nuts and bolts of next generaon sequencing analysis at accre · 2017-11-09 · nuts and bolts of...
TRANSCRIPT

NutsandBoltsofNextGenera1onSequencingAnalysisatACCRE
ThomasStrickerMD/PhDAssistantProfessor,PMI
11/09/2017

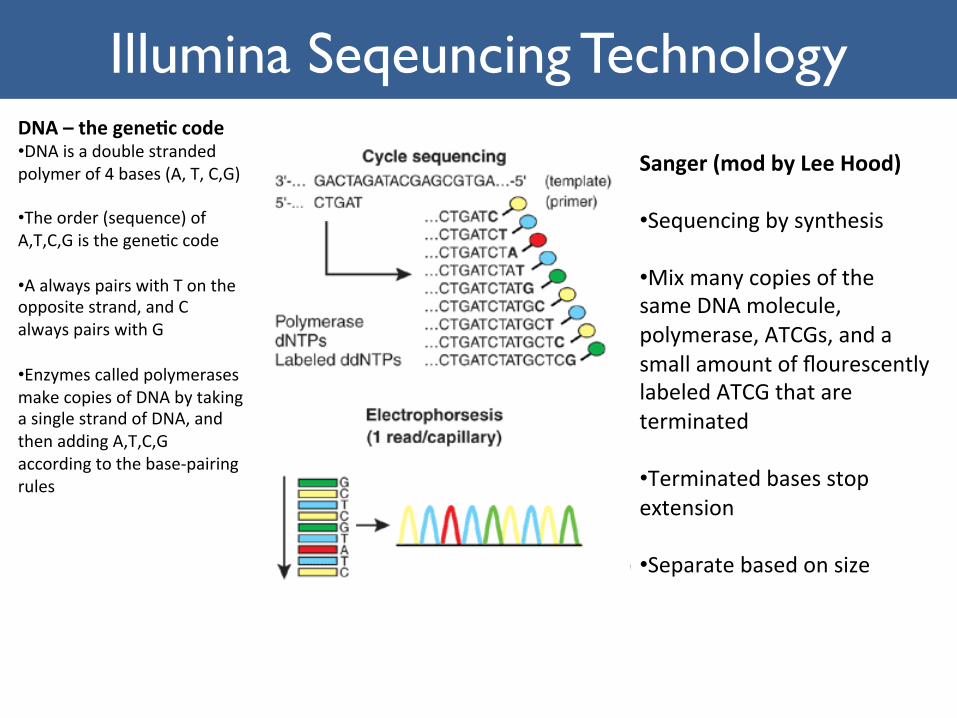
NextGenera1onSequencingIllumina Seqeuncing Technology
DNA–thegene+ccode• DNAisadoublestrandedpolymerof4bases(A,T,C,G)
• Theorder(sequence)ofA,T,C,Gisthegene1ccode
• AalwayspairswithTontheoppositestrand,andCalwayspairswithG
• EnzymescalledpolymerasesmakecopiesofDNAbytakingasinglestrandofDNA,andthenaddingA,T,C,Gaccordingtothebase-pairingrules
Sanger(modbyLeeHood)• Sequencingbysynthesis
• MixmanycopiesofthesameDNAmolecule,polymerase,ATCGs,andasmallamountofflourescentlylabeledATCGthatareterminated
• Terminatedbasesstopextension
• Separatebasedonsize
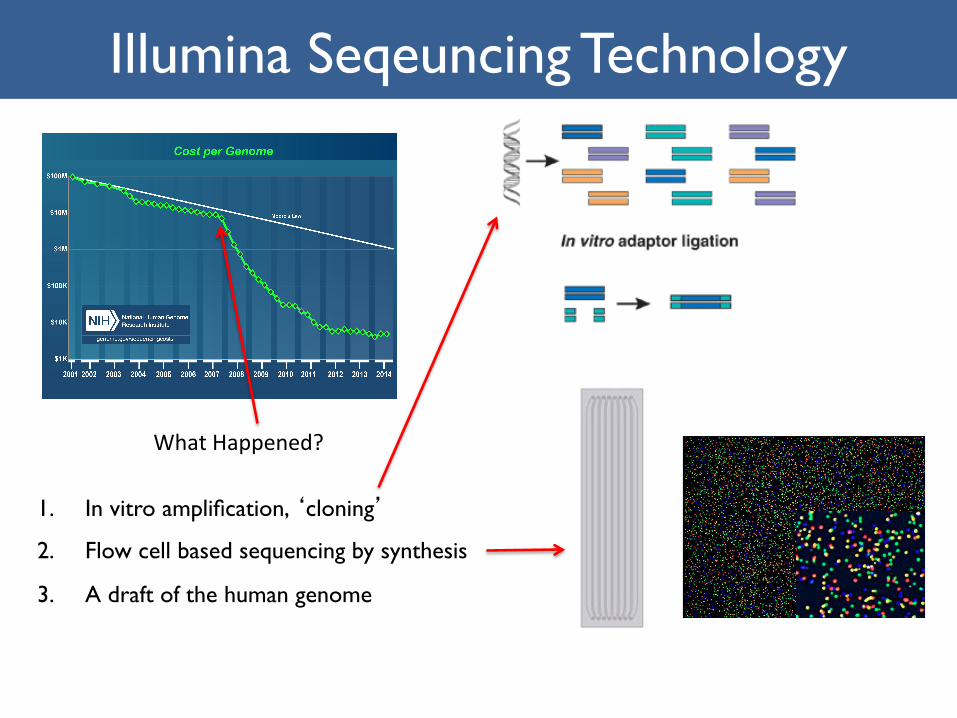
1. In vitro amplification, ‘cloning’
2. Flow cell based sequencing by synthesis
3. A draft of the human genome
Illumina Seqeuncing Technology
WhatHappened?

