author's personal copy - connecting repositoriesauthor's personal copy g. liciniet al. /...
TRANSCRIPT

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Coordination Chemistry Reviews 255 (2011) 2345– 2357
Contents lists available at ScienceDirect
Coordination Chemistry Reviews
jo ur n al homepage: www.elsev ier .com/ locate /ccr
Review
Recent advances in vanadium catalyzed oxygen transfer reactions
Giulia Licinia,∗, Valeria Conteb, Alessia Colettib, Miriam Mbaa, Cristiano Zontaa
a Dipartimento di Scienze Chimiche, Università di Padova, Via Marzolo 1, 35131 Padova, Italyb Dipartimento di Scienze e Tecnologie Chimiche, Università di Roma “Tor Vergata”, Via della Ricerca Scientifica snc, 00133 Roma, Italy
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23452. Sulfoxidations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2346
2.1. Di and trialkanolamine derivatives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23462.2. Di and triphenolamine derivatives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23462.3. Polyhedral oligomeric silsesquioxane trisilanolate (POSS) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23472.4. Schiff base systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2349
3. Epoxidations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23503.1. Epoxidation of allylic and homoallylic alcohols . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23513.2. Epoxidation of simple olefins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2354
4. Haloperoxidations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23545. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2356
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2356
a r t i c l e i n f o
Article history:Received 20 February 2011Accepted 1 May 2011Available online 7 May 2011
Keywords:VanadiumOxidationsEpoxidationSulfoxidationHaloperoxidationAlkyl hydroperoxidesHydrogen peroxide
a b s t r a c t
Vanadium complexes have proven to be effective catalysts for the activation of peroxides and the selectiveoxidation of substrates like bromides, sulfides and alkenes. Besides their capability to form metalloperoxospecies, which effectively transfer oxygen atoms to the substrate, these systems are synthetically usefulfor obtaining valuable oxidized molecules on a preparative scale, with a high degree of selectivity andTONs. Furthermore, the use of environmentally friendly oxidants like hydrogen and alkyl hydroperoxidesincreases significantly their potential application at an industrial level.
Here we report a critical survey on the most effective homogeneous vanadium catalysts reportedin the last decade concerning their synthetic application in oxygen transfer reactions (sulfoxidation,epoxidation, haloperoxidation) using hydrogen peroxide or alkyl hydroperoxides, demonstrating thedifferent classes of ligands and complexes, their catalytic performances, their reactivity, chemo, stereoand substrate selectivity. Some examples of the use of non conventional reaction media or techniquesand catalyst recycling studies will be also discussed.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
The capability of d0 metal complexes to catalyze oxygen transferreactions has received much attention during the past thirty years[1]. These metals are able to activate peroxides such as hydrogenperoxide, cumyl or t-butyl hydroperoxide for the oxidation to alarge variety of substrates, and, with the addition of proper chi-ral ligands, high degrees of stereoselectivity have been achieved.In our groups we have been largely interested in oxygen transferreactions involving zirconium [2], titanium [3], molybdenum [4]
∗ Corresponding author.E-mail address: [email protected] (G. Licini).
and vanadium [5] based catalysts. In particular vanadium catalyzedoxidations have attracted our attention by virtue of the interestingproperties of this metal in terms of selectivity, reactivity and stere-oselectivity. Indeed, vanadium mediated oxygen transfer reactionsare frequently applied in chemical research, and in recent years,important advances have been achieved. In this context, vanadiumcatalyzed oxygen transfer reactions have gained renewed inter-est due to the discovery of vanadium-dependent haloperoxidase(VHPO) enzymes able to activate hydrogen peroxide for the oxi-dation of halides with the consequent production of halogenatedcompounds and the stereoselective sulfoxidations [6].
Peroxovanadium complexes, depending on the nature of theligands coordinated to the metal and on the experimental con-ditions, can act either as electrophilic oxygen transfer reagents
0010-8545/$ – see front matter © 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.ccr.2011.05.004

Author's personal copy
2346 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Fig. 1. Synthesis of enantiopure bis-alkanolamines 1–3 derived from (R)-styrene oxide or (S)-propene oxide [8].
Scheme 1. Interconversion of V(V) diastereomeric complexes obtained from ligands 1–3, presenting an equatorial or an axial vanadium oxo function [8].
or as radical oxidants. Typical electrophilic processes are theoxidation of sulfides and tertiary amines and the epoxidation ofallylic alcohol or simple alkenes. The oxidation of alcohols andthe hydroxylation of aliphatic and aromatic hydrocarbons areexamples of homolytic reactivity. This contribution reviews thevanadium-catalyzed oxidations in which electrophilic oxygentransfer reactions are taking place. This translates to the oxidationof sulfides, alkenes, halides which have been studied in the pres-ence of terminal oxidants such as molecular oxygen, hydrogenperoxide, and alkyl hydroperoxides. Thus, Section 2 is devotedto sulfoxidations, while Section 3 presents epoxidations. In boththese sections great emphasis is given to the stereoselectiveprocesses. The oxidation of halides, haloperoxidation, is discussedin Section 4. All the processes in which vanadium catalysts reactin a heterolytic fashion have been considered. Several reviewarticles have appeared on vanadium-catalyzed oxidation [6,7],consequently, the present review emphasizes the work publishedduring the past 7–10 years covering studies up to 2010.
2. Sulfoxidations
Vanadium catalyzed sulfoxidation is an important transforma-tion both for the relevance of the products obtained (sulfoxides andsulfones) and for the information that this reaction can give on thenature of the active peroxo species involved into the process andthe mechanism of the oxygen transfer. Furthermore the process canbe chemo and stereoselective, yielding mainly sulfoxides with highenantiomeric excesses [7].
2.1. Di and trialkanolamine derivatives
In 2003 a series of enantiopure bis-alkanolamines were synthe-sized by Rehder and coworkers as models of vanadate-dependentperoxidases [8]. The ligands were prepared by the aminolysis ofenantiopure (R)-styrene oxide or (S)-propylene oxide with differentprimary amines (Fig. 1).
The coordination chemistry of the new class of ligands 1–3 hasbeen investigated by reaction with VO(O-i-Pr)3. Usually a mixtureof mononuclear complexes with a distorted trigonal bipyramidalgeometry was obtained, equilibrating among the possible diastere-omeric forms having an equatorial or an axial vanadium oxofunction (Scheme 1).
These complexes have been characterized both via X-ray diffrac-tion and, in solution, 1H and 51V NMR, also at variable temperature.The catalytic performances of the new complexes in sulfoxidationhave been tested using alkyl hydroperoxides as oxygen source [tert-butyl peroxide (TBHP) or cumyl peroxide (CHP)] in chloroform onmethyl p-tolyl sulfide 4 or benzyl phenyl sulfide 5. The best resultsobtained in these studies, with respect mainly to the reactivity andstereoselectivity of the process, are reported in Table 1.
Stereoselectivities up to 38% were obtained in satisfactorychemical yields and selectivity towards sulfoxide formation. Theabsolute stereochemistry of the dialkanolamine controls the stere-oselection of the oxygen transfer process [(S,S) ligands afford(S)-sulfoxides, (R,R) ligands give the (R)-ones].
More recently Martinez and Dutasta reported on the syn-thesis and catalytic performance in the sulfoxidation of methylp-tolyl sulfide 4 and benzyl phenyl sulfide 5 of enantiopurehemicryptophane–vanadium oxo complexes 6 and 7 (Fig. 2) [9].
The supramolecular catalysts 6 and 7 catalyze the sulfoxidationof both substrates in the presence of CHP with catalyst loadingdown to 0.5%, chemical yields up to 95%, high sulfoxide/sulfoneselectivity (98–100%) and TON up to 180. Interestingly, reactionscarried out with the model catalyst 8 give much lower yields(25–46%). As far as the stereoselectivity of the systems is concerned,the authors report enantiomeric excesses around 10% indepen-dently from the enantiopure vanadium complex employed andfrom the ratio of the two possible �/� diastereomers.
