pulsed-uv light inactivation of cryptosporidium parvum
TRANSCRIPT

ORIGINAL PAPER
Pulsed-UV light inactivation of Cryptosporidium parvum
Soo-Ung Lee & Migyo Joung & Dong-Jin Yang &
Soon-Ho Park & Sun Huh & Woo-Yoon Park &
Jae-Ran Yu
Received: 31 December 2007 /Accepted: 28 January 2008 /Published online: 20 February 2008# Springer-Verlag 2008
Abstract Cryptosporidium parvum is an organism thatthreatens public health in the water industry. It is critical todevelop improved detection methods as well as disinfectionmethods for protecting against cryptosporidiosis, which iscaused by C. parvum. In this study, we investigated theability of pulsed-light irradiation at 200–900 nm toinactivate C. parvum. Absolute quantitative real-time PCRwas performed with cDNA made from total RNA extractedfrom C. parvum oocysts or HCT-8 cells infected with C.parvum oocysts in vitro. C. parvum oocysts in 100-mLquartz flasks were positioned 20, 30, and 40 cm from thelight source, and the duration of irradiation was either 5 or60 s. The reductions in oocyst viability (4.9 log10) andinfectivity (6 log10) were maximal when the C. parvumoocysts were irradiated 20 cm from the pulsed-light sourcefor 60 s, for which the UV dose was 278 mJ/cm2. Theminimum dose of pulsed-UV light required for effectivereduction in C. parvum infectivity (2 log10) was 15 mJ/cm2,
which was achieved by 5 s of irradiation at 30 cm from thelight source. This study confirmed that short-duration pulsed-UV light is an effective disinfection measure for C. parvum.
Introduction
The parasitic coccidian Cryptosporidium parvum is animportant cause of diarrheal illness in immunocomprom-ized persons and animals. Most infections are acquiredfrom water or food contaminated with infectious oocysts(MacKenzie et al. 1994; Nichols 2000). C. parvumrepresents a threat to public health in the water industrydue to the very low infection dose of oocysts (DuPont et al.1995; Okhuysen et al. 1999) and the high resistance tovarious disinfectants at their commonly used concentrations(Korich et al. 1990). Thus it is critical to develop improveddisinfection measures as well as an accurate detectionmethod for assessing the inactivation.
Traditional methods for detecting cryptosporidia in watermostly rely on immunofluorescence staining with anti-bodies against oocysts (Simmons et al. 2001). However,this method is not only time consuming and labor intensivebut also is not suitable for identifying live oocysts. Theviability of C. parvum could be assessed by detectingmRNA using reverse transcriptase (RT) polymerase chainreaction (PCR; Stinear et al. 1996), and its infectivity couldbe assessed by combined cell culturing and PCR techniquesusing specific primers and probes (MacDonald et al. 2002;Keegan et al. 2003). In vitro cell culture systems areconsidered to be an appropriate alternative to animalinfectivity assays (Shin et al. 2001; Rochelle et al. 2002).A quantitative real-time PCR (qPCR) method was recentlyused to quantitatively detect C. parvum in environmentalfecal samples and clinical samples (MacDonald et al. 2002;
Parasitol Res (2008) 102:1293–1299DOI 10.1007/s00436-008-0908-5
S.-U. Lee :M. Joung : J.-R. Yu (*)Department of Environmental and Tropical Medicine,Konkuk University School of Medicine,Chungju 380-701, Republic of Koreae-mail: [email protected]
D.-J. Yang : S.-H. ParkEn-Bio R&D Technology Center, Green EnTech Co.,153-787 Seoul, Republic of Korea
S. HuhDepartment of Parasitology, College of Medicine,Hallym University,Chuncheon 200-702, Republic of Korea
W.-Y. ParkDepartment of Radiation Oncology, College of Medicine,Chungbuk National University,Cheongju 361-763, Republic of Korea

Fontaine and Guillot 2003; Verweij et al. 2004). qPCR hasadvantages over classical PCR, such as time saving,providing quantitative information, and removing thenecessity for running an agarose gel after PCR.
We previously obtained highly reliable and reproducibleresults using the novel CP2 gene as a viability marker of C.parvum in qPCR (Lee et al. 2008). The function of the CP2gene has not been clarified, but its expression patternsuggests that it is involved in the invasion process and/oraffects the integrity of the parasitophorous vacuole mem-brane (O’Hara et al. 2004).
