patología del desarrollo y 1...
TRANSCRIPT

Patología del Desarrollo y
Genéticas.
Los trastornos o alteraciones que se
producen en las etapas prenatales del
desarrollo embriológico humano constituye
un grupo heterogéneo de patologías.
Enfermedades Congénitas
Enfermedades Genéticas
Teratología.
LM
1

Malformación
Es el defecto morfológico o
anormalidad estructural de
un órgano, o región
anatómica debido a una
morfogénesis perturbada.
LM
2

Agentes Teratógenos
Químicos: talidomida, alcohol, antiepilepticos, flúor,
tetraciclinas, avitaminosis (A,B, D,E,K) nicotina,
sulfamidas, salicilatos, monoxido de carbono, etc.
Biológicos: toxoplasmosis, herpes simple, rubéola,
diabetes, hiper o hipotiroidismo, sífilis congénita,
etc.
Físicos: radiaciones, traumatismos o presiones,
temperaturas extremas, etc.
LM
3

Principios Generales de la
Teratología
1. La susceptibilidad varia según el individuo, los genotipos del niño en gestación y de la madre son determinantes primarios.
Ej. Síndrome del Alcoholismo Fetal solo afecta a algunos niños de madres alcohólicas y a otros NO.
2. La susceptibilidad es específica para cada etapa del desarrollo. La mayoría de los agentes son teratógenos en la etapa inicial del desarrollo ( tres meses de gestación).
LM
4

Principios Generales de la Teratología
3. Los mecanismos biológicos o farmacológicos son
específicos para cada agente teratógeno. La
teratogenicidad de cada sustancia depende de su
mecanismo de acción: a. inhibir la actividad enzimática
o receptores cruciales. b. Interfieren la formación del huso
mitotico. c. Bloqueo energético.
4. La respuesta se relaciona con la dosis.
5. El resultado de las influencias teratógenas incluye:
muerte, retardo del crecimiento, malformación y
malfunción. LM
5

Errores de la Morfogénesis
Tipo Mecanismo Ejemplo
Agenesia y falta precoz del Agenesia
Aplasia desarrollo Total / Parcial Renal
Hipoplasia acción teratógena Microcefalia
desarrollo durante fase crecimiento
incompleto
Disrafía falta de fusión completa Espina Bífida
primordio embrionario (Mielomeningocele)
LM
6

Errores de la Morfogénesis
Tipo Mecanismo Ejemplo
Falta de estructuras embrionarias Cond. Tirógloso
Involución transitorias que no desaparecen
Atresia
Falta formación cilindros celulares sólidos Atresia biliar
de luces sin apoptosis central- luz
Ectopia falla de la migración normal Puntos de
desplazamiento de de células durante su desarrollo Fordyce
Órganos o tejidos
LM
7

Anomalías de Cabeza y Cuello
Anomalías del Desarrollo de Huesos Maxilares
Micrognatismo
Macrognatismo-Prognatismo
Anomalías del Desarrollo de la Lengua
Microglosia-aglosia
Macroglosia (anomalía, tumores, enfermedades)
Anquiloglosia
Hendiduras de la Lengua: bífida, fisurada, escrotal.
Anomalías del Desarrollo de la Mucosa Bucal
Puntos de Fordyce
Nevo Esponja Blanco LM
8
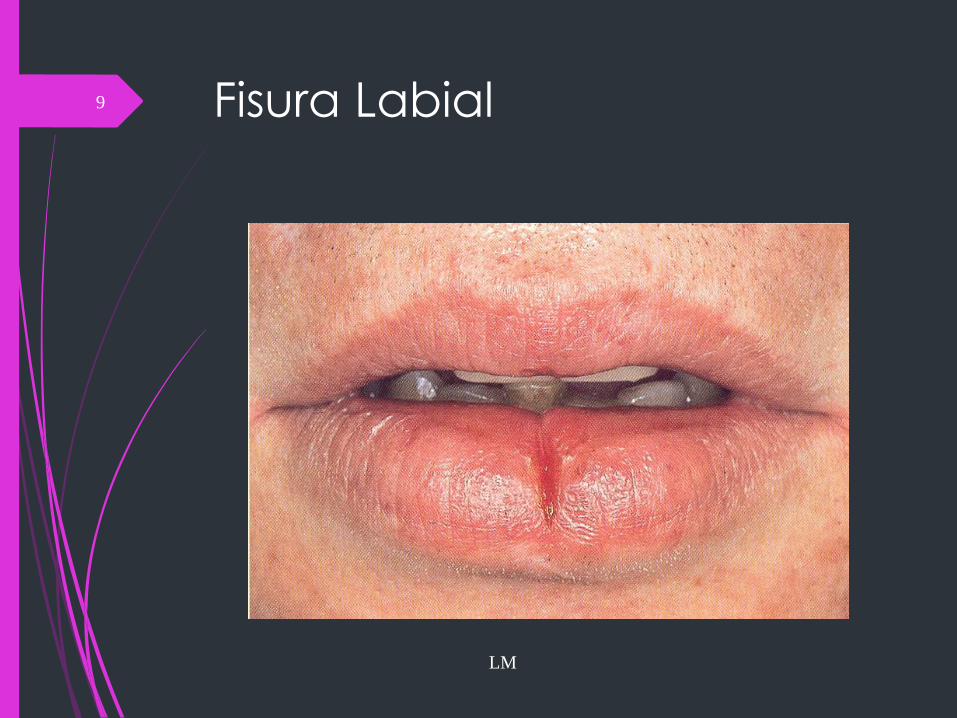
Fisura Labial
LM
9