2.2. Di and triphenolamine derivatives
C3 V(V) amino triphenolates have been prepared and testedas haloperoxidase structural and functional models by Licini and
Table 1Oxidation of methyl p-tolyl sulfide 4 or benzyl phenyl sulfide 5 by TBHP or CHP in the presence of V(V)/1–3 catalysts at 0–20 ◦C. Substrate:oxidant:V(V) = 10:10:1([sulfide]0 = 0.1 M) [8].
Entry Substrate Ligand Time (h) Conv (%) SO/SO2 Ees (conf) Ref.
1 4 2a 2.5 (0 ◦C) 100 85:15 31 (S) [8a]2 5 2a 4.5 (0 ◦C) 100 79:21 23 (S) [8a]3 4 1a 2.0 (−20 ◦C) 70 93:7 38 (R) [8b]4 5 1a 0.33 (−20 ◦C) 70 96:4 37 (R) [8b]5 4 3 3.0 (0 ◦C) 60 95:5 37 (R) [8c]

Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2347
Fig. 2. Dutasta enantiopure hemicryptophane–vanadium oxo complexes 6, 7 and the model system 8 [9].
Scheme 2. Amino triphenolate vanadium complexes 10a–c [10].
coworkers [10]. Triphenolamine 9 can be easily prepared via three-fold reductive amination of salicylic aldehyde derivatives [11].Reaction of 9 with VO(O-i-Pr)3 affords in high yields of the mononu-clear C3 complexes 10b (R = Me) and 10c (R = t-Bu) as deep redcrystalline solids, which were characterized by 1H and 51V NMR andX-ray diffractometric analysis (Scheme 2). Complex 10a derivedfrom the less hindered ligand 10a shows more complex NMR spec-tra consistent with the formation of aggregates/mixture of species.
The catalytic performances of complexes 10a–c (10%) weretested in the sulfoxidation of thioanisole 11 with hydrogen perox-ide as primary oxidant under homogeneous conditions (methanol).All three mononuclear catalysts gave fast and totally selective con-version into the sulfoxide and in the case of 10c (R = t-Bu) withquantitative yields. This system was further optimized decreasingthe catalyst loading down to 0.01%. Methyl phenyl sulfoxide 12was obtained in quantitative yields and very high selectivity withrespect to methyl phenyl sulfone 13, reaching TONs up to 9900and TOF up to 8000 h−1 working with [sulfide]0 = 1.0 M and moreconcentrated H2O2 (70%) (Table 2, entries 3 and 5).
The optimized procedure was used for determining the scopeof the reaction with a series of sulfides (Fig. 3). In all case chemicalyields over 94% were obtained with very high SO/SO2 selectivities,results that are in line with an electrophilic oxygen transfer processand prove the synthetic applicability of the method on a preparativescale.
Other V(V) catalysts containing amino phenolate-based ligandswere reported by Martins and coworkers [12]. Ligands 14a,b wereprepared via Mannich condensation from the corresponding o,p-di(tert-butyl)phenols and different bis-amines (Fig. 4) [13]. Theauthors prepared the vanadium(III) complexes 15a,b by reactionwith VCl3(THF)3. Oxidation by air gave the corresponding vana-dium(V) complexes 16a,b, whose 1H and 13C NMR are compatiblewith an average Cs octahedral geometry with trans phenolate coor-dination and an axial oxo group. Addition of i-PrOLi gave 17a as a
Fig. 3. Sulfoxides obtained by sulfoxidation by 30% aqueous H2O2 catalyzed by 9c(0.1%) (Chemical yields, sulfoxide, sulfone selectivity.) [10].
diastereomeric mixture having the vanadium oxo function equa-torial or axial. On the contrary, the chiral complex (R)-17b shows1H and 13C NMR spectra consistent with a C1 octahedral complex,with an axial oxo group. Most of these complexes have been char-acterized via X-ray analysis (Fig. 4).
The catalytic activity of these complexes has been investigatedusing H2O2 (20%) in slight excess in the presence of 1% catalyst usingdifferent solvents and reaction temperatures. The best results arereported in Table 3.
The most reactive and selective catalyst is (R)-16b in homo-geneous phase (acetone) at 0 ◦C (Table 3, entry 4). Completeconversion with high SO/SO2 selectivity was obtained already after4 h. The same system provided much lower conversions underbiphasic systems (DCE/H2O, Tables 3 and 4, entries 2–3). The bipha-sic system provides better results when the catalysts are preparedin situ from VO(acac)2 reaching 91% conversions after 24 h (Table 3,entries 6 and 7). In all the cases no enantiomeric excesses wereobtained using the chiral catalysts.
2.3. Polyhedral oligomeric silsesquioxane trisilanolate (POSS)
Polyhedral oligomeric silsesquioxane trisilanolate V(V) com-plex 18 catalyzes the sulfoxidation, as well as amine oxidation tonitrones or N-oxides (Scheme 3) [14].
Quantitative yields were obtained in the oxidation of methyl p-tolyl sulfide 4 with CHP working at room temperature with highsulfoxide/sulfone selectivity (Table 4, entry 1). Furthermore in thepresence of Lewis base coligands a significant increase of the reac-tivity (up to 24 fold) could be obtained associated with decreasedsulfoxide/sulfone selectivity (Table 4).

Author's personal copy
2348 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Table 2Oxidation of thioanisole 11 by aqueous hydrogen peroxide catalyzed by 10c.a Effect of concentration and catalyst loading [10].
Entry [11]0 (M) 9c (%) Yields (%) 12:13 Time (min) TON TOF (h−1)
1 0.1 1 97 96:4 25 97 2402 1.0 0.1 98 98:2 80 980 13303 1.0b 0.1 98 97:3 20 980 80004 1.0 0.01 97 97:3 850 9700 17905 1.0b 0.01 99 98:2 255 9900 2667
a Reactions were carried out at 28 ◦C in CD3OD using a 1:1 molar ratio of substrate/H2O2 (35% in water).b Reactions performed with H2O2, 70% in water.
Fig. 4. Amino bis-phenols 14 and the corresponding V(V) complexes 15–17 [13].
Table 3Oxidation of thioanisole (11) by H2O2 with catalysts VO(acac)2/14, 15–17a [13].
Entry Catalyst Solvent Temp. (◦C) Time (h) Conv (%) Sulfone 13 (%)
1 (R)-16b DCE 10 18 96 112 (R)-16b DCE 0 24 27 13 (R)-16b DCE 25 4 78 34 (R)-16b Acetone 0 4 >99 25 (R)-17b DCE 10 20 93 56 VO(acac)2/14a DCE 0 24 91 147 VO(acac)2/(R)-14b DCE 0 24 91 6
a Reactions carried out at 0–25 ◦C with a 1:1.2 molar ratio of substrate/aq H2O2; 1% catalyst on 0.55 mmol scale.
Table 4Oxidation of methyl p-tolyl sulfide 4 by CHP catalyzed by VO–POSS complex 18 (1%)a [14].
Entry Co-ligand t1/2 s (equiv. co ligand)b SO:SO2
1 None 6000 98:22 MeSOpTol 5580 (5) 98:24 HMPA 3042 (5) 93:75 HMPA 456 (150) 68:326 (nOct)3PO 290 (5) 95:57 HexMe2N+-O− 233 (5) 87:13
a Reactions were carried out in CHCl3 at 28 ◦C using [4]0 = [CHP]0 = 0.1 M and 1 mol% of 18.b Time required for a 50% decrease of the initial concentration of 4.

Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2349
Scheme 3. Synthesis of VO–POSS complex 18 [15].
The modified reactivity of the catalysts in the presence of Lewisbases can be ascribed to the formation of new catalytic speciesin solution via coordination of the Lewis base to the V(V) cen-ter. Experimental evidence of the coordination of the coligand tothe catalyst was obtained by 51V NMR studies. In fact, in chloro-form in the presence of only two equivalents of HexMe2NO thesignal of 18 (−690.9 ppm) evolves completely into a new species(−605.9 ppm). Furthermore the reaction performed in the pres-ence of an enantiopure phosphoric amide gave a stereoselectiveoxidation (ee = 14%).