Pulsed-UV light from a xenon (Xe) flash lamp at 200–300 nm is an efficient form of UV radiation for inactivatingEscherichia coli and is considered a strong candidate foruse in water purification systems (Wang et al. 2005).Pulsed-UV has advantages over continuous-UV sources,such as low- or medium-pressure UV light sources insituations where rapid disinfection is required because of itshigh peak power delivered over a period of several seconds(Wang et al. 2005). In the present study, we evaluated theefficacy of pulsed-UV light in inactivating C. parvum.
Materials and methods
C. parvum oocyst purification
Oocysts of C. parvum were obtained from the feces ofC57BL female mice that had been orally infected withoocysts (2×106 oocysts/mouse; KKU isolate, Yu et al.2002) after being immunosuppressed by dexamethasonephosphate disodium salt (Sigma, St. Louis, MO, USA) pro-vided ad libitum in drinking water at a dosage of 10 mg/mL(Yang and Healey 1993). Mouse feces were examined bymodified Ziehl-Neelsen staining to confirm the oocystshedding (Casemore et al. 1985), and they were collectedin 2.5% potassium dichromate at 4°C. The oocysts werepurified according to the method described by Petry etal. (1995).
Pulsed-UV light irradiation
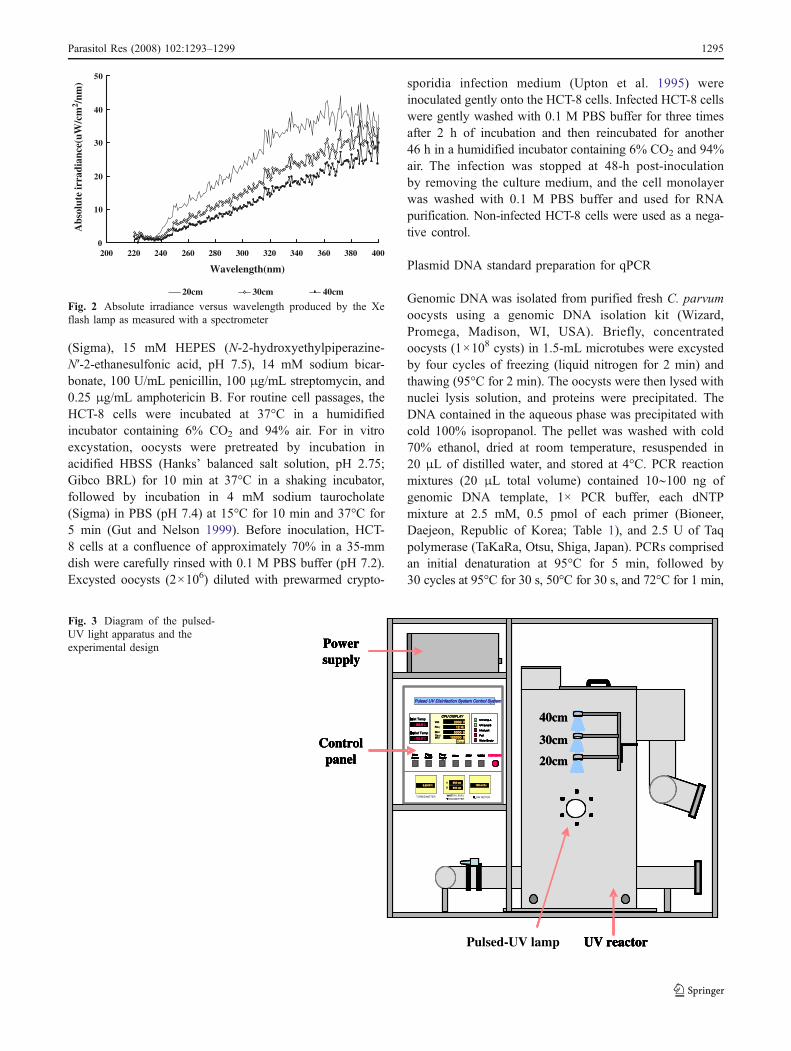
The pulsed-UV light source (PS2000, Green EnTech,Seoul, Republic of Korea) was a Xe-filled flash lampproducing a high-intensity diverging beam of polychromat-ic pulsed light. This pulsed-light source utilizes a solid-statepulsed power source with power compression technology totransfer stored electrical energy to the Xe flash lamp in ashort duration with a high peak power. The spectrum of theradiation from the Xe flash lamp as measured with aspectrometer (USB4000, Ocean Optics, Dunedin, FL,USA) ranged from 200 to 900 nm (Fig. 1), with emissionpeaks at 470 to 520 nm (Fig. 1). In these experiments, the
pulsed power source was operated at 2 kV, storing 200 J ofenergy in a 100-μF capacitor, with this energy beingdissipated in the flash lamp within 150 μs, therebyproducing a peak power of 2 MW per pulse.
