multiple approaches to enantiopure spirocyclic
TRANSCRIPT
S1
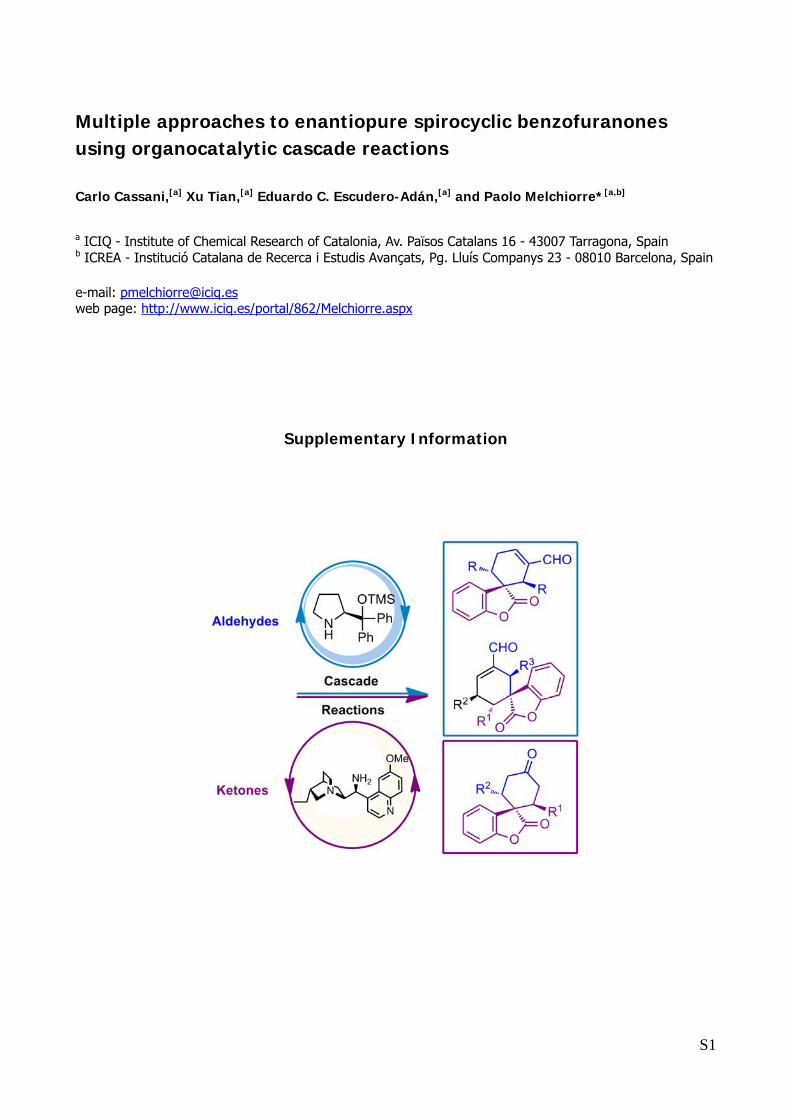
Multiple approaches to enantiopure spirocyclic benzofuranones using organocatalytic cascade reactions
Carlo Cassani,[a] Xu Tian,[a] Eduardo C. Escudero-Adán,[a] and Paolo Melchiorre*[a,b]
a ICIQ - Institute of Chemical Research of Catalonia, Av. Països Catalans 16 - 43007 Tarragona, Spain b ICREA - Institució Catalana de Recerca i Estudis Avançats, Pg. Lluís Companys 23 - 08010 Barcelona, Spain
e-mail: [email protected] web page: http://www.iciq.es/portal/862/Melchiorre.aspx
Supplementary Information
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S2
Table of Contents
General S3
Materials S3
Determination of Diastereomeric and Enantiomeric Ratios S4
Structural and absolute configuration determination S4
X-ray Data for compound 3d S4
X-ray Data for compound 6b S6
X-ray Data for compound 8b S7
Influence of the compound 4 geometry on the stereochemical outcome
of the cascade reactions S9
Experimental procedures S10
General Procedure for the Organocatalytic Triple Cascade of Aldehydes proceeding via an
Iminium-Iminium-Enamine Sequence (Table 1) S10
General Procedure for the Organocatalytic Triple Cascade proceeding via enamine – iminium – enamine
activation of aldehydes (Table 2) S14
General Procedure for the Organocatalytic Tandem double Michael additions
(enamine – iminium activation sequence) (Table 3) S17
NMR spectra S21
HPLC Traces S43
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S3
General Methods. The 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively. The
chemical shifts (δ) for 1H and 13C are given in ppm relative to residual signals of the solvents (CHCl3 @ 7.26
ppm 1H NMR, 77.0 ppm 13C NMR). Coupling constants are given in Hz. Carbon types were determined from
DEPT 13C NMR experiments. The following abbreviations are used to indicate the multiplicity: s, singlet; d,
doublet; t, triplet; q, quartet; m, multiplet; bs, broad signal. Chromatographic purification of products was
accomplished using force-flow chromatography (FC) on silica gel (230-400 mesh) according to the method
of Still.1 For thin layer chromatography (TLC) analysis throughout this work, Merck precoated TLC plates
(silica gel 60 GF254, 0.25 mm) were used, using UV light as the visualizing agent and an acidic mixture of
ceric ammonium molybdate or basic aqueous potassium permangante (KMnO4), and heat as developing
agents. High-resolution mass spectra (HRMS) were obtained from the ICIQ High Resolution Mass
Spectrometry Unit on Waters GCT gas chromatograph coupled time-of-flight mass spectrometer (GC/MS-
TOF) with electron ionisation (EI). X-ray data were obtained from the ICIQ X-Ray Unit using a Bruker-Nonius
diffractometer equipped with an APPEX 2 4K CCD area detector. Optical rotations are reported as follows:
[α]rtD (c in g per 100 mL, solvent). All reactions were set up in the air and using undistilled solvent, without
any precautions to exclude moisture unless otherwise noted.
Materials. Commercial grade reagents and solvents were used without further purification; otherwise,
where necessary, they were purified as recommended.2 Catalysts (S) and (R)-A were purchased from Aldrich
and used as received. Chiral primary amine catalysts, 9-Amino(9-deoxy)epi-hydroquinine B and its pseudo-
enantiomer 9-Amino(9-deoxy)epi-hydroquinidine were prepared from commercially available hydroquinine
and hydroquinidine, respectively, following the literature procedure.3 α,β-unsaturated enones (E)-4-(4-chlorophenyl)but-3-en-2-one 7b and (E)-4-(thiophen-3-yl)but-3-en-2-one
7g, were prepared according to the literature procedure,4 whereas the enal (E)-3-(4-
chlorophenyl)acrylaldehyde 2d was prepared using the Wittig olefination reaction from commercially
available 4-chloro benzaldehyde and (triphenylphosphoranylidene)acetaldehyde.
All the other enals 2, aldehydes 5 and unsaturated ketones 7 were purchased from Aldrich or Alfa Aesar and
used as received. 1-benzofuran-2(3H)-one 1 is commercially available. All the Michael acceptors bearing a benzofuranone moiety (4) were prepared according to literature
procedures.5 3-benzylidenebenzofuran-2(3H)-one 4a was obtained as a 3:1 mixture of E/Z isomer; isolation
of the pure E isomer has been accomplished by standard chromatography on silica gel.
(E)-3-(4-chlorobenzylidene)benzofuran-2(3H)-one, (E)-3-(4-nitrobenzylidene)benzofuran-2(3H)-one, and
(E)-3-butylidenebenzofuran-2(3H)-one were all synthesized as a mixture E/Z isomers, and used without
further purification.
(E)-ethyl 2-(2-oxobenzofuran-3(2H)-ylidene)acetate was synthesized as a single E isomer.
1 W. C. Still, M. Kahn, A. J. Mitra, J. Org. Chem. 1978, 43, 2923. 2 W. L. F. Armarengo, D. D. Perrin, In Purification of Laboratory Chemicals, 4th ed.; Butterworth Heinemann: Oxford, 1996. 3 S. H. McCooey, S. J. Connon. Org. Lett. 2007, 9, 599‐602. 4 K. Zumbansen, A. Döhring, B. List, Adv. Syn. Cat. 2010, 352, 1135‐1138. 5 M. Msaddek, M. Rammah, K. Ciamala, J. Vebrel, B. Laude, Synthesis, 1997, 1495‐1498.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S4
Determination of Enantiomeric Purity. HPLC analysis on chiral stationary phase was performed on an
Agilent 1200-series instrumentation. Daicel Chiralpak IA or IC columns with i-PrOH/hexane as the eluent
were used. HPLC traces were compared to racemic samples prepared by performing the reactions using a
1:1 mixture of (S)- and (R)-A as the catalyst for the synthesis of compounds 3 & 6. Racemic mixtures for
compounds 8 were obtained by mixing the two antipodes obtained performing distinct reactions with
catalyst 9-Amino(9-deoxy)epi-hydroquinine B and the pseudo-enantiomer 9-Amino(9-deoxy)epi-
hydroquinidine.
Determination of Diastereomeric Ratios
The diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture, and confirmed
by HPLC analysis on chiral stationary phase columns.
Absolute and Relative Configuration Determination
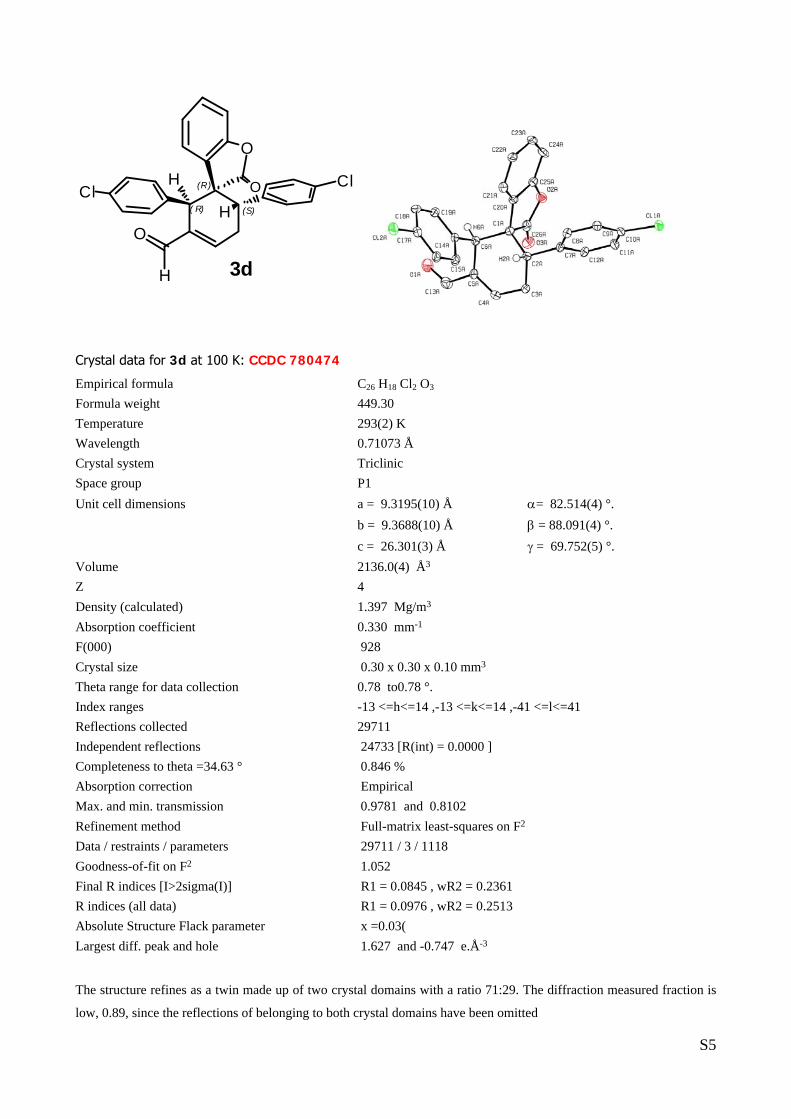
X-Ray Structure Analysis.
The absolute and relative configurations of compound 3d, 6b, and 8b (Scheme 1) were assigned by X-ray
crystallographic analysis. CCDC 780474, 781708, 781524, respectively, contain the supplementary
crystallographic data for those compounds. These data can be obtained free of charge from the Cambridge
Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
Single Crystal X-ray Diffraction Data for compound 3d (table 1, entry 5):
X-ray structure determinations: Crystals of compound 3d were obtained by slow evaporation in a mixture of
Hexane and Diethyl-ether in a 1:1 portion at room temperature. The measured crystals were stable under
atmosphere conditions; nevertheless they were prepared under inert conditions immersed in perfluoropoly-
ether as protecting oil for manipulation.
Data Collection. Measurements were made on a Bruker-Nonius diffractometer equipped with an APPEX 2 4K
CCD area detector, a FR591 rotating anode with MoKα radiation, Montel mirrors and a Kryoflex low
temperature device (T = -173 °C). Full-sphere data collection was used with ω and ϕ scans.
Programs used: Data collection Apex2 V2009 1.0 (Bruker-Nonius 2008), data reduction Saint + Version
7.60A (Bruker AXS 2008) and absorption correctionTWINABS.6
Structure Solution. SIR2007 program was used.7
Structure Refinement. SHELXTL-97-UNIX VERSION.
6 TWINABS Version 2008/4 Bruker AXS Blessing, Acta Cryst. (1995) A51 33‐38 7 R.Caliandro, B. Carrozzini, G.L.Cascarano, L. De Caro, C. Giacovazzo and D. Siliqi (2007) Advances in ab initio protein phasing by Patterson deconvolution techniques J. Appl. Cryst. 40,883‐890.
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S5
3d
( R) (S)
(R)
H
HCl
O
O
H
ClO
Crystal data for 3d at 100 K: CCDC 780474
Empirical formula C26 H18 Cl2 O3 Formula weight 449.30 Temperature 293(2) K Wavelength 0.71073 Å Crystal system Triclinic Space group P1 Unit cell dimensions a = 9.3195(10) Å α= 82.514(4) °. b = 9.3688(10) Å β = 88.091(4) °. c = 26.301(3) Å γ = 69.752(5) °. Volume 2136.0(4) Å3 Z 4 Density (calculated) 1.397 Mg/m3 Absorption coefficient 0.330 mm-1 F(000) 928 Crystal size 0.30 x 0.30 x 0.10 mm3 Theta range for data collection 0.78 to0.78 °. Index ranges -13 <=h<=14 ,-13 <=k<=14 ,-41 <=l<=41 Reflections collected 29711 Independent reflections 24733 [R(int) = 0.0000 ] Completeness to theta =34.63 ° 0.846 % Absorption correction Empirical Max. and min. transmission 0.9781 and 0.8102 Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 29711 / 3 / 1118 Goodness-of-fit on F2 1.052 Final R indices [I>2sigma(I)] R1 = 0.0845 , wR2 = 0.2361 R indices (all data) R1 = 0.0976 , wR2 = 0.2513 Absolute Structure Flack parameter x =0.03( Largest diff. peak and hole 1.627 and -0.747 e.Å-3
The structure refines as a twin made up of two crystal domains with a ratio 71:29. The diffraction measured fraction is
low, 0.89, since the reflections of belonging to both crystal domains have been omitted
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S6
6b
(R)
(S)(S)
(R) H
H
O HO
MeH
O
Cl
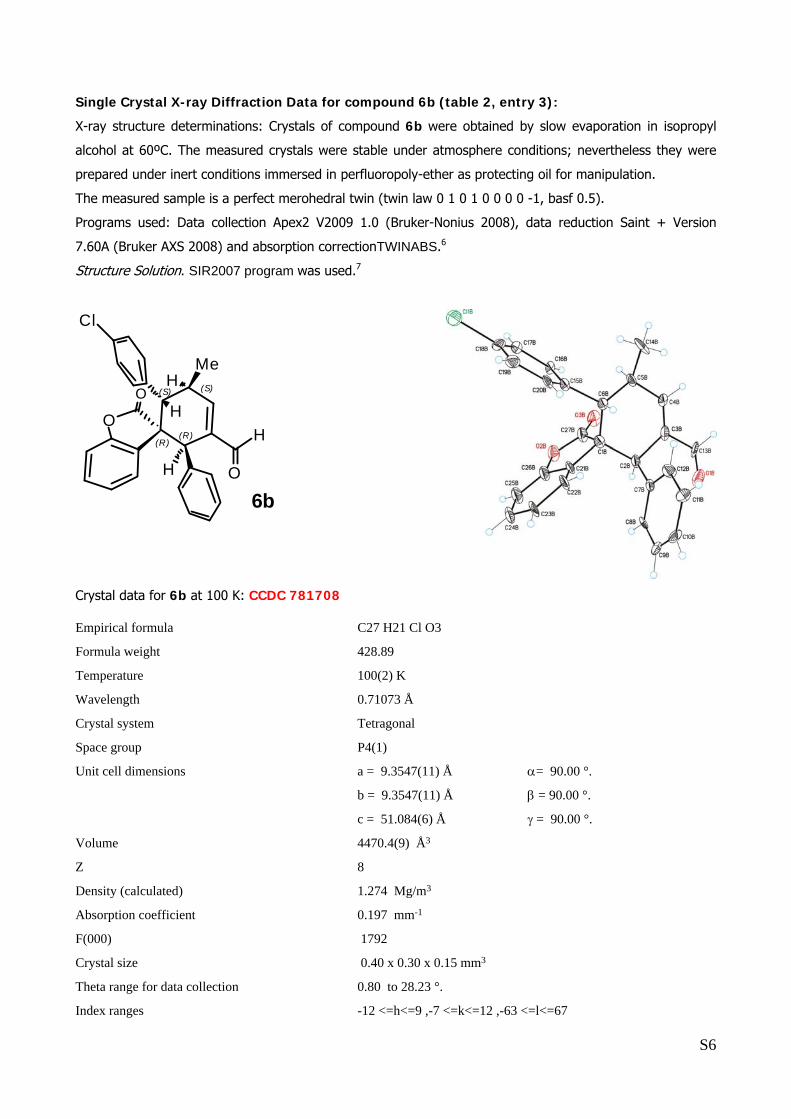
Single Crystal X-ray Diffraction Data for compound 6b (table 2, entry 3):
X-ray structure determinations: Crystals of compound 6b were obtained by slow evaporation in isopropyl
alcohol at 60ºC. The measured crystals were stable under atmosphere conditions; nevertheless they were
prepared under inert conditions immersed in perfluoropoly-ether as protecting oil for manipulation.
The measured sample is a perfect merohedral twin (twin law 0 1 0 1 0 0 0 0 -1, basf 0.5).
Programs used: Data collection Apex2 V2009 1.0 (Bruker-Nonius 2008), data reduction Saint + Version
7.60A (Bruker AXS 2008) and absorption correctionTWINABS.6
Structure Solution. SIR2007 program was used.7
Crystal data for 6b at 100 K: CCDC 781708
Empirical formula C27 H21 Cl O3
Formula weight 428.89
Temperature 100(2) K
Wavelength 0.71073 Å
Crystal system Tetragonal
Space group P4(1)
Unit cell dimensions a = 9.3547(11) Å α= 90.00 °.
b = 9.3547(11) Å β = 90.00 °.
c = 51.084(6) Å γ = 90.00 °.
Volume 4470.4(9) Å3
Z 8
Density (calculated) 1.274 Mg/m3
Absorption coefficient 0.197 mm-1
F(000) 1792
Crystal size 0.40 x 0.30 x 0.15 mm3
Theta range for data collection 0.80 to 28.23 °.
Index ranges -12 <=h<=9 ,-7 <=k<=12 ,-63 <=l<=67
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S7
(S)
(S)
(R)
H
Cl
OH
O
O
8b
Reflections collected 10668
Independent reflections 9044 [R(int) = 0.0714 ]
Completeness to theta =28.23 ° 0.989 %
Absorption correction Empirical
Max. and min. transmission 0.9711 and 0.9255
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 10668 / 631 / 562
Goodness-of-fit on F2 1.112
Final R indices [I>2sigma(I)] R1 = 0.1074 , wR2 = 0.2585
R indices (all data) R1 = 0.1222 , wR2 = 0.2669
Absolute Structure Flack parameter x =0.08(17)
Largest diff. peak and hole 0.564 and -0.795 e.Å-3 Single Crystal X-ray Diffraction Data for compound 8b (table 3, entry 3):
X-ray structure determinations: Crystals of compound 8b were obtained by slow evaporation in methanol at
room temperature. The measured crystals were stable under atmosphere conditions; nevertheless they were
prepared under inert conditions immersed in perfluoropoly-ether as protecting oil for manipulation.
Crystal data for 8b at 100 K: CCDC 781524
Crystallized from Methanol Empirical formula C25 H19 Cl O3 Formula weight 402.85 Temperature 100(2) K Wavelength 0.71073 Å Crystal system Monoclinic Space group P2(1)
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S8
Unit cell dimensions a = 9.3490(9) Å α= 90.00 °. b = 6.8640(6) Å β = 100.578(4) °. c = 15.7148(15) Å γ = 90.00 °. Volume 991.31(16) Å3 Z 2 Density (calculated) 1.350 Mg/m3 Absorption coefficient 0.217 mm-1 F(000) 420 Crystal size 0.40 x 0.30 x 0.15 mm3 Theta range for data collection 1.32 to 28.41 °. Index ranges -12 <=h<=11 ,-8 <=k<=7 ,-20 <=l<=20 Reflections collected 4166 Independent reflections 2922 [R(int) = 0.0658 ] Completeness to theta =28.41 ° 0.924 % Absorption correction Empirical Max. and min. transmission 0.9682 and 0.9183 Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 4166 / 1 / 262 Goodness-of-fit on F2 0.921 Final R indices [I>2sigma(I)] R1 = 0.0552 , wR2 = 0.1372 R indices (all data) R1 = 0.0958 , wR2 = 0.1601 Absolute Structure Flack parameter x =-0.06(10) Largest diff. peak and hole 0.378 and -0.327 e.Å-3
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S9
Influence of the compound 4 geometry on the stereochemical outcome of the
cascade reactions.
Partial equilibration of the double-bond geometry of 3-benzylidenebenzofuran-2(3H)-one 4a (bearing a β-
phenyl group) has been observed under the catalytic reaction conditions of the cascade processes reported
in table 2 and 3 (Figure S1).
NH Ph
OTMSPh
N
N
OMe
NH2
A
OO
Ph20 mol%
oF-C6H4CO2H (20 mol%)
toluene, 40 °C, 24 hsingle E isomer
OO
Ph
OO
Ph
3 : 1
E Z
B
OO
Ph
20 mol%oF-C6H4CO2H (30 mol%)
toluene, 60 °C, 24 hsingle E isomer
OO
Ph
OO
Ph
3 : 1
E Z
Figure S1. Scrambling Experiments
Notably, using a 3:1 mixture of E/Z isomers of compound 4 leads to the same stereochemical outcome as
employing geometrically pure E isomer (compare entries 1 & 2, Table 2, a single stereoisomer, de and ee
>99%).
At the same extent, the stereochemical outcome of cascade reactions depicted in Table 3 is not dependent
on the geometry of the starting compound 4.
The stereo-convergence of the triple cascade depicted in Table 2 might be rationalized on the basis of a
reversible first enamine-catalysed Michael addition step between aldehyde 5 and 4, leading to intermediate
III: a rapid equilibrium may account for a dynamic kinetic resolution process driven by the perfectly suited
matched-pair combination of the chiral iminium ion (generated by condensation of catalyst B and enals 2)
and only one out of the possible eight stereoisomers of intermediate III. Unfortunately, to date we were not
able to isolate this key intermediate.
At present, we cannot rule out a stereo-convergence path driven by a different reactivity of the E and Z
isomers of 4, where the double-bond geometry scrambling continuously regenerates the fast reacting
isomer.
Form a synthetic standpoint, the fact that the geometry of the starting material 4 is not reflected into the
stereochemistry of the final compounds accounts for a more practical synthetic protocol (a mixture of 4 can
be used).
