quand penser au syndrome des anti-synthétases...
TRANSCRIPT

Quand penser au Syndrome des Anti-Synthétases ?
Dr Baptiste Hervier Département de Médecine Interne et Immunologie Clinique
DU Maladies Systémiques et Autoimmunes
6 juin 2014
Hal$Pi'é)Salpêtrière$
$APHP$

Qu’est-ce que le SAS ? Pourquoi faut-il penser au SAS ?
Comment traiter un patient avec SAS ?

Pas de critère diagnostique ou de classification établi
Qu’est-ce que le SAS ?

Qu’est-ce que le SAS ?
1976 « John-P »: découverte 1er anticorps anti-synthétase (Σ) 1983 Identification cible antigénique Mathews M, 1983;26:604-11
1980 Corrélation des anti-Σ (Jo-1) avec PM (26p)
Nishikai M Arthritis Rheum 1980;23:881-8
1983 Corrélation des anti-Σ (Jo-1) avec PM associée à PID (9p) Yoshida S, Arthritis & Rheum 1983;26:604-11
1990 Caractérisation du SAS.
5/29 patients sans Myosite (83%) et 6/29 sans PID (79%) Marguerie C Q J Med 1990;77:1019-38
Myosite Myosite + PID Myosite ou PID

Qu’est ce que le SAS ??? Marguerie C Love LA Hervier B & al. Hervier B & al.
(Revue)
Q J Med 1990 Medicine 1991 Autoimmunity R Curr Rheum Rep 2013
29 p dt 19 Jo-1 47 p dt 35 Jo-1 233 p Jo1-PL 12-7 833 p
PID 79 89 77 (puis 83) 85
Myosite 83 100 57 (puis 73) 75
Articulations 90 94 63 68
Raynaud 93 62 42 45
Fièvre - 87 28 51
Hyperkératose mains - 71 19 25
S. Gougerot-Sjögren 59 - 35 42
S. Sclérodermie 72 - 28 40
%

Syndrome de chevauchement (Connectivite Mixte)
Alarcon-Segovia D, J Rheum 1989;16:328-34

Association d’atteintes cliniques mixtes et d’un marqueur sérologique particulier auto-immun
Qu’est-ce que le SAS ?
Venables PJW, Rheumatol 1996 ;35:305-8

Classification des Myopathies Inflammatoires
PID$ MDA'5!
TIF'1γ!Cancer$
Jo#1%
PL#7%
PL#12%
EJ%
OJ%
Ku$
PM'Scl$
U1'RNP$ SAE!
HMG'coA$R$
SRP$
Mi'2!
NXP2!
Myosite%de%
Chevau
chem
ent$ D
ermatom
yosite%
%
Myopathie%
nécrosante%AI%Myosite%à%Inclusion%
5’'NucleoAdase$
KS%
Zo%
YRS%
ARS%

Hervier B. Curr Rheum Rep 13
Avec anticorps anti-synthétases
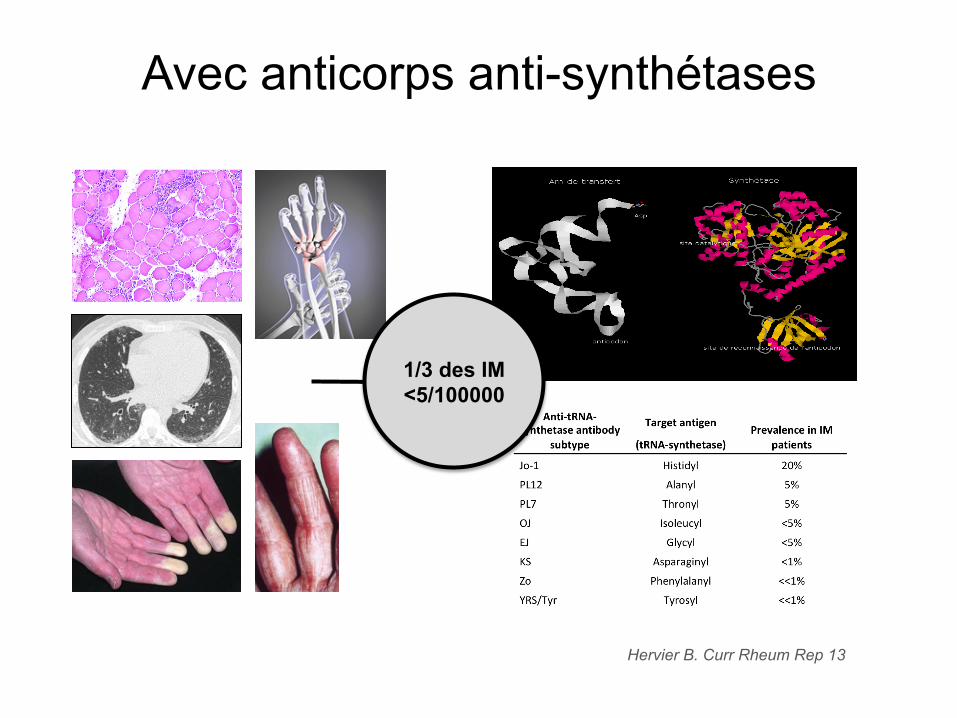
Hervier B. Curr Rheum Rep 13
1/3 des IM <5/100000
Avec anticorps anti-synthétases

