perfect mixing of immiscible macromolecules at fluid interfaces · 2017-04-18 · perfect mixing of...
TRANSCRIPT

LETTERSPUBLISHED ONLINE: 26 MAY 2013 | DOI: 10.1038/NMAT3651
Perfect mixing of immiscible macromolecules atfluid interfacesSergei S. Sheiko1*, Jing Zhou1, Jamie Arnold1, Dorota Neugebauer2†, Krzysztof Matyjaszewski2,Constantinos Tsitsilianis3, Vladimir V. Tsukruk4, Jan-Michael Y. Carrillo5, Andrey V. Dobrynin5
and Michael Rubinstein1
The difficulty of mixing chemically incompatible substances—in particular macromolecules and colloidal particles—is acanonical problem limiting advances in fields ranging fromhealth care to materials engineering1–4. Although the self-assembly of chemically different moieties has been demon-strated in coordination complexes, supramolecular structures,and colloidal lattices among other systems5–18, the mechanismsof mixing largely rely on specific interfacing of chemically,physically or geometrically complementary objects. Here, bytaking advantage of the steric repulsion between brush-likepolymers tethered to surface-active species, we obtained long-range arrays of perfectly mixed macromolecules with a varietyof polymer architectures and a wide range of chemistrieswithout the need of encoding specific complementarity. Thenet repulsion arises from the significant increase in the con-formational entropy of the brush-like polymers with increasingdistance between adjacent macromolecules at fluid interfaces.This entropic-templating assembly strategy enables long-range patterning of thin films on sub-100 nm length scales.
The phase behaviour of multicomponent systems is determinedby the free energy of mixing 1Fm = 1Hm − T1Sm, where1Hm and 1Sm are, respectively, the changes in enthalpy andentropy on mixing of the constituting species, and T is thetemperature. Blends phase-separate if 1Fm > 0, which typicallyoccurs for large species, such as particles and macromoleculesthat exhibit positive mixing enthalpy (1Hm > 0) and negligiblegains in translational entropy (T1ST � 1Hm). One exampleis shown in Fig. 1a,c, where a mixture of linear poly(n-butylacrylate) (PBA) and linear poly(dimethyl acrylamide) (PDMA)undergoes phase separation when deposited on a substrate (seeMethods). The marginal role of the translational entropy highlightsthe challenging problem of how to enhance a contribution ofconformational entropy and thus shift the free-energy balancetowards mixing. This can be achieved through modification of themacromolecular architecture by tethering multiple linear chainsto a finite-size core, resulting in a variety of densely-branchedspecies, including stars, micelles, bottlebrushes, and hairy particles.Dense grafting results in a conformationally frustrated system,where an increase in volume fraction of branched macromolecules(either in solution or at the interface) causes stretching ofpolymer chains and thus a significant decrease of conformationalentropy. In this case, mixing of the conformationally stressed
1Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290, USA, 2Department of Chemistry, Carnegie MellonUniversity, Pittsburgh, Pennsylvania 15213, USA, 3Department of Chemical Engineering, University of Patras, 26504 Patras, Greece, 4School of MaterialsScience and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332-0245, USA, 5Polymer Program, Institute of Materials Science andDepartment of Physics, University of Connecticut, Storrs, Connecticut 06268, USA. †Present address: Silesian University of Technology in Gliwice, 44-100Gliwice, Poland. *e-mail: [email protected]
macromolecules with conformationally relaxed species, such asnanoparticles and coiled polymer chains, allows an increase indistance between the branched macromolecules, which lessensthe extension of the polymer branches and therefore enhancesthe net entropy of mixing. We demonstrate such enhancementfor molecular bottlebrushes, in which every monomeric unitof a polymer backbone carries a long side-chain, resultingin an extremely densely branched system. Figure 1b shows aLangmuir–Blodget (LB) monolayer of a 50/50 mol/mol mixtureof PBA and PDMA bottlebrushes. Unlike phase separationof linear polymers (Fig. 1a), monolayers of the bottlebrusheswith identical chemical compositions exhibit a homogeneousstructure, suggesting molecular mixing. More surprisingly higherresolution imaging demonstrates that, in contrast to the featurelessstructure of the linear PBA/PDMA microdomains in Fig. 1c,the bottlebrush mixtures exhibit remarkable intercalation ofthe chemically different macromolecules. Figure 1d shows adense monolayer composed of alternating wormlike PBA andPDMA bottlebrushes with mutually aligned contours. Accurateidentification of individual PBA and PDMA molecules in Fig. 1dwas facilitated by the difference in the length of the side chains,which are shorter for the PBA brushes. For equally long side chains,molecular intercalation was verified by the material-sensitive phaseimaging mode, which clearly distinguished softer PBA moleculesand harder PDMAs (Supplementary Fig. S1).
