polymyosite/dermatomyosite: du diagnostic au traitement polymyosites.pdf · myopathies...
TRANSCRIPT

Eric Hachulla
Service de Médecine Interne, Hôpital Huriez, CHU Lille
Polymyosite/Dermatomyosite: du diagnostic au traitement

Myopathies inflammatoires idiopathiques
Polymyosite
- Syndrome des anti-synthétases
- Myopathie nécrosante immune (immune-mediated
necrotising myopathy)
Dermatomyosite
- Dermatomyosite amyopathique (Clinically amyopathic
dermatomyositis = CADM)
- Myosite associée au cancer (MAC)
Myosite à inclusion
Dermatomyosite juvénile
Myosite associée aux connectives
Divers: Granulomateuse, à éosinophile, focale, orbitaire,
macrophagique, myofasciite

• Prévalence en France : environ 3000 à 6000 patients (0,5 à
1/10000 hab)
• 2 pics de fréquence : 2ème enfance (5-15 ans, DM), 40-60 ans
• Forme familiale exceptionnelle
Myopathies inflammatoires idiopathiques

Syndrome musculaire
• Atteinte des ceintures scapulaires et pelviennes, bilatérales et
symétriques, sans spécificité musculaire
• Installation progressive ou brutale, au 1er plan ou au 2nd plan
• Simple gêne fonctionnelle ou déficit musculaire sévère (de la
réduction de l’activité sportive à l’impossibilité de lever les bras,
monter une marche d’escalier, soulever la tête de l’oreiller)

Myalgies
• Plus fréquentes dans les formes aiguës, parfois totalement
absentes, spontanées ou provoquées à la palpation
• Rare augmentation oedémateuse du volume musculaire (parfois
muscles fermes ou indurés)
• Formes évoluées : amyotrophie voire rétraction notamment chez
l’enfant
Syndrome musculaire

• Possible atteinte de la musculature distale dans les formes évoluées
• Musculature paravertébrale --> camptocormie
• Musculature respiratoire : muscles intercostaux, diaphragme
• Musculature striée du pharynx et 1/3 supérieur de l’œsophage (au moins
1/4 des cas, intérêt de la manométrie oesophagienne) : dysphonie,
dysphagie, troubles de la déglutition et fausses routes
• Muscles oculo-moteurs en principe épargnés sauf forme pseudo-
myasthénique ou myasthénie associée
Syndrome musculaire

Evaluation musculaire
• Echelle fonctionnelle
• Testing musculaire
• CPK
• EMG
• Scanner et surtout IRM musculaire
• Histologie musculaire


Atteinte cardiaque
• 10 à 15 % des cas sur des critères cliniques, 1/3 à 3/4 des cas selon
les méthodes d’exploration : ECG, holter, échocardiographie,
scintigraphie au thallium, IRM
- troubles du rythme, de la conduction, de la repolarisation
- atteinte vasculaire coronaire ou microcirculatoire
- myocardite
Troponine > CPK MB
Recherche systématique
par ECG, holter,
écho-cardiographie
IRM cardiaque si doute

Manifestations cutanées
• Erythro-œdème : territoire photosensible, paupières supérieures
coloration liliacée, érythème en lorgnette
• Papules de Gottron : face dorsale des mains en regard des
articulations, plus rarement coudes, genoux, tendon d’Achille
• Erythème péri-unguéale
• Signe de la manucure
• Erythro-œdème télangiectasique et pigmentaire avec atrophie
• Lésions purpuriques, nodulaires ou bulleuses
Evolution indépendante de l’atteinte musculaire




