mm-gbsa for calculating binding affinity a rank … for calculating binding affinity a rank-ordering...
TRANSCRIPT

MM-GBSA for Calculating Binding Affinity
A rank-ordering study for the lead optimization of Fxa
and COX-2 inhibitors
Thomas Steinbrecher
Senior Application Scientist

Databases ABCDE pocket of XYZ
Glide SP*
1. CACDB2010 lead/drug-like set.
2. Phase mining, with multiple hypotheses, of CACDB phase
database using ABCDE as queries (shape similarity > 0.6 or top
0.5%).* 3. Fingerprint-based similarity search of whole CACDB using
HTS hits as queries (Tanimoto >= 0.6 or top 0.5%).*
1. Receptor of ABCDE site with HB to Res123 (-C=O and –NH)
and/or Res 122 (-NH) on chain A.
2. Receptor of ABCDE site with HB to Res123 (-C=O and –NH)
and/or Res 122 (-NH) on chain B.
3. Receptor of ABCDE site with HB to Res123 (-C=O and –NH)
on both chain A and B
Glide XP*
Ranking (in a reasonable timeframe)?
(25 % top scoring)
•Three conformations of ligands for XP docking
•ConfGen/MM/multiple FFs
Glide HTVS
(15 % top scoring)
(post-processing of ensemble)
•Two conformations of ligands for SP docking
•Glide shows dependency on input conformations
Typical Docking Workflow
In molecular docking it is challenging to develop a scoring function which is accurate to conduct HTS, eliminate false positives, get good pose prediction and get good ranking. Approximations are built in for computational efficiency which sacrifice the accuracy of prediction. More rigorous methods such as MMGBSA are the natural follow on after docking in predicting good binding poses and estimating binding free energies

Typical Discovery Workflow
• Once a promising lead compound has been identified in a drug discovery program, chemical variations of the lead compound are usually synthesized and tested to identify a molecule that has optimized chemical properties. Molecules in this congeneric series generally have different substituents that are attached to a common molecular core.
• A key property in this congeneric series of ligands that needs to be optimized is the binding free energy, ∆G(binding) .
• Experimentally, binding free energies are obtained by evaluating the concentration of the compound that is required to inhibit the activity of the protein, e.g., Ki or IC50 data.
– Computationally, binding free energies for a compound to a particular protein can be calculated from
the difference between the free energy of each ligand bound to the protein and the free energies of the components of the complex, i.e.,:
∆G(binding) = ∆G(complex) - (∆ G(free receptor) - ∆ G(free ligand))

Intro to MM-GBSA
• The Molecular Mechanics‐Generalized Born Surface Area (MM‐GBSA) method
calculates binding free energies for molecules by combining molecular mechanics
calculations and continuum (implicit) solvation models.
– Implicit solvent models are often used to estimate free energies of solute‐solvent interactions
and significantly improve the computational speed and reduce errors in statistical averaging that
arise from incomplete sampling of solvent conformations.
• The molecular mechanics part estimates the enthalpic contributions for the
protein‐ligand interactions.
• In cases where the ligands in the congeneric series are very similar to one another
then, as a first approximation, the entropic contribution to the protein‐ligand
interactions are assumed to be similar across the series and can be neglected in
evaluating the relative binding free energies of the ligands.

Solvation Free Energy of Macromolecules
Macromolecules are charged, polar, irregular objects
+ + + -
-
+
+ + + -
-
+ DGSolv
DGelec, int DGelec
DGcavity
ElecSASnonpolarcavity
EleccavitySolv
GAkG
GGG
DD
DDD

Explicit solvent
pro contra
accurate large systems
standard approach boundary artifacts

Poisson Boltzmann Equation
Contains ionic contributions
The Gold standard of continuum models
but hard to solve
Widely applied but slow
)(2
)()(0 rkT
Irr
i
i
grid-based solutions
are practical

Generalized Born models
Fast and analytical (good for MD)
ji ij
GB
ji
Wji ij
ji
Elecrf
r
qqE
)(
11
2
1
4 0
vacuum energy solvation contribution
2
2
22
4exp)(
ij
ij
ijijijij
rrrf
There are many different "flavors" of GB
jiij

