plasmin(ogen) promotes renal interstitial fibrosis by promoting epithelial-to-mesenchymal
TRANSCRIPT

Plasmin(ogen) Promotes Renal Interstitial Fibrosis byPromoting Epithelial-to-Mesenchymal Transition: Role ofPlasmin-Activated Signals
Guoqiang Zhang,* Kelly A. Kernan,* Sarah J. Collins,* Xiaohe Cai,* Jesus M. Lopez-Guisa,*Jay L. Degen,† Yigal Shvil,‡ and Allison A. Eddy**Division of Nephrology, University of Washington and Children’s Hospital and Regional Medical Center, Seattle,Washington; †Division of Basic Science Research, Children’s Hospital Research Foundation, Cincinnati, Ohio; and‡Hebrew University-Hadassah School of Medicine, Jerusalem, Israel
Plasminogen (Plg) activator inhibitor-1 (PAI-1) is an important fibrosis-promoting molecule. Whether this effect can beattributed to PAI-1’s activity as an inhibitor of plasmin generation is debated. This study was designed to investigate the roleof Plg in renal fibrosis using in vivo and in vitro approaches. Plg-deficient (Plg�/�) and wild-type (Plg�/�) C57BL/6 micewere subjected to unilateral ureteral obstruction or sham surgery (n � 8/group; sham, days 3, 7, 14, and 21). Plg deficiency wasconfirmed by the absence of Plg mRNA, protein, and plasmin activity. After 21 d of unilateral ureteral obstruction, totalkidney collagen was significantly reduced by 35% in the Plg�/� mice. Epithelial-to-mesenchymal transition (EMT), astypified by tubular loss of E-cadherin and acquisition of �-smooth muscle actin, was also significantly reduced in Plg�/�mice, 76% and 50%, respectively. Attenuation of EMT and fibrosis severity in the Plg�/� mice was associated withsignificantly lower levels of phosphorylated extracellular signal–regulated kinase (ERK) and active TGF-�. In vitro, additionof plasmin (20 �g/ml) to cultures of murine tubular epithelial cells initiated ERK phosphorylation within minutes, followedby phenotypic transition to fibroblast-specific protein-1�, �-smooth muscle actin�, fibronectin-producing fibroblast-likecells. Both plasmin-induced ERK activation and EMT were significantly blocked in vitro by the protease-activated receptor-1(PAR-1) silencing RNA; by pepducin, a specific anti–PAR-1 signaling peptide; and by the ERK kinase inhibitor UO126.Plasmin-induced ERK phosphorylation was enhanced in PAR-1–overexpressing tubular cells. These findings support impor-tant profibrotic roles for plasmin that include PAR-1–dependent ERK signaling and EMT induction.
J Am Soc Nephrol 18: 846–859, 2007. doi: 10.1681/ASN.2006080886
P rogressive renal disease is characterized by tubular at-rophy and interstitial fibrosis (1). Interstitial myofibro-blasts that accumulate during renal fibrogenesis seem
to be the primary source of the extracellular matrix (ECM)proteins that accumulate and ultimately destroy nephron in-tegrity and function (2). The primary origin of interstitial myo-fibroblasts is still debated. The possibilities include residentinterstitial fibroblasts that activate and proliferate; tubular ep-ithelia that have undergone epithelial-to-mesenchymal transi-tion (EMT) into motile, fibroblast-like cells and migrate into theinterstitium; and stem cells that are recruited to the interstitium(3,4). Phenotypic features of renal tubular EMT include de-creased expression of the cell adhesion receptor E-cadherin andde novo expression of �-smooth muscle actin (�-SMA) andfibroblast-specific protein-1 (FSP-1) (3,5). The molecular path-ways that control tubular cell EMT and their interstitial migra-
tion during progressive kidney disease remain incompletelydelineated. TGF-� and tissue-type plasminogen (Plg) activator,a key component of the Plg activation system, have been shownto play important roles (6,7).
The active serine protease plasmin is generated from thezymogen Plg, a circulating protein that is synthesized in theliver and often extravasates to extravascular sites during in-flammation (8). The zymogen Plg is activated by tissue-typeand urokinase-type Plg activators (uPA). Plasmin activity hasbeen associated with a variety of processes that are involved intissue remodeling and/or cell migration, including embryonicdevelopment, macrophage recruitment, wound healing, angio-genesis, atherosclerosis, and tumor cell invasion (9). In additionto its fibrinolytic function, plasmin degrades a number of non-fibrin substrates to elicit diverse effects that may modulatefibrosis severity. These include its direct proteolytic activity forcertain extracellular matrix (ECM) proteins such as fibronectinand laminin (but not elastin or native helical collagen), and itsability to activate latent matrix metalloproteinases and to acti-vate latent TGF-� (10–14). By degrading ECM, plasmin mayalso liberate sequestered growth factors. Direct degradation ofE-cadherin in vitro by plasmin has also been reported (15).Given its pleiotropic effects in vitro, it has been unclear whether
Received August 22, 2006. Accepted December 29, 2006.
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Allison A. Eddy, Children’s Hospital & RegionalMedical Center, 4800 Sand Point Way NE, Division of Nephrology, Mail StopM1-5, Seattle, WA 98105. Phone: 206-987-2524; Fax: 206-987-2636; E-mail:[email protected]
Copyright © 2007 by the American Society of Nephrology ISSN: 1046-6673/1803-0846

the predominant effect of Plg in vivo promotes or inhibits tissuefibrosis severity. It has long been speculated that tissue plasminactivity would attenuate fibrosis as a result of its matrix-de-grading effects (16). However, a recent study by Edgtton et al.(14) challenged this hypothesis, although the underlying mech-anisms whereby plasmin promoted fibrosis were not deter-mined.
More recently, it has become apparent that in addition to itsextracellular effects, plasmin may regulate cellular responses.Cellular receptors for plasmin seem to be ubiquitous. A 41- to43-kD cell surface protein identical to glyceraldehyde-3-phos-phate dehydrogenase has been identified as a plasmin receptoron group A Streptococci, and this receptor is thought to partic-ipate in the pathogenesis of human acute poststreptococcalglomerulonephritis (17–19). Best known as a receptor forthrombin (20), protease-activated receptor-1 (PAR-1) can alsobe activated by plasmin, leading to signal transduction andgene expression in fibroblasts via p44/42 extracellular signal–regulated kinases (ERK)/mitogen-activated protein kinase(MAPK) pathways (21). In macrophages, plasmin has beenreported to initiate signaling via p38 MAPK and signal trans-ducers and activators of transcription (22). However, plasmin-dependent signal transduction pathways have not been exten-sively investigated in vivo, and their role in the pathogenesis ofrenal fibrosis unknown.
The aim of the present study was to investigate the role of theplasmin(ogen) signaling pathway in an experimental model ofprogressive kidney disease. The severity of renal fibrosis wasfound to be significantly reduced in Plg-deficient comparedwith wild-type C57Bl/6 mice after unilateral ureteral obstruc-tion (UUO). In vitro studies using a murine cortical tubular(MCT) epithelial cell line demonstrated that plasmin promotedEMT and tubular cell migration via the activated PAR-1–ERKsignaling pathway. These findings identify an important rolefor plasmin-dependent cellular effects in the induction of tubu-lar EMT and the pathogenesis of renal interstitial fibrosis.
Materials and MethodsAnimals and Experimental Design
Plg�/� and �/� mice on a C57BL6 background (8) were littermatesthat were bred in our animal facility. They were allowed to reach aminimum weight of 20 g before the initiation of the study. The geno-type of each mouse was confirmed by PCR analysis of DNA that wasextracted from tails using previously published primers (8). Groups ofage- and gender-matched Plg-deficient and wild-type mice were stud-ied 3, 7, 14, and 21 d after UUO and after sham surgery (n � 8 pergroup). All mice were female except for the day 7 UUO and sham timepoint. UUO surgery was performed under general anesthesia. The leftureter was ligated with 4.0 silk at two separate points in the UUOgroups. All mice were killed by exsanguination under general anesthe-sia. All procedures were performed in compliance with the guidelinesestablished by National Research Council Guide for the Care and Useof Laboratory Animals and were approved by our Institutional AnimalCare and Use Committee.
Kidney Tissue PreparationAfter exsanguination, the left kidney was procured, the capsule was
removed, and the kidney was weighed. The kidney was divided lon-
gitudinally and further subdivided. One half of the kidney was pre-pared for histologic studies: One piece (approximately one third) wasfixed in 10% buffered formalin and paraffin embedded; the remainingpiece (approximately two thirds) was imbedded in Tissue-Tek OCTcompound (Sakura Finetek, Torrence, CA) and snap-frozen. Pieces ofthe second half of the kidney were frozen at �80°C for total collagenmeasurement (approximately one fourth) and for the total RNA andprotein extraction (approximately three fourths).
Analysis of Tubulointerstitial FibrosisTotal renal collagen was measured biochemically as described pre-
viously (23). In brief, an accurately weighed portion of the kidney washomogenized in distilled water, hydrolyzed in 10 N HCl, and incu-bated at 110°C for 18 h. The hydrolysate was dried by speed-vacuumcentrifugation and redissolved in buffer (25 g of citric acid, 6 ml ofglacial acetic acid, 60 g of sodium acetate, and 17 g of sodium hydroxidein 500 ml [pH 6.0]). Total hydroxyproline in the hydrolysate wasdetermined according to the chemical method of Kivirikko et al. (24).Total collagen in the tissue was calculated on the assumption thatcollagen contains 12.7% hydroxyproline by weight. Final results wereexpressed as �g/mg kidney wet weight. UUO data were expressedrelative to wild-type sham samples (corrected to an average 5 �g/mgkidney wet weight) in each assay to compare all data together.
