research paper comparing alcohol dehydrogenase
TRANSCRIPT

Shadkam 1
Parvaneh Shadkam-Farrokhi Submission date: 12/2/16
Structure and Function Analysis of Alcohol Dehydrogenases
Introduction:
As the world population continues to increase, the demand for environmentally-friendly energy sources also
continue to increase. It has become imperative that we utilize renewable sources such as biofuels. As such, we aim to
engineer effective bacteria to produce higher order alcohols that can be used as substitutes for gasoline. Previously,
we tested the efficiency of isobutanol production in three-metabolically-engineered, bio-fuel producing Escherichia
coli (E. coli) strains via colorimetric enzyme-based assay. The three strains of E. coli produced isobutyraldehyde
through the overexpression of the genes encoding acetolactate synthase (alsS), acetohydroxy acid isomeroreductase
(ilvC), dihydroxy-acid dehydrogenase (ilvD), and 2-ketoacid decarboxylase (kivd), with their respective alcohol
dehydrogenase (ADH) (Atsumi). Strain 1 contained yqhD from E.coli, while strains 2 and 3 had ADH2 from
Saccaromyces cerevisiae and adhA from Lactcoccus lactis, respectively (Gober). Our studies indicated that AdhA
(Strain 3) exhibited the most efficient alcohol conversion/biofuel production while ADH2 (Strain 2) exhibited the
least catalytic efficiency. Our results coincide with previous studies done by Atsumi et al whom also found ADH2 to
be the least efficient enzyme for isobutanol production and AdhA and YqhD to be the most effective. We now aim to
analyze and compare the crystal protein structures of AdhA and ADH1 to determine structure-function relationships.
Liu et al. and Plapp et al. have determined the crystal structures of AdhA and ADH1, respectively. According to Liu
et al., AdhA contains many conserved amino acid residues where mutagenesis cannot improve the efficiency of
isobutanol conversion while the mutagenesis of select amino acids such as Y50F, I212T, and L264V increases
isobutanol production. Likewise, Plapp et al. have demonstrated the conformational changes ADH1 undergoes when
