quality risk management -...
TRANSCRIPT

Quality Risk Management
According to ASTM E2500: Standard Guide for Specification, Design and Verification for Pharmaceutical and Biopharmaceutical Manufacturing Systems and EquipmentBrian Evald Andreasen, May 2009
[email protected] telephone: +4530751381

Slide 2
Agendaen
Fra indbydelsen:•På mødet vil Brian Andreasen fra NNE Pharmaplan fortælle om hans erfaringer med at implementere standarden hos Pfizer globalt.
•Han vil give et overblik over indholdet i standarden sammenholdt med traditionel kvalificering,
•og der vil blive givet konkrete eksempler på RiskManagement og Verifikation.

Slide 3
NNE Pharmaplan, my workplace
• Over 80 years of experience in the pharma and biotech industries
• Spanned over 3 continents across Europe, North America and Asia
• Workforce 2008: More than 1500
• Turnover 2007: DKK 1.444M, €194M, $249M
• ISO 9001 certified since 1995; certified worldwide in 2008
• ISO 14001 certified since 2003

Slide 4
Global reach - local knowledge

Slide 5
Where is the best Quality?
Traditional
C&Q
New Risk Based Verification Approach
David Dolgin, Abbott2008 Washington ASTM C&Q

Slide 6
The benefits from E2500 are incredible……
Moves away from ‘paper quality’ and rigid documentation practices -to product quality and patient safety.
Faster project executionFocus on patient safetySignificantly reduces documentationEnhanced process understandingSaves time and resourcesHigher production yield (OEE)Improved quality at less costs - better use of expertiseCompliance with Health Authorities expectationsto risk based approach
Thus, improved quality, major cost reduction (above 40%), faster to market and longer time to take the right decisions for investments.
…and p
roba
bly tr
ue

Slide 7
The Pfizer case
Pfizer is the largest pharmaceutical company in the world
They had with support from NP implemented a risk based verification system globally in the company according to the new ASTM E2500 standard.(Before Pfizer had a traditional C&Q approach according to ISPE C&Q guide Vol 5 similar to NN)
•By adapting the new risk based approach…Pfizer expect to:
• save 40% of all expenses to Commissioning and Qualification • Eliminate between 2 and 6 month of project execution time.
• Get improved quality level

Slide 8
ASTM!!!!What is that….?
• Large voluntary standards development organization
• Not-for-profit organization
• Established in 1898
• Purpose is to develop standards in response to market needs
• Technical standards for materials, products, systems, and services
• HQ in Philadelphia,• Sub-offices in London, Mexico City &
Beijing

Slide 9
ASTM Committee E55
• Established Spring 2003
• Development of standardized nomenclature and definitions of terms recommended practices, guides, test methods, specifications, and performance standards for the manufacture of pharmaceutical products.

Slide 10
Issued Standards (Pharma) :
• E2363-06a Standard Terminology Relating to Process Analytical Technology in the Pharmaceutical Industry
• E2474-06 Standard Practice for Pharmaceutical Process Design Utilizing Process Analytical Technology
• E2500-07 Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment
• E2503-07 Standard Practice for Qualification of Basket and Paddle Dissolution Apparatus
• E2537-08 Standard Guide for Application of Continuous Quality Verification to Pharmaceutical and Biopharmaceutical Manufacturing

Slide 11
• WK5930 Standard Practice for Risk Assessment and Risk Control as it Impacts the Design, Development, and Operation of PAT Processes for Pharmaceutical Manufacture (Gawayne Mahboubian Jones)
• WK5935 Standard Guide to Process Understanding (Jean Marie Geoffroy)
• WK9192 Standard Practice/Guide for the Application of Continuous processing in the Pharmaceutical Industry (Trevor Page)
• WK15151 Standard Guide on Sampling (Joep Timmermans)
• WK9182 Standard Guide for Verification of Process Analytical Technology (PAT) Control Systems (Bruce Davis)
• WK9191 Standard Guide for Multivariate Data Analysis Related to Process Analytical Technology (Chun Cai)
• WK13538 Standard Practice for Identification of Critical Attributes of Raw Materials in Pharmaceutical Industry (Marino Nebuloni) WK11898 Standard Practice for Real-time Release of Pharmaceutical Water for the Total Organic Carbon Attribute (Rich Godec)
• WK15778 Guide for Science-based and Risk-based Cleaning Process Development and Validation (Andrew Walsh)
• WK16888 Guide for Validation of PAT Methods (Jim Rydzak)
Work Items in Progress (E55.01)

Slide 12
Essence of the E2500
The scope and extent of quality risk management forspecification, design, and verification activities and documentation should be based on the risk to product quality and patient safety.
The level of effort, formality and documentation of the quality risk management process should becommensurate with thelevel of risk.

