sep chez l'enfant
TRANSCRIPT

Sclérose en plaques chez l’enfant
S o u s l a d i r e c t i v e d u D r . O u b a ï c h e e t d u
D r . O u d r e r
S e r v i c e d e n e u r o l o g i e d u C H U d ’ O r a n
S e p t e m b r e 2 0 1 0
Mr Guennoun. Z Interne au service de neurologie du CHU d’Oran
République Algérienne démocratique et populaire Faculté de médecine d’Oran

Remerciements
Je tiens à remercier le Dr. Oubaïche, le Dr. Oudrer, le Dr. Bahmani, le Dr. Ider, le Dr. Aïdi, le Dr. Benlaredj, ainsi que tous les maître-assistants et résidents en neurologie pour la science et l'ouverture d'esprit qu'on a trouvé, mes collègues et moi, auprès d'eux. Nous savons que c'est grâce à leur savoir et à la réflexion qu'ils ont partagé avec nous que s'ouvrent devant nous des horizons nouveaux et que nous pouvons prétendre mener un travail de recherche aussi modeste soit-il.
Je remercie tout particulièrement mon encadrante Dr. Oudrer pour sa disponibilité, sa confiance, et pour les remarques et conseils qu'elle m'a prodigué au cours de l'élaboration de ce travail.

Planducours · Introduction et intérêt de la question
· Rappel sur les pathologies démyélinisantes primaires
· Aspects historiques
· Définition
· Incidence et prévalence
· Aspects démographiques
· Pathogénèse
· Facteurs environnementaux
· Facteurs immunologiques
· Facteurs génétiques
· AnatomoPathologie
a. Aspect macroscopique et Histopathologie
· Immunopathologie
· Manifestations cliniques
· Diagnostic positif
· Etudes immunologiques
· Neurophysiologie
· Neuroimagerie anatomique
· Neuroimagerie fonctionnelle
· Diagnostic différentiel
· Traitement
· Pronostic

Introductionetinteretdelaquestion
Au cours des 5 dernières années, l'intérêt et la connaissance de la sclérose en plaques (SEP) pédiatrique, y compris son traitement, sa pathogenèse, sa démographie, son histoire naturelle, et les signes à l'IRM et à la biologie, a augmenté considérablement.
Dans ce papier, nous examinerons les données actuellement disponibles dans la littérature sur ce désordre , dont les plus récentes avancées de la recherche.

Rappelsurlespathologiesdemyelinisantesprimaires
a SEP fait des partie des 3 pathologies démyélinisantes primaires du SNC, les 2 autres étant l'encéphalomyélite disséminée aiguë et la névrite optique.
Le concept de démyélinisation primaire implique la destruction de la myéline, des oligodendrocytes, des cellules de Schwann et de la préservation relative des autres composants du système nerveux central. Toutefois, les lésions axonales sont fréquemment rencontrées dans les lésions démyélinisantes, ce qui corréle avec les déficits fonctionnels permanents.
Les maladies de démyélinisation du système nerveux central peuvent être la conséquence de :(a) un processus auto-immun inflammatoire entraînant la destruction de la myéline normalement développée (maladies démyélinisantes ou myélinoclastique), (b) les troubles métaboliques et génétiques du métabolisme de la myéline, qui induisent une myéline anormale (maladies dysmyélinisantes) , ou (c) un processus de démyélinisation primaire qui survient à la suite d'insultes cérébrale hypoxique-ischémique et de certaines formes d'intoxication.
L'encéphalomyélite disséminée aigue (EMDA) est plus fréquente chez les enfants de moins de 12 ans; la sclérose en plaques est plus fréquente chez les adolescents et les adultes. La difficulté à distinguer l'EMDA du 1er épisode d'une SEP est l'une des raisons les plus importantes pour l'exigence d'un deuxième épisode distinct survenu au moins 1 mois après le premier pour le diagnostic de la SEP. Il reste controversé de savoir si la notion de « EMDA récurrente » doit être distinguée de la SEP, mais il semble probable que cette distinction est mal définie chez les enfants prépubères. La névrite optique et la combinaison de névrite optique + myélite transverse (maladie de Devic) se produit généralement en tant que manifestations de l'EMDA ou de la SEP, mais une myélite transverse

pure semble être une entité nosologique particulière, qui peut résulter d'autres types de maladies.
Il existe en sémiologie une zone de chevauchement entre l'EMDA et le syndrome de Guillain-Barré (GBS). Cette zone de chevauchement comprend une partie ou peut-être tous les patients qui manifestent des signes cliniques du syndrome de Miller Fisher (triade =ophtalmoplégie externe; ataxie; aréflexie). Il comprend également la minorité de cas d'EMDA qui se manifestent par l'absence ou la diminution des réflexes ostéotendineux en combinaison avec une faiblesse et des modifications sensorielles afférentes dues à un dysfonctionnement des nerfs périphériques. La désignation de :encéphalomyélo-radiculo-neuropathies (EMRN) peut être appliquée à des cas présentant ce chevauchement entre manifestations démyélinisantes centrales et périphériques.

Classification des pathologies de ma yéline du SNC

D'autres conditions démyélinisantes beaucoup plus rares peuvent survenir chez les enfants et sont souvent difficiles à classer exactement, ceci comprend la SEP aiguë (de type Marburg), la sclérose diffuse myelinoclastique (maladie de Schilder) et la sclérose concentrique (maladie de Bala). La maladie rencontrée chez les nourrissons de moins de 2 ans, qui peuvent manifester un seul épisode de démyélinisation sévère avec oedème, devraient être qualifiées soit de SEP aiguë, ou peut-être de manière plus appropriée, une EMDA sévère.
L'étiologie et la pathogenèse de ces différentes maladies démyélinisantes primaires sont encore mal comprises. En outre, le degré de chevauchement pathogénique entre l'EMDA, la SEP et d'autres maladies démyélinisantes comme la maladie de Devic et la myélite transverse est encore inconnu. Qua ce soit la SEP ou l'EMDA, ces 2 entités sont considérées comme des maladies auto-immunes qui impliquent des réponses cellulaires et humorales qui sont dirigées, au moins en partie, contre des antigènes de la myéline.
Le début de la SEP n'a pas de corrélation claire étiologique avec une infection précédente, et les épisodes cliniquement manifestes de la maladie sont généralement associées à la production d'immunoglobulines oligoclonales détectable dans le liquide céphalo-rachidien (LCR). L'EMDA apparaît dans de nombreux cas, être provoquée par un antécédent de maladie infectieuse et est accompagnée par des concentrations élevées d'immunoglobulines au LCR ou une oligoclonalité d'immunoglobuline seulement dans une minorité de cas. Ainsi un profil Normal d'immunoglobuline au LCR est caractéristique des récidives de l'EMDA, comparativement à une probabilité supérieure à 94% d'anomalies du LCR en liaison avec une récurrence de SEP.
Une petite minorité d'individus qui ont connu des cas typiques de l'EMDA dans la petite répondent aux critères cliniques pour le diagnostic de sclérose en plaques au cours de l'adolescence, tandis que d'autres

répondent à des critères pour le diagnostic de sclérose en plaques avec des manifestations soit rémittente-récurrente soit progressive de démyélinisation centrale primaire.
Il n'existe aucun test diagnostique spécifique ou de biomarqueurs pour faire la différence entre EMDA et SEP dans la population pédiatrique, et un certain nombre d'autres conditions doivent être exclues avant de poser l'un ou l'autre des 2 diagnostics. Il peut être particulièrement difficile de distinguer l'EMDA et certaines formes d'encéphalite. En effet, certaines formes d'encéphalite (tels que celles causées par l'herpès ou la rougeole) peuvent se confondre avec l'EMDA en terme d'anomalies clinicopathologiques.

Aspect d’EMDA à l’IRM cérébrale
Cette jeune fille a présenté une hémiparésie gauche d’installation progressive, après avoir eu une infection virale, une semaine auparavant. Cette IRM montre des lésions bilatérales asymétriques avec un aspect d’anneau ouvert typique de lésions de démyélinisation. Notez que la diffusion restreinte n'est pas perçu au centre (habituellement dans les abcès cérébraux, mais au niveau des bords de la démyélinisation. Elle a été traitée avec des corticostéroïdes, et son état s’est rapidement amélioré.

Aspect microscopique d’une biopsie cérébrale chez un patient ayant développé une EMDA post-vaccinale, notez les zones pâles de démyélinisation péri-veineuse

Aspectshistoriques
La SEP est la principale maladie auto-immune démyélinisante de l'homme. Les lésions pathologiques de la SEP ont été décrites par Cruveilhier et Carswell au début du XIXe siècle. Frerichs a été le premier à faire un diagnostic clinique de sclérose en plaques, en 1840. Alors que les études approfondies de Charcot sur les manifestations cliniques et l'histoire naturelle de la SEP résultèrent en l'établissement de critères diagnostiques pour une entité cohérente cliniques désignées sous le nom de :sclérose disséminée ou sclérose en plaques disséminée, ou maladie de Charcot . Cette condition a été reconnu dès le départ exclusivement chez les jeunes adultes. L'apparition de la SEP chez les enfants a été un sujet de discussion depuis plus de 50 ans
En 1922, Wechsler a rejeté la plupart des cas signalés dans des populations pédiatriques, mais a déclaré que « d'authentiques cas de SEP chez les enfants, en dépit de leur rareté, peuvent se produire », après que quelques cas isolés en pédiatrie concernant des petits groupes d'enfants touchés aient été signalés. Toutefois, durant cette ère pré-tomodensitométrique, il y avait peu de preuves pour identifier les caractéristiques distinctives de la condition chez les enfants. En 1948, Kabat et ses collègues signalèrent une augmentation des immunoglobulines oligoclonales dans le liquide céphalo-rachidien de patients atteints de SEP, en fournissant des preuves tangibles d'une nature immunologique inflammatoire de la maladie. En 1965, les critères diagnostiques de Schumacher et ses collègues établirent l'âge du début de la SEP comme étant entre 10 et 59 ans, en reconnaissant que cette condition peut en effet se produire en dessous de l'âge de 16 ans.
Une meilleure compréhension de l'histoire naturelle de la SEP au cours des 40 dernières années a été rendue possible par des progrès importants en neuro-immunologie, génétique moléculaire et la biochimie, ainsi que le développement de l'imagerie par résonance magnétique (IRM), qui ont permis une visualisation anatomique précise de la matière blanche in

vivo. En conséquence, les causes métaboliques et infectieuses, inflammatoires de démyélinisation ont été délimités et le diagnostic de SEP chez les enfants corroboré. Plusieurs études rétrospectives ont été publiées dans les années 1980 et le début des années 1990, y compris ceux des patients adultes atteints de SEP dont les symptômes avaient été initiés au cours de l'adolescence, la description des patients prépubères a été exceptionnelle.
Aujourd'hui, il est généralement admis que la SEP peut survenir chez les enfants et même des nourrissons, bien que les caractères distinctifs de la maladie à cet âge ne soient pas bien établis. Il a été estimé qu'entre 2,7% et 5,6% des patients présentent des symptômes de SEP attribuable à la maladie avant 16 ans.
La fréquence de la SEP à partir de la petite enfance a été calculée à 0,2% - 0,7%. Selon ces pourcentages, la prévalence correspondante de la SEP pédiatrique serait de 1,35 à 2,5 pour 100.000 personnes et celle de la forme infantile précoce serait de 0,4 à 1,4 pour 100.000, même si des prévalences plus larges, allant de 0,8 à 248 pour 100.000 habitants, ont été signalées au Japon et au Canada respectivement. Les filles et les femmes sont 1,5 fois à 2 fois plus à risque que les garçons ou les hommes.
Les personnes à peau claire sont plus à risque que les individus plus fortement pigmentés.
Toutefois, la SEP peut survenir chez les enfants noirs, où il semble avoir un cours de progression rapide. La résidence dans un climat nordique avant l'âge de 15 ans confère un risque accru de développer la maladie, bien que l'immigration à un climat sud à un âge précoce réduit considérablement le risque.

Definition
Selon la définition consensuelle du groupe d'étude international de la SEP pédiatriqe (IPMSSG), la SEP pédiatrique peut être diagnostiquée après deux épisodes cliniques de démyélinisation du SNC qui sont séparés par au moins 30 jours. Pas de limite d'âge inférieure n'est spécifiée. Selon ces définitions, les critères de Barkhof d'IRM cérébrale chez l'adulte peuvent être utilisés pour satisfaire l'exigence de dissémination des lésions dans l'espace. Trois des quatre caractéristiques suivantes doivent être démontrées: d'abord, neuf ou plusieurs lésions de la substance blanche ou une lésion rehaussées par le gadolinium ; en deuxième lieu, trois ou plusieurs lésions périventriculaires; en troisième lieu, une lésion juxtacorticale et la quatrième, une lésion infratentorielle. Ces critères IRM adultes n'ont cependant, pas été validés chez les enfants. La combinaison d'anomalies cytochimiques au liquide céphalo-rachidien (LCR) et deux lésions à l'IRM, dont l'une doit se situer dans le cerveau, peuvent aussi répondre aux critères de diffusion dans l'espace. Le LCR doit indiquer, soit au moins deux bandes oligoclonales (CCO) ou un index en IgG élevé.
L'IRM peut aussi être utilisée pour satisfaire des critères de dissémination dans le temps suivant la poussée clinique initiale, même en l'absence d'une nouvelle rechute démyélinisante cliniquement manifeste. De nouveaux foyers hypertenses en T2 ou rehaussés par le gadolinium doivent se développer 3 mois ou plus après l'épisode clinique initial.
De manière importante, un épisode compatible avec les caractéristiques cliniques de l'encéphalomyélite disséminée aiguë (EMDA) ne peut actuellement être considéré comme une première poussée de SEP . Des critères diagnostiques précis sont disponibles afin de clarifier la distinction clinique entre l'EMDA et une première poussée de sclérose en plaques, soulignant la nécessité de biomarqueurs précis qui permettent de distinguer les conditions démyélinisantes monophasiques des autres récurrentes.

Incidenceetprevalence
La prévalence mondiale de la SEP pédiatrique est inconnue, mais les données sont disponibles en provenance de pays individuels ou des centres de SEP. Une large étude de 17.934 adultes atteints de SEP à partir de 13 centres participants en France et en Belgique ont identifié 394 individus (2,2%) chez qui l'installation de la SEP s'est faite à l'âge de 16 ans ou moins. D'autres études utilisant de grands ensembles de données, y compris 4399 cas d'un centre des États-Unis à Boston, 3375 cas d'Italie, et 3.223 cas en provenance du Canada, ont trouvé des taux de prévalence de SEP à début précoce, allant de 3,1 à 4,4% de tous les cas de SEP. Les âges seuils dans ces études étaient de 18 ans dans l'étude de Boston et 16 ans dans les études italiennes et canadiennes.
L'incidence mondiale de la SEP pédiatrique est également inconnue, mais une étude du Canada évaluant les conditions démyélinisantes survenant avant l'âge de 18 ans, incluant la SEP, la neuromyélite optique, l'EMDA, la myélite transverse compléte, et la névrite optique récurrente, a rapporté une incidence de 0,9 pour 100.000 individus. Pris ensemble, les cas pédiatriques de SEP représentent un petit mais important pourcentage de la population SEP. Que l'incidence de la SEP chez les enfants ait augmenté au cours des dernières décennies, comme cela a été signalé chez les adultes, n'est pas claire.