The wavelength range of the power source covers theUV, visible, and infrared regions. The light source wasoperated at 10 pulses per second in each experiment. TheUV dose per pulse was measured from 220 to 400 nm in0.22-nm steps using a spectrometer (USB4000, OceanOptics) (Fig. 2). There was no discernable increase inwater temperature during each treatment.
C. parvum oocysts (3×107) were deposited into 40 mLof deionized water in a 100-mL quartz flask and placed in atap-water bath after tightly closing the cap that housed thepulsed-UV light flash lamp (Fig. 3). Quartz flasks wereplaced under the water at 20, 30, and 40 cm from theirradiation source and exposed to UV irradiation for 5 or60 s with continuous and gentle mixing at 25°C. Eachcondition of pulsed-UV light irradiation was repeatedthree times using three quartz flasks.
After the predetermined exposure durations, the sampleswere removed from the UV irradiation chamber andtransferred into 50 mL tubes (Corning, NY, USA). Eachquartz flask was rinsed with about 10 mL of deionizedwater, with the solution added to the same 50 mL tube andcentrifuged at 1,500×g for 10 min to concentrate theoocysts. Ten million oocysts were kept in deionized waterfor studying infectivity, and the remaining oocysts (2×107)were resuspended in RNAprotect cell reagent (Qiagen,Valencia, CA, USA) for qPCR.
In vitro cell culture and infection of C. parvum
Human ileocecal adenocarcinoma cells (HCT-8; KoreanCell Line Bank, Seoul, Republic of Korea) were incubatedin DMEM (Dulbecco’s modified Eagle’s medium, Sigma)with 10% heat-inactivated fetal bovine serum (Gibco BRL,Gaithersburg, MD, USA) supplemented with L-glutamine
0
10,000
20,000
30,000
40,000
50,000
60,000
0 100 200 300 400 500 600 700 800 900 1,000
Wavelength(nm)
Inte
nsit
y(co
unts
)
Fig. 1 Spectrum of the pulsed-UV light from the Xe flash lamp asmeasured with a spectrometer
1294 Parasitol Res (2008) 102:1293–1299
(Sigma), 15 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.5), 14 mM sodium bicar-bonate, 100 U/mL penicillin, 100 μg/mL streptomycin, and0.25 μg/mL amphotericin B. For routine cell passages, theHCT-8 cells were incubated at 37°C in a humidifiedincubator containing 6% CO2 and 94% air. For in vitroexcystation, oocysts were pretreated by incubation inacidified HBSS (Hanks’ balanced salt solution, pH 2.75;Gibco BRL) for 10 min at 37°C in a shaking incubator,followed by incubation in 4 mM sodium taurocholate(Sigma) in PBS (pH 7.4) at 15°C for 10 min and 37°C for5 min (Gut and Nelson 1999). Before inoculation, HCT-8 cells at a confluence of approximately 70% in a 35-mmdish were carefully rinsed with 0.1 M PBS buffer (pH 7.2).Excysted oocysts (2×106) diluted with prewarmed crypto-
sporidia infection medium (Upton et al. 1995) wereinoculated gently onto the HCT-8 cells. Infected HCT-8 cellswere gently washed with 0.1 M PBS buffer for three timesafter 2 h of incubation and then reincubated for another46 h in a humidified incubator containing 6% CO2 and 94%air. The infection was stopped at 48-h post-inoculationby removing the culture medium, and the cell monolayerwas washed with 0.1 M PBS buffer and used for RNApurification. Non-infected HCT-8 cells were used as a nega-tive control.