Slide 13
Important TermsCritical Quality Attributes (CQA)
Dissolution
Disintegration
Hardness
Contentuniformity
Apperance
A Critical Quality Attribute is defined as a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range or distribution to ensure the desired product quality (ICH Q8)
Slide 13
Slide 14
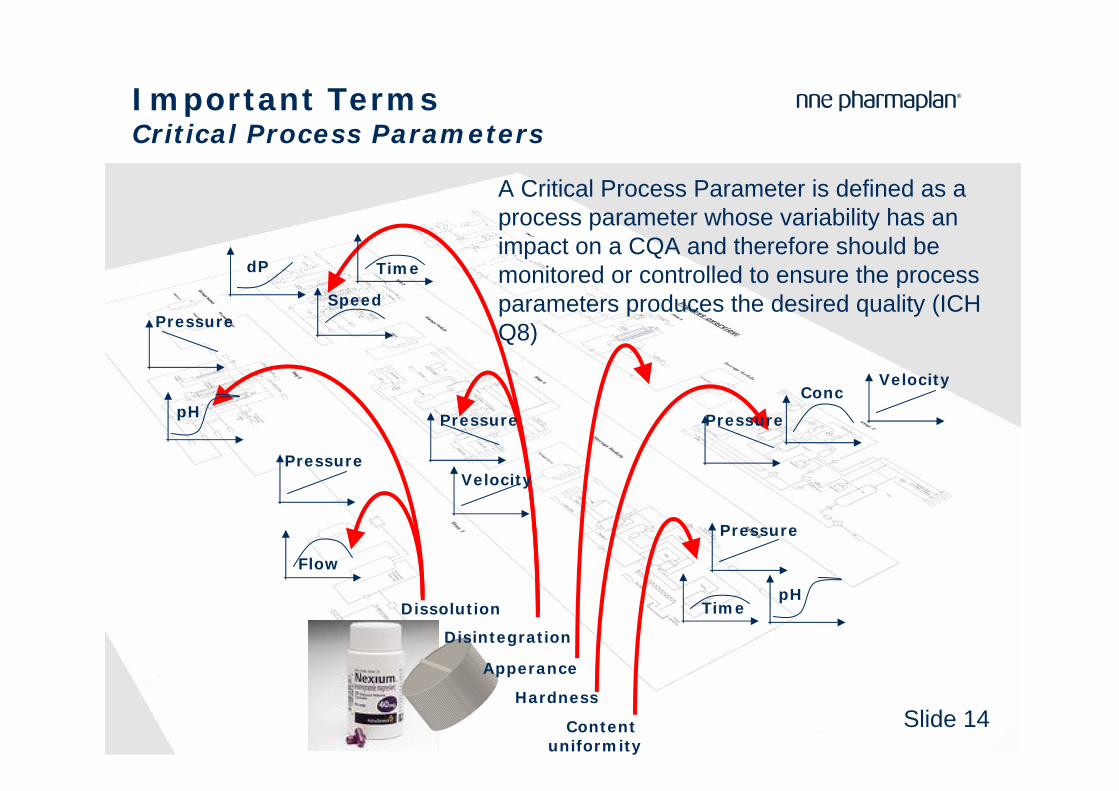
Important TermsCritical Process Parameters
Dissolution
Disintegration
Hardness
Contentuniformity
Apperance
Pressure
Flow
pH
Pressure
dP
Speed
Pressure
Velocity
Velocity
pHTime
Pressure
Pressure
Conc
Time
A Critical Process Parameter is defined as a process parameter whose variability has an impact on a CQA and therefore should be monitored or controlled to ensure the process parameters produces the desired quality (ICH Q8)
Slide 14

Slide 15
Important Terms Critical Aspects
Dissolution
Disintegration
Hardness
Contentuniformity
Apperance
Critical Aspects are defined as functions, features, abilities, and performance or characteristics necessary for the manufacturing process and systems to ensure consistent product quality and patient safety (ASTM E-2500-07)Sanitary
design
Vision system for monitoring
GMP
Batch documentation
Calibration
Filter integrityParameter alarms Non turbulent
agitor
Slide 15

Slide 16
Important TermsSubject Matter Experts (SME)
SME is defined by ASTM as:
• Individuals with specific expertise and responsibility in a particular area or field (for example, quality unit, engineering, automation, development, operations, and so forth).