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S10
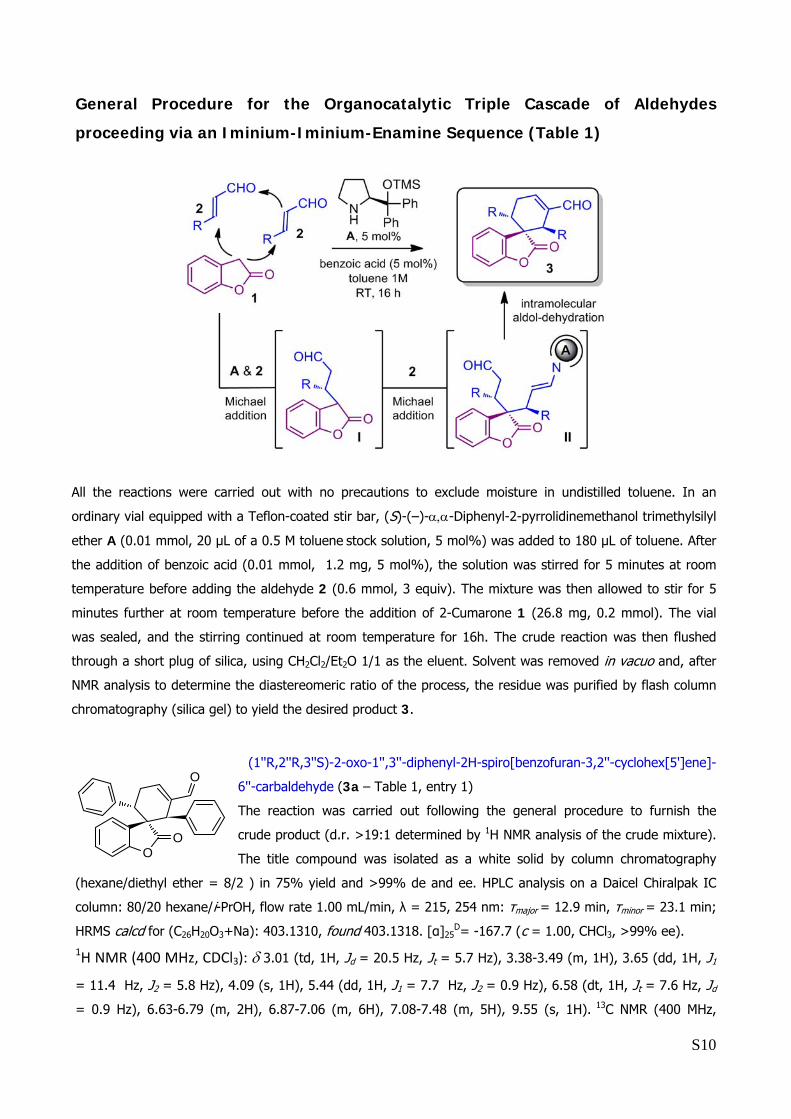
General Procedure for the Organocatalytic Triple Cascade of Aldehydes
proceeding via an Iminium-Iminium-Enamine Sequence (Table 1)
All the reactions were carried out with no precautions to exclude moisture in undistilled toluene. In an
ordinary vial equipped with a Teflon-coated stir bar, (S)-(–)-α,α-Diphenyl-2-pyrrolidinemethanol trimethylsilyl
ether A (0.01 mmol, 20 μL of a 0.5 M toluene stock solution, 5 mol%) was added to 180 μL of toluene. After
the addition of benzoic acid (0.01 mmol, 1.2 mg, 5 mol%), the solution was stirred for 5 minutes at room
temperature before adding the aldehyde 2 (0.6 mmol, 3 equiv). The mixture was then allowed to stir for 5
minutes further at room temperature before the addition of 2-Cumarone 1 (26.8 mg, 0.2 mmol). The vial
was sealed, and the stirring continued at room temperature for 16h. The crude reaction was then flushed
through a short plug of silica, using CH2Cl2/Et2O 1/1 as the eluent. Solvent was removed in vacuo and, after
NMR analysis to determine the diastereomeric ratio of the process, the residue was purified by flash column
chromatography (silica gel) to yield the desired product 3.
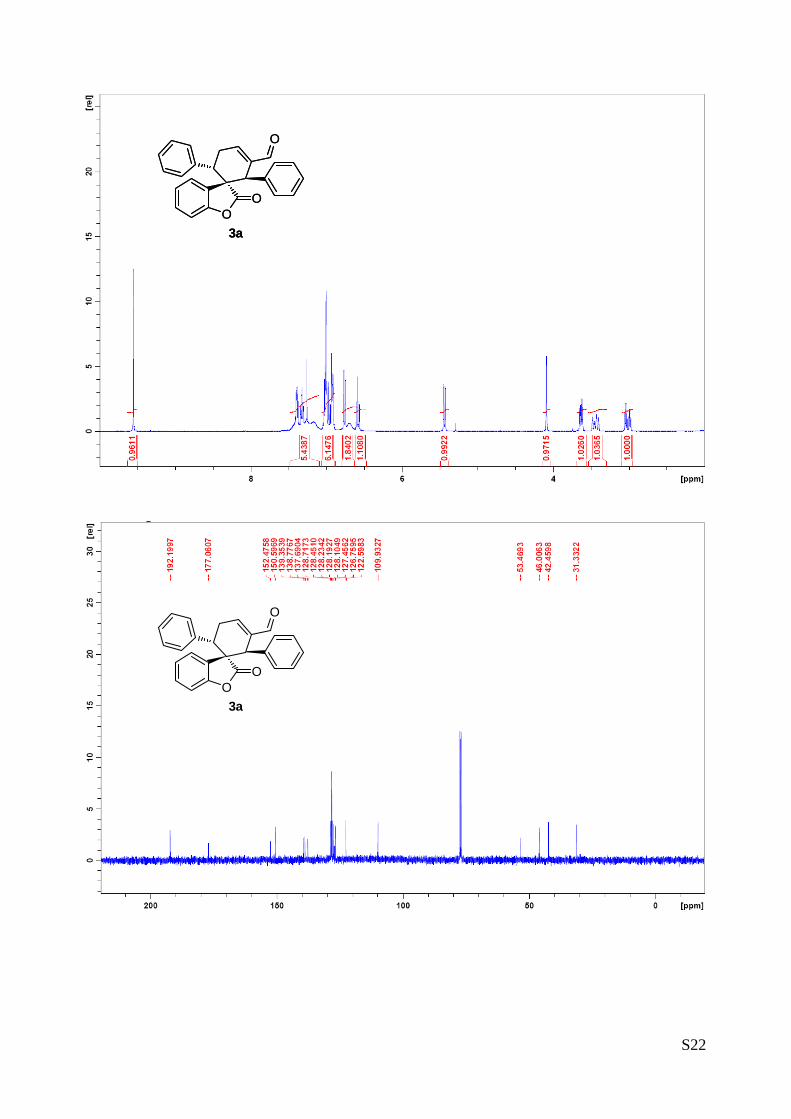
(1''R,2''R,3''S)-2-oxo-1'',3''-diphenyl-2H-spiro[benzofuran-3,2''-cyclohex[5']ene]-
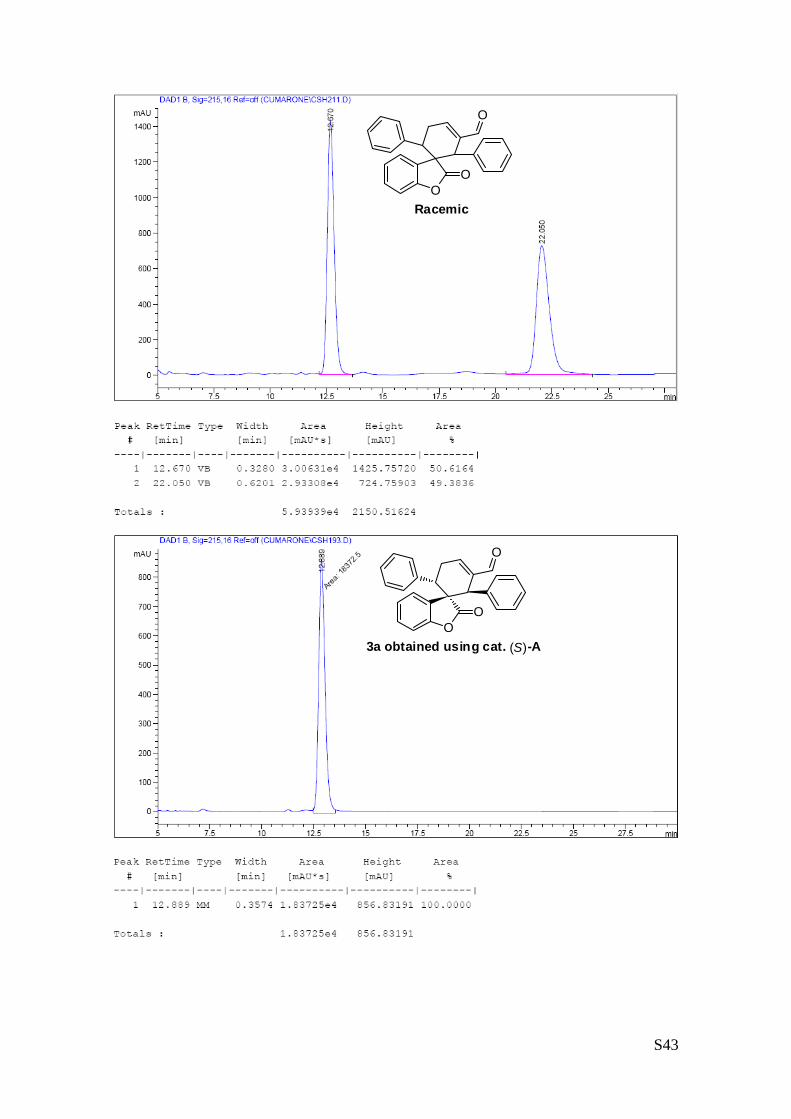
6''-carbaldehyde (3a – Table 1, entry 1)
The reaction was carried out following the general procedure to furnish the
crude product (d.r. >19:1 determined by 1H NMR analysis of the crude mixture).
The title compound was isolated as a white solid by column chromatography
(hexane/diethyl ether = 8/2 ) in 75% yield and >99% de and ee. HPLC analysis on a Daicel Chiralpak IC
column: 80/20 hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254 nm: τmajor = 12.9 min, τminor = 23.1 min;
HRMS calcd for (C26H20O3+Na): 403.1310, found 403.1318. [α]25D= -167.7 (c = 1.00, CHCl3, >99% ee).
1H NMR (400 MHz, CDCl3): δ 3.01 (td, 1H, Jd = 20.5 Hz, Jt = 5.7 Hz), 3.38-3.49 (m, 1H), 3.65 (dd, 1H, J1
= 11.4 Hz, J2 = 5.8 Hz), 4.09 (s, 1H), 5.44 (dd, 1H, J1 = 7.7 Hz, J2 = 0.9 Hz), 6.58 (dt, 1H, Jt = 7.6 Hz, Jd
= 0.9 Hz), 6.63-6.79 (m, 2H), 6.87-7.06 (m, 6H), 7.08-7.48 (m, 5H), 9.55 (s, 1H). 13C NMR (400 MHz,
OO
O
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S11
CDCl3):・δ 31.3 (CH2), 42.4 (CH), 46.0 (CH), 53.5 (C), 109.9 (CH), 122.9 (CH), 126.7 (CH), 127.3 (C), 127.4
(CH), 128.1 (CH), 128.2 (C), 128.2 (CH), 128.4 (CH), 128.7 (CH), 137.7 (CH), 138.8 (CH), 139.3 (C), 150.6
(CH), 152.5 (C), 177.0 (C), 192.2 (CH).
Carrying out the reaction in the presence of 1 mol% of the catalyst A at 40 degrees afforded similar results:
full conversion, 68% isolated yield, de & ee >99%
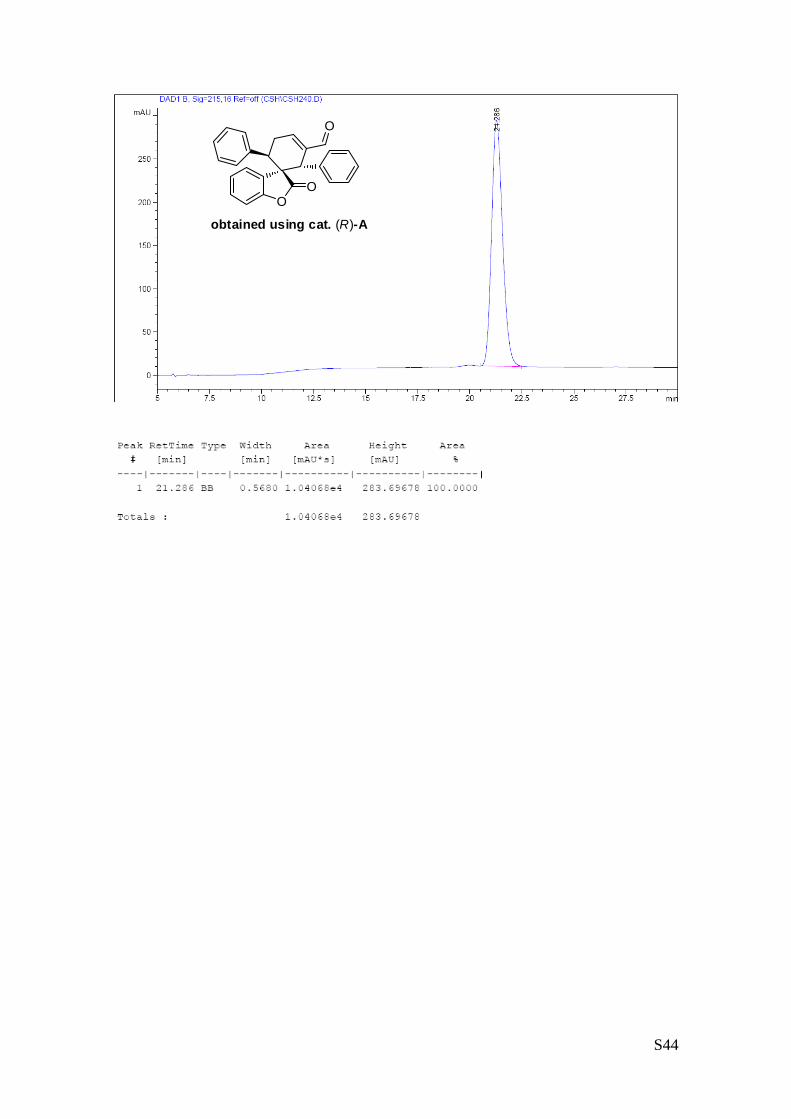
(1''S,2''S,3''R)-2-oxo-1'',3''-diphenyl-2H-spiro[benzofuran-3,2''-cyclohex[5']ene]-
6''-carbaldehyde.
The reaction was carried out following the general procedure using (R)-(+)-α,α-
Diphenyl-2-pyrrolidinemethanol trimethylsilyl ether as catalyst to furnish the
crude product (d.r. >19:1 determined by 1H NMR analysis of the crude mixture).
The title compound was isolated as a white solid by column chromatography (hexane/diethyl ether = 8/2 )
in 72% yield and >99% ee. HPLC analysis on a Daicel Chiralpak IC column: 80/20 hexane/i-PrOH, flow rate
1.00 mL/min, λ = 215, 254 nm: τ = minor12.9 min, τmajor = 23.1 min;. [α]25D= +132.9 (c = 1.06, CHCl3, >99%
ee).
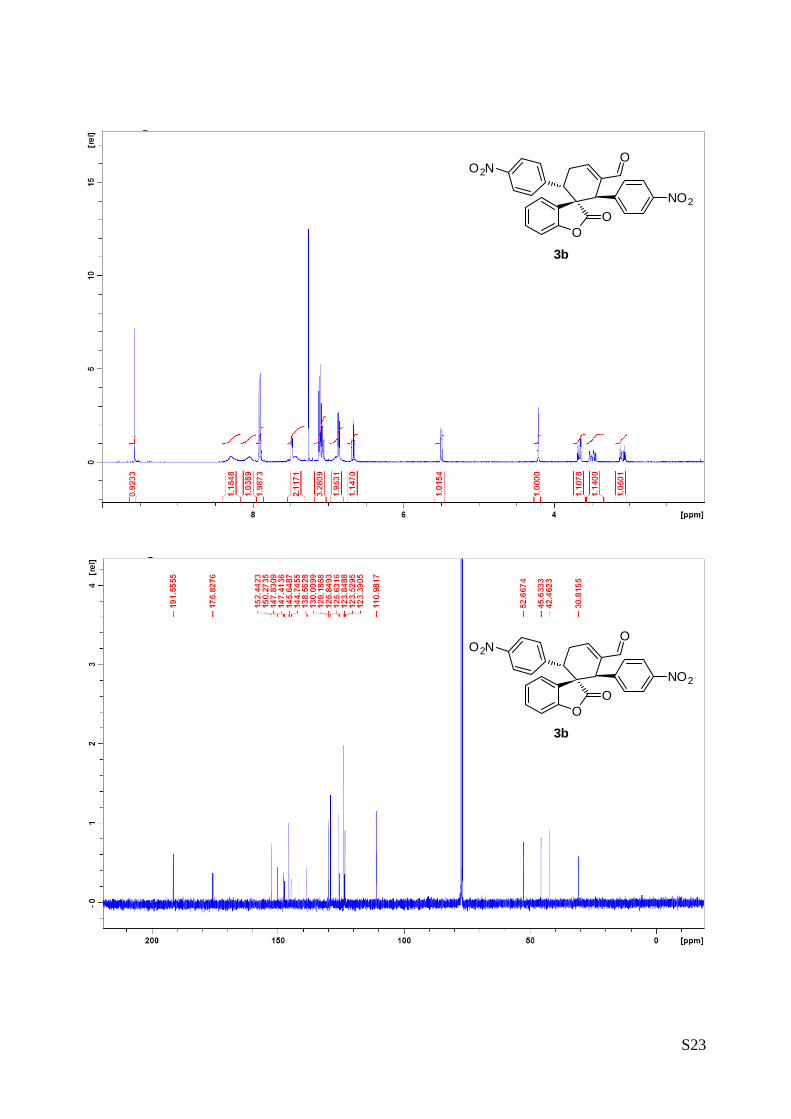
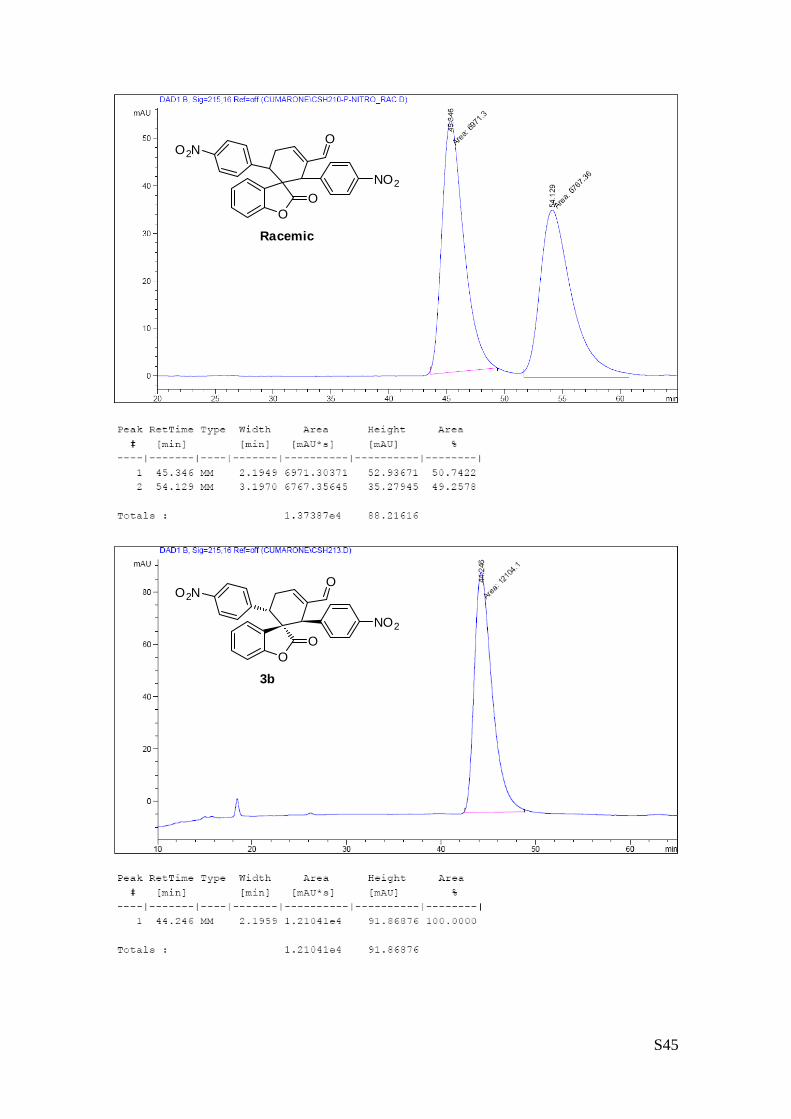
(1''R,2''R,3''S)-1'',3''-bis(4-nitrophenyl)-2-oxo-2H-spiro[benzofuran-
3,2''-cyclohex[5']ene]-6''-carbaldehyde (3b – Table 1, entry 3).
The reaction was carried out following the general procedure to
furnish the crude product (d.r. >19:1 determined by 1H NMR
analysis of the crude mixture). The title compound was isolated as
a yellow solid by column chromatography (hexane/diethyl ether = 6/4 ) in 59% yield and >99% de and ee.
HPLC analysis on a Daicel Chiralpak IC column: 70/30 hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254
nm: τmajor = 45.3 min, τminor = 54.1 min; HRMS calcd for (C26H17N2O7): 469.1036, found 469.1048. [α]26D= -
258.5 (c = 1.07, CHCl3, >99% ee).
1H NMR (400 MHz, CDCl3): δ 3.08 (td, 1H, Jd = 20.5 Hz, Jt = 5.4 Hz), 3.45-3.53 (m, 1H), 3.66 (dd, 1H, J1 =
11.3 Hz), 4.21 (s, 1H), 5.50 (dd, 1H, J1 = 7.6 Hz, J2 = 0.9 Hz), 6.67 (dt, 1H, Jt = 7.5 Hz, Jd 0.9= Hz), 6.83-
6.95 (m, 2H), 7.04-7.13 (m, 3H), 7.37-7.50 (m, 2H), 7.88-7.92 (m, 2H), 8.00-8.11 (m, 1H), 8.24-8.36 (m,
1H), 9.57 (s, 1H). 13C NMR (400 MHz, CDCl3): δ 30.8 (CH2), 42.5 (CH), 45.6 (CH), 52.6 (C), 111.0 (CH),
123.4 (CH), 123.5 (C), 123.8 (CH), 125.6 (C), 125.8 (CH), 129.2 (CH), 130.0 (CH), 138.6 (C), 144.7 (C),
145.6 (C), 147.4 (C), 147.8 (C), 150.3 (CH), 152.4 (C), 175.8 (C), 191.5 (CH).
