network querying algorithms
DESCRIPTION
Network Querying Algorithms. Roded Sharan Tel-Aviv University. Protein Interactions. Crucial to cell function. Measured by high-throughput technologies: yeast two-hybrid co-immunoprecipitation Systematic data available for several species. Network Querying Problem. - PowerPoint PPT PresentationTRANSCRIPT

Network Querying Algorithms
Roded Sharan
Tel-Aviv University
Protein Interactions
• Crucial to cell function.• Measured by high-throughput technologies:
– yeast two-hybrid– co-immunoprecipitation
• Systematic data available for several species.
0
1
2
3
4
5
6
7
8
9
19992000200120022003200420052006
Year
#sp
ecie
s
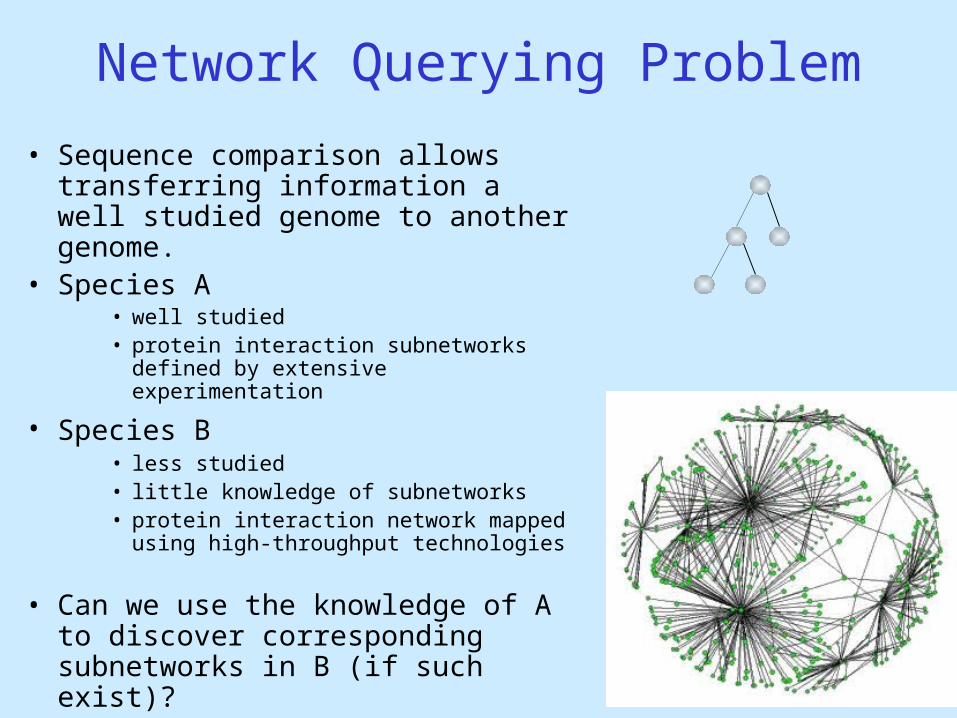
Network Querying Problem
• Sequence comparison allows transferring information a well studied genome to another genome.
• Species A• well studied• protein interaction subnetworks defined by
extensive experimentation
• Species B • less studied• little knowledge of subnetworks• protein interaction network mapped using
