of - inflibnetshodhganga.inflibnet.ac.in/bitstream/10603/368/10/10_chapter 3.pdf · santhosh, s....
TRANSCRIPT

Chapter 3
Synthesis and Characterization of Dinitramide Salts
Part of the results from this chapter has been published:
1. G. Santhosh, S. Venkatachalarn, M. Kanakavel, K.N. Ninan "Study On The Formation of Dinitram~de Using Mixed Acid Nitrating Agents" Indian Journal of Chemical Technology. 2002, 9. 223-226.

INITRAMIDE anion having the structural formula - N ( N 0 2 ) ? forms
new class of salts with a wide variety of cations. The ammonium
salt of this anion, ammonium dinitramide (ADN), is a candidate
oxidizer for solid propellants and it considerably exceeds ammonium
perchlorate in propellant performance. The acid form of this anion,
dinitramidic acid (DNA, HIV(N02)2) is one of the strongest inorganic acids
and hundreds of simple and complex salts can be synthesized on the basis
of reactions of well-known inorganic acids. There are different approaches
to the synthesis of dinitramide salts as reviewed in part 2 of chapter 1. The
focus of the present chapter is to explore in detail the chemistry of the
formation of dinltramide on the nitration of deactivated amines using mixed
acids. This chapter is divided into two parts. The first part deals with the
synthesis and elaborate studies on the formation of ADN and its
characterization using various analytical methods. The effect of variation of
ratio of reactants on the yield of ADN is explored in detail. The studies lead
to the selection of an optirnum ratio of reactants for maximum yield of ADN.
The second part of the chapter details the synthesis of few more
dinitramide analogues viz., potassium dinitramide (KDN), guanylurea
dinitramide (GUDN), tetramine Cu(ll) dinitramide and their characterization
by spectral and thermal methods. It also discusses a process for making
coarse ADN grains from as-synthesised ADN.

3.1. Synthesis and Characterization of Ammonium Dinitramide (ADN)
ADN is the ammorlium salt of 1 , I ,3,3-tetraoxo-I ,2,3-triazapropene
anion [ ' I . D~nitramide salts are useful oxidizers for high-energy materials
such as propellants, pyrotechnics and gas generating formulations [2,31. The
syntheses of dinitramide salts by various methods have been reported by
several authors 14-'21. Majority of the synthetic methods on the synthesis of
ADN are from patented literature. The dinitramide salt is generally prepared
by nitration of deactivated amines viz., NH2N02. NH2COONH4,
NH2COOC2H5, NH3 etc.. using very strong nitrating agents namely N02BF4
or N2O5 14-91. The ability of dinitramide anion to form stable oxygen rich salts
with a variety of cations to make compounds of high densities makes it a
promising candidate for the development of high performance solid
propellants. The present chapter discusses the low temperature nitration of
ammonium sulphamate (AS, NH2S03NH4) using mixed acids (HN03 &
H2S04) and the mechanism of formation of ADN.
This part of the chapter describes the synthesis, characterization and
the influence of reaction parameters like the variation of reactant ratios on
the formation of arnrnonium dinitramide. The effect of sulphuric acid and
nitric acid ratio in the mixed acid on the yield of dinitramide is studied in
detail. The chapter also highlights the kinetics and mechanism of the
nitration reaction and the influence of added water molecules on the
dinitramidic acid for ma ti or^. The separation of ADN by solvent extraction
and adsorption is highlighted. Characterization of ADN by spectroscopic
and therrnoanalytical methods is also described.

3.2. Experimental
3.2.1. Materials
Ammonium sulphamate AR (SRL, Bombay) was powdered in a
relative humidity of 50% and was further dried in vacuum to obtain a free
flowing powder (m.p 130-132°C). Con. HzS04 98% (Qualigens, Mumbai)
was used as received. Fuming HN03 of assay > 98% was distilled in the
laboratory trom a mixture of 1:l (by weight) of fuming HN03 (92% ) with
con. H2S04. The fraction between 83 - 85°C was used. Liquor ammonia
AR, about 25% NH3 (Qualigens, Mumbai) was used as received. The
solvents isopropanol and ethylacetate were distilled and dried over
molecular sieves prior to use.
3.2.2. Synthesis of Ammonium Dinitramide (ADN)
I n a typical experirrlent ammonium sulphamate (5.79, 0.05mol) was
added in s~nall portions to a mixture containing HN03 (assay-98%,18.9g,
0.3mol) and con.H2S04 1(9.89, 0.lmol) maintained at a temperature of
-35" C to -45" C in a 3-necked RB flask with stirring. The rate of addition
of AS was controlled in such a way that the temperature of the mixture
does not exceed --35" C. Formation of white precipitate was observed
during the course of the reaction and the viscosity of the mixture increases
as the reaction progresses. Stirring was continued for pre determined time
intervals (1 0 minutes to 40 minutes). The reaction mixture was then diluted
by pouring into about 1009 of crushed ice. The diluted acid solution was
neutralized immediately by addition of cold liquor ammonia solution while
maintaining the temperature below 0°C. The pH of the solution was
checked during the course of the neutralisation and it was continued till the
solution becomes slightly alkaline (pH - 7.5 to 8).
The dinitram~dic acid (DNA) formation (section 3.3.1) was measured
by taking samples from the reaction mixture at regular intervals and then
measuring the absorbance at 284nm. Figure 5.1 shows a typical case of
increase in absorbance wii.h time obtained during the nitration reaction.

Figure 5.1: Change in absorbance during the course of reaction
Figure 5.1 shows an increase in absorption at 284nm (characteristic
of -N (N02 )~ ) for the samples taken at different time intervals. However the
absorption curve at 40min showed a decrease in absorbance at 284
indicating the optimum time is achieved for the maximum conversion of
dinitramidic ac~d is betweell 30 and 40min. The reaction is discontinued at
this moment and further processed. Small weighed quantity of the
neutralized solution was diluted to a known concentration and analyzed by
UV spectroscopy. The yield of ADN was calculated by measuring the
absorbance at 284nm using the experimentally determined molar extinction
co-efficient as explained in chapter 2. section 2.6.
3.2.3. Separation of Ammonium Dinitramide
Two methods of separation of ADN from aqueous solution were
practiced. In the first method, the neutralized solution was evaporated in a
rotary evaporator under vacuum to completely remove water. The obtained

solid was further drled under vacuum. The dried solid was then extracted
with hot isopropanoi (500rnl) in batches, and the isopropanol extracts were
flltered through a filter paper, concentrated under vacuum and dried to give
a crystalline solid. The obtained solid was purified further by extraction with
hot ethyl acetate (200ml), filtered and evaporated under vacuum to yield
pale yellow crystals of pure ADN. The yield is 70%.
The second methotj of separation involves the use of different
adsorbents to selectively adsorb ADN. Different adsorbents are explored
for the effectiveness in adsorbing ADN. Adsorbents like activated charcoal,
granulated charcoal, silica gel, alumina etc., have ability to adsorb ADN
from aqueous solutions. The adsorbed ADN was then eluted using
solvents such as hot water, methanol, acetone etc., and then evaporated
under vacuum to obtain 23 free flowing pale yellow powder. A detailed
investigation on the use of powdered and granulated charcoal for the
adsorption of ADN from aqueous solutions was carried out and the results
are given in Chapter 4 (vlde supra).
3.3. Results and Discussion
3.3.1. Nitration of Ammonium Sulphamate (AS) using Mixed Acids
In the nitration, the first step is the formation of nitronium ion (N02' )
by ionization of HN03 by s'trong acids such as HF, HC104 , H2S04 or solid
acid catalysts 'I". The latter compounds promote ionization of HN03 to
NO>'. H2S04 IS one of the most frequently employed reagents during
nitrations. The sequence of reactions occurring with HzS04 and HN03 is
given in Equat~ons 3.1 to 3.4.