Slide 17
The SME role & responsibilities
From E2500
• Subject matter experts should take the lead role in the verification of manufacturing systems as appropriate within their area of expertise and responsibility.
• Subject matter expert responsibilities include planning and defining verification strategies, defining acceptance criteria, selection of appropriate test methods, execution of verification tests, and reviewing results.

Slide 18
Risk Based ApproachThe old way
Impact Assessment – (systems & Components)
• Evaluating design for impacting the product quality, but no evaluation of the level of potential risk for product and patient.
• No Risk Mitigation• System and component level focused • Component Impact Assessment is conducted after design development.
Direct Impact
Indirect Impact
C NC
Slide 19
Risk Based Approach The new way
Ongoing and iterative Risk Management process
From CQA and CPP are defined to ………handover for operation.
Risk 1 ..…
Slide 19
Quality Risk Management –
Slide 20
Risk Based Approach The new way
• Definition of CQA, CPP & Critical aspects• Risk ranking• Risk mitigation:
Design solutionsControl strategyDesign ReviewSuppliers selectionSME allocationTestDocumentationetc
Risk 1 ..…
Slide 20
Quality Risk Management –

Slide 21
Quality Risk Management Determination of Critical Aspects
Risk 1 ..…Critical Aspects must be identified during the risk
Management process.
Critical Aspects are derived directly from CQA and the general GMP compliance issues.
Critical Aspects are subject to risk assessment and risk mitigation; if possible and Verification testing.
Slide 22
Well knownvendor
ExactCopy
EquipmentSimple
EquipmentCustomized
Complex processand
many interfaces
Customized
Newtechnology
Customized
Well knowntechnology
NewVendor
BadVendor
ManualEquipment
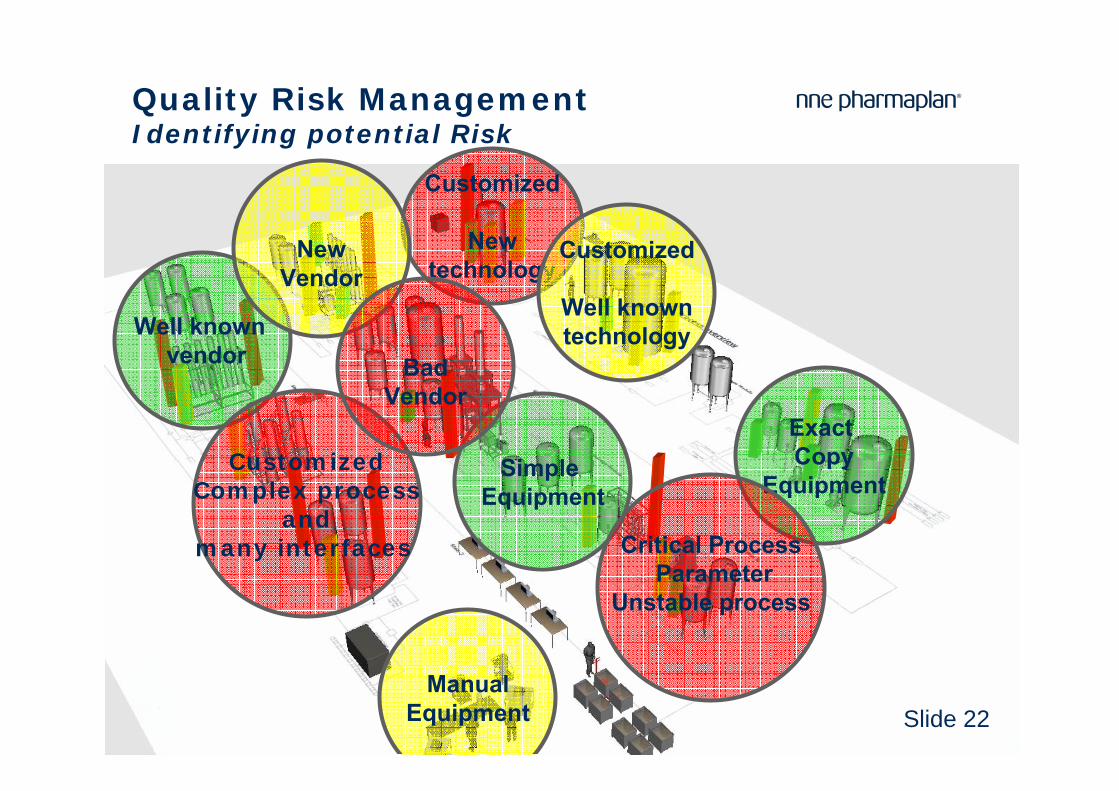
Quality Risk ManagementIdentifying potential Risk
Critical ProcessParameter
Unstable process
Slide 22