Note that the brush molecules in Fig. 1b,d have the samechemical composition as their linear counterparts in Fig. 1a,c,although showing the very distinct mixing behaviour. Apparently,the hairy architecture of the hydrophilic bottlebrushes plays akey role in stabilizing the molecular intercalation. To verifythis hypothesis, we have compared the mixing behaviour offour (2× 2) different combinations of the linear and brush-likepolymers. The micrographs in Fig. 1c,d correspond to the linear–linear and brush–brush combinations, whereas the micrographsin Fig. 1e,d depict morphologies of the linear-PDMA/brush-PBAand brush-PDMA/linear-PBA mixtures, respectively. As shown inFig. 1e, switching from brush to linear hydrophilic PDMA led toconventional phase separation of the hydrophilic and hydrophobicpolymers, similar to the linear chains mixture in Fig. 1c. However,mixing of hydrophilic PDMA bottlebrushes and hydrophobic PBAchains showed perfect mixing, confirmed by uniform dispersion ofthe chemically different macromolecules, a large distance between
NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials 735
© 2013 Macmillan Publishers Limited. All rights reserved

LETTERS NATURE MATERIALS DOI: 10.1038/NMAT3651
PBA
PBA
PDMA
PDMA
Linear A¬Linear B Brush A¬Brush B Brush A¬Star B
Brush A¬Linear BLinear A¬Brush B
Brush A¬Linear BLinear A¬Brush Ba
c
b
d
e
g
f
3 µm 1 µm 200 nm 200 nm
200 nm150 nm500 nm
Figure 1 | Miscibility and entropic templating. a–d, AFM height imaging demonstrates (a,c) microscopic domains due to phase separation of a 50/50monomer mol/mol mixture of linear PBA (Mn= 2.26× 105) and linear PDMA (Mn= 2.31× 105) deposited on a mica substrate, and (b,d) molecular mixingin a 50/50 mol/mol mixture of PBA (Mn=9.97× 105) and PDMA (Mn=6.36× 106) bottlebrushes prepared as LB films on mica at a film pressure of7 mN m−1 (see Methods). e–g, AFM height images of LB monolayers of PBA bottlebrushes (Mn= 3.26× 106) that phase-separate when mixed with linearPDMA (Mn= 1.99× 106) (e), a uniform mixture of linear PBA and brush-like PDMA (Mn=6.36× 106) (f), and a 50/50 wt% mixture of intercalatedPDMA bottlebrushes (Mn= 2.46× 106) and PSn(P2VP-b-PtBA)n heteroarm polymer stars (Mn= 7.17× 105) (g). In the micrograph titles, letters A and Bstand for PDMA and PBA, respectively. Arrows in c–e point to the corresponding type of polymer.
the PDMA bottlebrushes in excess of the two-fold contour lengthof their side chains, and strong materials contrast in atomicforce microscopy (AFM) phase images (Supplementary Fig. S1c,d).Moreover, we have also studied mixtures of polymer bottlebrushesand polymer stars19. Figure 1g shows AFM micrographs of anLB monolayer of hydrophilic PDMA bottlebrushes finely mixedwith hydrophobic PSn(P2VP-b-PtBA)n stars19, which prefer toconcentrate between the wormlike bottlebrushes. The results ofthese systematic experiments strongly suggest that the brush-likearchitecture of hydrophilic macromolecules is themain cause of thepeculiar molecular intercalation in polymer mixtures at a water/airinterface. Another important conclusion from these observationsis that the mixing behaviour is very tolerant with respect to thearchitecture and dimensions of the hydrophobic diluent species,as both linear-chain, brush-like and star-like macromoleculessupport intercalation.