2 HN0,--, NO,'+ NO,. + H 2 0
H2S04+ H 2 0 - H,O++ HSO;
HNO, + H,>O - ---+ H,O'+ NO,.
In a mixture of different ratios of H2S04/HN03, the mole % of NO2'
has been measured and well documented L'411. For a given concentration of
H2S04/HN03, maximum cc~ncentration of NOz' is obtained when the mole
ratio of H2S04 IHNO:{ is 2. During aromatic nitration using mixed acids it is
necessary that the nitronium ion concentration is as high as possible. The
attack of the latter with the aromatic substrate is the slow rate-determining
step. For the conversion of AS to dinitramidic acid (DNA) the required
stoichiometric ratio of the nitronium ion to AS is 2 as per Equation 3.5.
NH4S0,NH, + 2 NO,' + 2 HSO; + HzO - HN(N02), + 2 H2S04 + NH4HS04
3.3.2. Derivation of a Reaction Scheme
Unlike aromatic nitration, in the present case mono nitration on the
nitrogen of the substituted amine [A] to mono nitramine [B] followed by
further nitration of the latter to the dinitramine [Dl or the nitration of
nitramide [C] has to take place as shown in scheme 3.1. The formed mono
nitro interrned~ate should encounter another NO>' without undergoing
decomposition The mechanism of formation of mono and dinitramine can
be represented by the reaction scheme given in 3.1.

' NH,SO,NH; + NO,' H,N+-S03NH,
Scheme 3.1: Mechanism of formation of mono and dinitramide
The formation of dinitramidic acid is suggested as a network of
parallel and consecutive reactions. The dinitramidic acid is formed via two
intermediates, which are able to react in different manners. Apart from the
formation of nitramide [C] and dinitramidic acid, their decomposition should
also be considered as shown in Equations 3.6 and 3.7. The
autocatalyt~c decomposition of dinitramidic acid is shown in Equation 3.7.
Alkaline Catalyst NH,NO, P* N20 + H20
Ac~d Catalyst pH <I f
Acid Catalyst HN(NO,), - N20 + HNO,
In order to understand the reaction mechanism, two known reactions
for the formation of nitramide and dinitramidic acid are compared [16-181
The discovery of ADN led in 1994 to its synthesis by the reaction of
NH3 and N205 as shown in Equation 3.8.
4 NH, + 2 N,Oj - NH4N(N02), + 2 NH4N03
.,........ (3.8)

The reaction of N205 and NH3 and the formation of nitrarnide at
-78°C can be represented b.y Equation 3.9.
Schmitt and co-workers postulate a certain probability of forming
nitramide as an unstable intermediate at low temperatures in the course of
the formation of ADN. The reaction sequences shown in Equations 3.10 to
3.1 2 represent the formatiori of ADN.
The f~rst step of nitration of ammonia yields nitramide, whereby
nitramide reacts with a second equivalent of N205 to form the dinitramide.
A reference is hereby made to derive a possible mechanism of
dinitration of ammonium sulphamate to dinitramidic acid by comparing the
scheme postulated by Tellier-Pollon and C.Canis ''gl. The formation of
nitrarnide and dinitramidic acid shown in Equations 3.10 to 3.12 will not
give a probable mechanisrr~ of formation of dinitramidic acid in the nitration
of ammonium sulphamate. On the other hand, Tellier-Pollon has shown
that nitramide can be formed from fuming HN03 by reaction of sodium salt
of sulphamic acid [lbl (NaS03NH2). The reaction is found to follow the
scheme given in Equation 3.13.

The authors have isolated the formed nitramide at low temperatures
(-10 to -35°C) with yields ranging from 12-60%. The formation of nitramide
is further confirmed by carrying out potentiometric titration using a base.
Scheme 3.1 represented the probable mechanism of formation of
dinitramidic acid. The dinitramidic acid formation could occur via the
formation of nitramide, followed by subsequent nitration of the same to
DNA. The DNA formation could also be a concerted mechanism through
the formation of intermediate [Dl. There is no experimental data available
on the formation of either nitramide or the intermediate [Dl. Based on the
results of Tellier-Pollon, the mechanism of nitration of AS appears to
involve the formation of nitramide. Further studies are to be carried out in
order to ascertain the formation of nitramide as an intermediate in the
nitration of ammonium sulphamate.
3.3.3. Spectral and Thermal Analysis of Ammonium Dinitramide
The obtained ADN was characterized by means of UV, IR,
differential scanning calorimetry (DSC), simultaneous TG-DTA and
elemental analysis techniques.
UV spectrum of the sample was recorded in water solution.
Ammonium dinitramide shows UV maximum in water at 212 and 284nm ['I.
UV spectrum of ammonium dinitramide in water is given in Figure 3.2. The
absorbance at 284nm is characteristic of the -N(N02)- ion due to the low
energy n - n' transition and the absorption maximum at 212nm is attributed
to the high energy 0 - 0' !:ransition. The molar extinction coefficient, E284
obtained from the slope of the concentration versus absorbance plot is

5.258 x 10" mol~'cm~' and this value is comparable with the value
reported ['I by Bottaro et al., (~284 = 5.207 x 10% mol~'crn~'). The results on
the calculation of molar ext~nction co-efficient for ADN in water, methanol
and acetonitrile are given in chapter 2, section 2.6 (vide infra).
Wavelength (nm)
Figure 3.2: UV spectrum of ammonium dinitramide (ADN) in water
Infrared spectrum of ammonium dinitramide was recorded in KBr
pellet. The spectrurn shows characteristic peaks and the assignments for
the peaks are given in Table 3.1. A typical IR spectrum of ADN is given in
Figure 3.3. The peak assignments were made in comparison with the
calculated v~brational frequencies reported for ADN '20,211.