bound by a coenzyme or substrate. We will be utilizing these findings and Cn3D software to identify specific sites
where differences between AdhA and ADH1 can lead to varying degrees of isobutanol production efficiency. Once
these sites have been identified, we can possibly alter them in a manner to facilitate increased catalytic efficiency,
increased production of biofuels such as isobutanol.
Results:
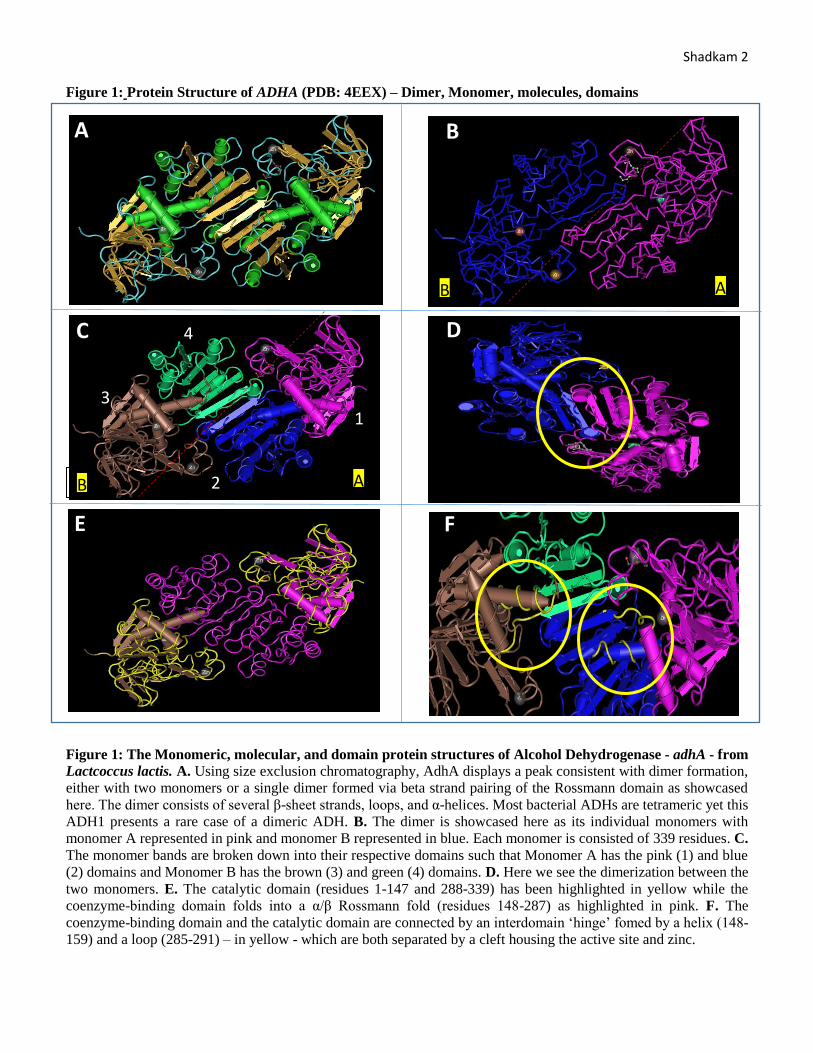
Shadkam 2 Figure 1: Protein Structure of ADHA (PDB: 4EEX) – Dimer, Monomer, molecules, domains
Figure 1: The Monomeric, molecular, and domain protein structures of Alcohol Dehydrogenase - adhA - from
Lactcoccus lactis. A. Using size exclusion chromatography, AdhA displays a peak consistent with dimer formation,
either with two monomers or a single dimer formed via beta strand pairing of the Rossmann domain as showcased
here. The dimer consists of several β-sheet strands, loops, and α-helices. Most bacterial ADHs are tetrameric yet this
ADH1 presents a rare case of a dimeric ADH. B. The dimer is showcased here as its individual monomers with
monomer A represented in pink and monomer B represented in blue. Each monomer is consisted of 339 residues. C.
The monomer bands are broken down into their respective domains such that Monomer A has the pink (1) and blue
(2) domains and Monomer B has the brown (3) and green (4) domains. D. Here we see the dimerization between the
two monomers. E. The catalytic domain (residues 1-147 and 288-339) has been highlighted in yellow while the
coenzyme-binding domain folds into a α/β Rossmann fold (residues 148-287) as highlighted in pink. F. The
coenzyme-binding domain and the catalytic domain are connected by an interdomain ‘hinge’ fomed by a helix (148-
159) and a loop (285-291) – in yellow - which are both separated by a cleft housing the active site and zinc.
A
B
C
C
D
E
F
A
B
A
B
4
3
2
1
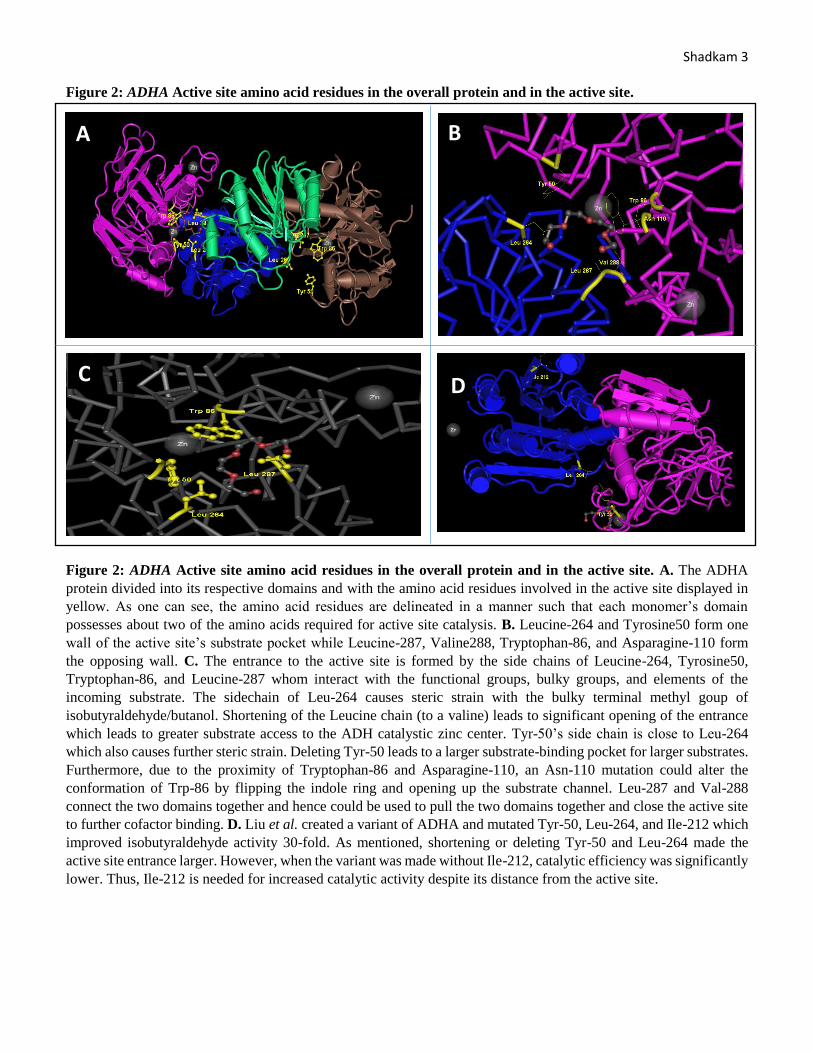
Shadkam 3 Figure 2: ADHA Active site amino acid residues in the overall protein and in the active site.