Aspectsdemographiques
Le ratio fille : garçon de la SEP pédiatrique varie selon l'âge. En dessous de l'âge de 6 ans, le ratio filles / garçons est de 0.8:1. Ce rapport augmente à 1,6:1 entre les âges de 6 et 10 ans, et de 2,1:1 pour les enfants âgés de plus de 10 ans, comparativement à un ratio d'approximativement 3: 1 chez les adultes adultes.
Il existe aussi une plus grande diversité dans la SEP pédiatrique par comparaison aux adulte en terme d'origine ethnique, et de race; un fait qui n'est très claire, mais qui pourrait représenter une combinaison de facteurs génétiques et environnementaux, ainsi que l'évolution des facteurs démographiques régionaux qui affectent cette tranche d'âge.

Pathogenese
La SEP est clinicopathologiquement définie comme une maladie inflammatoire démyélinisante primaires du SNC. Bien que son étiologie est inconnue la SEP est largement considérée comme une maladie auto-immune impliquant principalement une réponse immunitaire cellulaire anormale à de potentiels autoantigènes de la myéline centrale.
Au fil des années, deux modèles animaux expérimentaux de la SEP ont aidé à mieux comprendre les mécanismes pathogéniques de cette condition
Les données épidémiologiques soutiennent l'association de la SEP avec un/des facteur(s) environnemental (aux) jusque là inconnus rencontrés au cours de la petite enfance qui, après des années de latence déclenchent la maladie ou contribuent à son développement. Trois grands groupes de facteurs ont été avancés dans l'immunopathogenèse de la SEP: (a) les facteurs environnementaux, en particulier la persistance d'une infection virale, (b) des facteurs immunologiques impliquant des mécanismes auto-immunes avec une perte de tolérance à la myéline des antigènes ou mimétisme moléculaire des antigènes viraux et certaines protéines envers la myéline, et (c) les facteurs génétiques induisant une prédisposition génétique à un dysfonctionnement immunitaire.
Ainsi Peu de données sont disponibles sur l'immunophysiopathologie sous-jacents de la SEP pédiatrique, car aucune étude approfondie n'a été réalisée. En outre, aucune étude systématique n'a comparé l'immunophysiopathologie de la SEP pédiatrique avec celle des adultes . Une étude publiée en 2008 d'une large cohorte d'enfants atteints de démyélinisation inflammatoire du SNC, de diabète de type 1 ou de lésions du SNC a démontré que les enfants avec à ces conditions exhibaient des réponses accrues de la part cellules T à un large éventail d'autoantigenes. Les enfants atteints de maladies auto-immunes ou de lésions du SNC ont également montré des réponses anormales des lymphocytes T vis-à-vis

des protéines du lait de vache. Une étude plus petite évaluant les réponses des cellules T aux épitopes de la protéine basique de myéline (MBP) et la myéline oligodendrocyte glycoprotéine (MOG) chez l'adulte et les patients pédiatriques atteints de SEP ont trouvé des réponses similaires à certains peptides spécifiques- principalement MBP83-102, MBP139-153, MBP146-162, MOG1-26, MOG38-60 et-87-MOG63 dans les deux groups. De manière intéressante, les réponses aux MBP foetaux ont été minimes, et étaient encore une fois similaires dans les deux groupes. Une analyse tétramèrique, qui permet la détection d'auto-anticorps contre des protéines repliées, a révélé que 20% des enfants atteints d'EMDA, mais aucun avec une SEP pédiatrique, comportait une élévation des anticorps anti-MOG.
Les études du LCR ont démontré que les enfants de moins de 11 ans présentent un profil cellulaires distinct par rapport à leurs homologues adolescents (D. Chabas et al., travaux non publiés). Comparativement aux enfants de 11 ans ou plus, les jeunes enfants avec leur premier épisode de SEP étaient plus susceptibles d'avoir un LCR sans Bandes oligoclonales (BOC -) et sans index IgG élevé, et avaient un pourcentage plus élevé de neutrophiles dans leur LCR, suggérant l'activation prédominante de la réponse immunitaire innée, par opposition à l'activation typique de la réponse adaptative vue chez les patients plus âgés.
Des études examinant marqueurs d'atteinte axonale dans le LCR ont montré des changements minimes chez la plupart des enfants atteints de SEP; Cependant un sous-groupe avec des symptômes cliniques importants au moment de l'examen du LCR présentait des niveaux élevés de protein tau. Les implications de cette découverte ne sont pas claires.

Facteursenvironnementaux
Il a été démontré que les facteurs environnementaux jouent un rôle central dans la susceptibilité des adultes. Les facteurs pertinents comprennent la latitude de résidence du patient, l'exposition virale, en particulier, le virus d'Epstein-Barr (EBV), tabagisme, et le bilan de la vitamine D.
De nombreux chercheurs considèrent une infection virale comme le facteur environnemental le plus probable pouvant expliquer la pathogenèse de la sclérose en plaques. La preuve indirecte appuyant cette théorie provient de la distribution particulière de la maladie, avec des zones de risque élevé et faible, des études sérologiques, l'isolement des protéines virales et de matériel génomique du cerveau de patients atteints de SEP, et différents modèles expérimentaux viraux entraînant des lésions du SNC similaire à la SEP. Les virus probablement liée à la SEP comprennent l'herpèsvirus (en particulier l'herpès humain de type 6), virus d'Epstein-Barr, paramyxovirus, et rétrovirus. La récente découverte du rétrovirus endogène humain (HERV)-W a une particule extracellulaire, nommé HSRV, qui est associée à la SEP. Des études récentes ont montré que les niveaux de HSRV dans le LCR sont liées au degré d'inflammation du SNC, et, même si cela était un épiphénomène, il pourrait être utilisé comme marqueur pronostique clinique de la SEP précoce.
Détermination de l'importance de l'exposition virale chez l'adulte atteint de SEP est très difficile, étant donné le décalage entre l'exposition et l'apparition de la maladie. En outre, à l'âge adulte, la plupart des personnes ont été en contact avec les virus les plus courants, mais seulement une fraction minuscule de la population aura développé une SEP. La population pédiatrique offre une occasion unique d'étudier le rôle des virus dans la SEP , étant donné l'étroite relation temporelle entre l'infection et l'apparition de la SEP, et le fait que les enfants sont moins

susceptibles que les adultes à être exposés à un large éventail de virus. Banwell et al. A comparé 137 enfants avec une SEP établie en Amérique et en Europe avec sujets contrôles de même âge. Aucune différence n'a été observée entre les deux groupes en ce qui concerne la séropositivité au cytomégalovirus, au virus de l'herpès simplex de type 1, au virus varicelle-zona (VZV) et au parvovirus B19, mais la séropositivité à EBV a été associée à une probabilité presque trois fois plus élevée d'installation de la SEP. De même, Pohl et al. a étudié 147 patients avec une SEP pédiatrique et les a comparés à des sujets controles, il a constaté que la séropositivité pour l'EBV a été plus fréquente chez les patients que chez les témoins (99% versus 72%, P = 0,001) .
Les données du réseau de SEP pédiatrique des États-Unis d'Amérique confirment aussi l'association importante entre l'EBV et l'augmentation de la susceptibilité vis-à-vis de la SEP pédiatrique. Cette association semble être plus forte pour l'antigéne EBV nucléaire 1 (EBNA1) que pour l'antigéne EBV de la capside virale, et le demeurent après ajustement de l'âge et de la race.
Mikaeloff et al. a évalué 137 enfants atteints de SEP et 1.061 sujets contrôles pour des épisodes cliniques de varicelle. 77% de la population atteinte de SEP avait des antécédents de varicelle, contre 85% de la population contrôle (odds ratio 0,58, 95% IC 0,36 à 0,92), suggérant un possible effet protecteur de VZV.
Outre les expositions virales, des inquiétudes ont été soulevées quant à l'utilisation de la vaccination -plus récemment, le vaccin antihépatite B- et le développement ultérieur de la SEP. Mikaeloff et al. a conclu qu'il n'y avait pas de risque accru de développer un premier épisode de SEP durant l'enfance jusqu'à 3 ans après la vaccination dans la population françaiss, ces auteurs ont, cependant, montré une tendance pour le vaccin contre l'hépatite B à augmenter le risque de SEP dans le long cours. Ces résultats

doivent être confirmés dans des études plus larges. Une seconde étude des mêmes auteurs n'a observé aucune augmentation du taux de rechute après un premier épisode de démyélinisation inflammatoire du SNC dans l'enfance lorsque les patients étaient vaccinés contre l'hépatite B ou la tétanos. En outre, le risque d'installation de la SEP ne semble pas augmenter après la vaccination, bien que l'étude n'était pas suffisamment approfondie pour pouvoir détecter une légère augmentation du risque.
Le même groupe a évalué le risque d'apparition de la SEP par rapport à l'exposition à du tabagisme passif. Les chercheurs ont comparé 129 cas atteints de SEP pédiatrique en France avec 1.038 sujets contrôles appariés pour l'âge, le sexe et le lieu de résidence. Le risque d'un premier épisode de sclérose en plaques chez les personnes exposées au tabagisme parental a été trouvé à plus de deux fois supérieur que chez les individus dont les parents ne fument pas, et était encore plus élevé pour les personnes ayant une exposition prolongée de 10 ans ou plus.
Chez les adultes, de faibles niveaux de vitamine D ont été associés à une susceptibilité plus élevée de SEP. Cette relation n'a pas encore été confirmée chez les enfants, bien que les niveaux de la 25-hydroxyvitamine D3 soient inférieurs à la normale dans la grande majorité des patients pédiatriques atteints de SEP.
Des études complémentaires sont nécessaires pour étudier les mécanismes précis qui mènent de l'exposition environnementale à l'apparition de la maladie.

Facteursimmunologiques
Il est généralement admis que, en plus à une infection virale au début, il doit y avoir une réaction auto-immune qui attaque certains des composants de la myéline. La plupart des patients présentent une réactivité des lymphocytes T à un certain nombre d'antigènes de la myéline, ce qui suggère que le temps qu'un patient développe une la SEP, il ya eu épandage d'épitope avec réactivité à de multiples épitopes de myéline. Les cellules T capables de réagir contre les antigènes de myéline sont normalement présentes dans le système immunitaire. Ces cellules du thymus se sont échappées des mécanismes de contrôle tels que la délétion clonale.
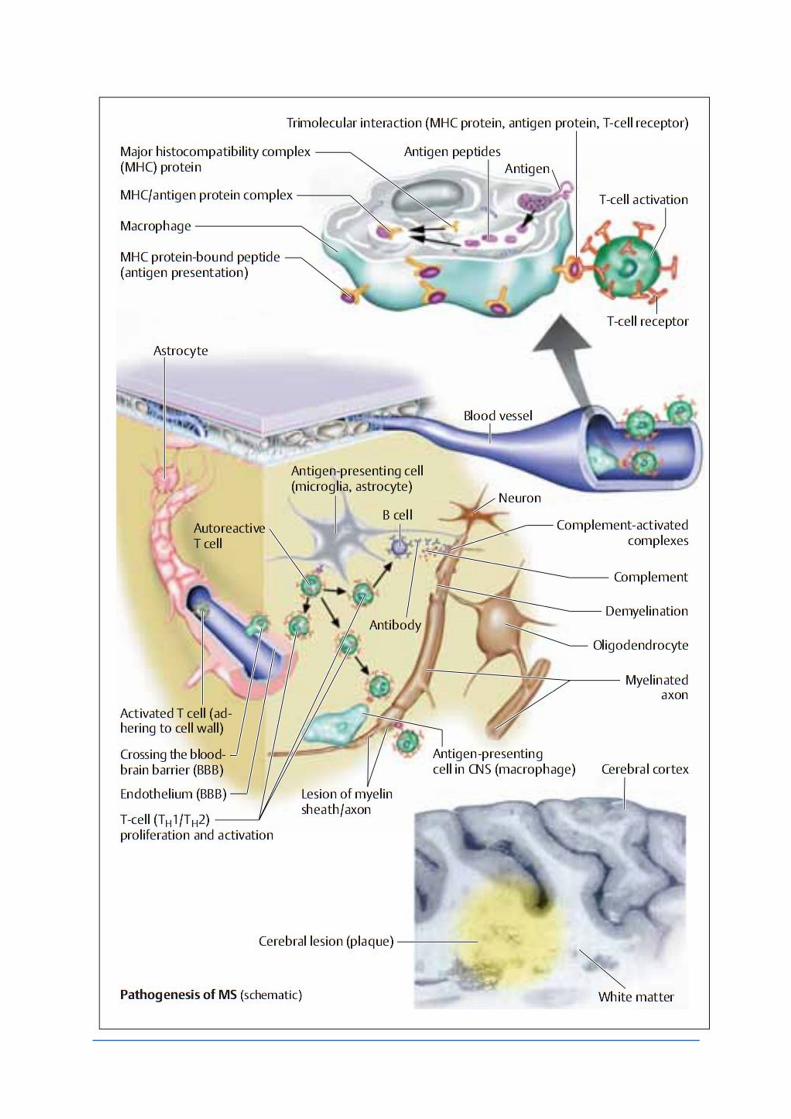
Une cellule CD4 + T nontolérante activée en périphérie reconnaît son autoantigène au sein du parenchyme du SNC grâce aux molécules de classe II du CMH exprimées et par les cellules locales gliales présentatrices d'antigène, et les cellules dendritiques , qui engagent les cellules T vers un phénotype Th1 . Les Cell Th1 activées, vont léser la myéline et induire la libération de nouveaux autoantigènes pour le CNS.
Les cytokines sécrétées pro-inflammatoires, comme l'IFN-gamma et le TNF-alpha, recruteent des chémokines supplémentaires non spécifique des cellules inflammatoires ainsi que des Cell B spécifiques formant des Ac antimyeline , exacerbant les lésions tissulaires.
Enfin, la clairance apoptotique des cellules T et leur conversion vers un phénotype Th2 module l'issue de la lésion. Des cellules supplémentaires sont nécessaires pour le développement de lésions de SEP typiques, telles que les cellules CD8+ cytotoxiques , qui montrent une expansion plus importante clonale au sein de plaques de SEP et une meilleure corrélation que les cellules CD4 + à l'étendue des lésions axonales aiguës.
La cascade d'événements inflammatoires qui aboutit à une démyélinisation des axones dépend de l'activation des lymphocytes T périphériques. Les Interactions des lymphocytes avec l'endothélium

vasculaire sont nécessaires pour le trafic des lymphocytes dans le SNC . Les molécules d'adhésion cellulaire jouent un rôle crucial dans ce processus et sont essentielles dans l'infiltration des lymphocytes dans le système nerveux central à travers la BHE. Une telle molécule est la molécule d'adhésion intercellulaire-1 (ICAM-1), une glycoprotéine qui interagit avec de nombreux alpha-intégrines telles que des lymphocytes functionnelle à antigène associé-1 (LFA-1) sur les cellules T et sur CD11b/CD18 monocytes . D'autres molécules d'adhérence médiant des interactions entre les lymphocytes et les cellules endothéliales sont de type very late antigen 4/molécule d'adhésion vasculo cellulaire-1 (VCAM-1), la L-sélectine (sur les lymphocytes) et E-sélectine (sur les cellules endothéliales).