Quels sont les AC anti-ARNt-synthétase ?

Quels sont les AC anti-ARNt-synthétase ?
40-75% des Myosites avec PID Fathi M, ARD 2004;63:297-301
Yoshifuji H, Autoimmunity 2006;39:233-41
Schnabel A, Semin Artritis & R 2003;32:273-84
Jusqu’à 6 fois > aux autres AC antisynthétases
vs PL7-PL12 Méta-Analyse JNLF
Brouwer R, ARD 2001;60:116-23

• Disease initiated in the lungs… Hystidyl-tRNA synthetase (HRS) expressed in the lungs > regenerating
muscle Casiola-Rosen L. JEM 05
Association of HLA DRB1*03, Smocking & Jo-1 Chinoy H. ARD 12
HRS cleaved by Granzyme B in the lung (new epitopes) Levine SM. AR 07
HRS acts as chemokine attracting CCR5+cells Howard X.JEM 02
• Are anti-Jo1-Ab deleterious ? Correlation of anti-Jo-1-Ab level & disease activity Richards TJ. AR 09
• ARS Σ & SLE similarities… Clinical association Hervier B Autoimmun Rev. 12
IFNα & BAFF Lundberg IE. Autoimmunity 10 Krystufkova O, ARD 09
Physiopathologie SAS

ARS Σ
Immunization of C57BL/6 mice by HRS triggers IM
HRS (60-90) binds to LPS These complexes syndergistically
activate TLR2 & TLR4
HRS-induced IM is MyD88-dependent HRS signals via TLR2 & TLR4
HRS signaling is redundant (immunization of TLR2-/-, TLR4-/- sKO vs dKO BL/6)
Fernandez I. JI 13 Harlow L. Innate Immun
12 Soejima M AR 11 Albrecht I.
In vitro Stimulation of PBMCs by HRS
Detection of specific LT4 cells (Blood & BAL)
TH1 profile (CCR5 & CXCR3) Epitope from HRS is a.a. 0-60

Portrait Robot Myosite de chevauchement (Connectivite Mixte) AC anti-Jo-1+

Quand penser au SAS ?
Myosite Signes
Extra-musculaires

Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue

Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue
Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue

Données immunologiques : Quand penser au SAS ?
Anticorps anti-nucléaires (Fluorescence Indirecte Hep-2)
Kit de Recherche de Spécificité (Anti-DNA, -ENA)
ELISA/ Luminex
Anti-Jo-1
Homogène Cytoplasmique
Anti-Ribosome
Anti-SRP
Anti-PL7/ PL12
Anti-SSA

Données immunologiques : DOT-Myosite Quand penser au SAS ?
EJ-OJ
Quand demander cet examen ?
Cet examen est-il à répéter ?
Et si cet examen est négatif ?