Slide 23
Quality Risk ManagementRisk Mitigation – Safe design solutions & Control strategy
PATMonitoring
EquipmentMonitoring
PATMonitoring
ManualMonitoring
Risk 1 ..…
Sanitary issue:Removal of pumps,
by using gravity
Slide 23

Slide 24
Quality Risk ManagementRisk Mitigation - Vendor Assessment
Documentation? Experienced employee?
New Technology?
Custom made
or bulk?
Interfaces
to other disciplines
or vendors ?
Risk 1 ..…

Slide 25
Quality Risk ManagementRisk Mitigation -Design Review (DR)
The DR is an important part of the risk management process, and DR must be focused on the risk aspects:
• Design meets the mitigated unacceptable riskderived from the risk assessment process.
• Planned Verification testing and acceptance criteria are sufficient and commensurate to level of quality risk.
• Design Meets relevant Critical aspects and ensure that Critical Process Parameters can be controlled to the desired level
• The Critical Quality Attributes (CQA) can be ensured by the proposed design
Risk 1 ..
…
√√√
√√
√√√√

Slide 26
Quality Risk ManagementUse of Vendors test results etc
Vendors test documentation may be used as part of the verification documentation, if the vendor is assessed, and there is evidence of:
• An acceptable vendor quality system,
• Vendor technical capability
• Vendor application of GEP such that information obtained from the vendor will be accurate and suitable to meet the purpose of verification.
If inadequacies are found then we have to mitigate potential risks by applying specific, targeted, additional verification checks or other controls rather than repeating vendor activities and replicating vendor documentation.
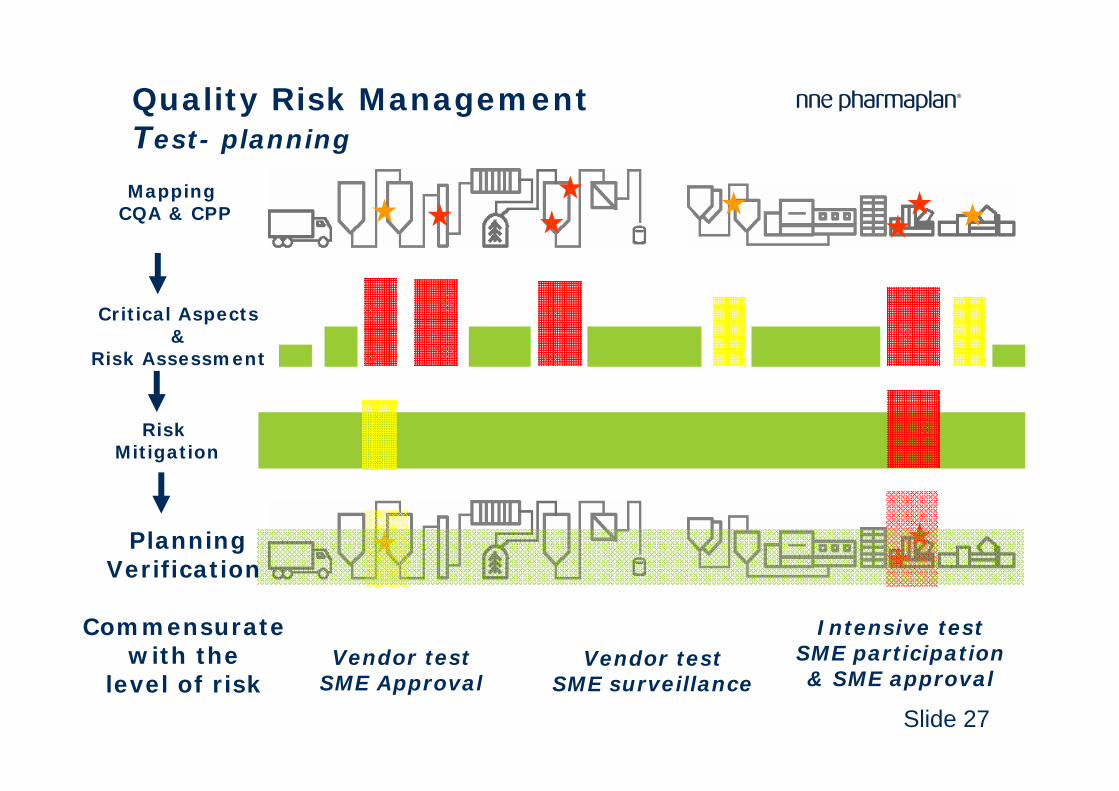
Slide 27
Quality Risk ManagementTest- planning
MappingCQA & CPP
Critical Aspects &
Risk Assessment
Risk Mitigation
PlanningVerification
Commensurate with the
level of risk Vendor test
SME Approval
Intensive testSME participation & SME approval
Vendor testSME surveillance
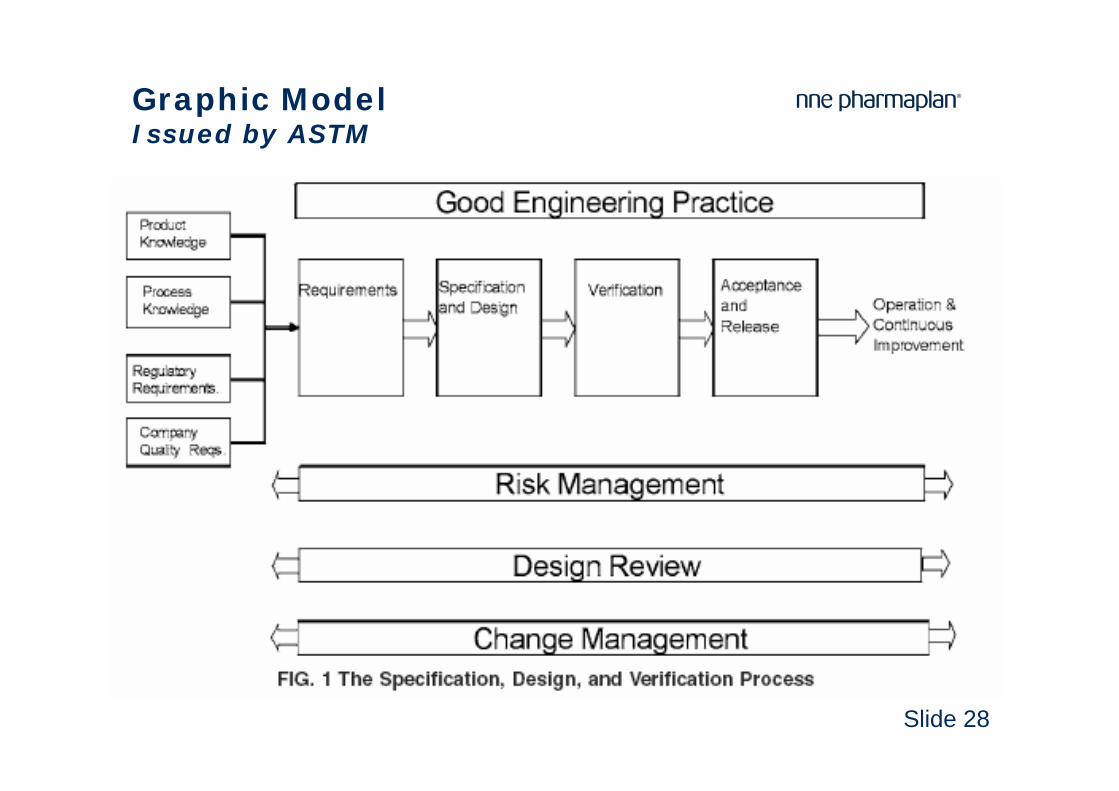
Slide 28
Graphic ModelIssued by ASTM

Slide 29
Quality Risk Management Process
CQACPP
Criticalaspects
RiskRanking
RiskMitigation
+New
ranking
Design+
DERissues
Vendorissues
Verification SMEInvolve-
ment
Link to illustration