Facteursgenetiques
Les facteurs génétiques ont également été supposée être contributeurs à la pathogenèse de la sclérose en plaques, basée sur différentes études. Les taux de prévalence de la SEP chez les parents au premier degré de personnes atteintes de SEP sont environ 20 fois supérieures à celles des autres individus de la même région. Le risque de développer une SEP dans la population générale de 1 pour 1.000 et augmente de 20 à 40 sur 1.000 pour les parents au premier degré . Dans une série pédiatrique de 44 enfants atteints de SP, 10 (23%) avaient une histoire familiale positive de la SEP chez un parent au premier degré (1 enfant), un parent au deuxième degré (5 enfants), ou leur famille élargie (4 enfants ). Ceci est très similaire aux 15-20% des antécédents familiaux positifs signalés dans une série pour des adultes atteints de SEP. Les vrais jumeaux ont 25-35% de taux de concordance pour la SEP, comparativement à 0,5% pour les enfants (peut-être beaucoup plus élevé pour les filles de mères atteintes de SEP), 0,6% pour les parents, 1,2% pour les frères et sœurs, et 2% à 4% pour les jumeaux dizygotes .
Guidé par les considérations liées à l'immunopathogenèse supposée de la maladie, les recherches de gènes candidats ont été axées sur les gènes delymphocytes T et des protéines de la myéline. Des résultats négatifs ont été trouvés pour les gènes des récepteurs des cellules T, complément, les récepteurs de chimiokines, interférons, d'autres récepteurs de cytokines, et l'antigène des lymphocytes T cytotoxiques 4 (CTL4): un gène situé dans la région HLA de classe I. Il existe une association très significative de la SEP avec HLA-DR2 et un affaiblissement de liaison avec HLA-A3 et B7 chez les Blancs. D'autres candidats tels que HLA de classe II ont donné des résultats positifs, surtout quand typés en HLA DRB1 * 1501, DQA * 0102, DQB1 * 0602 à haplotype étendu, bien que le risque relatif estimé est seulement 2 à 4 .

HLA-DR15 a été spécifiquement associée à une apparition plus précoce de la SEP dans une grande étude de plus de 900 patients.
L'analyse récente des marqueurs de polymorphisme microsatellites dans des populations de tailles et d'ethnies différentes a identifié des régions chromosomiques jouant un rôle dans la susceptibilité MS: chromosome 6 dans le CMH et les chromosomes 3q21; q24, 18p11, et 17q22; q24; une méta-analyse a cependant , indiqué que les éléments de preuve les plus élevés se situaient pour la liaison à 17p11 .
Toutes les études sont concordantes avec la conclusion que HLA contribue, quoique modestement, à la sensibilité générale et qu'un nombre relativement important d'autres génes CMH et non-HMC ayant des effets épistatiques individuellement petits peuvent être responsables d'une prédisposition à la SEP. Cela implique un certain nombre de gènes en interaction entre eux et suggère une hérédité polygénique de la maladie. Par exemple, l'haplotype ApoE4 est également un facteur génétique déterminant la gravité de la SEP; ces patients ont un risque deux fois plus élevé de développer un « trou noir » sur l'IRM et ont un taux environ cinq fois plus élevé de développer une atrophie cérébrale. Il est probable que l'expression des gènes impliqués dépende de facteurs environnementaux.
En résumé, il ya créance dans le principe plutot ancien de la pathogenèse de la SEP qui stipule que la maladie est produite par un agent de l'environnement agissant sur un individu génétiquement sensible dans lequel il ya des réponses immunitaires affaiblies.

Risk of Developing MS
1 sur 3 Si un jumeau identique à la SEP
1 sur 15 Si un jumeau dizygote a la SEP
1 sur 25 Si un des frères/soeurs a la SEP
1 sur 50 Si un parent ou un demi-frère/soeur a la SEP
1 sur 100 Si un cousin du 1er degree a la SEP
1 sur 1000 Si un conjoint a la SEP
1 sur 1000 Si ATCD familiaux négatifs
Risque de developer la SEP

Anatomo-Pathologie
La SEP est définie comme une maladie inflammatoire démyélinisante du système nerveux central caractérisée par une inflammation, démyélinisation, et une détérioration axonale. La principale caractéristique histologique de la maladie, la plaque de SEP, est considérée comme la lésion finale du processus inflammatoire. Les plaques de SEP reflètent un continuum de l'activité immunologique comprenant des modifications inflammatoires et cellulaires secondaire dans le cerveau touchées. Morphologiquement, les plaques sont classés comme actives, chronique active, inactive chronique, de type « ombre ». La variabilité histologique et l'âge des plaques peut faire partie d'un gradient temporospatial de modifications morphologiques qui reflète les mécanismes immunologiques différents à l'origine des lésions cellulaires.
La morphgénèse des Plaques et leur évolution sont la conséquence d'interactions de facteurs immunologiques et métaboliques, y compris les effets des cellules T cytotoxiques, des anticorps, des métabolites toxiques dérivés de monocytes activés / macrophages et troubles du métabolisme des oligodendrocytes.
Notre compréhension de la pathogenèse de la démyélinisation dans la SEP est en accord avec l'idée que les plaques pourraient représenter un point commun morphologiques où découle les différentes voies immunologique d'atteinte de la myéline et des axones.

Aspectmacroscopique
L'apparence externe du cerveau chez les patients atteints de SEP peut être normal. Dans les cas chroniques, des signes d'atrophie avec élargissement des sillons ainsi que du système ventriculaire peut être observée. Sur une coupe histologique du cerveau, il est possible d'identifier des lésions grises sur la surface du tronc cérébral, moelle épinière et les nerfs optiques et des plaques multiples de diamètre variable dans la substance blanche. Les lésions inflammatoires démyélinisantes sont généralement diffuses dans tout le névraxe, mais sont plus fréquemment rencontrées dans certains sites anatomiques corrélant à certains aspects reconnaissables de la sémiologie neurologique. Bien que la distribution des plaques varie selon les patients, les emplacements préférentiels sont les suivants: la substance blanche périventriculaire, le plancher de l'aqueduc et quatrième ventricule (tronc cérébral), pédoncules cérébelleux, la moelle épinière cervicale, et les nerfs optiques .
Des corrélats anatomiques distinctifs sont rencontrés dans certaines variantes, comme le neuromyélite optique ou la maladie de Devic. Même si dans les hémisphères cérébraux, les lésions ont une prédilection périventriculaire, une proportion importante de lésions peut être observée dans d'autres emplacements de la substance blanche centrale et la jonction substance blanche-grise. Il ya des cas où des lésions de la substance blanche dans le cortex s'étendent dans le cortex contigu fait de substance grise voire dans les noyaux gris centraux et le thalamus. Des plaques intracorticale et sous piales ont été décrites et pourraient représenter un corrélat important de séquelles neurologiques. Exceptionnellement, de volumineuses lésions pseudo-tumorales sphériques sont rencontrées, mais ces lésions sont le plus souvent considéré en relation avec la sclérose diffuse myelinoclastique (maladie de Schilder).

Histopathologie
Les plaques de MS peuvent être classés en quatre catégories selon les modifications cellulaires qui reflètent l'activité ou la quiescence de la maladie :
1. Lésions aiguës : Ce sont des plaques récentes « fraiches » caractérisées par une inflammation périvasculaire (comprenant principalement les lymphocytes et les monocytes / macrophages), de l'oedème, une infiltration de la myéline, et l'activation des cellules endothéliales . Une déplétion souvent prononcée des oligodendrocytes est présente aussi. Les cellules plasmatiques sont peu fréquentes. Il y a une préservation apparente des axones, mais un début de lésions axonales peut être présent.
2. Lésions chroniques actives: Ce sont des plaques complétement constituées marquées par l'inflammation et la démyélinisation active, généralement à leurs rebords . Les modifications morphologiques suivantes sont présentes: un infiltrat lymphocytaire périvasculaire, destruction de la myéline en cours, des macrophages chargés de myéline, déplétion des oligodendrocytes, et gliose astrocytaire. L'infiltrat lymphocytaire peut s'étendre au-delà des marges de la plaque. En apparence, les axones sont épargnés, mais des degrés variables de détérioration axonale peuvent être présents.
3. Lésions chroniques inactives: Ce sont de vieilles plaques ou des lésions de repos équivalant à des cicatrices gliales (d'où l'utilisation classique du terme sclérose). Ils sont nettement délimités de la partie de la substance blanche qui est normalement myélinisée. Il existe une importante gliose astrocytaire, une déplétion d'oligodendrocytes, et un degré variable de lésions axonales allant de la névrites dystrophique jusqu'à une transection

pure et simple des axones et des dendrites. Des touffes éparses périvasculaires de lymphocytes, monocytes et macrophages est focalement présente. Les parois des vaisseaux sanguins sont sclérosées et hyalinisées, confirmant l'antécédent inflammatoire des lésions vasculaires. Une mince bordure de la fibrose de collagène périvasculaire peut être présente dans les lésions anciennes. La barrière hémato-encéphalique est finalement perturbé .
4. Plaques d'ombre: Ils représentent une zone de taille variées et mal définies de tissu démyélinisé ou incomplètement remyélinisé entourant, voire parfois même se superposant à la plaque principale. Leur présence dans la SEP chronique est imprévisible. Il n'existe pour l'instant aucune corrélation clinique, mais ces plaques pourraient sous-tendre une voie distinctive pathogénique.
Parfois, les lésions sont si graves qu'elles se transforment en kystes. Il est classique d'observer chez un même patient des lésions à différents stades de progression (différents « âges »)

Démyélinisation dans la SEP. La coloration de Klüver-Barrera montre des tissus de couleur claire correspondant àune décoloration au niveau de de la lésion (échelle originale 1:100)

Démyélinisation dans la SEP. Le tissu de couleur brunâtre correspond aux macrophages CD68 au niveau des lésions de démyélinisation. échelle 1:100

Immunopathologie
Il y a deux éléments faisant partie intégrante des lésions de SEP: l'inflammation périvasculaire et la démyélinisation. On a longtemps émis l'hypothèse que la démyélinisation inflammatoire est le résultat d'une réponse immunitaire médiée par les antigénes myéliniques dans les gaines de myéline des axones et / ou au niveau de la myéline formant les oligodendrocytes. Les destruction de la myéline et des oligodendrocytes n'est pas uniforme dans les plaques de SEP. La morphogenèse des plaques n'est pas entièrement comprise, même si quelques preuves orientent vers une destruction précoce de la barrière hémato encéphalique et une infiltration par les monocytes et les lymphocytes, principalement les lymphocytes T. La perturbation de la barrière hémato encéphalique correspond au début des symptômes cliniques, mais une corrélation entre les symptômes et la démyélinisation inflammatoire n'est pas claire.
Quatre modèles distincts pathogénique de démyélinisation (I à IV) ont été proposées . Les deux premiers se concentrent sur la notion que l'inflammation provoque une démyélinisation par mécanismes directs et / ou indirects. Les lymphocytes contribuent au processus pathologique par réponse immunitaire à médiation humorale et cellulaire (supposés être des mécanismes directs) ou par la production de lymphokines et de cytokines (mécanismes indirects). Il s'ensuit donc que les modèles I et II présentent des similitudes remarquables aux encéphalomyélites auto-immunes à médiation cellulaire et/ou humorale. Les monocytes / macrophages contribuent au processus de démyélinisation d'une double manière. Tout d'abord, grâce à leur rôle traditionnel phagocytaires, les cellules de la lignée monocytes / macrophages, y compris les dérivés hématogène et activé des microglies du système nerveux central, sont des effecteurs puissants de la destruction de la myéline des axones et des dommages des oligodendrocytes. Les monocytes contribuent à une démyélinisation par voie de production de cytokines, de l'oxyde nitrique, et de protéases et / ou en ciblant directement les oligodendrocytes à la

frontière des lésions de SEP. Les microglies activées du SNC jouent un rôle dans les premiers stades de démyélinisation grâce à des intéractions cellulaires avec la myéline des axones sur les rebords des lésions chroniques actives de SEP.
Les deux autres modèles (III et IV) sont compatibles avec une atteinte primaire des oligodendrocytes (dystrophie) dues à une démyélinisation viro- ou toxino-induite directe, par opposition aux mécanismes auto-immuns. Au fil des ans, diverses théories ont été avancées concernant la nature des dommages des oligodendrocytes dans les lésions de SEP. On croit que ces dégâts sont occasionnés par une variété de mécanismes immunologiques, y compris des anticorps anti-MOG, cytokines produites par les monocytes / macrophages et les lymphocytes T, des immunoglobulines et des composants du complément activé, l'apoptose, et une variété d'autres facteurs cytotoxiques. Dans le modèle III il ya perte des protéines de la myéline dans les processus cellulaires des oligodendrocytes les plus distaux (périaxonaux), ce qui est associé à la mort cellulaire par apoptose. Ce mécanisme a déjà été défini comme un oligodendrogliopathie distale, et est assimilable à une blessure oligodendrogliale engagée au début de la démyélinisation hypoxique-ischémique de la substance blanche. Dans le modèle IV il existe une mort cellulaire des oligodendrocytes dans la substance blanche près des lésions actives. À cet égard, il a été démontré que les monocytes activés / microglies exprimant VCAM-1 ciblent sélectivement les oligodendrocytes à la frontière des lésions de SEP.
Les dommages axonaux sous forme de renflements axonaux (neurites dystrophiques) et transsections sont une composante majeure de la maladie. Les lésions axonales sont en corrélation avec certains paramètres de l'IRM fonctionnelle (réduction de N-acétylaspartate) et certains types de handicaps neurologiques dans la SEP. La pathologie axonale, attestée par la coloration immunohistochimique de la protéine précurseur de l'amyloïde (APP) dans des échantillons post mortem, est

plus marquée dans les lésions de SEP active que dans les plaques chroniques inactives.
Les dommages hypoxique-ischémique sont un aspect important, mais un peu sous-estimée de la neuropathologie de la SEP. Les lésions inflammatoires de la paroi vasculaire, de l'endothélium, et de la barrière hémato encéphalique induites par cellules T et monocyes ressemblent aux lésions causées par les infiltrats de cellules T angiocentriques associées aux formes du SNC du VIH-1 chez les enfants. Ce dernier étant composé par un oedème et des troubles de la microcirculation cérébrale, peut transmettre des dommages à la myéline, les axones et les oligodendrocytes.
Les perturbations dans le métabolisme oxydatif dans la SEP dans le cadre de lésions d'hypoxie-ischémie peuvent résulter de facteurs vasculaires (par exemple, l'inflammation vasculaire) et / ou la libération de métabolites toxiques associée à une hypoxie-ischémie. Fait intéressant, des lésions ischémiques manifestes ont été démontrées dans des cas graves de SEP aiguë de type Marburg, de sclérose concentrique de Bal, et de neuromyélite optique. On suppose que certaines lésions actives de SEP peuvent représenter une forme lésion hypoxique d'une zone de pénombre ischémique de la substance blanche. Et la preuve métabolique de ces lésions tissulaires hypoxiques dans la SEP telle que la libération d'excitotoxines, les espèces réactives de l'oxygène et l'oxyde nitrique accorde plus de crédibilité à ce postulat.
En résumé, la compréhension actuelle des mécanismes de neuropathogénétiques dans la SEP soutient l'hypothèse selon laquelle la démyélinisation de la substance blanche, l'atteinte axonale, la transection dendritique, et la perte de l'apoptose des neurones dans le cortex cérébral contribuent à un dysfonctionnement neurologique chez les patients atteints de SEP.


