p rogress in b iom olecular s tructure determinatio n … · p rogress in b iom olecular s tructure...
TRANSCRIPT

Vol. 12, No. 4 185
Progress In Biomolecular StructureDetermination by NMR
David E. Wemmer
Department of Chemistry and Chemical Biodynamics DivisionUniversity of California
Berkeley, CA 94720
ContentsI. Introduction 186
II. NMR Methods 186A. Homonuclear .186B. Heteronuclear NMR 190C. Three Dimensional NMR . . . . . . ' . . . . , . 191
III. Assignment Strategies 192A. Proteins . . 192B. Nucleic Acids 194C. Oligosaccharides . . . . . ' 195
IV. Constraint Determinations 195A. NOEs . . . 195B. J Couplings 196C. Paramagnetic ' . . . . . ' 197D. Exchange . 197
V. Structure Analysis 198A. Metric Space DG . . . . . . . . . . . . . . ; .198B. Variable Target Function 199C. Molecular Mechanics/Dynamics 199D. Exclusion method . . . . 200E. Representation of structures . . 200F. Quality of structures .201
VI. Examples 202
VII. Summary 203
VIII. References 205

186 Bulletin of Magnetic Resonance
I. Introduction
Over the past several years there has been re-markable progress in our ability to use NMR spec-troscopy to determine the three dimensional struc-ture of biomolecules in solution. The implemen-tation of two dimensional NMR experiments, andthe realization that systematic assignment of protonresonances could be achieved with a sequential ap-proach were crucial to the development of this struc-ture determination method. The initial work wasdone for proteins, but the ideas of the method weresoon extended to nucleic acids and polysaccharides.The first applications were in systems of sufficientlylow molecular weight that resonance overlap in the2D spectra was not extensive. New experiments forobtaining extended correlations have now improvedthe ability to assign more complex spectra, extend-ing the size range of molecules which can be ana-lyzed. Additional progress has been made throughincorporation of isotopic labels, using them to filterproton spectra. Applications of three dimensionalNMR have been increasing, and it appears that forlarge molecules an important improvement in reso-lution can be obtained. Modifications of the assign-ment strategy have also been developed, which lendthemselves to automation. With resonance assign-ments in hand, NOE cross peak intensities can beinterpreted to give interproton distance estimateswhich are used as restraints in structure calcula-tions. Back calculation of the NOESY spectrumfrom a particular model, for comparison with theoriginal data, has improved the distance interpreta-tion. Additional structural information can be ob-tained through determination of coupling constants,both homo- and heteroiniclear. The first calcula-tions to reconstruct atomic coordinates from theNMR data used the metric matrix distance geome-try approach. This method is still in use, but severalalternate algorithms have now been implemented,some with advantages in particular cases. In thepresent review we will concentrate on areas whichhave developed in the past few years, and presentexamples of the current state of this method. Thelimits of space and time require that this be donein a rather descriptive and qualitative way, which isfar from comprehensive, especially in terms of refer-ences to all of the original work. Where possible wewill comment on the areas where it appears caution
must still be exercised in interpretation of the NMRdata.
II. NMR Methods
The basic principles of two dimensional NMRhave been described in many different places, andwill not be discussed here. A basic knowledge of 2DNMR will be assumed. A recent compendium whichcovers a majority of the relevant pulse sequences hasbeen provided by Ernst and his coworkers (1). Avery good introduction to the necessary quantummechanical background has been given by Goldman(2), and a useful formalism for NMR calculationshas been described (3). For present purposes it willbe sufficient to simply list classes of experiments, tonote how relevant spectral information is obtained,and to describe how this can be used in obtaining as-signments or structural restraints. In addition manyof the experiments must be carried out in 1 # 2 0 so-lution, which requires solvent suppression of someform. The methods for achieving this will not bediscussed here, though the success of the NMR ex-periments often hinges critically on how well thiscan be done (4).
A. Homonuclear
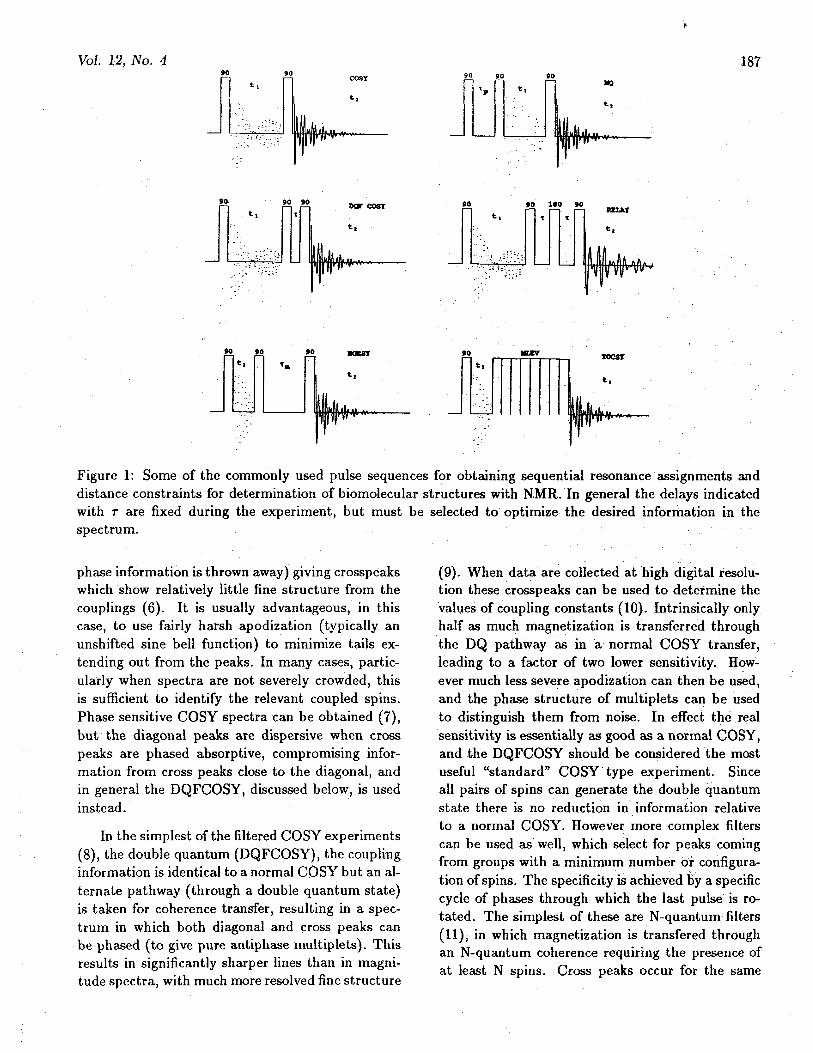
In all of the assignment methods, which will bedescribed below, it is important to identify groupsof J-coupled spins, which will generally correspondto a single amino acid, nucleoside or monosaccharideresidue. This is done with coherence transfer experi-ments; initially we restrict ourselves to homonuclearcases: COSY, filtered COSY, RELAY, TOCSY andMQ. The basic pulse sequences for these experi-ments are shown in Figure 1.
In a COSY spectrum a cross peak will appear forany particular pair spins only if there is a couplingbetween them such that the coupling constant is asubstantial fraction of the homogeneous linewidth.The transfer in all COSY type experiments yieldslines which are antiphase in character (5), causingcancellation for broad resonances. For moderate sizeproteins (with linewidths of a few Hz) couplingsbetween the C2H and C4H of histidine often giverise to observable crosspeaks although the couplingconstant is only about 1 Hz. In the past COSYspectra were run in the magnitude mode (that is all
Vol. 12, No. 4 18790 SO
t .COS*
t ,
90 ISO 90
90 90 TOCSt
Figure 1: Some of the commonly used pulse sequences for obtaining sequential resonance assignments anddistance constraints for determination of biomolecular structures with NMR. In general the delays indicatedwith T are fixed during the experiment, but must be selected to optimize the desired information in thespectrum.
phase information is thrown away) giving crosspeakswhich show relatively little fine structure from thecouplings (6). It is usually advantageous, in thiscase, to use fairly harsh apodization (typically anunshifted sine bell function) to minimize tails ex-tending out from the peaks. In many cases, partic-ularly when spectra are not severely crowded, thisis sufficient to identify the relevant coupled spins.Phase sensitive COSY spectra can be obtained (7),but the diagonal peaks are dispersive when crosspeaks are phased absorptive, compromising infor-mation from cross peaks close to the diagonal, andin general the DQFCOSY, discussed below, is usedinstead.
In the simplest of the filtered COSY experiments(8), the double quantum (DQFCOSY), the couplinginformation is identical to a normal COSY but an al-ternate pathway (through a double quantum state)is taken for coherence transfer, resulting in a spec-trum in which both diagonal and cross peaks canbe phased (to give pure antiphase multiplets). Thisresults in significantly sharper lines than in magni-tude spectra, with much more resolved fine structure
(9). When data are collected at high digital resolu-tion these crosspeaks can be used to determine thevalues of coupling constants (10). Intrinsically onlyhalf as much magnetization is transferred throughthe DQ pathway as in a normal COSY transfer,leading to a factor of two lower sensitivity. How-ever much less severe apodization can then be used,and the phase structure of multiplets can be usedto distinguish them from noise. In effect the realsensitivity is essentially as good as a normal COSY,and the DQFCOSY should be considered the mostuseful "standard" COSY type experiment. Sinceall pairs of spins can generate the double quantumstate there is no reduction in information relativeto a normal COSY. However more complex filterscan be used as well, which select for peaks comingfrom groups with a minimum number or configura-tion of spins. The specificity is achieved By a specificcycle of phases through which the last pulse is ro-tated. The simplest of these are N-quantum filters(11), in which magnetization is transfered throughan N-quantum coherence requiring the presence ofat least N spins. Cross peaks occur for the same