Binding Free Energies
"Corpora non agunt nisi fixata"
(No compound is active unless it is bound by a receptor)
Paul Ehrlich, 1913
DGBind

The MM-GBSA approach
- simulations or snapshots in implicit
solvent
- estimate solution contribution via GB
equation plus surface term
- internal energies via MM-forcefield
The MM-GBSA thermodynamic cycle
DG° (Bind) DG° (Vac)
DG°
(Solv1)
DG°
(Solv2)
21 SolvSolvVacuumBind GGGG DDDD
Resources
• www.schrodinger/kb
• Tasks, Prime MM-GSBA

Example Case: FXa

Software Demo

Quality of Results
R2 = 0.08 R2 = 0.68 R2 = 0.04 R2 = 0.75

Example Case: Cox-2

MM-GBSA: Considering Flexibility • Panel: Tasks, Prime MMGBSA
– Our COX-2 example: 16 ligands, 8A flex

MM-GBSA: View results
• Show relevant properties with Property Tree – pIC50 – Glide GScore – MM GBSA DG Binding

MMGSA: What can you expect to see?
There is a clear re-orientation of key residues to improve binding interactions in the MM-GBSA complex. Largest changes are seen in TYR355 with pyrazole nitrogen of ligand (middle right) and SER353 with amine of ligand (bottom right)
Comparison of a Glide docked pose (orange) with MMGBSA (cyan)
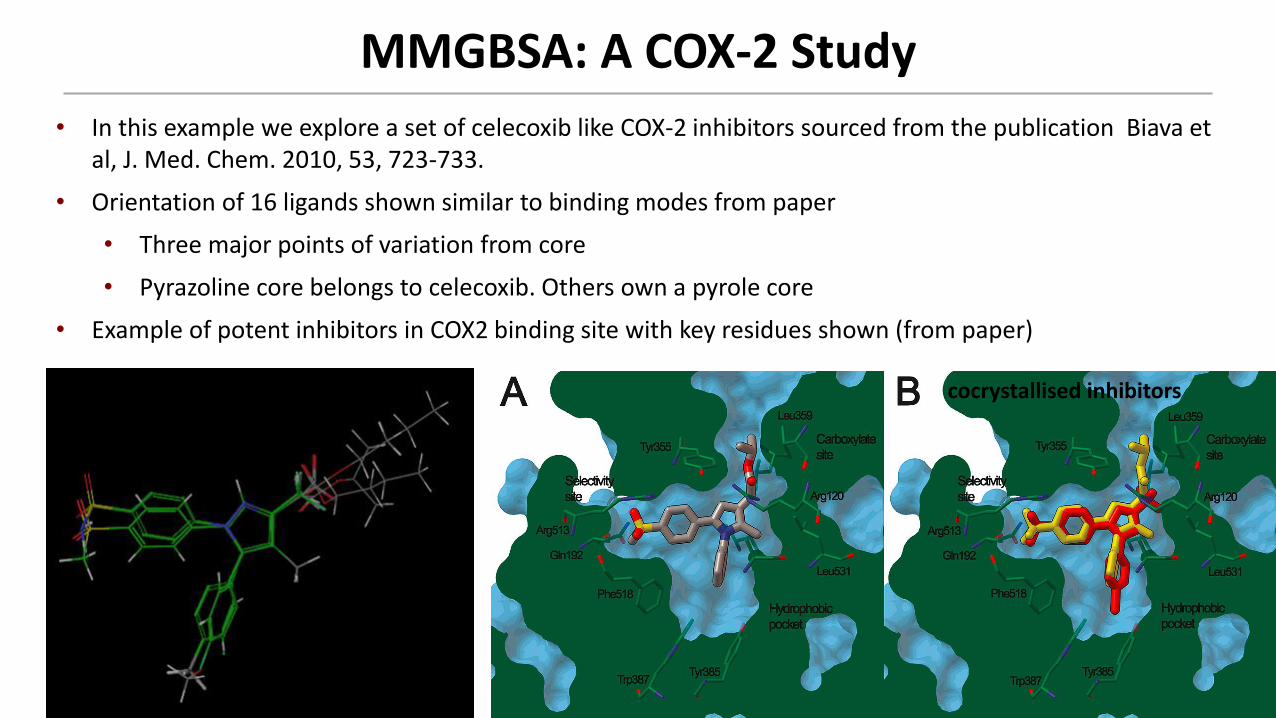
MMGBSA: A COX-2 Study
• In this example we explore a set of celecoxib like COX-2 inhibitors sourced from the publication Biava et al, J. Med. Chem. 2010, 53, 723-733.
• Orientation of 16 ligands shown similar to binding modes from paper
• Three major points of variation from core
• Pyrazoline core belongs to celecoxib. Others own a pyrole core
• Example of potent inhibitors in COX2 binding site with key residues shown (from paper)
cocrystallised inhibitors

