proteiinianalyysi 7 kolmiulotteisen rakenteen ennustaminen ...
TRANSCRIPT

Proteiinianalyysi 7
Kolmiulotteisen rakenteen ennustaminen
http://www.bioinfo.biocenter.helsinki.fi/downloads/teaching/spring2006/proteiinianalyysi

Sekvenssistä rakenteeseen
• komparatiivinen mallitus
• 1-ulotteinen tilan (luokan) ennustaminen sekvenssistä
• 3-ulotteisen rakenteen tunnistaminen annetusta kirjastosta (fold recognition)
• 3-ulotteisen rakenteen ennustaminen ab initio

Motivation
• Protein structure determines protein function
• For the majority of proteins the structure is not known
structures
sequences
0 250000 500000 750000 1000000 1250000 1500000
Structural coverage

0
0.5
1
1.5
2
2.5
3
0 0.2 0.4 0.6 0.8 1
fraction of mutated residues
rmsd
of
mai
n c
hai
n a
tom
s [A
]
Chothia & Lesk (1986)
Curve fitted to datafor homologous families
Divergence of common cores• fraction in coredecreases withincreasing sequencedivergence

Steps in comparative modelling
• Find suitable template(s)
• Build alignment between target and template(s)
• Build model(s)– Replace sidechains– Resolve conflicts in the structure– Model loops (regions without an alignment)
• Evaluate and select model(s)

State of the art in homology modelling
• Template search
– (iterative) sequence database searches (PSIBLAST)
• Alignment step
– multiple alignment of close to fairly distant homologues
• Modelling step
– rigid body assembly
– segment matching
– satisfaction of spatial constraints
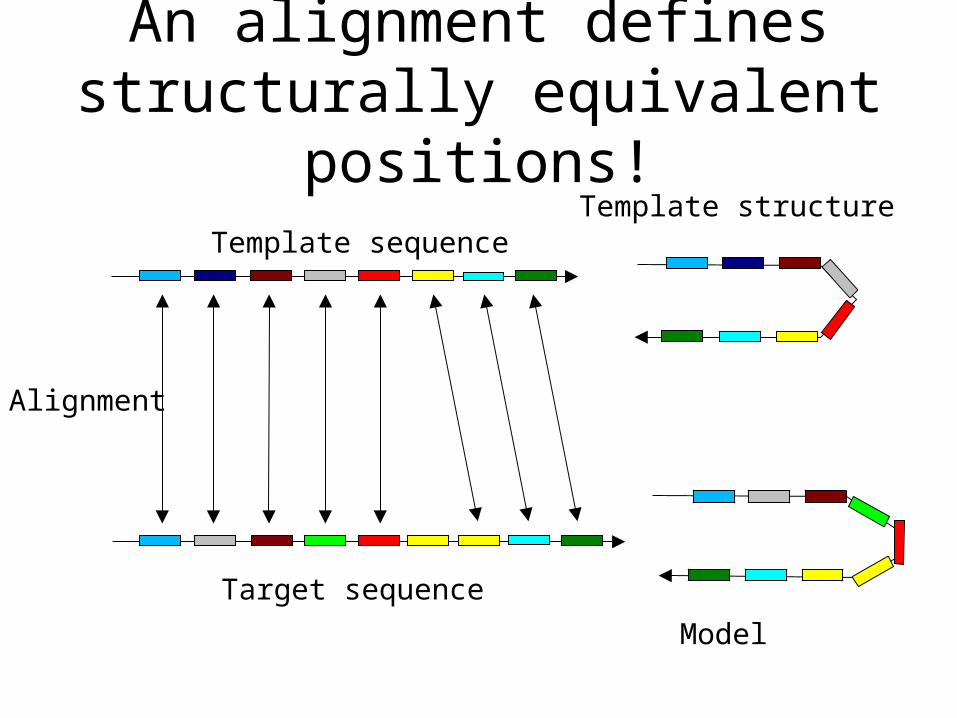
An alignment defines structurally equivalent positions!
Template sequenceTemplate structure
Target sequence
Alignment
Model

The crucial importance of the alignment
Template sequence
Template structure
Target sequence
Alignment
Model

Modelling by spatial restraints
• Generate many constraints:– Homology derived constraints
• Distances and angles between aligned positions should be similar
– Stereochemical constraints• Bond lengths, bond angles, dihedral angles,
nonbonded atom-atom contacts
• Model derived by minimizing restraints
Modeller: Sali & Blundell (1993)