OO
O
NO2
O2N
OO
O
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S12
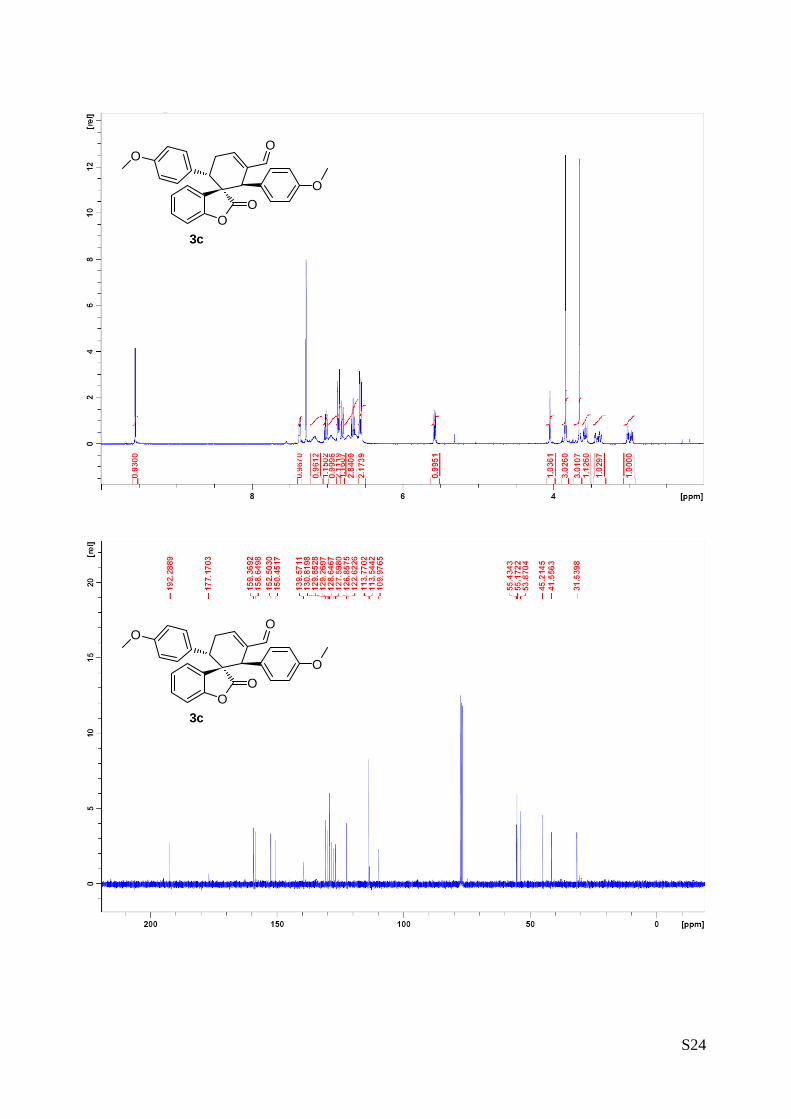
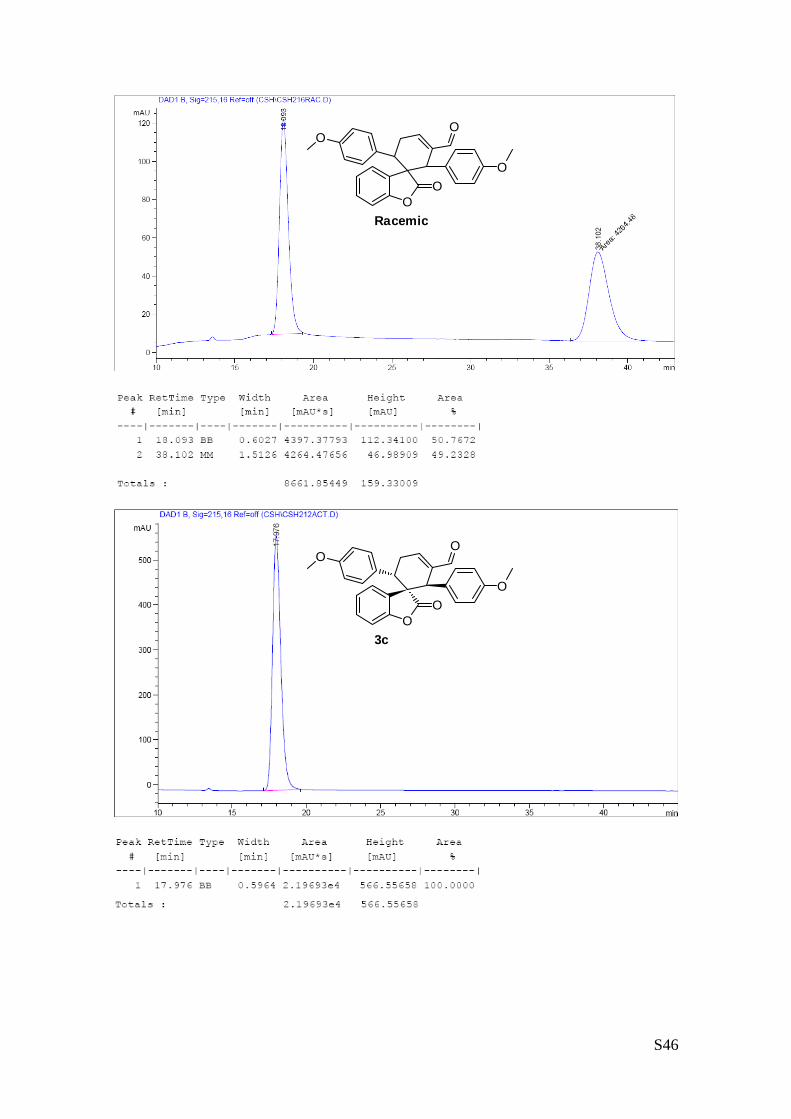
(1''R,2''R,3''S)-1'',3''-bis(4-methoxyphenyl)-2-oxo-2H-spiro[benzofuran-
3,2''-cyclohex[5']ene]-6''-carbaldehyde (3c – Table 1, entry 4).
The reaction was carried out following the general procedure to
furnish the crude product (d.r. >19:1 determined by 1H NMR analysis
of the crude mixture). The title compound was isolated as a pale
yellow solid by column chromatography (hexane/diethyl ether = 7/3 ) in 54% yield and >99% de and ee.
HPLC analysis on a Daicel Chiralpak IC column: 70/30 hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254
nm: τmajor = 18.0 min, τminor = 38.1 min; HRMS calcd for (C28H24O5+Na): 463.1521, found 463.1533. [α]26D= -
215.1 (c = 1.08, CHCl3, >99% ee).
1H NMR (400 MHz, CDCl3): δ 2.98 (td, 1H, Jd = 20.7 Hz, Jt 5.6= Hz), 3.32-3.44 (m, 1H), 3.58 (dd, 1H, J1 =
11.4 Hz, J2 = 5.8 Hz), 3.65 (s, 3H), 3.84 (s, 3H), 4.05 (s, 1H), 5.58 (dd, 1H, J1 = 7.5 Hz, J2 = 1.0 Hz),
6.54-6.58 (m, 2H), 6.59-6.78 (m, 3H), 6.80 (d, 1H, J = 8.0), 6.83-6.87 (m, 2H), 6.91-6.99 (m, 1H),7.02 (dt,
1H, Jd = 1.3 Hz, Jt = 7.7 Hz), 7.11-7.22 (m, 1H), 7.34-7.37 (m, 1H), 9.55 (s, 1H). 13C NMR (400 MHz,
CDCl3):・δ 31.4 (CH2), 41.4 (CH), 45.0 (CH), 53.7 (C), 55.0(CH3), 55.3 (CH3), 109.8 (CH), 113.4 (C), 113.6
(CH), 122.5 (CH), 126.7 (CH), 127.4 (C), 128.5 (CH), 129.1 (CH), 129.7 (C), 130.7 (C), 139.4 (C), 150.3
(CH), 152.4 (C), 158.5 (C), 159.2 (C), 177.0 (C), 192.2 (CH).
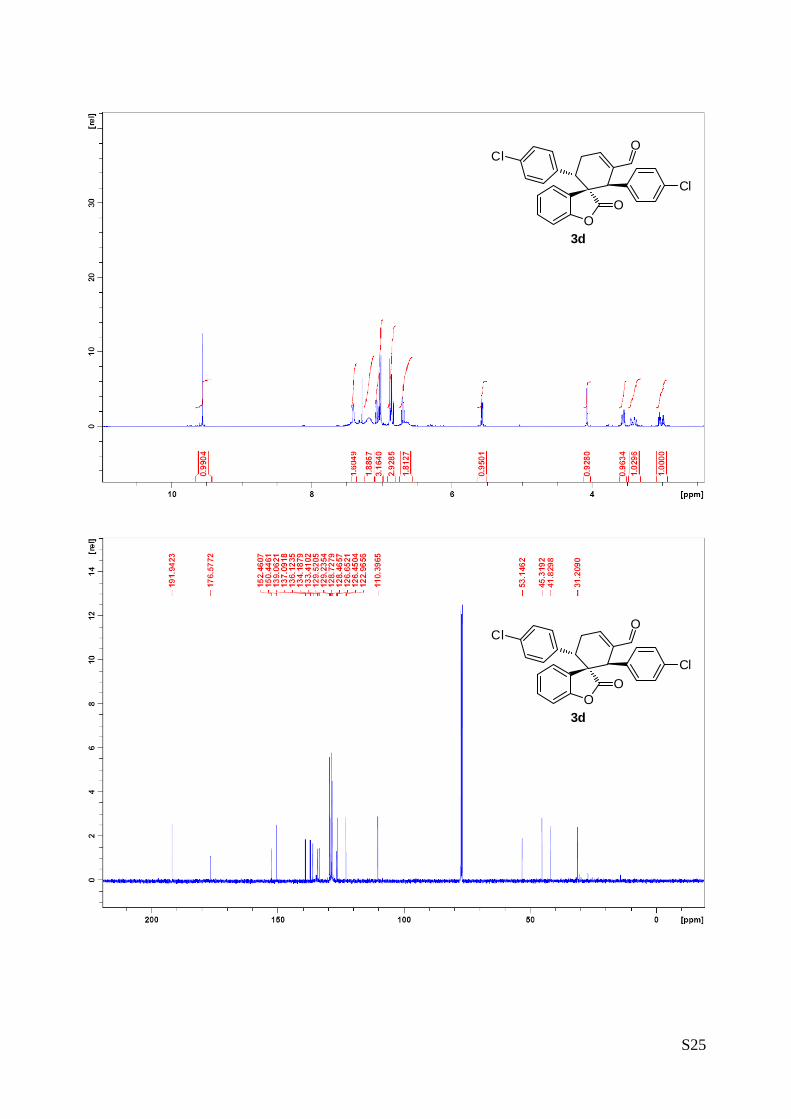
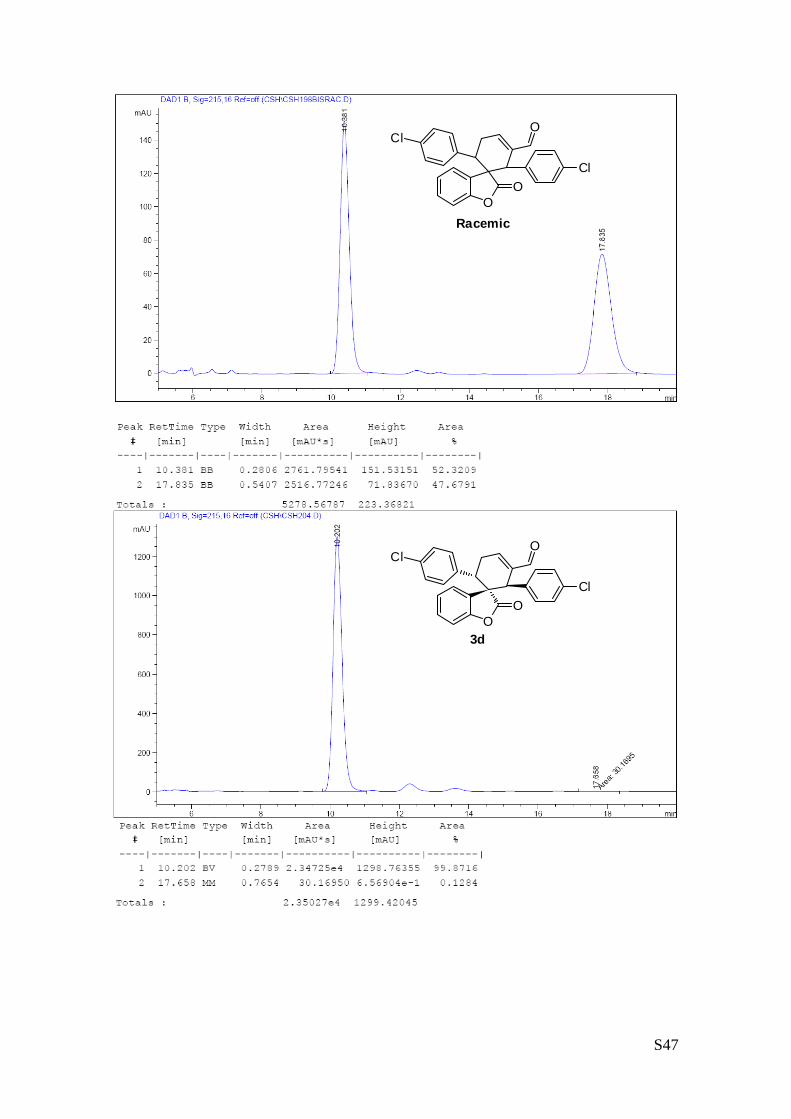
(1''R,2''R,3''S)-1'',3''-bis(4-chlorophenyl)-2-oxo-2H-spiro[benzofuran-3,2''-
cyclohex[5']ene]-6''-carbaldehyde (3d – Table 1, entry 5).
The reaction was carried out following the general procedure to furnish the
crude product (d.r. >19:1 determined by 1H NMR analysis of the crude
mixture). The title compound was isolated as a white solid by column
chromatography (hexane/diethyl ether = 7/3 ) in 80% yield and 99% ee. HPLC analysis on a Daicel
Chiralpak IC column: 70/30 hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254 nm: τmajor = 10.4 min, τminor
= 17.8 min; HRMS calcd for (C26H18Cl2O3+Na): 471.0531, found 471.0535. [α]25D= -198.1 (c = 1.32, CHCl3,
99% ee).
1H NMR (400 MHz, CDCl3): δ 3.01 (td, 1H, Jd = 20.4 Hz, Jt 5.2= Hz), 3.35-3.46 (m, 1H), 3.52 (dd, 1H, J1 =
11.4 Hz, J2 = 5.7 Hz), 4.07 (s, 1H), 5.57 (dd, 1H, J1 = 7.7 Hz, J2 = 1.2 Hz), 6.55-6.74 (m, 2H), 6.82-6.90
(m, 3H), 7.00-7.04 (m, 3H), 7.07(dt, 1H, Jd = 1.4 Hz, Jt = 8.0 Hz), 7.12-7.26 (m, 2H), 7.38-7.46 (m, 2H),
9.55 (s, 1H). 13C NMR (400 MHz, CDCl3): δ 31.2 (CH2), 41.8 (CH), 45.3 (CH), 53.1 (C), 110.4 (CH), 122.9
(CH), 126.4 (CH), 126.6 (C), 128.5 (CH), 128.7 (CH), 129.2 (CH), 129.5 (CH), 133.4 (C), 134.2 (C), 136.1
(C), 137.1 (C), 139.1 (C), 150.4 (CH), 152.5 (C), 195.5 (CH).
The absolute configuration of 3d was unambiguously determined by single crystal X-ray
analysis, see page S4.
OO
O
O
O
OO
O
Cl
Cl
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S13
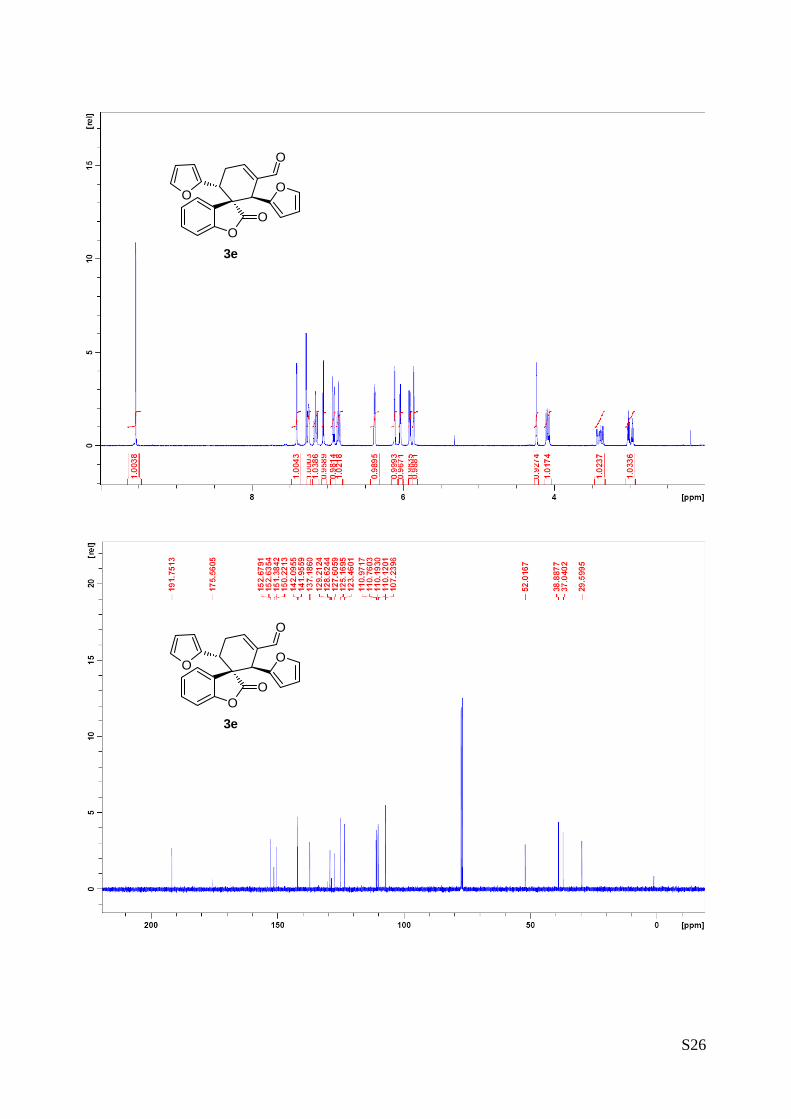
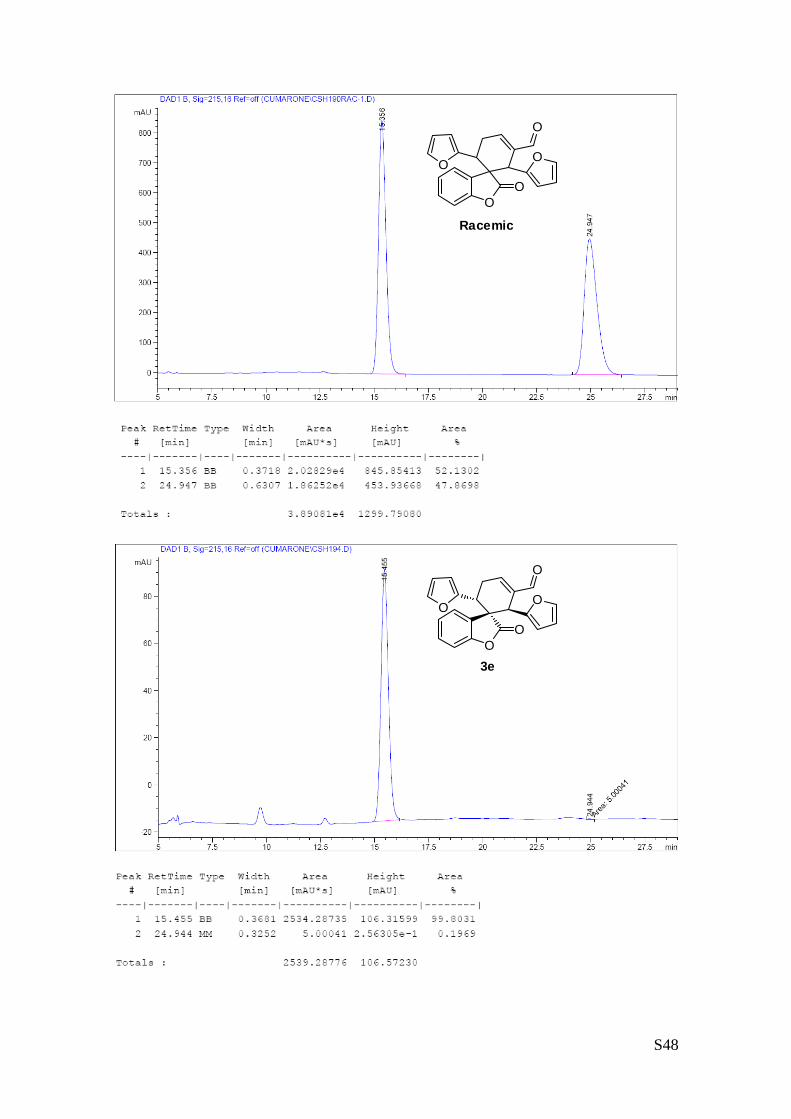
(1'R,2'R,6'R)-2',6'-di(furan-2-yl)-2-oxo-2H-spiro[benzofuran-3,1'-cyclohex[3]ene]-3'-
carbaldehyde (3e – Table 1, entry 6).
The reaction was carried out following the general procedure to furnish the crude
product (d.r. >19:1 determined by 1H NMR analysis of the crude mixture). The title
compound was isolated as a white solid by column chromatography (hexane/
diethyl ether = 7/3) in 49% yield and 99% ee. HPLC analysis on a Daicel Chiralpak IC column: 80/20
hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254 nm: τmajor = 15.3 min, τminor = 24.9 min; HRMS calcd for
(C22H16O5+Na): 383.0895, found 383.0913. [α]26D= -163.9 c = 0.84, CHCl3, 99% ee).
1H NMR (400 MHz, CDCl3): δ 2.96 (td, 1H, Jd = 20.9 Hz, Jt = 5.4 Hz), 3.31-3.42 (m, 1H), 4.06 (dd, 1H, J1 =
11.2 Hz, J2 = 5.8 Hz), 4.21 (s, 1H), 5.84 (d, 1H, J = 3.2 Hz), 5.86-5.90 (m, 1H), 6.01 (dd, 1H, J1 = 3.2 Hz,
J2 = 1.8 Hz), 6.08 (d, 1H, J = 3.2 Hz), 6.35 (dd, 1H, J1 = 3.2 Hz, J2 = 1.9 Hz), 6.83 (dt, 1 H, Jt = 7.6 Hz,
Jd = 0.9 Hz), 6.89 (d, 1H, J = 7.9 Hz), 7.02-7.04 (m, 1H), 7.13 (dt, 1H, Jt = 7.6 Hz, Jd = 1.0 Hz), 7.20-7.24
(m, 1H), 7.37-7.39 (m, 1H), 9.51(s, 1H). 13C NMR (400 MHz, CDCl3):・・ 29.6 (CH2), 37.0 (CH), 38.9 (CH),
52.0 (C), 107.2 (CH), 110.1(CH), 110.2 (CH), 110.8 (CH), 110.9 (CH), 123.4 (CH), 125.2 (CH), 127.6
(C), 129.2 (CH), 137.2 (C), 141.9 (CH), 142.1 (CH), 150.2 (CH), 151.4 (C), 152.6 (C), 152.7 (C), 191.7
(CH).
(1'R,2'R,6'R)-2',6'-dimethyl-2-oxo-2H-spiro[benzofuran-3,1'-cyclohex[3]ene]-3'-
carbaldehyde (3f– Table 1, entry 7).
The reaction was carried out following the general procedure to furnish the crude
product (d.r. >19:1 determined by 1H NMR analysis of the crude mixture). The title
compound was isolated as a yellowish oil by column chromatography (hexane/diethyl ether = 8/2 ) in 51%
yield and 99% ee. HPLC analysis on a Daicel Chiralpak IC column: 80/20 hexane/i-PrOH, flow rate 1.00
mL/min, λ = 215, 254 nm: τmajor = 13.3 min, τminor = 17.5 min; HRMS calcd for (C16H16O3+Na): 279.0997,
found 297.0994. [α]26D= -21.8 (c = 1.40, CHCl3, 99% ee).
1H NMR (400 MHz, CDCl3): δ 0.74 (d, 3H, J = 6.8 Hz), 1.21 (d, 3H, J = 6.9 Hz), 2.43-2.68 (m, 3H), 2.90 (q,
1H, J = 6.9 Hz), 6.92-6.96 (m, 1H), 7.10-7.22 (m, 3H), 7.30-7.35 (m, 1H), 9.48 (s, 1H).
13C NMR (400 MHz, CDCl3):δ 16.9 (CH3), 17.2 (CH3), 29.3 (CH), 32.1 (CH2), 33.5 (CH), 52.7 (C), 111.0
(CH), 123.7 (CH), 125.7 (CH), 128.9 (C), 129.0 (CH), 142.3 (C), 150.4 (CH), 153.4 (C), 176.6 (C), 193.1
(CH).