Top: Schematic depiction of the clinical evolution ofMS by a clinical scale
(EDSS, red line); the frequency of inflammatory events when studied by MRI (T1
lesions with contrast showing blood-brain barrier opening, blue arrows); T2 lesion
load documenting all tissue damage (blue line); brain atrophy (green line). Pathology:
Main pathological characteristics of MS. On the left, perivascular inflammation with
mononuclear cells and open blood-brain barrier (courtesy of H.F. McFarland, NIB,
NINDS, NIH); on the right, demyelinated areas shown in light blue and white, and,
on the far right, axonal transactions (blue onion bulb-like structure) and segmental
demyelination (from Reference 339, with kind permission of N. Engl. J. Med.). MRI:
TypicalMRI characteristics. On the left, T1-weighted image with Gadolinium contrast
enhancement.White lesions indicate areas of fresh inflammation and open blood-brain
barrier. T2-weighted image shows the CSF-filled ventricles in white andMS lesions in
the brain parenchyma. On the right, brain atrophy with widened lateral ventricles and
cortical sulci.

Schéma illustrant les différentes étapes pathogéniques et les facteurs contribuant aux lésions tissulaires dans la SEP


Manifestationscliniques
Bauer et Hanefeld ont classés la SEP en fonction de l'âge lors de la présentation: SEP infantile précoce (SEPIP), débutant entre 1 et 5 ans; SEP infantile retardée (SEPIR), entre 5,1 et 10 ans, et la SEP juvénile (SEPJ), entre 10,1 et 16 ans.
Les caractéristiques cliniques de 51 patients pédiatriques dans une étude de cohorte qui remplissaient les critères de diagnostic de la SEP établis par Poser et ses collègues sont présentés au tableau 8.2. Chez 13 enfants la maladie avait débuté avant 5 ans, et chez 13 autres elle a été initiée entre 5 et 10 ans. Le plus jeune patient était une fille de 18 mois qui a été suivie pendant 16 ans. Jusqu'à présent, le plus jeune patient décrit dans la littérature est un bébé de 11 mois.
La plus forte prévalence de la SEP infantile chez les filles a été établie dans une étude de Göttingen qui a inclus 20 enfants, avec un ratio de 2,3:1. Dans l'expérience d'une autre institution britannique, ce ratio était de 1:3.3 dans SEPIP, 1:1,2 dans SEPIR, et 1.8:1 dans SEPJ, ce dernier ratio étant presque similaire à ceux des adultes (qui vacille entre 1,5:1 à 1,9:1). Cette découverte suggère que les changements hormonaux liés à la puberté peuvent interagir avec le système immunitaire et neuroendocrinien et influer sur le cours de certaines maladies auto-immunes en modifiant la réponse immunitaire humorale et cellulaire.

les données cliniques et symptômes au moment de la présentation initiale
de la sclérose en plaques chez 51 enfants n = 13
SEPIP n = 13 SEPIR
n = 25 SEPJ
% Total
Données cliniques Âge (ans, moyenne) 3,4 7,1 13,5 Sexe (ratio féminin : masculin)
3:10 06:07:00 16:09:00
Monosymptomatique 2 2 19 45 Polysymptomatique 11 11 6 55 Symptômes syndrome pyramidal 12 9 16 73 Paresthésie 1 2 16 37 Myélopathie 4 5 6 29 Atteinte du tronc cérébral 6 5 3 29 Altération de la conscience 5 6 3 27 Ataxie 6 5 2 25 Perte de vision * 3 3 4 20 Signes méningés 5 3 2 20 Convulsions** 3 0 0 6 Aphasie 1 1 0 4 signes extrapyramidaux 0 1 1 4 Patients pédiatriques du Children's Hospital Dr. Garrahan JP, Buenos Aires, l'Argentine. SEPIP, SEP infantile précoce; SEPIR, sclérose en plaques rétardée; SEPJ, la sclérose multiple juvénile. * :une névrite optique, bilatérale chez 8 patients, unilatérale chez 2. ** : convulsions partielles secondairement généralisées.
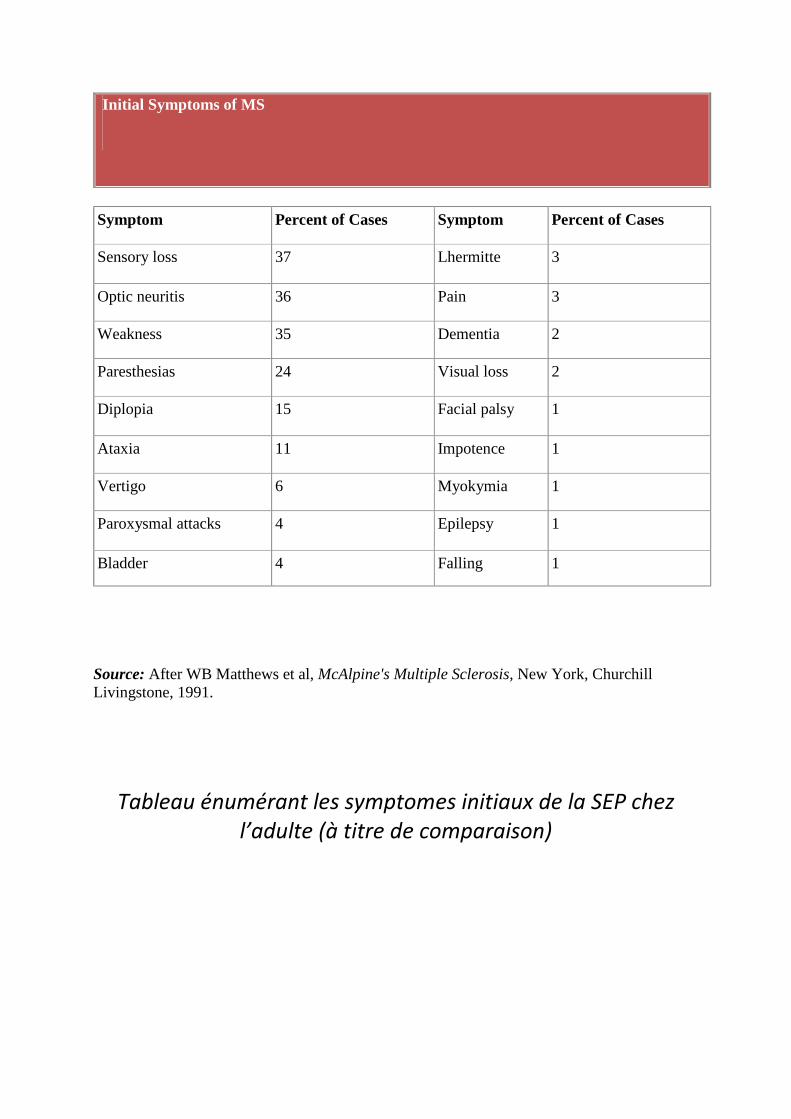
Initial Symptoms of MS
Symptom Percent of Cases Symptom Percent of Cases
Sensory loss 37 Lhermitte 3
Optic neuritis 36 Pain 3
Weakness 35 Dementia 2
Paresthesias 24 Visual loss 2
Diplopia 15 Facial palsy 1
Ataxia 11 Impotence 1
Vertigo 6 Myokymia 1
Paroxysmal attacks 4 Epilepsy 1
Bladder 4 Falling 1
Source: After WB Matthews et al, McAlpine's Multiple Sclerosis, New York, Churchill Livingstone, 1991.
Tableau énumérant les symptomes initiaux de la SEP chez l’adulte (à titre de comparaison)

La présentation clinique la plus fréquente chez les enfants atteints de SEPIP et SEPIR est une encéphalopathie aiguë avec des déficits multifocaux, le plus souvent une hémiparésie aiguë avec des signes pyramidaux uni- ou bilatéraux(81%). Les autres signes neurologiques et les symptômes présents chez 30% à 40% des patients comprennent un état mental altéré, des céphalées, des vomissements, une atteinte du tronc cérébral, une ataxie cérébelleuse, et signes méningés. La plupart des enfants se remettent de ce tableau dramatique après corticothérapie ou peuvent laisser de légers déficits résiduels.
Les patients atteints de SEPJ, toutefois, se présentent avec un syndrome démyélinisant isolé, le plus fréquent étant l'hémisyndrome sensoriel (64%), associé ou non à des signes moteurs, et le plus souvent sans signes d'encéphalopathie aiguë diffuse. La perte aiguë de la vision due à une névrite optique est observée plus fréquemment comme un premier symptôme chez les enfants de moins de 10 ans (23%) que chez ceux avec une SEPJ (16%). Les crises sont peu fréquentes (6%), et ils existent seulement chez les enfants de moins de 5 ans, dans le cadre de l'encéphalopathie aiguë. Néanmoins une fréquence des crises, aussi élevée que 22% a été rapporté dans les formes infantiles précoces de la SEP. Les constatations sensorielles, comme déjà indiqué, est une observation fréquente chez ces patients, et doit faire évoquer le diagnostic de la SEP. La paraparésie est souvent associée à des anomalies cordonales postérieures, et est souvent négligée chez les adolescents en raison d'un examen neurologique peu fouillé. Parfois, les patients sont témoins phénomènes sensitifs paroxystiques et transitoires comme la sensation d'une bande de constriction tronculaire ou une aggravation momentanée et inexpliquée de troubles sensoriels préexistants, associés ou non à une sensation de faiblesse musculaire, le signe de l'Hermitte (sensations douloureuses à type de choc électrique se propageant vers le bas le long de la colonne vertébrale et les extrémités) , et le phénomène d'Uhthoff [apparition transitoire ou aggravation d'un dysfonctionnement neurologique en liaison avec l'exercice ou l'exposition à des températures ambiantes chaudes (atmosphérique ou sous la douche ou le bain)] .
Les enfants atteints de SEP présentent plus fréquemment une forme polysymptomatique de la maladie (43%) que monosymptomatique (36%), alors que le contraire est vrai pour les adultes (35% et 65%,

respectivement). Les enfants et les adultes développent généralement des signes pyramidaux, des paresthésies ou une myélopathie, mais les adultes ont une incidence élevée de signes bulbaires et cérébelleux et manifestent rarement une encéphalopathie aiguë.
D'autres symptômes que les patients pédiatriques peuvent manifester à la suite de la SEP sont la fatigue, la spasticité, les difficultés scolaires, et la labilité émotionnelle. La fatigue est décrite comme un manque subjectif d'énergie physique ou mentale d'une gravité suffisante pour interférer avec la capacité de l'enfant pour compléter ses devoirs d'école, s'engager dans des activités extrascolaires, ou interagir socialement avec ses pairs. La spasticité est l'un des symptômes les plus courants de la SEP, elle freine la mobilité fonctionnelle, et est liée à l'évolution de la maladie. C'est pourquoi elle est plus importante chez les adultes que chez les enfants. Les troubles cognitifs ont été démontrés chez les patients pédiatriques atteints de SEP. Les adolescents avec la SEP rapportent des difficultés à gérer des concepts intellectuels complexes et le multitache. L'impact psychologique de la SEP sur l'enfant ou l'adolescent peut être profond, bien que la plupart des enfants gérent assez bien la situation à l'annonce de leur diagnostic. Les manifestations neuropsychiatriques les plus sérieuses sont observés chez les adultes. Les troubles sphinctériens peuvent être un réel problème, mais ils sont beaucoup moins fréquents chez les enfants que chez les patients adultes. L'épilepsie apparaît généralement tard dans l'évolution de la maladie et, par conséquent, n'est généralement pas observée chez les enfants atteints de SEP. Parfois, des convulsions peuvent annoncer l'apparition de la maladie ou une rechute, et dans ce cas ont un bon pronostic.


Structure du neurone et mécanisme de mdémyélinisation,un seul oligodendrocyte pour fournir des internoeuds pour 60
axones avoisinants . Le neurone du bas a été partiellement démyélinisé, et est entouré par des lymphocytes T qui
sécrétent des cytokines inflammatoires (interleukin-2 [IL-2], interferon gamma [IFN-γ], and tumor necrosis factor—α
[TNF-α]), et par des macrophages qui sont en train d’ôter la
myélinede l’axone ; les macrophages vont contenir des
débris myéliniques dans des vacuoles phagocytaires

Diagnosticpositif
Le diagnostic de sclérose en plaques chez l'enfant est essentiellement clinique. Il est soutenu par l'examen neurologique, qui révèle des signes d'atteinte de la substance blanche avec une dissémination temporo-spatiale bien définie, suivant les critères diagnostiques de Poser et ses collègues. Des études supplémentaires, y compris l'IRM, l'étude du LCR, et les potentiels évoqués cérébraux, complètent le processus diagnostique, conformément aux nouvelles recommandations du Groupe d'experts international sur le diagnostic de SEP
Critères de diagnostic de la sclérose en plaques
Présentation clinique Données supplémentaires requises pour le Diagnostic de SEP
Deux ou plusieurs poussées, preuves cliniques objectives de deux lésionsou plus
Aucun a
Deux ou plusieurs poussées, preuve clinique objective d'une lésion
Diffusion dans l'espace démontrée par l'IRM b ou Deux lésions IRM ou plus compatibles avec SEP + LCR positif c ou Attendre nouvelle poussée clinique sur un site différent
Une poussée; preuves cliniques objectives de deux lésions ou plus
Diffusion dans le temps, démontré par l'IRM d ou Deuxième poussée clinique
Une poussée; preuves cliniques objectives d'une lésion (présentation monosymptomatique; syndrome clinique isolé)
Diffusion dans l'espace démontré par l'IRM b ou deux lésions ou plus à IRM compatibles avec SEP + LCR positif et Diffusion dans le temps, démontré par l'IRM d ou Deuxième poussée clinique
progression neurologique Insidieuse évocatrice de la SEP
LCR positif c et Diffusion dans l'espace, démontré par

(a) neuf lésions ou plus en T2 dans le cerveau ou
(b) deux lésions ou plus de la moelle épinière, ou (c) quatre à huit lésions dans le cerveau, plus une lésion de la moelle épinière ou Anormale PEV e associée à
*quatre à huit lésions cérébrales,
*moins de quatre lésions cérébrales plus une lésion de la moelle épinière à l'IRM et Diffusion dans le temps, démontrée par l'IRM d ou progression continue pendant 1 an
SEP, la sclérose en plaques; IRM, imagerie par résonance magnétique; LCR, liquide céphalo-rachidien; PEV, potentiel évoqués visuels. a pas de tests supplémentaires sont nécessaires, mais si l'IRM et les études du LCR sont négatives, d'autres diagnostics doivent être considérés. b IRM doit satisfaire les critères de la diffusion spatiale (voir texte). c présence de bandes oligoclonales différente de tous les groupes du sérum, ou indice élevé d'immunoglobuline. d Présence d'une nouvelle lésion rehaussée par le gadolinium au moins 3 mois plus tard. e-tumoraux potentiels évoqués visuels: onde retardée à forme bien préservé. De WI McDonald, Compston A G Edan, et al. critères diagnostiques recommandés de la sclérose en plaques: des lignes directrices du Groupe d'experts international sur le diagnostic de la sclérose en plaques. Ann Neurol 2001; 50:121 - 127. Avec autorisation.