Aswill be discussed later in the paper, themolecular intercalationis reversible and therefore does not depend on the process usedfor preparation of the monolayers, suggesting its equilibriumnature. A separate study was also conducted to verify the stabilityof monolayers of the hydrophilic PDMA bottlebrushes duringcompression and prove that both the hydrophobic and hydrophilicmacromolecules stay at the water/air interface in the range of filmpressures studied in this paper (Supplementary Fig. S2).
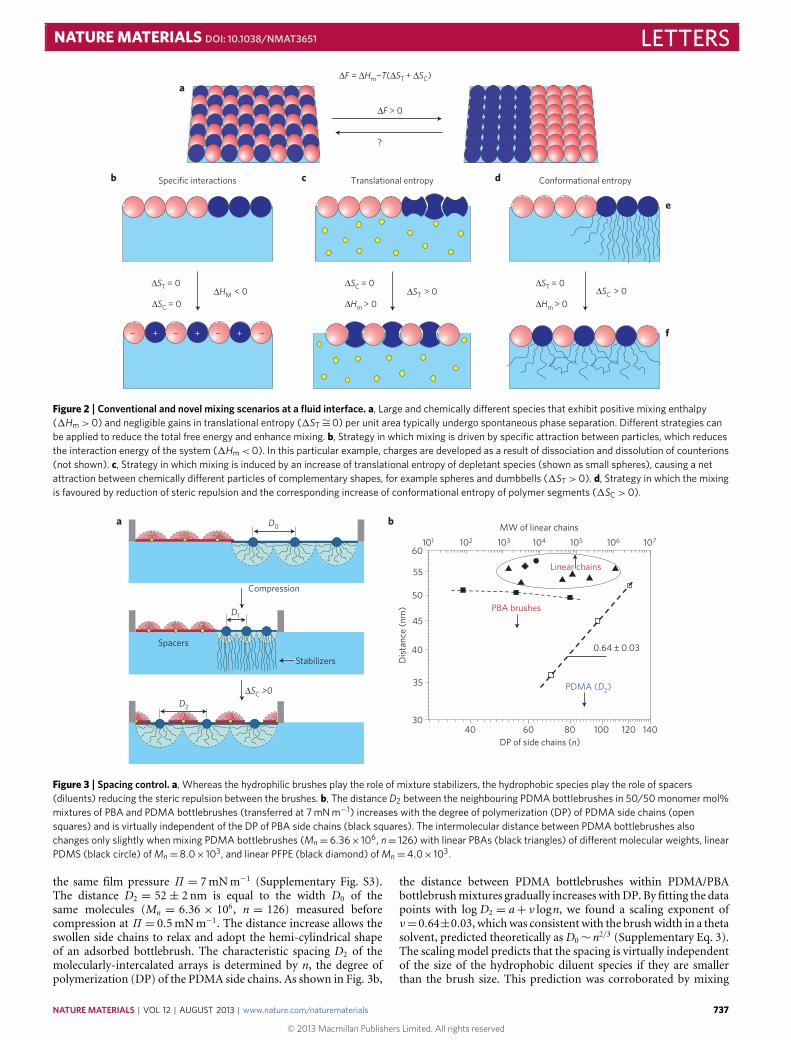
Figure 2 explains the concept of conformational enhancementof molecular mixing at liquid/air interfaces leading to interca-lation of chemically different species. This concept differs fromothers where the ineffectiveness of the translational entropy wasameliorated by short-range5–9 or long-range specific attractions10–12(Fig. 2b) and/or depletion forces13 (Fig. 2c) that require particularcomplementarity of the objects’ chemical composition, sizes andshapes. In the entropic templating (ET) mechanism exploited here,global segregation is prevented as the mixing process includes asignificant increase of one component’s conformational entropy.Figure 2 outlines the basic physical principles of ET involvinglyophilic brush-like species and spacer species of any nature. Bothspecies are assumed to be surface-active, that is, they reduce the totalsurface free energy by adsorbing to a liquid interface, such as an
interface between a gas and a liquid, or between two immiscibleliquids, such as oil and water. It is also assumed that one of theliquids, for example water, is a good solvent for the polymer brush.For example, polymer bottlebrushes, comprised of hydrophilicside chains emanating from a hydrophobic backbone, form stablemonolayers as they reconfigure while remaining pinned to thewater/air interface. In dense monolayers, steric repulsion betweenthe swollen bottlebrushes leads to extension of their side chains,resulting in a decrease of conformational entropy SC (Fig. 2e).However, if the hydrophilic bottlebrushes are mixed with diluentspecies, the steric repulsion can be reduced by intercalating thediluents between brush macromolecules (Fig. 2f). Juxtaposing ahydrophobic diluent next to the hydrophilic polymer bottlebrushresults in energetically unfavourable contacts (1Hm > 0), yet thisenergetic penalty is compensated by a decrease of steric repulsionand the corresponding increase of the conformational entropy(1SC > 0) (Supplementary Model). Effectively, the diluent speciesplay the role of spacers that may have different chemical com-positions or shapes, for example nanoparticles, linear chains, andbranched macromolecules. A similar principle has been appliedto promote microphase separation and ordering in lipid–polymermixtures20 and nanoparticles coated with monolayers of ligandmixtures21. In our systems, significant enhancement and control ofthe entropic contribution has been attained by dense tethering ofmultiple polymer chains to a single backbone.