Plasmid DNA standard preparation for qPCR
Genomic DNA was isolated from purified fresh C. parvumoocysts using a genomic DNA isolation kit (Wizard,Promega, Madison, WI, USA). Briefly, concentratedoocysts (1×108 cysts) in 1.5-mL microtubes were excystedby four cycles of freezing (liquid nitrogen for 2 min) andthawing (95°C for 2 min). The oocysts were then lysed withnuclei lysis solution, and proteins were precipitated. TheDNA contained in the aqueous phase was precipitated withcold 100% isopropanol. The pellet was washed with cold70% ethanol, dried at room temperature, resuspended in20 μL of distilled water, and stored at 4°C. PCR reactionmixtures (20 μL total volume) contained 10∼100 ng ofgenomic DNA template, 1× PCR buffer, each dNTPmixture at 2.5 mM, 0.5 pmol of each primer (Bioneer,Daejeon, Republic of Korea; Table 1), and 2.5 U of Taqpolymerase (TaKaRa, Otsu, Shiga, Japan). PCRs comprisedan initial denaturation at 95°C for 5 min, followed by30 cycles at 95°C for 30 s, 50°C for 30 s, and 72°C for 1 min,
PoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V VInIn
OuOu
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
WATT
FL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYPoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V VInIn
OuOu
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
WATER LEVEL T
FL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCY
Powersupply
20cm
30cm
40cm
10cm
Controlpanel
UV reactorPulsed-UV lamp
PoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V VInIn
OuOu
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYPoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V VInlet TempInlet Temp
OuOu
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCY
Powersupply
20cm
30cm
40cm
20cm
30cm
40cm
Controlpanel
UV reactor
PoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V V
OuOu
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYPoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V V
OutOutlet Templet Temp
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYPuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYEMERGENCY
Powersupply
20cm
30cm
40cm
10cm
20cm
30cm
40cm
10cm
Controlpanel
UV reactor
PoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V V
O
± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPCPU D DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCY
Pulsed UV Disinfection System Control System Pulsed UV Disinfection System Control System
PoPowerSupplpply
MaMainPoPower
UV Lamp A
UV Lamp B
Water Empty
12 H12 Hz
20002000 V V± 00.000.0 oC
InterLock
Fail
± 00.000.0 oC
CPU DISPLAYCPU DISPLAY
VolVolt.
FrFreq.
50005000 S SShShot
120000120000 S SToTotalshshot
Reset
PuPumpOnOn/ Off
LALAMP STSTBY WORK
TFL
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYPuPumpOnOn/ Off
LALAMP STSTBY WORK
TURBIDIMETERTRANSMITTER
FLOW METER
0.0.23 N23 NTU900 cm900 cm
100 m100 m3/h/h r900 cm900 cm
A
B
EMERGENCYEMERGENCY
Powersupply
20cm
30cm
40cm
20cm
30cm
40cm
Controlpanel
UV reactor
Fig. 3 Diagram of the pulsed-UV light apparatus and theexperimental design
0
10
20
30
40
50
200 220 240 260 280 300 320 340 360 380 400
Wavelength(nm)
Abs
olut
e ir
radi
ance
(uW
/cm
2 /nm
)
20cm 30cm 40cm
Fig. 2 Absolute irradiance versus wavelength produced by the Xeflash lamp as measured with a spectrometer
Parasitol Res (2008) 102:1293–1299 1295

with a final extension at 72°C for 10 min. PCR ampliconswere visualized by electrophoresis on 1.5% agarose gels withethidium-bromide staining. For cloning into the vector, theamplified gene fragment was cut out of the agarose gel with asharp knife and purified with a gel extraction kit (Qiagen).The PCR product was cloned using the pGEM-T EasyVector system (Promega) and transformed into JM109competent cells in accordance with the manufacturer’sinstructions. The inserted genes were confirmed usinggeneral restriction enzyme digestion, and the DNA sequencewas determined using a sequence detector (ABI 7700,Applied Biosystems, Foster City, CA, USA) with SDSsoftware (version 1.6.3, Applied Biosystems) at a sequencingfacility (Cosmo, Seoul, Republic of Korea). The constructedplasmid containing the CP2 gene fragment was amplifiedand purified using the QIAprep Spin Miniprep kit (Qiagen)in accordance with the manufacturer’s instructions. Plasmidwas serially diluted with DNase/RNase-free water (Bioneer)and used as external standards for qPCR. All samples werestored at −20°C until use.