Figure 3.3: IR spectrum of ammonium dinitramide (ADN)
Table 3.1: Characteristic peaks in IR spectrum of ADN
- ~ - ~.
v N-H of NH4' ~- - ~ ~
v, NO* in phase -
- ~ - ~ p ~ ~ ~ - - ~ ~
v, NO2 out of phase -. ~~ ~.~
v,, NO2 in phase - . . . .- ~~
v,, NO* out of phase -- -~~
-p-- ~- ~.
a,,,,, NO2 in phase - ~ ---- ~. .
6,ci,, NO2 out of phase ~ ~ --
~ . ... -
ijlOCk NO2 out of phase -- ~~~~~ ~ ~~- ~ ~ ~~-p~~--~
L.S N3 - ~- ~ ~ ~ ~ . ~ . -- -~~~
~ . a 5 N3
Wave number (cm")
31 24
1343
1178
1538
1403
827
76 1
731
952
1022
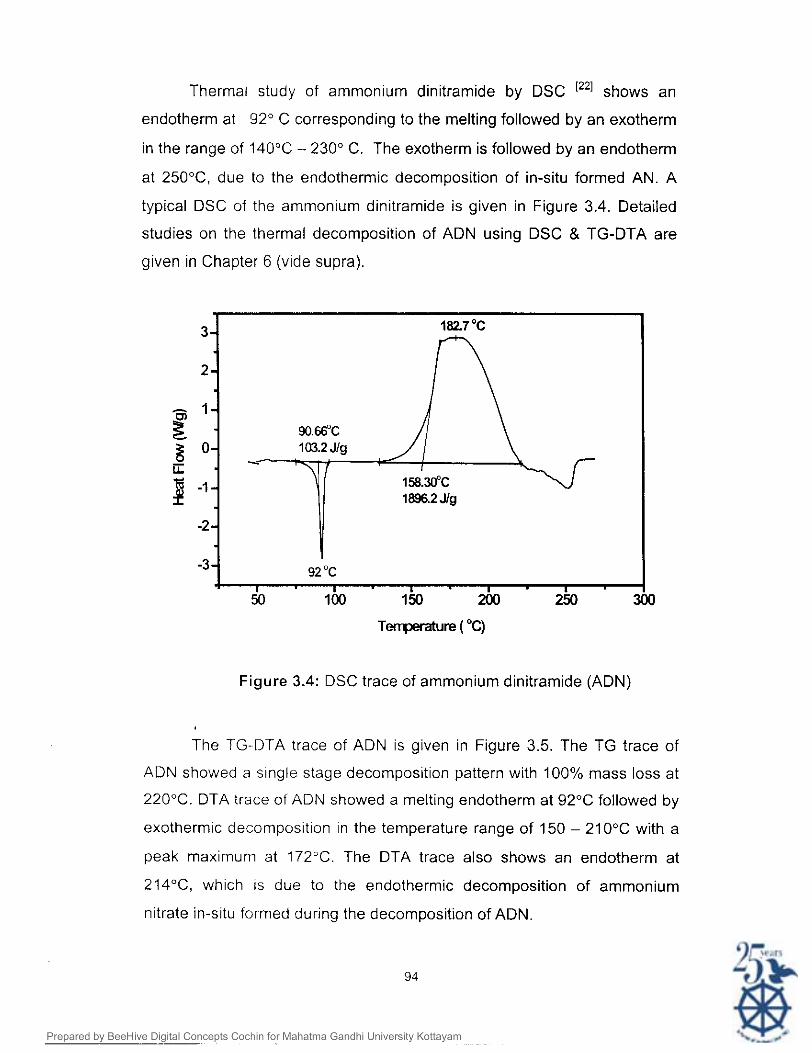
Thermal study of ammonium dinitramide by DSC shows an
endotherm at 92" C corresponding to the melting followed by an exotherm
in the range of 140°C - 230" C. The exotherm is followed by an endotherm
at 250°C, due to the endothermic decomposition of in-situ formed AN. A
typical DSC of the ammonium dinitramide is given in Figure 3.4. Detailed
studies on the thermal decomposition of ADN using DSC & TG-DTA are
given in Chapter 6 (vide supra).
Figure 3.4: DSC trace of ammonium dinitramide (ADN)
The TG-DTA trace of ADN is given in Figure 3.5. The TG trace of
ADN showed a s~ngle stage decomposition pattern with 100% mass loss at
220°C. DTA trace of ADN showed a melting endotherm at 92°C followed by
exothermic decomposition n the temperature range of 150 - 210°C with a
300
3 - 2-
- 1- -
O- q -,: -2 - -3 -
peak maximum at 172°C. The DTA trace also shows an endotherm at
214"C, which is due to the endothermic decomposition of ammonium
nitrate in-situ formed during the decomposition of ADN.
1827OC
9066°C: 1 W.2 Jlg -- Y I
158.30"C 1896.2 Jig
92 "C i I I I I I
50 100 150 200 250

Figure 3.5: TG-DTA trace of ammonium dinitramide (ADN)
The elemental analysis results of ADN agree with the calculated
values. The calculated and found values are given in Table 3.2.
Table 3.2: CHN analysis results of ADN
3.3.4. Effect of Variation of Acid Ratio on the Yield of Dinitramidic Acid
Calculated -~
C 1 Nil
H 3.23%
N 45.16%
When experiments were carried using the stoichiometric ratio of 2,
ie., with sulphuric acid (SA, H2S04) to nitric acid (NA, HN03) in the mole
Found
0.27%
2.87%
44.14%

ratio of 2:1, maximum yield of 45% was obtained in 20min. The acid
mixture, however, is very viscous at - 40 "C hence fast addition of the
reactant (AS) at a controlled rate becomes very difficult.
Based on the Zamen's Raman spectral measurements L231, the mole
% of nitronium Ion NO2' in the mixed acids containing 50 mole% H2S04
and 50 mole% HN03, is 14. Therefore, the mixture containing 0.1M each
of H2S04 and HNO3, the niolar concentration of nitronium ion would be
0.028M. When the mixture contains 0.2M each of HzS04 and HN03 the
molar concentration of NO2' would be 0.056M. It is seen that as the molar
concentration of the acids increased further, the NO; concentration also
would increase correspondingly. When equimolar mixture of acids was
taken in quantities as shown in Table 3.3, the yield of dinitramidic acid
increases from 6% to 37%. The yield of DNA for the mole ratio 1: l is 6%
and the mole ratio 3.5:3.5 is 37%. As the total acid concentration increased
further the reaction mixture becomes very viscous and hence further
increase of acid concentration was not attempted.
Table 3.3: Yield of din~tramidic acid after reaction time of 20 min
SI.No 1 Mole ratio I DNA yield * (%) SA:NA
The yield of dinitramidic acid during nitration depends on important
parameters like viscosity of the medium, temperature, agitation speed and
rate of addition of the reactants. To reduce the viscosity of the medium at
temperatures below -40°C, Inore nitric acid was taken in the system, which
will act as a solvent. The decomposition of the dinitramidic acid in acid
6
26
37
1 i i 1: l I
based on UV spectroscopy
2
3
2:2
3.5:3.5

medium also becomes a critical factor in determining the yield of the
product.
These factors can be overcome by increasing the rate of agitation of
the reaction m~xture, cooling the reactor at much faster rate and also by
making the system less viscous so that the transfer of nitronium ion to the
substrate takes place efft:ctively. In order to make the reactant well
dispersed in the medium, experiments were carried out using SA:NA in the
mole ratio of 2:6. In a typical experiment the yield of dinitramidic acid was
measured at d~fferent times and the results are shown in Table 3.4.
Table 3.4: Yield of dinitramidic acid at different times
I Time (min) I % Yield of DNA* (for ratio of SAINA 2:6)
As it is seen from Table 3.4, the yield of dinitramidic acid increases
from 17O/0 to 35%.
35
3.3.5. The Effect of Nitric Acid on the Dinitramidic Acid Yield
35
The effect of nitric acid on the dinitramidic acid yield was studied by
varying the nitric acid concentration from 2 moles to 12 moles keeping the
concentration of sulphuric acid at 2 moles. The results are given in Table
3.5 for the reaction carried out using 1 mole of AS at -45°C for a reaction
time of 30min.
* based on UV spectroscopy

Table 3.5: Yield of DNA for different ratios of SAINA
From Table 3.5 it is seen that as the mole percent of nitric acid in the
reaction mixture increases, the yield of dinitramidic acid shows an increase
from 26% to 35%. This is because the excess nitric acid present in the
system acts as a solvent and makes the system less viscous so that
effective nitration of the substituted arnine takes place thereby improving
the yield. The yield however has not gone beyond 35% even when the
nitric acid was increased above 12%.
Sulphuric acid /Nitric acid mole ratio
~~
2:2
2:6
2:lO
2:12
3.3.6. The Effect of Sulphuric Acid Ratio on the Rate of Formation of Dinitramidic Acid
% yield of DNA at 30min
26
30
3 1
35
In order to further study the effect of variation of sulphuric acid ratio
on the rate of formation of dinitrarnidic acid, experiments were carried out
for fixed quantities of AS and nitric acid, by varying the sulphuric acid. The
rate of formation of dinitrarnidic acid was monitored by taking in-process
samples of the reaction mixture and then analyzing it for the amount of
dinitramidic acid formed. The obtained rates for different ratios of sulphuric
acid and nitric acid are given in Table 3.6.