Figure 2: ADHA Active site amino acid residues in the overall protein and in the active site. A. The ADHA
protein divided into its respective domains and with the amino acid residues involved in the active site displayed in
yellow. As one can see, the amino acid residues are delineated in a manner such that each monomer’s domain
possesses about two of the amino acids required for active site catalysis. B. Leucine-264 and Tyrosine50 form one
wall of the active site’s substrate pocket while Leucine-287, Valine288, Tryptophan-86, and Asparagine-110 form
the opposing wall. C. The entrance to the active site is formed by the side chains of Leucine-264, Tyrosine50,
Tryptophan-86, and Leucine-287 whom interact with the functional groups, bulky groups, and elements of the
incoming substrate. The sidechain of Leu-264 causes steric strain with the bulky terminal methyl goup of
isobutyraldehyde/butanol. Shortening of the Leucine chain (to a valine) leads to significant opening of the entrance
which leads to greater substrate access to the ADH catalystic zinc center. Tyr-50’s side chain is close to Leu-264
which also causes further steric strain. Deleting Tyr-50 leads to a larger substrate-binding pocket for larger substrates.
Furthermore, due to the proximity of Tryptophan-86 and Asparagine-110, an Asn-110 mutation could alter the
conformation of Trp-86 by flipping the indole ring and opening up the substrate channel. Leu-287 and Val-288
connect the two domains together and hence could be used to pull the two domains together and close the active site
to further cofactor binding. D. Liu et al. created a variant of ADHA and mutated Tyr-50, Leu-264, and Ile-212 which
improved isobutyraldehyde activity 30-fold. As mentioned, shortening or deleting Tyr-50 and Leu-264 made the
active site entrance larger. However, when the variant was made without Ile-212, catalytic efficiency was significantly
lower. Thus, Ile-212 is needed for increased catalytic activity despite its distance from the active site.