high-throughput technologies
• Can we use the knowledge of A to discover corresponding subnetworks in B (if such exist)?
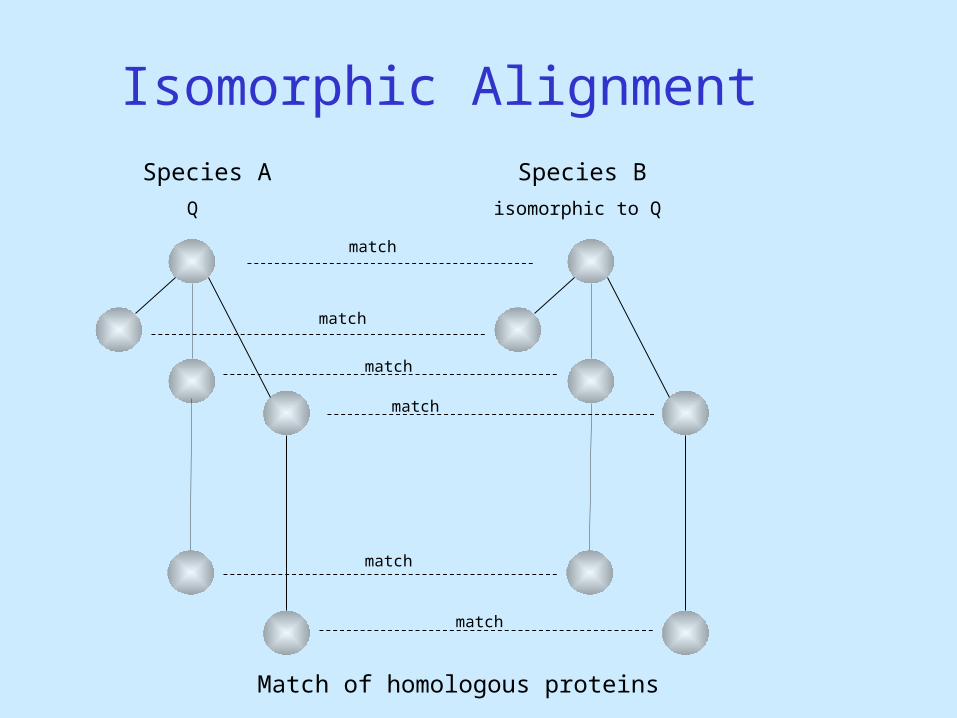
Isomorphic Alignment
isomorphic to Q
match
match
match
match
match
match
Match of homologous proteins
Species BSpecies A
Q
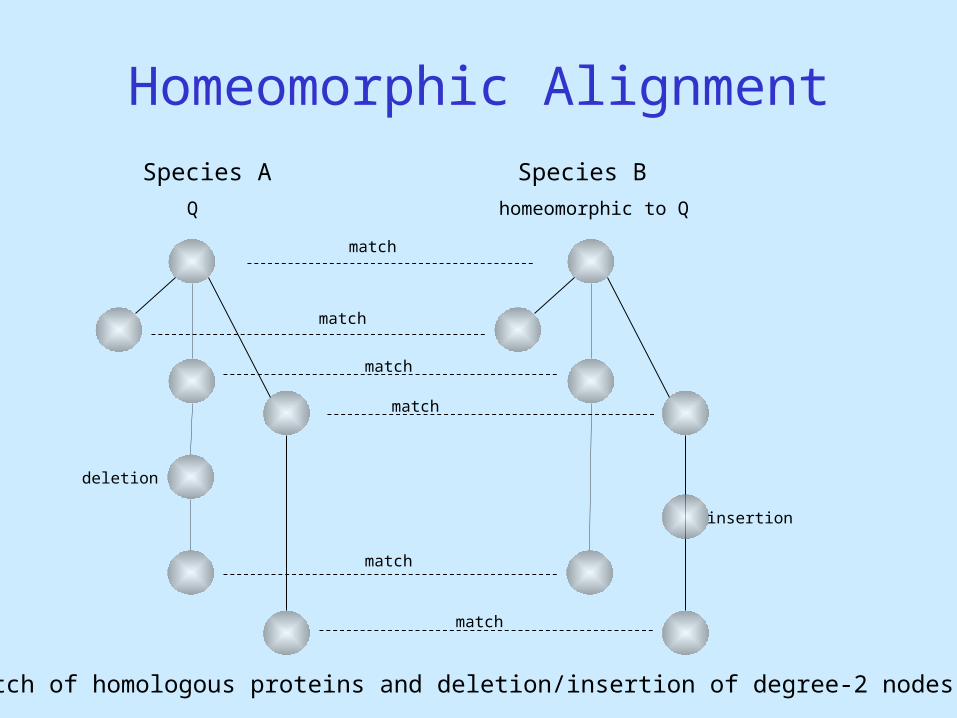
Homeomorphic Alignment
homeomorphic to Q
insertion
match
match
match
match
match
match
Match of homologous proteins and deletion/insertion of degree-2 nodes
deletion
Species BSpecies A
Q
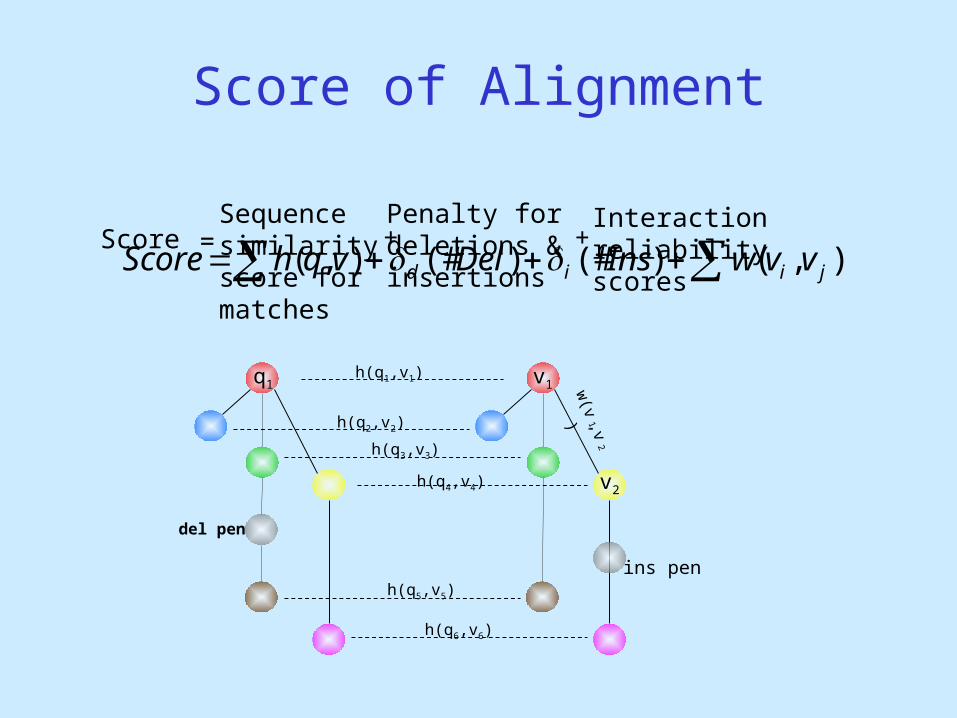
Score of Alignment
ScoreSequence similarity score for matches
Penalty for deletions &insertions
Interaction reliability scores
+ +=
h(q1,v1)q1 v1
h(q2,v2)
h(q3,v3)
h(q4,v4)
h(q6,v6)
h(q5,v5)
del pen
ins pen
v2
w(v
1,v2)
),()(#)(#),( jiid vvwInsDelvqhScore
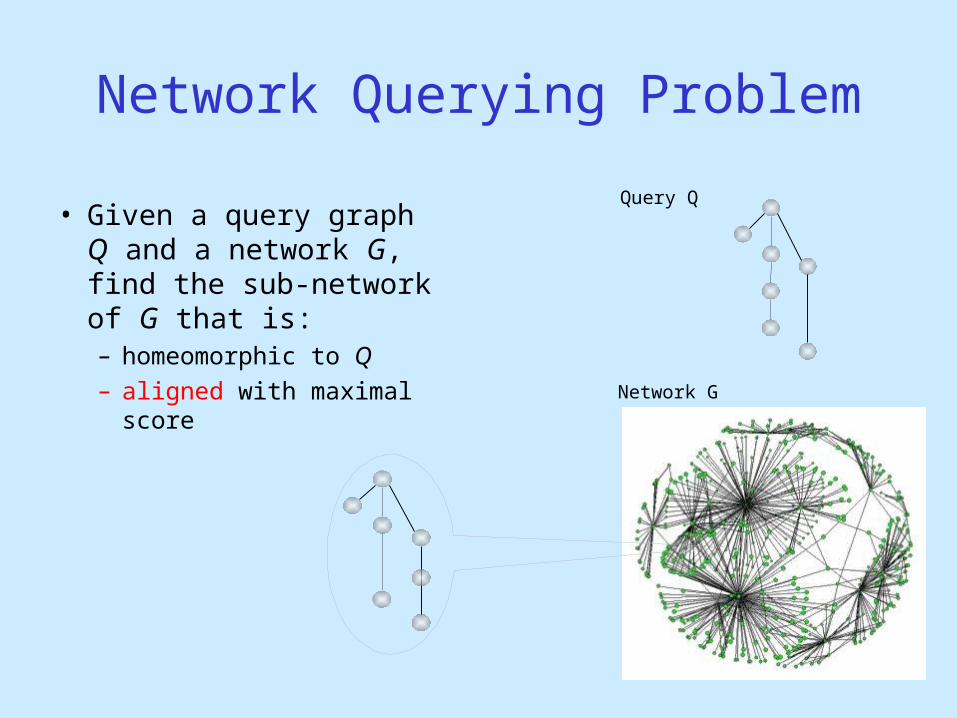
Network Querying Problem
• Given a query graph Q and a network G, find the sub-network of G that is:– homeomorphic to Q – aligned with maximal
score
Query Q
Network G