Données immunologiques : AC-non spé ? Quand penser au SAS ?
Anti-RNP
Anti-SSA 52 kDa
Anti-SSA 60
Anti-SSB
Anti-Scl-70/ Centromères
Anti-ADN et anti-Sm
Facteur Rhumatoïde*
Anti-CCP*
* Tous les patients n’ont pas été testés
3%
38%
17%
5%
5%
3%
15%
< 5%
Hervier B & al. Curr Rheum Rep 2013
Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue

Signes cutanés: Mains de Mécaniciens Quand penser au SAS ?
Bachmeyer C, B J Dermatol 2007;156:192-4
Sensibilité… ? 25%
Spécificité ?
Anti-PM/Scl Lega JC J Rheum 2010;37:1000-9
Diagnostics différentiels

Autres signes cutanés… Sclérodermie Quand penser au SAS ?
Doigts Boudinés
Sclérodactylie
Raynaud
Télangiectasies
Iconographie, Thanks to Pr E. Hachulla

Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue

Symptômes Rhumatologiques Quand penser au SAS ?
Arthralgies > Arthrites 68%
Non spécifiques, Petites et Moyennes articulations
Le plus souvent modérées Marguerie C, Q J Med 1990
Parfois érosives… Rarement luxantes Späth M, J Neurol 2004
Meyer O, ARD 2009;68:252-3
Associations aux anti-CCP
N articulations dlses >, Erosions >, Biologics > Meyer A & CRI, 17p (soumis)

Quand penser au SAS ?
Myosite
Dermatologue Immunologiste
Rhumatologue Pneumologue

Atteintes pulmonaires Quand penser au SAS ?
Voies aériennes
Pneumopathies d’inhalation
Atteintes des Muscles respiratoires
Pleurésie
HTAP
Pneumopathies infiltrantes diffuses

Pneumopathie infiltrante diffuse Quand penser au SAS ?
AIGUE
Insuffisance respi: 47%
Myosite + rare (31%)
Tillie-Leblond I, Thx 2008;63:53-9
CHRONIQUE 85%
Retardée 30-50%, dans les 2 ans
Asymptomatique
Toux, (RGO), Dyspnée graduelle !! Masquée si déficit moteur !!
PRONOSTIC +++ 17% 59% 24% Marie I. Arthr care Res 12

Pneumopathie infiltrante diffuse Quand penser au SAS ?
AIGUE
Insuffisance respi: 47%
Myosite + rare (31%)
Tillie-Leblond I, Thx 2008;63:53-9
CHRONIQUE 85%
Retardée 30-50%, dans les 2 ans
Asymptomatique
Toux, (RGO), Dyspnée graduelle !! Masquée si déficit moteur !!
PRONOSTIC +++
+ Sévères (NYHA/ EFR/ PaO2) LBA neutrophilique
TDM: Combinaisons (bilatéral, DAD) Amélioration initiale mais + progression à 1 an

PIC PINS PO
Associations étendues : Condensations + Réticulations + V. dépoli
Rayon de Miel Verre dépoli Opacités linéaires Condensations Alvéolaires
Bronchectasies par traction
Opacités réticulaires
DAD

Patterns radiologiques : Quel intérêt ?
Nicholson AG, AJRCCM 200; 162:2213-7
Devant une ILD apparemment isolée… FPI (UIP) est plus sévère A pattern identique, sévérité « idiopathique » > PID de connectivite

Facteur pronostique ? TDM : Répartition des pattern similaires PM/DM Pattern PIC - corrélé à la progression PID Marie I. Arthr Care R. 12
- non corrélé à la mortalité Hervier B. Autoimmunity Rev 12
Extension ? Mc Donald SL. Radiology 01 (Idiopathic)
Goh N. AJRCCM 08 (SSc) PINS corrélé aux modifications de traitement Stanciu R. J Rheum 12

