reviews meeting report - sage · pdf filerichard k. burt ronald busuttil carl j. cardella...
TRANSCRIPT

REVIEWS
Living Related Small Bowel Transplantation . . . . . 526Luca Cicalese, Pierpaolo Sileri, Cristiana Rastellini, Herand Abcarian, and Enrico Benedetti
Islet of LangerhansAutotransplantation: Rationale, Results, and New Developments . . . . . . . 535Thierry Berney, Aileen Caulfield, Jose Oberholzer, Leo Buhler, Christian Toso, and Philippe Morel
Pharmacoeconomic andOutcomes Analyses in SolidOrgan Transplantation . . . . . 544Kathleen D. Lake
“Engineering” MyoblastTransplantation . . . . . . . . . . 558Daniel Skuk and Jacques P. Tremblay
MEETING REPORT
TOLERANCE: DEFINING, ACHIEVING AND MEASURING IT
Report from “The Tolerance Assay: Where are we now?” Workshop,Transplant 2001 . . . . . . . . . . 571Anne M. VanBuskirk and Peter S. Heeger
MISMATCHES
The Unkindest Cut: Where Are All the TransplantPrograms Going? . . . . . . . . . 574Roger W. Evans
Index . . . . . . . . . . . . . . . . . 577
december 2001 volume 4 number 8
Cover art: Illustration created in Photoshop byRavi Balasuriya.
Graft (ISSN 1522-1628) is published 8 times
annually (January/February, March, April/May,
June, July/August, September, October/November,
and December) by Sage Science Press, 2455
Teller Road, Thousand Oaks, CA 91320, U.S.A.
© 2001 Sage Science Press, an imprint of
Sage Publications. No part of this publication
may be reproduced, stored in an information
retrieval system, or transmitted by any form or
by any means, electronic or otherwise, without
the prior written permission of the publisher.
EDITORIAL OFFICE
Landes Bioscience810 South Church StreetGeorgetown, Texas 78626512.863.7762 phone • 512.863.0081 fax
JOURNAL PUBLICATIONS DIRECTOR
Kimberly A. Mitchell
DESIGN/PRODUCTION
Kelli E. Palma
s a g e p u b . c o m g r a f t v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1
organ and cell transplantation
Randall MorrisFolk Cardiovascular ResearchStanford University School
of Medicine
Charles G. OroszDepartments of Surgery,Pathology and Medical
Microbiology/ImmunologyDivision of Transplantation
Ohio State University
Jeffrey L. PlattDepartments of Surgery,
Immunology and PediatricsTransplantation Biology
Mayo Clinic
Camillo RicordiDivision of Cellular
TransplantationDiabetes Research InstituteUniversity of Miami School
of Medicine
Amelia BartholomewUniversity of Illinois
(Stem Cells)
Laurine BowHartford Transplant Center
(Histocompatibility)
David BriscoeHarvard
(Pediatrics and Artificial Organs)
Nelson ChaoDuke University
(Bone Marrow Transplantation)
Francis L. DelmonicoHarvard
(Organ Donation and Allocation)
Roger W. EvansPrivate Investigative Consultant
(Mismatches)
Jay A. FishmanMassachusetts General Hospital
(Infectious Disease)
Bernhard J. HeringUniversity of Minnesota
(Islets)
Luca InverardiUniversity of Miami
(Literature Review—Cell)
Bruce KaplanUniversity of Michigan
(Pharmacology)
Stuart J. KnechtleUniversity of Wisconsin
(Literature Review—Clinical)
Jonathan R.T. LakeyUniversity of Alberta
(Cell Transplantation—Methods)
Bruce RosengardUniversity of Pennsylvania
(Heart and Lung Transplantation)
Russell H. WeisnerMayo Clinic
(Liver Transplantation)
Martin S. ZandUniversity of Rochester
(Kidney and Pancreas Transplantation)
EDITORS
Editors
Associate Editors
december 2001 volume 4 number 8
organ and cell transplantation
Michael M. AbecassisDavid H. AdamsPatrick AebischerRodolfo AlejandroJ. Wesley AlexanderNancy L. AscherFritz H. BachW. Henry BarberClyde F. BarkerAmelia BartholomewStephen BartlettMark R. BenfieldGilles BenichouD. Keith BishopHenri BismuthSteven F. BollingR. Randal BollingerKenneth L. BraymanReinhard G. BretzelChristoph E. BroelschJonathan S. BrombergWilliam J. BurlinghamRichard K. BurtRonald BusuttilCarl J. CardellaCharles B. CarpenterNelson J. ChaoThomas M. CoffmanDavid J. CohenDavid K.C. CooperA. Benedict CosimiDonald CramerDonald C. DafoeGabriel M. DanovitchIngemar J.A. DavidsonAchilles A. DemetriouRobert B. EttengerM. Roy FirstJay A. FishmanM. Wayne FlyeAdaani E. FrostJohn Fung
Ronald M. FergusonDenis GlotzThomas A. GonwaDavid GrantBartley P. GriffithCarl G. GrothScott A. GruberRainer GruessnerNadey HakimPhillip F. HalloranWayne W. HancockMark A. HardyWilliam E. HarmonAxel HaverichAlberto HayekPekka HäyryPeter S. HeegerJ. Harold HeldermanJeffrey HosenpudDonald E. HricikSharon A. HuntIan V. HutchinsonSuzanne T. IldstadSilviu ItescuStuart W. JamiesonAnthony M. JevnikarRahul M. JindalStanley C. JordanBarry D. KahanBertram L. KasiskeDixon B. KaufmanNorma Sue KenyonRonald H. KermanRaja B. KhauliJames KirklinGoran B. KlintmalmNorman M. KnetemanSheri M. KramsHenri KreisAlan M. KrenskyJ.W. Kupiec-WeglinskiJohn R. Lake
Fadi G. LakkisChristian P. LarsenGary LevyRichard M. LewisMarc I. LorberMichael LuceySteven V. LynchJoren C. MadsenMasimo F. MartelliOlivia M. MartinezArthur J. MatasSue V. McDiarmidEdgar L. MilfordGeraldine G. Miller Joshua MillerCharles M. MillerAnthony MonacoBarbara MurphyAli NajiPeter NeuhausJohn F. NeylanDouglas NormanAndrew C. NovickSoji F. OluwoleLeendert C. PaulThomas PearsonBrian J.G. PereiraJohn D. PirschRaymond PollakRay V. RajotteAbdul S. RaoDavid J. ReichBruno ReichartNancy L. ReinsmoenYair ReisnerBruce ReitzGiuseppe RemuzziDale G. RenlundEric A. RoseLawrence RosenbergJ. Thomas RosenthalDavid Roth
David M. RothsteinRobert H. RubinMary E. RussellDaniel R. SalomonPaul SanbergFred P. SanfilippoAntonio SecchiAbraham ShakedByers W. ShawHaval ShirwanDaniel A. ShoskesSara J. ShumwayHans W. SollingerVaughn A. StarnesThomas E. StarzlStanislaw M. StepkowskiPeter StockJeffrey S. StoffRobert J. Stratta Terry B. StromFrank P. StuartManikkam SuthanthiranDavid E.R. SutherlandMegan SykesAmir TejaniPaul I. TerasakiFrancis T. ThomasJudith M. ThomasAngus W. ThomsonNicholas L. TilneyJacques P. TremblayAndreas G. TzakisJoseph VacantiHannah A. ValantineAnne M. VanBuskirkHector O. VenturaFlavio VincentiHans-Dieter VolkBruno WatschingerE. Steve WoodleJames B. YoungAdriana Zeevi
Editorial Board
s a g e p u b . c o m g r a f t v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1

GUIDELINES
The introduction should describe the back-ground of the topic.
Acknowledgments should be kept to a minimum.
References
References for review articles are limited to30. Important references should be annotated.
References in the text are numbered consecutivelyas superscripts beginning with number 1.
When referring the reader to specific refer-ences as part of a sentence, cite as:
Example:
For a review see refs. 20 to 25.
not ...For a review see 20 to 25
The list of references should be numberedconsecutively according to the order in whichthey are mentioned within the article. Our pre-ferred style for reference listings is“Vancouver.” Abbreviate journal namesaccording to the style used in Index Medicus.Spell out foreign or less commonly knownjournal names.
Journals: [Author’s last name] [Author’s initials],[Other authors’ last names followed by theirinitials]. [Title of article with only first wordcapitalized]. [Journal’s standard abbreviatedname] [Year]; [Volume (number)]:[Pages].
Only the first 6 authors are listed. If there aremore than 6 authors, the first 6 names are fol-lowed by “et al.” Initials and abbreviations arenot followed by periods.
Example:
1. Knotts R, Finn W, Armstrong T. Psycho-social factors impacting patients, donors, andnon-donors involved in renal transplant eval-uation. Perspectives 1995;15:11-23.
Books: [Author’s last name] [Author’s initials],[Other authors’ last names followed byinitials]. [Chapter title]. In: [Editor’s last name][Editor’s initials], editor(s). [Book title].[Number of edition]. [City]: [Publisher];[Year]. [Pages].
Example:
1. Pitou AS, Barrett FG. Endocrine changesfollowing median sternotomy. In: KerkaportaA, editor. Textbook of endocrinologic sur-gery. 3rd ed. Austin: Landes Bioscience;1996. p. 511-65.
Unpublished data and personal communicationsare not listed as references but rather appearin parentheses in the text.
Production GuidelinesWe are an entirely Mac-based office. However,most IBM-compatible or Macintosh wordprocessing programs are acceptable.
How to prepare text files
Our preferred word processing program isMicrosoft Word; please save as version 6.0(please no “Fast-Save” format).
Article text files should be submitted on a 3.5inch, high-density computer disk. Save tablesand figures in a document separate from text.
Figure captions, however, can be at the end ofthe review as text. There is no need to make aunique file for captions.
Tables will be reformatted during production,and therefore they need only be minimally for-matted in your text file. Include printouts oftables with the manuscript.
How to prepare figures, illustrations, andphotos
When art is provided on disk, a single hardcopy should be included to verify the illustration.It is not desirable to embed graphics withinyour text documents.
Compatible computer graphics programs areAdobe Illustrator, Freehand, QuarkXpress,Pagemaker, and Photoshop.
Figures and illustrations may be provided byauthors as hard copy as well. Hard copiesshould be high-quality prints, with 2 copies ofeach illustration submitted. Figures will bereformatted by a graphic designer, not a medicalscientist, so an enclosed figure description forcomplex illustrations will be appreciated andwill result in improved quality.
Send only original artwork, no photocopies.Photography will be published only if thequality is reproducible. Please submit high-quality prints or slides for best quality.
All artwork should be labeled with the author’sname, the figure number, and the correctorientation of the figure, but be sure thatlabeling is clear of the image. Do not put thelabel directly behind the image. Do not writedirectly on the back of the photograph or onthe label after it has been applied. Indicate anyspecial cropping on a photocopy of the figure.
When illustrations are reproduced from othersources, acknowledge the copyright holder atthe end of the figure legend or as a footnote totables. Do not use superscripted referencenumbers in lieu of a full credit line.
Scope
Graft publishes reviews in all areas of organand cell transplantation. These include basicimmunologic topics relevant to clinical trans-plantation, such as tolerance induction,immunoprotection, and gene therapeuticmodulation of the immune response. Othertopics include xenotransplantation, tissuetyping, patient selection, and operative tech-niques in clinical transplantation, short- andlong-term graft follow-up, pharmacothera-peutic modulation of the immune response,and the physiology of grafted organs and cells.
Articles and ReviewsReviews will be brief (2000 to 4000 words).These will generally be invited, but unsolicitedproposals for reviews will be considered. Weencourage color illustrations. Assistance increating artwork can be provided by LandesBioscience on a limited basis if necessary.
Meeting reports will be invited. They are to be1000 to 2000 words.
Other feature articles (including specialforums; commentaries; and columns onethics, technology, and managed care) shouldbe 1000 to 2000 words in length.
Editorial GuidelinesSubmission
Two printed copies of the article, in English,should be submitted. Text should be double-spaced, with page numbers throughout.Figures and disk as described below must beincluded. Please supply telephone and faxnumbers, and email addresses if available.Send to:
Landes Bioscience810 South Church StreetGeorgetown, TX USA 78626
Language and Nomenclature
Abbreviations and acronyms should bedefined the first time they are used, and a listof all abbreviations should be provided.American spellings are preferred.
Organization
The title page must indicate correspondingauthor and include complete addresses for allauthors, as well as an abstract. The abstractshould be a maximum of 150 words. Pleaseprovide one key term definition used withinthe text per page of submitted article.
Example:
Reprinted with permission from: Fox N,Aparicio L. Postgrad Gen Surg 1994;24:611-765. ©1996 Landes Bioscience.
Label disks with author name(s), article title,files enclosed, and please name your file[main author’s surname] or a keywork fromthe title.
If you cannot submit your application thedescribed way or have any further questions,please get in touch with us before you sendyour work, and we will find a solution.
Page ProofsPage proofs should be returned within 2working days, preferably by overnight mail.Corrections should be marked on the actualproof; do not write a corrections list. Lengthyadditions should be avoided, but where neces-sary should be provided on disk with writteninstructions.
Offprints and ReprintsOffprints can be ordered before press time.Reprints can be ordered later, at additionalcost. Prices depend on the quantity orderedand length of article.
PoliciesPublication in Graft implies that authors of thepaper have read and agreed to its content, andthat readily replaceable material described inthe paper will be freely distributed to academiccolleagues. Atomic coordinates, nucleic acidsequences, and protein sequences must bedeposited in an appropriate data bank; papersshould state that this has been done, andwhere possible give the entry name or accessionnumber.
Peer ReviewsEach contribution to Graft is rigorously vettedby at least 2 expert reviewers who are eithermembers of the Editorial Board or arerecruited by Board members. Contributorsmay be requested to make additions and/orchanges to papers. Compliance with reviewers’recommendations is evaluated before a paperis accepted for publication.
s a g e p u b . c o m g r a f t v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1

Living Related Small Bowel TransplantationLuca Cicalese, Pierpaolo Sileri, Cristiana Rastellini, Herand Abcarian, and Enrico Benedetti
Intestinal transplantation recently became a valid therapeutic option for patients with ir-reversible intestinal failure. The vast majority of the intestinal transplants have been per-formed using whole intestinal grafts obtained from cadaveric donors, and fewer than10% have been performed using segmental grafts obtained from living related donors.Intestinal living donation offers several advantages, such as minimized preservation in-jury, eliminating waiting time, optimal donor quality and better HLA matching and pos-sibly reduced incidence of rejection, lower immunosuppression and side effects, possi-bility to decontaminate the graft prior to transplantation, and possibly reduced risk ofinfectious complications. In the last few years, a standardized technique has been pro-posed for living related small bowel transplantation (LR-SBTx). Utilizing such a tech-nique, the authors performed a series of LR-SBTx in their center and evaluated thesepotential advantages. In this review, the authors summarize the worldwide experiencewith LR-SBTx, including their own.
REVIEWS
5 2 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
ABBREVIATIONS:
BT Bacterial translocationCMV CytomegalovirusEBV Epstein Barr virusIF Irreversible intestinal failureLR-SBTx Living related small bowel
transplantatiosnPTLD Posttransplant lympho-
proliferative disorderSBTx Small bowel transplantationSBS Short bowel syndromeTPN Sotal parenteral nutrition
Luca Cicalese, M.D.Assistant Professor of SurgeryDirector Intestinal Transplant ProgramDivision of Transplant SurgeryUniversity of Illinois at ChicagoRoom 402 Clinical Science Building840 South Wood Street (MC 958)Chicago, Illinois, USA 60612Tel.: 312.996.6771Fax: 312.413.3483email: [email protected]
BackgroundRegardless of the etiology, irreversible intestinal
failure (IF) is the condition in which absorption offluids and nutrients from the small bowel is not ad-equate to sustain life. Although long-term total par-enteral nutrition (TPN) is adequate to support pa-tients with IF, it is associated with importantcomplications such as line sepsis, venous thrombo-sis, and hepatic dysfunction and cirrhosis.1 Thesecomplications are responsible for a significant mor-tality rate. In a recent study, patient survival onlong-term TPN for nonmalignant IF has been shownto be as low as 49% at 5 years.2 Furthermore, thequality of life of patients on TPN is suboptimalsince they often do not tolerate oral diet and arelimited in their activity during the infusions. Addi-tionally, TPN is associated with high costs. In 1992in the United States, the estimated cost per patientper year was approximately $100,000 for suppliesonly, not including home nursing, physician fees,laboratory costs, and expenses related to the treat-ment of TPN-related complications.3
Small bowel transplantation (SBTx) representsthe physiologic alternative to TPN. Recent ad-
vances in immunosuppression, surgical technique,and postoperative management made SBTx a validtherapeutic option for patients with IF—with a 5-year intestinal graft survival up to 70%.4
From a report of the International IntestinalTransplant Registry, approximately 300 intestinaltransplants have been performed worldwide since1985.5 However, the widespread application of thisprocedure is still limited by the relatively high rateof complications. Infections, surgical complica-tions, acute rejection, graft versus host disease(GVHD), and posttransplant lympho-proliferativedisorder (PTLD) are all observed following SBTx,with higher incidence when compared with thetransplant of other organs.6,7
The vast majority of the intestinal transplants hasbeen performed using whole intestinal grafts (aloneor in association with liver or pancreas) obtainedfrom cadaveric donors,5 with or without the inclu-sion of the colon.8 However, fewer than 10% havebeen performed using segmental grafts obtainedfrom living related (LR) donors.
Similarly to the transplant of other organs, intes-tinal living donation offers several advantages, such

as reduced preservation injury, better HLA match-ing, and optimal donor and graft conditions. How-ever, this procedure cannot be performed from liv-ing donors using the standardized techniques usedwith cadaver grafts, and a series of transplants usingLR donors has not been available to unequivocallydemonstrate such advantages. Moreover, LR-SBTxhas not encountered initial preference among theintestinal transplant surgeons since bowel grafts arewidely available from cadavers.
In the last few years, a standardized technique hasbeen proposed for LR-SBTx.9 Utilizing such a tech-nique, we performed in our center a series of LR-SBTx and we evaluated these hypothetical advan-tages. In this review, we summarize the worldwideexperience with LR-SBTx.
Worldwide Experience with LR-SBTxThe reported data on worldwide experience with
LR-SBTx are summarized in Table 1. Initial at-tempts were reported in the 1960s and 1970s fromBoston, Mississippi, and New York.10,11 In Boston, apediatric recipient was transplanted using a seg-ment of ileum donated from the mother and died12 h after the procedure. From the same group, asecond attempt was mentioned during the discus-sion of a scientific meeting, but neither of thesecases was ever published.
In Mississippi, 100 cm of distal ileum was trans-planted in a pediatric recipient. The graft was re-moved 9 days later for extensive necrosis, and thepatient died shortly thereafter.
The group in New York transplanted 170 cm ofjejunum and ileum between HLA identical sisters.The recipient survived 79 days, and she was able totolerate oral diet for approximately 6 weeks.12 Theimmunosuppression used has not been reported byall these centers with the exception of New Yorkand Mississippi where azathioprine, prednisone,and ALG were used. Although technically feasibleand promising, this procedure remained a uniquechallenge mostly because the immunosuppressionavailable at the time was inappropriate. The intro-duction of TPN in 1968 further reduced the inter-est in clinical SBTx.13 The intestine was consideredthe “untouchable” organ for transplant surgeons forapproximately 20 years, while other solid organswere transplanted worldwide with enormous inter-
est and impressive results in terms of graft and pa-tient survival.
The introduction of cyclosporine elicited a newburst of interest for this procedure in the 1980s. AGerman group led by Deltz was the first to report asuccessful clinical LR-SBTx in 1988. They used a60-cm segment of distal jejunum and proximalileum donated by the half sister of the recipientwho survived 4 years on oral diet.14 A previous un-successful attempt was performed 10 months earli-er by the same group in a pediatric recipient. The60-70 cm jejunum/ileum graft, obtained from themother, was unfortunately rejected 12 days afterthe procedure.15 The immunosuppressive regimensused in these cases were based on cyclosporine,steroids, and ATG.
In the 1990s, a new impulse for SBTx was givenby the introduction of FK-506, and LR-SBTxswere performed in 5 centers.16 Pollard in the Unit-ed Kingdom successfully transplanted a segment of180 cm of ileum from the mother to the daughter.This patient had several episodes of rejection anddied 18 months later from pneumonia.17 Morris, inCalifornia, reported the transplant of a segment of110 cm of distal ileum, ileocecal valve, and cecumbetween twin brothers. Survival has been reportedup to 1 year.18 The group in New Orleans, lead byJaffe, performed 2 transplants between mother andoffspring using 200 cm of jejunum. These patientshad rejection and infectious complications. Sur-vival up to 1 year has been reported.19 In Min-neapolis, Gruessner performed 2 successful LR-SBTx from parent to offspring using approximately200 cm of distal ileum. The author was the first todescribe in detail the donor work-up and the surgi-cal technique used to establish a standardized ap-proach for LR-SBTx.9 The Japanese group of Fuji-moto and Tanaka performed 2 pediatric transplantsbetween mother and offspring using 100 to 120 cmof terminal ileum. Both patients had severalepisodes of rejection. One of them died 16 monthsafter the transplant owing to Pneumocystis cariniipneumonia, whereas the other was reported alive ata 14-month follow-up.20
In 1998, the first successful transplant was per-formed in our institution. In the following years,we performed a total of 4 adult LR-SBTx (Table 2).21
In our experience, the graft used was always 180 to
REVIEWS
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 2 7

Table 1 LIVING RELATED SMALL BOWEL TRANSPLANTATION—WORLDWIDE EXPERIENCE
RECIPIENT AGE (YRS.)/SEX UTILIZED GRAFT
YEAR/PLACE/AUTHOR/REF. CAUSE OF IF DONOR HLA MATCH (COLD ISCHEMIA TIME) IMMUNOSUPPRESSION OUTCOME
1964 Boston (USA)10 • ? • Mother Ileum • ? • Death 12 h after Tx• Child • ?
1964 Boston (USA)10 • ? • ? • Death• ? • ? ? ?
1969 Jackson at • 8/male • Mother 100 cm distal ileum • AZA • Graft removed at POD 9 for extensiveMississippi (USA)11 • Illeal strangulation • Class B (Terasaki Scale) (75 min) • Antilymphocyte globulin ischemic necrosis
• Prednisone • Sepsis and death on POD 30
1972 New York (USA)12 • 37/female • Sister 170 cm lower jejunum and • AZA • 1 severe acute rejection• Gardner’s syndrome • Identical upper ileum (110 min) • Antilymphcyte globulin • Eating for 6 weeks
• Prednisone • Death 76 days after Tx with E. colisepsis
1987 Kiel (Germany)15 • 4/male • Mother 60 cm from the medium • ATG • Acute rejection after graft loss 12 days • Volvulus jejunum (80 min) • CsA after Tx
• Steroids
1988 Kiel (Germany)14 • 42/female • Half sister 60 cm lower part jejunum • ATG • 4 acute rejection episodes• SMV and IMV thrombosis • Haploidentical and upper ileum (75 min) • CsA • TPN free for 4 years when graft loss due
• Steroids to acute and chronic rejection• Died 5 yrs after Tx
1995 Leeds (UK)39 • 28/female • Mother 180 cm distal ileum • FK506 • 3 episodes of acute rejection at 1, 3, and• Gardner’s syndrome and • Haploidentical (less than 30 min) • Steroids 10 weeks after Tx
desmoid tumor • AZA • 1 episode of acute rejection was associatedto candida infection
• Death at 18 months from severe pneumonia
1995 Stanford • 34/male • Twin brother Distal ileum, ileocecal valve • None •Sepsis-like syndrome on POD 4California (USA)18 • Desmoid tumor • Identical and portion of the caecum • Alive and TPN free at 1-year follow-up
(110 min)
1995 New Orleans • 26/female • Mother 200 cm proximal jejunum • OKT3 • Loss of 20 cm of graft on POD 7Louisiana (USA)19 • Gardner’s syndrome • Haploidentical • FK506 (ischemic necrosis)
• MMF • Severe acute rejection 7 months after Tx• Prednisone • Need of night TPN after 6 months
1996 New Orleans • 29/male • Mother 180 cm jejunum • OKT3 • Jejunocolostomy leakage on POD 18Louisiana (USA)19 • Ganglioneuropathy • Haploidentical • FK506 • 2 episodes of rejection 3 months after Tx
• MMF • 4 episodes of bacterial overgrowth• Prednisone • 2 episodes of CMV infection
• 1 candida sepsis from invasive fungalduodentitis
• Need of TPN 7 months after Tx
REV
IEWS
52
8v
olu
me
4is
su
e 8
de
ce
mb
er 2
00
1g
raft
sa
ge
pu
b.c
om

REV
IEWS
sa
ge
pu
b.c
om
gra
ftd
ec
em
be
r 20
01
vo
lum
e 4
iss
ue
85
29
1996 Kyoto • 2.5/male • Mother 100 cm distal ileum • FK506 • 4 episodes of acute rejection (Japan)20 • Volvulus • Haploidentical • Steroids followed by line infection,
• AZA EB, CMV• Patient had been on TPN for
almost his entire post-Tx course• Death after 16 months due to
Pneumocyst carinii infection
1997 Minneapolis • 17/male • Father 200 cm distal ileum • OKT3 • Alive and TPN free at 18-monthMinnesota (USA)9 • SMA injury • 4 • FK506 follow-up
• MMF• Prednisone
1997 Minneapolis • ? • Mother 200 cm distal ileum • OKT3 • Alive and TPN free at 1-monthMinnesota (USA)9 • Chron • FK506 follow-up
• MMF• Prednisone
1997 Cambridge • 40/male • Twin brother 150 cm distal ileum • None • Alive and TPN free in 1997(UK)22 • SMV thrombosis • Identical
1999 Kyoto • 4.5/female • Mother 120 cm distal ileum • OKT3 • 4 episodes of acute rejection(Japan)20 • Midgut volvulus • Haploidentical • FK506 • Line infection during acute
• Steroids rejection• Cyclophosphamide • EBV and CMV enteritis
• Alive and TPN free at 14-monthfollow-up
1999 Geneva • 13/male • Twin brother 160 cm midileum • None • Line infection sustained by(Switzerland)23 • Midgut volvulus • Identical Staphylococcus aureus
• Alive and TPN free at 14-month follow-up
1999 Xi’an24 • 18/male • Father 150 cm distal ileum • PK506 • HSV infection, intestinal• MMF hemorrhage and line sepsis• Prednisone after 1 month
• 1 episode of acute rejection• Alive and TPN free at 4-month
follow-up

REVIEWS
200 cm of distal ileum, donated by a family mem-ber (brother, sister, father, and mother) with excel-lent HLA matching (3 to 6 antigens). Three ofthese patients are currently alive, TPN free, andback on regular daily activities with a follow-up of6, 21, and 36 months. No episodes of rejection orsevere infectious complications have been observed.Only 1 patient developed CMV enteritis and wastreated with IV ganciclovir. In the 4th patient, wehad to remove the graft 6 weeks after the transplantfollowing ischemia, probably due to octreotidetreatment for severe pancreatitis. The graft had patentblood vessels and did not present immunologic orinfectious complications. The patient returned toTPN and died 1 year later for TPN-induced liverfailure.
Three additional successful cases have been re-ported worldwide in the last few years. The Cam-bridge group performed 1 transplant between 2identical triplets, using a segment of 150 cm of dis-tal ileum and no immunosuppression.22 Morel’sSwiss group performed a transplant betweenmonozygotic twins using 160 cm of mid ileum.23
Also, a Chinese group, headed by Wang, performedan LR-SBTx between father and son using a seg-ment of distal ileum.24
Surgical Technique and ConsiderationsAs mentioned above, cadaveric intestinal trans-
plantation is performed using the whole intestine,whereas the LR intestinal transplant implies the useof a portion of the small bowel. It is possible to uti-lize segmental jejunal or ileal grafts, and both tech-
niques have been used. However, the vascular sup-ply of the terminal ileum offers a convenient pedi-cle for the graft, and this technique has been stan-dardized. In addition, the distal ileum allows theabsorption of vitamin B12, bile salts, and unlikethe jejunum, a better absorption of water andsolutes and is known to ensure adequate morpho-logic adaptation.25
The approach used in our experience for LR-SBTx implies a careful donor selection. Theseshould be young, healthy individuals for whompreoperative angiogram of the superior mesentericartery excludes abnormalities of the vascular supplyto the cecum, ileocecal valve, and terminal ileum.Furthermore, an optimal HLA matching betweendonor and recipient is recommended and donorsshould be selected, if possible, among multiple can-didates accordingly. The preoperative graft decont-amination is obtained with standard mechanicalbowel preparation and antibiotics. A segment of180 to 200 cm of ileum is resected 15 cm from theileocecal valve that is spared in the donor to reducethe risk of diarrhea and liposoluble vitamin absorp-tion impairment. In our experience, the length ofthe graft obtained is decided in relationship to thetotal length of the donor small bowel. The vascularpedicle of the graft is obtained dissecting the ileo-colic vessels immediately distal to the origin of theright colic artery that is carefully preserved to main-tain vascular flow to the right colon. The mesen-teric peritoneum is scored, and the vessels are iden-tified and dissected up to the origin of the ileocolicvessels. Once the segment of ileum is removed, the
5 3 0 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
Table 2 OUR EXPERIENCE AT THE UNIVERSITY OF ILLINOIS AT CHICAGO
RECIPIENT AGE DONOR/YEAR (YEARS/SEX) CAUSE OF IF HLA MATCHING GRAFT IMMUNOSUPPRESSION OUTCOME
1998 27/male Trauma Twin sister 200 cm distal ileum FK506, ATG, Steroids • 1 CMV gastritis episode6 antigens • Alive and TPN free at 36 months
1999 29/male Trauma Father 200 cm distal ileum FK506, ATG, Steroids • 1 CMV enteritis episode3 antigens • Alive and TPN free at 24 months
1999 46/male SMA Son 200 cm distal ileum FK506, ATG, Steroids • Acute pancreatitis, graft removed5 antigens 6 weeks after Tx
• Death after 12 months for TPN induced liver failure
2000 30/male Trauma Brother 200 cm distal ileum FK506, ATG, Steroids • Alive and TPN free at 6 months