Kidney sections were stained with picrosirius red and photographedwith a computerized SPOT camera under polarized light microscopy(25). The percentage of Sirius red–stained tubulointerstitial area wasmeasured using Image-Pro Plus software (Media Cybernetics, SilverSpring, MD).
RNA Isolation, Northern Blot Analysis, and ReverseTranscription–PCR/Real-Time PCR
Total RNA was isolated from each mouse kidney or cultures of MCTcells by the phenol/guanidine isothiocyanate extraction method usingTRIZOL-BRL reagent (Gibco BRL Life Technologies, Bethesda, MD).Total RNA (15 �g) was loaded into individual wells and separated bya 1.0% agarose formaldehyde gel electrophoresis. A photomicrographof the ethidium bromide–stained gel was obtained to determine RNAloading equality. The RNA was transferred to a hybridization mem-brane (GeneScreen Plus; New England Nuclear Life Science Products,Boston, MA) and ultraviolet light cross-linked (UV Crosslinker; HoefferScientific Instruments, San Francisco, CA). cDNA probes were radiola-beled with 32P dCTP (3000 Ci/mmol) by random priming with T7
Quick Prime kit (Pharmacia Biotech, Piscataway, NJ). The blots werehybridized with the radiolabeled cDNA probes using the QuickHybhybridization buffer (Stratagene, La Jolla, CA). Autoradiographs wereobtained, and the density of each band was quantified using the Na-tional Institutes of Health Image program. The 18S ribosomal bands ofthe ethidium bromide–stained gels or the band density of the house-keeping gene cyclophilin-A were used to adjust for RNA loadinginequality.
The cDNA probes used were mouse Plg (from American TissueCulture Collection, Rockville, MD), rat TGF-�1 (from Dr. S.W. Qian,National Cancer Institute, Bethesda, MD) (26), human PAR-1 (a giftfrom Dr. Shaun Coughlin, University of California at San Francisco, SanFrancisco, CA) (27), mouse fibronectin and �-SMA (both are reversetranscription–PCR [RT-PCR] products), and mouse cyclophilin-A (Am-bion, Austin, TX).
For RT-PCR/real-time PCR, 1 �g of total RNA was applied in reversetranscription using BioScript kit (Bioline USA, Randolph, MA). A total of 0.5�l of the first-strand cDNA was used in each semiquantitative PCR (for�-SMA) or real-time PCR (for fibronectin) reaction using iQ SYBR Green
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 847

Supermix (Bio-Rad Laboratories, Hercules, CA). The primers used were de-signed using PrimerBank (http://pga.mgh.harvard.edu/primerbank/in-dex.html): Fibronectin forward 5�-CTGGGGTCACGTACCTCTTCA-3� andreverse 5�-AGTCGGTAGCCTGCTATACGG-3�; and �-SMA forward 5�-TATCGAGCACGGCATCATCA-3� and reverse 5�-TAAATGGGCACGTT-GTGGGT-3�. The expected PCR products were 181 and 288 bp, respectively.Real-time surveillance of fluorescence intensity that was emitted from theamplified product was performed with an iCycler PCR machine (Bio-RadLaboratories). The amplification efficiency was 97%, and the expected singlepeak was confirmed in the melting curve.
Protein Extraction and Zymographic AnalysisProtein was isolated from kidney tissue that had been stored at
�80°C. Pieces were individually ground into a fine powder underliquid nitrogen conditions, using a mortar and pestle that had beenprechilled with dry ice. The powder was mixed with homogenizingbuffer (0.05 M Tris [pH 7.6], 0.2% Triton X-100, and 0.1% SDS). Indi-vidual samples were then centrifuged for 5 min (14,000 � g), and theprotein concentration was measured in the supernatants using the BCAprotein assay reagent kit (Pierce Biotechnology, Rockford, IL). Sampleswere aliquotted and stored at �80°C.
Casein urokinase zymography was performed to evaluate renal plas-min activity using previously published methods (8). This procedure issimilar to casein Plg zymography for PA detection that we previouslydescribed (23) except that 20 IU/ml human high molecular weight uPA(American Diagnostica, Greenwich, CT) was cast in the 10% SDS poly-acrylamide gels rather than human Plg. Molecular weight markers andhuman plasmin standards (Sigma-Aldrich, St. Louis, MO) were loadedinto the outer wells. Plasmin-specific bands were verified by theirdisappearance when re-run in an identical gel that lacked uPA.
Western Blot AnalysisProtein samples (40 �g) were separated by 7.5 to 15% SDS-PAGE.
The proteins were transferred to a polyvinylidene difluoride mem-brane, and the immunoreactive protein was visualized using ECLenhanced chemiluminescence (Amersham Pharmacia Biotech). The pri-mary antibodies were horseradish peroxidase–conjugated monoclonalanti-human �-SMA (Dako Corp., Carpinteria, CA), rabbit anti–FSP-1antiserum (a gift from Dr. Eric Neilson, Vanderbilt University, Nash-ville, TN) (28), goat anti-mouse Plg kringle 5 antibody (R&D Systems,Minneapolis, MN), rabbit anti-thrombin receptor PAR-1 (Santa CruzBiotechnology), goat anti-mouse E-cadherin antibody (R&D Systems),rabbit anti–TGF-�1 polyclonal antibody (Santa Cruz Biotechnology),and rabbit anti-murine phospho-p44/42 MAPK (Thr202/Tyr204) oranti-total p44/42 MAPK antibodies (Cell Signaling Technology, Bev-erly, MA). The secondary antibodies were a set of horseradish peroxi-dase–conjugated Ig that included goat anti-mouse IgG (Sigma-Al-drich), rabbit anti-rat or rabbit anti-goat IgG, and goat anti-rabbit IgG(all from Chemicon International, Temecula, CA). Protein loadingequality was determined by probing the same blots with FITC-conju-gated anti–�-actin mAb (Sigma) and scanning with a Typhoon 9410Variable Mode Imager (Amersham Biosciences) or by amido blackstaining as used previously (29).
ImmunohistologyImmunochemical staining was performed on 4-�m sections of par-
affin-embedded kidney tissue. Primary antibodies used were from thesame sources as were used for Western blot analysis, described above.Staining was performed using the ABC ELITE kit that included biotin-ylated secondary antibodies (Vector Laboratories, Burlingame, CA).Sections that were stained with the secondary antibodies alone were
confirmed to be negative. Images were captured with a SPOT camera,quantified using Image-Pro Plus software, and the results were ex-pressed as percentage of positive tubulointerstitial area (23).
PAI-1/Luciferase TGF-� Bioactivity AssayTGF-� bioactivity was measured in 21 d UUO and sham kidney
protein extracts using a TGF-�–responsive mink lung epithelial cell line(a gift from Dr. Daniel B. Rifkin, New York University Medical Center,New York, NY) (30) with minor modifications as described previously(23). Briefly, kidney samples (1 �l that contained approximately 20 �g
Figure 1. Plasmin(ogen) expression and activity. (A) Northernblot analysis of plasminogen (Plg) gene expression in the kid-ney and liver of Plg�/� and Plg�/� mice. The two specific Plgtranscript variants (2.9 and 5.2 kb, indicated by arrowheads)were detected only in the livers of wild-type mice. (B) Zymo-graphic analysis of plasmin activity (arrowhead) in kidneyhomogenates (40 �g of protein) from Plg�/� and Plg�/� mice7 and 14 d after unilateral ureteral obstruction (UUO) or shamsurgeries. (C) Western blot analysis identified a specific 85-kDPlg band in Plg�/� kidneys; the band density increased inresponse to UUO. *P � 0.05, UUO versus sham. A human Plgstandard (hPlg) was loaded in the outer lane as a positivecontrol. (D) Localization of renal Plg protein was determinedby indirect immunoperoxidase staining using a specific mousePlg antibody. Representative photomicrographs illustrate kid-ney morphology and focal Plg expression in tubules and theinterstitium of Plg�/� mice at 14 d of UUO. Staining wasnegative in sham kidneys and UUO kidneys from the Plg�/�mice. Magnification, �400.
848 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007

of protein, triplicate samples from five individual animals per group) inCelLytic MT Cell Lysis Reagent (Sigma-Aldrich) were added directly tomonolayers of confluent mink lung epithelial cell cultures in 96-wellplates in serum free media (SFM). A standard curve of TGF-� activitywas generated using serial dilutions of recombinant human activeTGF-�1 (Amersham Pharmacia Biotech). After overnight incubation at37°C in 5% CO2, cells were harvested and luciferase activity wasmeasured using the Enhanced Luciferase Assay Kit (Pharmingen, SanDiego, CA). Luminescence, expressed as counts per minute (cpm), wascorrected for protein loading of each sample.
Cell Culture and Plasmin TreatmentA mouse cortical tubular epithelial cell line (MCT) was provided by
Dr. Carolyn Kelly (University of California, San Diego, CA) (31). Cellswere maintained in DMEM/Ham’s F12 media (1:1) supplemented with5% FCS. The cells were seeded in 12-well culture plates, grown to 60 to70% confluence in full medium that contained 5% FCS overnight, andthen changed to SFM after two washes with SFM. Human plasmin(Sigma-Aldrich) dissolved in SFM was added to the culture medium ata final concentration of 10 or 20 �g/ml. Cells were incubated foranother 24 to 72 h. For control experiments, the cells were incubatedwith an equal volume of SFM alone. For evaluation of ERK signalactivation that was directly induced by plasmin, MCT cells were ren-dered quiescent in SFM overnight and stimulated with plasmin (20�g/ml SFM) for a period that ranged from 12 min to 24 h. Results areexpressed as the mean of two independent experiments.