In Fig. 3a, the ET concept has been further adapted to explainthe unique molecular intercalation observed in 50/50 mol/molPBA/PDMA bottlebrush mixtures deposited at an air/water in-terface (see Methods). Unlike hydrophobic PBAs, the hydrophilicside chains of the PDMA bottlebrushes swell and extend towardsthe water subphase. On lateral compression, the extension of thehydrophilic side chains increases, causing a decrease of confor-mational entropy. At a certain film pressure, a transition from aphase-separated to intercalated morphology occurs, resulting in adrastic increase of the distance between the PDMA bottlebrushes.This was corroborated by measuring an intermolecular distance ofD1 = 20± 1 nm in pure PDMA domains before intercalation andD2 = 52± 2 nm between the intercalated PDMA bottlebrushes at
736 NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials
© 2013 Macmillan Publishers Limited. All rights reserved
NATURE MATERIALS DOI: 10.1038/NMAT3651 LETTERSΔF = ΔHm¬T(ΔST + ΔSC)
ΔST = 0
ΔSC = 0ΔHM < 0
ΔSC = 0
ΔHm > 0ΔST > 0
ΔST = 0
ΔHm > 0ΔSC > 0
ΔF > 0
?
a
b c d
e
f¬ + ¬ + ¬ ¬+
Specific interactions Conformational entropyTranslational entropy
Figure 2 | Conventional and novel mixing scenarios at a fluid interface. a, Large and chemically different species that exhibit positive mixing enthalpy(1Hm >0) and negligible gains in translational entropy (1ST∼=0) per unit area typically undergo spontaneous phase separation. Different strategies canbe applied to reduce the total free energy and enhance mixing. b, Strategy in which mixing is driven by specific attraction between particles, which reducesthe interaction energy of the system (1Hm <0). In this particular example, charges are developed as a result of dissociation and dissolution of counterions(not shown). c, Strategy in which mixing is induced by an increase of translational entropy of depletant species (shown as small spheres), causing a netattraction between chemically different particles of complementary shapes, for example spheres and dumbbells (1ST >0). d, Strategy in which the mixingis favoured by reduction of steric repulsion and the corresponding increase of conformational entropy of polymer segments (1SC >0).
Dis
tanc
e (n
m)
DP of side chains (n)
PBA brushes
PDMA (D2)
Linear chains
0.64 ± 0.03
MW of linear chainsD0
D1
ΔSC >0D2
Compression
Stabilizers
Spacers
30
35
40
45
50
55
60101 102 103 104 105 106 107
40 60 80 100 120 140
a b
Figure 3 | Spacing control. a, Whereas the hydrophilic brushes play the role of mixture stabilizers, the hydrophobic species play the role of spacers(diluents) reducing the steric repulsion between the brushes. b, The distance D2 between the neighbouring PDMA bottlebrushes in 50/50 monomer mol%mixtures of PBA and PDMA bottlebrushes (transferred at 7 mN m−1) increases with the degree of polymerization (DP) of PDMA side chains (opensquares) and is virtually independent of the DP of PBA side chains (black squares). The intermolecular distance between PDMA bottlebrushes alsochanges only slightly when mixing PDMA bottlebrushes (Mn=6.36× 106, n= 126) with linear PBAs (black triangles) of different molecular weights, linearPDMS (black circle) of Mn=8.0× 103, and linear PFPE (black diamond) of Mn=4.0× 103.