2.4. Schiff base systems
One of the most efficient catalytic systems for stereoselectivesulfoxidation so far reported is the Bolm protocol, based on vana-dium complexes of chiral Schiff bases 19a,b and hydrogen peroxideas primary oxidant (Fig. 5) [16]. The reactions, under the stan-dard conditions, are carried out in a two-phase system (aqueoushydrogen peroxide/dichoromethane) at room temperature usingVO(acac)2 as vanadium source and a chiral ligand. The operativeconditions are simple, the catalyst (1%) is formed in situ and thestereoselectivities obtained are generally very high. The system ishighly modular and therefore, after its discovery in 1995, inten-sive investigations were carried out for further optimizations, alsowith the significant contribution of other research groups. Theinfluence of parameters like the nature of the Schiff base ligands19–23, the reaction protocol and scope have been examined indetail to allow further increasing of the stereoselectivity of theprocess (ees > 99.5%) [7a].
This particular aim was obtained via over oxidation to sulfone,taking advantage of the cooperative stereoselective kinetic resolu-tion process which builds up the same enantiomeric sulfoxide.
While the Bolm protocol did not indicate significant kinetic res-olution on the second oxidation step (oxidation of sulfoxide tosulfone), a report by Zeng et al. [17] showed that at lower tempera-tures (0 ◦C vs 20 ◦C) the preformed vanadium complexes (S)-24aand (S)-24b (Fig. 6), in the presence of increasing amounts ofhydrogen peroxide afford increased stereoselections in the methylphenyl sulfoxide formation (ees from ca. 65 to 99%) in reason-able chemical yields (40%). The selectivity of both in situ andpreformed catalysts was examined and compared, finding slightlybetter results in the second case. The preformed systems (S)-24aand (S)-24b have been tested with different substrates reachingees in the range of 75–99%
Jackson and coworkers reported a further modification of theBolm original oxidation protocol which increased the selectiv-ity factor (S) [18] of the kinetic resolution of the sulfoxide fromalmost one (S = 1.1) to much higher values (S = 28) [19]. In par-ticular the effect of the reaction temperature and the solvent arecritical. Initially the system was optimized using the VO(acac)2/3,5-diiodo salicylic aldehyde 19c (R = R′ = I)/H2O2 system. Working indichoromethane the kinetic resolution occurs only at 20 ◦C, whileat 0 ◦C the stereoselectivity of sulfide oxidation is higher. A protocol
with two subsequent additions of oxidant at different temperatures(1.2 equiv. at 0 ◦C + 0.3 equiv. at 20 ◦C.) was set up. This new protocolallows the recovery of the (R)-sulfoxide with ees = 97% (44%). How-ever, the best results were obtained working in chloroform, at 0 ◦Cwith only 1.2 equiv. of oxidant (catalyst:chiral ligand = 1%:1.5%):(R)-p-tolyl and 2-naphthyl methyl sulfoxides were obtained withees > 99.5% in very good chemical yields (70 and 73%). The methodcould be effectively used with a series of alkyl aryl sulfides with eesin the range of 87–99.5%. The reactions could be carried out also onmultigram scale.
Maguire and coworkers optimized the reaction protocol for thearyl benzyl sulfides [20]. Aryl benzyl sulfides can have a peculiarbehavior in stereoselective sulfoxidation [21] and this depends onthe occurrence of aromatic interactions between the substrates andthe catalysts. Also in this case the diiodo Schiff base 19c (R = R′ = I)affords the best stereoselectivities working in dichloromethane atroom temperature (ees > 99%, 40–50% yield) with a series of arylbenzyl sulfides with selectivity factors up to 17.6. Interestingly alsoin this case higher selectivities were obtained at room tempera-ture in dichloromethane (Srt = 4.6, S0 ◦C = 3.3) or at 0 ◦C in chloroform(Srt = 5.8, S0 ◦C = 7.2).
More recently Li and coworkers took advantage of the combinedstereoselective sulfoxidation/kinetic resolution for obtaining highees in the oxidation of aryl alkyl sulfides [22]. They examined aseries of different Schiff base ligands bearing two stereocenter atthe �-amino alcohol arm. Among the ligands tested 23 (1%) gavethe best results (ees ≥ 99%) with alkyl aryl sulfides in chloroformat 0 ◦C in the presence of 1.35 equiv. of H2O2. Also in this case thebest stereoselectivities were obtained in chloroform at low tem-perature.
Gau and coworkers [23] determined the X-ray structures ofvanadium(V) complexes 24b–f and the reactivity of both iso-lated and in situ formed complexes reactivity in sulfoxidationwas examined. The authors obtained comparable stereoselectivi-ties and this is a strong indication that the active species are thesame in both processes. All the isolated complexes have a squaredpyramidal geometry. Complexes with R3 = Bn (24c,e,f) adopt anendo-structure, in which the Bn substituent and the oxo groupare on the same side of the pyramidal base, while 24d with R3 = t-Bu adopts an exo structure with the t-Bu trans to the oxo moiety(Fig. 7).
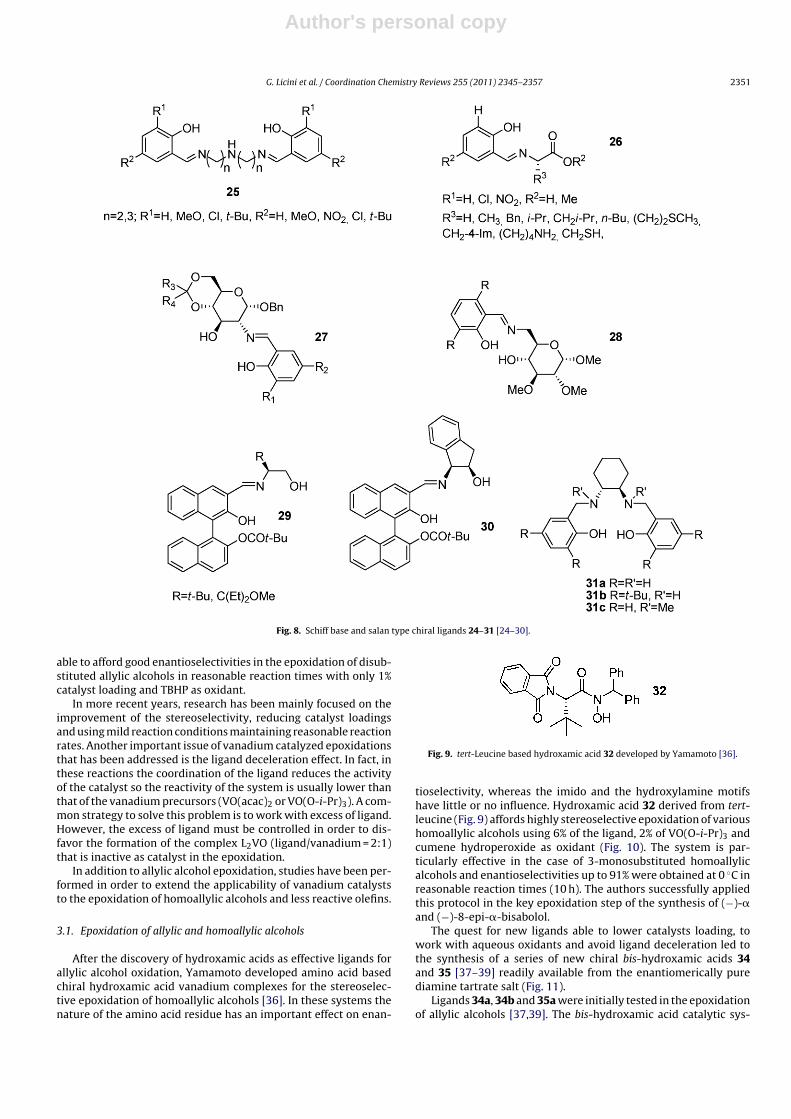
Pentadentate Schiff base 25 vanadium complexes have beensynthesized and tested in sulfoxidation by Maeda (Fig. 8) [24]. Theligands are obtained from bis(aminoalkyl)amines salicyl aldehydederivatives. The vanadium(IV) complexes displayed a distortedoctahedral coordination in solid state, and they afforded lower ratesof oxidation of the sulfide than the tetradentate salen ligands. Thesame group also reported on fourteen amino acids and amino acidester Schiff base 26 (Fig. 8) and the corresponding V(V) complexes[25]. Peroxo complexes prepared from Schiff-base complexes ofoxovanadium(V) converted methyl phenyl sulfide in high yield butlow ees (5–20%). Other Schiff bases have been obtained using sugars27, 28 [26,27]. In the oxidation of thioanisole with hydrogen per-oxide V(V)/27 complexes afforded ees up to 60% [26] while V(V)/28ees up 26% (Fig. 8) [27].