EFR: Profil Evolutif : 1 seul travail prospectif !
Fathi M. Arthr Care Res 08 (23 p)
EFR
« L’évolution de la PID ne peut être prédite au Diagnostic ! »
Test marche 6’
Formes progressives : Cinétique > 1e Exploration
Formes « stables ou lentes » Durée de suivie suffisante
Interstitial lung disease. ILD was defined as restrictivelung function impairment (TLC and DLCO !80% pre-dicted) and/or radiologic signs consistent with ILD onchest radiograph or HRCT.
Statistical analyses. Student’s t-test was used for com-parison of continuous variables (age, lung function, andtime since onset of symptoms) between 2 groups, andanalyses of variance were used for comparison of groupsimproving, deteriorating, or remaining unchanged. Maxi-mum likelihood chi-square test was used for discrete vari-ables (sex, PM/DM diagnosis, prevalence of pathologiclaboratory tests, and symptoms). For long-term evaluationof ILD prevalence, last observation for lung function andradiology was carried forward.
RESULTS
Patients’ characteristics. Fourteen patients (9 women)diagnosed with PM and 9 patients (6 women) with DMwith a mean " SD age of 56.2 " 14.8 years were included.The main clinical and laboratory characteristics are sum-marized in Table 1. Increased serum CK levels were notedin all patients.
All patients were treated with high doses of glucocorti-coids with slowly tapering doses during the first year.Additional immunosuppressive agents administered ac-cording to the treating physician’s decision are listed inTable 1. In some patients, combination therapy with !2immunosuppressive agents was used.
Two patients died 10 days and 18 months, respectively,after diagnosis. In both cases the cause of death was pro-gressive respiratory insufficiency with radiologic signs ofILD. At the time of death, disease activity was consideredto be high despite treatment with high doses of glucocor-ticoids and IV cyclophosphamide in both patients. At au-topsy, pneumonia, focal pulmonary hemorrhage, and
acute lung infarction were observed in one case, andchronic alveolitis in the other.
In the remaining 21 patients, disease activity and func-tional status according to physician’s global assessmenthad improved at the time of followup evaluation com-pared with the initial presentation. Serum CK and serumCRP levels had returned to normal in all tested patients.Relapses with temporary elevated serum CK levels werenoted during the followup period in 3 patients. At the finalevaluation, 15 patients were still receiving glucocorticoidtreatment with a prednisolone dosage of 2.5–30 mg/day,and 14 were treated with other immunosuppressive agentssuch as azathioprine (n # 5), cyclophosphamide (n # 3),methotrexate (n # 5), cyclosporine (n # 2), and IV immu-noglobulin monthly or every 3 months (n # 2). Completeremission without need of further immunosuppressivetreatment was achieved in 1 patient.
PFTs. PFTs were available in 21 patients; 2 declined toundergo the investigation. Repeated PFTs were performedin 18 patients (2 had died, 3 declined investigation). Thechanges in TLC and DLCO between the first and last fol-lowup examination are shown in Figure 1.
Eight patients had restrictive lung function impairmentat the initial investigation (TLC !80% predicted) with amean TLC of 68% predicted (range 53–79%), and they allhad reduced DLCO (mean 50% predicted, range 37–58%predicted). Followup investigation in 6 of these patientsshowed that TLC and DLCO had become normal in 4 pa-tients, and DLCO had normalized in 2. TLC and DLCO
remained unchanged in 1 and deteriorated in 1.Seven patients had isolated reduction of DLCO (!80%).
After correction for the reduced lung volume (DLCO cor-rected for alveolar volume), there was no significant dif-ference in DLCO between patients with restrictive lungimpairment and those with isolated reduction of DLCO
(mean 59% and 62%, respectively). Followup data were
Figure 1. Results of initial and followup total lung capacity (TLC) and diffusing capacity forcarbon monoxide (DLCO) measurements and total chest radiography score in patients withpolymyositis and dermatomyositis. Each line represents 1 individual patient.
680 Fathi et al

Facteur pronostique ?
EFR : données controversées DLCO initiale + basse dans les PID progressives Marie I. Arthr Care R. 12 DLCO initiale non corrélée - aux modifications de traitement (DMARD) Stanciu R. J Rheum 12 - à la mortalité Hervier B. Autoimmunity Rev 12