188 Bulletin of Magnetic Resonance
pairs of spins, and at the same positions as the ordi-nary COSY, but only for spin systems in which thereare N or more spins. As N grows it becomes moredifficult to generate the high order coherence, andthe sensitivity decreases. Unfortunately cross peaksmay not be seen for all spin systems with N spinsdue to linewidth or weak couplings, although thosepeaks which are present should be from groups N ormore spins. A more sophisticated version of this is"spin topology filtering" which selects for a particu-lar pattern of couplings rather than just a number ofspins (12). While DQFCOSY is now very routinelyused, both higher order filters and topological fil-ters have yet to see much practical use for complexbiopolymers.
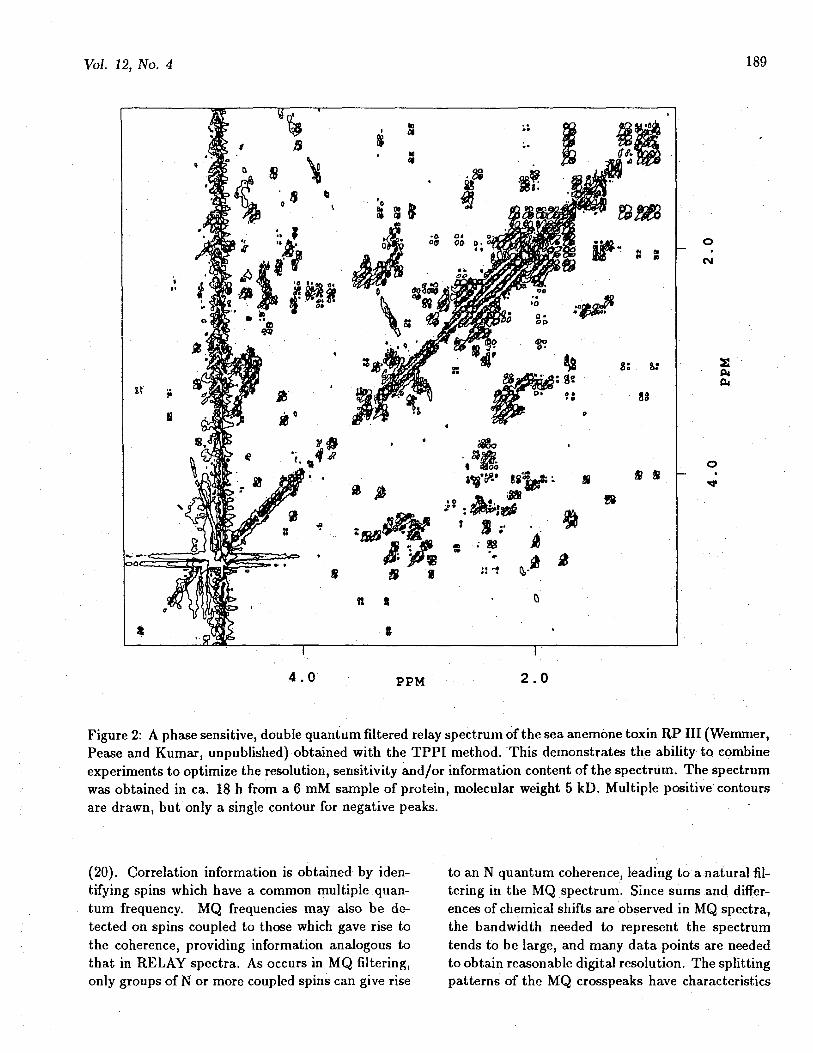
Degeneracies in chemical shift make interpreta-tion of COSY experiments difficult. The RELAY(or multiple RELAY) experiment (11) is really justa COSY which is repeated during the same exper-iment. Coherence is then passed not only to di-rectly coupled spins but also between spins with acommon coupling partner (1.3). Spin system iden-tifications then can skip over crowded regions. Theefficiency of the transfer depends on coupling con-stants, and is thus sensitive to the value of thedelay(s) used in the experiment. The behavior ofmany typical spin systems has been discussed, andthe experiment can be optimized for a particulartype of residue (14). Repeated experiments with dif-ferent delays can give relatively complete informa-tion about extended couplings. When the coherencetransfer process must be repeated, there is a loss ofsensitivity due to T2 relaxation, obviously increas-ingly severe for higher molecular weights and longerdelays. However good quality data have been ob-tained for relatively large proteins, such as ribonu-clease A (14 kD) and lysozyme (15). The RELAYexperiment transfers coherence in defined steps (un-like TOCSY discussed below) so that the numberof transfer steps which have occured is well defined(except in the case that strongly coupled spins arepresent). Having the extended coupling informationbecomes increasingly important in systems of highercomplexity. An example is shown in Figure 2.
The newest member of the coherence transferfamily of experiments is TOCSY (16) (aka H0-HAHA(17)). The basic mechanism for coherencetransfer is significantly different with this method,however the information provided is the same as
that in a combination of single and multiple RE-LAY experiments. In TOCSY coherence transfer isachieved during a mixing period in which the spinsare driven by an r.f. field. The pulse sequence (madeup of composite 180° pulses of some kind) removeschemical shifts during the mixing time, so that allspins within a coupled group evolve as though theywere strongly coupled. Coherence is transferredamong all of the spins of the group in a gradualbut continuous fashion (unlike RELAY which pro-cedes in discrete steps). With longer mixing timesmagnetization will be transfered through a largernumber of intermediate spins. As with other coher-ence transfer experiments, there is a damping whichoccurs with a time constant related to Ti and T ^rather than T2 as for COSY (18). In addition, oneof the pathways for coherence transfer leads to in-phase magnetization rather than antiphase (as oc-curs for all other coherence transfer experiments)reducing cancellation effects. These features makethe TOCSY experiment particularly useful for largemolecules.
In the filtered COSY experiments a multiplequantum coherence is generated, but is not allowedto evolve before reconversion to single quantum co-herence. If the fixed and variable periods of the ex-periment are reversed however, just the opposite istrue and one has a true multiple quantum (MQ) ex-periment (19). Spins are labelled in ti at their multi-quantum frequency, but are detected at their singlequantum position in t2. In the multiquantum periodthe evolution occurs at a frequency corresponding tothe sum of chemical shifts for spins involved in gen-erating the coherence. The order of the coherence isdefined by the difference in total quantum numberof the two energy levels which form the coherence.Experiments can be done with order selectivity dur-ing excitation, and/or detection. The magnitudeof any individual MQ line will depend on the fixedpreparation time rp in the experiment and on fre-quencies and coupling constants of spins involved.To assure excitation of all possible MQ lines sev-eral Tp values must be used. During detection (onlysingle quantum coherence is directly detectable) theMQ coherence can be transfered to either to oneof the spins involved in the MQ coherence itself, orto other spins coupled to these. The rules govern-ing transfer are known, and the pattern of transfercan be used as an aid in analysis of the spin systems
Vol. 12, No. 4 189
SI
* $
55*
o
4 . 0 PPM 2 . 0
Figure 2: A phase sensitive, double quantum filtered relay spectrum of the sea anemone toxin RP III (Wemmer,Pease and Kumar, unpublished) obtained with the TPPI method. This demonstrates the ability to combineexperiments to optimize the resolution, sensitivity and/or information content of the spectrum. The spectrumwas obtained in ca. 18 h from a 6 mM sample of protein, molecular weight 5 kD. Multiple positive contoursare drawn, but only a single contour for negative peaks. •
(20). Correlation information is obtained by iden-tifying spins which have a common multiple quan-tum frequency. MQ frequencies may also be de-tected on spins coupled to those which gave rise tothe coherence, providing information analogous tothat in RELAY spectra. As occurs in MQ filtering,only groups of N or more coupled spins can give rise
to an N quantum coherence, leading to a natural fil-tering in the MQ spectrum. Since sums and differ-ences of chemical shifts are observed in MQ spectra,the bandwidth needed to represent the spectrumtends to be large, and many data points are neededto obtain reasonable digital resolution. The splittingpatterns of the MQ crosspeaks have characteristics

190 Bulletin of Magnetic Resonance
similar to COSY peaks, although the multiplicitygoes down as the order of transition increases.
Once covalent connectivity has been establishedusing these coherence transfer experiments furtherinformation is needed about spatial proximity ofspins to carry through the sequential assignmentstrategy. This is available through two dimensionalversions of nuclear Overhauser effect (NOE) mea-surements; NOESY (21) in the laboratory frame orROESY (22) (originally called CAMELSPIN (23))in the rotating frame. In either case crosspeaksstem from cross relaxation arising from dipolar cou-pling between the spins. Although the basic NOESYpulse sequence contains three pulses, as do the fil-tered COSY and MQ experiments, in this case thez component of magnetization rather than the x-ycomponents is involved in generating cross peaks.The magnitude of the NOE changes with rotationalcorrelation time. For short correlation times (lowmolecular weight) saturation of one peak leads toan enhancement of up to 50% for the resonance ofa nearby spin. When the correlation time is suchthat UJTC =1.1 the effect vanishes altogether. In thelong correlation time limit (high molecular weight)saturation of one resonance leads to a decrease upto 100% in intensity for spins close in space. In thiscase the rate at which cross relaxation occurs variesinversely with the sixth power of the distance be-tween spins and linearly with the correlation time.For most practical cases the dramatic dependenceon distance means that proton pairs separated bymore than about 4 A (perhaps 5 A in favorablecases) will not give rise to observable cross peaks. Inthe long correlation time limit, the NOESY exper-iment may also suffer from a propagation of crossrelaxation through a chain of spins (24), which iscalled spin diffusion (this is well known and domi-nant in solid state NMR spectroscopy). For assign-ment purposes it is often important to know thatcross peaks arise from direct relaxation, requiringthat fairly short mixing times be used. For the ro-tating frame experiment, ROESY, the short corre-lation time limit behavior is always seen, regardlessof actual correlation time. This means that spindiffusion is reduced, and that spin diffusion peakswill be opposite in sign from direct peaks makingthem more easily identified. It is particularly valu-able for the intermediate size molecules (m.w. in therange 1-2 kD) for which lab frame NOEs are either
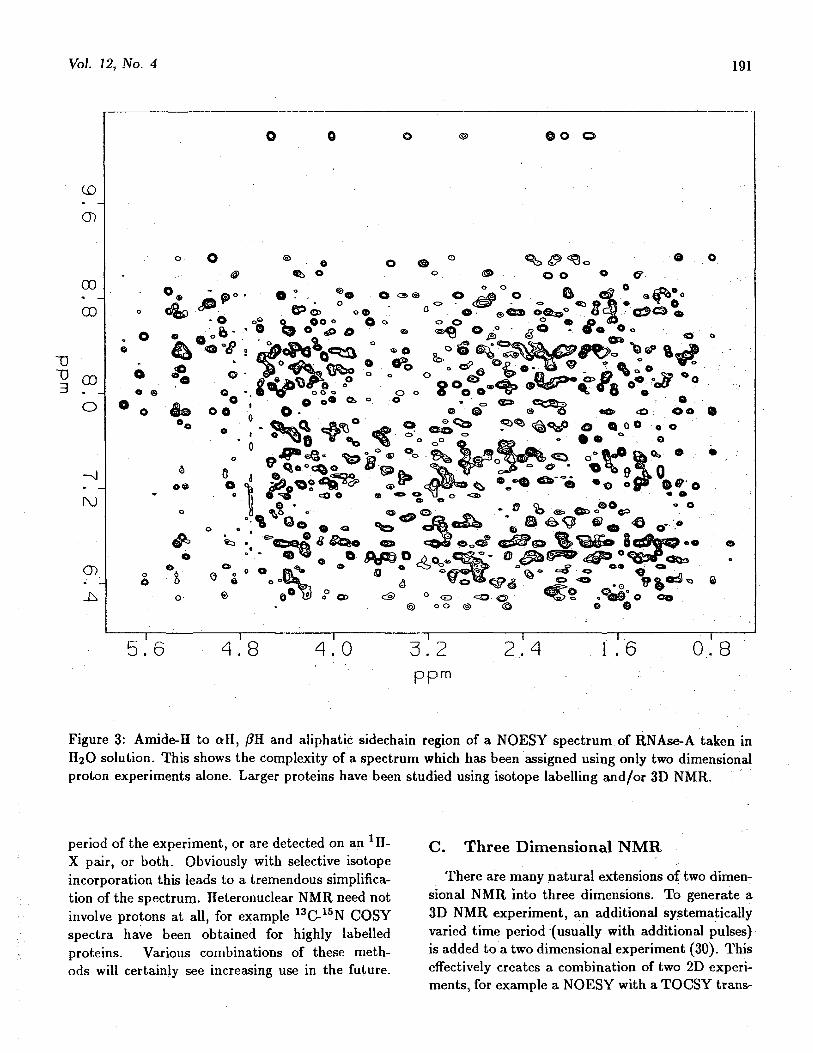
very weak or completely absent. An example of aNOESY spectrum is shown in Figure 3.
With the pulse sequences used in either experi-ment, there is a possibility of coherence transfer (aswell as the cross relaxation), which must be sup-pressed through a suitable selection of experimentalparameters and phase cycling (25). The methodsfor and importance of doing this have been doc-umented, are incorporated into the normally dis-tributed pulse sequences, and hence will not be dis-cussed here.
B. Heteronuclear NMRThe basic idea of transferring coherence to es-
tablish a correlation between coupled spins can beapplied in heteronuclear cases as well as homonu-clear. For both 13C and 15N the coupling constantsto directly bonded protons are quite large, and thetransfer of coherence between them is very efficient.Experiments were first done by detecting the X nu-cleus directly, however it has been shown that "in-direct" detection through the 1H is much more ef-ficient (26) (by two to three orders of magnitude)and hence this mode is typically used now. Exper-iments on biomolecules at a concentration of a fewmM for natural abundance 13C and 15N can thenbe collected within a day, and with enrichment canbe done even at 1 mM in a matter of a few hours.There are numerous modifications of experiments toachieve decoupling in either frequency domain, toachieve suppression of signals from 1H not coupledto an X atom, and to implement solvent suppres-sion when experiments are done in 1H2O solution.Many variations have been discussed in a recent re-view (27). Correlations can also be generated us-ing weaker two or three bond couplings, since thecoupling constants then approach the linewidth thesenstivity is significantly lower (28), but recent ap-plications have shown these may be used to obtainsequential connectivities (29). A second importantuse of the heteroatom is in filtering proton spec-tra. By using combinations of pulses on both the1H and X atoms, proton signals from H-X pairs canbe distinguished from isolated protons. These pulsecombinations can be applied within other pulse se-quences, such as COSY or NOESY, to give isotopeedited spectra - that is subspectra where cross peakseither arise from an 1H-X pair during the labelling
Vol. 12, No. 4 191
CD
O)
00
00
TS"0 003 . _
O
- s i
I\J
CD
0 o o
©
«= . * M J S < ^ O <s»
5 . 6 4 . 8 4 . 0 3 . 2ppm
2 . 4 1 . 6 0 . 8
Figure 3: Amide-H to all, /?H and aliphatic sidechain region of a NOESY spectrum of RNAse-A taken inH2O solution. This shows the complexity of a spectrum which has been assigned using only two dimensionalproton experiments alone. Larger proteins have been studied using isotope labelling and/or 3D NMR.
period of the experiment, or are detected on an 1H-X pair, or both. Obviously with selective isotopeincorporation this leads to a tremendous simplifica-tion of the spectrum. Heteronuclear NMR need notinvolve protons at all, for example 13C-15N COSYspectra have been obtained for highly labelledproteins. Various combinations of these meth-ods will certainly see increasing use in the future.
C. Three Dimensional NMRThere are many natural extensions of two dimen-
sional NMR into three dimensions. To generate a3D NMR experiment, an additional systematicallyvaried time period (usually with additional pulses)is added to a two dimensional experiment (30). Thiseffectively creates a combination of two 2D experi-ments, for example a NOESY with a TOCSY trans-