Protein was isolated from harvested cells in lysis buffer (50 mM Tris[pH 7.6] and 5% SDS). Western blot analysis was performed for PAR-1,p44/42 MAPK and phospho-p44/42 MAPK (Thr202/Tyr204), E-cad-herin, and FSP-1 proteins. In a separate set of experiments, cells weregrown on coverslips and immunostained in situ to identify changes incellular E-cadherin and FSP-1 expression after treatment with plasminfor 48 h. The FITC-conjugated goat anti-rabbit secondary antibody waspurchased from Organon Teknika (West Chester, PA).
Total RNA was isolated in additional independent experiments fromcells that were cultured in the presence or absence of plasmin in SFMfor 96 h (n � 3) using TRIZOL-BRL reagent (Gibco BRL Life Technol-ogies). RT-PCR or quantitative real-time PCR was performed to eval-uate �-SMA and fibronectin gene expression, respectively.
Manipulation of PAR-1 Gene Expression and FunctionFor determination of whether cellular PAR-1 is involved in plasmin-
induced EMT, MCT cells that were grown in six-well plates weretransfected with a vector that expresses human PAR-1 (pBJ-PAR1) orempty vector (mock transfection) using siPort Transfectant (Ambion)(27). Overexpression of the PAR-1 gene was confirmed by Northernblot analysis at 48 h after gene transfection. In addition to overexpress-ing PAR-1, a group of cells, designated to suppress PAR-1 function,were pretreated with 10 �M PAR-1–specific inhibitor pepducinP1pa1-12 (pa1-RCLSSSAVANRS peptide, synthesized, and purified bySigma-Genosys) for 5 min before plasmin addition. The P1pa1-12 pep-ducin that comprises the N-terminal portion of the i3 loop of PAR-1lacks agonist activity but is a full antagonist of PAR-1–dependentinositol phosphate production and Ca2� signaling (32–34). For the ERKsignaling study, cells were rendered quiescent in SFM overnight, thenstimulated with plasmin for 45 min. For the EMT studies, cells wereincubated with plasmin (20 �g/ml) in SFM for 48 h. Protein and totalRNA were then extracted and subjected to Western blot or Northernblot analysis.
For further determination of whether cellular PAR-1 is required forplasmin-induced EMT, a PAR-1 small interfering RNA (siRNA) strat-
Figure 2. Plg gene disruption attenuates renal fibrosis. (A) Rep-resentative photomicrographs of Sirius red (SR) staining illus-trates significantly fewer collagen fibrils 3, 7, and 21 d afterUUO in Plg�/� compared with the Plg�/� mice. (B) Thehistogram shows the SR-stained interstitial area (%) in shamand obstructed kidneys (3, 7, 14, and 21 d). (C) Total collagencontent in sham-operated kidneys or obstructed kidneys 3, 7,14, and 21 d after UUO was measured biochemically. Resultsare expressed as �g hydroxyproline/mg wet kidney weight.Data are means � 1 SD (n � 6 per group). *P � 0.05, Plg�/�versus Plg�/�. Magnification, �400.
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 849
egy was used to downregulate PAR-1 expression. MCT cells that weregrown in 12-well plates (n � 6 per group) were transfected with siRNAthat specifically targets the mouse PAR-1 gene sequence or a negativecontrol scrambled siRNA (10 �M; Santa Cruz Biotechnology) usingoligofectamine reagent (5 �g/ml; Invitrogen, Gaithersburg, MD).Thirty hours after transfection, three wells of cells from each groupwere harvested to evaluate PAR-1 expression by Western blotting. Theremaining groups of cells were incubated with plasmin (10 �g/ml) foran additional 16 h (overnight) and harvested for evaluation of PAR-1and FSP-1 expression. Three wells of cells in additional experimentswere harvested to evaluate E-cadherin expression by flow cytometry(FACSCalibur; Becton Dickinson, San Jose, CA). The primary E-cad-herin antibody used in flow cytometry was the same as was used forWestern blotting. E-cadherin was labeled with a phycoerythrin-conju-gated secondary antibody (R&D Systems). Experiments were repeatedtwice independently.
ERK Signaling Inhibition and Cell Migration AssayFor determination of the contribution of ERK activation to plasmin-
induced EMT, cells were preincubated with the MEK inhibitor UO126(10 or 20 �M; Cell Signaling Technology) 2 h before plasmin was addedfor 48 h. Western blots were then performed to evaluate levels ofphosphorylated ERK (pERK) and cellular FSP-1 expression.
Tubular cell migration was evaluated using 6.5-mm Transwell poly-carbonate filters of 8-�m pore size (Corning, Corning, NY) (35). Thelower side of the filter was coated with 50 �l of plasmin (20 �g/ml) orBSA (2%). Plasmin- or BSA-coated filters were placed into migrationassay medium (DMEM/F12 with 0.5% FCS), with or without 20 �MMEK inhibitor UO126 (n � 6), and MCT cells (1 � 104) in 100 �l of thesame medium were seeded onto the upper side of each filter. After 96 hof incubation at 37°C, filters were fixed with 4% paraformaldehyde inPBS and stained with 0.1% Coomassie blue in 10% methanol/10%acetic acid. Cells on the upper surface of the filters were gently re-moved with cotton-tipped applicators. Cells and cell extensions thatpassed through the pores were counted in five randomly selected,nonoverlapping �400 microscopic fields. Results are expressed asmean percentage of filter pores that were occupied by migrating cells intwo independent experiments.
Statistical AnalysesAll results are expressed as means � 1 SD. Results were analyzed by
the Mann Whitney U test. Correlation analysis was performed betweenthe percentage of �-SMA� interstitial area and E-cadherin proteinlevels. P � 0.05 was considered statistically significant.
ResultsPlasmin(ogen) Renal Expression
The Plg gene transcript was detected only in livers from thePlg�/� mice; it was undetectable in the liver of Plg�/� miceand the kidneys of mice of either genotype (Figure 1A). Plasminactivity (Figure 1B) and Plg protein (Figure 1C) were readilydetectable in UUO kidneys of Plg�/� mice, with significantincreases observed after UUO (1.6-, 1.9-, and 2.3-fold increasedon days 3, 7, and 14, respectively; all P � 0.05 versus sham). Byimmunostaining, Plg was identified in renal tubular and inter-stitial locations of the wild-type UUO mice (Figure 1D).
Severity of Interstitial FibrosisRenal fibrosis, as measured by Sirius red staining of collagen
fibrils, increased progressively after UUO (two-, three- and
four-fold increase in Plg�/� mice, days 3, 7, and 21, respec-tively; UUO versus sham, all time points P � 0.05; Figure 2, Aand B). The Sirius red–positive interstitial areas were signifi-cantly less in Plg�/� compared with the Plg�/� mice (e.g.,1.2 � 0.4 versus 2.3 � 0.4% and 1.9 � 0.3 versus 3.0 � 0.6%, �/�
versus �/�, days 7 and 21 of UUO). Reduced fibrosis in thePlg�/� mice on days 7 and 21 after UUO was reconfirmedbiochemically by measuring total kidney collagen (41 and 35%reduction, respectively; both P � 0.05; Figure 2C).
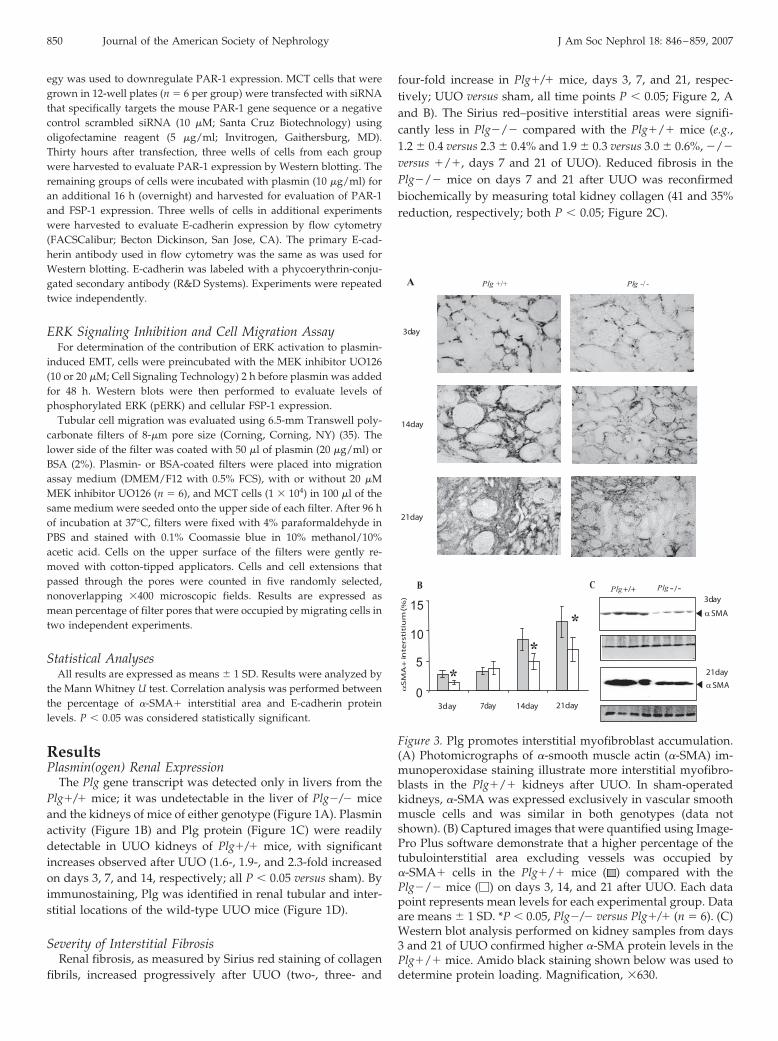
Figure 3. Plg promotes interstitial myofibroblast accumulation.(A) Photomicrographs of �-smooth muscle actin (�-SMA) im-munoperoxidase staining illustrate more interstitial myofibro-blasts in the Plg�/� kidneys after UUO. In sham-operatedkidneys, �-SMA was expressed exclusively in vascular smoothmuscle cells and was similar in both genotypes (data notshown). (B) Captured images that were quantified using Image-Pro Plus software demonstrate that a higher percentage of thetubulointerstitial area excluding vessels was occupied by�-SMA� cells in the Plg�/� mice ( ) compared with thePlg�/� mice (�) on days 3, 14, and 21 after UUO. Each datapoint represents mean levels for each experimental group. Dataare means � 1 SD. *P � 0.05, Plg�/� versus Plg�/� (n � 6). (C)Western blot analysis performed on kidney samples from days3 and 21 of UUO confirmed higher �-SMA protein levels in thePlg�/� mice. Amido black staining shown below was used todetermine protein loading. Magnification, �630.
