photocatalytic reduction of chromium and oxidation of organics by polyoxometalates
TRANSCRIPT

Photocatalytic reduction of chromium and oxidation
of organics by polyoxometalates
E. Gkika, A. Troupis, A. Hiskia, E. Papaconstantinou *
Institute of Physical Chemistry, NCSR Demokritos, 153 10 Athens, Greece
Received 18 May 2005; received in revised form 21 June 2005; accepted 23 June 2005
Available online 1 August 2005
Abstract
The photocatalytic reduction of Cr(VI) to the less toxic Cr(III) is presented in the presence of the polyoxometalates (POM) PW12O403� or
SiW12O404� as photocatalyst and an organic substrate (salicylic acid or propan-2-ol) as electron donor. Cr(VI), as dichromate, is reduced to
Cr(III), according to the 6:1 stoichiometry of PW12O404� versus Cr2O7
2� indicated from experiments in the dark. Increase of POM or salicylic
acid (SA) concentration accelerates, till a saturation value, both the reduction of metal and the oxidation of the organic, suggesting that these
two conjugate reactions act synergistically. The photocatalytic action of POM is not so important in the case of highly concentrated solutions
of organics that exhibit direct photochemical reduction of Cr(VI), i.e. propan-2-ol (i-prOH), while it becomes important at low concentrations
of i-prOH, especially for organics that do not react directly photochemically with Cr(VI), such as SA. Increase of Cr(VI) concentration
enhances consumption of SA and Cr(VI) till an optimum value, due to inner filter effect. The method is suitable for a range of chromium
concentration from 5–100 ppm achieving complete reduction of Cr(VI) to Cr(III) up to non-detected traces (>98%). The presence of oxygen
does not influence the efficiency of SA and Cr(VI) consumption. In contrast to the semiconductor-based heterogeneous photocatalysis, the
POM-based homogeneous process seems superior in the frame that: (i) it remains catalytic throughout illumination by providing more active
sites and (ii) among the two POM used, the one that is more efficient in the degradation of the organic, that is PW12O403� compared to
SiW12O404�, is also more efficient in reducing Cr(VI), due to a kinetic effect, and a compromise is not needed.
# 2005 Elsevier B.V. All rights reserved.
Keywords: Photocatalytic reduction; Chromium; Polyoxometallates
www.elsevier.com/locate/apcatb
Applied Catalysis B: Environmental 62 (2006) 28–34
1. Introduction
Environmental, whereas samples are usually contami-
nated by both organic and inorganic pollutants, i.e. metal
ions. Therefore, it is of interest to develop processes that are
efficient in removing both categories of pollutants.
In conventional methods, the presence of one species
usually impedes the removal of the other. For instance,
hydrometallurgy, a classical process to recover metals, is
inhibited by the presence of organic compounds and a pre-
treatment step, to remove or destroy organics, is generally
required, pyrometallurgy which is able to decontaminate
systems from organic pollutants and recover metals suffers
* Corresponding author. Tel.: +30 2106503642; fax: +30 2106511766.
E-mail address: [email protected] (E. Papaconstantinou).
0926-3373/$ – see front matter # 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcatb.2005.06.012
from lack of controllability, demanding extremely high
temperatures (beyond 2000 8C) [1].The most promising methods to treat such complex
systems are the photocatalytic ones, e.g. those based either
on semiconductors, such as TiO2, or polyoxometalates
(POM), which consume cheap photons from the UV–near
visible region. These photocatalysts serve as electron relays,
from the organic substrates to metal ions. Thus, they induce
degradation of organic pollutants and recovery of metals in
one-pot systems, operable at traces of the target compounds
(less than ppm).
Hexavalent chromium exhibits remarkably toxic activity
in most organics, being carcinogenic in animals. Conse-
quently, it is no surprise that Cr(VI) is included in the list of
priority pollutants of the US EPA, while European Union has
determined a maximum concentration limit for chromium at

E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–34 29
50 ppb for drinking water. Cr(VI) is being introduced in the
environment mainly by its industrial uses, which span from
electroplating to leather tanning and paints applications.
Efforts to remove Cr(VI) involve mainly its reduction to the
100 times less toxic and less mobile Cr(III), accompanied by
neutralization or alkalization of the solution in order to
precipitate Cr(OH)3, while in some cases the more difficult
complete reduction to Cr0 has been achieved [2,3].
The photocatalytic reductive recovery of Cr(VI) using
TiO2 dates back to 1979 [4] and since then is continued [5–7]
and extended using various other semiconducting material,
such as ZnO [8,9], WO3 [10,11], ZnS [10], CdS [10,11]
among others. All these semiconductor-based processes,
although highly efficient even for traces of Cr(VI), they
exhibit some disadvantages: (i) the photocatalyst is masked
from the adsorbed reactants at prolonged illumination times,
rendering the process non-catalytic and (ii) onemore step, this
of separation of the metal product from the catalyst is needed.
The above-mentioned drawbacks are overcome in the
POM-based homogeneous photocatalysis. POM constitutes
a large variety of well defined oxygen bridged metal clusters
anions of mainly tungsten and molybdenum [12,13]. In a
photocatalytic mode, the UV–near visible light excitation of
POM, creates a powerful oxidant able to mineralize a variety
of organic species, including organic pollutants [14,15].