Etudesimmunologiques
Les anomalies immunologiques peuvent être trouvées aussi bien dans le sérum que dans le LCR. Le dysfonctionnement de l'immunité cellulaire est représenté par une augmentation dans la circulation du ratio CD4 + T-helper/inducteur : CD8 + à cellules T-suppresseur/cytotoxique au cours des poussées de SEP. Les marqueurs moléculaires de l'apoptose (CD95-régulateur, les caspases 8 et 10), et les cytokines IL-10 et TNF-alpha présentes dans les cellules mononucléées du sang corrélent inversement avec activité inflammatoire détectée à l'IRM, ce qui indique que la voie CD95-dépendante joue un rôle complexe dans la régulation de la survie des cellules immunitaires activées durant la SEP.
Les anomalies du LCR des patients atteints de SEP sont caractéristiques, bien que ni spécifique ni pathognomomiques. Pendant la phase aiguë de démyélinisation, il existe une pléocytose lymphocytaire de degré variable (30% à 70%), généralement pas plus de 50 cellules / mm 3. Le dysfonctionnement de l'immunité humorale est une constante chez les patients atteints de SEP et est représenté par la présence quasi universelle dans le LCR des (a) immunoglobulines oligoclonales détectable par électrophorèse, (b) des taux élevés de synthèse et de concentrations dans le LCR d'immunoglobuline G (IgG) générées en intrathécale et d'IgM à spécificité épitopiques inconnue ou variée, et (c) des niveaux accrus de composants des immunoglobulines tel que les chaînes kappa. La découverte de bandes oligoclonales (BOC) d'IgG présentes dans le LCR et absentes dans le sang a été décrite dans 65% à 95% des patients adultes atteints de SEP. La fréquence des BOC chez les enfants atteints de SEP est variable, ce qui est probablement le reflet des différentes méthodes de mesure utilisées. Avec l'approche

isoélectrique suivie par une immunofixation, Tenembaum et ses collègues ont détecté un taux de LCR BOC positif dans 27 des 51 (53%) des enfants atteints de SEP. dans leur étude Le taux de détection pour les trois formes cliniques, SEPIP, SEPIR, SEPJ, ont été, respectivement, 46%, 38% et 64%. Récemment, l'analyse dans le sérum d'anticorps antimyeline, notamment anti-MOG et anti-MBP, a été montré relativement efficace pour prédire la progression de syndromes démyélinisants isolés en une SEP totalement établie. Rejdak et collaborateurs avaient signalé une augmentation dans le LCR des niveaux de métabolites d'oxyde nitrique (taux de nitrites et de nitrates) dans les adultes atteints de SEP, qui corrélaient avec la progression clinique et IRM de la maladie sur une période de 3 ans. Sueoka et ses collègues ont trouvé que la synthèse sélective dans le LCR d'anticorps dirigés contre la ribonucléoprotéiques B1 chez les adultes atteints de SEP rémittente, suggérant qu'ils pourraient être un marqueur de maladie pour la SEP.
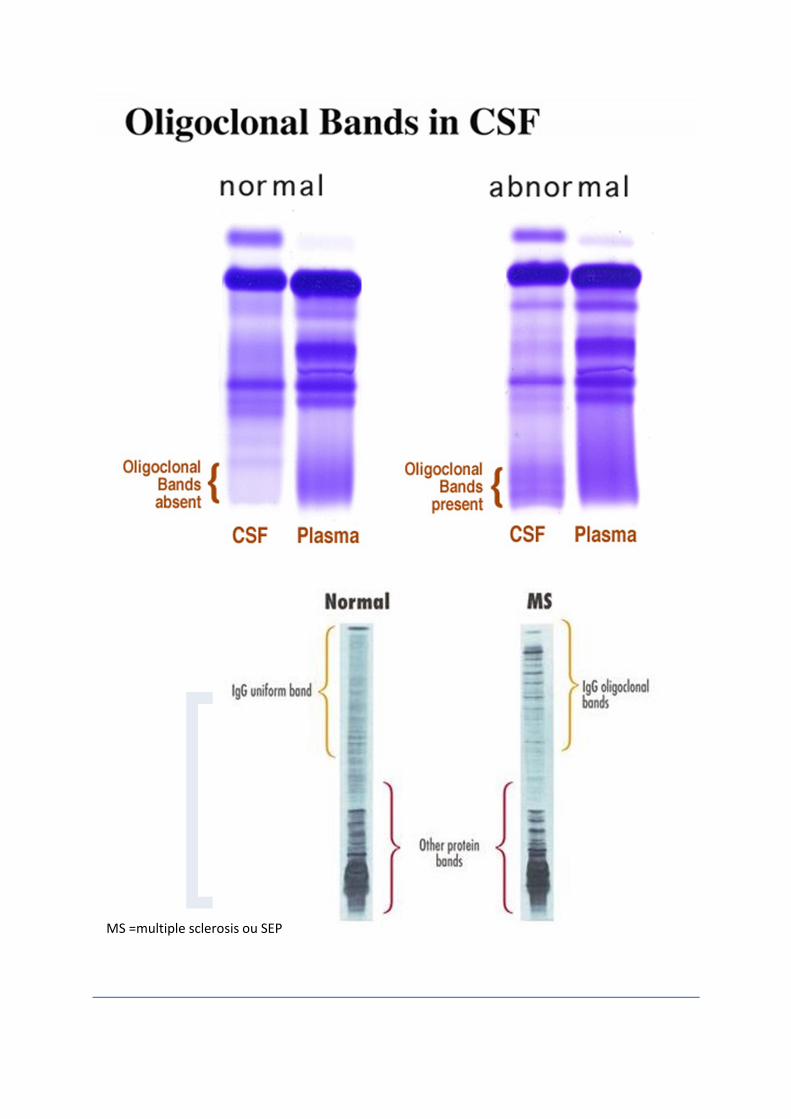
MS =multiple sclerosis ou SEP

Neurophysiologie
Les études neurophysiologiques ne sont pas spécifiques. L'électroencéphalogramme (EEG) peut montrer des changements correspondant à l'épilepsie diagnostiquée chez certains patients. Un état de mal épileptique aphasiques, une épilepsie partielle continue, et des décharges épileptiformes latéralisées périodiques ont été rapportés. Le traitement prolongé par corticoïdes induit des modifications du tracé de sommeil de l'EEG chez les patients atteints de SEP similaires aux changements observés chez les patients présentant un épisode dépressif aigu. Les potentiels évoqués (PE) fournissent des informations sur la diffusion de maladie démyélinisante dans le SNC . Les PEV Visuels et somatosensoriel peuvent démontrer une lésion passée jusque-là inaperçue sur le plan clinique ou IRM. Bien que leur utilité dans la SEP pédiatrique n’ait pas encore été formellement évaluée, des anomalies dans les PEV et PES ont été démontrées, et sont susceptibles d'être d'une importance diagnostique similaire aux formes adultes de SEP.

Neuroimagerieanatomique
L'IRM cérébrale et médullaire sont les modalités de neuro-imagerie de choix pour évaluer les enfants avec des troubles de démyélinisation en général, et la SEP en particulier. Les lésions typiques de l'IRM décrites chez des patients adultes atteints de SEP rémittente sont des plaques rondes ou ovales, brillantes ou hypersignal en image de pondération T2 et images en FLAIR (Fluid attenuated inversion recovery). Elles sont en nombre variable et distribuées dans la substance blanche du centre semiovale, à côté des ventricules, et dans le corps calleux, le tronc cérébral, le cervelet, les nerfs optiques et la moelle épinière. Ces lésions sont perpendiculaires aux ventricules latéraux, et sont généralement de petite taille (moins de 5 mm), et montrent un réhaussement nonuniforme et incomplet après injection de gadolinium. Dans une étude faite analysant les images cérébrales de patients atteints de SEP juvénile, l'on a trouvé qu'elles n'étaient pas différentes de cette description classique au moment de la présentation initiale ou lors de rechutes.
En revanche, l'IRM cérébrale réalisée au cours de la démyélinisation d'un épisode initial chez des enfants de moins de 10 ans montrait de grandes plaques de démyélinisation multifocale avec une tendance à se regrouper dans 80% des cas, une constatation qui est quelque peu sous-estimée. Ces lésions peuvent avoir un aspect pseudo-tumoral avec effet de masse variable. Habituellement, ce type de lésions peut être différencié des tumeurs par la quantité moindre d'oedème autour des lésions souvent -mais pas toujours- en association avec d'autres lésions plus petites et atypiques. Cela dit, il ya des cas signalés de SEP infantile avec une plaque cellulaire associée à un œdème périlésionnel. Le réhaussement au gadolinium peut être utile pour établir cette différence parce que la plaque de démyélinisation montre généralement un réhaussement incomplet de la lésion prenant un aspect d'anneau ouvert. Jusqu'à 15% des patients atteints de SEP juvénile peuvent avoir des lésions pseudo-tumorale à l'IRM au moment de la poussée initiale. La présence de trous

noirs sur des desimages pondérées T1 de l'IRM cérébrale non réhaussées par le gadolinium, ainsi que des signes d'atrophie cérébrale, tel que des espaces sous-arachnoïdiens élargis, une ventriculomégalie, et un amincissement du corps calleux, n'ont pas été fréquemment décrits dans la littérature pédiatrique, mais ces résultats sont clairement présents chez les enfants atteints de SEP à forme secondairement progressive.
Les images IRM observées chez les enfants avec une première poussée de SEPIP ou SEPIR rappelent les images des patients atteints d'EMDA. Les lésions caractéristiques sont grandes et diffuses dans la substance blanche sous corticale, mais aussi dans le cortex et les noyaux gris profonds, sans la distribution et la morphologie observée dans la SEP juvénile et adulte. En règle générale, le diagnostic de SEP ne peut être confirmé que si des études ultérieures au décours du suivi clinique des récurrences montrent de nouvelles lésions réhaussées au gadolinium. Avec ceci en tête, il faut savoir que l'EMDA peut être accompagnée généralement par un ou plusieurs épisodes de rechute (EMDA biphasique ou multiphasiques), mais les IRM successives révèlent des lésions actives uniquement dans le contexte d'une poussée clinique concomittante; en d'autres termes, les lésions « infracliniques » ou « silencieuses » exhibant des modifications actives à l'IRM (qui sont typiques de la SEP) ne sont pas vues dans l'EMDA.
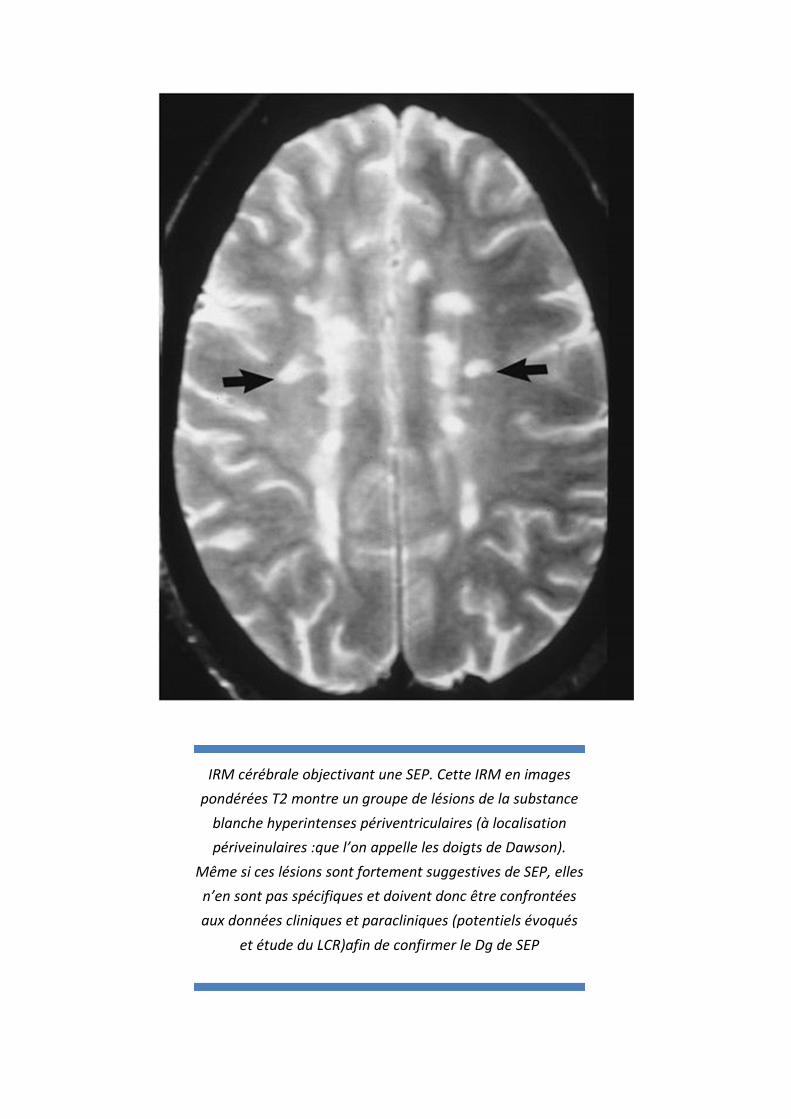
IRM cérébrale objectivant une SEP. Cette IRM en images pondérées T2 montre un groupe de lésions de la substance
blanche hyperintenses périventriculaires (à localisation périveinulaires :que l’on appelle les doigts de Dawson).
Même si ces lésions sont fortement suggestives de SEP, elles n’en sont pas spécifiques et doivent donc être confrontées
aux données cliniques et paracliniques (potentiels évoqués
et étude du LCR)afin de confirmer le Dg de SEP

Selon les nouvelles recommandations du Groupe d'experts international sur le diagnostic de sclérose en plaques, , dans certaines situations cliniques, les lésions IRM doivent remplir les critères diagnostiques de la dissémination spatiale, qui comprennent trois des quatre critères suivants: (a) une lésion réhaussée au gadolinium ou neuf lésions hyperintenses en T2 s'il n'y a pas de réhaussement au gadolinium, (b) au moins une lésion infratentorielle, (c) au moins une lésion juxtacorticale, et (d) au moins trois lésions périventriculaires. Une lésion de la moelle épinière peut être substituée à une lésion cérébrale. Toutefois, ces critères peuvent ne pas s'appliquer aussi bien à la population pédiatrique atteinte de SEP comme chez les adultes. Les enfants atteints de SEP semblent avoir moins de lésions de la substance blanche au moment de leur diagnostic par comparaison aux adultes nouvellement diagnostiqués. En outre, parce que la myélinogenese est incomplète dans l'enfance, ceci peut influencer l'aspect des lésions, leur taille et leur distribution dans le SNC. Des études supplémentaires sont nécessaires pour élaborer les critères de diagnostic IRM qui seront validées dans la population pédiatrique de SEP.
L'IRM et la spectroscopie à résonnance magnétique (SRM) chez les patients atteints de SEP âgés de moins de 5 ans montrent une atrophie cérébrale et une perte de l'intégrité axonale. Bien que les mécanismes exacts qui sous-tendent cette atrophie du SNC soient largement inconnues, il est prouvé que l'atrophie peut être secondaire aux effets répétés de démyélinisation inflammatoire, et de lésions axonales, y compris les modifications dystrophiques dues à la transsection axonale franche, la dégénérescence wallérienne, et la perte neuronale.