D’apres Dalakas M et al Lancet 2003
Dermatomyosites: physiopathologie

DM
certaine DM probable
DM
amyopathique
DM sine
dermatitis
possible 1. Critères cliniques :
Critères positifs :
•a. Age de début : enfant ou adulte
b. Déficit moteur bilatéral symétrique prédominant proximal, touchant
plus les fléchisseurs que les extenseurs de la nuque OUI OUI NON OUI
c. Rash cutané caractéristique de dermatomyosite: éruption liliacée (±
oedémateuse) des paupières supérieures, signe de la manucure,
éruption érythémato-squameuse de la face d’extension des MCP et
IPP, coude, genoux (papules de Gottron), érythème des zones photo-
sensibles. OUI OUI OUI NON
Critères ne faisant pas retenir le diagnostic de dermatomyosite
a. Déficit moteur évoquant une myosite à inclusions (asymétrique,
sélectif, biceps, cubito-antérieurs, fléchisseurs des doigts, quadriceps
et tibio-antérieurs)
b. Déficit des muscles oculo-moteurs, dysarthrie isolée, atteinte
prédominante sur les extenseurs de la nuque.
c. Myopathie toxique, endocrinopathie (hypo ou hyperthyroïdie,
hyperparathyroïdie), amylose, histoire familiale de dystrophie ou de
neuropathie OUI OUI NON OUI
2. Elévation des CPK
OUI OUI NON OUI
3. Autres critères paracliniques :
EMG : activité de fibrillation de repos, réduction de la durée et de la
contraction volontaire plus ou moins diminution de l’amplitude des
potentiels d’unité motrice, présence d’indentations sur les potentiels
d’unité motrice (critères d’exclusion : salves pseudo-myotoniques,
augmentation de la durée ou de l’amplitude des potentiels moteurs,
diminution du recrutement des potentiels moteurs) OUI OUI NON OUI
ou IRM musculaire : hypersinaux intramusculaires (oedèmes diffus
ou focaux en séquence STIR ou T2 saturation de graisse) ou OUI NON ou OUI
ou Auto-anticorps spécifiques des myosites dans le sérum Parfois Parfois NON Parfois
4. Biopsie musculaire :
a. atrophie périfasciculaire OUI NON OUI
b. Dépôts du complexe d’attaque membranaire (MAC) dans les
capillaires musculaires, OU réduction de la densité capillaire, OU
inclusions tubulo-réticulaires dans les cellules endothéliales en
microscopie électronique, OU expression du MHC-I sur les fibres
périfasciculaires OUI OUI NON ou OUI
c. infiltrat inflammatoire T périvasculaire, périmysial OUI OUI NON
4. Biopsie cutanée :
Diminution de la densité en capillaires, dépôts de MAC au niveau de
la paroi des vaisseaux capillaires de la jonction dermo-épidermique et
au niveau des keratinocytes. OUI
Critères de classification ENMC 2004 (Hoogendijk JE. Neuromuscul Disord 2004 ;14 :337-45)
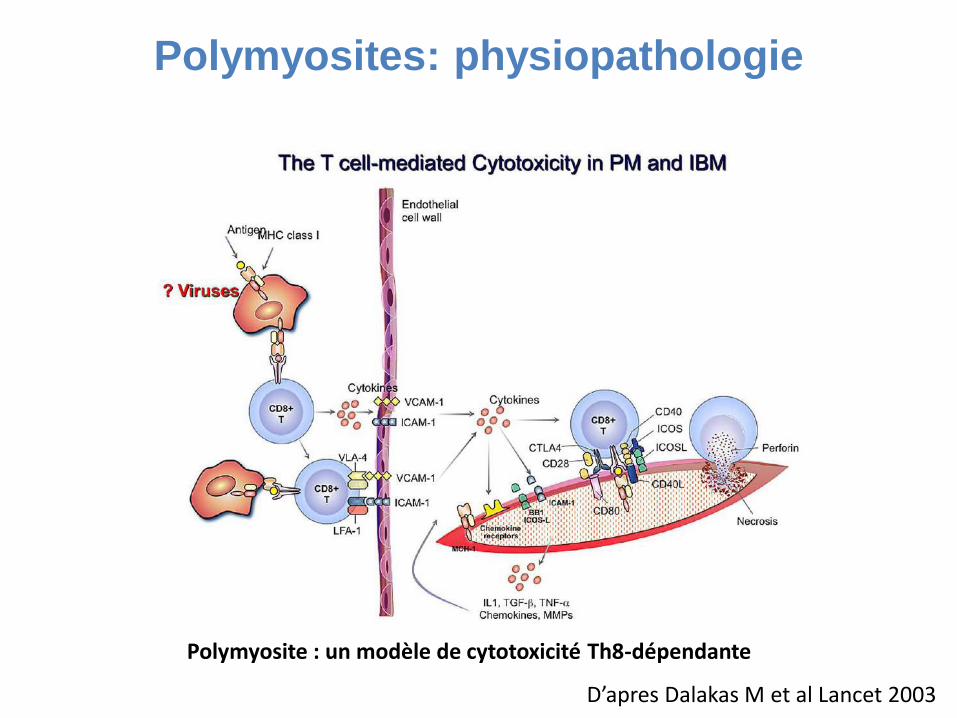
Polymyosite : un modèle de cytotoxicité Th8-dépendante
Polymyosites: physiopathologie
D’apres Dalakas M et al Lancet 2003