Loop modelling
• Exposed loop regions usually more variable than protein core
• Often very important for protein function
• Loops longer than 5 residues difficult to built
• Mini-protein folding problem

Model evaluation
• Check of stereochemistry– bond lengths & angles, peptide bond planarity,
side-chain ring planarity, chirality, torsion angles, clashes
• Check of spatial features– hydrophobic core, solvent accessibility, distribution
of charged groups, atom-atom-distances, atomic volumes, main-chain hydrogen bonding
• 3D profiles/mean force potentials– residue environment

Knowledge-based mean force potentials
Melo & Feytmanns (1997)
• Compute typical atomic/residue environments based on known protein structures

• Sequence from different species
• Is binding to ligand conserved?
Ligand
DNA
Modelling a transcription factor

Ligand binding domain
hydrogen bonds to ligand homo-serine lactone moiety binding acyl moiety binding

DNA binding domain
Linker DNA binding domain

Template
Target
Variable loops
New Loop
MODELLER output
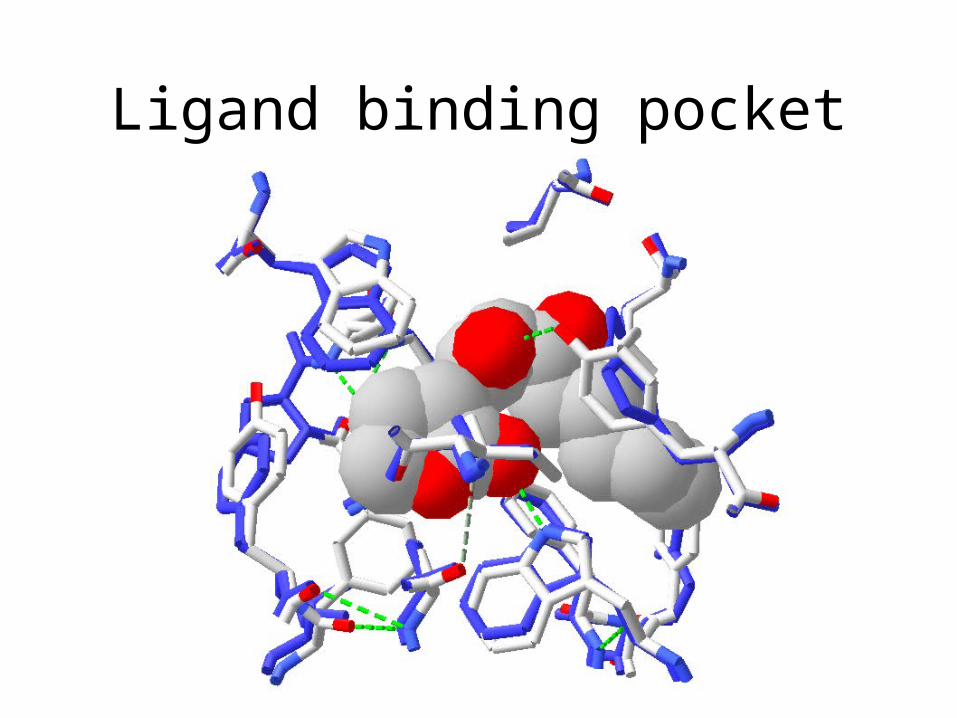
Ligand binding pocket

Errors in comparative modelling
Marti-Renom et al. (2000)
a)Side chain packing
b)Distortions and shifts
c)Loops
d)Misalignments
e)Incorrect template
TemplateModel
True structure

Modelling accuracy
Marti-Renom et al. (2000)

Applications of homology modelling
Marti-Renom et al. (2000)

Structural genomics
• Post-genomics:– many new sequences, no function
• Aim: a structure for every protein
• High-throughput structure determination– robotics– standard protocols for
cloning/expression/crystallization

Structural coverage
Vitkup et al. (2001)
high quality models
Complete models
Total = 43 %

Target selection

Fold recognition - Assumption
• Native structure is the global minimum energy conformation
• So, need– Discriminating energy function– Conformation generator
• Backbone from homologous template (comparative modelling)
• Backbone from analogous template (fold recognition)
• Comprehensive sampling (ab initio)