REVIEWS
remaining intestinal segments are primarily re-anastomosed in end-to-end fashion using 4-0polyglyconate for the mucosal layer and 4-0polypropylene for the seromuscular layer. Follow-ing vascular flush with chilled University of Wis-consin solution, the segmental graft is transplantedsuturing the ileocolic vessels in an end-to-side fash-ion to the infrarenal aorta and inferior vena cava ofthe recipient using 6-0 polypropylene. Using thistechnique, the cold ischemia time is approximatelyless than 10 min and the warm ischemia time is 30-40 min. The intestinal continuity is immediatelyreestablished anastomosing the graft to the recipi-ents’ intestinal stumps using 4-0 polyglyconate forthe mucosal layer and 4-0 polypropylene for theseromuscular layer. A temporary distal loop ileosto-my is performed to monitor graft output and toperform endoscopic biopsies to evaluate rejectionor viral infections. Perioperative recipient prophy-laxis for infectious complications is accomplishedwith vancomycin (1 g IV at induction of anesthe-sia), piperacillin (3 g IV 6-8 times a day, adjustedfor renal function, for 3 days), and ganciclovir (5mg/kg IV every 12 h for 14 days) followed by acy-clovir (800 mg PO 4 times a day for 3 months).
Our immunosuppressive protocol consists of oraltacrolimus and prednisone. Intravenous inductionwith atgam is used until therapeutic blood levels oftacrolimus are achieved.
DiscussionLR-SBTx offers several advantages compared with
cadaveric SBTx. This is an elective procedure andcan be performed when the donor and recipientconditions are optimal and donor bowel decontam-ination can be easily performed. This should resultin a decreased risk of early infectious complications.In a previous study on recipients of cadaveric grafts,we showed that the length of preservation was a sig-nificant factor in inducing perioperative bacterialtranslocation (BT).26 With cadaveric intestinaltransplant, such risk cannot be avoided since he-modynamic instability of the donor and subse-quent splancnic hypoperfusion can trigger ischemicdamage even before the intestine is procured.27 Fur-thermore, bowel decontamination in the donor isnot feasible and these grafts are often subject toprolonged cold preservation while specific preserva-
tion solutions designed for intestinal grafts are notyet available. In a recent study, we also showed thatischemic injury induces chronic morphologic alter-ations of the intestinal mucosa.28 An additional ad-vantage of LR-SBTx is that the availability of a liv-ing related donor allows minimization of transplantwaiting time, thus reducing the evolution of TPN-related complications, such as liver damage.
An immunologic advantage is also obtained withLR-SBTx, since optimal HLA tissue matching canbe obtained between donor and recipient that arerelated. It is a common belief that HLA matchingis not important in SBTx, and this is possibly con-sequent to the frequent association of bowel-livertransplantation. However, no data are availablefrom cadaveric SBTx to confirm such a belief—anda high rate of rejection, approximately 90%, havebeen reported in these patients.29-31 In our opinion,liver and intestinal grafts behave differently from animmunologic standpoint. In our experience withwell-matched donor-recipient combinations, wehave not seen rejection using an immunosuppres-sive regimen based on tacrolimus and prednisone.Furthermore, other groups reported LR-SBTx suc-cessfully performed between twins with low or noimmunosuppression. This seems to confirm theimportance of tissue matching in intestinal trans-plantation and, thus, should also be obtained in ca-daveric SBTx since intestinal graft donors are wide-ly available. From this experience, we adopted thestrategy in our cadaveric intestinal transplant pro-gram to minimize the preservation time and to usewell-matched, hemodynamically stable donors.
This strategy allows a reduction of the immuno-suppression, with the consequent benefit of fewerrelated complications. This is of particular impor-tance since cadaveric SBTx is reportedly burdenedby a high rate of PTLD up to 20%, which is high-er than observed in any other organ transplant.32
Although unlikely in cadaveric SBTx, no cases ofPTLD have been reported in LR-SBTx recipients.
An additional advantage of segmental grafts isthat their smaller size allows them to be transplant-ed in patients with a retracted abdominal cavity. Thiscan be due to multiple laparotomies, loss of ab-dominal wall, or severe intra-abdominal adhesions.
A potential disadvantage of LR-SBTx is the surgicalrisk for the donor. However, this is low if associated
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 3 1

REVIEWS
with elective small bowel resection and primaryanastomoses in otherwise healthy individuals, espe-cially when the procedure is performed by experi-enced surgeons. To date, no surgical complicationsor deaths have been reported for LR intestinaldonors. Furthermore, according to the available lit-erature, it does not appear that the donor will suf-fer long-term absorption problems with ileal resec-tion limited to approximately 200 cm.9,14,17,19-21 Mildoccasional diarrhea can be observed only in the ear-ly postoperative time and is well controlled withmedical therapy, with no evidence of vitamin B
12
absorption deficit or weight loss, in our experience.Additionally, to our knowledge, no long-term im-pairment of intestinal absorption in bowel donorshas ever been reported.
An additional disadvantage of using intestinalgrafts obtained from living donors rather than ca-davers is the technical difficulty in using smaller di-ameter vessels for the vascular anastomoses. This isparticularly true if the segment used is jejunum. Asreported in the literature, the use of jejunum oftenrequires multiple vessels as vascular pedicle, makingthe operation more challenging and increasing therisk of thrombosis or chronic hypoperfusion of thegraft.19 In our experience, we utilized a single ileocol-ic artery and vein, performing the arterial anastomo-sis with interrupted technique to minimize such risksand did not witness any of these complications.
It can be argued that the use of a shorter segmentof bowel in LR-SBTx may not be sufficient to pro-vide an adequate absorption of nutrients. From theliterature, most of the surgeons performing LR-SBTx have used segmental grafts of 160 to 200 cm.The decision on how to select an optimal length ofbowel is purely empiric. However, it is based on theknowledge that a segment of 50 cm of small intes-tine will not allow sustaining of life with enteral al-imentation.33-35 Considering that the graft can un-dergo injury for manipulation, preservation, andrejection, we believe that it is safe to use a segmentof 180 to 200 cm of ileum. The choice of thislength also ensures that the donor is left with a seg-ment of at least 300 cm of native small bowel andterminal ileum that are not subject to similar dam-ages. Furthermore, the preservation of the ileocecalvalve in the donor contributes to reducing postre-section dehydration. In our experience, the seg-
mental grafts underwent complete functional adap-tation within 6 months. These patients were TPNfree immediately after the transplant and able to re-gain—and maintain—preintestinal failure bodyweight and serum albumin levels with oral diet.36
After cadaveric SBTx, bacterial, fungal, and viralinfections are quite common. The incidence ofsuch complications is higher than any other organtransplant, probably due to the need for more vig-orous immunosuppression. Infectious complica-tions are the most common cause of death and graftloss, accounting for up to 69% of patient loss aftercadaveric SBTx.37 Line infections, sepsis, abdomi-nal fungal infections, and viral infection or reinfec-tions (EBV and CMV) are also reported after LRintestinal transplantation. Although less frequentthan cadaveric SBTx, severe infections leading torecipient death have been reported.19,20 Several au-thors speculate that some of these infectious com-plications originate from bacterial translocation ofenteric flora during rejection episodes.17 Recently,we analyzed the number of bacterial translocationepisodes (evaluated by the simultaneous presence ofa specific microorganism in the stool and othersites) in 50 pediatric SBTx recipients.26 This analy-sis showed that 44% of patients had at least oneepisode of BT associated with rejection and coldpreservation. In a recent analysis of our LR-SBTxexperience, we observed a very low rate of infec-tions and no episodes of BT.38 It is difficult to ex-trapolate any conclusion since our experience islimited, but the absence of bacterial infections andthe low rate of viral complications observed suggestan advantage to this approach. Several factorsmight have contributed in this regard, such as he-modynamic stability of donors and recipients, opti-mal graft decontamination, minimization of preser-vation injury, and reduced immunosuppression.However, it is impossible to identify which of thesefactors plays a dominant role and probably they allcontribute in part to reducing BT and infectiouscomplications in LR-SBTx.
Despite all these considerations, several attemptsperformed worldwide with LR-SBTx have beenunsuccessful. However, long-term patient and graftsurvival were achieved with LR-SBTx, even in thepre-tacrolimus era in some patients, probably dueto some degree of immunologic advantage obtained
5 3 2 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
with the tissue matching. Another limitation of thereported experience with LR-SBTx is the dishomo-geneity of the cases. Often, these were performed asisolated attempts by each group, making it impos-sible for the surgeon to overcome an unavoidablelearning curve. Furthermore, different surgicaltechniques were often used as well as different im-munosuppressive regimens (Table 1). In our expe-rience, we used a standardized approach to evaluatethe potential advantages of LR compared with ca-daveric SBTx.
In conclusion, intestinal living donation offersseveral advantages, such as minimized preservationinjury, eliminating waiting time, optimal donorquality, and better HLA matching and reduced in-cidence of rejection, lower immunosuppression andreduced associated side effects, possibility to decon-taminate the graft prior to transplantation, and re-duced risk of infectious complications. From thereported cumulative experience, LR-SBTx reacheda 1-year survival rate of approximately 50%. Evalu-ating the reported cases in the tacrolimus era, thesurvival rate at 1 year goes up to approximately70%. In our opinion, these rates are not reflectingthe real potential of the procedure. As we alreadydiscussed, different groups utilized many differentapproaches, creating confusion without gaining ex-tensive experience. We suggest that a standardizedapproach should be used for LR-SBTx. In our lim-ited but significant experience with this procedure,we are confident that LR-SBTx is a valid alternativeto cadaveric SBTx.
Significant advantages offered by this approachsuch as short ischemia time, HLA match, and se-lection of hemodynamically stable donors shouldbe, in our opinion, adopted for cadaveric intestinaltransplantation as well.
References
1. Robinson MK., Ziegler TR., Wilmore DW. Overview of intestinal adaptationand its stimulation. Eur J Pediatr Surg 1999;9:200-6.
2. Messing B, Crenn P, Beau P, et al. Long-term survival and parenteral nutri-tion dependence in adult patients with the short bowel syndrome. Gas-troenterology 1999;117:1043-50.
3. Howard L., Malone M. Current status of home parenteral nutrition in theUnited States. Transplant Proc 1996;28:2691-5.
4. Abu-Elmagd K, Reyes J, Fung JJ, et al. Evolution of clinical intestinal trans-plantation: improved outcome and cost effectiveness. Transplant Proc1999;31:582-4.
5. Grant D. Intestinal transplantation: 1997 report of the international registry.Transplantation 1999;6:1061-4.
6. Abu-Elmagd K, Reyes J, Todo S, et al. Clinical intestinal transplantation:new perspectives and immunologic considerations. Am Coll Surg1998;186:512-25; discussion 525-7.
7. Kusne S, Furukawa H, Abu-Elmagd K, et al. Infectious complications aftersmall bowel transplantation in adults: an update. Transplant Proc1996;28:2761-2.
8. Todo S, Tzakis A, Reyes J, et al. Small intestinal transplantation in humanswith or without the colon. Transplantation 1994;57(6):840-8.
9. Gruessner RW, Sharp HL. Living-related intestinal transplantation: firstreport of a standardized surgical technique. Transplantation 1997;64:1605-7.
10. Margreiter R. The history of intestinal transplantation. Transplant Rev1997;11:9-21.
11. Alican F, Hardy JD, Cayirli M, et al. Intestinal transplantation: laboratory ex-perience and report of a clinical case. Am J Surg 1971;121:150-9.
12. Fortner JG, Sichuk G, Litwin SD, et al. Immunological response to an in-testinal allograft with HL-A identical donor-recipient. Transplantation1972;14(5):531-5.
13. Dudrick SJ, Wilmore DW, Vars HM, et al. Long-term total parenteral nutri-tion with growth, development, and positive nitrogen balance. Surgery1968;64:134-42.
14. Deltz E, Schroeder P, Gebbart H, et al. Successful clinical small boweltransplantation: report of a case. Clin Transplant 1989;3:89-91.
15. Hansmann ML, Deltz E, Gundlach M, et al. Small bowel transplantation ina child. Morphologic, immunohistochemical, and clinical results. Am JClin Pathol 1989;92:686-692.
16. Tzakis AG, Reyes J, Todo S, et al. Two-year experience with FK 506 in pe-diatric patients. Transplant Proc 1993;25:619-21.
17. Pollard SG. Intestinal transplantation: living related. Br Med Bull1997;53:868-78.
18. Morris JA, Johnson DL, Rimmer JA, et al. Identical-twin small-bowel trans-plant for desmoid tumour. Lancet 1995;345:1577-8.
19. Jaffe BM, Beck R, Flint L, et al. Living-related small bowel transplantationin adults: a report of two patients. Transplant Proc 1997;29(3):1851-2.
20. Fujimoto Y, Uemoto S, Inomata Y, et al. Small bowel transplantation usinggrafts from living-related donors. Two case reports. Transpl Int2000;13(Suppl 1):S179-84.
21. Cicalese L, Rastellini C, Sileri P, et al. Segmental living related small bow-el transplantation in adults. J Gastrointest Surg 2001;5:168-73.
22. Calne RY, Friend PJ, Middleton S, et al. Intestinal transplant between two ofidentical triplets. Lancet 1997;350:1077-8.
23. Morel P, Kadry Z, Charbonnet P, et al. Paediatric living related intestinaltransplantation between two monozygotic twins: a 1-year follow-up. Lancet2000;355:723-4.
24. Wu GS, Wang WZ, Song WL, et al. The living-related small bowel trans-plant: the first case in China. Transplant Proc 2000;32:1218.
25. Ferguson DC, Thompson JS. Structural adaptation in intestinal transplants.Transplant Proc 2000;32:1249.
26. Cicalese L, Sileri P, Green M, et al. Bacterial translocation in clinical intes-tinal transplantation. Transplantation 2001;71:1414-17.
27. Kane TD, Johnson SR, Alexander JW, et al. Bacterial translocation in organdonors: clinical observations and potential risk factors. Clin Transplant1997;11:271-4.
28. Cicalese L, Kuddus R, Yacoub W, et al. Ischemia/reperfusion injury induceschronic changes in the small bowel. Transplant Proc 2000;32:1315.
29. Ghanekar A, Grant D. Small bowel transplantation. Curr Opin Crit Care2001;7:133-7.
30. Sudan DL, Kaufman S, Horslen S, et al. Incidence, timing, and histologicgrade of acute rejection in small bowel transplant recipients. TransplantProc 2000;3:1199.
31. Reyes J, Bueno J, Kocoshis S, et al. Current status of intestinal transplan-tation in children. J Pediatr Surg 1998;33:243-54.
32. Finn L, Reyes J, Bueno J, et al. Epstein-Barr virus infections in children af-ter transplantation of the small intestine. Am J Surg Pathol 1998;22(3):299-309.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 3 3

REVIEWS
33. Wasa M, Takagi Y, Sando K, et al. Intestinal adaptation in pediatric patientswith short-bowel syndrome. Eur J Pediatr Surg 1999;9:207-9.
34. Bianchi A.. Longitudinal intestinal lengthening and tailoring: results in 20children. J R Soc Med 1997;90:429-32.
35. Fisher JE. Metabolism in surgical patients. In: Sabiston textbook of surgery.15th ed. WB Saunders; 1997. p. 137-75.
36. Benedetti E, Baum C, Cicalese L, et al. Progressive functional adaptation ofsegmental bowel graft from living related donor. Transplantation2001;71:569-71.
37. Roberts CA, Radio SJ, Markin RS, et al. Histopathologic evaluation of pri-mary intestinal transplant recipients at autopsy: a single-center experience.Transplant Proc 2000;32:1202-3.
38. Cicalese L, Sileri P, Asolati M, et al. Low infectious complications in seg-mental living related small bowel transplantation in adults. Clin Transplant2000;14:567-71.
39. Pollard, SG. Intestinal transplantation: living related. BR Med Bull 1997;53:868-78.
5 3 4 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

Islet of Langerhans Autotransplantation:Rationale, Results, and New DevelopmentsThierry Berney, Aileen Caulfield, Jose Oberholzer, Leo Buhler, Christian Toso, and Philippe Morel
Autotransplantation of islets of Langerhans should be offered to patients undergoing ex-tensive pancreatic resection for chronic pancreatitis. Results of clinical trials of islet au-totransplantation (in which allorejection and recurrence of autoimmunity do not exist ascauses of graft destruction) have been superior to those of allotransplantation, with in-sulin independence for more than 1 year achieved in 47% of recipients. The number ofislets transplanted is a major indicator of outcome, since insulin independence at 1 yearincreases to 71% in recipients of more than 300,000 islets. Importantly, long-term paincontrol after extensive pancreatic resection is excellent and reaches 82% to 100%. Evenin patients who achieve insulin independence, responses to intravenous glucose chal-lenge are depressed and functional insulin secretory reserve is markedly decreased, in-dicating that only a reduced mass of islets engrafts. New indications for islet autotrans-plantation are emerging and include benign pancreatic tumors, blunt trauma, and, morecontroversially, malignant tumors of the pancreas.
REVIEWS
ABBREVIATIONS
CP Chronic pancreatitisDIC Disseminated intravascular
coagulationIEQ Islet equivalentITR International islet transplant
registryIVGTT Intravenous glucose tolerance
testPP Pancreatic polypeptide
Thierry Berney, M.D., Ph.D.Visceral and Transplantation SurgeryDivision of Diabetes and EndocrinologyGeneva University HospitalGeneva, SwitzerlandTel.: 4122.372.77.02Fax: 4122.372.77.55email: [email protected]
IntroductionIslet of Langerhans transplantation is in the lime-
light, thanks to remarkable results recently ob-tained by the Edmonton group after islet allotrans-plantation in type 1 diabetes mellitus patients.1 Anew surge of interest has been generated and is like-ly to benefit other domains of islet transplantation,notably autologous transplantation for the preven-tion of surgical diabetes. This is an interesting rolereversal, since autotransplantation was recentlyviewed from a technical standpoint as a criticalmodel for studying the determinants for successfulislet transplantation in the absence of immunolog-ical mechanisms of graft loss, and thus as a first stepto master before successful islet allotransplanta-tion.2-4 Indeed, successful results of functional isletautotransplantation after extensive pancreatectomywere frequently obtained, as compared with thedismal outcome of a vast majority of allogeneictransplantation procedures.4,5 A number of factorsdoubtless account for the differences observed, in-
cluding the absence of administration of diabeto-genic drugs (steroids and calcineurin inhibitors), al-logeneic rejection, and the recurrence of autoim-munity. Other not-as-well-defined mechanisms,such as the result of the interaction between theislet graft and the microenvironment at the site ofimplantation, might also be involved in islet graftloss.6
Surgical diabetes, provoked by extensive pancreat-ic resection, is a condition comparable in severity totype 1 diabetes. Chronic pancreatitis is the mostcommon indication for extensive pancreatic resec-tion. Such patients are hyperglycemic and at risk ofketosis in the absence of exogenous insulin. Theysuffer frequent hypoglycemic episodes, resultingfrom a lack of counterregulatory mechanisms (i.e.,absence of glucagon), and of poor compliance inthe context of chronic alcohol abuse.7 On the oth-er hand, extensive pancreatic resection is often re-quired for patients with intractable pain due tochronic pancreatitis, and islet autotransplantation
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 3 5

REVIEWS
has emerged as a valuable solution for the preven-tion of surgical diabetes.
Chronic Pancreatitis: to Resect or Not to Resect?
Patients suffering from chronic pancreatitis (CP)are usually referred to the surgeon for chronic in-tractable abdominal pain. The type of surgicaltreatment is a matter of controversy, but it is gener-ally accepted that pancreatic duct drainage shouldbe performed in the presence of a dilated duct,whereas resection should be offered to patients with“small duct disease.”8,9 However, this principle hasbeen challenged by the failure to obtain pain reliefby pancreaticojejunostomy in a number of patientswith “enlarged duct” CP.9-11 The notion that thepancreatic head might be the “pacemaker” of thedisease in alcohol-induced chronic pancreatitis12
and the fact that damage to nerves located aroundand within the pancreatic inflammatory massplays a significant role in the generation of pain13
are likely explanations for failed duct drainage pro-cedures. Suspicion of carcinoma or local complica-tions, such as thrombosis, pseudoaneurysms,pseudocysts, and compression of the biliary or di-gestive tracts, also indicate the performance of a re-section procedure.
Both distal pancreatectomy and pancreatoduo-denectomy have been associated with growingsafety—with mortality rates under 1%.14,15 Goodquality of life16 and satisfactory long-term pain con-trol are achieved by an appropriate resection proce-dure in about 90% of cases.17 A significant numberof patients have a long-lasting history of pain andundergo multiple surgical procedures before pan-creatic resection is decided on, suggesting that re-section is often considered and performed too latein the course of disease.18,19
In the extreme, total or near-total pancreatectomyis the most effective procedure in relieving pain,but it invariably results in insulin-dependent dia-betes. Surgical diabetes is severe and difficult tomanage: patients develop hyperglycemia and are atrisk of ketoacidosis in the absence of insulin thera-py. They may also develop long-term diabetic com-plications if they live long enough. Moreover, theypresent frequent hypoglycemic episodes because ofpoor compliance in a context of continued alcohol
abuse, and because of a lack of the counterregulato-ry mechanisms provided by glucagon.7,20 Therefore,the possibility of preserving endocrine functionthrough islet autotransplantation would be a signif-icant asset for pancreatectomized patients.
Another important consideration when balancingthe metabolic risks and symptomatic benefits of ex-tended pancreatic resection resides in the naturalhistory of chronic pancreatitis. A prospective seriesof 245 patients reported a 74% incidence of dia-betes with a median time of 5.7 years from diagno-sis.21 We have reported a 26% diabetes-free survivalat 10 years after pancreatic resection for CP, withno difference regarding type (duodenopancreatec-tomy vs. distal pancreatectomy) or extent of resec-tion.18 These findings illustrate the relentless char-acter of the disease with an almost inexorableprogression toward total glandular destruction.They might provide a rationale for the performanceof earlier and more extensive pancreatic resectionand islet autotransplantation in order to providethese patients, who are inexorably headed towarddiabetes, with a larger number of healthier islets.Although these considerations remain controver-sial, islet autotransplantation should nonetheless beoffered to any patient with CP undergoing exten-sive pancreatic resection.18
Experimental Islet AutotransplantationThe door to successful clinical islet autotransplan-
tation was opened with the description of newmethods for the isolation and transplantation ofislets of Langerhans in rodents, and the demonstra-tion of diabetes reversal after the transplantation ofsyngeneic islets in animals with “chemical pancrea-tectomy” induced by streptozotocin injection.22
However, the experiments conducted in inbred ro-dents did not reflect the technical difficulties thatare encountered when applying the method for ap-plication in larger mammals, including the human.Studies performed on large animals to demonstratethe feasibility of diabetes reversal by islet autotrans-plantation have been instrumental in applying theconcept to the clinical situation. Reinfusion ofislets isolated after total pancreatectomy into theportal system was shown to result in consistentlong-term correction of surgical diabetes in subhu-man primates (dogs and pigs) and enabled the
5 3 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
quantification of the critical mass of islet tissue nec-essary to revert diabetes in each species.23-26
Much research was conducted in the search for anoptimal implantation site for the islets. The liver(by intraportal infusion) and the spleen (by retro-grade infusion into the splenic vein) were consis-tently identified as the most favorable sites for im-plantation of purified autologous islets in largemammals, as demonstrated by rate of engraftmentor posttransplant metabolic studies.23,27-29 The theo-retically more physiological insulin secretion, di-rectly into the portal vein of the splenic location,does not seem to offer significant advantages. In-terestingly, free intraperitoneal islet autotransplan-tation showed better engraftment and long-termendocrine function when unpurified dispersed pan-creatic tissue was compared with purified islets incanine models.28,30 Moreover, long-term autograftfunction of intraperitoneal unpurified tissue wassimilar to that of intrahepatic purified islets.30
Omental pouches were designed in a canine modelas ideal transplant sites, combining the advantagesof insulin secretion into the portal flow with easyretrievability for biopsy purposes. However, thismethod required a significantly larger autologousislet mass to reverse diabetes than did the in-trasplenic site.31 The kidney capsule is a highly fa-vored transplantation site in rodents because of thetechnical simplicity of the procedure and the possi-bility of demonstrating graft function by observinga return to diabetes after nephrectomy. Analysis ofnonimmunologic mechanisms of graft failure canbe performed in murine models of transplantationof a marginal mass of syngeneic islets under thekidney capsule.32,33 However, largely because of lackof engraftment, which is likely due to poor vascu-larization of the graft site,25,28,34 poor functional out-come is achieved after transplantation of purifiedautologous islets under the kidney capsule of largemammals.
With islets isolated from healthy animals, autotrans-plantation falls short of the situation encounteredwhen dealing with patients with chronic pancreati-tis, in which islets must be isolated from a fibrousand scarred pancreas. In an attempt to reproducethe clinical situation, islet isolation and autotrans-plantation in canine models of chronic pancreatitisinduced by duct ligation achieved diabetes reversal
in, at best, 50% of recipients, a result of low yields,but demonstrated the feasibility of the method.35-38
Animal models have allowed extensive studies ofthe metabolic function of the autotransplantedislets. Such studies pointed out that, in spite of aeuglycemic status, autotransplanted animals hadimpaired glucose responses to glucose tolerancetests39 and markedly reduced insulin responses toglucose and arginine, the latter parameter being adirect measure of the islet secretory capacity, that is,the engrafted islet mass.29,40 The defective glucagonresponse to hypoglycemia, observed after humanintrahepatic islet autotransplantation,41 could be re-produced in a canine model but was restored whenislets were transplanted intraperitoneally.42 Thisfinding suggested that the defective glucagon re-sponse may not solely be the result of an isolation-induced destruction of α-cells or a lack of au-tonomous innervation, and was tentatively explainedby the lack of a proper hypoglycemic stimulus inthe hepatic site because of high glucose concentra-tions in the microenvironment.42,43 Interestingly,basal pancreatic polypeptide (PP) levels were con-sistently low, suggesting a loss of the vagally medi-ated PP response to hypoglycemic stimuli.39,42
Technical Considerations for Human Islet Autotransplantation
When autologous islet transplantation is consid-ered, the surgeon must preserve the vascularizationof the pancreas until its final removal to minimizethe ischemic injury to the gland. The pancreas isimmediately transported to the isolation laboratory,and the islets are isolated with a collagenase diges-tion method. Liberated by enzymatic digestion, theislets are traditionally not purified from the dis-persed ductal and exocrine tissue, mainly to maxi-mize yield.44 This also reduces the processing timeof the pancreatic tissue, which can be ready to in-fuse in less than 2 h, during which pancreatic sur-gery can be completed.45 Transplantation of unpu-rified dispersed pancreatic islet tissue wasintroduced by the Minneapolis group after theyhad shown that it could successfully reverse dia-betes in pancreatectomized dogs.46,47 However, theextra volume of tissue to be transplanted, and thepotential presence of activated pancreatic enzymesin the absence of purification, carries an increased
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 3 7