Slide 30
Risk Mitigation
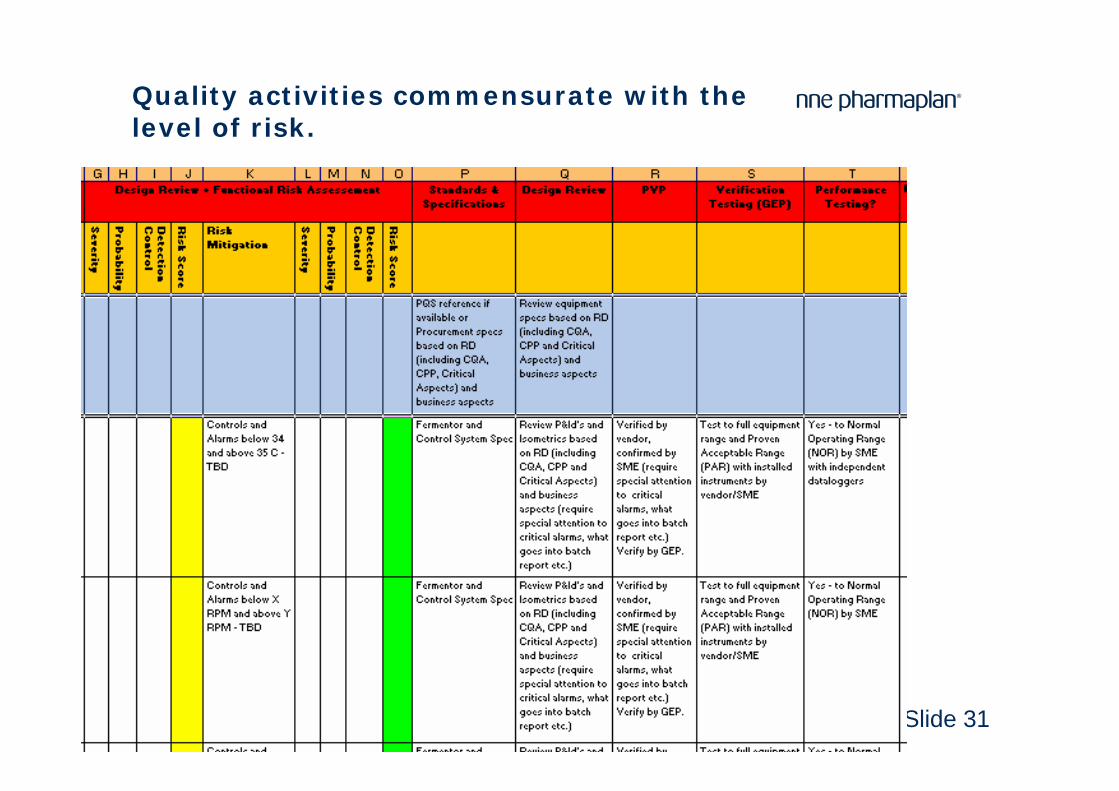
Slide 31
Quality activities commensurate with the level of risk.

Pfizer ASTM E2500 Transformation

Slide 33
Pfizer CaseASTM E2500 Transformation
• Transformation from C&Q to Verification based on ASTM E2500
• Part of a Lean project to streamline current practicesand enable more productioncapacity for the same investment capital
• Transformation project done by global Engineering & Quality team together withNNE Pharmplan
• 2 project phases:• Gap mapping & Scope
Setting• Document writing, review
and approval• Pilot implementation ongoing
Pfizer C&Q
Pfizer Verification

Slide 34
Pfizer CaseASTM E2500 Transformation - 2
• Combines E2500 with Quality by Design principles and Lean methods
• Integrates• Engineering• Automation• Quality• Maintenance etc.
• Based on Quality by Design Quality Risk Management principles (ICH Q8, Q9, Q10)
• Applies to new and legacymanufacturing systems
• Global Engineering Rollout on all sites is ongoing
Pfizer Verification

Slide 35
ASTM E2500 Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment
Quality by Design in NNE Pharmaplan:Transform C&Q to ASTM VerificationClient
• Pfizer• Global Engineering - for global use on all
manufacturing sites
Service• Transformation of Pfizer C&Q program to
the ASTM E2500 standard for Verification, for use globally
• Combined with a Pfizer Lean Program to streamline current practices and getmore value of investments – both onmoney and time
• 3 months delivery time• 5 consultants in NP Consulting Denmark
and USA plus global roll-out assistance
Deliverables• Full update of existing procedures to
ensure the harvesting of benefits of theVerification approach instead of thetraditional validation approach ofCommissioning and Qualification
What’s in it• Faster and more cost-effective
verification compared to traditional’validation’
DissolutionDisintegration
HardnessContent uniformity
Apperance
Customer Quote:”We selected NNE Pharmaplan for the workbecause these guys really understands the QbD principles in practice. You guys really got it!”
Client Case 2008

Slide 36
C&Q vs Verification Opportunities
C&
Q C
ost
#1 Fewer systems in Quality Unit scope
#5 Reduce Periodic Review Scope
100%#2 Leverage GEP & Subject Matter Experts (Less details)
#3 Leverage Supplier Documentation#4 Avoid repetition of tests etc.
Verifica
tion
Co
st
#6 Reduce number of docs retained etc.#7 Flexibility and Scalability
#8 Other