Subsequently, the photoreduced POM deliver the electrons
to a great variety of chemical species including metal ions
[16,17]. This way both decontamination of aqueous
solutions from organic pollutants and recovery of metal
ions can take place in a one-pot system. Up to now, we have
reported the photocatalytic recovery of several metals, i.e.
copper [18], silver [19], palladium [20] and mercury [21] in
the presence of POM. In this paper we present the ability of
POM, namely PW12O403� and SiW12O40
4� to photocatalyze
reduction of Cr(VI) in aqueous solutions [22,22a]. The
process involves addition of POM in a Cr2O72� solution, in
the presence of salicylic acid or propan-2-ol as the target
organic and irradiation with near visible and UV light. The
influence of the kind and concentration of POM catalyst, the
kind and concentration of organic and the concentration of
Cr(VI) on the efficiency of both organic degradation and
Cr(VI) reduction are addressed. We show that the two
conjugated reactions (the photooxidation of organic and the
reduction of metal) act synergistically. The influence of
dioxygen on the efficiency of Cr(VI) reduction is also
examined. A comparison of the POM-based process with the
semiconductor-based one is also reported.
2. Experimental
H3PW12O40 and H4SiW12O40 were provided by Panreac
Spain (98%) and Aldrich, respectively, and were used as
received.
Potassium dichromate was obtained from Merck with a
purity of 99.8%. Propan-2-ol (i-prOH) was analytical grade
while ultra-purewaterwas obtained froma Purelab apparatus.
Salicylic acid (SA) was analytical grade and was provided by
Carlo Erba, Milan. HClO4 70% and CsCl was obtained from
Riedel et Haen and Koch-Light Laboratories Ltd., respec-
tively. Extra pure argon (99.999%) and dioxygen (>99.95%)
were used for deaeration oroxygenationof solutionswherever
necessary.
A typical experiment was as follows: 4 ml of aqueous
K2Cr2O7 solution containing SA or i-prOH and H3PW12O40
or H4SiW12O40 catalyst was added to a spectrophotometer
cylindrical cell about 4 cm diameter and covered with a
cerum cap. The pH was adjusted at pH 1 with HClO4
whenever necessary.
Photolysis was performed with an illumination box
consisted of five lamps with an illumination peak at 350 nm.
Alternatively, photolysis was performed with an Oriel
1000 W Xe arc lamp equipped with cool water circulating
filter to absorb the near IR radiation and a 320 nm cut-off
filter in order to avoid direct photolysis of substrates, using a
quartz cell of 1 cm path length. The total photonic flux (320–
345 nm) determined by ferrioxalate actinometer was
7.9 � 10�6 Einstein min�1. As a matter of comparison,
for typical experiments (PW12O403� 0.7 mM, i-prOH 0.2 M
at pH 1) the quantum yield of formation of the one-
equivalent reduced POM at 254 nm is ca. 12% for
PW12O404� as has been reported previously [23]. The
quantum yield is independent of wavelength below ca.
350 nm, as has been recently verified [24].
The degree of reduction of POM in photolyzed deaerated
solutionswas calculated from the knownextinction coefficient
of reduced catalyst at ca. 750 nm (for the one-electron reduced
PW12O403�, e752 nm = 2000 M�1 cm�1 and for the one-
electron reduced SiW12O404�, e730 nm = 2100 M�1 cm�1)
using a Perkin-Elmer Lambda19 Spectrometer. The concen-
tration of Cr(VI) ions and SA was determined spectro-
metrically monitored at 350 and 300 nm, respectively, after
addition of 100 ml CsCl 5% (w/v) into the photolyzed solution
and subsequent filtration with a 0.45 mm Millipore filter.
The initial rate of Cr(VI) reduction or SA degradation
were determined by monitoring spectrometrically their
concentration in the photolyzed filtered solutions and
calculating the slope of the curve obtained until about
30% of Cr(VI) or SA had been consumed and using linear
regression fit. Following the comments of reviewer, we note
that to a very good approximation the results were similar to
those obtained under stricter kinetic treatment, i.e. using the
tangent d[Cr(VI)]/dt for the initial rates of Cr(VI) reduction.
3. Results
3.1. Photocatalytic reduction of chromium and
simultaneous oxidation of SA
In order to examine whether or not a direct photoreaction
between SA and Cr(VI) ions takes place, blank experiments
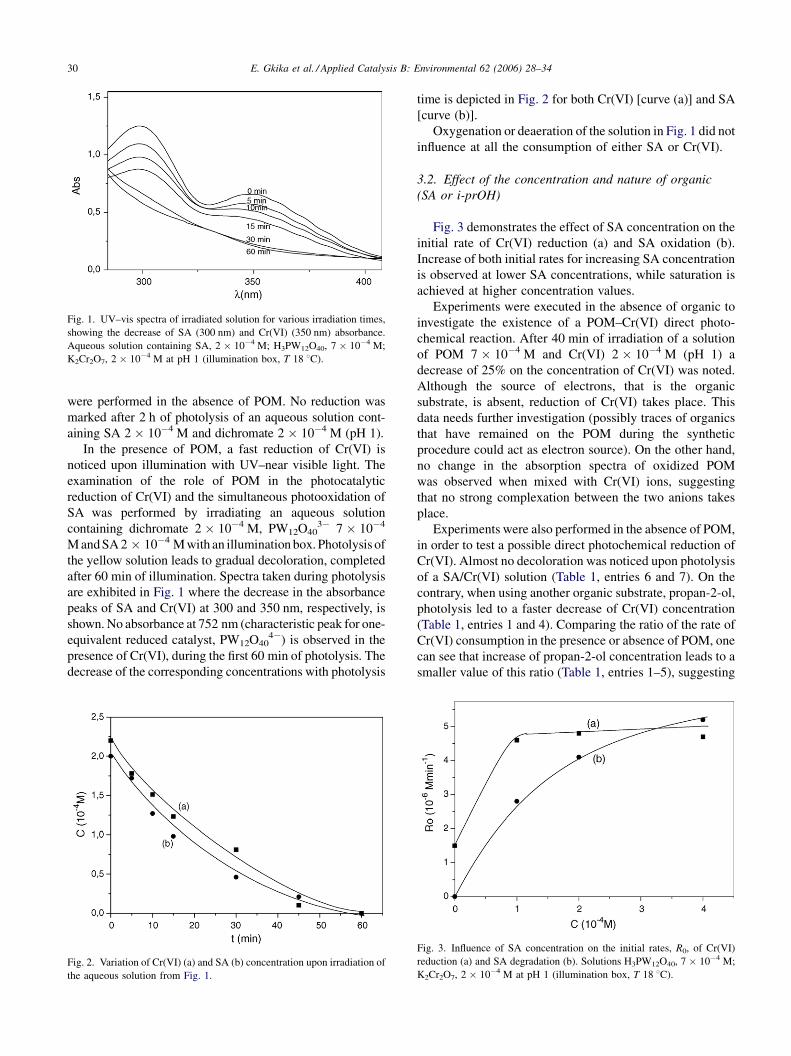
E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–3430
Fig. 1. UV–vis spectra of irradiated solution for various irradiation times,
showing the decrease of SA (300 nm) and Cr(VI) (350 nm) absorbance.