Complexity
• Network querying problem is NPC by reduction from subgraph isomorphism
• Naïve algorithm has O(nk) complexity– n = size of the PPI network, k = size of the query
– Intractable for realistic values of n and k
– n ~5000, k~10
• Reduction in complexity can be achieved by:– Constraining the network [Pinter et al., Bioinformatics’05]
– Constraining the query (fixed parameter algs.)
– Allowing vertex repetitions

Path Querying
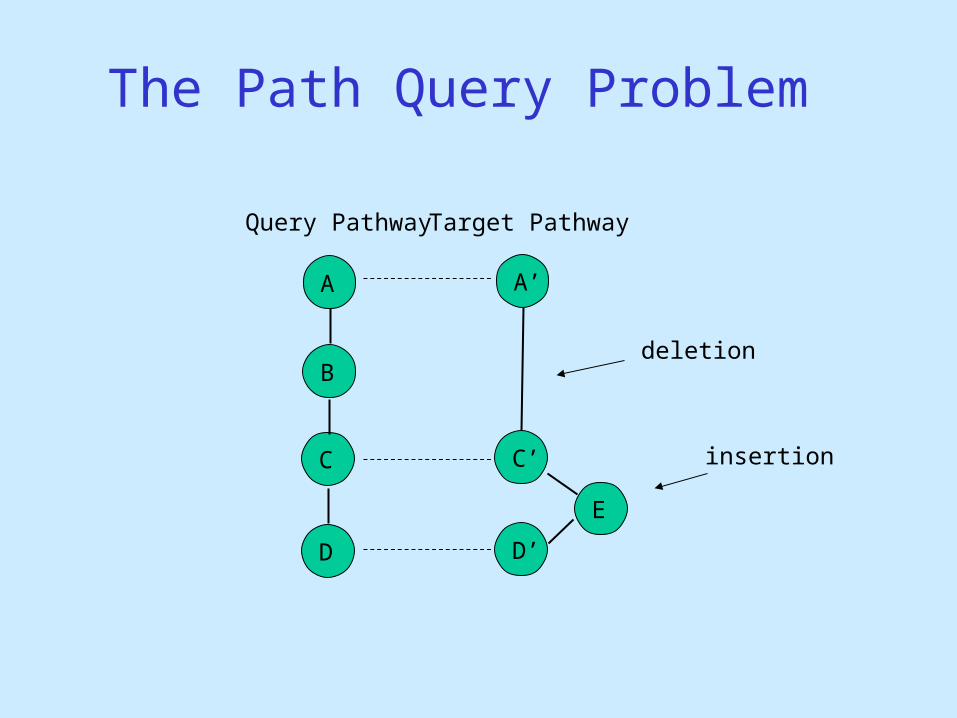
The Path Query Problem
A
B
C
D
A’
C’
D’
E
Query Pathway Target Pathway
deletion
insertion

Pe random
Pv random
q
eq
p
vpPS
log
log
p(v) – sequence similarity
q(e) – interaction reliability
PathBLAST
Kelley et al., PNAS’03

Alignment-Based Approach
Pros:• Conceptually simple.• Extensible to general queries (using any network
alignment program).
Cons:• No general treatment of indels.• Protein Repetitions.

DP-Based Approach
• Use dynamic programming (a la sequence alignment):
W(i,j) is the maximal score of a partial alignment of query
nodes {1…i} that ends at vertex j of the network.
d
imj
mj
jiW
EjmwmiW
EjmwjihmiW
jiW
),1(
),(,),(
),(,),(),1(
max),(
• But this may introduce protein repetitions along the path.
match
insertion
deletion
Shlomi et al., BMC Bioinformatics ’06; Yang & Sze, JCB’07

Color Coding [AYZ’95]Problem: Given a graph G=(V,E) and a parameter k, find a
simple path of length k in G.
Algorithm: Randomly color vertices with k colors, and find a
colorful path (distinct colors).
Complexity: – Colorful path found by DP in O(km2k).– Prob. of success (path is colorful): k!/kk e-k.– Overall: m2O(k).
1)})({,(max),(
2];,1[:
)}({)(,),(:
],1[
vcSuPSvP
SkVc
vcSucEvuu
k
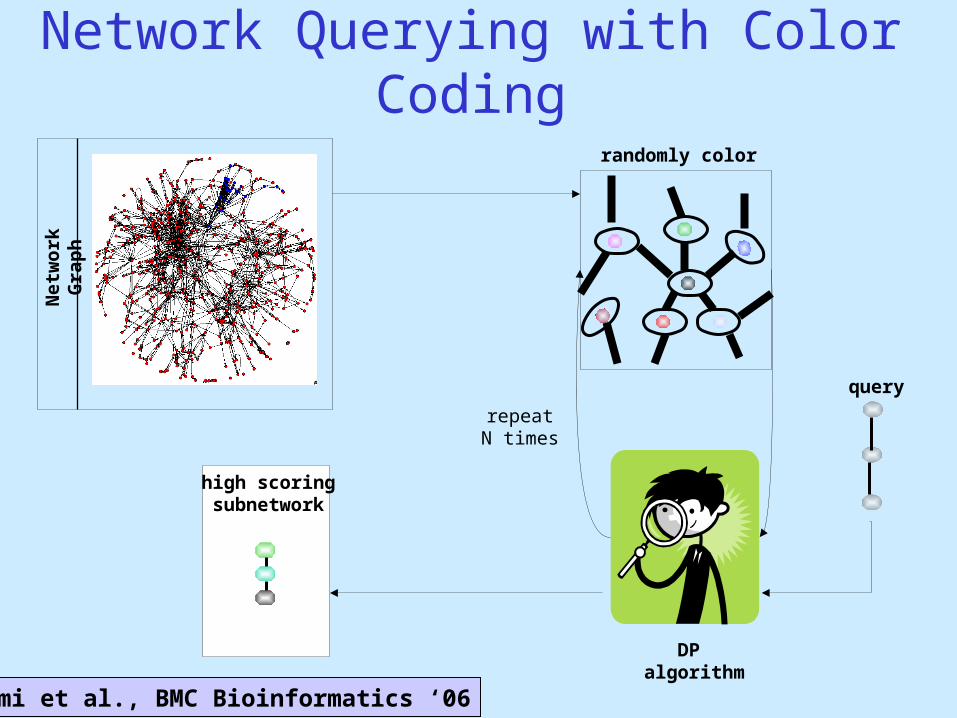
Network Querying with Color Coding
Net
wo
rk
Gra
ph
high scoringsubnetwork
query
randomly color
DP algorithm
repeatN times
Shlomi et al., BMC Bioinformatics ‘06

Yeast & Fly PPI Networks
Number of pathways
Functional enrichment
Expressioncoherency(p-value)
Yeast27180%<1e-300
Fly13239%2.7e-3
S. cerevisiae• 4,726 proteins• 15,166 interactions
D. melanogaster• 7,028 proteins• 22,837 interactions

Yeast-Fly Queries
• Applied QPath to 271 yeast queries spanning the yeast network.
• 63% of queries were matched, most requiring protein indels.

The Scoring Module
• Functional enrichment of a matched path correlates with:– Its interaction reliabilities– Its sequence similarities– Its numbers of protein
insertions and deletions (anti-correlation).
Goal: score matched pathways by their prob. to be functionally enriched.Method: logistic regression on path attributes – PPI reliabilities, sequence similarities, #insertions, #deletions.