PM certaine PM probable 1. Critères cliniques :• Critères d’inclusion :
- Début généralement après 18 ans
- Déficit moteur bilatéral symétrique prédominant proximal, touchant plus les fléchisseurs
que les extenseurs de la nuque
OUI OUI
Rash cutané caractéristique de dermatomyosite NON NON
• Critères d’exclusion :
- Déficit moteur évoquant une myosite à inclusions (asymétrique, sélectif, biceps, cubito-
antérieurs, fléchisseurs des doigts, quadriceps et tibio-antérieurs)
- Déficit des muscles oculo-moteurs, dysarthrie isolée, atteinte prédominante sur les
extenseurs de la nuque
- Myopathie toxique, endocrinopathie, amylose, histoire familiale de dystrophie ou de
neuropathie OUI OUI
2. Elévation des CPK
OUI OUI
3. Autres critères paracliniques :
EMG : activité de fibrillation de A11 réduction de la durée et de la contraction volontaire
plus ou moins diminution de l’amplitude des potentiels d’unité motrice, présence
d’indentations sur les potentiels d’unité motrice (critères d’exclusion : salves pseudo-
myotonique, augmentation de la durée ou de l’amplitude des potentiels moteurs,
diminution du recrutement des potentiels moteurs) OUI
ou IRM musculaire : hypersignaux intramusculaires (œdèmes diffus ou focaux en
séquence STIR ou T2 saturation de graisse) OUI
ou Auto-anticorps spécifiques des myosites dans le sérum Parfois
4. Biopsie musculaire :
(a) Infiltrat inflammatoire lymphocytaire T endomysial entourant et envahissant les fibres
musculaires non nécrotiques (tunnellisation myocytaire) OUI
(b) Lymphocytes CD8 entourant les fibres musculaires non nécrotiques sans les envahir
ou expression myocytaire diffuse du CMH de classe 1 OUI
(c) Atrophie périfasciculaire NON NON
(d) Dépôt du complexe d’attaque membranaire C5b9 dans les capillaires musculaires ou
réduction de la densité capillaire ou inclusion tubulo-réticulaire dans les cellules
endothéliales en ME ou expression du CMH de classe 1 par les fibres périfasciculaires NON NON
(e) Infiltrat inflammatoire T périvasculaire périmysial NON NON
(f) Nécrose musculaire prédominante, les cellules inflammatoires sont rares, possible
dépôt du complexe d’attaque membranaire NON NON
(g) Vacuole bordé, fibres rouges déchiquetées, fibres cytochromoxydase négative,
suggérant une myosite à inclusions NON NON
(h) Dépôt du complexe d’attaque membranaire C5b9 dans le sarcolemme des fibres
musculaires non nécrotiques et autres éléments en faveur d’une dystrophie musculaire NON NON On retient le diagnostic de polymyosite de manière certaine si :
- tous les critères cliniques sont présents à l’exception du rash cutané
- élévation des CPK
- critères histologiques incluant a et excluant c, d, g, h
On retient le diagnostic de polymyosite probable si :
- tous les critères cliniques sont présents à l’exception du rash cutané
- s’il y a élévation de CPK
- si présence d’autres critères paracliniques (un des trois)
- si critères histologiques incluant b et excluant c, d, f, g, h
Critères de classification ENMC 2004

Détection des auto-anticorps
Screening par l’immunofluorescence sur lignées Hep-2
Identification de l’auto-anticorps par technique complémentaire
Immunodiffusion ELISA Immunodot Immuno-empreinte
Image : Labodia http://www.labodia.com/fr/ana/Atlas/index.htm

Autoanticorps spécifiques des myosites
identification des cibles par immunodot

(n = 384) p140: 4,2%
P155/140: 17,2%
p140/56:
1,8%
MAA: 14,2%%
Undiferencited16,9%
Negative
JO-1: 13,5%
Non Jo-1 ASA 2,7%
SRP: 1,4%%
Mi-2: 13,1%
SAE: 6,6%
Dermatomyosite et auto-anticorps
Betteridge ZE, Gunawardena H, McHugh NJ. Arthritis Res Ther. 2011;13:209