Au total Facteurs pronostic progression PID
CLINIQUE Age >
PID clinique* Aiguë*
Marie I. AR 11 Marie I. AR 11
Marie I. J Rheum 98
EFR CVF & DLCO + basses*
Marie I. AR 11 Kang EH. Rheum 05
TDM Verre dépoli* Schnabel A. SAR 03
LBA Neutrophilique* Schnabel A. SAR 03 Marie I. J Rheum 98
Une seule et même entité ? 4 études
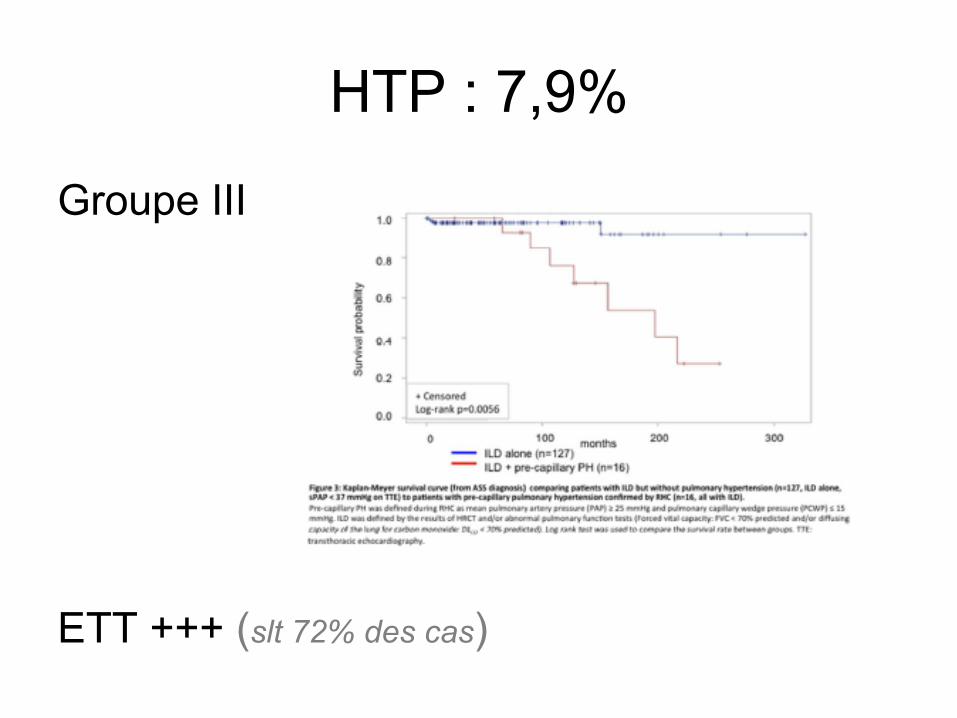
HTP : 7,9%
Groupe III ETT +++ (slt 72% des cas)

Myosite Particulière ?
Quand penser au SAS ?

Myosite du SAS Sévérité ?
Sémiologie myogène aspécifique
Moins sévère que les Myosites non SAS ? Love LA, Medicine 1991
Spectre très étendu: Absente Asymptomatique Déficitaire
Moins fréquente et/ou moins sévère selon l’AC anti-Σ ? PL7: Yamasaki Y, A&R 2004, PL12: Kalluri M, Chest 2009

Myosite du SAS Histologie particulière ?
HISTOLOGIE 1 seule série comparative, 11p Mozaffar T, J Neurol Neurosurg Psychiatry 2000;68:472-8
Infiltrat Périmysial prédominant et quasi-exclusif
Atrophie périfasciculaire
Nécrose et régénération discrète
IF: non spécifique
DM ou PM ??? Marguerie C, Q J Med 1990 Love LA, Medicine 1991
Hirakata M, A&R 1991 Brouwer R, ARD 2001
Ni DM ni PM !

Aspect de polymyosite avec infiltration des éléments cellulaires
musculaires par des Lymphocytes CD4+ et CD8+. Répartition
inhabituelle des sous-types lymphocytaires et macrophagiques.
Cette étude histologique confirme l’existence d’un processus inflammatoire atypique. Il n’existe aucun argument habituel pour une dermatomyosite. Néanmoins, le tableau n’est pas celui habituellement rencontré au cours des polymyosites primitives, et évoque donc la possibilité d’une connectivite beaucoup plus diffuse.!
Myopathie inflammatoire et nécrosante non typable
d’après les élément histo-enzymologiques et
immunochimiques habituels.
Aspect histologique inhabituel fait de lésions
inflammatoires lymphocytaires essentiellement
périmysiales et de phénomènes de myopathie
nécrosante.
Confirmation du diagnostic de polymyosite floride,
active, avec formule nécrose régénération et des
infiltrats lymphocytaires perivasculaires et dans le fascia.
Conclusion: P r o c e s s u s i n fl a m m a t o i r e m u s c u l a i r e mé l a n g e a n t d e s a s p e c t s i m m u n o -pathologiques classiquement décrits dans les dermatomyosites mais également dans les polymyosites.

Myosite du SAS Histologie particulière ?
HISTOLOGIE 1 seule série comparative, 11p Mozaffar T, J Neurol Neurosurg Psychiatry 2000;68:472-8
Infiltrat Périmysial prédominant et quasi-exclusif
Atrophie périfasciculaire
Nécrose et régénération discrète
IF: non spécifique
DM ou PM ??? Marguerie C, Q J Med 1990 Love LA, Medicine 1991
Hirakata M, A&R 1991 Brouwer R, ARD 2001
Ni DM ni PM !