Best Matches
• 171 best matches identified.• 51% were functionally enriched.• Best matches were significantly more functionally enriched
and expression coherent than arbitrary pathways (p<1e-4).
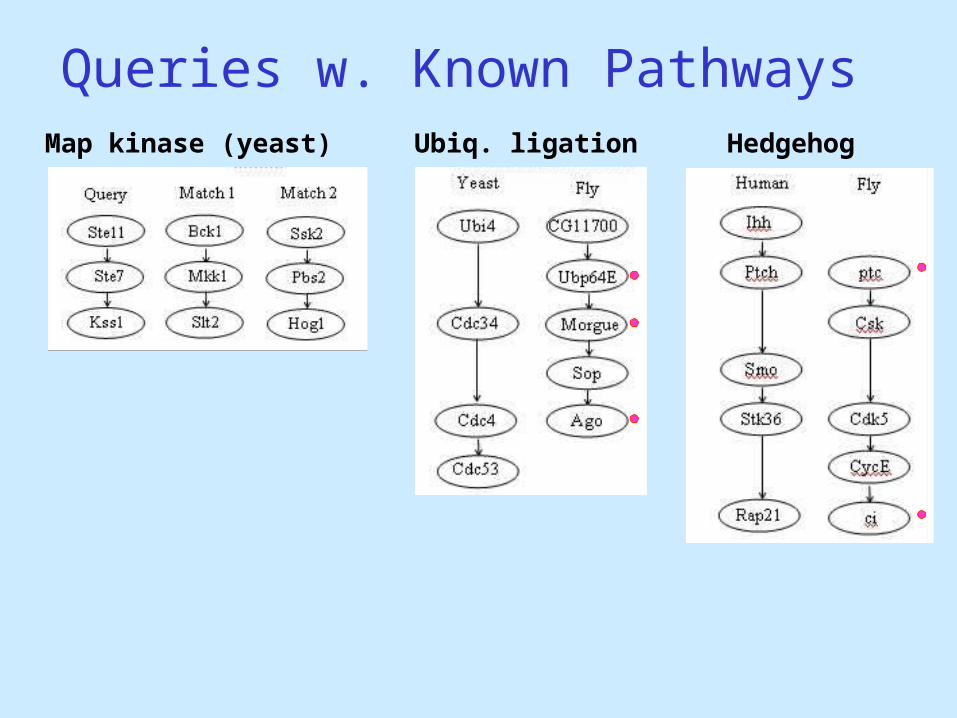
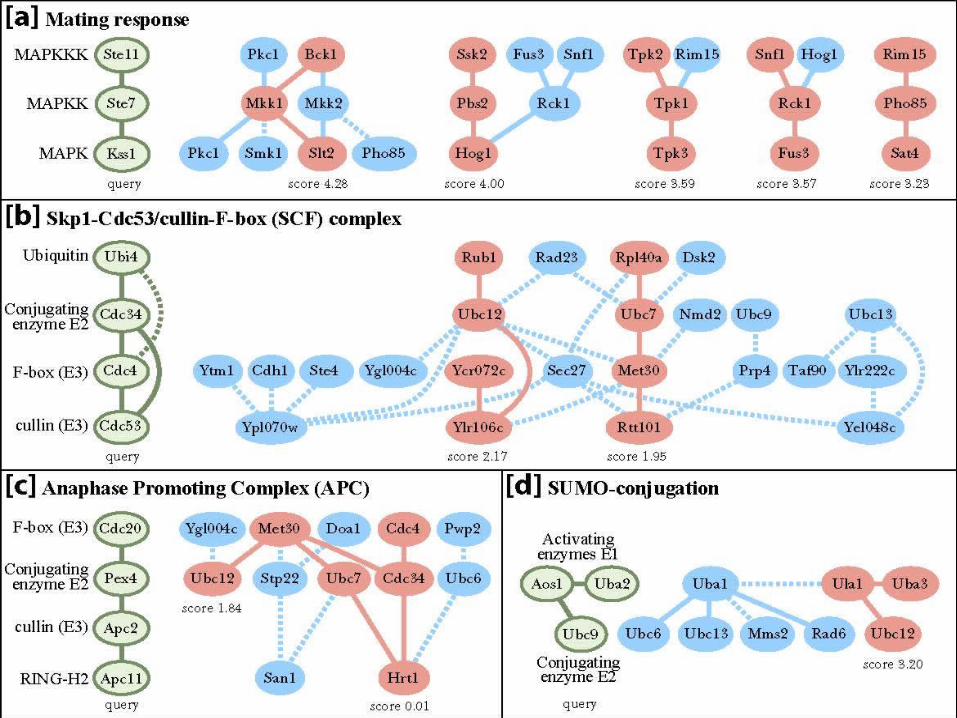
Queries w. Known PathwaysHedgehogUbiq. ligationMap kinase (yeast)

Function Conservation
• 69 best matches had an enriched function in both species.
• 64% preserved their function; significantly more than the random expectation (31%).
• In comparison, sequence best matches preserve their function in only 40% of the cases!
Pathway homology can be used to predict function!

Fly Conserved Pathway Map
• Predicted annotations were significantly prevalent.• Map exhibits modularity (cc=0.26).

Querying for Trees & General Graphs

Query
QNet: Tree QueriesNetwork
Query has k nodes.
Dost et al., RECOMB’07
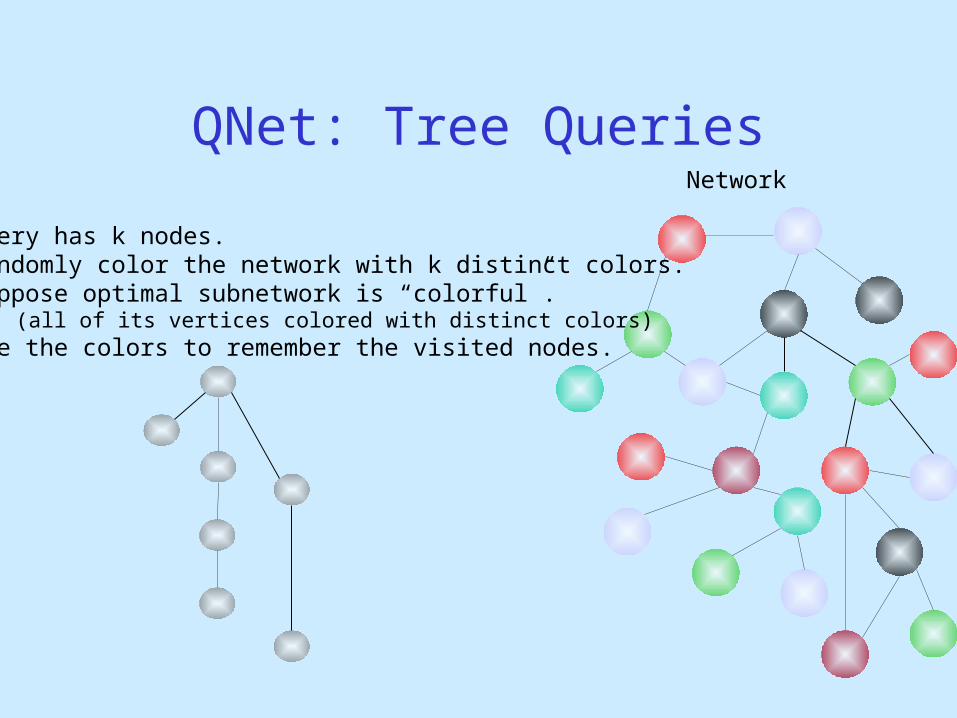
Network
Query has k nodes.Randomly color the network with k distinct colors.Suppose optimal subnetwork is “colorful”.
(all of its vertices colored with distinct colors)Use the colors to remember the visited nodes.
QNet: Tree Queries
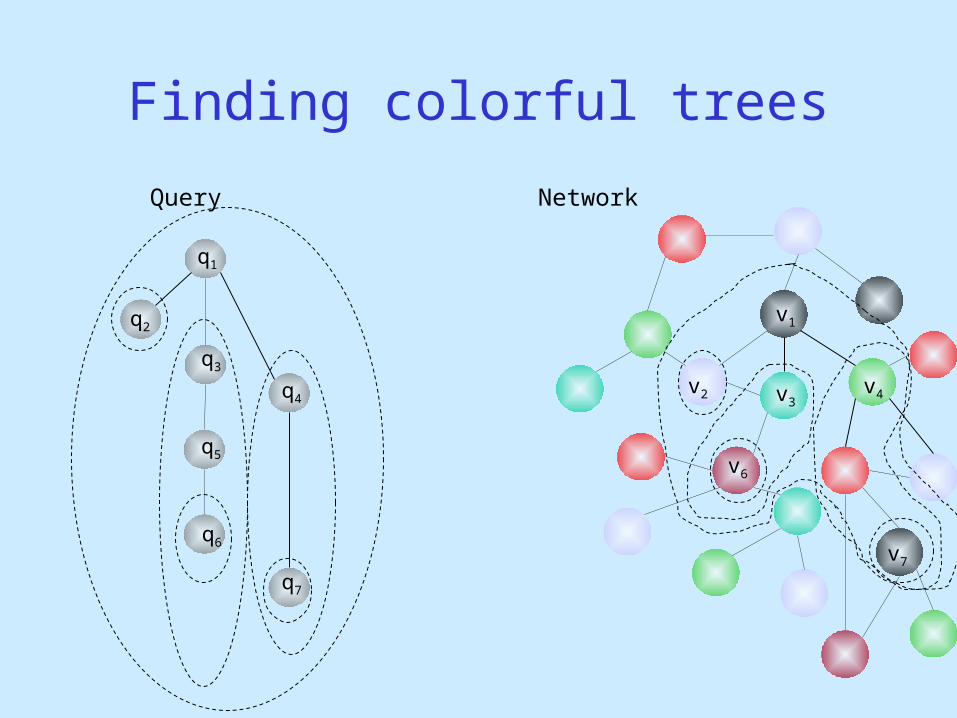
Query Network
Finding colorful trees
q1
q2
q3
q4
q5
q6
q7
v1
v2 v3
v6
v7
v4