Interesting results have been obtained by Ahn and coworkersusing Schiff bases of �-amino alcohols 29 and 30 [28]. The vanadiumcomplexes obtained from these ligands are very effective in termof stereoselectivity of aryl benzyl sulfides (ees up to 96%).
Zhu and coworkers reported high enantioselectivities in theasymmetric oxidation of sulfides using chiral vanadium complexeswith salan ligands 31 in chloroform at 0 ◦C [29]. The complex with31a (R = R′ = H) shows superior results in term of enantioselectivitythan the corresponding salen analogue and provides the sulfox-ides with opposite configuration. The presence of substituents inortho–para positions of the aromatic ring, N-methylation or the

Author's personal copy
2350 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Fig. 5. Schiff base ligands 19–23 for V-catalyzed stereoselective sulfoxidation [16–23].
Fig. 6. Isolated V(V) catalysts 24a–f [17].
Fig. 7. Isolated complexes endo-24c, exo-24d, endo-24e and endo-24f [23].
use of different bis-amine backbones, solvents or higher reactiontemperatures significantly decrease the selectivity of the system.More recently other research groups like Bryliakov and Talsi [30]or Correia and coworkers [31] explored the reactivity of analogoussystem finding much lower stereoselections. In particular Correia[31], working with isolated salan-complexes, reported oppositestereoselectivities with respect to the Zhu system.
Polymer bound vanadium complexes have been synthesizedand used in sulfoxidation, in particular for the desulfurization ofsulfur containing aromatic compounds like thiophene derivatives[32].
Schiff base ligands containing an histamine unit or 3-formylsalicylic acid have been covalently bonded to chloromethy-lated polystyrene [32b] in some cases cross linked withdivinylbenzene (5%) [32a]. Treatment with VO(acac)2 eventually inthe presence of oxygen gave the polymeric catalysts. The catalyticdesulfurization has been carried out in apolar solvents at 60 ◦Cwith 3–15% catalyst loading in the presence of three fold excessof hydrogen peroxide. Depending on the substrate, desulfurization
of 70–99% was achieved in 2 h. The systems were recycled up tothree times without lost of activity.
3. Epoxidations
Chiral epoxides are key building blocks for the synthesis ofnatural products and biologically active compounds. Among thedifferent methodologies available for their synthesis, the Ti(IV)catalyzed oxidation of allylic alcohols developed by Katsuki andSharpless is, to date, the most efficient one [33]. On the other hand,this methodology presents some limitations, mainly the ratherhigh catalysts loadings and anhydrous working conditions. Alter-natively, vanadium catalyzed epoxidations have been explored.Vanadium is less moisture sensitive than titanium and permittedprocedures with lower catalyst loading. After the pioneering workof Sharpless, who reported enantioselectivities up to 80% using aproline-based hydroxamic acid [34], another important achieve-ment was reported by Yamamoto and coworkers in 2000 [35]. Theydesigned a new chiral hydroxamic acid derived from tert-leucine
Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2351
Fig. 8. Schiff base and salan type chiral ligands 24–31 [24–30].
able to afford good enantioselectivities in the epoxidation of disub-stituted allylic alcohols in reasonable reaction times with only 1%catalyst loading and TBHP as oxidant.
In more recent years, research has been mainly focused on theimprovement of the stereoselectivity, reducing catalyst loadingsand using mild reaction conditions maintaining reasonable reactionrates. Another important issue of vanadium catalyzed epoxidationsthat has been addressed is the ligand deceleration effect. In fact, inthese reactions the coordination of the ligand reduces the activityof the catalyst so the reactivity of the system is usually lower thanthat of the vanadium precursors (VO(acac)2 or VO(O-i-Pr)3). A com-mon strategy to solve this problem is to work with excess of ligand.However, the excess of ligand must be controlled in order to dis-favor the formation of the complex L2VO (ligand/vanadium = 2:1)that is inactive as catalyst in the epoxidation.
In addition to allylic alcohol epoxidation, studies have been per-formed in order to extend the applicability of vanadium catalyststo the epoxidation of homoallylic alcohols and less reactive olefins.
3.1. Epoxidation of allylic and homoallylic alcohols
After the discovery of hydroxamic acids as effective ligands forallylic alcohol oxidation, Yamamoto developed amino acid basedchiral hydroxamic acid vanadium complexes for the stereoselec-tive epoxidation of homoallylic alcohols [36]. In these systems thenature of the amino acid residue has an important effect on enan-
Fig. 9. tert-Leucine based hydroxamic acid 32 developed by Yamamoto [36].
tioselectivity, whereas the imido and the hydroxylamine motifshave little or no influence. Hydroxamic acid 32 derived from tert-leucine (Fig. 9) affords highly stereoselective epoxidation of varioushomoallylic alcohols using 6% of the ligand, 2% of VO(O-i-Pr)3 andcumene hydroperoxide as oxidant (Fig. 10). The system is par-ticularly effective in the case of 3-monosubstituted homoallylicalcohols and enantioselectivities up to 91% were obtained at 0 ◦C inreasonable reaction times (10 h). The authors successfully appliedthis protocol in the key epoxidation step of the synthesis of (−)-�and (−)-8-epi-�-bisabolol.
The quest for new ligands able to lower catalysts loading, towork with aqueous oxidants and avoid ligand deceleration led tothe synthesis of a series of new chiral bis-hydroxamic acids 34and 35 [37–39] readily available from the enantiomerically purediamine tartrate salt (Fig. 11).
Ligands 34a, 34b and 35a were initially tested in the epoxidationof allylic alcohols [37,39]. The bis-hydroxamic acid catalytic sys-

Author's personal copy
2352 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Fig. 10. Homoallylic epoxides 33a–i by oxidation with VO/hydroxamic acid 32 catalyst. Conditions: 32 (6%), VO(O-i-Pr)3 (2%), cumene hydroperoxide (1 equiv.) in tolueneat 0 ◦C [36].
Fig. 11. Yamamoto’s bis-hydroxamic acids 34 and 35 [37–39].
tems were superior to the ones based on monohydroxamic acids. Infact (1) higher enantioselectivities than Ti(IV)-based systems wereobtained, (2) aqueous TBHP could be used as oxidant, (3) the reac-tions could be run with as little as 0.2% of catalyst without decrease
in enantioselectivity or reactivity, and (4) no ligand decelerationeffect was observed when a ligand/vanadium ratio 3:1 was used.Under the optimized conditions (2% ligand, 1% vanadium, DCM,−20 ◦C), trans-monosubstituted and disubstituted allylic alcohols
Fig. 12. Epoxidation of allylic alcohols using VO/bis-hydroxamic acid 34a (a) and 35a (b). Reaction conditions: 1 mol% catalyst, TBHP (70% aq), CH2Cl2 [37,39].

Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2353
Fig. 13. Epoxidation of homoallylic alcohols using the bis-hydroxamic acid ligand 30c. Reaction conditions: 1 mol% catalyst, CHP, toluene, rt [38].