TermsandDefini1ons
• Singleorpairedend• Lane• Barcodeorindexes
• Library–Thebaseunitofprepara1on.PoolofDNAmoleculesthatareseq.
• Sample–Asingleindividual

TermsandDefini1ons• FASTA=sequencefile• Fastq=sequence+qualityfile• BAM=alignmentfileinbinary• VCF=variantcallfile• Index=allowsrapidlookupoffasta,bam,vcf• MAF=muta1onannota1onfile• GTF/GFF=informa1onaboutgenestructure• Intervallist=likeBED,uniquePicard(usepicardBedteoIntercalto
generate)• BED=

Contents of the human genome [CC-BY-SA-3.0] Steve Cook
SequencingandGenomeSize
S. cerevisiae = 12 million bp C. elegans = 100 million bp Drosophila m. = 130 million bp D. rerio = 1.4 billion bp M. musculus = 2.1 billion bp H. sapiens = 3 billion bp

Genomic DNA
Pros: Large capture space Copy Number and Translocations Variants outside baits might be sampled Cons: Larger amount of DNA necessary (>250ng) Sequencing inefficient Library builds slow, add to turnaround time
TargetEnrichment–HybridCaptureApproach
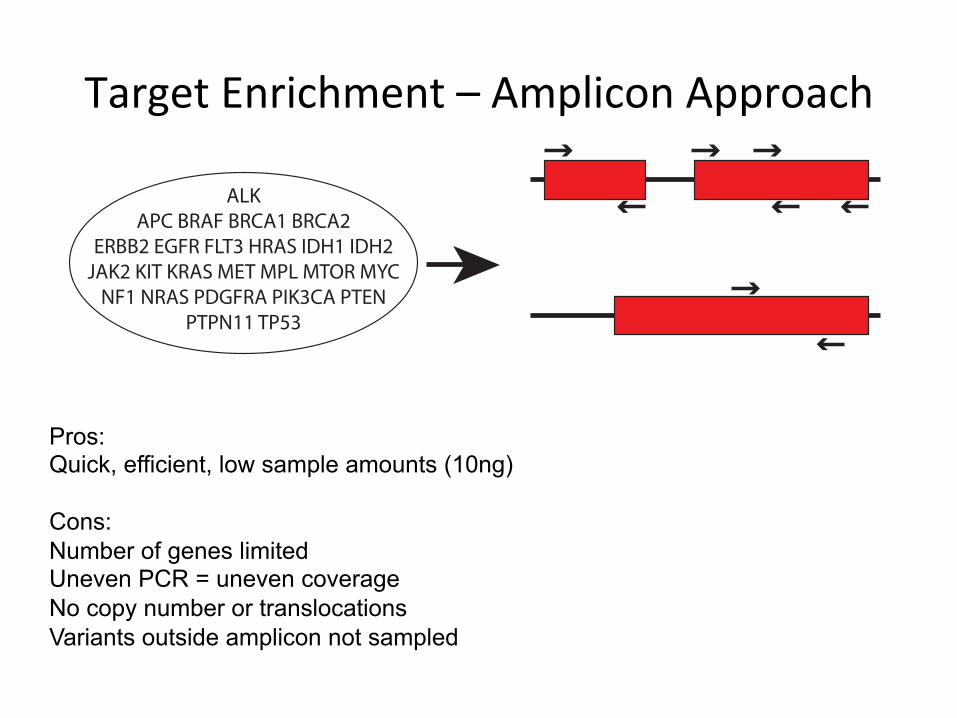
ALK APC BRAF BRCA1 BRCA2
ERBB2 EGFR FLT3 HRAS IDH1 IDH2 JAK2 KIT KRAS MET MPL MTOR MYC
NF1 NRAS PDGFRA PIK3CA PTEN PTPN11 TP53
Pros: Quick, efficient, low sample amounts (10ng) Cons: Number of genes limited Uneven PCR = uneven coverage No copy number or translocations Variants outside amplicon not sampled
TargetEnrichment–AmpliconApproach
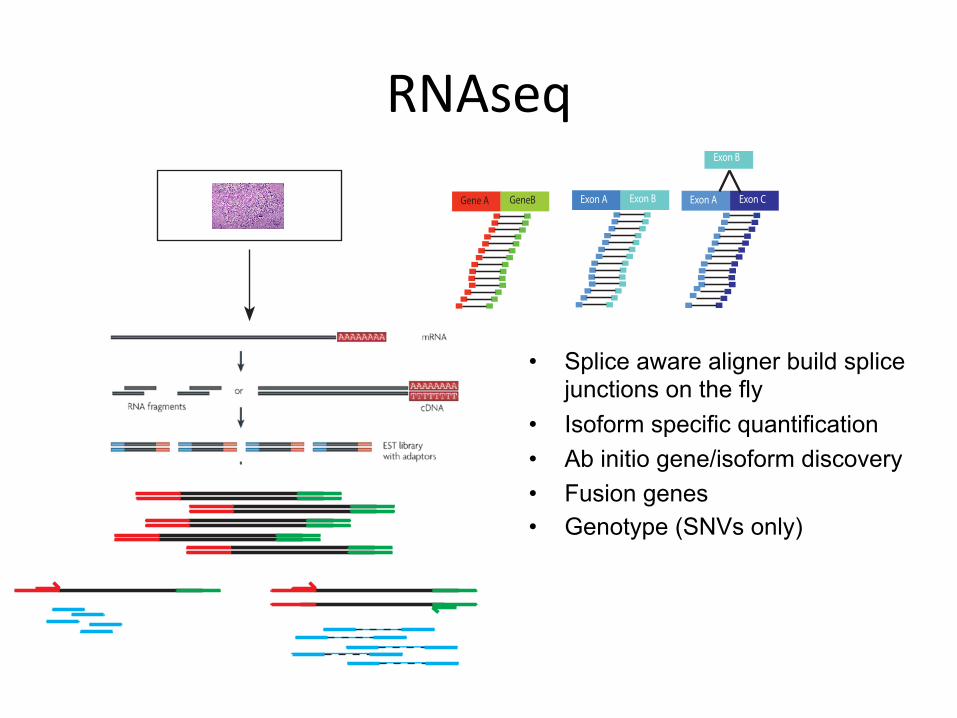
RNAseq
• Splice aware aligner build splice junctions on the fly
• Isoform specific quantification • Ab initio gene/isoform discovery • Fusion genes • Genotype (SNVs only)
Gene A GeneB Exon A Exon B Exon A Exon C
Exon B
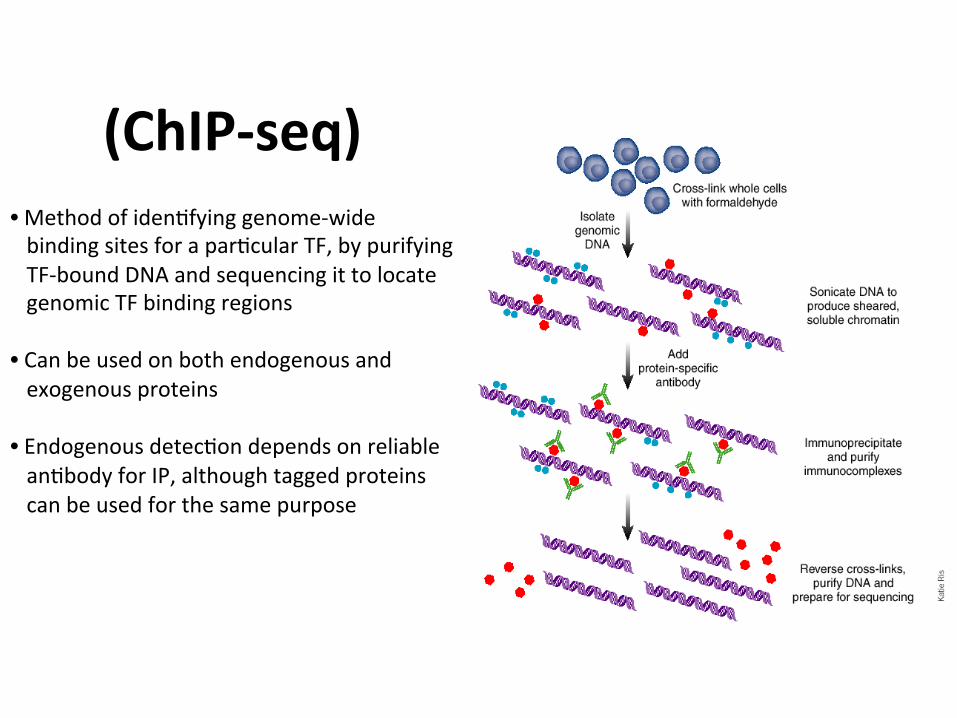
(ChIP-seq)• Methodofiden1fyinggenome-widebindingsitesforapar1cularTF,bypurifyingTF-boundDNAandsequencingittolocategenomicTFbindingregions• Canbeusedonbothendogenousandexogenousproteins• Endogenousdetec1ondependsonreliablean1bodyforIP,althoughtaggedproteinscanbeusedforthesamepurpose