Preparation of cDNA for qPCR
Pulsed-UV light irradiated C. parvum oocysts (2×107)treated with four cycles of freezing (liquid nitrogen for 2min) and thawing (95°C for 2 min) and C. parvum infectedHCT-8 cells were lysed in Trizol® Reagent (Invitrogen,Carlsbad, CA, USA), and total RNA was extracted inaccordance with the manufacturer’s instructions. Briefly,after homogenization and centrifugation at 12,000×g for 15min, the supernatant was precipitated with isopropylalcohol and washed with 75% ethanol. After centrifugation,the RNA pellet was briefly dried at room temperature andthen resuspended in 10 μL of RNase/DNase-free water(Bioneer). To eliminate contaminating DNA, 1 U ofDNaseI (Promega) and 1 U of recombinant RNasin ribo-nuclease inhibitor (Promega) were placed in a total volume
of 15 μL and incubated at room temperature for 15 min(Hallier-Soulier and Guillot 2003). DNase activity wasstopped by adding 1 μL of 25 mM EDTA and heating at65°C for 10 min. Reverse transcription was performedwith 0.5∼1 μg of total RNA. Reaction mixtures included10 pmol of oligo(dT)20 primer (Bioneer), 1× M-MLV(Moloney Murine Leukemia Virus Reverse Transcriptase)RT reaction buffer, 0.25 mM dNTP mixture (TaKaRa), 1 Uof recombinant RNasin ribonuclease inhibitor (Promega),and 10 U of M-MLV RT enzyme (Enzynomics, Daejeon,Republic of Korea). RT reactions were carried out at 42°Cfor 1 h and were stopped by incubation at 70°C for 10 min.Total RNA extracted from non-UV-irradiated C. parvumoocysts, HCT-8 cells infected with non-UV-irradiated C.parvum, and non-infected HCT-8 cells were used aspositive or negative controls, respectively.
Absolute qPCR
Absolute qPCR reactions were performed in a 20-μLvolume using cDNA as a template. Reaction mixturesincluded 0.1× LightCycler® FastStart HybProbe master mix(Roche, Mannheim, Germany), each C. parvum primer set(Table 1) at 0.5 μM (Bioneer), and the probe set at 0.1 μM(TIB MOLBIO, Berlin, Germany). The probes included adonor probe labeled with fluorescent dye FL at the 3′ endand a reporter probe labeled at the 5′ end with Red LC 705.qPCR was carried out with a LightCycler® device (Roche).Each qPCR mixture was subjected to an initial denaturationat 95°C for 10 min, followed by 55 cycles of denaturationat 95°C for 5 s, annealing at 55°C for 15 s, and extension at72°C for 8 s, with a final cooling step at 40°C for 30 s. Theresults were analyzed by LightCycler® software (version4.05, Roche). DNase/RNase-free water was included as anegative control.
Results
We amplified a PCR product of approximately 223 bpcorresponding to the CP2 gene. Plasmid pGEMT-CP2 wasconstructed containing the CP2 gene, and the minimumdetection limit of the qPCR assay when using the Light-Cycler® device was ten copies (as determined using seriallydiluted pGEMT-CP2). Figure 4 shows a linear regressioncurve for the relationship between serially diluted plasmidDNA and crossing-point values. Four plasmid DNA copieswere considered to correspond to one C. parvum oocyst inqPCR because there are four sporozoites in each oocyst,and there were no available data on the number of copies ofthe CP2 gene in C. parvum.
The degree of C. parvum oocyst inactivation induced byirradiation with pulsed-UV light was quantified by oocyst
Table 1 Primer-probe sets for the CP2 gene (GenBank no.AY471868) for detecting C. parvum by qPCR
Name (type) Sequence (5′–3′) Nucleotide position
CP2-F (F) AAAACAAAAACTACTCAGACTCAAG
1520–1544
CP2-R (R) GGGTTGTAAACTTCATTGACTTT
1743–1721
CP2FL (DP) TCAATATCAGAGGTTACTTCAACTTCGC-FL
1632–1605
CP2LC (AP) 705-TGGGACTGGTAAAACTATGACGTCTTC-P
1603–1577
Expected PCR product size is 223 bp.F Forward primer, R reverse primer, DP donor probe for hybridiza-tion, AP acceptor probe for hybridization
1296 Parasitol Res (2008) 102:1293–1299

viability as evaluated by absolute qPCR involving totalRNA extracted from C. parvum oocysts with or without invitro cell infection. We quote here the base-10 logarithmicreduction calculated as log10 (number of oocysts prior topulsed-UV light irradiation/number of oocysts followingpulsed-light irradiation) as described by Zimmer et al.(2003). Oocyst inactivation as measured by the oocystviability was less than 1 log10 after 5 s of pulsed-UV lightirradiation regardless of the distance (20–40 cm; Fig. 5).Oocyst inactivation was effective (more than 2 log10) for60 s of irradiation at 20 cm from the light source (Fig. 5),for which the pulsed-UV light dose was 278 mJ/cm2.Oocyst inactivation was less than 2 log10 at distancesgreater than 30 cm, even with 60 s of irradiation (Fig. 5).