850 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007

Interstitial �-SMA Expression and EMTIn normal kidneys, �-SMA expression is limited to vessels
with no significant difference observed between Plg�/� and�/� mice. After UUO, �-SMA– expressing cells begin topopulate the interstitium in mice of both genotypes, but theinterstitial area that is occupied by �-SMA� cells was sig-nificantly less in the Plg�/� mice (e.g., 1.3 � 0.3 versus 2.7 �
0.5% and 6.8 � 2 versus 11.4 � 2.6%, on days 3 and 21, �/�
versus �/�; P � 0.05; Figure 3, A and B). Days 3 and 21Western blot analysis reconfirmed higher �-SMA levels inthe Plg�/� mice (2.4- and 1.8-fold, respectively; both P �
0.05; Figure 3C). The �-SMA� interstitial area was inverselycorrelated with tubular E-cadherin protein levels in kidneyswith advanced obstructive injury (days 14 and 21; mice ofboth genotypes combined; r � �0.95, R2 � 0.91, P � 0.05),indicating EMT as one likely source of these �-SMA� inter-stitial cells.
The loss of E-cadherin and de novo FSP-1 expression is an-other morphologic feature of tubular cells that undergo EMT.As evaluated by immunoblot analysis and immunohistochem-istry, tubular epithelial cells of the Plg�/� mice gradually lostE-cadherin in response to UUO (Figure 4). By day 21, E-cad-herin protein levels had decreased by seven-fold comparedwith the sham kidneys (P � 0.05). In the Plg�/� mice, tubularE-cadherin protein was preserved to a significantly greaterextent (two- to four-fold higher) on days 7, 14, and 21 of UUO,despite initially similar expression levels in sham and day 3UUO kidneys (Figure 4).
Significantly more FSP-1 protein was detected from day 7 today 21 in the kidneys of Plg�/� compared with the �/� mice(Figure 4). Whereas in the Plg�/� mice FSP-1 expression wasremarkably increased by day 7, this did not happen in thePlg�/� mice until day 14, and the amount of FSP-1 actuallydeclined slightly on day 21 in the Plg�/� mice. This dynamicchange resulted in a greater difference in FSP-1 expressionbetween Plg�/� and the �/� mice on days 7 and 21 (12-foldand two-fold reduced in the �/�, respectively; P � 0.05;Figure 4).
PAR-1 and ERK ActivationPlasmin is known to activate/deactivate PAR-1 by proteoly-
sis (21,36). PAR-1 protein is abundantly expressed by normalcortical tubules. By both Western blotting and immunohisto-chemical staining, PAR-1 protein (50 kD) levels were reduced
Figure 4. Plg deficiency reduces tubular epithelial-to-mesenchy-mal transition (EMT). (Top) The graph illustrates the results ofE-cadherin protein levels as measured by Western blotting.Representative immunoblots from days 7 and 14 of UUO illus-trate higher E-cadherin protein (120 kD) levels in the Plg�/�mice. Amido black staining shown below was used to correctfor differences in protein loading. Representative photomicro-
graphs from day 7 of UUO illustrate stronger E-cadherin stain-ing in kidneys of the Plg�/� mice compared with the Plg�/�mice. (Bottom) The graph illustrates the results of fibroblast-specific protein-1 (FSP-1) protein levels as analyzed by Westernblotting. Representative immunoblots from days 7 and 21 ofUUO illustrate significantly reduced FSP-1 protein (11.5 kD,arrowhead) levels in the Plg�/� mice. Amido black stainingshown below was used to correct for differences in proteinloading. FSP-1 immunostaining of a kidney section from day 7of UUO illustrates tubular cells (arrow) and cells. Data aremeans � 1 SD. *P � 0.05, Plg�/� versus Plg�/� (n � 4). ,Plg�/�; �, Plg�/�. Magnification, �630.
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 851

compared with sham kidneys 7 d after UUO (168 � 21 versus83 � 3 arbitrary units in Plg�/� mice; P � 0.05; Figure 5A).PAR-1 levels were similar in the Plg�/� and the Plg�/�
groups, indicating that PAR-1 was activated/degraded by var-ious proteases during UUO. Tubules seemed to be the primarysite of PAR-1 expression in the kidneys (Figure 5A).
Plasmin has been reported to initiate MAPK ERK signalingvia activation of the PAR-1 receptor (21,36). Inactive ERK wasabundant in the cytoplasm of many tubular cells, whereas ERKthat was activated by phosphorylation was rarely detected byimmunostaining in tubules of sham kidneys from both geno-types (data not shown). In response to UUO, a significantamount of ERK was Thr202/Tyr204 phosphorylated to agreater extent in the wild-type mice (Figure 5B). By Westernblot analysis, the ratio of pERK/total ERK was significantlyhigher in the kidneys of Plg�/� mice (0.82 � 0.14 versus 0.26 �
0.04; 0.94 � 0.14 versus 0.42 � 0.16 units, Plg�/� versus Plg�/�,days 7 and 21 of UUO, respectively; Figure 5B).
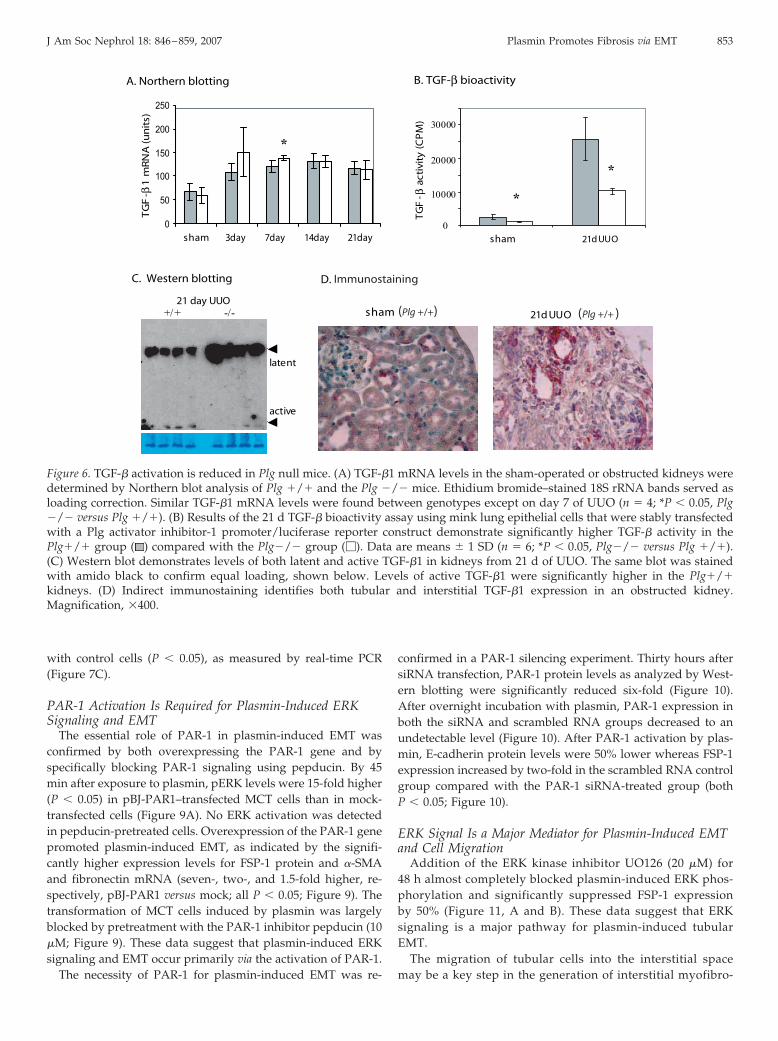
TGF-� Gene Expression and Protein ActivationTGF-�1 mRNA levels were significantly elevated (2�) com-
pared with the sham-operated kidneys of both genotypes at alltime points (Figure 6A). The differences between the Plg �/�
and the �/� groups were NS except on day 7 of UUO (139 �
4.6 versus 122 � 13 units, �/� versus �/�; P � 0.05). However,on day 21 of UUO, TGF-� activity was significantly lower in Plg�/� mice (10303 � 840 versus 25896 � 6507 cpm, Plg �/�
versus Plg �/�) as measured by the PAI-1/luciferase assay(Figure 6B). TGF-�1 Western blot analysis confirmed that theactive form of the protein was significantly higher (5�; P �
0.05), whereas levels of latent TGF-�1 protein were three-foldlower in the Plg�/� mice (Figure 6C), indicating less TGF-�activation in vivo in the absence of plasmin activity. Immuno-staining identified tubular and interstitial locations of TGF-�1in obstructed kidneys of both genotypes (Figure 6D).