Contents of the human genome [CC-BY-SA-3.0] Steve Cook
SequencingandGenomeSize
S. cerevisiae = 12 million bp C. elegans = 100 million bp Drosophila m. = 130 million bp D. rerio = 1.4 billion bp M. musculus = 2.1 billion bp H. sapiens = 3 billion bp

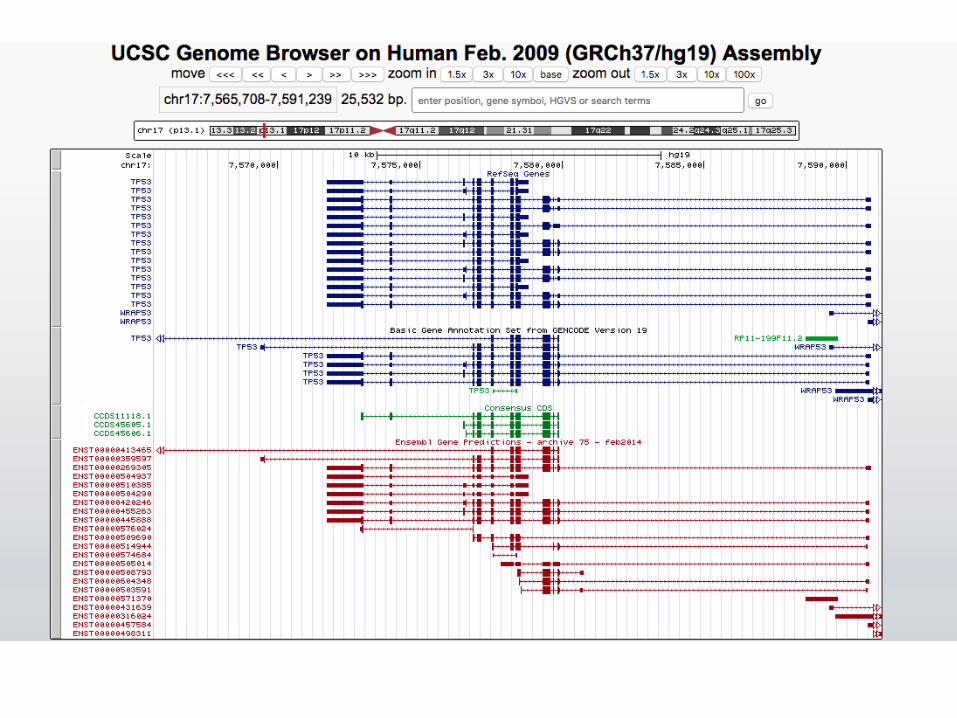
GenomeandGeneVersions
• Hg19/GRCh37orGRCh38• Refseq,Genbank,Ensembl…