OO
O
OO
OO
O
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S14
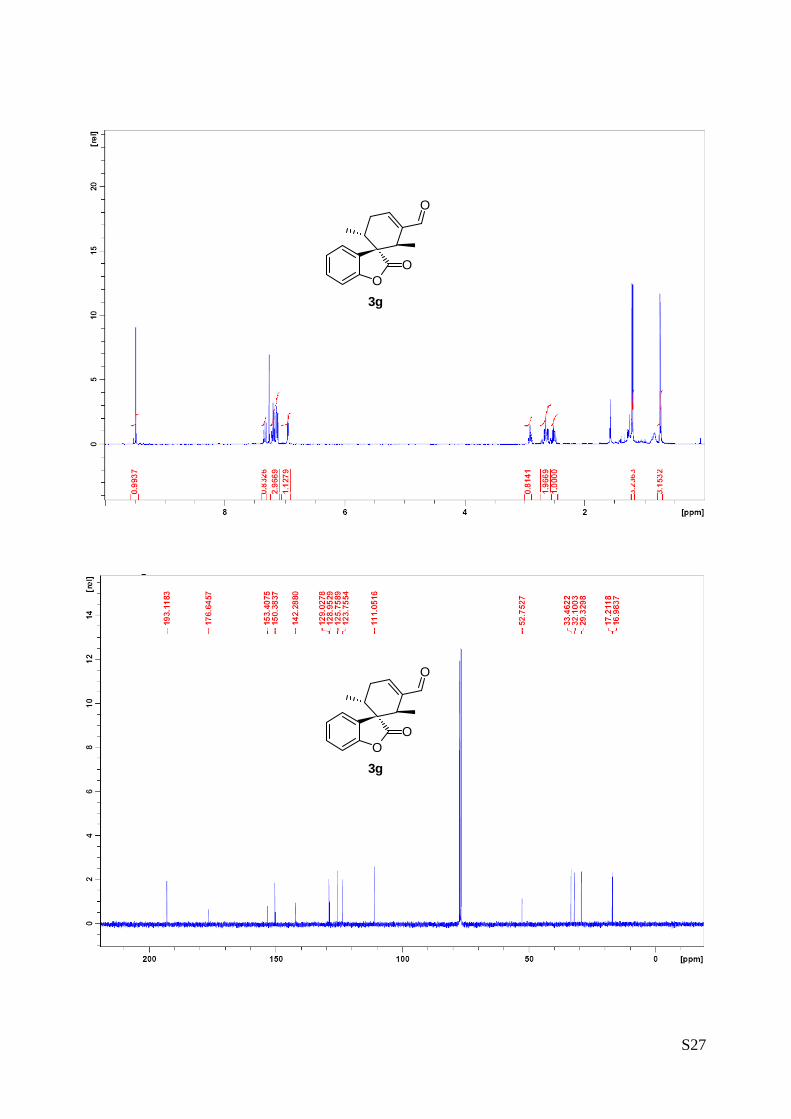
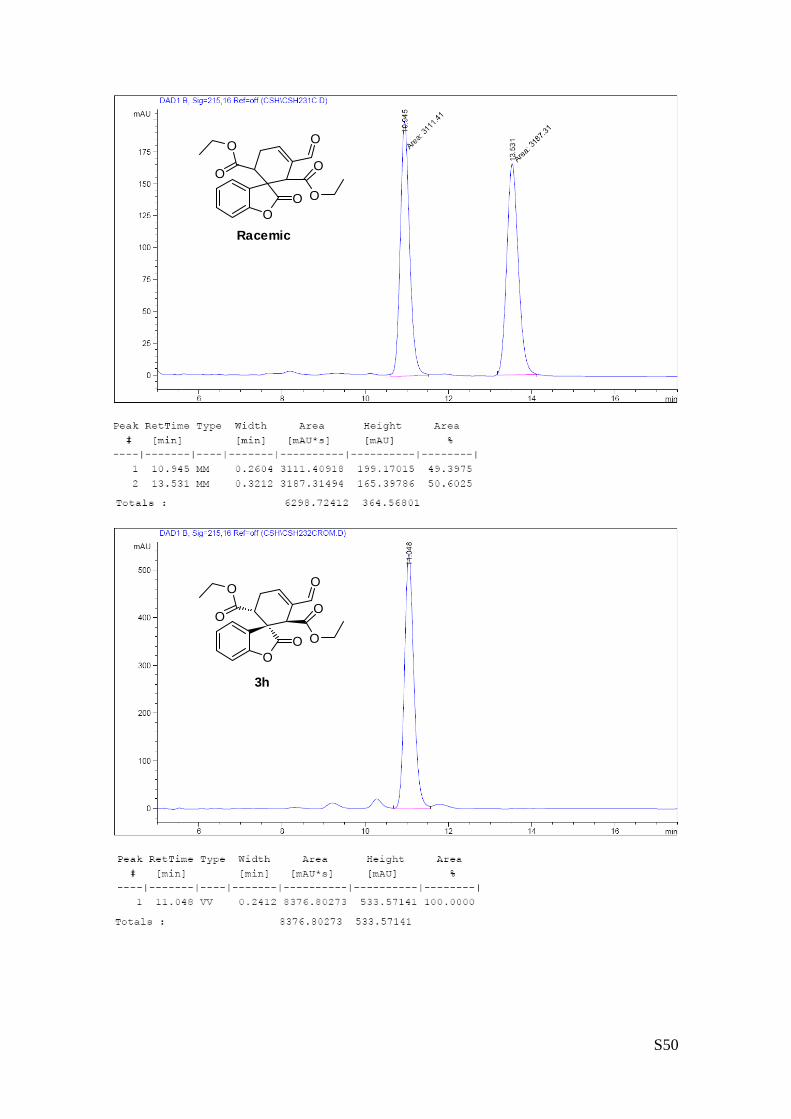
(1'R,2'R,6'R) - diethyl 3'-formyl-2-oxo-2H-spiro [benzofuran-3,1'-cyclohex[3]ene]
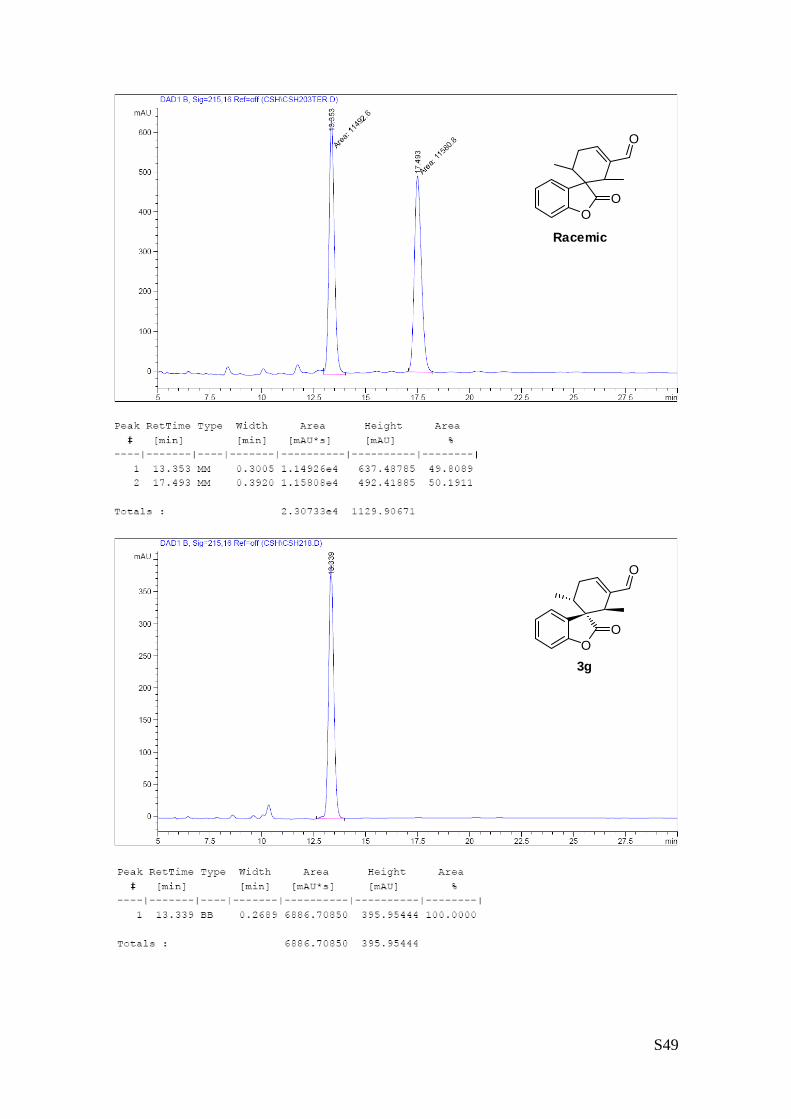
-2',6'-dicarboxylate (3g – Table 1, entry 8).
The reaction was carried out following the general procedure to furnish the crude
product (d.r. >19:1 determined by 1H NMR analysis of the crude mixture). The
title compound was isolated as a colourless oil by column chromatography
(hexane/acetone = 8/2 ) in 38% yield and 99% ee. HPLC analysis on a Daicel Chiralpak IA column: 80/20
hexane/i-PrOH, flow rate 1.00 mL/min, λ = 215, 254 nm: τmajor = 10.9 min, τminor = 13.5 min; HRMS calcd for
(C20H20O7+Na): 395.1107, found 395.1111. [α]25D= -75.5 (c = 1.23, CHCl3, 99% ee).
1H NMR (400 MHz, CDCl3): δ 0.97 (t, 3, J = 7.2 Hz), 1.10 (t, 3H, J = 7.2 Hz ), 3.02 (td, 1H, Jd = 20.0 Hz, Jt =
5.7 Hz), 3.12-3.23 (m, 1H), 3.71 (s, 1H), 3.85-4.04 (m, 4H), 4.08-4.17 (m, 1H), 7.02-7.18 (m, 3H), 7.24-
7.27 (m, 1H), 7.37 (dt, 1H, Jt = 7.8 Hz, Jd 1.3 = Hz), 9.54 (s, 1H).
13C NMR (400 MHz, CDCl3): δ 13.8 (CH3), 13.9 (CH3), 27.2 (CH2), 42.6 (CH), 45.2 (CH), 46.25 (C), 61.5
(CH2), 61.7 (CH2), 111.2 (CH), 123.9 (CH), 124.0 (CH), 127.1 (C), 130.2 (CH), 135.7 (C), 150.7 (CH), 153.6
(C), 170.2 (C), 171.4 (C), 174.78 (C),191.7 (CH).
General Procedure for the Organocatalytic Triple Cascade proceeding via
enamine – iminium – enamine activation of aldehydes (Table 2)
All the reactions were carried out in an ordinary vial equipped with a Teflon-coated stir bar, with no
precautions to exclude moisture in undistilled toluene. Catalyst A (0.02 mmol, 20 mol %), aldehyde 5 (0.2
mmol) and o-fluoro-benzoic acid (20 mol %) were stirred in toluene (0.2 mL) at room temperature for 10
minutes. The cascade was started by adding a solution of benzofuranone derivative 4 (0.15 mmol) and
aldehyde 2 (0.1 mmol), dissolved in 0.3 mL of toluene. The vial was sealed and heated up to 40 degree, and
stirring was continued till completion of the reaction, as monitored by TLC (generally 48 hours). The crude
reaction was then flushed through a short plug of silica, using CH2Cl2/Et2O 1/1 as the eluent. Solvent was
removed in vacuo and, after NMR analysis to determine the diastereomeric ratio of the process, the residue
was purified by flash column chromatography (silica gel) to yield the desired spiro-compounds 6.
OO
O
O
O
O
O
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S15
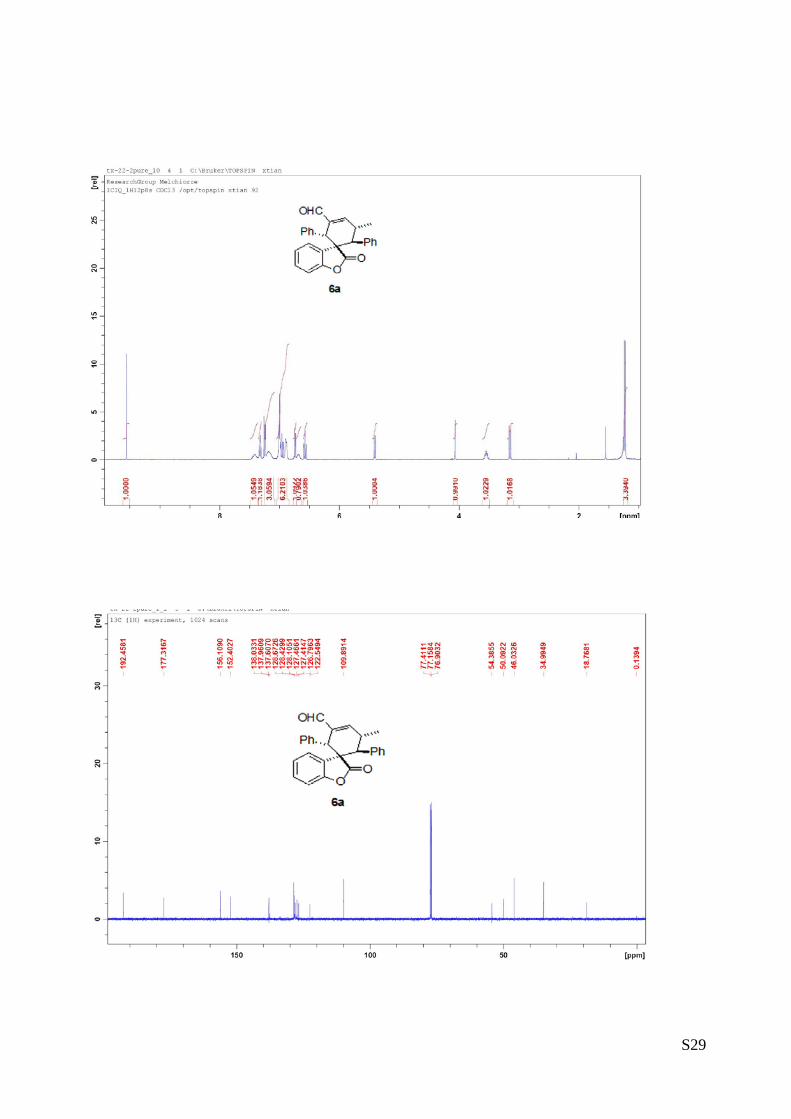
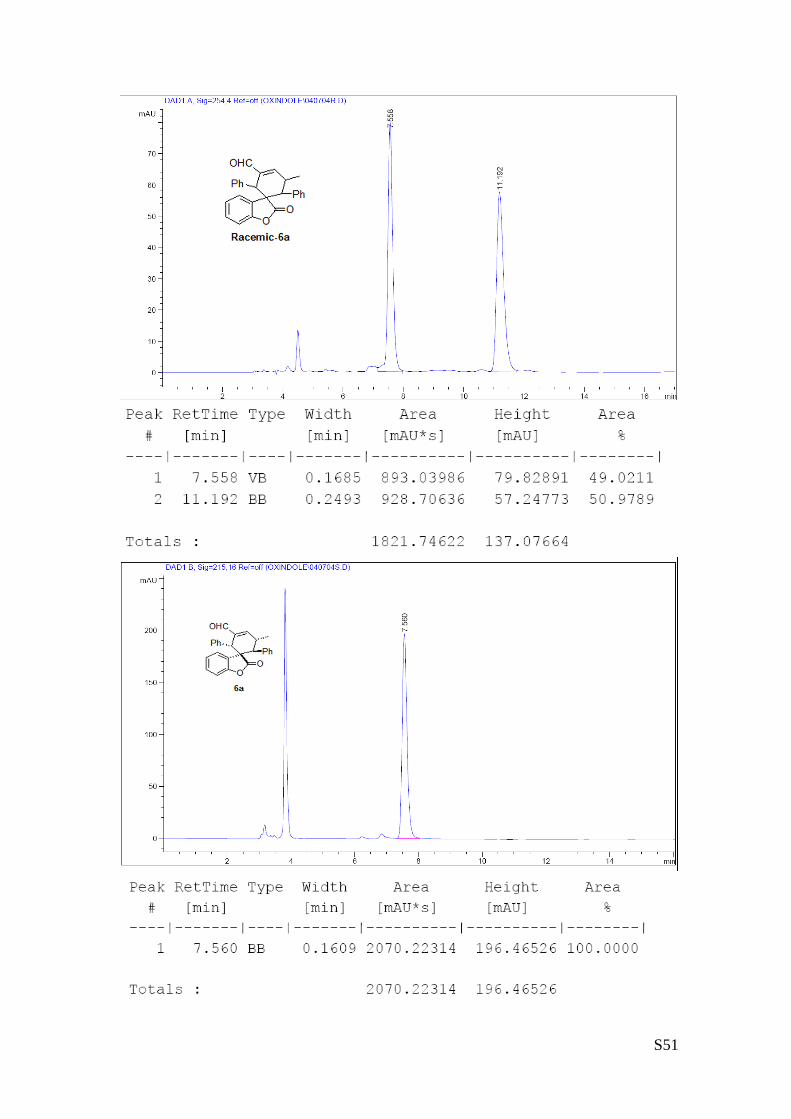
(6a - Table 2, entry 1). The reaction was carried out at 40°C following the general
procedure to furnish the crude product (d.r. >19:1 determined by 1H NMR analysis of
the crude mixture). The title compound was isolated as a white solid by column
chromatography (hexane/AcOEt = 20/1) in 56% yield and >99% ee. HPLC analysis on a
Daicel Chiralpak IA column: 90/10 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τmajor = 7.56 min, τminor
= 11.2 min. HRMS calcd for (C27H22O3+Na): 417.1467, found 417.1483. [α]rtD= -180.9 (c = 0.57, CHCl3).1H
NMR (400 MHz, CDCl3): δ 1.23 (d, J = 7.2, 3H), 3.15 (d, J = 11.2, 1H), 3.53-3.57 (m, 1H), 4.07 (s, 1H), 5.41
(d, J = 7.2, 1H), 6.57 (t, J = 7.2, 1H), 6.68 (bs, 1H), 6.74 (d, J = 8.4, 1H), 6.88-7.02 (m, 6H), 7.17-7.24 (m,
3H), 7.32 (t, J = 7.2, 1H), 7.42 (bs, 1H), 9.56 (s, 1H).13C NMR (100 MHz, CDCl3): δ 18.7, 34.9, 46.0, 50.0,
54.3, 109.8, 122.5, 126.7, 127.41, 127.46, 128.1, 128.4, 128.6, 137.9, 138.0, 152.4, 156.1, 177.3, 192.4.
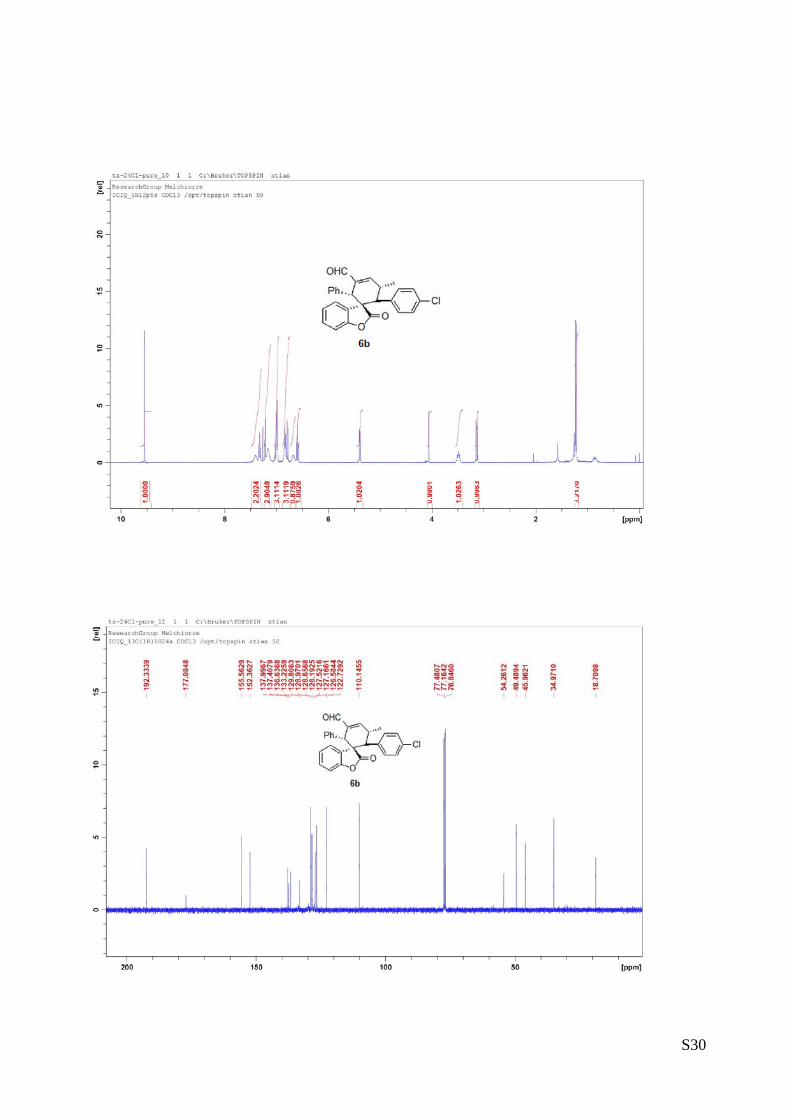
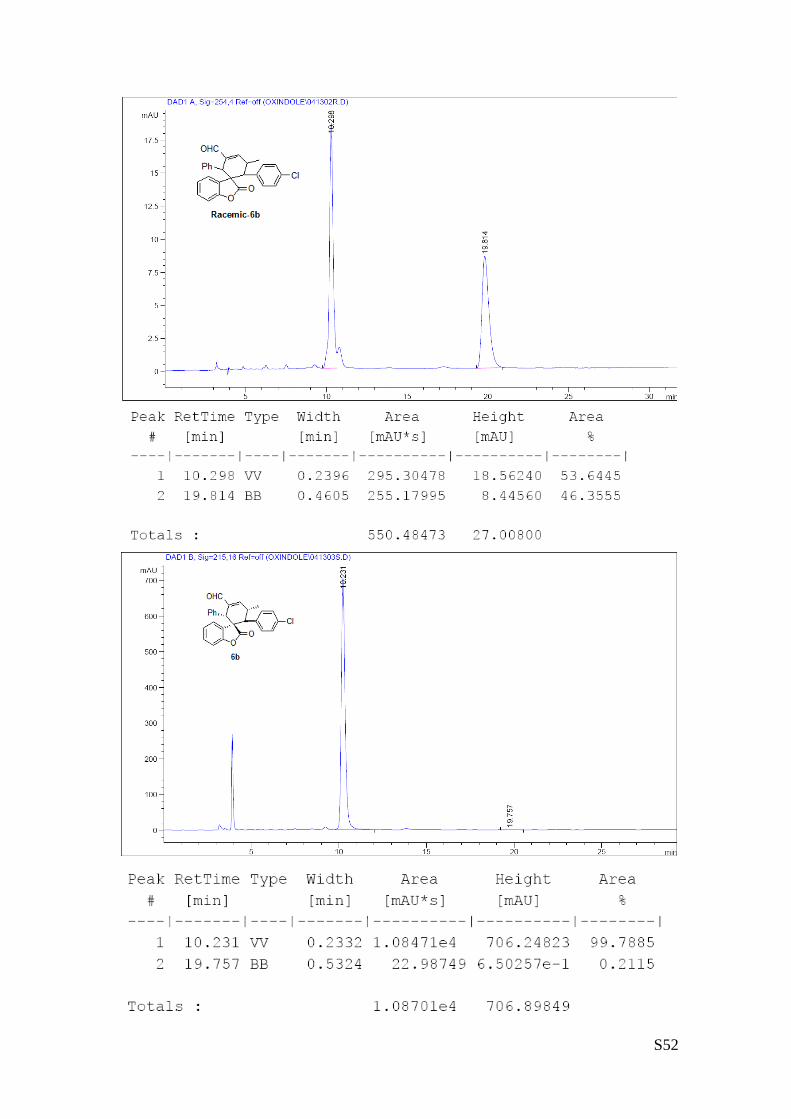
(6b - Table 2, entry 3). The reaction was carried out at 40°C following the
general procedure to furnish the crude product (d.r. >19:1 determined by 1H
NMR analysis of the crude mixture). The title compound was isolated as a white
solid by column chromatography (hexane/AcOEt = 20/1) in 57% yield and 99%
ee. HPLC analysis on a Daicel Chiralpak IA column: 95/5 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm:
τmajor = 10.23 min, τminor = 19.75 min. HRMS calcd for (C27H21ClO3+Na): 451.1077, found 451.1089. [α]rtD= -
148.5 (c = 0.76, CHCl3).1H NMR (400 MHz, CDCl3): δ 1.22 (d, J = 7.2, 3H), 3.13 (d, J = 10.4, 1H), 3.45-3.53
(m, 1H), 4.06 (s, 1H), 5.38 (d, J = 8, 1H), 6.59 (t, J = 7.6, 1H), 6.67 (bs, 1H), 6.77-7.02 (m, 6H), 7.16-7.22
(m, 3H), 7.32 (t, J = 7.2, 1H), 7.40 (bs, 1H), 9.55 (s, 1H).13C NMR (100 MHz, CDCl3): δ 18.7, 34.9, 45.9,
49.4, 54.2, 110.1, 122.7, 126.5, 127.1, 127.5, 128.6, 128.9, 133.2, 136.6, 137.4, 137.9, 152.3, 155.5,
177.0, 192.3.