Slide 37
1st Generation – and the next:Face the Learning Curve
• Every new paradigm has its learning curve• 1st Generation is not always right• Example: From the Horse Cap to the 1st Generation Taxi
1st Generation Taxi 2nd Generation Taxi


Slide 39
Back up slides

A Brief Validation History

Slide 41
First FDA Validation Guideline1987
• “Process validation is
establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a
product meeting its pre-determined specifications and quality characteristics” GUIDELINE ON GENERAL PRINCIPLES OF
PROCESS VALIDATIONMAY, 1987 FDA CDER, CBER, CDRH

Slide 42
ISPE C&Q Baseline Guide 2001
• “The application of this C&Q process helps to ensure that the appropriate resources are applied to those systems and components within a facility that have the potential to affect product quality; and secondly, provides the rationale to reduce the scope of work for the qualification of the plant, while still ensuring compliance for the product(s).”
• Commissioning (i.e. GEP)• Qualification (i.e. QA)
ISPE Baseline Engineering Guide Commissioning & Qualification(Baseline Guide 5) 2001

Slide 43
New FDA Process Validation GuideDRAFT Nov. 2008
• “Effective process validation contributes significantly to assuring drug quality.
• The basic principle of quality assurance is that a drug should be produced that is
fit for its intended use
• This principle incorporates the understanding that the following conditions exist:
• Quality, safety, and efficacy are designed or built into the product.
• Quality cannot be adequately assured merely by in-process and finished-product inspection or testing.
• Each step of a manufacturing process is controlled to assure that the finished product meets all design characteristics and quality attributes including specifications. “
DRAFT
Stage 1 – Process DesignStage 2 – Process QualificationStage 3 – Continued Process Verification

Slide 44
C&Q vs. ASTM E2500
Old C&Q
New VerificationRisk Based ApproachASTM E2500
ProcessValidation
Engineering Change Management
QA Change Control
IQ & OQ
CommissioningEnhanced Design Review
Design Development
PQ
ProcessValidation
Engineering Change Management QA ChangeControl
Performance Testing
CommissioningDesign Review
Design Development
PT
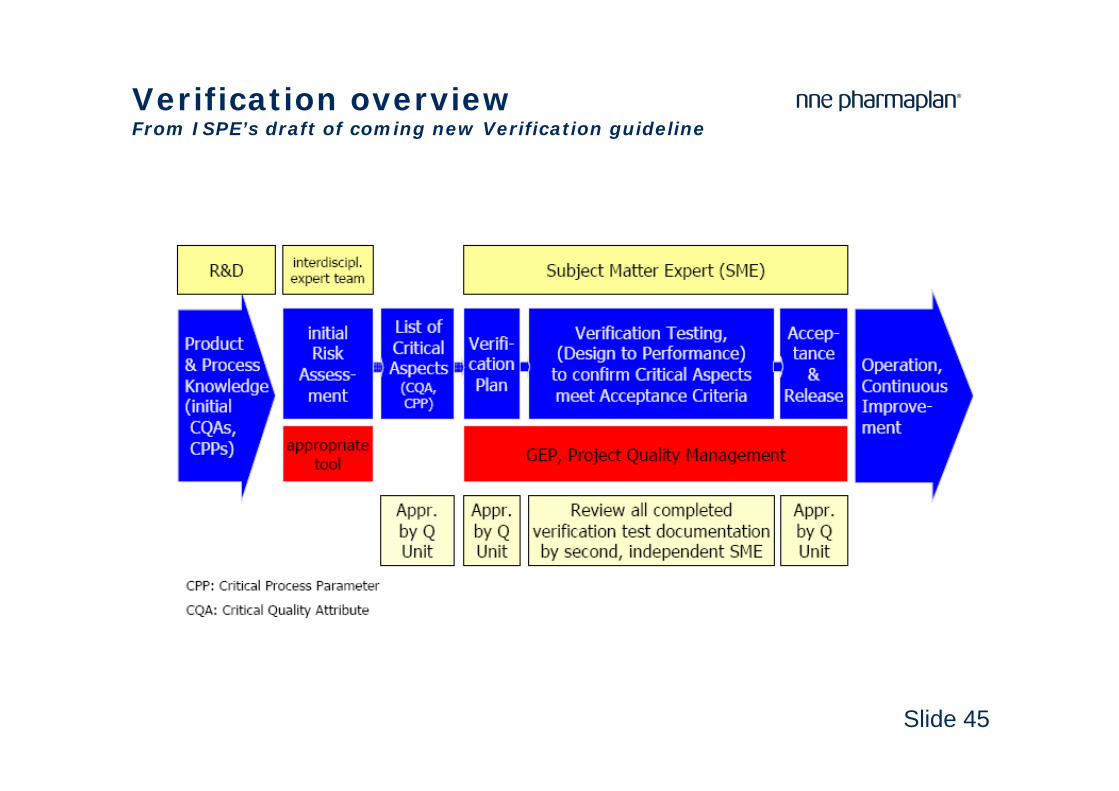
Slide 45
Verification overviewFrom ISPE’s draft of coming new Verification guideline

