharides (22, 23). The synthetic properties of Ly-man- nosidase were examined by incubation of 0.25 ml of various monosaccharides (1 M) and 0.25 ml of the enzyme solution containing 100 units of the enzyme. Both the monosaccharides and the enzyme were dissolved in 0.05 M citrate buffer, pH 4.5. The incubation was maintained at 25” for 6 hours. After the incubation, the enzyme was inactivated by heat.ing in a boiling water bath for 5 min. After removal of precipitated protein by centrifugation, 10 al of the reaction mixture were applied to Whatman No. 1 paper, and the products were resolved by de- scending paper chromatography, with Solvent a. The synthesis of new oligosaccharides from monosaccharides is very specific. The enzyme synt,hesized new oligosaccharides only from mannose, not from glucose or galactose. cY-Mannosidase synthesized two disaccharides from mannose when examined by paper chromatography with Solvent b. The two disaccharides were separated from mannose by means of a gel filtration with a Seph- adex G-25 (fine mesh) column (2 X 150 cm) as described by Lee and Ballou (5). These two disaccharides were further separated in pure form by preparative paper chromatography with What- man No. 3MM paper. One of the disaccharides (R,,,,,,, 0.29 with Solvent b) was found to have the same chromato- graphic mobility as an authentic O-or-n-mannopyranosyl- (1 --) 6)-O-D-mannopyranose. When this disaccharide was subjected to periodate oxidation in an alkaline medium, by the procedure of ‘Hough and Perry (24), no production of foxmalde- hyde was detected by the method of O’Dea and Gibbons (25). The other disaccharide (Rmannose 0.38 with Solvent b) and the authentic 0-a-n-mannopyranosyl-(1 -+ 2)-n-mannopyranose had the same chromatographic mobility. The alkaline periodate oxidation of this disaccharide resulted in detection of formalde- hyde. Due to the limited amount of disaccharides obtained, no attempt was made to quantitatively estimate the production of formaldehyde at different time intervals of periodate oxida- tion.
The ability of a-mannosidase to synthesize oligosaccharides from various pentoses was also examined by incubation of 0.25 ml of enzyme (contained 100 units of ar-mannosidsse) with 0.25 ml of various pentoses (1 M) at 25” for 6 hours. ar-Mannosidase synthesized three new oligosaccharides from lyxose, as de- tected by paper chromatography. No formation of new oligosaccharides was observed from ribose, xylose, or arabinose.
DISCUSSION
Classical sources for the cY-mannosidases were the vegetable materials, such as almond emulsin which contains a mixture of several glycosidases. Jack bean meal has been widely used for the preparation of urease and can be easily obtained in large quantities from commercial sources. Probably because of the rareness of the naturally occurring cr-mannosides, the purifi- cation of ar-mannosidase has not received the attention it deserves. The major cr-mannoside in nature is yeast mannan, and the structure of this polysaccharide is still not established. Re-
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of December 10, 1967 Y..T. G
cently, various glycoproteins and glycopeptides were found to contain cu-mannosides (3). The availability of cr-mannosidase free from other glycosidases adds another useful tool for the structural studies of glycoproteins and oligosaccharides con- taining ar-mannosidic linkage.
The activity of a-mannosidase in jack bean meal varied with the source of jack bean meal. In addition to cr-mannosidsse, jack bean meal also contains several other glycosidases, i.e. /3-N-acetylglucosaminidase, P-glucosidase, and (Y- and @-pa- lactosidases. It is, therefore, advantageous to check the levels of various glycosidase activities before isolation. In general, @-glucosidase and (Y- and @galactosidase activities in jack bean meal are much lower than a-mannosidase or /3-N-acetylglucos- aminidase when the enzyme activities are assayed with nitro- phenylglycosides as substrate. The presence of several glycosi- dases along with cY-mannosidase causes considerable difficulty in obtaining cu-mannosidase free from other glycosidases. Among several methods tested and developed, the method described above, although it involves a substantial sacrifice in the quantita- tive yield of the enzyme, is a relatively simple and reproducible one for the isolation of cY-mannosidase free from other glycosi- dases.