A
B
C
D
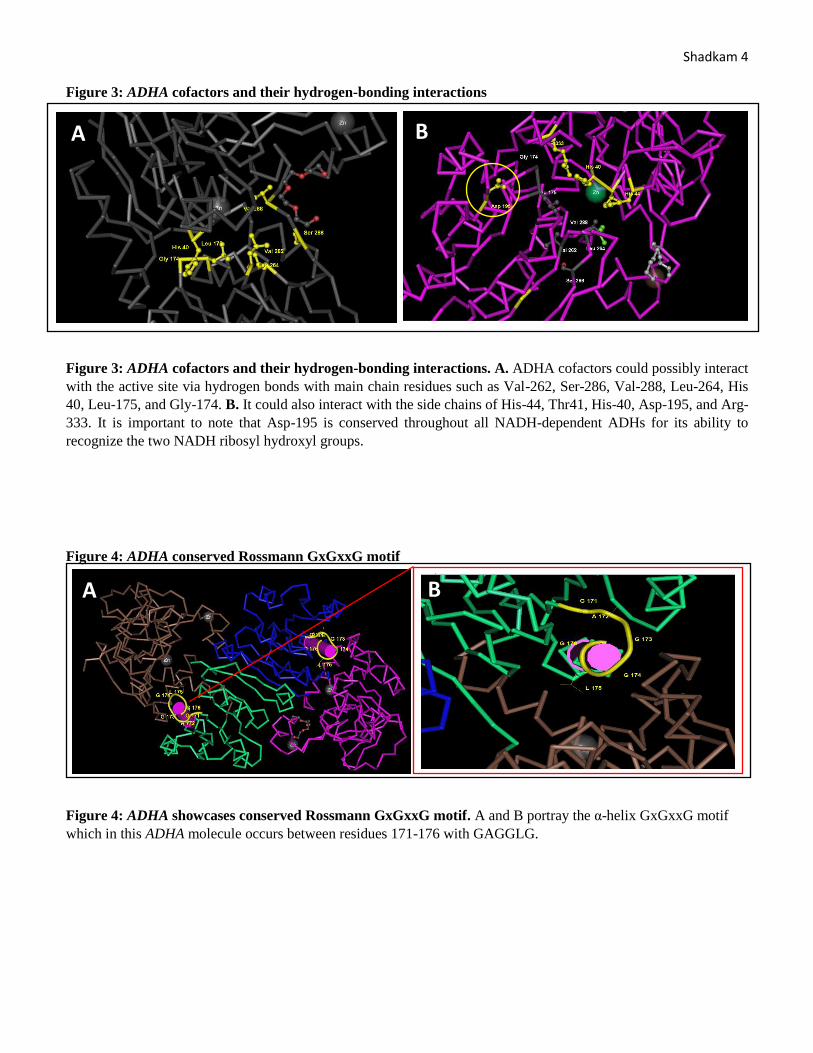
Shadkam 4 Figure 3: ADHA cofactors and their hydrogen-bonding interactions
Figure 3: ADHA cofactors and their hydrogen-bonding interactions. A. ADHA cofactors could possibly interact
with the active site via hydrogen bonds with main chain residues such as Val-262, Ser-286, Val-288, Leu-264, His
40, Leu-175, and Gly-174. B. It could also interact with the side chains of His-44, Thr41, His-40, Asp-195, and Arg-
333. It is important to note that Asp-195 is conserved throughout all NADH-dependent ADHs for its ability to
recognize the two NADH ribosyl hydroxyl groups.
Figure 4: ADHA conserved Rossmann GxGxxG motif
Figure 4: ADHA showcases conserved Rossmann GxGxxG motif. A and B portray the α-helix GxGxxG motif
which in this ADHA molecule occurs between residues 171-176 with GAGGLG.
A
B
A
B

Shadkam 5 Figure 5: ADHA, its two Zinc ions, and coordination with amino acid residues
Figure 5: ADHA, its two Zinc ions, and coordination with amino acid residues. A. The ADHA monomer contains
two zinc ions – one structural and one catalytic. B. The structural zinc is coordinated by Cys-91, Cys-94, Cys-97, and
Cys-105. C. The catalytic zinc is coordinated by Cys-39, His-60, Cys-147, and finally by a water molecule on the co-
factor-free enzyme subunit. These variable coordination amino acid residues could play variable roles in the opening
and closing of the active site.