Querying General Graphs
• We have extended the algorithm also for general graphs.
• Idea:– Map the original graph into a tree, i.e. tree
decomposition.
– Solve the querying problem on this tree using DP.
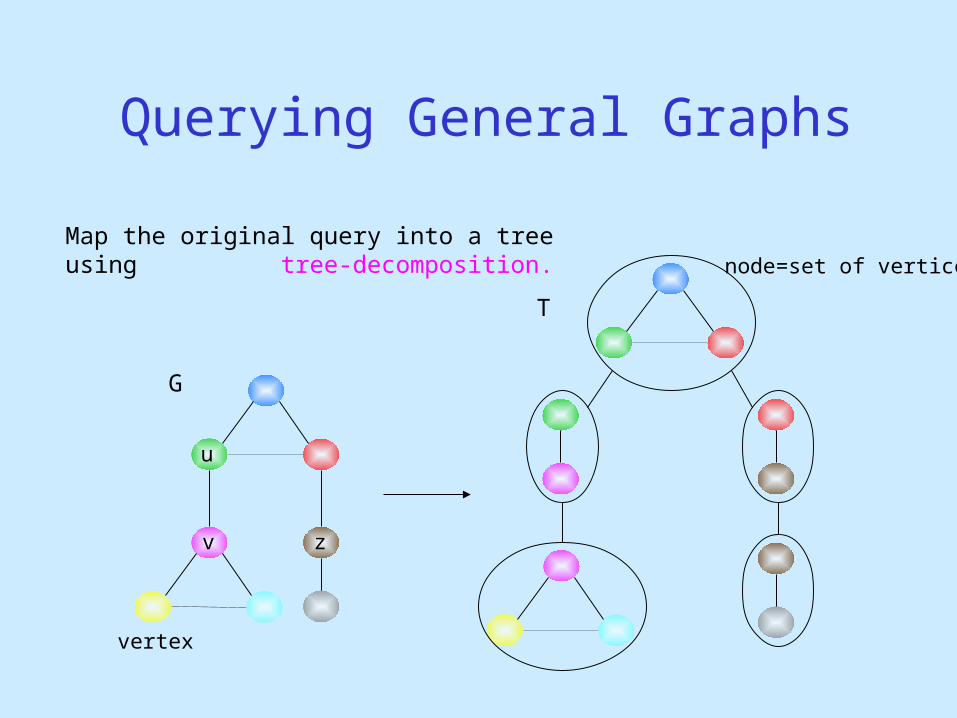
Querying General Graphs
Map the original query into a tree using tree-decomposition.
G
T
vertex
node=set of vertices
u
v z
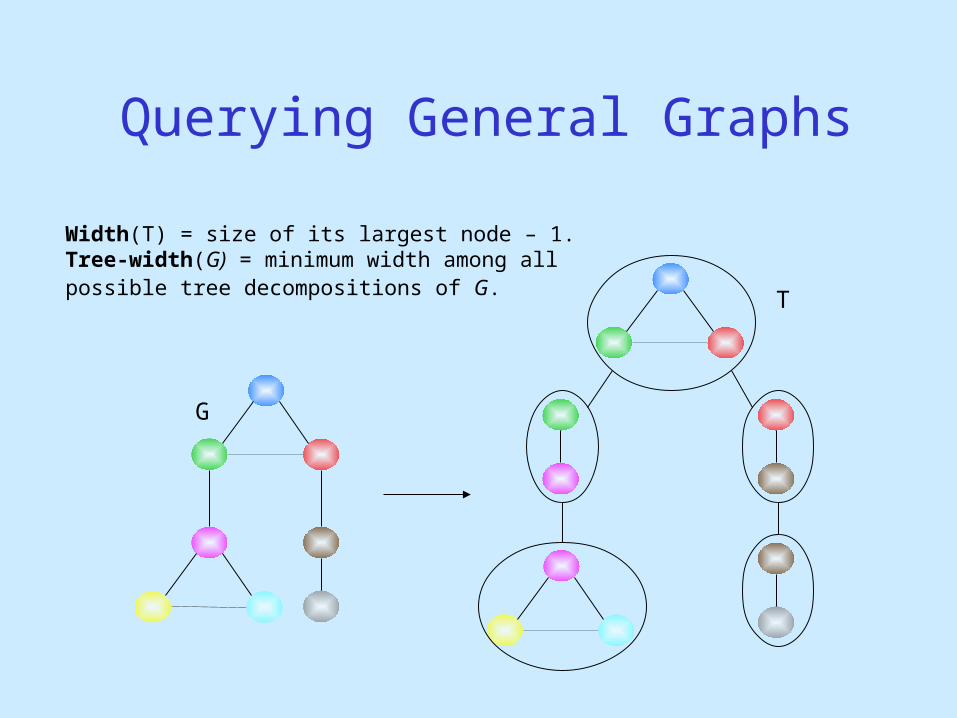
Querying General Graphs
Width(T) = size of its largest node – 1.Tree-width(G) = minimum width among all possible tree decompositions of G.
G
T
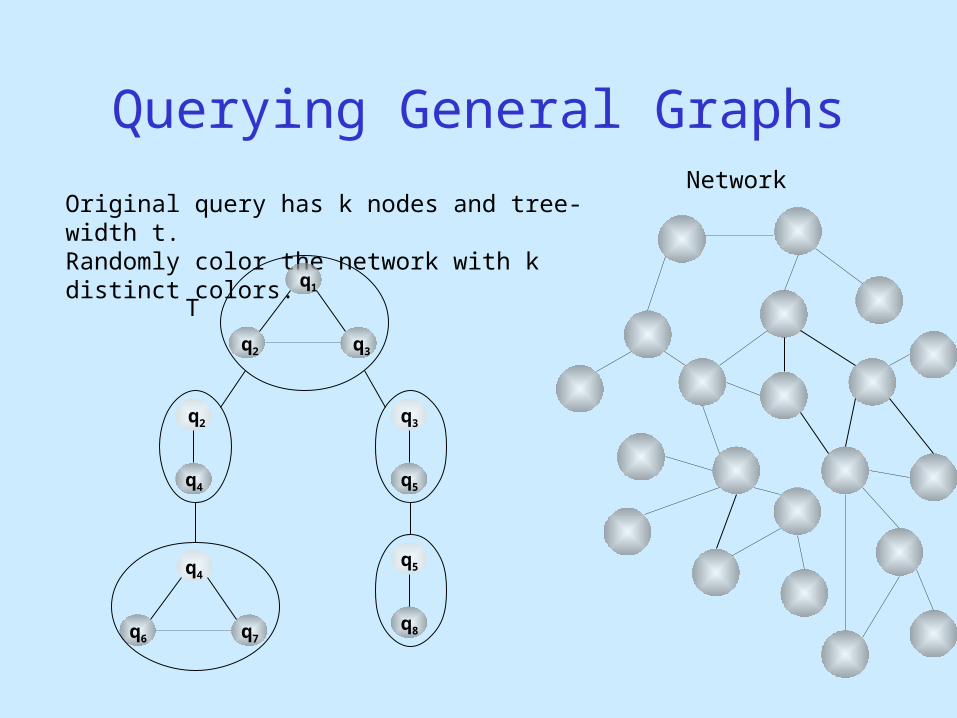
Network
Querying General Graphs
Original query has k nodes and tree-width t.Randomly color the network with k distinct colors.
q1
q2
q2
q3
q3
q4
q4q5
q5
q6 q7q8
T
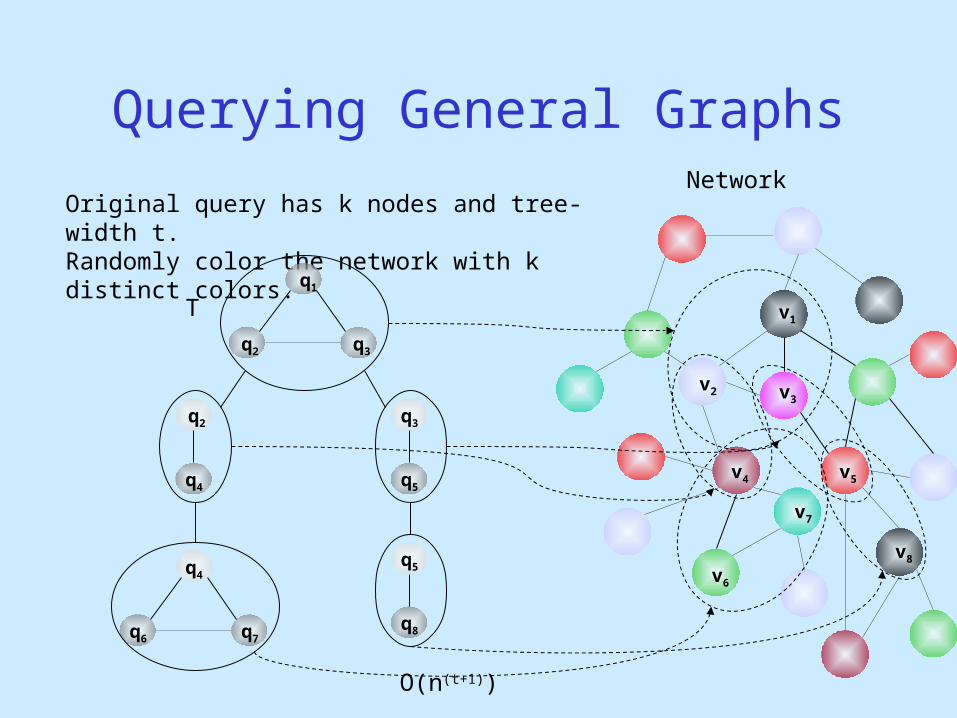
Network
Querying General Graphs
Original query has k nodes and tree-width t.Randomly color the network with k distinct colors.
q1
q2
q2
q3
q3
q4
q4q5
q5
q6 q7q8
v2 v3
v7
v6
v5
v8
v1
v4
O(n(t+1))
T