The C. parvum oocyst inactivation as measured by invitro cell infectivity was more than 2 log10 in allexperimental conditions except for the condition of 5 s ofirradiation at 40 cm from the light source (Fig. 6). The UVdose necessary for more than 2 log10 inactivation was15 mJ/cm2 (Table 2). C. parvum oocysts infectivity wasreduced by more than 3 log10 for 60 s of irradiation at 20–40 cm from the light source. The reduction in oocyst
infectivity (6 log10) was maximal with 60 s of irradiationat 20 cm from the light source (Fig. 6), for which the UVdose was 278 mJ/cm2. Table 2 lists the UV doses at eachlocation and exposure duration at 220∼400 nm. Irradiationdoses in each of the UVA, UVB, and UVC regions werenot measured. The target gene of qPCR (CP2) was notdetected in the total RNA extracted from uninfected HCT-8 cells.
Discussion
The qPCR using fluorescent hybridization probes specificto the target gene is known to be highly sensitive andspecific to detect C. parvum (MacDonald et al. 2002). Thenumber of cycles in the qPCR when the reaction productwas first detected indicated that the crossing-point valuedecreased as the amount of cDNA increased. This valuecould be converted to the number of C. parvum oocyststhrough a standard curve determined from the plasmidDNA containing the C. parvum CP2 gene fragment. TheCP2 gene has been shown to be a reliable viability markerof C. parvum in qPCR (Lee et al. 2008).
The results of this study suggested that at least 60 s ofexposure at 20 cm from the pulsed light was necessary foran effective reduction (more than 2 log10) in oocystviability. The corresponding UV dose was 278 mJ/cm2,
0
1
2
3
4
5
6
7
8
20/5 30/5 40/5 20/60 30/60 40/60
Cm/sec
Log
10(N
o/N
d)
Fig. 6 C. parvum oocyst inactivation shown as log10(N0/Nd). Thereduction in infectivity was measured using cDNA of the CP2 genefrom HCT-8 cells infected with C. parvum by qPCR with aLightCycler® device
-1
0
1
2
3
4
5
6
7
8
20/5 30/5 40/5 20/60 30/60 40/60
Cm/sec
Log
10(N
o/N
d)
Fig. 5 C. parvum oocyst inactivation shown as log10(N0/Nd), whereN0 and Nd are the numbers of live oocysts in the non-irradiated andirradiated groups, respectively. The reduction in viability wasmeasured using cDNA of the CP2 gene from C. parvum oocysts byqPCR with a LightCycler® device. Data are mean and SD values
R2 = 0.9873
0
5
10
15
20
25
30
35
40
1.E+01 1.E+02 1.E+03 1.E+04 1.E+05 1.E+06 1.E+07
No. of plasmid DNA
CP
val
ue
Fig. 4 Standard curve obtained using serially dilutions of plasmidDNA containing the CP2 gene fragment of C. parvum. The crossing-point (CP) value was inversely proportional to the amount of plasmidDNA
Table 2 UV dose for each location and duration condition in theexperiments
Distance (cm) UV light irradiance (mW/cm2) UV dose (mJ/cm2)
5 s 60 s
20 4.6 23.2 278.430 3.0 15.0 180.140 2.2 11.1 133.7
UV irradiance was measured over the total range of UV wavelengths(220∼400 nm).
Parasitol Res (2008) 102:1293–1299 1297

and the reduction in viability was 4.9 log10. The reductionin viability did not reach 2 log10 in all the otherexperimental conditions tested, indicating that UV dosesof less than 180 mJ/cm2 were not effective at reducing C.parvum oocyst viability.
The reduction in oocyst infectivity was more than 2log10 for all experimental conditions except for 5 s ofirradiation at 40 cm. The data showed that a UV dose ofmore than 15 mJ/cm2 could effectively reduce (i.e., by morethan 2 log10) the infectivity of C. parvum and with morethan 23 mJ/cm2 being necessary for a 3-log10 reduction.
Previous studies have demonstrated that UV lighteffectively inactivate cryptosporidia. Most of the publisheddata were obtained using low- and medium-pressure UVlight sources. Craik et al. (2001), Keegan et al. (2003),Mofidi et al. (2001), and Rochelle et al. (2005) reported thatUV doses of 6, 5.8, 10, and 20 mJ/cm2, respectively,provided an average of 2-log10 reduction in the infectivityof C. parvum. Our data are consistent with these data. It hasbeen previously reported that the dose required to reducethe infectivity of C. parvum by 3 log10 was 3 mJ/cm2 (Shinet al. 2001; Zimmer et al. 2003) and 25 mJ/cm2 (Craik et al.2001), and our results are more consistent with those ofCraik et al. (2001). This variation is probably due todifferences in the C. parvum strain, UV irradiation method,or type of UV lamp used.