For the gel filtration of crude enzyme fraction by Bio-Gel P- 200, it was found that the pH greatly altered the elution pattern. In order to obtain the elution pattern shown in Fig. 1, the pH of the eluent and protein solution should be kept between 5 and 6. If the pH of the eluent was above 7.0, the protein came off the column as a broad peak. Sephadex G-200 could not replace Bio-Gel P-200, although these two gels have about the same exclusion limits. For some unknown reason, a-mannosidase was strongly bound to Sephadex G-200 gel. The binding of a jack bean protein, concanavalin A, to cross-linked dextran gels has been described by Agrawal and Goldstein (26).
The enzyme deteriorates at its pH optimum with a half-life of about 20 hours. Consideration should be given to the in- activation of the enzyme if prolonged incubation is necessary.
The optimal pH for jack bean cr-mannosidase is rather broad, between pH 4 to 5, whereas the sharp optimal pH curves with optimal pH of 5.0 and 8.0 have been reported for rat epididymis and Streptomyces grbeus cr-mannosidase, respectively (27, 28).
The substrate saturation curves and the Lineweaver-Burk plots indicate that the affinity of the enzyme for three synthetic ar-mannosides differs considerably. When the purified enzyme was subjected to heat inactivation studies at 55”, identical thermal inactivation curves were observed for p-nitrophenyl-, methyl-, and benzyl-a-n-mannoside. Thus, the difference in K,,, among those three mannosides appeared to be attributable to aglycone influence.
Jack bean ar-mannosidase was competitively inhibited by mannono-(1 + 4)- and mannono-(1 + 5)-lactone. Mannono- (1 -+ 5)-lactone is much the more potent inhibitor of the two. Similar results have been reported for rat epididymis cr-man- nosidase (8).
Among different cr-mannosides, jack bean cr-mannosidase was able to cleave a-1,2’-, a-1,3’-, and a-l ,6’-linked oligo- mannosides. The a-l ,4’-linked mannoside is currently not available.
Jack bean a-mannosidase was not able to hydrolyze the polysaccharide (0 antigen) isolated from Salmonella typhimutium which is a long polymer based on a repeating (-mannosyl-
rhamnosyl-galactosyl-) linear sequence. On the other hand, the mannosyhhamnose obtained by partial hydrolysis of this polysaccharide is readily split by the enzyme to mannose and rhamnose.’
When yeast mannan was incubated with cu-mannosidase, the amount of free mannose liberated varied with different mannan preparations. In most cases about 5% of the total mannose present in mannan could be set free by a-mannosidase after prolonged incubation. The fact that no sugar other than mannose was detected in the mannan digests suggests that the enzyme is not a polysaccharidase (endoenzyme) in nature. The partial liberation of mannose from yeast mannan by QI- mannosidase may suggest that yeast mannan contains oligoman- nosidic chains branched from the main polysaccharide chain, that these branched oligomannosidic chains are susceptible to jack bean a-mannosidase, and that the main polysaccharide chain is resistant. The structure proposed by Peat, Whelan, and Edwards (29) and Lee and Ballou (5) suggests that yeast mannan is highly branched with an CY-1,6’-linked chain which forms the backbone of the polysaccharide, and with the a-1,2’ linkage being at branch points.
Like other glycosidases, jack bean cr-mannosidase cata- lyzes hydrolysis, glycosyl transfer, and synthesis of glycoside. The specificity of the jack bean cr-mannosidase on the glycosidic cleavage is very strict. @Mannosides, LY- or &glucosides, and o- or @-galactosides were not hydrolyzed by this enzyme. The strict specificity was also noticed when the synthesis of glycosides was catalyzed by this enzyme. The fact that cz-mannosidase synthesized oligosaccharides from lyxose is also noteworthy. Although lyxose is not a naturally occurring sugar, the structure of lyxose and mannose is identical except for the group attached to carbon atom 5. It is interesting to note that Pigman (30) has shown that almond emulsin was able to hydrolyze cu-lyxoside as well as cr-mannoside. By examination of chromatographic mobility and periodate oxidation, one of the two disaccharides synthesized from mannose by cu-mannosidase was identified as O-a-n-mannopyranosyl-(1 --f 6)-O-n-mannopyranose.
Acknowledgments-The author is indebted to Dr. Su-Chen Li for her valuable suggestions and discussions. It is also a pleasure to acknowledge the skilled assistance of Mr. Robert A. Bridges, Jr., and Mr. Tokutaro Kato in some phases of this work.