A
B
C
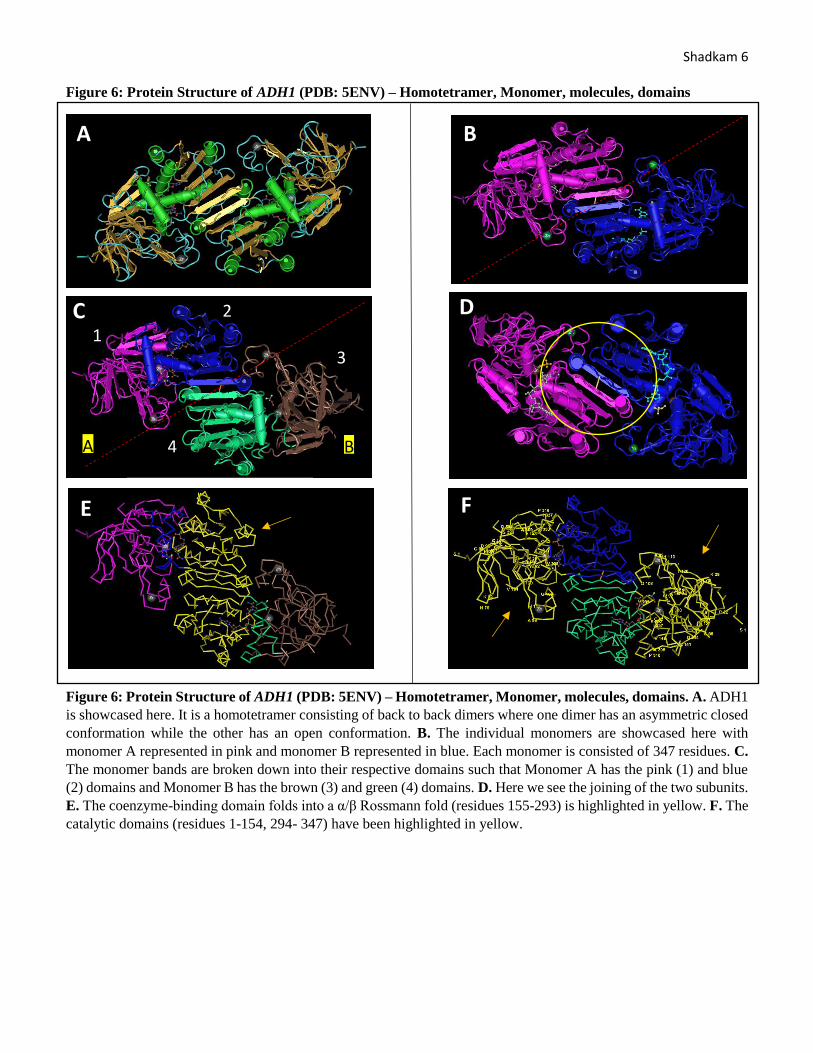
Shadkam 6 Figure 6: Protein Structure of ADH1 (PDB: 5ENV) – Homotetramer, Monomer, molecules, domains
Figure 6: Protein Structure of ADH1 (PDB: 5ENV) – Homotetramer, Monomer, molecules, domains. A. ADH1
is showcased here. It is a homotetramer consisting of back to back dimers where one dimer has an asymmetric closed
conformation while the other has an open conformation. B. The individual monomers are showcased here with
monomer A represented in pink and monomer B represented in blue. Each monomer is consisted of 347 residues. C.
The monomer bands are broken down into their respective domains such that Monomer A has the pink (1) and blue
(2) domains and Monomer B has the brown (3) and green (4) domains. D. Here we see the joining of the two subunits.
E. The coenzyme-binding domain folds into a α/β Rossmann fold (residues 155-293) is highlighted in yellow. F. The
catalytic domains (residues 1-154, 294- 347) have been highlighted in yellow.