Slide 46
Planning Verification
From E-2500:
• The acceptance criteria and verification strategy should be documented in appropriate verification plans.
• The verification plan should define what constitutes acceptable documentation of subsequent verification activities.
• The verification plan should be developed and approved by subject matter experts.
• Verification plans for systems containing critical aspects should be approved by the quality unit.
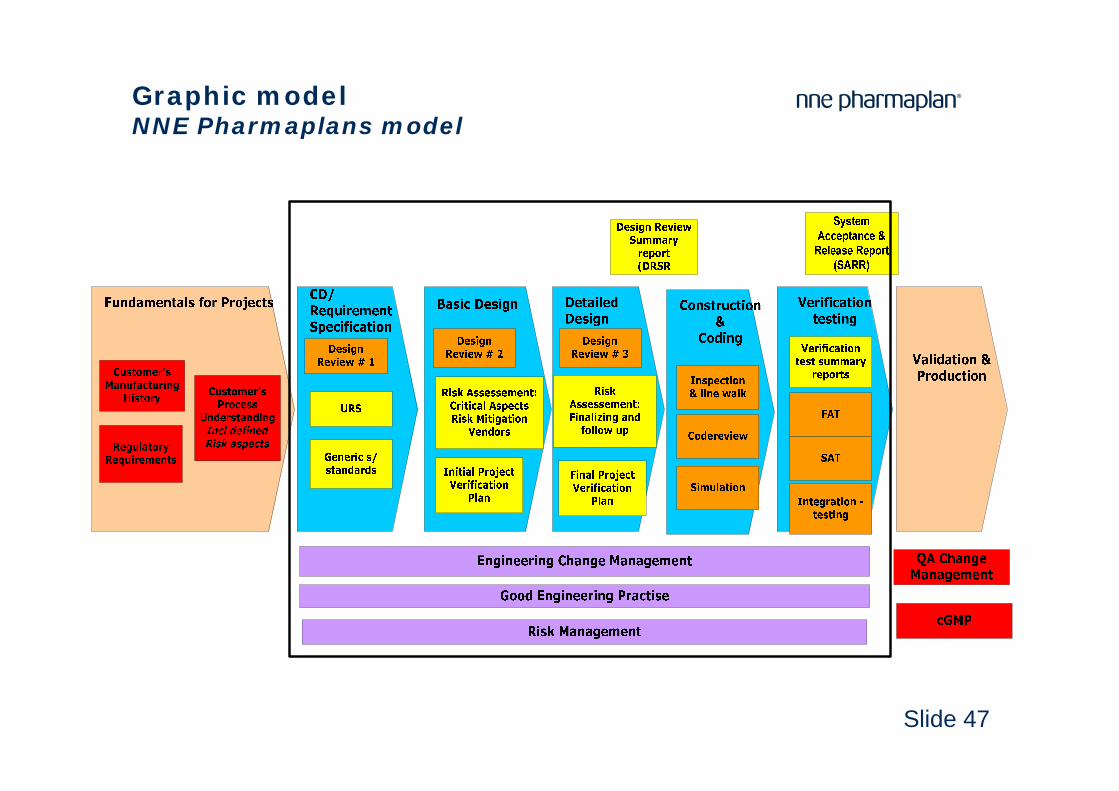
Slide 47
Graphic modelNNE Pharmaplans model

Slide 48
NNE Pharmaplan is involved in thenew Pharma Standard Settings
• NNE Pharmaplan is closely involved in thesetting of the new standards of thepharmaceutical industry
• ASTM Committee E55 on Manufacture of Pharmaceutical Products
• ISPE• PDA• We have participated in setting the new
standards of the pharmaceutical industryin ISA, GAMP, ISPE, PDA and ASTM for 20 years
• We call it ”More than Engineering…”