REVIEWS
risk of portal hypertension and/or thrombosis andintravascular coagulation.48-51 For these reasons, cer-tain groups prefer to purify the pancreatic digest ondensity gradients prior to transplantation.51,52 Theautomated method for islet isolation,53 in which thepancreas is fully immersed in a chamber with a 400to 500 µm screen filtering the outlet where it un-dergoes continuous enzymatic digestion by a 37 °Ccollagenase solution circulating in a closed circuit,can be used effectively to separate islets from glandswith CP. It also offers the advantage of a partial pu-rification because the fibrous components of thepancreas are retained in the chamber.54
The dispersed islet tissue is brought back to theoperating room for intraportal infusion. Islets areinfused via a catheter inserted inside a branch of themesenteric vein after systemic heparinization.45,51,55
Since the volume of the unpurified digest can be ashigh as 35 to 45 ml, the infusion is performed slow-ly and under constant monitoring of the portal veinpressure. Peak portal pressures, as high as 70cmH
2O (50 mmHg), have been recorded during
islet infusion.45,55 The upper safety limit at whichinfusion should stop is not well defined and obvi-ously depends on the pretransplantation value. TheMinneapolis group has opted to inject the remain-ing tissue freely into the peritoneal cavity when por-tal vein pressure reaches 40 cmH
2O (30 mmHg).45 In
this regard, it was shown in canine models that un-purified pancreatic tissue survived better than puri-fied islets in the peritoneal cavity.28,30
The spleen has been explored as an alternate sitefor islet autotransplantation.51 It has the theoreticaladvantage of a more physiological location upstreamfrom the liver and is able to sustain islet function incanine models.29 The islets are transplanted by ret-rograde venous infusion, generally into a short gas-tric vein. However, even if this solution is feasibleand can lead to insulin independence, it has beenassociated with an increased rate of thromboticcomplications, which implies that the performanceof spleen preservation during pancreatic resectionin an inflammatory terrain may be difficult.51
Interestingly, the lack of an in-house islet isolationfacility is not an obstacle for the performance ofislet autotransplantation after pancreatectomy. Agroup in Portland, Oregon, has reported on 5 pa-tients, for whom resected pancreata were shipped in
cold preservation solution to Minneapolis for pro-cessing and the dispersed tissue was shipped backfor infusion. Islet transplantation was performed af-ter a 16- to 24-h delay via a percutaneous mesen-teric vein catheter positioned during surgery andcontinuously flushed with low-volume dilute he-parin solution. Satisfactory long-term results interms of insulin requirements demonstrate that dis-tant processing of islet tissue for autotransplanta-tion is a feasible and reasonable option.56
Results of Clinical Islet AutotransplantationThe latest newsletter of the International Islet
Transplant Registry (ITR) reports 240 autologousislet transplant procedures performed through De-cember 2000 in 15 institutions worldwide.5 Earlyexperience in the 1970s and early 1980s, under thepioneering leadership of the Minneapolis group,demonstrated the feasibility of islet autotransplan-tation after near total or total pancreatectomy, withsome success in preserving metabolic func-tion.7,20,35,57-60 Results of these small series of selectedcases are difficult to interpret, but an exhaustiveanalysis of the published early experience showedthat, overall, 32% to 57% of patients achieved atleast transient insulin independence, depending onthe extent of pancreatectomy.61
Between 1990 and 1999, the ITR reports that64% of patients were insulin independent for morethan 1 week and 47% for more than 1 year. If morethan 300,000 islet equivalents (IEQ: number ofislets if all had an idealized diameter of 150 µm)were transplanted, this proportion rose to 71%,5
with a longest insulin independence follow-up ofmore than 13 years (Fig. 1).62 The most active centersin the past decade have been Minneapolis, MN;Leicester, UK; Geneva, Switzerland; and Indianapo-lis, IN.45,51,52,55,63 Recently published results by theseinstitutions are summarized in Table 1 and show amarked improvement in the achievement of sustainedinsulin independence. In the Minneapolis series,islet yields and probability of insulin independenceafter islet autotransplantation were significantly in-creased after the introduction of the automatedmethod for islet isolation in 1991.45 Unsurprising-ly, the major determinant of success (i.e., insulin in-dependence) for islet autotransplantation is the num-ber of islets infused, either calculated as the number
5 3 8 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
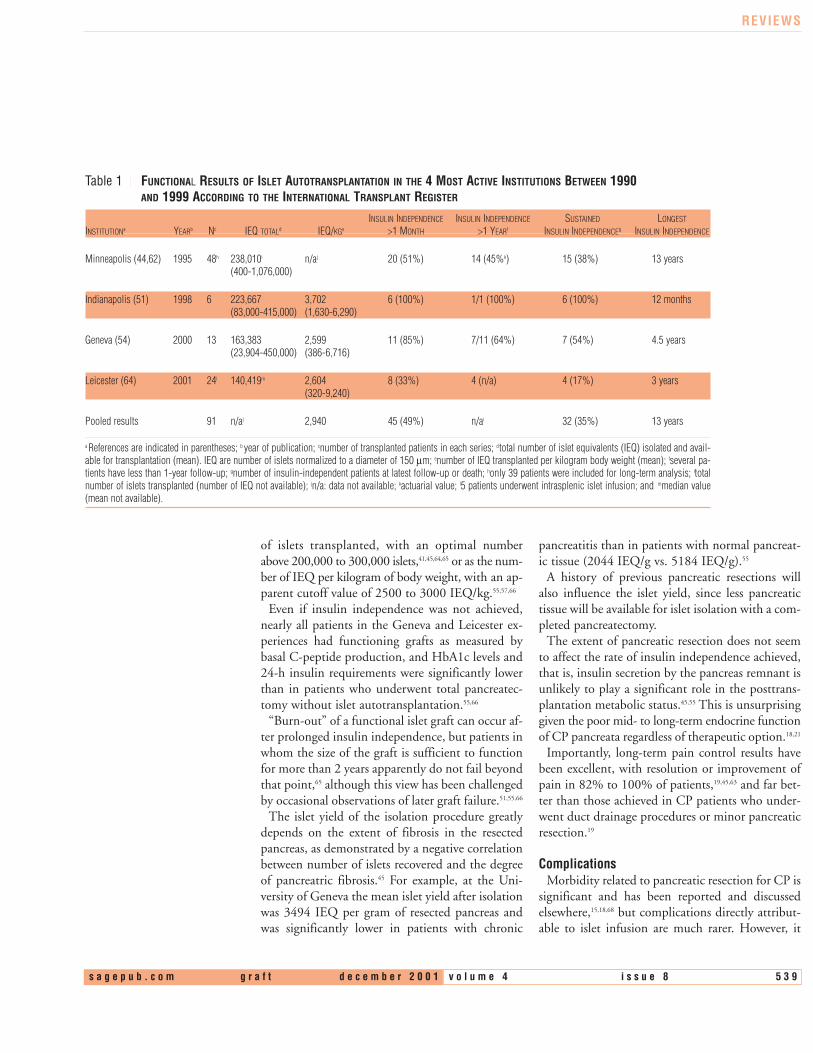
Table 1 FUNCTIONAL RESULTS OF ISLET AUTOTRANSPLANTATION IN THE 4 MOST ACTIVE INSTITUTIONS BETWEEN 1990 AND 1999 ACCORDING TO THE INTERNATIONAL TRANSPLANT REGISTER
INSULIN INDEPENDENCE INSULIN INDEPENDENCE SUSTAINED LONGEST
INSTITUTIONa YEARb Nc IEQ TOTALd IEQ/KGe >1 MONTH >1 YEARf INSULIN INDEPENDENCEg INSULIN INDEPENDENCE
Minneapolis (44,62) 1995 48h 238,010i n/aj 20 (51%) 14 (45%k) 15 (38%) 13 years(400-1,076,000)
Indianapolis (51) 1998 6 223,667 3,702 6 (100%) 1/1 (100%) 6 (100%) 12 months(83,000-415,000) (1,630-6,290)
Geneva (54) 2000 13 163,383 2,599 11 (85%) 7/11 (64%) 7 (54%) 4.5 years(23,904-450,000) (386-6,716)
Leicester (64) 2001 24l 140,419m 2,604 8 (33%) 4 (n/a) 4 (17%) 3 years(320-9,240)
Pooled results 91 n/aj 2,940 45 (49%) n/ai 32 (35%) 13 years
a References are indicated in parentheses; b year of publication; cnumber of transplanted patients in each series; dtotal number of islet equivalents (IEQ) isolated and avail-able for transplantation (mean). IEQ are number of islets normalized to a diameter of 150 µm; enumber of IEQ transplanted per kilogram body weight (mean); fseveral pa-tients have less than 1-year follow-up; gnumber of insulin-independent patients at latest follow-up or death; honly 39 patients were included for long-term analysis; itotalnumber of islets transplanted (number of IEQ not available); jn/a: data not available; kactuarial value; l5 patients underwent intrasplenic islet infusion; and mmedian value(mean not available).
REVIEWS
of islets transplanted, with an optimal numberabove 200,000 to 300,000 islets,41,45,64,65 or as the num-ber of IEQ per kilogram of body weight, with an ap-parent cutoff value of 2500 to 3000 IEQ/kg.55,57,66
Even if insulin independence was not achieved,nearly all patients in the Geneva and Leicester ex-periences had functioning grafts as measured bybasal C-peptide production, and HbA1c levels and24-h insulin requirements were significantly lowerthan in patients who underwent total pancreatec-tomy without islet autotransplantation.55,66
“Burn-out” of a functional islet graft can occur af-ter prolonged insulin independence, but patients inwhom the size of the graft is sufficient to functionfor more than 2 years apparently do not fail beyondthat point,65 although this view has been challengedby occasional observations of later graft failure.51,55,66
The islet yield of the isolation procedure greatlydepends on the extent of fibrosis in the resectedpancreas, as demonstrated by a negative correlationbetween number of islets recovered and the degreeof pancreatric fibrosis.45 For example, at the Uni-versity of Geneva the mean islet yield after isolationwas 3494 IEQ per gram of resected pancreas andwas significantly lower in patients with chronic
pancreatitis than in patients with normal pancreat-ic tissue (2044 IEQ/g vs. 5184 IEQ/g).55
A history of previous pancreatic resections willalso influence the islet yield, since less pancreatictissue will be available for islet isolation with a com-pleted pancreatectomy.
The extent of pancreatic resection does not seemto affect the rate of insulin independence achieved,that is, insulin secretion by the pancreas remnant isunlikely to play a significant role in the posttrans-plantation metabolic status.45,55 This is unsurprisinggiven the poor mid- to long-term endocrine functionof CP pancreata regardless of therapeutic option.18,21
Importantly, long-term pain control results havebeen excellent, with resolution or improvement ofpain in 82% to 100% of patients,19,45,63 and far bet-ter than those achieved in CP patients who under-went duct drainage procedures or minor pancreaticresection.19
ComplicationsMorbidity related to pancreatic resection for CP is
significant and has been reported and discussedelsewhere,15,18,68 but complications directly attribut-able to islet infusion are much rarer. However, it
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 3 9

REVIEWS
should be remembered that the early days of isletautotransplantation were marked by reports of seri-ous, and often fatal, complications, seemingly in-volving a chain of events that began with acute por-tal hypertension and led to disseminatedintravascular coagulation (DIC), and occasionallywas accompanied by portal vein thrombosis andhepatic infarction.48,49,69,70 Pancreatic enzymes,trypsin in particular, have long been known fortheir thrombogenic properties and ability to lead toDIC if released into the bloodstream, an effect thatcan be blocked by heparinization.71 In addition,commercial crude collagenase preparations wereshown to activate proteolytic pancreatic enzymesduring the digestion process.72 These factors maywell explain the development of DIC after infusionof unpurified pancreatic digest into the portal sys-tem. Indeed, since the advent of the automatedmethod of islet isolation, in which partial purifica-tion of the pancreatic tissue is achieved,54 and withthe availability of a new generation of gentler en-zyme blends73 and the routine administration of he-parin,45,48 DIC has no longer been reported after in-fusion of autologous islets into the portal vein.
The only significant complications of islet auto-transplantation recently reported have been 2 casesof partial portal vein thrombosis and 1 wedge splenicinfarct (all 3 without functional consequence), 1 caseof splenic vein thrombosis after intrasplenic infu-
sion, and 2 cases of splenic hilar bleeding after in-traportal infusion (all 3 leading to splenectomy).45,51,66
One case of fatal DIC also occurred after intrasplenicislet infusion, secondary to microembolization intothe lungs of pancreatic tissue fragments that mi-grated through portosystemic collaterals.50
The invariable elevation of intraportal pressurethat occurs during islet infusion may understand-ably lead to a marked decrease of the portal bloodvelocity, with ensuing thrombosis. However, theremay be more to these thrombotic events than thesheer effect of a large mass of tissue carrying acti-vated proteolytic enzymes. Interestingly, it was re-cently shown in allogeneic and xenogeneic in vitromodels that isolated islets infused into the blood-stream could activate the coagulation and comple-ment cascades, thus leading to clot formation andplatelet consumption.74,75 This phenomenon is like-ly of significance in an autologous situation aswell.76 Finally, for reasons that mostly remain un-clear, intraportal infusion has been associated withfewer complications than intrasplenic infusion andshould therefore be the preferred site for autologousislet infusion.50,51,66
Metabolic Studies in Recipients of Autologous Islet Transplants
Preoperative assessment of the pancreatic en-docrine function should be obtained by oral and/or
5 4 0 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
Figure 1. Insulin independence in recipients of islet autografts (squares) for the prevention of pancreatectomy-induced diabetesmellitus (PIDM) transplanted between 1990 and 1999 compared with that observed in recipients of islet allografts (circles) as atreatment of type 1 diabetes mellitus transplanted over the same period. When only autograft recipients transplanted with morethan 300,000 IEQ are considered (triangles), sustained insulin independence is markedly increased. Only well-documented caseswere included in this figure. (Reproduced from Newsletter No. 9 from the International Islet Transplant Registry.5)

REVIEWS
intravenous glucose tolerance tests (IVGTT) andintravenous glucagon challenge55,66 because it is easyto foresee that a patient with impaired metabolictests, let alone established diabetes, is unlikely tobecome euglycemic after islet autotransplantation.Indeed, in the Minneapolis series, almost all pa-tients had normal or near-normal pretransplantglucose tolerance tests.44
Normal IVGTTs, defined by a K value (glucosedisposal rate) greater than 1% per minute, are oftenobserved when insulin independence is achieved af-ter islet autotransplantation and correlate signifi-cantly with the number of islets infused. However,K values are usually higher before isolation, al-though they have occasionally improved after trans-plant.3,41,44,61,62 Similarly, acute insulin responses tointravenous glucose or to arginine are consistentlylower in euglycemic islet autograft recipients withnormal HbA1c, as compared with healthy controlsor pretransplant values.66,67
Functional insulin secretory reserve, measured byglucose-potentiated, arginine-induced insulin se-cretion 3 years after pancreatectomy and autotrans-plantation in 8 patients with sustained insulin in-dependence, correlated highly to the mass of isletstransplanted. Despite insulin independence andnormoglycemia, the response was markedly de-creased in all patients when compared withmatched controls, indicating that only a reducedmass of islets had engrafted.67
In further metabolic studies, this group of pa-tients had no glucagon response to insulin-inducedhypoglycemia, and a depressed but positiveglucagon response to arginine.41 Similar observa-tions were made in autografted patients after 2.5years of insulin independence and normoglycemiaduring hypoglycemic hyperinsulinemic clampstudies.77 The fact that a glucagon response is ob-tained after arginine stimulation indicates that lossof α-cells is not responsible for this observation.These findings have been verified in animal modelsand are discussed above.42 The defective glucagonresponse was not observed in recipients of wholeorgan pancreatic allografts after pancreatectomy.41
PP responses to insulin-induced hypoglycemia orto the high-protein meal are completely absent,whereas recipients of pancreatic allografts had a PPresponse only to the high-protein meal, but not to
insulin. No definite explanation has been offeredfor this observation.41
New IndicationsThe increasing success of islet autotransplantation
after pancreatectomy for CP has prompted theGeneva group to expand the indications for theprocedure.78 We have transplanted islets isolatedfrom 6 pancreata resected for other benign patholo-gies (3 cystadenomas, 2 insulinomas, 1 blunt trau-ma to the pancreas). Median percentage of resectedtissue was 80%. Five of these 6 patients have sus-tained insulin independence after a median follow-up of 35 months.55 Caution must be applied whentransplanting islets isolated from supposedly be-nign tumors, and a diagnosis of malignancy mustbe unequivocally ruled out before making the deci-sion to perform the transplant, especially if the de-cision for tumor removal arises from preoperativediagnostic uncertainty. However, this approach canbe useful for benign lesions whose size and/or loca-tion (neck and body of the pancreas) require theperformance of an extended pancreatic resection toachieve complete extirpation.
More controversially, total pancreatectomy, com-bined with islet autotransplantation, was recentlyproposed as an option for the treatment of pancre-atic adenocarcinoma. This was reported in one pa-tient who underwent completion of a proximalpancreatoduodenectomy for a life-threateninganastomotic leakage, and who is alive with a func-tional islet graft 1 year after the procedure.79 Obvi-ously, a curative pancreatic resection and the infu-sion of islets uncontaminated by tumoral cells areprerequisites for the performance of such a proce-dure. Detection of the K-ras mutation by PCR inthe islet preparation might be a useful technique toprevent infusion of contaminated islets.77
ConclusionsNumerous advances in understanding mecha-
nisms of islet graft loss at the cellular and molecu-lar levels, in the development of new reagents forislet isolation and purification, and in the clinicalmanagement of islet graft recipients, have led tosignificant improvement and success in the func-tional results of islet of Langerhans transplantation.As a result, an increasing number of centers are
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 4 1

REVIEWS
launching islet transplantation programs. This islikely to lead to an increase in the number of auto-transplant procedures after pancreatic resection forchronic pancreatitis or other indications. The islettransplant community will have to take advantageof this ideal situation for the implementation ofmulticenter, prospective randomized trials, aimedat validating the concept of pancreatic resection/islet autotransplantation. Regarding long-termmetabolic results, such studies should focus on de-termining the optimal timing for, and extent of,pancreatic resection, as well as identifying selectioncriteria and providing guidelines for pancreatic re-section and islet autotransplantation, in compari-son to more conservative approaches.
REFERENCES
1. Shapiro AMJ, Lakey JRT, Ryan EA, et al. Islet transplantation in seven pa-tients with type 1 diabetes mellitus using a glucocorticoid-free immuno-suppressive regimen. N Engl J Med 2000;343:230-8.
2. Fontana I, Arcuri V, Nocera A, et al. Islet autotransplantation in humans—amodel for allotransplantation. Transplant Proc 1989;21:2621.
3. Wahoff DC, Papalois BE, Najarian JS, et al. Clinical islet autotransplanta-tion after pancreatectomy: determinants of success and implications for al-lotransplantation? Transplant Proc 1995;27:3161.
4. Hering BJ, Schultz AO, Schultz B, Brendel MD, Bretzel RG, eds. Interna-tional Islet Transplant Registry. 1995;Newsletter No. 6.
5. Brendel MD, Hering BJ, Schultz AO, Bretzel RG, eds. International IsletTransplant Registry. 2001;Newsletter No. 9.
6. Hering BJ, Ricordi C. Results, research priorities, and reasons for opti-mism: slet transplantation for patients with type 1 diabetes. Graft1999;2:12-27.
7. Najarian JS, Sutherland DER, Baumgartner D, et al. Total or near total pan-createctomy and islet autotransplantation for treatment of chronic pancre-atitis. Ann Surg 1980;192:526-39.
8. Rao R, Prinz RA. Surgical therapy in chronic pancreatitis. Curr Opin GenSurg 1993;287-93.
9. Bell RH Jr. Surgical options in the patient with chronic pancreatitis. CurrGastroenterol Rep 2000;2:146-51.
10. Markowitz JS, Rattner DW, Warshaw AL. Failure of symptomatic relief afterpancreaticojejunal decompression for chronic pancreatitis. Strategies forsalvage. Arch Surg 1994;129:374-9.
11. Buhler L, Schmidlin F, de Perrot M, et al. Long-term results after surgicalmanagement of chronic pancreatitis. Hepatogastroenterology1999;46:1986-9.
12. Beger HG, Schlosser W, Friess HM, et al. Duodenum-preserving head re-section in chronic pancreatitis changes the natural course of the disease. Asingle-center 26-year experience. Ann Surg 1999;230:512-9.
13. Bockman DE. Chronic pancreatitis: morphology and the role of nerves. DigSurg 1994;11:261-6.
14. Lillemoe KD, Kaushal S, Cameron JL, et al. Distal pancreatectomy: indica-tions and outcomes in 235 patients. Ann Surg 1999;229:693-8.
15. Barens SA, Lillemoe KD, Kaufman HS, et al. Pancreaticoduodenectomy forbenign disease. Am J Surg 1996;171:131-4.
16. Huang JJ, Yeo CJ, Sohn TA, et al. Quality of life and outcomes after pan-creaticoduodenectomy. Ann Surg 2000;231:890-6.
17. Falconi M, Valerio A, Caldiron E, et al. Changes in pancreatic resection forchronic pancreatitis over 28 years in a single institution. Br J Surg2000;87:428-33.
18. Berney T, Rudisuhli T, Oberholzer J, et al. Long-term metabolic results afterpancreatic resection for severe chronic pancreatitis. Arch Surg2000;135:1106-11.
19. Hinshaw DB, Jolley WB, Hinshaw DB, et al. Islet autotransplantation afterpancreatectomy for chronic pancreatitis with a new method of islet prepara-tion. Am J Surg 1981;142:118-21.
20. Morrow CE, Cohen JI, Sutherland DER, et al. Chronic pancreatitis:long-term surgical results of pancreatic duct drainage, pancreatic resection,and near-total pancreatectomy and islet autotransplantation. Surgery1984;96:608-14.
21. Ammann RW, Akovbiantz A, Largiader F, et al. Course and outcome ofchronic pancreatitis. Longitudinal study of a mixed medical-surgical seriesof 245 patients. Gastroenterology 1984;86:820-8.
22. Bretzel RG, Hering BJ, Stroedter D, et al. Experimental islet transplantationin small animals. In: Ricordi C, editor. Pancreatic islet cell transplantation.Austin, TX: RG Landes; 1992. p. 249-60.
23. Gray DWR, Warnock GL, Sutton R, et al. Successful autotransplantation ofisolated islets of Langerhans in the cynomolgus monkey. Br J Surg1986;73:850-3.
24. Alejandro R, Cutfield RG, Shienvold FL, et al. Natural history of intrahepat-ic canine islet cell autografts. J Clin Invest 1986;78:1339-48.
25. Kaufman DB, Morel P, Field MJ, et al. Purified canine islet autografts. Func-tional outcome as influenced by islet number and site of implantation.Transplantation 1990;50:385-91.
26. Mellert J, Hering BJ, Liu X, et al. Successful islet auto- and allotransplan-tation in diabetic pigs. Transplantation 1998;66:200-4.
27. Warnock GL, DeGroot T, Untch D, et al. The natural history of pure canineislet autografts in hepatic and splenic sites. Transplant Proc 1989;21:2617.
28. Nelson L, Wahoff D, Papalois B, et al. Comparison of various sites of isletautotransplantation in the canine model. Transplant Proc 1997;29:2095.
29. van der Burgh MPM, Guicherit OR, Jansen JB, et al. Function and survivalof intrasplenic islet autografts in dogs. Diabetologia 1996;39:37-44.
30. Wahoff DC, Sutherland DER, Hower CD, et al. Free intraperitoneal islet au-tografts in pancreatectomized dogs—impact of islet purity and posttrans-plantation exogenous insulin. Surgery 1994;116:742-50.
31. Ao Z, Matayoshi K, Lakey JRT, et al. Survival and function of purified isletsin the omental pouch site of outbred dogs. Transplantation 1993;56:524-9.
32. Kaufman DB, Gores PF, Field MJ, et al. Effect of 15-deoxyspergualin onimmediate function and long-term survival of transplanted islets in murinerecipients of a marginal islet mass. Diabetes 1994;43:778-83.
33. Berney T, Molano RD, Cattan P, et al. Endotoxin-mediated delayed islet graftfunction is associated with increased intra-islet cytokine production andislet cell apoptosis. Transplantation 2001;70:125-32.
34. Mellert J, Hering BJ, Hopt UT, et al. Functional outcome after porcine isletautotransplantation beneath the kidney capsule and into the portal vein.Transplant Proc 1994;26:682-3.
35. Mehigan DG, Zuidema GD, Eggleston JC, et al. Pancreatic islet autotrans-plantation: results in dogs with chronic duct ligation. Am J Surg1980;139:170-4.
36. Grodsinsky C, Malcom S, Goldman J, et al. Islet cell autotransplantation af-ter pancreatectomy for chronic pancreatitis. Arch Surg 1981;116:511-6.
37. Horaguchi A, Cobb L, Marincola F, et al. Islet recovery in chronic pancre-atitis. J Surg Res 1983;35:277-82.
38. Yamauchi H, Kakizaki K, Maeda M, et al. Effect of islet cell autograft fromthe highly scarred pancreas on pancreatectomized dogs. Tohoku J Exp Med1985;147:379-87.
39. Watt PC, Ricordi C, Zeng Y, et al. Impaired glucose response following to-tal pancreatectomy and islet cell autotransplantation in dogs. TransplantProc 1992;24:3026-28.
40. Tobin BW, Lewis JT, Tobin BL, et al. Markedly reduced β-cell function doesnot result in insulin resistance in islet autografted dogs. Diabetes1992;41:1172-81.
5 4 2 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
41. Pyzdrowski KL, Kendall DM, Halter JB, et al. Preserved insulin secretionand insulin independence in recipients of islet autografts. N Engl J Med1992;327:220-6.
42. Gupta V, Wahoff DC, Rooney DP, et al. The defective glucagon responsefrom transplanted intrahepatic pancreatic islets during hypoglycemia istransplantation site-determined. Diabetes 1997;46:28-33.
43. Hamilton-Wessler M, Bergman RN, Halter JB, et al. The role of liver glu-cosensors in the integrated sympathetic response induced by deep hypo-glycemia in dogs. Diabetes 1994;43:1052-60.
44. Farney AC, Najarian JS, Nakhleh RE, et al. Autotransplantation of dispersedpancreatic islet tissue combined with total or near-total pancreatectomy fortreatment of chronic pancreatitis. Surgery 1991;110:427-39.
45. Wahoff DC, Papalois BE, Najarian JS, et al. Autologous islet transplantationto prevent diabetes after pancreatic resection. Ann Surg 1995;222:562-79.
46. Sutherland DER, Matas AJ, Goetz FC, et al. Transplantation of dispersedpancreatic islet tissue in humans. Autografts and allografts. Diabetes1980;29(Suppl 1):31-44.
47. Kretschmer GJ, Sutherland DER, Matas AJ, et al. Autotransplantation ofpancreatic islets without separation of exo and endocrine tissue in totallypancreatectomized dogs. Surgery 1977;82:74-81.
48. Mehigan DG, Bell WR, Zuidema GD, et al. Disseminated intravascular co-agulation and portal hypertension following pancreatic islet autotransplan-tation. Ann Surg 1980;191:287-93.
49. Mittal VK, Toledo-Pereyra LH, Sharma M, et al. Acute portal hypertensionand disseminated intravascular coagulation following pancreatic islet auto-transplantation after subtotal pancreatectomy. Transplantation1981;31:302-4.
50. Froberg MK, Leone JP, Jessurun J, et al. Fatal disseminated intravascularcoagulation after autologous islet transplantation. Hum Pathol1997;28:1295-8.
51. White SA, London NJM, Johnson PRV, et al. The risks of total pancreatec-tomy and splenic islet autotransplantation. Cell Transplant 2000;9:19-24.
52. Jindal RM, Fineberg SE, Sherman S, et al. Clinical experience with autolo-gous and allogeneic pancreatic islet transplantation. Transplantation1998;66:1836-41.
53. Ricordi C, Lacy PE, Finke EH, et al. Automated method for isolation of hu-man pancreatic islets. Diabetes 1988;37:413-20.
54. Fontes PA, Rilo HLR, Carroll PB, et al. Human islet isolation and trans-plantation in chronic pancreatitis using the automated method. TransplantProc 1992;24:2809.
55. Oberholzer J, Triponez F, Mage R, et al. Human islet transplantation.Lessons from 13 autologous and 13 allogeneic transplantations. Trans-plantation 2000;69:1115-23.
56. Rabkin JM, Olyaei AJ, Orloff SL, et al. Distant processing of pancreas isletsfor autotransplantation following total pancreatectomy. Am J Surg1999;177:423-7.
57. Valente U, Ferro M, Campisi C, et al. Report of clinical cases of islet auto-transplantation. Transplant Proc 1980;12(Suppl 2):202-4.
58. Traverso LW, Abou-Zamzam AM, Longmire WP. Human pancreatic cell au-totransplantation following total pancreatectomy. Ann Surg1981;193:191-5.
59. Cameron JL, Mehigan DG, Broe PJ, et al. Distal pancreatectomy and isletautotransplantation for chronic pancreatitis. Ann Surg 1981;193:312-7.
60. Toledo-Pereyra LH. Islet cell autotransplantation after subtotal pancreatec-tomy. Arch Surg 1983;118:851-8.
61. Farney AC, Sutherland DER. Islet autotransplantation. In: Ricordi C, editor.Pancreatic islet cell transplantation. Austin, TX: RG Landes, 1992:291-312.
62. Robertson RP, Lanz KL, Sutherland DER, et al. Prevention of diabetes for upto 13 years by autoislet transplantation after pancreatectomy for chronicpancreatitis. Diabetes 2001;50:47-50.
63. White SA, Dennison AR, Swift SM, et al. Intraportal and splenic human isletautotransplantation combined with total pancreatectomy. Transplant Proc1998;30:312-13.
64. White SA, Davies JE, Pollard C, et al. Pancreas resection and islet auto-transplantation for end-stage chronic pancreatitis. Ann Surg2001;233:423-31.
65. Fontana I, Arcuri V, Tommasi GV, et al. Long-term follow-up of human isletautotransplantation. Transplant Proc 1994;26:581-2.
66. Farney AC, Hering BJ, Nelson L, et al. No late failures of intraportal humanislet autografts beyond 2 years. Transplant Proc 1998;30:420.
67. Teuscher AU, Kendall DM, Smets YFC, et al. Successful islet autotrans-plantation in humans. Functional insulin secretory reserve as an estimate ofsurviving islet cell mass. Diabetes 1998;47:324-30.
68. Trede M, Schwall G. The complications of pancreatectomy. Ann Surg1998;207:39-47.
69. Walsh TJ, Eggleston JC, Cameron JL. Portal hypertension, hepatic infarc-tion, and liver failure complicating pancreatic islet autotransplantation.Surgery 1982;91:485-7.
70. Toledo-Pereyra LH, Rowlett AL, Cain W, et al. Hepatic infarction followingintraportal islet cell autotransplantation after near-total pancreatectomy.Transplantation 1984;38:88-9.
71. Kwaan HC, Anderson MC, Gramatica L. A study of pancreatic enzymes as afactor in the pathogenesis of disseminated intravascular coagulation duringacute pancreatitis. Surgery 1971;69:663-72.
72. Traverso LW, Abou-Zamzam AM. Activation of pancreatic proteolytic en-zymes by commercial collagenases. Transplantation 1978:25:226-7.
73. Linetsky E, Bottino R, Lehmann R, et al. Improved human islet isolation us-ing a new enzyme blend, liberase. Diabetes 1997;46:1120-3.
74. Bennet W, Sundberg B, Groth CG, et al. Incompatibility between humanblood and isolated islets of Langerhans: a finding with implications for clin-ical intraportal islet transplantation? Diabetes 1999;48:1907-14.
75. Bennet W, Sundberg B, Lundgren T, et al. Damage to porcine islets ofLangerhans after exposure to human blood in vitro, or after intraportaltransplantation to cynomologus monkeys: protective effects of sCR1 andheparin. Transplantation 2000;69:711-9.
76. Ricordi C, Inverardi L. Towards protection of the islands in the (blood)stream. Transplantation 2000;69:708-9.
77. Kendall DM, Teuscher AU, Robertson RP. Defective glucagon secretion dur-ing sustained hypoglycemia following successful islet allo- and autotrans-plantation in humans. Diabetes 1997;46:23-7.
78. Fournier B, Andereggen E, Buhler L, et al. Human islet autotransplantations:new indications. Transplant Proc 1997;29:2420-2.
79. Liu X, Forster S, Adam U, et al. Islet autotransplantation combined with to-tal pancreatectomy for treatment of pancreatic adenocarcinoma. TransplantProc 2001;33:662-3.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 4 3