Lengua Fisurada
LM
10

Glositis Romboidal Media(Malformación Lingual o Candidiasis)
LM
11

LM
12

Gránulos de
Fordyce
LM
13

Nevo Esponja Blanco
LM
14

Malformaciones del Cuello
Quiste del Conducto Tirogloso
LM
15
Asintomatico, ubicado en la línea media. Delimitado por epitelio remanente del conducto tirogloso.
Este epitelio puede transformarse en quistes o tumores.
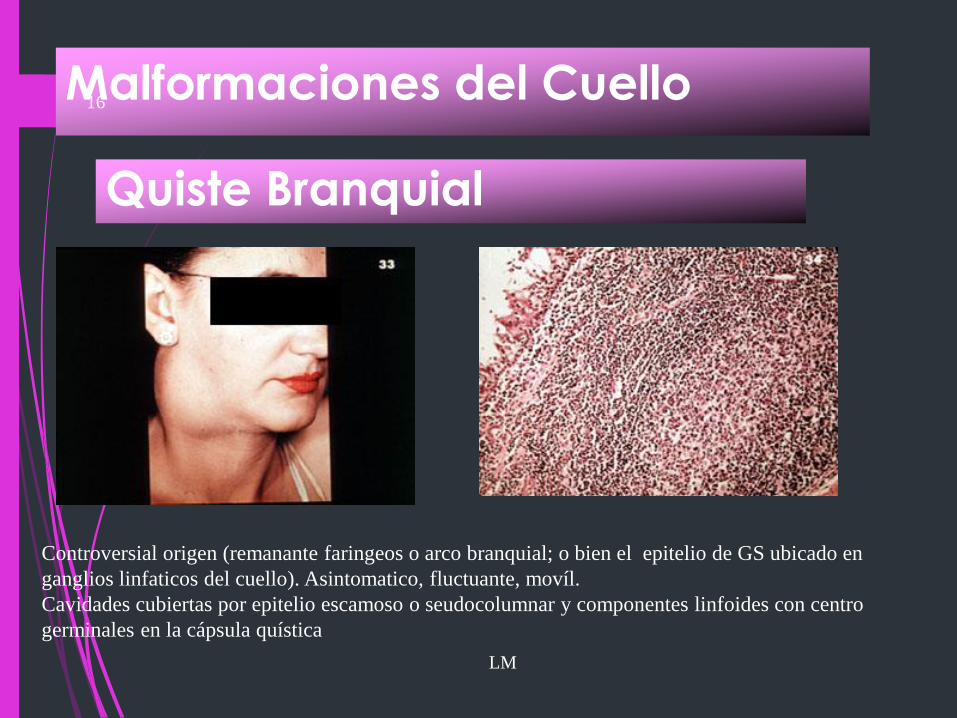
Malformaciones del Cuello
Quiste Branquial
LM
16
Controversial origen (remanante faringeos o arco branquial; o bien el epitelio de GS ubicado en
ganglios linfaticos del cuello). Asintomatico, fluctuante, movíl.
Cavidades cubiertas por epitelio escamoso o seudocolumnar y componentes linfoides con centro
germinales en la cápsula quística