Running time
• n=size of network, k=size of query.
• Tree queries:– m2O(k).
• Tractable for realistic values of m and k.
• E.g.: n ~5000, k=9 =< 11 seconds
• Bounded-tree-width graphs:– t : tree-width– n(t+1)2O(k)
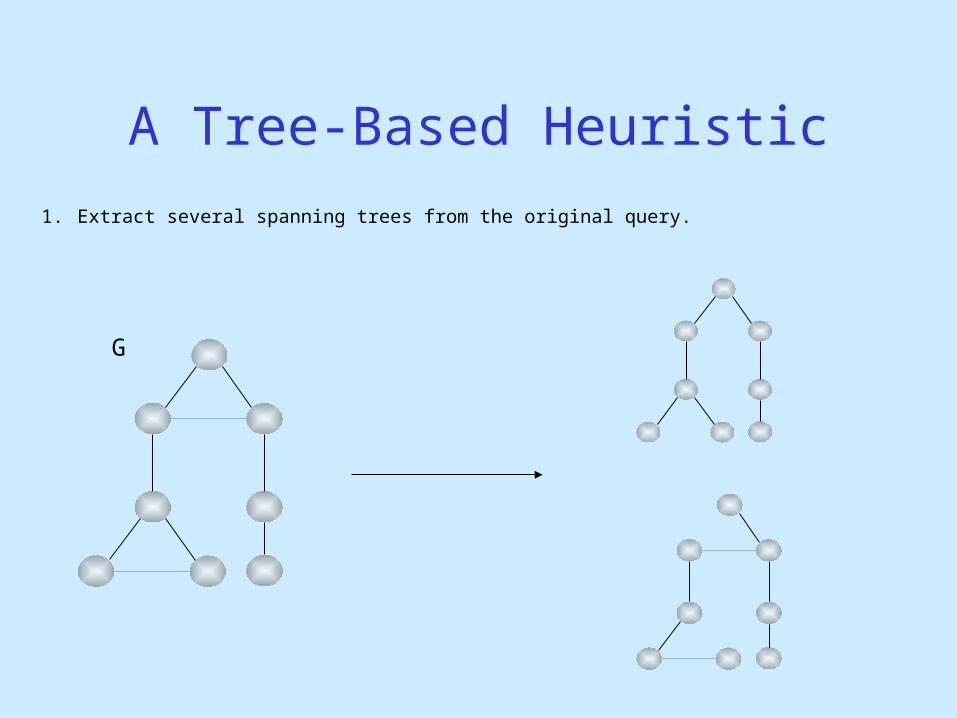
A Tree-Based Heuristic
G
1. Extract several spanning trees from the original query.
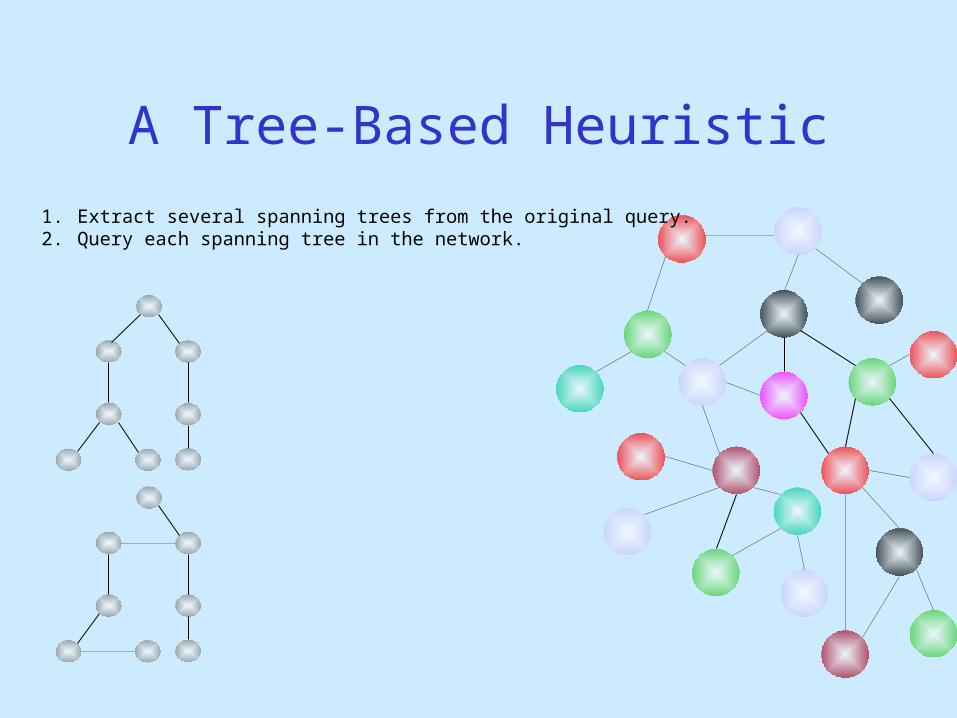
1. Extract several spanning trees from the original query.2. Query each spanning tree in the network.
A Tree-Based Heuristic
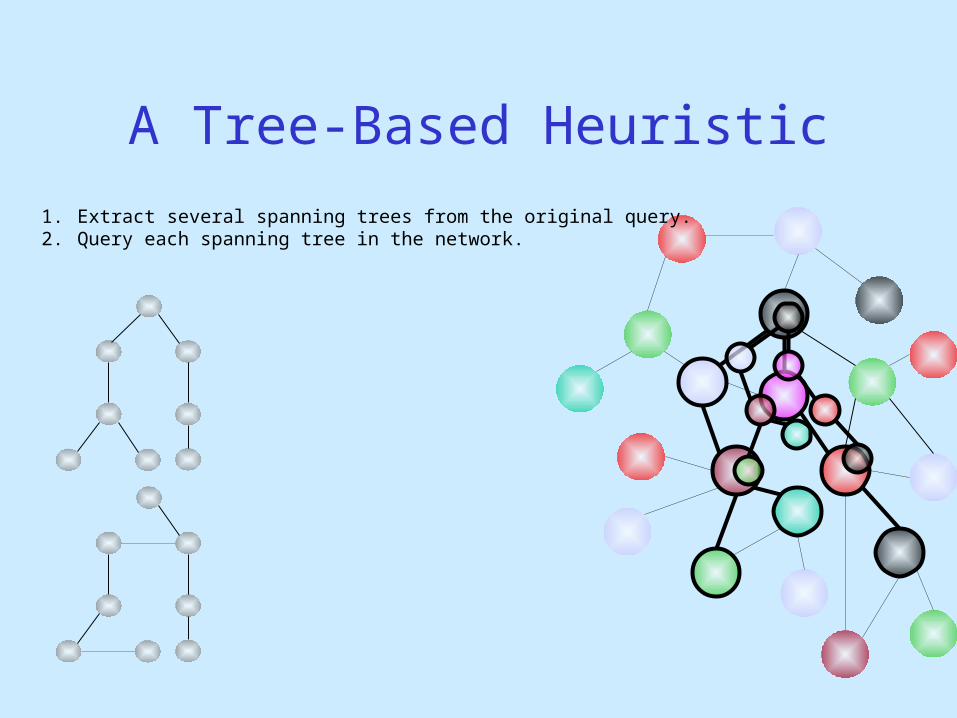
1. Extract several spanning trees from the original query.2. Query each spanning tree in the network.
A Tree-Based Heuristic
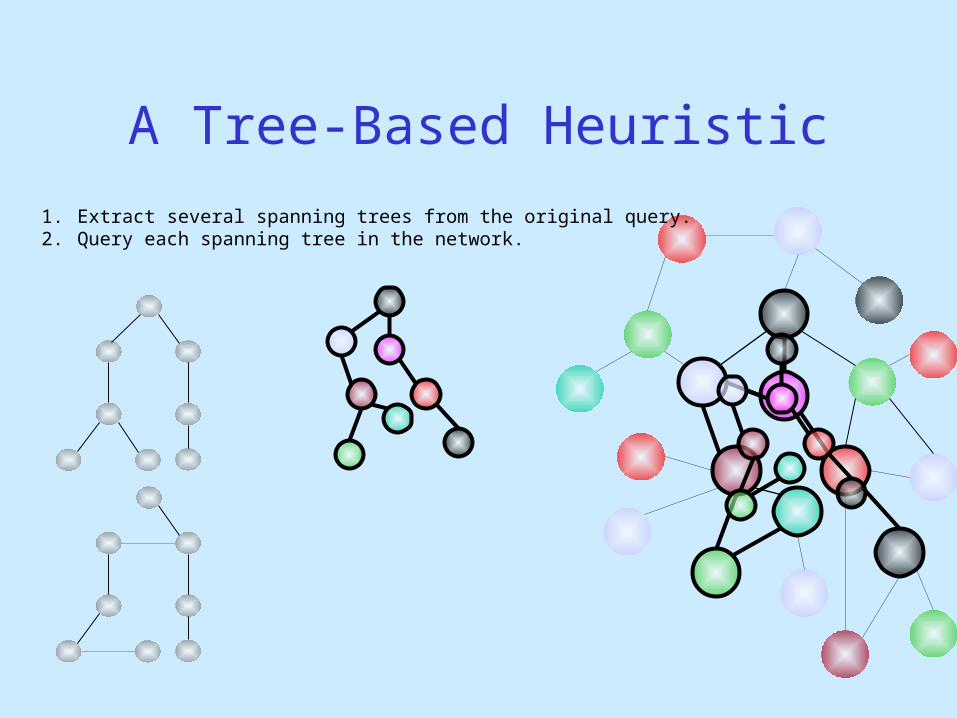
1. Extract several spanning trees from the original query.2. Query each spanning tree in the network.
A Tree-Based Heuristic
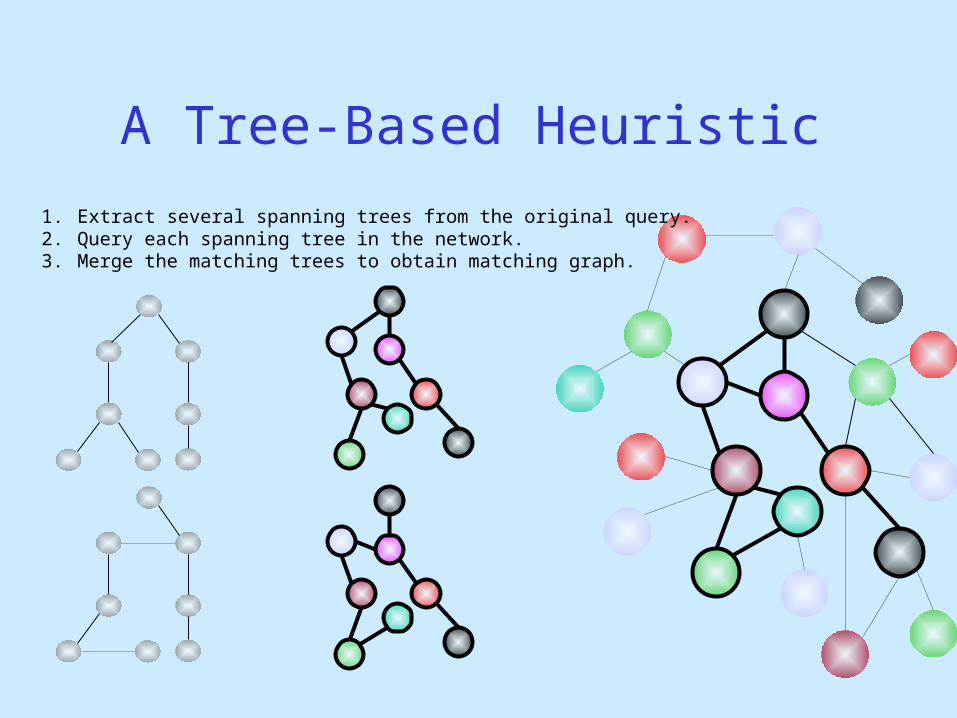
1. Extract several spanning trees from the original query.2. Query each spanning tree in the network. 3. Merge the matching trees to obtain matching graph.
A Tree-Based Heuristic