In Vitro Studies: Plasmin Activated PAR-1 Receptor andInduced ERK Signaling and EMT
PAR-1, which can be activated by plasmin proteolytic activ-ity, was degraded within 30 min after exposure to plasmin (20�g/ml), coincident with the initiation of ERK phosphorylation(Figure 7A). Plasmin strongly induced ERK phosphorylation inMCT cells as early as 30 min. By 48 h, pERK had increasedsix-fold in plasmin-treated compared with control MCT cells(P � 0.05). Within 2 h, E-cadherin protein levels began todecrease (data not shown) and had completely disappeared48 h after the addition of plasmin (P � 0.05, compared withmedia alone), suggesting possible degradation of E-cadherin byplasmin. At 48 h, de novo FSP-1 expression was readily detectedby immunoblotting and in situ immunocytochemistry (Figures7 and 8). �-SMA, the myofibroblast hallmark, was activelytranscribed as shown in semiquantitative RT-PCR (Figure 7C).By 96 h, the cells that were transformed by plasmin exposureexpressed 10-fold higher levels of fibronectin mRNA compared
Figure 5. Protease-activated receptor-1 (PAR-1) expression and extracellular signal–regulated kinase (ERK) signaling in Plg nullmice. (A) Immunohistochemical photomicrographs illustrate strong tubular PAR-1 expression (Fast red) in Plg�/� sham kidneysthat decreased with time after UUO. Not shown, a similar pattern was observed in the Plg�/� kidney. The Western blotillustrates reduced expression of PAR-1 protein (50 kD), a plasmin-activated and signaling receptor, at 7 d of UUO in kidneys ofboth genotypes. (B) Immunoperoxidase staining for active phosphorylated ERK (pERK) illustrates lower levels in the Plg�/�kidney on day 7 of UUO. Immunoblots from kidneys from days 7 and 21 of UUO illustrate significantly higher pERK levels (p44and p42 bands indicated by double arrowheads) in the Plg�/� mice (n � 4 mice per group). Magnification, �500.
852 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007
with control cells (P � 0.05), as measured by real-time PCR(Figure 7C).
PAR-1 Activation Is Required for Plasmin-Induced ERKSignaling and EMT
The essential role of PAR-1 in plasmin-induced EMT wasconfirmed by both overexpressing the PAR-1 gene and byspecifically blocking PAR-1 signaling using pepducin. By 45min after exposure to plasmin, pERK levels were 15-fold higher(P � 0.05) in pBJ-PAR1–transfected MCT cells than in mock-transfected cells (Figure 9A). No ERK activation was detectedin pepducin-pretreated cells. Overexpression of the PAR-1 genepromoted plasmin-induced EMT, as indicated by the signifi-cantly higher expression levels for FSP-1 protein and �-SMAand fibronectin mRNA (seven-, two-, and 1.5-fold higher, re-spectively, pBJ-PAR1 versus mock; all P � 0.05; Figure 9). Thetransformation of MCT cells induced by plasmin was largelyblocked by pretreatment with the PAR-1 inhibitor pepducin (10�M; Figure 9). These data suggest that plasmin-induced ERKsignaling and EMT occur primarily via the activation of PAR-1.
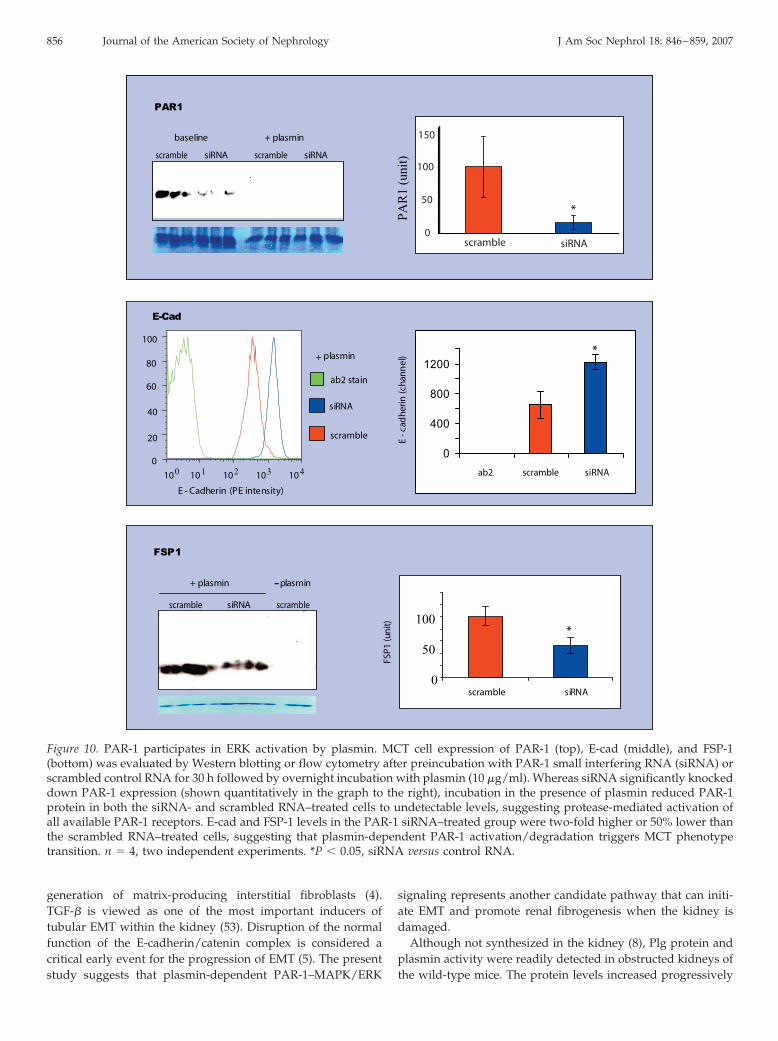
The necessity of PAR-1 for plasmin-induced EMT was re-
confirmed in a PAR-1 silencing experiment. Thirty hours aftersiRNA transfection, PAR-1 protein levels as analyzed by West-ern blotting were significantly reduced six-fold (Figure 10).After overnight incubation with plasmin, PAR-1 expression inboth the siRNA and scrambled RNA groups decreased to anundetectable level (Figure 10). After PAR-1 activation by plas-min, E-cadherin protein levels were 50% lower whereas FSP-1expression increased by two-fold in the scrambled RNA controlgroup compared with the PAR-1 siRNA-treated group (bothP � 0.05; Figure 10).
ERK Signal Is a Major Mediator for Plasmin-Induced EMTand Cell Migration
Addition of the ERK kinase inhibitor UO126 (20 �M) for48 h almost completely blocked plasmin-induced ERK phos-phorylation and significantly suppressed FSP-1 expressionby 50% (Figure 11, A and B). These data suggest that ERKsignaling is a major pathway for plasmin-induced tubularEMT.
The migration of tubular cells into the interstitial spacemay be a key step in the generation of interstitial myofibro-
Figure 6. TGF-� activation is reduced in Plg null mice. (A) TGF-�1 mRNA levels in the sham-operated or obstructed kidneys weredetermined by Northern blot analysis of Plg �/� and the Plg �/� mice. Ethidium bromide–stained 18S rRNA bands served asloading correction. Similar TGF-�1 mRNA levels were found between genotypes except on day 7 of UUO (n � 4; *P � 0.05, Plg�/� versus Plg �/�). (B) Results of the 21 d TGF-� bioactivity assay using mink lung epithelial cells that were stably transfectedwith a Plg activator inhibitor-1 promoter/luciferase reporter construct demonstrate significantly higher TGF-� activity in thePlg�/� group ( ) compared with the Plg�/� group (�). Data are means � 1 SD (n � 6; *P � 0.05, Plg�/� versus Plg �/�).(C) Western blot demonstrates levels of both latent and active TGF-�1 in kidneys from 21 d of UUO. The same blot was stainedwith amido black to confirm equal loading, shown below. Levels of active TGF-�1 were significantly higher in the Plg�/�kidneys. (D) Indirect immunostaining identifies both tubular and interstitial TGF-�1 expression in an obstructed kidney.Magnification, �400.
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 853

blasts that are derived by EMT in vivo. With the use of a cellmigration assay, the movement of MCT cells was signifi-cantly enhanced by 31% in the presence of plasmin (Figure11, C through G). Suppression of ERK signal transductionwith UO126 blocked MCT cell migration in both the presenceand the absence of plasmin. These data suggest that the ERK
signaling pathway regulates both plasmin-dependent and-independent tubular cell migration.