Table 3.6: Rates of formation of DNA for different ratios of sulphuric acid
Ratio of sulphuric acid I nitric acid
0.0183
It is seen from Table :3.6, that for the system with no sulphuric acid
the rate is 0.018:3, while other systems with varying ratios of sulphuric acid,
show a phenomenal increase in the rate indicating the catalytic effect of
sulphuric acid. Maximum rate was obtained for the ratio of 4:8. However,
on increasing the sulphuric acid concentration further to 8 moles, the rate
has drastically come down. The reason for this is the heterogeneity of the
mixture occurring due to solidification of the sulphuric acid rich reaction
mixture at low temperature. This prevents the effective attack of nitronium
ion to the substrate in the nitrating mixture.
3.3.7. The Effect o f Water on the Yield of Ammonium Dinitramide
Having studied the effect of sulphuric acid and nitric acid on the
formation of dinitramidic acid, and to improve the yield of dinitramidic acid,
experiments were carried out by adding different quantities of water to the
nitrating mixture. The water concentration was varied from 1 to 2.3M, and
the formation of ammonuum dinitramide was studied. In a typical
experiment for lmole of AS and SNNA ratio 2:12, the increase in water
concentration upto 1.7M showed an increase in the yield of ammonium
dinitramide from 30% to 5O0/0. This is because the added water molecule

facilitates formation of dinitramidic acid from the nitrated intermediate as
shown in Equations 3.14 to 3.18.
I NO, [ Bl [ Cl .......... (3.15)
[B] + NO,' -.- (N02)2NS03NH4 [Dl .......... (3.16)
[ C] + NO,' .- HN(N02), ......... .(3.17)
When water content is very low, the conversion of the intermediates
B and D to dinitrarnidic acid is poor, thereby reducing the yield. When the
water content is about 1.33M for 1M of AS, Equation 3.14 is a predominant
step thereby resulting n the favourable NH~NOZ. AS the water
concentration is increased beyond that, the mono nitro compound itself
undergoes hydrolysis leading to the formation of hydrolyzed products
(equation 3.19), which are unstable under these reaction conditions.
Hence large concentration of water is detrimental to get maximum yield.
Table 3.7 glves the results for the reaction carried out for 1M of AS and
with 2 moles of sulphuric acid and 12 moles of nitric acid.
excess H20 NH,N02 N 2 0 + H 2 0

Table 3.7: Effect of water on the yield of ADM
. .:
I . . .. . .
Yield of ADN * (%) ~-. .- .-
Water (mole) I
I ; 42.5
52.0
2 3 I 46.0
* By UV spectroscopy
With the add~tion of 1.7M of water, the yield of ammonium
dinitramide was ~ncreased to 52%. The reproducibility of the results at the
same level was tested and the results obtained show good reproducibility.
In all the experiments the obtained yield was above 50% for a water
concentration of 1 7M.
3.3.8. The Effect of Variation of Temperature
The effect of variatior of temperature was studied for the reaction
carried out using a SAINA mole ratio of 2:6 for l M of AS at -65", -55", and
-45°C. The reaction rate was found to be very slow at -65°C and slowly
increase at -55°C and a maxlmum rate is obtained at -45°C.
A$ seen from Figure :3.6, at above -45"C, run-away reaction takes
place because of the high exothermic nature of the reaction, the instability
of the intermediates and also due to the instability of the formed
dinitramidic acid at this temperature. At temperatures below -65"C, the
reaction becomes too slow due to high viscosity of the medium and also it
does not provide the required activation for the formation of mono nitro
intermediate leading to very poor yield.

Time
Figure 3.6: Rate of reaction at different temperatures
3.3.9. The Effect of Using Solvents as Nitrating Medium
In order to promote better stirring and effective heat transfer, inert
solvent namely CH2CI2 was used as a nitrating medium. In all the
experiments, the obtained yield of dinitramidic acid was very low. The yield
was less than 5%. This is because the reaction mixture was totally
insoluble in the solvent, it becomes heterogeneous and the solvent layer
separates from the reaction mixture thereby making the system highly in-
homogeneous and effective reaction does not takes place.
3.3.10. Summary of Yield of ADN for Different Ratios of SAlNA
Different ratios of SA/NA were studied in the nitration of ammonium
sulpharnate at a temperature of -45°C. The results obtained from the study
are given as a bar chart in Figure 3.7.

SPJNA ratio
Figure 3.7: Yield of ammonium dinitrarnide for different ratios of SAINA
As it is seen from Figure 3.6, the yield of ADN increases from 16%
to 35% as the concentration of nitric acid increases. Further improvement
in yield was achieved by addition of water. Increasing the sulphuric acid
concentration to 4 moles, the yield of ADN decreases as in the case of
ratios 4:2 to 4:12

PART-II
3.4. Synthesis of Potassium Dinitrarnide (KDN)
Potassium dinitramide KN(NO& has attracted a wide interest as a
promising new class of energetic oxidizer, which finds applications as an
energetic phase stabilizer in ammonium nitrate (AN) based propellants
[24,251, a dinitramide transfer reagent '261. It also finds applications in various
pyrotechnic formulations. The synthesis of potassium dinitramide was
carried out by two methods. In the first method, potassium sulphamate
KS03NH2 was nitrated using mixed acids under the experimental conditions
given in chapter 3, section 3.2.2. The second method of synthesis involves
the double decomposition of ADN with potassium hydroxide in a solvent.
3.5. Experimental
3.5.1. Materials
Potassium sulphamate was prepared by neutralization of sulphamic
acid NH2S03H with KOH. Sulphamic acid (48.59, 0.5mol) was dissolved in
50ml of water. KOH (289, 0.5mol) was dissolved in 50ml of water and both
the solutions were mixed at 0°C. The resultant solution (pH 7+1) was
precipitated in methanol. The precipitated potassium sulphamate was
filtered, washed with methanol and dried in an oven at 70°C for 2 hrs.
KOH (NICE chemicals, Cochin) was used as received. Solvents
viz., isopropanol. acetone, methanol were distilled and dried over molecular
sieves prior to use. ADN with purity > 98% was synthesized as explained
in section 3 2.2. Con. H2S04 98% (Qualigens, Mumbai) was used as
received. Fuming HN03 of assay > 98% was distilled in the laboratory from
a mixture of 1 : I (by welght:) of fuming HN03(92% ) with con. H2S04. The
fraction between 83 - 85°C was used.

3.5.2. Method 1
In a typical experiment potassium sulphamate (6.759, 0.05mol) was
added in small portions to a mixture containing HN03 (assay-98%, 18.9g,
0.3mol) and con.H2S04 (9 89, 0.lmol) at a temperature of -35" C to
-45" C in a 500ml jacketed reactor with stirring. The rate of addition was
controlled in such a way that the temperature of the mixture does not
exceed -35" C Formation of white precipitate was o b s e ~ e d during the
course of the reaction due to the precipitation of potassium sulphamate and
the viscosity of the mixture increases as the reaction progresses. Stirring
of the contents was continued for a pre determined time and then the
reaction mixture was diluted by pouring into about 1009 of crushed ice.
The diluted acid solution was neutralized immediately by adding cold KOH
solution while maintaining the temperature below 0°C. The pH of the
solution was checked during the course of the neutralisation and it was
continued till the solution becomes alkaline (pH - 7.5 to 8). The reaction
sequences are given in Equations 3.20 and 3.21.
KS03NH2 + 2 NO,' + 2 HSO; + H20 - HN(N02), + 2 H,SO, + KHSO,
HN(N02), + KOH ---* KN(NO,), + H20 .......... (3.21)
The neutralized solution was diluted to known concentration and
analyzed by UV spectroscopy. The yield of the ammonium dinitramide was
calculated by measuring the absorbance at 284nm and then calculating the
concentration using the calc:ulated molar extinction co-efficient ( ~ ~ ~ 4 = 5.379
x l o 3 L mol-'cm'). The calculation of molar extinction co-efficient for KDN
was given in chapter 2 section 2.6.6 (vide infra)