Aqueous solution containing SA, 2 � 10�4 M; H3PW12O40, 7 � 10�4 M;
K2Cr2O7, 2 � 10�4 M at pH 1 (illumination box, T 18 8C).
were performed in the absence of POM. No reduction was
marked after 2 h of photolysis of an aqueous solution cont-
aining SA 2 � 10�4 M and dichromate 2 � 10�4 M (pH 1).
In the presence of POM, a fast reduction of Cr(VI) is
noticed upon illumination with UV–near visible light. The
examination of the role of POM in the photocatalytic
reduction of Cr(VI) and the simultaneous photooxidation of
SA was performed by irradiating an aqueous solution
containing dichromate 2 � 10�4 M, PW12O403� 7 � 10�4
MandSA2 � 10�4 Mwith an illuminationbox. Photolysis of
the yellow solution leads to gradual decoloration, completed
after 60 min of illumination. Spectra taken during photolysis
are exhibited in Fig. 1 where the decrease in the absorbance
peaks of SA and Cr(VI) at 300 and 350 nm, respectively, is
shown. No absorbance at 752 nm (characteristic peak for one-
equivalent reduced catalyst, PW12O404�) is observed in the
presence of Cr(VI), during the first 60 min of photolysis. The
decrease of the corresponding concentrations with photolysis
Fig. 2. Variation of Cr(VI) (a) and SA (b) concentration upon irradiation of
the aqueous solution from Fig. 1.
time is depicted in Fig. 2 for both Cr(VI) [curve (a)] and SA
[curve (b)].
Oxygenation or deaeration of the solution in Fig. 1 did not
influence at all the consumption of either SA or Cr(VI).
3.2. Effect of the concentration and nature of organic
(SA or i-prOH)
Fig. 3 demonstrates the effect of SA concentration on the
initial rate of Cr(VI) reduction (a) and SA oxidation (b).
Increase of both initial rates for increasing SA concentration
is observed at lower SA concentrations, while saturation is
achieved at higher concentration values.
Experiments were executed in the absence of organic to
investigate the existence of a POM–Cr(VI) direct photo-
chemical reaction. After 40 min of irradiation of a solution
of POM 7 � 10�4 M and Cr(VI) 2 � 10�4 M (pH 1) a
decrease of 25% on the concentration of Cr(VI) was noted.
Although the source of electrons, that is the organic
substrate, is absent, reduction of Cr(VI) takes place. This
data needs further investigation (possibly traces of organics
that have remained on the POM during the synthetic
procedure could act as electron source). On the other hand,
no change in the absorption spectra of oxidized POM
was observed when mixed with Cr(VI) ions, suggesting
that no strong complexation between the two anions takes
place.
Experiments were also performed in the absence of POM,
in order to test a possible direct photochemical reduction of
Cr(VI). Almost no decoloration was noticed upon photolysis
of a SA/Cr(VI) solution (Table 1, entries 6 and 7). On the
contrary, when using another organic substrate, propan-2-ol,
photolysis led to a faster decrease of Cr(VI) concentration
(Table 1, entries 1 and 4). Comparing the ratio of the rate of
Cr(VI) consumption in the presence or absence of POM, one
can see that increase of propan-2-ol concentration leads to a
smaller value of this ratio (Table 1, entries 1–5), suggesting
Fig. 3. Influence of SA concentration on the initial rates, R0, of Cr(VI)
reduction (a) and SA degradation (b). Solutions H3PW12O40, 7 � 10�4 M;
K2Cr2O7, 2 � 10�4 M at pH 1 (illumination box, T 18 8C).