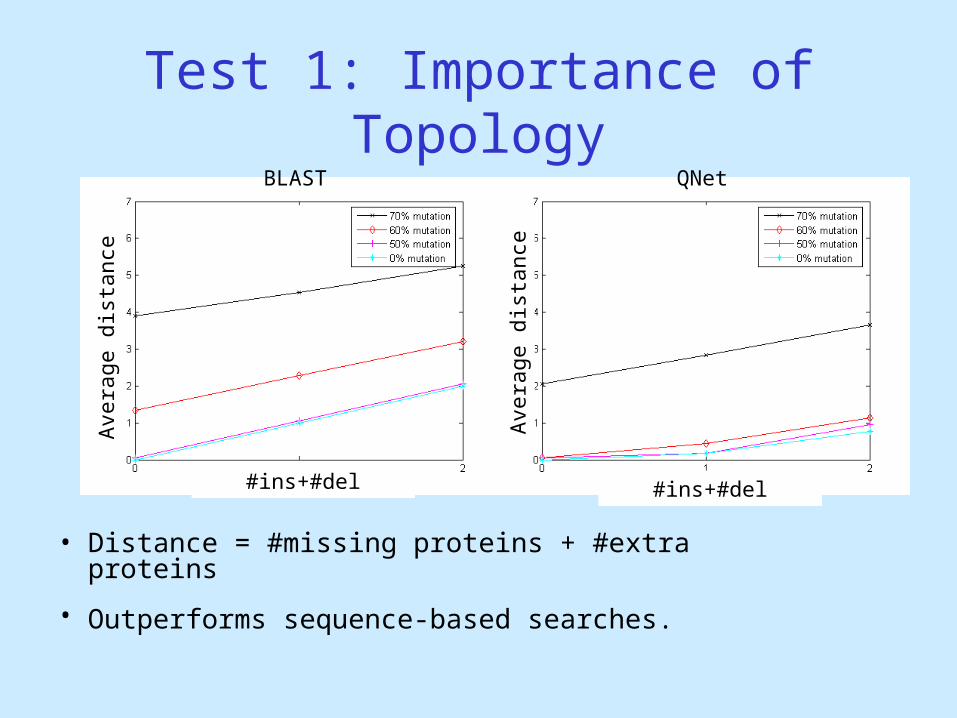
Test 1: Importance of Topology
• Motivation: Is sequence similarity enough to find corresponding sub-network?
• Queries:– Random tree queries from yeast DIP network [Salwinski,
2004]– Topology perturbed (≤2 ins-dels).
• Network:– Yeast PPI – Protein sequences mutated (50-70 percent)
• How distant is the result from the original extracted tree?
Test 1: Importance of Topology
Ave
rage
dis
tanc
e
Ave
rage
dis
tanc
e
#ins+#del #ins+#del
QNetBLAST
• Distance = #missing proteins + #extra proteins
• Outperforms sequence-based searches.