192 Bulletin of Magnetic Resonance
fer into the third dimension before detection. Thusall NOESY cross peaks will be detected on all peakscoupled to either of the spins involved in the TOCSYcross peak. Doing this improves resolution, since thecross peaks will now be spread along three frequencyaxes. In addition the problems of degeneracy are re-duced since there is effectively multiple detection ofeach peak. Essentially any pair of normal 2D exper-iments can be combined to create a 3D experiment(the burden this will create on the pulse sequenceliterature is staggering if any significant fractionof those possible is actually described in papers).Some of these combinations contain new informa-tion which is not present in a single 2D experiment,for example the NOESY-NOESY contains informa-tion about spin diffusion pathways and rates. Obvi-ously heteronuclear versions can also be done, andin fact use of NOESY-heteroCOSY ^H^H NOESYfollowed by XH-15N correlation for example) is likelyto be particularly important because of the high ef-ficiency of the hetero transfer step, and the needfor only modest resolution in the third dimension(31). The time and computational requirements forthese experiments are substantial: a modest resolu-tion 256 x 256 x 256 matrix (with a typical 5000 Hzspectral width this is 20 Hz/pt) occupies 64 Mbytesof disk space (64 more to process it), and with 8scans averaged per ti ,t2 point at a rate of 1/sec thisrequires 6 days of data collection. These facts ap-pear somewhat intimidating, but a variety of suchexperiments have already been successfully done onproteins and will undoubtably become more com-mon. The high information content offsets the longacquisition times, and will probably be best usedwhen more sophisticated computerized analysis pro-grams become available.
III. Assignment Strategies
The basic concepts in carrying out systematicresonance assignments in proteins using 2D spectracame from Wiithrich (32). He has described theoriginal approach in detail in a book (33), only anoutline of which will be presented here. The ba-sic combined COSY/NOESY assignment strategiesall depend on the existence of a covalent backbonealong the molecule of interest, which assures thatsome protons on neighboring residues are close inspace. Groups of spins within each residue are iden-
tified in the correlated spectra, and these are con-nected to neighboring residues using the NOESYdata. When the groups of spins are distinguish-able then the molecule can actually be sequencedin this fashion. In practice degeneracies in chem-ical shift are often present, and only some of theresidues are distinguishable through their couplingpattern, so that sequence information from tradi-tional chemical methods is required to complete theanalysis. An alternate possibility is available usingonly coherence transfer experiments. In this casea coupling between residues must be used in addi-tion to those within residues. In all major classes ofbiopolymers there are no sufficiently strong 1H-1Hcouplings between residues to accomplish this. How-ever, if one includes heteronuclear couplings from XHto 13C and/or 15N for proteins, 13C for oligosaccha-rides, and 31P for oligonucleotides then interresiduecouplings are generally present, and can be used toestablish the backbone connectivity, and hence alsothe resonance assignments. All of the assignmentmethods will be discussed by category of molecule.
A. Proteins
In the sequential COSY/NOESY assignmentmethod the first stage is to identify groups of cou-pled spins arising from individual amino acids (34).In so far as is possible the number of protons, thepattern of coupling and the grouping of chemicalshifts are used to identify the type of amino acid.Some are often uniquely identifiable (G, A, T, V,I, often P and L, and aromatic sidechains), whileothers can only be grouped as short sidechain (S,C, D, N) or long sidechain (E, Q, M, R, and K). Incrowded spectra the use of RELAY and/or TOCSYis often quite important for grouping all spins ofthe same system unambiguously. NOESY data alsohelp in this respect, but must be used with cautionsince there is no clear way to distinguish intraresidueNOEs from interresidue. The clear association ofamide, alpha and beta protons is particularly im-portant. Establishing sequential connectivities re-lies on NOEs involving the amide (NH) proton. Astatistical analysis of protein structures showed thatthe distance from an NH to one proton among theNH, aH and /?Hs of the preceding residue (towardthe N-terminus) will be shor^ enough to give rise toan observable NOE (35). When several residues canbe sequentially connected, and some of these are of

Vol. 12, No. 4 193
established amino acid type, then a correspondencewith a particular specific sequence in the proteincan be made and the resonances are then assigned.(It may be noted that for several proteins such acorrespondence was initially not possible (36), andit was later determined through chemical methodsthat the originally reported sequence was in error.)Only a portion of the NOEs present are sequentialin nature, much of the complexity in the assign-ment process is in identifying the sequential ones,and eliminating ambiguities from degeneracies. Thisis tedious, and takes a good deal of time. The re-maining NOEs arise from folding of the protein intodefined secondary and tertiary structure and will bediscussed further below.
An alternate, but somewhat related, "mainchain directed" (MCD) approach has been sug-gested recently (37). In this case spin system identi-fications are initially carried through to group HN,aH and /?H for as many amino acids as possible. Se-quential connectivities are then sought, but in addi-tion NOEs which can be interpreted to correspondto particular secondary structures are also used.The reasoning is that there are many pathways(comprised of combinations of COSY and NOESYconnectivities) through any element of secondarystructure which could be followed to establish the re-lationship of a pair of resonances. In the case whereany degeneracy in resonance position occurs, differ-ent pathways could then give different results. If thenumber of degeneracies is not too large, then thereshould only be one (or at worst a small number) ofpossible assignments which are consistent with all ofthe observed data. The search for such a solution iscomputerized, and can be carried out very rapidly.When data are complete, all of the backbone reso-nances can be assigned in this way, these can thenbe extended to the sidechains through further anal-ysis of the coherence transfer data. Identificationof correct sidechain types at the appropriate back-bone positions confirms the assignments. In the casethat backbone data are not complete, the inclusionof some sidechain data can eliminate some assign-ment possibilities during the search. This approachhas worked very well for several proteins of moder-ate size (38), prospects are good for application inlarger proteins as well. The inclusion of 3D NMRdata will improve resolution, and hence the abilityto identify peaks, but will not significantly change
the 1H NOE based sequential assignment strategy.For proteins whose genes have been cloned and
expressed, it is possible to incorporate isotopic la-bels 2H, 13C, and 15N. The uses of each are severalfold. It has been realized for many years that inclu-sion of 2H in a specific type of amino acid will re-move the signals in the *H spectrum. This obviouslyfunctions well in all sorts of 2D spectra, and resultsin assignment of resonances to a specific amino acidtype, even for those amino acids which are in thedifficult to distinguish classes. More recently it hasbeen shown that random fractional deuteration (in-clusion of 75-80% 2H at all sites through growth ona partially deuterated medium) leads to a reductionin multiplicity of lines (essentially all resonances aresinglets or doublets) and to a reduction in spin dif-fusion in NOESY spectra (39). Although there isa large direct loss in signal, this is offset by the re-duction in linewidth even for 2D spectra in whichonly pairs of neighboring protons are observed. Itappears that this scheme may be particularly usefulfor large proteins.
13C and 15N can also be used to identify pro-ton resonances by using heteronuclear filtering ex-periments, reducing the spectrum to only thosecrosspeaks involving a proton attached to the het-eroatom. By feeding a single type of labelled aminoacid, subspectra can be generated specific to thatamino acid type (40). This has been used partic-ularly with 1SN amino acids to identify NH to aHcrosspeaks. A single label alone, however, does notdirectly aid in assignments. Double labelling with15N (amide) and 13C (in the carbonyl position) ondifferent amino acids leads to a distinct splitting inthe 15N filtered proton spectrum if a 13C label isin any amino acid preceding one with art 15N label(41). Thus selection of two particular amino acidswill generally lead to assignment of a single aminoacid's resonances (effective but expensive). More re-cently uniform 13C incorporation to a level of 25%has been done (42). In this case the carbons in eachamino acid comprise a coupled network, and COSYtype experiments can be used to establish connec-tivities between neighboring carbons. In practice adouble quantum experiment (INADEQUATE) hasbeen used instead to obtain equivalent information(43). To achieve complete assignments double la-belling with both 13C and 15N has been done, andthe 13C-15N correlations are then used to establish