DiscussionThis study confirms that in vivo, the net effect of plasmin in
chronic kidney disease (CKD) that is induced by complete UUOis to enhance interstitial fibrosis, as recently reported by Edgt-ton et al. (14). This study provides new mechanistic insights,indicating that plasmin promotes the transition of tubular ep-ithelial cells into cells of a mesenchymal phenotype, an eventthat was thought to be involved in the genesis of active matrix-producing renal interstitial fibroblasts. The data demonstratethat this pro-EMT effect of plasmin can be attributed at least inpart to PAR-1–mediated ERK activation. Although thrombin isconsidered to be the primary PAR-1–activating protease, thisstudy provides additional evidence that plasmin is an alterna-tive and biologically important receptor activating protease, aspreviously suggested (21). Other known PAR-1 activators in-clude activated protein C (37), Factor Xa (38), matrix metallo-
A
0’ 12’ 30’ 1h 2h 24h 48h
pERK
ERK
SFM + plasmin
0’ 30’ 2h
PAR1
1h 15’ 5’
β-actin
PAR1
β-actin
0’ 15’ 30’ 2h
SFM alone
durationduration
duration0’ 12’ 30’ 1h 2h 24h 48h duration
pERK
ERK
PAR1 and pERK
B
FSP1
media alone plasmin 48h
E-Cad
*
*
E-Cad FSP1
Pro
tein
leve
l (ar
bit
ary
un
its)
plasmin
SFM
120
80
40
0
E-cad and FSP1
SFM plasmin
2
6
10*
FN /β
-act
in m
RNA
SMAβ-actinα
media alone plasmin 48h
C
αSMA and FN
Figure 7. Plasmin induces ERK signaling and EMT in vitro. Afterquiescence in serum free media (SFM) overnight, mouse corti-cal tubular epithelial (MCT) cells were treated with 20 �g/mlactive plasmin in SFM or SFM alone for variable periods of timeas indicated. (A) Western blots illustrate changes in expressionof the receptor PAR-1, total ERK, and pERK. (B) Western blotson the left illustrate that MCT cells begin to express FSP-1whereas E-cadherin (E-cad) expression declines when culturedwith plasmin for 48 h. Below each Western blot is the amidoblack staining used for protein loading correction. The graphsto the right represent the results of immunoblot band densi-tometry analysis at 48 h, expressed in arbitrary units, with MCTcultured in SFM alone shown in blue and in SFM plus plasminshown in red. Data are means � 1 SD; n � 4 experiments. *P �0.05. (C) The reverse transcription–PCR gel on the left demon-strates de novo expression of �-SMA mRNA in the cells after48 h of incubation with plasmin. The �-actin mRNA levelsserved as loading control. Real-time PCR data on the rightshow that fibronectin (FN) mRNA expression is increased 10-fold compared with control cells (blue bar) in the MCT cells thatwere exposed to plasmin for 96 h (red bar). Data are means �1 SD; n � 3, each sample assayed in triplicate. *P � 0.05.
Figure 8. Immunohistochemical evidence of plasmin-inducedEMT in vitro. Representative photomicrographs illustrate E-cad(A and B) and FSP-1 (C through E) MCT cell immunostaining.Normal MCT cells express E-cad but not FSP-1 proteins (A andC). When cultured for 48 h with plasmin, MCT cells lose E-cad(B) and begin to express FSP-1 (D and E). Staining with sec-ondary antibodies alone revealed no signal. The arrowheadhighlights a transformed cell undergoing mitosis. Magnifica-tions: �630 in A, B, and E; �400 in C and D.
854 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007
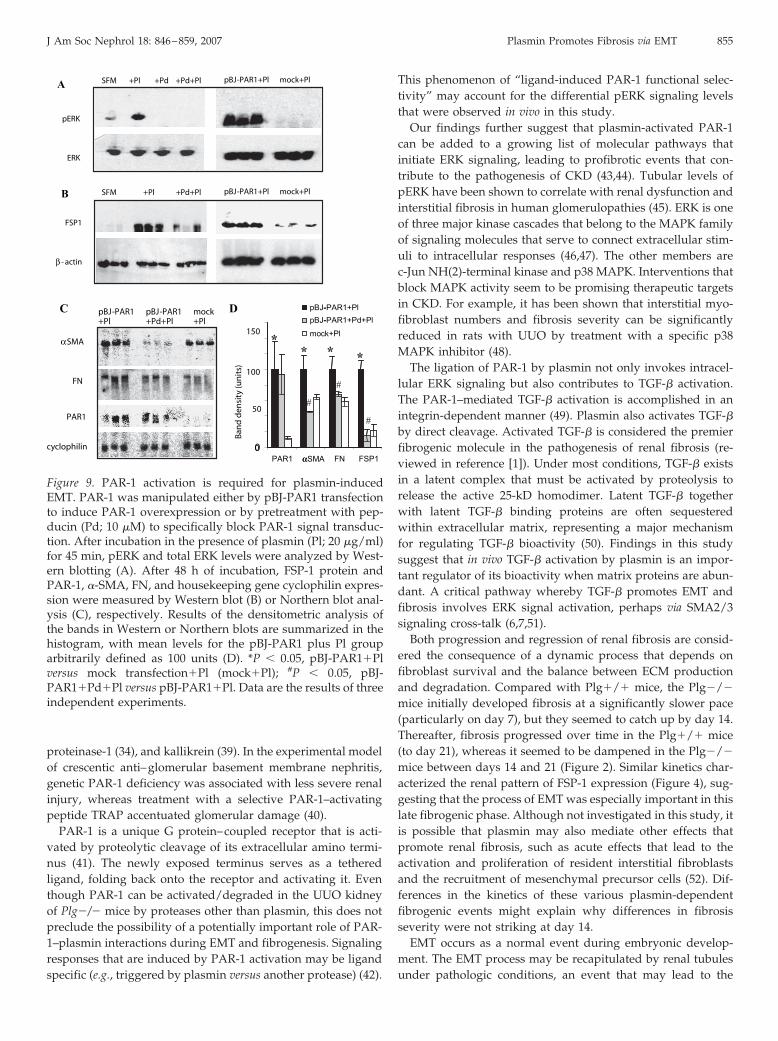
proteinase-1 (34), and kallikrein (39). In the experimental modelof crescentic anti–glomerular basement membrane nephritis,genetic PAR-1 deficiency was associated with less severe renalinjury, whereas treatment with a selective PAR-1–activatingpeptide TRAP accentuated glomerular damage (40).
PAR-1 is a unique G protein–coupled receptor that is acti-vated by proteolytic cleavage of its extracellular amino termi-nus (41). The newly exposed terminus serves as a tetheredligand, folding back onto the receptor and activating it. Eventhough PAR-1 can be activated/degraded in the UUO kidneyof Plg�/� mice by proteases other than plasmin, this does notpreclude the possibility of a potentially important role of PAR-1–plasmin interactions during EMT and fibrogenesis. Signalingresponses that are induced by PAR-1 activation may be ligandspecific (e.g., triggered by plasmin versus another protease) (42).
This phenomenon of “ligand-induced PAR-1 functional selec-tivity” may account for the differential pERK signaling levelsthat were observed in vivo in this study.
Our findings further suggest that plasmin-activated PAR-1can be added to a growing list of molecular pathways thatinitiate ERK signaling, leading to profibrotic events that con-tribute to the pathogenesis of CKD (43,44). Tubular levels ofpERK have been shown to correlate with renal dysfunction andinterstitial fibrosis in human glomerulopathies (45). ERK is oneof three major kinase cascades that belong to the MAPK familyof signaling molecules that serve to connect extracellular stim-uli to intracellular responses (46,47). The other members arec-Jun NH(2)-terminal kinase and p38 MAPK. Interventions thatblock MAPK activity seem to be promising therapeutic targetsin CKD. For example, it has been shown that interstitial myo-fibroblast numbers and fibrosis severity can be significantlyreduced in rats with UUO by treatment with a specific p38MAPK inhibitor (48).
The ligation of PAR-1 by plasmin not only invokes intracel-lular ERK signaling but also contributes to TGF-� activation.The PAR-1–mediated TGF-� activation is accomplished in anintegrin-dependent manner (49). Plasmin also activates TGF-�by direct cleavage. Activated TGF-� is considered the premierfibrogenic molecule in the pathogenesis of renal fibrosis (re-viewed in reference [1]). Under most conditions, TGF-� existsin a latent complex that must be activated by proteolysis torelease the active 25-kD homodimer. Latent TGF-� togetherwith latent TGF-� binding proteins are often sequesteredwithin extracellular matrix, representing a major mechanismfor regulating TGF-� bioactivity (50). Findings in this studysuggest that in vivo TGF-� activation by plasmin is an impor-tant regulator of its bioactivity when matrix proteins are abun-dant. A critical pathway whereby TGF-� promotes EMT andfibrosis involves ERK signal activation, perhaps via SMA2/3signaling cross-talk (6,7,51).
Both progression and regression of renal fibrosis are consid-ered the consequence of a dynamic process that depends onfibroblast survival and the balance between ECM productionand degradation. Compared with Plg�/� mice, the Plg�/�
mice initially developed fibrosis at a significantly slower pace(particularly on day 7), but they seemed to catch up by day 14.Thereafter, fibrosis progressed over time in the Plg�/� mice(to day 21), whereas it seemed to be dampened in the Plg�/�
mice between days 14 and 21 (Figure 2). Similar kinetics char-acterized the renal pattern of FSP-1 expression (Figure 4), sug-gesting that the process of EMT was especially important in thislate fibrogenic phase. Although not investigated in this study, itis possible that plasmin may also mediate other effects thatpromote renal fibrosis, such as acute effects that lead to theactivation and proliferation of resident interstitial fibroblastsand the recruitment of mesenchymal precursor cells (52). Dif-ferences in the kinetics of these various plasmin-dependentfibrogenic events might explain why differences in fibrosisseverity were not striking at day 14.