E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–34 31
Table 1
Variation of Cr(VI) photoreduction rates from solutions containing either
i-prOH or SA in the presence, and absence, of PW12O403�
Entry [Organic]0 (M) R0 (POM)
(10�6 M
min�1)
R0 (blank)
(10�6 M
min�1)
R0
(POM)/R0
(blank)
1 i-prOH 2 � 10�4 a 8.1 1 8.1
2 i-prOH 0.1a 12.5 3.6 3.5
3 i-prOH 0.5a 38.5 32.8 1.2
4 i-prOH 2 � 10�4 b 4.5 0.5 9
5 i-prOH 0.5b 7.5 2.1 3.5
6 SA 2 � 10�4 a 6.7 0.02 335
7 SA 2 � 10�4 b 3.4 0.02 170
Other conditions the same. i-prOH or SA/PW12O403� 7 � 10�4 M/K2Cr2O7
2 � 10�4 M, pH 1.a Xe lamp, l > 320 nm.b Illumination box.
Fig. 5. Influence of Cr(VI) concentration on the initial rates, R0, of Cr(VI)
reduction (a) and SA degradation (b). Solutions SA, 2 � 10�4 M;
H3PW12O40, 7 � 10�4 M; K2Cr2O7, 2 � 10�4 M at pH 1 (illumination
box, T 18 8C).
that the direct photochemical reduction of Cr(VI) becomes
important at higher concentrations of the organic.
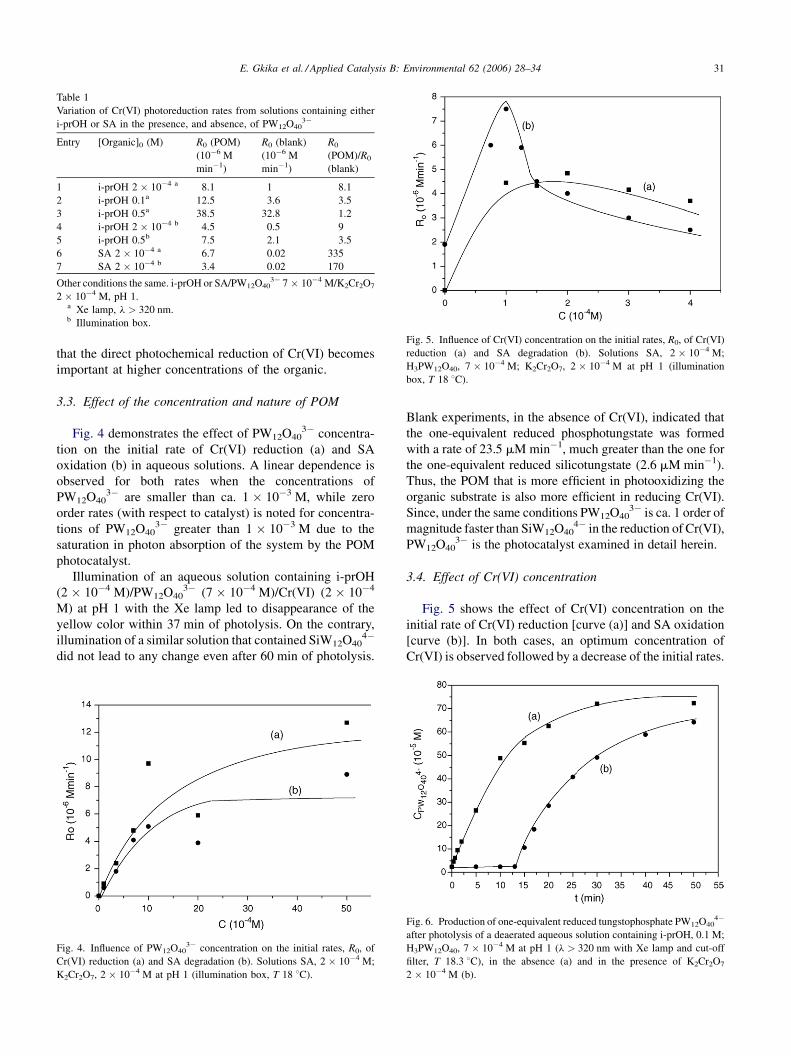
3.3. Effect of the concentration and nature of POM
Fig. 4 demonstrates the effect of PW12O403� concentra-
tion on the initial rate of Cr(VI) reduction (a) and SA
oxidation (b) in aqueous solutions. A linear dependence is
observed for both rates when the concentrations of
PW12O403� are smaller than ca. 1 � 10�3 M, while zero
order rates (with respect to catalyst) is noted for concentra-
tions of PW12O403� greater than 1 � 10�3 M due to the
saturation in photon absorption of the system by the POM
photocatalyst.
Illumination of an aqueous solution containing i-prOH
(2 � 10�4 M)/PW12O403� (7 � 10�4 M)/Cr(VI) (2 � 10�4
M) at pH 1 with the Xe lamp led to disappearance of the
yellow color within 37 min of photolysis. On the contrary,
illumination of a similar solution that contained SiW12O404�
did not lead to any change even after 60 min of photolysis.
Fig. 4. Influence of PW12O403� concentration on the initial rates, R0, of
Cr(VI) reduction (a) and SA degradation (b). Solutions SA, 2 � 10�4 M;
K2Cr2O7, 2 � 10�4 M at pH 1 (illumination box, T 18 8C).
Blank experiments, in the absence of Cr(VI), indicated that
the one-equivalent reduced phosphotungstate was formed
with a rate of 23.5 mMmin�1, much greater than the one for
the one-equivalent reduced silicotungstate (2.6 mMmin�1).
Thus, the POM that is more efficient in photooxidizing the
organic substrate is also more efficient in reducing Cr(VI).
Since, under the same conditions PW12O403� is ca. 1 order of
magnitude faster than SiW12O404� in the reduction of Cr(VI),
PW12O403� is the photocatalyst examined in detail herein.