REVIEWS
Pharmacoeconomic and Outcomes Analyses in Solid Organ TransplantationKathleen D. Lake
A number of new immunosuppressive agents have been introduced within the pastdecade. Each of these agents has produced impressive results in Phase III clinical tri-als, with acute rejection rates declining from the 40% to 50% range to well under 15%to 25% with newer immunosuppressive combinations. However, with the addition ofeach agent comes an incremental increase in the cost of therapy, resulting in mainte-nance regimens that vary in price from $1,700 with azathioprine and prednisone to wellover $16,000 per year for some of the newer, more potent combinations. Pharma-coeconomic and outcomes analyses can assist practitioners in identifying optimalstrategies for patients when selecting among a number of highly effective but costlyagents. Utilization of these techniques, in combination with the evidence-based medicalliterature, allows healthcare decision makers to make both scientifically and economi-cally sound decisions. The intent of this article is to provide a review of the current phar-macoeconomics literature for transplantation.
Kathleen D. Lake, Pharm.D., B.C.P.S.,F.C.C.P.Director, Clinical Research and TransplantTherapeuticsUniversity of Michigan Medical CenterDivision of Nephrology3914 Taubman Center, Box 0364Ann Arbor, Michigan, USA 48109-0364Tel.: 734.615.0349Fax: 734.647.3417
IntroductionOver the past decade, progress in the field of
transplantation has been accompanied by an in-creased emphasis on controlling the overall costs as-sociated with it. The average billed charges for thevarious transplantation procedures in 1999 were$111,400 for kidney, $303,300 for heart, and$244,600 for liver (Table 1).1-2 Discounted contractreimbursement and Medicare/Medicaid reimburse-ment typically run much less for any given proce-dure. Managed care organizations have also imple-mented the use of contracts based on capitated orglobal payments inclusive of the transplant hospi-talization, physician fees, certain periods of follow-up care (first 90 days to 1 year), and in some cases,also include consultant fees. These types of reim-bursement strategies have placed an increased bur-den on transplant centers to share the risk and hasforced them to evaluate both the cost and the ef-fectiveness of various treatment regimens and pro-cedures. Patients also feel the increased pressures of
healthcare reform with higher copays, limited life-time maximums on insurance coverage, and insur-ers dictating where patients may have their trans-plants performed (i.e., “centers of excellence”).Costly maintenance immunosuppressive regimensmay “spend down” the allocated resources morequickly for a given patient, but this apparent disad-vantage must be weighed against the cost of expen-sive complications, including the possible return todialysis or need for retransplantation.
To complicate the financial issues further, a num-ber of new immunosuppressive agents have beenintroduced during the past decade. Many of themulticenter trials have reported impressive results,with acute rejection rates declining from the 40%to 50% range, with cyclosporine and prednisone,with or without azathioprine, to well under 15% to25% with the newer 3 or 4 drug combination cock-tails.3-9 However, the addition of each agent is asso-ciated with an incremental increase in the overallcost of immunosuppressive therapy.10 Maintenance
5 4 4 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
regimens vary in price from $1,700 with azathio-prine and prednisone to more than $16,000 per yearfor some of the more potent regimens (Table 2).When reviewing the various multicenter clinicaltrials, it is apparent that similar reductions in theincidence of acute rejection can be achieved withdifferent regimens. The following question is thencalled for: Is it possible to achieve the same out-come at a lower cost or a better outcome at thesame cost? Certainly, drug therapy for transplanta-tion is expensive; however, this is overshadowed bythe costs associated with treating the consequencesof failed immunosuppressive therapy. Even though
there is a wide variation in reported costs associat-ed with major complications following solid organtransplantation, it is well recognized that the loss ofa kidney graft and the return to dialysis and/or thetransplant waiting list is neither cost-effective norbeneficial to the patient’s quality of life.11-13
In the early days of economic analyses, a com-mon, albeit shortsighted, approach was to lookonly at the actual cost of the given medications, as-sume outcomes were equivalent, and then use thecheapest product. If that practice were in use today,immunosuppressive regimens consisting of azathio-prine and prednisone might still be the mainstay of
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 4 5
TABLE 1 CHARGES FOR ORGAN ACQUISITION AND TRANSPLANT PROCEDURES (IN DOLLARS)
1999 MEAN LOCAL
STANDARD ACQUISITION
CHARGES BY OPOS* ESTIMATED U.S. AVERAGE BILLED CHARGES FOR TRANSPLANTATION (AS OF JULY 1, 1999 ENDING 1 YEAR AFTER TRANSPLANT)**
ACQUISITION EVAL CANDIDACY PROC HOSP MD F/U IMMUNO TOTAL
Kidney 16,734 10,000 0 20,500 39,200 8,100 20,000 13,600 111,400Kidney/Pancreas - 10,000 0 28,700 56,800 7,900 20,000 14,900 138,300Liver 17,321 15,000 8,900 25,100 107,600 30,300 45,000 12,700 244,600Heart 21,908 15,000 8,900 23,700 181,000 23,300 40,000 11,400 303,300Heart/Lung 21,908 15,000 8,900 22,400 174,900 27,700 40,000 12,300 301,200Lung 16,977 15,000 8,900 22,400 145,800 12,900 40,000 12,700 257,700Double Lung 20,195 - - - - - - - -Pancreas 16,502 10,000 0 17,900 64,400 7,900 7,500 6,000 113,700Small Intestine 15,288 - - - - - - - -
*1999 AOPO Annual Report; **Milliman and Robertson's 1999 report.
TABLE 2 TYPICAL IMMUNOSUPPRESSIVE REGIMEN COST (AVERAGE WHOLESALE PRICE)
NEORAL
CYCLOSPORINE SANDIMMUNE MYCOPHENOLATE AZATHIPRINE TACROLIMUS PREDNISONE
(MICROEMULSION) CYCLOSPORINE MOFETIL 2.5 MG/KG 0.10 MG/KG SIROLIMUS 0.15 MG/KG
4.25 MG/KG PER DAY 5 MG/KG PER DAY 1 G EVERY 12 H PER DAY PER DAY 4 MG/DAY PER DAY ANNUAL COSTS (AWP)
X X X 13,100-13,400X X X 8,400-8,600X X 6,100-6,700
X X X 15,100-15,400X X X 8,600-8,700X X X 8,600
X 7,300-8,000X X 1,700-1,900
X X X 13,800-14,800X X X 14,900
X X X 15,500-16,200X X X 14,700-15,000

REVIEWS
therapy. Fortunately, the focus has gradually shiftedfrom using the acquisition cost of the agents toevaluating the overall benefits derived from thetherapy. Short-term benefits of the various im-munosuppressive regimens are typically measuredin terms of avoidance of acute rejection and adverseeffects. Ideally, economic comparisons should con-sider not only these short-term resource savings butalso potential long-term benefits, such as improvedpatient and graft survival, as well as improvementsin health-related quality of life. It is important torecognize that the more successful the immunosup-pressive regimen is in extending both patient andgraft survival, the more cost-effective it will be. Im-proved long-term outcomes will ultimately benefitsociety in several ways, including the following:
1. reducing the number of retransplant proce-dures, allowing the existing organ supply tobe used for first-time transplants;
2. reducing the time spent on dialysis and thewaiting list; and
3. improving the overall efficiency of the trans-plantation system.
Types of Pharmacoeconomic AnalysesPharmacoeconomics is typically considered a sub-
set of outcomes research that deals specifically withpharmaceutical interventions. The therapy can becompared with other drugs, invasive and noninva-sive therapy, or even watchful waiting. Pharma-coeconomic analyses can be divided into 2 cate-gories: economic evaluations and humanisticevaluations (Table 3). These studies can be viewedfrom a number of perspectives, including that ofsociety, the payer, the patient, the provider, or theproducer. Specific methods for performing thesestudies are reviewed elsewhere.14-16
Pharmacoeconomic studies attempt to examinetotal resource consumption, or all costs associatedwith monitoring a given therapy, including the ac-quisition cost of the drugs, the cost of providingfollow-up services, the cost of side effects, and anyother costs such as concomitant medications. Uti-lization of charge data is often misleading becauseof cost-shifting that may occur in an institution.17
Costs can be defined further as either direct (e.g.,pharmacy products and services), indirect (e.g., lostproductivity), those based on clinical outcomes(e.g., reductions in symptoms), or those based onhumanistic outcomes (e.g., QOL). However, costsneed to also be viewed relative to the potential sav-ings associated with a diminution in either diseaseprogression or new disease onset, and in light ofany complications, which might arise with a stan-dard treatment protocol.
Costs can be divided temporally, into those thatoccur either in a pretransplant environment (e.g.,evaluation and managing the patient’s chronic dis-ease), those that occur during the actual transplantitself (e.g., hospitalization-related costs), or thosethat occur subsequently (e.g., immunosuppres-sants, rejection therapy 1 year posttransplant, etc.).Clinical outcomes specific to transplant, whichneed to be accounted for in cost-consequence mod-eling, include the clinical disease features of rejection,infection, and chronic rejection. Also to be consid-ered are adverse events such as nephrotoxicity, hy-pertension, hyperlipidemia, and steroid-relatedcomplications, and the need for retransplantationalong with the attendant possible consequence ofmortality. Some of the pertinent variables in cost-consequence modeling for transplantation are de-scribed in Table 4. Most of the existing pharma-coeconomic analyses have limited their focus to 1 or2 of the major drivers of the transplant process (re-
5 4 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
TABLE 3 TYPES OF PHARMACOECONOMIC ANALYSES
ECONOMIC EVALUATIONS HUMANISTIC EVALUATIONS
Cost of illness Quality of lifeCost-minimization Quality-adjusted-life-yearsCost-identification Patient preferencesCost-effectiveness Patient satisfactionCost-utilityCost-benefit

REVIEWS
jection and readmissions during the 1st year) andrarely provide a comprehensive analysis of all as-pects listed in this table.
Exclusion of the procedure costs, organ procure-ment fees, or even initial hospitalization may be ap-propriate, assuming that the use of a given agentwill have little impact on certain factors. Othershave highlighted the importance of focusing on theimmunologically relevant variables most likely tobe affected by a regimen or a given procedure andwould exclude those aspects unrelated to transplan-tation (e.g., hospitalization for a motor vehicle ac-cident is unlikely to be related to immunosuppres-sive regimen but could dramatically increase lengthof stay or charge/readmission for a given patient).17
Humanistic EvaluationsEconomic advantages have been well documented
for renal transplantation as compared with dialysisand other healthcare interventions.1-13,18-20 Health-related quality-of-life (HR-QOL) benefits havebeen described for various types of organ trans-plantation.21 Shield et al. showed that patients whowere receiving dialysis for end-stage renal dysfunc-tion had a significant improvement in HR-QOLfollowing kidney transplantation. This study alsoshowed a lower perceived QOL in patients who ex-perienced an acute rejection episode.22
To date, very few studies have compared human-istic outcomes of the various immunosuppressiveregimens, but as additional agents become availablethis will become more relevant.
Application of Pharmacoeconomic Methods
Resource Utilization MethodsThere are 2 primary methods for collecting data
to be used in the economic evaluations of drugs.One way is to collect all the healthcare resourcesused for any given outcome. Clinical trials are oftenused as a way to collect major items of resource uti-lization such as hospitalizations and in-patient re-sources (drugs, lab tests, etc.). Some studies have at-tempted to collect actual financial data from eachparticipating center; however, this method is limit-ed by the interinstitution variability of charges/procedure or medication.23 A better method is tocollect actual resource utilization data and then applystandard costs for the various items (i.e., Medicarereimbursement rates, etc.). This eliminates the vari-ability in charges that exists from institution to in-stitution and also the challenge posed by accuratelycollecting financial data from multiple centers. Thismethod also allows for standardization of charges, asif all of the procedures were performed in one center.
Advantages of piggybacking these studies onto ex-isting trials are that a large number of patients arerandomized to the various treatments, the study hasbeen powered to determine whether a statistically sig-nificant difference exists in predetermined endpoints,and the majority of the data are already being col-lected. If designed correctly, the financial or resourceuse data can be collected in a prospective manner.
The major limitation of piggybacking pharma-coeconomic and outcomes research onto Phase III
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 4 7
TABLE 4 VARIABLES IN COST-CONSEQUENCE MODELING
DIRECT MEDICAL COSTS INDIRECT MEDICAL COSTS CLINICAL OUTCOMES HUMANISTIC OUTCOMES
Drug therapy Noncompliance Clinical disease features Functional statusPhysician visits Work days missed • rejection Quality of LifeAncillary services Family assistance • infection Satisfaction• drug assays Equipment/maintenance • chronic rejection• nephrotoxicity Transportation costs Diagnoses and cures
Hospitalizations/readmissions/LOS Adverse effects• nephrotoxicity• hypertension• hyperlipidemia• steroid-related complications• others
RetransplantationMortality

REVIEWS
multicenter trials (MCT) is the fact that the studiesare conducted under highly controlled conditions(i.e., best-case scenario) designed to measure safetyand efficacy of a regimen in ideal patients. High-risk patients who typically require more frequentmonitoring and dosage adjustments are usually ex-cluded during the screening process. These types ofanalyses would be better termed cost-efficacy,rather than cost-effectiveness analyses. It is not un-til the drug is used in the real-world setting that onecan truly evaluate its effectiveness or cost-effective-ness. Additionally, the Phase III study has a strin-gent protocol for monitoring the drug therapy, andonce practitioners learn how to use the drug, themonitoring frequency may be different than in theinitial phase of the study. This latter factor makes itvery challenging because pharmacoeconomic analy-sis is based on comparing a new medication to onewith which practitioners have far more experience.A pharmacoeconomic analysis performed on aPhase III MCT may find there is no additionalcost-benefit with the new agent, but it is importantto remember the learning curve effect may have animpact on subsequent costs. Another limitation isthat the actual cost of the study drug and its mon-itoring is not known during a Phase III MCT, andin some trials this differential can be sufficient tosway the economic analysis in one direction or theother.
Comparisons of drug regimens, both for effec-tiveness and economics, should be conducted inlarge, randomized, prospective multicenter trials,and ideally performed 3 to 5 years after the drughas been approved, when everyone has experienceusing the new medication. Realistically, it is unlike-ly that the pharmaceutical industry would fund astudy of such magnitude once the drug is approvedand in widespread use.
Pharmacoeconomic Modeling TechniquesAs described above, prospective pharmacoeco-
nomic studies can be very complicated and takeyears to complete. Administrators typically want toknow what impact a new medication, device, orprocedure will have on their institution’s financialstatus in real time rather than waiting for actual re-sults. Therefore, alternative strategies using statisti-cal techniques are frequently used to predict future
implications based on existing data and certain as-sumptions. Pharmacoeconomic studies commonlyemploy one or more of the following techniques toanswer economic questions in a timely manner:modeling, decision analysis, or meta-analysis.
Modeling data have become a popular way of ap-plying pharmacoeconomic analyses to varioussources of data available within and outside health-care organizations.24 Sources of data include med-ical records, financial and administrative databases,expert panels, randomized clinical trials, medicalclaims databases (e.g., Blue Cross and Blue Shield),government or other databases (e.g., Medicare,Medicaid, USRDS, UNOS), and private consult-ants. These types of studies typically use existingclinical and epidemiologic data to project the effectof a clinical, policy, or medication decision on a pa-tient, population, or organization.
Advantages of modeling include that it is a rela-tively inexpensive and timely means of obtainingpharmacoeconomic data (i.e., utilizes existing datarather than repeating the study or collecting newdata). Modeling can also serve as a bridge betweenefficacy data and effectiveness data, allowing one topopulate the model with local or internal datarather than only using data from Phase III trials.
Modeling studies are also inherently disadvanta-geous, largely because they are approximations thatare only as good as the assumptions made and thesensitivity of the model. Modeling also has the po-tential to introduce bias into its findings. It hasbeen suggested that models can be designed to sup-port any results desired by a researcher, sponsor, ordecision maker. If a stakeholder sponsors the study,a degree of skepticism exists with any conclusions.It is also unlikely that a negative pharmacoeconom-ics study will be published if it reflects poorly onthe sponsor’s product. Another limitation of mod-eling involves the quality of data incorporated intothe model. The quality can vary greatly dependingon the source and the rigor under which the datawere collected. Finally, because of a lack of famil-iarity with modeling techniques, practitioners mayquestion the value of data derived this way.
The easiest way to model one’s own data is toadapt an existing model to one’s specific institutionby substituting outcomes data and institutionalcosts. This is not always possible because some of the
5 4 8 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
models published in the literature do not provideadequate detail to allow you to perform your owncalculations, nor do they always share the same keyvariables for your setting. Certainly, institutionswith a heavy managed-care influence may have dif-ferent priorities than those with less capitation.
Clinical Decision AnalysisDecision analysis is a modeling technique used
under conditions of uncertainty. It quantitativelydescribes a problem in terms of multiple possiblecourses of action, probabilities that certain eventsand outcomes will occur, and the value of the ex-pected outcomes resulting from those differentcourses of action. By combining the probabilitiesthat events will occur with the value of each possi-ble outcome, decision analysis determines whichoption to select to maximize the outcome of a giv-en decision. A commonly used component of deci-sion analysis is called the decision tree that incor-porates the various outcomes. A variety of softwarepackages exist to aid the clinician in performing de-cision analyses.
The main advantage of decision analysis is that itforces the user to structure a decision as well asidentify the consequences of the possible decisionoutcomes. It is quantitative in that it forces the userto assign probability estimates and outcome valua-tions to identify the best outcome. Decision treesallow a therapeutic management problem to beseparated into discrete manageable steps and workbest for problems involving events or interventionsthat occur once over a short period of time. Fur-thermore, a treatment decision model can be basedon the relative nature and degree of costs incurredunder different treatment scenarios. Unfortunately,the majority of treatment decisions made today arenot based on such models, largely because of thelack of suitable comparative published studies andbecause of the natural bias toward the selection ofstudies with positive findings for publication.
The main disadvantage of decision trees is thatthey can become very complex (i.e., multiple se-quential branches) when trying to deal with eventsthat occur repeatedly (e.g., acute rejection and in-fection) or over a prolonged period of time (e.g.,chronic rejection). In these situations, it is better touse an alternative method, such as Markov model-
ing, which allows a patient to move from one con-dition to another.
Markov ModelingDepending on the circumstances, a simple deci-
sion tree may not be adequate to address complexissues that can be characterized by the recurrence ofvarious conditions. Conventional decision trees de-scribe the various ways a group of patients in onestate of health may end up in other states over afixed period.24 Markov models, alternatively, focuson transitions among a number of possible healthstates (e.g., healthy, diseased, diseased with compli-cation, and dead) during a series of time cycles.25-26
The general idea behind Markov modeling is that apatient can be in one state of health at any giventime and that the patient’s health status can changefrom that state to another and in some situationsback again, depending on a set of transition-relatedprobabilities.24-26 Potential transplant “states,” inwhich the patient might be categorized, includewell, rejection, CMV infection, other infection,chronic rejection, malignancy, renal failure, anddeath. Markov modeling is the most commonlyused method, but other multistate models are re-viewed elsewhere.27
Advantages of Markov modeling include its utili-ty for more accurately reflecting the various statesin the clinical course of transplant patients. It canalso be used to predict the impact of a change inimmunosuppressive therapy on the expected sur-vival and frequency of other events (e.g., what im-pact does a 50% reduction in rejection that resultsin a 50% increase in CMV have on survival and onlong-term costs?). A limitation of this method in-cludes using data from clinical trials, which may ormay not provide information regarding new im-munosuppressive regimens. For instance, it wouldbe difficult to model the efficacy of different CMVprophylactic regimens in a sirolimus regimen ifthere are no data reporting the efficacy of asirolimus-based regimen. One could make the as-sumption that the antiviral regimens are equally ef-ficacious as in azathioprine or mycophenolatemofetil regimens, but this assumption would com-pletely influence the outcome of the model. Simi-larly, if controlled trials are lacking for regimenscurrently in use, it is not possible to populate a
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 4 9

REVIEWS
model with such data unless it is available in someother setting. Another limitation of the Markovmodel is the assumption that clinical events orstates are mutually exclusive when, in fact, it is pos-sible for a patient to have a rejection episode and aCMV infection at the same time or an acute rejec-tion episode superimposed on chronic rejection.The model is only as good as the assumptions uponwhich it is based.
Markov modeling has primarily been used in trans-plantation for predicting trends such as number ofpatients requiring renal replacement therapy and trans-plantation in Denmark,28 Canada,29 and Australia;30
distribution of donor hearts to maximize recipientsurvival;31 progression of allograft vasculopathy afterheart transplantation;32-33 and analyzing the cost ofmain clinical events after cardiac transplantation.34
Cost of Transplant-Related Complications
Acute RejectionThe cost of maintenance immunosuppression is
high (Table 2). However, the cost of treating an acuterejection episode is also expensive if it does not re-spond to pulse steroid therapy. The cost to treat anepisode of acute rejection is approximately $3,300with a course of steroids and $14,500 with a courseof antilymphocyte therapy, but may be even higherdepending on the number of courses and durationof therapy needed to reverse the process.18,35
CMV InfectionCMV infection is well recognized for increasing
length of stay and hospitalization charges followingboth kidney and liver transplantation.36-38 A num-ber of economic studies have supported the use ofganciclovir38-40 in organ transplant patients, whereasvalacyclovir was studied in another.41 Two of thestudies supported antiviral prophylaxis only in thehighest risk groups.39,41 The economic results fromthe valacyclovir study, using the French healthcaresystem perspective, were difficult to apply to U.S.centers since the length of stay was much longerthan currently reported in this country.41
Das constructed a Markov model to compare thecost-effectiveness of different prophylactic strate-gies for CMV in a hypothetical cohort of 1000 liv-er transplant patients.42 Seven possible posttrans-
plantation states of health were included in theanalysis: healthy, those undergoing acute rejection,those with chronic rejection, patients with CMVinfection but no disease, patients with CMV dis-ease, those with CMV disease complicated by op-portunistic infections, and the 7th state was deathrelated or unrelated to CMV. The model was limit-ed to the 1st year after liver transplantation tosimulate the usual period of CMV-related morbid-ity and mortality and because of the lack of litera-ture using CMV prophylaxis beyond this time pe-riod. Antiviral strategies included providingprophylaxis to all patients or to high-risk patientsonly (D+R-, steroid-resistant rejection, OKT3)and consisted of 5 different regimens (IV ganci-clovir x 100 days, oral ganciclovir x 100 days,CMV immune globulin up to 16 weeks, acyclovirx 6 months, acyclovir x 3 months). In the initialanalysis, all patients received some type of prophy-laxis, with IV ganciclovir and oral ganciclovir iden-tified as being the 2 best strategies. These 2 agentswere then used in the 2nd stage of analysis to de-termine whether universal prophylaxis or selectiveadministration to high-risk patients was preferable.Based on the incremental cost-effectiveness ratio,universal oral ganciclovir was the most favoredstrategy.
This outcome is not surprising considering themost effective strategies were the 2 different ganci-clovir regimens (IV vs. PO); however, the model islimited in that it assumed IV ganciclovir would beadministered for the full 3 months, which is morecostly as compared with oral therapy for 3 months.Another limitation of the study is that some cur-rently used combinations of CMV prophylacticagents (e.g., IV ganciclovir followed by PO ganci-clovir, CMV-Ig in combination with ganciclovir)were not included. Similarly, the analysis of univer-sal versus selective prophylaxis only compared thesestrategies against using no prophylaxis whatsoever,rather than against other contemporary regimenssuch as targeted preemptive therapy.
Steroid-Related ComplicationsVeenstra et al. used Markov modeling to predict
the incidence and long-term cost of steroid-relatedside effects after renal transplantation.43 Data onthe incidence of steroid-related complications (e.g.,
5 5 0 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
hypertension, posttransplant diabetes, peripheralbone fractures, avascular necrosis, cataracts) wereobtained from the transplant literature and werelimited to studies using cyclosporine-based im-munosuppression. If data were not available in thetransplant literature, other sources from the med-ical literature were used. A 10-year time frame wasselected for capturing the costs of steroid-relatedside effects as it would reflect the average graft sur-vival of a kidney transplant recipient. The mostcostly side effects were hypertension and posttrans-plant diabetes. The cost of treating steroid-relatedside effects over 10 years ranged from $2,500 to$7,500 per patient or $265,900 for the 50-patientcohort. Limitations of this analysis include the factthat not all steroid-related side effects were includ-ed such as lipid disorders and cardiovascular com-plications, hip fractures, glycemic control in pa-tients with preexisting diabetes and diabetes-relatedcomplications, and stunted growth, nor werechanges in quality of life related to steroids consid-ered. These additional adverse effects may have in-creased the overall cost per patient.
This study highlights the importance of consider-ing the costs and long-term consequences of im-munosuppressant-related side effects. Certainly, asthe economics of the various new immunosuppres-sive regimens are evaluated, it will be important tofactor in the cost of using steroids when making de-cisions between equally effective but possiblysteroid-free regimens.
Pharmacoeconomic Evaluations of Current Immunosuppressive Regimens
Economic studies evaluating immunosuppressiveregimens have used the various procedures de-scribed above, although most have focused on theshort-term impact of immunosuppressive therapiesand limited their analysis to hospitalization costsand/or readmissions during the 1st year posttrans-plant. Some have included out-patient data, but ona limited basis.
Cyclosporine (Sandimmune, Neoral)The introduction of cyclosporine dramatically in-
creased the cost of maintenance immunosuppres-sion for transplant patients. However, previousstudies have shown that the cost of adding cy-
closporine to the regimen was offset by decreasedreadmissions for treatment of acute rejection dur-ing the 1st year after transplantation, making trans-plantation more cost-effective than dialysis.12,44-46
More recently, a number of economic analysesbased on resource utilization have been conductedcomparing the 2 cyclosporine formulations. Mostwere simple cost analyses that compared the directmedical costs of immunosuppressive therapy dur-ing the short term (e.g., 12 weeks to 1 year post-transplant) after renal or hepatic transplantation.Two preliminary economic studies in Canada per-formed on the data from a stable conversion studyand a de novo trial compared Neoral with the old-er cyclosporine (Sandimmune).47,48 These studiesdid not produce any statistically significant cost dif-ferences as resource utilization was similar in the 2treatment groups, although there was a trend in fa-vor of Neoral. Both studies enrolled a small num-ber of patients, 30 and 41, respectively, and the du-ration was only 12 weeks. Another study in Europeenrolled 68 patients into a de novo trial, and thesepatients were followed for 12 months. From a soci-etal perspective, potential savings of 27% from theuse of Neoral was identified when compared withSandimmune.49 In 3 other economic analyses, therewas an overall cost advantage for Neoral in de novolivers of about 8% to 10% at 4 months,50 an ad-vantage for Neoral versus IV in liver patients withrespect to costs associated with acute rejection,51
and a cost savings from dosage reduction in a con-version trial at 6 months posttransplant.52 A limita-tion of the above studies was that the studies werenot primarily designed to test economic hypothe-ses. Most were not powered to detect a statisticallysignificant difference in clinical outcome, and thusit is no surprise there were not statistically signifi-cant cost differences other than the savings pro-duced by the differential pricing of Neoral versusSandimmune.
Lewis et al. used Markov modeling to evaluate thecost-effectiveness of de novo Sandimmune cyclos-porine versus the modified solution Neoral.53 The 2Neoral cohorts were composed of 35 primary CADrenal transplant recipients participating in U.S. tri-al OLM 103 (Neoral-US) and an aggregate of 77patients studied in European trials OLM 103,OLM 104, and OLM 105 (Neoral-EUR). Each tri-
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 5 1