The absolute configuration of 6b was unambiguously determined by single crystal X-ray
analysis, see page S6.
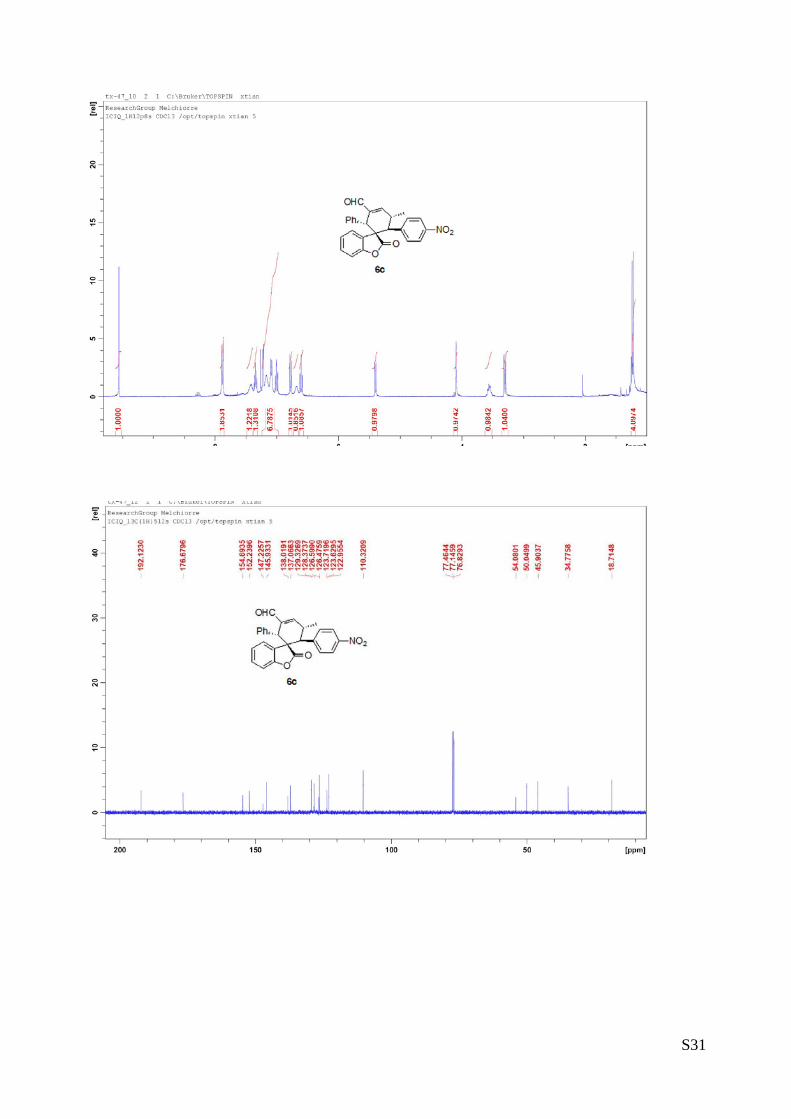
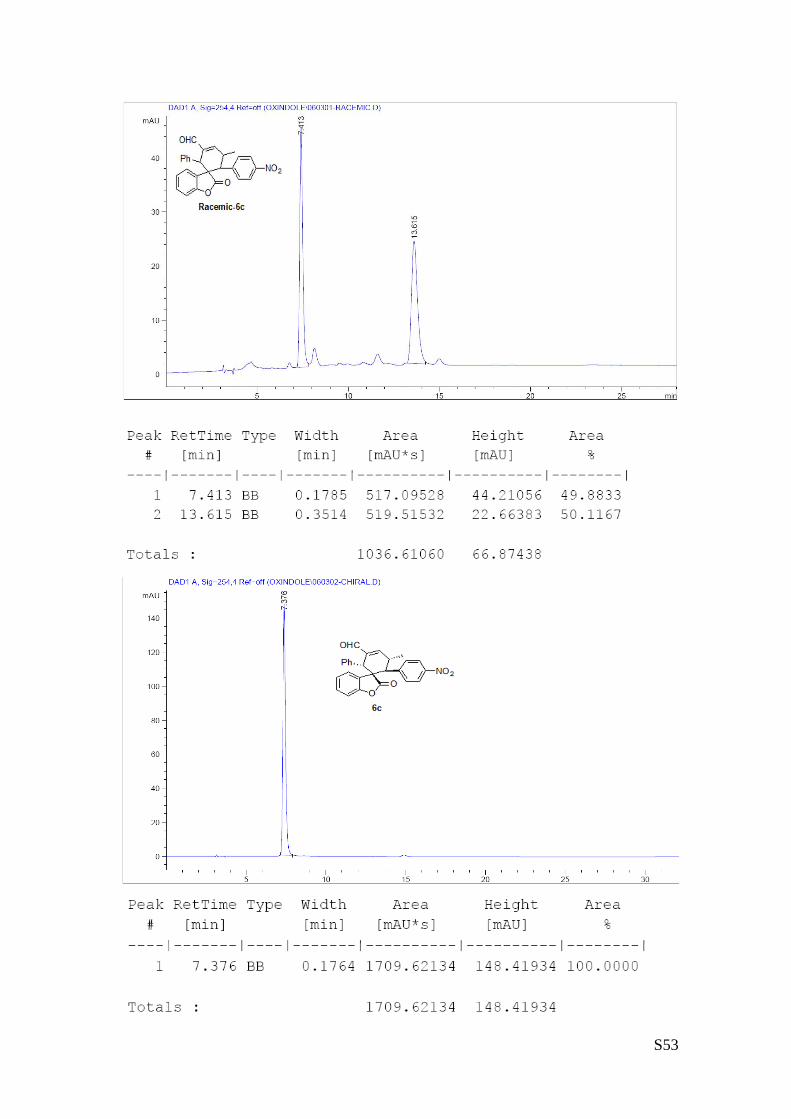
(6c - Table 2, entry 4). The reaction was carried out at 40°C following the
general procedure to furnish the crude product (d.r. >19:1 determined by 1H
NMR analysis of the crude mixture). The title compound was isolated as a
white solid by column chromatography (hexane/AcOEt = 7/1) in 52% yield
and >99% ee. HPLC analysis on a Daicel Chiralpak IA column: 80/20 hexane/i-PrOH, flow rate 1.0 mL/min, λ
= 254 nm: τmajor = 7.37 min, τminor = 13.6 min.. HRMS calcd for (C27H21NO5+Na): 462.1317, found 462.1331.
[α]rtD= -199.9 (c = 0.61, CHCl3).
1H NMR (400 MHz, CDCl3): δ 1.23 (d, J = 7.2, 3H), 3.30 (d, J = 10.4, 1H), 3.54-3.59 (m, 1H), 4.09 (s, 1H),
5.40 (d, J = 8.4, 1H), 6.60 (t, J = 8, 1H), 6.68 (bs, 1H), 6.77 (d, J = 8, 1H), 6.98-7.23 (m, 6H), 7.34 (t, J =
7.2, 1H), 7.41(bs, 1H), 7.88 (d, J = 9.6, 2H), 9.56 (s, 1H).13C NMR (100 MHz, CDCl3): δ 18.7, 34.7, 45.9,
50.0, 54.0, 110.3, 122.9, 123.6, 123.7, 126.4, 126.5, 128.3, 129.3, 137.0, 138.0, 145.9, 147.2, 152.2,
154.6, 176.6, 192.1.
O
Ph
OHC
PhO
O
Ph
OHC
OCl
O
Ph
OHC
ONO2
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S16
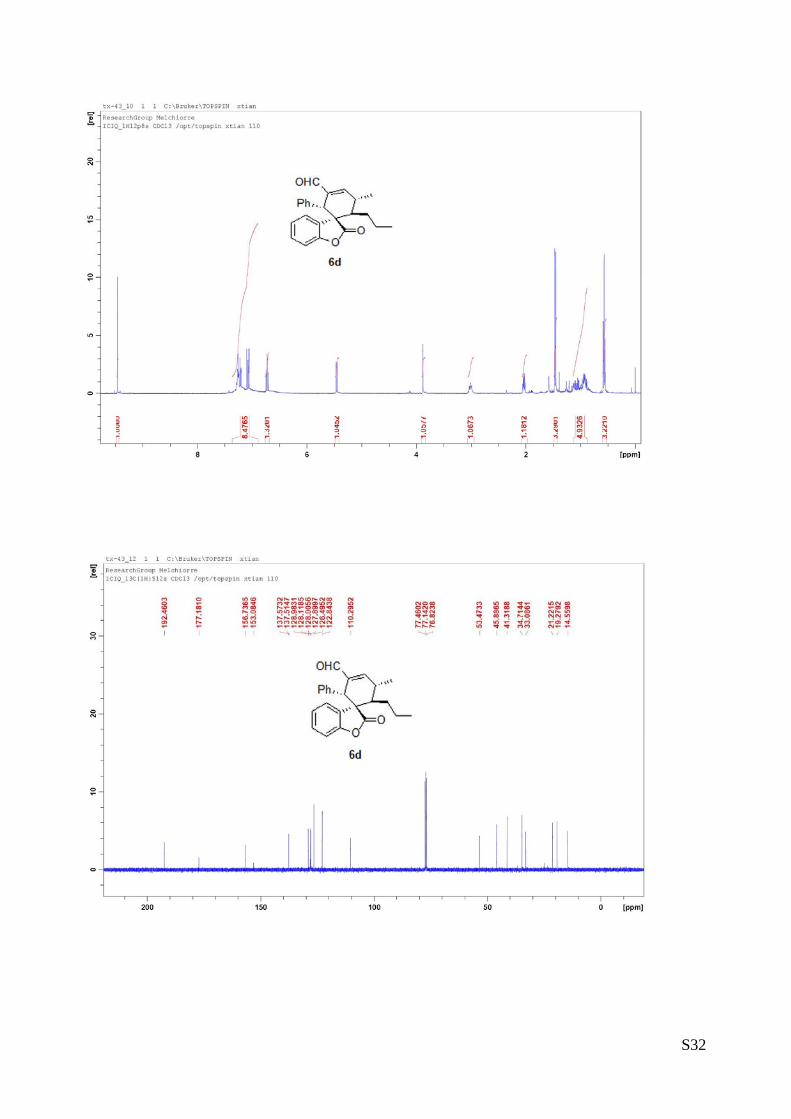
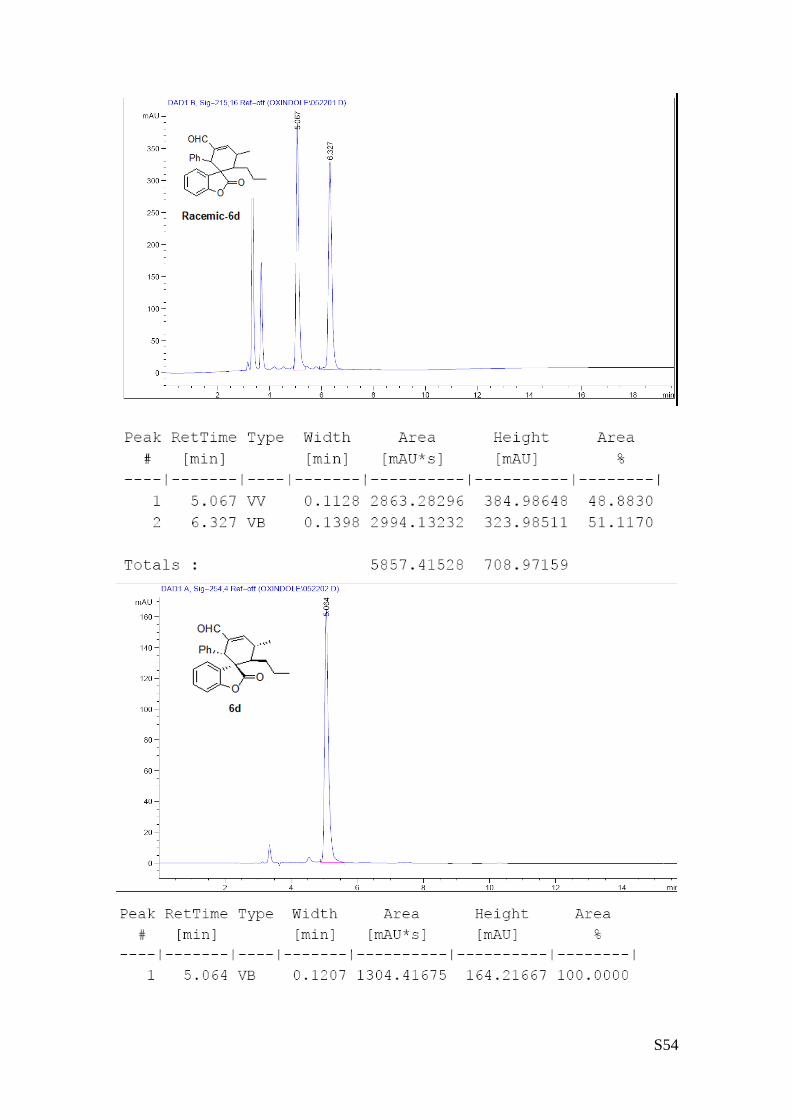
(6d - Table 2, entry 5). The reaction was carried out at 40°C following the general
procedure to furnish the crude product (d.r. >19:1 determined by 1H NMR analysis of
the crude mixture). The title compound was isolated as a white solid by column
chromatography (hexane/AcOEt = 20/1) in 56% yield and >99% ee. HPLC analysis
on a Daicel Chiralpak IA column: 80/20 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τmajor = 5.06 min,
τminor = 6.3 min. HRMS calcd for (C24H24O3+Na): 383.1623, found 383.1605. [α]rtD= -148.2 (c = 0.55, CHCl3).
1H NMR (400 MHz, CDCl3): δ 0.57 (t, J = 8, 3H), 0.88-1.14 (m, 4H), 1.46 (d, J = 7.6, 3H), 2.01-2,06 (m,
1H), 2.96-3.05 (m, 1H), 3.89 (s, 1H), 5.46 (d, J = 7.6, 1H), 6.73 (t, J = 8, 1H), 7.05-7.28 (m, 8H), 9.47 (s,
1H).13C NMR (100 MHz, CDCl3): δ 14.5, 19.2, 21.2, 33.0, 34.7, 41.3, 45.8, 53.4, 110.2, 122.8, 126.4, 127.8,
128.0, 128.1, 128.9, 137.5, 153.0, 156.7, 177.1, 192.4.
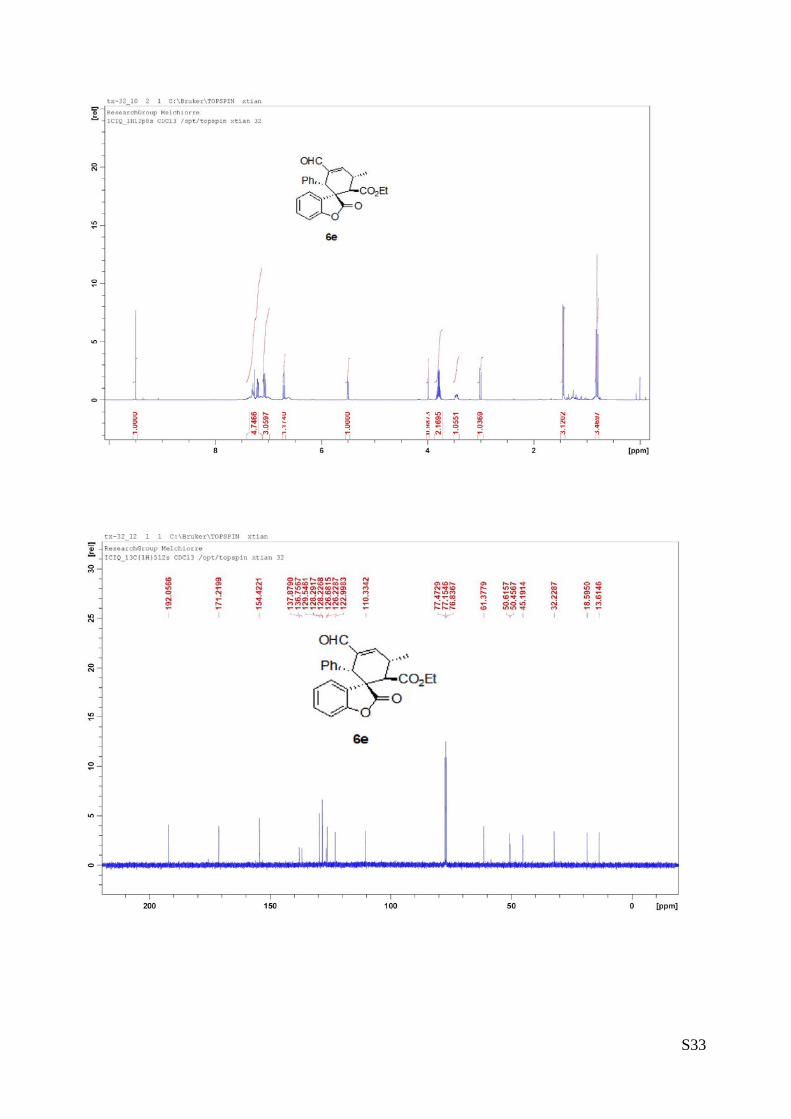
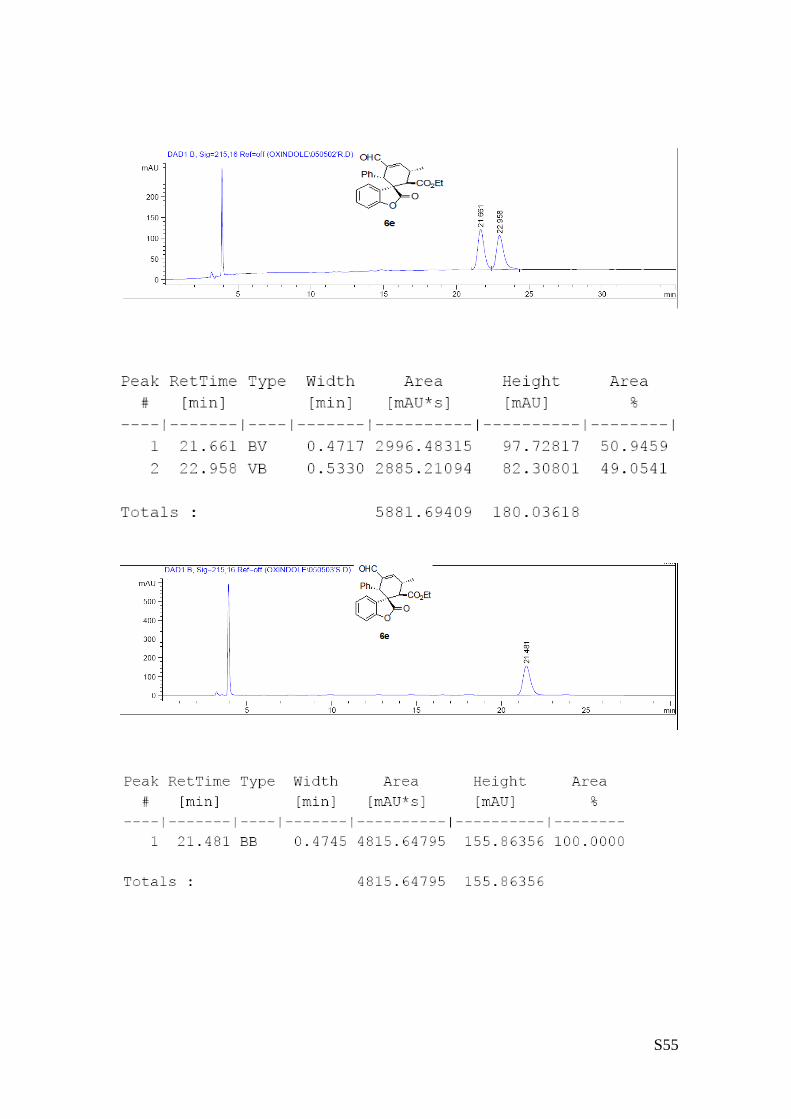
(6e - Table 2, entry 6). The reaction was carried out at 40°C following the general
procedure to furnish the crude product (d.r. >19:1 determined by 1H NMR analysis of
the crude mixture). The title compound was isolated as a white solid by column
chromatography (hexane/AcOEt = 10/1) in 54% yield and >99% ee. HPLC analysis
on a Daicel Chiralpak IA column: 95/5 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τmajor = 21.48 min,
τminor = 22.9 min. HRMS calcd for (C24H22O5+Na): 413.1365, found 413.1366. [α]rtD= -97.6 (c = 0.55, CHCl3).
1H NMR (400 MHz, CDCl3): δ 0.81 (t, J = 7.2, 3H), 1.44 (d, J = 8, 3H), 3.01 (d, J = 11.2, 1H), 3.43-3.48
(m, 1H), 3.75-3.83 (m, 2H), 3.99 (s, 1H), 5.50 (d, J = 8, 1H), 6.71 (t, J = 8, 1H), 7.05-7.09 (m, 3H), 7.19-
7.31 (m, 5H), 9.50 (s, 1H).13C NMR (100 MHz, CDCl3): δ 13.6, 18.5, 32.2, 45.1, 50.4, 50.6, 61.3, 110.3,
122.9, 126.2, 126.6, 128.22, 128.29, 129.5, 136.7,137.8, 154.4, 171.2, 192.0.
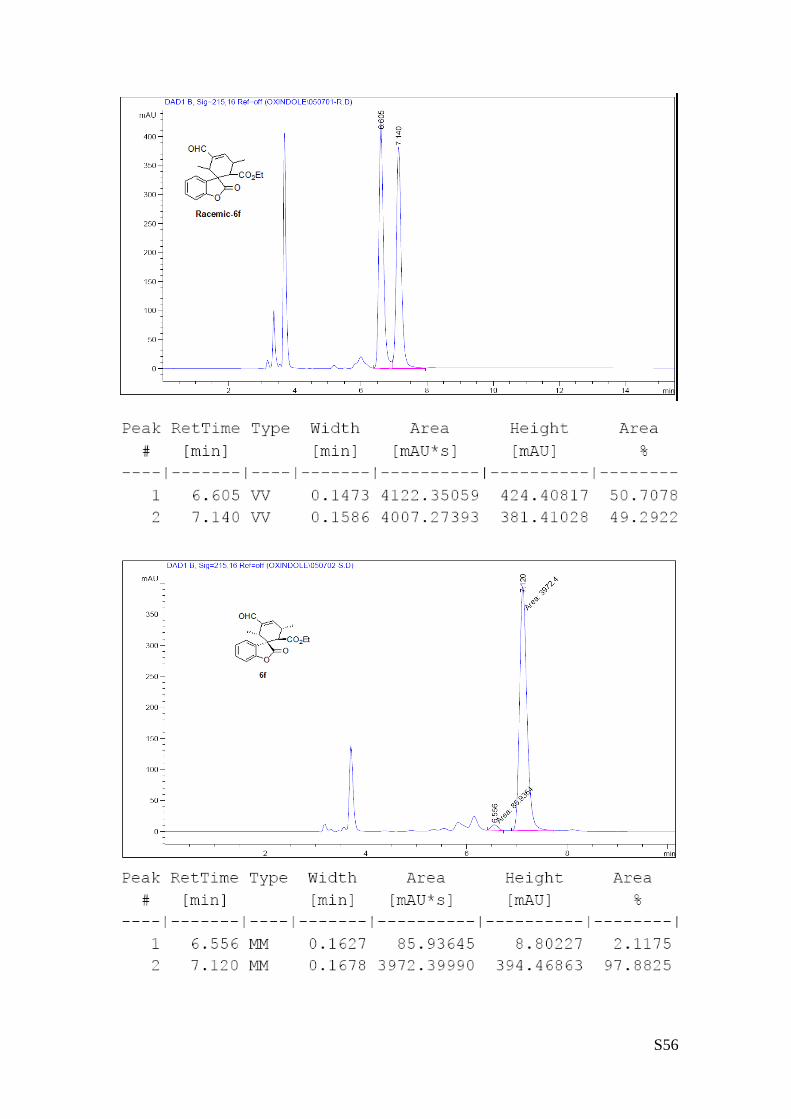
(6f - Table 2, entry 7). The reaction was carried out at 40°C following the general
procedure to furnish the crude product (d.r. >19:1 determined by 1H NMR analysis of
the crude mixture). The title compound was isolated as a colorless oil by column
chromatography (hexane/AcOEt = 10/1) in 70% yield and 95% ee. HPLC analysis on
a Daicel Chiralpak IC column: 80/20 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τminor = 6.55 min,
τmajor = 7.12 min. HRMS calcd for (C19H20O5+Na): 351.1208, found 351.1215.[α]rtD= +69.3 (c = 0.61, CHCl3).