A
B
1
2
3
4
A B
C D
E F
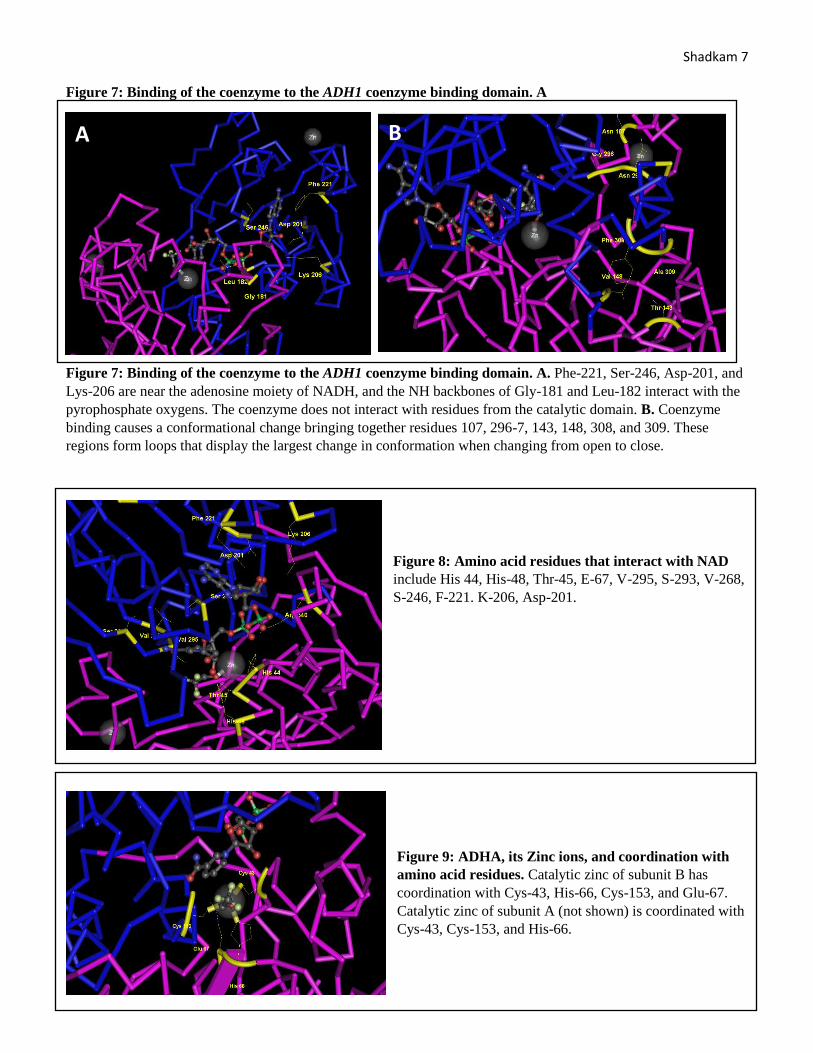
Shadkam 7 Figure 7: Binding of the coenzyme to the ADH1 coenzyme binding domain. A
Figure 7: Binding of the coenzyme to the ADH1 coenzyme binding domain. A. Phe-221, Ser-246, Asp-201, and
Lys-206 are near the adenosine moiety of NADH, and the NH backbones of Gly-181 and Leu-182 interact with the
pyrophosphate oxygens. The coenzyme does not interact with residues from the catalytic domain. B. Coenzyme
binding causes a conformational change bringing together residues 107, 296-7, 143, 148, 308, and 309. These
regions form loops that display the largest change in conformation when changing from open to close.
Figure 8: Amino acid residues that interact with NAD
include His 44, His-48, Thr-45, E-67, V-295, S-293, V-268,
S-246, F-221. K-206, Asp-201.
Figure 9: ADHA, its Zinc ions, and coordination with
amino acid residues. Catalytic zinc of subunit B has
coordination with Cys-43, His-66, Cys-153, and Glu-67.
Catalytic zinc of subunit A (not shown) is coordinated with
Cys-43, Cys-153, and His-66.