We found a large discrepancy between the UV dosesnecessary for reducing oocyst viability and infectivity,which has also been reported previously. Morita et al.(2002) described that the UV dose for a 2-log10 reductionof viability was approximately 200 times higher than thedose required for the same reduction of animal infectivity.In our study, we found that the ratio was approximately18 times, although our infectivity assay involved cell cul-tures. Irradiation with pulsed UV light at a dose of 15–278 mJ/cm2 would remove the infectivity of C. parvumoocysts but not kill them. These findings suggest thatdeterminations of C. parvum oocyst viability should in-clude in vitro cell infectivity measurements rather thanperforming qPCR using C. parvum oocyst mRNA.
Wang et al. (2005) found that the germicidal efficiencydid not differ between Xe flash lamps and continuous-UVlow-pressure mercury lamps operating at 254 nm. Pulsedlight at 200–300 nm exerts large germicidal effects againstmicrobial species such as Listeria monocytogenes, E. coli,Salmonella enteritidis, Pseudomonas aeruginosa, Bacilluscereus, and Staphylococcus aureus (Rowan et al. 1999;Wang et al. 2005). Our data confirmed that pulsed light canalso strongly inactivate C. parvum. As Wang et al. (2005)mentioned, the short pulse width and high doses of a pulsedUV source may provide practical advantages over contin-uous-UV sources in those situations where rapid disinfec-tion is required.
In conclusion, our assay combining qPCR with an invitro cell culture of C. parvum is a valuable tool forevaluating disinfection systems for drinking-water treat-ment. The pulsed-UV light used in this study was highlyeffective at rapidly disinfecting C. parvum oocysts in water.The data showed that UV doses of 15, 23, and 278 mJ/cm2
were necessary for 2-, 3-, and 6-log10 reductions in C.parvum infectivity, respectively, with 278 mJ/cm2 beingnecessary for an effective reduction in oocyst viability(more than 2 log10).
Acknowledgment This paper was supported by the Korea Ministryof Environment as “The Eco-technopia 21 project” in 2006 and by theprogram of Basic Atomic Energy Research Institute (BAERI), whichis a part of the Nuclear R & D Programs funded by the Ministry ofScience & Technology (MOST) of Korea in 2007.
References
Casemore DP, Armstrong M, Sands RL (1985) Laboratory diagnosisof cryptosporidiosis. J Clin Pathol 8:1337–1341
Craik SA, Weldon D, Finch GR, Bolton JR, Belosevic M (2001)Inactivation of Cryptosporidium parvum oocysts using mediumand low-pressure ultraviolet radiation. Water Res 35:1387–1398
DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB,Jakubowski W (1995) The infectivity of Cryptosporidiumparvum in healthy volunteers. N Engl J Med 332:855–859
Fontaine M, Guillot E (2003) An immunomagnetic separation-real-time PCR method for quantification of Cryptosporidium parvumin water samples. J Microbiol Methods 54:29–36
Gut J, Nelson RG (1999) Cryptosporidium parvum: synchronizedexcystation in vitro and evaluation of sporozoite infectivity witha new lectin-based assay. J Eukaryot Microbiol 46:56S–57S
Hallier-Soulier S, Guillot E (2003) An immunomagnetic separation-reverse transcription polymerase chain reaction (IMS-RT-PCR)test for sensitive and rapid detection of viable waterborneCryptosporidium parvum. Environ Microbiol 5:592–598
Keegan AR, Fanok S, Monis PT, Saint CP (2003) Cell culture-Taqman PCR assay for evaluation of Cryptosporidium parvumdisinfection. Appl Environ Microbiol 69:2505–2511
Korich DG, Mead JR, Madore MS, Sinclair NA, Sterling CR (1990)Effects of ozone, chlorine dioxide, chlorine, and chloramine onCryptosporidium parvum oocyst viability. Appl Environ Micro-biol 56:1423–1428
Lee SU, Jung M, Ahn MH, Huh S, Song H, Park WY, Yu JR (2008)CP2 gene as a useful viability marker for Cryptosporidiumparvum. Parasitol Res 102:381–387