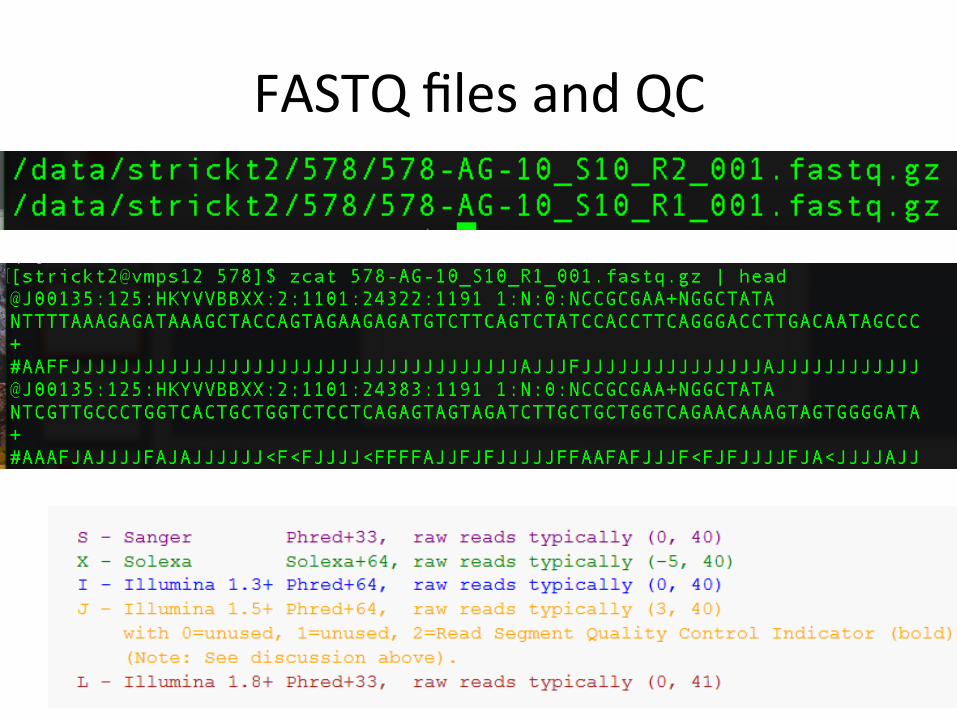
FASTQfilesandQC

Pre-processingSteps
• 1)QCrawdatawithfastqc• 2)Trimtoremoveadaptors/lowqualitysequencing
• 3)QCclippeddatawithfastqc• 4)Summarizeoutputwithmul1qc

FASTQandQC
• FASTQC• /home/strickt2/scripts/qc/fastqc.pl
• GotoFASTQCandMul1QCsummaries

RNAseqExpressionAnalysis
• Splice-awarealignment–bamfile– Tophat2,STAR,RSEM
• PicardandRSeQCalignmentQC• SummarizeQC• Assignreadstotranscripts,producecountfiles
– HtSeq,featurecounts,cufflinks• Collectintomatrix• Differen1alExpressionwithDESeq2,limma/voom,EdgeR,others,mostlyinR

Splice-awarealignment
• STAR--2step– /home/strickt2/scripts/rnaseq/star.step1.pl
• BamFile– /home/strickt2/SEQanalysis/samtools/bin/samtoolsview-H/data/strickt2/578/clip/578-AG-49.clip/Aligned.sortedByCoord.out.bam
• SummarizedmetricQC• Featurecounts
– /data/strickt2/578/clip/new_count/final.transcript.table.txt