A B

Shadkam 8 Results Summary:
Our previous studies indicated that AdhA (Strain 3) exhibited the most efficient alcohol conversion/biofuel
production while ADH2 (Strain 2) exhibited the least catalytic efficiency. Our results coincide with previous studies
done by Atsumi et al whom also found ADH2 to be the least efficient enzyme for isobutanol production and AdhA
and YqhD to be the most effective. We analyzed and compared the protein structures of AdhA and Adh1 using Cn3D
software to identify specific sites where similarities and differences between AdhA and Adh1 occur. We found that
AdhA was composed of a single dimer formed via beta strand pairing of the Rossmann domain. The dimer consists
of several β-sheet strands, loops, and α-helices. Most bacterial ADHs are tetrameric yet this ADH1 presents a rare
case of a dimeric ADH. Each monomer is consisted of 339 residues. Each AdhA monomer possesses 2 dimers with a
catalytic domain (residues 1-147 and 288-339) and a coenzyme-binding domain that folds into a Rossmann fold
(residues 148-287). These domains are connected by an interdomain ‘hinge’ formed by a helix (148-159) and a loop
(285-291) which are both separated by a cleft housing the active site and zinc. Adh1 is a homotetramer consisting of
back to back dimers where one dimer had an asymmetric closed conformation while the other had an open
conformation. Each monomer is consisted of 347 residues. Each Adh1 molecule has two domains with one being
the coenzyme-binding domain which folds into a α/β Rossmann fold (residues 155-293), and a catalytic domain
(residues 1-154, 294- 347). Therefore, although AdhA and Adh1’s molecules both possess two domains, Adh1 has a
few more residues than AdhA.
AdhA’s active site is consisted of Leucine-264 and Tyrosine-50 forming one wall of the active site’s
substrate pocket while Leucine-287, Valine288, Tryptophan-86, and Asparagine-110 forming the opposing wall.
The entrance to the active site is formed by the side chains of Leucine-264, Tyrosine-50, Tryptophan-86, and
Leucine-287. Meanwhile, Adh1’s active site is consisted of Phe-221, Ser-246, Asp-201, and Lys-206, Gly-181, and
Leu-182. His-44, His-48, Thr-45, Glu-67, Val-295, Ser-293, and Val-268 also interact with NAD+ when it enters
the active site. Similarly, ADHA cofactors could interact with the active site via hydrogen bonds with main chain
residues such as Val-262, Ser-286, Val-288, Leu-264, His-40, Leu-175, and Gly-174. AdhA’s structural zinc is
coordinated by Cys-91, Cys-94, Cys-97, and Cys-105 while the catalytic zinc is coordinated by Cys-39, His-60,
Cys-147, and finally by a water molecule on the co-factor-free enzyme subunit. In Adh1, the catalytic zinc of
subunit B has coordination with Cys-43, His-66, Cys-153, and Glu-67 and the catalytic zinc of subunit A is
coordinated with Cys-43, Cys-153, and His-66. Therefore, AdhA and Adh1’s zinc ions are coordinated by differently
located amino acid residues.
These similarities and differences in structure relate to their function and their catalytic efficiency in making
isobutanol.
Discussion:
Upon analyzing and comparing the protein structures of AdhA and Adh1, there were more differences
between the two strains than similarities. AdhA is a single dimer composed of two monomer whom are consisted of
a catalytic domain (residues 1-147 and 288-339) and a coenzyme-binding domain (residues 148-287). On the other
hand, Adh1 is a homotetramer with back to back dimers that are also consisted of a catalytic domain (residues 1-
154, 294- 347) and a coenzyme-binding domain (residues 155-293). Furthermore, each AdhA monomer is composed
of 339 residues while each Adh1 subunit is composed of 347 residues. Nevertheless, the boundary residues for each
alcohol dehydrogenase is in relatively the same spot. Furthermore, AdhA has a much lower Km and higher catalytic
efficiency than Adh1 (Atsumi, 2010). This suggests that perhaps the folding pattern and amino acid interactions
between substrates, other amino acids, and cofactors is responsible for the variable bio-efficiency rate. This is
supported by Liu et al. and Plapp et al. whom have determined the crystal structures of AdhA and ADH1,
respectively. According to Liu et al., AdhA contains many conserved amino acid residues where mutagenesis cannot
improve the efficiency of isobutanol conversion while the mutagenesis of select amino acids such as Y50F, I212T,
and L264V increases isobutanol production. Likewise, Plapp et al. have demonstrated the conformational changes
ADH1 undergoes when bound by a coenzyme or substrate.