Jo-1; 24,2%
Non Jo-1; 3,4%
Negative; 13,1%
Unidentified; 22,1%
MAA; 30,7%
p155/140; 1,0%
p140/56; 0,1%
p140; 0,3%SAE; 0,1% Mi-2; 0,2%
EIF; 0,9%
SRP; 2,8%
N = 458
Betteridge ZE, Gunawardena H, McHugh NJ. Arthritis Res Ther. 2011;13:209
Polymyosite et autoanticorps

Myosites associées aux cancers
- Femme 32 ans opérée d’un cancer du sein - mutations dans le gène BRCA1

Le plus souvent, il s’agit de DM avec un risque x 2,4 à 7,7
Ovaire, poumon, tube digestif, sein et nasopharyngien
Présence d’anticorps anti-p155/140
Spécificité : 89%
Sensitivité : 70%
Valeur prédictive négative : 93%
OR pour le diagnostic : 23.2
Présence d’anticorps anti-p155/140 chez un patient avec DM justifie
un dépistage systématique large à la recherche d’un cancer qui doit
être renouvelé si le bilan initial est négatif.
Myosites associées aux cancers
Selvat-O’Callaghan. Curr Opin Rheumatol 2010

Autoanticorps spécifiques des myosites
identification des cibles par immuno-
empreinte (western blot)
Gunawardena H. Curr Opin Rheumatol 2008; 20: 675 Gunawardena H. Rheumatology 2008;47:324

Myosites associées aux cancers 61 patients (issus d’une cohorte de 565 patients) 9 PM et 52 DM
p155/140
40,2%
p140/56
4,1%MAA : 8,2%
Unidentified 13,1%
Negative
8,2%
Jo-1 : 4,1%
Non Jo-1 ASA
0,8%
SRP : 1,6%
Mi-2
11,5%
SAE
8,2%
Betteridge ZE, Gunawardena H, McHugh NJ. Arthritis Res Ther. 2011;13:209

Myosites associées aux cancers

Myosites associées aux cancers
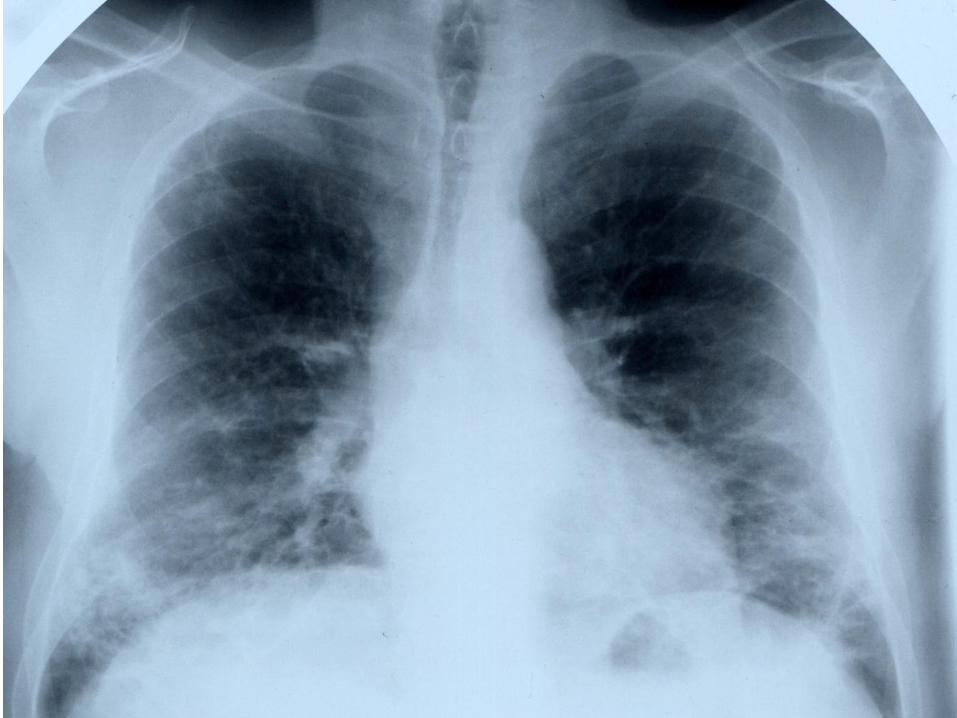

• Dyspnée progressive aiguë ou subaiguë
• Toux sèche et persistante
• Râles crépitants prédominant aux bases
• Hippocratisme digital peu fréquent
• Possible pneumothorax ou pneumomédiastin par rupture de
lésions bulleuses
Pneumopathie interstitielle diffuse