Scheme 4. Desymmetrization of allylic alcohols with ligand 34a [40].
Fig. 14. Sulfonamide-based hydroxamic acid ligands 37 and 38 developed byMalkov [41–43].
were epoxidized with excellent enantioselectivities using ligand34a, whereas ligand 35a was used for the enantioselective epoxida-tion of cis-substituted allylic alcohols [37] (Fig. 12). Low molecularweight allylic alcohols were also epoxidized with high enantiose-lectivities using the catalysts derived from 34a, 34b or 35a [37,39]and the products could be obtained after extraction from the aque-ous phase.
In the case of homoallylic alcohols ligand 35c was the most effec-tive in terms of yields and enantioselectivities in the oxidation ofsubstrates with highly hindered phenyl groups [38,39]. Excellentstereoselectivities were systematically obtained in the epoxidationof both cis- and trans-substituted homoallylic alcohols with CHP;for example in the synthesis of epoxide 33d the use of ligand 35cincreases the ees from 46% (obtained with ligand 32, see Fig. 9 [36])to 96% (Fig. 13). In addition, the system works at room tempera-ture and allows the use of lower catalyst loading (2 mol% ligand,ligand/metal ratio 2:1).
bis-Hydroxamic acid ligands were then used for the kineticresolution of racemic allylic and homoallylic alcohols [37,38].Working under the conditions optimized for the stereoselectiveepoxidation of achiral substrates it was possible to obtain boththe starting alcohol and the epoxide with good stereoselectivi-ties. The desymmetrization of meso secondary allylic alcohols andhomoallylic alcohols was also successfully achieved [40]. Using 1%catalyst, the oxidizing system VO(O-i-Pr)3/34a/CHP transformedtrans-substituted and 1,1-disubstituted allylic alcohols with highyields and enantioselectivities (Scheme 4), while 35a was more effi-cient for cis-substituted substrates. In the case of meso homoallylicsecondary alcohols it was observed that the system derived fromligand 35c (1 mol% VO(O-i-Pr)3, 2 mol% ligand) was effective for cisalkenes but not for the trans ones.
Contemporarily to the development of bis-hydroxamic acid-based ligands, Malkov described a new series of sulfonamide-basedhydroxamic acids (Fig. 14) [41].
Reactions carried out in the presence of ligand 37 affordedfrom moderate to good enantioselectivities in the epoxidation oftrans monosubstituted allylic alcohols in toluene at −20 ◦C usingTBHP as oxidant, but much lower reactivity and enantioselectiv-
Scheme 5. Synthesis of epoxy-alcohol 36b via epoxidation with V(V)/sulfonamidebased hydroxamic acids 37 and 38 [41–43].
ity in the epoxidation of nerol, a cis allylic alcohol, were obtained[41]. Although in terms of enantioselectivities is less efficient thanthe bis-hydroxamic ligand-based systems reported by Yamamoto[35,37], this system exhibits a high reactivity even comparable tothat of VO(acac)2 alone. The observed accelerating effect of the lig-and was attributed to the presence of hydrogen bonds between thesulfonamide group and the incoming allylic alcohol that tether thetwo reactants together.
An important improvement of this methodology came from theuse of aqueous reaction medium [42]. In this case, VOSO4·H2O wasused as metal source and 70% aqueous TBHP as oxidant. Thougha general decreased reactivity was observed, the enantioselectivi-ties were not affected and the hydrolysis of the resulting epoxidescounted less than 5% at low temperatures (Scheme 5). The reac-tion carried out with only VOSO4·H2O is very slow and the additionof 0.5 equiv. of ligand led to complete reagent conversion. A fur-ther improvement in enantioselectivity and reactivity was achievedusing ligand 38, bearing a N-chiral substituent [43]. Optimizationof the reaction conditions (5% catalyst loading, carefully addition oftoluene) allowed obtaining the epoxide 36b in short reaction timeswith 98% yield and 90% ee (Scheme 5) [43].
Terpenoid type ligands have been also studied by Bryliakov andcoworkers in the oxidation of (−)-(R)-linalool [44], with stereose-lectivities not exceeding 50% ee.
An interesting strategy in stereoselective epoxidation is theuse of chiral alkyl hydroperoxides. Zhang and coworkers achievedthe stereoselective epoxidation of allylic alcohols catalyzed bysandwich type polyoxometalates (POMs) using a chiral hydroper-oxide [45]. First, a screening of various transition metal substitutedsandwich type POMs (V(V), Mn(II), Fe(III), Zn(II), Pd(II), Pt(II))demonstrated the superiority of oxovanadium-substituted polyox-ometalate [ZnW (VO)2(ZnW9O34)2]12− in terms of reactivity andselectivity in the oxidation of a model substrate. A survey of chi-ral hydroperoxides indicated a TADDOL derived hydroperoxide 39(TADOOH) as the best choice, enantioselectivities up to 91% couldbe obtained working at rt (Scheme 6). Thanks to the high robustnessof POM catalyst, loadings down to 0.002% could be employed andTONs up to 42,000 were achieved without any decrease in enan-tioselectivity. The protocol has an additional value: TADDOL canbe easily recovered without loss of enantiopurity and recycled intothe chiral hydroperoxide [46].
TADOOH has been also effective in stereoselective epoxida-tions (up to 72% ee) when VO(O-i-Pr)3 was used as catalyst in thepresence of hydroxamic acid 40 (Scheme 8) [47]. More recently,Lattanzi and coworkers described the use of the chiral hydroper-oxide derived from (S)-norcamphor 41 (Scheme 6) in reactionscatalyzed by both VO(acac)2 and VO(O-i-Pr)3 using N-hydroxy-N-

Author's personal copy
2354 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Scheme 6. Vanadium catalyzed epoxidation of allylic alcohols using chiral hydroperoxides 39 and 41 [45,47,48].
phenylbenzamide 42 as ligand [48]. Stereoselectivities up to 61%were achieved with epoxide 36b using 10% of catalyst and 15% ofligand (Scheme 6). Good stereoselectivities were obtained in forsome selected substrates, however both systems are less active andstereoselective than the POM system described by Zhang.
Stereoselective vanadium catalyzed epoxidation of enantiopureallylic alcohols bearing an � sulfinyl moiety (VO(acac)2/TBHP) hasbeen reported by Fernández de la Pradilla and coworkers [49].
3.2. Epoxidation of simple olefins
Polyoxometalates (POMs) have been extensively studied as oxi-dation catalyst due to their robustness, ease of preparation andcompatibility with different oxygen sources. Mizuno and cowork-ers reported that vanadium-based �-Keggin type polyoxometalate[�-1,2-H2SiV2W10O40]4− which has a {VO-(�-OH)2-VO} core is aneffective catalyst for the epoxidation of alkenes [50]. A series of lin-ear and cyclic alkenes were efficiently oxidized using the tetrabutylammonium POM salt as catalyst (5%) and H2O2 as oxidant (1 equiv.)(Fig. 15). Various non conjugated dienes were also oxidized withhigh regioselectivity for the less substituted double bond.
The epoxidation of cyclohexene with sandwich type polyox-ometalates using chiral TADOOH as oxidant was reported by Zhang[45b]. Much lower reactivities and basically no stereoselectivitiescompared with the allylic alcohols were obtained (27% yield withcyclohexene).
The oxidation of alkenes with oxygen using vanadium/Schiffbase type ligands gave reasonable results only in the case ofcyclooctene, while for other olefins very low conversions andselectivities were observed [51]. The catalytic activity of 4-acyl-5-pyrazolone ligands in the oxidation of styrene, �-methylstyreneand cis-�-methylstyrene with TBHP was also investigated [52],but as in the case of Schiff base ligands, very poor conver-sion and selectivity were obtained. Vanadium complexes derivedfrom 2-(2-butoxyethoxy)ethanolate epoxidize cyclooctene with68% conversions [53]. Amine triphenolate vanadium complexeshave a scarce catalytic activity in styrene and trans-stilbene epoxi-dation [54].