Protocol for COX-2 study
• Usual steps of preparing ligands and protein
• Explicit conformational sampling of the ligands with ConfGen followed by Glide for ligand docking
• Use of MacroModel to refine poses post-Glide and pre-mmgbsa
• The best scoring poses are explored through MMGBSA and the correlations to experimental activities examined
• Examine the improved correlation from the MMod refined poses. – Run with zero flexibility and 8A flexibility around the ligand
• See pre-generated graphs

Software Demo
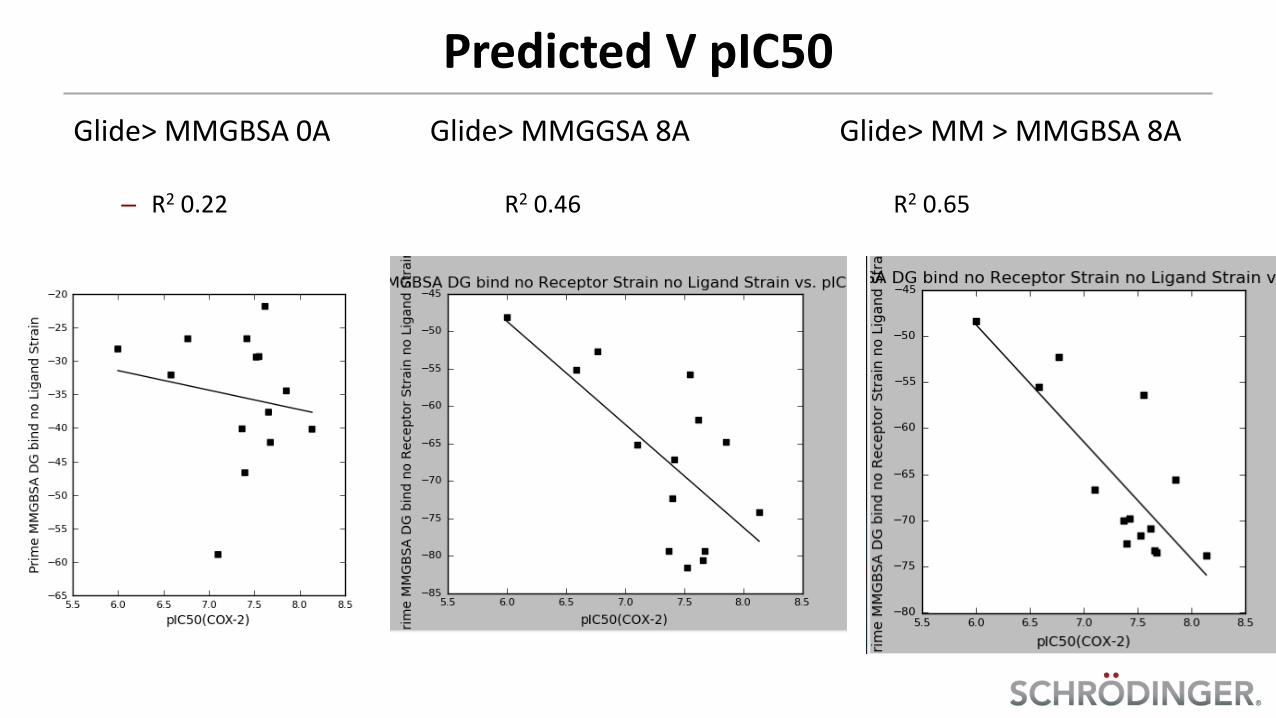
Predicted V pIC50
Glide> MMGBSA 0A Glide> MMGGSA 8A Glide> MM > MMGBSA 8A – R2 0.22 R2 0.46 R2 0.65

Thank You For Yor Attention!