the same film pressure Π = 7mNm−1 (Supplementary Fig. S3).The distance D2 = 52± 2 nm is equal to the width D0 of thesame molecules (Mn = 6.36 × 106, n = 126) measured beforecompression at Π = 0.5mNm−1. The distance increase allows theswollen side chains to relax and adopt the hemi-cylindrical shapeof an adsorbed bottlebrush. The characteristic spacing D2 of themolecularly-intercalated arrays is determined by n, the degree ofpolymerization (DP) of the PDMA side chains. As shown in Fig. 3b,
the distance between PDMA bottlebrushes within PDMA/PBAbottlebrushmixtures gradually increaseswithDP. By fitting the datapoints with log D2 = a+ ν logn, we found a scaling exponent ofν=0.64±0.03, whichwas consistent with the brushwidth in a thetasolvent, predicted theoretically as D0∼ n2/3 (Supplementary Eq. 3).The scaling model predicts that the spacing is virtually independentof the size of the hydrophobic diluent species if they are smallerthan the brush size. This prediction was corroborated by mixing
NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials 737
© 2013 Macmillan Publishers Limited. All rights reserved

LETTERS NATURE MATERIALS DOI: 10.1038/NMAT3651
S (f
ract
ion
of c
onta
cts)
P (mN M¬1)
S (f
ract
ion
of c
onta
cts)
Time, h
400 nm
Compression
Expansionannealing
Compression Time Time
Expansion
Phase separation
Rela
tive
devi
atio
n, Δ
A/A
Surface pressure (mN m¬1)
Surf
ace
pres
sure
(m
N m
¬1 )
Monomeric area (nm2)
PBA
PDMA
50/50
Exp. Interp.
Intercalation
Random mixture
0.0 0.1 0.2 0.3 0.4 0.5¬0.20
¬0.15
¬0.10
¬0.05
0.00
0 1 2 3 4 5 6 7 8 9
¬0.4
¬0.2
0.0
0.2
0.4
1 2 3 4 5 6 7
a
b
d
f g
e
c
0
2
4
6
8
¬1.0
¬0.8
¬0.6
¬0.4
¬0.2
0.0
0.2
0.4
1050 15 20
Figure 4 | Equilibrium and kinetics. a, Schematics of lateral compression followed by equlibration at constant pressure. b, Film-pressure versus moleculararea isotherms of three systems: pure PBA bottlebrushes, pure PDMA botlebrushes, 50/50 monomer mol% mixture of the PBA and PDMA brushes. Thedashed line corresponds to an isotherm of a phase-separated system calculated by linear interpolation of the molecular area at any given film pressure asA0=φAPBA+(1−φ)APDMA for φ=0.5-monomer molar fraction of PBA bottlebrushes. c, Relative difference (A−A0)/A0 between the experimental (exp.)and interpolated (interp.) isotherms measured for the 50/50 mixture and shown in b as solid and dashed lines, respectively. d,e, Intercalation parameterS= nAB−nAA−nBB was measured for monolayers of a 50/50 monomer mol.% mixture of PBA bottlebrushes (Mn= 3.26× 106) and PDMA bottlebrushes(Mn=6.36× 106) prepared at different film pressures (d) and different equilibration times at a constant pressure (e). In d, the length fraction ofPBA/PDMA pairs notably increases at film pressures above 5 mN m−1. The dashed line corresponds to a random mixture of cylindrical objects(Supplementary Fig. S4). In e, the length fraction of PBA/PDMA pairs increases with time after separate deposition of the PBA and PDMA species.f,g, PDMA bottlebrushes (Mn= 2.46× 106) and PSn(P2VP-b-PtBA)n heteroarm stars (Mn= 7.17× 105) mix at a higher pressure of 7 mN m−1 (g) andphase-separate at a lower film pressure of 1 mN m−1 and/or on annealing of LB monolayers on a mica substrate in a humid environment (98% RH; f).
PDMA bottlebrushes with diluents of different sizes and chemicalcompositions. Figure 3b shows that the distance between PDMAbottlebrushes (n= 121) depends neither on the side-chain lengthof PBA bottlebrushes nor the DP of linear PBAs in a broad rangeof n= 103–106 (see Methods). Furthermore, mixtures with linearpoly(dimethylsiloxane)s (PDMS) and linear perfluoro-polyethers(PFPE) showed a similar distance of 54±3 nm (Fig. 3b).