Fold recognition steps
• Template library– Known structures from Protein Data Bank– Fold classification suggests a limited number of fold
types• Score = sequence-structure fitness
– Environmental preferences of amino acids– Boltzmann engine
• Search problem = alignment– Complicated with pair potentials
• Significance of best score in database search– Reference state
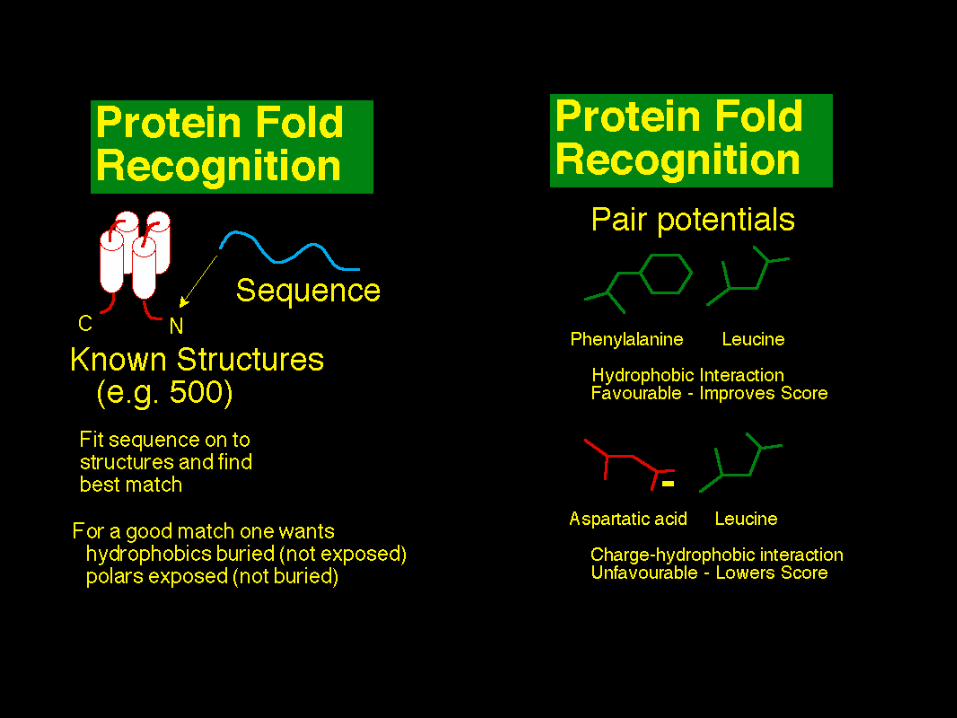

Potentials of mean force
• “Boltzmann engine”
• In thermodynamic equilibrium, particles are partitioned between states proportionally to exp(-G)
• Effective energy = negative logarithm of the equilibrium constant– Count occurrences per state– Radial distribution of aa pairs (Sippl)

Structural environment
• Single-residue preferences 20 x 3 x 3 x 3– Helix, strand, coil– Accessibility– Contact area (indirectly codes for aa type)
• Contact pair potentials– Atomic contacts within 4 A – C-beta atoms within 7 A– Secondary structure of residues i and j
• 3 x 20 x 3 x 20 = 3600 preferences

Information content
Arg-Asp helix-helix(dashed)
Arg-Asp strand-strand(solid)
Arg-Asp (dotted)

Threading algorithms
• Dynamic programming– Simple– “frozen approximation”
• Read sequence-dependent environment from template (1st round), then from aligned target sequence
• Stochastic optimization (Monte Carlo)– Pair potentials
• Exhaustive search– Simplify search space (e.g., ignore loops)

Prospect model (Xu & Xu)
Etotal = vmutateEmutate x vsingleEsingle x vpairEpair x vgapEgap
Weights v optimized on training set

Prospect - segmentation
- Finds optimal threading fairly efficiently- Topological complexity - No gaps in secondary structure elements- Pair energy term only evaluated between
secondary structure elements