3.5.3. Method 2
This involves a sirnple procedure. Potassium hydroxide (6.69,
0.lmol) was dissolved in 100ml of dry CH30H. Ammonium dinitramide
(12.49, b . lmo~ ) was dissolved in another 100ml of CH30H. The two
solutions were combined and kept in a freezer for 2-3 hrs, the resulting
crystalline solid of KDN was collected by filtration and dried under vacuum
for 1-2 hrs. The yield is 90-92%.
3.5.4. Separation of Potassium Dinitramide
The neutralized solution (from method 1) was evaporated under
vacuum to remove water. The evaporated solid was further dried in
vacuum to a dry powder. The dried solids were then extracted with
acetone in portions, concentrated under vacuum and precipitated in
isopropanol to get a crystalline white solid. This was purified further by
recrystallization from methanol to get pure potassium dinitramide. The yield
is 75-80%.
3.6. Results and Discussion
3.6.1. Characterization of Potassium Dinitramide
KDN was characterized by FTIR, simultaneous thermogravimetry
differential thermal analysis (TG-DTA) and elemental analysis. FT-IR
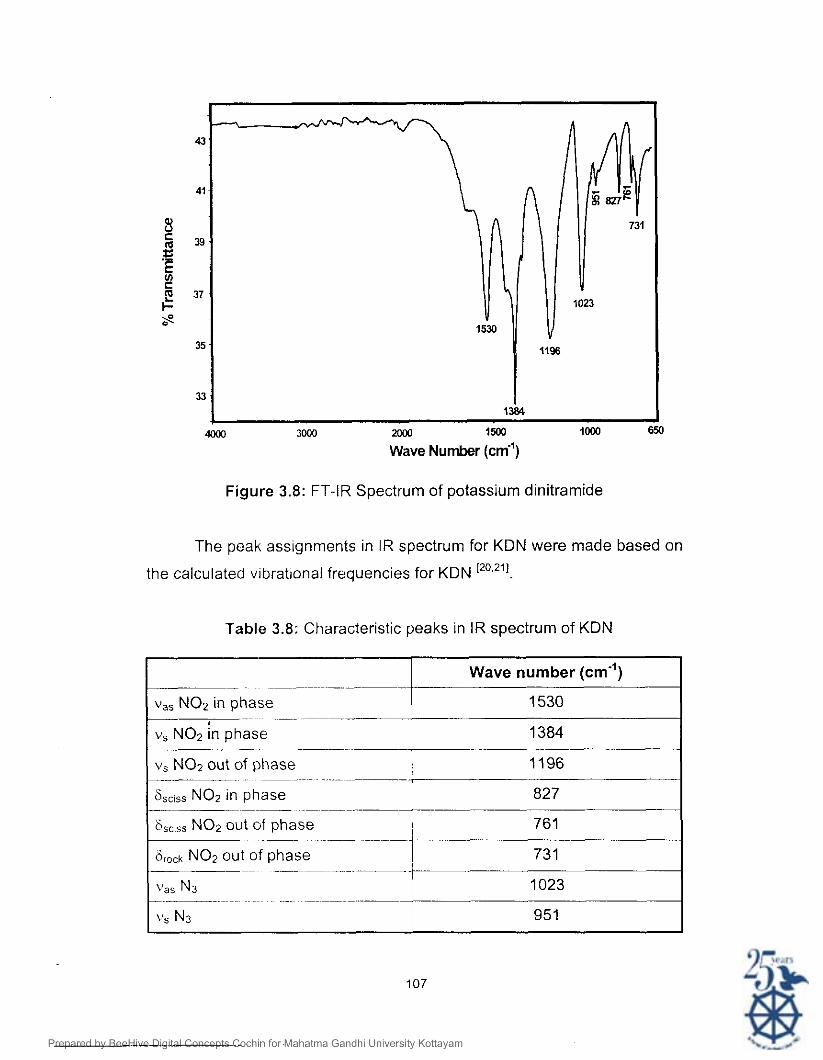
spectrum of potassium dinitramide IS given in Figure 3.8. The characteristic
IR peaks for KDN are given in Table 3.8.
Wave Number (cm')
Figure 3.8: FT-IR Spectrum of potassium dinitrarnide
The peak assignments in IR spectrum for KDN were made based on
the calculated vibrational frequencies for KDN L20.211
Table 3.8: Characteristic peaks in IR spectrum of KDN
- . ~ - - - ~ - ~
v,, NO2 in phase -~ ~ ~~~
V, NO2 /n phase -~ .
Wave number (cm")
1530
1384
V, NO2 out of phase ! 1196 . . ~
6,,,,, NO2 in phase : ~
827 +-- 6,,,,s NO2 out of phase I 761
.. ~
firock NO2 out of phase f 731 ~ ~ I
vas N3 1023 ~~ ~- ~~ ~ ~ t-
vs N3 951
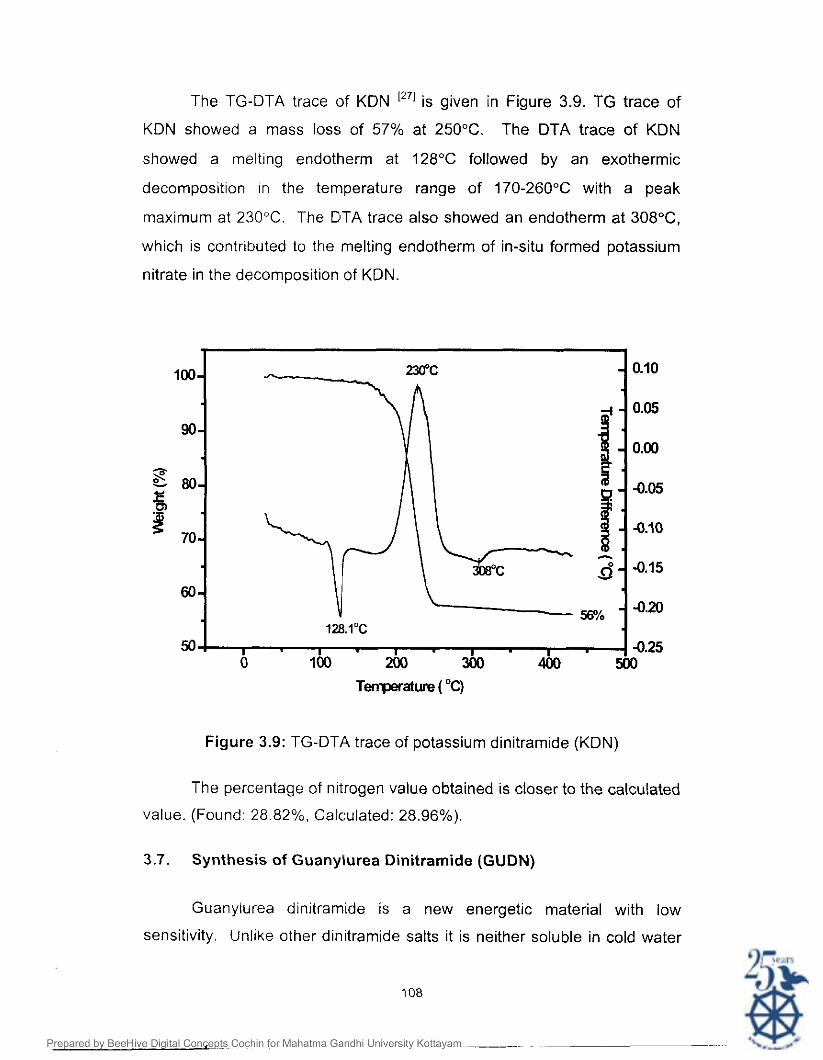
The TG-DTA trace of KDN [271 is given in Figure 3.9. TG trace of
KDN showed a mass loss of 57% at 250°C. The DTA trace of KDN
showed a melting endotherm at 128°C followed by an exothermic
decomposition in the temperature range of 170-260°C with a peak
maximum at 230°C. The DTA trace also showed an endotherm at 308"C,
which is contributed to the melting endotherm of in-situ formed potassium
nitrate in the decomposition of KDN.
Figure 3.9: TG-DTA trace of potassium dinitramide (KDN)
The percentage of nitrogen value obtained is closer to the calculated
value. (Found: 28.82%. Calc:ulated: 28.96%).
3.7. Synthesis of Guanylurea Dinitramide (GUDN)
Guanylurea dinitramide is a new energetic material with low
sensitivity. Unlike other dinitramide salts it is neither soluble in cold water