The model outlined in Supplementary Fig. S7 predicts thatintercalation of molecular bottlebrushes is caused by stericrepulsion between the PDMA side chains, which increases onlateral compression of a bottlebrush monolayer on a Langmuirtrough (Fig. 4a). To demonstrate the effect of compression, Fig. 4bdepicts three pressure–area isotherms measured for pure PDMAbottlebrushes, pure PBA bottlebrushes, and a 50/50 monomer
738 NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials
© 2013 Macmillan Publishers Limited. All rights reserved

NATURE MATERIALS DOI: 10.1038/NMAT3651 LETTERSmol% mixture of PBA and PDMA bottlebrushes. The dashedline corresponds to a hypothetical isotherm calculated assuminga PBA/PDMA phase separation. Onset of intercalation in the50/50 mixture was observed by AFM at a film pressure ofΠ = 5mNm−1. Using the Delaunay triangulation22, we countedthe nearest neighbours in AFM micrographs and quantified thelength of contact between PBA and PDMA bottlebrushes in termsof an intercalation parameter S = nAB − nAA − nBB, where nAB,nAA, and nBB are number fractions of line contacts between unlikePDMA/PBA pairs and like pairs (PDMA/PDMA and PBA/PBA).Figure 4d shows the evolution of the intercalation parameter as afunction of the film pressure. At lower pressures, <5mNm−1, thePBA and PDMA molecules are phase-separated, as evidenced by alarge negative S value due to a dominating fraction of like pairs.At higher pressures, approaching 7mNm−1, S sharply increases,exceeding the S∼=−0.17 that is characteristic of random mixturesof rod-like objects of finite size generated by the Monte Carlomethod (Supplementary Fig. S4b). The increase in the fractionof unlike-pair contacts is consistent with the visual observationof intercalated macromolecules. The highest S value approachesS∼= 0.43, expected for perfect intercalation of finite-size rods thathave an aspect ratio similar to the bottlebrushes studied in thispaper (Supplementary Fig. S4c). Imperfect intercalation and thussmaller S values are ascribed to intrinsic polymer limitations (sizepolydispersity and molecular curvature) and a slow equilibrationprocess, as demonstrated in Fig. 4e. This results in small domainsof like molecules of the order of the molecular size, as observedby AFM. The AFM observation evidence is consistent with themacroscopic measurements of pressure–area isotherms. As shownin Fig. 4c, the experimentally measured isotherm of a 50/50mixture deviates from the hypothetical phase-separation isotherm,suggesting an enhancement of the interaction between the PBAand PDMA macromolecules. A significant decrease of area wasobserved at pressures >5mNm−1, which was consistent withthe onset of molecular intercalation observed by AFM in LBmonolayers (Fig. 4b).
The data points in Fig. 4d were obtained after equilibration ofmonolayers at constant film pressure. The duration of the equili-bration process depended on the sample preparation conditions.Particularly long equilibration times were required for monolayersprepared by separate/sequential deposition of PBA and PDMAsolutions, which initially led to the formation of macrodomains ofthe PBA and PDMA bottlebrushes. In this case, the equilibrationtook more than 20 h (Fig. 4e). However, if premixed solutionswere used, equilibration was relatively quick: stabilization of theintermolecular distance took several minutes. Independently of thesample preparation, we observed identical isotherms and the onsetof mixing at 5mNm−1. Reversibility of the mixing process wasalso tested throughmultiple compression–equilibration–expansioncycles and long-term annealing of LB monolayers on solid sub-strates. As shown in Fig. 4f, after one day, the once-mixed PDMAbottlebrushes and PSn(P2VP-b-PtBA)n heteroarm stars becamephase-separated again, indicating the equilibrium nature of themolecular intercalation observed at the fluid interface.
In summary, we report a new strategy for mixing of chemicallydifferent polymers—entropic templating—that is driven by con-formational entropy and steric repulsion enhanced by the hairyarchitecture of molecular brushes. Furthermore, owing to the smallcontribution of translational entropy, this strategy leads to per-fect intercalation of lyophilic and lyophobic macromolecules andhence the formation of long-range chemically patterned arrays. Theentropic templating strategy opens the possibility for the targeteddesign ofmacromolecules with well-defined dimensions and chem-ical functionalities, thus creatingmolecular-scale ordered substrateswith potential applications in photovoltaic films, multifunctionalcoatings, and enzymatically active surfaces.