EMT occurs as a normal event during embryonic develop-ment. The EMT process may be recapitulated by renal tubulesunder pathologic conditions, an event that may lead to the
SFM +Pl +Pd +Pd+Pl pBJ-PAR1+Pl mock+PlA
B
pERK
ERK
FSP1
pBJ-PAR1+Pl mock+Pl
β - actin
SFM +Pl +Pd+Pl
pBJ-PAR1 +Pl
mock +Pl
pBJ-PAR1 +Pd+Pl
C
αSMA
PAR1
cyclophilin
FN
0PAR1 αSMA FN FSP1
pBJ-PAR1+Pl
pBJ-PAR1+Pd+Pl
mock+Pl
0α
-
-
50
150
100
** * *
#
#
#
Ban
d d
ensi
ty (u
nit
s)
D
Figure 9. PAR-1 activation is required for plasmin-inducedEMT. PAR-1 was manipulated either by pBJ-PAR1 transfectionto induce PAR-1 overexpression or by pretreatment with pep-ducin (Pd; 10 �M) to specifically block PAR-1 signal transduc-tion. After incubation in the presence of plasmin (Pl; 20 �g/ml)for 45 min, pERK and total ERK levels were analyzed by West-ern blotting (A). After 48 h of incubation, FSP-1 protein andPAR-1, �-SMA, FN, and housekeeping gene cyclophilin expres-sion were measured by Western blot (B) or Northern blot anal-ysis (C), respectively. Results of the densitometric analysis ofthe bands in Western or Northern blots are summarized in thehistogram, with mean levels for the pBJ-PAR1 plus Pl grouparbitrarily defined as 100 units (D). *P � 0.05, pBJ-PAR1�Plversus mock transfection�Pl (mock�Pl); #P � 0.05, pBJ-PAR1�Pd�Pl versus pBJ-PAR1�Pl. Data are the results of threeindependent experiments.
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 855
generation of matrix-producing interstitial fibroblasts (4).TGF-� is viewed as one of the most important inducers oftubular EMT within the kidney (53). Disruption of the normalfunction of the E-cadherin/catenin complex is considered acritical early event for the progression of EMT (5). The presentstudy suggests that plasmin-dependent PAR-1–MAPK/ERK
signaling represents another candidate pathway that can initi-ate EMT and promote renal fibrogenesis when the kidney isdamaged.
Although not synthesized in the kidney (8), Plg protein andplasmin activity were readily detected in obstructed kidneys ofthe wild-type mice. The protein levels increased progressively
Figure 10. PAR-1 participates in ERK activation by plasmin. MCT cell expression of PAR-1 (top), E-cad (middle), and FSP-1(bottom) was evaluated by Western blotting or flow cytometry after preincubation with PAR-1 small interfering RNA (siRNA) orscrambled control RNA for 30 h followed by overnight incubation with plasmin (10 �g/ml). Whereas siRNA significantly knockeddown PAR-1 expression (shown quantitatively in the graph to the right), incubation in the presence of plasmin reduced PAR-1protein in both the siRNA- and scrambled RNA–treated cells to undetectable levels, suggesting protease-mediated activation ofall available PAR-1 receptors. E-cad and FSP-1 levels in the PAR-1 siRNA–treated group were two-fold higher or 50% lower thanthe scrambled RNA–treated cells, suggesting that plasmin-dependent PAR-1 activation/degradation triggers MCT phenotypetransition. n � 4, two independent experiments. *P � 0.05, siRNA versus control RNA.
856 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007

over time, possibly as a result of persistently enhanced vascularpermeability after UUO (9). Once deposited and activatedwithin the kidney, plasmin may ligate tubular PAR-1. Plasminbinding to the cellular receptor PAR-1 was shown to activateMAPK/ERK signal transduction in cultured MCT cells and toinduce phenotypic alterations to FSP-1 and �-SMA–positive/E-cadherin–negative transitional cells. In addition, plasmin wasshown to enhance epithelial cell migration, suggesting a mech-anism that facilitates the translocation of transdifferentiatedtubular epithelia into the interstitial space. The ERK signalingpathway seems to be downstream of PAR-1 activation and iscritical for plasmin-induced EMT. Blockade of plasmin-acti-vated ERK signaling, with either the pepducin-based PAR-1inhibitor or the ERK kinase inhibitor UO126, significantly in-hibited tubular cell EMT and migration. In vivo, targeted dis-ruption of the Plg gene also resulted in significantly less ERKactivation and interstitial myofibroblast recruitment in re-sponse to chronic UUO. These observations support the viewthat plasmin-dependent ERK activation is a significant path-way for signaling tubular EMT and interstitial fibrosis.
Although the finding of reduced fibrosis in Plg�/� mice onday 21 is in agreement with the findings of Edgtton et al. (14),only our study detected Plg genotype–dependent differences in�-SMA (and FSP-1) expression. Although the reason for thediscrepancy is not clear, it may be partially due to differences ingenetic backgrounds of the mice that were used in these twostudies (75 versus 99% C57BL6), given that strain-dependentphenotypic variation has been recognized in the gene-targetedmice (54,55).
ConclusionThe findings in this study confirm that Plg deficiency signif-
icantly attenuates tubular EMT and renal fibrosis in mice re-sponding to UUO and that this outcome seems to be due atleast in part to the reduced activation of the fibrogenic signalsERK and TGF-�. It is tempting to speculate that new strategiesthat are designed to antagonize the proteolytic activity of plas-min and PAR-1 activation might be effective at intercepting theprofibrotic actions of TGF-� and EMT, central pathways inrenal fibrogenesis.
Pl (µ Pl+UO126 SFM
pERK
loading
g/ml)
10 20 10 20 µM
FSP1
48h treatment
UO126 - +
0
10
20
30
40
50
60
70
Plasmin +
- +
0
10
20
30
40
50
60
70
Buffer + + + +
- - +
+
* *Po
res
wit
h ce
lls (%
)
C GD
E F
A B
0
100
200
SFM Pl(20ug/ml) Pl+UO126(20uM)
FSP1
p ERK
+ +
*
*
Band
de
nsit
y (u
nit
s)
Figure 11. Plasmin-induced EMT and cell migration are ERK dependent. (A) After incubation in SFM supplemented with plasmin(P) for 48 h, Western blot analysis of MCT cell protein demonstrates dosage-dependent ERK activation and FSP-1 expressioncompared with cells that were cultured in SFM alone. Addition of the ERK inhibitor UO126 (20 �M) suppressed FSP-1 expression.The blots were stained with amido black dye for protein loading correction. (B) The graph illustrates the relative abundance ofpERK and FSP-1 proteins under various cultured conditions (n � 4). (C through G) Boyden chambers migration assays wereperformed using lower chamber filter membranes that were coated with plasmin (20 �g/ml; D and F) or BSA (C and E) in thepresence (E and F) or absence (C and D) of UO126 (20 �M). The photomicrographs illustrate migrating cells stained withCoomassie blue. Data are means � 1 SD; n � 3 per group in two independent experiments. �P � 0.05, plasmin versus media alone;*P � 0.05, U0126 versus no U0126.
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 857

AcknowledgmentsThis work was funded by National Institutes of Health grants
DK54500 and DK44757 (A.A.E.) and a Young Investigator Award (G.Z.)from Children’s Hospital & Regional Medical Center in Seattle.
Part of this work was presented at the annual meeting of the Amer-ican Society of Nephrology and published in abstract form (J Am SocNephrol 15: 36A, 2004).
Our sincere thanks to Dr. Carolyn Kelly, University of California atSan Diego, for providing the MCT cell line and to Dr. David Plieth,Vanderbilt University, for the purified FSP-1 rabbit antiserum.
DisclosuresNone.