194 Bulletin of Magnetic Resonance
lli
sequential connectivities. Proton resonances canthen be assigned using 1H-13C heteronuclear cor-relations. This approach is rather new, and the fullpossibilities have not yet been explored, but it seemsquite powerful.
Regardless of the method used to assign thebackbone resonances, there is usually a remainingambiguity in the assignment of the proR and proSprotons at the /? carbon (similarly further out thesidechain, but these are less often resolved), andsimilarly for the methyls of valine (44) and leucine.When distances to these protons are defined forstructure analysis, it is helpful to have the stereospe-cific assignments (45). This can be accomplishedby using a combination of coupling and NOE infor-mation, which taken together rule out one of theassignment possibilities. This procedure can be au-tomated (46), and can result in stereospecific assign-ments for 40-90% of residues (depending in part towhat extent distance information is treated quanti-tatively).
B. Nucleic AcidsOnce the pattern for sequential assignments
was established for proteins the extensions to nu-cleic acids were straight-forward (47). For DNAthere are only four types of base, which (as forammo acids) can be identified by coupling pattern:T with a methyl to aromatic coupling, and C with anaromatic-aromatic coupling are easily distinguishedfrom the purines A and G, which have aromaticprotons which show no coupling. In RNA U andC are identical in couplings, but still distinguish-able from G and A. For DNA each sugar is a groupof seven coupled spins, couplings can be identifiedfrom COSY, RELAY and/or TOCSY data. TheHI', H2', H2" and H3' are usually well separated,while the H4', H5' and 115" are close in chemicalshift and often cannot be resolved. For RNA thereis no H2" proton, and only the chemical shifts ofthe Hi's are well dispersed. Unfortunately for thetypical C3' endo sugar conformation of "A" formRNA the Hl'-H2' coupling is very weak, making itdifficult to identify protons on the same sugar: Al-though resolution is not particularly good it is pos-sible to identify some of the couplings among otherprotons in the sugars. For proteins the amide pro-ton plays a central role since it is near the back-bone and hence is near protons on the neighboring
residue. For right handed helical nucleic acids theequivalent role is played by the sugar HI' and H2'protons. The sense of the helix rotation causes thebase of the residue to the 3' side to stack abovea particular sugar. The distance from an aromaticproton to the sugar proton in the same residue is alsosufficiently short to give rise to an NOE. Thus thearomatic-Hl' and similarly the aromatic-H2'(H2")regions contain all information required for sequen-tial assignment. If there are no degeneracies thisserves to sequence the nucleic acid; when chemi-cal shift degeneracies occur, then a known sequencecan be used to eliminate some of the assignmentpossibilities. The two regions can be-assigned in-dependently, then the information compared to ver-ify assignments. Additional checks on assignmentsare provided by matching H1'-H2',H2" crosspeaks tothe positions predicted from other regions, and veri-fying that all connectivities determined from COSYor TOCSY data (48) are in agreement. Other sugarprotons (extending even to the crowded H5',H5" insimple cases) can then be assigned using any com-bination of intrasugar NOEs and couplings. Thespacings between the relevant protons are differentin RNA vs. DNA, but this general method worksfor both. If the nucleic acid is not in a regular sec-ondary structure, such as near the standard "A" or"B" form, then some or all of the sequential con-nectivities may be missing. In addition some non-sequential NOEs may be seen. However for shortsequences (loops of four or five bases, or bulges ofsimilar size) it is generally possible to assign reso-nances anyway relying on the fact that NOEs aremost likely between neighboring bases, and that thefinal assignments must correspond with the knownprimary sequence and all observed NOEs.
An alternate approach was suggested very earlywhich uses only through bond correlation informa-tion. In the case of nucleic acids the residues arelinked through phosphates, and significant couplingsare seen from the H4', H5' and H5" to the 31Pof the 5' phosphate, and from the H3' to the 3'phosphate. When appropriate connectivities areestablished through a combination of homo- andheteronuclear correlation experiments then assign-ments of the sugar resonances follow immediately.The extension of these to the bases must still useNOE information. The first application of this typewas to a single stranded DNA (49), which had sharp

Vol. 12, No. 4 195
resonances and fairly strong couplings. However re-cently it has also been applied to double strandedoligomers as well (50). Resolution in the phospho-rous spectrum is not particularly good, and the di-rect proton method is probably more applicable forlonger oligomers. Operator DNAs with 34-46 nu-cleotides (51) (m.w. >11 kD) and RNAs with 20nucleotides have been assigned.
Unlike the case for proteins the exchangeableprotons of nucleic acids are not crucial to the as-signment of the backbone. The imino and aminoprotons for a separate group, which can also be sys-tematically assigned with NOESY spectra (52). Theimino protons are particularly useful probes of sec-ondary structure since there is only one per basepair, and upon melting their exposure to solventleads to dramatic broadening. There is an exten-sive literature on base pair lifetimes derived from ex-change data, base catalysis has recently been shownto be important (53) and some care must be used ininterpretation of older papers on this subject.
Isotope labelling has found less use for nucleicacids than for proteins to date. However several sim-ple types of labelling have been explored. Deuter-ation of the purine H8 position can be achieved(sometimes inadvertantly) simply by heating in2H2O (the rate increases with pH). The H5 positionof cytosine and uracil can be exchanged by heatingin the presence of bisulfite (54) in 2H2O. For severaldifferent RNAs, produced in vivo, biosynthetic in-corporation of 13C and 15N has been done (26,55),either uniformly or in a specific type of base. Suchlabels have been useful for dynamic studies, and forassignment of imino proton resonances.
C. OligosaccharidesThe identification of groups of coupled protons
is again done with coherence transfer experments,COSY, RELAY and TOCSY (56). In many sugarsall of the protons on a residue can be identified usingTOCSY correlations to the anomeric protons, whichtypically have the best resolution. Further assign-ments are complicated in two ways. First, the fullprimary structure is often not known from chemicalmethods (unlike proteins or nucleic acids), and ispart of the information desired from the NMR ex-periment. Second, the branching is complex, anddifferent chemical linkages between sugars occur.Some contacts between sugars can be established
with NOE data (either NOESY or ROESY), but of-ten gaps remain which have been filled to a largeextent by heteronuclear correlations. The associ-ation of 13C with *H resonances is achieved usingthe one bond couplings in an ordinary heterocorre-lated spectrum. The experiment is then adjusted tobe optimized for long range couplings (three bond*H to 13C), and the spectrum is analyzed for inter-residue couplings. These can establish connectiv-ity across ether linkages which are difficult to seewith other methods, and then define the primarystructure. The positions of acetyl groups, and othermodifications can be established in this way as well.
IV. Constraint Determinations
A necessary source of distance information for re-construction of a molecular structure is the primarycovalent linkage: which atoms are bonded to which,standard bond lengths and geminal bond angles.These are derived primarily from crystallographicstudies of small molecules (where such parameterscan be determined with great accuracy), e.g. aminoacids, nucleosides, monosaccharides, etc. This in-formation is tabulated and is used in solving crystalstructures of biological macromolecules, as well asfor NMR. van der Waals radii for the atoms are alsoincluded, at least in a hard sphere approximation.
A. NOEsThe primary source of experimental structural in-
formation in the NMR studies is the NOESY spec-trum. For an isolated pair of spins, the buildup ofcrosspeak intensity is simple (57), for short mixingtimes it is linear with a slope proportional to rc/r6.Even when proton pairs are not isolated it is oftenpossible use short mixing times and keep the linearapproximation. The most conservative interpreta-tion of NOE peaks is to assign an estimated upperbound based on the intensity (33) - a strong peakcorresponding to a bound <2.5A, a moderate inten-sity peak to a bound of <3.0A and a weak peak toa bound <4.0A. Lower bounds are left at the vander Waals distance of about 2A. In principle a morequantitative estimate of the distance can be madeby using the buildup curve (58), crosspeak inten-sity vs. mixing time. The shape of the buildupcan then be seen, and models are checked to see

196 Bulletin of Magnetic Resonance
whether they correctly predict the buildup. Sincethe correlation time is not known in general, dis-tances between spins (the r values) are calculatedfrom ratios of crosspeak intensities using one peakfor which the distance is fixed by covalent bonds(e.g. geminal proton pairs on the /?C, ortho posi-tions on an aromatic ring or the aH to methyl onalanine). The unknown distance is then the sixthroot of the ratio of cross peak intensities times theknown distance. This practice works well in manycases, but can be severely compromised by spin dif-fusion in real cases where more than two spins arepresent (59). In certain arrangements of spins theindirect path (spin diffusion) for transfer of magne-tization is more efficient than the direct one, leadingto a large error in distance estimate if no correctionis applied. For three spins A, B and C if strong A-Band B-C crosspeaks are present then the A-C inten-sity should be suspect, especially when the distanceappears to be fairly long. For a known arrange-ment of spins it is possible to calculate the full NOEbuildup behavior accurately, including spin diffusionin a natural way (21). There are now procedures inwhich initial distance estimates are derived using anapproximation, or an initial model is selected, butthe NOE intensities are reanalyzed iteratively to in-clude spin diffusion corrections, and hence to givebetter distance estimates (60). These are then recy-cled into the structure calculation, and the processis repeated until it converges.
There are some cases in which the distance in-formation cannot be interpreted as simply as onewould like. If geminal hydrogens e.g. /?Hs in pro-teins, or H2',H2" in DNA, are not stereospecificallyassigned, either because insufficient information isavailable to do so or because they are degenerate infrequency, an uncertainty in a distance constraintmust included since either proton could have givenrise to the NOE. A similar case is generated by aro-matic rings of Y and F residues in proteins, whichoften flip rapidly about their symmetry axis. Inthat case any particular NOE could arise from ei-ther side of the ring. Pseudostructures have beendeveloped for amino acids (61) whicfy reflect theseuncertainties, with rules for how to modify distanceestimates appropriately. There are fewer such prob-lems in nucleic acids, but whenever degeneracies inchemical shifts are present caution must be used ininterpreting intensities to give distances.
An implicit assumption in the NOE distanceanalysis is that the effective correlation time is thesame for all pairs of protons, and that there is no av-eraging of spatial positions through motion on anytimescale (62). These assumptions are certainly notcorrect, it is well known from molecular dynamicscalculations that any biomolecule undergoes fast,small amplitude fluctuations. In many cases theremust be other larger amplitude motions as well,which may be difficult to detect. It has been shownthat if the small amplitude, fast (relative to the Lar-mor frequency) fluctuations are uncorrelated, whichseems likely in most cases, then they do not dis-tort distance estimates significantly (63). Relax-ation measurements (both Ti and T2) can be usedto try to detect the presence of other motions, ev-idence for localized motion has been obtained forseveral proteins.
B. J CouplingsA second source of structural constraints comes
from analysis of coupling constant values using aKarplus type relation (64). These can be deter-mined from any of a number of experiments, theDQFCOSY being the most common for 1H-1H cou-plings (65). Some care must be used in the anal-ysis since peak separations do not reflect couplingconstant values accurately when the linewidth iscomparible to the coupling constant. There are ex-periments, such as ECOSY (66) or P.E.COSY (67)which get around this problem by creating line-shapes in which the antiphase cancellation is re-duced. Direct simulation of cross peaks is also effec-tive for analysis of coupling constants (68) especiallyif effects of strong coupling are present. Heteronu-clear couplings can be measured from 2D experi-ments as well. Recently an elegant method for de-termining heteronuclear couplings in enriched sam-ples has been described (69). In this approach thedisplacement of crosspeaks arising from the two spinstates of the heteroatom are measured, this can bedone with any 2D experiment (TOCSY and NOESYare probably best in most cases). The advantageis that cancellation and overlap problems are min-imized, so that even rather weak couplings can bedetermined. Some care must also be used in theinterpretation of the J value, even when careful cor-rections for linewidths, etc. have been done. The in-formation is intrinsically a restraint on angles, but