Critères de Dalakas 03:
DM/PM : 50/50 !
Critères 118th ENMC workshop 04:
>>> DM
Correspondance diagnostique entre les 2 classifications : 50% !!
Résultats

1. Modifications histologiques correspondant au pattern Dermatomyosite
Atteinte Perifasciculaire: • Atrophie périfasciculaire (45%) • Nécrose / Régéneration (66%)
Résultats

MAIS… association avec un infiltrat endomysial entourant voire envahissant les fibres musculaires (polymyosite)
Résultats

Critères primaires 1- Lésions myocytaires de topographie péri-fasciculaire prédominante (incluant en particulier la nécrose, la régénération et l’atrophie)
2- Fragmentation périmysiale (visible sur une coloration par le trichrome et l’histoenzymologie pour les PAL) 3- Signes d’activation locale du complément ( marquage sarcolemmique de C5b9 de topographie péri-fasciculaire)
Critères secondaires 4- Marquage myocytaire diffus pour HLA-I, avec un renforcement péri-fasciculaire 5- Inflammation périmysiale pouvant s’étendre dans l’endomysium péri-fasciculaire

Myosite du SAS: Quand penser au SAS ?
« En l’absence de myosite franche clinique/ EMG»
En cas d’Histologie « atypique »/ « frustre »
Quelle signification physiopathologique ? Formes plus précoces ?
Quel Intérêt : Pronostique ?

Autres Symptômes
• Digestifs : RGO – Œsophage sclérodermique
• Cardiaques : 2,7% = idem autres IIM C. Dieval (in preparation), 10p
Pas lié à un Ab particulier Péricardite ?
Labirua-Iturburu, Medicine 2013 Hervier B, ERJ 2011
• Sclérodermie… • Gougerot-Sjögren • Cancer ? Troyanov I, 2005 & Hervier B, 2012 vs Marie I, 2012

Comment expliquer l’hétérogénéité clinique ?
Existe t’il des différences
selon l’AC ?
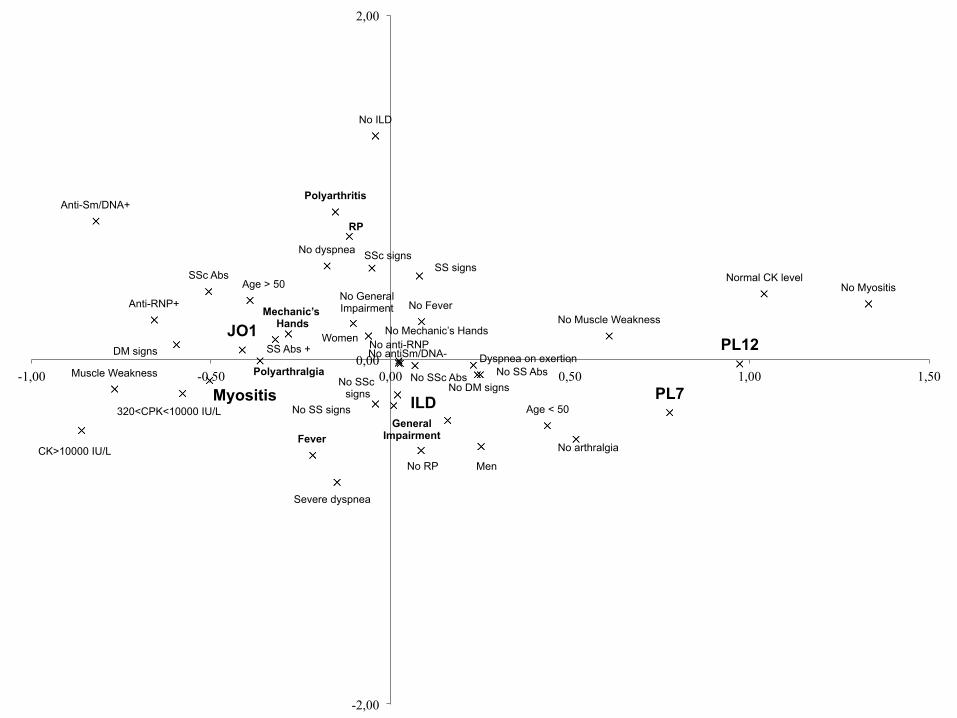
No arthralgia
Polyarthralgia
Polyarthritis
Age < 50
Age > 50
Men
Women
Muscle Weakness
No Muscle Weakness
Severe dyspnea
Dyspnea on exertion
No dyspnea
RP
No RP
Mechanic’s Hands
No Mechanic’s Hands
No Fever
Fever General
Impairment
No General Impairment
No SS signs
SS signs
SS Abs +
No SS Abs
SSc signs
No SSc signs
SSc Abs
No SSc Abs
DM signs
No DM signs
320<CPK<10000 IU/L
CK>10000 IU/L
Normal CK level
JO1 PL12
PL7
Anti-RNP+
No anti-RNP
Anti-Sm/DNA+
No antiSm/DNA-
No Myositis
Myositis ILD
No ILD
-2,00
0,00
2,00
-1,00 -0,50 0,00 0,50 1,00 1,50