REVIEWS
al was a prospective, parallel group, randomized,double-blind comparative study of de novoSandimmune (SIM) versus Neoral conducted dur-ing 1992 and 1993. Follow-up in each of the trialcohorts was limited to 12 weeks at the time of dataanalysis. The Sandimmune-treated patients consist-ed of the current controls participating in the U.S.de novo Neoral trial (SIM-US, n = 32) and a cohortof 4737 Sandimmune-treated, 1st-CAD transplantrecipients selected from the U.S. Health Care Fi-nancing Administration (HCFA) databases (SIM-HCFA).
A Markov decision analytic model was construct-ed for each study cohort by assigning one of the fol-lowing 4 health states to each patient: no previousrejection, one or more previous rejectionepisode(s), return to permanent dialysis because ofgraft failure, and death.53
Patients remained in the same health state orwere transmitted to another health state at the endof arbitrarily selected, discrete-time intervals re-ferred to as Markov cycles. The present model wasrun for 6 cycles, each of 15 days duration, to en-compass an observation period of 3 months. Prob-abilities of rejection, graft loss due to rejection,graft loss due to other causes, and death were cal-culated for each 15-day Markov cycle. The cumu-lative probabilities of these events were then calcu-lated and, together with itemized cost data, used tocalculate the costs per functioning graft and per re-jection-free clinical course for the first 3 monthsfollowing transplantation.
Because the rejection rates within the various Ne-oral and Sandimmune cohorts varied so greatly andoverlapped (32% to 45% and 26% to 61%, re-spectively), the data did not demonstrate a conclu-sive difference with respect to cost-effectiveness.The major limitations of the study were the smallsample sizes in each of the de novo clinical trials,protocol-driven patient management and resourceutilization in the clinical trial patients, and differ-ences in European versus U.S. practice patternsthat were not characterized in the de novo studydatabases. Another major limitation of the studywas that the HCFA database was unable to distin-guish between an antibody-treated versus corticos-teroid-treated rejection episode, and a mean costfor all rejection episodes was calculated. Certainly
the use of the actual cost for either polyclonal ormonoclonal rejection therapy might have swayedthe financial analysis.
TacrolimusSeveral studies have been conducted evaluating
the short-term data comparing tacrolimus and cy-closporine based on studies in Europe and theUnited States.23,54-60 The majority of the studies fo-cused on direct medical costs during the short term(e.g., 1st year) after renal or hepatic transplantationand were associated with immunosuppressive ther-apy and readmissions for acute rejection. In someof the studies, an overall cost advantage for tacrolimusof about 10% to 20% was reported,23,57,61 whereasothers reported specific cost advantages (e.g., costsassociated with acute rejection,23,60 immunosuppres-sive regimen,54,57,59 and subsequent rehospitaliza-tions55,57,58). Most of the cost benefits of tacrolimusover cyclosporine were the result of lower rates ofacute rejection reported with tacrolimus mainte-nance therapy.
It is always important to evaluate all of the datapresented within an economic study. A good exam-ple of this is in a recent U.K. study that used a ret-rospective design to analyze resources used in themanagement of adult cadaveric renal transplant pa-tients with Neoral or tacrolimus as primary im-munosuppression.56 Eighty-nine patients with atleast 6 months of follow-up were included in a costanalysis of hospital expenditures for that time peri-od. The authors concluded that there were similaroverall direct medical costs, with mean costs being13,200 pounds for Neoral and 12,982 fortacrolimus patients; however, key factors includingdeath, graft loss, and return to dialysis, which werehigher in the Neoral group, were not included inthe financial analysis.
Short-term and long-term benefits for tacrolimuswere reported in a study by Gjertson and col-leagues reviewing the data on 38,057 first cadaver-ic kidney recipients in the UNOS Kidney Trans-plant Registry from 1988 through 1994. One-yeargraft survival rates of 91.1% ± 1.3% versus 86.6%± 0.2% were reported for tacrolimus versus cy-closporine, respectively. They estimated a signifi-cantly longer graft half-life of 14.5 years for thetacrolimus and 8.8 years for the cyclosporine
5 5 2 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
group.62 If these figures are accurate, the implica-tion is that the cyclosporine group will incur the ex-tra cost of returning to dialysis or need for retrans-plantation 5 years sooner than the tacrolimuspatients. Another interesting finding in this analy-sis was that 60% of the tacrolimus patients were re-ported to be steroid free by 1 year as compared withonly 15% of the cyclosporine-treated patients. Thegraft half-life in the tacrolimus patients success-fully withdrawn from steroids was 26 ± 10 years.The primary limitation of this study was that only24 (11%) of the centers contributed thetacrolimus patients. It is difficult to discernwhether the improvement in graft survival is a re-flection of the primary immunosuppressant orwhether these patients, the majority of whom weresteroid free, represent an immunologically privi-leged population, or whether steroids contributedto the decreased graft half-life seen with the otherpatients.
Mycophenolate MofetilAn economic analysis based on the mycopheno-
late mofetil (MMF) multicenter clinical trial evalu-ated the costs of quadruple therapy involving in-duction, cyclosporine, corticosteroids, and MMFor azathioprine in the 1st year after transplanta-tion.63 Treated acute rejection rates, graft failurerates, and medical care utilization data obtained di-rectly from the U.S. trial were used as inputs to theeconomic analysis. Additional data were obtainedfrom American Hospital Association annual reports(hospital per diem cost estimates), Medicare End-Stage Renal Disease program reports (annualdialysis and functioning graft expenditures), andliterature-base patient preference (utility) estimates.Data from a U.S. quadruple therapy induction tri-al demonstrated a statistically and clinically signifi-cant reduction in the incidence of biopsy-provenacute rejection or treatment failure at 6 months(47.6% in the control group vs. 31.1% in theMMF 2-g treatment group [P = 0.0015]).6 Theclinical results showed a much lower incidence ofrejection, better graft survival, and no difference inthe incidence of opportunistic infections withMMF therapy. Even though MMF was more ex-pensive than azathioprine, the cost of MMF wasoffset by the lower 1st-year treatment costs for re-
jection, dialysis, and graft failure. MMF wasdeemed to be more cost-effective from a societalperspective than azathioprine, and even in theworst-case scenario, with sensitivity analysis ap-plied, MMF was cost-neutral at the end of 1 year.
Two other economic analyses with MMF wereperformed in Canada but provided conflictingdata, with one reporting slightly higher costs withMMF therapy64 and the other finding MMF to bemore cost-effective.65 Limited data are available asboth were only reported in abstract form. Threeother single-center analyses reported early econom-ic benefits from the health system perspective, pri-marily related to the decreased incidence of rejec-tion 3 to 6 months posttransplantation and lessneed for expensive antilymphocyte therapy.66-69
SirolimusLimited pharmacoeconomic data are available for
sirolimus. A recent abstract described an econom-ic analysis using Medicare claims data for the 1styear charges from the recent U.S. sirolimus safetyand efficacy trial.7 The analysis showed lower inpa-tient and physician/supplier charges ($4600) forthe sirolimus 2 mg/day arm as compared with aza-thioprine; however, the cost of the study drugs wasexcluded.70
Induction RegimensMuch controversy has existed regarding the bene-
fits of induction therapy as the randomized trialshave failed to show improved allograft survival.Szczech et al.71 recently conducted a meta-analysisof these trials, which showed a benefit of inductionat 2 years, particularly among presensitized pa-tients, and in the latter population, the patientscontinued to have a benefit at 5 years.
This controversy also exists for pharmacoeconom-ic analyses of the various products as conflictingdata exist for the comparative studies and reflectthe differences that may occur at single centers ver-sus pooled data from multicenter trials.
Shield et al. compared the cost of induction ther-apy with OKT3 versus no induction therapy withcyclosporine, azathioprine, and prednisone bymodeling clinical trial results with financial datafrom separate sources.22 Cost estimates were basedon results from a 5-center randomized trial com-
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 5 3

REVIEWS
paring OKT3 induction with conventional tripledrug therapy in 207 patients. Financial data wereobtained from the National Cooperative Transplan-tation Study, the Medicare Provider and AnalysisReview database, and other sources. The compara-tive measures included costs incurred betweentransplantation and graft failure, the effectivenessof the 2 regimens as defined by length of graft sur-vival, and cost-effectiveness ratios through 5 yearsof observed follow-up, and modeled beyond 5 yearsby assuming a graft failure rate of 4% annually. Theauthors concluded that the initial cost of the OKT3induction therapy was almost offset by savings as-soc- iated with a lower acute rejection rate and atrend for better graft survival. However, depend-ing on which parameter is evaluated, one couldconclude that OKT3 is more expensive, less expen-sive, or cost-neutral. Another single-center studyreported favorable results and improved cost-effectiveness with a shorter course of OKT3 thera-py, but they did not perform a formal economicanalysis.72
Schommer et al.73 performed a retrospectiveanalysis comparing the economics of ATG andOKT3 in a retrospective, multicenter study usingcharge data obtained from the HCIA “ClinicalPathways Data Base.” Five hundred fifty-two pa-tients who had received either OKT3 or ATG wereselected from 22 hospitals. The authors concludedthat the increased pharmacy charges for ATG werepartially offset by reductions in ancillary charges. Ina subsequent publication, the authors pointed outthe limitations of using secondary databases andthat significant variations between hospitals’ clini-cal practices and charging policies made interpreta-tion of the results difficult.74
Brennan et al. conducted a retrospective analysisof their single-center experience of 183 patients re-ceiving induction therapy with either ATG orOKT3.75 There were some demographic differencesbetween the 2 groups as the ATG patients wereolder, which might have contributed to the lowerincidence of rejection, but more extended donorswere also used in that group. The 1-year posttrans-plant rejection was lower for ATG (34% vs. 47%)than for OKT3, and graft survival was better inthe ATG group (93% vs. 85%). The overall hos-pital-related costs for ATG ($39,937 ± $17,014)
and OKT3 ($42,850 ± $20,923 for OKT3) weresimilar.
Schnitzler et al.76,77 demonstrated cost savings forthymoglobulin as compared with ATG in the treat-ment of acute rejection. This pharmacoeconomicstudy was conducted from the perspective ofMedicare and performed on the data from 163 pa-tients enrolled in the randomized double-blind 25-center trial evaluating the safety and efficacy ofthese agents in reversing acute rejection. The studyfocused on the first 90 days following initiation ofrejection therapy and assessed differences in im-munosuppression, therapy for refractory rejection,CMV treatment, and return to dialysis, and com-plications requiring hospitalization were includedin the analysis. Thymoglobulin was associated witha significantly lower cost (overall $5277 savings)during the 90 days posttherapy, with a cost differenceof $7133 in recipients of cadaveric donors. Savingsranged from $6,581 to $12,509 in other high-risksubpopulations. It is important to note that the costof both study agents was excluded, as thymoglobu-lin had not yet been priced and inclusion of this in-formation could change the savings differentials.
Other Methods to Reduce the Cost of Immunosuppressants
Other efforts that have been used to reduce thecostly nature of immunosuppressants include theintentional administration of interacting medications(e.g., ketoconazole, diltiazem, itraconazole, eryth-romycin) or food products (e.g., grapefruit juice).78-
80 These strategies for reducing dosages, necessary toachieve therapeutic concentrations, are dependenton the competitive inhibition of cytochrome P-450IIIA4 enzymes and p-glycoprotein to improvethe absorption of agents such as cyclosporine,tacrolimus, and sirolimus. A dosage decrease andcost savings can be achieved by these strategies, butthe added monitoring costs need to be considered.
SummaryAs more and more immunosuppressive agents are
introduced to the market, practitioners need toscrutinize both the reported clinical results and thesubsequent economic analyses. A number of so-called pharmacoeconomic studies have been pub-lished in the literature, but most are limited by
5 5 4 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
their narrow focus. The majority of these analyseshave shown favorable or at least neutral results.Close attention must be paid to determiningwhether the results are applicable in the clinical en-vironment or whether the healthcare decision mak-ers need to recalculate the anticipated cost benefitsbased on their own data. In these situations, mod-eling techniques can be employed to ensure thatproduct selection is cost-effective. Scientificallysound economic analyses should be performed on aroutine basis; however, rather than piggybackingeconomic analyses onto Phase III clinical trials,these studies should be conducted after practition-ers have adequate experience with the new agents.Guidelines for such studies have been publishedelsewhere.81
REFERENCES
1. 11999 Mean Local Standard Acquisition Charges for All Organ Types. 1999Association of Organ Procurement Organizations (AOPO) Annual Report.
2. Estimated US average billed charges per transplantation as of July 31,1999. Millman and Robertson, Inc.
3. European Mycophenolate Mofetil Cooperative Study Group. Placebo-con-trolled study of mycophenolate mofetil combined with cyclosporine andcorticosteroids for prevention of acute rejection. Lancet 1995;345(8961):1321-5.
4. Mayer AD, Dmitrewski J, Squifflet JP, et al. Multicenter randomized trialcomparing tacrolimus (FK506) and cyclosporine in the prevention of renalallograft rejection: a report of the European Tacrolimus Multicenter RenalStudy Group. Transplantation 1997;64(3): 436-43.
5. Miller J, Mendez R, Pirsch JD, Jensik, SC. Safety and efficacy of tacrolimusin combination with mycophenolate mofetil (MMF) in cadaveric renal trans-plant recipients. Transplantation 2000;69(5): 875-879.
6. Sollinger HW. Mycophenolate mofetil for the prevention of acute rejectionin primary cadaveric renal allograft recipients. U.S. Renal Transplant My-cophenolate Mofetil Study Group. Transplantation 1995;60(3): 225-32.
7. Kahan BD. Efficacy of sirolimus compared with azathioprine for reduction ofacute renal allograft rejection: a randomised multicentre study. The Rapa-mune US Study Group. Lancet 2000;356(9225):194-202.
8. Brennan DC, Flavin K, Lowell JA, et al. A randomized, double-blinded com-parison of Thymoglobulin versus Atgam for induction immunosuppressivetherapy in adult renal transplant recipients [published erratum appears inTransplantation 1999. May 27;67(10):1386]. Transplantation 1999;67(7):1011-8.
9. Johnson C, Ahsan N, Gonwa T. Randomized trial of Tacrolimus (Prograf) incombination with azathioprine or mycophenolate mofetil vs. Cyclosporine(Neoral) with mycophenolate mofetil after kidney transplantation. Trans-plantation 2000;69: 834-841.
10. Kasiske BL, Cohen D, Lucey MR, Neylan, JF. Payment for immunosup-pression after organ transplantation. J Am Med Assoc 2000(May 10);283(18): 2445-2450.
11. Eggers PW, Kucken LE. Cost issues in transplantation. Surg Clin N Am1994;74(5):1259-67.
12. Simon DG. A cost-effectiveness analysis of cyclosporine in cadaveric kid-ney transplantation. Med Dec Making 1986;6(4):199-207.
13. Schneider T, Fagnani F, Lanoe JL, Hourmant M, Soulillou JP. Economicanalysis of an immunosuppressive strategy in renal transplantation. HealthPolicy 1988;9:75.
14. Bootman JL, Townsend RJ, McGhan WF, eds. Principles of pharma-coecomnomics. 2nd ed. Cincinnati: Harvey Whitney; 1996:150-77.
15. Sanchez LA. Applied pharmacoeconomics: evaluation and use of pharma-coeconomic data from the literature. American Journal of Health-SystemPharmacy 1999;56(16): 1630-8.
16. Sanchez LA, Lee JT. Applied pharmacoeconomics: Modeling data from in-ternal and external sources. Am J Health-Syst Pharm 2000; 57(2);146-155.
17. Evans RW, Naessens JM, Rademacher DM, et al. A new approach to thepharmacoeconomic analysis of immunosuppressive agents. Presented atthe American Society of Transplant Physicians 15th Annual Meeting, Chica-go, IL May, 1996.
18. Best JH, Sullivan SD. The changing cost-effectiveness of renal transplan-tation: The impact of improvements in immunosuppressive therapy. Trans-plant Rev 1998;12:34-41.
19. Laupacis A, Keown P, Pus N, et al. A study of the quality of life and cost-utility of renal transplantation. Kidney Int 1996;50:235-242.
20. Evans RW. Kitzmann DJ. An economic analysis of kidney transplantation.Surg Clin N Am. 1998;78(1):149-74.
21. Dew MA, Switzer GE, Goycoolea JM, et al. Does transplantation producequality of life benefits? A quantitative analysis of the literature. Transplanta-tion 1997;64(9): 1261-73.
22. Shield III IC, McGrath MM, Goss TF. Assessment of health-related qualityof life in kidney transplant patients receiving tacrolimus (FK506)-based ver-sus cyclosporine-based immunosuppression. Transplantation 1997;64:1738-43.
23. Lake JR, Gorman KJ, Esquivel CO, et al. The impact of immunosuppres-sive regimens on the cost of liver transplantation – results from the U.S.FK506 multicenter trial. Transplantation 1995;60(10):1089-1095.
24. Keeler E. Decision trees and Markov models in cost-effectivness research.In: Sloan FA, ed. Valuing health care: costs, benefits, and effectiveness ofpharmaceuticals and other medical technologies. Cambridge, England:Cambridge Univ. Press; 1995:185-205.
25. Sonnenberg FA. Beck JR. Markov models in medical decision making: apractical guide. Med Dec Making. 1993;13(4):322-38.
26. Naimark D, Krahn MD, Naglie G, Redelmeier, DA, Detsky, AS. Primer onmedical decision analysis: part 5 – working with Markov processes. MedDec Making 1997; 17:152-9.
27. Hougaard P. Multi-state models: a review. Lifetime Data Analysis.1999;5(3):239-64.
28. Vestergaard P, Lokkegaard H. Predicting future trends in the number of pa-tients on renal replacement therapy in Denmark. Neph Dial Transplant1997;12(10):2117-23.
29. Schaubel DE, Morrison HI, Desmeules M, Parsons D, Fenton SS. End-stage renal disease projections for Canada to 2005 using Poisson andMarkov models. Int J Epidemiol 1998;27(2):274-81.
30. McBride AJ. A forcast of requirements for the treatment of chronic renal fail-ure in Victoria. Aust & New Zealand J Med 1975;5(5):401-7, 1975.
31. Stevenson LW, Warner SL, Hamilton MA, et al. Modeling distribution ofdonor hearts to maximize early candidate survival. Circ 1992;86(5 Sup-pl):II224-30.
32. Sharples LD. Use of the Gibbs sampler to estimate transition rates betweengrades of coronary disease following cardiac transplantation. Stat Med.1993;12(12):1155-69.
33. Sharples LD, Mullins PA, Cary NR, Large SR, Schofield PM, Wallwork J. Amethod of analyzing the onset and progression of coronary occlusive dis-ease after transplantation and its effect on patient survival. JHeart LungTransplant 1993;12(3):381-7.
34. Sharples LD, Briggs A, Caine N, McKenna M, Buxton M. A model for an-alyzing the cost of main clinical events after cardiac transplantation. Trans-plantation. 1996;62(5):615-21.
35. Chan GLC, Hodge EE. The pharmacoeconomics of antirejection therapy inrenal transplant recipients. Pharmacotherapy 1990;10:248-9.
36. McCarthy J. Cost impact of CMV disease in renal transplant recipients.Transplantation 1993;55(6): 1277-82.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 5 5

REVIEWS
37. Falagas ME, Arbo M, Ruthazer R, et al. Cytomegalovirus disease is asso-ciated with increased cost and hospital length of stay among orthotopic liv-er transplant recipients. Transplantation. 1997;63(11):1595-601.
38. Kim WR, Badley AD, Wiesner RH, et al. The economic impact of cy-tomegalovirus infection after liver transplantation. Transplantation2000;69(3): 357-61.
39. Speich R, Thurnheer R, Gaspert A, Weder W, Boehler A. Efficacy and costeffectiveness of oral ganciclovir in the prevention of cytomegalovirus dis-ease after lung transplantation. Transplantation. 1999;67(2):315-20.
40. Wright FH Jr, Banowsky LH. Cytomegalovirus infection and prophylaxis inrenal transplantation: financial considerations. Transplant Proc.1998;345(8955):955-956.
41. Legendre CM, Norman DJ, Keating MR, Maclaine GD, Grant DM. Valaci-clovir prophylaxis of Cytomegalovirus infection and disease in renal trans-plantation: an economic evaluation. Transplantation 2000;70(10): 1463-1468.
42. Das A. Cost-effectiveness of different strategies of cytomegalovirus pro-phylaxis in orthotopic liver transplant recipients. Hepatology.2000;31(2):311-7.
43. Veenstra DL, Best JH, Hornberger J, Sullivan SD, Hricik DE. Incidence andlong-term cost of steroid-related side effects after renal transplantation. AmJ Kid Dis 1999;33(5): 829-39.
44. Showstack J, Katz P, Amend W, et al. The association of cyclosporine withthe 1-year costs of cadaver-donor kidney transplants. J Am Med Assoc1990;264: 1818–1823.
45. Manninen DL. Evans RW. The costs and outcomes of kidney transplantationaccording to initial immunosuppressive drug protocol. Clin Transplant1987: 269-75.
46. Canafax DM, Gruber SA, Chan GL, et al. Pharmacoeconomics of renaltransplantation: increased drug costs with decreased hospital costs. Phar-macotherapy 1990;10: 205-10.
47. Keown P, Lawen JG, Landsberg D, et al. Economic analysis of SandimmuneNeoral in Canada in stable renal transplant patients. Transplant Proc1995;27(2): 1845-8.
48. Kingma I, Ludwin D, Dandavino R,. et al. Economic analysis of Neoral in denovo renal transplant patients in Canada. Clin Transplant 1997;11(1): 42-8.
49. Abella I. Pharmacoeconomics of Neoral, a new formulation of cyclosporine,in renal transplantation. Transplant Proc 1996;28(6):3131-4.
50. Peeters P, Kazek M, Abella I, Noble I. Economic evaluation of Neoral ver-sus Sandimmune maintenance therapy for de novo liver transplant patients:results from an International Randomized Controlled Trial. Milton StudyGroup. Transplant Proc 1998;30(5):1838-42.
51. Hemming AW, Cattral MS, Greig PD, et al. A pharmacoeconomic analysisof Neoral without intravenous cyclosporine in liver transplantation in Cana-da. Clin Transplant 1998;12(5): 425-9.
52. Cogny-Van Weydevelt F, Ngohou C, Bacquaert-Dufour K, et al. Economicrelevance of cyclosporine microemulsion in kidney transplanted patients.Transplant Proc1998;30(6): 2802-3.
53. Lewis RL, Canafax DM, Pettit KG, et al. Use of Markov modeling for eval-uating the cost-effectiveness of immunosuppressive therapies in renaltransplant recipients. Transplant Proc 1996;28(4):2214-7.
54. Tchervenkov JI, Metrakos P, Barkun J, et al. Rejection rate and cost ofmaintenance immunosuppression is reduced when using tacrolimus com-pared to Neoral based immunosuppression in liver transplant recipients[abstract]. Hepatology 1998(Oct);28(Pt 2) Suppl.: 355A.
55. Pirsch JD, Miller J, Deierhoi MH, Vincenti F, Filo RS. A comparison oftacrolimus (FK506) and cyclosporine for immunosuppression after cadav-eric renal transplantation. FK506 Kidney Transplant Study Group. Trans-plantation 1997(Apr 15);63(7): 977-83.
56. Morris-Stiff G, Richards T, Singh J, et al. Pharmaco-economic study of FK506 (Prograf) and cyclosporine A Neoral in cadaveric renal transplantation.Transplant Proc 1998;30(4): 1285-6.
57. McAlister VC, Peltekian K, Bitter-Suemann H, MacDonald AS, Beaudry L.Cost of liver transplantation using tacrolimus. Transplant Proc 1998;30(4):1502.
58. Neylan JF, Sullivan EM, Steinwald B, Goss TF. Assessment of the fre-quency and costs of posttransplantation hospitalizations in patients re-ceiving tacrolimus versus cyclosporine. Am J Kid Dis 1998;32(5): 770-7.
59. Everson GT, Shrestha R, Trouillot T, et al. Costs of cyclosporine (Neoral)and tacrolimus (Prograf) in the first year after liver transplantation [ab-stract]. Transplantation 1998(Jun 27);65: S51.
60. Olivera D. Economic analysis of Prograf (tacrolimus) and cyclosporin in theprevention of kidney allograft rejection. N Horiz Kidney Transpl 1997;1: 12-5.
61. McKenna M, Alexander G, Jones M, et al. Economic analysis of tacrolimus(FK506) and cyclosporin in prevention of liver allograft rejection. Eur HospPharm 1996;2(4): 181-8.
62. Gjertson DW, Cecka JM, Terasaki PI. The relative effects of FK506 and cy-closporine on short- and long-term kidney graft survival. Transplantation1995;60(12): 1384-8.
63. Sullivan SD, Garrison LP Jr, Best JH. The cost effectiveness of mycophe-nolate mofetil in the first year after primary cadaveric transplant. U.S. RenalTransplant Mycophenolate Mofetil Study Group. J Am Soc Nephrol1997;8(10): 1592-8.
64. Keown PA, Sullivan SD, Best JH, et al. Economic evaluation of mycophe-nolate mofetil for prevention of acute graft rejection after cadaveric renaltransplantation. Presented at the American Society of Transplant Physicians16th Annual Meeting, Chicago, IL May, 1997.
65. Kiberd BA. Use of an economic model to estimate the cost effectiveness:The case of mycophenolate mofetil. Presented at the American Society ofTransplant Physicians 15th Annual Meeting, Chicago, IL May, 1996.
66. Baker GM, Martin JE, Jang R, et al. Pharmacoeconomic analysis of my-cophenolate mofetil versus azathioprine in primary cadaveric renal trans-plantation. Transplant Proc 1998;30(8): 4082-4.
67. Khosla UM, Martin JE, Baker GM, Schroeder TJ, First MR. One-year, sin-gle-center cost analysis of mycophenolate mofetil versus azathioprine fol-lowing cadaveric renal transplantation. Transplant Proc 1999;31(1-2): 274-5.
68. Shaffer D, Madras PN, Conway P, Davis C, Simpson MA, Monaco AP. My-cophenolate mofetil eliminates the rationale for antilymphocyte inductiontherapy in nonhaploidentical living-donor kidney transplants. TransplantProc 1997;29(1-2): 342-3.
69. Wuthrich RP, Weinreich T, Schwarzkopf AK, Candinas D, Binswanger U.Postmarketing evaluation of mycophenolate mofetil-based triple therapyimmunosuppression compared with a conventional azathioprine-basedregimen reveals enhanced efficacy and early pharmacoeconomic benefit af-ter renal transplantation. Transplant Proc 1998;30(8): 4096-7.
70. Manninen D, Dong F, Wang F. Economic evaluation of sirolimus therapy inkidney transplantation – results from the US study. XVIII International Con-gress of the Transplantation Society 2000.
71. Szczech LA, Berlin JA, Feldman HI. The effect of antilymphocyte inductiontherapy on renal allograft survival. A meta-analysis of individual patient-level data. Anti-Lymphocyte Antibody Induction Therapy Study Group. AnnInt Med 1998;128(10): 817-26.
72. Alsina J, Bover J, Grinyo JM. Cost-effectiveness of OKT3 induction thera-py in cadaveric kidney transplantation. Am J Kid Dis 1996;28(6): 958.
73. Schommer JC, Pleil AM, Pathak DS. Two approaches to comparing hospi-tal charges between cadaveric renal transplant patients who received ortho-clone OKT 3 sterile solution or Atgam sterile solution for induction therapy.Clin Therapeutics 1995;17(4): 749-69; discussion 748.
74. Schommer JC, Pathak DS, Grauer DW. Economic evaluation of immuno-suppressive drugs: an empirical example using a secondary database ofhospital charges. Transplant Proc 1996;28(2): 906.
75. Brennan DC, Schnitzler MA, Baty JD, et al. A pharmacoeconomic compar-ison of antithymocyte globulin and muromonab CD3 induction therapy inrenal transplant recipients. Pharmacoeconomics 1997;11(3): 237-45.
76. Schnitzler MA, Woodward RS, Lowell JA, et al. Costs savings associatedwith thymoglobulin for treatment of acute renal transplant rejection in pa-tient subsets. Transplant Proc 1999;31(3B Suppl): 7S-8S.
5 5 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 5 7
77. Schnitzler MA, Woodward RS, Lowell JA, et al. High risk kidney transplantrejection treatment: cost savings from thymoglobulin. Transplant Proc1999;31(1-2): 269-71.
78. Jones TE. The use of other drugs to allow a lower dosage of cyclosporin tobe used. Therapeutic and pharmacoeconomic considerations. Clin Phar-macokinet 1997;32(5):357-67.
79. Yee GC, Stanley DL, Pessa LJ, et al. Effect of grapefruit juice on blood cy-closporin concentration. Lancet 1995;345(8955): 955-6.
80. Edwards DJ, Fitzsimmons ME, Schuetz EG, et al. 6’,7’-Dihydroxyberg-amottin in grapefruit juice and Seville orange juice: effects on cyclosporinedisposition, enterocyte CYP3A4, and P-glycoprotein. Clin Pharmacol Ther-ap 1999;65(3):237-44.
81. Whiting JF. Standards for economics and quality of life studies in trans-plantation. Transplantation 2000; 70: 1115-21.