Vol. 12, No. 4 197
can be converted to distances. In doing so uncer-tainty in the Karplus relation (calibration constants,etc.), its multivalued nature, and possible averagingeffects must be considered.
C. ParamagneticParamagnetic relaxation effects can also be used
to derive distances of spins from the unpaired elec-tron (70). Many studies of this type have beendone, although relatively few with 2D NMR (71).Effects on both Ti and T2 can be observed, andin general it is best to use both to estimate dis-tances. Either metal ions (which can be intrinsicto the protein's function, or adventitious binders)or stable organic radicals (generally nitroxides) canbe used. The relaxation effects can be determinedthrough broadening of crosspeaks (in almost anykind of 2D spectrum), or in more sophisiticated in-version recovery 2D experiments. When metal ionsare used great care must be taken to establish thenumber, positions and occupations of various avail-able binding sites (72). This is often done in partthrough titration behavior. The analysis of organicradicals which are covalently linked (preferably ina rigid fashion), or which are attached to a sub-strate or other small molecule which binds tightly,is simpler (73). The distances which can be ob-tained can be much longer than those determinedfrom NOEs, thus providing new information. Therehas not yet been much systematic study to deter-mine how much the inclusion of longer distances(generally with lower precision) affects the calcu-lated structures.
D. ExchangeA final source of structural information is through
identification of hydrogen bonds. It is well estab-lished in proteins that the participation of an amideproton in a hydrogen bond greatly slows its ex-change rate with solvent (74). Once all amide res-onances are assigned, it is a straightforward matterto lyophilize a sample from XH2O and redissolve itin 2H2O. Following the spectrum (usually a COSY)as a function of time after dissolving the sample al-lows the exchange rates of many amides to be deter-mined (75). It is then assumed that those which ex-change slowly are participating in hydrogen bonds.For proteins whose crystal structures have been de-
termined there is a good correlation between slowexchange and presence of hydrogen bonds. Whilethe NMR experiment defines the hydrogen bonddonors, the acceptors are generally oxygen or ni-trogen, and hence cannot be identified directly. Inmany cases the hydrogen bonds arise in regions ofregular secondary structure, and the combinationsof NOEs present leave little doubt about the ac-ceptor. In such cases it is reasonable to include aconstraint defining such a hydrogen bond, althoughthe allowable length should be conservatively cho-sen, and a range of angles should be allowed to in-clude the range of hydrogen bonds seen in crystalstructures. However some caution must be used.The NOE patterns for a-helix and 3io-helix are verysimilar, and a 3io-bend in an a-helix may be dif-ficult to identify. Thus if normal a-helical hydro-gen bonds were defined where a 3io-helix was reallypresent the correct structure might never be found.In addition, there are numerous slowly exchangingamide protons which are involved in hydrogen bondswith sidechain atoms, or even water molecules, forwhich the acceptor is even more difficult to iden-tify. Finally there may even be slowing exchang-ing protons arising from histidine and tryptophanNHs, sidechain amides, or even sidechain hydroxyls.There has been recent evidence that cross relaxationfrom water molecules associated with the proteincan be seen (76). These exchange rapidly with thebulk solvent, and hence have same chemical shifts asthe bulk, but as work progresses with larger proteinsit is possible that distinct resonances may be seen forsome bound waters. The exchange rate of an iminoproton in DNA or RNA is also significantly slowedwhen it is involved in a hydrogen bond (74). Whenthe imino is protected, and NOE evidence points tonormal Watson-Crick base pairing, it is reasonableto include a hydrogen bond constraint. For aminoprotons it is more difficult to determine exchangerates, and recent crystal structures have shown thathigh propeller twist can lead to bifurcated hydrogenbonds involving neighboring bases. Thus data fromamino protons should be treated conservatively. Anumber of alternate hydrogen bonding schemes areknown for nucleic acids, these should only be usedas structural constraints when it is clear that boththe donor and acceptor are clearly identified.

198 Bulletin of Magnetic Resonance
V. Structure Analysis
During the course of obtaining resonance assign-ments one usually obtains a good qualitative idea ofthe secondary structure of the molecule (77). Thiscomes from the pat tern of connectivities seen be-tween adjacent residues, regardless of the type ofbiopolymer. In proteins, for example, an antiparal-lel /?-sheet is easily recognized by a series of aH(i)-N H ( i + l ) connectivities, and aH-aH cross s trandNOEs, and is distinguished from helix which hascharacteristic NH(i)-NH(i+l) NOEs (although o>helix and 3io-helix may be difficult to distinguish),both are distinct from turns and loops where no re-peating connectivity pattern is seen. In nucleic acidsthe sugar pucker can be determined, and the spac-ing of certain protons defines "A" type or "B" typehelix, while regions of irregular structure are againusually distinct (78).
|li i This qualitative analysis is only descriptive, andit is difficult to reach conclusions about structureand function from it. Thus there has been extensivework to reconstruct sets of coordinates which areconsistent with all NMR data, and other "known"structural information, which then can be used inthe same ways tha t models developed from crys-tallography are. There are now quite a number ofdifferent methods for carrying out this reconstruc-tion, which have various advantages and problems.Several will be discussed including: metric space dis-tance geometry, variable target function minimiza-tion, pseudo- and real molecular dynamics, and anexclusion method. Of course there are also hybridapproaches (combining features of more than one ofthese) which are also used, and may well be the bestoption. In general when the structure generationprocess is repeated (either with the same method oranother) the final results are similar, but not iden-tical. Comparison of the range of structures foundgives some idea of the quality of the structure de-termination. To date the most work has been donefor reconstructing structures of proteins, and theleast for oligosaccharides. For most of the meth-ods the ideas are quite general, but the implemen-tation requires specific information about the typeof molecule to be built into the program. As suchmany of the current implementations will functiononly for certain types of systems, the range will cer-tainly increase as time goes on.
A. Metric Space DGThe general mathematical aspects of convert-
ing a set of distances between points to coordi-nates in space were treated long ago. In the late1970s Crippen, Havel and Kuntz applied these ideasto reconstruction of molecular structures from ex-perimental distance information, though not origi-nally with NMR in mind. Once resonance assign-ments were obtained, and distance estimates werederived from NOESY data, programs applying thisapproach were developed (79). The basic processis to collect all known distance information (fromall possible sources) into a distance matrix, repre-sented as the best estimates for upper and lowerbounds for the internuclear distances (heavy atomsare included in addition to the hydrogens). Thismatrix is then smoothed using triangle inequality(and sometimes higher order extensions of it), anda trial matrix is generated by choosing distanceswithin the allowed range. This trial matrix thenis transformed into a metric matrix (distances froman effective center of mass), and finally to a set oftrial ("embedded") coordinates. If the informationin the distance matrix is complete and accurate, thisprocess produces a set of coordinates which satisfyall of the original constraints. However this is nevertrue with real data (life is incomplete and inaccu-rate), and in the coordinates there are violations ofthe input constraints, which are used to generate anerror value. Tests have been done to analyse the ef-fects of limited information (80} Most of the workin the calculation is to minimize the error function(making the coordinates and the constraints agree)by varying the coordinates of each atom. This canbe done with pure minimization routines (conjugategradient, etc.), and variable weights can be assignedto errors from different sorts of data. Varying theweights will change local minima, making it easierto avoid trapping in local minima during refinement.The general problem of finding the global minimumin a complex function is a difficult one, and it isnot possible to prove that the real global minimumhas been found. Many recent applications have usedthe initial coordinates from DG, and subsequentlyrefined them using a dynamics-like (or simulated an-nealing) algorithm. This process will be discussedfurther below. In all structure generation methodsan important question is whether the process willsample (hopefully fairly uniformly) all of the possi-

Vol. 12, No. 4 199
ble conformations which have very low error. Forlarge structures and random choices of distances forthe trial matrix, it has been found recently that theDG approach does not effectively sample the allowedsolutions, at least in the initially embedded struc-tures (81). In spite of this it appears that for manystructures the process of refinement is sufficientlyrandom that the final results do not grossly mis-represent the true range of allowed structures, al-though there does seem to remain some bias in thesampling. Several groups are aware of this problem,and there appear to be solutions possible throughjudicious choice of the distribution functions in thetrial distance selection phase. Time (mostly on com-puters) will tell how satisfactory such fixes will be.When a dynamics refinement procedure is used thesampling is better than with minimization alone.
B. Variable Target FunctionIn any minimization procedure, the major prob-
lem is local minima which prevent the algorithmfrom finding a global minimum. During a refine-ment an incorrectly folded segment of the moleculecan effectively block the path to the correct struc-ture, making a deep local minimum. Braun and Gorealized that one solution would be to initially re-strict constraints to those involving residues whichare very close in the primary structure, allowing themolecule to fold the correct local structure whileeven passing through itself in more distant parts(82). Then gradually during the refinement therange of constraints is increased until finally all areused. Their implementation of this procedure usesan initial structure which has correct covalent ge-ometry, but which has all undetermined angles ran-domly chosen. The minimization is done using allnon-fixed bond angles as the variables (requiringvariation of far fewer parameters than the three peratom required in metric space DG). The changes inangles at each step are calculated using the gradi-ent in the error function. The procedure is quiteefficient and many, if not most, of the initial struc-tures converge to low residual error. By startingwith different random angles and repeating the cal-culation the range of allowed structures can againbe evaluated. Since angles are the natural variablein the problem, the random initial structures aretruly random. A more recently developed alterna-tive, termed the ellipsoidal algorithm, has been de-
scribed recently (83)- Rather than working with asingle structure, this approach begins with a largeellipsoidal region of conformation space, and thensuccessively reduces the region which contains theoptimal (minimum error and/or energy) solution.This is done by selecting at each step a constraintwhich is violated at the center of the current elip-soid. Then the center is moved, and the volume isreduced in a way such that the selected constraintis satisfied. The process is repeated until each con-straint is satisfied, at which point another function,such as energy, can be evaluated to further reducethe allowed region of conformational space. The re-duction in size of the ellipsoid at each step dependson the number of variables in the problem. As suchit is more efficient to define the structure with theminimum number of variables, normally the allowedvariable angles. While the minimization is nonphys-ical, it has been shown that large and discontinuoussteps in conformation are generated, avoiding prob-lems of local minima. This approach is rather new,and has not been applied in many cases yet.
C. Molecular Mechanics/DynamicsA significant number of variants in structure re-
finement methods come under this heading. Thefirst is a full restrained molecular dynamics cal-culation (84). In this case a full potential energyfunction is used, which is supplemented by restraintterms derived from experimental distances. The ex-act forms of the potential terms, their parameteri-zation and calibration, and the relative weights ofterms during the calculation vary somewhat in dif-ferent implementations. In principle just minimiza-tion of this full potential function will give a goodstructure, both in terms of satisfying distance re-straints and with respect to energy. However thereis again the major problem of finding the globalminimum (and knowing that the potential functionsare good enough to give a correct minimum, espe-cially in the absence of solvent in the calculations,as they are usually done). The molecular dynam-ics approach assigns velocities to all of the atoms,then carrys out classical trajectory calculations us-ing the potential function. During such a run, var-ious conformations can be extracted and energyminimized. This effectively improves the searchingand, when distance restraints are included, also as-sures that the structure obtained agrees with exper-