4. Haloperoxidations
Selective bromination of organic compounds is a very importanttool in organic synthesis. Organic bromo-derivatives are impor-tant precursors for various selective and efficient transformationsin organic synthesis, as industrial intermediates and pharmaceuti-cals. Hazards and environmental problems correlated with classicalbromination methods have encouraged the attention towards thedevelopment of safer and environmentally friendlier methods [55].In this contest vanadium haloperoxidases (VHPOs), which havebeen isolated mainly from marine algae and fungi, have receivedmuch attention because they are able to catalyze the oxidation ofhalides to the corresponding hypohalous acid using hydrogen per-oxide as primary oxidant [56]. Studies for determining the structureand activity of active sites in VHPOs and the synthesis of structuralVHPOs models have stimulated the research on biomimetic vana-dium systems as catalysts for haloperoxidation. Attention has beenmainly focused on complexes which mimic the active sites of theenzyme [57,58] and, more recently, interest has moved towardsthe use of non conventional solvents, new oxidants or the heteroge-nization of the catalytic systems. This part of the review is dedicatedto the recent advancements in this field.
Plass [59] reported that a N-salicylidene alkoxo vanadium(V)oxoperoxo complex which has, in the solid state, a pentagonal-bipyramidal coordination geometry, with a side-on bonded peroxoligand in the equatorial plane, is able to brominate, under stoichio-metric conditions, 1,3,5-trimethoxybenzene (TMB) in the presenceof tetrabutylammonium bromide (90% yield). More recently Xinget al. reported that oxovanadium(IV)-carboxylate complexes areeffective systems in the bromination reaction of phenol red to bro-mophenol blue in phosphate buffer [60].
Licini and coworkers reported the C3 vanadium(V) amino triph-enolate complexes 10a–c as structural and functional model ofvanadium haloperoxidases [10]. These complex oxidize bromideand chloride ions by hydrogen peroxide, leading to the correspond-ing mono-halogen TMB. The catalyst loading has been lowered to0.05% with respect to the limitation agent leading to the recoveryof the BrTMB product in 63% yield with TONs up to 1260. Reac-

Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2355
Fig. 15. Epoxidation of alkenes using �-Keggin type vanadium polyoxometalates [50].
Scheme 7. Halocyclization of bis-homoallylic alcohols in the presence of bromide,ROOH, and vanadium catalysts [63].
tions were also carried out in the presence of chloride ions. Slowchlorination of TMB could be achieved obtaining the correspondingchlorinated product in 40% yields after 2 days.
Conte et al. explored the possibility to perform haloperoxida-tion of styrene in ionic liquids using NH4VO3 as metal source,and hydrogen peroxide as oxidant [61]. Good to excellent yieldsof brominated products were obtained with styrene using imida-zolium salts as solvent, much higher than in the correspondingtwo-phase H2O/CHCl3 system [5]. Ionic liquids have also been suc-cessfully used in the oxidation of sulfides with the same systems[62].
The possibility to oxidize bromide ion has been used not onlyfor bromoperoxidations but also for the combined halocyclization[63].
Hartung used (Schiff-base)vanadium(V) complexes to catalyzethe oxidation of bromide for the stereoselective synthesis offunctionalized cyclic ethers (Scheme 7). This methodology waseffective for the synthesis of the 2,2,3,5,6,6-substituted tetrahy-dropyran nucleus of the marine natural product aplysiapyranoid A46 (Scheme 8). bis-Homoallylic alcohol 44 was treated with TBHP,pyridinium hydrobromide and 5 mol% of VOL(OEt) (EtOH) [L = N-(2-hydroxyphenyl)salicylidene imine dianion] to furnish 43% of6-endo-bromocyclized product 45 [63].
The mechanism of this bromination reaction consists of TBHPactivation by mean of the vanadium complex, via in situ formationof the corresponding tert-butylperoxo complex. The metalloperoxocomplex oxidizes bromide to Br2 as the active brominating reagent,which is released into the solution and serves as reagent for thehalocyclization.
Heterogenization of vanadium complexes has been object ofstudies as well. New peroxovanadate complexes anchored to
soluble polymers of the type, Na3[V2O2(O2)4(carboxylate)]–Pol[Pol = poly(acrylate) or poly(methacrylate)] have been synthesizedfrom the reaction of V2O5 with H2O2 and the sodium salts ofthe respective macromolecular ligands [64]. Carboxylate groupsof polymer chains are able to coordinate to V(V) centers. Thesesupported peroxo species have been tested in bromination of aro-matics compounds in acetonitrile–water media using tetraethylammonium bromide as bromide source. Activated aromatics, suchas aniline, were brominated to produce predominantly p-bromoproducts (yields 75–95%).
Heterogenization of vanadium complexes has been alsoreported by the group of Maurya and coworkers. In particular vana-dium salts have been incorporated in polymeric Schiff bases [65],inserted in Na-Y zeolites [66], anchored to polystyrene resins [67]and reacted with polymer bound imidazole [68]. The latter systemfurnished the best results in term of product yield and capabilityto be recycled in the haloperoxidation of salicylic aldehyde. Underthe optimized conditions, using aqueous 30% H2O2, the supportedcatalyst, KBr and perchloric acid, 81.4% conversion of salicyl alde-hyde was achieved. The supported catalyst can be reused withoutlost of activity (77.1% of conversion after four cycles).
Vanadium complexes bearing organic ligands are not the onlycatalysts for haloperoxidation. Vanadium pentoxide (V2O5) alsovery efficiently promotes the bromination of organic substratesin the presence of hydrogen peroxide [69]. However the methoddeveloped by Khan requires high catalyst loadings and a largeexcess of oxidant: typical conditions require 50% of V2O5 and16 equiv. of H2O2. This methodology is useful for a wide rangeof substrates such as aromatic rings, olefins, chalcones, �-ketoesters and 1,3-diketones Interestingly, the vanadium content canbe reduced if a mineral acid, such as sulfuric acid, is used [70].
While all of the systems described use of peroxides as oxida-tion source, more recent catalytic systems capable of using oxygenas oxidant have been reported. Hirao and coworkers developed acatalytic system which performs oxidative bromination of arenesusing ammonium metavanadate (NH4VO3) in the presence of a bro-mide salt (Bu4NBr) and a Brønsted or Lewis acid in the presence ofmolecular oxygen [71]. This catalytic system has been applied tothe bromination of alkenes and alkynes to give the correspond-ing vic-bromides, and to ketones to give �-bromination. All thesereactions furnish the products with excellent yields.

Author's personal copy
2356 G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357
Scheme 8. Vanadium(V)-catalyzed oxidation of bromide in the 6-endo-selective bromocyclization of styrene-derived alcohol for the synthesis of aplysiapyranoid A 46 [63].
5. Conclusions
In this contribution we have shown that, in the last decade,important results have been obtained in oxygen transfer reactionscatalyzed by vanadium complexes. In the field of stereoselectiveoxidation of sulfides, and epoxidation of allylic and homoallylicalcohols important results have been achieved. Even though itis still not possible always to use hydrogen peroxide in aqueoushomogenous conditions, phase transfer systems or alkylperoxideshave been used successfully in terms of yield and stereoselectivity.The capability of POM vanadium complexes to transfer oxygen tounfunctionalized olefins has been a major achievement. Haloper-oxidations have seen also an increasing interest in the scientificcommunity. Important results have been achieved in the bromina-tion of organic substrates and in the field of halocyclization.
Important challenges remain. For example, the development ofa vanadium catalyst able to transfer oxygen stereoselectively tounfunctionalized olefins or simple sulfides like dialkyl sulfides, andthe development of catalyst able to work efficiently in the haloper-oxidations. For the existing catalytic systems, the possibility toimmobilize the catalyst in solid supports or to work in unconven-tional solvents in order to provide a “greener” process remains animportant challenge.