Anti-Jo1 (n=160)
Anti-PL12 (n=48)
Anti-PL7 (n=25) p -value
Demographic dataMean Age at onset (years) 46.6 ± 14.6 51.8 ±16,7 50.5 ± 17.6 0.09Men 45 (28%) 15 (31%) 8 (32%) 0.87Mean Follow-up (months) 90 ± 72 44 ± 35 56 ± 65 < 0.001
Phenotype at DiagnosisMyositis 118 (74%) 19 (40%) 11 (44%) < 0.001ILD 107 (67%) 42 (88%) 20 (80%) 0.014Polyarthralgia 113 (71%) 22 (46%) 11 (44%) < 0.001Polyarthritis 38 (24%) 6 (13%) 3 (12%) 0.13Raynaud's phenomenon 68 (43%) 23 (48%) 8 (32%) 0.43Mechanic's hands 36 (23%) 4 (8%) 5 (20%) 0.09Fever 38 (24%) 18 (38%) 10 (40%) 0.07General Impairment 32 (20%) 20 (42%) 13 (52%) < 0.001
Prévalence PID selon AC anti-synthétases
Hervier B. Autoimmunity Rev 12
PID + fréquente et plus souvent « Isolée »

Sévérité PID selon AC anti-synthétases
Hervier B. Autoimmunity Rev 12
Anti-Jo1 (n=160)
Anti-PL12 & anti-PL7
(n=73) p -value
Characteristics of the ILD# n=128 n=65Dyspnea on exertion 84 (66%) 46 (71%) 0.47Severe Dyspnea (NYHA = III/IV) 34 (27%) 19 (29%) 0.69UIP 10 (9%) 7 (11%) 0.67NSIP 87 (77%) 50 (77%) 0.86OP 13 (12%) 5 (8%) 0.42Mean Forced Vital Capacity (%) 75 ± 21 66 ± 18 0.012Mean DLCO (%) 52.7 ± 18 49 ± 21 0.24
n=178°
n=163

Sévérité PID selon AC anti-synthétases
Hervier B. Autoimmunity Rev 12
Anti-Jo1 (n=160)
Anti-PL12 & anti-PL7
(n=73) p -value
Characteristics of the ILD# n=128 n=65Dyspnea on exertion 84 (66%) 46 (71%) 0.47Severe Dyspnea (NYHA = III/IV) 34 (27%) 19 (29%) 0.69UIP 10 (9%) 7 (11%) 0.67NSIP 87 (77%) 50 (77%) 0.86OP 13 (12%) 5 (8%) 0.42Mean Forced Vital Capacity (%) 75 ± 21 66 ± 18 0.012Mean DLCO (%) 52.7 ± 18 49 ± 21 0.24
n=178°
n=163
Survie PL7/PL12 < Jo1
months
Patients with anti-Jo1 Patients with anti-PL7/12

Gravité MYOSITE ? Anti-Jo1 (n=160)
Anti-PL12 &
anti-PL7 (n=73) p -value
Characteristics of the Myositis + n=135 n=34Muscle w eakness 87 (64%) 15 (44%) 0.03Mean CK level (IU/L) 5248 ± 92831292 ± 1691 0.015Patients w ith CK > 10000 IU/L 21 (16%) 0 (0%) 0.014Myopathic changes on EMG n=154 72 (64%) 14 (56%) 0.15Myofiber Necrosis/Regeneration on Muscular Biopsy
n=118 58 (64%) 13 (59%) 0.59
Characteristics of the ILD# n=128 n=65Dyspnea on exertion 84 (66%) 46 (71%) 0.47Severe Dyspnea (NYHA = III/IV) 34 (27%) 19 (29%) 0.69UIP 10 (9%) 7 (11%) 0.67NSIP 87 (77%) 50 (77%) 0.86OP 13 (12%) 5 (8%) 0.42Mean Forced Vital Capacity (%) 75 ± 21 66 ± 18 0.012Mean DLCO (%) 52.7 ± 18 49 ± 21 0.24
n=178°
n=163