Pneumopathie interstitielle diffuse
• Prévalence variable selon les méthodes d’exploration utilisées, environ
40 % sur les données autopsiques
• Peut précéder de plusieurs mois voire de plusieurs années les
manifestations musculaires ou cutanées
• Une fois/3 concomitante
• Une fois/3 retardée parfois de quelques mois (réévaluation pulmonaire à
6 et 12 mois)


Syndrome des anti-synthetases

Syndrome des anti-synthetases Données cliniques Myosite (PM>DM) Pneumopathie interstitielle (50-80 %) Arthrites (50 – 90 %) Phénomène de Raynaud (60 %) Mechanics hands (70 %) Fièvre (80 %)

Syndrome des anti-synthetases Données cliniques Myosite (PM>DM) Pneumopathie interstitielle (50-80 %) Arthrites (50 – 90 %) Phénomène de Raynaud (60 %) Mechanics hands (70 %) Fièvre (80 %)
Auto-anticorps Aminoacyl-tRNA synthetase ciblé
Prévalence
Jo-1 Histidine 25 – 30 %
EJ Glycérine < 2 %
PL-7 Thréonine 3 – 4 %
KS Asparagine < 2 %
OJ Isoleucine < 2 %
PL -12 Alanine 3 – 4 %
Zo Phénylalanine < 2 %

- Patients with anti-SRP antibodies usually develop severe myopathy
with high CK levels, specific pathological features upon muscle biopsy
and usually poor response to corticosteroids
- Association aux atteintes cardiaques
non-inflammatory necrotizing myopathy
MHC class I expression to necrotic myocytes
C5b-9 onto endomysial capillaries
Arlet JB. Neuromuscular Disorders 2006; 16:334
Myopathies nécrosantes
(immune-mediated necrotising myopathy)

Images : Professeur René louis Humbel et l’équipe du LLIP, Luxembourg
Myopathies nécrosantes à anti-SRP
Autoantibodies Target autoantigen Autoantigen function Clinical phenotype
Anti-SRP SRP 6 polypeptides and ribonucleoprotein 7SLRNA
Intracytoplasmic protein translocation (endoplasmic reticulum)
Severe necrotizing myopathy

Mammen AL. Arthritis Rheum. 2011;63:713
Myopathies nécrosantes aux statines

• Le plus souvent les symptômes musculaires régressent rapidement en quelques semaines ou quelques mois après l’arrêt des statines
• l’exposition aux statines induit l’expression des auto-antigènes de 100 et 200 Kd sur des cultures cellulaires
• HMG CoA reductase =auto-antigène 100 Kd
Mammen AL. Arthritis Rheum. 2011;63:713
Myopathies nécrosantes aux statines

• 45/750 (6%) des myopathies inflammatoires de la cohorte du John’s
Hopkins Myositis Centre
• déficit musculaire proximal, une élévation des CPK (jusque 30 000
et plus)
• myopathie nécrosante immune (immune-mediated necrotizing
myopathy – IMNN)
• expression CMH classe I sur les fibres musculaires non nécrotiques,
auto-anticorps et réponse habituelle aux IS
Mammen AL. Arthritis Rheum. 2011;63:713
Myopathies nécrosantes aux statines

Auto-anticorps et myosites
Auto-anticorps spécifiques
des myosites (Myositis
« Specific » Autoantibodies)
- Anti-tRNA synthetases
- Anti-Mi-2
- Anti-signal recognition
particle (SRP)
- Anti-SAE
- Anti-p155
- Anti-p140
- Anti-CADM-140
- Anti-HMGCR
- Anti-EIF-3
Auto-anticorps associés
aux myosites (Myositis
Associated Autoantibodies)
- Anti-PM-Scl
- Anti-U1RNP
- Anti-Ku
- Anti-U3RNP

Anti-PM/Scl
Image : Labodia http://www.labodia.com/fr/ana/Atlas/index.htm
Myosites à anticorps anti-PM-Scl