IRM cérébrale. séquence FLAIR rapide dans le plan sagittal chez un patient présentant un
premier épisode clinique avec probable origine démyélinisante du trouble. L’IRM montre des
lésions focales hyperintenses visibles au niveau périventriculaire et de la zone sous-
épendymaire du corps calleux.

IRM cérébrale. séquences pondérées en T2 avec de longues écho rapide FLAIR dans le plan transverse chez un patient atteint de sclérose en plaques cliniquement définie. Il ya une blessure gauche juxtacorticale, qui affecte les fibres en U. La blessure est mieux objectivée en séquence rapide FLAIR (à droite) avec la séquence rapide T2 (flèche).

IRM cérébrale. séquences pondérées en T2 (à gauche),
la densité à protons (au centre) et fast-FLAIR (à
droite) dans le plan transverse chez un patient
atteint de sclérose en plaques cliniquement définie.
Il existe de multiples petites lésions démyélinisantes,
beaucoup d'entre elles n'étant pas visible dans la
séquence FLAIR rapide.

IRM du rachis. séquences pondérées en densité à protons (à gauche) et T2 (à droite) obtenus dans le plan sagittal chez un patient atteint de sclérose en plaques cliniquement définie, montrant de multiples lésions démyélinisantes dans la moelle épinière. Les séquences en densité à protons sont très sensibles pour détecter les lésions, ainsi qu’objectiver leur caractère hyperintense par rapport à la moelle épinière normale des et le liquide céphalo-rachidien. Les séquences T2 facilitent quant à elles leur localisation dans la moelle épinière.
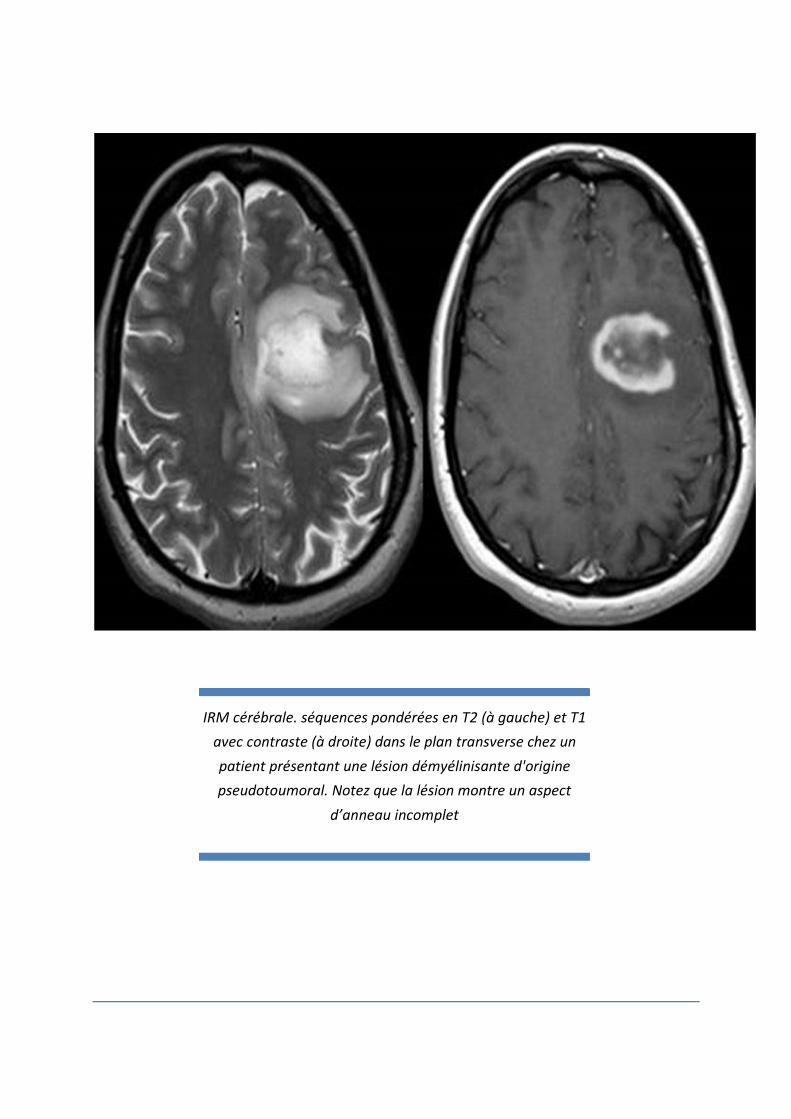
IRM cérébrale. séquences pondérées en T2 (à gauche) et T1 avec contraste (à droite) dans le plan transverse chez un
patient présentant une lésion démyélinisante d'origine pseudotoumoral. Notez que la lésion montre un aspect
d’anneau incomplet

IRM cérébrale. séquences pondérées en T2 (à gauche) et densité à protons (à droite) dans le plan transverse chez un
patient présentant une lésion démyélinisante d'origine
pseudotumoral. Notez la configuration des bandes
concentriques dans la lésion (modèle Balo-like).

IRM cérébrale. séquences pondérées en T2 obtenus dans le plan transverse chez un sujet sain (à gauche) et chez un patient atteint de sclérose en plaques (à droite). Notez l’hypersignal discret et diffus de la substance blanche hémisphérique chez les patients atteints de SEP (aspect de substance blanche « sale »).
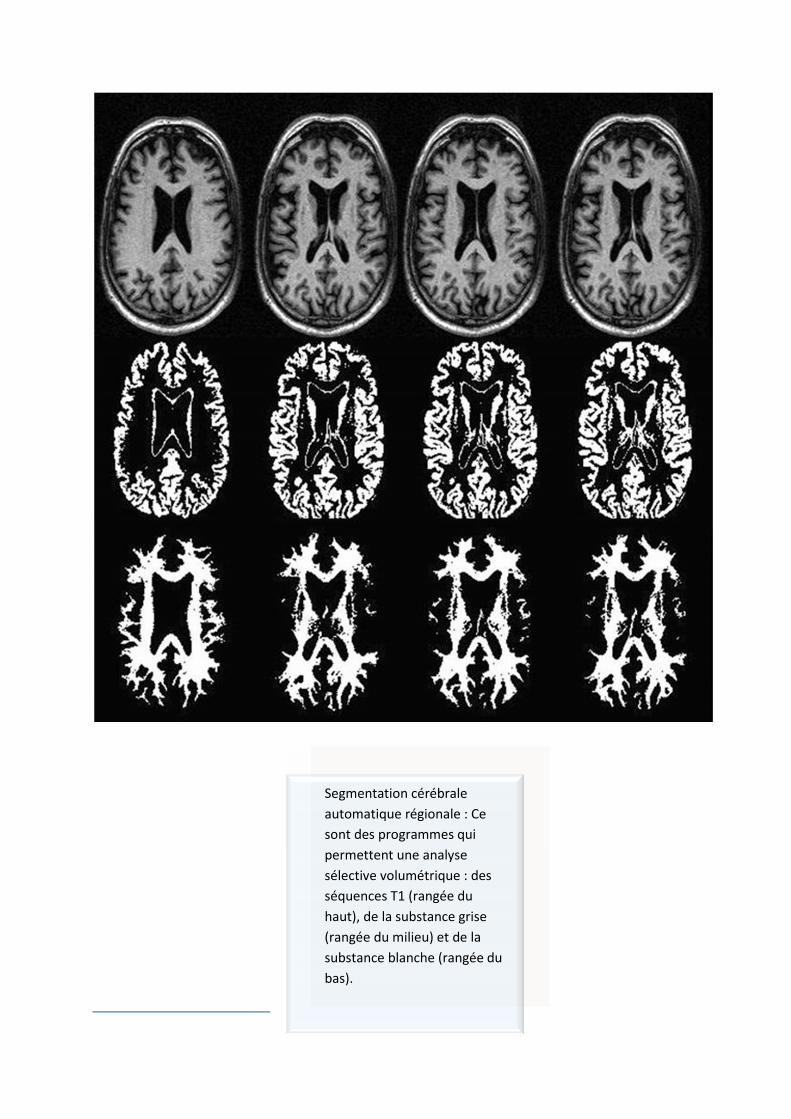
Segmentation cérébrale automatique régionale : Ce sont des programmes qui permettent une analyse sélective volumétrique : des séquences T1 (rangée du haut), de la substance grise (rangée du milieu) et de la substance blanche (rangée du bas).

Neuroimageriefonctionnelle
Bien que l'IRM soit le pilier du diagnostic de la SEP, il existe un intérêt croissant dans l'utilisation de méthodes quantitatives d'IRM pour mieux comprendre la pathologie dans la substance grise et dans la substance blanche d'aspect normal. L'IRM par aimentation de transfert et l'IRM par diffusion pondérée sont des techniques qui fournissent des informations supplémentaires utiles sur le processus de démyélinisation et de remyélinisation dans la SEP. En outre, des études de neuro-imagerie fonctionnelle comme SRM, l'IRM fonctionnelle (IRMf), la tomographie par émission de photons unique (SPECT) et la tomographie par émission de positrons (PET) permettent une meilleure étude de la dysfonction du SNC secondairement à l'évolution pathologique de la maladie.
La SRM des plaques de démyélinisation cérébrale chez les enfants montre un spectre de modifications incluant la réduction de la N-acétylaspartate (NAA) et de la créatine et l'augmentation de la choline et du myo-inositol par rapport aux sujet contrôles spécifiques à l'âge . La SRM chez les adultes atteints de SEP peuvent aussi montrer l'élévation du lactate dans la plaque aiguë démyélinisante, mais cette constatation n'a pas été rapportés chez des enfants . La substance blanche adjacente à la plaque qui a un aspect normal à l'IRM a aussi un profil métabolique normal sur les SRM, mais la substance grise adjacente montre généralement une diminution du NAA. Ces données sont similaires aux résultats chez les patients adultes, et sont la conséquence d'atteintes neuronales (axones inclus), en plus de la destruction myélinique qui se produisent précocément en raison des atteintes répétées inflammatoires démyélinisantes. L'IRMf est actuellement utilisée dans la recherche clinique pour étudier les mécanismes neuronaux qui sous-tendent la fonction du SNC et ainsi définir les profils anormaux d'activation du cerveau qui découlent de la maladie.
Une modification du profil d'activation corticale, principalement caractérisée par une activation accrue du cortex sensitivomoteur primaire

controlatéral, a été trouvée chez les patients atteints de SEP avec des syndromes cliniquement isolés quand ils accomplissent une tâche motrice simple. Une forte corrélation a également été trouvée entre le degré d'activation du cortex sensitivomoteur primaire controlatéral et la réduction de la concentration du cerveau entier en NAA, ce qui suggère que la réorganisation corticale fonctionnelle pourrait contribuer au maintien de la fonction corticale normale dans les premiers stades de la SEP. Une augmentation de l'activation des plusieurs zones sensorielles, principalement dans l'hémisphère cérébral homolatéral à l'extrémité utilisé pour accomplir une tâche donnée, a également été rapportée chez des patients à un stade précoce de SEP et précédé par un épisode d'hémiparésie. Chez les patients présentant des caractéristiques similaires mais avec une névrite optique comme manifestation clinique initiale, les régions sensorimotrices principalement situés dans le l'hémisphère cérébral controlatéral étaient recrutés. L'activation cérébrale anormale du lobe préfrontal / frontal a été démontrée chez les patients avec une SEP à l'IRMf lors de tâches spécifiques, la dysfonction se normalise avec la rivastigmine, un inhibiteur central de la cholinestérase.
Les Nouvelles techniques d'IRM fonctionnelle sont en cours d'élaboration pour étudier in vivo les pathologies du SNC au niveau moléculaire. Des études expérimentales par le virus d'encéphalomyélite murin de Theiler, ont étudié les techniques d'IRM en utilisant des anticorps liés à des particules superparamagnétiques immunitaire dirigée contre des déterminants spécifiques immunitaire. Cela permet d'obtenir une imagerie sélective des cellules T CD4 +, lymphocytes T CD8 +, et les cellules MAC1 + du SNC. Être en mesure de contrôler dynamiquement l'activité de catégories spécifiques de cellules inflammatoires dans la SEP fournira un moyen important de comprendre l'évolution de la pathologie.
Dans une étude de 17 patients atteints de SEP, la tomographie à émission de photons unique (SPECT) utilisant le 99m Tc-D, hexamethylpropylene amine oxime-L (99m Tc-HMPAO) a montré une réduction du ratio activité régionale cérébrale : activité cérébrale globale surtout au niveau des

lobes frontaux et temporaux gauche. Une relation a été trouvée entre une anomalie du lobe temporal gauche et le déficit de fluence verbale et de la mémoire verbale. Le SPECT est également utile dans l'évaluation des patients atteints de SEP ayant des troubles dépressifs, et dans l'établissement du diagnostic différentiel des lésions pseudo-tumorales.
Des études de TEP (tomographie à émission de positrons) ont démontré que les niveaux global et régional du métabolisme du glucose cortical sont significativement réduits chez les patients atteints de SEP par rapport aux sujets contrôles normaux. Cette diminution est corrélée avec le nombre de rechutes, la surface atteinte totale sur l'IRM, et les troubles cognitifs, ce qui indique que la charge lésionnelle quantifiée sur l'IRM provoque une détérioration de la fonction neuronale du cortex cérébral. Hypométabolisme est très répandu, notamment au niveau du cortex cérébral (frontal, pariétal, occipital), de la substance blanche supratentorielle (pariétal), et des structures infratentorielle (Pons). En utilisant un radioligand pour le récepteur périphérique aux benzodiazépines, la TEP a permis la visualisation de la microglie et son implication dans les processus inflammatoires causant la SEP.
Les techniques de neuroimagerie continueront à se développer à l'avenir non seulement pour fournir un diagnostic plus précis de la SEP, mais aussi pour apporter d'importantes informations pronostiques.

spectroscopie par résonance magnétique du cerveau d'un patient atteint de la sclérose en plaques centré sur le semiovale centrum et objectivant des changements temporaires dans l'apparence
normale de la substance blanche, y compris l'annulation d'une dépression du niveau du N-acétylaspartate (NAA) niveau. Les spectres ont été obtenus montré durant les jours 98 (A), 147 (B),
189 (C) et 259 (D) d'une étude longitudinale, les deuxième et troisième de la série montrent une baisse du pic de NAA (à 2,0 ppm ), mais la 4ème série, montre une récupération.