3.4. Effect of Cr(VI) concentration
Fig. 5 shows the effect of Cr(VI) concentration on the
initial rate of Cr(VI) reduction [curve (a)] and SA oxidation
[curve (b)]. In both cases, an optimum concentration of
Cr(VI) is observed followed by a decrease of the initial rates.
Fig. 6. Production of one-equivalent reduced tungstophosphate PW12O404�
after photolysis of a deaerated aqueous solution containing i-prOH, 0.1 M;
H3PW12O40, 7 � 10�4 M at pH 1 (l > 320 nm with Xe lamp and cut-off
filter, T 18.3 8C), in the absence (a) and in the presence of K2Cr2O7
2 � 10�4 M (b).
E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–3432
3.5. Stability tests of the catalyst—reproduction of the
process
Fig. 6 presents another indication that the catalyst is
not affected by the reduction of Cr(VI). In this figure,
the formation of the one-equivalent tungstophosphate
(PW12O404�) is monitored as a function of photolysis time.
In one experiment, a deaerated i-prOH/PW12O403� is
photolyzed, and the formation of PW12O404� is observed
[curve (a)]. In the other experiment Cr(VI) has been added to
the deaerated solution [propan-2-ol/PW12O403�/Cr(VI)] and
the formation of PW12O404� is also observed as a function of
photolysis time. After a period in which all Cr(VI) has been
reduced, the rate of PW12O404� formation [curve (b)] is the
same with the one in the preceding experiment without
Cr(VI). Thus, the process remains catalytic throughout
photolysis, without any sign of poisoning of the POM
photocatalyst.
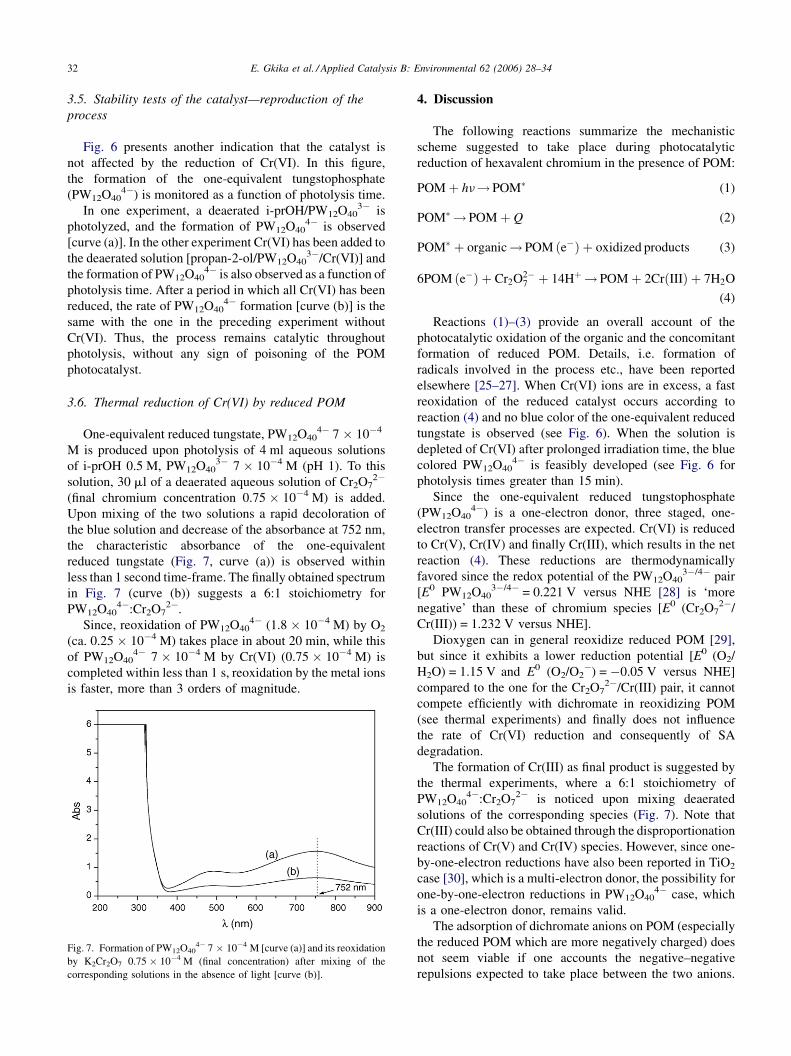
3.6. Thermal reduction of Cr(VI) by reduced POM
One-equivalent reduced tungstate, PW12O404� 7 � 10�4
M is produced upon photolysis of 4 ml aqueous solutions
of i-prOH 0.5 M, PW12O403� 7 � 10�4 M (pH 1). To this
solution, 30 ml of a deaerated aqueous solution of Cr2O72�
(final chromium concentration 0.75 � 10�4 M) is added.
Upon mixing of the two solutions a rapid decoloration of
the blue solution and decrease of the absorbance at 752 nm,
the characteristic absorbance of the one-equivalent
reduced tungstate (Fig. 7, curve (a)) is observed within
less than 1 second time-frame. The finally obtained spectrum
in Fig. 7 (curve (b)) suggests a 6:1 stoichiometry for
PW12O404�:Cr2O7
2�.
Since, reoxidation of PW12O404� (1.8 � 10�4 M) by O2
(ca. 0.25 � 10�4 M) takes place in about 20 min, while this
of PW12O404� 7 � 10�4 M by Cr(VI) (0.75 � 10�4 M) is
completed within less than 1 s, reoxidation by the metal ions
is faster, more than 3 orders of magnitude.