MacDonald LM, Sargent K, Armson A, Thompson RC, ReynoldsonJA (2002) The development of a real-time quantitative-PCRmethod for characterisation of a Cryptosporidium parvum in vitroculturing system and assessment of drug efficacy. Mol BiochemParasitol 121:279–282
MacKenzie WR, Hoxie NJ, Proctor ME, Gradus MS, Blair KA,Peterson DE, Kazmierczak JJ, Addiss DG, Fox KR, Rose JB,Davis JP (1994) A massive outbreak in Milwaukee of Crypto-sporidium infection transmitted through the public water supply.N Engl J Med 331:161–167
Mofidi AA, Baribeau H, Rochelle PA, De Leon R, Coffey BM, GreenJF (2001) Disinfection of Cryptosporidium parvum with poly-chromatic UV light. J Am Water Works Assoc 93:95–109
1298 Parasitol Res (2008) 102:1293–1299

Morita S, Namikoshi A, Hirata T, Oguma K, Katayama H, Ohgaki S,Motoyama N, Fujiwara M (2002) Efficacy of UV irradiation ininactivating Cryptosporidium parvum oocysts. Appl EnvironMicrobiol 68:5387–5393
Nichols GL (2000) Food-borne protozoa. Br Med Bull 56:209–235O’Hara SP, Yu JR, Lin JJ (2004) A novel Cryptosporidium parvum
antigen, CP2, preferentially associates with membranous struc-tures. Parasitol Res 92:317–327
Okhuysen PC, Chappell CL, Crabb JH, Sterling CR, DuPont HL(1999) Virulence of three distinct Cryptosporidium parvumisolates for healthy adults. J Infect Dis 180:1275–1281
Petry F, Robinson HA, McDonald V (1995) Murine infection modelfor maintenance and amplification of Cryptosporidium parvumoocysts. J Clin Microbiol 33:1922–1924
Rochelle PA, Marshall MM, Mead JR, Johnson AM, Korich DG,Rosen JS, DeLeon R (2002) Comparison of in vitro cell cultureand a mouse assay for measuring infectivity of Cryptosporidiumparvum. Appl Environ Microbiol 68:3809–3817
Rochelle PA, Upton SJ, Montelone BA, Woods K (2005) Theresponse of Cryptosporidium parvum to UV light. TrendsParasitol 21:81–87
Rowan NJ, Macgregor SJ, Anderson JG, Fouacre RA, McIlcilvaney L(1999) Pulsed-light inactivation of food-related microorganisms.Appl Environ Microbiol 65:1312–1315
Shin GA, Linden KG, Arrowood MJ, Sobsey MD (2001) Low-pressure UV inactivation and DNA repair potential of Cryp-tosporidium parvum oocysts. Appl Environ Microbiol 67:3029–3032
Simmons OD, Sobsey MD, Heaney CD, Schaefer FW, Francy DS(2001) Concentration and detection of cryptosporidium oocystsin surface water samples by method 1622 using ultrafiltration andcapsule filtration. Appl Environ Microbiol 67:1123–1127
Stinear T, Matusan A, Hines K, Sandery M (1996) Detection of asingle viable Cryptosporidium parvum oocyst in environmentalwater concentrated by reverse transcription PCR. Appl EnvironMicrobiol 62:3385–3390
Upton SJ, Tilley M, Brillhart DB (1995) Effects of select mediumsupplements on in vitro development of Cryptosporidiumparvum in HCT-8 cells. J Clin Microbiol 33:371–375
Verweij JJ, Blange RA, Templeton K, Schinkel J, Brienen EA, vanRooyen MA, van Lieshout L, Polderman AM (2004) Simulta-neous detection of Entamoeba histolytica, Giardia lamblia, andCryptosporidium parvum in fecal samples by using multiplexreal-time PCR. J Clin Microbiol 42:1220–1223
Wang T, MacGregor SJ, Anderson JG, Woolsey GA (2005) Pulsedultra-violet inactivation spectrum of Escherichia coli. Water Res39:2921–2925
Yang S, Healey MC (1993) The immunosuppressive effects of dexa-methasone administered in drinking water to C57BL/6N miceinfected with Cryptosporidium parvum. J Parasitol 79:626–630
Yu JR, O’Hara SP, Lin JL, Dailey ME, Cain G, Lin JL (2002) Acommon oocyst surface antigen of Cryptosporidium recognizedby monoclonal antibodies. Parasitol Res 88:412–420
Zimmer JL, Slawson RM, Huck PM (2003) Inactivation and potentialrepair of Cryptosporidium parvum following low- and medium-pressure ultraviolet irradiation. Water Res 37:3517–3523
Parasitol Res (2008) 102:1293–1299 1299