200 Bulletin of Magnetic Resonance
iments. Fully random starting structures, such atthose used in variable target function minimization,are satisfactory for this approach. However, struc-tures can be used to start the calculation, such asthose obtained from model building with secondarystructure elements, or from DG embeds. It is pre-sumed that satisfactory sampling is generated bythe combination of initial structure and calculationof many trajectories with different random startingvelocities. In this method it is clear that the ex-perimental distance information plays a dominantrole in defining the correct structure (unrestraineddynamics has not been successful in correctly pre-dicting a protein structures). An alternate, relatedapproach, referred to as simulated annealing (85),carries out trajectory calculations using just thebonding, hard sphere van der Waals and distanceconstraint terms in the "potential" function (elim-inating attractive dispersive forces and electrostat-ics, which are numerous and slow to calculate). Theaverage random initial velocities assigned define a"temperature", after evolution for some time thesystem is cooled (velocities are damped), and fi-nally the "potential" is minimized. The process isrepeated with new random velocities until the finalresult yields a structure of sufficiently low error tobe acceptable. This process tends to be much fasterthan full MD calculations, and can be supplementedwith full potential minimization or short, full MDruns to assure that the energy of the final struc-tures is low. The initial structures used to beginthis process seem not to be at all crucial, extendedstructures, crudely embedded structures, and evenrandomly assigned coordinates (preserving nothingof even the correct covalent geometry initially) seemto give comparable results.
D. Exclusion methodA final approach has been to try to define the
space of correct solutions using exclusion of struc-tures which are not consistent with the expermentalinformation (86). All of the other methods defineindividual structures which are consistent with ob-served data, while the exclusion method defines re-gions of conformation space in which correct struc-tures can or cannot lie. The initial implementationof this method has first established segments of reg-ular secondary structure, based on NOE connectiv-ity patterns. One such segment is then taken as an
anchor, and for each successive segment all possiblerelative positions and orientations are searched ina grid mode, and all which do not agree with theexperimental data are excluded from the solutionspace. Finally loops or other regions with irregularsecondary structure are added in the same fashion.Thus the result is comprised of regions of coordinatespace which contain structures agreeing with the in-put data. Individual structures may be generatedfrom this solution space by a "threading" algorithmwhich uses a local least squares fitting procedure toselect the local coordinates. The advantage of themethod is that it systematically samples the solu-tion space, rather than relying on a randomizationin the generation of individual structures to sampleall allowed conformations. To date this approachhas been used only for proteins.
E. Representation of structuresA structure derived from x-ray diffraction is a
best fit to the electron density, which is tested di-rectly by calculating the agreement with the originaldiffraction intensities. This is supplemented withB-factors, which provide a description of the uncer-tainty in atomic positions, which may arise eitherthrough motion or static disorder. Because most ofthe NMR based structure calculation methods cal-culate individual structures which agree with theNMR data, but may be (at least in part) quite dif-ferent from one another, there is no really equivalentsingle structure. To date the majority of structureshave been represented as a "bundle", that is a draw-ing of several least squares superimposed structures(87). When the spread in the bundle is very smallfor part of the structure it is implied that the localstructure there is well defined, while a large spreadmeans that the structure is less well defined. How-ever, since any of the calculated structures agreeswith the NMR data equally well it is not possi-ble (without additional information) to decide whichbest represents reality, or whether in fact many ofthese structures are sampled by the molecule. Inregions which are well defined it seems reasonableto define an average structure (though it may notbe physically meaningful) to use in fashions similarto "the" structure determined crystallographically.The exclusion method directly calculates an allowedconformation space. When the structure is quitewell defined a representation of this "space" may be

Vol. 12, No. 4 201
useful, but for more poorly defined structures or re-gions it will be difficult to visualize directly withoutthreading structures through it. The volume of theallowed solution space defined with this approachmay provide a good basis for defining a "degree ofdetermination" factor.
F. Quality of structures
During the evolution of the NMR based structuredetermination method a question which consistentlyarose was "to what accuracy can a structure be de-termined from NMR data?". This has been testedboth through model calculations, avoiding the prob-lems inherent in quantitative derivation of distancesfrom NOEs, and through actual determination ofstructures of many sorts. Unlike the general casefor crystallographically determined structures, thequality of an NMR structure may depend highly onthe nature of the structure. That is, for compactglobular structures the chain fold may be defined ac-curately and equally well throughout the molecule.In other molecules there may be loops, or extendedregions which are less well defined than a core re-gion. The worst case is an extended structure inwhich the local structure may be quite well defined,but overall shape is poorly defined. This appearsto be the case for some peptides, and for helicalnucleic acids. Since there may be problems withsampling in some of the structure generation meth-ods, it is important to be aware of the potentialproblems, and to use intuition when considering theresults of calculations. In spite of this warning theprogress in quality of NMR structures has been im-pressive. Early structures showed a spread of ca.2 A in backbone atom positions (88). Even usingonly semiquantitative distance information, globu-lar protein structures have now been defined whichhave ca. 1 A RMS variation in backbone atom posi-tions (89), and 1.4 A variation when sidechains areincluded. This makes them roughly equivalent tocrystal structures at 2-2.5 A resolution. An impres-sive test of the NMR method was the simultaneousbut independent solution of the structure of the a-amylase inhibitor tendamistat by both NMR anddiffraction methods by Wiithrich (90) and Huber(91) respectively. Not only are the overall struc-tures similar, they are essentially identical in detailthroughout most of the protein. Some of the re-gions where differences are seen can clearly be at-
tributed to intermolecular interactions which arisefrom packing in the crystal (92).
Quite recently there have been a number ofNMR studies of small peptides and protein frag-ments in solution (93), for which one cannot de-fine a single structure. In this case the NOE pat-terns indicate conformational preferences, showingpropensities for turn or helix formation. However, inthese cases conformational averaging is certainly oc-curring, and it is not possible to associate observedNOEs with specific conformations (especially glob-ally). In this case it is not possible to carry outmeaningful structure calculations, such as those de-scribed above, although the information obtainedis valuable to understanding protein folding. Suchunstructured regions also occur as part of a largerprotein structure, and care must be taken to cor-rectly interpret the NOE data. Such an example isshown in Figure 5. These regions may show abnor-mal linewidths, or NOE patterns which are not con-sistent with a single conformation (94), indicatingthe presence of dynamic processes, but it is likely insome proteins that more subtle effects of averagingwill be present, but not obvious.
The situation for nucleic acids is somewhat morecomplex. DNA and RNA oligomers are rather ex-tended and few if any tertiary contacts occur. Thismeans that the global structure is determined onlythrough short distances which, as has been dis-cussed, are of limited accuracy. This implies thatglobal features, such as curvature, are not likely tobe well defined. As in the case of proteins sometest calculations have been done. A set of calcu-lations done with distances derived from a DNAcrystal structure were done using the DSPACE pro-gram (95). It was shown that there is considerablevariation in the ability to reproduce local structuralparameters from distances which could be derivedfrom NMR. In spite of the variation there are sev-eral local parameters of interest which are fairly welldetermined in the structures. Other calculations onslightly longer models derived from idealized B-formDNA showed that there can be significant globalvariation in the structure, such as gentle curvature,even when very accurate distances are used for theinput (96). As is the case for proteins the inclusionof hydrogen bonds between base pairs improves thequality of the structures, but there is some ambigu-ity in how to identify the donors and acceptors, par-

202 Bulletin of Magnetic Resonance
ticularly for amino protons. In addition to strictlydistance based methods, molecular mechanics meth-ods have also been applied. This includes both con-strained minimization (97) and molecular dynamics(98). Structures which have been generated withthese methods are "regularized", that is they tend tolook more like regular B-form DNAs since the poten-tial functions used have been calibrated to achievethis end. It is then primarily the distance con-straints which introduce nonuniform local structure(99). The problem with this approach, as for pro-teins, is to determine which aspects of the structurewere really experimentally determined, and whichresult from application of the potential functions.An advantage is that reasonble structures can beobtained in cases where there is limited distanceinformation (in either quantity or quality). Suchstructures are not fully "determined" by NMR, butcertainly the energy calculations are strongly guidedby even the limited distance information.
These methods can also be applied to more com-plex nucleic acids, and as the folding becomes morecomplex there may be new tertiary constraints in-troduced. In this case the quality of "global" aspectsof the structure may improve. There are a numberof studies underway which may demonstrate this.Tinoco and coworkers have analysed the structureof an RNA pseudoknot (100), showing that the ba-sic proposed structure is correct. Current work willextend the structure analysis. Other larger RNAsare also being studied, although to date their com-plexity has made progress slow.
Oligosaccharides have also been studied. Muchof the useful information has been in the determi-nation of the primary structure. Although mostoligosaccharides do not fold into compact structures,the conformation at branching points can be deter-mined, and a few long range constraints can some-times be obtained. In such cases the general shapeof the molecules can be then be described. Againthe direct distance information is often being agu-mented with potential energy calculations.
VI. Examples
Finally we present several examples of the meth-ods discussed above. The system studied first and indetail using both models and real data was BPTI,but the structure was well known from crystallo-
graphic work before the NMR work was done. Fairlydetailed distance geometry calculations have beendone with distances derived from the crystal struc-ture which showed clearly that distance constraintswhich could realistically be derived from NMR dodefine the structure to reasonable accuracy. It wasalso shown that increasing either the number of con-straints or the accuracy of the constraints improvesthe quality of the final structure (101). Recently astudy of a previously uncharacterized protein struc-ture, the a-amylase inhibitor known as tendamistat,was done in parallel using both NMR and diffrac-tion. The assignment procedure followed the stan-dard sequential method (102), and from the NOEpatterns a basic secondary structure a Greek-key /?-barrel was deduced (103). Further characterizationsof NOEs and calculations using the variable targetfunction method (DISMAN) resulted in a moder-ate resolution structure (90). This gave a clear in-dication of the shape of the protein, and regionswhich were best defined by the NMR data. Finallyextensive analysis of the NOESY data was done,including determination of many stereospecific as-signments, and assigning initially ambiguous NOEsusing information from the preliminary structures.This has resulted in a very high resolution structurefor this protein (91,104), a representation of whichis shown in Figure 4.
Considerable care was taken to include all of theavailable distance information, although it is worthnoting that the distances used were still only semi-quantitative, that is distance ranges from 2.0 A to2.5 A (strong) to 3.0 A (medium) or 3.5 A (weak)were used. The definition of the backbone is verygood, somewhat more spread is seen in the loop re-gions where fewer constraints are present. A major-ity of sidechains, particularly those on the inside ofthe protein are also in well defined positions. A com-plete discussion of the structure has been given. Asa comparison of two structure determination meth-ods, NMR data on BPTI were used with both theDISGEO and DISMAN programs, and the resultswere compared (105). Very similar structures wereobtained in that case.
The structure is not always so well definedthroughout the entire structure. In the case ofsea anemone toxins, there are a number of naturalvariants which have been studied by NMR (106).Initial problems of impure proteins, and incorrect