Prospect- observations
• Mutation energy is the most important
• Single-residue terms with profile information generate reasonably good alignments for ~2/3 of test cases
• The pairwise energy term can thus be ignored during the search for optimal alignment, but is used in evaluating the fold recognition
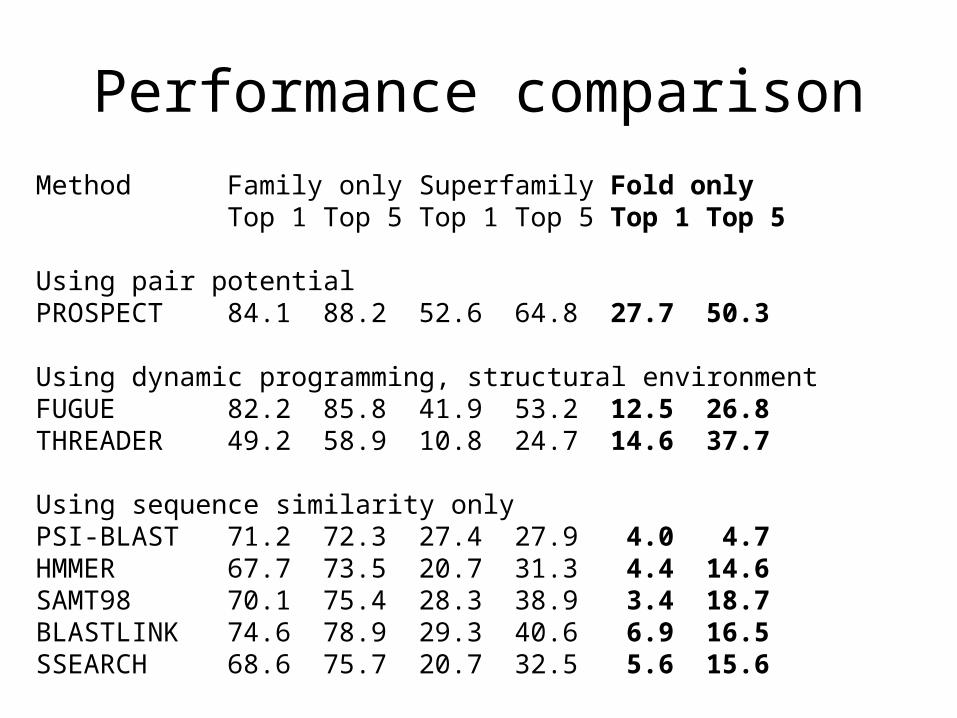
Method Family only Superfamily Fold onlyTop 1 Top 5 Top 1 Top 5 Top 1 Top 5
Using pair potentialPROSPECT 84.1 88.2 52.6 64.8 27.7 50.3
Using dynamic programming, structural environmentFUGUE 82.2 85.8 41.9 53.2 12.5 26.8THREADER 49.2 58.9 10.8 24.7 14.6 37.7
Using sequence similarity onlyPSI-BLAST 71.2 72.3 27.4 27.9 4.0 4.7HMMER 67.7 73.5 20.7 31.3 4.4 14.6SAMT98 70.1 75.4 28.3 38.9 3.4 18.7BLASTLINK 74.6 78.9 29.3 40.6 6.9 16.5SSEARCH 68.6 75.7 20.7 32.5 5.6 15.6
Performance comparison

Threading score - significance
• Target sequence – fold library– Each threading aligns a different sub-
sequence• Compute Z-score for each by ungapped threading
on large decoy (Sippl)
• “Reverse threading”– Design optimal sequence for a given fold