1H NMR (400 MHz, CDCl3): δ 0.86 (t, J = 7.6, 3H), 1.21 (d, J = 7.2, 3H), 1.30 (d, J = 7.2, 3H), 2.88 (q, J =
7.2, 1H), 2.99 (d, J = 10.8, 1H), 3.32-3.37 (m, 1H), 3.79-3.89 (m, 2H), 6.78 (d, J = 3.2, 1H), 7.08-7.34 (m,
4H), 9.50 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 13.6, 17.3, 18.5, 32.4, 33.7, 49.9, 50.0, 61.4, 111.0, 123.5,
125.8, 127.5, 129.6, 140.9, 154.1, 171.3, 192.9.
OO
Ph
OHC
OO
CO2EtPh
OHC
OO
CO2Et
OHC
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S17
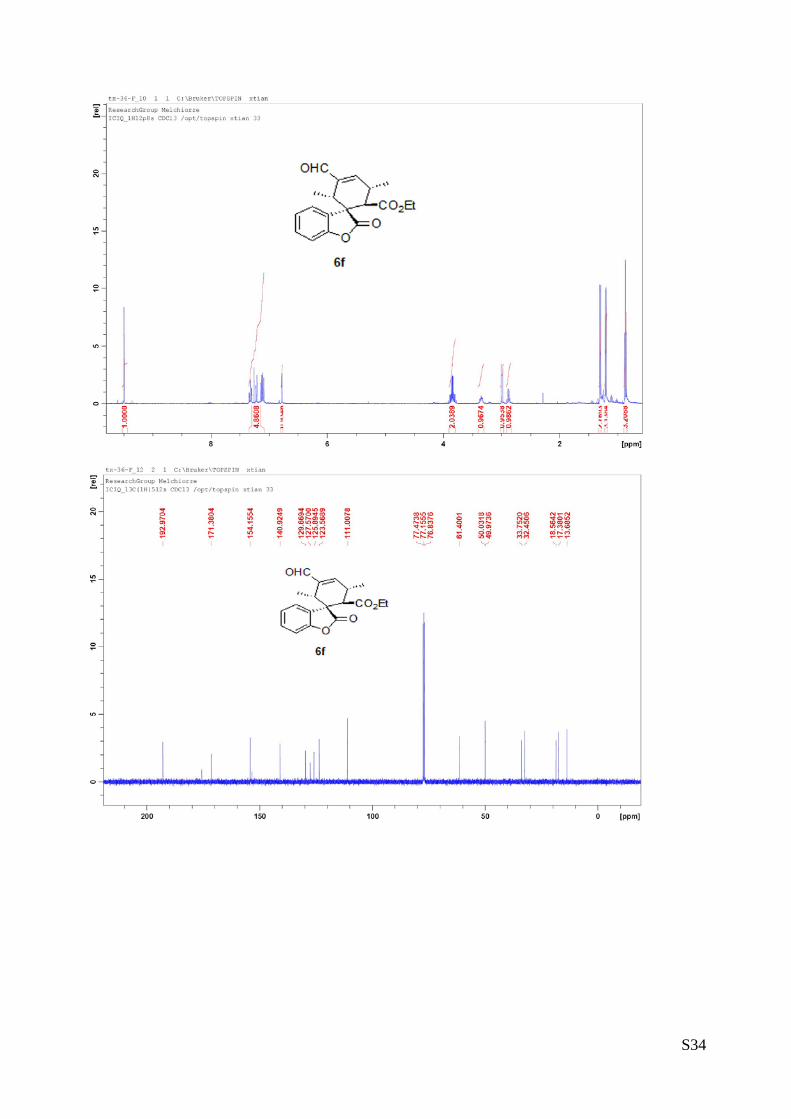
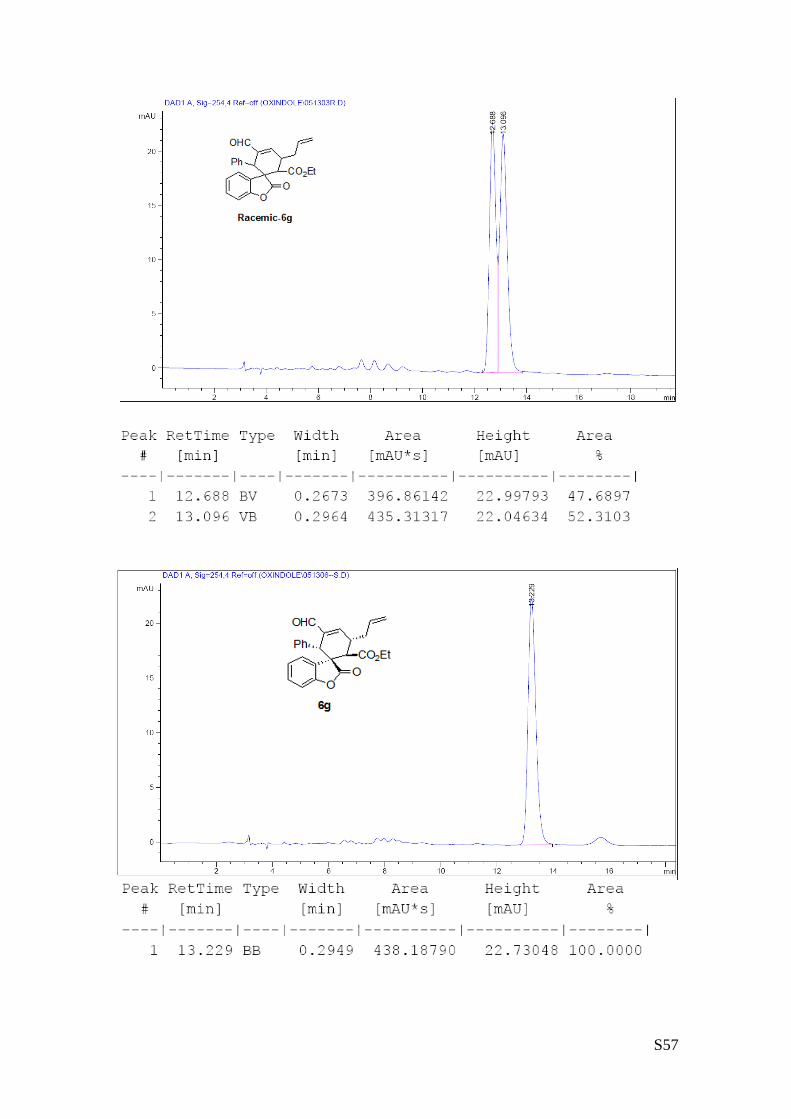
(6g - Table 2, entry 8). The reaction was carried out at 40°C following the general
procedure to furnish the crude product (d.r. >19:1 determined by 1H NMR analysis of
the crude mixture). The title compound was isolated as a white solid by column
chromatography (hexane/AcOEt = 10/1) in 57% yield and >99% ee. HPLC analysis
on a Daicel Chiralpak IA column: 90/10 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm : τminor = 12.7
min, τmajor = 13.22 min. HRMS calcd for (C26H24O5+Na): 439.1521, found 439.1507. [α]rtD= -99.5 (c = 0.77,
CHCl3). 1H NMR (400 MHz, CDCl3): δ 0.8 (t, J = 7.2, 3H), 2.33-2.41 (m, 1H), 2.65-2.71 (m, 1H), 3.16 (d, J = 12,
1H), 3.50-3.56 (m, 1H), 3.78 (q, J = 7.2, 2H), 3.97 (s, 1H), 5.30-5.35 (m, 2H), 5.51 (d, J = 8.4, 1H), 5.93-
6.03 (m, 1H), 6.7(t, J = 8, 1H), 7.06 (d, J = 7.2, 2H), 7.18-7.31 (m, 6H), 9.50 (s, 1H).13C NMR (100 MHz,
CDCl3): δ 13.5, 36.5, 36.6, 45.1, 48.0, 50.5, 61.3, 110.3, 119.7, 122.9, 126.4, 126.5, 128.20, 128.23, 129.5,
133.6, 136.9, 138.8, 152.2, 153.0, 171.2, 191.9.
General Procedure for the Organocatalytic Tandem double Michael additions
(enamine – iminium activation sequence) (Table 3)
All the reactions were carried out in an ordinary vial equipped with a Teflon-coated stir bar, with no
precautions to exclude moisture in undistilled toluene. Catalyst A (0.02 mmol, 20 mol %), aldehyde 5 (0.2
mmol) and o-fluoro-benzoic acid (20 mol %) were stirred in toluene (0.2 mL) at room temperature for 10
minutes. Catalyst B (0.02 mmol, 20 mol %), enone 7 (0.2 mmol) and o-fluoro-benzoic acid (30 mol %) were
stirred in toluene (0.1 mL) at room temperature for 10 minutes. The cascade was started by adding a
solution of benzofuranone derivative 4 (0.15 mmol) dissolved in 1 mL of toluene. The vial was sealed and
heated up to 60 degrees, and stirring was continued till completion of the reaction, as monitored by TLC
(generally 72 hours). The crude reaction was then flushed through a short plug of silica, using CH2Cl2/Et2O
1/1 as the eluent. Solvent was removed in vacuo and, after NMR analysis to determine the diastereomeric
ratio of the process, the residue was purified by flash column chromatography (silica gel) to yield the desired
spiro-compounds 8.
OO
CO2EtPh
OHC
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S18
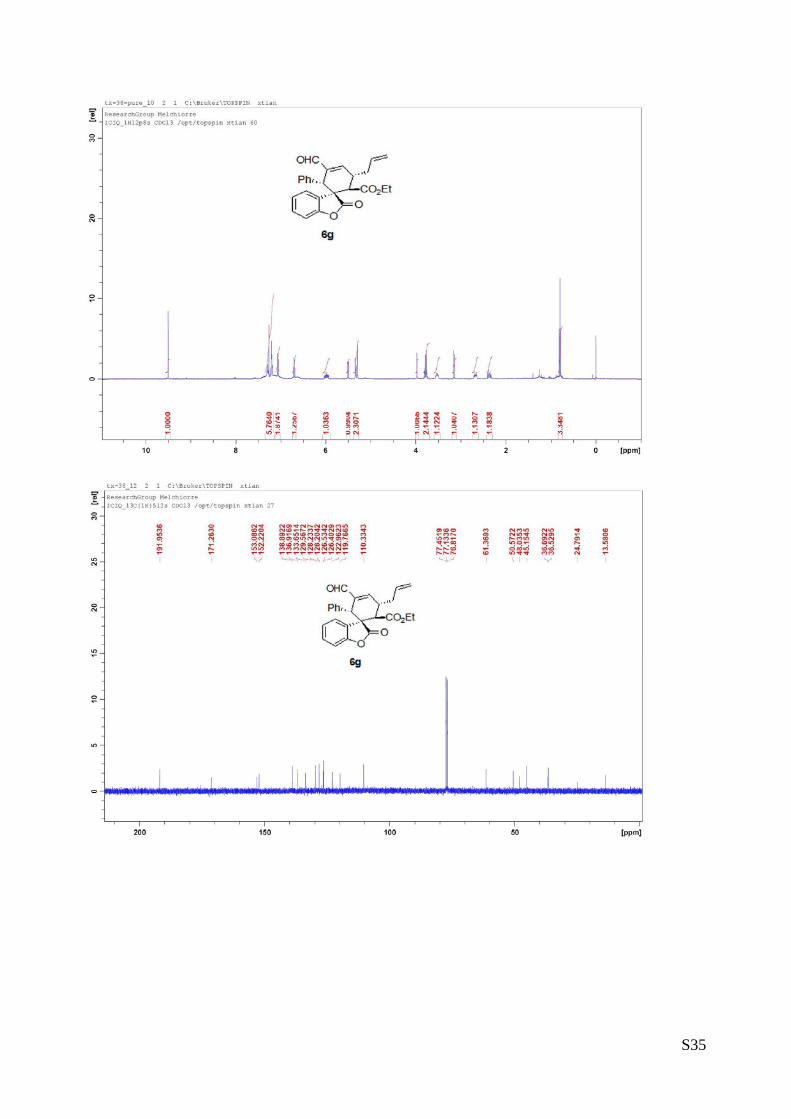
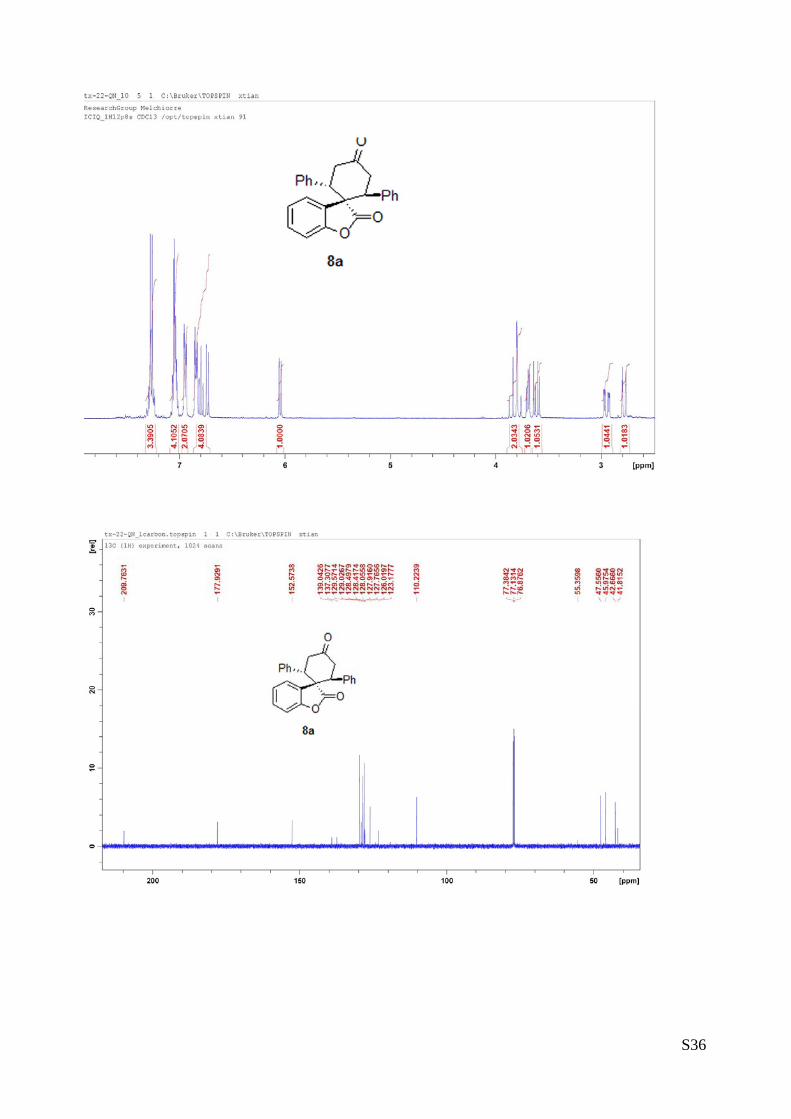
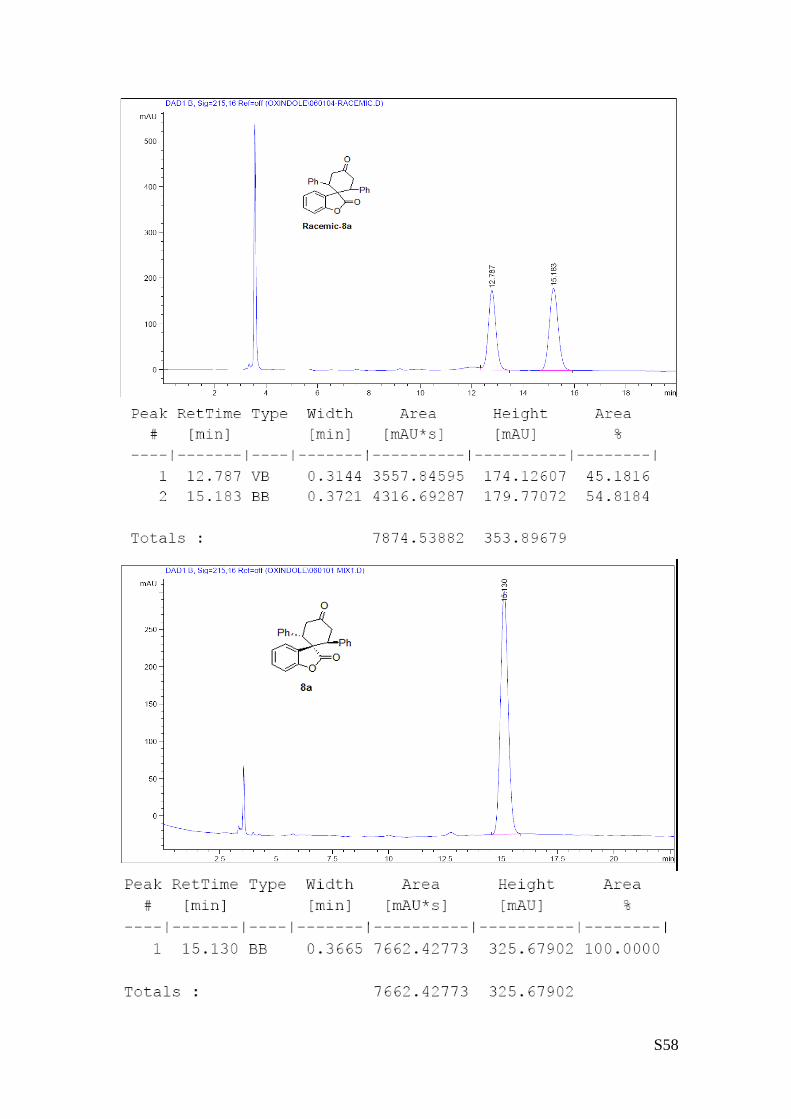
(8a - Table 3, entry 1).The reaction was carried out at 60°C following the general
procedure to furnish the crude product as a 6.3/1 mixture of diastereoisomers (dr. 6.3:1
determined by integration of 1H NMR signal: δminor 6.45 d, δmajor 6.06 ppm, d). The title
compound was isolated as a white solid by column chromatography (hexane/AcOEt =
90/10) in 70% yield (as a single of diastereoisomer) and >99% ee. HPLC analysis on a
Daicel Chiralpak IC column: 80/20 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τminor = 12.8 min,
τmajor = 15.13 min. HRMS calcd for (C25H20NO3+Na): 391.1310, found 391.1329. [α]rtD= -195.8 (c = 0.76,
CHCl3).1H NMR (400 MHz, CDCl3): δ 2.76 (d, J =13.2, 1H), 2.95 (dd, J = 5.2, 16, 1H), 3.61 (dd, J = 6, 15.6,
1H), 3.68-3.71 (m, 1H), 3.76-3.87 (m, 1H), 6.04 (d, J = 7.2, 1H), 6.73-6.86 (m, 4H), 6.93-6.96 (m, 2H),
7.03-7.07 (m, 4H), 7.24-7.29 (m, 3H).13C NMR (100 MHz, CDCl3): δ 41.8, 42.6, 45.9, 47.5, 110.2,
123.1,126.0, 127.7, 127.9, 128.0, 128.41, 128.49, 129.0, 129.5, 137.3, 139.0, 152.5, 177.9, 209.7.
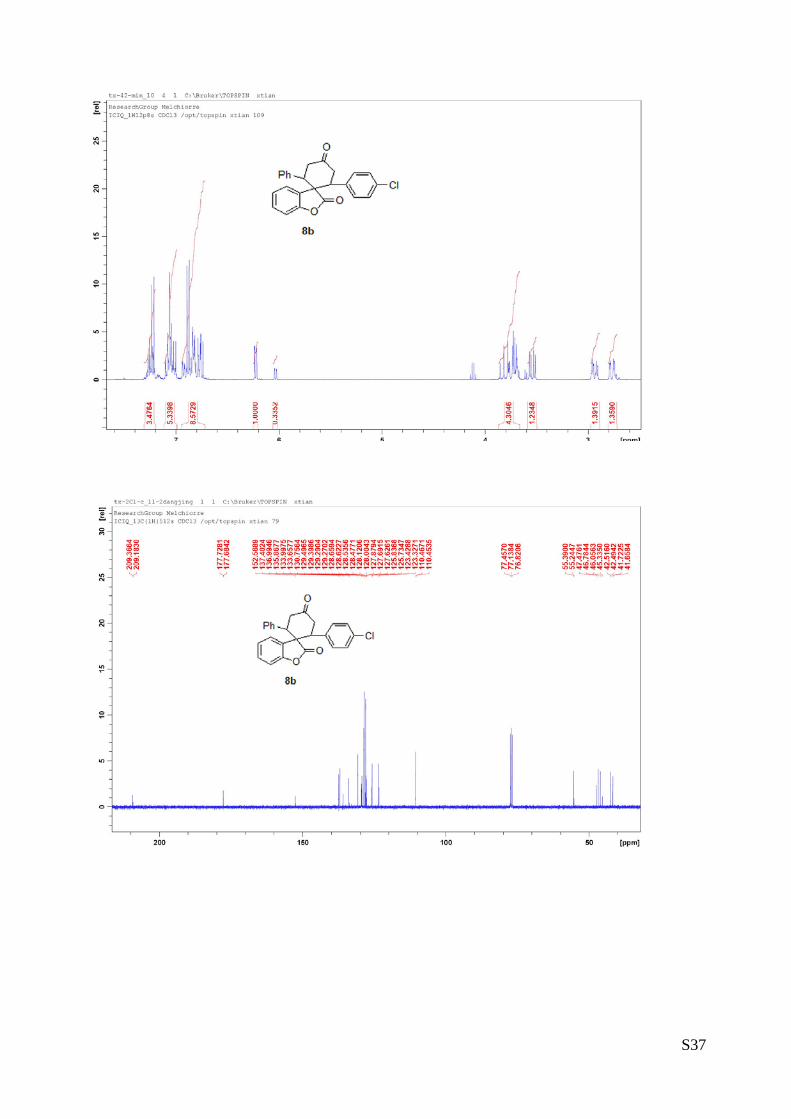
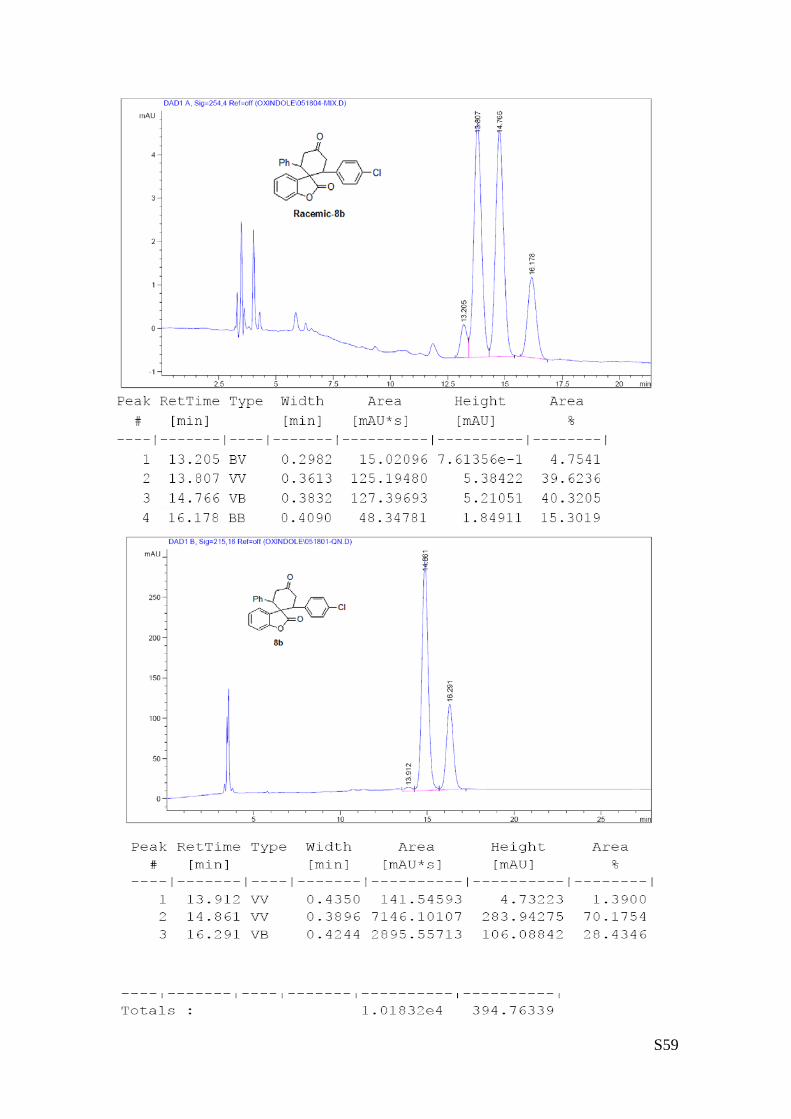
(8b - Table 3, entry 3). The reaction was carried out at 60°C following the
general procedure to furnish the crude product as a 3.5/1 mixture of
diastereoisomers (d.r. 3.5:1 determined by integration of 1H NMR signal: δmajor
6.25 d, δminor 6.05 ppm. d). The title compound was isolated as a white solid
by column chromatography (hexane/AcOEt = 90/10) in 75% yield (as a 3:1
mixture of diastereoisomers) and 97% ee. HPLC analysis on a Daicel Chiralpak IC column: 80/20 hexane/i-
PrOH, flow rate 1.0 mL/min, λ = 254 nm: Major diastereomer (97% ee): τminor = 13.91 min, τmajor = 14.86
min; Minor diastereomer (>99% ee): τminor = 13.20 min, τmajor = 16.29 min. HRMS calcd for (C25H19ClO3+Na):
425.0920, found 425.0937. [α]rtD= -155.7 (c = 0.78, CHCl3, dr 3.5:1, 97% eemajor, >99% eeminor).