Genotyping--Germline
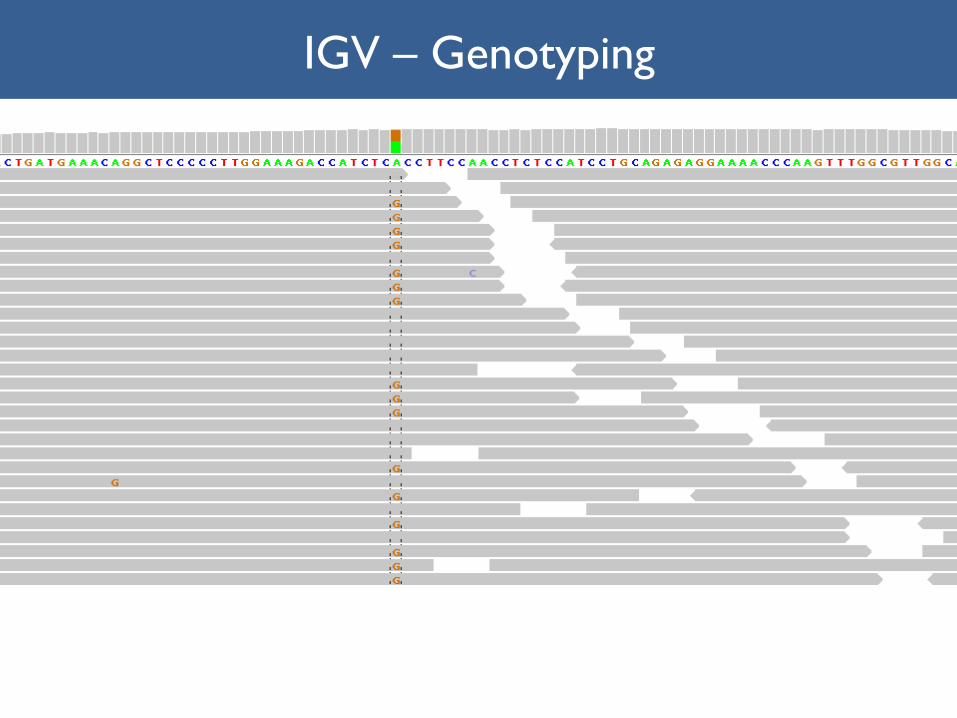
IGV – Genotyping

AddReadGroups
• ReadGroups=setofreadsgeneratedfromasinglesequencingrun
• ID=flowcellID+Lane• PU=plahormunit=flowcellID+Lane+SampleID
• PL=Illumina,IonTorrent,etc.• LB=LibraryID

/home/strickt2/scripts/rnaseq_genotyping/final.stricker.merge.rg.mdup.pl
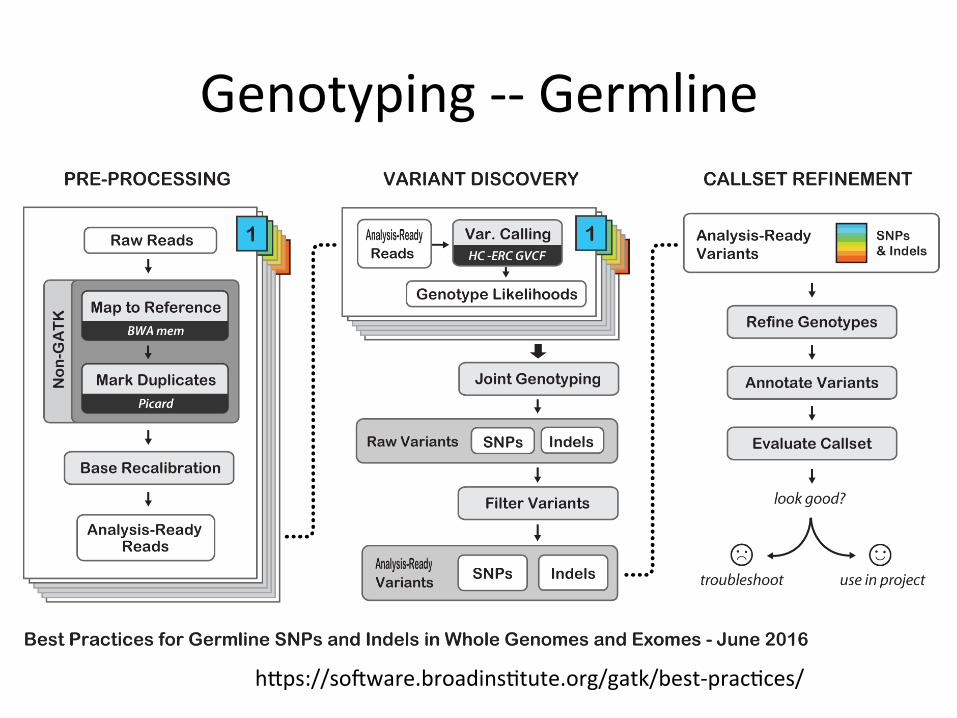
Genotyping--Germline
hips://sokware.broadins1tute.org/gatk/best-prac1ces/