Incorrect self-threading





Fold recognized

Fold recognizedPoor alignment of residues
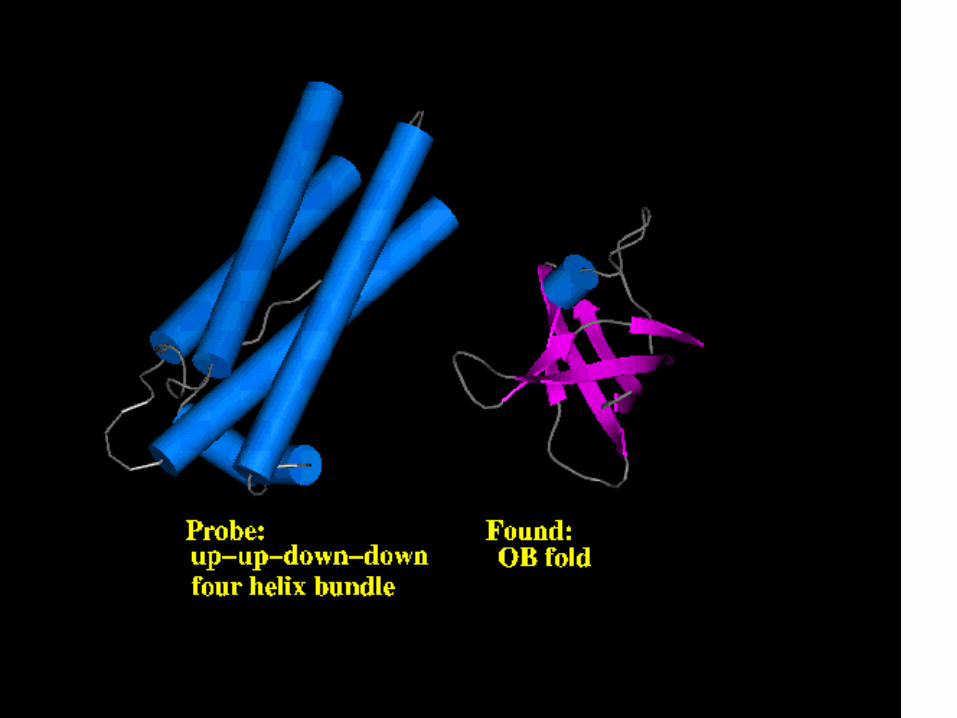

Ab initio prediction
• HMMSTR/I-sites/RosettaHMMSTR is a Hidden Markov Model based on protein STRucture. Each Markov state in this model represents a position in one of the I-sites motifs. HMMSTR can predict local structure (as backbone angles), secondary structure, and supersecondary structure (edge versus middle strand, hairpin versus diverging turn).
• I-sites LibraryI-sites is a library of folding initiation site motif, which are sequence motifs that correlate with particular local structures such as beta hairpins and helix caps. I-sites can be used to predict local structure, or to predict which parts of a protein are likely to fold early, initiating folding.

Folding is 2-stateUnfolded Folded
Intermediates are not observed, but

Nucleation sites
something happens first...

Early folding events might be recorded in the database
HDFPIEGGDSPMQTIFFWSNANAKLSHGY CPYDNIWMQTIFFNQSAAVYSVLHLIFLT IDMNPQGSIEMQTIFFGYAESAELSPVVNFLEEMQTIFFISGFTQTANSD INWGSMQTIFFEEWQLMNVMDKIPSIFNESKKKGIAMQTIFFILSGR PPPMQTIFFVIVNYNESKHALWCSVD PWMWNLMQTIFFISQQVIEIPS MQTIFFVFSHDEQMKLKGLKGA
Short, recurrent sequence patterns could be folding Initiation sites
Nature has selected for these patterns because they speed folding.
Non
-hom
olog
ous
pro
tein
s
recurrent part