MethodsMaterials. Linear poly(n-butyl acrylate)s (PBA) with molecular weights rangingfromMn=1.44×103 to 1.13×106 (PDI=1.15), linear poly(dimethyl acrylamide)s(PDMA) with molecular weights ranging from Mn = 2.31×105 to 1.99×106(PDI=1.9), and PDMSwith amolecular weight ofMn=8.0×103 (PDI=1.3) werepurchased from Polymer Source. The linear PFPE diol Fomblin ZDOL4000 with amolecular weight ofMn=4.0×103 was purchased from Solvay Solexis.
The bottlebrush polymers were synthesized by grafting PBA (or PDMA) sidechains from a poly(2-(2-bromopropionyloxy)ethyl methacrylate) macroinitiator,using atom transfer radical polymerization23 (ATRP). In total, six polymerswere prepared, including three PDMA bottlebrushes (n= 70, 97 and 121;Mn= 2.46×106, 5.12×106 and 6.36×106;Mw/Mn= 1.2, 1.22 and 1.17) and threePBA bottlebrushes (n= 38, 55 and 80;Mn = 9.97×105, 3.26×106 and 4.69×106;Mw/Mn= 1.27, 1.14 and 1.21) with n-degree of polymerization (DP) of side chains;Mn—number averagemolecular weight, andMw/Mn—dispersity.
The polystyrene/poly(2-vinylpyridine)/poly(tert -butyl acrylate)PSn(P2VP−PtBA)n heteroarm polymer stars were prepared through a multistepsequential anionic living polymerization19. PS arms were formed by reactingsBuLi with styrene, followed by reaction with DVB, forming a star polymer withpolyDVB core and PS arms (DP=34). A second generation of arms was introducedby polymerization of 2VP (DP= 136). The living ends of P2VP arms initiatepolymerization of PtBA (DP= 119), forming a PSn(P2VP−PtBA)n heteroarmstar block terpolymer with a total of 44 arms and a weight average molecularweight of Mw = 717,000.
LB monolayers. Dilute solutions of the studied polymers and their mixtures inchloroform were deposited onto the surface of water (Milli-Q double-distilled,ρ = 18.2M� cm) using a Langmuir–Blodgett trough (KSV-5000 instrumentequipped with a Wilhemy plate balance) at room temperature. 20min wereallowed for the solvents to evaporate before compression of the monolayers. Thecompression rate was 10mmmin−1. The monolayers were transferred onto afreshly-cleaved mica substrate at a controlled constant pressure and a pullingrate of 5mmmin−1.
Atomic force microscopy. Height and phase micrographs of individual moleculeswere recorded using a Multimode atomic force microscope (Bruker) in tappingmode and silicon cantilevers with a resonance frequency of about 160 kHz and aspring constant of about 5Nm−1. The analysis of molecular images was performedusing a custom software program developed in-house. Molecular dimensions weremeasured from two 2 µm×2 µm AFM images, including more than 400 moleculesto ensure a relative standard deviation of themean below 5%.
Received 21 December 2012; accepted 10 April 2013;published online 26 May 2013
References1. Freed, K. F. & Dudowicz, J. Influence of monomer molecular structure on the
miscibility of polymer blends. Adv. Polymer Sci. 183, 63–126 (2005).2. Higgins, J. S., Lipson, J. E. G. & White, R. P. A simple approach to polymer
mixture miscibility. Phil. Trans. R. Soc. A 368, 1009–1025 (2010).3. Wolf, B. A. Making Flory–Huggins Practical: Thermodynamics of
polymer-containing mixtures. Adv. Polymer Sci. 238, 1–66 (2011).4. Ganesan, V., Ellison, C. J. & Pryamitsyn, V. Mean-field models of structure and
dispersion of polymer-nanoparticle mixtures. Soft Matter 6, 4010–4025 (2010).5. Park, S. Y. et al. DNA-programmable nanoparticle crystallization. Nature 451,
553–556 (2008).6. Nykypanchuk, D., Maye, M. M., van der Lelie, D. & Gang, O. DNA-guided
crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008).7. Valignat, M., Theodoly, O., Crocker, J., Russel, W. & Chaikin, P. Reversible
self-assembly and directed assembly of DNA-linked micrometer-sized colloids.Proc. Natl Acad. Sci. USA 102, 4225–4229 (2005).