Vol. 12, No. 4 203
RP 18
C U SIALA 28
GIT SI
( 0 ) ( b )
Figure 4: NMR derived structures of tendemistat determined by Braun et al. (1989) from NMR data, (a)shows the backbone position of the average of 9 structures determined using the DISMAN algorithm (heavyline) compared with the backbone determined by x-ray diffraction (light line). On the bottom is shownthe alpha carbon positions of the 9 NMR structures, superimposed on elipsoids representing the B-factorsdetermined crystallographically.
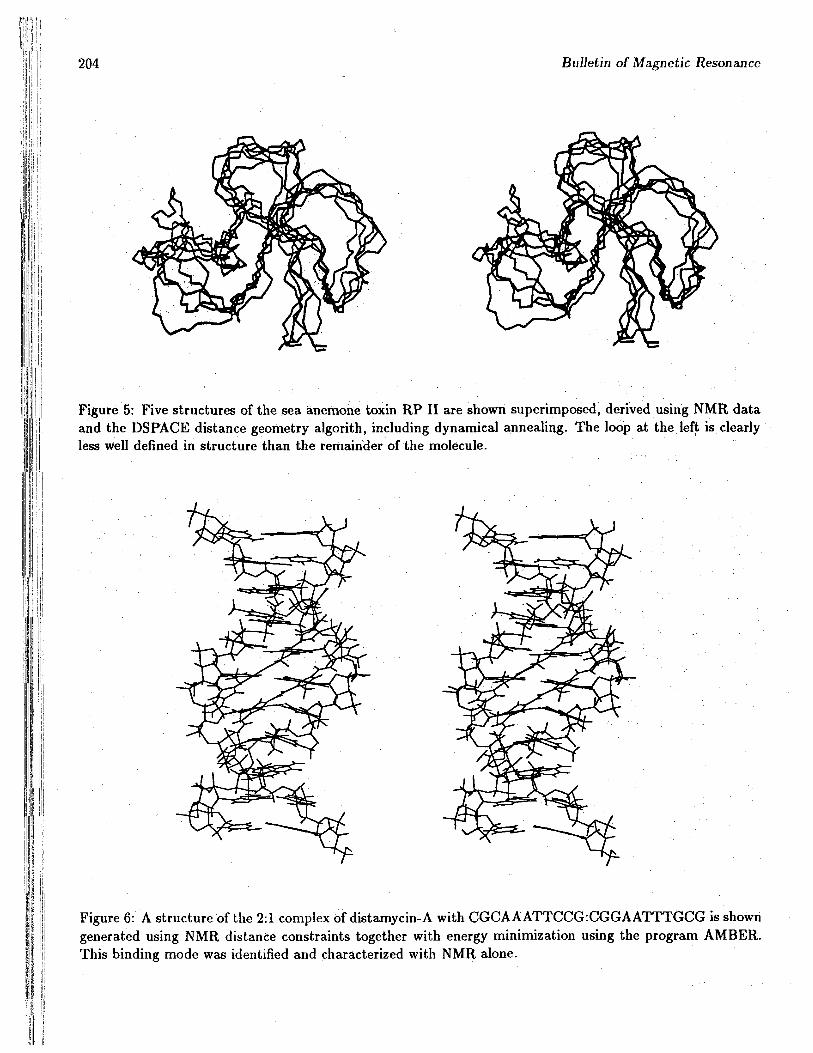
sequences were overcome, and essentially completeresonance assignments done for at least four vari-ants. In all of these there is a well defined corewhich contains disulfide cross-links, but there is arather large loop whose structure is poorly defined.Figure 5 shows several structures for the toxin RPIII, which were generated using a distance geom-etry program which includes dynamic refinement(DSPACE). In spite of the lack of NOEs to de-fine the structure in this region, the linewidthsfrom residues in this loop do not seem significantlysharper than those from other regions of the pro-tein. Thus the nature and dynamics aspects of thisdisorder remain unclear.
The structures of a number of nucleic acids havebeen analyzed as well. One of the first was a DNAhairpin loop, for which the loop structure was quitewell defined (107), with the NOE data even defininga few tertiary contacts. Other DNA oligomers con-taining extra bases, or base mismatches, have alsobeen analyzed (108). In these cases the local struc-ture is probably fairly well determined, but becauseof the problems discussed above there remains somequestion about the global structure. NMR distanceconstrained structure calculations have also beencarried out for drug-DNA complexes, with good re-sults (109). In these cases there was no previous
information about the detailed geometry of the com-plexes, and in spite of its limitations NMR has pro-vided a very clear picture of the binding modes, fromwhich further studies can be designed. An exampleis one of the previously unknown 2:1 distamycin-Acomplexes with a DNA oligomer containing the se-quence AAATT, shown in Fig. 6.
VII. Summary
In the past few years there has been dramaticprogress in our ability to assign and analyze spec-tra from biomolecules. The size range has beenincreased by combinations of pulse sequences, iso-tope labelling, and higher magnetic fields. Meth-ods for quantification of NOEs have been developed,making possible more accurate distance determina-tions. Several methods are available for reconstruct-ing structures based on covalent primary structureand NMR derived constraints. Although these dif-fer somewhat in amount of work required and themethod for refinement, they seem to give quite com-parible results. It is clear that structures really arebeing determined from NMR data, and that themethod hold even greater future promise as furtheradvances in the methodology are implemented.
Acknowledgments: I would like to thank the
204 Bulletin of Magnetic Resonance
Figure 5: Five structures of the sea anemone toxin RP II are shown superimposed, derived using NMR dataand the DSPACE distance geometry algorith, including dynamical annealing. The loop at the left is clearlyless well defined in structure than the remainder of the molecule.
« I1
Figure 6: A structure of the 2:1 complex of distamycin-A with CGCAAATTCCG:CGGAATTTGCG is showngenerated using NMR distance constraints together with energy minimization using the program AMBER.This binding mode was identified and characterized with NMR alone.

Vol. 12, No. 4 205
members of my group for the wide variety of dis-cussions which contributed to my familiarity withassignment methods, and especially Joseph Pease,Jeffrey Pelton and Dr. Vasant Kumar, who gen-erated the data and structures for Figures 2, 3, 5,and 6. This work was supported by the Office ofHealth and Environmental Research, fiealth EffectsResearch Division of the U.S.Department of Energyunder Contract No. DE-AC03-76SF00098.
VIII. References1K. R. Ernst, J. Bodenhausen k A. Wokaun,
Principles of Nuclear Magnetic Resonance in Oneand Two Dimensions, Oxford Univ. Press, London,1988.
2M. Goldman, Quantum Mechanical Descriptionof High Resolution NMR in Liquids, Oxford Univ.Press, London, 1988.
3O. W. S0rensen, G. W. Eich, M. H. Levitt,G. Bodenhausen k R. R. Ernst, Progress in NMRSpectroscopy 16, 163 (1983); K.J. Packer & K. M.Wright, Mol. Phys. 6, 225 (1983).
4G. Wider, R. V. Hosur & K. Wiithrich, /.Magn. Reson. 52, 130 (1983).
5W. P. Aue, E. Bartholdi k R. R. Ernst, /.Chem. Phys. 64, 2229 (1976).
6G. Wider, S. Macura, A. Kumar, R. R. Ernstk K. Wiithrich, / . Magn. Reson. 56, 207 (1984).
7D. Marion k K. Wiithrich, Biochemistry Bio-phys. Res. Comm. 113, 967 (1983).
8U. Piantini, 0. W. S0rensen k R. R. Ernst, /.Am. Chem. Soc. 104, 6800 (1982).
9M. Ranee, O. W- S0rensen, G. Bodenhausen,G. Wagner, R. R. Ernst k K. Wiithrich, Biochem-istry Biophys. Res. Comm. 117, 479 (1983).
10H. Widmer k K. Wiithrich, /. Magn. Reson.74, 316 (1987).
11G. Eich, G. Bodenhausen k R. R. Ernst, / .Am. Chem. Soc. 104, 3731 (1982).
12M. H. Levitt k R. R. Ernst J. Chem. Phys.83, 3297 (1985).
13G. King & P. E. Wright, J. Magn. Reson. 54,328 (1983). G. Wagner, J. Magn. Reson. 55, 151(1983).
14A. Bax k G. Drobny, /. Magn. Reson. 61,306 (1985). W. J. Chazin k K. Wiithrich, J. Magn.Reson. 72, 358 (1987).
1 5C Redfield k C. M. Dobson, Biochemistry 27,122(1988).
16L. Braunschweiler k R. R. Ernst, /. Magn.Reson. 53, 521 (1983).
17D. G. Davis k A. Bax, / . Am. Chem. Soc.107, 2821 (1985). D. G. Davis k A. Bax, /. Magn.Reson. 65, 355 (1985).
18A. Bax and D. G. Davis in "Advanced Mag-netic Resonance Techniques in Systems of HighMolecular Complexity," N. Nicolai k G. Valensin,Eds., Birkhauser, Basel, 1986.
19G. Wagner k E. R. P. Zuiderweg, BiochemistryBiophys. Res. Comm. 113, 854 (1983).
WM. Ranee k P. E. Wright, /. Magn. Reson.66, 372 (1986).
21 J. Jeener, B. H. Meier, P. Bachmann k R. R.Ernst, J. Chem. Phys. 71, 4546 (1979). S. Macurak R. R. Ernst, Mol. Phys. 41, 95 (1980).
22A. Bax k D. G. Davis, J. Magn. Reson. 63,207 (1985).
^A. A. Bothner-By, R. L. Stephens, J. Lee, C.D. Warren k R. Jeanloz, / . Am. Chem. Soc. 106,811 (1984).
24A. Kalk k H. J. C. Berendsen, / . Magn. Re-son. 24,343(1976).
25S. Macura, Y. Huang, D. Suter k R. R. Ernst,J. Magn. Reson. 43, 259 (1981).
^A. Bax, R. H. Griffey & B, L. Hawkins, J. Am.Chem. Soc. 105, 7188 (1983).
27R. H. Griffey and A. G. Redfield, Quart Rev.Biophys. 19, 51 (1987).
28J. Ashcroft, S. R. LaPlante, P. N. Borer k D.Cowburn, /. Am. Chem. Soc. I l l , 363 (1989),
MD. A. Torchia, S. W- Sparks & A. Bax, Bio-chemistry 28, 5509 (1989).
^G. W. Vuister k R. J. Boelens,.-/. Magn. Re-son. 73, 328 (1987). C. Griesinger, O. W. S0rensenk R. R. Ernst, J. Magn. Reson. 73, 574 (1987); C.Griesinger, O. W. S0rensen k R. R. Ernst, J. Am..Chem. Soc. 109, 7227 (1987); H. Oschkinat, C.Griesinger, P. J. Kraulis, O. W. S0rensen k R. R.Ernst, A. Gronenbom, k G. M. Clore, Nature 332,374 (1988); G. W. Vuister, P. de Waard, R. Boelens,J. F. G. Vliegenthart k R. Kaptein, /. Am. Chem.Soc. I l l , 772 (1989).
31S. W. Fesik k E. R. P. Zuiderweg, J. Magn.Reson. 78, 588 (1988). S. W. Fesik, R. T. Gampek E. R. P. Zuiderweg, J. Am. Chem. Soc. I l l , 770(1989).