Diagnosticdifferentiel
Le diagnostic différentiel de la SEP est large. Bien que les signes et symptômes cliniques, les modifications IRM, et les résultats du LCR sont caractéristiques de la SEP, ils ne sont pas spécifiques et peuvent être trouvés dans d'autres conditions inflammatoires ou infectieuses, comme indiqué dans le tableau ci-dessous.
Dans de nombreux cas, les atteintes monophasiques démyélinisantes sont difficiles à distinguer d'un premier épisode démyélinisant de SEP pédiatrique, surtout chez les jeunes patients. EMDA a été définie comme un premier épisode clinique, affectant le système nerveux central, d'étiologie inflammatoire ou démyélinisante avec un début aigu ou subaigu et impliquant une présentation polysymptomatique. Selon les définitions proposées par un consensus publié en 2007, l'encéphalopathie -définie par l'IPMSSG comme un trouble du comportement, tels qu'une confusion ou une irritabilité excessive, ou une altération de la conscience, tels que coma ou léthargie- est nécessaire pour parvenir à un diagnostic d'EMDA. Les études appliquant les définitions établies par l'IPMSSG dans des études des cohortes rétrospectives suggèrent que l'encéphalopathie est utile pour distinguer le syndrome clinique isolé (SCI-le premier épisode clinique de SEP) de l'EMDA. Certains patients présentant initialement un phénotype EMDA, ont, toutefois, des épisodes récurrents de démyélinisation sans encéphalopathie, parfois avec l'accumulation de lésions hypertenses en T2 dans le cerveau, et sont par la suite diagnostiqués comme ayant une SEP ou une Neuromyélite optique. De plus en plus, les données suggèrent que l'encéphalopathie peut être associée à un premier épisode de sclérose en plaques, particulièrement chez les jeunes patients. Plutôt que d'être une maladie spécifique, par conséquent, la présence de l'encéphalopathie pourrait être liée à l'immaturité du cerveau ou le système immunitaire chez les patients jeunes.

Aucun marqueur prédictif fiable clinique, biologique ou IRM n'est actuellement disponibles pour identifier un premier épisode de sclérose en plaques chez les enfants qui se présentent avec un épisode démyélinisant initial EMDA-like. Les patients avec une présentation semblable à l'EMDA, dont l'IRM est évocateur de SEP auront, toutefois, un risque accru de développer un deuxième épisode.
Les diagnostics différentiels de la sclérose en plaques Infection aiguë du SNC L'encéphalite virale aiguë (HSV, entérovirus) * infection à HTLV-1 (paraparésie spastique tropicale) la tuberculose du SNC La leucoencéphalopathie multifocale progressive Neuro-sida Neuroborréliose (maladie de Lyme) Panencéphalite sclérosante subaiguë Conditions postinfectieuses encéphalomyélite aiguë disséminée * encéphalomyélite bi- ou multiphasiques * Myélite transverse cerebellite postinfectieuse Encéphalite du tronc cérébral postinfectieuse Vascularite lupus érythémateux systémique * maladie de Behçet syndrome Sjôgren vascularite isolée du CNS * États prothrombotiques Syndrome des antiphospholipides Maladie vasculaire ischémique la maladie de Moya-Moya Les tumeurs intracrâniennes Gliomatose cérébrale* Troubles métaboliques héréditaires Leucodystrophies (adréno-, métachromatique- ) encéphalomyopathie mitochondriale * troubles de bêta-oxydation

Atteinte médullaire aiguë, subaiguë et chronique Tumeur Syringomyélie moelle fixée Malformation artérioveineuse Sclérose combinée de la moelle ( déficit en B12) malformation d'Arnold-Chiari Céphalée vasculaire (migraine) Polyneuropathie inflammatoire démyélinisante chronique Atrophie optique de Leber * L'astérisque indique les diagnostics différentiels les plus fréquents.
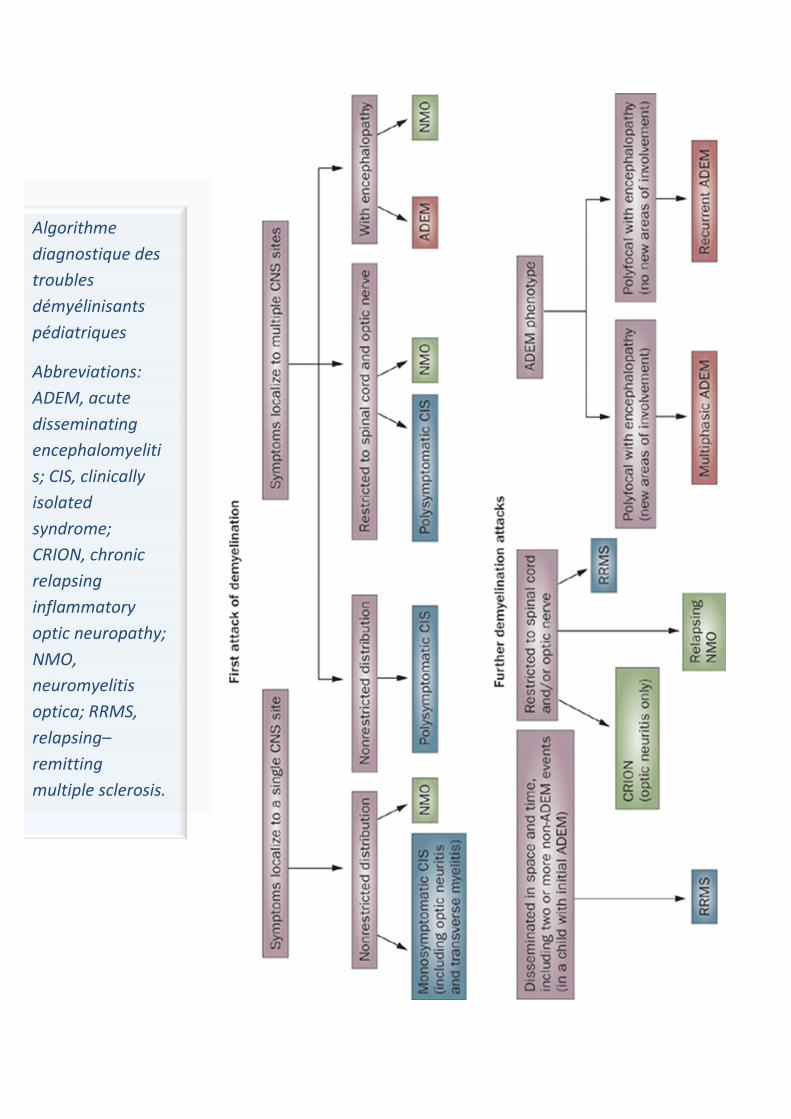
Algorithme diagnostique des troubles démyélinisants pédiatriques
Abbreviations: ADEM, acute disseminating encephalomyelitis; CIS, clinically isolated syndrome; CRION, chronic relapsing inflammatory optic neuropathy; NMO, neuromyelitis optica; RRMS, relapsing–remitting multiple sclerosis.

Traitement
Le traitement de la SEP chez les enfants, comme chez les adultes, doit inclure la suppression des réponses immunitaires inflammatoires pendant les poussées et l'amélioration de symptômes associés entre ces crises (la fatigue, la spasticité, les infections des voies urinaires).
Le traitement initial consiste en l'administration de corticostéroïdes, que ce soit oralement ou par voie intraveineuse. L'utilisation de bolus par voie intraveineuse (IV) de méthylprednisolone est indiquée dans des poussées sévères, caractérisées par une atteinte marquée de l'état mental, des nerfs optiques, ou de la moelle épinière, ou dans les cas se manifestant par des lésions pseudo-tumorales à l'IRM. La dose recommandée est de 30 mg / kg / jour à 1 g / jour, administrée en une perfusion de 1 heure sur 3 à 5 jours consécutifs, suivis par l'administration orale de méthylprednisolone, 1 mg / kg par jour le matin pendant les 10 jours suivants, suivie d'une dégression de la corticothérapie sur une période de 3 semaines. L'administration de la méthylprednisolone IV permet d'accélérer la récupération des exacerbations aiguës de la maladie. Des études récentes ont montré que la méthylprednisolone supprime l'expression de gènes associés à la différenciation des cellules T-et l'activation, ce qui peut contribuer à son effet bénéfique dans les rechutes de la SEP . Le traitement par corticoïdes nécessite une surveillance attentive de la pression artérielle, de la glycosurie et de la kaliémie, et l'administration d'agents gastroprotecteurs.
Les poussées qui ne sont pas aussi graves peuvent être traitées avec méthylprednisolone par voie orale à une dose de 1 à 2 mg / kg par jour pendant 10 à 15 jours. En général, les exacerbations légères ne se manifeste que par des symptômes sensitifs comme des paresthésies ne nécessitant aucun traitement. L'administration chronique de corticoïdes n'est pas indiqué car il a été démontré qu'elle ne modifiait pas l'histoire naturelle de la maladie chez les patients adultes et peut provoquer des effets secondaires graves chez les enfants.

Certains enfants qui ne répondent pas aux corticoïdes IV peuvent bénéficier d'immunoglobulines IV (IgIV). La dose recommandée est de 2 mg / kg réparties sur 2 à 5 jours. Il n'existe aucune étude évaluant l'efficacité des IgIV dans la SEP pédiatrique. Son efficacité chez les adultes a été récemment revue dans une méta-analyse, qui avait suggéré un rôle potentiel des IgIV chez les patients avec une fréquence de rechute élevé. Similairement,un traitement par IgIV dès la première année du début de troubles neurologiques suggestifs de SEP, a comme effet de diminuer significativement l'incidence d'une seconde poussée, et permet aussi de baisser l'activité de la maladie attestée par l'IRM. Dans une étude en simple aveugle impliquant adolescents et adultes, les IgIV avaient montré une efficacité similaire par rapport aux INF-bêta 1 pour diminuer le taux de poussées dans une SEP de type poussée -rémission.
En revanche, les IgIV n'ont pas montré de bénéfice clinique dans un groupe de patients adultes atteints de SEP secondairement progressive.
L'efficacité et les avantages à long terme des autres agents immunosuppresseurs dans la SEP ne sont pas claires. À cet égard, il ya eu seulement quelques essais cliniques chez l'adulte et aucun chez les enfants. L'azathioprine a été utilisé seul et en combinaison avec les IgIV ou IFN-bêta 1b. Même si cet agent immunosuppresseur semble réduire le taux de rechute chez les patients atteints SEP, son effet sur la progression de la maladie et le handicap neurologique n'a pas encore été établi. Le méthotrexate peut modifier favorablement le cours de la maladie chez les patients atteints de SEP progressive, mais la preuve est mince.
La Cyclophosphamide a été utilisé chez l'adulte avec un succès variable. Il semble être plus efficace dans le stade précoce de la sclérose en plaques progressive indépendemment de l'âge, des rechutes, ou de l'ampleur des séquelles neurologiques. L'analyse préliminaire d'une étude randomisée, en simple aveugle, sur groupes parallèles, et multicentrique chez des patients atteints de SEP mis sous bolus de cyclophosphamide avait démontré qu'il s'agissait d'une option thérapeutique fiable dans le

traitement de seconde intention pour les patients qui sont résistants aux IFN. En plus d'avoir un effet immunosuppresseur général, la cyclophosphamide a des effets immunosuppresseurs sélectifs dans la SEP en supprimant les réponses de type Th1 via IL-12 et en améliorant les réponses Th2/Th3 (via IL-4, IL-10). La Cyclophosphamide a été utilisé chez un nombre d'enfants avec une SEP très agressive après l'échec des IgIV et des thérapies à base d'IFN. Les potentiels risques de l'immunosuppression, des cancers et de l'infertilité devraient être prudemment mis en balance avec les potentiels avantages et doivent être examinés en détail avec le patient et la famille .
Le Mitoxantrone est un autre puissant agent immunosuppresseur utilisé pour traiter la SEP chez les adultes. Dans une étude de 94 patients suivis pendant 3 ans, il a été efficace dans l'amélioration ou la stabilisation de l'évolution de la maladie, particulièrement chez les patients avec un faible taux de rechutes.
La plasmaphérèse a montré son efficacité dans certaines études impliquant des adultes atteints de SEP. Toutefois, son efficacité dans le traitement de la SEP est controversée.
L'utilité des traitements immunomodulateurs, qui ont été démontrées améliorer l'évolution naturelle de la SEP chez les adultes, comme les interférons et l'acétate de glatiramère (copolymère 1, ou Copaxone), n'a pas été étudiée par des études contrôlées chez les enfants, Bien qu'il y ait des preuves de succès dans des cas isolés . IFN-bêta 1b (Betaseron, Betaferon) est bien toléré et a démontré un certain bénéfice clinique chez les enfants atteints de SEP. Tenembaum et ses collègues ont déclaré que leur expérience du traitement de 31 enfants par l'IFN-bêta 1a (Avonex) 30 µg une fois par semaine, l'IFN-bêta 1a (Rebif) 22 µg trois fois par semaine, l'IFN-bêta 1b (Betaseron), 250 µg tous les deux jours , et l'acétate de glatiramère (Copaxone) 20 mg par jour. La tolérance aux quatre traitements était similaire à celle rapportée chez les patients adultes. Les enfants présentaient des symptômes grippaux dans 61% des cas, des

réactions au site d'injection chez 39%, des migraines dans 29%, et une réaction systémique transitoire dans 6% des cas; ces symptômes ont diminué progressivement jusqu'à leur disparition en quelques mois. Les doses ont été ajustées selon l'âge, le poids et la réponse clinique. L'analyse de l'efficacité a montré une réduction significative du taux de rechutes dans 94% des patients atteints de SEP à poussée-rémission et dans 33% des patients atteints de SEP secondairement progressive .

Mechanisms of action of glatiramer acetate (GA) in MS. Reprinted from Autoimmun Rev, Vol. 6, Schrempf W, Ziemssen T, Glatiramer acetate: mechanisms of action in multiple sclerosis, 469–475.Copyright © 2007, with permission from Elsevier. 1. GA exhibits competitive binding at the MHC-II complex and TCRantagonism. In addition, GA is able to displace MBP from the binding site on MHC-II molecules. Treatment with GA leads to the induction of antigen specifc TH2 T cells in the periphery. 2. In addition CD8+ and CD4+ CD25+ regulatory T cells are induced by GA therapy. 3. The constant activation seems to have an important impact on the induction and maintenance of the regulatory/suppressive immune cells. 4. Because of the daily activation, GA T cells are believed to be able to cross the blood–brain barrier. 5. Inside the CNS, some GA-specifc T cells cross-react with products of local myelin turnover presented by local APCs. 6. In response anti-infammatory cytokines are secreted which dampen the local infammatory process (bystander suppression). 7. Furthermore GA specifc T cells secrete neurotrophic factors which might favor remyelination and axonal protection. Abbreviations: APC, antigen-presenting cells; MHC, major histocompatibility complex; TCR, T cell receptor.