REVIEWS
“Engineering” Myoblast TransplantationDaniel Skuk and Jacques P. Tremblay
Myoblast transplantation (MT) designates the intramuscular implantation of myogeniccells as a treatment for muscle diseases. As a therapeutic tool, MT can act in 2 com-plementary manners: It can be a vehicle for a normal genome and it can increase themyogenic capacity of the host muscle. Although many experiments in rodents demon-strate these properties, the experiments in nonhuman primates allow for a better defini-tion of the parameters that allow for making MT an applicable strategy in humans. In thepresent review, special attention is given to the clinical possibilities of MT. Two chal-lenging factors are especially analyzed: the strategy of cell delivery and the control ofrejection. The 3 issues that the authors identify as requiring further study to introduce im-provements in MT design are intramuscular donor-cell migration, early donor-cell sur-vival, and methods to avoid allograft rejection (development of specific tolerance or au-totransplantation of genetically corrected myoblasts).
DYSTROPHIN:
A fibrilar protein that connects sarcomericactin with a complex of proteins related tothe sarcolemma.
Jacques P. TremblayUnité de Recherche en Génétique Hu-maineCentre Hospitalier de l’Université Laval2705 Boulevard LaurierSte-Foy, Québec, Canada G1V 4G2Fax: 418.654.2207email: [email protected]
IntroductionThe principal targets of myoblast transplantation
(MT) are muscular dystrophies, a group of diseasesin which molecular defects of genetic origin pro-duce progressive muscle wasting. Among these, theseverity and frequency of Duchenne muscular dys-trophy (DMD) make it the prime target for MT.DMD is caused by the absence of dystrophin, a fib-rilar protein that connects sarcomeric actin with acomplex of proteins related to the sarcolemma. Thelack of dystrophin produces progressive muscle de-generation, expressed clinically by loss of muscleforce early in childhood, severe weakness by the ageof 10, and death by the age of 20. As clinical trialsconducted in the 1990s were unsuccessful,1-8 subse-quent skepticism arose in regard to MT. However,some groups continued animal research, allowingfor a better understanding of the factors implicatedin MT success. Although mice experiments sup-ported the potential benefits of MT and answeredsome basic questions, it is the experience obtainedin nonhuman primates9-12 that allows for an appli-cable approach in humans.
The importance of nonhuman primates overmouse models for clinical MT research is basedupon 3 elements:
1. In contrast to the differences between humanand murine immune systems, monkeys andhumans share similarities such as the struc-tural homology of major histocompatibilitycomplex (MHC) class I and class II loci.13
2. Muscle regeneration can be different in miceand primates.14
3. The anatomy and size of monkey muscles area better, more applicable to human, model totest the strategies of donor cell injection.12
In the present review, we examine a strategy forclinical applications, mainly based upon the obser-vations of the nonhuman primate model.
The Basis of Myoblast TransplantationAmong the basic issues that must be defined be-
fore designing a cell transplantation strategy are thecharacteristics of the target tissue, the type of donorcell, the mechanisms allowing the efficient incor-poration of the donor cells in the target tissue, andthe effects of cell transplantation on the target tis-sue. We will briefly consider these issues in the con-text of MT.
The target tissue in MT is the skeletal muscle, themajor tissue component of the human body (40%-50% of the total body weight). The function of
5 5 8 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
skeletal muscle is to generate voluntary mechanicalwork, which is done by polynucleated syncytiacalled myofibers (Fig. 1). Myofibers are longenough to join the points that must be reached formechanical work, and parallel to each other, allowa sum capacity to generate force. Myofibers arejoined by a delicate connective tissue (the endomy-sium), are grouped into fascicles joined by the per-imysium, and are surrounded by the epimysium.This connective network ensures the coordinatedwork of the ensemble.
Since myofibers are differentiated syncytia with ahighly specialized structure, they cannot enter mi-tosis and, thus, cannot be expanded in culture orused for transplantation. For MT, a precursor myo-genic cell capable of proliferating in vitro and dif-ferentiating into myofibers is needed. During post-natal life, the skeletal muscle ensures growth,hypertrophy, and regeneration of necrosed my-ofibers through the presence of mononucleatedmyogenic stem cells named “satellite cells” (for a re-view, see ref. 15). The denomination of “satellite” isdue to their placement, occupying a depression onthe periphery of the myofiber, lying between thesarcolemma and the basal lamina (Fig. 1). An in-jury causing myofiber necrosis stimulates satellitecells to be drawn out of their quiescent state, pro-liferating and fusing, either to fill the defect pro-duced by segmental necrosis (Fig. 2) or to restorean entire myofiber after total necrosis. Satellite cellscan be isolated enzymatically from muscle and canbe proliferated in vitro (Fig. 1), maintaining theircapacity to fuse and differentiate into myofibers. Asmononucleated and undifferentiated cells with thecapacity to differentiate in skeletal muscle, thesecells are referred to as “myoblasts.”
The transplantation of these cells into hostmuscles has 2 complementary properties: Donormyoblasts are vehicles of normal genome, andthey can increase the myogenic capacity of the hostmuscle.
Donor Myoblasts as a Vehicle of Normal GenesAfter fusing with host myofibers, donor myoblasts
induce “gene complementation”; that is, myofiberswill express genes of both donor and host nuclei, al-lowing the expression of the protein that is mutat-ed in the host nuclei (Fig. 2). These myofibers are
referred to as “hybrid” myofibers. Gene complemen-tation was demonstrated after MT in myopathicmouse models. MT in the mdx mouse, a model ofDMD, led to dystrophin expression in host my-ofibers.16-19 There is evidence that this expression ofdonor-dystrophin protects mdx myofibers from thepathological process.16,20,21 In the dy/dy mice, a mod-el of congenital dystrophy with lack of merosin,MT leads to hybrid fibers expressing merosin.22
MT in the SJL/J mice, a model for limb-girdlemuscular dystrophy with lack of dysferlin, restoreddysferlin expression in hybrid myofibers.23
Donor Myoblasts Increase the Myogenic Capacity of the Muscle
A characteristic of DMD is the progressive ex-haustion of satellite cells after recurrent cycles ofdegeneration-regeneration. The injection of donormyoblasts can act as reinforcement to the myogeniccapacity in these muscles. Restoration of musclemass and force after MT was observed in many ro-dent experiments.24-27
Another potential benefit of MT would be the ca-pacity to reconstitute a pool of donor satellite cellsable to participate in later regeneration. Donor my-oblasts remaining as quiescent satellite cells wereobserved in biopsies of DMD patients that partici-pated in an MT clinical trial.28 In rodents, there isevidence that some donor myoblasts persist as pre-cursors able to participate in later muscle regenera-tion,29,30 specifically, as functional satellite cells.31
The Strategy of Cell DeliveryThe most efficient method to deliver donor my-
oblasts to muscles is direct injection. Intramuscularinjections ensure sufficient quantities of donor cellsinto the host tissue. They produce muscle injury,allowing myofiber regeneration (that incorporatesefficiently the donor cells in the preexisting my-ofibers), and the breaking of endomysial tubes (anevent that could allow the movement of the donormyoblasts into them). Two parameters must be de-fined for an efficient strategy of donor-cell injec-tion: the optimal distance between injections andthe optimal number of cells to be delivered per in-jection trajectory. These parameters will dependon a previous definition of the objective to bereached.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 5 9

REVIEWS
5 6 0 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
Figure 1. (1) As a treatment of muscular dystrophies, the target of MT is the striated skeletal muscle. (2) Skeletal muscles are composed by longsyncytia (myofibers) parallel-arranged. (3) Satellite cells, the quiescent myogenic stem-cells situated at the periphery of the myofibers, are the prin-cipal source of donor cells for MT. (4) Satellite cells can be isolated from muscle and proliferated in vitro as myogenic mononucleated precursors(myoblasts). (5) For monkey experiments, these myoblasts are labeled in vitro by retroviral transfection with the LacZ gene and injected in a bi-ceps brachii using a grid to define the inter-injection distance. (6) A cross section of the whole bicep shows high percentages of myofibers ex-pressing β-galactosidase (dark staining). (1: detail of Rembrandt’s Anatomy Lesson of Dr. Tulp, 1632.)

REVIEWS
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 6 1
Figure 2. (1) In monkeys, donor myoblasts are injected by parallel injections, perpendicular to the muscle axis. (2) The needle producesmyofiber damage during introduction (2a), and cells are delivered in the injection trajectory during needle withdrawal (2b). (3) The in-jection produces myofiber damage (3a), triggering a process of segmental regeneration in the presence of donor cells. Muscle regen-eration involves proliferation and fusion of myogenic cells (3b), which in this case is done with the participation of donor and host cells.After fusion (3c), donor-cells are incorporated in a “hybrid” myofiber (3d). The expression of those proteins present exclusively in thedonor cells (e.g., dystrophin in the case of DMD) is obtained in a region of the hybrid myofiber: the nuclear domain of the donor-nuclei(3d). (4) One month after MT, expression of the reporter protein is observed in those myofibers reached by the injections. (5) This isevident in cross sections of monkey muscles, where the trajectories of myoblast-injection are seen as tracts of β-galactosidase-positivemyofibers (in dark). Original magnification (5): 25X.

REVIEWS
Defining an ObjectiveThe ultimate objective of MT is to improve the
muscle function in severe myopathic patients andto ensure an acceptable quality of life. Since thereare no monkey models of myopathies, experimentsto test the functional effects of MT cannot be con-ducted in nonhuman primates. However, injectionof β-galactosidase-labeled myoblasts in normalmonkeys allows for an understanding of the pa-rameters that produce high numbers of hybrid my-ofibers in the muscles. To extrapolate these data forDMD, it is necessary to know the percentage ofdystrophin-positive fibers that must be obtained fora clinical effect. This can be estimated by the ob-servations in DMD carriers. DMD carriers have amosaic of dystrophin-positive and dystrophin-neg-ative myofibers and can be asymptomatic or exhib-it different degrees of severe to mild myopathy.Some authors have observed a correlation betweenthe percentage of dystrophin-positive fibers and theclinical phenotype,32,33 as illustrated in Figure 3. Itcan be supposed schematically that less than 25%of hybrid fibers (as a result of MT in a DMD pa-tient) may not significantly ameliorate the clinicalpicture, between 50% and 80% may significantlyslow down the clinical evolution (leading to a mildmyopathy), and between 80% and 100% will stopthe evolution. Since most of our recent monkey ex-periments reached the second category (Fig. 3), thismodel allows us to advance toward the goal of set-ting parameters for clinical MT. It must be notedthat some differences can be expected when com-paring β-galactosidase and dystrophin expression,considering the different nuclear domains of bothproteins.34
The Inter-Injection DistanceThe distance between injections may be deter-
mined by the efficacy of each individual injection,that is, the volume of muscle that will express thereporter gene introduced by the donor myoblasts(e.g., dystrophin or β-galactosidase) after a singleinjection. The more efficacy of a single injection,the more distributed the injections can be to reachthe predetermined objective. The efficacy of an in-dividual injection depends on
1. the capacity of donor myoblasts to migrateand fuse with distant myofibers,
2. the extent of the muscle damage aroundinjections, and
3. the length of the nuclear domain for the pro-tein restored.
The intramuscular injection of myoblasts in mon-keys (the donor-cells homogeneously delivered dur-ing the needle withdrawal) produces defined tracksof hybrid myofibers (Figs. 2, 3, and 4). This indi-cates that donor myoblasts are incorporated mostlyby myofibers along the injection trajectory. Theseobservations concur with mouse experiments thatshow primary cultured myoblasts do not migratethrough nondamaged muscle.35-37 Since a single my-oblast injection produces a defined track of hybridmyofibers, the percentage of hybrid myofibers in amuscle section depends on the density of thesetracks, and, therefore, on the density of injections.In monkey experiments, an inter-injection distanceof 1 mm resulted in 25% to 70% of myofibers ex-pressing a reporter gene present in the donor my-oblasts, whereas an inter-injection distance of 2 mmproduced only 6% to 12% of myofibers expressingthe reporter gene (Fig. 3).
The inter-injection distance could be increased ifthe volume of muscle expressing the reporter genefollowing a single injection is increased. This po-tentially could be achieved by
1. increasing the capacity of the donor myoblaststo migrate and fuse with distant myofibers,
2. producing more tissue damage to providemore regenerating myofibers able to incorpo-rate the donor myoblasts and initiate moreextracellular matrix breakdown, and
3. increasing the nuclear domain of the thera-peutic donor-protein.
Increasing the Intramuscular Migration of Donor Myoblasts
Our experimental data support the idea that in-ducing the secretion of metalloproteinases (the en-zymes that migrating cells, such as leukocytes, useto degrade the extracellular matrix) could increasethe migration of myoblasts through the muscle tis-sue.36-38 Incubation of donor myoblasts with con-canavalin A (an inducer of metalloproteinase ex-pression) increases the dissemination of donormyoblasts in the host muscle.35
5 6 2 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
SYNCYTIA:
Plural of syncytium: a multinucleated massof protoplasm produced by the merging ofcells.

REVIEWS
Increasing the Fusion of Donor Myoblasts with Host Fibers
A larger number of regenerating myofibers in-creases the opportunities of donor myoblasts to beincorporated into most host myofibers. One of themost frequently used methods to produce this ef-fect was the injection of notexin, a potent myotox-ic phospholipase from the venom of the Australiantiger snake Notechis scutatus scutatus.18,19,39,40 In-hibiting the participation of host satellite cells inmuscle regeneration will also favor donor my-oblast incorporation after muscle damage, and themost used method to obtain this effect in experi-mental MT is ionizing radiation.18,19,39,40 Interven-tions that combined myonecrosis and host satel-lite cell elimination, such as cryodamage, werealso used to improve MT in rodents.27,41 Although
it is doubtful whether these methods can be appli-cable to improve MT in humans, this must ulti-mately be evaluated in a cost-benefit balance, thatis, how much a difficult intervention will benefitthe patient.
The Number of Donor Myoblasts Delivered per Injection Trajectory
It was shown that the number of hybrid my-ofibers increased (within a given range) along withthe number of donor myoblasts until reaching aplateau.42 Monkey experiments showed that withina given range, the success of transplantation in-creased with the number of myoblasts injected (fora similar inter-injection distance),12 whereas aplateau is reached at higher donor-cell concentra-tions.11 Defining the optimal number of myoblasts
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 6 3
Figure 3. The histologic success of MT in monkeys (as percentage of hybrid fibers expressing β-galactosidase) is compared with the histologic find-ings in DMD carriers (with a mosaic of dystrophin-positive and dystrophin-negative myofibers). Examples of monkey muscle cross sections ex-hibiting different percentages of β-galactosidase-positive myofibers are shown in the bottom (original magnification: 25X).
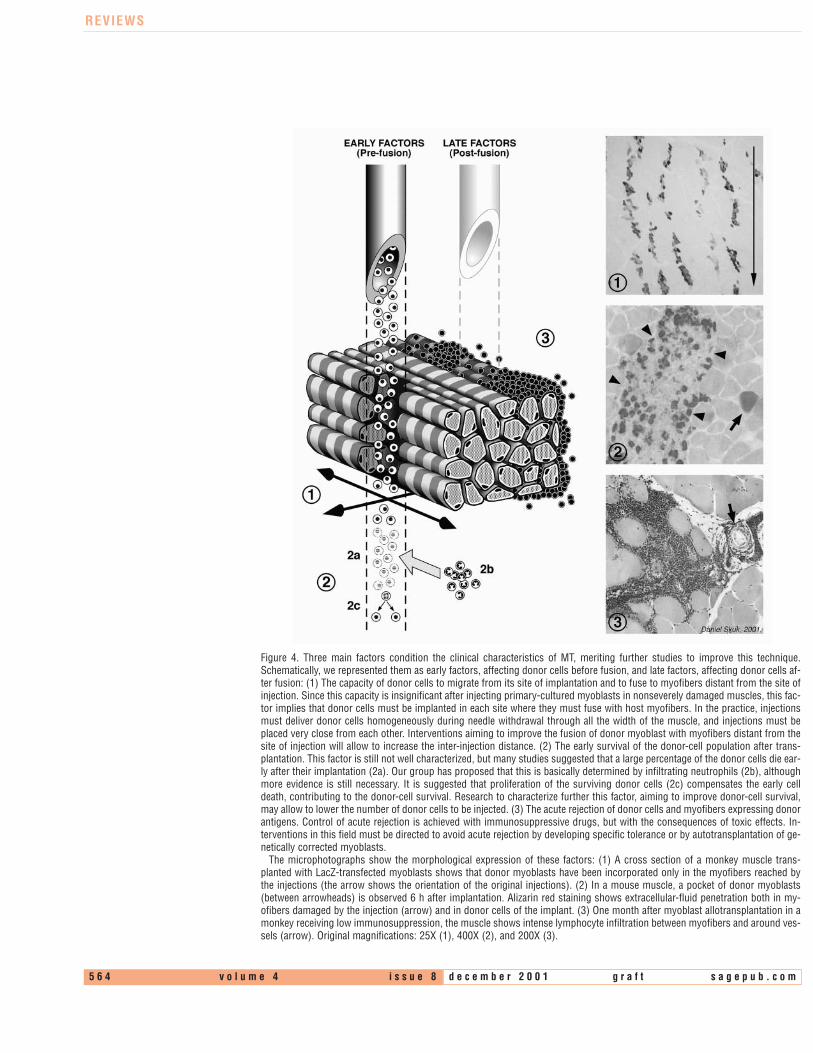
REVIEWS
5 6 4 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
Figure 4. Three main factors condition the clinical characteristics of MT, meriting further studies to improve this technique.Schematically, we represented them as early factors, affecting donor cells before fusion, and late factors, affecting donor cells af-ter fusion: (1) The capacity of donor cells to migrate from its site of implantation and to fuse to myofibers distant from the site ofinjection. Since this capacity is insignificant after injecting primary-cultured myoblasts in nonseverely damaged muscles, this fac-tor implies that donor cells must be implanted in each site where they must fuse with host myofibers. In the practice, injectionsmust deliver donor cells homogeneously during needle withdrawal through all the width of the muscle, and injections must beplaced very close from each other. Interventions aiming to improve the fusion of donor myoblast with myofibers distant from thesite of injection will allow to increase the inter-injection distance. (2) The early survival of the donor-cell population after trans-plantation. This factor is still not well characterized, but many studies suggested that a large percentage of the donor cells die ear-ly after their implantation (2a). Our group has proposed that this is basically determined by infiltrating neutrophils (2b), althoughmore evidence is still necessary. It is suggested that proliferation of the surviving donor cells (2c) compensates the early celldeath, contributing to the donor-cell survival. Research to characterize further this factor, aiming to improve donor-cell survival,may allow to lower the number of donor cells to be injected. (3) The acute rejection of donor cells and myofibers expressing donorantigens. Control of acute rejection is achieved with immunosuppressive drugs, but with the consequences of toxic effects. In-terventions in this field must be directed to avoid acute rejection by developing specific tolerance or by autotransplantation of ge-netically corrected myoblasts.
The microphotographs show the morphological expression of these factors: (1) A cross section of a monkey muscle trans-planted with LacZ-transfected myoblasts shows that donor myoblasts have been incorporated only in the myofibers reached bythe injections (the arrow shows the orientation of the original injections). (2) In a mouse muscle, a pocket of donor myoblasts(between arrowheads) is observed 6 h after implantation. Alizarin red staining shows extracellular-fluid penetration both in my-ofibers damaged by the injection (arrow) and in donor cells of the implant. (3) One month after myoblast allotransplantation in amonkey receiving low immunosuppression, the muscle shows intense lymphocyte infiltration between myofibers and around ves-sels (arrow). Original magnifications: 25X (1), 400X (2), and 200X (3).

REVIEWS
to be delivered per injection will allow for the bestMT results, without wasting large quantities ofdonor cells. Although further work to define thisparameter will be necessary, recent monkey experi-ments showed that 50% to 70% of hybrid my-ofibers were observed after injecting 10x106 β-galactosidase-labeled myoblasts in 1 cm3 of muscle,using an inter-injection distance of 1 mm.
Can the Number of Injected Myoblasts Be Reduced?
There is some evidence that the intramuscular in-jection of donor myoblasts can be followed by arapid donor-cell death.43-46 (Fig. 4). At present, wecannot say that the problem of early survival of thedonor myoblasts is well understood. Indeed, thesuccess of MT in monkeys11,12 shows that the earlysurvival of the donor myoblasts is not a limitingfactor for MT in primates. In spite of this, a betterdefinition of this problem may help to lower thenumber of myoblasts to be injected.
Can Myogenic Cells Be Systemically Delivered?The possibility of delivering myogenic cells
through the blood stream was tested because of theobvious advantage of this route: accessibility tomany muscles by a single injection, including im-portant muscles (such as the diaphragm), which areinappropriate for direct injections. Injection of pri-mary cultured myoblasts, intraperitoneally and in-travenously, was negative, even when extensivemuscle damage was produced to favor the incorpo-ration of the donor myoblasts.47 Some success wasobtained by intra-arterial administration of my-oblasts from a cell line, but only when the hostmuscle was mechanically injured.48 Extracorporealcirculation was also used to infuse donor myoblastsdirectly into muscles, but the success of this ap-proach was dependent also on inducing muscle in-jury.49 The possibility of delivering pluripotent stemcells with myogenic capacities by the blood stream(such as unfractionated bone marrow,50 haema-topoietic stem cells,51 and muscle-derived stemcells52) was tested, but it was consistently observedthat donor cells fuse with host myofibers after mus-cle damage. Therefore, if host muscles must be ex-tensively injured to incorporate donor myogenic-cells infused in the blood stream, systemic cell
delivery will not be more advantageous but, indeed,more complex than direct intramuscular cell injec-tions.
Control of RejectionEven if an efficient donor-cell delivery is achieved,
donor cells and hybrid myofibers must survive themechanisms of allograft rejection. In organ trans-plantation, 3 types of rejection menace the survivalof the graft: hyperacute, acute, and chronic. Hy-peracute rejection occurs within minutes to hoursafter transplantation and is dependent on comple-ment activation by preformed antibodies. Thisproblem is usually avoided by the routine pretrans-plant screening of antidonor alloantibodies. Inacute rejection, alloantigen-reactive cytolytic T-cellsdestroy the graft by a mechanism implicatingMHC recognition on the donor cells. Chronic re-jection, although not examined in long-term MTexperiments, is basically produced by different vas-cular endothelial injuries in the graft, and it is notclear whether it can be produced in the recipientvessels of muscles implanted with myoblasts.
Acute Rejection in MTAlthough MHC expression is not observed in
normal mature myofibers, they exhibit MHC ex-pression during inflammation, muscle regenera-tion, and in DMD.53,54 MHC is also expressed inmyotubes.55 Acute rejection following MT is welldocumented and was observed in mice,41,56-58 dogs,59
and monkeys9-12 (Fig. 4). Infiltration by CD8+ andCD4+ lymphocytes was observed in host musclesafter allogeneic MT in immunocompetent, nonim-munosuppressed mice.56 Expression of IL-2 recep-tors, Th-1 cytokine, and granzyme B confirmedthat these infiltrating cells are activated lympho-cytes.60,61 Rejection due to minor antigens was alsoobserved after syngeneic MT from males to fe-males.62 Another factor that can trigger immune re-actions is the expression of the therapeutic proteinthat is absent in the host (e.g., dystrophin in DMDpatients), although whether or not this factor trig-gers rejection was controversial.63,64
ImmunosuppressionThe degree of control of the humoral response af-
ter MT in mouse experiments varied among differ-
SATELLITE CELLS:
Myogenic stem cells present in the periph-ery of mature myofibers.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 6 5

REVIEWS
5 6 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
Figure 5. Transplantingwith myoblasts manyregions of the bodywould be especiallychallenging because ofthe anatomical com-plexity and the presenceof many noble struc-tures (such as in theneck). The forearm isrepresented because ofthe complex superposi-tion of muscles with pe-ripheral nerves and ves-sels. The figuresummarizes the possi-ble stages in a hypothet-ical modus operandi of adevice for whole-musclestereotaxic cell- delivery,aiming to reach theforearm from the anteri-or and posterior sides:(1) The limb is scanned.(2) The operator individ-ualizes the differentanatomic structures. (3)The operator selects thetargets for cell deliveryand the anatomic struc-tures that must beavoided. (4) The auto-mated device for cellinjection delivers thedonor cells only in se-lected regions notblocked by protectedstructures.