Myosites à anticorps anti-PM-Scl
Anticorps dirigés contre un complexe macromoléculaire du
nucléole
Retrouvés chez 5 à 25 % des patients avec myopathies
inflammatoires ; s’observent chez les PM et/ou DM
Présents chez 43 à 88 % des patients avec PM/DM et syndrome
de chevauchement sclérodermie
Sévérité de l’atteinte pulmonaire
85 % de formes chroniques, rechute fréquente
Marie I. Br J Dermatol 2010;162:337

Seulement 10 % des patients ont des anticorps anti-PM-Scl
Augmentation du risque d’atteinte cardiaque (OR = 3,0 ; IC 95 %
= 1,3 – 34,9) ; risque d’insuffisance cardiaque congestive,
fraction d’éjection IVG < 60 %, d’arythmie ou de troubles
conductifs
Augmentation du risque de crise rénale : OR = 3,0 [1,3 – 34,9]
Deux types d’anticorps plus fréquemment retrouvés : anti-PM-
Scl et anti-RNP
Ranque B. Scand J Rheumatol 2010;39:498
Myopathies associées à la SSc

• Lésions inflammatoires:
– Infiltrat de cellules mononuclées et/ou nécrose
• Perimysial, périvasculaire et/ou endomysial
• Lésions de sclérodermies:
– Fibrose (périmysiale)
– Microangiopathie
– Parfois isolées: pronostic plus sombre
Histologie: hétérogène
Myopathies associées à la SSc

Anti-RNP
Myosites à anticorps anti-U1-RNP
Image : Labodia http://www.labodia.com/fr/ana/Atlas/index.htm

Myosites à anticorps anti-U1-RNP
Anticorps classiquement associés au syndrome
de Sharp ou connectivites mixtes, mais retrouvés
aussi au cours du syndrome de Sjögren, de la
sclérodermie
Retrouvés chez 5 à 60 % des patients ayant une
myosite et une connectivite associée
Association particulière avec l’atteinte pulmonaire
interstitielle, la polyarthrite et l’atteinte
neurologique

• 20 600 avec AAN +
• 32 patients anti-Ku (détection + puis confirmation par immunoblot)
• 28 dossiers analysables : 4 H et 24 F, âge moy 49 ans
• 9 patients (32%) = myosite inflammatoire dont 7 syndromes de chevauchement
– 4 sclérodermie-PM
– 2 LES-PM
– 1 Sjögren-PM
– 1 Sclérodermie-Myosite à inclusion
– 1 PM-Myosite à inclusions
Rigolet, A. Revue de Médecine Interne 30, 2009
Myosites avec anti-Ku

• Taux de mortalité PM et DM avant les corticoïdes =
50 % à 1 an
• Depuis l’ère des corticoïdes, survie
globale à 10 ans de l’ordre de 80 à 90 %
MAIS
Corticothérapie = Traitement de référence
Durée de suivi (mois)
Probabilité de survie
JDM = Dermatomyosite juvénile (n = 9) ;
OM = Myosite sur syndrome de
chevauchement (n = 29) ;
PM = Polymyosite (n = 75) ;
DM = Dermatomyosite (n = 42) ;
CAM = Myosite associée à un cancer (n = 7)
Fujisawa T. J Rheumatol 2005;32:58

• 25 % des patients ne répondent pas à une corticothérapie par voie générale
• 50 % des patients sont dépendants des corticoïdes
• 25 à 50 % des patients développent des complications liées à l’utilisation des corticoïdes
• 2/3 des patients ont une évolution chronique et la moitié d’entre eux ont une forme évolutive à rechute
Callen JP. Lancet 2000;355:53 ;
Marie I. J Rheumatol 2001;28:2230
Corticothérapie = Traitement de référence

• Sont considérées comme corticorésistantes les formes n’étant
pas améliorées au plan clinique et biologique après 6 semaines d’un
traitement corticoïde 1 mg/kg
Se discute alors un traitement immunosuppresseur
Méthotrexate, azathioprine, cyclophosphamide, ciclosporine,
chlorambucil : 50 à 75 % de répondeurs sur la base d’une
amélioration de la force musculaire ou sur la base d’une épargne
cortisonique
PM/DM corticorésistantes
Villalba L. Curr Opin Rheumatol 1996;8:544

Rider LG. JAMA 2011;305:183
Traitement des PM corticorésistantes :
alternatives thérapeutiques