Des essais cliniques préliminaires suggèrent que les combinaisons thérapeutiques dans la SEP n'augmentaient pas les effets secondaires par rapport à une monothérapie approuvée; son efficacité vis-à-vis de la monothérapie se doit donc d'être testée .
Le natalizumab (Tysabri) est un anticorps monoclonal anti-alpha-4-intégrine qui bloque la migration des leucocytes médiée par l'alpha-4-integrine. Cette migration permet la liaison aux molécules d'adhésion des cellules vasculaires -1 (VCAM-1), qui est exprimée à des taux élevés dans les vaisseaux sanguins du SNC au cours des poussées de SEP. Une étude comportant 38 patients atteints de la forme à poussée-rémission de la SEP, stable sous traitement par l'IFN-bêta, a démontré l'innocuité de cette association, et une large étude multicentrique a fourni des résultats suffisants pour que l'administration des médicaments et des aliments des USA « US Food and Drug » approuve le Tysabri en 2004, sous forme injectable IV pour le traitement de la sclérose en plaques (et la maladie de Crohn) chez l'adulte.
D'autres approches thérapeutiques sont actuellement à l'étude et pourraient offrir de nouvelles alternatives thérapeutiques pour la SEP à l'avenir : (a) une immunosuppression sélective contre les lymphocytes T résidents; (b) immunomodulation utilisant de nouveaux médicaments comme les statines, ou le ciblage de nouveaux mécanismes comme les récepteurs de chimiokines ou les cellules dendritiques qui inhibent les cellules T, des agents neuroprotecteurs comme l'érythropoïétine , ou la migration des cellules T avec antimetalloproteinases, ou des antioxydants; (c) les interventions visant à la production de radicaux libres médiée par le fer; (d) la vaccination des cellules T; (e) transplantation de cellules souches, et (f) la thérapie génique
Un traitement symptomatique est primordial afin d'améliorer la qualité de vie des patients atteints de SEP. La Modafinil et l'amantadine sont efficaces pour traiter la fatigue. L'exercice et le yoga ont également

démontré un effet bénéfique. La dépression répond bien à la pharmacothérapie. La spasticité et l'épilepsie doivent être traités avec des médicaments spécifiques appropriées.
Une approche multidisciplinaire pour les soins de ces enfants et leurs familles à travers l'évaluation, le soutien, l'éducation, la sensibilisation, l'orientation et la coordination des soins est idéal pour un meilleur contrôle de la maladie et une meilleure qualité de vie.

Récapitulatif du traitement de la sclérose en plaques
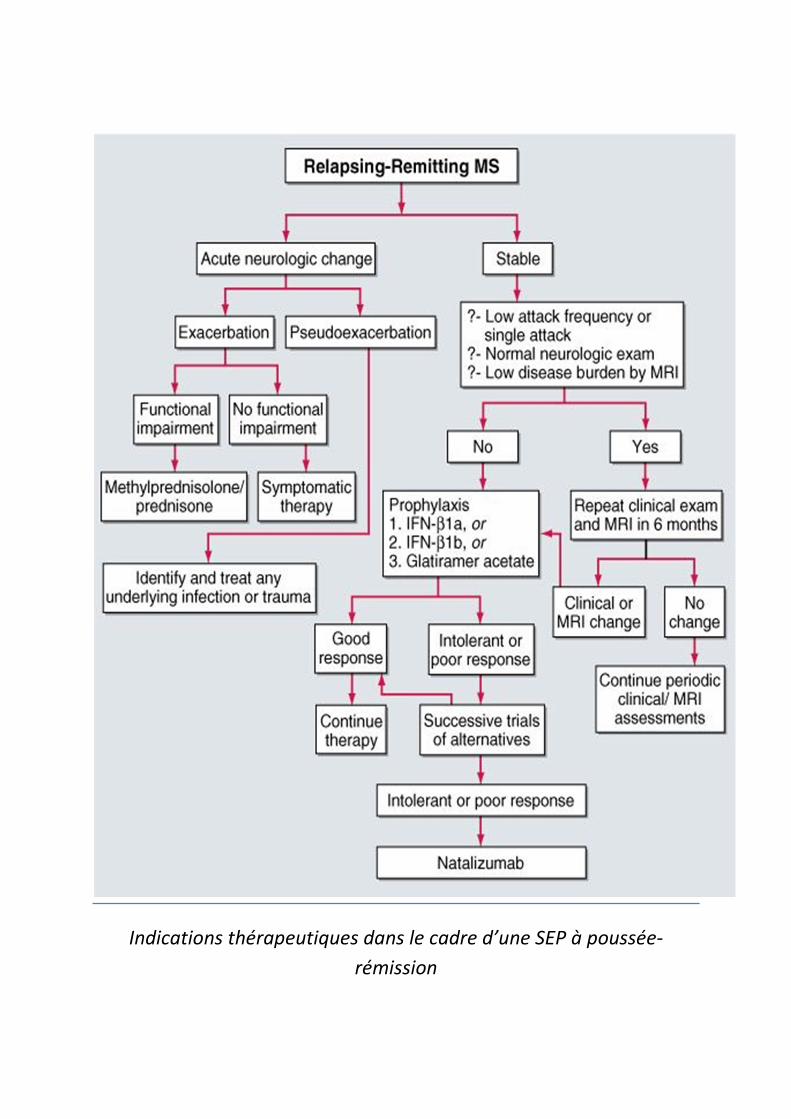
Indications thérapeutiques dans le cadre d’une SEP à poussée-
rémission

Indications thérapeutiques dans le cadre d’une SEP progressive

Pronostic
Chez les patients adultes, l'évolution clinique de la maladie est très variable. La sclérose en plaques à poussées-rémissions est la forme la plus fréquente de la progression (80%), suivie de la SEP secondairement progressive après une période initiale de fréquentes poussées (26%) et la SEP progressive primaire (6% à 14%).

Les différents types d’évolution cliniques de la SEP
Il ya très peu d'informations sur l'évolution clinique et le pronostic de la SEP chez les enfants. Dans une étude de 296 enfants ayant eu un premier épisode de démyélinisation inflammatoire aiguë du SNC, la fréquence d'une seconde poussée était plus élevée chez les patients de plus de 10 ans, si une première IRM était suggestive de SEP, s'il y avait existence d'une lésion du nerf optique ou médullaire,et chez les patient ayant un état mental éltéré. Dans une autre étude l'évolution clinique d'un groupe d'enfants avec la SEP comme diagnostic établi était :forme poussée-rémission (73%), secondairement progressive (25%). Aucun patient n'avait développé de SEP progressive primaire. D'autres auteurs ont publié des conclusions similaires
L'évolution naturelle de la SEP chez les adultes montre que dans le long terme, la fréquence des exacerbations de la maladie diminue spontanément. Dans l'expérience de Tenembaum et ses collègues, le nombre moyen de poussées par an était de 2, 2,3, et 2,2 pour SEPIP, SEPIR,et SEPJ respectivement, au cours de la première année de la maladie. Ces chiffres ont diminué à 0,9, 1,4 et 1,5, respectivement, sur une période de 4 à 8 ans de suivi. Ces résultats indiquent que les enfants atteints de SEP ont tendance à subir plus de poussées de démyélinisation que les adultes, mais ils montrent également une réduction spontanée de la fréquence des rechutes avec le temps.
Les poussées peuvent être provoquées par des viroses aiguës fébriles. Les infections bactériennes, généralement celles qui impliquent les voies urinaires des filles ou des femmes atteintes de SEP, peuvent provoquer une poussée, mais il est plus probable qu'elles aggravent le niveau d'activité de la maladie déjà existante. Il n'existe aucune preuve que des vaccins à virus vivants ou le vaccin anti-grippal provoque des rechutes. D'autres facteurs qui peuvent être associés à une rechute comprennent le stress, les traumatismes physiques, la chirurgie et la rachianesthésie.

Avec l'augmentation de la fréquence des poussées, la récupération peut s'en trouver entravée et les déficits peuvent devenir cumulatifs. En règle générale, plus la poussée sera longue, et plus grande est la probabilité que la récupération sera incomplète. L'évaluation du handicap neurologique est habituellement effectuée en utilisant l'échelle d'état de handicap étendu : Expanded Disability Status Scale (EDSS) de Kurztke. Toutefois, cette méthode, qui est largement utilisée chez l'adulte, montre des limites claires chez les enfants. Dans la série de Tenembaum et ses collègues, après un suivi moyen de 6,6 ans, 70% des enfants atteints de SEP avaient un score EDSS de 3,5, ce qui signifie qu'ils étaient indépendants en terme d'ambulation sans aide, 9% avaient un score entre 4 et 4,5 , indiquant une incapacité modérée avec des limitations, mais toujours ambulatoires, 15% ont obtenu des scores allant de 6 à 9,5, ce qui signifie qu'ils nécessitaient pour leur ambulation une assistance ou un fauteuil roulant. Ces données démontrent que la SEP chez les enfants, à la fois les variantes infantile et juvénile, ne peut pas être considérée comme une maladie bénigne en raison de son évolution clinique qui peut être agressive et provoquer des handicaps physiques.

Systéme de scoring de la SEP
Kurtzke Expanded Disability Status Score (EDSS)
0.0 = Normal neurologic exam [all grade 0 in functional status (FS)]
1.0 = No disability, minimal signs in one FS (i.e., grade 1)
1.5 = No disability, minimal signs in more than one FS (more than one grade 1)
2.0 = Minimal disability in one FS (one FS grade 2, others 0 or 1)
2.5 = Minimal disability in two FS (two FS grade 2, others 0 or 1)
3.0 = Moderate disability in one FS (one FS grade 3, others 0 or 1) or mild disability in three or four FS (three/four FS grade 2, others 0 or 1) though fully ambulatory
3.5 = Fully ambulatory but with moderate disability in one FS (one grade 3) and one or two FS grade 2; or two FS grade 3; or five FS grade 2 (others 0 or 1)
4.0 = Ambulatory without aid or rest for 500 m
4.5 = Ambulatory without aid or rest for 300 m
5.0 = Ambulatory without aid or rest for 200 m
5.5 = Ambulatory without aid or rest for 100 m
6.0 = Unilateral assistance required to walk about 100 m with or without resting
6.5 = Constant bilateral assistance required to walk about 20 m without resting
7.0 = Unable to walk beyond about 5 m even with aid; essentially restricted to wheelchair; wheels self and transfers alone
7.5 = Unable to take more than a few steps; restricted to wheelchair; may need aid to transfer
8.0 = Essentially restricted to bed or chair or perambulated in wheelchair, but out of bed most of day; retains many self-care functions; generally has effective use of arms
8.5 = Essentially restricted to bed much of the day; has some effective use of arm(s); retains some self-care functions

9.0 = Helpless bed patient; can communicate and eat
9.5 = Totally helpless bed patient; unable to communicate or eat
10.0 = Death due to MS
Score de l’état fonctionnel
A. Pyramidal functions 0 = Normal 1 = Abnormal signs without disability 2 = Minimal disability 3 = Mild or moderate paraparesis or hemiparesis, or severe monoparesis 4 = Marked paraparesis or hemiparesis, moderate quadriparesis, or monoplegia 5 = Paraplegia, hemiplegia, or marked quadriparesis 6 = Quadriplegia B. Cerebellar functions 0 = Normal 1 = Abnormal signs without disability 2 = Mild ataxia 3 = Moderate truncal or limb ataxia 4 = Severe ataxia all limbs 5 = Unable to perform coordinated movements due to ataxia C. Brainstem functions 0 = Normal 1 = Signs only 2 = Moderate nystagmus or other mild disability 3 = Severe nystagmus, marked extraocular weakness, or moderate disability of other cranial nerves 4 = Marked dysarthria or other marked disability 5 = Inability to swallow or speak D. Sensory functions 0 = Normal 1 = Vibration or figure-writing decrease only, in 1 or 2 limbs 2 = Mild decrease in touch or pain or position sense, and/or moderate decrease in vibration in 1 or 2 limbs, or vibratory decrease alone in 3 or 4 limbs 3 = Moderate decrease in touch or pain or position sense, and/or essentially lost vibration in 1 or 2 limbs, or mild decrease in touch or pain, and/or moderate decrease in all proprioceptive tests in 3 or 4 limbs 4 = Marked decrease in touch or pain or loss of proprioception, alone or combined, in 1 or 2 limbs or moderate decrease in touch or pain and/or severe proprioceptive decrease in more than 2 limbs 5 = Loss (essentially) of sensation in 1 or 2 limbs or moderate decrease in touch or pain and/or loss of proprioception for most of the body below the head 6 = Sensation essentially lost below the head E. Bowel and bladder functions 0 = Normal 1 = Mild urinary hesitancy, urgency, or retention 2 = Moderate hesitancy, urgency, retention of bowel or bladder, or rare urinary incontinence

3 = Frequent urinary incontinence 4 = In need of almost constant catheterization 5 = Loss of bladder function 6 = Loss of bowel and bladder function F. Visual (or optic) functions 0 = Normal 1 = Scotoma with visual acuity (corrected) better than 20/30 2 = Worse eye with scotoma with maximal visual acuity (corrected) of 20/30 to 20/59 3 = Worse eye with large scotoma, or moderate decrease in fields, but with maximal visual acuity (corrected) of 20/60 to 20/99 4 = Worse eye with marked decrease of fields and maximal acuity (corrected) of 20/100 to 20/200; grade 3 plus maximal acuity of better eye of 20/60 or less 5 = Worse eye with maximal visual acuity (corrected) less than 20/200; grade 4 plus maximal acuity of better eye of 20/60 or less 6 = Grade 5 plus maximal visual acuity of better eye of 20/60 or less G. Cerebral (or mental) functions 0 = Normal 1 = Mood alteration only (does not affect EDSS score) 2 = Mild decrease in mentation 3 = Moderate decrease in mentation 4 = Marked decrease in mentation 5 = Chronic brain syndrome—severe or incompetent
Source: After JF Kurtzke: Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 33:1444, 1983.

Des enfants dans une étude de Mikaeloff et ses collaborateurs, ont été suivis pendant une moyenne de 3 ans. Quatre-vingt dix pour cent n'avaient pas de handicap ou seulement un handicap mineur. La présence d'un handicap grave était associée à un début polysymptomatique, persistance de séquelles après la première poussée, la survenue de poussées fréquentes par la suite, et de la constitution d'une forme progressive de la SEP.
Le taux de mortalité dans la SEP pédiatrique a été rapporté dans une fourchette de 10% à 40% pendant les 5 premières années de la maladie. Dans une étude faite à cet égard sur le suivi de 51 enfants atteints de SEP, 3 des 51 enfants (6%) avec une SEP secondairement progressive sont décédés après une période de 3 à 12 ans de la maladie .