How to read an I-sites motif profile

Backbone angles and sequence pattern for Amphipathic alpha-helix

Superposition of the top scoring 30 true-positives

Conserved polar (green) and non-polar (purple) sidechains

Serine alpha-N-cap

A Markov state. A hidden Markov modelconsists of Markov states connected by directed transitions. Each state emits an output symbol, representing sequence or structure. There are four categories of emission symbols in our model: b, d, r, and c, corresponding to amino acid residues, three-state secondary structure, backbone angles (discretized into regions of phi-psispace) and structural context (e.g. hairpin versus diverging turn, middle versus end-strand), respectively.
Bystroff C, Thorsson V & Baker D. (2000). HMMSTR: A hidden markov model for local sequence-structure correlations in proteins. Journal of Molecular Biology 301, 173-90.
HMMSTR
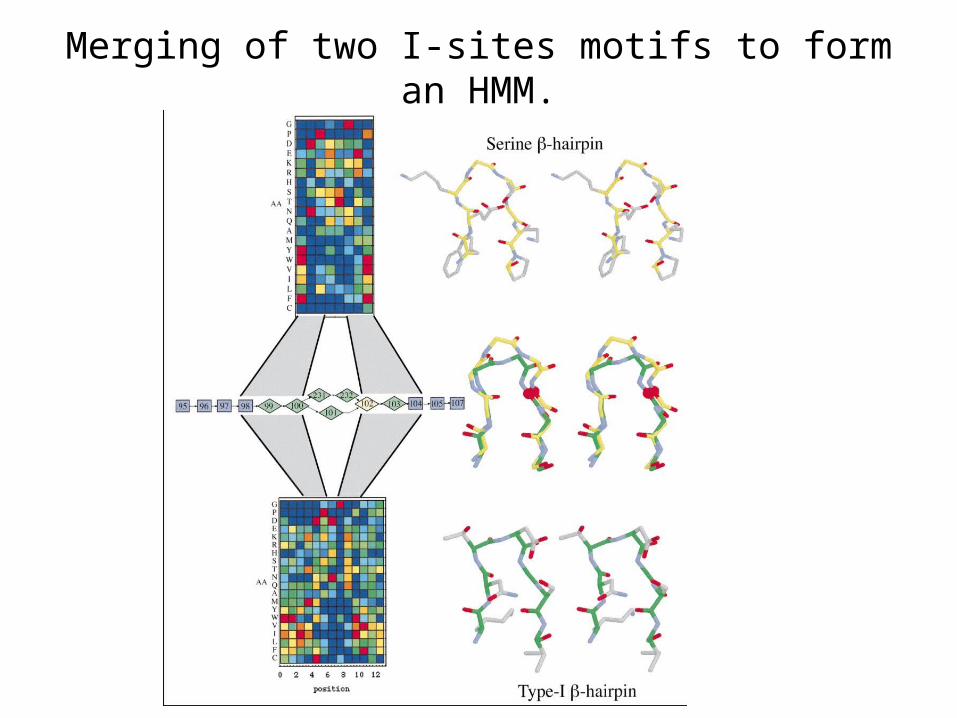
Merging of two I-sites motifs to form an HMM.

VIVAANRSAVIVSAARTAVIASAVRTAVIVDAGRSAVIASGVRTAVIVAAKRTAVIVSAVRTPVIVSAARTAVIVSAVRTPVIVDAGRTAVIVDAGRTAVIVSGARTPVIVDFGRTPVIVSATRTPVIVSATRTPVIVGALRTPVIVSATRTPVIVSATRTPVIASAARTAVIVDAIRTPVIVAAYRTAVIVSAARTPVIVDAIRTPVIVSAVRTAVIVAAHRTA
••••••
Sequence alignment
Sequence profile
Pij wk skj aai
kseqs
wkkseqs
Sequence Profiles
Red = high prob ratio (LLR>1)Green = background prob ratio (LLR≈0)Blue = low prob ratio (LLR<-1)
aa

diverging type-2 turn
Serine hairpin
Proline helix C-capalpha-alpha corner glycine helix N-cap
Frayed helix
Type-I hairpin
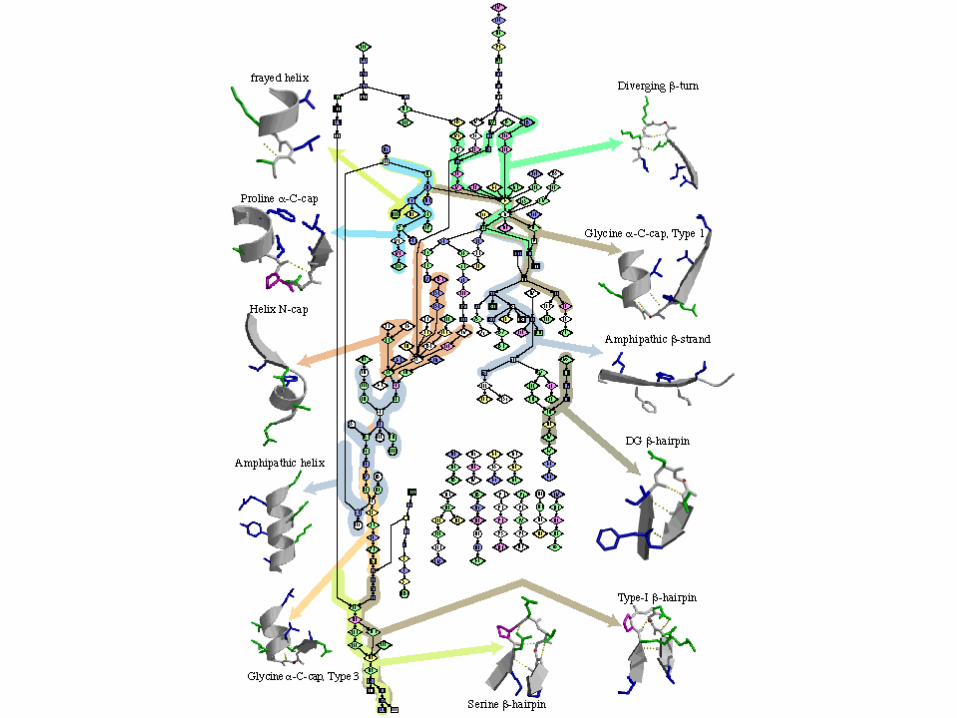
I-sites motifs
Amino acids arranged from non-polar to polar
Backbone angles: =green, =red

Why do I-sites exist?
1. They are ancient conserved regions?
2. They fold independently?