Trastornos del Desarrollo de
Maxilares
Consideraciones Quirúrgicas
Muchos defectos de los maxilares tales como fisuras labio-palatinas, defectos de los arcos branquiales, dientes supernumerarios pueden tener una característica genética. Sin embargo en pocos casos la historia familiar fue informativa.
Algunos se desarrollan desde una nueva mutación genética y luego pueden ser potencialmente heredable.
Hereditarios: ej. Síndrome de Down.
Ambientales: ej. Síndrome de Pierre Robin
Una vez que la Patología se ha estabilizado en el manejo depende mas de la magnitud del defecto primario que de su etiología
LM
17

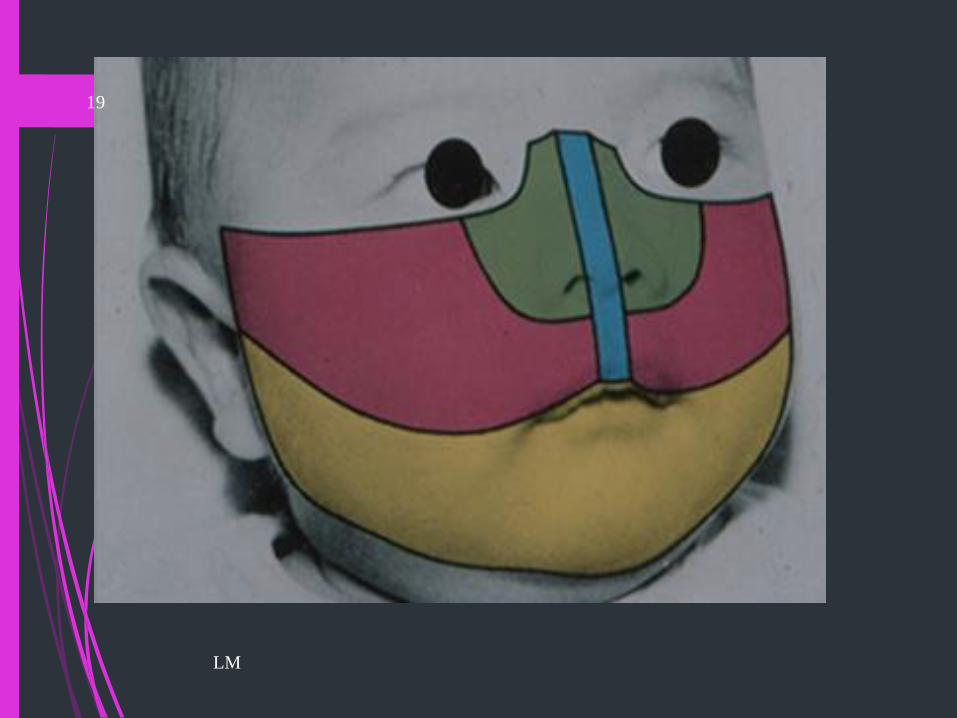
Anomalías de Cabeza y Cuello
Anomalías de Cara
Fisuras Faciales
Fisura Facial o Macrostomía o MicrosomiaHemifacial o Síndrome Goldenhar : fisura desde tragus a comisura
Fisura Oblicua o Coloboma: ala de nariz al ángulo externo del ojo.
Fisuras Labiales
Labial, labio-alveolar, labio-alveolo-palatino
(uni-bilateral) labio-alveolo-velo-palatino(uni-bilateral)
Fisuras Palatinas
Paladar duro y velo-palatina. Úvula bífidaLM
18
LM
19

Fisuras Faciales
Hay una fuerte influencia familiar, el riesgo de tener una fisura es mayor en pacientes con padres o hermanos fisurados
Se observa una asociación con pits labiales (gen autosómico dominante 80%). Otras anomalías asociadas como pie equinovarus, sindactilia y defectos oculares.
Pero hay una gran variedad de síndromes craneofaciales donde la fisura es característica (Gorlin y col. 1971)
LM
20