nor hygroscopic [28,'91. This prompted us to study its synthesis and
characterization. The synthesis of guanylurea dinitramide involves two
steps. The first step is the formation of guanylurea sulphate. The treatment
of guanylurea sulphate in water with a water solution of ADN results in the
formation of guanylurea dinitramide as fine powder.
3.8. Experimental
3.8.1. Materials
Dicyandiamide (CDH, Mumbai) & Con.H2S04 (Qualigens, Mumbai)
were used as received. lsopropanol (Qualigens, Mumbai) was distilled and
dried over molecular sieves. Ammonium dinitramide (ADN) was prepared
by the procedure described in chapter 3, section 3.2.2. The purity of ADN
used is >98%.
3.8.2. Preparation of Guanylurea Sulphate
Dicyandiamide (16.89, 0.2mol) was treated with aqueous sulphuric
acid (9.89, 0.1 mol in 50ml of water) and kept over a hot water bath for 3-4
hrs. The resulting solut~on was kept in an ice bath for 1 hr, the formed
white crystals were filtered off. The rest of the filtrate was concentrated
over a hot water bath and then cooled in ice. The resulting crystals were
filtered, combined, washed with isopropanol and then dried in an hot air
oven. The yield is 88-90%. The reaction sequence is given in Equation
3.22.

3.8.3. Preparation of Guanylurea Dinitramide
Guanylurea sulphate (30.29, 0.lmol) was dissolved in 50ml of water
with slight warming and stirring. When the solution is clear it was taken out
and cooled to room temperature. ADN (24.89, 0.2mol) dissolved in 10ml of
water was added to the above solution in portions. A fine white crystals
were formed. This was filtered, washed several times with cold water and
finally dried under vacuum for an hour. The yield is of the same is 90-95%.
The reaction is glven in Equation 3.23.
3.9. Results and Discussion
3.9.1. Characterization of GUDN
The purity of GUDN was determined from elemental analysis, the
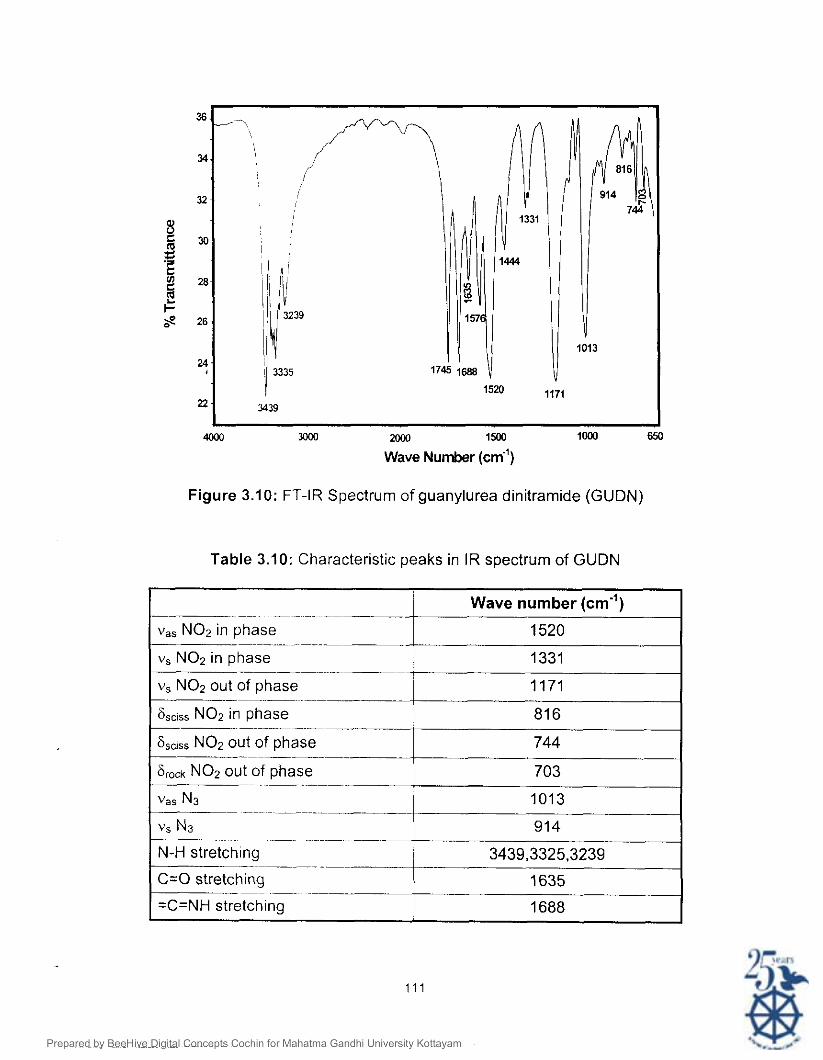
values are given in Table 3.9. IR spectrum of GUDN is given in Figure 3.10.
The IR peak assignments for GUDN are given in Table 3.10.
Table 3.9: CHN analysis of guanylurea dinitramide
C
H
N
Calculated ~
11.48%
3.35%
46.89%
Found
11.70%
2.80%
46.90%
2WO 1500
Wave Number (cm")
Figure 3.10: FT-IR Spectrum of guanylurea dinitramide (GUDN)
Table 3.10: Characteristic peaks in IR spectrum of GUDN
-- ~ ~~~
v,, NO2 in phase
vs NO2 in phase ~ -
v, NO2 out of phase
6sciss NO2 in phase >
6,,is, NO2 out of phase ~
Srock NO2 out of phase -- ~
vas N3 - ~ .---
vs N3 .. ~ ~ - - ~ ~~~ ~
N-H stretching .
C=O stretching - --
=C=NH stretching
Wave number (cm")
1520
1331
1171
81 6
744
703
1013
- 914
3439,3325.3239
1635
1688