Shadkam 9 Upon carefully analyzing the amino acids located in the substrate, the two ADHs possess different residues
in their active sites. Furthermore, aside from the fact that both of their zinc ions are coordinated by cysteine
molecules, their interactions do not occur at the same residue number. However, upon analyzing the cysteine’s
molecules location relative to their protein peptide length, AdhA’s catalytic zinc and Adh1’s subunit A zinc share
cysteine and histidine molecules in the same relative location. Furthermore, they also share similarities as to what
their cofactors such as NADH could bind to such as His-44 (Adh1) versus His-40 (AdhA), Val 295 (Adh1) versus
Val-288 (AdhA), Ser-293 (Adh1) versus Ser-286 (AdhA), Val-268 (Adh1) versus Val-262 (AdhA), Asp-195
(Adh1) versus Asp-201 (AdhA), Gly-174 (AdhA) versus Gly-181 (Adh1), and Leu-175 (AdhA) versus Leu-182
(Adh1). While these residues do not numerically match, they have the same interactions in the similar area of their
respective residues. These similarities yet differences between Adh1 and AdhA indicate that specific amino acid
residues are possibly affecting their biofuel production efficiency.
We could test these ideas by genetically engineering a variety of strains with specific mutations in certain
areas while maintaining all other genes unaffected. This idea was tested by Liu et al. whom shortened the leucine-
264 of AdhA’s active site to a valine and found it to lead to significant opening of the active site entrance which
lead to greater substrate access to the ADH catalystic zinc center. They also deleted Tyr-50, removing the steric
strain is created with Leu-264’s side chain, and leading to a larger substrate-binding pocket for larger substrates.
They then created a variant of ADHA and mutated Tyr-50, Leu-264, and Ile-212 which improved isobutyraldehyde
activity 30-fold. However, when the variant was made without Ile-212, catalytic efficiency was significantly lower.
Thus, Ile-212 is needed for increased catalytic activity despite its distance from the active site. By performing such
experiments, it would be possible to specificy which amino acid residues are needed for catalysis and which ones
improve biofuel production.
Citations:
Atsumi, Shota, and James C. Liao. "Metabolic Engineering for Advanced Biofuels Production from Escherichia
Coli." Current Opinion in Biotechnology 19.5 (2008): 414-19.
Atsumi, Shota, Tung-Yun Wu, Eva-Maria Eckl, Sarah D. Hawkins, Thomas Buelter, and James C. Liao. "Engineering the
Isobutanol Biosynthetic Pathway in Escherichia Coli by Comparison of Three Aldehyde Reductase/alcohol
Dehydrogenase Genes." Applied Microbiology and Biotechnology 85.3 (2009): 651-57.
Gober, James W. "Laboratory 8: Structure and Function Analysis of Alcohol Dehydrogenase" Chemistry and
Biochemistry 153L - Biochemical Methods I: Introduction to Protein Science - Laboratory Manual. 10th ed. Los
Angeles: Course Reader Material, 2016. 58-61. Print.
Liu, Xiang, Sabine Bastian, Christopher D. Snow, Eric M. Brustad, Tatyana E. Saleski, Jian-He Xu, Peter Meinhold, and
Frances H. Arnold. "Structure-guided Engineering of Lactococcus Lactis Alcohol Dehydrogenase LlAdhA for Improved
Conversion of Isobutyraldehyde to Isobutanol." Journal of Biotechnology 164.2 (2013): 188-95.
Plapp, Bryce V., Henry A. Charlier, and S. Ramaswamy. "Mechanistic Implications from Structures of Yeast Alcohol
Dehydrogenase Complexed with Coenzyme and an Alcohol." Archives of Biochemistry and Biophysics 591 (2016): 35-
42.
Sulzenbacher, Gerlind, Karine Alvarez, Robert H.h. Van Den Heuvel, Cees Versluis, Silvia Spinelli, Valérie
Campanacci, Christel Valencia, Christian Cambillau, Hans Eklund, and Mariella Tegoni. "Crystal Structure of E.coli
Alcohol Dehydrogenase YqhD: Evidence of a Covalently Modified NADP Coenzyme." Journal of Molecular
Biology 342.2 (2004): 489-502. Web.