Patterns of conservation suggest independent folding
1. backbone angle constraints
3. negative design
2. sidechain contacts

(a)
(b)
(c)
(d)
color scale
1.0.80.60.40.20.0-.2-.4-.6-.8Š-1
AA AA
26272829303132position
CFLIVWYMAQNTSHRKEDPG
AA
26272829303132
CFLIVWYMAQNTSHRKEDPG
1 2 3 4 5 6 7position
CFLIVWYMAQNTSHRKEDPG
AA
1 2 3 4 5 6 7
CFLIVWYMAQNTSHRKEDPG
NMR structure of a 7-residue I-sites motif in isolation (Yi et al, J. Mol. Biol, 1998)
diverging turn
motif
NMR structures confirm independent folding

Fold prediction – Rosetta method
• Knowledge based scoring function
P(structure) * P(sequence|structure)
P(sequence)P(structure|sequence) =
P(structure) = probability of a protein-like structure(no clashes, globular shape)
P(sequence|structure) = f(residue contacts in native structures)
Simons et al. (1997)
Bayes' law:
protein-likestructures
sequence consistentlocal structure
near-native structures

Rosetta
(1) A stone with three ancient languages on it.
(2) A program (David Baker) that simulates the folding of a protein, using statistical energies and moves.

The “Folding Problem”
Two parts:
(1) The “Search Problem”
Is the true structure one of my 2 million guesses?
(2) The “Discrimination Problem”
If it’s one of these 2 million, which one is it?

Fragment insertion Monte Carlo
Energyfunctionchange backbone
angles
Convert angles to 3D coordinates
accept or reject
Choose fragment from moveset
mov
eset
backbone torsion angles
Rosetta

Backbone angles are restrained in I-sites regions
mov
eset
backbone torsion angles
Generally, about one-third of the sequence has an I-sites prediction with
confidence > 0.75, and is restrained.
Fragments that deviate from the paradigm (>90° in or ) are removed from the moveset.
regions of high-confidence I-sites prediction
Rosetta
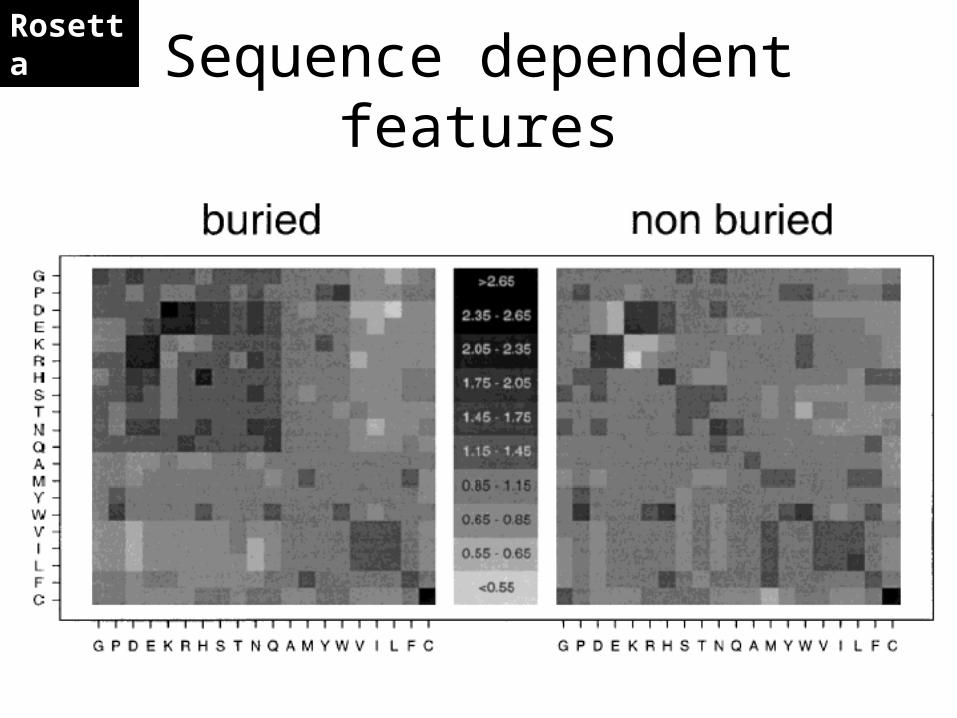
Sequence dependent featuresRosetta