Fig. 7. Formation of PW12O404� 7 � 10�4 M [curve (a)] and its reoxidation
by K2Cr2O7 0.75 � 10�4 M (final concentration) after mixing of the
corresponding solutions in the absence of light [curve (b)].
4. Discussion
The following reactions summarize the mechanistic
scheme suggested to take place during photocatalytic
reduction of hexavalent chromium in the presence of POM:
POMþ hn! POM� (1)
POM� ! POMþ Q (2)
POM� þ organic! POM ðe�Þ þ oxidized products (3)
6POM ðe�Þ þ Cr2O2�7 þ 14Hþ ! POMþ 2CrðIIIÞ þ 7H2O
(4)
Reactions (1)–(3) provide an overall account of the
photocatalytic oxidation of the organic and the concomitant
formation of reduced POM. Details, i.e. formation of
radicals involved in the process etc., have been reported
elsewhere [25–27]. When Cr(VI) ions are in excess, a fast
reoxidation of the reduced catalyst occurs according to
reaction (4) and no blue color of the one-equivalent reduced
tungstate is observed (see Fig. 6). When the solution is
depleted of Cr(VI) after prolonged irradiation time, the blue
colored PW12O404� is feasibly developed (see Fig. 6 for
photolysis times greater than 15 min).
Since the one-equivalent reduced tungstophosphate
(PW12O404�) is a one-electron donor, three staged, one-
electron transfer processes are expected. Cr(VI) is reduced
to Cr(V), Cr(IV) and finally Cr(III), which results in the net
reaction (4). These reductions are thermodynamically
favored since the redox potential of the PW12O403�/4� pair
[E0 PW12O403�/4� = 0.221 V versus NHE [28] is ‘more
negative’ than these of chromium species [E0 (Cr2O72�/
Cr(III)) = 1.232 V versus NHE].
Dioxygen can in general reoxidize reduced POM [29],
but since it exhibits a lower reduction potential [E0 (O2/
H2O) = 1.15 V and E0 (O2/O2�) = �0.05 V versus NHE]
compared to the one for the Cr2O72�/Cr(III) pair, it cannot
compete efficiently with dichromate in reoxidizing POM
(see thermal experiments) and finally does not influence
the rate of Cr(VI) reduction and consequently of SA
degradation.
The formation of Cr(III) as final product is suggested by
the thermal experiments, where a 6:1 stoichiometry of
PW12O404�:Cr2O7
2� is noticed upon mixing deaerated
solutions of the corresponding species (Fig. 7). Note that
Cr(III) could also be obtained through the disproportionation
reactions of Cr(V) and Cr(IV) species. However, since one-
by-one-electron reductions have also been reported in TiO2
case [30], which is a multi-electron donor, the possibility for
one-by-one-electron reductions in PW12O404� case, which
is a one-electron donor, remains valid.
The adsorption of dichromate anions on POM (especially
the reduced POM which are more negatively charged) does
not seem viable if one accounts the negative–negative
repulsions expected to take place between the two anions.

E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–34 33
This is also in accordance to spectrometric measurements
where no absorbance change is seen. Thus, reaction (4) can
take place in the bulk upon collisions between reduced POM
and dichromate anions.
4.1. Direct photochemical reduction of Cr(VI)—effect of
the organic and POM
Propan-2-ol is a substrate that is able to induce direct
photochemical reduction of Cr(VI) to Cr(III) [31]. This is
depicted in Table 1, where a high propan-2-ol concentra-
tion is effective to reduce Cr(VI) in the absence of POM
with a rate only three times slower than in the presence of
POM. However, the ratio of the rate of Cr(VI) reduction in
the presence of POM versus the rate in the absence of
POM increases at lower propan-2-ol concentrations. Thus,
in the case of substrates that can trigger direct photo-
reduction of Cr(VI), the presence of POM is more
favorable for traces of the organic. This is important from
a practical aspect, since photocatalysis is usually the right
process to mineralize traces, but still toxic remnants, of
pollutants, which cannot be treated by the conventional
processes.
4.2. Synergism of the two conjugated reactions
In the POM-based integrated system, the two conjugated
processes [reactions (3) and (4)] were directly correlated
acting synergistically the one for the other. Enhancement of
the part of photooxidation of the organic by: (i) increasing
POM or SA concentration or (ii) using more efficient
photooxidants POM (PW12O403� instead of SiW12O40
4�)
favors Cr(VI) reduction, by favoring the production of
reduced POM. In an analogous way, enhancement of the rate
of reduction of Cr(VI) by increasing the concentration of
Cr(VI), favors the photooxidation of SA (Fig. 5, curve (b))
through replenishing of the oxidized-photoactive form of the
POM catalyst. This is in agreement with the fact that the rate
determining step in the photocatalytic cycle with POM is the
regeneration-reoxidation of catalyst which relates to the fact
that the oxidized form of the catalyst (for instance,
PW12O403�) acts as photooxidant more than one order of
magnitude faster than the corresponding one-equivalent
reduced form (PW12O404�) [23].
4.3. Comparison of POM-based with the
semiconductor-based photocatalysis in treating complex
systems
The ability to treat complex systems is of paramount
practical importance since it is the actual case in
environment. Although both POM and semiconductor
photocatalysis are able to treat complex systems since they
both produce highly oxidizing and reducing species in one-
pot system [14], they present some practical differences
when breaking into details:
(1) A
lthough in both POM-based and semiconductor-basedprocesses [32–35] the two reactions act synergistically
during the initial stages, the latter show signs of
deactivation during the final stages, which are important
for the complete mineralization of the treated solution.