References
[1] (a) G. Strukul (Ed.), Catalytic Oxidations with Hydrogen Peroxide as Oxidant,Kluwer Academic Publishers, Dordrecht, 1992;(b) W. Adam (Ed.), Peroxide Chemistry. Mechanistic and Preparative Aspectsof Oxygen Transfer, Wiley-VCH Verlag GmbH, Weinheim, 2000;(c) V. Conte, O. Bortolini, in: Z. Rappoport (Ed.), The Chemistry of Peroxides –Transition Metal Peroxides Synthesis and Role in Oxidation Reactions, WileyInterscience, 2006.
[2] (a) M. Bonchio, G. Licini, F. Di Furia, S. Mantovani, G. Modena, W.A. Nugent, J.Org. Chem. 64 (1999) 1326;(b) G. Licini, M. Bonchio, G. Modena, W.A. Nugent, Pure Appl. Chem. 71 (1999)463.
[3] (a) C. Zonta, E. Cazzola, M. Mba, G. Licini, Adv. Synth. Catal. 350 (2008) 2503;(b) M. Forcato, M. Mba, W.A. Nugent, G. Licini, Eur. J. Org. Chem. (2010) 740;(c) M. Mba, L.J. Prins, C. Zonta, M. Cametti, A. Valkonen, K. Rissanen, G. Licini,Dalton Trans. 39 (2010) 7384;(d) G. Santoni, M. Mba, M. Bonchio, W.A. Nugent, C. Zonta, G. Licini, Chem. Eur.J. 16 (2010) 645.
[4] F. Romano, A. Linden, M. Mba, C. Zonta, G. Licini, Adv. Synth. Catal. 352 (2010)2937.
[5] (a) V. Conte, B. Floris, Inorg. Chim. Acta 363 (2010) 1935;(b) V. Conte, B. Floris, Dalton Trans. 40 (2011) 1419.
[6] (a) H. Vilter, Phytochemistry 23 (1984) 1387;(b) M.A. Andersson, A. Willetts, S.G. Allenmark, J. Org. Chem. (1997) 8455;(c) A.G.J. Ligtenbarg, R. Hage, B.L. Feringa, Coord. Chem. Rev. 237 (2003) 98.
[7] (a) C. Bolm, Coord. Chem. Rev. 237 (2003) 245;(b) E. Wojaczyska, J. Wojaczyski, Chem. Rev. 110 (2010) 4303.
[8] (a) G. Santoni, G. Licini, D. Rehder, Chem. Eur. J. 9 (2003) 4700;(b) C. Wikete, W. Pingsong, G. Zampella, L. De Gioia, G. Licini, D. Rehder, Inorg.Chem. 46 (2007) 196;(c) W. Pingsong, C. C elik, G. Santoni, J. Dallery, D. Rehder, Eur. J. Inorg. Chem.(2008) 5203.
[9] A. Martinez, J.-P. Dutasta, J. Catal. 267 (2009) 188.[10] M. Mba, M. Pontini, S. Lovat, C. Zonta, G. Bernardinelli, E.P. Kündig, G. Licini,
Inorg. Chem. 47 (2008) 8616.[11] L.J. Prins, M. Mba, A. Kolarovic, G. Licini, Tetrahedron Lett. 47 (2006) 2735.[12] S. Barroso, P. Adão, MadeiraF P., M.T. Duarte, J. Costa Pessoa, A.M. Martins, Inorg.
Chem. 49 (2010) 7452.[13] E.Y. Tshuva, M. Versano, I. Goldberg, M. Kol, H. Weitman, Z. Goldschimidt, Inorg.
Chem. Commun. 2 (1999) 371.
[14] S. Lovat, M. Mba, H.C.L. Abbenhuis, D. Vogt, C. Zonta, G. Licini, Inorg. Chem. 48(2009) 4724.
[15] F. Carniato, E. Boccaleri, L. Marchese, A. Fina, D. Tabuani, G. Camino, Eur. J. Inorg.Chem. 4 (2007) 585.
[16] C. Bolm, F. Bienewald, Angew. Chem. Int. Ed. Engl. 34 (1995) 2640.[17] Q. Zeng, H. Wang, T. Wang, Y. Cai, W. Weng, Y. Zhao, Adv. Synth. Catal. 347
(2005) 1933.[18] (a) H.B. Kagan, J.C. Fiaud, in: E.L. Eliel, S.H. Wilen (Eds.), Topics in Stereochem-
istry, vol. 18, John Wiley & Sons, 1988, p. 249;(b) C.J. Sih, S.H. Wu, in: E.L. Eliel, S.H. Wilen (Eds.), Topics in Stereochemistry,vol. 18, John Wiley & Sons, 1988, p. 63, vol. 19.
[19] (a) C. Drago, L. Caggiano, R.F.W. Jackson, Angew. Chem. Int. Ed. 44 (2005) 7221;(b) M. Hinch, O. Jacques, C. Drago, L. Caggiano, R.F.W. Jackson, C. Dexter, M.S.Anson, S.J.F. Macdonald, J. Mol. Chem. A: Chem. 251 (2006) 123.
[20] P. Kelly, S.E. Lawrence, A.R. Maguire, Eur. J. Org. Chem. (2006) 4500.[21] (a) F. Di Furia, G. Licini, G. Modena, R. Motterle, W.A. Nugent, J. Org. Chem. 61
(1996) 5175;(b) M.I. Donnoli, S. Superchi, C. Rosini, J. Org. Chem. 63 (1998) 9392;(c) P. Axe, S.D. Bull, M.G. Davidson, M.D. Jones, D.E.J.E. Robinson, W.L. Mitchell,J.E. Warren, Dalton Trans. 46 (2009) 10169.
[22] Y. Wu, J. Liu, X. Li, A.S.C. Chan, Eur. J. Org. Chem. (2009) 2607.[23] S.-H. Hsieh, Y.-P. Kuo, H.-M. Gau, Dalton Trans. (2007) 97.[24] R. Ando, S. Mori, M. Hayashi, T. Yagyu, M. Maeda, Inorg. Chim. Acta (2004) 1177.[25] R. Ando, H. Inden, M. Sugino, H. Ono, D. Sakaeda, T. Yagyu, M. Maeda, Inorg.
Chim. Acta (2004) 1337.[26] M.E. Cucciolito, R. Del Litto, G. Roviello, F. Ruffo, J. Mol. Catal. A: Chem. 236
(2005) 176.[27] I. Lippold, J. Becher, D. Klemm, W. Plass, J. Mol. Catal. A: Chem. 299 (2009) 12.[28] (a) Y.-C. Jeong, S. Choi, Y.D. Huang, K.-H. Ahn, Tetrahedron Lett. 45 (2004) 9249;
(b) Y.-C. Jeong, Y.D. Huang, S. Choi, K.-H. Ahn, Tetrahedron: Asymmetry 16(2005) 3497.
[29] J. Sun, C. Zhu, Z. Dai, M. Yang, Y. Pan, H. Hu, J. Org. Chem. 69 (2004) 8500.[30] K.P. Bryliakov, E.P. Talsi, Eur. J. Org. Chem. (2008) 3369.[31] P. Adão, J. Costa Pessoa, R.T. Henriques, M.L. Kuznetsov, F. Avecilla, M.R. Maurya,
U. Kumar, I. Correia, Inorg. Chem. 48 (2009) 3542.[32] (a) M.R. Maurya, A. Arya, A. Kumar, J. Costa Pessoa, Dalton Trans. (2009) 2185;
(b) M.R. Maurya, A. Arya, A. Kumar, M.L. Kuznetsov, F. Avecilla, J. Costa Pessoa,Dalton Trans. 49 (2010) 6586.
[33] T. Katsuki, K.B. Sharpless, J. Am. Chem. Soc. 102 (1980) 5974.[34] (a) R.C. Michaelson, R.E. Palermo, K.B. Sharpless, J. Am. Chem. Soc. 99 (1977)
1990;(b) K.B. Sharpless, T.R. Verhoeven, Aldrichim. Acta 12 (1979) 63.