1H NMR (400 MHz, CDCl3): δ 2.73-2.79 (m, 1H), 2.91-2.97 (m, 1H), 2.53-2.67 (m, 3H), 3.54 (dd, J = 6.12,
16.2, 1H), 3.67-3.85 (m, 4H), 6.03 (d, J = 7.6, 1H minor), 6.23 (d, J = 8, 1H major), 6.74-6.93 (m, 8H),
7.0-7.10 (m, 5H), 7.21-7.27 (m, 3H).13C NMR (100 MHz, CDCl3): δ 41.6, 41.7, 42.4, 42.5, 45.3, 46.0, 46.7,
47.4, 55.2, 55.3, 110.45, 110.46, 123.3, 123.4, 125.7, 125.8, 127.62, 127.69, 127.8, 128.0, 128.1, 128.4,
128.5, 128.62, 128.65, 129.27, 129.29, 129.3, 129.4, 130.7, 133.9, 135.8, 136.9, 137.4, 152.5, 177.6,
177.7, 209.1, 209.3.
The absolute configuration of 8b was unambiguously determined by single crystal X-ray
analysis, see page S7.
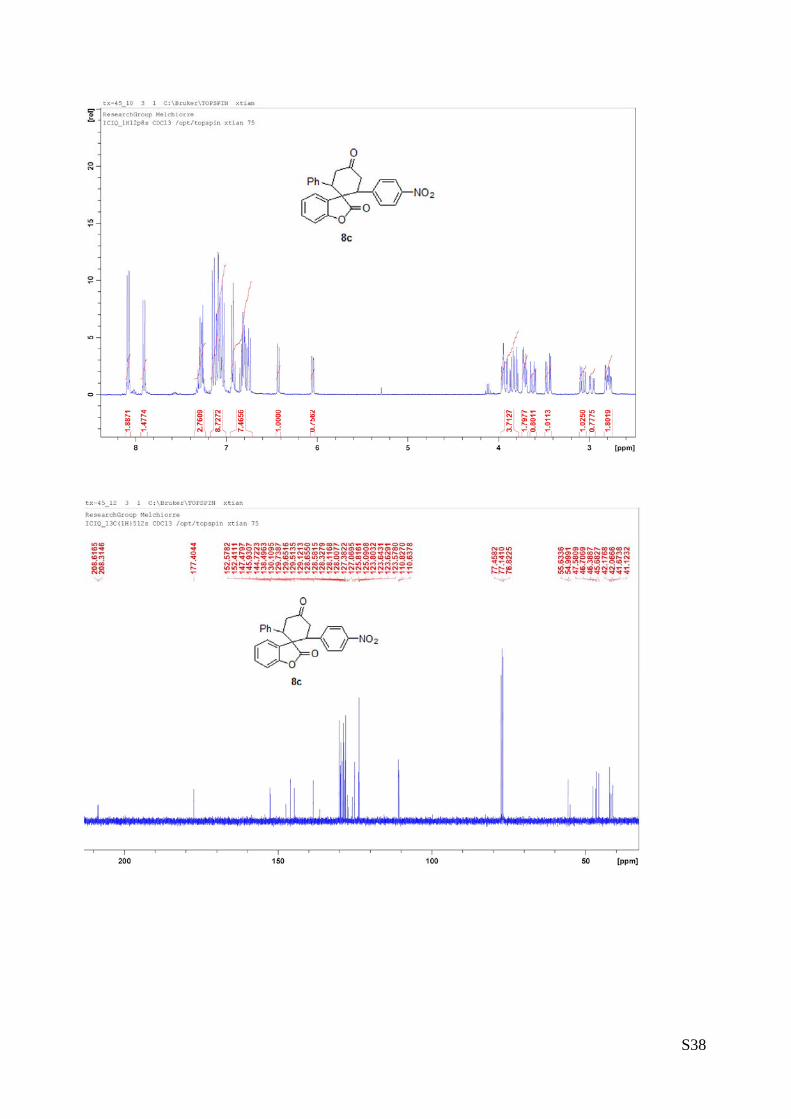
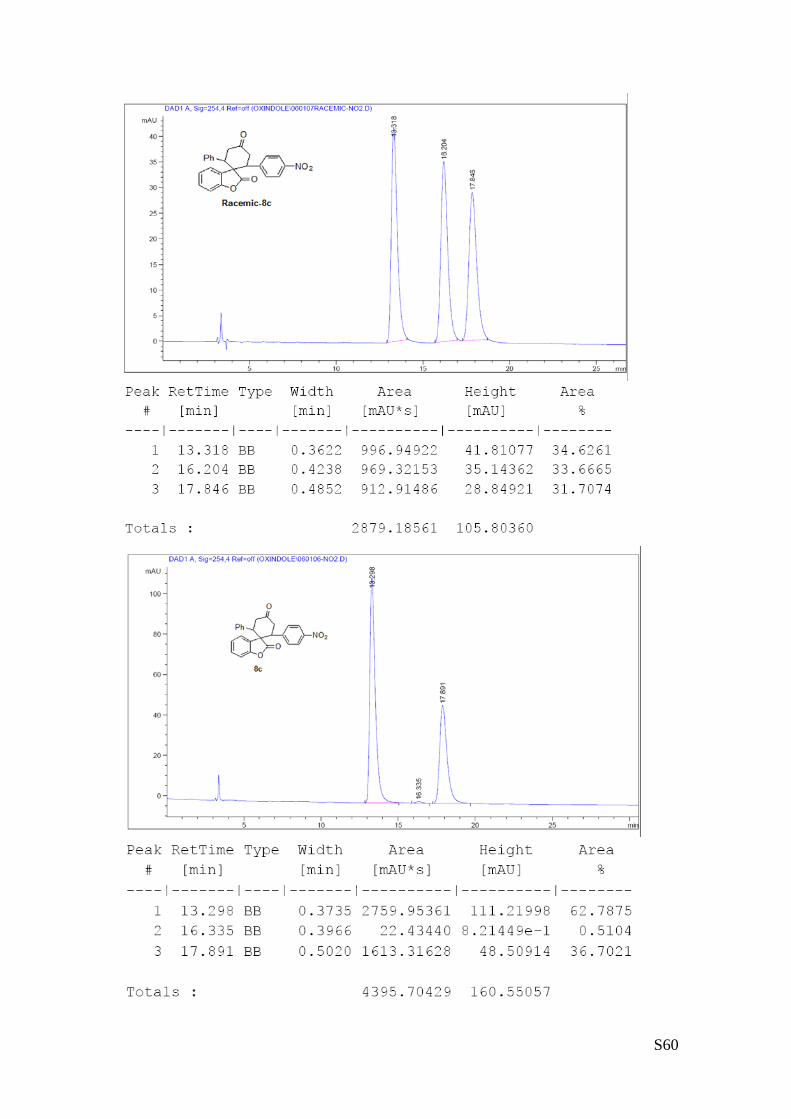
(8c - Table 3, entry 4).The reaction was carried out at 60°C following the
general procedure to furnish the crude product as a 2/1 mixture of
diastereoisomers (d.r. 2:1 determined by integration of 1H-NMR signal: δmajor
6.46 d, δminor 6.08 ppm. d). The title compound was isolated as a white solid
by column chromatography (hexane/AcOEt = 7/1) in 82% yield (as a 2:1
mixture of diastereoisomers) and 98% ee. HPLC analysis on a Daicel Chiralpak IA column: 80/20 hexane/i-
PrOH, flow rate 1.0 mL/min, λ = 254 nm: Major diastereomer (98% ee): τmajor = 13.29 min, τminor = 16.33
OO
O
PhPh
OO
O
PhCl
OO
O
PhNO2
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S19
min; Minor diastereomer can’t be separated. HRMS calcd for (C25H19NO5+Na): 436.1161, found 436.1173.
[α]rtD= -240.2 (c = 0.8, CHCl3, dr 2:1, 98% eemajor).
1H NMR (400 MHz, CDCl3): δ 2.75-2.82 (m, 1H), 2.97 (dd, J = 4.76, 16.2, 1H, minor), 3.07 (dd, J = 7.76,
16.56, 1H, major), 3.62 (dd, J = 6.4, 16, 1H, major), 3.45 (dd, J = 5.6, 16.32, 1H, minor), 3.69-3.73 (m,
2H), 3.78-3.96 (m, 4H), 6.05 (d, J = 8, 1H, minor), 6.42 (d, J = 8, 1H, major), 6.73-6.93 (m, 7H), 7.02-
7.05(m, 8H), 7.24-7.30 (m, 3H), 7.90 (d, J = 8.4, 2H, minor), 8.09 (d, J = 8.8, 2H, major).13C NMR (100
MHz, CDCl3): δ 41.1, 41.6, 42.0, 42.1, 45.6, 46.3, 46.7, 47.5, 54.9, 55.6, 110.6, 110.8, 123.5, 123.62,
123.64, 123.8, 125.0, 125.8, 127.0, 127.3, 128.0, 128.1, 128.3, 128.5, 128.6, 129.1, 129.5, 129.6, 129.7,
130.1, 138.4, 144.7, 145.9, 152.4, 152.5, 177.4, 208.3, 208.6.
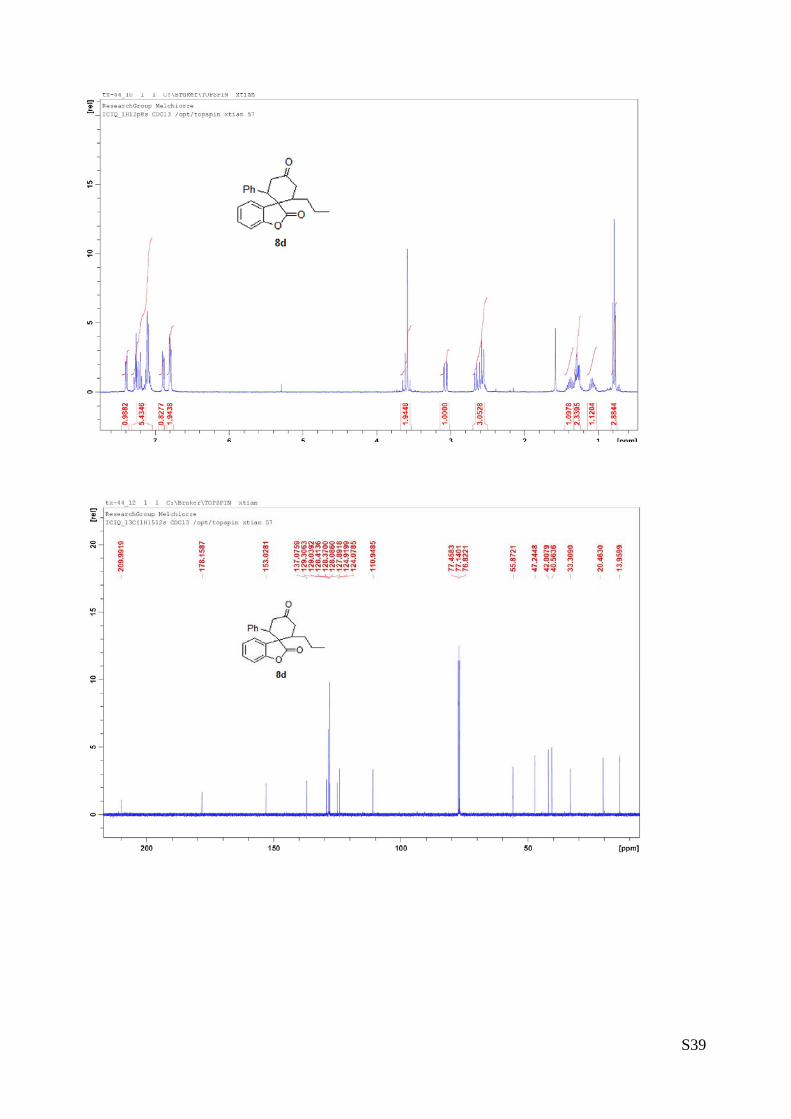
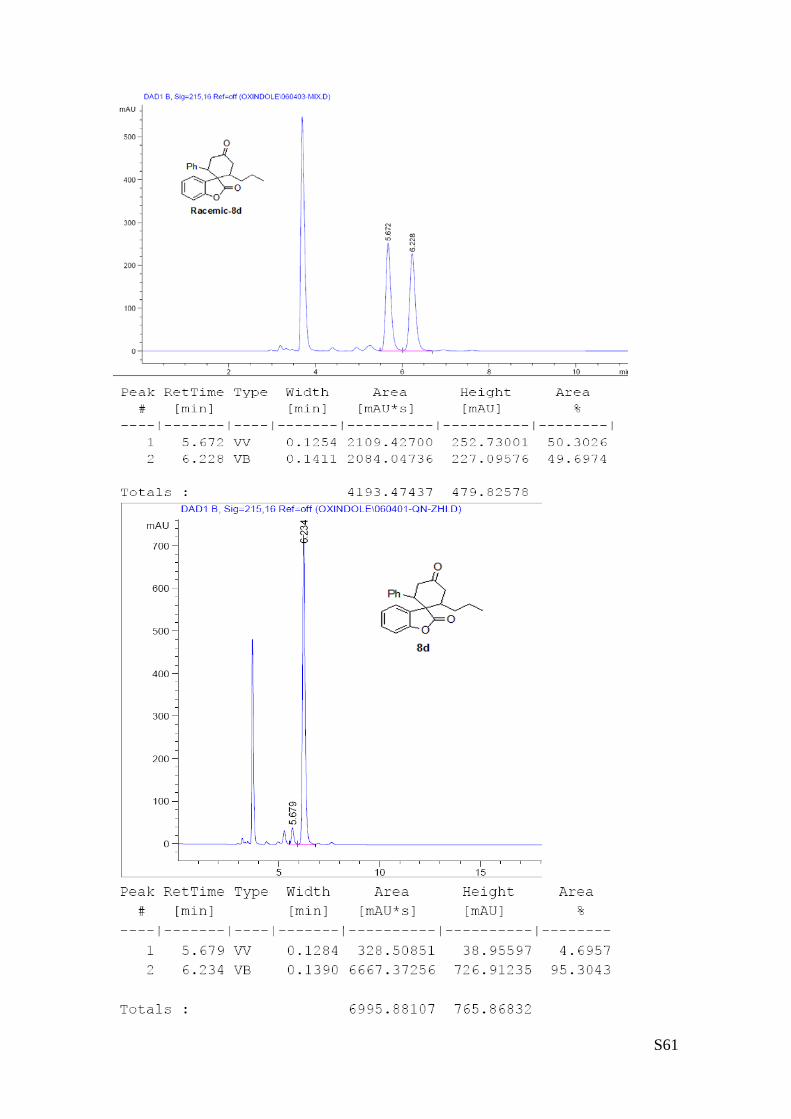
(8d - Table 3, entry 5).The reaction was carried out at 60°C following the general
procedure to furnish the crude product as a single diastereoisomer. The title
compound was isolated as a white solid by preparative TLC (toluene/AcOEt = 100/1)
in 40% yield (as a single diastereoisomer) and 91% ee. HPLC analysis on a Daicel
Chiralpak IA column: 80/20 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: τminor
= 5.67 min, τmajor = 6.23 min; HRMS calcd for (C22H22O3+Na): 357.1467, found 357.1456. [α]rtD= -118.2 (c =
0.55, CHCl3, dr 1.5:1, 98% eemajor, 88% eeminor). 1H NMR (400 MHz, CDCl3): δ 0.78 (t, J = 7.2, 3H), 1.03-1.12 (m, 1H), 1.25-1.43 (m, 3H), 2.53-2.67 (m, 3H),
3.07 (dd, J = 4, 15.6, 1H), 3.55-3.65 (m, 2H), 6.78-6.81 (m, 2H), 6.89 (d, J = 8, 1H), 7.07-7.28 (m, 5H),
7.39 (d, J = 8, 1H).13C NMR (100 MHz, CDCl3): δ 13.9, 20.4, 33.3, 40.5, 42.0, 47.2, 55.8, 110.9, 124.0,
124.9, 127.8, 128.0, 128.3, 128.4, 129.0, 129.3, 137.0, 153.0, 178.1, 209.9.
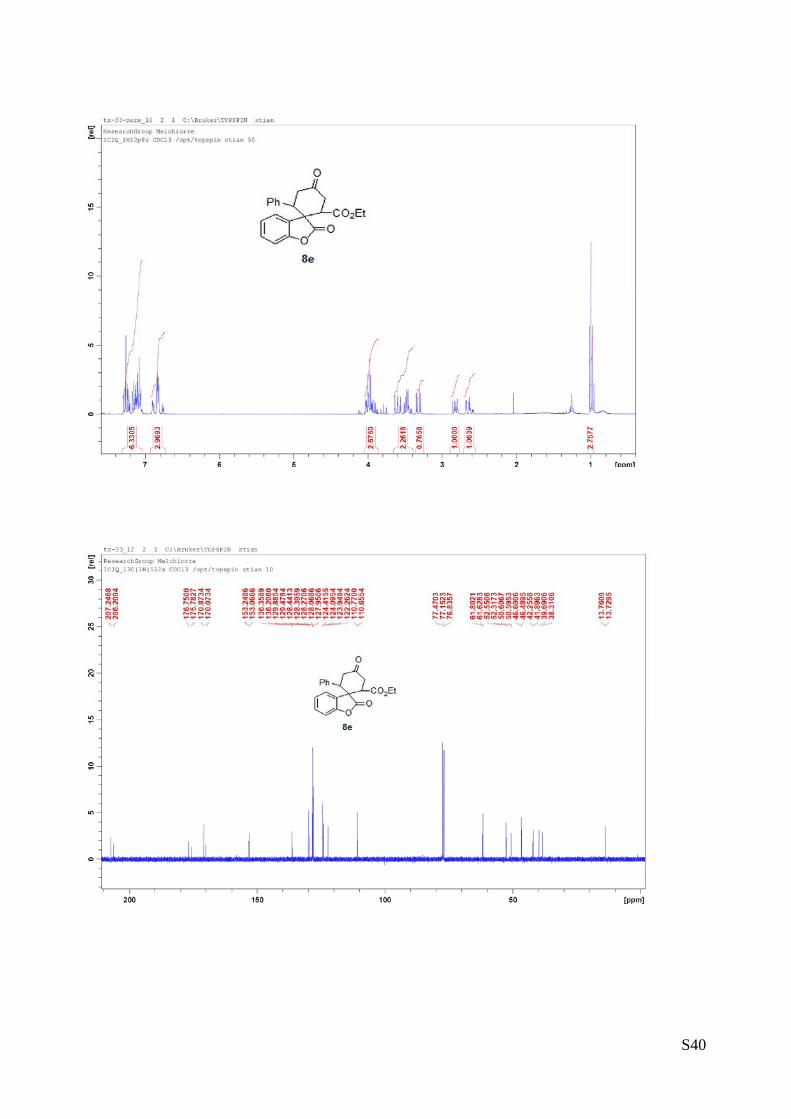
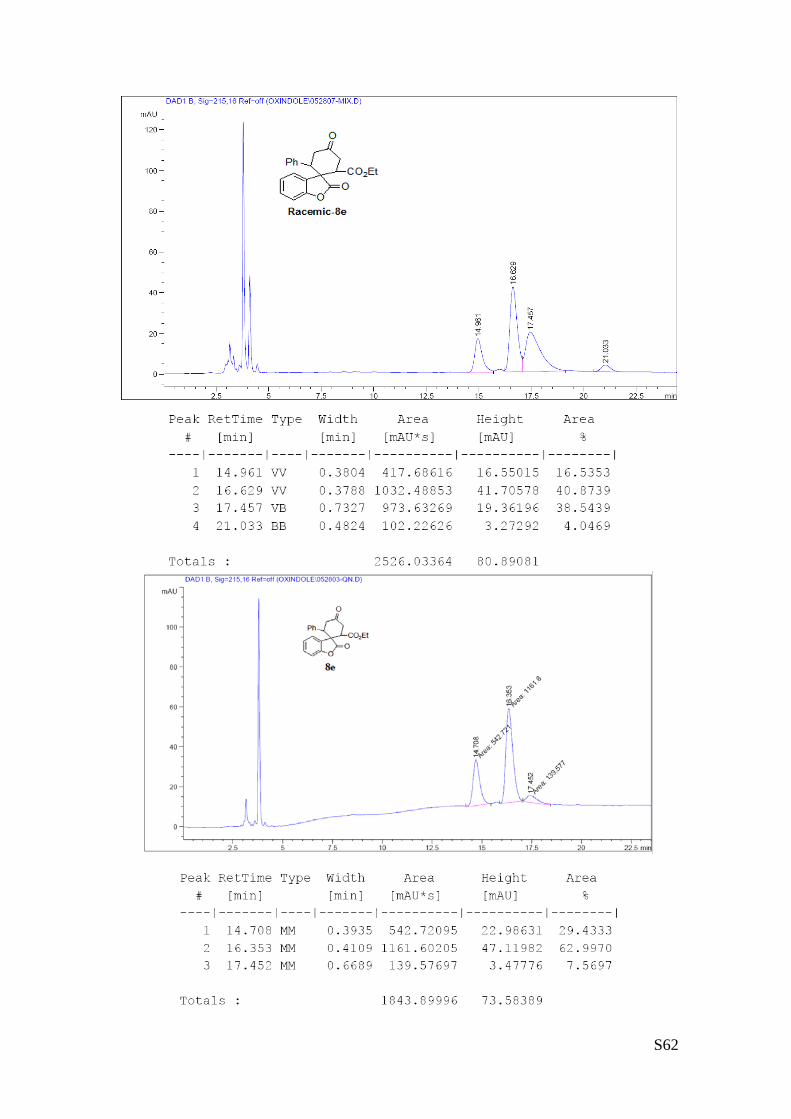
(8e - Table 3, entry 6).The reaction was carried out at 60°C following the general
procedure to furnish the crude product as a 2.2/1 mixture of diastereoisomers (d.r.