Test 2: Cross-species Comparison of MAPK
Pathways• Motivation: finding
conserved pathways.
• Query: human MAPK pathway involved in cell proliferation and differentiation.
• Network: fly PPI network– ~7K proteins – ~20K interactions
• Match: a known fly MAPK pathway involved in dorsal pattern formation.
Query from
human
Match in fly

Test 3: Cross-species Comparison of Protein
Complexes• Motivation: conserved protein complexes between
yeast and fly.
• Queries:– Hand-curated yeast MIPS complexes.– Project onto yeast DIP network.– Extract several spanning trees.

Test 3: Cross-species Comparison of Protein
Complexes• Motivation: conserved protein complexes between yeast
and fly.
• Queries:– Hand-curated yeast MIPS complexes.– Project onto yeast DIP network. – Extract several spanning trees.
• Network:– Fly DIP network
• Match– Consensus matching graph for each query complex.

Test 3: Cross-species Comparison of Protein
Complexes
Result: • ~40 of the queries resulted in a match with <1 protein.• 72% of the consensus matches were functionally enriched. • In comparison, 17% of the random trees extracted from network
are functionally enriched.
YeastCdc28p complex
Fly

Conclusions
• Fixed parameter algorithms for querying paths and trees.
• Definition of a match: homeomorphism
• General queries: – Yang & Sze JCB’07: branch-and-bound
– Alignment-based

Acknowledgments
Vineet Bafna, UCSD
Banu Dost
Nitin Gupta
Eytan Ruppin
Tomer Shlomi
Danny Segal, TAU
Trey Ideker, UCSD
Richard Karp, ICSI