[35] Y. Hoshino, H. Yamamoto, J. Am. Chem. Soc. 122 (2000) 10452.[36] N. Makita, Y. Hoshino, H. Yamamoto, Angew. Chem. Int. Ed. 42 (2003) 941.[37] W. Zhang, A. Basak, Y. Kosugi, Y. Hoshino, H. Yamamoto, Angew. Chem. Int. Ed.
44 (2005) 4389.[38] W. Zhang, H. Yamamoto, J. Am. Chem. Soc. 128 (2007) 286.[39] A.U. Barlan, W. Zhang, H. Yamamoto, Tetrahedron 63 (2007) 6075.[40] Z. Li, W. Zhang, H. Yamamoto, Angew. Chem. Int. Ed. 47 (2008) 7520.[41] A.V. Malkov, Z. Bourhani, P. Kocovsky, Org. Biomol. Chem. 3 (2005) 3194.[42] Z. Bourhani, A.V. Malkov, Chem. Commun. (2005) 4592.[43] A.V. Malkov, L. Czemerys, D.A. Malyshev, J. Org. Chem. 74 (2009) 3350.[44] K.P. Bryliakov, E.P. Talsi, S.N. Stas’ko, O.A. Kholdeeva, S.A. Popov, A.V. Tkachev,
J. Mol. Catal. A: Chem. 194 (2003) 79.[45] (a) W. Adam, P.L. Alster, R. Neumann, C.R. Saha-Möller, D. Seebach, R. Zhang,
Org. Lett. 5 (2003) 725;(b) W. Adam, P.L. Alster, R. Neumann, C.R. Saha-Möller, D. Seebach, A.K. Beck,R. Zhang, J. Org. Chem. 68 (2003) 8222.
[46] M. Aoki, D. Seebach, Helv. Chim. Acta 84 (2001) 187.[47] W. Adam, A.K. Beck, A. Pichota, C.R. Saha-Möller, D. Seebach, N. Vogl, R. Zhang,
Tetrahedron: Asymmetry 14 (2003) 1355.[48] A. Lattanzi, S. Piccirillo, A. Scettri, Eur. J. Org. Chem. (2005) 1669.[49] R. Fernández de la Pradilla, A. Castellanos, J. Fernández, M. Lorenzo, P. Manzano,
P. Méndez, J. Priego, A. Viso, J. Org. Chem. 71 (2006) 1569.[50] (a) Y. Nakagawa, K. Kamata, M. Kotami, K. Yamaguchi, N. Mizuno, Angew. Chem.
Int. Ed. 44 (2005) 5136;(b) N. Mizuno, Y. Nakagawa, K. Yamaguchi, J. Mol. Catal. A: Chem. 251 (2006)286.
[51] (a) D.M. Boghaei, A. Bezaatpour, M. Behzad, J. Mol. Catal. A: Chem. 245 (2006)12;(b) S. Mohebbi, A.H. Sarvestani, Transit. Met. Chem. 31 (2006) 749;(c) S. Rayati, N. Torabi, A. Ghaemi, S. Mohebbi, A. Wojtczak, A. Kozakiewicz,Inorg. Chim. Acta 361 (2008) 1239;

Author's personal copy
G. Licini et al. / Coordination Chemistry Reviews 255 (2011) 2345– 2357 2357
(d) S. Rayati, M. Koliaei, F. Ashouri, S. Mohebbi, A. Wojtczak, A. Kozakiewicz,Appl. Catal. A: Gen. 346 (2008) 65;(e) H.H. Monfared, R. Bikas, P. Mayer, Inorg. Chim. Acta 363 (2010) 2574.
[52] F. Marchetti, C. Pettinari, C. Di Nicola, R. Pettinari, A. Crispini, M. Crucianelli, A.Di Giuseppe, Appl. Catal. A: Gen. 378 (2010) 211.
[53] E.C.E. Rosenthal, H. Cui, M. Hummert, Inorg. Chem. Commun. 11 (2008) 918.[54] S. Groysman, I. Goldberg, Z. Goldschmidt, M. Kol, Inorg. Chem. 44 (2005) 5073.[55] A. Podgorsek, M. Zupan, J. Iskra, Angew. Chem. Int. Ed. 48 (2009) 8424.[56] (a) A. Butler, Coord. Chem. Rev. 187 (1999) 17;
(b) V.M. Dembitsky, Tetrahedron 59 (2003) 4701.[57] (a) R. de La Rosa, M.J. Clague, A. Butler, J. Am. Chem. Soc. 114 (1992) 760;
(b) M.J. Clague, N.L. Keder, A. Butler, Inorg. Chem. 32 (1993) 4754.[58] (a) D.C. Crans, J.J. Smee, E. Gaidamauskas, L.Q. Yang, Chem. Rev. 104 (2004) 849;
(b) G.J. Colpas, B.J. Hamstra, J.W. Kampf, V.L. Pecoraro, J. Am. Chem. Soc. 118(1996) 3469;(c) A. Butler, M.J. Clague, G.E. Meister, Chem. Rev. 94 (1994) 625.
[59] S. Nica, A. Pohlmann, W. Plass, Eur. J. Inorg. Chem. (2005) 2032.[60] (a) Z.-P. Li, Y.-H. Xing, Y.-Z. Cao, X.-Q. Zeng, M.-F. Ge, S.-Y. Niu, Polyhedron 28
(2009) 865;(b) Z.-P. Li, Y.-H. Xing, C.-G. Wang, J. Li, Y.-Z. Cao, X.-Q. Zeng, M.-F. Ge, S.-Y. Niu,Z. Anorg. Allg. Chem. 63 (2009) 2601.
[61] V. Conte, B. Floris, P. Galloni, A. Silvagni, Pure Appl. Chem. 77 (2005) 1575.
[62] V. Conte, F. Fabbianesi, B. Floris, P. Galloni, D. Sordi, I.C.W.E. Arends, M. Bonchio,D. Rehder, D. Bogdal, Pure Appl. Chem. 81 (2009) 1265.
[63] (a) M. Greb, J. Hartung, F. Kohler, K. Spehar, R. Kluge, R. Csuk, Eur. J. Org. Chem.(2004) 3799;(b) J. Hartung, Pure Appl. Chem. 77 (2005) 1559.
[64] D. Kalita, S. Sarmah, S. Prasad Das, D. Baishya, A. Patowary, S. Baruah, N.S. Islam,React. Funct. Polym. (2008) 876.
[65] M.R. Maurya, A. Kumar, P. Manikandan, S. Chand, Appl. Catal. A: Gen. 277 (2004)45.
[66] M.R. Maurya, H. Saklani, S. Agarwal, Catal. Commun. 5 (2000) 563.[67] (a) M.R. Maurya, S. Sikarwar, T. Joseph, P. Manikandan, S.B. Halligudi, React.
Funct. Polym. 63 (2005) 71;(b) M.R. Maurya, U. Kumar, P. Manikandan, Dalton Trans. (2006) 3561.
[68] M.R. Maurya, M. Kumar, A. Arya, Catal. Commun. 10 (2008) 187.[69] (a) U. Bora, G. Bose, M.K. Chaudhuri, S.S. Dhar, R. Gopinath, A.T. Khan, B.K. Patel,
Org. Lett. 2 (2000) 247;(b) A.T. Khan, P. Goswami, Tetrahedron Lett. 46 (2005) 4937;(c) A.T. Khan, P. Goswami, L.H. Choudhury, Tetrahedron Lett. 47 (2006) 2751.
[70] G. Rothenberg, J.H. Clark, Org. Process Res. Dev. 4 (2000) 270.[71] (a) K. Kikushoma, T. Moriuchi, T. Hirao, Asian J. Chem. 4 (2009) 1213;
(b) K. Kikushima, T. Moriuchi, T. Hirao, Tetrahedron Lett. 51 (2010) 340;(c) K. Kikushima, T. Moriuchi, T. Hirao, Tetrahedron 66 (2010) 6906.