REVIEWS
ent immunosuppressive agents.65 Cyclophos-phamide showed negative results in mice, and itwas suggested that this drug kills the transplantedcells because of its antiproliferative properties.66 Cy-closporine A was administered in some MT clinicaltrials4-6 and was effective in mouse experiments.41,57
Nevertheless, cyclosporine A was less effective thansirolimus in controlling the humoral responseagainst allografted myoblasts.65 Sirolimus was veryeffective as an immunosuppressant for MT inmice,19 although the best success was obtained us-ing tacrolimus immunosuppression.17 Undertacrolimus immunosuppression, up to 95% of themyofibers in mdx mice expressed dystrophin afterMT.17 Tacrolimus was effective in controlling theacute rejection after MT in monkeys9-12 and was ob-served up to 1 year following transplantation in thismodel.12 Control of acute rejection after MT wasalso obtained using monoclonal antibodies directedagainst lymphocyte adhesion molecules.67
Potential Strategies to Avoid ImmunosuppressionAlthough the new immunosuppressive drugs have
significantly improved the results of clinical trans-plantation, these drugs have side effects. The long-term need for administration of immunosuppres-sants increases the risks of life-threateninginfections and cancer. An important field in MT re-search is the development of strategies to avoidlong-term immunosuppression, either develop-ment of tolerance or autotransplantation of cells ge-netically corrected in vitro.
Development of ToleranceAlthough immunologic tolerance to allogeneic
grafts can be induced with different strategies inmice, it was more difficult to achieve in nonhumanprimates.13 The 3 main strategies tested in the non-human primate model were mixed allogeneicchimerism, T-cell depletion, and costimulationblockage (for a review, see ref. 13). Development ofimmunologic tolerance in the context of MT iscurrently being studied in our laboratory. Weshowed that a short course of anti-CD154, in com-bination with donor-specific transfusion, pro-longed the survival of the hybrid myofibers, al-though a slow rejection was observed at longperiods.68
Autotransplantation of Genetically Corrected Myoblasts
Autologous MT requires genetic correction ofdonor cells in vitro. Both the human mini-dys-trophin and the full-length dystrophin genes weretransferred with adenoviral vectors in vitro to mdxmyoblasts, and these genetically corrected my-oblasts produced dystrophin-positive myofibers af-ter transplantation in mdx mice.69,70 The mini-dystrophin gene was also introduced in humanDMD myoblasts, and these genetically correctedDMD-myoblasts were transplanted in SCID mice,producing myofibers expressing the human mini-dystrophin.71 However, as explained before, the in-troduction of dystrophin could be potentially im-munogenic in DMD patients.63,64
In addition to the problems of efficiently intro-ducing the correct gene in the donor cells, anotherdifficulty of this approach is the limited capacity ofautologous DMD myoblasts to proliferate in cul-ture. As a consequence of recurrent cycles of my-ofiber degeneration-regeneration, senescence ofDMD myoblasts in culture occurs early.72-74 Somestrategies are being tried to increase the proliferativecapacity of DMD myoblasts. One of them is theexpression of the SV40 large T antigen, which de-layed senescence in myoblasts but failed to inducecell immortality.72 The other one was the introduc-tion of the telomerase gene, although telomeraseexpression alone did not significantly extend lifespan of human myoblasts.73 It was the coexpressionof telomerase and T antigen that allowed DMDmyoblasts to divide more than 55 doublings, pre-serving the ability to fuse and differentiate.74
Is MT Attainable in Humans?A clinical MT strategy based in our monkey ob-
servations implies some complexities.
1. Each injection must be as efficient as possi-ble; thus, it must be made across the wholemuscle, delivering the donor myoblasts ho-mogeneously throughout its trajectory.
2. Injection trajectories must be placed veryclose to each other.
3. Delivery of donor myoblasts in subcutaneoustissue and nonmuscle organs must be avoided.
4. Some anatomic structures (e.g., peripheralnerves and large vessels) must be spared fromrepetitive needle damage.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 6 7

REVIEWS
Taking into account the complexity of efficientlyinjecting myoblasts through 40% to 50% of thebody with the premises exposed, MT becomes achallenging task. If we desire to move to clinical ap-plications, we must propose a method extrapolatingfrom the experience obtained in a single monkeymuscle to most of the skeletal muscle system of ahuman. In fact, if the previous premises will be theonly ones allowing a significant genetic correctionin the muscles of dystrophic patients, we believethat the future of MT will be determined by the useof automated systems for whole-muscle stereotaxiccell-delivery. This is because making 100 injectionsper cm2 of muscle surface may be more rapidly andefficiently performed by an automated device capa-ble of injecting simultaneously with several needles.Furthermore, to reach as much muscle tissue aspossible, avoiding donor-cell delivery in nontarget-ed tissues and avoiding repetitive lesions of otherorgans, requires accurate topographical precisionfor cell delivery (even if the inter-injection distancecan be increased). The principle of stereotaxic celldelivery to muscles (as we envision this possibility)is illustrated in Figure 5. The principal factor of riskassociated with repetitive and close muscular injec-tions is the extensive muscle damage associatedwith the release of intracellular metabolites (mainlymyoglobin and potassium), although this factorcan be managed by controlling the volume of mus-cle injected per MT session.12 As in other fields ofmedicine, MT technology (and possibly othertypes of cell transplantation) may need the interdis-ciplinary cooperation of different specialties, notonly immunology and molecular biology but alsomedical imaging, data processing, and robotics.Whether this complex technology will be devel-oped may depend on the results of better-plannedMT clinical trials and also on a cost-benefit balancecompared with the potential success of other exper-imental approaches to the treatment of musculardystrophies.
REFERENCES
1. Gussoni E, Pavlath GK, Lanctot AM, Sharma KR, Miller RG, Steinman L, etal. Normal dystrophin transcripts detected in Duchenne muscular dystrophypatients after myoblast transplantation. Nature 1992;356:435-8.
2. Huard J, Bouchard JP, Roy R, Malouin F, Dansereau G, Labrecque C, et al.Human myoblast transplantation: preliminary results of 4 cases. MuscleNerve 1992;15:550-60.
3. Karpati G, Ajdukovic D, Arnold D, Gledhill RB, Guttman R, Holland P, et al.Myoblast transfer in Duchenne muscular dystrophy. Ann Neurol 1993;34:8-17.
4. Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, et al. My-oblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl JMed 1995;333:832-8.
5. Miller RG, Sharma KR, Pavlath GK, Gussoni E, Mynhier M, Lanctot AM, etal. Myoblast implantation in Duchenne muscular dystrophy: the San Fran-cisco study. Muscle Nerve 1997;20:469-78.
6. Morandi L, Bernasconi P, Gebbia M, Mora M, Crosti F, Mantegazza R, et al.Lack of mRNA and dystrophin expression in DMD patients three months af-ter myoblast transfer. Neuromuscul Disord 1995;5:291-5.
7. Tremblay JP, Bouchard JP, Malouin F, Theau D, Cottrell F, Collin H, et al.Myoblast transplantation between monozygotic twin girl carriers ofDuchenne muscular dystrophy. Neuromuscul Disord 1993;3:583-92.
8. Neumeyer AM, Cros D, McKenna-Yasek D, Zawadzka A, Hoffman EP, Pe-goraro E, et al. Pilot study of myoblast transfer in the treatment of Beckermuscular dystrophy. Neurology 1998;51:589-92.
9. Kinoshita I, Vilquin JT, Gravel C, Roy R, Tremblay JP. Myoblast allotrans-plantation in primates [letter]. Muscle Nerve 1995;18:1217-18.
10. Kinoshita I, Roy R, Dugre FJ, Gravel C, Roy B, Goulet M, et al. Myoblasttransplantation in monkeys: control of immune response by FK506. J Neu-ropathol Exp Neurol 1996;55:687-97.
11. Skuk D, Roy B, Goulet M, Tremblay JP. Successful myoblast transplantationin primates depends on appropriate cell delivery and induction of regener-ation in the host muscle. Exp Neurol 1999;155:22-30.
12. Skuk D, Goulet M, Roy B, Tremblay JP. Myoblast transplantation in wholemuscle of nonhuman primates. J Neuropathol Exp Neurol 2000;59:197-206.
13. Knechtle SJ. Knowledge about transplantation tolerance gained in primates.Curr Opin Immunol 2000;12:552-6.
14. Borisov AB. Regeneration of skeletal and cardiac muscle in mammals: dononprimate models resemble human pathology? Wound Repair Regen1999;7:26-35.
15. Bischoff R. The satellite cell and muscle regeneration. In: Engel AG, Fran-szini-Armstrong C, editors. Myology. New York: McGraw-Hill; 1994. p. 97-118.
16. Brussee V, Merly F, Tardif F, Tremblay JP. Normal myoblast implantation inMDX mice prevents muscle damage by exercise. Biochem Biophys ResCommun 1998;250:321-7.
17. Kinoshita I, Vilquin JT, Guerette B, Asselin I, Roy R, Tremblay JP. Very effi-cient myoblast allotransplantation in mice under FK506 immunosuppres-sion. Muscle Nerve 1994;17:1407-15.
18. Kinoshita I, Vilquin JT, Guerette B, Asselin I, Roy R, Lille S, et al. Immuno-suppression with FK 506 insures good success of myoblast transplantationin mdx mice. Transplant Proc 1994;26:3518.
19. Vilquin JT, Asselin I, Guerette B, Kinoshita I, Roy R, Tremblay JP. Success-ful myoblast allotransplantation in mdx mice using rapamycin. Transplan-tation 1995;59:422-6.
20. Morgan JE, Pagel CN, Sherratt T, Partridge TA. Long-term persistence andmigration of myogenic cells injected into pre- irradiated muscles of mdxmice. J Neurol Sci 1993;115:191-200.
21. Morgan JE, Hoffman EP, Partridge TA. Normal myogenic cells from new-born mice restore normal histology to degenerating muscles of the mdxmouse. J Cell Biol 1990;111:2437-49.
22. Vilquin JT, Kinoshita I, Roy B, Goulet M, Engvall E, Tome F, et al. Partiallaminin alpha2 chain restoration in alpha2 chain-deficient dy/dy mouse byprimary muscle cell culture transplantation. J Cell Biol 1996;133:185-97.
23. Leriche-Guérin K, Anderson LVB, Wrogemann K, Roy B, Goulet M, Trem-blay JP. Dysferlin expression after normal myoblast transplantation in SCIDand SJL mice. Neuromuscul Disord. In press.
24. Arcila ME, Ameredes BT, DeRosimo JF, Washabaugh CH, Yang J, JohnsonPC, et al. Mass and functional capacity of regenerating muscle is enhancedby myoblast transfer. J Neurobiol 1997;33:185-98.
5 6 8 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

REVIEWS
25. DeRosimo JF, Washabaugh CH, Ontell MP, Daood MJ, Watchko JF, WatkinsSC, et al. Enhancement of adult muscle regeneration by primary myoblasttransplantation. Cell Transplant 2000;9:369-77.
26. Alameddine HS, Louboutin JP, Dehaupas M, Sebille A, Fardeau M. Func-tional recovery induced by satellite cell grafts in irreversibly injured mus-cles. Cell Transplant 1994;3:3-14.
27. Irintchev A, Langer M, Zweyer M, Theisen R, Wernig A. Functional im-provement of damaged adult mouse muscle by implantation of primary my-oblasts. J Physiol (Lond) 1997;500:775-85.
28. Gussoni E, Blau HM, Kunkel LM. The fate of individual myoblasts aftertransplantation into muscles of DMD patients. Nat Med 1997;3:970-7.
29. Gross JG, Morgan JE. Muscle precursor cells injected into irradiated mdxmouse muscle persist after serial injury. Muscle Nerve 1999;22:174-85.
30. Yao SN, Kurachi K. Implanted myoblasts not only fuse with myofibers butalso survive as muscle precursor cells. J Cell Sci 1993;105:957-63.
31. Heslop L, Beauchamp JR, Tajbakhsh S, Buckingham ME, Partridge TA, Za-mmit PS. Transplanted primary neonatal myoblasts can give rise to func-tional satellite cells as identified using the Myf5(nlacZl+) mouse. Gene Ther2001;8:778-83.
32. Bonilla E, Schmidt B, Samitt CE, Miranda AF, Hays AP, de Oliveira AB, et al.Normal and dystrophin-deficient muscle fibers in carriers of the gene forDuchenne muscular dystrophy. Am J Pathol 1988;133:440-5.
33. Di Blasi C, Morandi L, Barresi R, Blasevich F, Cornelio F, Mora M. Dys-trophin-associated protein abnormalities in dystrophin-deficient musclefibers from symptomatic and asymptomatic Duchenne/Becker musculardystrophy carriers. Acta Neuropathol (Berl) 1996;92:369-77.
34. Kinoshita I, Vilquin JT, Asselin I, Chamberlain J, Tremblay JP. Transplanta-tion of myoblasts from a transgenic mouse overexpressing dystrophin pro-duced only a relatively small increase of dystrophin-positive membrane.Muscle Nerve 1998;21:91-103.
35. Ito H, Hallauer PL, Hastings KE, Tremblay JP. Prior culture with con-canavalin A increases intramuscular migration of transplanted myoblast.Muscle Nerve 1998;21:291-7.
36. El Fahime E, Torrente Y, Caron NJ, Bresolin MD, Tremblay JP. In vivo mi-gration of transplanted myoblasts requires matrix metalloproteinase activi-ty. Exp Cell Res 2000;258:279-87.
37. Torrente Y, El Fahime E, Caron NJ, Bresolin N, Tremblay JP. Intramuscularmigration of myoblasts transplanted after muscle pretreatment with metal-loproteinases. Cell Transplant 2000;9:539-49.
38. Caron NJ, Asselin I, Morel G, Tremblay JP. Increased myogenic potentialand fusion of matrilysin-expressing myoblasts transplanted in mice. CellTransplant 1999;8:465-76.
39. Huard J, Verreault S, Roy R, Tremblay M, Tremblay JP. High efficiency ofmuscle regeneration after human myoblast clone transplantation in SCIDmice. J Clin Invest 1994;93:586-99.
40. Skuk D, Furling D, Bouchard JP, Goulet M, Roy B, Lacroix Y, et al. Trans-plantation of human myoblasts in SCID mice as a potential muscular mod-el for myotonic dystrophy. J Neuropathol Exp Neurol 1999;58:921-31.
41. Wernig A, Irintchev A, Lange G. Functional effects of myoblast implantationinto histoincompatible mice with or without immunosuppression. J Physi-ol (Lond) 1995;484:493-504.
42. Rando TA, Blau HM. Primary mouse myoblast purification, characterization,and transplantation for cell-mediated gene therapy. J Cell Biol1994;125:1275-87.
43. Beauchamp JR, Morgan JE, Pagel CN, Partridge TA. Dynamics of myoblasttransplantation reveal a discrete minority of precursors with stem cell-likeproperties as the myogenic source. J Cell Biol 1999;144:1113-22.
44. Guerette B, Skuk D, Celestin F, Huard C, Tardif F, Asselin I, et al. Preventionby anti-LFA-1 of acute myoblast death following transplantation. J Immunol1997;159:2522-31.
45. Huard J, Acsadi G, Jani A, Massie B, Karpati G. Gene transfer into skeletalmuscles by isogenic myoblasts. Hum Gene Ther 1994;5:949-58.
46. Qu Z, Balkir L, van Deutekom JC, Robbins PD, Pruchnic R, Huard J. De-velopment of approaches to improve cell survival in myoblast transfer ther-apy. J Cell Biol 1998;142:1257-67.
47. Partridge TA. Invited review: myoblast transfer: a possible therapy for in-herited myopathies? Muscle Nerve 1991;14:197-212.
48. Neumeyer AM, DiGregorio DM, Brown RH Jr. Arterial delivery of myoblaststo skeletal muscle. Neurology 1992;42:2258-62.
49. Torrente Y, D’Angelo MG, Del Bo R, DeLiso A, Casati R, Benti R, et al. Ex-tracorporeal circulation as a new experimental pathway for myoblast im-plantation in mdx mice. Cell Transplant 1999;8:247-58.
50. Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cos-su G, et al. Muscle regeneration by bone marrow-derived myogenic pro-genitors [see comments] [published erratum appears in Science 1998 Aug14;281(5379):923]. Science 1998;279:1528-30.
51. Gussoni E, Soneoka Y, Strickland CD, Buzney EA, Khan MK, Flint AF, et al.Dystrophin expression in the mdx mouse restored by stem cell transplan-tation. Nature 1999;401:390-4.
52. Torrente Y, Tremblay J-P, Pisati F, Belicchi M, Rossi B, Sironi M, et al. In-traarterial injection of muscle-derived CD34+Sca-1+ stem cells restores dy-strophin in mdx mice. J Cell Biol 2001;152:335-48.
53. Appleyard ST, Dunn MJ, Dubowitz V, Rose ML. Increased expression ofHLA ABC class I antigens by muscle fibres in Duchenne muscular dystro-phy, inflammatory myopathy, and other neuromuscular disorders. Lancet1985;1:361-3.
54. Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major his-tocompatibility complex products in human skeletal muscles. Ann Neurol1988;23:64-72.
55. Michaelis D, Goebels N, Hohlfeld R. Constitutive and cytokine-induced ex-pression of human leukocyte antigens and cell adhesion molecules by hu-man myotubes. Am J Pathol 1993;143:1142-9.
56. Guerette B, Asselin I, Vilquin JT, Roy R, Tremblay JP. Lymphocyte infiltra-tion following allo- and xenomyoblast transplantation in mdx mice. MuscleNerve 1995;18:39-51.
57. Irintchev A, Zweyer M, Wernig A. Cellular and molecular reactions in mousemuscles after myoblast implantation. J Neurocytol 1995;24:319-31.
58. Pavlath GK, Rando TA, Blau HM. Transient immunosuppressive treatmentleads to long-term retention of allogeneic myoblasts in hybrid myofibers. JCell Biol 1994;127:1923-32.
59. Ito H, Vilquin JT, Skuk D, Roy B, Goulet M, Lille S, et al. Myoblast trans-plantation in non-dystrophic dog. Neuromuscul Disord 1998;8:95-110.
60. Guerette B, Roy R, Tremblay M, Asselin I, Kinoshita I, Puymirat J, et al. In-creased granzyme B mRNA after alloincompatible myoblast transplantation.Transplantation 1995;60:1011-16.
61. Guerette B, Tremblay G, Vilquin JT, Asselin I, Gingras M, Roy R, et al. In-creased interferon-gamma mRNA expression following alloincompatiblemyoblast transplantation is inhibited by FK506. Muscle Nerve1996;19:829-35.
62. Boulanger A, Asselin I, Roy R, Tremblay JP. Role of non-major histocom-patibility complex antigens in the rejection of transplanted myoblasts.Transplantation 1997;63:893-9.
63. Vilquin JT, Wagner E, Kinoshita I, Roy R, Tremblay JP. Successful histo-compatible myoblast transplantation in dystrophin-deficient mdx mousedespite the production of antibodies against dystrophin. J Cell Biol1995;131:975-88.
64. Ohtsuka Y, Udaka K, Yamashiro Y, Yagita H, Okumura K. Dystrophin acts asa transplantation rejection antigen in dystrophin- deficient mice: implica-tion for gene therapy. J Immunol 1998;160:4635-40.
65. Vilquin JT, Asselin I, Guerette B, Kinoshita I, Lille S, Roy R, et al. Myoblastallotransplantation in mice: degree of success varies depending on the ef-ficacy of various immunosuppressive treatments. Transplant Proc1994;26:3372-3.
66. Vilquin JT, Kinoshita I, Roy R, Tremblay JP. Cyclophosphamide immuno-suppression does not permit successful myoblast allotransplantation inmouse. Neuromuscul Disord 1995;5:511-17.
67. Guerette B, Wood K, Roy R, Tremblay JP. Efficient myoblast transplantationin mice immunosuppressed with monoclonal antibodies and CTLA4 Ig.Transplant Proc 1997;29:1932-4.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 6 9

REVIEWS
68. Caminard G, Caron NJ, Turgeon NA, Rossini AA, Tremblay JP. Treatmentwith anti-CD154 antibody and donor-specific transfusion prevents acute re-jection of myoblast transplantation. In press.
69. Floyd SS Jr, Clemens PR, Ontell MR, Kochanek S, Day CS, Yang J, et al. Exvivo gene transfer using adenovirus-mediated full-length dystrophin deliv-ery to dystrophic muscles. Gene Ther 1998;5:19-30.
70. Moisset PA, Gagnon Y, Karpati G, Tremblay JP. Expression of human dys-trophin following the transplantation of genetically modified mdx my-oblasts. Gene Ther 1998;5:1340-6.
71. Moisset PA, Skuk D, Asselin I, Goulet M, Roy B, Karpati G, et al. Success-ful transplantation of genetically corrected DMD myoblasts following exvivo transduction with the dystrophin minigene. Biochem Biophys ResCommun 1998;247:94-9.
72. Simon LV, Beauchamp JR, O’Hare M, Olsen I. Establishment of long-termmyogenic cultures from patients with Duchenne muscular dystrophy byretroviral transduction of a temperature-sensitive SV40 large T antigen. ExpCell Res 1996;224:264-71.
73. Seigneurin-Venin S, Bernard V, Moisset PA, Ouellette MM, Mouly V, DiDonna S, et al. Transplantation of normal and DMD myoblasts expressingthe telomerase gene in SCID mice. Biochem Biophys Res Commun2000;272:362-9.
74. Seigneurin-Venin S, Bernard V, Tremblay JP. Telomerase allows the immor-talization of T antigen-positive DMD myoblasts: a new source of cells forgene transfer application. Gene Ther 2000;7:619-23.
5 7 0 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

MEETING REPORT
Transplantation tolerance remains the goalof clinical transplantation and the focus of alarge amount of research. Despite anecdotalreports,1 a rational scheme for reliably induc-ing and maintaining clinical transplant toler-ance has not been defined. Nonetheless, cli-nicians and researchers realize the need fordesigning and testing assays capable of defin-ing the immunologic fingerprint of a toler-ant state. At the recent Transplant 2001meeting, the 2nd joint meeting of the Amer-ican Society of Transplantation (AST) andAmerican Society of Transplant Surgeons(ASTS), we were charged with moderating aworkshop entitled, The Tolerance Assay:Where Are We Now? A very lively discussionresulted in the consensus, which we reporthere.
Prior to the workshop, we both agreed thatone of the great challenges in developing areliable tolerance assay is the lack of a uni-versal definition of “tolerance.” The abilityto measure tolerance using a laboratory testclearly requires a “gold standard” definitionof tolerance itself. The workshop startedwith a fairly textbook definition of trans-plantation tolerance:
The normal function of a transplanted or-gan, without exogenous immunosuppres-sive therapy, in the absence of a pathologicdonor-specific immune response, but ac-companied by an otherwise fully compe-tent immune system.
The 60 to 100 people attending the work-shop initially agreed that this was a reasonabledefinition. However, upon probing a bit fur-ther, there were many different opinions on thedetails of that definition. In an effort to facil-itate the discussion, we used a series of ques-tions originally formulated by Dr. CharlesOrosz regarding how to best model humantolerance in rodents. It was thought that de-veloping criteria for defining tolerance in ro-dents, where controlled testing can be per-formed, would be easier than initiallyattempting to define tolerance in humans.The questions are printed below, and the re-sulting consensus was quite surprising to usall.
To model human allograft tolerance, allo-graft tolerant mice (or rats):
(a) must, (b) should, (c) may, (d) may not,or (e) should never:
1. Display micro-chimerism,2. Display altered T cell responses to
donor antigen,3. Accept a similar allograft without fur-
ther therapy,4. Accept a donor-matched skin graft,5. Display chronic-rejection like histo-
logic changes,6. Display persistent inflammation with-
in the graft,7. Require intermittent immunosup-
pression,
Tolerance: Defining, Achieving, and Measuring ItReport from The Tolerance Assay: Where Are We Now?Workshop, Transplant 2001
Anne M. VanBuskirk and Peter S. Heeger
8. Lose an allograft 200 to 300 days aftertransplantation,
9. Require preparative irradiation,10. Display clonal deletion of graft-reactive
T cells,11. Produce donor-reactive IgG,12. Require preparative injection of donor
bone marrow.
Results1. A few individuals thought micro-
chimerism must or should be present.In contrast, the majority thought thatmicro-chimerism may or may not bedisplayed.
2. All participants thought that donor-reactive T cell responses should be al-tered, either through deletion of thereactive cells or through regulation(suppression, cytokine immune devia-tion, etc).
3. All participants thought that a 2nd al-lograft of the same type must/should beaccepted without further treatment.
4. Most participants thought that donor-matched skin graft should be accepted.This is the ideal situation, in which thestrongest stimulus is accepted.
5. All participants thought that chronicrejection-like histologic changes shouldnever be present.
6. Mononuclear cell infiltration withinthe graft is acceptable, but tissue dam-age resulting from that infiltration isnever acceptable. One point made wasthat the presence of an infiltrate maybe related to the underlying disease,rather than due to alloantigenicity.
7. Participants thought that intermittentimmunosuppression may be used, al-though the absence of intermittentimmunosuppression is preferable.
8. Graft loss after 200 to 300 days may beacceptable, especially given that theaverage life span of a mouse is notmuch more than 365 days.
9. Participants did not think that prepar-ative irradiation must be required, butits use is acceptable as long as the re-
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 7 1

MEETING REPORT
cipients are immuno-competent aftertransplantation.
10. Clonal deletion of graft-reactive T cellsis considered necessary by some partic-ipants, but not by the majority. Mostparticipants thought that regulation orblockade of the donor-reactive T cellsis sufficient.
11. In general, donor-reactive IgG shouldnot be present, according to most par-ticipants. However, a significant pro-portion raised the issue of protectiveantibodies.
12. Preparative injection of donor bone mar-row is not required, but is acceptable.
Overall, the consensus opinion was muchmore vague than the original proposed defi-nition. An animal model for graft tolerancethat best models tolerance in humans will ex-hibit long-term, but not necessarily indefi-nite, graft survival and will exhibit no evidenceof chronic histopathologic changes attributa-ble to chronic rejection. The animals shouldaccept a 2nd allograft of the original donorstrain and, ideally, accept a skin graft of theoriginal donor strain. In terms of immunefunction, donor-reactive T cells and alloanti-bodies should either be absent or should ex-hibit a protective or regulatory phenotype.
The participants were then asked an addi-tional question: To be considered a viablemodel of the human allograft tolerance, doesthe rodent model need to be operative in oneinbred strain, in several inbred strains, or inoutbred mice? The consensus was that ideal-ly the model would be operative in outbredmice but that showing the tolerance using atleast 2 disparate recipient strains was suffi-cient (for example, BALB/c and C57BL/6).
The group next considered whether any ofthe existent rodent models actually meet allof these criteria. The 3 models most dis-cussed were those introduced and initiallystudied by Megan Sykes (irradiation andpreparative bone marrow transplant, re-viewed in ref. 2) and by Kathryn Wood(DST and anti-CD4 antibody treatment3,4),along with Anita Chong’s modification of the
protocol originally described by theLarsen/Pearson group at Emory (anti-CD154 antibody and donor bone fragmentat the time of transplantation5). In the lattersystem, femur fragments are placed under thekidney capsule on day 0 and antibody is ad-ministered on days 0-3, 5, 7, 9, 11, and 13relative to heart transplantation. A challengewith a 2nd heart is performed on days 60-90.The tolerant animals accept donor, but not3rd party, grafts. The tolerance is associatedwith a peripheral loss of donor-reactive IFNγproduction by T cells, but central T cell dele-tion does not occur. Allograft tolerance hasbeen induced by this approach in inbredmice (C57BL/6) and in mice on a mixedbackground (129 × B/6 × DBA/2). TheChong adaptation at this time appears tomeet the criteria of only peri-operative im-munosuppression: an absence of alloanti-body, no chronic rejection, acceptance of a2nd graft, as well as donor-matched skingraft, and rejection of 3rd party allografts asa measure of immuno-competence. The oth-er tolerance-inducing protocols discussedhave not been fully evaluated in multiplestrains, although a variation of the Sykesmodel has been used successfully in larger an-imals and nonhuman primates. However,concerns were raised by some participantsabout immuno-competence of the treatedanimals following the Sykes model, and/orthe presence of chronic histo-pathologicchanges in the graft in some situations fol-lowing the Wood model. Based on this dis-cussion, there is still a significant amount ofbasic science experimentation required be-fore we have an ideal model of tolerance inrodents. It is only when such a model is es-tablished that we can perform a variety of rig-orously controlled laboratory tests to deter-mine which one(s) best predict or correlatewith the tolerant state.
Despite the above deficiencies, the groupnext discussed how one would go aboutmeasuring tolerance in humans. It is obviousthat certain experimental protocols, includ-ing placement of 2nd grafts or skin grafts,could not be performed in clinical trials and
that other surrogate markers need to be es-tablished. It was further noted that long-termgraft survival with normal graft function inthe presence of minimal immunosuppressionmight suggest a clinically tolerant state butthat more stringent immunologic criteriamight help define truly tolerant individuals.Eventual, total discontinuation of immuno-suppression with maintenance of normal or-gan function is the ultimate goal. The pro-posed tolerance assays would be used toevaluate the alloimmune repertoire in an ef-fort to confirm tolerance and to define thepresence of a potentially pathologic immuneresponse prior to graft dysfunction.
A variety of assays capable of measuring im-mune reactivity were discussed with thecaveat that there are no definitive data andonly preliminary information about any ofthe proposed assays. Proliferative hypo-re-sponsiveness to donor cells in an Mixed Lym-phocyte Reaction (MLR), the lack of killingin Cytolytic T Lymphocyte (CTL) assays, theabsence of donor-reactive recall Enzyme-Linked Immuno-SPOTs (ELISPOTs) fortype 1 cytokines (i.e., IFNγ), and the absenceof mRNAs for perforin/granzyme in graftbiopsies, and the absence of alloantibodieswere considered consistent with, but not di-agnostic of, a tolerant state. In some situa-tions, the presence of micro-chimerism wasalso considered by the group to be consistentwith tolerance. An additional intriguing as-say discussed by the group was the trans vivoDelayed-Type Hypersensitivity (DTH) assay.In this assay, control antigens and solubilizeddonor antigen are mixed with recipient cellsand tested for their ability to mediate DTHin mice by simply measuring skin thickness.Recent studies have shown that PeripheralBlood Leukocytes (PBLs) from some tolerantpatients can specifically suppress otherwisepotent immune responses as measured bythis assay, providing a readout that correlateswell with tolerance.6 Whether or not this ap-proach or any of the proposed assays will beuseful for defining or identifying tolerance inthe clinic remains to be determined inprospective trials.
5 7 2 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