2.2:1 determined by HPLC). The title compound was isolated as a white solid by
column chromatography (hexane/AcOEt = 10/1) in 77% yield (as a 2.2:1 mixture of
diastereoisomers) and 85% ee. HPLC analysis on a Daicel Chiralpak IA column:
90/10 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: Major diastereomer (85% ee): τmajor = 16.35 min,
τminor = 17.45 min; Minor diastereomer (>99% ee):τmajor = 14.7 min, HRMS calcd for (C22H20O5+Na):
387.1208, found 428.1225. [α]rtD= -57.2 (c = 0.58, CHCl3,).
1H NMR (400 MHz, CDCl3): δ 1.0 (t, J = 7.2, 3H), 2.63-2.68 (m, 1H), 2.80-2.86 (m, 1H), 3.32 (dd, J = 5.6,
16, 1H), 3.42-3.63 (m, 2H), 6.75-6,91 (m, 3H), 7.06-7.29 (m, 6H).13C NMR (100 MHz, CDCl3): δ 13.73,
13.76, 38.3, 39.6, 41.9, 42.2, 46.4, 46.6, 50.60, 50.61, 52.3, 52.5, 61.6, 61.8, 110.6, 110.7, 122.2, 123.9,
124.1, 124.4, 127.9, 128.0, 128.2, 128.40, 128.44, 129.4, 129.8, 136.2, 136.3, 153.0, 153.2, 170.0, 170.8,
175.7, 176.7, 206.2, 207.2.
OO
Ph
O
OO
CO2EtPh
O
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010

S20
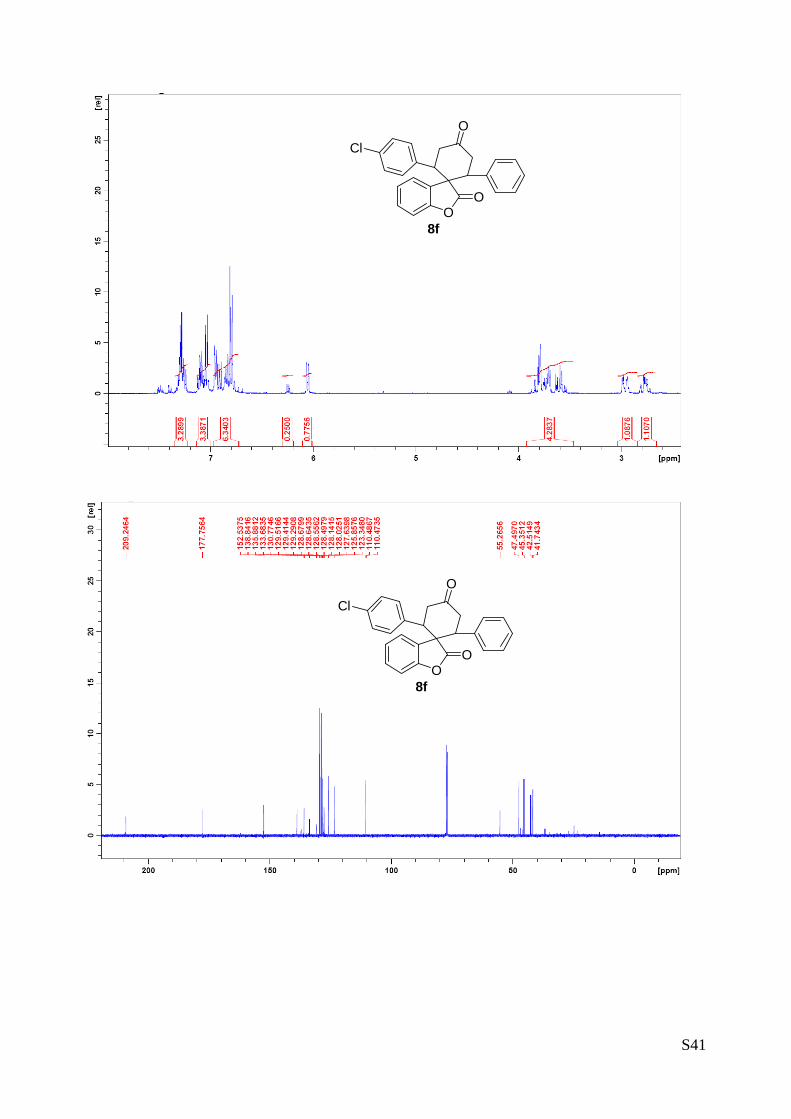
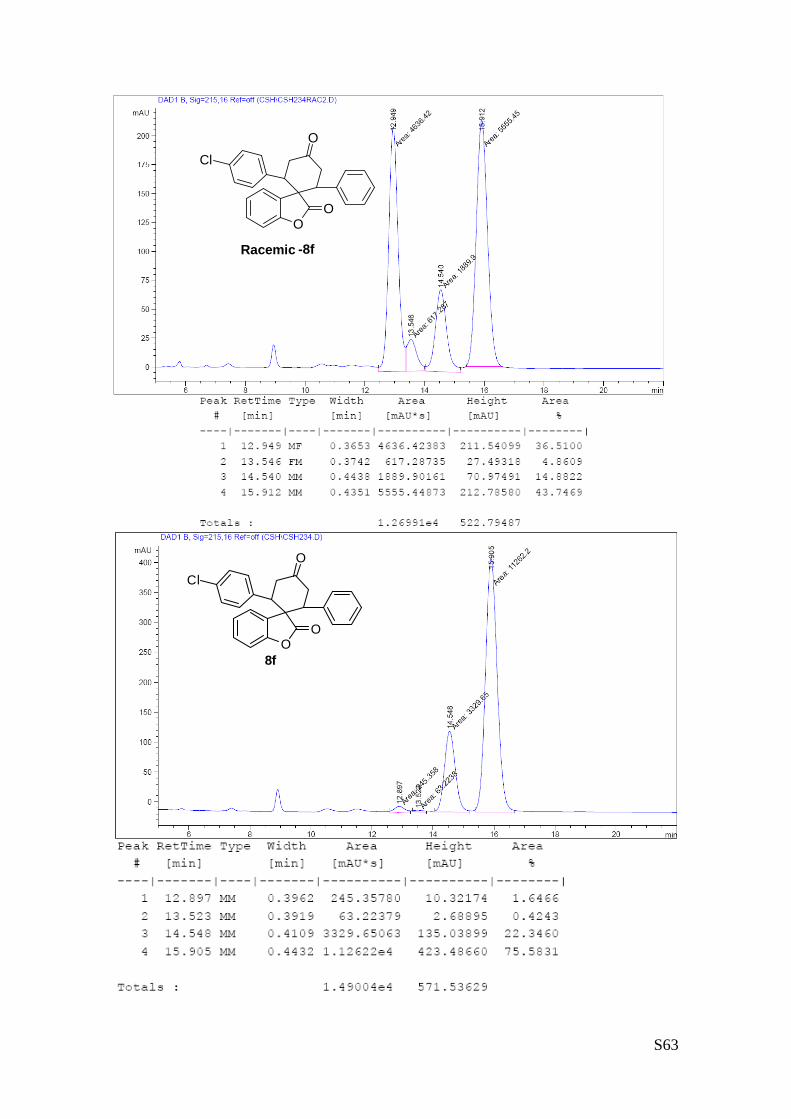
(8f - Table 3, entry 7).The reaction was carried out at 60°C following the
general procedure to furnish the crude product as a 4/1 mixture of
diastereoisomers (d.r. 3.5:1 determined by integration of 1H NMR signal:
δmajor 6.05 d, δminor 6.24 ppm. d). The title compound was isolated as a
white solid by column chromatography (hexane/AcOEt = 85/15) in 85%
yield (as a 3.4:1 mixture of diastereoisomers) and 97% ee. HPLC analysis on a Daicel Chiralpak IC column:
8/2 hexane/i-PrOH, flow rate 1.0 mL/min, λ = 254 nm: Major diastereomer (97% ee): τminor = 12.9 min,
τmajor = 15.9 min; Minor diastereomer (96% ee): τminor = 13.5 min, τmajor = 14.5 min. HRMS calcd for
(C25H19Cl O3+Na): 425.0920, found 425.0938. [α]rtD= -130.6 (c = 1.07, CHCl3, dr 4:1, 97% eemajor, 96%
eeminor). 1H NMR (400 MHz, CDCl3): δ 2.71-2.84 (m, 1H), 2.91-3.01 (m, 1H), 3.53.3.88 (m, 4H), 6.05 (d, 1H, J = 7.5
Hz major), 6.24 (d, 1H, J = 7.5 Hz minor), 6.75.-6.98 (m, 6H), 7.01-7.13 (m, 3H), 7.22-7.34 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 41.7 (CH2), 42.5 (CH2), 45.3 (CH), 47.5 (CH), 55.3 (C), 110.5 (CH), 123.3
(CH), 125.8 (CH), 127.6 (C), 128.0 (CH), 128.1 (CH), 128.5 (CH), 128.6 (C), 128.6 (C), 128.7 (C), 129.3
(CH), 129.4 (CH), 129.5 (CH), 130.8 (C), 133.7 (C), 135.9 (C), 138.8 (C), 152.5 (C), 177.7 (C), 209.2 (C)
ppm.
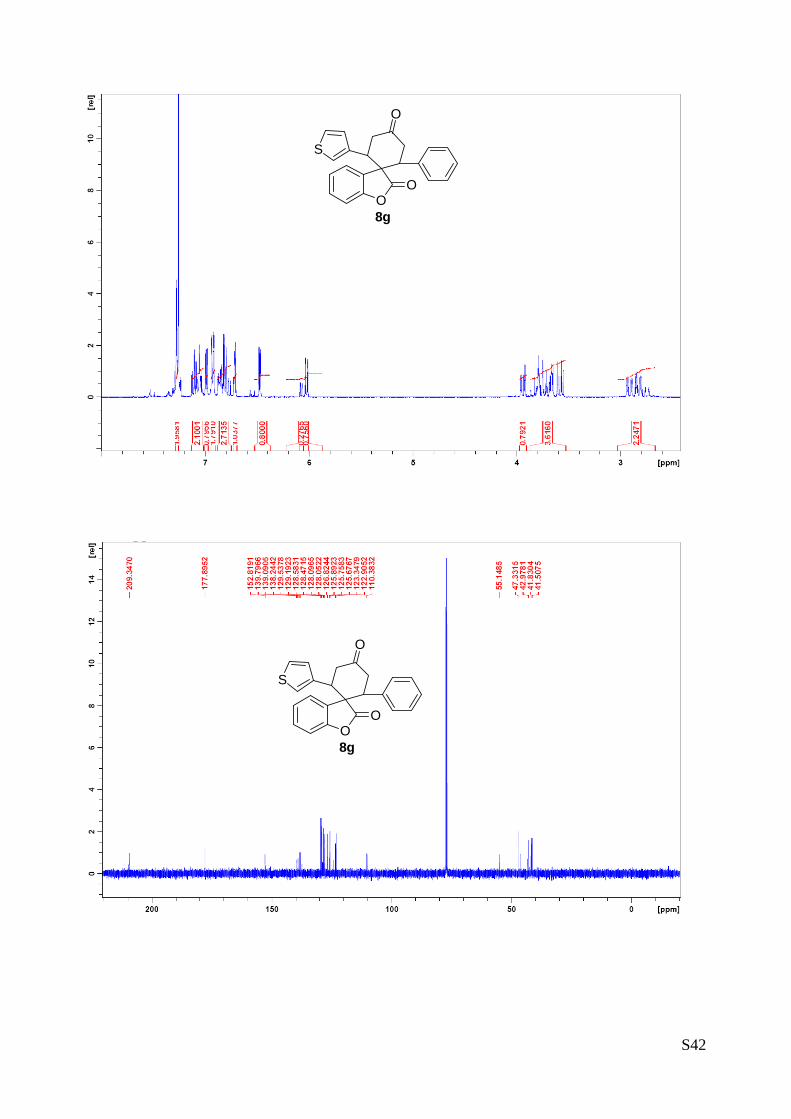
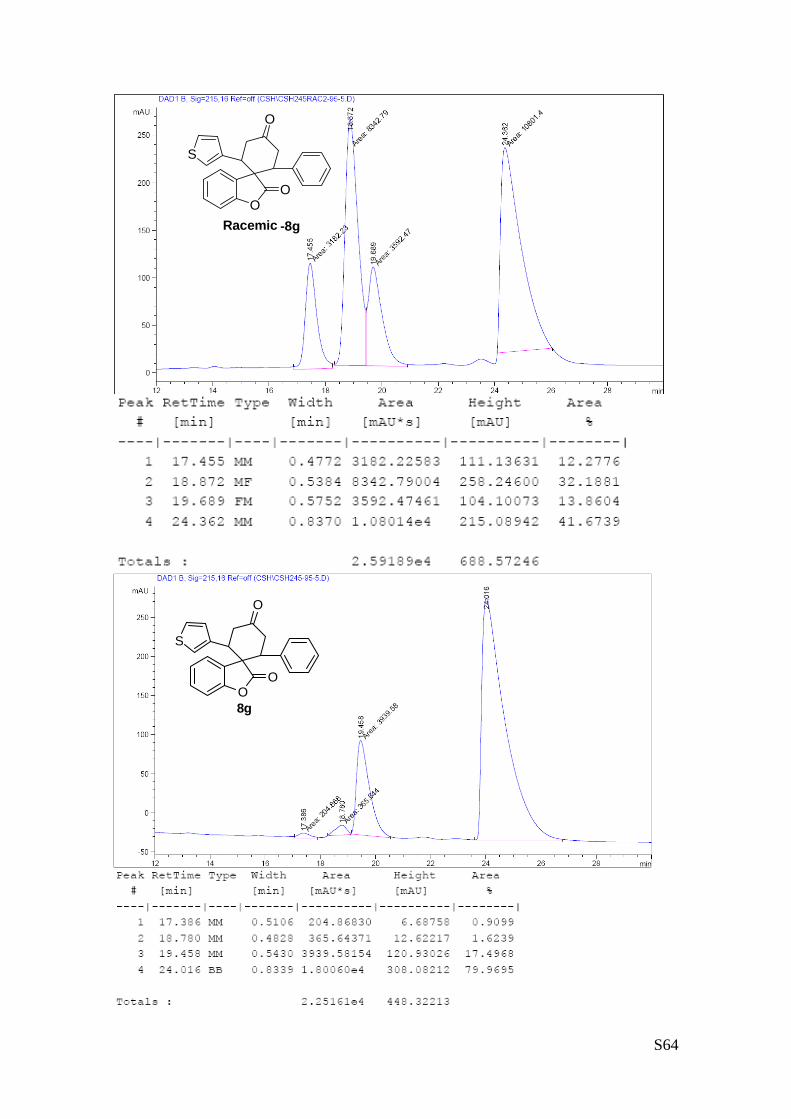
(8g - Table 3, entry 8).The reaction was carried out at 60°C following the
general procedure to furnish the crude product as a 4/1 mixture of
diastereoisomers (d.r. 4:1 determined by integration of 1H-NMR signal: δmajor 6.02
d, δminor 6.07 ppm. d). The title compound was isolated as a white solid by
column chromatography (hexane/Acetone = 95/5) in 91% yield (as a 4:1 mixture
of diastereoisomers) and 96% ee. HPLC analysis on a Daicel Chiralpak IA column: 95/5 hexane/i-PrOH, flow
rate 1.0 mL/min, λ = 254 nm: Major diastereomer (96% ee): τminor = 18.7 min, τmajor = 24.4 min; Minor
diastereomer (93% ee): τminor = 17.3 min, τmajor = 19.6 min. [α]rtD= -99.1 (c = 0.78, CHCl3, dr 4:1, 96%
eemajor, 93% eeminor). 1H NMR (400 MHz, CDCl3): δ 2.72-2.95 (m, 2H), 3.54.3.84 (m, 4H), 3.94 (dd, 1H, J1 = 13.7, J2= 4.2 Hz),
6.02 (d, 1H, J = 7.5 Hz major), 6.07 (d, 1H, J = 7.5 Hz minor), 6.47 (dd, 1H, J1= 5.2, J2= 1.4 Hz ), 6.70-
6.73 (m, 1H), 6.75-6.88 (m, 2H), 6.91-6.94 (m, 2H), 6.99 (dd, 1H, J1= 4.9, J2= 2.9 Hz), 7.02-7.16 (m, 2H),
7.26-7.30 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ 41.5 (CH), 41.8 (CH2), 42.9 (CH2), 47.3 (CH), 55.1 (C), 110.4 (CH), 122.9
(CH), 123.3 (CH), 125.7 (C), 125.8 (CH), 125.9 (CH), 126.8 (CH), 128.0 (C), 128.1 (CH), 128.5 (CH), 128.6
(CH), 129.1 (CH), 129.5 (CH), 138.2 (C), 139.1 (C), 139.7 (C), 152.8 (C), 177.9 (C), 209.3 (C) ppm.
OO
O
Cl
OO
O
S
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
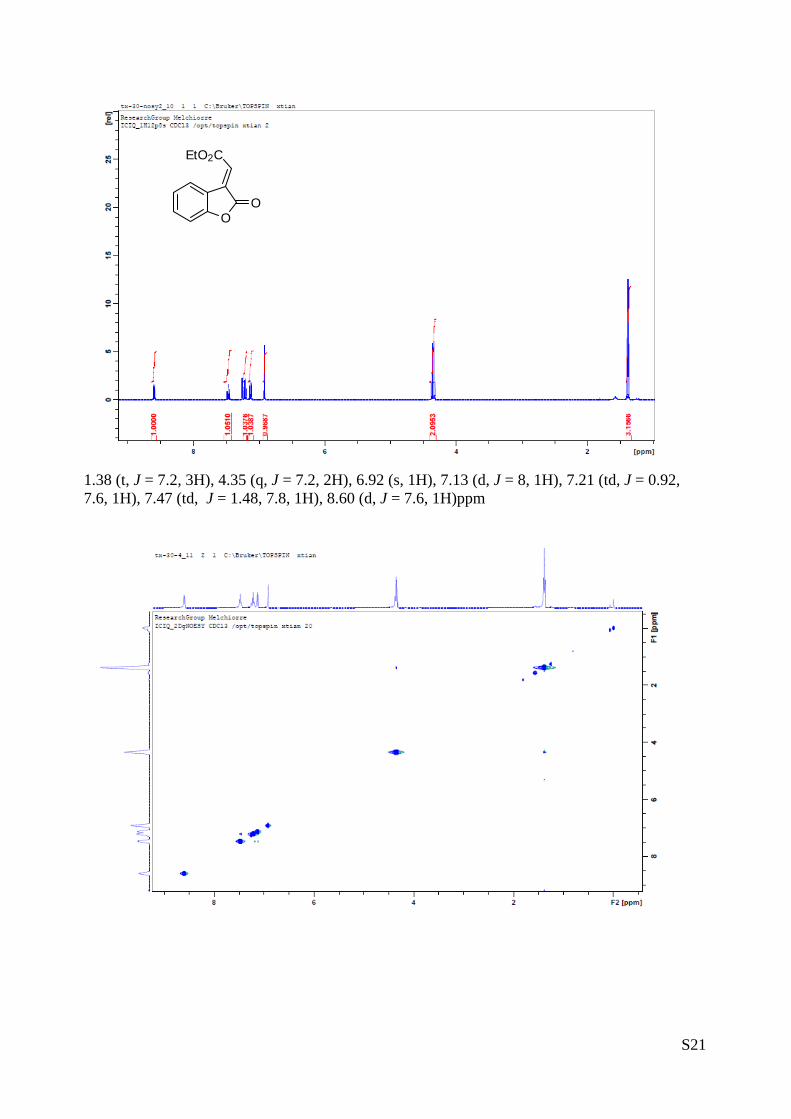
S21
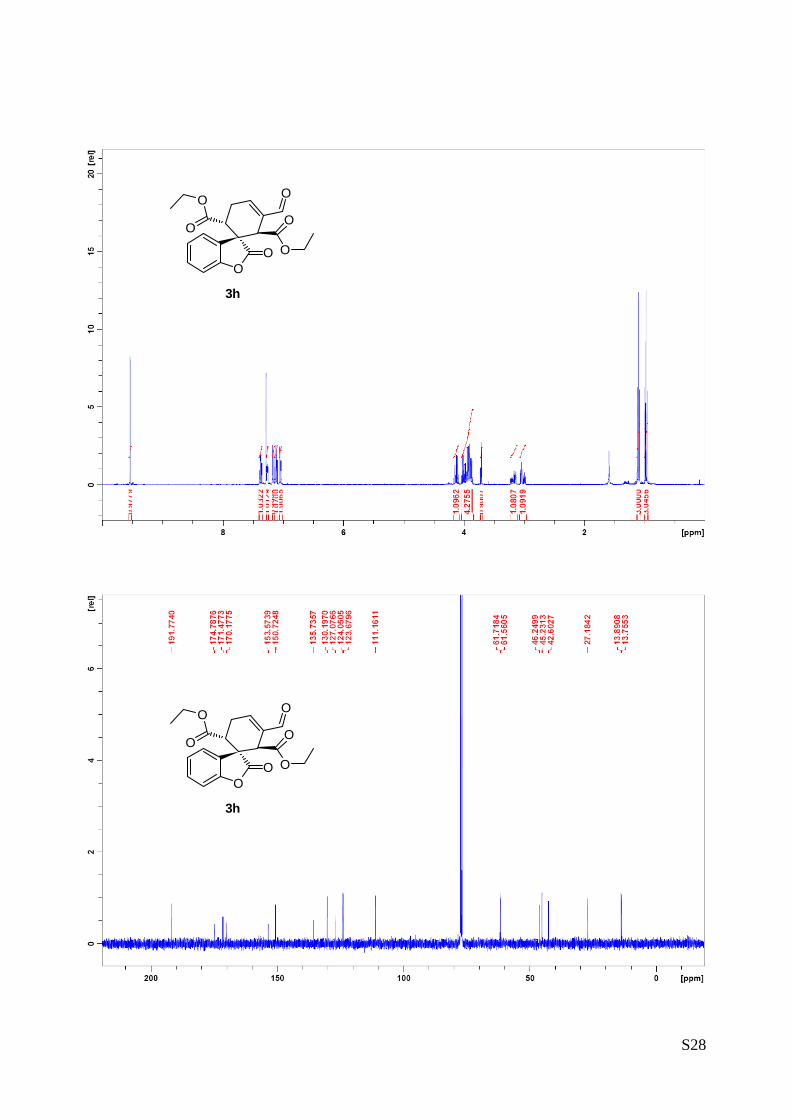
1.38 (t, J = 7.2, 3H), 4.35 (q, J = 7.2, 2H), 6.92 (s, 1H), 7.13 (d, J = 8, 1H), 7.21 (td, J = 0.92, 7.6, 1H), 7.47 (td, J = 1.48, 7.8, 1H), 8.60 (d, J = 7.6, 1H)ppm
OO
EtO2C
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S22
OO
O
3aO
O
O
3a
OO
O
3a
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S23
OO
O
NO2
O2N
3b
OO
O
NO2
O2N
3b
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S24
OO
O
O
O
3c
OO
O
O
O
3c
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S25
OO
O
Cl
Cl
3d
OO
O
Cl
Cl
3d
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S26
OO
OO
O
3e
OO
OO
O
3e
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S27
OO
O
3g
OO
O
3g
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S28
OO
O
O
O
O
O
3h
OO
O
O
O
O
O
3h
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S29
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S30
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S31
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S32
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S33
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S34
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S35
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S36
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S37
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S38
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S39
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S40
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S41
OO
OCl
8f
OO
OCl
8f
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S42
OO
O
8g
S
OO
O
8g
S
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S43
OO
O
Racemic
OO
O
3a obtained using cat. (S)-A
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S44
OO
O
obtained using cat. (R)-A
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S45
OO
O
NO2
O2N
Racemic
OO
O
NO2
O2N
3b
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S46
OO
O
O
O
Racemic
OO
O
O
O
3c
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S47
OO
O
Cl
Cl
Racemic
OO
O
Cl
Cl
3d
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S48
OO
OO
O
Racemic
OO
OO
O
3e
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S49
OO
O
Racemic
OO
O
3g
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S50
OO
O
O
O
O
O
Racemic
OO
O
O
O
O
O
3h
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S51
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S52
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S53
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S54
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S55
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S56
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S57
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S58
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S59
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S60
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S61
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S62
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S63
OO
OCl
-8fRacemic
OO
O
Cl
8f
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010
S64
-8gO
O
O
Racemic
S
OO
O
8g
S
Supplementary Material (ESI) for Chemical CommunicationsThis journal is (c) The Royal Society of Chemistry 2010