3.10. Preparation of Tetramine Cu(ll) Dinitramide
A complex salt of dinitramide was prepared employing ADN, copper
sulphate and ammonia solution ["I. The isolation of [ C U ( N H ~ ) ~ ] [ N ( N ~ ~ ) ~ ] ~
was achieved by mixing of ADN, CuS04.5H20 and liquor ammonia in
water.
3.1 1. Experimental
3.1 1 . I . Materials
CuS04.5H20 (Purex Laboratories, Bangalore) and liquor ammonia
(Qualigens, Mumbai) were used as received. ADN was synthesized by the
procedure described in chapter 3, section 3.2.2. The purity of ADN used in
the study is >98%.
3.11.2. Synthesis of Tetramine Cu(ll) Dinitramide
A saturated solution of CuS04.5H20 (3.7g, 0.015mol) in aqueous
NH3 was reacted with a saturated solution of ADN (3.79, 0.03mol) in H20
and cooled to 0°C producing violet crystals of tetramine Cu(ll) dinitramide
[CU(NH~)~] [N(NO~)Z]~. The formed crystals were filtered and were dried in a
desiccator. The y~eld is 9045%.
3.12. Results and Discussion
IR and elemental analysis of tetramine Cu(ll) dinitramide was
performed and the results are given Figure 3.11 and Table 3.11
respectively.
Table 3.11: CHN analysis of tetramine Cu(ll) dinitramide
Calculated
t ~
Nil
H 3.49%
N 40.76%
Found
Nil
2.9 %
40.3%
28
26
24
P
18
14
10
6
40M) 2WO 1500
Wave Number (cm")
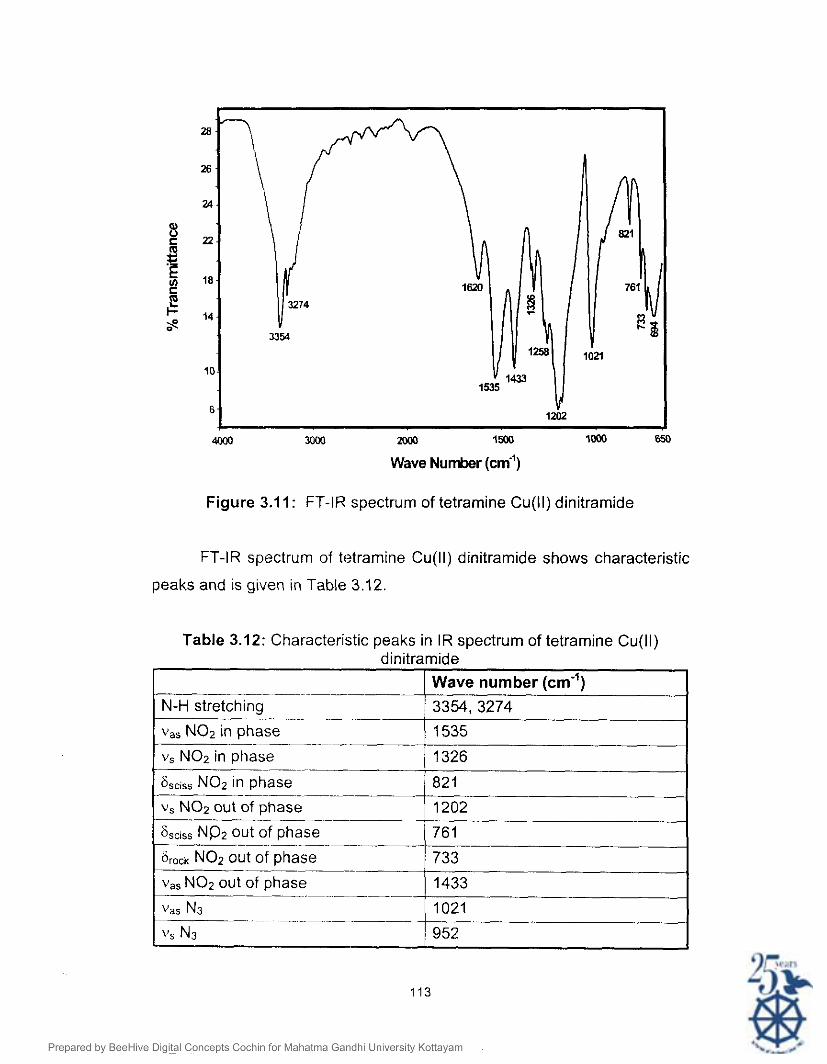
Figure 3.1 1: FT-IR spectrum of tetramine Cu(ll) dinitramide
FT-IR spectrum of tetramine Cu(ll) dinitramide shows characteristic
peaks and is given in Table 3.12.
Table 3.12: Characteristic peaks in IR spectrum of tetramine Cu(ll) dinitramide
I Wave number fcrn-ll
v, NO2 in phase ~
6,,i,, NO2 in phase
v, NO2 out of phase -- 6,,,,, NP2 out of phase
~
6,,,1, NO2 out of phase 733

3.13. Emulsion Crystallization of Ammonium Dinitramide (ADN)
Solid propellants often consist of heavily laden polymer systems. In
these, the particulate components that can comprise upto 90% by weight of
the total mass are especially important. The formation of these spherical
particles by recrystallization or other means is a production step, which
follows after the synthesis of the compound to give a finished solid
propellant or explosive charge. Emulsion crystallization is particularly suited
to recrystallization of fusible propellant and explosive materials[301.
Ammonium dinitramide crystallizes naturally in the form of needles
or plates, which are not readily amenable to subsequent processing. For
use in propellant it is necessary to use solid particulate ADN of controlled
size to obtain predictable results. Particles in the range of about 50 to 500
pm are considered useful [3'-331. Efforts to control crystallization or to
physically process solid ADN to obtain a selected particle size have been
unsuccessful. Hence a suitable method for preparing spherical grains of
ADN was worked out. The process by which spherical ADN can be
produced is explained below.
3.14. Experimental
3.14.1. Materials
Ammonium dinitrarr~ide with purity >98% and a mean particle size of
40-501rrn was used. The synthetic procedure was described in chapter 3.
section 3.2.2.
Paraffin oil (XCELTHERM 600, Radco Industries lnc., USA) was
used as received. The specific gravity is 0.82 to 0.88. Methylene chloride
(AR) (SRL laboratories, Mumbai) was used as received.

Fumed silica (Cab-0-31) with a mean particle size of 50nm was
used as a protective colloid.
3.15. Emulsion Crystallization Process
The emulsion crystallization process involves the melting of ADN in
paraffin oil maintained above the melting temperature of ADN.
Subsequently, the molten ADN in the paraffin oil is stirred to get uniform
spherical grains of ADN. Finally cooling the paraffin oil to room
temperature and isolation of spherical grains by washing with a solvent.
The process diagram for the emulsion crystallization process is
given in Figure 3.12 .In a typical experiment 209 of dried ADN was slowly
introduced into hot paraffin oil maintained at 92-93°C in a jacketed reactor
(JR), along with 0.1% of fumed silica (Cab-0-Sil) as a protective colloid to
prevent particle-particle adhesion. Temperature of the paraffin oil was
controlled using the heating and cooling exchanger (HCE). The hot paraffin
oil along with the molten ADN was mechanically stirred using a propeller
stirrer (M) at 400-500 rpm.
Ethylene glycol -i- H20
Paraffin Oil
tieatlng and i--~---J [JRI Cooling Exchanger
Figure 3.12: Schematic representation of emulsion crystallization process

Afler the formation of visible spherical molten ADN grains, the
paraffin oil was slowly cooled to room temperature by circulating water
through the jacketed reactor. The molten ADN spherical grains will
crystallize as the temperature is lowered. The spherical ADN particles
were then filtered off to remove the paraffin oil and washed twice or thrice
with methylene chloride to remove adhered paraffin oil on the surface of
ADN. The methylene chloride washed ADN was dried further under
vacuum and stored in a desiccator.
3.16. Results and Discussion
Table 3.1 3 shows the results obtained in the emulsion crystallization
process of ADN with different quantities of starting material.
Table 3.13: Batch-wise results on the emulsion crystallization of ADN
' with a battle
It is seen from Table 5.2. for batches EC-I to Ill, the particle size
obtained was above 300prn. Further improvement to control the particle
size was made by using a baffle in the reactor which promotes uniform
stirring and avoids the turbulence of paraffin oil. The results shown in
Table 5.2 for EC-IV and EC-V are obtained using a baffle. The results
show that very good particle size distribution is achieved by using a baffle.
I
Batch ADN (g) Cab-0-Sil 4 (g)
. -- . -
EC-I a , 0 027
Paraffin oil (ml)
200
200
400
500
600
EC-II 15
EC-Ill , 25 I
EC-IV a 25
EC-V a 1 30 I
Q -.
0 2
0.050
0 056
0 093
Stirring time (min)
10
5
5
15
10
Particle size (pm)
>300
2500
>500
100-400
200-600

3.16.1. Particle Size Analysis
The particle size of emulsion crystallized ADN was analysed by
optical microscopy. Figures 3.13 and 3.14 show the optical microscopic
images of fine and coarse ADN respectively. It is seen that uneven needle
shaped crystals are observed for fine ADN, while the emulsion crystallized
coarse ADN showed smooth spherical surfaces.
Figure 3.13: Optical microscopic image of as-synthesized ADN
Figure 3.14: Optical microscopic image of emulsion crystallized ADN
117

3.16.2. Analysis of Emulsion Crystallized ADN
The influence of emulsion crystallization on ADN is studied by IR
and differential scanning calorimetry (DSC). The IR spectra and DSC of as-
synthesised ADN was compared with that of the emulsion crystallized ADN.
Figure 3.15 shows the overlay of IR spectrum of as-synthesised ADN and
emulsion crystallized ADN. It is seen from Figure 3.15, that all the
characteristic peaks for ADN are present in both the spectra. The ADN
particles formed by the emulsion crystallization process show no significant
changes.
I I I I I 4WO 3WO
I 2WO 1m 10M 650
Wavenurrber (uri')
Figure 3.15: Overlay of IR spectra of ADN (as-synthesised) and ADN (Emul. Cryst)
Figure 3.16 shows the DSC overlay of ADN (as-synthesised) and
ADN (emul.cryst). Comparison of the DSC curves shows no difference in
the melting and decomposition pattern. The emulsion crystallization
process does not seem to affect the properties of ADN.