MEETING REPORT
The group consensus was that many of theproposed assays need to be evaluated in thecontext of animal models in which stringentcriteria can be used to define tolerant versusnontolerant states. Until this is done, it willbe difficult to interpret any results of studiesperformed in humans. Nonetheless, attemptsat human tolerance induction are being per-formed in the clinical setting, and immunemonitoring is therefore desirable. It seemsthat under these circumstances, the best ap-proach to evaluate the usefulness of any ofthe proposed assays is to simultaneouslystudy the same tolerant and nontolerant in-dividuals (defined based on clinical criteria)using as many of the available assays as possi-ble. In this way, it might be possible to corre-late responsiveness, lack of responsiveness, orthe presence of regulatory immunity with aparticular clinical outcome. This is indeedthe approach being championed by theNIH-funded Immune Tolerance Network, amulticenter program focusing on inductionand measurement of tolerance in human dis-ease. It is hoped that this effort, among manyothers, will provide us with better insightinto the best ways to define and measure tol-erance in the laboratory—and at the bedside.
REFERENCES
1. Spitzer TR, Delmonico F, Tolkoff-Rubin N, McAfee S, Sackstein R,Saidman S, et al. Combined histocompatibility leukocyte antigen-matched donor bone marrow and renal transplantation for multi-ple myeloma with end stage renal disease: the induction of allo-graft tolerance through mixed lymphohematopoietic chimerism.Transplantation 1999;68:480.
2. Wekerle T, Sykes M. Mixed chimerism and transplantation toler-ance. Annu Rev Med 2001;52:353.
3. Bushell A, Niimi M, Morris PJ, Wood KJ. Evidence for immuneregulation in the induction of transplantation tolerance: a condi-tional but limited role for IL-4. J Immunol 1999;162:1359.
4. Saitovitch D, Bushell A, Mabbs DW, Morris PJ, Wood KJ. Kinet-ics of induction of transplantation tolerance with a nondepletinganti-Cd4 monoclonal antibody and donor-specific transfusionbefore transplantation. A critical period of time is required for de-velopment of immunological unresponsiveness. Transplantation1996;61(11):1642.
5. Bingaman AW, Waitze SY, Alexander DZ, Cho HR, Lin A, Tucker-Burden C, et al. Transplantation of the bone marrow microenvi-ronment leads to hematopoietic chimerism without cytoreductiveconditioning. Transplantation 2000;69:2491.
6. VanBuskirk A, Burlingham W, Jankowska-Gan E, Chin T, KusakaS, Geissler F, et al. Human allograft acceptance is associated withimmune regulation. J Clin Invest 2000;106:145.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 7 3

The Unkindest Cut: Where Are All the Transplant Programs Going?Roger W. Evans
5 7 4 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m
MISMATCHES
The “cost” of transplantation has been, andremains to some extent, a matter of consid-erable controversy. Although debate con-cerning insurance coverage of specific organtransplant procedures has all but vanished,there is still the odd skirmish over the level ofreimbursement that providers receive, andthe financial obligations patients are forcedto endure. Although the picture may not beas attractive as Marilyn Monroe, it is morehandsome than Cary Grant.
But times remain both troubled and uncer-tain. Increasingly, amid a sea of red ink,transplant programs are running afoul of in-stitutional efforts to either maintain orachieve financial solvency. Once consideredto be the cornerstone of high technology, thefoundation for research and innovation, aharbinger of success, and a marketer’s posterchild, transplant programs are hitting thechopping block with about the same enthu-siasm as a Perdue chicken headed to Popeye’s.
In anticipation of this inevitable state of af-fairs, I have, on many occasions, climbedatop a soapbox and pontificated at lengthabout the economics of transplantation.Sobering thoughts have often been sand-wiched between humorous sentences. How-ever, most people preferred to chuckle thanto knuckle, and a few got downright mad. Asa result, despite my efforts to impart knowl-edge, I entertained, and absorbed the oddcomment disparaging my ancestry. In theend, I must confess, people prefer to ignorewhat they don’t want to hear.
The level of institutional self-promotionassociated with transplantation has oftenamused me. Unfortunately, like “New Coke”in 1985, great marketing concepts some-times lack business sense. In health care, thisis usually apparent when hospital boards callin management consultants and other dubi-ous characters to “turn around” failing med-
ical centers. Popeye’s “little helper” typicallyexercises a “one-size-fits-all” mentality—chop, chop, chop—and, as their critics pointout, when it comes to economics, these folksare savages.
In my opinion, terminology is the root ofthe problem. Words typically get in the way
of understanding. This is certainly true inany attempt to appreciate the nuances associ-ated with the economics of transplantation.Although most people now realize there is adifference between a cost and a charge, whenmore detailed discussion ensues, my level ofamusement is soon on par with a Sanford andSon or Monty Python rerun.
Over the past decade, hospitals and healthcare systems have periodically grappled withthe problem of controlling “costs.” In an ef-fort to do so, most hospitals and health sys-tems have managed to cobble together ac-counting systems with the versatility of afour-function calculator. This is not surpris-ing. In more lucrative times, it wasn’t evennecessary to understand “production” or ac-tual costs since reimbursement typicallyequaled or, at the very least, approximatedbilled charges. Frankly, there was more gravyin the system than the entire chain of Crack-er Barrel restaurants has served over thecourse of its corporate life. In this era, costcontainment meant increasing charges to en-hance margins, without the faintest under-standing of true costs.
“Expense management” is a concept thathas now achieved buzzword status. It hascaptured the imagination of the proverbialbean counters. A new fetish has evenemerged—a clinically morbid fascinationwith the bottom line. To relieve anxiety,there has been a proliferation of “decisionsupport systems.” These mindless con-trivances enable accountants and “financialanalysts,” with varying degrees of expertise,to allocate costs, set charges, project reim-bursement, and speculate about margins. Inshort, for clinical “product lines,” decisionsupport systems abstractly identify the “win-ners” and the “losers”—those services thatmake and lose money. Transplantation is of-ten considered suspect because it has a ten-dency to generate a lot of revenue, but little,if any, margin.
In reality, decision support systems consistof nothing more than computer software—ifyou will, metaphorically like a cowboy with a bighat and no cattle. The software manipulates
Over the past decade, hospitals
and health care systems have
periodically grappled with
the problem of controlling
“costs.” In an effort to do so,
most hospitals and health
systems have managed to
cobble together accounting
systems with the versatility of
a four-function calculator.

extant data according to various accountingprinciples—principles that are often modi-fied to meet the needs of a particular external“client,” or internal “customer.” This featureis necessary since few hospitals and health-care systems follow precisely the same rules inderiving their costs, allocating their expenses,setting their fees, and determining theircharges. Ultimately, a diversity of accountingprinciples has its benefits when accountantsfail to adequately explain results.
Unlike science, accounting seems to placelittle value on replication. Therefore, differ-ent accountants and financial analysts essen-tially use the same system to provide widelydiscrepant results and divergent conclusions,all of which depend on the desired answer.(This isn’t hypothesis testing, it is belief con-firming.) Thus, when a transplant program isabout to face the chopping block, there areusually endless analyses by different personswho offer inconsistent interpretations of thesame data, all in a predictable direction.
Now that decision support systems haveseemingly metastasized throughout the entirehealthcare system, a whole host of folks havebecome expert financial analysts. Many nowbear the in vogue credential—a master’s inbusiness administration (M.B.A.). Theseweekend warriors, unlike the U.S. NationalGuard, have spent a bit of their rest and re-laxation time earning some administrative“stripes,” thus enabling them to function as“physician managers” or, worse yet, managersof physicians. When approached by one ofthese painful characters, I reach for the oldstandby—Preparation H—to relieve my he-morrhoidal symptoms.
I believe we have finally reached a point wherehospital officials are beginning to reconsiderthe market value of what they now confess are“loss leaders”—services that have marketingappeal, but for which there is little expecta-tion of a favorable margin. And, as my fore-going remarks imply, while many argumentshave been used to justify transplant centers,there remain some very outspoken critics ofthe alleged “proliferation” of centers. In addi-
tion, analogies with the space program have,thus far, proven to be wide of the mark. As aresult, perhaps the chicken has come home toroost as transplant volumes have moderatedin response to donor organ constraints, andtransplant iatrogenesis has become increasing-ly expensive, in both medical and humanterms.
As currently practiced, transplantation isanything but a growth industry. Based on ex-isting technology, it is a service that has
reached its market potential. And, as financ-ing has become a more pressing and wide-spread concern for most segments of thehealthcare industry, transplantation pro-grams are falling victim to both the successand the inherent limitations of their owntechnological underpinnings.
Admittedly, it is no fun being fiscally re-sponsible when the goal is to save lives. Ig-noring evidence to the contrary, physiciansand surgeons continue to erroneously believethat life has no price. Thus, national policydictates that we first transplant those patientswho will have the poorest outcome at thegreatest expense. This practice ultimatelyshortens the life of patients, compromisesany benefits they may enjoy while living, andwill almost certainly undermine the survivalof transplant centers as well. Ethical argu-ments to the contrary are as bogus as theproponents of cost-ineffective health care.
There are many ways to assess the econom-ic toll of transplantation. It can be examinedfrom the perspective of the individual proce-dure, evaluated in terms of aggregate expen-ditures, or, in the case of a health plan, theimpact can be computed on a per-member-
per-month basis. Each perspective yields adifferent picture. Individually, transplantprocedures are very expensive. In the aggre-gate, expenditures associated with transplan-tation are unremarkable. And, from the per-spective of a large health plan, the expense oftransplantation is almost trivial.
I have always maintained that a paucity ofdonor organs has favorably limited the eco-nomic implications of transplantation. Ifevery person who might benefit from a trans-plant received one, the economic burdenwould be much greater, and the impactwould be felt at every level, regardless of per-spective or participant. In short, consideringthe possibilities, the debate concerning re-source allocation could easily be far morevigorous than it has been.
I have also insisted that, in the UnitedStates, managed care has done more to mod-erate the economic costs of transplantation,both individually and in the aggregate, thanany drug, medical, or surgical innovation ofthe past 20 years. It is foolhardy to think oth-erwise. When insurers moved from generouspayments based on discounted billed chargesto case rates, transplantation actually becamea far less expensive intervention. No im-munosuppressive drug has even come closeto producing a similar effect.
However, depending on one’s perspective,the situation I describe here has been bothgood and bad. In reality, in addition to cap-ping prices, managed care has had another ef-fect. There has been a reallocation of increas-ingly scarce resources, much to the detrimentof transplant centers. In effect, margins andprofits within the system have simply beenreallocated among the participating parties.Providers are making less money than they wereand writing off more; insurers are less gener-ous in their reimbursement and limiting thesize of their annual premium increases; patientsenjoy fewer benefits and are paying more outof pocket; pharmaceutical companies are ex-periencing record profits and are enduringserious criticism for doing so. In the end,transplant centers have been the big losers.
MISMATCHES
Ignoring evidence to the con-
trary, physicians and surgeons
continue to erroneously believe
that life has no price.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 7 5

Clearly, if it is not obvious by now, what wehave here is the proverbial shell game whereno one seems intelligent enough to grasp andconvey the big picture. I have tried to do so,but I am convinced that clinical judgmentoften stands in the way of prudent healthcarepolicy. The problem of seeing the forest forthe trees is unavoidable.
The picture I have painted is dismal, but Ifirmly believe that institutional pressures ontransplant programs to cover their costs andto turn a profit will persist. Programs can nolonger be sustained at or below cost. Intra-institutional “charity programs” or “loss lead-ers,” where there is little or no expectation ofa payoff relative to investment, are less ap-pealing when the technology has become es-tablished and the service routine.
“Cutting edge” technology has a short shelflife, and in the case of transplantation, therehas been a relentless pursuit of new “gim-micks” that will enable transplant centers todistinguish themselves among their competi-tors. Recent examples include living-relatedlung transplantation, living donor liver trans-plantation, and laparoscopic nephrectomy.In reality, these are not new technologies atall. They are merely variations on old themes,and not particularly persuasive ones at that.
Thus, when the roosters start crowing, thedecision support systems begin churning,and the hospital officials call in the manage-ment consultants, transplant programs willexperience firsthand the lesson I havepreached—life does have a price, and someclinical programs intended to ensure humanlongevity are more valuable than others.Consequently, most transplant centers woulddo well to build their case now to minimizethe discomfort associated with a stretchedneck and to avoid the disquieting sound ofyet another devastating hatchet job—chop,chop, chop.
Roger W. Evans, Ph.D.2251 Baihly Hills Drive SouthwestRochester, Minnesota, USA 55902-1311Tel: 507.281.0886email: [email protected]
MISMATCHES
5 7 6 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

INDEX
Author Index
Abcarian, H., 526.Abecassis, M., 398, 474.Abouna, G.M., 120. Abtahi, P., 266.Adams, D.H., 355.Alwayn, I.P.J., 23, 50.Anaizi, N., 232.Awwad, M., 23, 36.Ayares, D., 80.Azimzadeh, A., 10.
Baden, L., 276.Barreau, N., 135.Barth, R.N., 105. Basker, M., 23.Bedard, E., 96.Benedetti, E., 526.Berney, T., 535.Bishop, D.K., 508.Blancho, G., 135.Bloom, E.T., 160.Bollinger, R.R., 416.Bracy, J.L., 102.Buell, J.F., 205.Buhler, L.. 36, 99, 535.
Cao, S., 202.Caulfield, A., 535.Cassileth, B.R., 146.Chen, R., 355. Chen, Z.-c., 32.Cicalese, L., 526.Clark, D.A., 338Clark-Borre, L., 391, 392.Cohen, I.R., 369.Colman, A., 80.
Colvin, R.B., 44.Cooper, D.K.C., 6, 23, 36, 94, 137.Cosimi, A.B., 146.Cowan, P.J., 47, 76, 78.Cozzi, E., 66.
Daar, A.S., 164.Daha, M.R., 188, 220.Dahlberg, R., 449.Dai, Y., 80.Dalmasso, A.P., 53.d’Apice, A.J.F., 47, 76, 78.Demme, R.A., 2478, 307.Davis, R.S., 300.de Haij. S., 188, 220.Dorian, R., 491.Dorling, A., 72.
Edge, A.S.B., 118.Erturk, E., 300.Evans, R.W., 154, 467, 574.
Farivar, R.S., 355.Fischer,S., 481.Forrest, S., 369.Friend, P., 418.Friend, P.J., 66, 111.
Galili, U., 32.Gasser, M., 346.Gianello, P., 18.Gock, H., 76, 78.Goddard, M., 66.Gollackner, B., 23, 137.Goodman, D.J., 47.Gorcyzynski, R.M., 338.Graca, L., 174.Grassi, J.S., 445.
Groth, C.G., 115.
Hammer, C., 108.Hanaway, M.J., 205.Heeger, P.S., 195,. 571.Heinrichs, D.F., 407.Hijazi, F., 307.Hofmeyr, S., 369.Howard, R.J., 424.Huang, S., 326.
Iacomini, J., 102.Imagawa, D.K., 202.Inverardi, L., 209, 519.
Jacobs, C.L., 410.Jamieson, I.R., 424.Ji, P., 202.Joyce, J., 452.
Kalayoglyu, M., 205.Katz, J., 276.Kaufman, D.B., 398.Khabbaz, G.R., 435.Keshavjee, S., 481.Kim, D.Y., 500.King, S.R., 491.Klintmalm, G.B., 421.Kobayashi, T., 27.Koffron, A., 474.Korsgren, O., 115.Koski, G., 146.
Lake, K.D., 427, 544.Larson, T.S., 500.Link, C.J., 32.Lipori, P., 424.Lorenzini, R., 68.
Lucey, M.R., 223.Lundin, S., 150.
Macchiarini, P., 10.Magee, J.C., 508.Martinez, D.M, 180.McEwan, R.N., 432.McKenzie, I.F.C., 83.Michaels, M.G., 129.Moore, M., 80.Morel, P., 535.Morgan, B.P., 63.Morgan, M.F., 40.Mullon, C., 126.Musat, A., 205.
Nickels, M.W., 290.
Oberholzer, J., 535.Oh, S., 383.Olivier, D.L., 435.Orosz, C.G., 365, 369.Ortiz, J., 202.
Patience, C., 133.Paul, L.C., 188, 220.Pierson III, R.N., 10.Pino-Chavez, G., 60, 66.Platt, J., 8.Podnos, Y.D., 202.
Rastellini, C., 526.Remensnyder, J.P., 146.Robson, S.C., 50.Rose, A.G., 14.Rose, M.L., 57.Rothblatt, M., 143.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 7 7
Index to Graft
Volume 4Issue 1, (January/February, 2001), pp. 1-88.Issue 2, (March, 2001), pp. 89-150.Issue 3, (April/May, 2001), pp. 151-224.Issue 4, (June, 2001), pp. 225-320.Issue 5 (July/August, 2001), pp. 321-384.Issue 6, (September, 2001), pp. 385-468.Issue 7, (October/November, 2001), pp. 469-520.Issue 8, (December, 2001), pp. 521-580.

Title Index
Articles“A Comprehensive Approach to Outpatient Transplant Pharmacy,” McEwan, 432.“A History of the LifeLink Transplant Institute,” Heinrichs, 407.“A Map for the Journey: Seeing Patients as People and as Your Customers,” Joyce, 452.“Academia and Industry: Partners or Protagonists? A Review of Critical Business Issues in Academic Med-
icine,” Clark-Borre, 392.“Alternative Approaches to Reducing Gal Expression in Pig Organs,” Gock, Cowan, and d’Apice, 78.“Apoptosis in Lung Transplantation,” Fischer and Keshavjee, 481.
“Building and Effective Data Collection System to Support the Transplant Infrastructure “ Khabbaz and Olivi-er, 435.
“Choosing Risk-Benefit Analysis or Precautionary Principle as Our Approach to Clinical Xenotransplanta-tion, Daar, 164.
“Cloning Pigs Deficient in Alpha 1,3 Galactosyltransferase” Ayares, Colman, Dai, Shiels, and Moore, 80.“CMV in Solid Organ Transplantation,” Koffron and Abecassis, 474.“Common Psychiatric Problems in the Well Transplant “ Nickels, 290.“Deleting the Gal Epitope from the Donor Pig “ Gock, Cowan, and d’Apice, 76.“Detecting Non-Gal Epitopes of Importance” Kobayashi, 29.“Determining and Preventing the Potential PERV Problem” Patience, 133.“Determining Compatibility of Adhesion Molecules” Morgan and Warrens, 40.“Determining Significant Physiologic Incompatibilities” Hammer and Thein, 108.“Determining the Histologic and Immunopathologic Features of Acute Humoral Xenograft Rejection”
Shimizu and Colvin, 44.“Determining the Histopathology of Hyperacute Rejection” Rose, 14.“Determining the Rate of Growth of a Pig Organ in a Primate” Friend and Soin, 111.“Determining the Real Cost of Keeping Transplant Patients Well: Evaluating Pharmacoeconomic Studies in
Transplantation Medicine” Lake, 427.“Determining the Risks of Xenozoonoses” Michaels,129.“Determining Whether Allotransplantation Can Be Safely Carried out After a Porcine Organ Bridge” Gol-
lackner and Cooper, 137.“Determining Whether HLA—Sensitive Patients Will be Suitable Xenotransplant Candidates” Blancho, Bar-
reau, Soulillou, and Gilles, 135.“Differentiating Acute Humoral from Acute Cellular Rejection Histopathologically,” Pino-Chavez, 60.“Doctors Marketing Excellence: The Duke Experience in Transplantation” Bollinger, 416.“Does Successful Allopregnancy Mimic Transplatation Tolerance?” Gorczynski, Yu, Clark, 338.“Dominant Transplantation Tolerance,” Graca and Waldmann,174.“Drug Interactions Involving Immunosuppressive Agents” Anaizi, 232.“’Engineering’ Myoblast Transplantation,” Shuk and Tremblay, 558.“Erectile Dysfunction in the Transplant Recipient” Erturk and Davis, 300.“Evolving Infrastructural Issues in Blood and Marrow Transplant Center Development” “Klingemann,
Schumer, Friend, 418.“’Excellence Is Available to All Who Are Willing…’:A Manifesto for Today’s Leaders in Health Care” Clark-
Borre, 390.“Expanding Organ Donor Options and Financial Resources” Jacobs, 410.
“Expression and Function of CD40 on Hematopoietic Cells” van Kooten, de Haij, Paul, and Daha,188.“Financial Sustainability in Transplant Programs” Jamieson, Lipori, and Howard, 424.“Genetically Engineering a Pig that Minimizes Coagulation Incompatibilities” Dorling, 72.“Graft-Versus-Host Disease in Solid Organ Transplantation” Hanaway, Buell, Musat, and Kalayoglu, 205.“How Grassroots Efforts Turned Tobacco Tax Money into Transplants: Social Workers as Advocates”
Thomas, 459.“Hypertension in the Kidney Transplant Patient,” Demme, 248.“Immunobiology of CD30+ T Lymphocytes,” Martinez, 180.“Immunosuppression in Pancreas Transplantation,” Stegall, Kim and Larson
“Inducing Tolerance by Mixed Hematopoietic Cell Chimerism,” Buhler, Sachs, and Cooper,99.“Inducing Tolerance by Molecular Chimerism,” Iacomini and Bracy, 102. “Inducing Tolerance by Thymus Transplantation” Yamada, Barth, and Sachs,105.“Islet of Langerhans Autotransplantation: Rationale, Results and New Development,” Berney, Caulfield,
Oberholzer, Buhler, Toso, and Morel, 535.“Infectious Disease Issues in the Well Transplant Patient” Baden and Katz, 276.“Key Legal Issues: Determining the Rise of Geoethics as the Optimum Analytical Paradigm” Rothblatt, 143.“Living Related Small Bowel Transplantation,” Cicalese, Sileri, Rastellini, Abcarian, and Benedetti, 526.“Long-Term Management of Post-Transplant Anemia and Erythrocytosis” Hijazi, Zand, and Demme, 307.“Management of Diabetes Mellitus After Solid Organ Transplantation” Schwimmer and Zand, 256.“Management of Hyperlipidemia in the Stable Solid Organ Transplant Recipient” Abtahi and Zand, 266.“Marketing—What is it?” Klintmalm, 421.“Matching the Results Obtained in Concordant Primate Xenotransplantation Models” Zhong and Bedard, 96.“Minimizing the Risk of Xenozoonosis” Yamanouchi, 131.“Overcoming the Risks of ‘Ethical Rejection’?” Vanderpool, 140.“Pharmacoeconomic and Outcomes Analyses in Solid Organ Transplantation,” Lake, 544.“Post-Genomic Biology: Gene Expression Profiles, Cluster Analysis and Beyond” Huang, 326.“Post-Transplantation Osteoporosis: Prevention and Treatment” Suresh, 316.“Preparing Your Transplant Center to Work with a Consultant” Grassi, 445.“Preventing Acute Vascular Rejection” Goodman, d’Apice, and Cowan, 47.“Preventing Hyperacute Rejection” Gianello, 18.“Preventing the Induced Anti-Pig Antibody Response” Buhler, Awwad, Sachs,and Cooper, 36.“Regulating Clinical Xenotransplantation in Europe” Tibell, 157.“Regulating Clinical Xenotransplantation in the United States” Bloom, 160.“Regulatory and Fiscal Relationships between Transplant Centers and Transplant Surgeons/Physicians”
Stuart, Abecassis, and Kaufman, 398.“Requirements for Encapsulation Technology and the Challenges for Transplantation of Islets of Langer-
hans,” King, Dorian, and Storrs, 491.“Sperm-Mediated Transgenesis: Practical Implications of a Bilogical Process” Spadafora and Lorenzini, 68.“Studies with T Cell lines and Clones and Their Implications for the Understanding of Mechanisms in Pe-
diatric Graft Rejection and Tolerance” Kist-van Hoelthe, Gasser, and Waaga, 346.“Suppressing Natural Antibody Production” Alwayn, Basker, Teranishi, Gollackner, Sachs, Awwad, and
Cooper, 23.“The Expanding Role of Case Management” Dahlberg, 449.“The National Kidney Foundation of Illinois: A Case of a Collaborative Effort in Transplant Education” Wight-
man, 464.
INDEX
Sachs, D.H., 23, 36, 99, 105.Sandrin, M.S., 83.Schmoeckel, M., 66.Schumer, M., 418.Schwimmer, J., 256.Segel, L.A., 370.Seregina, T., 32.Skuk, D., 558.Shiels, P., 80.Shizmizu, A., 44.Sileri, P., 526.Soin, B., 111.Souillou, J.P., 135.
Spadafora, C., 68.Stegall, M.D., 500.Storrs, R.W., 491.Stuart, F.P., 398.Suresh, U., 316.Surman, O.S., 146.
Tanemura, M., 32.Teranishi, K., 23.Thein, E., 108.Thomas, C., 459.Thomson, A., 403.Tibell, A., 157.
Toso, C., 535.Tremblay, J.P., 558.
Valujskikh, A., 195.Van Buskirk, A.M., 571.van den Berg, C.W., 63.van Kooten, C., 188, 220.Vanderpool, H.Y., 140.Waaga, A.M., 346.Waldmann, H., 174.Wallwork, J., 66.Warrens, A.N., 40.Weenberg, L., 115.
White, D.J.G., 66.Wightman, R.L., 464.
Yamada, K., 105.Yamanouchi, K., 131.Yu, G., 338.Young, C.J., 514.
Zand, M.S., 230, 256, 266.Zavala, E.Y., 412.Zhong, R., 96.
5 7 8 v o l u m e 4 i s s u e 8 d e c e m b e r 2 0 0 1 g r a f t s a g e p u b . c o m

INDEX
“The Use of Ex Vivo Xenogeneic Whole Liver Perfusion as a Bridge to Liver Regeneration or Liver Trans-plantation” Abouna, 120.
“The Well Transplant Patient” Zand, 230.“Transplant Center Marketing” Zavala, 412.“Transplant Infrastructure” Thomson, 403.“Transplanting Organs from Pigs Transgenic for a Single Human Complement Regulator Protein”
Schmoeckel, Cozzi, Dunning, Goddard, Pino-Chavez, Friend, Wallwork, and White, 66.“Treating Acute Liver Failure with an Extracorporeal Liver-Assist Device“ Mullon, 126.“Treatment of Groin Lymphocele Following Liver Transplantation with Fibrin Glue” Podnos, Ortiz, Ji, Cao,
and Imagawa, 202.“Understanding and Achieving Accomodation” Dalmasso, 53.“Understanding and Preventing the Coagulation Disorders Associated with Xenograft Rejection” Alwayn and
Robson, 50.“Understanding Cultural Perspectives on Clinical Xenotransplantation” Lundin, 150.“Understanding Hyperacute Rejection of the Lung: Is This a Special Case?” Pierson III, Macchiarini, and Az-
imzadeh, 10.“Understanding Public Attitudes to Clinical Xenotransplantation” Cassileth, Remensnyder, Koski, Surman,
and Cosimi, 146.“Understanding the Immune Protection Afforded by Endogenous Complement Regulatory Molecules” van
den Berg and Morgan, 63.“Understanding the Induced Antibody Response” Galili, Chen, Tanemura, Seregina, and Link, 32.“Understanding the Mechanism of Acute Cellular Rejection” Rose, 57.“Understanding the Mechanism of Hyperacute Rejection” Platt, 8.“Understanding Xenophobia Induced by Economics” Evans, 154.“Xenotransplantation and Endothelium” Farivar, Chen, and Adams, 355.“Xenotransplantation—A Closer Look pt 1” Cooper, 6.“Xenotransplantation: A Closer Look pt 2” Cooper, 94.“Xenotransplanting Neural Cells” Edge, 118.“Xenotransplanting Pancreatic Islets” Korsgren, Wennberg, and Groth, 115.
Biotoon“Expression and Function of CD40 on Various Cell Types,” van Kooten, de Haij, Paul, and Daha, 220.
Endpage/Mismatches“Living Donor Liver Transplantation: Balancing Donor Risk with Recipient Need” Lucey, 223.“Taking Aim: Reflections on Quality-of -Life ‘Research’—A Sinner’s Plea for Salvation” Evans, 467.
“The American Way,” Oh, 383.“The Unkindest Cut: Where Are All the Transplant Programs Going?” Evans, 574.
Forum Articles: Complexity“Engineering an Immune System” Forrest and Hofmeyr, 369.“How Complexity Helps To Shape Alloimmunity” Orosz, 365.“How Does the Immune System See To It That It Is Doing a Good Job?” Segel, 370.“Immunity as a Swarm Function,” Orosz, 369.“Immunity, Set Points, Reactive Systems and Allograft Rejection,” Cohen, 369.
Literature Reviews“Cell Transplantation,” Inverardi, 209.“Cell Transplantation,” Inverardi, 519.“Disparities in Access to Renal Transplantation for African Americans,” Young and Gaston, 514.
Meeting Reports“Recent Advances in Pig-to-Primate and Related Xenotransplantation: A Brief Review of Presentations Re-
lating to Xenotransplantation at the 18th International Congress of The TransplantationSociety,Rome,August 2000,” McKenzie and Sandrin, 83.
“Tolerance: Defining, Achieving, and Measuring It. Report from the ‘Tolerance Assay: Where are we now?’Workshop, Transplant 2001,” VanBuskirk and Heeger, 571.
Methods“Enzyme-Linked Immunosorbent Spot (ELISPOT) Assay for Detection of Alloreactive Cytokine-Reacting
Cells-Detailed Method” Valujskikh and Heeger, 195.
Miscellaneous“Pharmaceutical Clinical Trials—All Phases,” 216.“Pharmaceutical Clinical Trials—Phase Three,” 212.
State of the Art“Considerations Regarding the Strengths and Weaknesses of Experimental Approaches Employed in Basic
Transplant Research: TGFβ1 Gene Transfer in Transplantation,” Bishop and Magee, 508.
s a g e p u b . c o m g r a f t d e c e m b e r 2 0 0 1 v o l u m e 4 i s s u e 8 5 7 9