References1. Eddy AA: Progression in chronic kidney disease. Adv
Chronic Kidney Dis 12: 353–365, 20052. Chai Q, Krag S, Chai S, Ledet T, Wogensen L: Localisation
and phenotypical characterisation of collagen-producingcells in TGF-beta1-induced renal interstitial fibrosis. Histo-chem Cell Biol 119: 267–280, 2003
3. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, NeilsonEG: Evidence that fibroblasts derive from epithelium dur-ing tissue fibrosis. J Clin Invest 110: 341–350, 2002
4. Kalluri R, Neilson EG: Epithelial-mesenchymal transitionand its implications for fibrosis. J Clin Invest 112: 1776–1784, 2003
5. Wheelock MJ, Johnson KR: Cadherins as modulators ofcellular phenotype. Annu Rev Cell Dev Biol 19: 207–235,2003
6. Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, KneitzS, Piek E, Bottinger EP: Genetic programs of epithelial cellplasticity directed by transforming growth factor-beta. ProcNatl Acad Sci U S A 98: 6686–6691, 2001
7. Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C,Nejak K, Liu Y: Disruption of tissue-type plasminogenactivator gene in mice reduces renal interstitial fibrosis inobstructive nephropathy. J Clin Invest 110: 1525–1538, 2002
8. Bugge TH, Flick MJ, Daugherty CC, Degen JL: Plasmino-gen deficiency causes severe thrombosis but is compatiblewith development and reproduction. Genes Dev 9: 794–807, 1995
9. Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL,Dano K: Plasminogen and wound healing. Nat Med 2: 725,1996
10. Nagase H: Activation mechanisms of matrix metallopro-teinases. Biol Chem 378: 151–160, 1997
11. Kasai S, Arimura H, Nishida M, Suyama T: Primary struc-ture of single-chain pro-urokinase. J Biol Chem 260: 12382–12389, 1985
12. Eddy AA: Plasminogen activator inhibitor-1 and the kid-ney. Am J Physiol Renal Physiol 283: F209–F220, 2002
13. Matsushima K, Taguchi M, Kovacs EJ, Young HA, Oppen-heim JJ: Intracellular localization of human monocyte as-sociated interleukin 1 (IL 1) activity and release of biolog-ically active IL 1 from monocytes by trypsin and plasmin.J Immunol 136: 2883–2891, 1986
14. Edgtton KL, Gow RM, Kelly DJ, Carmeliet P, Kitching AR:Plasmin is not protective in experimental renal interstitialfibrosis. Kidney Int 66: 68–76, 2004
15. Ryniers F, Stove C, Goethals M, Brackenier L, Noe V,Bracke M, Vandekerckhove J, Mareel M, Bruyneel E: Plas-min produces an E-cadherin fragment that stimulates can-cer cell invasion. Biol Chem 383: 159–165, 2002
16. Eddy AA, Fogo AB: Plasminogen activator inhibitor-1 inchronic kidney disease: Evidence and mechanisms of ac-tion. J Am Soc Nephrol 17: 2999–3012, 2006
17. Winram SB, Lottenberg R: The plasmin-binding protein Plrof group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 142: 2311–2320,1996
18. Yamakami K, Yoshizawa N, Wakabayashi K, Takeuchi A,Tadakuma T, Boyle MD: The potential role for nephritis-associated plasmin receptor in acute poststreptococcal glo-merulonephritis. Methods 21: 185–197, 2000
19. Oda T, Yamakami K, Omasu F, Suzuki S, Miura S, SugisakiT, Yoshizawa N: Glomerular plasmin-like activity in rela-tion to nephritis-associated plasmin receptor in acute post-streptococcal glomerulonephritis. J Am Soc Nephrol 16: 247–254, 2005
20. Coughlin SR: How the protease thrombin talks to cells.Proc Natl Acad Sci U S A 96: 11023–11027, 1999
21. Pendurthi UR, Ngyuen M, Andrade-Gordon P, PetersenLC, Rao LV: Plasmin induces Cyr61 gene expression infibroblasts via protease-activated receptor-1 and p44/42mitogen-activated protein kinase-dependent signalingpathway. Arterioscler Thromb Vasc Biol 22: 1421–1426, 2002
22. Burysek L, Syrovets T, Simmet T: The serine proteaseplasmin triggers expression of MCP-1 and CD40 in humanprimary monocytes via activation of p38 MAPK and januskinase (JAK)/STAT signaling pathways. J Biol Chem 277:33509–33517, 2002
23. Zhang G, Kim H, Cai X, Lopez-Guisa J, Alpers C, Liu Y,Carmeliet P, Eddy A: Urokinase receptor deficiency accel-erates fibrosis in obstructive nephropathy. J Am Soc Nephrol14: 1254–1271, 2003
24. Kivirikko KI, Laitinen O, Prockop DJ: Modifications of aspecific assay for hydroxyproline in urine. Anal Biochem 19:249–255, 1967
25. Zhang G, Kim H, Cai X, Lopez-Guisa J, Carmeliet P, EddyA: Urokinase receptor modulates cellular and angiogenicresponses in obstructive uropathy. J Am Soc Nephrol 14:1234–1253, 2003
26. Qian SW, Kondaiah P, Roberts AB, Sporn MB: cDNA clon-ing by PCR of rat transforming growth factor beta-1. Nu-cleic Acids Res 18: 3059, 1990
27. Ishii K, Hein L, Kobilka B, Coughlin SR: Kinetics of throm-bin receptor cleavage on intact cells. Relation to signaling.J Biol Chem 268: 9780–9786, 1993
28. Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM,Neilson EG: Conditional abatement of tissue fibrosis usingnucleoside analogs to selectively corrupt DNA replicationin transgenic fibroblasts. Mol Ther 3: 149–159, 2001
29. Zhang G, Cai X, Lopez-Guisa JM, Collins SJ, Eddy AA:Mitogenic signaling of urokinase receptor-deficient kidneyfibroblasts: Actions of an alternative urokinase receptorand LDL receptor-related protein. J Am Soc Nephrol 15:2090–2102, 2004
30. Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, RifkinDB: An assay for transforming growth factor-beta usingcells transfected with a plasminogen activator inhibitor-1
858 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 846–859, 2007

promoter-luciferase construct. Anal Biochem 216: 276–284,1994
31. Haverty TP, Kelly CP, Hines WH, Amenta PS, WatanabeM, Harper RA, Kefalides NA, Neilson EG: Characteriza-tion of a renal tubular epithelial cell line which secretes theautologous target antigen of autoimmune experimentalinterstitial nephritis. J Cell Biol 107: 1359–1368, 1988
32. Covic L, Misra M, Badar J, Singh C, Kuliopulos A: Pepdu-cin-based intervention of thrombin-receptor signaling andsystemic platelet activation. Nat Med 8: 1161–1165, 2002
33. Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC,Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McCom-sey DF, Oksenberg D, Reynolds EE, Santulli RJ, Scarbor-ough RM, Smith CE, White KB: Design, synthesis, andbiological characterization of a peptide-mimetic antagonistfor a tethered-ligand receptor. Proc Natl Acad Sci U S A 96:12257–12262, 1999
34. Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopu-los A: PAR1 is a matrix metalloprotease-1 receptor thatpromotes invasion and tumorigenesis of breast cancercells. Cell 120: 303–313, 2005
35. Tarui T, Majumdar M, Miles LA, Ruf W, Takada Y: Plas-min-induced migration of endothelial cells. A potentialtarget for the anti-angiogenic action of angiostatin. J BiolChem 277: 33564–33570, 2002
36. Majumdar M, Tarui T, Shi B, Akakura N, Ruf W, Takada Y:Plasmin-induced migration requires signaling throughprotease-activated receptor 1 and integrin alpha(9)beta(1).J Biol Chem 279: 37528–37534, 2004
37. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W:Activation of endothelial cell protease activated receptor 1by the protein C pathway. Science 296: 1880–1882, 2002
38. Blanc-Brude OP, Archer F, Leoni P, Derian C, Bolsover S,Laurent GJ, Chambers RC: Factor Xa stimulates fibroblastprocollagen production, proliferation, and calcium signal-ing via PAR1 activation. Exp Cell Res 304: 16–27, 2005
39. Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I,Blaber M, Blaber SI, Scarisbrick I, Andrade-Gordon P, Cot-trell GS, Bunnett NW, Diamandis EP, Hollenberg MD:Proteinase-activated receptors (PARs), targets for kal-likrein signaling. J Biol Chem 281: 32095–32112, 2006
40. Cunningham MA, Rondeau E, Chen X, Coughlin SR, Hold-sworth SR, Tipping PG: Protease-activated receptor 1 me-diates thrombin-dependent, cell-mediated renal inflamma-tion in crescentic glomerulonephritis. J Exp Med 191: 455–462, 2000
41. Vu TK, Hung DT, Wheaton VI, Coughlin SR: Molecularcloning of a functional thrombin receptor reveals a novelproteolytic mechanism of receptor activation. Cell 64: 1057–1068, 1991
42. McLaughlin JN, Shen L, Holinstat M, Brooks JD,Dibenedetto E, Hamm HE: Functional selectivity of G pro-tein signaling by agonist peptides and thrombin for theprotease-activated receptor-1. J Biol Chem 280:25048–25059, 2005
43. Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB:Role of reactive oxygen species in TGF-beta1-induced mi-togen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells.J Am Soc Nephrol 16: 667–675, 2005
44. Yang M, Huang H, Li J, Li D, Wang H: Tyrosine phosphor-ylation of the LDL receptor-related protein (LRP) and ac-tivation of the ERK pathway are required for connectivetissue growth factor to potentiate myofibroblast differen-tiation. FASEB J 18: 1920–1921, 2004
45. Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Ni-kolic-Paterson DJ: Activation of the extracellular-signalregulated protein kinase pathway in human glomerulopa-thies. J Am Soc Nephrol 15: 1835–1843, 2004
46. Platanias LC: Map kinase signaling pathways and hema-tologic malignancies. Blood 101: 4667–4679, 2003
47. Roux PP, Blenis J: ERK and p38 MAPK-activated proteinkinases: A family of protein kinases with diverse biologicalfunctions. Microbiol Mol Biol Rev 68: 320–344, 2004
48. Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF,Nikolic-Paterson DJ: The role of p38alpha mitogen-acti-vated protein kinase activation in renal fibrosis. J Am SocNephrol 15: 370–379, 2004
49. Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, LaurentGJ, Davis GE, Chambers RC, Matthay MA, Sheppard D:Ligation of protease-activated receptor 1 enhancesalpha(v)beta6 integrin-dependent TGF-beta activationand promotes acute lung injury. J Clin Invest 116: 1606 –1614, 2006
50. Tamaki K, Okuda S, Miyazono K, Nakayama M, FujishimaM: Matrix-associated latent TGF-beta with latent TGF-betabinding protein in the progressive process in adriamycin-induced nephropathy. Lab Invest 73: 81–89, 1995
51. Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, MenkeA, Ruhland C, Adler G, Gress TM: Transforming growthfactor beta1 treatment leads to an epithelial-mesenchymaltransdifferentiation of pancreatic cancer cells requiring ex-tracellular signal-regulated kinase 2 activation. Cancer Res61: 4222–4228, 2001
52. Roztocil E, Nicholl SM, Galaria II, Davies MG: Plasmin-induced smooth muscle cell proliferation requires epider-mal growth factor activation through an extracellular path-way. Surgery 138: 180–186, 2005
53. Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W,Atkins RC, Lan HY: Transforming growth factor-beta reg-ulates tubular epithelial-myofibroblast transdifferentiationin vitro. Kidney Int 56: 1455–1467, 1999
54. Dominguez-Salazar E, Bateman HL, Rissman EF: Back-ground matters: The effects of estrogen receptor alphagene disruption on male sexual behavior are modified bybackground strain. Horm Behav 46: 482–490, 2004
55. Morice E, Denis C, Giros B, Nosten-Bertrand M: Pheno-typic expression of the targeted null-mutation in the do-pamine transporter gene varies as a function of the geneticbackground. Eur J Neurosci 20: 120–126, 2004
J Am Soc Nephrol 18: 846–859, 2007 Plasmin Promotes Fibrosis via EMT 859