Tenperature ec)
Figure 3.16: DSC overlay of ADN (as-synthesised) and ADN (EmuLCryst.)
3.17. Conclusions
The nitration of ammonium sulphamate using mixed acids was
studied. In the nitration of ammonium sulphamate, the formation of
dinitramidic acid depends on the mole ratio of the sulphuric acidlnitric acid
taken and the time of the reaction. Maximum yield of dinitramidic acid 45%
was obtained in 20mts when the ratio of SAINA is 2:l. The addition of
sulphuric acid to the nitric acid catalyses the reaction. The effect of
variation of nitric acid and sulphuric acid on the formation of dinitramidic
acid was studied. Maximum rate was obtained for SNNA ratio 4:8. The
addition of water to the nitrating medium helps in improving the yield of
ammonium dinitramide upto 50%. None of the solvents were proved to be
an efficient nitration medium. The study on the effect of temperature on the
formation of dinitramidic acid shows that for optimum reaction, a
temperature of -45°C is necessary, above which the reaction is highly

exothermic and below which the reaction is too slow. The study given in
this chapter helped optimizing the reaction conditions for getting a higher
yield of ammonium dinitrarnide.
Dinitramide salts viz., potassium dinitramide, guanylurea dinitramide
ar~d tetramine Cu(1l) dinitramide were synthesized and characterized by IR
and elemental analysis. The synthetic routes either involve a nitration
reaction or a simple double decomposition. The synthesized dinitramide
salts were thermally characterized by thermo analytical methods.
Elaborate thermal studies are given in chapter 6 (vide supra). The above
salts will find applications in inorganic and organic synthesis. A process for
the formation of spherical grains of ADN from fine ADN was achieved by an
emulsion crystallization process. The described process enables
production of spher~cal grains of ADN with a mean particle size of 100-
500pm. The analytical results by IR and DSC show no significant
difference between the as-synthesized ADN and the emulsion crystallized
ADN.

3.18. References
1. Bottaro J.C, Penwell P.E, Schmitt R.J., J.Am. Chem.Soc., 1997, 1 1 9. 9405-941 0.
2. Pak Z.P, American Institute of Aeronautics Inc., 1993, paper 93- 1755.
3. Stu Borman, Chem. & Engg. News, 1994, 18-22
4. Schmitt R.J., Bottaro J.C, Penwell P.E, Bornberger D.C, (SRI International) U.S. Patent 5316749, May 31, 1994.
5. Schmitt R.J., Bottaro J.C, Penwell P.E, Bomberger D.C, (SRI International) U.S. Patent 5316749A, 1994.
6. Schmitt R.J., Bottaro J.C, Penwell P.E, Bomberger D.C, (SRI International) U.S. Patent 5198204, 1993.
7 . Schmitt R.J., Bottaro J.C, Penwell P.E. Bomberger D.C. (SRI International), U.S. Patent 5415852, 1995.
8 . Suzuki S, Miazaki S, Hatano H, Shiino K, Onda T, (Nissan Motor company, Kanagawa and Hosaya fireworks Co., Tokyo, Japan) U.S. Patent 5659080,1997.
9. Schrnitt R.J., Bottaro J.C, Penwell P.E, Bomberger D.C, (SRI International) U.S. Patent, 5254324, 1993.
10. Langlet Abraham, Ostmark Henric, Wingborg Niklas, (Forsvarets Forskningsanstalt, Stockholm, Sweden) U.S. Patent 5976483, 1999.
11. Vladirnir A. Tartakovsky, Oleg A. Luk'yanov, 25'h lnt. Annu. Conf ICT, 1994, 13-1 t013-9.
12. Robert J. Schmitt, Jeffrey C. Bottaro, Paul E. Penwell, SRI International Report No. AD-A 263 271, April 13. 1993.
13. George A. Olah, Nitration - methods and mechanisms. VCH publishers, New York. 1989.
14. Lyle F. Albright, Sood M.K, Modeling nitronium ion concentrations in HzSO~-HNO~-HZO mixtures, Nitration-recent laboratory and industrial developments : ACS symposium series 623. Chapter 18, 201-213,1996.

15. Christian Frenck, Werner Weisweiler, Chem.Eng.Technol., 2002, 25, 123-128.
16. Pierre Vast, Joseph Heubel, C.R. Acad. Sc. Paris, 1965, t 260, 5799-5801.
17. Pierre Vast, Joseph Heubel, C.R.Acad.Sc.Paris, 1967, t 264, 1697- 1699
18. Sylvaine Tellier-Pollon, Joseph Heubel, Revue de Chimie Minerale, 1967,4, 413-423.
19. Canis C, Revue Chimie Minerale, 1964, 1, 521-528.
20. Karl.O.Christe, William W Wilson, Mark A Petrie, Harvey H Michels, Jeffrey C. Bottaro, Richard Gillardi, lnorg.Chem., 1996, 35, 5068- 5071
Shlyapochnikov, Oleneva G.1, Cherskaya N.0, Lukyanov O.A, Gorelik V.P, Anikin O.V, Tartakovsky V.A, Russ.Chem.Bull., 1995, 44(8). 1449-1453.
Sergey Vyazovkin, Ciharles A. Wight, J.Phys.Chem., A ,1997, 101 5653-5658.
Zamen M.5, Nitronium ions in nitrating acid mixtures. Ph.D. Thesis, University of Bradford, Bradford, UK, 1972.
Ming Lei, Zhi-Zhong Zhang, Yang-Hui Kong, Zi-Ru Liu, Chun-Hua Zhu, Ying-Hui Shao, Pei Zhang, Therm.Chim.Acta, 1999, 335, 105- 112.
Ming Lei, Zi-Ru Liu, Yang-Hui Kong, Cui-mei Yin, Bo-Zhou Wang, Yuan Wang, Pei Zhang, Therm.Chim.Acta, 1999, 335, 113-120.
How-Ghee Ang, Wolfgang Fraenk, Konstantin Karaghiosoff, Thomas M. Klapotke, Peter Mayer, Heinrich Noth, Joanna Sprott, Marcus Warchhold, Z. Anorg. Allg. Chem., 2002, 628, 2894-2900.
Oxley J.C, Smith J.L, Zheng W, Rogers E, Coburn M.D, J. Phys. Chem., A, 1997,101, 5646-5652.
Ostmark H, Bernm U. Bergman H, Langlet A, Therm.Chim.Acta., 2002, 384, 253-259.

29. Abraham Langlet, L!S. publication No. US2001/0007913A1,2001
30. Ulrich Teipel, Prop.Expl.Pyro., 1999, 24, 134-1 39.
31. Ulrich Teipel, Thomas Heintz, Horst H. Krause, Prop.Expl.Pyro., 2000, 25, 81-85.
32. Ulrich Teipel, Krause Horst, Leisinger Karlfred, Heintz Thomas, European Patent No EP 0953555A7,1999.
33. Teipel U, Heintz T, Leisinger K, Krause H.H. 29" lnt. Annu. Conf. ICT, Germany, 1998, 6311 to 63/14.