In semiconductor-based photocatalysis the adsorp-
tion of the reactants on the catalyst surface is important
for the redox process to proceed. For instance, slower or
no reduction of Cr(VI) takes place at pH higher than the
isoelectric point of TiO2 due to the repelling action of
the negatively charged TiO2 surface for dichromates
[36,37]. Moreover, since semiconductor molecules form
aggregates-suspensions in aqueous solutions that leave
only a fraction of the catalyst molecules exposed, they
suffer from lower surface areas compared to soluble
POM and possess a restricted number of adsorbing sites.
As a result, the coexistance of SA and Cr(VI) in a
complex system leads finally to deactivation of the
catalyst due to competition of both reactants for the
adsorbing sites [2,38]. In addition, the oxidation
products of SA [39], the precipitated Cr(OH)3 which
masks the catalyst [2,40], proton starvation due to their
consumption upon Cr(VI) reduction [41] (in this case
Cu2+ [42] or NH4+ has to be added in order [41] to
facilitate Cr (VI) reduction) or the photocorrosion of
TiO2 in acidic solutions where Cr(VI) photoetches the
catalyst surface [43], can induce poisoning of the
catalyst.
The solution of increasing drastically the concentra-
tion of catalyst in order to provide more available
adsorbing sites [34] is not promising not only due to
economical reasons but also due to the detrimental effect
observed at higher concentrations (attributed to shadow-
ing of light or extended aggregation of the catalyst
particles) [8]. Thus, there is continuous need for
refreshment of the catalyst in order to achieve miner-
alization and complete the reduction of Cr(VI) [32].
The use of POM, which are ideally dispersed in the
bulk and guarantee a huge surface area, would be a
satisfactory solution to the problem of poisoning the
photocatalyst. Indeed, Fig. 6 shows that no change in the
photooxidizing efficiency of PW12O403� is marked even
after all Cr(VI) has been reduced.
(2) B
y examining the influence of POM on the rate ofCr(VI) reduction we noticed that PW12O403� which is
more efficient in the photooxidation of SA than
SiW12O404�, is also more efficient in the subsequent
reduction of Cr(VI). This has also been shown in the
case of reduction of palladium with various POM [20]
and is one more advantage compared to the semi-
conductor-based processes, where due to the strong
interference of adsorption phenomena, things are not so
clear. According to Rajeshwar, a given semiconductor
may not be simultaneously optimal for both photo-
oxidation and reduction and a compromise is usually the
best solution [44].

E. Gkika et al. / Applied Catalysis B: Environmental 62 (2006) 28–3434
4.4. Practical aspects
Some aspects of practical concern are exhibited in the
POM-based photocatalytic reduction of chromium. These
are summarized as follows:
� T
he two conjugated reactions, that is photooxidation of theorganic and reduction of metal act synergistically rather
than competitively, under our experimental conditions.
� T
he POM catalyst which is more efficient in photoox-idizing the organic is also more effective in reducing
Cr(VI) and a compromised solution is avoided.
� T
he process is effective in a range of chromiumconcentration varying from 5 to 100 ppm, whereas
prolonged irradiation leads to complete decontamination
from dichromate ions up to non-detected traces (>98%).
The range of chromium concentration, at which the POM
method is applied, as well as the low final concentration of
Cr(VI) ions left in solution, can account for the
environmental assessment of the process.
� T
his method remains effective even in the presence ofoxygen, a result that simplifies the whole procedure since
a pre-deaeration step is not required.
� T
he soluble POM catalyst is not poisoned-saturated byadsorbing species, since it provides many adsorbing sites,
retaining its photocatalytic activity throughout irradiation.
� W
hen the solution is depleted of Cr(VI) ions, the bluecolor of reduced POM appears promptly. This is a highly
interesting aspect since the process is rendered self-
indicating and could be automated.
Acknowledgments
We thankMinistry of Development, General Secretariat of
Research and Technology of Greece, for supporting part of
this work. A.T. is grateful to Institute of Physical Chemistry,
NCSR Demokritos, for a post-Doctoral fellowship.
References
[1] L.A. Smith, J.L. Means, A. Chen, B. Alleman, C.C. Chapman, J.S.
Texier Jr., S.E. Brauning, A.R. Gavaskar, M.D. Royer, Remedial
Options For Metals-Contaminated Sites, CRC Lewis Publishers, Boca
Raton, 1995 , pp. 55, 109, 15755.
[2] X. Wang, S.O. Pehkonen, A.K. Ray, Ind. Eng. Chem. Res. 43 (2004)
1665.
[3] T. Kanki, H. Yoneda, N. Sano, A. Toyoda, Ch. Nagai, Chem. Eng. J. 97
(2004) 77.
[4] H. Yoneyama, Y. Yamashita, H. Tamura, Nat. (Lond.) 282 (1979) 817.
[5] M.I. Litter, Appl. Catal. B: Environ. 23 (1999) 89.
[6] J.A. Navio, J.J. Testa, P. Djedjeian, J.R. Padron, D. Rodriguez, M.I.
Litter, Appl. Catal. A: Gen. 178 (1999) 191.
[7] A. Navio, G. Colon, M. Trillas, J. peral, X. Domenech, J.J. Testa, P.
Djedjeian, J.R. Padron, D. Rodriguez, M.I. Litter, Appl. Catal. B:
Environ. 16 (1998) 187.