• Jo1 +++, Polymorphe • PL7 et PL12 proches, Poumon +++
• AC RNP et Sm/DNA: groupe à part, Raynaud +++ Alors que SS et SSc, également répartis Comment les classer ???

Conclusion
• PL7 = PL12 = Maladie du Poumon et PID plutôt plus sévère que Jo1
• Jo1 = Myosite plus fréquente et plus sévère
Confirmation: Hamaguchi M, PLOS ONE 2013
Influence sur le pronostic Aggarwal R, ARD 2014
Autres explications à l’hétérogénéité ?


Portrait Robot
2006
2002
2005
2003
Renouveler les évaluations
Myosite de chevauchement (Connectivite Mixte) AC anti-Jo-1+

2001
1992
1999
1998
Et alors ?

Pourquoi penser au SAS ?
1. Connaissance, GHM 2. Mesures symptomatiques (RHD, TTT) 3. Prédire 4. Traiter de façon adéquate

Pourquoi penser au SAS ? Prédire: Survie
Le SAS n’est pas statistiquement associé aux Cancers Douglas WW, AJRCCM 01 Troyanov YT, Medicine 05
Le pronostic est pulmonaire (pas de = Jo-1+ vs Jo-1-)
Myosite PID
Marie I, Arthritis Care Research 02 Park JH, AJRCCM 07

Pourquoi penser au SAS ? Prédire: Réponse thérapeutique
Meilleure réponse initiale aux Corticoïdes, Indépendamment de l’AC
Myosite « chronique » en cas d’AC anti-Σ, donc Corticothérapie longue Troyanov Y, Medicine 2005;84:231-49

SAS: Comment traiter ?
Aucune étude prospective comparative 68 à 100% des patients reçoivent IS
Kalluri M, Chest 09, Gomard-Menneson Ann NY acad Sci 07 Späth M, J Neurol 04, Tillie-Leblond I, Thorax 08
Prise en charge peu spécifique Gravité Bollus Corticoïdes IV/ IV Ig
Corticothérapie Longue durée + IS d’emblée ? Troyanov Y, Medicine 2005
Choix de l’IS: Classique > Biothérapie Myo
site

SAS: Comment traiter la PID ?
Sévérité ? NON
+/- Corticothérapie p.os
Bilan à 3 mois
Cinétique
OUI
Corticothérapie IV 6x Endoxan IV
Amélioration ?
NON OUI
+IS p.os: AZA/ MMF > MTX Roos N, J Ph E T 07
Saravanan V, Rheumatol 06
Traiter tôt Marie I, J Rheum 2001
Score Extension TDM > 20%
CVF < 70%
Goh NSL, AJRCCM 08
Progression
Cf Sclérodermie Hoyles RK, AR 06
Même ttt
Bilan /6mois
Amélioration

Questions Thérapeutiques 1. IS d’emblée ? 2. Tacrolimus > Endoxan ?
Petites séries rétrospectives…(n=15-20) Labirua-Iturburu A, Clin Exp Rheum 2013
3. RTX ? RIM study: réponse musculaire = 83% des réfractaires. Mauvais design…
Oddis CV, A&R 2013 Les patients réfractaires avec anti-Jo1 font partie de ceux qui
répondent le mieux (HR 3.08, p<0.01) Aggarwal R, AR 2014
4. Moins de Corticoïdes ??? Condon MB ARD 2013

Synthèse Pour le diagnostic de SAS
Etre attentif aux Myosites frustres
Signes extra-musculaires (interrogatoire):
Toux, Dyspnée / RGO, Arthralgies
(inspection): Mains ++
Rénouveler car métachrone
Lire attentivement les résultats AAN
Demander un « DOT-Myosite »
TDM thorax « systématique » + EFR
et ETT
Réévaluer car métachrone
Pronostic
Pulmonaire ++
Traitements agressifs, rapidement
? Corticoïdes + IS d’emblée