206 Bulletin of Magnetic Resonance
32K. Wiithrich, G. Wider, G. Wagner & W.Braun, /. Mol. Biol. 155, 311 (1982).
fflK.. Wuthrich, "NMR of Protein and NucleicAcids," Wiley, N.Y. 1986.
MG. Wagner k K. Wiithrich, J. Mol Biol 155,347 (1982).
KM. Billeter, W. Braun & K. Wiithrich, / . MolBiol 155, 321(1982).
" P . Strop, G. Wider k K. Wiithich, /. MolBiol 166, 641 (1983); D. E. Wemmer, N. V. Ku-mar, R. M. Metrione, M. Lazdunski, G. Drobny kN. R. Kallenbach, Biochemistry 25, 6842 (1986).
37S. W. Englander k A. J. Wand, Biochemistry26,5953(1987).
a D . L. DeStefano k A. J. Wand, Biochemistry26, 7272 (1987); A. J. Wand, D. L. DiStefano, Y.Feng, H. Rpder k S. W. Englander, Biochemistry28, 186 (1989); Y. Feng, H. Roder, S. W. Englander,A. J. Wand & D. L. DiStefano, Biochemistry 28,195(1989).
mD. M. LeMaster k F. M. Richards, Biochem-istry 27, 142(1988).
*D. M. LeMaster k F. M. Richards, Biochem-istry 24, 7263 (1985).
41R. H. Griffey, A. G. Redfield, L. Mclntosh, T.Oas k F. W. Dahlquist J: Am. Chem. Soc. 108,6816(1986).
42B. H. Oh, W. M. Westler, P. Darba & J. L.Markley, Science 240, 908 (1988).
43B. J. Stockman, M. D. Reily, W. M. Westler,E. L. Ulrich k J. L. Markley Biochemistry 28, 230(1989).
^E . R. P. Zuiderweg, R. Boelens & R. Kaptein,Biopolymers 24, 601 (1985),
45G. Wagner, W. Braun, T. F. Havel, T. Schau-mann, N. Go & K. Wiithrich,/. Mol Biol 196,611 (1987); S. G. Hyberts, W. Marki k G. Wagner,Eur. J. Biochemistry 164, 625 (1987); P. L. Weber,R. Morrison k D. Hare, J. Mol Biol 204, 483(1988); P. C. Driscoll, A. M. Gronenborn k G. M.Clore, FEBS Lett. 243, 223 (1989).
46P. Giintert, W. Braun, M. Billeter k K.Wuthrich, J. Am. Chem. Soc. I l l , 3997 (1989).
47J. Feigon, J. M. Wright, W. Leupin, W. A.Denny k D. R. Kearns, /. Am. Chem. Soc. 104,5540 (1982); R. M. Scheek, R. Boelens, N. Russo, J.H. vanBoom k R. Kaptein, J. Am. Chem. Soc.105, 2914 (1983); R. M. Scheek, R. Boelens, N.Russo, J. H. vanBoom k R. Kaptein, Biochemistry
23, 1371 (1984); D. R. Hare, D. E. Wemmer, S. HChou, G. Drobny k B. R. Reid J. Mol Biol 171,319 (1983);
^ P . F. Flynn, A. Kintanar, B. R. Reid k G.Drobny, Biochemistry 27, 1191(1988).
49A. Pardi, R. Walker, H. Rapoport, G. Widerk K. Wuthrich, J. Am. Chem. Soc. 105, 1652(1983).
^D. Marion k G- Lancelot, Biochemistry Bio-phy. Res. Comm. 124, 774 (1984); M. H. Frey, W.Leupin, O. W. S0rensen, W. A. Denny, R. R. Ernstk K. Wuthrich, Biopolymers 24, 2371 (1985).
S1D. E. Wemmer, S.-H. Chou k B R. Reid, /.Mol. Biol 180, 41 (1984); M. A. Weiss, D. J. Patel,R. T. Sauer & M. Karplus, Proc. Natl. Acad. Sci.,U.S.A. 81, 130 (1984); R. Grutter, G. Otting, K.Wuthrich k W. Leupin, Eur. Biophys. Jon. 16,279(1988).
52R. Boelens, P. Gros, R. M. Scheek, J. A. Ver-poorte k R. Kaptein, J. Magn. Reson. 62, 378(1985).
^ J . L. Leroy, D. Broseta k M. Gueron, J. Mol.Biol 184, 165 (1985).
MC. K. Brush, M. P. Stone k T. M. Harris, Bio-chemistry 27, 115 (1988).
" R H . Griffey, C. D. Poulter, Z. Yamaizumi, S.Nishimura k R. E. Hurd, J. Am. Chem. Soc. 104,5810 (1982); C. Smith, P. F. Agris, P. G. Schmidtk J. Petch, Biochemistry 24, 1434 (1984).
^ C . A. Bush, 5a//. Magn. Reson. 10, 73(1988); K. Bock k H. Th0gersen, Ann. Rep. NMRSped. 13, 1 (1983).
57G. Wagner k K. Wuthrich, J. Magn. Reson.33,675(1979).
58A. Kumar, G. Wagner, R. R. Ernst & K.Wuthrich, J. Am. Chem. Soc. 103, 3654 (1981).
59J. W. Keepers k T. L. James, J. Magn. Reson.57, 404 (1984); E. T. Olejniczak, R. T. Gampe, Jr.k S. W. Fesik, /. Magn. Reson. 67, 28 (1986).
^R. Boelens, T. M. G. Koning k R. Kaptein, /.Molec. Structure 173, 299 (1988); B. A. Borgias kT. L. James, J. Magn. Reson. 79, 493 (1988).
61K. Wuthrich, M. Billeter k W. Braun, J. MolBiol 169, 949 (1983).
62O. Jardetzky k G. C K. Roberts, "NMR inMolecular Biology," Academic Press, N.Y., 1981.
^ D . M. LeMaster, L. E. Kay, A. T. Brunger kJ.H.Prestegard, FEBS Lett. 236, 71 (1988).

Vol. 12, No. 4 207
MV. F. Bystrov, Prog. NMR Spectrosc. 10, 41(1976).
•"A. Pardi, M. Billeter k K. Wuthrich, /. Mol.Biol. 180, 741 (1984).
^ C . Griesinger, 0 . W. S0rensen k R. R. Ernst,/. Am. Chem. Soc. 107, 6394 (1985).
67L. Mueller, / . Magn. Reson. 72, 191 (1987).WH. Widmer k K. Wuthrich, J. Magn. Re-
son. 70, 270 (1986); H. Widmer k K. Wuthrich,J. Magn. Reson. 74, 316 (1987).
^G. T. Montelione k G. Wagner, /. Am. Chem.Soc. I l l , 5474(1989).
TOR. W. Wein, J. D. Morrisett k H. M. Mc-Connell Biochemistry 11, 3707 (1972).
71P. A. Kosen, R. M. Scheek, H. Naderi, V. J.Basus, S. Manogaran, P.G. Schmidt, N. J. Oppen-heimer k I. D. Kuntz Biochemistry 25, 2356 (1986).
72R. A. Dwek, "Nuclear Magnetic Resonance inBiochemistry: Applications to Enzyme Systems,"Oxford University Press, Oxford, 1975.
raJ. Anglister, T. Frey k H. M. McConnell, Bio-chemistry 23, 1138 (1984); J. Anglister, T. Frey kH. M. McConnell, Biochemistry 23, 5372 (1984); T.Frey, J. Anglister k H. M. McConnell, Biochemistry23, 6470 (1984).
74S. W. Englander k N. R. Kallenbach, Quart.Rev. Biophys. 16, 521 (1984).
75G. Wagner, Quart. Rev. Biophys. 16, 1(1983).
76G. Otting k K. Wuthrich, /. Am. Chem. Soc.I l l , 1871 (1989).
77K. Wuthrich, M. Billeter k W. Braun, /. Mol.Biol. 180,715 (1984).
78F. J. M. van de Ven k C. W. Hilbers, Eur. J.Biochemistry 178, 1 (1988).
79T. F. Havel, I. D. Kuntz k G. M. CrippenBull. Math. Biol. 45, 665 (1983); T. F. Havel k K.Wuthrich, Bull. Math. Biol. 46, 673 (1984).
WT. F. Havel k K. Wuthrich, /. Mol. Biol. 182,281 (1985).
81W. J. Metzler, D. R. Hare k A. Pardi, Bio-chemistry 28, 7045 (1989).
82W. Braun & N. Go, /. Mol. Biol. 186, 611(1985).
MM. Billeter, T. F. Havel k K. Wuthrich, J.Comp. Chem. 8, 132 (1987).
MR. Kaptein, E. R. P. Zuiderweg, R. M. Scheek,R. Boelens, W. F. van Gunsteren, /. Mol. Biol.182, 71 (1985); L. Nilsson, G. M. Clore, A. M. Gro-
nenborn, A. T. Brunger k M. Karplus, J. Mol. Biol.188, 455 (1986); A.T. Brunger, G. M. Clore, A. M.Gronenborn k M. Karplus, Proc. Natl. Acad. Sci.,tf.S.A 83, 3801 (1986).
85M. Nilges, A. M. Gronenborn, A. T. Brungerk G. M. Clore, Prot. Eng. 2, 27 (1988).
^R. B. Altman k O. Jardetzky, /. Biochemistry100, 1403(1986).
87K. Wuthrich, Science 243, 45 (1989).^M. P. Williamson, T. F. Havel k K. Wuthrich,
J. Mol. Biol. 182, 295 (1985).WP. C Driscoll, A. M. Gronenborn, L. Beress k
G. M. Clore, Biochemistry 28, 2188 (1989)." A D . Kline, W. Braun & K, Wuthrich, /, Mol.
Biol. 189, 377 (1988).91 J. Pflugrath, E. Wiegand, R. Huber k L
Vertesy, / . Mol. Biol. 189, 383 (1986).92M. Billeter, A. D. Kline, W. Braun, R. Huber
k K. Wuthrich, /. Mol. Biol. 206, 677 (1989).^H. J. Dyson, M. Ranee, R. A. Houghten, R.
A. Lerner k P. E. Wright, J. Mol. Biol. 201, 161(1988); H. J. Dyson, R. M. Ranee, R. A. Houghten,P. E. Wright & R. A. Lerner, /. Mol. Biol. 201, 201(1988); P. E. Wright, H. J. Dyson k R. A. Lerner,Biochemistry 27, 7168 (1988).
M P. L. Weber, D. E. Wemmer k B. R. Reid,Biochemistry 24, 4553 (1985).
95A. Pardi, D. R. Hare k C Wang, Proc. Natl.Acad. Sci., U.S.A. 85, 8785 (1988).
^ D . Wemmer, unpublished results.97G. M. Clore, A. M. Gronenborn, D. S. Moss k
I. J. Tickle, J. Mol. Biol. 185, 219 (1985); G. M.Clore k A. M. Gronenborn, EMBO J. 4, 829 (1985).
^L. Nilsson, G. M. Clore, A. M. Gronenborn, A.T. Brunger k M. Karplus, /. Mol. Biol. 188, 455(1986); G. M. Clore, H. Oschkinat, L. W. McLaugh-lin, F. Benseler, C. Scalfi Happ, E. Happ k A. M.Gronenborn, Biochemistry 27, 4185 (1988).
"A. M. Gronenborn k G. M. Clore, Biochem-istry 28, 5978 (1989).
100J. Puglisi, Ph.D. thesis, Univ of California,Berkeley, 1989.
101T. F. Havel k K. Wuthrich, J. Mol. Biol.182, 281 (1985).
102A. D. Kline k K. Wuthrich, J. Mol. Biol.192, 869 (1986).
103A. D. Kline k K. Wuthrich, J. Mol. Biol.183, 503 (1985).

i208 Bulletin of Magnetic Resonance
H
104A. D. Kline, W. Braun & K. Wiithrich, J.Mol. Biol. 204, 675 (1988).
105G. Wagner, W. Braun, T. F. Havel, T. Schau-mann, N. Go & K. Wiithrich, / . Mol. Biol. 196,611(1987).
1 0 6P. R. Gooley k R. S. Norton, Biopolymers25, 489 (1986); D. E. Wemmer, N. V. Kumar, R.M. Metrione, M. Lazdunski, G. Drobny & N. R.Kallenbach, Biochemistry 25, 6842 (1986); H. Wid-mer, G. Wagner, H. Schweitz, M. Lazdunski & K.Wiithrich, Eur. J. Biochemistry 171, 177(1988);A. E. Torda, B.C. Mabbutt, W. F. van Gunsteren& R. S. Norton FEBS Lett. 239, 266 (1988); R. H.Fogh, B. C. Mabbutt, W, R. Kern & R. S. Norton,Biochemistry 28, 1826 (1989); 3. H. B. Pease, N.V. Kumar, H. Schweitz, N. R. Kallenbach & D. E.Wemmer, Biochemistry 28, 2199 (1989).
107D. R. Hare & B. R. Reid, Biochemistry 25,5341(1986).
108D. J. Patel, L. Shapiro & D. R. Hare, Ann.Rev. Biophys. Biophys. Chem. 16, 423 (1987).
109X. Gao & D. J. Patel Biochemistry 2, 751(1989); J. G. Pelton & D. E. Wemmer, Biochemistry27, 8088 (1988); J. G. Pelton & D. E. Wemmer,Proc. Natl. Acad. Sci., USA 86, 5723 (1989).