Current structure
Sequence-independent features
The energy score for a contact between secondary structures is summed using database statistics.
vector representationProbabilities from the database
Rosetta

CASP4 predictions
31 target sequences. Ab initio prediction
i.e. Sequence homolog data was ignored if present.
61% “topologically correct”
60% “locally correct”
73% secondary structure correct
Rosetta

T0116 262-322 (61 residues) prediction true structure
Topologically correct (rmsd=5.9Å) but helix is mis-predicted as loop.
Rosetta

T0121 126-199 (66 residues)prediction true structure
Topologically correct (rmsd=5.9Å) but loop is mis-predicted as helix.
Rosetta

T0122 57-153 (97 residues)
...contains a 53 residue stretch with max deviation = 96°
prediction true structure
Rosetta

T0112 153-213
Low rmsd (5.6Å) and all angles correct ( mda = 84°), but topologically wrong!
prediction true structure
(this is rare)
Rosetta

CO 1
L NSij
N
8% of the residues in the targets have > 0.
44% of these are at Glycine residues.
7% of the residues in the predictions have > 0.
but only 16% of these are at Glycines.
True structure: 0.252
Predictions: 0.119
What needs to be fixed?Turns
Contact order
Rosetta

Prediction of protein structure• ROSETTA program most famous• different models to treat the local and nonlocal interactions. • sequence-dependent local interactions bias segments of the chain
to sample distinct sets of local structures– turn to in known three-dimensional structures as an approximation to the
distribution of structures sampled by isolated peptides with the corresponding sequences.
• nonlocal interactions select the lowest free-energy tertiary structures from the many conformations compatible with these local biases.– The primary nonlocal interactions considered are hydrophobic burial,
electrostatics, main-chain hydrogen bonding and excluded volume. • minimizing the nonlocal interaction energy in the space defined by
the local structure distributions using Monte Carlo simulated annealing.

Using NMR to guide Rosetta
• We have extended the ROSETTA ab initio structure prediction strategy to the problem of generating models of proteins using limited experimental data. By incorporating chemical shift and NOE information and more recently dipolar coupling information into the Rosetta structure generation procedure, it has been possible to generate much more accurate models than with ab initio structure prediction alone or using the same limited data sets with conventional NMR structure generation methodology. An exciting recent development is that the Rosetta procedure can also take advantage of unassigned NMR data and hence circumvent the difficult and tedious step of assigning NMR spectra.

Rosetta in comparative modelling
• We have also developed a method for comparative modeling that was one of the top performing methods in the CASP4 experiment. The method utilizes a new protein sequence structure alignment method and structurally variable regions such as long loops not present in the structure of a homologue are built using a modification of the rosetta ab initio structure prediction methodology. Both the ab initio and the comparative modeling methods have been implemented in a server called ROBETTA which was one of the best all around fully automated structure prediction servers in the CASP5 test.

Prediction algorithms have Underlying principles
Darwin = protein evolution.
Principle: Proteins that evolved from common ancestor have the same fold.
Boltzmann = protein folding
Principle: Proteins search conformational space, minimizing the free energy.

Summary•Most prediction methods depend on sequence homology.(Darwin)
•Folding predictions combine statistics and simulations.
•Putative folding initiation sites can be found using database statistics.
•Knowledge-based energy functions are derived from database statistics.
•The folding problem is really two problems: the search problem and the discrimination problem.
•If we knew how proteins fold, we could predict their structures.
•We don’t know how proteins fold.

CASP6 – current status
• Comparative modelling extended to distant homologues– Easy: PSI-Blast neighbours– Hard: indirect PSI-Blast neighbours
• Fold recognition merged with comparative modelling
• Ab initio methods based on fragment assembly generate models (among top N predictions) that have some resemblance to the real structure