HaplotypeCaller/JointCaller
hips://sokware.broadins1tute.org/gatk/best-prac1ces/

VCF–variantcallfile• /data/strickt2/3102/3102-CLA-67.3102.split.vcf

FilterGenotypes• Recalibrate(VQSR)orhard-filter
– Recalibra1onisrecommendedforgermlinehumanvariantsfor30ormoreexomes,ormodelorganismsforwhichtheappropriatetrainingsetsexist
• Recalibra1onhastoberuntwice–onceforSNPsandonceforpolymorphisms
• Trainamodelofvariantsta1s1cs(QD(qualityscoreoverdepth),MQ(RMSofmappingquality),SB(strandbias),etc.)forknownvariants
• Usethatmodeltodeterminetheprobabilitythatothervariantsaretrue.
hips://sokware.broadins1tute.org/gatk/best-prac1ces/

VQSR–Recalibrate
hips://sokware.broadins1tute.org/gatk/best-prac1ces/
SNPs
Indels

VQSR--Recalibrate
hips://sokware.broadins1tute.org/gatk/best-prac1ces/
SNP Indel

HardFilteringhips://sokware.broadins1tute.org/gatk/best-prac1ces/

HardFiltering• QD=QualByDepth=Variantconfidence/Depth• FS=FisherStrand=Phred-scaledp-valuefromFETstrandbias• MQ=RMSMappingQuality• MQRankSum=MappingQualityRankSumTest=CompareMQforreadswithrefvs.
readswithaltviaMann-Whitney• ReadPosRankSum=ReadPosRankSumTest=Mann_Whitneyfordistanceofvariant
fromendofread.Variantsonlyseenatendofreadslikelyerrors.• SOR=StrandOddsRa1o=strandbias.
• READ:• hips://sokware.broadins1tute.org/gatk/documenta1on/ar1cle.php?id=3225
• hips://sokware.broadins1tute.org/gatk/documenta1on/ar1cle.php?id=6925and
hips://sokware.broadins1tute.org/gatk/best-prac1ces/

Annota1ons
• Annovar,VEP,others• Availabledataresources
– Humanvaria1on(dbSNP,ExAC,gnomAD,EVS,etc.)
– Clinicalsignificance– ClinSig,CancerHotspots,others
– Measuresofconserva1onordeleteriousness• GERP,CADD,SIFT,Polyphen,Provean,etc
• Annota1ontablesandRadarplots
Somatic Genotyping –Tumor-Normal Contamination

Tumor-Normal Contamination
Framptonet.al.hips://www.nature.com/ar1cles/nbt.2696.

Soma1cMuta1onCalling• Tumor-NormalPairs

OtherAnalyses
• ChIPseq/ATAC-seq/etc.• Methyla1on/Bisulfitesequencing• FusionIden1fica1on• Noveltranscript/isoformiden1fica1on• CopyNumberAltera1ons
– BothGermlineandSoma1c
• Allele-Specificexpression

FusionIden1fica1onCCL14:PCGF2USP22:PTPRDNR2C2:RFTN1ESR1:AKAP12ESR1:C6orf211PAK1:RP11-807H22.7ESR1:C6orf97THSD4:LRRC49FOXK2:ZBTB40CES7:FANCD2ESR1:C6orf97PBRM1:NKIRAS1PTPRN2:CYP3A5SPOP:SWAP70DNAJA3:PTPN2NR3C2:FSTL5OSBPL2:CDH4ROR1:LRP8SSH1:FOXN4NRIP1:AF127936.7NCOR1:WDR16CCL4L2:CCL4ERLIN1:PTENFAM188A:PIP4K2APBX1:RP11-705O24.1STK32B:CLEC16AMLL3:ANKRD36
A B
Gene A GeneB Exon A Exon B Exon A Exon C
Exon B

TheFuture
• SingleCellRNAseq– ddSEQ,10XChromium(JeffRathmell),InDrop(LauLab),andSmartSeq2(Mallal)
• Synthe1cLongReads– haplotypingandcopynumber/structuralvariantanalysis
• Spark-enabledGATK4• Costeffec1veexomeandwholegenomesequencing=datadeluge