Microsomía Hemifacial (Síndrome Goldenhar,
Oculo-auriculo vertebral displasia, Síndrome del 1º y
2º arco branquial)
Este síndrome no ha demostrado predisposición familiar, se da 1/3000 nacimientos, igualdad de sexo. 70% es unilateral, si es bilateral es asimétrica. El 7 % de los casos tiene fisuras labiales y palatinas.
Los defectos pueden ser leves o severos cuando se extiende hasta mandíbula incluyendo defectos de aurícula, oído medio, malar, maxilar, temporal, tejidos blandos relacionados, parótida y músculos masticadores.
Anormalidades de la columna cervical y costillas
Poswillo(1973) en modelos experimentales y clínicos observo ruptura del sistema arterial del estribo a 35 días intrauterina, necrosis focal en vecindad de rama mandibular. La severidad depende del grado de destrucción primaria y de reparación compensada
LM
21

LM
22

Fisuras Labiales y Palatinas
Fisura Labial con o sin Fisura Palatina
La frecuencia es de 1/1000 nacimientos (caucásicos), 0.4/1000 nacimientos (afro-americanos) y 1.7/1000 nacimientos (japoneses) En varones, unilateral o bilateral (70% izquierdo).
La asociación de fisura palatina (duro y blando) con fisuras labiales bilaterales (85%) y unilaterales el 70%
Fisura Palatina Aislada
La frecuencia es de 1/2000 nacimientos en caucásicos y afro-americanos. Hay una relación 2 mujeres :1 hombre para paladar duro. En paladar duro y blando no hay diferencias de sexos. La úvula bífida esta presente en 1/80 caucásicos y suele presentar fisura palatina submucosa con unión imperfecta del músculo del velo
LM
23

Fisuras Labial, Alveolar y Palatina
Fisuras de los labios, reborde alveolar y paladar son anomalías congénitas que ocurren como resultado de defectos de fusión de procesos embriológicos
Fisura Labial Maxilar resulta de la ausencia o deficiencia del componente mesodérmico y degeneraciones del componente ectodérmico en el proceso nasal medio y proceso maxilar que se fusionan durante la 6ta semana
Fisura Labial Media resulta de la ausencia o defecto de una porción del proceso medio
Etiología hereditaria o factores ambientales: presión intracraneal normal, anormalidad del ancho de base de cráneo, rubéola, drogas.
LM
24

Niveles de Riesgo de Fisuras con
Parientes Afectados
Fisura Labial Fisura Palatina
c/s Palatina
Padres normales
Un hermano afectado 4% 3,5%
Un padre afectado
Hermanos no afectado 4% 3,5%
Un hermano afectado 12% 10%
Ambos padres afectados
Hermanos no afectado 35% 25%
Un hermano afectado 45% 40%LM
25

LM
26

Fisura Labial
LM
27

Fisuras Labio- Alveolo- Palatinas
LM
28

LM
29

Fisuras Palatinas
LM
30

LM
31

Aspectos Quirúrgicos de Fisuras Labiales
y o PalatinasSe acepto por mucho tiempo que la micrognasia
en los tres planos era una característica intrínseca de las fisuras. Sin embargo pacientes con fisuras sin operar demostraron un crecimiento casi normal de las estructuras afectadas.
Parece que la cirugía pos-natal al cerrar la fisura labial y o palatina inhibe el crecimiento posterior de esas áreas
Dos factores son importantes: a) no dañar el centro de crecimiento del vómer, a fin de permitir el crecimiento del maxilar b) la presencia de una cicatriz que limita el crecimiento. Von Langenbeckpermite que el colgajo deje desnudo áreas del paladar para permitir la granulación
Disección en piso anterior nasal inhibe el crecimiento hueso alveolar y resulta a menudo en infra-oclusión LM
32

Anomalía de Pierre Robin
Esta formada por fisura palatina, micrognasia y glosoptosis
Aparentemente se debe a que el feto no puede extender el cuello debido a una deficiencia de liquido amniótico. Lengua no desciende y evita la fusión del paladar. El mentón presiona sobre el esternón con micrognasia y obstrucción de vías respiratorias.
El potencial de crecimiento es normal
LM
33