[8] L.B. Khalil, W.E. Mourad, M.W. Rophael, Appl. Catal. B: Environ. 17
(1998) 267.
[9] J. Domenech, J. Munoz, Electrochim. Acta 32 (1987) 1383.
[10] J. Domenech, J. Munoz, J. Chem. Res. Synopses 4 (1987) 106.
[11] S. Wang, Z. Wang, Q. Zhuang, Appl. Catal. B: Environ. 1 (1992) 257.
[12] M.T. Pope, in: C.K. Jorgensen, et al. (Eds.), Heteropoly and Isopoly
Oxometalates, Inorganic Chemistry Concepts, vol. 8, Springer Verlag,
West Berlin, 1983.
[13] T. Yamase, Catal. Surv. Asia 7 (2003) 226.
[14] A. Hiskia, A. Mylonas, E. Papaconstantinou, Chem. Soc. Rev. 30
(2001) 62.
[15] Y.H. Guo, C.W. Hu, J. Cluster Sci. 14 (2003) 505.
[16] A. Troupis, A. Hiskia, E. Papaconstantinou, New J. Chem. 25 (2001)
361.
[17] A. Troupis, A. Hiskia, E. Papaconstantinou, Angew. Chem. Int. Ed. 41
(2001) 1911.
[18] A. Troupis, A. Hiskia, E. Papaconstantinou, Environ. Sci. Technol. 36
(2002) 5355.
[19] A. Troupis, A. Hiskia, E. Papaconstantinou, Appl. Catal. B: Environ.
42 (2003) 305.
[20] A. Troupis, A. Hiskia, E. Papaconstantinou, Appl. Catal. B: Environ.
52 (2004) 41.
[21] E. Gkika, A. Troupis, A. Hiskia, E. Papaconstantinou, Environ. Sci.
Technol. 39 (2005) 4242.
[22] Recently, polyoxomolybdates–CrIII complexes have been used as
precursors, which at elevated temperatures (>500 8C) form mixed
molybdenum-chomium oxides able to photocatalyze reduction of
Cr(VI) to Cr(III) [22a]. Herein we use directly polyoxotungstates
as photocatalysts for the reduction of Cr(VI), avoiding the painstaking
thermal treatment.
[22a] W.G. Hanna, Appl Catal. B: Environ. 28 (2000) 259.
[23] E. Papaconstantinou, Chem. Soc. Rev. 16 (1989) 1, and references
therein.
[24] R.R. Ozer, J.L. Ferry, J. Phys. Chem. 106 (2002) 4336.
[25] E. Papaconstantinou, A. Hiskia, in: J.J. Borras-Almenar, E. Coronado,
A. Muller, M. Pope (Eds.), Polyoxometalate Molecular Science,
Kluwer Academic Publishers, Dordrecht, 2003, p. 381.
[26] A. Hiskia, A. Mylonas, E. Papaconstantinou, Chem. Soc. Rev. 30
(2001) 62.
[27] A. Mylonas, A. Hiskia, E. Androulaki, D. Dimotikali, E. Papacon-
stantinou, Phys. Chem. Chem. Phys. 1 (1999) 437.
[28] M.T. Pope, G.M. Varga, Inorg. Chem. 5 (1966) 1249.
[29] A. Hiskia, E. Papaconstantinou, Inorg. Chem. 31 (1992) 163.
[30] J.L. Testa, M.A. Grela, M.I. Litter, Langmuir 17 (2001) 3515.
[31] P. Mytych, A. Karocki, Z. Stasicka, J. Photochem. Photobiol. A:
Chem. 160 (2003) 163.
[32] G. Colon, M.C. Hidalgo, J.A. Navio, J. Photochem. Photobiol. A:
Chem. 138 (2001) 79.
[33] M.R. Prairie, L.R. Evans, B.M. Stange, S.L. Martinez, Environ. Sci.
Technol. 27 (1993) 1776.
[34] S.G. Schrank, H.J. Jose, R.F.P.M. Moreira, J. Photochem. Photobiol.
A: Chem. 147 (2002) 71.
[35] S.M. Lee, T.W. Lee, B.J. Choi, J.K. Yang, J. Environ. Sci. Health A 38
(2003) 2219.
[36] F.U. Hu, G.X. Lu, S.B. Li, Adv. Sci. Technol. 16 (1998) 117.
[37] M.L.G. Gonzalez, A.M. Chaparro, P. Salvador, J. Photochem. Photo-
biol. A: Chem. 73 (1993) 221.
38] (a) Thisdetrimental effect is amatter of the organic. For example, it is not
observed when 4-cp is used instead of SA (b)
(b) H. Ju,G.Lu, S.Li, J. Photochem. Photobiol.A:Chem. 114 (1998) 81.
[39] G. Colon, M.C. Hidalgo, J.A. Navio, Langmuir 17 (2001) 7174.
[40] J. Gimenez, M.A. Aguado, S. Cervera-March, J. Mol. Catal. A: Chem.
108 (1996) 67.
[41] E.J. Wolfrum, C.R. Chentharamakshan, K. Rajeshwar, Langmuir 16
(2000) 2715.
[42] S. Goeringer, C.R. Chentharamakshan, K. Rajeshwar, Electrochem.
Commun. 3 (2001) 290.
[43] M.L.G. Gonzalez, P. Salvador, J. Electroanal. Chem. 326 (1992) 323.
[44] K. Rajeshwar, C.R. Chenthamarakshan, S. Goeringer, M. Djukic, Pure
Appl. Chem. 73 (2001) 1849.