REFERENCES
1. VEIBEL, S., in J. B. SUMNER AND K. MYRB;~CK (Editors), The enzymes, Fool. I, Part 1, Academic Press, New York, 1959, p. 630.
2. CONCHIE. J.. FINDLAY. J.. AND LEVVY. G. A.. Biochem. J.. 71. 318 (1959):
, ,
3. LI, Y.-T., j. Biol. Chem., 241, 1010 (1966). 4. MURAMATSU. T.. Arch. Biochem. Biovhus.. 116. 427 (1966). 6. LEE, Y.-C., END’ BALLOU, C. E., Bio>h~mstry; 4, 25i (1965). 6. JONES, J. K. N., AND NICHOLBON, W. H., J. Chem. Sot., 27
(1958). 7. REESE, E. T., AND SHIBATA, Y., Can. J. Microbial., 11, 167
(1965). 8. LEVVY, G. A., HAY, A. J., AND CONCHIE, J., Biochem. J., 91,
378 (1964). 9. Report of the Commission on Enzymes of the International
Union of Biochemistry, Pergamon Press, New York, 1961. LO. WOLLEN, J. W., HEYWORTH, R., AND WALKER, P. G., Biochem.
J., 78, 111 (1961).
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from

5480 Studies on Glycosidases in Jack Bean Med. I Vol. 242, No. 23
11. SOMOQYI, M., J. Biol. Chem., 196, 19 (1952). 12. LOWRY, 0. Hi, ROSEBROUQH, N. J., FAF&, A; L., AND RANDALL,
R. J.. J. Biol. Chem.. 193. 266 (1951). 13. KALCK& H. M., J. Biol. &em.,‘167,‘461 (1947). 14. DUBOIS, &I., GIL~ES, K. A., HAMILTON, J. k., ~EBER~, P. A.,
AND SMITH. F.. Anal. Chem.. 26. 350 11956). 16. GORDON, H. k., ‘THORNBURO, ‘W.,- AND ~WER&, L. N., Anal.
Chem., 26, 349 (1956).
17. K&N, j.,
16. WOLFROM, M. L., AND MILLER, J. B., Anal. Chem., a8, 1037
Nature, 183, (1956).
1055 (1959). 18. SMITHIES. 0.. Biochem. J.. 61. 629 (1955). 19. LINEWEA~ER; H., AND BARK; D., >. Amer. Chem. Sot., 66,
658 (1934). 20. DEDONDER, R. A., Annu. Rev. Biochem., 30, 347 (1961). 21. HASSID, W. Z., AND NEUFELD, E. F., in P. D. BOYER, H. LARDY,
AND K. MYRB%CK (Editors), The enzymes, Vol. 6, Academic Press, New York, 1962, p. 277.
22. HILL, A. C., J. Chem. Sot., 635 (1898). 23. BOURQUELOT, E., AND AUBRY, A., Compt. Rend. Acad. Sci.
Paris, 165, 60 (1916). 24. HOUGH. L.. AND PERRY. M. B.. Chem. Ind. (London). 768
(1956). ’ 25. O’DEA, J. F., AND GIBBONS, R. A., Biochem. J., 66, 580 (1953). 26. AQRAWAL, B. B. L., AND GOLDSTEIN, I. J., Biochem. J., 96,
23c (1966).
WHITEHEAD, B. K., kochem. j., 67, 9;3 (1954).- ’ 29. PEAT, S., WHELAN, W. J., AND EDWARDS, T. E., J. Chem. Sot.,
29 (1961).
27. CONCHIE, J., AND HAY, A. J., Biochem. J., 73, 327 (1959). 28. HOCKENHULL. I>. J. D.. ASHTON. G. C.. FANTES, K. H.. AND
30. PIC+MAN, W. W., J. Amer. Chem. Sot., 62, 1371 (1940).
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from

Yu-Teh Li-MANNOSIDASEαPROPERTIES OF
Studies on the Glycosidases in Jack Bean Meal: I. ISOLATION AND
1967, 242:5474-5480.J. Biol. Chem.
http://www.jbc.org/content/242/23/5474Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/242/23/5474.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on June 16, 2018http://w
ww
.jbc.org/D
ownloaded from