8. Chen, Q., Bae, S. C. & Granick, S. Directed self-assembly of a colloidal kagomelattice. Nature 469, 381–385 (2011).
9. Liu, J., Zhang, Y. & Zhang, J. M. Stereocomplexation and monolayermorphologies of a stereoregular poly(methyl methacrylate) mixture formed atthe air/water surface. J. Phys. Chem. C 111, 6488–6494 (2007).
10. Shevchenko, E. V., Talapin, D. V., Kotov, N. A., O’Brien, S. &Murray, C. B. Structural diversity in binary nanoparticle superlattices. Nature439, 55–59 (2006).
11. Leunissen, M. E. et al. Ionic colloidal crystals of oppositely charged particles.Nature 437, 235–240 (2005).
12. Bartlett, P. & Campbell, A. I. Three-dimensional binary superlattices ofoppositely charged colloids. Phys. Rev. Lett. 95, 128302–128302 (2005).
13. Sacana, S., Irvine, W. T. M., Chaikin, P. M. & Pine, D. J. Lock and key colloids.Nature 464, 575–578 (2010).
14. Glotzer, S. C. & Solomon, M. J. Anisotropy of building blocks and theirassembly into complex structures. Nature Mater. 6, 557–562 (2007).
15. Mann, S. Self-assembly and transformation of hybrid nano-objects andnanostructures under equilibrium and non-equilibrium conditions. NatureMater. 8, 781–792 (2009).
NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials 739
© 2013 Macmillan Publishers Limited. All rights reserved

LETTERS NATURE MATERIALS DOI: 10.1038/NMAT3651
16. Liu, K. et al. Step-growth polymerization of inorganic nanoparticles. Science329, 197–200 (2010).
17. Aiba, N., Sasaki, Y. & Kumaki, J. Strong compression rate dependence ofphase separation and stereocomplexation between isotactic and syndiotacticpoly(methyl methacrylate)s in a Langmuir monolayer observed by atomicforce microscopy. Langmuir 26, 12703–12708 (2010).
18. Mayer, A. C. et al. Bimolecular crystals of fullerenes in conjugated polymersand the implications of molecular mixing for solar cells. Adv. Funct. Mater. 19,1173–1179 (2009).
19. Choi, I., Gunawidjaja, R., Suntivich, R., Tsitsilianis, C. & Tsukruk, V. V.Surface behavior of PSn(P2VP-b-PtBA)n heteroarm stars.Macromolecules 43,6818–6828 (2010).
20. Zhang, D., Carignano, M. A. & Szleifer, I. Cluster structure and corralling effectdriven by interaction mismatch in two dimensional mixtures. Phys. Rev. Lett.96, 028701 (2006).
21. Singh, C. et al. Entropy-mediated patterning of surfactant-coated nanoparticlesand surfaces. Phys. Rev. Lett. 99, 226106 (2007).
22. Delaunay, B. N. Sur la sphére, vide. Izv. Akad. Nauk SSSR, Otd. Matemat.Estestv. Nauk 7, 793–800 (1934).
23. Matyjaszewski, K. & Xia, J. Atom transfer radical polymerization. Chem. Rev.101, 2921–2990 (2001).
AcknowledgementsThe authors thank E. T. Samulski for insightful discussions and reviewing the paper.S.S.S., A.V.D., V.V.T. and M.R. acknowledge financial support from the NationalScience Foundation DMR-0906985, DMR-1004576, DMR-1122483, DMR-1002810and DMR-0907515.
Author contributionsS.S.S. wrote the manuscript and supervised the project, J.Z. and J.A. performed theexperiments, D.N. and C.T. synthesized materials, J-M.Y.C. performed Monte Carlomodelling of 2D patterns, A.V.D. and M.R. developed theoretical analysis of interfacialmixing, andK.M. andV.V.T. discussed results and contributed to revisions.
Additional informationSupplementary information is available in the online version of the paper. Reprints andpermissions information is available online at www.nature.com/reprints. Correspondenceand requests for materials should be addressed to S.S.S.
Competing financial interestsThe authors declare no competing financial interests.
740 NATURE MATERIALS | VOL 12 | AUGUST 2013 | www.nature.com/naturematerials
© 2013 Macmillan Publishers Limited. All rights reserved